Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения бисфенола А. Более конкретно, оно относится к способу получения бисфенола А, который делает возможным ингибирование потери активности катионообменной смолы в качестве катализатора, используемого на стадии реакции, с увеличением срока службы катализатора снижением содержания низших спиртов, таких как метанол, содержащихся в качестве примесей в ацетоне, который используется в качестве одного из исходных веществ.
Предпосылки создания изобретения
Бисфенол А обычно получают взаимодействием фенола и ацетона в присутствии кислотного катализатора, такого как катионообменная смола. Получаемая реакционная смесь содержит, кроме целевого бисфенола А, непрореагировавший фенол, непрореагировавший ацетон, воду, образовавшуюся в процессе реакции, и другие побочные продукты реакции, такие как окрашенные вещества. Для выделения целевого бисфенола А из реакционной смеси обычно используется способ дистилляции. Согласно данному способу дистилляция проводится в дистилляционной колонне при температуре ниже точки кипения фенола с извлечением низкокипящих веществ, таких как непрореагировавший ацетон, вода, образовавшаяся в процессе реакции, и части непрореагировавшего фенола из верхней части колонны. Концентрированный смешанный раствор из нижней части колонны охлаждают с кристаллизацией бисфенола А в виде кристаллического аддукта с фенолом, затем указанный кристаллический аддукт отделяют от маточного раствора, содержащего побочные продукты реакции, и фенол удаляют из кристаллического аддукта с извлечением бисфенола А.
Непрореагировавший ацетон содержится в низкокипящих веществах, получаемых из верхней части колонны. Обычно указанный ацетон отделяют и извлекают обработкой, такой как дистилляция, с использованием колонны отделения ацетона и возвращают на стадию реакции вместе с заново подаваемым ацетоном (который далее может называться свежим ацетоном).
Небольшое количество метанола содержится в виде примеси в свежем ацетоне. Поскольку указанный метанол не влияет на реакцию получения бисфенола А, он накапливается в выделенном и извлеченном непрореагировавшем ацетоне (который может далее называться извлеченным ацетоном). Когда указанный извлеченный ацетон повторно подают на стадию реакции со свежим ацетоном, концентрация метанола в реакционной смеси повышается.
Сильно кислотные катионообменные смолы обычно используются в качестве кислотного катализатора для вышеуказанной реакции, но указанные сильно кислотные катионообменные смолы имеют проблему в том, что они теряют активность под воздействием метанола, содержащегося в качестве примеси в ацетоне, подаваемом на стадию реакции, что приводит в результате к их укороченному сроку службы в качестве катализатора.
В качестве решения указанной проблемы был предложен способ, в котором регулируется концентрация метанола в ацетоне, подаваемом на стадию реакции, и, когда концентрация метанола возрастает выше определенного заданного уровня, обогащенный метанолом ацетон должным образом выводят из колонны отделения ацетона из ее средней части, и подачу свежего ацетона соответственно увеличивают с регулированием концентрации метанола в ацетоне, подаваемом на стадию реакции, с поддержанием ниже 10000 мас.ч./млн (см., например, выложенную Японскую заявку на патент (KOKAI) № 6-92889).
Однако когда концентрация метанола в свежем ацетоне является высокой, концентрация метанола не может быть снижена, как желательно, регулированием количества извлеченного ацетона, что в результате приводит к преждевременной потере активности сильно кислотной катионообменной смолы, используемой в качестве катализатора. Такая метанольная дезактивация катализатора является сильной, особенно когда в качестве катализатора используется кислотная ионообменная смола, частично нейтрализованная серусодержащим аминным соединением. Кроме того, почти невозможно предотвратить дезактивацию сильно кислотной катионообменной смолы в качестве катализатора только регулированием концентрации метанола в ацетоне, подаваемом на стадию реакции, с установлением на уровне "ниже 10000 мас.ч./млн"; необходимо регулировать концентрацию метанола в подаваемом ацетоне более точно на более низком уровне.
Также известны способы регулирования концентрации метанола в исходных веществах, подаваемых на стадию реакции (см., например, Японский перевод международной заявки РСТ (KOHYO) № 2001-503377). В указанных способах, однако, не приводится рассмотрение повторного использования непрореагировавшего ацетона, содержащегося в низкокипящих веществах.
Описание изобретения
Проблема, решаемая изобретением
Настоящее изобретение было сделано ввиду вышеуказанных обстоятельств, и его целью является создание способа получения бисфенола А, который дает возможность длительное время стабильно получать бисфенол А при ингибировании потери активности катионообменной смолы в качестве катализатора, используемого в реакции.
Способ решения проблемы
В результате серьезного исследования авторов настоящего изобретения по решению вышеуказанной проблемы было установлено, что вышеуказанная проблема может быть решена путем снижения концентрации метанола, содержащегося в качестве примеси в ацетоне, подаваемом на стадию реакции, ниже некоторого определенного уровня и осуществления настоящего изобретения на основе вышеуказанной находки.
В первом аспекте настоящего изобретения предусматривается способ получения бисфенола А взаимодействием исходных веществ - ацетона и фенола в присутствии катализатора,
причем концентрация низшего спирта в полном количестве ацетона, подаваемого на стадию реакции, регулируется так, чтобы она составляла не более 100 мас.ч./млн при подаче на стадию реакции указанных исходных веществ и ацетона, извлеченного на стадии реакции.
Во втором аспекте настоящего изобретения предусматривается способ получения бисфенола А взаимодействием исходных веществ - ацетона и фенола в присутствии катализатора,
причем концентрация низшего спирта в реакционном растворе, выходящем со стадии реакции, регулируется так, чтобы она составляла не более 30 мас.ч./млн при подаче на стадию реакции указанных исходных веществ и веществ, извлеченных со стадии реакции.
В третьем аспекте настоящего изобретения предусматривается способ получения бисфенола А взаимодействием исходных веществ - ацетона и фенола в присутствии катализатора,
причем, по меньшей мере, ацетон, извлеченный со стадии реакции из полной подачи ацетона на стадию реакции, очищается перед подачей на стадию реакции с удалением низших спиртов, содержащихся как примеси, при подаче на стадию реакции указанных исходных веществ и ацетона, извлеченного на стадии реакции.
Эффект изобретения
В соответствии со способом получения бисфенола А настоящего изобретения можно ингибировать дезактивацию ионообменной смолы в качестве катализатора, используемого в реакции, поскольку снижается концентрация метанола, содержащегося в качестве примеси в ацетоне, подаваемом на стадию реакции. Эффект ингибирования дезактивации является особенно заметным, когда в качестве катализатора используется кислотная ионообменная смола, частично модифицированная серусодержащим аминным соединением. Также обеспечивается стабильное получение бисфенола А с постоянным качеством, так как количество ацетона и фенола, присутствующих на стадии реакции, и их соотношение являются установившимися.
Краткое описание чертежа
На чертеже представлена технологическая схема, показывающая способ получения бисфенола А настоящего изобретения, когда способ (i) используется для стадии разделения.
Наилучший вариант осуществления изобретения
Ниже приводится подробное описание настоящего изобретения. В способе получения бисфенола А в аспектах настоящего изобретения с первого по третий исходные вещества - ацетон и фенол взаимодействуют в присутствии катализатора с получением бисфенола А. Способ получения настоящего изобретения обычно содержит, кроме указанной стадии реакции, стадию разделения, на которой реакционная смесь, получаемая на стадии реакции, разделяется на компонент, содержащий бисфенол А, и низкокипящий компонент, содержащий непрореагировавший ацетон, стадию рециркуляции для отделения и извлечения непрореагировавшего ацетона из низкокипящего компонента и рециркуляции его на стадию реакции, стадию кристаллизации, стадию извлечения бисфенола А и стадию циркуляции маточного раствора.
Сначала для того чтобы облегчить понимание настоящего изобретения, схема способа получения бисфенола А поясняется со ссылкой на чертеж, но способ получения согласно настоящему изобретению может быть осуществлен различными способами. Технологическая схема на чертеже показывает вариант, где способ (i) используется на стадии разделения, описанной далее.
Ацетон и фенол, которые являются исходными веществами, подаются в реактор 2 по линии 1. Реакционная смесь из реактора 2 подается в дистилляционную колонну 4 по линии 3. Низкокипящий компонент, содержащий ацетон, воду и фенол, выходящий из верхней части колонны, подается в систему разделения 20 и разделяется на ацетон, воду и фенол дистилляцией или другим способом. Ацетон транспортируется в устройство удаления метанола 1а, такое как рафинер, описанный далее, по линии 21 и линии А. Вода выходит из оборудования системы разделения 20. Фенол подается в рафинер 20а из системы разделения 20 и после очистки подается в емкость хранения фенола 22.
Кубовые продукты из дистилляционной колонны 4 транспортируются в кристаллизатор 5, где кристаллизуется кристаллический аддукт бисфенола А и фенола. Указанный кристаллический аддукт отделяется от маточного раствора сепаратором «твердое вещество/жидкость» 6, затем повторно растворяется в устройстве повторного растворения 7, перекристаллизуется в перекристаллизаторе 8, подвергается разделению твердое вещество/жидкость центрифугой или другим устройством и обрабатывается в системе промывки 9. Промывка кристаллического аддукта проводится очищенным фенолом, подаваемым из емкости 22. Промывочную воду направляют в устройство повторного растворения 7 по линии 10 (или подают в сепаратор «твердое вещество/жидкость» 6 по линии 17 для повторного использования в качестве промывки). После промывки кристаллический аддукт нагревается и разлагается на бисфенол А и фенол в устройстве разложения аддукта 11, причем бисфенол А, обработанный затем в рафинере 12, становится конечным продуктом бисфенолом А. Фенол, выходящий из устройства разложения аддукта 11 и рафинера 12, транспортируется в емкость 22.
Жидкая часть (маточный раствор), отделенная в сепараторе «твердое вещество/жидкость» 6, пропускается в емкость маточного раствора 14 по линии 13. Маточный раствор из емкости маточного раствора 14 транспортируется в линию 1 по линии 15, смешивается с ацетоном и подается в реактор 2. Маточный раствор из емкости маточного раствора 14 также транспортируется в линию 3 по линии 16.
Подача маточного раствора по линии 15 в линию 1 регулируется так, что соотношение ацетон/маточный раствор в смешанном растворе ацетона и маточного раствора, подаваемом в реактор 2, поддерживается в определенном интервале. Поддержание соотношения ацетон/маточный раствор приводит к стабильной реакции в реакторе 2 и последующей стабилизации качества и выхода продукта бисфенола А. Емкость маточного раствора 14 может действовать как буферная емкость для выравнивания колебаний скорости циркуляции по линии 13.
Стадии способа получения настоящего изобретения пояснены подробно ниже.
Стадия реакции
На стадии реакции, осуществляемой в реакторе 2, исходные вещества - фенол и ацетон взаимодействуют при стехиометрическом избытке фенола. Мольное соотношение фенол/ацетон обычно находится в интервале от 3 до 30, предпочтительно от 5 до 20. Реакцию проводят при температуре обычно от 50 до 100°C под давлением обычно от нормального давления до 600 кПа.
В качестве катализатора обычно используются сильно кислотные катионообменные смолы, такие как катионообменные смолы типа сульфоновой кислоты, предпочтительно частично нейтрализованные серусодержащим аминным соединением. В качестве серусодержащего аминного соединения могут использоваться обычные промоторы, используемые для синтеза бисфенола А, такие как, например, 2-(4-пиридил)этантиол, 2-меркаптоэтиламин, 3-меркаптопропиламин, N,N-диметил-3-меркаптопропиламин, N,N-ди-н-бутил-4-меркаптобутиламин и 2,2-диметилтиазолидин. Такой промотор используется в количестве обычно 2-30 мол.%, предпочтительно 5-20 мол.%, по отношению к кислотной группе (сульфогруппе) в кислотном ионообменнике.
Реакция конденсации фенола и ацетона проводится в проточной системе с неподвижным слоем, которая является системой с неразрывным течением или с поршневым течением, или периодической системой с взвешенным слоем. В случае проточной системы с неподвижным слоем пространственная скорость жидкости смеси исходных веществ, подаваемой в реактор, составляет обычно 0,2-50 ч-1. В случае периодической системы с взвешенным слоем используемое количество сильно кислотной ионообменной смолы, хотя варьируется в зависимости от температуры и давления реакции, обычно составляет 20-100 мас.% по отношению к смеси исходных веществ. Время обработки составляет обычно 0,5-5 ч.
Стадия разделения
Реакционная смесь, полученная на стадии реакции, разделяется на компонент, содержащий бисфенол А, и низкокипящий компонент, содержащий непрореагировавший ацетон. Имеются обычно три следующих способа разделения (i)-(iii).
Способ (i)
Как описано выше со ссылкой на технологическую схему на чертеже, реакционная смесь, полученная на стадии реакции, дистиллируется в дистилляционной колонне 4, и низкокипящий компонент, содержащий непрореагировавший ацетон, отделяется из верхней части колонны. Кубовый продукт представляет собой жидкость, содержащую бисфенол А. В качестве дистилляционной колонны могут использоваться известные дистилляционные колонны. Когда дистилляция проводится при нормальном давлении, работа осуществляется при температуре ниже точки кипения фенола. Дистилляция при пониженном давлении (вакуумная дистилляция) является предпочтительной. Вакуумную дистилляцию обычно проводят при температуре 50-150°C при давлении 50-300 мм рт.ст. Поскольку непрореагировавший фенол, содержащийся в реакционной смеси, образует аддукт с бисфенолом на последующей стадии кристаллизации, дистилляцию предпочтительно проводят при условии, что указанный непрореагировавший фенол выгружается в определенном количестве из нижней части колонны. Веществами, выделяемыми из верхней части колонны, являются непрореагировавший ацетон, вода, метанол, содержащийся в качестве примеси, непрореагировавший фенол и т.д.
Концентрация бисфенола А в кубовом продукте после отделения указанных низкокипящих веществ из верхней части колонны составляет обычно 20-50 мас.%. Когда концентрация бисфенола А составляет меньше 20 мас.%, выход бисфенола А снижается, а когда концентрация бисфенола А превышает 50 мас.%, кажущаяся вязкость концентрированного смешанного раствора повышается, что делает трудным транспортировку раствора. Кубовый продукт транспортируется на стадию кристаллизации для извлечения целевого бисфенола А. Стадия кристаллизации будет пояснена далее.
Способ (ii)
Реакционную смесь, полученную на стадии реакции, подают прямо в кристаллизатор 5 (линия подачи не показана). В кристаллизаторе 5 низкокипящие вещества, включающие непрореагировавший ацетон, выпариваются и отделяются при пониженном давлении с получением суспензии кристаллического аддукта бисфенола А и фенола. При необходимости в реакционную смесь могут быть введены вода и ацетон. На операции кристаллизации концентрация бисфенола А составляет обычно 20-50 мас.%, концентрация воды составляет обычно 1-10 мас.%, и концентрация ацетона составляет обычно 0,5-5 мас.%.
Давление в кристаллизаторе 5 сбрасывают, чтобы вызвать испарение воды, ацетона и небольшого количества фенола. Кроме того, кристаллизуемые вещества (реакционная смесь) с температурой 70-140°C охлаждается обычно до 35-60°C, и кристаллический аддукт образуется и превращается в суспензию. Предпочтительным является сброс давления до 50-550 мм рт.ст. Полученная суспензия разделяется на твердое вещество и жидкость в сепараторе «твердое вещество/жидкость» 6, содержащем фильтр или другое устройство. С другой стороны, низкокипящие вещества, а именно воду, ацетон и сопровождающий фенол, выпаренные в кристаллизаторе 5, подают на стадию рециркуляции ацетона, содержащую систему разделения 20, где ацетон фракционируется и подается обратно в устройство удаления метанола 1а, установленное перед стадией реакции.
Способ (iii)
Данный способ содержит стадию кристаллизации, на которой образуется и суспендируется кристаллический аддукт бисфенола А и фенола, стадию разделения твердое вещество/жидкость, на которой полученная суспензия разделяется на кристаллический аддукт бисфенола А и фенола и жидкую часть, и стадию отделения низкокипящих веществ, включающих непрореагировавший ацетон, от жидкой части, полученной на стадии разделения твердое вещество/жидкость.
Таким образом, в данном способе реакционную смесь, полученную на стадии реакции, подают прямо в кристаллизатор 5 (линия подачи не показана), и кристаллизацию проводят при введении, если необходимо, воды и ацетона. На операции кристаллизации концентрация бисфенола А устанавливается обычно равной 20-50 мас.%, концентрация воды - обычно равной 1-10 мас.%, и концентрация ацетона - обычно равной 0,5-5 мас.%. Способ кристаллизации специально не определен, но предпочтительно осуществляют кристаллизацию при охлаждении кристаллизуемых веществ в теплообменнике. Также можно осуществлять охлаждение за счет испарения при сбросе давления, как используется в способе (ii). Кристаллизуемые вещества, имеющие температуру 70-140°C, охлаждаются обычно до 35-60°C, в результате чего кристаллизуется и суспендируется кристаллический аддукт. Полученная суспензия разделяется на твердое вещество и жидкость сепаратором «твердое вещество/жидкость» 6.
Кристаллизационный маточный раствор из сепаратора «твердое вещество/жидкость» 6 или смесь маточного раствора и отработанной промывки из сепаратора «твердое вещество/жидкость» 6 транспортируется на стадию отделения низкокипящих веществ. В том случае, когда кристаллизацию проводят за счет испарения при сбросе давления, как используется в способе (ii), выпаренные низкокипящие вещества также транспортируются на стадию отделения низкокипящих веществ.
На стадии отделения низкокипящих веществ обычно используется дистилляционная колонна, и низкокипящие вещества, включающие непрореагировавший ацетон, воду и фенол, отделяются из верхней части колонны таким способом, как вакуумная дистилляция. Вакуумную дистилляцию обычно проводят при температуре 50-150°C под давлением 50-300 мм рт.ст. Выделенные низкокипящие вещества, включающие непрореагировавший ацетон, воду и фенол, транспортируют на стадию рециркуляции ацетона, содержащую систему разделения 20, где ацетон фракционируется и подается обратно в устройство удаления метанола 1а, установленное перед стадией реакции.
В том случае, когда используется один из способов (i)-(iii), в низкокипящем компоненте имеются алкилмеркаптаны, полученные из катализатора, дезактивированного метанолом, присутствующие в небольшом количестве в реакционной смеси. Поэтому при отделении низкокипящего компонента предпочтительно предусмотреть средство удаления и выведения алкилмеркаптанов в поток газа. Способ удаления алкилмеркаптанов специально не оговаривается; можно использовать различные способы, такие как способ промывки, способ окисления (озонирования), способ адсорбции, обработка микроорганизмами, сжигание, фотокаталитическая дезодорация, плазменная дезодорация и распыление дезодоранта. В том случае, когда алкилмеркаптан представляет собой диметилсульфид или подобное, и используется способ промывки, алкилмеркаптан окисляется в кислотное соединение, такое как серная кислота, при использовании гипохлорита натрия в качестве окислителя, и затем рН полученного вещества корректируется гидроксидом натрия или подобным. В способе адсорбции для адсорбции обычно используют активированный уголь. Данный способ является пригодным даже для обработки веществ в низкой концентрации менее нескольких ч./млн, и его эффективность улучшается, если активированный уголь, используемый для обработки с поглощением запаха, регенерируется для повторного использования. В способе сжигания предпочтительно использовать систему десульфуризации для обработки отработанного газа после сжигания.
Стадия рециркуляции ацетона
На данной стадии непрореагировавший ацетон выделяют и извлекают из низкокипящего компонента, выделенного на указанной стадии разделения, и рециркулируют обратно на стадию реакции. Отделение непрореагировавшего ацетона от низкокипящего компонента проводится в системе разделения 20. В качестве системы разделения 20 обычно используют многоступенчатую дистилляционную колонну, и вода, ацетон и фенол разделяются. Метанол содержится в качестве примеси в выделенном и извлеченном ацетоне. Выделенный и извлеченный ацетон пропускается по линии 21 и смешивается со свежим ацетоном, и смесь подается на стадию реакции. Выделенный и извлеченный фенол очищается вместе со свежим фенолом рафинером 20а, затем хранится в емкости хранения промывочного фенола 22 и используется для промывки кристаллического аддукта, описанной далее.
Стадия кристаллизации
В том случае, когда на стадии разделения используется способ (i), стадию кристаллизации осуществляют с кубовым продуктом (жидкая часть, содержащая бисфенол А), оставшимся после отделения низкокипящих веществ. На стадии кристаллизации, как в случае способов (ii) и (iii), вещества охлаждают от 70-140°С до 35-60°С в кристаллизаторе 5, затем кристаллический аддукт кристаллизуется и суспендируется, и указанная суспензия разделяется на твердое вещество и жидкость в сепараторе «твердое вещество/жидкость» 6.
В качестве кристаллизатора 5 в способах (i)-(iii) обычно используют кристаллизационную емкость, имеющую множество наружных холодильников, способных к переключению режима работы. Можно предотвратить обмерзание наружных холодильников обеспечением множества таких наружных холодильников и их работы по очереди. Поскольку характеристики кристаллизации в кристаллизационной емкости изменяются при переключении наружного холодильника, количество маточного раствора, выделенного в сепараторе «твердое вещество/жидкость» 6, соответственно колеблется. Для того чтобы предотвратить колебание количества маточного раствора, подаваемого от линии 15 к линии 1, обычно часть маточного раствора отводится к нижней стороне реактора 2 по линии 16. В случае использования способа (ii) при использовании кристаллизационной емкости, оборудованной холодильником типа рубашки, как кристаллизатор 5, предпочтительно использовать емкость, оборудованную скребком для удаления осадка, отложившегося на поверхности теплообмена.
Стадия извлечения бисфенола А
Кристаллический аддукт, извлеченный в сепараторе «твердое вещество/жидкость» 6 после стадии разделения, использующей любой из описанных выше способов, повторно растворяется в феноле в устройстве повторного растворения 7, затем перекристаллизовывается в перекристаллизаторе 8 с увеличением чистоты, разделяется на твердое вещество и жидкость системой разделения «твердое вещество/жидкость» и промывки 9, промывается промывочным фенолом и затем транспортируется в систему разложения кристаллического аддукта 11.
Лучшая часть жидкой фракции, выделенной в системе разделения «твердое вещество/жидкость» и промывки 9, и отработанная промывка используются в качестве фенола для повторного растворения кристаллического аддукта в устройстве повторного растворения 7 и в качестве промывки, подаваемой в сепаратор «твердое вещество/жидкость» 6. Часть жидкой фракции, выделенной системой разделения «твердое вещество/жидкость» и промывки 9, рециркулируется в емкость 14 вместе с циркулирующим маточным раствором.
В устройстве разложения кристаллического аддукта 11 кристаллический аддукт обычно нагревается до 100-160°C и поэтому растворяется, и большая часть фенола выпаривается из полученного раствора аддукта. В рафинере 12 остаточный фенол обычно удаляют подходящим способом, таким как десорбция паром с получением очищенного бисфенола А. Указанный способ описан, например, в выложенной Японской заявке на патент (KOKAI) № 2-28126 и 63-132850.
Стадия рециркуляции маточного раствора
Жидкая часть (маточный раствор), выделенная в сепараторе «твердое вещество/жидкость» 6, рециркулируется к верхней стороне и нижней стороне стадии реакции по линиям 13, 15 и 16.
Состав маточного раствора, полученного из сепаратора «твердое вещество/жидкость» 6, представляет собой 65-85 мас.% фенола, 10-20 мас.% бисфенола А и 5-15 мас.% побочных продуктов, таких как 2,4'-изомер. Таким образом, маточный раствор содержит примеси, такие как 2,4'-изомер, с высоким процентным содержанием. Для того чтобы предотвратить накопление таких примесей в системе циркуляции, предпочтительно все количество маточного раствора, полученного из сепаратора «твердое вещество/жидкость» 6, не рециркулируется, но часть маточного раствора отбирают и подвергают обработке с удалением примесей с повышением чистоты, и затем подают ее в систему циркуляции.
В качестве обработки с удалением примесей обычно используют способ, в котором часть маточного раствора из линии 13 подают в устройство удаления примесей 13а, где в подаваемый маточный раствор вводят катализатор разложения, такой как гидроксид натрия, и смесь подвергают дистилляции, причем фенол, бисфенол А и изопропилфенол извлекают в качестве продуктов из верхней части колонны с возвращением извлеченных продуктов по линии 13. Кубовые продукты выгружают из системы.
Соотношение маточного раствора, выделяемого и подаваемого в устройство удаления примесей 13а по линии 13, составляет обычно 4-15 мас.%. Благодаря указанному устройству удаления примесей 13а состав маточного раствора, циркулирующего в емкость 14 по линии 13, стабилизируется и остается по существу в интервале, определенном выше.
Небольшое количество остатка катионообменной смолы, полученного от стадии реакции, содержится в части подаваемого маточного раствора, так что предпочтительно предусмотреть средство удаления остатка катионообменной смолы из маточного раствора и затем снова возвратить его по линии 13.
Маточный раствор из линии 13 подается путем емкости 14 и по линиям 15 и 16 в линии 1 и 3 соответственно. Маточный раствор, подаваемый насосом из емкости 14, регулируется клапаном, предусмотренным на линии 15, с поддержанием постоянной скорости потока по линии 16.
Работа насоса (не показано) для подачи маточного раствора из емкости 14 в линию 15 регулируется так, что мольное отношение количества фенола к общему количеству (a+b) свежего ацетона (а), подаваемого по линии 1, и количества извлеченного ацетона (b), подаваемого по линии 21 (фенол/ацетон), поддерживается постоянным в интервале, указанном выше.
Далее поясняются отличительные признаки первого аспекта настоящего изобретения. Первый аспект настоящего изобретения отличается тем, что концентрация низшего спирта во всем количестве ацетона, подаваемого на стадию реакции, регулируется так, чтобы она составляла не более 100 мас.ч./млн, предпочтительно 20-100 мас.ч./млн, более предпочтительно 30-80 мас.ч./млн. Термин "низшие спирты" относится здесь к обозначению спиртов с числом углеродных атомов 1-8, типичным примером которых является метанол. Далее описываются случаи, включающие метанол в качестве низшего спирта. (То же самое распространяется на описание отличительных признаков второго и третьего аспектов, приведенное далее.)
Концентрация метанола в свежем ацетоне составляет обычно 50-400 мас.ч./млн (далее указывается просто как ч./млн), и концентрация метанола в извлеченном ацетоне составляет примерно 70-700 ч./млн. Отношение количества извлеченного ацетона (b) к количеству свежего ацетона (а) (b/a) составляет обычно примерно 0,05-0,2. Поэтому в том случае, когда обработка по удалению метанола не проводится, концентрация метанола в смеси свежего ацетона и извлеченного ацетона составляет обычно около 50-500 ч./млн. Однако даже если концентрация метанола в свежем ацетоне составляет менее 100 ч./млн, концентрация метанола в извлеченном ацетоне возрастает при проведении реакции, обуславливая то, что концентрация метанола в смешанном ацетоне превышает 100 ч./млн. Также в том случае, когда концентрация метанола в свежем ацетоне составляет более 100 ч./млн, имеется возможность того, что концентрация метанола в смешанном ацетоне будет превышать 100 ч./млн с самого начала.
В каждом случае метанол из ацетона удаляется на стадии, когда концентрация метанола превышает 100 ч./млн. В случае, когда концентрация метанола в свежем ацетоне составляет ниже 100 ч./млн, может использоваться, например, способ, в котором после удаления метанола из извлеченного ацетона последний смешивается со свежим ацетоном, или способ, в котором регулируется отношение количества извлеченного ацетона (b) к количеству свежего ацетона (а) (b/a). Когда концентрация метанола в свежем ацетоне превышает 100 ч./млн, метанол в смешанном ацетоне удаляется. В каждом способе пытаются поддерживать концентрацию метанола во всем ацетоне, подаваемом на стадию реакции, ниже 100 ч./млн. Концентрация метанола в ацетоне может быть определена газохроматографическим анализом. Способ удаления метанола из ацетона рассмотрен далее.
Теперь рассматриваются отличительные признаки второго аспекта настоящего изобретения. Второй аспект настоящего изобретения отличается тем, что концентрация низшего спирта в реакционном растворе, выходящем со стадии реакции, устанавливается не выше 30 ч./млн, предпочтительно не выше 20 ч./млн, более предпочтительно не выше 10 ч./млн. Другими словами, поскольку низшие спирты не принимают участие в реакции, концентрация низших спиртов, таких как метанол, присутствующих в качестве примеси в смеси веществ, включающей фенол, свежий ацетон и извлеченный ацетон, подаваемой на стадию реакции, поддерживается не выше 30 ч./млн.
В случае кислотной ионообменной смолы, частично нейтрализованной серусодержащим аминным соединением, помимо образования сульфида при взаимодействии метанола и меркаптогрупп промотора реакции позволяют протекать далее с образованием сульфониевых катионов со снижением активности на кислотном участке. В данном случае образуются одорантные вещества алкилмеркаптаны, такие как метилмеркаптан и диметилсульфид (продукты выделения разложения промотора), сопутствующие отравлению катализатора. Поэтому в случае кислотной ионообменной смолы, частично нейтрализованной серусодержащим аминным соединением, особенно важно снизить концентрацию метанола, подаваемого в реактор. Концентрация метанола в реакционном растворе может быть определена газохроматографическим анализом.
Для снижения концентрации метанола в указанной смеси веществ, подаваемой в реактор, может использоваться, например, описанный ниже способ, в котором концентрация метанола в ацетоне снижается.
Отличительные признаки третьего аспекта настоящего изобретения рассматриваются здесь. Третий аспект настоящего изобретения отличается тем, что во всем количестве ацетона, подаваемого на стадию реакции, по меньшей мере, ацетон, извлеченный со стадии реакции, очищается перед подачей на стадию реакции с удалением низших спиртов, содержащихся в качестве примесей в указанном ацетоне. Ацетоном, из которого должны быть удалены низшие спирты, такие как метанол, могут быть либо извлеченный ацетон, либо смесь свежего ацетона и извлеченного ацетона. В случае, когда концентрация метанола в свежем ацетоне является низкой, целесообразно пытаться удалить метанол из извлеченного ацетона. В случае, когда концентрация метанола в свежем ацетоне является высокой, целесообразно удалять метанол из смеси свежего ацетона и извлеченного ацетона.
Удаление метанола из ацетона осуществляют с использованием устройства удаления метанола 1а так, что концентрация метанола, присутствующего в качестве примеси в ацетоне, подаваемом в качестве реакционного вещества, снижается обычно ниже 10 ч./млн, предпочтительно до уровня 20-100 ч./млн, более предпочтительно 30-80 ч./млн.
Типовым способом удаления метанола, использующим устройство удаления метанола 1а, является способ, рассмотренный в выложенной Японской заявке на патент (KOKAI) № 9-278703. Предпочтительно используется дистилляционная колонна, и метанолсодержащий ацетон перегоняется в данной колонне с рабочим давлением, которое регулируется с установлением его не менее 200 кПа, предпочтительно в интервале 200-1100 кПа, более предпочтительно 300-600 кПа (как, например, описано в выложенной Японской заявке на патент (KOKAI) № 9-278703). В качестве дистилляционной колонны используется насадочная колонна или тарельчатая колонна с теоретическим числом тарелок обычно не менее 20, предпочтительно 25-50. Температуру верхней части колонны и температуру нижней части колонны выбирают в соответствии с рабочим давлением верхней части колонны. В предпочтительном варианте ацетон, содержащий небольшое количество метанола, подают в среднюю часть дистилляционной колонны, и колонна работает под давлением, определенным выше, с фракционированием ацетона, обогащенного метанолом, из верхней части колонны при получении из нижней части раствора ацетона с заметно сниженной концентрацией метанола. Указанный кубовый продукт подают как реакционное вещество по линии 1.
ПРИМЕРЫ
Настоящее изобретение будет описано более подробно со ссылкой на его примеры, но должно быть понятно, что настоящее изобретение не ограничивается указанными примерами, но может быть осуществлено по-другому, а также без отступления от объема изобретения. В последующем описании все проценты (%) даются по массе, если не указано иное. Количество бисфенола А и концентрация метанола определяются газохроматографическим анализом с использованием промышленного прибора "GC-17A" фирмы Shimadzu Corp. и детектора ионизации пламени.
Пример 1
Бисфенол А получают на пилотной установке, имеющей технологическую схему, показанную на чертеже. Концентрация метанола в свежем ацетоне составляет 300 ч./млн. Дистилляционную колонну, имеющую упорядоченную насадку ("MC Pack MC350S" фирмы Ryoka Forward Co., Ltd.) с числом теоретических тарелок 35, используют в качестве устройства удаления метанола 1а, и при давлении 410 кПа смесь свежего ацетона и извлеченного ацетона по линии 21 подают от 25-й тарелки сверху.
В качестве катализатора в реакторе 2 используют катионообменную смолу типа сульфокислоты (AMBERLIST 31 фирмы Rohm & Haas Company) с 20 мол.% сульфогрупп, частично нейтрализованную 2-(4-пиридил)этантиолом. Смолу набивают с объемом 3,4 м3. Свежее питание ацетона составляет 60 кг/ч. Подача извлеченного ацетона по линии составляет в среднем 10 кг/ч. Расход по линии 16 регулируют так, что подача маточного раствора от линии 15 к линии 1 становится постоянной при 1608,8 кг/ч.
Температуру нижней части дистилляционной колонны 4 устанавливают при 135°С, а давление - при 100 мм рт.ст. В качестве кристаллизатора 5 используют кристаллизационную емкость, обеспеченную тремя наружными холодильниками, и стадию кристаллизации проводят при приостановлении работы одного из трех наружных холодильников по очереди. Температура кристаллизационной емкости варьируется в интервале 49-51°С.
В результате концентрация метанола в извлеченном ацетоне из линии 21 составляет в среднем 515 ч./млн. Конверсия ацетона катализатора постепенно падает от 99,5% в начале реакции. Количество извлеченного ацетона из линии 21 также постепенно увеличивается в течение данного периода, но концентрация метанола в ацетоне после обработки устройством удаления метанола 1а поддерживается при 90-109 ч./млн. Концентрация метанола в реакционном растворе после смешения с маточным раствором, подаваемым по линии 1, поддерживается при 3-4 ч./млн. Конверсия ацетона после прохождения 12 месяцев составляет 81%. Можно продолжать работу после 12 месяцев без необходимости замены катализатора в реакторе 2.
Сравнительный пример 1
Осуществляют такие же операции, как в примере 1, за исключением того, что устройство удаления метанола 1а устанавливают на линии 21, извлеченный ацетон только из линии 21 подвергают дистилляции при нормальном давлении при использовании устройства удаления метанола 1а, затем обработанный таким образом извлеченный ацетон смешивают со свежим ацетоном и подают в реактор 2 по линии 1, и подачу маточного раствора от линии 15 к линии 1 изменяют на 500 кг/ч. Концентрация метанола в извлеченном ацетоне после обработки по удалению метанола падает только до уровня 900-1000 ч./млн. Конверсия ацетона катализатора постепенно снижается от 99,9% в начале реакции, причем количество извлеченного ацетона также постепенно увеличивается, и концентрация метанола в смеси извлеченного ацетона и нового питания ацетона увеличивается до 300-325 ч./млн, выше, чем концентрация метанола в свежем ацетоне. Также концентрация метанола в реакционном растворе после смешения с маточным раствором увеличивается до 32-34 ч./млн. Приблизительно через 6 месяцев после начала работы содержание ацетона падает до 69%, и катализатор в реакторе 2 стареет, требуя замены катализатора на новый. Таким образом, невозможно проводить процесс в течение длительного времени.
Описание ссылочных номеров
1а: Устройство удаления метанола
2: Реактор
4: Дистилляционная колонна
5: Кристаллизатор
6: Сепаратор «твердое вещество/жидкость»
7: Устройство повторного растворения
8: Перекристаллизатор
9: Система разделения «твердое вещество/жидкость» и промывки
11: Устройство разложения кристаллического аддукта
12: Рафинер
13а: Система удаления примесей
14: Емкость
20: Система разделения
20а: Рафинер
22: Емкость хранения фенола
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА А ВЫСОКОЙ ЧИСТОТЫ И ПРОИЗВОДСТВЕННАЯ УСТАНОВКА | 2007 |
|
RU2422429C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА А | 2013 |
|
RU2637311C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА | 2013 |
|
RU2627266C2 |
| ОБЕЗВОЖИВАНИЕ ЦИРКУЛИРУЮЩИХ ПОТОКОВ ПРИ ПОЛУЧЕНИИ БИСФЕНОЛА А | 2005 |
|
RU2392261C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА-А | 1994 |
|
RU2119906C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО ПОЛИКАРБОНАТА | 2004 |
|
RU2326133C2 |
| Способ получения бисфенола-А | 2019 |
|
RU2799337C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАНУЛИРОВАННОГО БИСФЕНОЛА А ВЫСОКОГО КАЧЕСТВА | 2006 |
|
RU2426718C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА-А | 2005 |
|
RU2342356C1 |
| МНОГОСТАДИЙНЫЙ СПОСОБ СУСПЕНЗИОННОЙ РЕАКЦИОННОЙ ОТПАРКИ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1994 |
|
RU2126706C1 |
Изобретение относится к способу получения бисфенола А взаимодействием исходных веществ - ацетона и фенола в присутствии катализатора, представляющего собой катионообменную смолу, в котором, по меньшей мере, ацетон, извлеченный со стадии рециркуляции ацетона в полном количестве ацетона на стадии реакции, очищают перед подачей на стадию реакции с удалением метанола, содержащегося в качестве примеси, при подаче на стадию реакции указанных исходных веществ и ацетона, извлеченного со стадии рециркуляции ацетона, причем обработка по удалению метанола представляет собой дистилляцию, проводимую при давлении 200-1100 кПа. Применение настоящего способа позволяет длительное время стабильно получать бисфенол А при ингибировании потери активности катионообменной смолы в качестве катализатора, используемого в реакции. 5 з.п. ф-лы, 1 ил.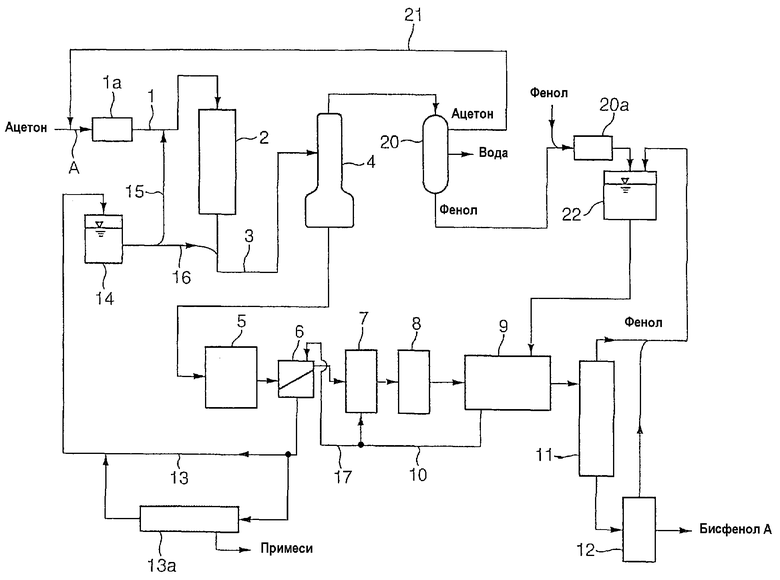
1. Способ получения бисфенола А взаимодействием исходных веществ - ацетона и фенола в присутствии катализатора, представляющего собой катионообменную смолу,
в котором, по меньшей мере, ацетон, извлеченный со стадии рециркуляции ацетона в полном количестве ацетона на стадии реакции, очищают перед подачей на стадию реакции с удалением метанола, содержащегося в качестве примеси, при подаче на стадию реакции указанных исходных веществ и ацетона, извлеченного со стадии рециркуляции ацетона, причем обработка по удалению метанола представляет собой дистилляцию, проводимую при давлении 200-1100 кПа.
2. Способ по п.1, в котором концентрация метанола в полном количестве ацетона, подаваемого на стадию реакции, регулируют так, чтобы она составляла не более 100 мас.ч./млн.
3. Способ по п.1, в котором концентрация метанола в реакционном растворе, выходящем со стадии реакции, регулируется так, чтобы она составляла не более 30 мас.ч./млн.
4. Способ по п.1, в котором свежий ацетон и ацетон, извлеченный со стадии рециркуляции ацетона, смешивают, и смесь очищают перед подачей на стадию реакции с удалением метанола, содержащегося в качестве примеси.
5. Способ по п.3, в котором ацетон, извлеченный со стадии рециркуляции ацетона, очищают с удалением метанола, содержащегося в качестве примеси, причем концентрация метанола, содержащегося в извлеченном ацетоне, становится ниже концентрации метанола, содержащегося в свежем ацетоне.
6. Способ по любому из пп.1-5, в котором катализатор, используемый на стадии реакции, представляет собой кислотную ионообменную смолу, частично нейтрализованную серосодержащим аминным соединением.
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Способ получения бисфенола А | 1988 |
|
SU1799376A3 |

