Область техники, к которой относится изобретение
Настоящее изобретение относится к новым кристаллическим солям 1,2-этандисульфоновой кислоты бифенильного соединения, которые, как предполагается, являются полезными в качестве терапевтических агентов для лечения легочных расстройств. Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие кристаллические соединения или полученным из таких кристаллических соединений, способам и промежуточным соединениям для получения таких кристаллических соединений и способам применения таких кристаллических соединений для лечения легочных расстройств.
Уровень техники
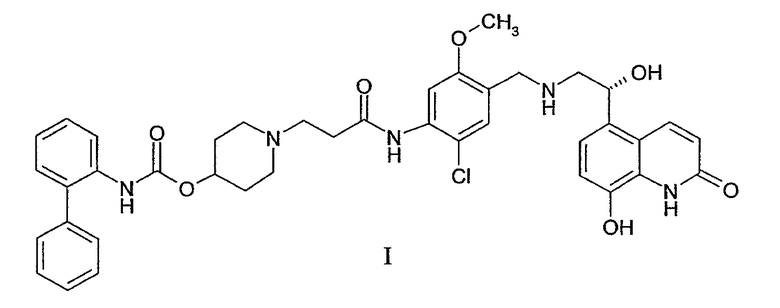
В заявке на патент США №10/779,157, поданной 13 февраля 2004 г., раскрыты новые бифенильные соединения, которые являются полезными в качестве терапевтических агентов для лечения легочных расстройств, таких как хроническое обструктивное заболевание легких (ХОЗЛ) и астма. В частности, соединение 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты раскрыто в этой заявке, как обладающее как мускариновой антагонистической активностью, так и агонистической активностью к β2-адренергическим рецепторам. Химическая структура 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты представлена в виде формулы I:
Терапевтические агенты, полезные для лечения легочных расстройств, преимущественно вводят непосредственно в дыхательные пути при помощи ингаляции. В этом отношении, некоторые типы фармацевтических ингаляционных устройств разработаны для введения терапевтических агентов посредством ингаляции, включая порошковые ингаляторы (ПИ, DPI), дозирующие ингаляторы (ДАИ, MDI) и ингаляторы-небулайзеры. При изготовлении фармацевтических композиций и лекарственных форм для использования в таких устройствах, очень желательно иметь кристаллическую форму терапевтического агента, которая не является ни гигроскопичной, ни расплывающейся и которая имеет относительно высокую точку плавления (т.e. выше чем примерно 150°C), таким образом, позволяя веществу находиться в микронизированной форме без значимого разложения или потери кристалличности.
Ранее не встречались публикации о кристаллических формах солей соединения формулы I. Следовательно, существует необходимость в стабильной, нерасплывающейся кристаллической форме соли соединения формулы I, которая имеет допустимый уровень гигроскопичности и относительно высокую точку плавления.
Сущность изобретения
Настоящее изобретение предоставляет кристаллические соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват.
С интересом было обнаружено, что такие кристаллические соли 1,2-этандисульфоновой кислоты соединения формулы I не расплываются даже под воздействием влажности окружающей среды. Кроме того, такие кристаллические соли имеют допустимый уровень гигроскопичности и очень высокую точку плавления, например выше примерно 215°C. В конкретном варианте осуществления кристаллическая соль настоящего изобретения имеет точку плавления выше примерно 230°C.
Среди других применений кристаллическая соль 1,2-этандисульфоновой кислоты соединения формулы I полезна в изготовлении фармацевтических композиций, которые, как ожидается, будут полезными в лечении легочных расстройств. Следовательно, в других аспектах этой композиции настоящее изобретение предоставляет фармацевтическую композицию, содержащую фармакологически приемлемый носитель и соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольваты.
В конкретном варианте осуществления фармацевтическая композиция настоящего изобретения дополнительно содержит стероидный противовоспалительный агент, такой как кортикостероид; или ингибитор фосфодиэстеразы-4; или их комбинацию.
В другом конкретном варианте осуществления настоящее изобретение предоставляет фармацевтическую композицию, содержащую водный изотонический солевой раствор, содержащий соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, причем раствор имеет pH в пределах от примерно 4 до примерно 6.
В другом варианте осуществления настоящее изобретение предоставляет комбинацию, содержащую:
(a) кристаллическую соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват; и
(b) стероидный противовоспалительный агент.
Соединение формулы I имеет как мускариновую антагонистическую активность, так и агонистическую активность к β2-адренергическим рецепторам. Следовательно, предполагается, что соль 1,2-этандисульфоновой кислоты настоящего изобретения является полезной в качестве терапевтического агента для лечения легочных расстройств, таких как астма, и хронического обструктивного заболевания легких.
Следовательно, в одном из аспектов указанного способа настоящее изобретение предоставляет способ лечения легочного расстройства, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата.
Кроме того, в другом из аспектов указанного способа настоящее изобретение предоставляет способ вызова у пациента бронходилатации, включающий введение пациенту посредством ингаляции такого количества соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата, которое вызывает бронходилатацию.
Настоящее изобретение также предоставляет способ лечения хронического обструктивного заболевания легких или астмы, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата.
Настоящее изобретение также относится к способу получения кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I. Следовательно, в других аспектах указанного способа настоящее изобретение предоставляет способ получения 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата, включающий приведение в контакт 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты с 1,2-этандисульфоновой кислотой.
В других аспектах указанного способа настоящее изобретение предоставляет способ получения кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I, включающий:
(а) приведение в контакт соединения формулы II:
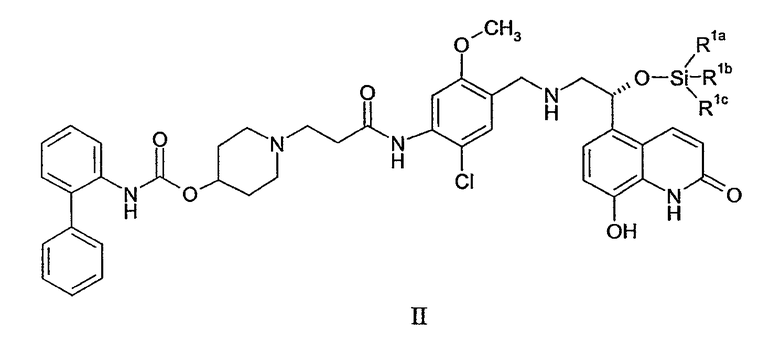
где R1a, R1b и R1c независимо выбирают из C1-4алкила, фенила, -C1-4алкил-(фенила), или один из R1a, R1b и R1c представляет собой -О-(C1-4алкил); с фторид-ионом; и
(b) приведение в контакт продукта из стадии (b) с 1,2-этандисульфоновой кислотой или ее гидратом; для образования кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I, причем стадии (a) и (b) выполняют в одном и том же реакционном сосуде без выделения продукта стадии (a).
В других аспектах указанного способа настоящее изобретение предоставляет способ получения кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I, имеющей точку плавления выше чем примерно 230°C, включающий добавление затравочного кристалла кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I в раствор, содержащий соль 1,2-этандисульфоновой кислоты соединения формулы I, растворенную в инертном растворителе, в котором затравочный кристалл имеет точку плавления выше чем примерно 230°C.
Этот способ также может быть использован для перекристаллизации кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I для получения кристаллической формы, имеющей точку плавления выше, чем примерно 230°C. Следовательно, настоящее изобретение дополнительно предоставляет способ получения кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I, имеющей точку плавления выше чем примерно 230°C, включающий:
(a) растворение кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I в инертном растворителе при первой температуре;
(b) охлаждение продукта стадии (a) до второй температуры; и
(c) добавление затравочного кристалла соли 1,2-этандисульфоновой кислоты соединения формулы I;
причем затравочный кристалл имеет точку плавления выше чем примерно 230°C, при этом первая температура представляет собой температуру, достаточную для растворения соли 1,2-этандисульфоновой кислоты, а вторая температура ниже температуры, при которой затравочный кристалл полностью растворяется при добавлении продукта стадии (b).
Кроме того, настоящее изобретение относится к способу очистки 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, включающему образование кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты. Настоящее изобретение также относится к продуктам, изготовленным при помощи способов, раскрытых в настоящем описании.
Настоящее изобретение также относится к кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвату для применения в терапии или в качестве лекарственного препарата.
Кроме того, настоящее изобретение относится к применению кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата для производства лекарственного средства; особенно для производства лекарственного средства для лечения легочного расстройства.
Настоящее изобретение также относится к применению:
(a) кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата; и
(b) стероидного противовоспалительного агента;
для производства лекарственного средства для лечения легочного расстройства.
Настоящее изобретение также относится к кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвату, в микронизированной форме; и фармацевтическим композициям, содержащим фармацевтически приемлемый носитель и кристаллическую соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват, в микронизированной форме.
Краткое описание чертежей
Различные аспекты настоящего изобретения проиллюстрированы со ссылкой на прилагаемые чертежи.
На фиг.1 показан график дифференциальной сканирующей калориметрии (DSC) и график дифференциального термического анализа (TGA), а на фиг.2 показан график DSC образцов кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты настоящего изобретения.
На фиг.3 и 4 показаны порошковые рентгенограммы (PXRD) кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты настоящего изобретения.
На фиг.5 показан инфракрасный (ИК) спектр поглощения кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты настоящего изобретения.
На фиг.6 показан график динамики поглощения влаги (DMS) кристаллической солью 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты настоящего изобретения.
Подробное описание настоящего изобретения
Настоящее изобретение предоставляет кристаллические соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват. Активный терапевтический агент в этих солях (т.e. соединение формулы I) содержит один хиральный центр, имеющий (R) конфигурацию. Однако специалистам в данной области техники очевидно, что в композициях настоящего изобретения может быть представлено небольшое количество (S) стереоизомера, если явно не указано противное, при условии, что наличие такого изомера в целом не влияет негативно на полезные свойства композиции настоящего изобретения.
Название соединения формулы I было составлено, используя коммерчески доступное программное обеспечение AutoNom (MDL, San Leandro, California). Кроме того, 1,2-этандисульфоновые соли иногда также называют эдизилатами.
Определения
При описании соединений, композиций, способов и процессов настоящего изобретения нижеприведенные термины имеют следующие значения, если явно не указано противное.
Термин "точка плавления", как используется в настоящем описании, означает температуру, при которой наблюдают максимальный эндотермический тепловой поток при помощи дифференциальной сканирующей калориметрии.
Термин "микронизированная форма" означает форму частиц, при которой, по меньшей мере, примерно 90% частиц имеют диаметр, меньший чем примерно 10 мкм.
Термин "сольват" означает комплекс или агрегат, образованный одной или несколькими молекулами растворенного вещества, т.e. соль 1,2-этандисульфоновой кислоты соединения формулы I, и одной или несколькими молекулами растворителя. Такие сольваты обычно имеют по существу молярное отношение растворенного вещества и растворителя. Этот термин также включает клатраты, включая клатраты с водой. Репрезентативные растворители включают, в качестве примера, воду, метанол, этанол, изопропанол, уксусную кислоту и т.п. Когда растворитель представляет собой воду, образованный сольват является гидратом.
Термин "терапевтически эффективное количество" означает количество, достаточное для эффективного лечения при введении нуждающемуся в лечении пациенту.
Термин "лечение" или "терапия", как используется в настоящем описании, означает лечение или терапию заболевания или медицинского состояния (такого как ХОЗЛ) у пациента, например млекопитающего (в частности, человека), которое включает:
(a) предупреждение проявления заболевания или медицинского состояния, т.e. профилактическая терапия пациента;
(b) облегчение заболевания или медицинского состояния, т.e. устранение или ослабление симптомов заболевания или медицинского состояния у пациента;
(c) сдерживание заболевания или медицинского состояния, т.e. замедление или купирование развития заболевания или медицинского состояния у пациента; или
(d) ослабление симптомов заболевания или медицинского состояния у пациента.
Термин "единичная дозированная форма" относится к физически дискретной единице, подходящей для дозировки пациенту, т.e. каждая единица содержит заданное количество соли настоящего изобретения, рассчитанное для получения желательного терапевтического эффекта либо отдельно, либо в комбинации с одной или несколькими дополнительными единицами. Например, такие единичные дозированные формы могут представлять собой капсулы для ингалятора с сухим порошком, отмеренные дозированные формы, отмеренные дозы для ингалятора, капсулы, таблетки, пилюли и т.п.
Соли 1,2-этандисульфоновой кислоты настоящего изобретения
Кристаллическая соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты настоящего изобретения может быть получена из 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты и 1,2-этандисульфоновой кислоты или ее гидрата.
Соль 1,2-этандисульфоновой кислоты настоящего изобретения обычно содержит от примерно 0,90 до примерно 1,10 молярных эквивалентов 1,2-этандисульфоновой кислоты на молярный эквивалент соединения формулы I; включая от примерно 0,95 до примерно 1,05 молярных эквивалентов 1,2-этандисульфоновой кислоты на молярный эквивалент соединения формулы I. В конкретном варианте осуществления соль 1,2-этандисульфоновой кислоты настоящего изобретения содержит примерно 1 молярный эквивалент 1,2-этандисульфоновой кислоты на молярный эквивалент соединения формулы I.
Молярное отношение 1,2-этандисульфоновой кислоты к 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты может быть легко определено различными способами, доступными специалистам в данной области техники. Например, такое молярное отношение может быть легко определено 1H ЯМР. В качестве альтернативы, для определения молярного отношения могут быть использованы способы элементного анализа и ВЭЖХ.
1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты, используемый в настоящем изобретении, может быть легко получен из коммерчески доступных исходных веществ и реагентов при помощи процедур, описанных в нижеприведенных примерах; или при помощи процедур, описанных в опубликованной заявке на патент США в разделе "Уровень техники, к которому относится изобретение".
1,2-этандисульфоновая кислота коммерчески доступна от, например, Alfa Chemicals Ltd., Berkshire, UK. В одном из вариантов осуществления 1,2-этандисульфоновая кислота, используемая для получения солей настоящего изобретения, представляет собой дигидрат. В конкретном варианте осуществления дигидрат 1,2-этандисульфоновой кислоты имеет чистоту, превышающую или равную 97% (как определено ВЭЖХ). При желании дигидрат 1,2-этандисульфоновой кислоты, используемый в настоящем изобретении, до применения может быть перекристаллизован, например, из уксусной кислоты или уксусного ангидрида.
Для получения кристаллической соли настоящего изобретения 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты обычно приводят в контакт с примерно 0,75-1,3 молярными эквивалентами 1,2-этандисульфоновой кислоты или ее гидрата. Обычно эту реакцию выполняют в инертном разбавителе при температуре, находящейся в пределах от примерно 0°С до примерно 60°C; включая примерно 20°C до примерно 55°C, например, от примерно 25°C до примерно 50°C. Подходящие инертные разбавители для этой реакции включают, без ограничений, метанол, этанол, изопропанол, изобутанол, этилацетат, дихлорметан и т.п., необязательно содержащие воду. В конкретном варианте осуществления раствор дигидрата 1,2-этандисульфоновой кислоты в этаноле добавляли в примерно в пять раз больший объем 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты в смеси изопропанола и дихлорметана (64:1). В конкретном варианте осуществления раствор дигидрата 1,2-этансульфоновой кислоты включает воду или этанол в качестве разбавителя, а раствор 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты включает изопропанол или этанол в качестве разбавителя.
В качестве альтернативы, кристаллическую соль 1,2-этандисульфоновой кислоты соединения формулы I можно получить путем приведения в контакт силил-защищенного производного соединения формулы I (т.e. соединения формулы II) с источником фторид-иона и затем, в том же реакционном сосуде, путем приведения в контакт с 1,2-этандисульфоновой кислотой или ее гидратом. В конкретном варианте осуществления силил-защитная группа представляет собой трет-бутилдиметилсилиловую группу. Другие подходящие силил-защитные группы включают трет-бутилдифенилсилил, дифенилметилсилил, ди-трет-бутилметилсилил, трет-бутоксидифенилсилил и т.п. Источник фторид-иона, используемый в этом способе, может представлять собой любой реагент, включающий или содержащий фторид-ион или фтороводород. В конкретном варианте осуществления источник фторид-иона представляет собой триэтиламина тригидрофторид. Другие подходящие источники фторид-иона включают тетрабутиламмония фторид, фторид калия с 18-краун-6, фтороводород, пиридин гидрофторид и т.п.
В общем случае этот способ выполняют в инертном разбавителе при температуре, находящейся в пределах от примерно 0°C до примерно 50°C; включая от примерно 20°C до примерно 35°C, например, от примерно 25°C до примерно 30°C. Подходящие инертные разбавители для этой реакции включают, без ограничений, дихлорметан, метанол и их смеси. В конкретном варианте осуществления раствор 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты оставляют в контакте с примерно 2,5-3,0 молярными эквивалентами триэтиламина тригидрофторида в дихлорметане при температуре окружающей среды в течение от примерно 12 до примерно 24 часов или до тех пор, пока по существу силильная группа не будет полностью удалена. В полученный раствор без выделения продукта реакции добавляют от примерно 0,9 до примерно 1,1 молярных эквивалентов дигидрата 1,2-этандисульфоновой кислоты в метаноле и эту смесь нагревают при температуре от примерно 25°C до примерно 35°C в течение от примерно 2 до примерно 6 часов. При полном завершении реакции кристаллическую соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты выделяют из реакционной смеси обычными средствами, такими как осаждение, концентрация, центрифугирование и т.п.
Необязательно, кристаллическая соль 1,2-этандисульфоновой кислоты настоящего изобретения может быть дополнительно очищена путем перемешивания или смешивания соли с изопропанолом, содержащим от примерно 15% до примерно 25%, включая примерно 20% воды по объему. В конкретном варианте осуществления используют от примерно 10 мл смеси изопропанол/вода на грамм соли 1,2-этандисульфоновой кислоты.
Способ получения кристаллической соли 1,2-этандисульфоновой кислоты настоящего изобретения необязательно может включать использование затравочного кристалла для преимущественного получения конкретной кристаллической соли. Например, путем использования затравочного кристалла кристаллической соли, имеющей более высокую точку плавления (например, выше чем примерно 230°C), может быть получена кристаллическая соль 1,2-этандисульфоновой кислоты соединения формулы I, которая имеет по существу такую же точку плавления, что и у затравочного кристалла. Такие затравочные кристаллы могут быть использованы для первоначального образования кристаллической соли, или они могут быть использованы для перекристаллизации кристаллической или частично кристаллической соли.
Обычно затравочные кристаллы получают в небольших количествах путем медленной кристаллизации без перемешивания и без применения охлаждения. Например, для получения затравочных кристаллов кристаллическую соль обычно растворяют в инертном разбавителе при температуре, достаточной для растворения. Обычно в начальном способе получения затравочных кристаллов используется небольшое количество, обычно меньше чем 10 г, включая меньше чем 5 г, например, меньше чем 1 г, кристаллической соли. В конкретном варианте осуществления метанол, содержащий от примерно 12% до примерно 20% воды, включая от примерно 13% до примерно 15% воды, используется в качестве разбавителя при температуре, находящейся в пределах от примерно 60°C до примерно 70°C, например, от примерно 60°C до примерно 65°C. Раствор охлаждают до комнатной температуры. Спустя примерно 1-3 дня полученные кристаллы выделяют фильтрованием или другим обычным способом. В качестве альтернативы, затравочные кристаллы могут быть получены из предыдущего способа получения кристаллического вещества.
В способе перекристаллизации при помощи затравочных кристаллов кристаллическую соль 1,2-этандисульфоновой кислоты настоящего изобретения растворяют в инертном разбавителе, как в способе получения затравочных кристаллов, обычно в метаноле, содержащем 15% воды, при температуре, находящейся в пределах от примерно 60°C до примерно 65°C. Раствор охлаждают до температуры, при которой затравочные кристаллы не растворяются, например до температуры, находящейся в пределах от примерно 30°C до примерно 40°C, и затем добавляют затравочные кристаллы. Обычно, отношение веса затравочных кристаллов к весу кристаллической соли в растворе находится между примерно 1:5 и примерно 1:35. Раствор охлаждают до температуры, при которой происходит кристаллизация, например до примерно 20°C, и перемешивают в течение от примерно 2 часов до примерно 24 часов. Полученные кристаллы выделяют обычными способами. Для получения достаточного количества затравочных кристаллов для приготовления больших партий вещества способ перекристаллизации может быть успешно выполнен с использованием кристаллов, полученных при помощи первой перекристаллизации, в качестве затравочных кристаллов для следующей стадии перекристаллизации. Очевидно, что определенные температуры, при которых выполняют стадии способа перекристаллизации, выбирают в зависимости от характера разбавителя и концентрации кристаллической соли в растворе. Кроме того, способ перекристаллизации может быть выполнен либо путем испарения, либо при помощи антирастворителя для облегчения кристаллизации, вместо охлаждения.
Помимо других преимуществ, было обнаружено, что образование кристаллической соли 1,2-этандисульфоновой кислоты соединения формулы I полезно для очистки соединения формулы I. Обычно, кристаллическая соль 1,2-этандисульфоновой кислоты настоящего изобретения имеет чистоту, превышающую 95%; и обычно превышающую 98%, как определено при помощи высокоэффективной жидкостной хроматографии.
Кристаллическая соль 1,2-этандисульфоновой кислоты настоящего изобретения характеризуется высокой точкой плавления, что подтверждено графиками дифференциальной сканирующей калориметрии (DSC), которые имеют пик в эндотермическом тепловом потоке в диапазоне от примерно 215°C до примерно 240°C. Обнаружено, что температура точки плавления кристаллической соли зависит от способа, при котором образуется кристаллическая соль. Затравочные кристаллы, образованные медленной кристаллизацией без перемешивания и без применения охлаждения, показывают точки плавления, превышающие примерно 230°C. Кристаллические соли, образованные способом, включающим перекристаллизацию с применением таких затравочных кристаллов, обычно показывают точки плавления в пределах от примерно 230°C до примерно 245°C, как показано, например, на фиг.1. Кристаллические соли, образованные без затравочных кристаллов, имеющих точку плавления, превышающую примерно 230°C, обычно показывают точки плавления в пределах от примерно 215°C до примерно 229°C, как показано, например, на фиг.2. В конкретных вариантах осуществления, следовательно, настоящее изобретение предоставляет кристаллическую соль 1,2-этандисульфоновой кислоты соединения формулы I, имеющую график DSC в диапазоне температур, превышающих примерно 200°C, что по существу соответствует графику, показанному на фиг.1, или графику, показанному на фиг.2.
В другом варианте осуществления кристаллическая соль 1,2-этандисульфоновой кислоты настоящего изобретения характеризуется порошковой рентгенограммой (PXRD), имеющей по существу пики дифракции при 2θ значениях от 5,0 ±0,3 до 15,0 ±0,3. Между положениями пиков в спектре PXRD кристаллической соли, полученной перекристаллизацией из затравочных кристаллов с высокой точкой плавления, как показано на фиг.3, и положениями пиков соли, полученной без использования такого затравочного кристалла, как показано на фиг.4, могут наблюдаться трудноуловимые различия. Соответственно, в отдельных вариантах осуществления кристаллическая соль 1,2-этандисульфоновой кислоты соединения формулы I характеризуется порошковой рентгенограммой, в которой положения пиков по существу соответствуют положениям пиков, показанным на фиг.3, или положениям пиков, показанным на фиг.4.
В другом варианте осуществления кристаллическая соль 1,2-этандисульфоновой кислоты соединения формулы I характеризуется инфракрасным (ИК) спектром поглощения, который показывает значимые полосы поглощения примерно при 704, 748, 768, 841, 900, 1055, 1104, 1166, 1218, 1294, 1408, 1522, 1609, 1655 и 1701 см-1, как показано на фиг.5.
Показано, что кристаллическая соль 1,2-этандисульфоновой кислоты соединения формулы I имеет взаимообратные профили сорбции/десорбции с допустимым со средним уровнем гигроскопичности (т.e. меньше чем примерно 2,5% увеличение веса в диапазоне влажности от 40% относительной влажности до 75% относительной влажности).
Эти свойства солей настоящего изобретения дополнительно проиллюстрированы нижеприведенными примерами.
Фармацевтические композиции и лекарственные формы
Соль 1,2-этандисульфоновой кислоты соединения формулы I обычно вводят пациенту в виде фармацевтической композиции или лекарственной формы. Такие фармацевтические композиции могут быть введены пациенту любым подходящим способом введения, включая, без ограничения, ингаляцию, пероральный, назальный, топический (включая трансдермальный) и парентеральный способы введения. Однако специалистам в данной области техники известно, что после образования кристаллической соли настоящего изобретения она может больше не находиться в кристаллической форме, т.e. соль может быть растворена в подходящем носителе.
Следовательно, в одном из аспектов композиций настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель или вспомогательное вещество и соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват. Необязательно, такие фармацевтические композиции могут содержать другие терапевтические агенты и/или агенты для лекарственных форм, если это необходимо.
Фармацевтические композиции настоящего изобретения обычно содержат терапевтически эффективное количество соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата. Обычно, такие фармацевтические композиции могут содержать от примерно 0,01 до примерно 95% по весу активного агента; включая от примерно 0,01 до примерно 30% по весу; например, от примерно 0,01 до примерно 10% по весу активного агента.
В фармацевтических композициях настоящего изобретения может быть использован любой обычный носитель или вспомогательное вещество. Выбор фармацевтического носителя или вспомогательного вещества или комбинаций носителей или вспомогательных веществ будет зависеть от режима введения, используемого для лечения конкретного пациента, или типа медицинского состояния, или состояния заболевания. В этом отношении изготовление растворимой фармацевтической композиции для конкретного режима введения хорошо известно специалистам в области фармацевтики. Кроме того, ингредиенты для таких композиций коммерчески доступны от, например, Sigma, P.O. Box 14508, St. Louis, MO 63178. В качестве дополнительной иллюстрации обычные техники составления лекарственных форм описаны у Remington: The Science and Practice of Pharmacy, 20-th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); and H.C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7-th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Характерные примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают, без ограничений, следующие: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлоза; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) вспомогательные вещества, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные вещества, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) апирогенная вода; (17) изотонический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) растворы фосфатного буфера; (21) сжатые газы-пропелленты, такие как хлорфторуглеводороды и гидрофторуглеводороды; и (22) другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции настоящего изобретения обычно получают путем тщательного перемешивания до однородной массы или путем смешивания соли настоящего изобретения с фармацевтически приемлемым носителем и одним или несколькими дополнительными ингредиентами. При необходимости или по желанию полученная смешанная до однородной массы смесь затем может быть формована или введена в таблетки, капсулы, пилюли, контейнеры, картриджи, диспенсеры и т.п., используя обычные процедуры и оборудование.
В одном из вариантов осуществления фармацевтические композиции настоящего изобретения являются подходящими для ингаляционного введения. Подходящие фармацевтические композиции для ингаляционного введения обычно могут находиться в виде аэрозоля или порошка. Такие композиции обычно вводятся при помощи хорошо известных устройств для введения, таких как ингалятор-небулайзер, дозирующий ингалятор (ДИ), порошковый ингалятор (ПИ), или аналогичных устройств подачи.
В специфическом варианте осуществления настоящего изобретения фармацевтическую композицию, содержащую активный агент, вводят путем ингаляции при помощи ингалятора-небулайзера. Такие устройства-небулайзеры обычно создают высокоскоростной поток воздуха, который распыляет фармацевтическую композицию, содержащую активный агент, в виде аэрозоля, который поступает в дыхательные пути. Следовательно, при составлении лекарственной формы для использования в ингаляторе-небулайзере активный агент обычно растворяют в подходящем носителе для образования раствора. Подходящие устройства-небулайзеры являются коммерчески доступными, например, от PARI GmbH (Starnberg, German). Другие устройства-небулайзеры включают Respimat (Boehringer Ingelheim) и раскрыты, например, в патенте США №6123068 и заявке на патент WO 97/12687.
Характерная фармацевтическая композиция для использования в ингаляторе-небулайзере содержит водный раствор, содержащий от примерно 0,05 мкг/мл до примерно 10 мг/мл соли 1,2-этандисульфоновой кислоты соединения формулы I или ее сольвата. В одном из вариантов осуществления водная аэрозольная лекарственная форма является изотонической. В одном из вариантов осуществления водная аэрозольная лекарственная форма имеет pH в пределах от примерно 4 до примерно 6. В конкретном варианте осуществления pH водной аэрозольной лекарственной формы доводят цитратным буфером до примерно 5. В другом конкретном варианте осуществления водная аэрозольная лекарственная форма содержит от примерно 0,1 мг/мл до примерно 1,0 мг/мл эквивалентов свободного основания 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты.
В другом специфическом варианте осуществления настоящего изобретения фармацевтическую композицию, содержащую активный агент, вводят путем ингаляции при помощи порошкового ингалятора. Такие порошковые ингаляторы обычно вводят активный агент в виде сыпучего порошка, который при вдохе диспергируется в воздушном потоке, создаваемом пациентом. В другом варианте осуществления для получения сыпучего порошка активный агент обычно составляют в лекарственную форму с подходящим вспомогательным веществом, таким как лактоза, крахмал, маннит, декстроза, полимолочная кислота (PLA), сополимер лактида и гликолида (ПЛГ), или их комбинацией. Обычно активный агент является микронизированным или объединенным с подходящим носителем для образования смеси микронизированных частиц, имеющих пригодный для вдыхания размер, причем "микронизированные частицы" или "микронизированная форма" означает, что, по меньшей мере, примерно 90% частиц имеют диаметр, меньший чем примерно 10 мкм.
Характерная фармацевтическая композиция для использования в порошковом ингаляторе содержит лактозу, имеющую размер частиц в пределах от примерно 1 мкм до примерно 100 мкм, и микронизированные частицы соли 1,2-этандисульфоновой кислоты соединения формулы I или ее сольвата.
Такая порошковая лекарственная форма может быть изготовлена, например, путем объединения лактозы с активным агентом и затем путем сухого смешивания компонентов. В качестве альтернативы при желании активный агент может быть составлен в лекарственную форму без вспомогательного вещества. Затем фармацевтическую композицию обычно загружают в диспенсер для сухого порошка или в картриджи или в капсулы для ингаляции для использования с порошковым устройством.
Примеры порошковых ингаляционных устройств подачи включают Diskhaler (GlaxoSmithKline, Research Triangle Park, NC) (см., например, патент США № 5035237); Diskus (GlaxoSmithKline) (см., например, патент США № 6378519); Turbuhaler (AstraZeneca, Wilmington, DE) (см., например, патент США № 4524769); Rotahaler (GlaxoSmithKline) (см., например, патент США №4353365) и Handihaler (Boehringer Ingelheim). Дополнительные примеры подходящих устройств ПИ описаны в патентах США №5415162, 5239993, 5715810 и ссылках, процитированных в них.
В другом специфическом варианте осуществления настоящего изобретения фармацевтическую композицию, содержащую активный агент, вводят путем ингаляции при помощи дозирующего ингалятора. Такие дозирующие ингаляторы обычно выдают отмеренное количество активного агента или его фармацевтически приемлемой соли, используя сжатый газ-пропеллент. Следовательно, фармацевтические композиции, вводимые при помощи дозирующего ингалятора, содержат раствор или суспензию активного агента в сжиженном пропелленте. Может быть использован любой сжиженный пропеллент, включая хлорфторуглеводороды, такие как CCl3F, и гидрофторалканы (HFA), такие как 1,1,1,2-тетрафторэтан (HFA 134a) и 1,1,1,2,3,3,3-гептафтор-н-пропан (HFA 227). Поскольку хлорфторуглеводороды влияют на озоновый слой, обычно предпочтительны лекарственные формы, содержащие HFA. Дополнительные необязательные компоненты HFA лекарственных форм включают вспомогательные растворители, такие как этанол или пентан, и поверхностно-активные вещества, такие как сорбитана триолеат, олеиновая кислота, лецитин и глицерин. См., например, патент США №5225183, EP 0717987 A2 и WO 92/22286.
Характерная фармацевтическая композиция для использования в дозирующем ингаляторе содержит от примерно 0,01% до примерно 5% по весу соли 1,2-этандисульфоновой кислоты соединения формулы I или ее сольвата; от примерно 0% до примерно 20% по весу этанола; от примерно 0% до примерно 5% по весу поверхностно-активного вещества; с оставшейся частью, которая представляет собой HFA пропеллент.
Такие композиции обычно получают путем добавления охлажденного или сжатого гидрофторалкана в подходящий контейнер, содержащий активный агент, этанол (если присутствует) и поверхностно-активное вещество (если присутствует). Для получения суспензии активный агент тонко измельчают и затем объединяют с пропеллентом. После этого состав лекарственной формы загружают в аэрозольный контейнер, который формирует часть дозирующего ингаляционного устройства. Примеры дозирующих ингаляционных устройств, специально разработанных для использования с HFA пропеллентами, предоставлены в патентах США №6006745 и 6143277. В качестве альтернативы, лекарственная форма в виде суспензии может быть изготовлена при помощи распылительной сушки поверхностно-активного вещества на микронизированных частицах активного агента. См., например, заявки на патент WO 99/53901 и WO 00/61108.
Дополнительные примеры способов получения пригодных для вдыхания частиц, составов лекарственных форм и устройств, подходящих для дозирующей ингаляции, см. в патентах США №6268533, 5983956, 5874063 и 6221398 и заявках на патент WO 99/55319 и WO 00/30614.
В другом примере фармацевтические композиции настоящего изобретения являются подходящими для перорального введения. Подходящие фармацевтические композиции для перорального введения могут находиться в виде капсул, таблеток, пилюль, лепешек, крахмальных капсул, драже, порошков, гранул; или в виде раствора или суспензии в водной или неводной жидкости; или в виде жидкой эмульсии масло-в-воде или вода-в-масле; или в виде эликсира или сиропа и т.п.; каждая из которых содержит заданное количество соли настоящего изобретения в виде активного агента.
В случае перорального введения в твердой дозированной форме (т.e. в виде капсул, таблеток, пилюль и т.п.) фармацевтические композиции настоящего изобретения обычно содержат соль настоящего изобретения в качестве активного агента и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия или дикальция фосфат. Необязательно или в качестве альтернативы, такие твердые дозированные формы также могут содержать: (1) наполнители или разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; (2) связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или камедь; (3) смачиватели, такие как глицерин; (4) диспергирующие вещества, такие как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и/или карбонат натрия; (5) замедлители растворения, такие как парафин; (6) ускорители абсорбции, такие как соединения четвертичного аммония; (7) увлажнители, такие как цетиловый спирт и/или глицеринмоностеарат; (8) абсорбенты, такие как каолин и/или бентонитовая глина; (9) лубриканты, такие как тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия и/или их смеси; (10) красители и (11) буферные вещества.
В фармацевтических композициях настоящего изобретения также могут присутствовать высвобождающие агенты, увлажнители, покровные вещества, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты. Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, цистеина гидрохлорид, натрия бисульфат, натрия метабисульфат, натрия сульфат и т.п.; (2) маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (БГА), бутилированный гидрокситолуол (БГТ), лецитин, пропилгалат, альфа-токоферол и т.п.; и (3) металлохелатные агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота (ЭДТА), сорбитол, тартаровая кислота, фосфорная кислота и т.п. Покровные вещества для таблеток, капсул, пилюль и т.п. включают такие вещества, которые используются для энтеросолюбильных покрытий, такие как целлюлозы ацетат фталат (ЦАФ), поливинилацетат фталат (ПВАФ), фталат гидроксипропилметилцеллюлозы, сополимеры метакриловая кислота-эфир метакриловой кислоты, тримелитата ацетат целлюлозы (CAT), карбоксиметилэтилцеллюлоза (CMEC), ацетата сукцинат гидроксипропилметилцеллюлозы (HPMCAS) и т.п.
При желании фармацевтические композиции настоящего изобретения также могут быть составлены в лекарственные формы для обеспечения медленного или управляемого высвобождения активного ингредиента, используя, например, гидроксипропилметилцеллюлозу в различных пропорциях; или другие полимерные матрицы, такие как полимолочная кислота (PLA) или сополимер лактида и гликолида (PLGA), липосомы и/или микросферы.
Кроме того, фармацевтические композиции настоящего изобретения необязательно могут содержать замутнители и могут быть составлены в лекарственные формы таким образом, чтобы они высвобождали активный агент только, или предпочтительно, в некоторой части желудочно-кишечного тракта, необязательно, замедленным способом. Примеры вариантов осуществлений композиций, которые могут быть использованы, включают полимерные вещества и воски. Активный ингредиент также может находиться в микроинкапсулированном виде, если это целесообразно, с одним или несколькими из вышеуказанных вспомогательных веществ.
Подходящие жидкие дозированные формы для перорального введения включают, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Такие жидкие дозированные формы обычно содержат активные ингредиенты и инертный разбавитель, такой как, например, вода или другие растворители, солюбилизаторы и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (особенно, хлопковое, арахисовое, кукурузное масло, масло пшеничных зародышей, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры сорбитана с жирными кислотами и их смеси. Суспензии дополнительно к активному ингредиенту могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтилен сорбитол и сложные эфиры сорбитана, микрокристаллическую целлюлозу, алюминия метагидроксид, бентонит, агар-агар и трагакант и их смеси.
В случае перорального введения фармацевтические композиции настоящего изобретения предпочтительно пакуют в разовые дозированные формы. Например, такие разовые дозированные формы могут представлять собой капсулы, таблетки, пилюли и т.п.
Соли настоящего изобретения и вспомогательные вещества также могут вводиться трансдермально при помощи известных трансдермальных систем подачи. Например, соединение настоящего изобретения может быть смешано с усилителями проникновения, такими как пропиленгликоль, полиэтиленгликоля монолаурат, азациклоалкан-2-оны и т.п., и включено в пластыри или аналогичные системы подачи. По желанию в таких трансдермальных композициях могут быть использованы дополнительные вспомогательные вещества, включающие желирующие агенты, эмульгаторы и буферы.
Фармацевтические композиции настоящего изобретения могут также содержать другие терапевтические агенты, которые вводятся совместно с солью 1,2-этандисульфоновой кислоты соединения формулы I или ее сольватом. Например, фармацевтические композиции настоящего изобретения могут дополнительно содержать один или несколько терапевтических агентов, выбранных из противовоспалительных агентов (например, стероидных противовоспалительных агентов, таких как кортикостероиды; и нестероидных противовоспалительных агентов (NSAID), ингибиторов фосфодиэстеразы IV, антиинфекционных агентов (например, антибиотиков или противовирусных агентов), антигистаминов, агонистов β2-адренергических рецепторов, агонистов мускариновых рецепторов (т.e. антихолинэргических агентов) и т.п. Другие терапевтические агенты могут быть использованы в виде фармацевтически приемлемых солей или сольватов. Кроме того, если это целесообразно, могут быть использованы другие терапевтические агенты в виде оптически чистых стереоизомеров.
При желании соли настоящего изобретения также могут быть введены в комбинации с другим терапевтическим агентом или агентами, такими как вышеописанные агенты. В этом варианте осуществления компоненты физически не смешивают вместе, но вводят одновременно или последовательно в виде отдельных компонентов. Например, соль настоящего изобретения может быть введена путем ингаляции одновременно или последовательно со стероидным противовоспалительным агентом, таким как кортикостероид, при помощи ингаляционного устройства подачи, которое использует отдельные компоненты (например, блистерные упаковки) для каждого терапевтического агента. В качестве альтернативы, комбинация может быть введена в виде множества устройств подачи, т.e. для каждого терапевтического агента по одному устройству подачи.
Характерные агонисты β2-адренергических рецепторов, которые могут использоваться в комбинации с соединениями настоящего изобретения, включают, без ограничения, сальметерол, сальбутамол, формотерол, сальмефамол, фенотерол, тербуталин, альбутерол, изоэтарин, метапротеренол, битолтерол, пирбутерол, левалбутерол и т.п. или их фармацевтически приемлемые соли. Другие агонисты β2-адренергических рецепторов, которые могут быть использованы в комбинации с соединениями настоящего изобретения, включают, без ограничений, 3-(4-{[6-({(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этил}амино)-гексил]окси}бутил)
бензолсульфонамид и 3-(-3-{[7-({(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этил}амино)гептил]окси}пропил)
бензолсульфонамид и родственные соединения, раскрытые в заявке на патент WO 02/066422, опубликованной 29 августа 2002; 3-[3-(4-{[6-([(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этил}амино)гексил]-окси}бутил)фенил]
имидазолидин-2,4-дион и родственные соединения, раскрытые в заявке на патент WO 02/070490, опубликованной 12 сентября 2002; 3-(4-{[6-({(2R)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид, 3-(4-{[6-({(2S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)-бензолсульфонамид, 3-(4-{[6-({(2R/S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2R)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]-окси}бутил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2R/S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид и родственные соединения, раскрытые в заявке на патент WO 02/076933, опубликованной 3 октября 2002; 4-{(1R)-2-[(6-{2-[(2,6-дихлорбензил)окси]этокси}гексил)амино]-1-гидроксиэтил}-2-(гидроксиметил)фенол и родственные соединения, раскрытые в заявке на патент WO 03/024439, опубликованной 27 марта 2003; N-{2-[4-((R)-2-гидрокси-2-фенилэтиламино)фенил]этил}-(R)-2-гидрокси-2-(3-формамидо-4-гидроксифенил)этиламин и родственные соединения, раскрытые в патенте США №6576793 B1, выданном 10 июня 2003; N-{2-[4-(3-фенил-4-метоксифенил)аминофенил]этил}-(R)-2-гидрокси-2-(8-гидрокси-2(1H)-хинолинон-5-ил)этиламин и родственные соединения, раскрытые в патенте США №6653323 B2, выданном 25 ноября 2003; и их фармацевтически приемлемые соли. В конкретном варианте осуществления агонист β2-адренергических рецепторов представляет собой кристаллическую соль моногидрохлорида N-{2-[4-((R)-2-гидрокси-2-фенилэтиламино)фенил]этил}-(R)-2-гидрокси-2-(3-формамидо-4-гидроксифенил)этиламина. При использовании агонист β2-адренергических рецепторов может находиться в фармацевтической композиции в терапевтически эффективном количестве. Обычно, агонист β2-адренергических рецепторов может находиться в количестве, достаточном для предоставления, от примерно 0,05 мкг до примерно 500 мкг на дозу.
Характерные стероидные противовоспалительные агенты, которые могут быть использованы в комбинации с соединениями настоящего изобретения, включают, без ограничений, метилпреднизолон, преднизолон, дексаметазон, флутиказона пропионат, S-фторметиловый эфир 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты, S-(2-оксотетрагидрофуран-3S-ил) эфир 6,9-дифтор-11-гидрокси-16-метил-3-оксо-17-пропионилокси-андроста-1,4-диен-17-
карботиокислоты, эфиры беклометазона (например, эфир 17-пропионата или эфир 17,21-дипропионата), эфиры будезонида, флунизолида, мометазона (например, эфир фуроата), триамцинолон ацетонид, рофлепонид, циклезонид, бутиксокорта пропионат, RPR-106541, ST-126 и т.п. или их фармацевтически приемлемые соли. В конкретном варианте осуществления стероидный противовоспалительный агент представляет собой S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиокислоты или его фармацевтически приемлемую соль или сольват. При использовании противовоспалительный агент может находиться в фармацевтической композиции в терапевтически эффективном количестве. Обычно, стероидный противовоспалительный агент может находиться в количестве, достаточном для предоставления, от примерно 0,05 мкг до примерно 500 мкг на дозу.
Другие подходящие комбинации включают, например, другие противовоспалительные агенты, например NSATD (такие, как кромогликат натрия; недокромил натрия; ингибиторы фосфодиэстеразы (PDE) (например, теофиллин, ингибиторы PDE4 или смешанные ингибиторы PDE3/PDE4); антагонисты лейкотриенов (например, монтелукаст); ингибиторы синтеза лейкотриенов; ингибиторы iNOS; ингибиторы протеаз, такие как ингибиторы триптазы и эластазы; антагонисты бета-2 интегрина и агонисты или антагонисты рецепторов аденозина (например, 2a агонисты аденозина); антагонисты цитокина (например, антагонисты хемокина, такие как антитело к интерлейкину (IL антитело), в особенности, при IL-4 терапии, IL-13 терапии, или их комбинации); или ингибиторы синтеза цитокинов.
Например, характерные ингибиторы фосфодиэстеразы-4 (PDE4) или смешанные ингибиторы PDE3/PDE4, которые могут быть использованы в комбинации с соединениями настоящего изобретения, включают, без ограничений, цис-4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновую кислоту, 2-карбометокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-он; цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-ол]; цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновую кислоту и т.п. или их фармацевтически приемлемые соли. Другие характерные ингибиторы PDE4 или смешанные ингибиторы PDE4/PDE3 включают AWD-12-281 (elbion); NCS-613 (INSERM); D-4418 (Chiroscience и Schering-Plough); CI-1018 или PD-168787 (Pfizer); соединения бензодиоксола, раскрытые в заявке на патент WO 99/16766 (Kyowa Hakko); K-34 (Kyowa Hakko); V-11294A (Napp); рофлумиласт (Byk-Gulden); соединения фталазинона, раскрытые в заявке на патент WO 99/47505 (Byk-Gulden); пумафентрин (Byk-Gulden, now Altana); арофиллин (Almirall-Prodesfarma); VM554/UM565 (Vernalis); T-440 (Tanabe Seiyaku); T2585 (Tanabe Seiyaku).
Характерные мускариновые антагонисты (т.e. антихолинэргические агенты), которые могут быть использованы в комбинации и дополнительно к соединениям настоящего изобретения, включают, без ограничений, антропин, антропина сульфат, атропина оксид, метилатропина нитрат, гоматропина гидробромид, гиосциамина (d,l) гидробромид, скополамина гидробромид, ипратропиум бромид, окситропиум бромид, тиотропиум бромид, метантелин, пропантелина бромид, анизотропина метилбромид, клидиниум бромид, копирролат (Робинул), изопропамида йодид, мепензолата бромид, тридигексетила хлорид (Pathilone), гексоциклия метилсульфат, циклопентолат гидрохлорид, тропикамид, тригексифенидила гидрохлорид, пирензепин, телензепин, AF-DX 116 и метоктрамин и т.п. или их фармацевтически приемлемые соли; или, для указанных соединений, которые перечислены как соли, их альтернативные фармацевтически приемлемые соли.
Характерные антигистамины (т.e. антагонисты H1-рецепторов), которые могут быть использованы в комбинации с соединениями настоящего изобретения, включают, без ограничения, этаноламины, такие как карбиноксамина малеат, клемастина фумарат, дифенилгидрамина гидрохлорид и дименгидринат; этилендиамины, такие как пириламина малеат, трипеленамина гидрохлорид и трипеленамина цитрат; алкиламины, такие как хлорфенирамин и акривастин; пиперазины, такие как гидроксизина гидрохлорид, гидроксизина памоат, циклизина гидрохлорид, циклизина лактат, меклизина гидрохлорид и цетиризина гидрохлорид; пиперидины, такие как астемизол, левокабастина гидрохлорид, лоратадин или его дескарбоэтокси аналоги, терфенодин и фексофенадина гидрохлорид; азеластина гидрохлорид и т.п. или их фармацевтически приемлемые соли; или, для этих перечисленных в качестве соли, их альтернативные фармацевтически приемлемые соли.
Подходящие дозы для других терапевтических агентов, вводимых в комбинации с соединением настоящего изобретения, находятся в пределах от примерно 0,05 мг/сутки до примерно 100 мг/сутки.
Нижеследующие лекарственные формы иллюстрируют характерные фармацевтические композиции настоящего изобретения.
Пример A лекарственной формы
Сухой порошок для введения путем ингаляции получают следующим образом.
Характерная процедура. Соединение настоящего изобретения микронизируют и затем смешивают с лактозой. Затем эту перемешанную смесь загружают в желатиновый ингаляционный картридж. Содержимое картриджа вводят, используя порошковый ингалятор.
Пример В лекарственной формы
Порошковую лекарственную форму для использования в порошковом ингаляционном устройстве получают следующим образом.
Характерная процедура. Фармацевтическую композицию готовят в виде лекарственной формы с объемным отношением микронизированной соли настоящего изобретения к лактозе, равным 1:200. Композицию пакуют в порошковое ингаляционное устройство, способное доставлять от примерно 10 мкг до примерно 100 мкг соединения настоящего изобретения на дозу.
Пример С лекарственной формы
Сухой порошок для введения путем ингаляции, отмеренный дозирующим ингалятором, получают следующим образом.
Характерная процедура. Суспензию, содержащую 5 вес.% соли настоящего изобретения и 0,1 вес.% лецитина, готовят путем диспергирования 10 г соединения настоящего изобретения до микронизированных частиц, средний размер которых меньше чем 10 мкм, в растворе, образованном из 0,2 г лецитина, растворенного в 200 мл деминерализованной воды. Суспензию сушат распылением и полученное вещество микронизируют на частицы, имеющие средний диаметр меньше чем 1,5 мкм. Частицы загружают в картриджи со сжатым 1,1,1,2-тетрафторэтаном.
Пример D лекарственной формы
Фармацевтическую композицию для использования в дозирующем ингаляторе получают следующим образом.
Характерная процедура. Суспензию, содержащую 5% соли настоящего изобретения, 0,5% лецитина и 0,5% трегалозы, готовят путем диспергирования 5 г активного ингредиента в виде микронизированных частиц со средним размером меньше чем 10 м в коллоидном растворе, образованном из 0,5 г трегалозы и 0,5 г лецитина, растворенного в 100 мл деминерализованной воды. Суспензию сушат распылением и полученное вещество микронизируют на частицы, имеющие средний диаметр меньше чем 1,5 мкм. Частицы загружают в канистры со сжатым 1,1,1,2-тетрафторэтаном.
Пример Е лекарственной формы
Фармацевтическую композицию для использования в аэрозольном ингаляторе получают следующим образом.
Характерная процедура. Водную аэрозольную лекарственную форму для использования в ингаляторе-небулайзере готовят путем растворения 0,5 мг соли настоящего изобретения в 1 мл 0,9% раствора хлорида натрия, подкисляют лимонной кислотой. Смесь перемешивают и обрабатывают ультразвуком до тех пор, пока не растворится активный ингредиент. pH раствора доводят до значения, равного примерно 5, путем добавления NaOH.
Пример F лекарственной формы
Твердые желатиновые капсулы для перорального введения получают следующим образом.
Характерная процедура. Ингредиенты тщательно перемешивают и затем загружают в твердую желатиновую капсулу (460 мг композиции на капсулу).
Пример G лекарственной формы
Суспензию для перорального введения получают следующим образом.
Характерная процедура. Ингредиенты смешивают для образования суспензии, содержащей 100 мг активного ингредиента на 10 мл суспензии.
Пример Н лекарственной формы
Инъецируемую лекарственную форму получают нижеследующим способом.
Характерная процедура. Вышеприведенные ингредиенты смешивают и доводят рН до 4 ±0,5, используя 0,5 н. HCl или 0,5 н. NaOH.
Полезность
Соединение формулы I обладает как агонистической активностью β2-адренергических рецепторов, так и антагонистической активностью мускариновых рецепторов, и, следовательно, предполагается, что соль 1,2-этандисульфоновой кислоты соединения формулы I настоящего изобретения будет полезной в качестве терапевтического агента для лечения медицинских состояний, опосредованных β2-адренергическими рецепторами или мускариновыми рецепторами, т.e. медицинских состояний, которые улучшаются благодаря лечению агонистом β2-адренергических рецепторов или антагонистом мускариновых рецепторов. Такие медицинские состояния включают, например, легочные расстройства или заболевания, включая заболевания, связанные с обратимой обструкцией дыхательных путей, такие как хроническое обструктивное заболевание легких (например, хронический и влажный бронхит и эмфизема), астма, легочный фиброз, аллергический ренит, ринорея и т.п. Другие состояния, которые могут быть излечены, включают преждевременные роды, депрессию, острую сердечную недостаточность, кожные заболевания (например, воспалительные, аллергенные, псориатические и пролиферативные заболевания кожи), состояния, при которых желательна пониженная кислотность желудка (например, язва желудка и кишечника) и заболевания атрофии мышц.
Следовательно, в одном из вариантов настоящее изобретение относится к способу лечения легочного расстройства, включающему введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата. При использовании для лечения легочного расстройства, соль настоящего изобретения обычно может вводиться путем ингаляции в виде многократных доз в сутки, в виде единичной дневной дозы или единичной недельной дозы. Обычно доза для лечения легочного расстройства находится в пределах от примерно 10 мкг/сутки до примерно 200 мкг/сутки.
При введении путем ингаляции соединения настоящего изобретения обычно вызывают эффект бронходилатации. Следовательно, в другом аспекте способа, настоящее изобретение относится к способу вызова бронходилатации у пациента, нуждающегося в бронходилатации, который включает введение пациенту соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата в количестве, вызывающем бронходилатацию. Обычно, доза, приводящая к бронходилатации, находится в пределах от примерно 10 мкг/сутки до примерно 200 мкг/сутки.
В одном из вариантов настоящее изобретение относится к способу лечения хронических обструктивных заболеваний легких или астмы, который включает введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата. При использовании в лечении ХОЗЛ или астмы соль настоящего изобретения обычно вводят путем ингаляции в виде многократных доз в сутки или в виде единичной суточной дозы. Обычно, доза для лечения ХОЗЛ или астмы находится в пределах от примерно 10 мкг/сутки до примерно 200 мкг/сутки. Как используется в настоящем изобретении, ХОЗЛ включает хронический обструктивный бронхит и эмфизему (см., например, Barnes, Chronic Obstructive Pulmonary Disease, N Engl J Med 2000: 343:269-78).
При использовании для лечения легочного расстройства соль настоящего изобретения необязательно вводят в комбинации с другими терапевтическими агентами. Следовательно, в конкретном варианте осуществления фармацевтические композиции и способы настоящего изобретения дополнительно содержат терапевтически эффективное количество стероидного противовоспалительного агента. Свойства и польза солей 1,2-этандисульфоновой кислоты настоящего изобретения могут быть продемонстрированы посредством множества анализов in vitro и in vivo, хорошо известных специалистам в данной области техники. Например, репрезентативные виды анализа показаны более подробно далее в нижеприведенных примерах.
Примеры
Нижеследующие процедуры получения и примеры приведены для иллюстрации специфических вариантов осуществления настоящего изобретения. Эти специфические варианты осуществления, однако, не предназначены для ограничения объема настоящего изобретения любым способом, если только это не указано специально. Нижеприведенные аббревиатуры имеют следующие значения, если явно не указано противное, и любые другие аббревиатуры, использованные и не определенные в настоящем описании, имеют свои стандартные значения:
Если явно не указано противное, реагенты, исходные вещества и растворители приобретали у коммерческих поставщиков (таких, как Aldrich, Fluka, Sigma и т.п.) и использовали без дополнительной очистки.
В нижеописанных примерах ВЭЖХ анализ проводили, используя прибор Agilent (Palo Alto, CA) серии 1100, оборудованный колонками Zorbax Bonus RP 2,1 x 50 мм, производства Agilent, (колонка C14), имеющими размер частиц 3,5 микрон. Детектирование проводили, используя УФ-поглощение при 214 нм. Данные ВЭЖХ 10-70 получали при скорости потока 0,5 мл/мин 10%-70% по объему в течение 6 минут. Подвижная фаза A составляла 2%-98% - 0,1% ACN-H2O-TFA; и подвижная фаза B составляла 90%-10% - 0,1% ACN-H2O-TFA. Используя подвижные фазы A и B, описанные выше, данные ВЭЖХ 5-35 и данные ВЭЖХ 10-90 получали при 5-минутном градиенте.
Данные жидкостной хроматографии с масс-спектрометрией (LCMS) получали, используя прибор Applied Biosystems (Foster City, CA) модели API- 150EX. Данные LCMS 10-90 получали при помощи 10%-90% мобильной фазы B в 5-минутном градиенте.
Очистку в небольших объемах проводили, используя систему препаративной рабочей станции API 150EX от Applied Biosystems. Мобильные фазы представляли собой, A: вода+0,05 вес.% TFA; и B: ацетонитрил+0,05 вес.% TFA. Для матриц (обычно примерно 3-50 мг восстановленного образца) использовали нижеследующие условия: скорость потока 20 мл/мин; 15 мин градиенты и колонка 20 мм x 50 мм Prism RP с размером частиц 5 микрон (Thermo Hypersil-Keystone, Bellefonte, PA). Для очистки в больших масштабах (обычно больше 100 мг неочищенного образца) использовали нижеследующие условия: скорость потока 60 мл/мин; 30 мин градиенты и колонка 41,4 мм x 250 мм Microsorb BDS с размером частиц 10 микрон (Varian, Palo Alto, CA).
Специфическое вращение для хиральных соединений (указанных как [α]20 D) измеряли, используя поляриметр Jasco (модель P-1010) с вольфрам-галогеновым источником света и фильтром 589 нм при 20°C. Образцы тестируемых соединений обычно измеряли при 1 мг/мл воды.
Процедура получения 1
Метил-4-амино-S-хлор-2-метоксибензоат
В раствор 4-амино-5-хлор-2-метоксибензойной кислоты (1,008 г, 5,0 мМ) в смеси толуола (9 мкл) и метанола (1 мл) при 0°C добавляли по каплям (триметилсилил)диазометан (2,0 M в гексане, 3,0 мл, 6,0 мМ). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч. Избыток (триметилсилил)диазометана гасили добавлением уксусной кислоты до тех пор, пока не исчезла светло-желтая окраска реакционной смеси. Затем смесь концентрировали под вакуумом для получения указанного в заголовке соединения в виде твердого грязно-белого вещества, которое использовали без дополнительной очистки.
Процедура получения 2
Метил-4-акрилоиламино-5-хлор-2-метоксибензоат
К неочищенному продукту из процедуры получения 1 добавляли дихлорметан (10 мл, 0,5 M) и триэтиламин (2,1 мл, 15 мМ). Эту смесь охлаждали до 0°C и при перемешивании добавляли по каплям акрилоил хлорид (812 мкл, 10 мМ). После 2 ч реакционную смесь гасили добавлением метанола (примерно 2 мл) при 0°C и полученную смесь перемешивали при комнатной температуре в течение 15 мин, а затем концентрировали под вакуумом. К остатку добавляли дихлорметан (30 мл) и воду (30 мл) и эту смесь тщательно перемешивали. Слои разделяли и водный слой экстрагировали дихлорметаном (20 мл). Органические слои объединяли, сушили (Na2SO4), фильтровали и под вакуумом удаляли растворитель, получая указанное в заголовке соединение в виде твердого коричневого пенообразного вещества, которое использовали без дополнительной очистки.
Процедура получения 3
Пиперидин-4-иловый эфир бифенил-2-карбаминовой кислоты
Бифенил-2-изоцианат (97,5 г, 521 мМ) и 4-гидрокси-1-бензилпиперидин (105 г, 549 мМ), оба коммерчески доступные от Aldrich, Milwaukee, WI, нагревали вместе при 70°C в течение 12 ч, в течение этого времени отслеживали образование 1-бензилпиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты при помощи LCMS. Затем реакционную смесь охлаждали до 50°C и добавляли этанол (1 л), после чего медленно добавляли 6 M соляную кислоту (191 мл). Затем реакционную смесь охлаждали до температуры окружающей среды и добавляли аммония формат (98,5 г, 1,56 М), через раствор энергично пропускали пузырьки газообразного азота в течение 20 мин. Затем добавляли палладий (10 вес.% (сухая масса) на активированном угле) (20 г). Реакционную смесь нагревали при 40°C в течение 12 ч, затем фильтровали через слой целита. После этого растворитель удаляли при пониженном давлении и к неочищенному остатку добавляли 1 M соляную кислоту (40 мл). После этого доводили pH раствора гидроксидом натрия (10 н.) до 12. Водный слой экстрагировали этилацетатом (2 x 150 мл) и сушили (сульфатом магния), затем при пониженном давлении удаляли растворитель для получения указанного в заголовке соединения (155 г, 100%). ВЭЖХ (10-70) Rt=2,52; MS m/z: [M+H4] вычисленный для C18H20N2O2 297,15; найденный 297,3.
Процедура получения 4
Метил-4-{3-[4-(бифенил-2-илкарбамоилокси)пиперидин-1-ил]пропиониламино}-5-хлор-2-метоксибензоат
К неочищенному продукту из процедуры получения 2 добавляли продукт из процедуры получения 3 (1,33 г, 4,5 мМ) и смесь THF (22,5 мл) и метанола (2,5 мл). Эту смесь нагревали при 50°C при перемешивании в течение 16 ч, затем растворитель удаляли под вакуумом. Остаток хроматографировали (силикагель; EtOAc) для получения указанного в заголовке соединения (0,82 г; Rf=0,4, выход 29% за 3 стадии) в виде грязно-белого твердого пенообразного вещества. MS m/z 566,4 (M+H, 565,20 ожидаемый для C30H32ClN3O6).
Процедура получения 5
1-[2-(2-хлор-4-гидроксиметил-5-метокси-фенилкарбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В раствор продукта из процедуры получения 4 (0,82 мг, 1,45 мМ) в смеси THF (4,5 мл) и метанола (0,5 мл) при 0°C добавляли лития бромид (32 мг, 1,45 мМ). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 41 ч. Затем реакцию гасили добавлением 1 н. водного раствора соляной кислоты при 0°C до прекращения появления пузырьков и эту смесь перемешивали в течение 10 мин. Растворитель удаляли под вакуумом и остаток растворяли в ацетонитриле (примерно 2 мл). Этот раствор очищали препаративным разделением методом обращенно-фазовой ВЭЖХ (градиент: 2-50% ацетонитрила в воде с 0,05% TFA). Подходящие фракции собирали, объединяли и лиофилизировали, получая указанное в заголовке соединение в виде соли трифторуксусной кислоты. Эту соль обрабатывали изопропилацетатом (10 мл) и 1 н. водным раствором гидроксида натрия (10 мл) и органический слой собирали, сушили (Na2SO4), фильтровали и растворитель удаляли под вакуумом, получая указанное в заголовке соединение (161 мг, выход 21%) в виде белого твердого пенообразного вещества. MS m/z 538,4 (M+H, 537,20 ожидаемый для C29H32ClN3O5).
Процедура получения 6
1-[2-(2-хлор-4-формил-5-метоксифенил-карбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В раствор продукта процедуры получения 5 (161 мг, 0,3 мМ) в дихлорметане (3 мл) добавляли диметилсульфоксид (213 мкл, 3,0 мМ) и диизопропилэтиламин (261 мкл, 1,5 мМ). Эту смесь охлаждали до -20°C и медленно добавляли пиридиновый комплекс триоксида серы (238 мг, 1,5 мМ). После 30 мин реакционную смесь гасили добавлением воды (примерно 3 мл). Слои разделяли и органический слой сушили (Na2SO4), фильтровали и растворитель удаляли под вакуумом, получая указанное в заголовке соединение в виде светло-желтого твердого вещества. MS m/z 536,3 (M+H, 535,19 ожидаемый для C29H30ClN3O5).
Процедура получения 7
8-бензилокси-5-(2-бромацетил)-1Н-хинолин-2-он
(a) 8-ацетокси-1Н-хинолин-2-он
8-гидроксихинолин-N-оксид (160,0 г, 1,0 М), коммерчески доступный от Aldrich, Milwaukee, WI, и уксусный ангидрид (800 мл, 8,4 М) нагревали при 100°C в течение 3 ч и затем охлаждали льдом. Продукт собирали в воронке Бюхнера, промывали уксусным ангидридом (2 x 100 мл) и сушили при пониженном давлении, получая 8-ацетокси-1H-хинолин-2-он (144 г) в виде твердого вещества.
(b) 5-ацетил-8-гидрокси-1Н-хинолин-2-он
Суспензию алюминия хлорида (85,7 г, 640 мМ) в 1,2-дихлорэтане (280 мл) охлаждали льдом и добавляли продукт из стадии (a) (56,8 г, 280 мМ). Смесь нагревали до комнатной температуры и затем нагревали при 85°C. После 30 мин добавляли ацетил хлорид (1,5 мл, 21 мМ) и смесь нагревали еще 60 мин. Затем реакционную смесь охлаждали и добавляли 1 н. раствор соляной кислоты (3 л) при 0°, хорошо перемешивая. После перемешивания в течение 2 ч твердые вещества собирали в воронке Бюхнера, промывали водой (3 x 250 мл) и сушили при пониженном давлении. Неочищенный продукт, выделенный из нескольких партий (135 г), объединяли и растирали с дихлорметаном (4 л) в течение 6 ч. Полученное твердое вещество собирали в воронке Бюхнера и сушили при пониженном давлении, получая указанное в заголовке соединение (121 г).
(c) 5-ацетил-8-бензилокси-1Н-хинолин-1-он
В продукт из этапа (b) (37,7 г, 186 мМ) добавляли N,N-диметилформамид (200 мл) и карбонат калия (34,5 г, 250 мМ), затем бензил бромид (31,8 г, 186 мМ). Смесь перемешивали при комнатной температуре в течение 2,25 часов, затем выливали в насыщенный раствор хлорида натрия (3,5 л) при 0°C и перемешивали в течение 1 ч. Продукт охлаждали и сушили в воронке Бюхнера в течение 1 часа, и полученные твердые вещества растворяли в дихлорметане (2 л), и смесь сушили над сульфатом натрия. Раствор фильтровали через слой целита, который затем промывали дихлорметаном (5 x 200 мл). Объединенный фильтрат затем концентрировали досуха и полученные твердые вещества растирали с эфиром (500 мл) в течение 2 ч. Продукт охлаждали в воронке Бюхнера, промывали эфиром (2 x 250 мл) и сушили при пониженном давлении, получая указанное в заголовке соединение (44 г) в виде порошка.
(d) 8-бензилокси-5-(2-бромацетил)-1Н-хинолин-2-он
Продукт из этапа (c) (20,0 г, 68,2 мМ) растворяли в дихлорметане (200 мл) и охлаждали до 0°C. Бора трифториддиэтиловый эфир (10,4 мл, 82,0 мМ) добавляли, используя шприц, и смесь нагревали до комнатной температуры, получая густую суспензию. Суспензию нагревали при 45°C (масляная баня) и в течение 40 мин добавляли раствор брома (11,5 г, 72,0 мМ) в дихлорметане (100 мл). Смесь держали при 45°C еще в течение 15 мин и затем охлаждали до комнатной температуры. Смесь концентрировали при пониженном давлении и затем растирали с 10% водным раствором карбоната (200 мл) в течение 1 часа. Твердые вещества собирали в воронке Бюхнера, промывали водой (4 x 100 мл) и сушили при пониженном давлении. Продукты двух прогонов объединяли для очистки. Неочищенный продукт (52 г) растирали с 50% метанолом в хлороформе (500 мл) в течение 1 часа. Продукт собирали воронкой Бюхнера и промывали 50% метанолом в хлороформе (2 x 50 мл) и метанолом (2 x 50 мл). Твердое вещество сушили при пониженном давлении, получая указанное в заголовке соединение (34,1 г) в виде порошка.
Процедура получения 8
8-бензилокси-5-[(R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил]-1H-хинолин-2-он
(a) 8-бензилокси-5-((R)-2-бром-1-гидроксиэтил)-1H-хинолин-2-он
(R)-(+)-α, α-дифенилпролинол (30,0 г, 117 мМ) и триметилбороксин (11,1 мл, 78 мМ) объединяли в толуоле (300 мл) и перемешивали при комнатной температуре в течение 30 мин. Смесь помещали в масляную баню при температуре 150°C и отгоняли жидкость. Добавляли толуол в 20 мл аликвоты и продолжали отгонку в течение 4 ч. Добавляли толуол общим количеством 300 мл. Затем смесь охлаждали до комнатной температуры. 500 мкл аликвоты выпаривали досуха и взвешивали (246 мг), определив, что концентрация катализатора составляла 1,8 M.
8-бензилокси-5-(2-бромацетил)-1H-хинолин-2-он (90,0 г, 243 мМ) помещали в атмосферу азота и добавляли тетрагидрофуран (900 мл), за которым добавляли описанный выше катализатор (1,8 M в толуоле, 15 мл, 27 мМ). Суспензию охлаждали до -10 ±5°C в бане лед/изопропанол. В течение 4 ч добавляли боран (1,0 M в THF, 294 мл, 294 мМ). Затем реакцию перемешивали в течение 45 мин при -10°C и после этого медленно добавляли метанол (250 мл). Смесь концентрировали под вакуумом и остаток растворяли в кипящем ацетонитриле (1,3 л), фильтровали в горячем состоянии и затем охлаждали до комнатной температуры. Кристаллы фильтровали, промывали ацетонитрилом и сушили под вакуумом для получения указанного в заголовке соединения (72,5 г, 196 мМ, выход 81%, 95% ee (энантиомерный избыток), 95% частоты по данным ВЭЖХ).
(b) 8-бензилокси-5-[(R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил]-1Н-хинолин-2-он
В продукт из этапа (b) (70,2 г, 189 мМ) добавляли N,N-диметилформамид (260 мл) и смесь охлаждали в водяной бане в атмосфере азота. В течение 5 мин добавляли 2,6-лутидин (40,3 г, 376 мМ) и затем медленно добавляли трет-бутилдиметилсилил трифторметансульфонат (99,8 г, 378 мМ), при этом поддерживали температуру ниже 20°C. Смесь нагревали до комнатной температуры в течение 45 мин. В смесь по каплям добавляли метанол (45 мл) в течение 10 мин и затем смесь разделяли между слоями метилацетат/циклогексан (1:1, 500 мл) и вода/рассол (1:1, 500 мл). Органические части промывали более двух раз смесью вода/рассол (1:1, по 500 мл каждый). Объединенные органические части выпаривали при пониженном давлении, получая светло-желтое масло. Две отдельные части циклогексана (400 мл) добавляли в масло и продолжали отгонку до образования густой белой суспензии. В суспензию добавляли циклогексан (300 мл) и полученные белые кристаллы фильтровали, промывали циклогексаном (300 мл) и сушили при пониженном давлении, получая указанное в заголовке соединение (75,4 г, 151 мМ, выход 80%, 98,6% ee).
Процедура получения 9A
8-бензилокси-5-[(R)-2-(N-бензиламино)-1-(трет-бутилдиметилсиланилокси)этил]-1Н-хинолин-2-он
Перемешиваемый раствор продукта из процедуры получения 8 (1,00 г, 2,05 мМ) и бензиламина (493 мкл, 4,51 мМ) в ДМСО (1,7 мл) нагревали при 105°C в течение 4 ч. Реакционную смесь охлаждали и затем разбавляли EtOAc (10 мл), органический слой промывали насыщенным водным раствором хлорида аммония (5 мл) и 1 н. раствором гидроксида натрия (5 мл), сушили (MgSO4) и растворитель удаляли при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией (50% EtOAc/гексаны), получая указанное в заголовке соединение (700 мг, 67%). MS m/z: [M+H4] вычисленный для C31H38N2O3Si 515,27; найденный 515,5.
Процедура получения 9B
8-бензилокси-5-[(R)-2-(N-бензиламино)-1-(трет-бутилдиметилсиланилокси)этил]-1Н-хинолин-2-он
В 500 мл трехгорлую круглодонную колбу добавляли 8-бензилокси-5-[(R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил]-1Н-хинолин-2-он (43 г, 0,124 М, примерно 95% хиральной чистоты), 1-метил-2-пирролидинон (210 мл) и бензиламин (28,3 г, 0,37 М). Полученную смесь продували азотом и затем перемешивали при 90°C в течение 6 часов. Затем смесь охлаждали до комнатной температуры, добавляли воду (300 мл) и этилацетат (300 мл). Слои разделяли и органический слой промывали водой (200 мл), смесью 1:1 воды и насыщенного водного раствора хлорида натрия (200 мл) и водой (200 мл). Затем органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде оранжевого масла.
К оранжевому маслу добавляли гептан (200 мл) и этилацетат (200 мл) и полученную смесь нагревали до 65°С для получения прозрачного раствора. Этот раствор охлаждали до комнатной температуры и отстаивали в течение ночи (примерно 16 часов), во время которой образовывался преципитат.Преципитат собирали фильтрованием, получая стереохимически-нечистое указанное в заголовке соединение (8,85 г, 79,6% ee). Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (38,6 г, 99,4% ee). Это вещество объединяли с ранее полученными партиями вещества (19,2 г, 99,5% ee) и добавляли гептан (250 мл) и этилацетат (100 мл). Эту смесь нагревали до 80°C (от мутного до прозрачного раствора), затем охлаждали до комнатной температуры и отстаивали в течение ночи. Полученный преципитат собирали фильтрацией для получения указанного в заголовке соединения в виде белого твердого вещества (36,8 г, 98,4% ee, 99,9% химической чистоты). Фильтрат концентрировали при пониженном давлении и остаток растворяли в гептане (100 мл). Полученное твердое вещество собирали, получая указанное в заголовке соединение в виде желто-коричневого твердого вещества (24 г, 100% хиральной чистоты, 95% химическая чистота).
Процедура получения 10А
5-[(R)-2-амино-1-(трет-бутилдиметилсиланилокси)этил]-8-гидрокси-1Н-хинолин-2-он
Насыщенный раствор продукта из процедуры получения 9A (3,16 г, 6,15 мМ) и палладий (10 вес.% (сухой вес) на активированном угле) (1,58 г) в этаноле (62 мл) помещали в атмосферу водорода на 24 ч. Реакционную смесь фильтровали через целит, промывали метанолом (15 мл) и затем удаляли растворитель при пониженном давлении, получая указанное в заголовке соединение в виде твердого вещества (1,52 г, 4,55 мМ, 74%).
Процедура получения 10B
5-[(R)-2-амино-1-(трет-бутилдиметилсиланилокси)этил]-8-гидрокси-1H-хинолин-2-он соль уксусной кислоты
8-бензилокси-5-[(R)-2-(N-бензиламино)-1-(трет-бутилдиметилсиланилокси)этил]-1Н-хинолин-2-он (100 г, 194 мМ) и уксусную кислоту (17,5 мл, 291 мМ) растворяли в метаноле (1 л). Прозрачный раствор продували азотом и затем добавляли гидроксид палладия на углероде (20 г, 20 вес.% Pd (сухой вес), влажность (примерно 50% воды)). Через перемешиваемый раствор пропускали пузыри газообразного водорода при комнатной температуре в течение 6 часов, в течение которых образовывалась густая суспензия. Затем реакционную смесь продували азотом и добавляли метанол (1 л). Реакционную смесь перемешивали в течение 30 минут (до растворения продукта) и затем смесь фильтровали через слой целита. Фильтрат концентрировали при пониженном давлении до объема примерно 500 мл и к полученной суспензии добавляли этанол (500 мл). Полученную смесь снова концентрировали при пониженном давлении до объема примерно 500 мл, полученный преципитат собирали фильтрованием и сушили для получения указанного в заголовке соединения в виде желто-белого твердого вещества (65 г, выход 85%, чистота>98%).
Процедура получения 11
1-[2-(4-{[(R)-2-(трет-бутилдиметилсиланилокси)-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-2-хлор-5-метокси-фенилкарбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В продукт из процедуры получения 6 в смеси дихлорметан (0,5 мл) и метанол (0,5 мл) добавляли продукт из процедуры получения 10A (124,1 мг, 3,1 мМ) и полученную смесь перемешивали при комнатной температуре в течение 1,5 ч. Добавляли триацетоксиборгидрид натрия (190,7 мг, 0,9 мМ) и полученную смесь перемешивали при комнатной температуре в течение 15 ч. Реакцию гасили добавлением воды (примерно 0,2 мл) и смесь концентрировали под вакуумом, получая указанное в заголовке соединение, которое использовали дальше без дополнительной очистки. MS m/z 854,5 (M+H, 853,36 ожидаемый для C46H56ClN5O7Si).
Процедура получения 12
1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В суспензию продукта из процедуры получения 11 в дихлорметане (1,0 мл, 0,3 M) добавляли триэтиламина тригидрофторид (245 мкл, 1,5 мМ). Эту смесь перемешивали при комнатной температуре в течение 45 ч, затем смесь концентрировали под вакуумом. Остаток растворяли в смеси DMF (0,5 мл), ацетонитрил/вода (1:1, с 0,1% TFA, 0,6 мл), TFA (0,3 мл) и ацетонитрила (примерно 1 мл), эту смесь очищали препаративным разделением методом обращенно-фазовой ВЭЖХ (градиент: 2-50% ацетонитрила в воде с 0,05% TFA). Соответствующие фракции собирали, объединяли и лиофилизировали, получая дитрифторацетат соли указанного в заголовке соединения (100 мг, выход 34%, 98,7% чистоты по данным ВЭЖХ) в виде грязно-белого твердого вещества. MS m/z 740,5 (M+H, 739,28, ожидаемый для C40H42ClN5O7).
Пример 1
Соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты
Раствор дигидрата 1,2-этандисульфоновой кислоты (3,8 мг, 0,02 мМ) в этаноле (0,2 мл) медленно добавляли в раствор продукта процедуры получения 12 (14,3 мг, 0,02 мМ) в смесь 64:1 по весу изопропанола и дихлорметана (1 мл). Полученный раствор нагревали при 45°C-50°C в течение примерно 30 минут. Затем смесь медленно охлаждали до комнатной температуры, при которой раствор слегка мутнел. Раствор отстаивали при температуре окружающей среды в течение ночи в слабом потоке азота. Полученный преципитат охлаждали фильтрованием и сушили, получая указанное в заголовке соединение в виде белого кристаллического вещества (13 мг, выход 72%).
Пример 2
Соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-карбаминовой кислоты
Раствор 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты (26,8 мг, 0,0362 мМ) получали в этаноле (5,36 мл) и перемешивали при комнатной температуре до полного растворения (5 мин). Раствор дигидрата 1,2-этандисульфоновой кислоты (8,2 мг, 0,0362 мМ) в этаноле (0,2 мл) медленно добавляли в первый раствор в течение примерно одной минуты. Полученную суспензию перемешивали в течение пяти минут, затем разделяли при помощи фильтрования в атмосфере азота. Полученный преципитат сушили, получая указанное в заголовке соединение в виде белого твердого вещества (28,5 мг, выход 85%).
Пример 3
Соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты
1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты (5 г, 6,75 мМ, чистота>99%) растворяли в изопропаноле (100 мл), с последующим добавлением дигидрата этандисульфоновой кислоты (1,525 мг, 6,75 мМ), растворенного в воде (20 мл). Полученную суспензию перемешивали при комнатной температуре в течение 1 часа и затем в течение ночи при температуре ~30°C. Указанное в заголовке соединение (6,0 г) выделяли в виде белого порошка. Продукт нагревали в 20% воды в изопропаноле (100 мл) при 30°C в течение 48 часов. После охлаждения до комнатной температуры полученный преципитат выделяли фильтрованием и сушили на воздухе в течение 2 часов до получения указанного в заголовке соединения (5,4 г).
Процедура получения 13
Метил-4-акрилоиламино-5-хлор-2-метоксибензоат
В однолитровую трехгорлую круглодонную колбу, снабженную мешалкой, термометром и воронкой для добавления, добавляли метил-4-амино-5-хлор-2-метоксибензоат (44,2 г, 200 мМ), дихлорметан (500 мл) и диизопропилэтиламин (104,5 мл, 600 мМ). Полученную смесь перемешивали при комнатной температуре до растворения ингредиентов, затем смесь охлаждали до 0°C. Затем добавляли по каплям акрилоилхлорид (16,25 мл, 200 мМ), при этом поддерживали температуру внутри реакционной смеси ниже 10°C. Общее время, в течение которого происходило добавление, составляло 30 мин. Затем реакционную смесь медленно нагревали от 0°C до комнатной температуры в течение примерно 2 часов. После этого добавляли насыщенный водный раствор бикарбоната натрия (200 мл) и дихлорметана (200 мл) и смесь перемешивали в течение 15 мин, затем слои разделяли. Слой дихлорметана промывали 1 M раствором соляной кислоты (200 мл), затем концентрировали при пониженном давлении до примерно одной третьей от ее первоначального объема, получая густую суспензию. Суспензию фильтровали, осадок на фильтре промывали дихлорметаном (100 мл) и сушили для получения указанного в заголовке соединения в виде грязно-белого твердого вещества (36 г, выход 67%, >98% чистоты по данным ВЭЖХ).
Процедура получения 14
Метил-4-{3-[4-(бифенил-2-илкарбамоилокси)пиперидин-1-ил]пропиониламино}-5-хлор-2-метоксибензоат
В однолитровую трехгорлую круглодонную колбу, снабженную мешалкой, термометром и обратным холодильником, добавляли пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты (36,3 г, 122 мМ), дихлорметан (500 мл) и изопропанол (100 мл). Полученную смесь перемешивали при комнатной температуре до растворения ингредиентов и затем добавляли продукт из процедуры получения 10 (30 г, 111,5 мМ). Перемешивание продолжали при комнатной температуре до растворения ингредиентов и затем смесь кипятили с обратным холодильником (50°C-55°C) в течение 18 часов. После этого полученную смесь охлаждали до комнатной температуры и добавляли этанол (200 мл). Смесь концентрировали при пониженном давлении до получения густой суспензии объемом примерно 150 мл. Суспензию фильтровали, затем осадок на фильтре промывали этанолом (50 мл) и сушили, получая указанное в заголовке соединение в виде белого твердого вещества (58 г, выход 92%, 99,5% чистоты по данным ВЭЖХ).
Процедура получения 15
1-[2-(2-хлор-4-гидроксиметил-5-метокси-фенилкарбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В двухлитровую круглодонную колбу добавляли продукт из процедуры получения 14 (40 г, 70,8 мМ) и THF (400 мл). Полученную смесь перемешивали при комнатной температуре до растворения ингредиентов и колбу продували азотом в течение 5 минут. Затем смесь охлаждали до 0°C (внутренняя температура) и по каплям добавляли 1 M раствор алюмогидрата лития в THF (106 мл, 106 мМ) через дополнительную воронку, при этом поддерживая внутреннюю температуру реакционной смеси ниже 10°С. Общее дополнительное время составляло 40 мин. Затем реакционную смесь перемешивали в течение 1 часа при температуре 0°C и после этого добавляли 1 M раствор гидроксида натрия (200 мл), при этом поддерживая внутреннюю температуру реакционной смеси ниже 15°C. Затем слои разделяли и слой THF промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде белого твердого вещества (38 г, выход 100%, 94% чистоты по данным ВЭЖХ).
Процедура получения 16
1-[2-(2-хлор-4-формил-5-метоксифенил-карбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В однолитровую круглодонную колбу добавляли продукт из процедуры получения 15 (28 г, 52 мМ) и дихлорметан (500 мл). Полученную реакционную смесь перемешивали при комнатной температуре до растворения ингредиентов и затем добавляли активированный оксид магния (IV) (45 г, 520 мМ). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 12 часов и затем фильтровали через слой целита. После этого смесь концентрировали при пониженном давлении и остаток сушили в течение ночи под вакуумом, получая указанное в заголовке соединение в виде желтого твердого вещества (26 г, выход 93%, примерно 93% чистоты по данным ВЭЖХ).
Процедура получения 17
1-[2-(4-{[(R)-2-(трет-бутилдиметилсиланилокси)-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-2-хлор-5-метокси-фенилкарбамоил)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В 500 мл круглодонную колбу добавляли продукт из процедуры получения 16 (6 г, 11,2 мМ) и дихлорметан (50 мл). Полученную смесь перемешивали при комнатной температуре до растворения ингредиентов и затем добавляли продукт из процедуры получения 7 (6 г, 15,0 мМ) и сухой метанол (50 мл). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 2 часов (до получения прозрачного раствора желто-оранжевого цвета) и затем смесь охлаждали до 0-5°C. В течение 10 минут добавляли частями твердый триацетоксиборгидрид натрия (7,2 г, 34 мМ) и затем реакционную смесь медленно нагревали от 0°C до комнатной температуры в течение 2 часов. После этого смесь охлаждали до 0°C и добавляли 1 M водный раствор гидроксида натрия (50 мл) и дихлорметан (150 мл). Смесь тщательно перемешивали и затем слои разделяли. Органический слой промывали насыщенным водным раствором хлорида натрия (50 мл), фильтровали, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде желтого твердого вещества (10,1 г, выход 100%, 87% чистоты по данным ВЭЖХ).
Пример 4
Соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты
В 50 мл круглодонную колбу добавляли продукт из процедуры получения 17 (2,0 г, 2,5 мМ) и дихлорметан (10 мл). Полученную смесь перемешивали при комнатной температуре до растворения ингредиентов, и затем добавляли триэтиламина тригидрофторид (1,2 мл, 7,5 мМ), и полученную смесь перемешивали при 25°C в течение 20 часов. Затем добавляли раствор дигидрата 1,2-этандисульфоновой кислоты (0,56 г, 2,5 мМ) в метаноле (10 мл) и эту смесь перемешивали при 30°C в течение 2 часов, в течение которых образовывалась белая густая суспензия. Суспензию медленно фильтровали и осадок на фильтре промывали метанолом (10 мл), в течение 2 часов сушили на воздухе и затем сушили в течение ночи под вакуумом, получая указанное в заголовке соединение в виде микронизированного белого порошка (1,5 г, >98% чистоты по данным ВЭЖХ).
Пример 5
Очистка соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты
К соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбомоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, полученной, как описано в примере 4 (80 г), добавляли раствор 20% воды в изопропаноле по объему (800 мл). Полученную суспензию отстаивали в течение ночи при комнатной температуре и затем фильтровали для получения указанного в заголовке соединения, имеющего улучшенную степень кристалличности и более высокую чистоту (74 г).
Процедура получения 18
Затравочные кристаллы соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты
Стадия (a)
Соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, полученную в примере 4 (100 мг), растворяли в метаноле (20 мл), содержащем 13% воды, при ~60°C. Полученный прозрачный раствор охлаждали до комнатной температуры в закрытом контейнере. После 48 часов полученные пластинчатые кристаллы отделяли фильтрованием.
Стадия (b)
Соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, полученную в примере 4 (1,0 г), растворяли в метаноле (100 мл), содержащем 15% воды, при 60-65°C. Прозрачный перемешанный раствор охлаждали до 30°C и затем добавляли кристаллический продукт стадии (a) (4,2 мг). Раствор охлаждали до 20°C и перемешивали в течение 2 часов. Полученный преципитат отделяли фильтрованием и сушили на воздухе в течение 1 часа, получая указанное в заголовке соединение (680 мг).
Стадия (c)
Процедуру стадии (b) повторяли, используя вместо продукта стадии (a) продукт стадии (b) (20 мг), для получения указанного в заголовке соединения (690 мг).
Стадия (d)
Соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, полученную в примере 4 (10 г), растворяли в метаноле (1 л), содержащем 15% воды, при 60-65°C. Прозрачный перемешанный раствор охлаждали до 30°C и затем добавляли кристаллический продукт из стадии (c) (4,2 мг). Раствор охлаждали до 20°C и перемешивали в течение 18 часов. Полученный преципитат отделяли фильтрованием и сушили на воздухе в течение 2 часов, получая указанное в заголовке соединение (5,5 г).
Пример 6
Перекристаллизация соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты
Использование затравочных кристаллов
В 12-литровую круглодонную колбу добавляли продукт примера 5 (60 г, 64,5 мМ), воду (0,9 л) и метанол (5,1 л). Полученную смесь нагревали с 25°C до 61-65°C, помешивая, до растворения ингредиентов и дополнительно перемешивали в течение 20 минут при температуре 60-65°C. Смесь остужали до 30°C и затем добавляли продукт из процедуры получения 18 (2 г, 2,15 мМ). Эту смесь медленно охлаждали до 20°C, полученную суспензию перемешивали в течение 2 часов при 30°C. Продукт фильтровали с метанолом (500 мл) и сушили на воздухе в течение 2 часов и затем под вакуумом при температуре 25-30°C в течение 18 часов до получения указанного в заголовке соединения (43 г, выход 72%, 99,2% чистоты).
Пример 7
Термический анализ
Выполняли дифференциальную сканирующую калориметрию (DSC), используя модуль TA Instruments Model Q-10 с контроллером Thermal Analyst. Данные собирали и анализировали, используя программное обеспечение TA Instruments Thermal Solutions. Образец, весом примерно 1 мг, взвешивали с высокой точностью в алюминиевом тигле с крышкой. Образец оценивали, используя линейное нагревание со скоростью 5°C/мин от температуры окружающей среды до температуры примерно 300°C. Во время использования DSC ячейку продували сухим азотом. Репрезентативный DSC график для образца кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты примера 6 показан на фиг.1, и репрезентативный DSC график для образца примера 2 показан на фиг.2.
Термогравиметрический анализ (TGA) выполняли, используя модуль TA Instruments Model Q-50 с высокой разрешающей способностью. Данные собирали и анализировали, используя программное обеспечение TA Instruments Thermal Solutions. Образцы, весящие примерно 10 мг, помещали в платиновую чашку и сканировали с высоким разрешением по скорости нагревания от температуры окружающей среды до 300°C. Камеры взвешивания и отжига во время использования продували азотом. Репрезентативный TGA график для образца кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты примера 6 показан на фиг.1. DSC график показывает, что соль 1,2-этандисульфоновой кислоты настоящего изобретения имеет прекрасную термическую стабильность с точками плавления при температуре примерно 239°C и примерно 219°C соответственно и термически не разлагается при температуре ниже примерно 200°C.
Пример 8
Дифракция рентгеновских лучей на порошке
Порошковые рентгенограммы получали, используя модель X'TRA рентгеновского дифрактометра ARL фирмы Thermo (Thermo ARL SA, Switzerland), используя излучение Cu K α при 1,542 Å (45 кВ, 40 мА), с Si(Li) твердотельным детектором. Обычно анализ проводили со скоростью сканирования 2°/мин с шагом 0,03° на точку в диапазоне от 2 до 30° по углу 2 тета. Образцы порошка, либо в том виде, в котором они получены, либо в микронизированном виде, аккуратно упаковывали в сделанную по заказу вставку небольшого объема, разработанную таким образом, чтобы она помещалась в тигле прибора с верхней загрузкой для анализа образца. Инструмент еженедельно калибровали на стандарте металлического кремния, в пределах ±0,02° по углу 2 тета. Характерные порошковые рентгенограммы для образца кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты примера 6 показаны на фиг.3, и порошковая рентгенограмма для образца примера 2 показана на фиг.4.
Пример 9
Инфракрасный анализ
Спектр инфракрасного (ИК) поглощения определяли в области частот от 4000 до 675 см-1, используя спектрометр Avatar 360 FT-IR, оборудованный держателем для образцов Nicolet с ослабленным полным внутренним отражением (ATR). Характерный ИК спектр поглощения для образца кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты для примера 6 имел значимые полосы поглощения при 704 ±1, 748 ±1, 768 ±1, 841 ±1, 900 ±1, 1055 ±1, 1104 ±1, 1166 ±1, 1218 ±1, 1294 ±1, 1408 ±1, 1522 ±1, 1609 ±1, 1655 ±1 и 1701 ±1, как показано на фиг.5.
Пример 10
Оценка динамики поглощения влаги
Оценку динамики поглощения влаги (DMS) (известное как профиль сорбции-десорбции влаги) выполняли для измельченного вручную образца кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты процедуры получения 18 при помощи VTI атмосферных микровесов, система SGA-100 (VTI Corp., Hialeah, FL 33016). Использовали образцы весом приблизительно 10 мг и в начале анализа устанавливали влажность, равную значению влажности окружающей среды. Обычный DMS анализ состоял из трех сканирований: 2% относительная влажность (RH) окружающей среды, 2% RH до 90% RH, 90% RH до 5% RH со скоростью сканирования 5% RH/стадия. Массу измеряли каждые две минуты и RH изменяли до следующего значения (+/-5% RH), если масса образца была стабильной в пределах 0,01% для 5 последующих точек. Характерный DMS график показан на фиг.6.
DMS график показывает, что соль 1,2-этандисульфоновой кислоты настоящего изобретения имеет взаимообратный профиль сорбции/десорбции со средней (<9%) гигроскопичностью. Вес соли увеличивается менее чем на 2,5% в диапазоне влажности от 40% RH до 75% RH. Взаимообратный профиль сорбции/десорбции влаги демонстрирует, что кристаллическая соль настоящего изобретения обладает допустимой гигроскопичностью и не расплывается.
Пример 11
Элементный анализ и отношение противоиона
Нижеприведенные проценты содержания углерода, водорода, азота и серы в образце кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)-этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты примера 6 определяли анализом сжигания, используя элементный анализатор Flash EA 1112 (CE Elantech, Lakewood, NJ): углерод 52,95%, водород 5,43%, азот 6,83% и сера 6,87%. Весовой процент 1,2-этандисульфоновой кислоты в кристаллическом образце, вычисленный из измеренного весового процента серы, составляет 20,4%, который равен ее теоретическому весовому проценту 20,4%, давая отношение противоионов 1:1.
Препарат A
Клеточная культура и мембранный препарат из клеток, экспрессирующих человеческие β1, β2 или β3-адренергические рецепторы
Клеточные линии яичников китайского хомячка (CHO), стабильно экспрессирующие клонированные человеческие β1, β2 или β3-адренергические рецепторы соответственно, выращивали до монослоя из тесно прилегающих друг к другу клеток в Hams F-12 среде с 10% FBS в присутствии 500 мкг/мл генетицина. Монослой клеток отслаивали при помощи 2 мМ ЭДТА в PBS. Клетки осаждали центрифугированием при 1000 об/мин и клеточные осадки либо хранили в замороженном виде при -80°C, либо из них сразу готовили мембранные препараты для использования. Для получения мембран, экспрессирующих β1 и β2-рецепторы, клеточные осадки ресуспендировали в лизисном буфере (10 мМ HEPES/HCl, 10 мМ ЭДТА, pH 7,4 при 4°C) и гомогенизировали, используя плотно пригнанный стеклянный гомогенизатор Dounce (30 встряхиваний) на льду. Для более восприимчивых к протеазам мембран, экспрессирующих β3-рецепторы, клеточные осадки гомогенизировали в лизисном буфере (10 мМ трис/HCl, pH 7,4), дополненном одной таблеткой "таблеток полного коктейля ингибиторов протеаз с 2 мМ ЭДТА" на 50 мл буфера (Roche Catalog No. 1697498, Roche Molecular Biochemicals, Indianapolis, IN). Гомогенаты центрифугировали при 20000g и полученный осадок промывали один раз лизисным буфером, ресуспендируя и центрифугируя, как описано выше. Затем конечный осадок ресуспендировали в охлажденном льдом буфере для анализа связывания (75 мМ трис/HCl, pH 7,4, 12,5 мМ MgCl2, 1 мМ ЭДТА). Концентрацию белка мембранной суспензии определяли способами, описанными у Lowry et al., 1951, Journal of Biological Chemistry, 193, 265; и Bradford, Analytical Biochemistry, 1976, 72, 248-54. Все мембраны хранили в замороженном виде в аликвотах при температуре -80°С или сразу использовали.
Препарат B
Клеточная культура и мембранный препарат из клеток, экспрессирующих человеческие M1, M2, M3 и M4-мускариновые рецепторы
CHO клеточные линии, стабильно экспрессирующие клонированные подтипы человеческих hM1, hM2, hM3 и hM4-мускариновых рецепторов соответственно, выращивали до монослоя из тесно прилегающих друг к другу клеток в HAMs F-12 среде, дополненной 10% FBS и 250 мкг/мл генетицина. Клетки выращивали в 5% CO2 инкубаторе при 37°C и отслаивали 2 мМ ЭДТА в dPBS. Клетки собирали 5-минутным центрифугированием при 650g и клеточные осадки либо хранили замороженными при -80°C, либо сразу готовили мембранные препараты для использования. Для мембранных препаратов клеточные осадки ресуспендировали в лизисном буфере и гомогенизировали тканевым дезинтегратором Polytron PT-2100 (Kinematica AG; 20 секунд x 2 импульса). Неочищенные мембраны центрифугировали при 40000g в течение 15 минут при 4°C. Затем мембранный осадок ресуспендировали буфером для ресуспендирования и снова гомогенизировали тканевым дезинтегратором Polytron. Концентрацию белка мембранной суспензии определяли способом, описанным у Lowry et al., 1951, Journal of Biochemistry, 193, 265. Все мембраны хранили в замороженном виде в аликвотах при -80°C или сразу использовали. Аликвоты полученных мембран hM5-рецепторов очищали непосредственно из Perkin Elmer и хранили при -80° до использования.
Процедура А лабораторного теста
Анализ связывания радиолигандов для человеческих β1, β2 или β3-адренергических рецепторов
Анализ связывания проводили в 96-луночной микротитровальной планшете общим анализируемым объемом 100 мкл с 10-15 мкг мембранного белка, содержащего человеческие β1, β2 или β3-адренергические рецепторы в буфере для анализа (75 мМ трис/HCl, pH 7,4 при 25°C, 12,5 мМ MgCl2, 1 мМ ЭДТА, 0,2% BSA). Насыщение связывания изучали для определения значений Kd радиолиганда там, где он был, используя [3H]-дигидроалпренолол (NET-720, 100 кюри/мМ, PerkinElmer Life Sciences Inc., Boston, MA) для β1 и β2-рецепторов и [125I]-(-)-йодоцианопиндолол (NEX-189, 220 кюри/мМ, PerkinElmer Life Sciences Inc., Boston, MA) при 10 или 11 различных концентрациях, лежащих в пределах от 0,01 нМ до 20 нМ. Анализ смещения для определения значений Ki тестируемых соединений проводили, используя [3H]- дигидроалпренолол при 1 нМ и [125I]-(-)-йодоцианопиндолол при 0,5 нМ для 10 или 11 различных концентраций тестируемого соединения, находящихся в пределах от 10 пМ до 10 мкМ. Неспецифическое связывание определяли в присутствии 10 мкМ пропранолола. Пробы инкубировали в течение 1 часа при 37°C и затем реакции связывания завершали быстрым фильтрованием через GF/B для β1 и β2-рецепторов или GF/C стекловолоконные фильтровальные пластины для β3-рецепторов (Packard BioScience Co., Meriden, CT), предварительно пропитанные 0,3% полиэтиленимином. Фильтровальные пластины промывали три раза буфером для фильтрования (75 мМ трис/HCl, pH 7,4 при 4°, 12,5 мМ MgCl2, 1 мМ ЭДТА) для удаления несвязанной радиоактивности. Затем пластины сушили на воздухе и добавляли 50 мкл Microscint-20 сцинтилляционной жидкости (Packard BioScience Co., Meriden, CT), результаты с планшетов подсчитывали жидкостным сцинтилляционным счетчиком Packard Topcount (Packard BioScience Co., Meriden, CT). Данные связывания анализировали при помощи нелинейного регрессионного анализа, используя пакет программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и используя конкурентную модель с одним участком. Минимальное значение на кривой было зафиксировано для неспецифического связывания, как было определено в присутствии 10 мкМ пропранолола. Значения Ki для тестируемых соединений вычисляли из наблюдаемых значений IC50 и значения Kd радиолиганда, используя уравнение Cheng-Prusoff (Cheng Y, and Prusoff WH., Biochemical Pharmacology, 1973, 22, 23, 3099-108).
В этом анализе более низкое значение Ki указывает, что тестируемое соединение имеет высокую аффинность связывания для тестируемого рецептора. В этом анализе при тестировании было обнаружено, что соединение формулы I имеет значение Ki для человеческого β2-адренергического рецептора меньше чем 10 нМ.
Процедура В лабораторного теста
Анализ связывания радиолигандов для мускариновых рецепторов
Анализ связывания радиолигандов для клонированных человеческих мускариновых рецепторов проводили в 96-луночных микротитровальных планшетах общим анализируемым объемом 100 мкл. CHO клеточные мембраны, стабильно экспрессирующие либо hM1, hM2, hM3, hM4 либо hM5 подтип мускариновых рецепторов, разбавляли в буфере для анализа при следующих концентрациях специфических целевых белков (мкг/лунку): 10 мкг для hM1, 10-15 мкг для hM2, 10-20 мкг для hM3, 10-20 мкг для hM4 и 10-12 мкг для hM5 для получения аналогичных сигналов (cpm). Мембраны быстро гомогенизировали, используя тканевый дезинтегратор Polytron (10 секунд) перед добавлением в планшеты для анализа. Изучение насыщения связывания для определения значений Kd радиолигандов выполняли, используя L-[N-метил-3H]скополаминметилхлорид ([3H]-ЯМР) (TRK666, 84,0 кюри/ммоль, Amersham Pharmacia Biotech, Buckinghamshire, England) в концентрациях, находящихся в пределах от 0,001 нМ до 20 нМ. Анализ сдвигов для определения значений Ki тестируемых соединений выполняли при помощи [3H]-ЯМР при 1 нМ и одиннадцати различных концентраций тестируемых соединений. Тестируемые соединения сначала растворяли в концентрации 400 мкМ в буфере для разбавления и затем проводили последовательное разбавление 5x, используя буфер для разбавления до конечных концентраций, находящихся в области от 10 пМ до 100 мкМ. Порядок добавления и объемы для тестируемых планшет были следующими: 25 мкл радиолиганда, 25 мкл разбавленного тестируемого соединения и 50 мкл мембран. Тестируемые планшеты инкубировали в течение 60 минут при 37°C. Реакции связывания завершали быстрым фильтрованием через GF/B стекловолоконные фильтровальные пластины (PerkinElmer Inc., Wellesley, MA), предварительно обработанные 1% BSA. Фильтровальные пластины промывали три раза буфером для промывки (10 мМ HEPES) для удаления несвязанной радиоактивности. Затем планшеты сушили на воздухе и в каждую лунку добавляли 50 мкл Microscint-20 сцинтилляционной жидкости (PerkinElmer Inc., Wellesley, MA). Затем результаты с планшетов подсчитывали на PerkinElmer Topcount жидкостном сцинтилляционном счетчике (PerkinElmer Inc., Wellesley, MA). Данные связывания анализировали при помощи нелинейного регрессионного анализа, используя пакет программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и одностороннюю модель сравнения. Значения Ki для тестируемых соединений вычисляли из наблюдаемых значений IC50 и значения Kd радиолиганда, используя уравнение Cheng-Prusoff (Cheng Y; Prusoff WH. (1973) Biochemical Pharmacology, 22(23):3099-108). Значения Ki были преобразованы в значения pKi для определения среднего геометрического и 95% доверительных интервалов. Затем проводили обратное преобразование этих суммарных статистик в значения Ki для опубликованных данных.
В этом анализе более низкое значение Ki указывает на то, что тестируемое соединение имеет более высокую аффинность связывания для тестируемых рецепторов. При тестировании в этом анализе было обнаружено, что соединение формулы I для человеческих M2 и M3-мускариновых рецепторов имеет значение Ki, меньшее чем 10 нМ.
Процедура С лабораторного теста
Flashplate анализ клеточной цАМФ в CHO клеточных линиях, экспрессирующих гетерологические человеческие β1, β2 или β3-адренергические рецепторы
Анализ цАМФ проводили в формате радиоиммунного анализа, используя Flashplate систему анализа активации аденилатциклазы с [125I]-цАМФ (NEN SMP004, PerkinElmer Life Sciences Inc., Boston, MA), согласно инструкции производителя. Для определения агонистической силы (ЕС50) β-рецептора СНО-К1 клеточные линии, стабильно экспрессирующие клонированные человеческие β1, β2 или β3-адренергические рецепторы, выращивали до монослоя из тесно прилегающих друг к другу клеток в HAMs F-12 среде, дополненной 10% FBS и генетицином (250 мкг/мл). Клетки отслаивали при помощи PBS и разъединяли в PBS (забуференный фосфатом физиологический раствор Дульбекко, без CaCl2 и MgCl2), содержащий 2 мМ ЭДТА или раствор трипсин-ЭДТА (0,05% трипсин/0,53 мМ ЭДТА). После подсчета клеток в счетчике клеточных культур в цитомере Coulter клетки осаждали центрифугированием при 1000 об/мин и ресуспендировали в буфере для стимуляции, содержащем IBMX (PerkinElrner Kit), предварительно нагретом до комнатной температуры, в концентрациях от 1,6·106 до 2,8·106 клеток/мл. В анализе использовали примерно 60000-80000 клеток на лунку. Тестируемые соединения (10 мМ в ДМСО) разбавляли PBS, содержащем 0,1% BSA в Beckman Biomek-2000, и тестировали в 11 различных концентрациях, находящихся в пределах от 100 мкМ до 1 пМ. Реакционные смеси инкубировали в течение 10 мин при 37°C и реакцию останавливали добавлением 100 мкл холодного буфера для детектирования, содержащего [125I]-цАМФ (NEN SMP004, PerkinElmer Life Sciences, Boston, MA). Количество продуцированного цАМФ (пМ/лунку) вычисляли на основании количества отсчетов, наблюдаемых для образцов, и цАМФ стандартов, как описано в инструкции производителя. Данные анализировали нелинейным регрессионным анализом, используя пакет программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и сигмоидальное уравнение. Для вычисления значений EC50 использовали уравнение Cheng-Prusoff (Cheng Y, and Prusoff WH., Biochemical Pharmacology, 1973, 22, 23, 3099-108).
В этом анализе более низкое значение EC50 указывает, что тестируемое соединение имеет более высокую функциональную активность в тестируемом рецепторе. При тестировании в этом анализе было обнаружено, что для человеческого β2-адренергического рецептора соединение формулы I имеет значение EC50 меньше чем 10 нМ.
Процедура D лабораторного теста
Функциональный анализ антагонизма для подтипов мускариновых рецепторов
A. Блокада связывания [35S]GTPγS, опосредованного агонистом
Функциональную силу тестируемого соединения определяли измерением способности соединения блокировать [35S]GTPγS связывание, стимулированное оксотреморином, в CHO-Kl клетках, экспрессирующих hM2-рецептор.
Во время использования замороженные мембраны размораживали и затем разбавляли буфером для анализа до конечной целевой концентрации ткани 5-10 мкг белка на лунку. Мембраны быстро гомогенизировали, используя тканевый дезинтегратор Polytron PT-2100, и затем добавляли в планшеты для анализа.
В каждом эксперименте определяли значение EC50 (эффективная концентрация для 90% максимального ответа) для стимуляции [35S]GTPγS связывания при помощи агониста, оксотреморина.
Для определения способности тестируемого соединения ингибировать [35S]GTPγS связывание, стимулируемое оксотреморином, в каждую лунку 96-луночной планшеты добавляли следующие ингредиенты: 25 мкл буфера для анализа с [35S]GTPγS (0,4 нМ), 25 мкл оксотреморина(EC90) и GDP (3 uM), 25 мкл разбавленного тестируемого соединения и 25 мкл клеточных мембран CHO, экспрессирующих hM2-рецептор. Затем планшеты для анализа инкубировали при 37°C в течение 60 минут. Планшеты для анализа фильтровали через GF/B фильтры, обработанные 1% BSA, используя 96-луночный харвестер PerkinElmer. Планшеты промывали охлажденным льдом буфером для промывки в течение 3x3 секунд и затем сушили на воздухе или под вакуумом. В каждую лунку добавляли сцинтилляционную жидкость Microscint-20 (50 мкл), каждую планшету плотно закрывали и подсчитывали радиоактивность на Topcounter (PerkinElmer). Данные анализировали при помощи нелинейного регрессионного анализа, используя пакет программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и уравнение нелинейной регрессии для конкурентного связывания на одном участке. Для вычисления Ki использовали уравнение Cheng-Prusoff, используя значения IC50 кривой концентрация-ответ для тестируемого соединения и концентрации оксотреморина в анализе, в виде KD и [L], концентрации лиганда соответственно.
В этом анализе более низкое значение Ki указывает, что тестируемое соединение имеет более высокую функциональную активность в тестируемом рецепторе. При тестировании в этом анализе было обнаружено, что соединение формулы I имеет значение Ki меньше чем примерно 10 нМ для блокады [35S]GTPγS связывания, стимулированного оксотреморином, в CHO-K1 клетках, экспрессирующих hM2-рецептор.
B. Блокада агонист-опосредованного высвобождения кальция при помощи FLIPR анализа
Подтипы (M1, M3 и M5-рецепторы) мускариновых рецепторов, которые соединяются с Gq протеинами, активируют фосфолипазный (PLC) путь при связывании агонистов с рецептором. В результате активированный PLC гидролизует фосфатилинозитол дифосфат (PIP2) до диацилглицерина (DAG) и фосфатидил-1,4,5-трифосфат (IP3), который в свою очередь генерирует высвобождение кальция из внутриклеточных депо, т.e. эндоплазматического и саркоплазмического ретикулума. FLIPR (Molecular Devices, Sunnyvale, CA) анализ использует такое увеличение внутриклеточного кальция, используя чувствительный к кальцию краситель (Fluo-4AM, Molecular Probes, Eugene, OR), который фруоресцирует, когда связывается со свободным кальцием. Такое событие флуоресценции измеряют в реальном времени при помощи FLlPR, который детектирует заряд при флуоресценции из монослоя клеток, клонированных человеческими M1 и M3-рецепторами и M5-рецептором шимпанзе. Сила антагониста может быть определена благодаря способности антагонистов ингибировать опосредованное агонистом увеличение внутриклеточного кальция.
Для FLIPR анализа стимуляции кальция CHO клетки, стабильно экспрессирующие hM1, hM3 и cM5-рецепторы, засевали в 96-луночные FLIPR планшеты за ночь до проведения анализа. Засеянные клетки промывали два раза Cellwash (MTX Labsystems, Inc.) с FLIPR буфером (10 мМ HEPES, pH 7,4, 2 мМ хлорида кальция, 2,5 мМ пробенецида в сбалансированном солевом растворе Хэнкса (HBSS) без кальция и магния) для удаления среды для выращивания, оставляя 50 мкл/лунку FLIPR буфера. Затем клетки инкубировали с 50 мкл/лунку 4 мкМ FLUO-4AM (был приготовлен 2X раствор) в течение 40 минут при 37°C, в присутствии 5% двуокиси углерода. После инкубации с краской клетки промывали два раза буфером FLIPR, сохраняя конечную концентрацию 50 мкл/лунку.
Для определения силы антагониста зависящую от дозы стимуляцию высвобождения внутриклеточного Ca2+для оксотреморина сначала определяли таким образом, чтобы можно было позже измерить силу антагониста относительно стимуляции оксотреморина при EC90 концентрации. Сначала клетки инкубировали с буфером для разбавления соединения в течение 20 минут, затем добавляли антагонист, используя FLIPR. Значение EC90 для оксотреморина получали согласно способу, подробно описанному в нижеприведенном разделе "FLIPR измерения и сокращение объема данных", совместно с формулой ECF=((F/100-F)^1/H)*EC50. Оксотреморин с концентрацией 3xECF готовили в планшетах для стимуляции, исходя из условия, чтобы в каждую лунку в планшетах для анализа ингибирования антагониста был добавлен оксотреморин в концентрации EC90.
Параметры, используемые для FLIPR, представляли собой: длительность экспозиции 0,4 секунды, мощность лазера 0,5 ватт, длина волны возбуждения 488 нм и длина волны излучения 550 нм. Была определена базовая линия путем изменения флуоресценции в течение 10 секунд перед добавлением антагониста. После стимуляции антагонистом FLIPR непрерывно измеряли изменение флуоресценции каждые 0,5-1 секунду в течение 1,5 минуты для регистрации максимального изменения флуоресценции.
Изменение флуоресценции для каждой лунки выражали в виде максимальной флуоресценции минус базовая линия флуоресценции. Необработанные данные анализировали в зависимости от логарифма концентрации лекарственного вещества, используя нелинейную регрессию при помощи GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и встроенную модель для сигмоидальной зависимости доза-ответ. Значения Ki антагониста определяли при помощи Prism, используя значение EC50 оксотреморина в качестве KD и EC90 оксотреморина для концентрации лиганда согласно уравнению Cheng-Prusoff (Cheng & Prusoff, 1973).
В этом анализе более низкие значения Ki указывают, что тестируемое соединение имеет более высокую функциональную активность в тестируемом рецепторе. При тестировании в этом анализе было обнаружено, что соединение формулы I имеет значение Ki меньше чем примерно 10 нМ для блокады высвобождения кальция, опосредованного агонистом, в CHO клетках, стабильно экспрессирующих hM1, hM3 и cM5-рецепторы.
Процедура Е анализа тестирования
Flashplate анализ клеточного цАМФ с линией клеток легочного эпителия, экспрессирующей эндогенный человеческий β2-адренергический рецептор
Для определения силы и эффективности агониста (характерные активности) в клеточной линии, экспрессирующей эндогенные уровни β2-адренергического рецептора, использовали линию клеток легочного эпителия человека (BEAS-2B) (ATCC CRL-9609, Американская коллекция типовых культур, Manassas, VA) (January B., et al., British Journal of Pharmacology, 1998, 123, 4, 701-11). Клетки росли до 75-90% монослоя из прилегающих друг к другу клеток в полнокомпонентной среде без сыворотки (LHC-9 MEDIUM, содержащей эпинефрин и ретиноидную кислоту, cat # 181-500, Biosource International, Camarillo, CA). За день до анализа среду меняли на LHC-8 (без эпинефрина или ретиноидной кислоты, cat # 141-500, Biosource International, Camarillo, CA).
Анализ цАМФ выполняли в формате радиоиммунного анализа, используя Flashplate систему анализа активации аденилатциклазы со [125I]-цАМФ (NEN SMP004, PerkinElmer Life Sciences Inc., Boston, MA), согласно инструкции производителя.
За день до анализа клетки промывали PBS, отделяли соскобом, используя 5 мМ ЭДТА в PBS, и подсчитывали. Клетки центрифугировали при 1000 об/мин и осадок ресуспендировали в буфере для стимуляции, предварительно нагретом до 37°C, до конечной концентрации 600000 клеток/мл. В этом анализе клетки использовали при конечной концентрации 100000-120000 клеток/лунку. Тестируемое соединение последовательно разбавляли в буфере для анализа (75 мМ трис/HCl, pH 7,4 при 25°C, 12,5 мМ MgCl2, 1 мМ ЭДТА, 0,2% BSA) в Beckman Biomek-2000. Анализ тестируемых соединений проводили для 11 различных концентраций, находящихся в пределах от 10 мкМ до 10 пМ. Реакционные смеси инкубировали в течение 10 мин при 37°С и останавливали добавлением 100 мкл охлажденного льдом буфера для детектирования. Планшеты плотно закрывали, инкубировали в течение ночи при 4°C и на следующее утро производили подсчет при помощи сцинтилляционного счетчика Topcount (Packard BioScience Co., Meriden, CT). Количество цАМФ, продуцированного на мл реакционной смеси, вычисляли на основании количества отсчетов, наблюдаемых для образцов, и цАМФ стандартов, как описано в инструкции производителя. Данные анализировали нелинейным регрессионным анализом, используя пакет программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и 4-параметрическую модель для сигмоидальной зависимости доза-ответ.
В этом анализе более низкие значения ЕС50 указывают, что тестируемое соединение имеет более высокую функциональную активность в тестируемом рецепторе. При тестировании в этом анализе было обнаружено, что для β2-адренергического рецептора соединение формулы I имеет значение ЕС50 меньше чем примерно 10 нМ.
Процедура F анализа тестирования
Продолжительность бронхопротекции при бронхопротекции, индуцируемой ацетилхолином или гистамином, в моделях морских свинок
Этот анализ in vivo использовали для оценки влияния на бронхопротекцию тестируемых соединений, демонстрирующих как антагонистическую активность к мускариновым рецепторам, так и агонистическую активность к β2-адренергическим рецепторам. Для выделения антагонистической мускариновой активности в модели бронхопротекции, индуцируемой ацетилхолином, животным вводили пропанолол, соединение, которое блокирует активность β2-рецептора, перед введением ацетилхолина. Продолжительность бронхопротекции в модели бронхопротекции, индуцируемой гистамином, отражает агонистическую активность к β2-адренергическому рецептору.
Группы морских свинок из 6 самцов (Duncan-Hartley (HsdPoc:DH) Harlan, Madison, WT), весящих от 250 до 350 г, идентифицировали при помощи ярлыков, размещенных на их клетках. Во время исследования животным предоставлялся свободный доступ к пище и воде.
Тестируемые соединения вводили путем ингаляции в течение 10 минут в дозирующую камеру, в которой находилось животное (R&S Molds, San Carlos, CA). Дозирующие камеры располагали таким образом, чтобы аэрозоль из центрального манифолда мог одновременно попадать в 6 отдельных камер. На морских свинок воздействовали аэрозолем тестируемого соединения или наполнителя (WFI). Эти аэрозоли получали из водных растворов, используя набор LC Star Nebulizer (Model 22F51, PARI Respiratory Equipment, Inc. Midlothian, VA), приводимый в действие смесью газов (CO2=5%, O2=21% и N2=74%) под давлением 22 фунта/кв.дюйм. Поток газа через распылитель при таком рабочем давлении составлял приблизительно 3 л/минуту. Получаемые аэрозоли вводились в камеры избыточным давлением. Во время подачи растворов в виде аэрозолей разбавление воздухом не использовалось. В течение 10 минут распыления распылялось примерно 1,8 мл раствора. Это значение измеряли гравиметрически путем сравнения весов заполненных распылителей до и после распыления.
Бронхопротективное влияние тестируемых соединений, вводимых посредством ингаляции, оценивали, используя плетизмографию всего тела через 1,5, 24, 48 и 72 часа после получения дозы.
За сорок пять минут до начала оценки пульмонарной функции каждую морскую свинку анестезировали путем внутримышечной инъекции кетамина (43,75 мг/кг), ксилазина (3,50 мг/кг) и ацепромазина (1,05 мг/кг). Затем хирургический участок обрили и очистили 70% спиртом, был сделан срединный надрез, размером 2-3 см, от шеи в брюшном направлении. После этого была отделена яремная вена, и в нее была вставлена канюля с катетером, заполненным физиологическим раствором (PE-50, Becton Dickinson, Sparks, MD) для возможности внутривенной инфузии ацетилхолина (Ach) или гистамина в физиологическом растворе. Затем трахею рассекли на части, свободную и снабженную канюлей из 14G тефлоновой трубки (#NE-014, Small Parts, Miami Lakes, FL). При необходимости анестезию поддерживали дополнительными внутримышечными инъекциями вышеуказанной смеси анестетиков. Глубину анестезии контролировали и регулировали, если животное реагировало на защемление лапы или если скорость дыхания начинала превышать 100 вдохов/минуту.
После завершения введения канюли животное помещали в плетизмограф (#PLY3114, Buxco Electronics, Inc., Sharon, CT) и канюлю для измерения пищеводного давления (PE-160, Becton Dickinson, Sparks, MD) вставляли для измерения давления, создаваемого легкими (давление). К отверстию плетизмографа прикрепляли тефлоновую трахейную трубку, чтобы дать возможность морской свинке дышать комнатным воздухом, который находится вне камеры. Затем камеру плотно закрывали. Для поддержания температуры тела использовали лампу нагрева и легкие морской свинки наполняли 3 раза 4 мл воздуха, используя 10 мл калибровочный шприц (#5520 Series, Hans Rudolph, Kansas City, MO) для обеспечения гарантии, что расположенные ниже воздушные пути не сомкнутся и что животное не пострадает от гипервентиляции.
После определения того что значение базовой линии находится внутри диапазона 0,3-0,9 мл/см H2O при расслаблении и внутри диапазона 0,1-0,199 см H2O/мл на секунду при сопротивлении, начинали проводить оценку пульмонарной функции. Компьютерная программа измерения состояния пульмонарной функции Buxco обеспечивает возможность сбора и дифференцировки значений пульмонарной функции.
Сначала программа инициировала экспериментальный протокол и сбор данных. Изменения в объеме относительно времени, которые происходили внутри плетизмографа с каждым вдохом, измеряли при помощи датчика давления Buxco. Путем интегрирования этого сигнала по времени вычисляли показатель потока для каждого вдоха. Этот сигнал вместе с изменениями давления, вызываемыми легкими, которые собирали, используя датчик давления Sensym (#TRD4100), соединяли через предварительный усилитель Buxco (MAX 2270) с интерфейсом сбора данных (#'s SFT3400 and SFT3813). Все другие параметры пульмонарной функции получали из этих двух входных сигналов.
Значения базовой линии собирали в течение 5 минут, спустя это время морских свинок стимулировали Ach или гистамином. Во время оценки мускаринового антагонистического влияния пропанолол (5 мг/кг, i.v.) (Sigma-Aldrich, St. Louis, MO) вводили за 15 минут до стимуляции Ach. Ach (Sigma-Aldrich, St. Louis, MO) (0,1 мг/мл) вводили внутривенно инфузией в течение 1 минуты при помощи шприца (sp210iw, World Precision Instruments, Inc., Sarasota, FL) следующими дозами и в течение заданных промежутков времени от начала эксперимента: 1,9 мкг/минуту за 5 минут, 3,8 мкг/минуту за 10 минут, 7,5 мкг/минуту за 15 минут, 15,0 мкг/минуту за 20 минут, 30 мкг/минуту за 25 минут и 60 мкг/минуту за 30 минут.В качестве альтернативы, бронхопротекцию тестируемых соединений определяли в модели стимуляции ацетилхолином без предварительной обработки соединением β-блокирования.
При оценке агонистического влияния к β2-адренергическому рецептору тестируемых соединений внутривенно инфузией вводили гистамин (25 мкг/мл) (Sigma-Aldrich, St. Louis, MO) в течение 1 минуты из шприца в следующих дозах и в течение заданных промежутков времени от начала эксперимента: 0,5 мкг/минуту за 5 минут, 0,9 мкг/минуту за 10 минут, 1,9 мкг/минуту за 15 минут, 3,8 мкг/минуту за 20 минут, 7,5 мкг/минуту за 25 минут и 15 мкг/минуту за 30 минут. Если расслабление или сопротивление не возвращалось к значениям базовой линии за 3 минуты после каждой дозы Ach или гистамина, легкие морской свинки продували 3 раза 4 мл воздуха из 10 мл калибровочного шприца. Записанные параметры пульмонарной функции включают частоту дыхания (вздохов/минуту), расслабление (мл/см H2O) и легочное сопротивление (см H2O/мл в секунду). После завершения сбора показателей пульмонарной функции за 35 минут такого протокола морскую свинку выносили из плетизмографа и подвергали эвтаназии асфиксацией двуокисью углерода.
Данные оценивали одним из двух способов.
(a) Легочное сопротивление (RL, см H2O/мл в секунду) вычисляли из отношения "изменение давления" к "изменению потока". RL ответ на ACh (60 мкг/мин, IH) вычисляли для наполнителя и группы тестируемых соединений. Средний ACh ответ у животных, обработанных наполнителем, за каждое заданное время обработки, вычисляли и использовали для вычисления % ингибирования ACh ответа, за соответствующее заданное время обработки для каждой дозы тестируемого соединения. Кривые доза ингибирования-ответ для 'RL' соответствовали четырехпараметрическому логистическому уравнению, используя GraphPad Prism, версия 3,00 для Windows (GraphPad Software, San Diego, California) для оценки бронхопротективной ID50 (доза, требуемая для ингибирования ACh (60 мкг/мин) бронхоконстрикторного ответа при 50%). Использовали следующее уравнение:
Y=Min+(Max-Min)/(1+10((logID50-Х)*Hillslope))
где X представляет собой логарифм дозы, Y представляет собой ответ (% ингибирования увеличения в RL, индуцированное ACh). Y начинается в Min и достигает асимптотически Max в форме сигмоиды.
(b) Количество PD2, которое определяется в виде количества Ach или гистамина, необходимого, чтобы вызвать удвоение базовой линии легочного сопротивления, вычисляли, используя значения легочного сопротивления, полученные из потока и давления во время Ach или гистаминной стимуляции, используя следующее уравнение (полученное из уравнения, использованного для вычисления значений PC20 в клинических условиях (см. Am. Thoracic Soc, 2000):
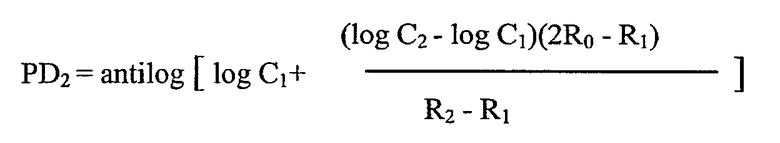
где:
C1=концентрация Ach или гистамина перед C2,
C2=концентрация Ach или гистамина, приводящая, по меньшей мере, к двукратному увеличению легочного сопротивления (RL),
R0=значение RL базовой линии,
R1=значение RL после C1,
R2=значение RL после C2.
Статистический анализ данных выполняли, используя двухфакторный t-тест Стьюдента. Значение P<0,05 считалось значимым.
При тестировании в этом анализе соединение формулы I оказывало бронхопротективное влияние, зависящее от дозы, относительно MCh-индуцированной бронхоконстрикции и His-индуцированной бронхоконстрикции. Кроме того, соединение формулы I имело в анализе продолжительность (PD T1/2) бронхопротективной активности, по меньшей мере, примерно 24 часа.
Поскольку настоящее изобретение было описано со ссылкой на его специфические аспекты или варианты осуществления, специалистам в данной области техники очевидно, что могут быть сделаны различные изменения или различные эквиваленты могут быть заменены без отступления от сущности и объема настоящего изобретения. Кроме того, до степени, допускаемой соответствующим патентным законодательством, все публикации, патенты и заявки на патент, процитированные в настоящем описании, включены в настоящее описание во всей своей полноте в качестве ссылки так, как если бы каждый документ был отдельно включен в настоящее описание при помощи ссылки.
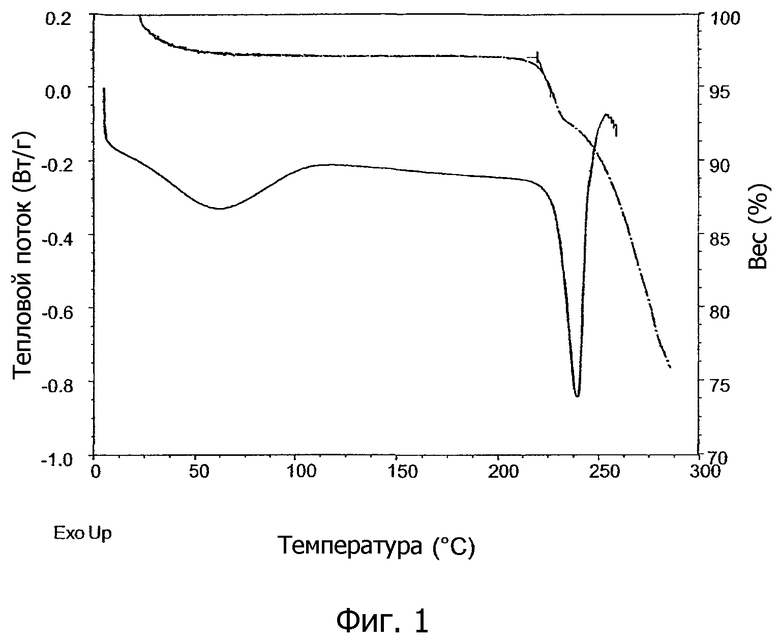
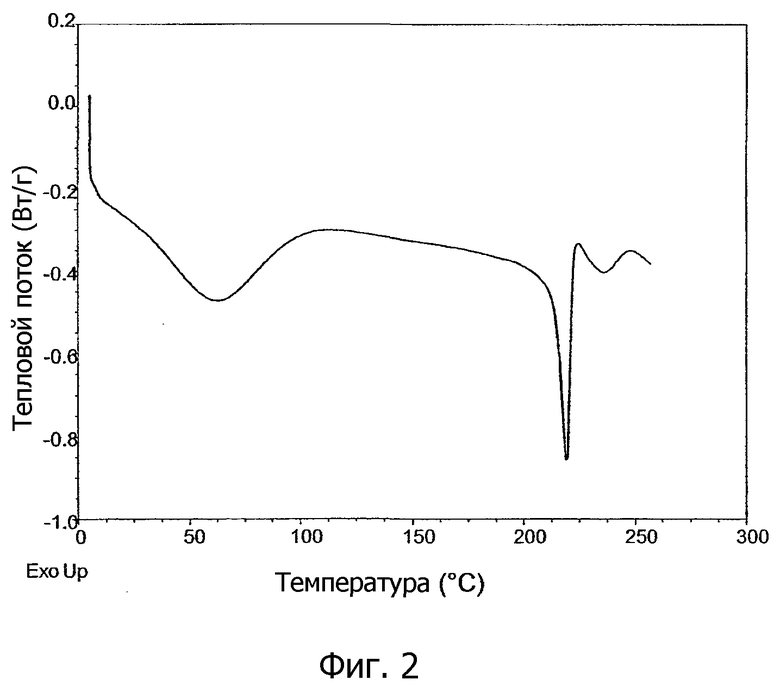
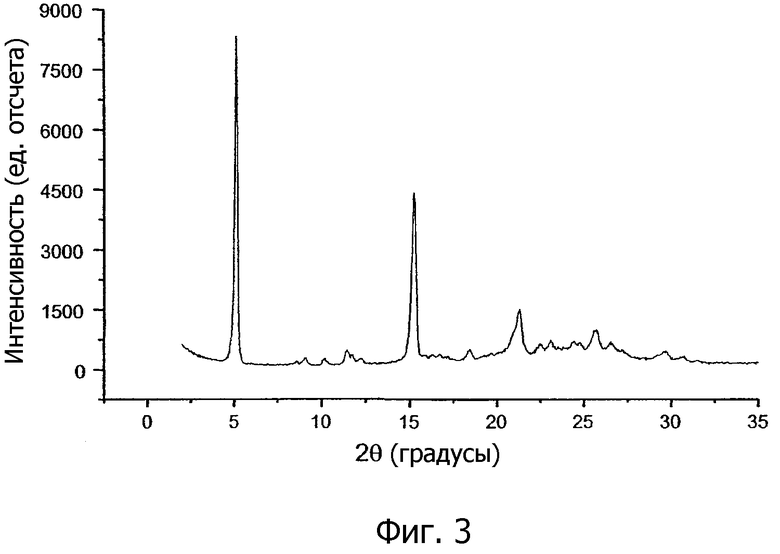
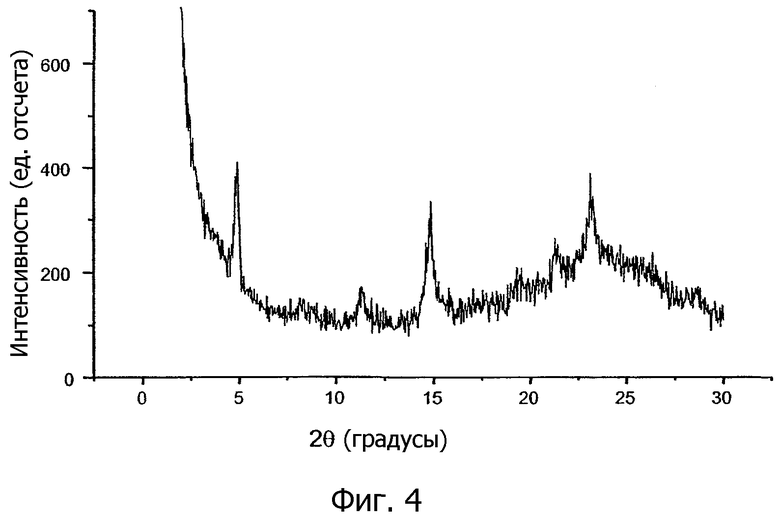
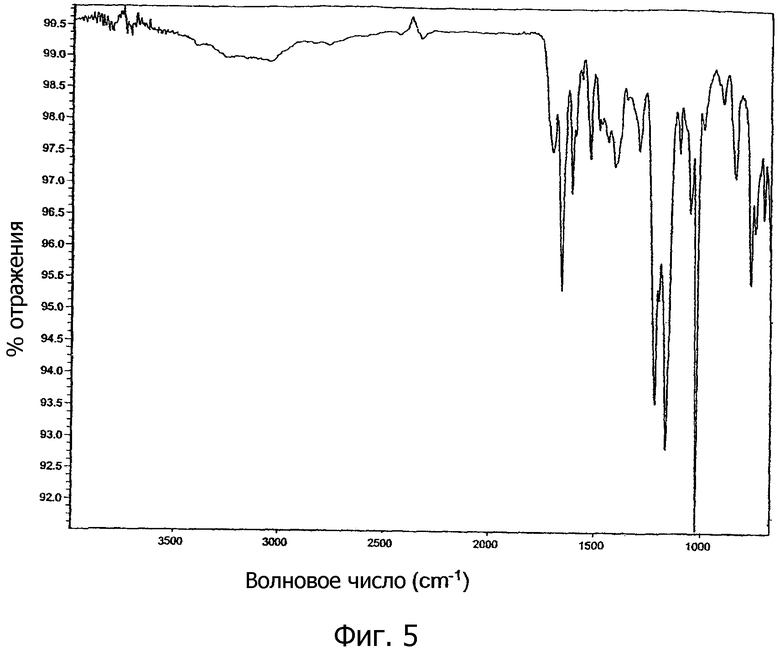
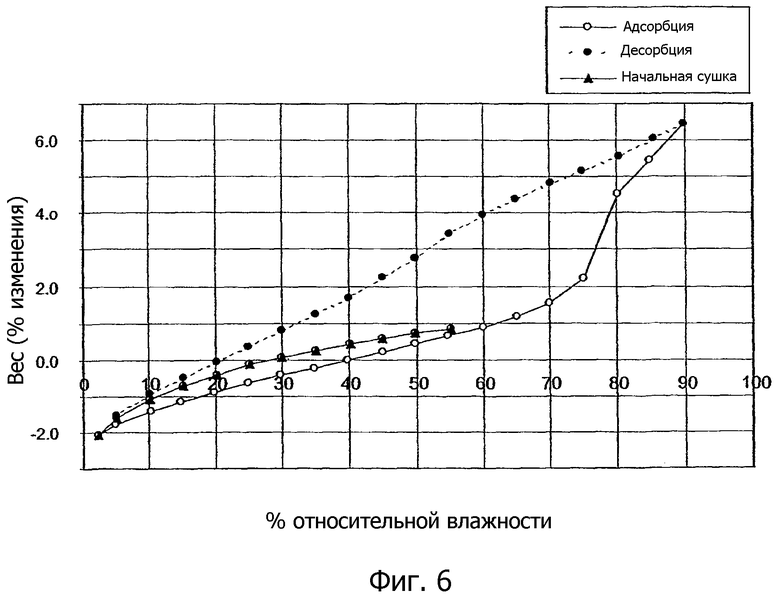
Настоящее изобретение относится к кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)
этиламино]метил}-
5-метоксифенилкарбамоил)этил]пиперидин-4-
илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвату. Также изобретение относится к фармацевтической композиции, содержащей такую соль, комбинации, содержащей соль по п.1 и стероидный противовоспалительный агент, способу лечения легочного расстройства, способу получения соли по п.1 и применению соли по п.1 для производства лекарственного средства. Технический результат: получена новая кристаллическая соль указанного выше соединения, обладающая полезными биологическими свойствами. 15 н. и 19 з.п. ф-лы, 6 ил., 4 табл.
1. Кристаллическая соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват.
2. Соединение по п.1, которое характеризуется графиком дифференциальной сканирующей калориметрии, показывающим максимальный эндотермический тепловой поток в диапазоне от примерно 215 до примерно 240°С.
3. Соединение по п.1, которое характеризуется графиком дифференциальной сканирующей калориметрии, показывающим максимальный эндотермический тепловой поток в диапазоне от примерно 215 до примерно 229°С.
4. Соединение по п.1, которое характеризуется графиком дифференциальной сканирующей калориметрии, показывающим максимальный эндотермический тепловой поток в диапазоне от примерно 230 до примерно 240°С.
5. Соединение по п.1, которое характеризуется графиком дифференциальной сканирующей калориметрии, который по существу соответствует графику, показанному на фиг.1, в диапазоне температур выше 200°С.
6. Соединение по п.1, которое характеризуется графиком дифференциальной сканирующей калориметрии, который по существу соответствует графику, показанному на фиг.2, в диапазоне температур выше 200°С.
7. Соединение по п.1, которое характеризуется порошковой рентгенограммой, имеющей дифракционные пики в значениях 2θ 5,0±0,3 и 15,0±0,3.
8. Соединение по п.1, которое характеризуется порошковой рентгенограммой, на которой положения пиков по существу соответствуют положениям пиков на рентгенограмме, показанной на фиг.3.
9. Соединение по п.1, которое характеризуется порошковой рентгенограммой, на которой положения пиков по существу соответствуют положениям пиков на рентгенограмме, показанной на фиг.4.
10. Соединение по п.1, которое имеет инфракрасный спектр поглощения со значимыми полосами поглощения при примерно 704, 748, 768, 841, 900, 1055, 1104, 1166, 1218, 1294, 1408, 1522, 1609, 1655 и 1701 см-1.
11. Соединение по п.1, у которого инфракрасный спектр поглощения по существу соответствует спектру поглощения, показанному на фиг.5.
12. Соединение по п.1 в микронизированной форме.
13. Фармацевтическая композиция для лечения легочного расстройства, содержащая фармацевтически приемлемый носитель и соединение по любому из пп.1-12.
14. Фармацевтическая композиция по п.13, которая дополнительно содержит стероидный противовоспалительный агент.
15. Фармацевтическая композиция по п.14, в которой стероидный противовоспалительный агент представляет собой S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиокислоты или его сольват.
16. Фармацевтическая композиция по п.13, которая составлена в лекарственную форму в виде порошка или суспензии для введения посредством ингаляции.
17. Фармацевтическая композиция по п.13 в микронизированной форме.
18. Фармацевтическая композиция по п.16, в которой носитель представляет собой лактозу, крахмал, маннит, декстрозу, полимолочную кислоту, сополимер лактида и гликолида или их комбинацию.
19. Комбинация, содержащая:
(a) соединение по любому из пп.1-12; и
(b) стероидный противовоспалительный агент.
20. Комбинация по п.19, в которой стероидный противовоспалительный агент представляет собой S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиокислоты или его сольват.
21. Способ лечения легочного расстройства, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соединения по любому из пп.1-12.
22. Способ вызова бронходилатации у пациента, включающий введение пациенту посредством ингаляции соединения по любому из пп.1-12 в виде порошка или суспензии, в количестве, приводящем к бронходилатации.
23. Способ лечения хронического обструктивного заболевания легких или астмы, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соединения по любому из пп.1-12.
24. Способ получения соединения по п.1, включающий приведение в контакт 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]-пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты с 1,2-этандисульфоновой кислотой или ее гидратом.
25. Способ получения соединения по п.1, включающий:
(а) приведение в контакт соединения формулы II:
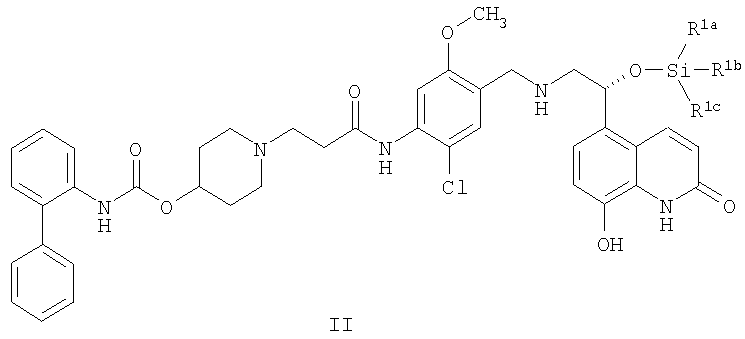
где R1a, R1b и R1c независимо выбирают из С1-4алкила, фенила, -C1-4алкил-(фенил), или один из R1a, R1b и R1c представляет собой -O-(C1-4алкил); с фторид-ионом; и
(b) приведение в контакт продукта, полученного на стадии (а), с 1,2-этандисульфоновой кислотой или ее гидратом; для образования кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата, причем стадии (а) и (b) проводят в одном и том же реакционном сосуде без выделения продукта на стадии (а).
26. Способ получения соединения по п.1, имеющего точку плавления выше, чем примерно 230°С, включающий добавление затравочного кристалла кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата в раствор, содержащий соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват, растворенные в инертном разбавителе, причем затравочные кристаллы имеют точку плавления выше, чем примерно 230°С.
27. Способ получения соединения по п.1, имеющего точку плавления выше, чем примерно 230°С, включающий:
(a) растворение кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты в инертном разбавителе при первой температуре;
(b) охлаждение продукта стадии (а) до второй температуры; и
(c) добавление затравочного кристалла соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
причем затравочный кристалл имеет точку плавления выше, чем примерно 230°С, первая температура представляет собой температуру, достаточную для растворения соли 1,2-этандисульфоновой кислоты, а вторая температура ниже температуры, при которой затравочный кристалл полностью растворяется при добавлении его к продукту, полученному на стадии (b).
28. Способ очистки 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты, включающий образование соединения по п.1.
29. Кристаллическая соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват по п.1 для применения в терапии или в качестве лекарственного средства для лечения легочного расстройства.
30. Применение кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата по п.1 для производства лекарственного средства для лечения легочного расстройства.
31. Применение по п.30, в котором легочное расстройство представляет собой хроническое обструктивное заболевание легких или астму.
32. Лекарственное средство для лечения легочного расстройства, содержащее кристаллическую соль 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил}-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольват по п.1.
33. Применение:
(a) кристаллической соли 1,2-этандисульфоновой кислоты 1-[2-(2-хлор-4-{[(R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этиламино]метил)-5-метоксифенилкарбамоил)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или ее сольвата по п.1; и
(b) стероидного противовоспалительного агента;
для производства лекарственного средства для лечения легочного расстройства.
34. Применение по п.33, в котором стероидное противовоспалительное средство представляет собой S-фторметиловый эфир 6α, 9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиокислоты или его сольват.
| US 6653323 B2, 25.11.2003 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| БИФЕНИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ | 1993 |
|
RU2122540C1 |

