Изобретение касается производных 4-замещенных 3-(3-диалкиламинометил-индол-1-ил)малеимидов, способов их получения и использования на основе выявленной активности как ингибиторов протеинкиназы С в качестве лекарственных средств, применяемых для лечения патологических состояний, в механизме возникновения которых участвует протеинкиназа С, или на основе их цитотоксического действия в качестве противоопухолевых препаратов.
Уровень техники
Опухолевые заболевания являются одной из основных причин смерти в России и во всем мире. Для раковых клеток характерны быстрое деление и инвазивность, т.е. способность мигрировать и метастазировать. В опухолевых клетках дефектны механизмы клеточной смерти (апоптоза), которые в противном случае могли бы регулировать их уровень, а также пролиферацию и миграцию. Напротив, в опухолевых клетках задействованы механизмы выживания, защищающие их от апоптоза. В некоторых случаях для оказания терапевтического эффекта при опухолевых заболеваниях с успехом применялись такие ингибиторы протеинкиназ как производные индолокарбазола - стауроспорин, ребеккамицин, UCN-01, NB-506 и другие. Одной из проблем, возникающих при химиотерапии опухолей, является развитие в опухолевых клетках множественной лекарственной устойчивости, индуцируемой химиотерапевтическими препаратами. Множественная лекарственная устойчивость (МЛУ) злокачественных новообразований - одна из основных причин клинического прогрессирования болезни, когда опухоль становится нечувствительна к химиотерапии независимо от комбинации применяемых лекарств. МЛУ может возникать в клетках, подвергнутых стрессовым воздействиям, прежде всего под действием химиотерапевтических препаратов. До этих воздействий механизмы защиты в таких клетках экспрессированы слабо или отсутствуют; но выживая после обработки одним токсином, клетки приобретают резистентность ко многим веществам (Gottesman MM, Fojo Т, Bates SE. Nature Rev. Cancer 2002; 2:48-58).
Одной из важнейших причин МЛУ является пониженное накопление токсинов в клетке, обусловленное их активным выведением в межклеточную среду. Такой транспорт осуществляется интегральным белком плазматической мембраны Р-гликопротеином (Pgp, масса 140-170 кДа). У человека Pgp кодируется геном MDR1 (multidrug resistance 1).
Причина клинической МЛУ - гиперэкспрессия гена MDR1 за счет индукции внеклеточными стимулами, в частности противоопухолевыми препаратами (Gottesman MM, Fojo Т, Bates SE. Nature Rev. Cancer 2002, 2, 48-58; Ambudkar SV et al. Annu. Rev. Pharmacol. Toxicol. 19996 39, 361-398.). Индуцибельность гена MDR1 предполагает активацию путей передачи сигналов к ядру клетки. Получены данные об участии протеинкиназ (ПКС) в срочном становлении МЛУ; фармакологическая инактивация этого механизма отменяла повышение иРНК MDR1, индуцированное антагонистами ПКС и химиопрепаратами. Блокирование ПКС с помощью фармакологических воздействий позволило бы предотвратить развитие МЛУ в процессе химиотерапии (Chaudhary P., Roninson I. Oncol. Res. 1992, 4, 281-290; Chaudhary P., Roninson I. J. Natl. Cancer Inst. 1993, 85, 632-639).
Известны производные бис(индол-3-ил)малеимидов (BIS-I, BIS-II, BIS-IV. BIS-VI, ВIS-VII и другие), являющиеся ингибиторами протеинкиназы С; исследованы и способы их получения (Davis et al. J.Med.Chem. 1992, v.35, N.1, p.177-184, US 6133452, US 5545636, US 2005004201, EP 0657458 A1, WO 02/38561 A1). Известна способность бис(индол-3-ил)малеимидов (например, BIS-I) [3-(1-(3-диметиламинопропил)индол-3-ил)-4-(индол-3-ил)пиррол-2,5-дион] предотвращать развитие МЛУ у опухолевых клеток за счет ингибирования ПКС-альфа. (V.Gekeler et al., Br.J.Cancer 1996, 74, 897-905).
Протеинкиназа С (серин-треониновая протеинкиназа) - семейство ферментов (более 10 изоформ), участвующих в регуляции таких процессов, как апоптоз, генная экспрессия, дифференцировка клеток, ангиогенез, активный транспорт химиотерапевтических веществ из клетки и возникновение множественной лекарственной устойчивости, инвазия и старение клеток (Hoffman, Curr. Cancer Drug Targets, 2004, 4, 2, 125-146). Таким образом, ингибиторы протеинкиназы С перспективны для применения в химиотерапии опухолей.
Известно, что серин-треониновые протеинкиназы эукариотического типа могут также играть роль в возникновении устойчивости микроорганизмов к антибиотикам (Elizarov S.M., et al, J. Molecular Biology, 2005 Vol.35, Ns 2,226), в том числе путем образования биопленок (Hussain H, et al, J.Bacteriol, 2006, 188, №4, 1628). Известна роль серии - треониновых протеинкиназ в развитии вирулентности у Streptomyces pneumoniae (Eshenique J. et al, J. Infection and Immunity, 2004, Apr., 2434). Поэтому специфические ингибиторы протеинкиназ можно использовать в комплексной терапии заболеваний, вызываемых такими микроорганизмами (Drews S.J.,et al, FEMS Microbiol Lett. 2001, 18, 205 (2), 369).
Известно, что препараты LY-333531, LY-379196, LY-317615, созданные на основе бис-(индол-3-ил)малеимида, являются селективными ингибиторами ПКС-бета и могут применяться как средства против ретинопатии и нефропатии при диабете (Jirousek M.R., Gillig J.R., Gonzalez C.M., et al., J. Med. Chem. 1996 39 2664. Paul M., Krumrich C.A., J. Org. Chem. 2001, 66, 2024). Известно, что ингибиторы протеинкиназ могут применяться при воспалительных и нейродегенеративных процессах (Goekjian P.G., Jirousek M.R., Curr. Med. Chem. 1999 6, 877, WO 02/38561 A1, WO 03/082859 A1).
Таким образом, бис-индолилмалеимиды, их аналоги и родственные структуры являются классами соединений, перспективными для поиска и разработки новых лекарственных препаратов (ингибиторов протеинкиназ), обладающих высокой селективностью действия и имеющих меньше побочных и общетоксических эффектов, что особенно важно при химиотерапии рака.
Наиболее близкими прототипами веществ, раскрываемых в настоящем изобретении, являются производные бис(индол-3-ил)малеимида.
Раскрытие изобретения.
Сущность изобретения состоит в том, что получены новые химические соединения - производные 4-замещенных 3-(3-диалкиламинометилиндол-1-ил)малеимидов, обобщенные структурные формулы (формула I и формула II), которых представлены на схеме 1.
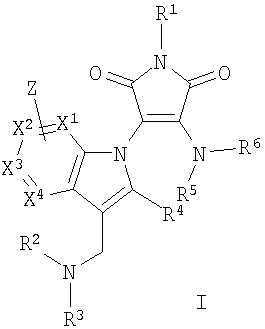

Схема 1.
где X1-X4 могут быть С или N, Y-S или О, Z - галоид, алкил, арил, в том числе замещенный, ОН, алкокси, NH2, NO2, СООН, CN или любой другой допустимый заместитель. R1 - алкил, арил, в том числе содержащие различные функциональные группы, а также ОН, алкокси, NH2, СООН, CN или любой другой допустимый заместитель. R2 и R3 - алкил, арил, в том числе замещенный, или любой другой допустимый заместитель. R4 - алкил, арил, в том числе содержащие различные функциональные группы, такие как ОН, алкокси, NH2, СООН, CN. В формуле I R5 и R6 могут быть алкильными, арильными заместителями, которые могут содержать различные функциональные группы. NR5R6 могут быть также производными индолина, тетрагидрохинолина или другого насыщенного гетероцикла, а также индола, пиррола. В формуле II R5 - алкил или арил, которые могут содержать различные функциональные группы.
В отличие от известных бис-индолилмалеимидов предложенные соединения имеют ряд структурных особенностей. А именно: оба заместителя в положениях 3 и 4 малеимидного фрагмента соединены с ним связью С-гетероатом (C-N в случае индольного заместителя в положении 3 малеимидного цикла или же связями C-N, C-S или С-O в случае заместителя в положении 4 малеимидного цикла). Далее, в известных прототипах ингибиторов протеинкиназы бис-индолилмалеимидного типа (препараты БИС) гетероатом в боковой цепи (в положении 1 или 3 индольного фрагмента) отделен от гетероцикла цепью из трех метиленовых групп. В отличие от этого, соединения, представленные в настоящей заявке, содержат в положении 3 индольного фрагмента диалкиламинометильный заместитель, таким образом, экзоциклический гетероатом (азот) отделен от индольного гетероцикла одной метиленовой группой. Раскрываются также методы получения этих соединений.
Осуществление изобретения
Вещества, представленные в настоящем изобретении, могут быть синтезированы с использованием известных химических превращений. Для синтеза всех соединений, представленных в изобретении, пригодны общие методы, изложенные ниже.
Исходный 3-бромо-4-(индол-1-ил)малеимид был получен, как описано ранее (Lakatosh S.A., Luzikov Y.N., Preobrazhenskaya M.N., Org. Biomol. Chem. 2003 1 826). Атом брома в молекуле легко замещается в присутствии оснований различными нуклеофильными реагентами, такими как амины (в том числе 2,3-дигидроиндол (индолин), тетрагидрохинолин и другие насыщенные азотсодержащие гетероциклы), тиолы или фенолы. Алкилирование по малеимидному атому азота осуществляется соответствующими алкилирующими агентами в присутствии основания (схема 2). Заместитель в положении 1 малеимидного цикла может быть также введен путем переаминирования нагреванием исходного вещества с соответствующим первичным амином.

Схема 2.
4-Замещенные 3-(индол-1-ил)малеимиды были превращены в соответствующие диалкиламинометильные производные с использованием реакции Манниха (аминометилирования) (схема 2).
Примерами целевых соединений, полученных по этой схеме, могут быть представленные ниже (схема 3) структуры:



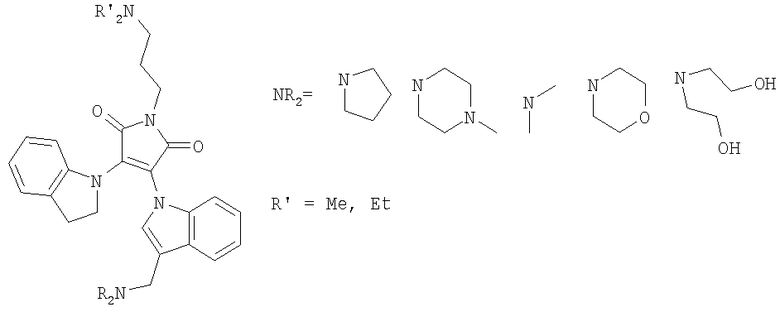
Схема 3.
3-(2,3-Дигидроиндол-1-ил)-4-(индол-1-ил)малеимиды легко превращаются в соответствующие бис-3,4(индол-1-ил)малеимиды под действием окислителей (например, действием дихлородицианобензохинона или активной МnО2 в толуоле или о-ксилоле). Диалкиламинометилирование таких ди(индол-1-ил)малеимидов в условиях реакции Манниха давало в зависимости от соотношения исходных реагентов смеси моно- и дизамещенных производных или же только дизамещенные производные. Монозамещенные производные отделяли от прочих продуктов реакции хроматографически (схема 4.).

Схема 4
Получение заявленных соединений поясняется следующими примерами.
Синтез индолилмалеимида из 3,4-дибромамалеимида в случае, когда в качестве нуклеофилов выступают индолин или N-алкил-N-арил- или диалкиламины, описан ранее (Lakatosh S.A., Luzikov Y.N., Preobrazhenskaya M.N., Org. Biomol. Chem. 2993 1 826). Прочие соединения могут быть получены с использованием других нуклеофильных реагентов по той же методике.
Пример 1.
1-(3-Бромопропил)-3-(2,3-дигидроиндол-1-ил)-4-(индол-1-ил)малеимид. К раствору 3-(2,3-дигидроиндол-1-ил)-4-(индол-1-ил)малеимида (1 г) в диоксане (30 мл) добавили 5-кратный избыток дибромопропана и К2СО3 (10 г) смесь кипятили при интенсивном перемешивании 12 ч, отфильтровали, фильтрат упарили. Остаток растворили в EtOAc (100 мл), промыли насыщенным раствором NaCl (50 мл), высушили и упарили.
Остаток хроматографировали (силикагель, система н-гексан - EtOAc 5:1).
Получили продукт в виде масла красного цвета.
1Н-ЯМР (400 MHz, DMSO-d6) 0.96 (2H, t, J=7.14 NEt2), 2.17 (2H, t, J=6.59 -CH2-), 3.08 (2H, t, J=8.05 -СН2 индолин), 3.61 (2H, t, J=6.73 -CH2-), 3.67 (2H, t, J=6.63 -CH2-), 4.23 (2H, t, J=8.01 -CH2 индолин), 6.08 (1H, d, J=8.02 индол), 6.49 (1H, t, J=7.58 индол), 6.60 (1H, d, J=3.3 индол), 6.64 (1H, t, J=7.32 индол), 6.95 (1H, t, J=7.04 индол), 7.00 (1H, t, J=7.04 индол), 7.04 (1H, d, J=7.29 индол), 7.20 (1H, d, J=8.10 индол), 7.37 (1H, d, J-3.29 индол), 7.46 (1H, d, J=7.65 индол);
Пример 2.
1-(3-Диэтиламиноопропил)-3-(2,3-Дигидроиндол-1-ил)-4-(индол-1-ил)малеимид. К раствору 1-(3-бромопропил)-3-(2,3-дигидроиндол-1-ил)-4-(индол-1-ил)малеимида (500 мг) в ДМФА (5 мл) добавили диэтиламин (1 мл) и К2СО3 (3 г). Смесь оставили при перемешивании на ночь, отфильровали. Фильтрат разбавили EtOAc, промыли насыщенным раствором NaCl (3×20 мл), высушили и упарили. Остаток хроматографировали (силикагель, система СНСl3-МеОН-Et3N 6:1:0,1)
Получили 1-(3-диэтиламиноопропил)-3 -(2,3 -дигидроиндол-1-ил)-4-(индол-1-ил)малеимид в виде твердого вещества оранжевого цвета.
1Н ЯМР (400 MHz, DMSO-d6) 0.96 (2H, t, J=7.14 NEt2), 1.76 (2H, t, J=7.14 -CH2-), (2.44-2.50) (6H, m, NEt2, -CH2-), 3.07 (2H, t, J=7.88 -CH2 индолин), 3.60 (2H, t, J=6.95 -CH2-), 4.26 (2H, t, J=7.92 -СН2 индолин), 6.04 (1H, d, J=8.02 индол), 6.48 (1H, t, J=7.83 индол), 6.62 (1H, t, J=3.34 индол), 6.64 (1H, t, J=7.50 индол), 6.96 (1H, t, J=7.18 индол), 7.00 (1H, t, J=8.02 индол), 7.03 (1H, d, J=7.36 индол), 7.19 (1H, d, J=8.05 индол), 7.37 (1H, d, J=3.3 индол), 7.48 (1H, d, J=7.33 индол);
Пример 3 (Аминометилирование)
1-Метил-3-(2,3-Дигидроиндол-1-ил)-4-(3-диметиламинометилиндол-1-ил)малеимид LCTA-1183. К раствору исходного индолилмалеимида 3 (R=Me, Nuc=индолин) (1.5 ммоль) в СН3СООН (50 мл) добавили параформ (500 мг) и NН(СН3)2 (40% водный раствор, 1 мл, 7.9 ммоль). Смесь перемешивали на 36 часов при 50°С, упарили при пониженном давлении, разбавили. EtOAc (100 мл), промыли насыщенным раствором NaHCO3. Органический слой отделили, водную часть повторно экстрагировали EtOAc (50 мл). Экстракты объединили и промыли насыщенным раствором NaCl (50 мл), высушили и упарили. Остаток хроматографировали (силикагель, система ЕtOАс: изопропанол: водный раствор аммиака (25%) 12:4:0,5) получили продукт LCTA-1183 (0,9 ммоль), в виде темно-красного кристаллического порошка. Выход 70%
Данные 1H ЯМР приводятся ниже в Таблице 1.
Другие диалкиламинометильные производные индолилмалеимидов могут быть получены из соответствующих интермедиатов и вторичных аминов в описанных условиях с выходом 50-75%.
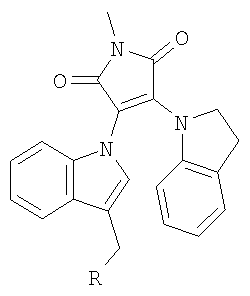



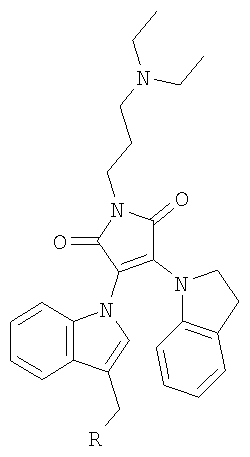




Окисление 3-(2,3-дигидроиндол-1-ил)-4-(индол-1-ил)малеимидов в соответствующие ди(индол-1-ил)малеимиды было описано ранее (Lakatosh S.A., Luzhikov Y.N., Preobrazhenskaya M.N., Org. Biomol. Chem. 2003 1 826).
Пример 4.
3-(3-Диметиламинометилиндол-1-ил)-4-(индол-1-ил)-1-метилмалеимид и 3,4-бис-(3-диметиламинометилиндол-1-ил)-1-метилмалеимид. К раствору исходного 1-метил-3,4-бис(индол-1-ил)малеимида (1 г, 2,9 ммоль) в СН3СООН (50 мл) добавили параформ (1 г) и NН(СН3)2 (40% водный раствор, 6 мл, 46 ммоль). Смесь перемешивали 36 часов при 80°С, упарили, разбавили EtOAc (100 мл) и промыли насыщенным раствором Na2CO3. Органический слой отделили и промыли насыщенным раствором NaCl (2×40 мл), высушили и упарили. Остаток подвергли хроматографичесому разделению (силикагель, н-гептан: EtOAc: АсОН 5:5:1) получили индивидуальные соединения.
Данные ЯМР для соединений, полученных по этой схеме, приводятся в Таблице 2


Соединения, раскрываемые в настоящем изобретении, проявили активность в отношении протеинкиназы С α человека. Они могут применяться для предотвращения и лечения патологических состояний, в генезе которых участвует протеинкиназа С. Помимо этого, предлагаемые соединения - ингибиторы протеинкиназы могут быть использованы при лечении инфекционных заболеваний, вызываемых микроорганизмами, в становлении вирулентности и устойчивости к антибиотикам которых, основную роль играют бактериальные аналоги протеинкиназы С.
Соединения, раскрываемые в настоящем изобретении, могут применяться в качестве активного компонента фармакологических композиций или в комбинации с другими активными компонентами, известными на момент раскрытия изобретения; которые могут вводиться одним из известных на настоящий момент способов (например, перорально, внутривенно, внутримышечно и.т.д.) в соответствующей лекарственной форме.
Соединения, раскрываемые в настоящем изобретении, могут применяться в виде свободных оснований или же в виде фармакологически приемлемых солей (например, сульфат, гидрохлорид, метансульфонат, фумарат и других).
Изучение биологической активности поясняется следующими примерами.
Пример 5.
Изучение ингибирования Альфа-изоформы ПКС.
Альфа-ПКС инкубировали в оптимизированной протеинкиназной системе в присутствии ее активаторов (фосфатидилсерина, диацилглицерина, Са2+) с [γ-32Р]АТФ и белковым субстратом (H1 гистон), после чего определяли катализируемое киназой включение 32Р-фосфата в H1 гистон без и в присутствии возрастающих концентраций ингибиторов (за вычетом фона сорбции). IC50 определяли графически по полученным кривым ингибирования. Полученные данные для некоторых веществ группы ЛХТА приведены в таблице 3.
Структурные формулы соединений приведены в таблицах 1 и 2.
Пример 6.
Подавление МЛУ определяли по отсутствию в клетках мРНК гена MDR1 методом обратной транскрипции и полимеразной цепной реакции (ПЦР). При исследованиях использовали метод, описанный ранее (Chaudhary P., Roninson I. J. Natl. Cancer Inst. 85,632-639; Shtil A.A., Azare J. Int. Rev. Cytol., 2005, 246, 1-29.)
Основной эффект, исследуемый в данном разделе, - гиперэкспрессия MDR1 в клетках Н9 в ответ на воздействие противоопухолевых лекарств. В серии экспериментов клетки Н9 обрабатывали цитозаром (Аrа С) - препаратом, применяемым в химиотерапии рака молочной железы и лейкозов. Концентрации цитозара определены в предварительных опытах по выживанию клеток Н9 в присутствии этого агента. Установлено, что минимальная доза, вызывающая гибель 100% клеток в течение 48 часов, составляет около 10 мкМ. Экспрессия MDR1 в необработанных клетках Н9 не выявляется и после 25 циклов ПЦР, тогда как в обработанных 10 мкМ цитозара клетках повышение количества иРНК MDR1 отмечали уже через 3-6 часов после воздействия цитозара - концентрацией, сопоставимой с применяемой в клинике.
Однократная обработка клеток Н9 доксорубицином и цитозаром приводила к индукции гена MDR1. Такой же эффект вызывал форбол миристат ацетат (ТФА), использованный как контрольный агент-агонист ПКС. Специфические ингибиторы ПКС - хелеритрин, кальфостин С и бисиндолилмалеимид I - предотвращали активацию гена MDR1. Аналогичные данные получены на клетках К562, обработанных цитозаром, доксорубицином или форболовым эфиром в присутствии каждого из указанных ингибиторов ПКС. Таким образом, фармакологическое блокирование ПКС отменяет активацию гена MDR1 противоопухолевыми препаратами.
Полученные результаты позволяют разделить вещества на три группы:
1. Нецитотоксичные соединения, с выраженным эффектом подавления развития МЛУ (таблица 4).
2. Цитотоксичные соединения, с выраженным эффектом подавления развития МЛУ (таблица 5).
3. Умеренно цитотоксичные соединения с выраженным эффектом подавления развития МЛУ (таблица 6).
Структурные формулы соединений приведены в таблицах 1 и 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР PIM1-КИНАЗЫ 6-[(4-МЕТИЛ-1-1-ПИПЕРАЗИНИЛ)МЕТИЛ]-ИНДОЛО[1',7':1,2,3]ПИРРОЛО[3',4':6,7]АЗЕПИНО[4,5-b]ИНДОЛ-1,3(2Н, 10Н)-ДИОН, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2011 |
|
RU2466132C1 |
| ЗАМЕЩЕННЫЕ В ИНДОЛЬНОМ ЯДРЕ ПРОИЗВОДНЫЕ ТРИИНДОЛИЛМЕТАНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ АНТИБАКТЕРИАЛЬНАЯ И ПРОТИВОГРИБКОВАЯ АКТИВНОСТЬ | 2008 |
|
RU2388749C2 |
| ЦИТОТОКСИЧЕСКИЕ ЛИНЕЙНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ АНТРАЦЕНДИОНА, СОДЕРЖАЩИЕ В БОКОВОЙ ЦЕПИ ЦИКЛИЧЕСКИЕ ДИАМИНЫ, АКТИВНЫЕ В ОТНОШЕНИИ ОПУХОЛЕВЫХ КЛЕТОК С МНОЖЕСТВЕННОЙ ЛЕКАРСТВЕННОЙ УСТОЙЧИВОСТЬЮ | 2009 |
|
RU2412166C1 |
| ЧЕТВЕРТИЧНЫЕ АММОНИЙНЫЕ ПРОИЗВОДНЫЕ 2-АМИНОТИОФЕН-3-КАРБОКСИЛАТОВ, ОБЛАДАЮЩИЕ ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2629369C1 |
| 4-(1-(4-(4-МЕТОКСИФЕНИЛТИО)-2,5-ДИОКСО-2,5-ДИГИДРО-1Н-ПИРРО-3-ИЛ)-1Н-ИНДОЛ-3-ИЛ)БУТИЛКАРБАМИМИДОТИОАТ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2009 |
|
RU2441000C2 |
| Способ синтеза индоло[1',7':1,2,3]пирроло[3',4':6,7]азепино[4,5-b]индол-1,3(2Н,10Н)-диона | 2017 |
|
RU2648039C1 |
| 1-ОКСО-3-(1Н-ИНДОЛ-3-ИЛ)-1,2,3,4-ТЕТРАГИДРОИЗОХИНОЛИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, КОМБИНАТОРНАЯ БИБЛИОТЕКА И ФОКУСИРОВАННАЯ БИБЛИОТЕКА | 2006 |
|
RU2302417C1 |
| СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ФИЗИОЛОГИЧЕСКИХ И/ИЛИ ПАТОФИЗИОЛОГИЧЕСКИХ СОСТОЯНИЙ, ОПОСРЕДУЕМЫХ РЕЦЕПТОРАМИ, СТИМУЛИРУЮЩИМИ СЕКРЕЦИЮ ГОРМОНА РОСТА, ТРИАЗОЛЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2006 |
|
RU2415134C9 |
| ЗАМЕЩЕННЫЕ ПИРИДО[4',3':5,6]ПИРАНО[2,3-d]ПИРИМИДИНЫ И КОМБИНАТОРНАЯ БИБЛИОТЕКА | 2004 |
|
RU2269538C1 |
| 7-АЦИЛАМИНОЦЕФАЛОСПОРИНЫ, ЗАМЕЩЕННЫЕ В ПОЛОЖЕНИИ 3 ЦИКЛИЧЕСКИМИ АМИНОГУАНИДИНОВЫМИ ГРУППАМИ | 1999 |
|
RU2245884C2 |
Изобретение относится к новым производным 4-замещенных 3-(3-диалкиламинометил-индол-1-ил)малеимида общей формулы I и II:
где Х1-Х4 означают С; Z означает Н; R1 означает алкил, Н, -(CH2)3-N-(С2H5)2; R2 и R3 означают алкил, либо вместе с атомом азота, с которым они связаны образуют С4-7-моноциклическое кольцо, содержащее 1 или 2 гетероатома, выбираемых из О или N, возможно замещенное алкилом; R4 означает Н; Y означает S, -N-(С2Н5); где в соединении формулы I R5 и R6 вместе с атомом азота, с которым они связаны, образуют C9-10 конденсированное бициклическое кольцо, содержащее в качестве гетероатома N возможно замещенное R, где R означает -N-(R2)-R3; в соединении формулы II R5 означает фенил, необязательно замещенный ОСН3; Соединения I и II могут быть использованы в качестве средств, ингибирующих протеинкиназу. 2 н.п. ф-лы, 6 табл.
1. Соединения, описываемые общими формулами I и II:


где X1-X4 означают С; Z означает Н; R1 означает алкил, Н, -(CH2)3-N-(С2Н5)2; R2 и R3 означают алкил, либо вместе с атомом азота, с которым они связаны, образуют С4-7- моноциклическое кольцо, содержащее 1 или 2 гетероатома, выбираемых из О или N, возможно замещенное алкилом;
R4 означает Н; Y означает S, -N-(С2Н5)-; где в соединении формулы I R5 и R6 вместе с атом азота, с которым они связаны, образуют С9-10 конденсированное бициклическое кольцо, содержащее в качестве гетероатома N, возможно замещенное R, где R означает -N-(R2)-R3;
а в соединении формулы II R5 означает фенил, необязательно замещенный ОСН3.
2. Применение соединения формулы I и II по п.1 в качестве средств, ингибирующих протеинкиназу.
| US 6133452, 17.10.2000 | |||
| US 5545636, 13.08.1996 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Известия АН Серия химическая, 2006, т.55, №5, с.754-761 | |||
| Химико-фармацевтический журнал, 2006, т.40, №8, с.29-34. | |||

