Изобретение относится к медицине и фармакологии и касается стабильной при хранении наносистемы с размером частиц до 10-30 нм, получаемой на основе растительных фосфолипидов и предназначенной для включения в фосфолипидную наночастицу биологически активных соединений, в частности лекарственных средств, и способа ее получения.
Одной из наиболее актуальных проблем современной фармакологии является создание лекарственных форм, которые при введении в организм обладают достаточной биодоступностью, чтобы достигать области поражения. Биодоступность лекарств определяется среди многих факторов их растворимостью.
Около 60% фармацевтических препаратов, находящихся в стадии разработки, и многие из широко используемых лекарственных препаратов относятся к плохо растворимым соединениям.
Для создания растворимых лекарственных форм могут использоваться солюбилизаторы, но их применение часто требует решения дополнительных проблем в связи с их возможной токсичностью.
Известно, что большинство лекарственных препаратов ограничено и медленно проникают в клетки через естественный барьер - биологическую мембрану. Для преодоления этого барьера лекарственное соединение должно обладать определенной степенью липофильности, а, как было уже отмечено выше, липофильные трудно растворимые лекарства требуют решений проблемы солюбилизации.
К числу других нежелательных свойств лекарственных препаратов можно отнести быструю потерю активности при введении в организм, высокую скорость сорбции и элиминации. Это приводит к необходимости увеличения дозы и/или частоты введения препарата, что, в свою очередь, также повышает побочные эффекты. Особенно этот эффект выражен для большинства цитостатиков и лекарств, действующих на центральную нервную систему.
В связи с этим в последнее десятилетие исследователи уделяют существенное внимание не только поиску новых биологически активных веществ, но и повышению эффективности уже созданных лекарств путем конструирования систем для их транспорта в организме, повышению биодоступности и эффективности специфического действия.
Интерес к разработкам систем доставки обусловлен многими причинами и, прежде всего, огромной потенциальной выгодой как с экономической, так и с медицинской стороны. Лекарства, снабженные системой доставки, имеют ряд преимуществ по сравнению со свободными препаратами: а) повышается растворимость гидрофобных лекарств; б) улучшается их проникновение в клетки; в) улучшается фармакокинетика, у многих лекарств появляется способность пересекать мембранные и гематоэнцефалический барьеры.
Примером лекарственных препаратов, включенных в биодеградируемые наносферы на основе полилактатов, полиглютамилаланина, полиалкилцианоакрилата и др., могут служить лидокаин (патент США №5543158), циклоспорин (патент США №6800296), тамоксифен (De Kozak Y., et all, Eur. J. ImmunoL, 2004, 34, 3702-3712) и др. Использование наносистем для транспорта лекарственных препаратов позволяет не только увеличить биодоступность последних, но и обеспечить пролонгированное поступление препарата в определенные органы и клетки-мишени. Наночастицы на основе полимеров обладают высокой стабильностью в биологических жидкостях и при хранении, но могут вызвать побочные эффекты при введении в организм.
В этом отношении большим преимуществом обладают фосфолипидные наночастицы, в эффективность которых существенный вклад вносят размеры - 10-20 нм. Фософлипидные наночастицы (липосомы, мицеллы) биодеградируемы, биологически инертны, не вызывают аллергических, антигенных или пирогенных реакций. Поверхность липидных наночастиц, в сравнении с другими частицами, легко модифицируется для обеспечения направленности доставки.
Наиболее распространенными наносистемами на современном фармацевтическом рынке в настоящее время являются липосомы. Основное их преимущество заключается в том, что липосомы, создавая лекарству как гидрофильное, так и гидрофобное окружение, повышают его растворимость, также защищают лекарство от преждевременной деградации, снижают его клиренс и действуют как система, ограничивающая его высвобождение. Существенное влияние липосом на фармакокинетику лекарства дает возможность использования его в более низких дозах. Благодаря инкапсулированию лекарства в липосоме существенно снижается объем распределения, и возрастает концентрация лекарства в органе-мишени.
Основные свойства липосом и характер их взаимодействия с клетками обусловлены их фосфолипидной основой, сходной со строением биомембран.
В настоящее время в мире существует 10-15 сертифицированных наносистем, используемых в качестве переносчиков лекарств. На мировом фармацевтическом рынке присутствует несколько десятков препаратов, в основном противоопухолевых (дауномицин, доксорубицин, винкристин, аннамицин и третиноин), снабженных липосомальной системой транспорта. Диаметр липосом, как правило, составляет 100-400 нм, а форма выпуска - раствор для инъекций.
Необходимо отметить, что такой размер частиц делает препарат «доступным» для лизиса, ретикулоэндотелиальной системой (РЭС) клетки, что существенно снижает эффективность лекарств, а жидкая лекарственная форма усложняет их хранение и транспортировку.
Лизис препарата чаще всего преодолевается путем стабилизации поверхности полиэтиленгликолем (ПЭГ). Так, препарат келикс представляет собой пегилированную липосомальную форму доксорубицина с размером частиц порядка 100 нм. Пегилированные липосомы имеют липидную матрицу с низкой проницаемостью и внутреннюю водную буферную систему, что позволяет удерживать доксорубицин внутри липосомы во время циркуляции ее в кровотоке, исключая его контакт с компонентами плазмы. В то же время использование ПЭГ может привести к дополнительным побочным действиям.
Существует способ, снижающий подверженность фосфолипидных частиц клеткам РЭС и тем самым способствующий продлению циркуляции - это снижение размера частиц.
Например, при сравнении липосом трех размеров с диаметрами 400, 200 и 50 нм оказалось, что соотношение времени их клиренса существенно превышает соотношение размеров и составляет 1: 5: 38, т.е. снижение размера в 8 раз продлевает их циркуляцию в кровотоке почти в 40 раз (Drummond D. Et al., Pharmacol.Rev.,1999, 51, 691-743). Снижение размеров частиц повышает и растворимость в них инкапсулированного гидрофобного вещества: антимикробный гидрофобный агент растворяется вдвое активнее при переходе от частиц 2,4 микрон (2400 нм) к частицам в 800 нм (Muller R., Peters Int J. Pharm. 1998, 160, 229-237), что обусловлено повышением общей площади поверхности частиц и поверхностного натяжения (Kesisoglou F. et al., Adv Drug Delivery Rew, 2007, 59, 631-644).
Естественным логическим продолжением подобных разработок является переход от липосомальной лекарственной формы к композиции лекарственного средства на основе фосфолипидных наночастиц диаметром до 100 нм, т.е. к разработке наносистемы для включения биологически активных соединений, чему в настоящее время уделяется большое внимание.
Эффективность фосфолипидных наночастиц как коллоидальных переносчиков лекарств определяется в основном их размерами - оптимально 10-20 нм, что обеспечивает большую «рабочую поверхность» частицы и за счет этого - высокую емкость.
Фосфолипидные наночастицы представляют собой лиотропные жидкие кристаллы, которые, благодаря своей химической структуре, способны служить переносчиками как для растворимых, так и для нерастворимых в биологических жидкостях (гидрофобных) лекарственных препаратов. Встраивание лекарственных соединений в липидную матрицу наночастиц позволяет получить новые наноформы лекарственных препаратов с высокой эффективностью, биодоступностью и сниженными побочными действиями.
Высокая общая площадь поверхности, в сочетании с наноразмерами, создает оптимальные условия для взаимодействия таких частиц с клеткой. Кроме того, наноразмер создает уникальную возможность внедрения частиц в области щелевых межклеточных контактов, ширина которых в некоторых участках может составлять 30-50 нм. Благодаря этому появляется возможность доставки терапевтических агентов к недоступным для других лекарственных форм участкам пораженной например, опухолевой, ткани.
Близкие размеры, наряду с общим характером поверхности, фосфолипидных наночастиц и липопротеинов создают также оптимальные условия для их взаимодействия друг с другом. При этом фосфолипидные частицы, несущие лекарство, включаются в систему липопротеинов плазмы крови, с участием липид-транспортных белков, в результате чего молекулы липофильного лекарства вместе с фосфолипидами могут транспортироваться к частицам липопротеинов.
Проникновение лекарства, вместе с липопротеиновой или фосфолипидной частицей в клетку создает условия для его цитозольной доставки и доступа к субклеточным структурам, являющихся часто мишенью действия многих лекарств (Wasan, et.al., Antimicrob Agents Chemother. 1998, 42 (7):1646-53; Wasan, et.al., Nat Rev Dmg Discov. 2008, 7 (1):84-99).
Все выше сказанное убедительно иллюстрирует перспективность разработок фосфолипидных наносистем, инкорпорирующих лекарственные соединения.
Из существующего уровня техники известны диспергируемые стабилизированные фосфолипидом микрочастицы, представляющие собой быстродиспергируемую твердую дозированную форму, состоящую из нерастворимого в воде соединения в виде наномерных или микромерных твердых частиц, поверхность которых стабилизирована поверхностными модификаторами, например фосфолипидом, при этом частицы диспергированы в создающей объем матрице (патент РФ 2233654). Размер получаемых частиц составляет 0,66-10,6 мкм (660-10000 нм).
Известен также способ получения субмикронных частиц водонерастворимого или плохо растворимого органического фармацевтически активного соединения, включающий стадии растворения этого соединения в смешиваемом с водой первом растворителе, смешивания этого раствора со вторым растворителем, в который может быть добавлен фосфатидилхолин, и гомогенизации или гомогенизации в противотоке полученной предсуспензии или воздействия на нее ультразвуком (патент РФ 2272616). Размер получаемых частиц составляет 0,1-2,46 мкм (100-2500 нм).
В Европейском патенте ЕР 0556394 А1 описан способ получения лиофилизированного препарата для доставки лекарственных субстанций, на основе рафинированного соевого масла и рафинированного яичного фосфатилилхолина. Препарат легко растворяется в воде с образованием частиц с размером 10-100 нм и представляет собой жировую эмульсию.
Задачей настоящего изобретения является разработка технологии получения нежировой фосфолипидной композиции со средним диаметром липосомально-мицеллярных частиц 10-30 нм, обладающей низкой токсичностью, способной выдерживать длительное хранение и осуществлять транспорт лекарственных средств в организме.
В соответствии с изобретением описывается фосфолипидная композиция лекарственного средства в виде фосфолипидных наночастиц размером 10-30 нм, включающая фосфатидилхолин, мальтозу и лекарственное средство при следующем соотношении компонентов, мас.%:
Фосфатидилхолина 20-43
Мальтозы 55-78
Лекарственного средства 2-8
Описывается также способ получения фосфолипидной композиции лекарственного средства в форме фосфолипидных наночастиц размером 10-30 нм, включающей 20-43 мас.% фосфатидилхолина, 55-78 мас.% мальтозы 2-8 мас.% лекарственного средства, заключающийся в том, что в случае гидрофобного лекарственного средства фосфолипид и гидрофобное лекарственное средство растворяют в этаноле, отгоняют этанол, добавляют воду, суспензируют, добавляют мальтозу и полученную суспензию подвергают нескольким циклам гомогенизации под высоким давлением 800-1500 бар при температуре 40-50°С с последующей лиофилизацией.
В случае гидрофильного лекарственного средства фосфолипид, гидрофильное лекарственное средство и мальтозу суспензируют в воде и полученную суспензию подвергают нескольким циклам гомогенизации под высоким давлением 800-1500 бар при температуре 40-50°С с последующей лиофилизацией.
Количество циклов гомогенизации составляет, как правило, 10-25 циклов, и рН используемой воды находится в пределах 6,0-7,5.
Описывается также композиция для встраивания лекарственного средства в липидную матрицу в форме лиофильно высушенных фосфолипидных наночастиц размером 10-30 нм, включающая растительный фосфолипид с содержанием фосфатидилхолина 78-95% и мальтозу при следующем соотношении компонентов, мас.%
Фосфатидилхолина 20-43
Мальтозы 57-80
Описывается также способ получения фосфолипидной композиции в форме фосфолипидных наночастиц размером 10-30 нм, включающей 20-43 мас.%. Фосфатидилхолина и 57-80 мас.% мальтозы, заключающийся в том, что фосфолипид и водный раствор мальтозы суспензируют, затем суспензию подвергают нескольким циклам гомогенизации под высоким давлением 800-1500 бар при температуре 40-50°С с последующей лиофилизацией. Количество циклов гомогенизации составляет, как правило, 10-30 циклов, и рН используемой воды находится в пределах 6,0-7,5. Разработанная фармацевтическая композиция может быть использована для получения новых лекарственных форм как гидрофильных, так и гидрофобных лекарств разного спектра действия (противовоспалительные, противо-опухолевые и т.д.).
Предлагаемая технология позволит внедрить в медицинскую практику высокоэффективные лекарственные препараты нового поколения и, прежде всего, для лечения онкологических заболеваний.
Используемый фосфатидилхолин является основным компонентом высокоочищенного растительного соевого фосфолипида, содержание фосфатидилхолина в котором не менее 78-95 мас.%. Другие фосфолипидные компоненты могут содержаться в количествах, не превышающих допустимые (лизофосфатидилхолина до 4 мас.%, следовые количества других фосфолипидов).
В качестве вспомогательных фармакологически приемлемых веществ композиция содержит мальтозу для возможности получения лиофилизата, способного после растворения в физиологическом растворе или воде полностью восстанавливать свою структуру (в частности, размер частиц).
Согласно изобретению предложенная технология позволяет получать фармацевтические препараты с большей биодоступностью по сравнению с исходным лекарством.
Материалы и методы
В работе использовались следующие материалы:
1. Соевый фосфолипид марки Липоид С 100 фирмы Липоид, Германия.
2. Мальтозы моногидрат фирмы MERCK, Германия.
3. Вода для инъекций (по ФС №42-4587-95).
Технологические этапы получения:
I. Композиция для встраивания лекарственного средства в липидную матрицу в форме лиофильно высушенных фосфолипидных наночастиц
Навеску соевого фосфолипида добавляют к водному раствору мальтозы, гомогенизируют методом роторно-статорной гомогенизации в течение 3-х мин при температуре не выше 40°С до получения однородной мелкодисперсной первичной суспензии.
II. Для гидрофобных лекарственных субстанций
Навески фосфолипидов и гидрофобной лекарственной субстанции растворяют в достаточном объеме этанола (96%), затем спирт отгоняют на роторном испарителе. К полученной массе добавляют 10% водный раствор мальтозы, гомогенизируют методом роторно-статорной гомогенизации в течение 3-х мин при температуре не выше 40°С до получения однородной мелкодисперсной первичной суспензии. Затем суспензию помещают в приемную воронку гомогенизатора высокого давления. Гомогенизируют циклически при давлении 1000 бар ± 10%. Раствор между последующими циклами пропускают через холодильник с водяным охлаждением, не допуская его нагрева выше 55°С. Процесс гомогенизации продолжают до достижения прозрачности >60% (контролируют по светопропусканию при 660 нм в кювете с длиной оптического пути 1 см).
III. Для гидрофильных лекарственных субстанций
Навески исходных компонентов смешивают в воде, гомогенизируют методом роторно-статорной гомогенизации в течение 3-х мин при температуре не выше 40°С до получения однородной мелкодисперсной первичной суспензии.
IV. Получение фосфолипидных наночастиц
Получение фосфолипидных наночастиц осуществляют двумя способами: гомогенизацией высокого давления и флуодизацией.
Гомогенизацию проводят с помощью гомогенизатора высокого давления Ranie MiniLab 7.30 VH (Дания) или микрофлуодайзера Microfluidizer Processor, M110 EN-30K (США).
Первичную суспензию, полученную на предыдущей стадии (I, II или III), пропускают через гомогенизатор/микрофлуодайзер под давлением 1000 бар ± 10%. Температуру суспензии поддерживают в пределах 40-50°С.
Процесс гомогенизации (5-7 циклов) или микрофлуодизации (4-5 циклов) повторяют до достижения величины светопропускания рабочего раствора >60% (контролируют по светопропусканию при 660 нм в кювете с длиной оптического пути 1 см).
V. Фильтрация и стерильный розлив во флаконы
Полученный раствор фильтруют через фильтр 5 мкм, затем через стерильный фильтр 0,22 мкм (стерилизующая фильтрация) и разливают по 10 мл во флаконы в стерильных условиях.
VI. Лиофилизация
Флаконы с препаратом предукупоривают резиновыми пробками, помещают на полки лиофильной сушки. Продукт замораживают до -40°С, затем полки нагревают до +10°С. При вакууме 150 мТорр продукт сублимируют в течение 30 часов. Затем температуру полок повышают до +50°С и досушивают препарат при вакууме 50 мТорр в течение 8-10 часов. Высушенный препарат укупоривают под вакуумом, закатывают алюминиевыми колпачками, этикетируют и отбирают пробы для контроля качества аналитическими и микробиологическими методами.
Определение размера частиц фосфолипидной композиции для встраивания лекарственного средства и фосфолипидной композиции с включенной субстанцией.
1. 20 мкл препарата после гомогенизации (либо после растворения лиофильно высушенного препарата) добавляют к 3 мл воды.
2. Полученный раствор дегазируют (ультразвуком или под вакуумом), фильтруют через фильтр с размером пор 0, 45 мкм в измерительную стеклянную кювету.
3. Анализ размера частиц осуществляют методом фотонно-корреляционной спектроскопии на субмикронном анализаторе размера частиц N5 Beckman.
Определение содержания лекарственных субстанций в липосомальной фракции фосфолипидной композиции.
1. Фосфолипидный препарат с лекарственной субстанцией количественно вносят в патрон для ультрафильтрации с мембраной, пропускающей соединения с мол. весом менее 3000 дальтон и центрифугируют при 3000 об/мин в течение 5-10 мин. В результате чего свободное лекарство (с растворителем) проходит через мембрану в нижнюю часть - приемник-патрона, а лекарство в составе нанофосфолипидных частиц задерживается фильтром и остается в растворе над мембраной.
2. Содержание свободного лекарства во внесенном в патрон объеме определяют с помощью ВЭЖХ на хроматографе Миллихром А-02, анализируя его в растворе, прошедшем через мембрану. Для определения количества субстанции сопоставляют результаты ВЭЖХ с коллибровочной кривой для стандартных растворов препарата. Отношение этой величины к таковой для раствора, не подвергшегося ультрафильтрации, свидетельствует о соотношении свободного и встроенного в фосфолипидные наночастицы лекарства. Отсутствие субстанции в фильтрате свидетельствует о ее 100%-ном встраивании.
Изобретение иллюстрируется следующими примерами.
Пример 1. Получение фосфолипидной композиции с растворимой субстанцией (диклофенаком)
Смешивают 0,625 г диклофенака, 6,25 г фосфатидилхолина, 25 г мальтозы, доводят объем до 250 мл водой для инъекций. Гомогенизируют в течении 6 минут для получения первичной суспензии с помощью бытового блендера Braun. Полученную мелкодерсперсную суспензию помещают в микрофлуодайзер Microfluidizer Processor, MHO EN-30K (США), флуодизируют циклически (4-5 циклов) до достижения прозрачности >60% (контролируют по светопропусканию при 660 нм в кювете с длиной оптического пути 1 см), при давлении 1000 бар ± 10%, при температуре не выше 50°С. После стерилизации раствора путем последовательного пропускания через фильтр 0,45 мкм и стерильный фильтр 0,22 мкм раствор разливают в стерильных условиях во флаконы по 10 мл. По данным лазерного корреляционного спектрометра Beckman-Coulter №5 размер частиц составляет (11±5) нм. Встраивание диклофенака в фосфолипидные наночастицы составляет 96,4±0,3%. Затем препарат лиофильно высушивают. Для внутривенного введения животным содержимое одного флакона растворяют в 10 мл воды для инъекции. Размер частиц после регидратации лиофильно высушенного порошка сохраняется.
Пример 2. Получение фосфолипидной композиции с гидрофобной лекарственной субстанцией (будесонидом или преднизолоном)
0,2 г субстанции и 2,0 г фосфолипида растворяют в 80 мл этанола (96%). Спирт отгоняют на роторном испарителе. К полученной массе добавляют 150 мл 10% раствора мальтозы в воде и гомогенизируют в течении 8 минут для получения первичной суспензии с помощью бытового блендера Braun. Полученную мелкодерсперсную суспензию помещают в приемную воронку гомогенизатора высокого давления Ranie MiniLab 7.30 VH (Дания). Гомогенизируют циклически (6-7 циклов) до достижения прозрачности >60% (контролируют по светопропусканию при 660 нм в кювете с длиной оптического пути 1 см), при давлении 1000 бар ± 10%, при температуре не выше 50°С. После стерилизации раствора путем последовательного пропускания через фильтр 0,45 мкм и стерильный фильтр 0,22 мкм раствор разливают в стерильных условиях во флаконы по 10 мл. По данным лазерного корреляционного спектрометра Beckman-Coulter №5 размер частиц составляет 22±5 нм для будесонида и 18±3 нм для преднизолона. Встраивание в фосфолипидные наночастицы составляет 100% для будесонида и 95% для преднизолона. Затем препарат лиофильно высушивают. Для внутривенного введения животным содержимое одного флакона растворяют в 10 мл воды для инъекции. Размер частиц после регидратации лиофильно высушенного порошка сохраняется.
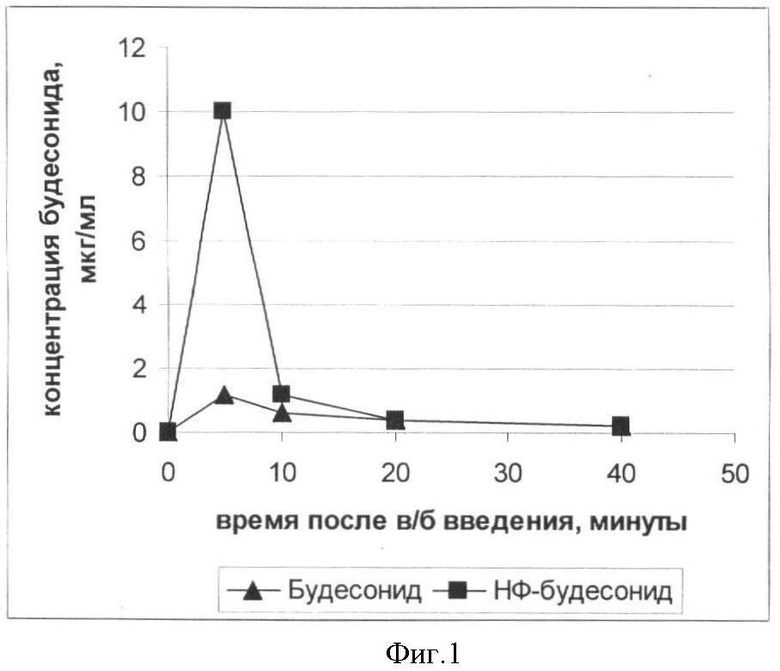
Пример 3. Сравнение биодоступности Будесонида свободного и Будесонида в виде нанофосфолипидных частиц (НФ-Будесонид) в эксперименте на крысах
Будесонид свободный и НФ-Будесонид готовят в виде 1%-ного спиртового раствора. Концентрация по будесониду составляет 2 мг/кг. Вводимый объем - 1 мл.
Животным весом 400-500 г вводят внутрибрюшинно растворы Будесонида и НФ-Будесонида в дозе 2 мг/кг. Через 5, 10, 20 и 40 минут осуществляют забор крови из хвостовой вены в пробирки с гепарином (5:1). Затем к 120 мкл гепаринизированной крови добавляют 880 мкл метанола, интенсивно перемешивают, центрифугируют, осаждают белки и другие компоненты крови, не растворимые в метаноле, и анализируют с помощью ВЭЖХ. Количественные измерения проводят, сопоставляя полученные данные с калибровочной кривой.
Приведенные на фиг.1 данные свидетельствуют о том, что будесонид в составе фосфолипидных наночастиц обладает значительно лучшей биодоступностью, чем его свободный аналог. Таким образом, нанофосфолипидная композиция на основе будесонида и фосфолипидов сои может быть рекомендована для дальнейших испытаний с целью создания новой лекарственной формы будесонида.
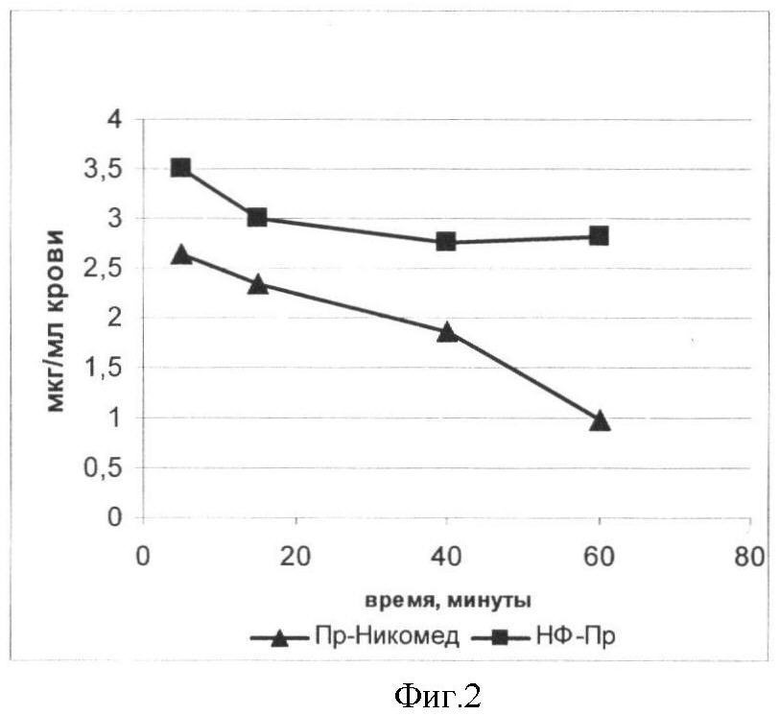
Пример 4. Сравнение биодоступности внутривенных препаратов Преднизолона-Никомед (Пр-Никомед) и Преднизолона в составе нанофосфолипидных частиц (НФ-Пр) в эксперименте на крысах
Пр-Никомед и НФ-Пр готовят в виде 1%-ного спиртового раствора. Концентрация по преднизолону составляет 2 мг/кг. Вводимый объем 1 мл.
Животным весом 400-500 г вводят внутривенно растворы НФ-Пр и Пр-Никомед в дозе 2 мг/кг. Через 5, 10, 20 и 40 минут осуществляют забор крови из хвостовой вены в пробирки с гепарином (5:1). Затем к 120 мкл гепаринизированной крови добавляют 880 мкл метанола, интенсивно перемешивают, центрифугируют, осаждают белки и другие компоненты крови, не растворимые в метаноле, и анализируют с помощью ВЭЖХ. Количественные измерения проводят, сопоставляя полученные данные с калибровочной кривой.
При внутривенном введении обоих препаратов для Преднизолона в составе нанофосфолипидных частиц элиминация из системного кровотока происходит значительно медленнее, чем для его свободного аналога (фиг.2).
В табл.1 представлено накопление в печени крыс Преднизолона и Преднизолона в составе фосфолипидных частиц через 15 мин. после их внутривенного введения. Показано, что накопление Преднизолона в составе фосфолипидных частиц меньше, чем свободного.
Накопление в печени крыс преднизолона и преднизолона в составе фосфолипидных частиц
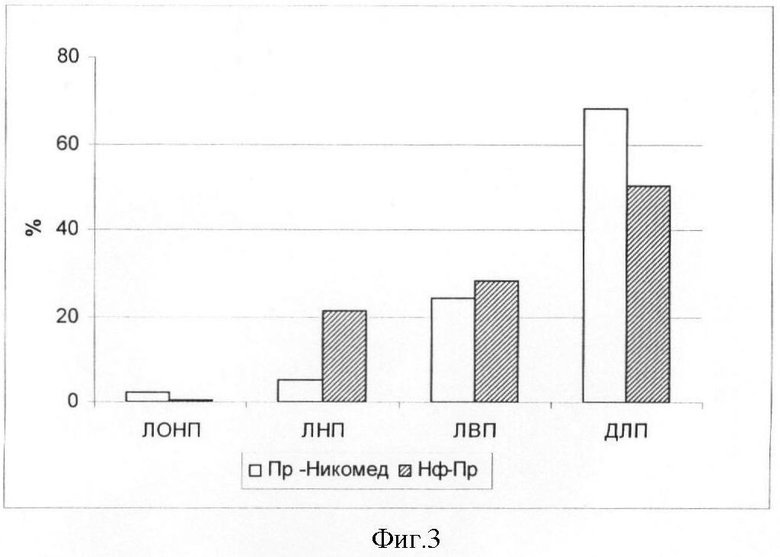
Пример 5. Изучение распределения Преднизолона-Никомед (Пр-Никомед) и Преднизолона в составе нанофосфолипидных частиц (НФ-Пр) по компонентам плазмы крови в эксперименте in vitro.
В экспериментах in vitro показано, что при инкубации Нф-Пр и Пр-Никомед в концентрации (0,5 мг/мл) с 3,7 мл плазмы крови человека при температуре 37°С в течение 30 мин наблюдается перераспределение лекарственной субстанции по компонентам крови (фиг.3). Наибольшее количество преднизолона в составе фосфолипидных частиц содержится в липидах низкой и высокой плотности. Наибольшее количество свободного преднизолона содержится в дилипидированной плазме.
Краткое содержание чертежей.
На фиг.1 представлена кинетика элиминации будесонида из крови подопытных животных при внутрибрюшинном введении свободного будесонида и будесонида в составе фосфолипидных частиц (НФ-Будесонид).
На фиг.2 изображена элиминация преднизолона после его внутривенного введения в виде свободного преднизолона (Пр-Никомед) и преднизолона в составе фосфолипидных частиц (Нф-Пр).
На фиг.3 представлена инкубация свободного преднизолона (Пр-Никомед) и преднизолона в составе фосфолипидных частиц (Нф-Пр) с плазмой крови в опыте in vitro. Условные сокращения:
ЛОНП - липопротеины очень низкой плотности
ЛНП - липопротеины низкой плотности
ЛВП - липопротеины высокой плотности
ДЛП - делипидированная плазма
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИЯ ДЛЯ ВСТРАИВАНИЯ ЛЕКАРСТВЕННЫХ СУБСТАНЦИЙ В ЛИПИДНУЮ МАТРИЦУ, КОМПОЗИЦИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА С ФОСФОЛИПИДНО-ЖИРНОКИСЛОТНОЙ СИСТЕМОЙ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2463056C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ АРБИДОЛ В СОСТАВЕ ФОСФОЛИПИДНЫХ НАНОЧАСТИЦ | 2009 |
|
RU2411942C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ДОКСОРУБИЦИНА И ФОСФОЛИПИДНЫХ НАНОЧАСТИЦ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2009 |
|
RU2411935C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РЕВМАТИЧЕСКИХ И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ НА ОСНОВЕ ИНДОМЕТАЦИНА, ВКЛЮЧЕННОГО В ФОСФОЛИПИДНЫЕ НАНОЧАСТИЦЫ | 2009 |
|
RU2417079C1 |
| Фармацевтическая композиция на основе глюкокортикостероида будесонида и фосфатидилхолина для сухой ингаляции | 2019 |
|
RU2730488C1 |
| КОМПОЗИЦИЯ НА ОСНОВЕ ДИГИДРОКВЕРЦЕТИНА, ВКЛЮЧЕННОГО В ФОСФОЛИПИДНЫЕ НАНОЧАСТИЦЫ | 2013 |
|
RU2536208C1 |
| ФОСФОЛИПИДНАЯ ЛЕКАРСТВЕННАЯ КОМПОЗИЦИЯ С НАНОРАЗМЕРОМ ЧАСТИЦ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ ЛИПИДНОГО ОБМЕНА И КОМАТОЗНЫХ СОСТОЯНИЙ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2010 |
|
RU2448715C1 |
| ИНДОМЕТАЦИН НА ОСНОВЕ ФОСФОЛИПИДНЫХ НАНОЧАСТИЦ ДЛЯ ПРИМЕНЕНИЯ В ОФТАЛЬМОЛОГИИ | 2011 |
|
RU2456979C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ ДЛЯ ФОТОДИНАМИЧЕСКОЙ ТЕРАПИИ В ФОРМЕ ФОСФОЛИПИДНЫХ НАНОЧАСТИЦ НА ОСНОВЕ ГЛЮКАМИНОВОЙ СОЛИ ХЛОРИНА Е6, МАЛЬТОЗЫ И ФОСФАТИДИЛХОЛИНА | 2014 |
|
RU2576025C1 |
| Липидное производное сарколизина в составе фосфолипидных наночастиц | 2017 |
|
RU2662086C1 |
Изобретение относится к медицине и фармакологии и касается стабильной при хранении наносистемы с размером частиц до 10-30 нм, включающей фосфатидилхолин растительного происхождения и мальтозу, предназначенной для включения в фосфолипидную наночастицу лекарственных средств, и способа ее получения и фосфолипидной композиции лекарственного средства в форме фосфолипидных наночастиц размером 10-30 нм, включающей фосфатидилхолин, мальтозу и лекарственное средство, и способа ее получения. 7 н. и 5 з.п. ф-лы, 3 ил., 1 табл.
1. Фосфолипидная композиция лекарственного средства в форме фосфолипидных наночастиц размером 10-30 нм, включающая фосфатидилхолин растительного происхождения (78-95%), мальтозу и лекарственное средство при следующем соотношении компонентов, мас.%:
2. Фосфолипидная композиция по п.1, отличающаяся тем, что лекарственным средством являются диклофенак, будесонид или преднизолон.
3. Способ получения фосфолипидной композиции по п.1, заключающийся в том, что в случае гидрофобного лекарственного средства фосфатидилхолин и гидрофобное лекарственное средство растворяют в этаноле, отгоняют этанол, добавляют воду, суспензируют, добавляют мальтозу и полученную суспензию подвергают нескольким циклам гомогенизации под высоким давлением 800-1500 бар при температуре 40-50°С с последующей лиофилизацией.
4. Способ по п.3, отличающийся тем, что лекарственным средством является будесонид или преднизолон; количество циклов гомогенизации составляет 10-25 циклов и используют воду с рН 6,0-7,5.
5. Способ получения фосфолипидной композиции по п.1, заключающийся в том, что в случае гидрофильного лекарственного средства фосфатидилхолин, гидрофильное лекарственное средство и мальтозу суспензируют в воде и полученную суспензию подвергают нескольким циклам гомогенизации под высоким давлением 800-1500 бар при температуре 40-50°С с последующей лиофилизацией.
6. Способ по п.5, отличающийся тем, что лекарственным средством является диклофенак; количество циклов гомогенизации составляет 10-25 циклов и используют воду с рН 6,0-7,5.
7. Фосфолипидная композиция для встраивания лекарственного средства в липидную матрицу в форме лиофильно высушенных фосфолипидных наночастиц размером 10-30 нм, включающая фосфатидилхолин растительного происхождения (78-95%) и мальтозу при следующем соотношении компонентов, мас.%:
8. Способ получения фосфолипидной композиции по п.7, заключающийся в том, что фосфатидилхолин и мальтозу суспензируют в воде и полученную суспензию подвергают нескольким циклам гомогенизации под высоким давлением 800-1500 бар при температуре 40-50°С с последующей лиофилизацией.
9. Способ получения фосфолипидной композиции лекарственного средства по п.1, заключающийся в том, что композицию для встраивания лекарственного средства в липидную матрицу по п.7 в случае гидрофобного лекарственного средства растворяют в воде и добавляют к растворенной в спирте и упаренной досуха на роторном испарителе лекарственной субстанции, затем суспензируют, полученную суспензию подвергают нескольким циклам гомогенизации под высоким давлением 800-1500 бар при температуре 40-50°С с последующей лиофилизацией.
10. Способ по п.9, отличающийся тем, что лекарственными средствами являются будесонид и преднизолон.
11. Способ получения фосфолипидной композиции лекарственного средства по п.1, заключающийся в том, что композицию для встраивания лекарственного средства в липидную матрицу по п.7 в случае гидрофильного лекарственного средства растворяют в воде вместе с лекарственной субстанцией, затем суспензируют, полученную суспензию подвергают нескольким циклам гомогенизации под высоким давлением 800-1500 бар при температуре 40-50°С с последующей лиофилизацией.
12. Способ по п.11, отличающийся тем, что лекарственным средством является диклофенак.
| СПОСОБ ПОЛУЧЕНИЯ ИНЪЕКЦИОННОЙ ЛЕКАРСТВЕННОЙ ФОРМЫ ФОСФОЛИПИДНОГО ПРЕПАРАТА "ФОСФОГЛИВ" ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ОСТРЫХ И ХРОНИЧЕСКИХ ЗАБОЛЕВАНИЙ ПЕЧЕНИ | 2005 |
|
RU2304430C2 |
| КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВАМИ РЕПАРИРОВАТЬ БИОЛОГИЧЕСКИЕ МЕМБРАНЫ | 1998 |
|
RU2133122C1 |
| ДИСПЕРГИРУЕМЫЕ СТАБИЛИЗИРОВАННЫЕ ФОСФОЛИПИДОМ МИКРОЧАСТИЦЫ | 1999 |
|
RU2233654C2 |
| Имитатор помех | 1973 |
|
SU556394A1 |

