Область техники, к которой относится изобретение
Область настоящего изобретения относится к способу разделения амлодипинов по оптической активности («оптического разделения») путем использования изопропанольного растворителя и оптически активной O,O'-дибензоил-винной кислоты в качестве стереоспецифичного (хирального) реагента и более конкретно относится к способу, включающему: (а) образование (R)- или (S)-дибензоил-виннокислой соли амлодипина или ее сольвата путем проведения реакции (R,S)-амлодипинов с оптически активной O,O'-дибензоил-винной кислотой в изопропанольном растворителе и (б) обработку соли (R)- или (S)-амлодипина основанием, что в результате дает оптически активный амлодипин.
Уровень техники
Амлодипин (это общепринятое название, относящееся к соединению приведенной ниже формулы 1) хорошо известен как блокатор кальциевых каналов длительного действия и поэтому он полезен для лечения сердечно-сосудистых расстройств, таких как стенокардическая гипертензия и застойная сердечная недостаточность.
Формула 1:
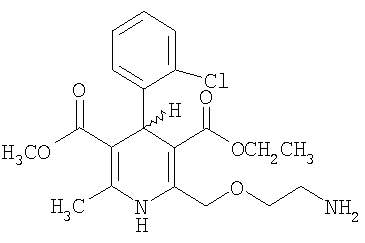
Амлодипин - это стереоспецифическое (хиральное) соединение с асимметричным центром. Как правило, энантиомерно чистый изомер обнаруживает фармацевтическую активность, превосходящую активность рацемической смеси. Его фармакологическая активность может быть различной, в зависимости от его стереоконформации и типа солей. (S)-(-)-изомер является более мощным блокатором кальциевых каналов, и было также установлено, что (R)-(+)-изомер эффективен в лечении или предотвращении атеросклероза.
Поэтому необходимо разработать способ получения энантиомерно чистого изомера из рацемических соединений, таких как амлодипин.
В качестве способа разделения амлодипина был описан способ разделения диастереомерного азидного эфира (a) [J.Е.Arrowsimth и др. // J. Med. Chem. 1986. Т.29. С.1696], способ разделения промежуточного соединения (б) с помощью карбоксилата цинхонидина [европейский патент № ЕР 0,331,315] и способ разделения диастереоизомерного амидного эфира (в) с помощью хроматографии [S.Goldman и др. // J. Med. Chem. 1992. Т.35. С.3341]. Однако считается, что ни один из этих способов непригоден для промышленного применения.

Ранее сообщалось о ряде усовершенствованных способов, применимых в промышленных условиях. Большинство этих способов включают процесс получения диастереомерной соли амплодипина с помощью D- или L-винной кислоты с последующим разделением с помощью подходящего растворителя. Эти способы могут быть полезны, поскольку возможно разделение с помощью только физического процесса, и соли также можно легко превратить обратно с помощью основания. Например, патент США №6046338 раскрывает способ разделения путем получения виннокислых солей в присутствии диметилсульфоксида (ДМСО). Патент США №6646131 раскрывает способ разделения через получение виннокислой соли с помощью замещенного дейтерием диметилсульфоксида (ДМСО-d6). Заявка на патент США №2003/0130321 А1 раскрывает способ разделения, основанный на получении виннокислой соли в присутствии диметилацетамида. В указанных выше способах в качестве хиральных агентов для получения (S)-амплодипина и (R)-амплодипина использованы соответственно D-винная кислота и L-винная кислота. Между тем заявка на патент США №2003/0176706 А1 и корейский патент №2004-23474 раскрывают способ получения (S)-амплодипина с помощью L-винной кислоты путем обработки профильтрованного раствора.
Хотя эти последние способы разделения обеспечивают относительно высокую оптическую чистоту, они не очень легки в промышленном применении, поскольку основаны на применении таких растворителей, как ДМСО, замещенный дейтерием ДМСО и диметилацетамид, которые довольно дороги, имеют высокую точку кипения и, скорее всего, остаются после завершения процесса разделения.
Авторы настоящего изобретения провели обширные исследования, чтобы разработать применимый в промышленности способ выделения каждого из оптических изомеров из рацемической смеси (R,S)-амлодипинов. В итоге было обнаружено, что у оптически активных солей амлодипина, приготовленных из (R,S)-амлодипинов и оптически активной O,O'-дибензоил-винной кислоты, обнаруживается значительное различие растворимостей в обычном растворителе, таком как изопропанол, что позволило завершить настоящее изобретение.
Таким образом, настоящее изобретение имеет целью предложить способ выделения оптически активных изомеров из (R,S)-амлодипинов.
Детальное описание изобретения
Настоящее изобретение относится к способу получения оптически активного амлодипина из (R,S)-амлодипинов оптическим разделением путем использования изопропанольного растворителя и O,O'-дибензоил-винной кислоты.
Далее представлено детальное описание настоящего изобретения.
Представленный здесь способ включает:
(а) приготовление оптически активной соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата (или ее сольвата) или (S)-(-)-амлодипин-геми-дибензоил-D-тартрата (или ее сольвата) путем проведения реакции (R,S)-амлодипинов с хиральным реагентом, соответственно дибензоил-L-винной кислотой или дибензоил-D-винной кислотой, в изопропанольном растворителе и
(б) получение оптически активного (R)-(+)-амлодипина или (S)-(-)-амлодипина путем обработки оптически активной соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата (или ее сольвата) или (S)-(-)-амлодипин-геми-дибензоил-D-тартрата (или ее сольвата) основанием.
Таким образом, в представленном здесь способе в качестве реакционного растворителя используется изопропанол, а в качестве хирального реагента - оптически активная O,O'-дибензоил-винная кислота.
По сравнению с обычно используемыми растворителями, такими как ДМСО, замещенный дейтерием ДМСО и диметилацетамид, изопропанольный растворитель является дешевым, имеет низкую точку кипения, практически не остается после проведения реакции и легко подвергается рециркуляции или очистке, что делает его чрезвычайно полезным в упрощении послеоперационных стадий.
Кроме того, оптически активная O,O'-дибензоил-винная кислота является хиральным соединением, и ее диастереозимерные соли имеют намного более высокую растворимость в изопропаноле. Поэтому два оптических изомера могут быть легко разделены на основании разницы в растворимости без использования каких-либо обычно применяемых растворителей, таких как ДМСО.
Далее приведено более детальное описание заявленного здесь способа. Согласно одному из вариантов осуществления изобретения, показанному на схеме 1, предусматривается способ, в котором проводится реакция (R,S)-амлодипинов с дибензоил-L-винной кислотой в изопропанольном растворителе с получением соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата, которую затем обрабатывают основанием, получая (R)-(+)-амлодипин.
Схема 1

После получения соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата в фильтрате имеется соль (S)-(-)-амлодипин-геми-дибензоил-L-тартрата или ее сольват. Таким образом, настоящее изобретение включает способ получения одного вида оптически активного амлодипина из фильтрата после получения другого оптически активного амлодипина. Конкретно, после получения, например, соли (S)-(-)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата путем перекристаллизации фильтрата соль обрабатывают основанием, чтобы получить (S)-(-)-амлодипин.
Согласно другому варианту осуществления изобретения, показанному на схеме 2, предусматривается способ, в котором проводится реакция (R,S)-амлодипинов с дибензоил-D-винной кислотой в изопропанольном растворителе с получением соли (S)-(-)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата, которую затем обрабатывают основанием для получения (S)-(-)-амлодипина.
Схема 2

В фильтрате после получения соли (S)-(-)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата имеется соль (R)-(+)-амлодипин-геми-дибензоил-D-тартрата или ее сольват. Таким образом, настоящее изобретение включает способ получения одного вида оптически активного амлодипина из фильтрата после получения другого оптически активного амлодипина. Конкретно, после получения, например, соли (R)-
(+)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата путем перекристаллизации оставшегося раствора соль обрабатывают основанием для получения (S)-(-)-амлодипина.
В схемах 1 и 2 дибензоил-L-винная кислота или дибензоил-D-винная кислота могут быть использованы в количестве от 0,2 до 0,6 моля на 1 моль (R,S)-амлодипинов. Когда какой-нибудь один изомер (R)- или (S)-амлодипина является целевым материалом, подлежащим выделению, предпочтительно использовать хиральный реагент в количестве от 0,2 до 0,4 моля, более предпочтительно от 0,2 до 0,3 моля. Однако если оба изомера: и (R)-, и (S)-амлодипины - являются подлежащим выделению целевым материалом, предпочтительно использовать хиральный реагент в количестве от 0,4 до 0,6 моля, более предпочтительно - от 0,5 до 0,6 моля.
В то же время возможно одновременное получение и (R)-, и (S)-амлодипина из (R,S)-амлодипинов в соответствии со схемами 3 и 4.
Как показано на схеме 3, проводится реакция (R,S)-амлодипинов с дибензоил-D-винной кислотой в изопропанольном растворителе с получением соли (S)-(-)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата путем фильтрации. Фильтрат подвергают реакции с дибензоил-L-винной кислотой с получением соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата путем фильтрации. Каждую оптически активную соль амлодипина или ее сольват обрабатывают основанием для одновременного получения (R)-амлодипина и (S)-амлодипина.
Схема 3

Как показано на схеме 4, проводится реакция (R,S)-амлодипинов с дибензоил-L-винной кислотой в изопропанольном растворителе с получением соли (R)-(+)-амлопидин-геми-дибензоил-L-тартрата или ее сольвата путем фильтрации. Фильтрат подвергают реакции с дибензоил-D-винной кислотой с получением соли (S)-(-)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата путем фильтрации. Каждую оптически активную соль амлопидина или ее сольват обрабатывают основанием для одновременного получения (R)-амлодипина и (S)-амлодипина.
Схема 4

В схемах 3 и 4 дибензоил-L-винная кислота или дибензоил-D-винная кислота предпочтительно должны быть использованы в количестве от 0,2 до 0,4 моля, более предпочтительно от 0,2 до 0,3 моля на 1 моль (R,S)-амлодипинов. Если количество хирального агента ниже или выше значений этого интервала, трудно оптимизировать выход и оптическую чистоту оптически активных солей.
В предложенном здесь способе в качестве реакционного растворителя использован изопропанол. Изопропанольным растворителем может быть сам изопропанол или смесь изопропанола и сорастворителя. В качестве сорастворителя могут быть использованы вода, кетоны, спирты, простые эфиры, амиды, сложные эфиры, углеводороды, хлоруглеводороды и нитрилы.
Предпочтительные примеры кетонов включают (но не ограничиваются ими) ацетон и метилэтилкетон (МЭК). Предпочтительные примеры спиртов включают (но не ограничиваются им) С1-С7 насыщенный спирт, такой как изопропанол. Предпочтительные примеры простых эфиров включают (но не ограничиваются ими) диэтиловый эфир и тетрагидрофуран (ТГФ). Предпочтительные примеры амидов включают (но не ограничиваются ими) N,N-диметилформамид (ДМФ), N,N-диметилацетамид (ДМАЦ) и N,N'-диметилпропилен-мочевину (ДМПМ). Предпочтительные примеры сложных эфиров включают (но не ограничиваются им) этилацетамид (EtOAc). Предпочтительные примеры углеводородов включают (но не ограничиваются им) C5-С10 углеводород, такой как гексан и толуол. Предпочтительные примеры хлоруглеводородов включают (но не ограничиваются ими) хлороформ, дихлорметан, 1,2-дихлорэтан и 1,1,1-трихлорэтан. Предпочтительные примеры нитрилов включают (но не ограничиваются ими) С2-С7 нитрилы, такие как ацетонитрил и пропиононитрил.
Конкретные примеры сорастворителей включают (но не ограничиваются ими) воду, ацетон, ацетонитрил, пропиононитрил, диметилсульфоксид, диметилацетамид, метилэтилкетон, тетрагидрофуран, этилацетат, дихлорметан, диметилформамид, гексан, толуол, метанол, этанол, трет-бутанол и N,N'-диметилпропилен-мочевину.
Кроме того, выбор сорастворителя зависит от его вида и может быть легко сделан специалистом в данной области. Предпочтительно, чтобы сорастворитель добавлялся в объеме менее 50% от объема изопропанола. Избыточное количество совместного растворителя, превышающее 50%, может вызвать заметное снижение оптической чистоты вследствие снижения различий в растворимости.
Здесь в ходе процесса разделения получают дибензоил-тартратную соль амлодипина или ее сольват. Оптически активные соли амлодипина, а именно (R)-(+)-амлодипин-геми-дибензоил-L-тартрат, (S)-(-)-амлодипин-геми-дибензоил-L-тартрат, (S)-(-)-амлодипин-геми-дибензоил-D-тартрат и (R)-(+)-амлодипин-геми-дибензоил-D-тартрат, все находятся в пределах охвата настоящего изобретения.
Соли амлодипина могут быть получены из реакционного раствора обычным способом, например фильтрацией, центрифугированием и декантацией. Среди них предпочтительно использовать фильтрацию или центрифугирование, а предпочтительнее - фильтрование. Как хорошо известно в данной области, способ выделения одного оптического изомера может быть также применен для выделения других оптических изомеров.
Далее, оптически чистые изомеры амлодипина могут быть получены путем обработки оптически активных солей амлопидина или их сольватов основанием. Перед обработкой основанием для повышения оптической чистоты можно провести перекристаллизацию оптически активных солей амлопидина или их сольватов.
В качестве растворителя для перекристаллизации может быть использован реакционный растворитель, то есть только изопропанол или смесь изопропанола с совместным растворителем. В качестве основания можно использовать гидроксид, оксид, карбонат, бикарбонат или амид щелочного или щелочноземельного металла. Предпочтительно можно использовать гидроксид или оксид щелочного металла, а наиболее предпочтительно - гидроксид натрия.
Кроме того, в пределах охвата настоящего изобретения находится разделение и фильтрование оптически активной соли амлодипина или ее сольвата из раствора, оставшегося после приготовления другой оптически активной соли амлодипина или ее сольвата путем фильтрации, центрифугирования или декантации. Конкретно, оставшийся после отделения или частичного отделения определенного оптического изомера раствор содержит также его оптический антипод. Поэтому к оставшемуся раствору добавляют оптический антипод использованной оптически активной O,O'-дибензоил-винной кислоты, что дает возможность получить другую оптически активную соль амлопидин-дибензоил-винной кислоты или ее сольват. Таким путем в настоящем изобретении могут быть одновременно получены два оптических изомера.
Примеры
Настоящее изобретение более конкретно описано в следующих примерах. Подразумевается, что примеры здесь только иллюстрируют настоящее изобретение, но ни в коей мере не ограничивают заявленное в формуле изобретение.
В нижеследующих примерах степень оптической чистоты определяли методом хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ). Для разделения использовали следующие условия ВЭЖХ:
- Колонка: Ultron ES-OVM (Ovomucoid Corp.), 15 см;
- Скорость протока 0,1 мл/мин.;
- Детектирование при длине волны 360 нм;
- Мобильная фаза: буфер двузамещенного фосфата натрия (20 нМ, pH 7)/ацетонитрил (80/20 по объему);
- Образцы растворяли при концентрации 0,1 мг/мл в смеси ацетонитрила с водой (50/50 по объему).
Пример 1
Получение (S)-(-)-амлодипин-геми-дибензоил-D-тартрата из (R,S)-амлодипинов
163,6 г (R,S)-амлодипинов растворяли в 3 л смеси ацетонитрила с изопропанолом (1/9) и перемешивали при 55°С. К раствору добавляли 35,8 г (0,25 молярного эквивалента) дибензоил-D-винной кислоты в 1 л смеси ацетонитрила с изопропанолом (1/9 по объему) и дополнительно перемешивали в течение 10 мин. Добавляли 0,2 г (S)-(-)-амлодипин-геми-дибензоил-D-тартрата (>99,5% d.e.) и перемешивали раствор в течение ночи при комнатной температуре. Твердый осадок отделяли фильтрованием, собирали, промывали 500 мл смеси ацетонитрила с изопропанолом (1/9 по объему) и сушили в течение ночи под вакуумом при 50°С, получая 97,6 г (8)-(-)-амлодипин-геми-дибензоил-D-тартрата (теоретический выход 83%).
Т.пл.: 116-118°С; состав установленный: С 59,12%, Н 5,50%, N 4,62%; состав рассчитанный для C20H25N2O5Cl·0,5[C18H14O8]: С 59,23%, Н 5,49%, N 4,76%; данные хиральной ВЭЖХ: 99,2% d.e.
Пример 2
Получение (S)-(-)-амлодипина из (S)-(-)-амлодипин-геми-дибензоил-D-тартрата
9 г полученного в примере 1 (S)-(-)-амлодипин-геми-дибензоил-D-тартрата растворяли в смеси 90 мл CH2Cl2 и 90 мл 2 н водного раствора NaOH и перемешивали в течение 30 мин. полученный органический слой отделяли и промывали один раз водой. CH2Cl2 выпаривали в вакууме и добавляли гексан, получая шламм. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 5,76 г (S)-(-)-амлодипина (92%).
Т.пл.: 107-109°С; состав установленный: С 58,64%, Н 6,25%, N 6,79%; состав рассчитанный для C20H25N2O5Cl: C 58,75%, Н 6,16%, N 6,85%; данные хиральной ВЭЖХ: 99,2% е.е.
Пример 3
Получение (S)-(-)-амлодипин-геми-дибензоил-D-тартрата из (R,S)-амлодипинов
163,6 г (R,S)-амлодипинов растворяли в 3 л смеси ацетонитрила с изопропанолом (1/9 по объему) и перемешивали при 55°С. К раствору добавляли 71,6 г (0,5 молярного эквивалента) дибензоил-D-винной кислоты, растворенной в 1 л смеси ацетонитрила с изопропанолом (1/9 по объему) и перемешивали в течение 10 мин. Добавляли 0,2 г (S)-(-)-амлодипин-геми-дибензоил-D-тартрата (>99,5% d.e.) и оставляли раствор при комнатной температуре в течение 18 ч, чтобы достигнуть равновесия. Твердое вещество отделяли фильтрованием, собирали и промывали 500 мл смеси ацетонитрила с изопропанолом (1/9 по объему). Твердый осадок сушили в течение ночи в вакууме при 50°С, получая 96,4 г (теоретический выход 78%) (S)-(-)-амлодипин-геми-дибензоил-D-тартрата. Данные хиральной ВЭЖХ: 90,0% d.e.
Пример 4
Перекристаллизация (S)-(-)-амлодипин-геми-дибензоил-D-тартрата 96,4 г, полученного в примере 3 (S)-(-)-амлодипин-геми-дибензоил-D-тартрата, растворяли в 4 л изопропанола при нагревании. Добавляли 0,2 г (S)-(-)-амлодипин-геми-дибензоил-D-тартрата (>99,5% d.e.), раствор оставляли на 4 ч для достижения равновесия при комнатной температуре. Твердое вещество отделяли фильтрованием, собирали и промывали 500 мл изопропанола. Твердый осадок сушили в течение ночи в вакууме при 50°С, получая 81,8 г (теоретический выход 89%) (S)-(-)-амлодипин-геми-дибензоил-D-тартрата. Данные хиральной ВЭЖХ: 99,2% d.e.
Пример 5
Извлечение (R)-(+)-амлопидин-геми-дибензоил-D-тартрата из фильтрата
Оставшийся после извлечения (S)-(-)-амлодипин-геми-дибензоил-D-тартрата в примере 3 фильтрат обрабатывали следующим образом.
К фильтрату добавляли 0,2 г (R)-(+)-амлодипин-геми-дибензоил-D-тартрата (>99,5% d.e.) и перемешивали при комнатной температуре в течение 2 ч. Раствор концентрировали упариванием растворителя до приблизительно 1/5 его исходного объема. Добавляли 2 л изопропанола и оставляли раствор для достижения равновесия на 4 ч при 5°С. Твердое вещество отделяли фильтрованием, собирали и сушили в вакууме при 50°С, получая 78,9 г (теоретический выход 67%) (R)-(+)-амлодипин-геми-дибензоил-D-тартрата.
Т.пл.: 116-118°С; состав установленный: С 59,15%, Н 5,54%, N 4,58%, состав рассчитанный для C20H25N2O5Cl·0,5[C18H14O8]: С 59,23%, Н 5,49%, N 4,76%; данные хиральной ВЭЖХ: 97,4% d.e.
Пример 6
Получение (R)-(+)-амлодипина из (R)-(+)-амлодипин-геми-дибензоил-D-тартрата
9 г полученного в примере 5 (R)-(+)-амлодипин-геми-дибензоил-D-тартрата добавляли к смеси 90 мл CH2Cl2 и 90 мл 2 н водного раствора NaOH и перемешивали в течение 30 мин. Органический раствор отделяли и промывали один раз водой. CH2Cl2 выпаривали в вакууме и добавляли гексан, получая шламм. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 5,69 г (выход 91%) (R)-(+)-амлодипина. Данные хиральной ВЭЖХ: 97.4% е.е.
Пример 7
Получение (R)-(+)-амлодипин-геми-дибензоил-L-тартрата из (R,S)-амлодипинов
163,6 г (R,S)-амлодипинов растворяли в 3 л смеси ацетонитрила и изопропанола (1/9 по объему) и перемешивали при 55°С. Добавляли раствор 35,8 г (0,25 молярного эквивалента) дибензоил-L-винной кислоты в 1 л смеси ацетонитрила и изопропанола (1/9 по объему) и перемешивали смесь в течение 10 мин. Добавляли 0,2 г (R)-(+)-амлодипин-геми-дибензоил-L-тартрата (>99,5% d.e.) и перемешивали раствор в течение ночи при комнатной температуре. Твердое вещество отделяли, собирали, промывали 500 мл смеси ацетонитрила и изопропанола (1/9 по объему) и сушили в течение ночи в вакууме при 50°С, получая 90,0 г (теоретический выход 77%) (R)-(+)-амлодипин-геми-дибензоил-L-тартрата.
Т. пл.: 115-117°С; состав установленный: С 59,17%, Н 5,65%, N 4,64%; состав рассчитанный для C20H25N2O5Cl·0,5[C18H14O8]: С 59,23%, Н 5,49%, N 4,76%; данные хиральной ВЭЖХ: 98,5% d.e.
Пример 8
Получение (R)-(+)-амлодипина из (R)-(+)-амлодипин-геми-дибензоил-L-тартрата
9 г полученного в примере 7 (R)-(+)-амлодипин-геми-дибензоил-L-тартрата вносили в смесь 90 мл CH2Cl2 и 90 мл 2 н водного раствора NaOH и перемешивали в течение 30 мин. Органический раствор отделяли и промывали один раз водой. Отгоняли CH2Cl2 и добавляли гексан для получения шламма. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 5,81 г (93%) (R)-(+)-амлодипина.
Т.пл.: 108-110°С; состав установленный: С 58,57%, Н 6,37%, N 6,76%; состав рассчитанный для C20H25N2O5Cl: С 58,75%, Н 6,16%, N 6,85%; данные хиральной ВЭЖХ: 98,7% е.е.
Пример 9
Получение (R)-(+)-амлодипин-геми-дибензоил-L-тартрата из (R,S)-амлодипинов
4,09 г (R,S)-амлодипинов растворяли в 100 мл смеси ацетонитрила и изопропанола (1/9 по объему) и перемешивали при 50°С. Добавляли раствор 1,79 г (0,5 молярного эквивалента) дибензоил-L-винной кислоты в 50 мл смеси ацетонитрила и изопропанола (1/9 по объему) и перемешивали смесь в течение 10 мин. Добавляли 0,002 г(R)-(+)-амлодипин-геми-дибензоил-L-тартрата (>99,5% d.e.) и перемешивали раствор при комнатной температуре в течение 18 ч. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 2,88 г (теоретический выход 98%) (R)-(+)-амлодипин-геми-дибензоил-L-тартрата; данные хиральной ВЭЖХ: 97,6% d.e.
Пример 10
Получение (R)-(+)-амлодипина из (R)-(+)-амлодипин-геми-дибензоил-L-тартрата
2 г полученного в примере 9 (R)-(+)-амлодипин-геми-дибензоил-L-тартрата растворяли в смеси 20 мл CH2Cl2 и 20 мл 2 н водного раствора NaOH и перемешивали в течение 30 мин. Органический раствор отделяли и промывали один раз водой. Выпаривали CH2Cl2 в вакууме и добавляли гексан для получения шламма. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 1,28 г (92%) (R)-(+)-амлодипина.
Т.пл.: 108-110°С; данные хиральной ВЭЖХ: 97,8% е.е.
Пример 11
Извлечение (S)-(-)-амлодипин-геми-дибензоил-L-тартрата из оставшегося раствора (фильтрата)
Фильтрат, оставшийся после сбора (R)-(+)-амлодипин-геми-дибензоил-L-тартрата в примере 9, обрабатывали следующим образом.
Добавляли 0,002 г (S)-(-)-амлодипин-геми-дибензоил-L-тартрата (>99,5% d.e.) и перемешивали раствор при комнатной температуре в течение 2 ч. Раствор концентрировали выпариванием растворителя до приблизительно 1/5 его первоначального объема. Добавляли 50 мл изопропанола и раствор оставляли в течение 4 ч при 5°С для установления равновесия. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 1,76 г (теоретический выход 60%) (S)-(-)-амлодипин-геми-дибензоил-L-тартрата.
Т.пл.: 114-116°С; состав установленный: С 59,10%, Н 5,52%, N 4,59%, состав рассчитанный для C20H25N2O5Cl·0,5[C18H14O8]: С 59,23%, Н 5,49%, N 4,76%; данные хиральной ВЭЖХ: 97,5% d.e.
Пример 12
Получение (S)-(-)-амлодипина из (S)-(-)-амлодипин-геми-дибензоил-L-тартрата
1 г полученного в примере 11 (S)-(-)-амлодипин-геми-дибензоил-L-тартрата добавляли к смеси 10 мл CH2Cl2 и 10 мл 2 н водного раствора NaOH и перемешивали раствор в течение 30 мин. Образовавшийся органический слой отделяли и промывали один раз водой. Выпаривали CH2Cl2 в вакууме и добавляли гексан для образования шламма. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 0,64 г (92%) (S)-(-)-амлодипина.
Т.пл.: 107-109°С; данные хиральной ВЭЖХ: 97,5% е.е.
Пример 13
Одновременное получение (S)-(-)-амлодипин-геми-дибензоил-D-тартрата и (R)-(+)-амлодипин-геми-дибензоил-L-тартрата из (R,S)-амлодипинов 163,6 г (R,S)-амлодипинов растворяли в 2 л смеси ацетонитрила и изопропанола (1/9 по объему) и перемешивали при 55°С. Добавляли раствор 35,8 г (0,25 молярного эквивалента) дибензоил-D-винной кислоты в 1 л смеси ацетонитрила и изопропанола (1/9 по объему) и перемешивали смесь в течение 10 мин. Добавляли 0,05 г (S)-(-)-амлодипин-геми-дибензоил-D-тартрата (>99,5% d.e.), перемешивали при комнатной температуре в течение 16 ч и дополнительно перемешивали в течение 6 ч при 0-5°С. Твердое вещество отделяли фильтрованием, собирали, промывали 500 мл смеси ацетонитрила и изопропанола (1/9 по объему) и сушили в течение ночи в вакууме при 50°С, получая 114,7 г (теоретический выход 97,5%) (S)-(-)-амлодипин-геми-дибензоил-D-тартрата.
Т.пл.: 116-118°С; состав установленный: С 59,17%, Н 5,51%, N 4,70%; состав,рассчитанный для C20H25N2O5Cl·0,5[C18H14O8]: С 59,23%, Н 5,49%, N 4,76%; данные хиральной ВЭЖХ: >98% d.e.
Оставшийся фильтрат обрабатывали следующим образом. Добавляли 35,8 г (0,25 молярного эквивалента) дибензоил-L-винной кислоты и перемешивали раствор при 60°С в течение 10 мин. Добавляли 0,05 г (R)-(+)-амлодипин-геми-дибензоил-L-тартрата (>99,5% d.e.) и перемешивали смесь в течение 5 ч, охлаждая ее от 60°С до 30°С. Твердое вещество отделяли фильтрованием, собирали, промывали 500 мл смеси ацетонитрила и изопропанола (1/9 по объему) и сушили в течение ночи в вакууме при 50°С, получая 98,9 г (теоретический выход 84,1%) (R)-(+)-амлодипин-геми-дибензоил-L-тартрата.
Т.пл.: 115-117°С; состав установленный: С 59,14%, Н 5,56%, N 4,63%; состав, рассчитанный для C20H25N2O5Cl·0,5[C18H14O8]: С 59,23%, Н 5,49%, N 4,76%; данные хиральной ВЭЖХ: >99% d.e.
Пример 14
Получение (S)-(-)-амлопидина из (S)-(-)-амлопидин-геми-дибензоил-D-тартрата
9 г полученного в примере 13 (S)-(-)-амлодипин-геми-дибензоил-D-тартрата перемешивали в смеси 90 мл CH2Cl2 и 90 мл 2 н водного раствора NaOH в течение 30 мин. Образовавшийся органический слой отделяли и промывали один раз водой. Выпаривали CH2Cl2 в вакууме и добавляли гексан, чтобы получить шламм. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 5,69 г (91%) (S)-(-)-амлодипина.
Т.пл.: 107-109°С; данные хиральной ВЭЖХ: >98% е.е.
Пример 15
Получение (R)-(+)-амлодипина из (R)-(+)-амлодипин-геми-дибензоил-L-тартрата
9 г полученного в примере 13 (R)-(+)-амлодипин-геми-дибензоил-L-тартрата перемешивали в смеси 90 мл CH2Cl2 и 90 мл 2 н водного раствора NaOH в течение 30 мин. Образовавшийся органический слой отделяли и промывали один раз водой. Выпаривали CH2Cl2 в вакууме и добавляли гексан, чтобы получить шламм.
Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 5,63 г (90%) (R)-(+)-амлодипина.
Т.пл.: 108-110°С; данные хиральной ВЭЖХ: >99% е.е.
Пример 16
Одновременное получение (R)-(+)-амлодипин-геми-дибензоил-L-тартрата и (S)-(-)-амлодипин-геми-дибензоил-D-тартрата из (R,S)-амлодипинов
163,6 г (R,S)-амлопидинов растворяли в 2 л смеси ацетонитрила и изопропанола (1/9 по объему) и перемешивали при 55°С. К раствору добавляли 35,8 г (0,25 молярного эквивалента) дибензоил-L-винной кислоты в 1 л смеси ацетонитрила и изопропанола (1/9 по объему) и перемешивали в течение 10 мин. Добавляли 0,05 г (R)-(+)-амлодипин-геми-дибензоил-L-тартрата (>99,5% d.e.), перемешивали при комнатной температуре в течение 16 ч и дополнительно перемешивали в течение 6 ч при 0-5°С. Твердое вещество отделяли фильтрованием, собирали, промывали 500 мл смеси ацетонитрила и изопропанола (1/9 по объему) и сушили в течение ночи в вакууме при 50°С, получая 113,2 г (теоретический выход 96,2%) (R)-(+)-амлодипин-геми-дибензоил-L-тартрата.
Фильтрат обрабатывали следующим образом. Добавляли 35,8 г (0,25 молярного эквивалента) дибензоил-D-винной кислоты и перемешивали при 60°С в течение 10 мин. Добавляли 0,05 г (S)-(-)-амлодипин-геми-дибензоил-D-тартрата (>99,5% d.e.) и перемешивали в течение 5 ч, охлаждая от 60°С до 30°С. Твердое вещество отделяли фильтрованием, собирали, промывали 500 мл смеси ацетонитрила и изопропанола (1/9 по объему) и сушили в течение ночи в вакууме при 50°С, получая 96,5 г (теоретический выход 82,0%) (S)-(-)-амлодипин-геми-дибензоил-D-тартрата.
Пример 17
Получение (R)-(+)-амлодипина из (R)-(+)-амлодипин-геми-дибензоил-L-тартрата
9 г полученного в примере 16 (R)-(+)-амлодипин-геми-дибензоил-L-тартрата перемешивали в смеси 90 мл CH2Cl2 и 90 мл 2 н водного раствора NaOH в течение 30 мин. Образовавшийся органический слой отделяли и промывали один раз водой. Выпаривали CH2Cl2 в вакууме и добавляли гексан, чтобы получить шламм. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 5,70 г (91,1%) (R)-(+)-амлодипина.
Пример 18
Получение (S)-(-)-амлодипина из (S)-(-)-амлодипин-геми-дибензоил-D-тартрата
9 г полученного в примере 16 (S)-(-)-амлодипин-геми-дибензоил-D-тартрата перемешивали в смеси 90 мл CH2Cl2 и 90 мл 2 н водного раствора NaOH в течение 30 мин. Образовавшийся органический слой отделяли и промывали один раз водой. Выпаривали CH2Cl2 в вакууме и добавляли гексан, чтобы получить шламм. Твердое вещество отделяли фильтрованием, собирали и сушили в течение ночи в вакууме при 50°С, получая 5,65 г (90,3%) (S)-(-)-амлодипина.
Экспериментальный пример
Степени оптической чистоты полученных солей в зависимости от растворителей и хиральных реагентов
Описанный в примере 1 способ получения повторяли, получая соли амлодипина с использованием различных растворителей, как показано в таблице. Составы смешанных растворителей указаны в объемных процентах.
Таблица показывает, что оптическая чистота остается сравнительно высокой, если в качестве реакционного растворителя используется один изопропанол или смесь изопропанола и сорастворителя, а в качестве хирального агента используется дибензоил-D-винная кислота или дибензоил-L-винная кислота.
Кроме того, из нее также видно, что использование этанола вместо изопропанола заметно снижает оптическую чистоту, что ясно указывает на значительную разницу растворимостей в изопропаноле.
Таким образом, подтверждено, что в настоящем изобретении очень важен выбор реакционного растворителя и хирального реагента.
Как указано выше, в настоящем изобретении для разделения (R,S)-амлодипинов применены изопропанол с низкой точкой кипения в качестве реакционного растворителя и O,O'-дибензоил-винная кислота в качестве хирального реагента, что обеспечивает эффективное разделение оптических изомеров на основании их различной растворимости. Кроме того, к оставшемуся раствору (фильтрату) добавляют оптический антипод примененной оптически активной O,O'-дибензоил-винной кислоты, благодаря чему извлекается другая оптически активная соль амлодипина или ее сольват.
Главное то, что оптически активная дибензоил-виннокислая соль амлодипина или ее сольват, которая является промежуточным продуктом, может быть подвергнута перекристаллизации до обработки основанием, что позволяет получить высокую степень оптической чистоты.
Таким образом, способ разделения (R,S)-амлодипинов согласно настоящему изобретению в высокой степени пригоден для промышленного применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНОГО ГЕНТИЗАТА АМЛОДИПИНА | 2006 |
|
RU2393150C2 |
| СПОСОБ ВЫДЕЛЕНИЯ R - /+/ - И S - /-/ - ИЗОМЕРОВ АМЛОДИПИНА ИЗ ИХ СМЕСЕЙ, СОЛЬВАТЫ И МОНОГИДРАТЫ R - /+/ - И S - /-/ - АМЛОДИПИН - ГЕМИ-L- ИЛИ D -ТАРТРАТА | 1995 |
|
RU2132845C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРА АМЛОДИПИНА ВЫСОКОЙ ОПТИЧЕСКОЙ ЧИСТОТЫ | 2005 |
|
RU2376288C2 |
| (S)-(-)-АМЛОДИПИНА КАМЗИЛАТ ИЛИ ЕГО ГИДРАТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ИХ | 2007 |
|
RU2403241C1 |
| ТРАНСИЗОМЕРНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПОЛУЧЕНИЕ | 2018 |
|
RU2759443C2 |
| АМЛОДИПИНА ГЕНТИЗАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2330843C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО ЧИСТЫХ 1,4-ДИГИДРОПИРИДИНОВ, ЭНАНТИОМЕРНО ЧИСТЫЕ 1,4-ДИГИДРОПИРИДИНЫ И ИЗОТИОУРЕИДЫ | 1990 |
|
RU2069658C1 |
| СПОСОБ ПОЛУЧЕНИЯ (4S)-4-(4-ЦИАНО-2-МЕТОКСИФЕНИЛ)-5-ЭТОКСИ-2,8-ДИМЕТИЛ-1,4-ДИГИДРО-1,6-НАФТИРИДИН-3-КАРБОКСАМИДА ПОСРЕДСТВОМ РАЗДЕЛЕНИЯ РАЦЕМАТА ПРИ ПОМОЩИ ДИАСТЕРЕОМЕРНОГО СЛОЖНОГО ЭФИРА ВИННОЙ КИСЛОТЫ | 2019 |
|
RU2805573C2 |
| СПОСОБ РАЗДЕЛЕНИЯ ОПТИЧЕСКИХ ИЗОМЕРОВ АМЛОДИПИНА | 2008 |
|
RU2357781C1 |
| ХИРАЛЬНОЕ РАЗДЕЛЕНИЕ СМЕСИ ЭНАНТИОМЕРОВ НИКОТИНА | 2018 |
|
RU2753492C1 |
Изобретение относится к способу получения оптически активного амлодипина путем оптического разделения (R,S)-амлодипинов, при котором (R,S)-амлодипин подвергают взаимодействию с оптически активной O,O'-дибензоил-винной кислотой в изопропанольном растворителе с получением оптически активной соли амлодипин-геми-дибензоил-L-тартрата или ее сольвата, и обрабатывают оптически активную соль амлодипин-геми-дибензоил-L-тартрата или ее сольват основанием с получением оптически активного амлодипина. Изобретение также относится к (R)-(+)-амлодипин-геми-дибензоил-L-тартрату, (S)-(-)-амлодипин-геми-дибензоил-D-тартрату, (S)-(-)-амлодипин-геми-дибензоил-L-тартрату, (R)-(+)-амлодипин-геми-дибензоил-D-тартрату или к их сольватам. Технический результат - получение энантиомерно чистых изомеров амлодипина, обладающих активностью блокатора кальциевых каналов. 5 н. и 18 з.п. формулы, 1 таблица.
1. Способ получения оптически активного амлодипина путем оптического разделения (R,S)-амлодипинов, при котором (R,S)-амлодипин подвергают взаимодействию с оптически активной O,O'-дибензоил-винной кислотой в изопропанольном растворителе с получением оптически активной соли амлодипин-геми-дибензоил-L-тартрата или ее сольвата, и обрабатывают оптически активную соль амлодипин-геми-дибензоил-L-тартрата или ее сольват основанием с получением оптически активного амлодипина.
2. Способ по п.1, при котором:
(а) получают оптически активную соль (R)-(+)-амлодипин-геми-дибензоил-L-тартрат или ее сольват путем проведения реакции (R,S)-амлодипинов с дибензоил-L-винной кислотой в изопропанольном растворителе и
(б) получают оптически активный (R)-(+)-амлодипин путем обработки оптически активной соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата основанием.
3. Способ по п.2, отличающийся тем, что дибензоил-L-винную кислоту используют в количестве от 0,2 до 0,6 моля на 1 моль (R,S)-амлодипинов.
4. Способ по п.2, отличающийся тем, что обработку основанием осуществляют после перекристаллизации соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата.
5. Способ по п.2, при котором также:
(в) осуществляют перекристаллизацию оптически активной соли (S)-(-)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата из фильтрата и (г) получают оптически активный (S)-(-)-амлодипин путем обработки оптически активной соли (S)-(-)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата основанием.
6. Способ по п.1, при котором:
(а) получают оптически активную соль (S)-(-)-амлодипин-геми-дибензоил-D-тартрат или ее сольват путем проведения реакции (R,S)-амлодипинов с дибензоил-D-винной кислотой в изопропанольном растворителе и
(б) получают оптически активный (S)-(-)-амлодипин путем обработки оптически активной соли (S)-(-)-амлодипина или ее сольвата основанием.
7. Способ по п.6, отличающийся тем, что дибензоил-D-винную кислоту используют в количестве от 0,2 до 0,6 моля на 1 моль (R,S)-амлодипинов.
8. Способ по п.6, отличающийся тем, что обработку основанием осуществляют после перекристаллизации соли (S)-(-)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата.
9. Способ по п.6, при котором также:
(в) осуществляют перекристаллизацию оптически активной соли (R)-(+)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата из фильтрата и
(г) получают оптически активный (R)-(+)-амлодипин путем обработки оптически активной соли (R)-(+)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата основанием.
10. Способ по п.1, при котором:
(а) проводят реакцию (R,S)-амлодипинов с дибензоил-L-винной кислотой в изопропанольном растворителе, отделяют фильтрованием и получают соль (R)-(+)-амлодипин-геми-дибензоил-L-тартрат или ее сольват, и
(б) проводят реакцию фильтрата с дибензоил-D-винной кислотой, отделяют фильтрованием и получают соль (S)-(-)-амлодипин-геми-дибензоил-D-тартрат или ее сольват,
(в) обрабатывают соль (R)-(+)-амлодипин-геми-дибензоил-L-тартрат или ее сольват основанием и получают (R)-амлодипин, и
(г) обрабатывают соль (S)-(-)-амлодипин-геми-дибензоил-D-тартрат или ее сольват основанием и получают (S)-амлодипин.
11. Способ по п.10, отличающийся тем, что дибензоил-L-винную кислоту и дибензоил-D-винную кислоту используют в количестве от 0,2 до 0,3 моля каждой на 1 моль (R,S)-амлодипинов.
12. Способ по п.10, отличающийся тем, что обработку основанием осуществляют после перекристаллизации соответственно соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата или соли (S)-(-)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата.
13. Способ по п.1, при котором:
(а) проводят реакцию (R,S)-амлодипинов с дибензоил-D-винной кислотой в изопропанольном растворителе, отделяют фильтрованием и получают соль (S)-(-)-амлодипин-геми-дибензоил-D-тартрат или ее сольват, и
(б) проводят реакцию фильтрата с дибензоил-L-винной кислотой, отделяют фильтрованием и получают соль (R)-(+)-амлодипин-геми-дибензоил-L-тартрат или ее сольват,
(в) обрабатывают соль (S)-(-)-амлодипин-геми-дибензоил-D-тартрат или ее сольват основанием и получают (S)-амлодипин, и
(г) обрабатывают соль (R)-(+)-амлодипин-геми-дибензоил-L-тартрат или ее сольват основанием и получают (R)-амлодипин.
14. Способ по п.13, отличающийся тем, что дибензоил-D-винную кислоту и дибензоил-L-винную кислоту используют в количестве от 0,2 до 0,3 моля каждой на 1 моль (R,S)-амлодипинов.
15. Способ по п.13, отличающийся тем, что обработку основанием осуществляют после перекристаллизации соответственно соли (S)-(-)-амлодипин-геми-дибензоил-D-тартрата или ее сольвата или соли (R)-(+)-амлодипин-геми-дибензоил-L-тартрата или ее сольвата.
16. Способ по любому из пп.1, 2, 6, 10 и 13, отличающийся тем, что реакционный растворитель представляет собой одиночный растворитель, состоящий только из изопропанола, или смесь изопропанола и сорастворителя, выбранного из группы, состоящей из воды, кетонов, спиртов, простых эфиров, амидов, сложных эфиров, углеводородов, хлоруглеводородов и нитрилов.
17. Способ по п.16, отличающийся тем, что сорастворитель выбран из группы, состоящей из воды, ацетона, ацетонитрила, пропиононитрила, диметилсульфоксида, диметилацетамида, метилэтилкетона, тетрагидрофурана, этилацетата, дихлорметана, диметилформамида, гексана, толуола, метанола, этанола, трет-бутанола и N,N'-диметилпропилен-мочевины.
18. Способ по любому из пп.4, 5, 8, 9, 12 и 15, отличающийся тем, что перекристаллизацию проводят в растворителе для кристаллизации, которым является одиночный растворитель, состоящий только из изопропанола, или смесь изопропанола и сорастворителя, выбранного из группы, состоящей из воды, кетонов, спиртов, простых эфиров, амидов, сложных эфиров, углеводородов, хлоруглеводородов и нитрилов.
19. Способ по любому из пп.2, 4-6, 8-10 и 12-14, отличающийся тем, что основание выбирают из группы, состоящей из гидроксида, оксида, карбоната, бикарбоната и амида щелочного металла или щелочноземельного металла.
20. (R)-(+)-амлодипин-геми-дибензоил-L-тартрат или его сольват.
21. (S)-(-)-амлодипин-геми-дибензоил-D-тартрат или его сольват.
22. (S)-(-)-амлодипин-геми-дибензоил-L-тартрат или его сольват.
23. (R)-(+)-амлодипин-геми-дибензоил-D-тартрат или его сольват.
| KR 20040023474 A, 18.03.2004 | |||
| WO 2004024689 A1, 25.03.2004 | |||
| US 2003130321 A1, 10.07.2003 | |||
| СПОСОБ ВЫДЕЛЕНИЯ R - /+/ - И S - /-/ - ИЗОМЕРОВ АМЛОДИПИНА ИЗ ИХ СМЕСЕЙ, СОЛЬВАТЫ И МОНОГИДРАТЫ R - /+/ - И S - /-/ - АМЛОДИПИН - ГЕМИ-L- ИЛИ D -ТАРТРАТА | 1995 |
|
RU2132845C1 |

