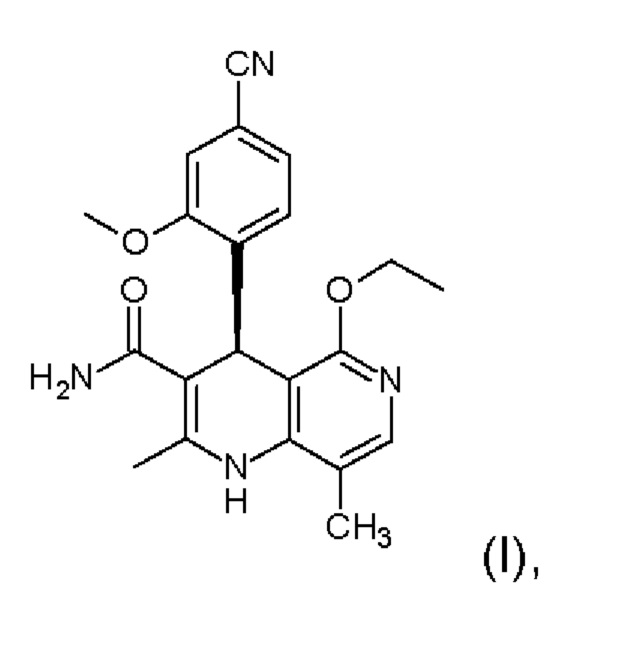
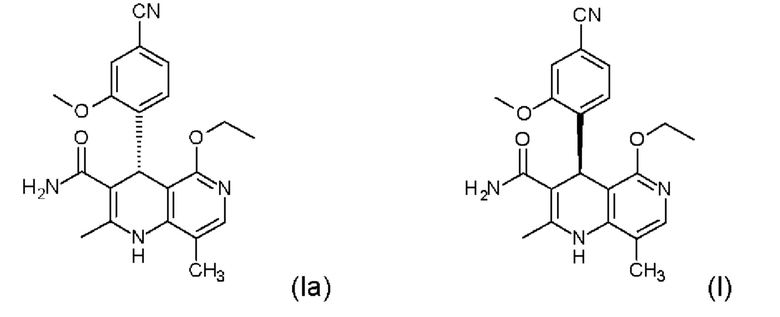
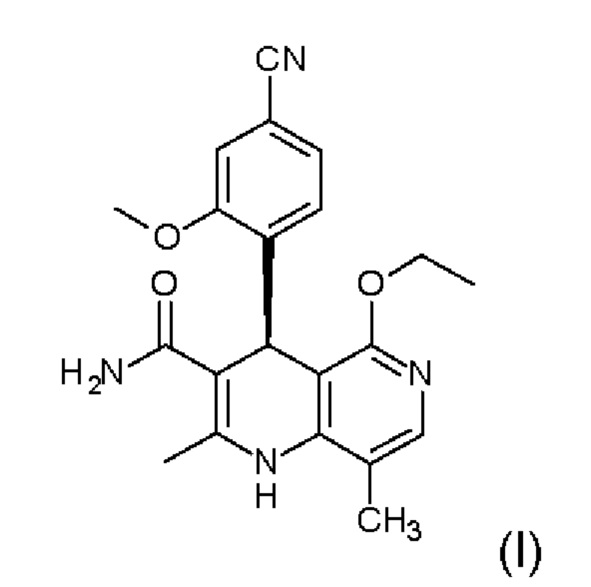
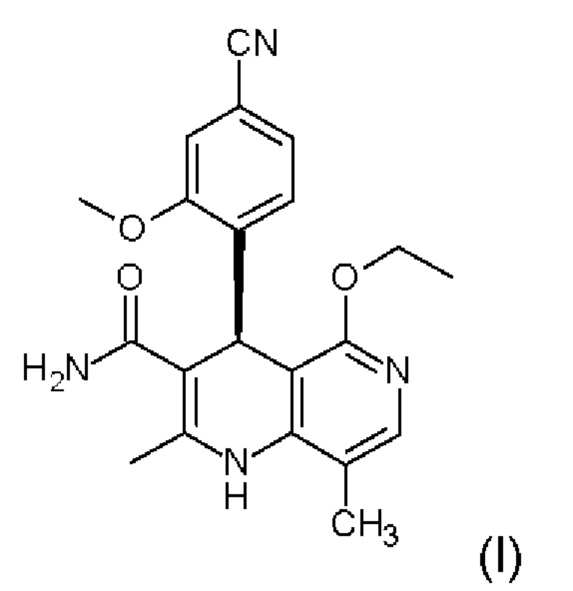
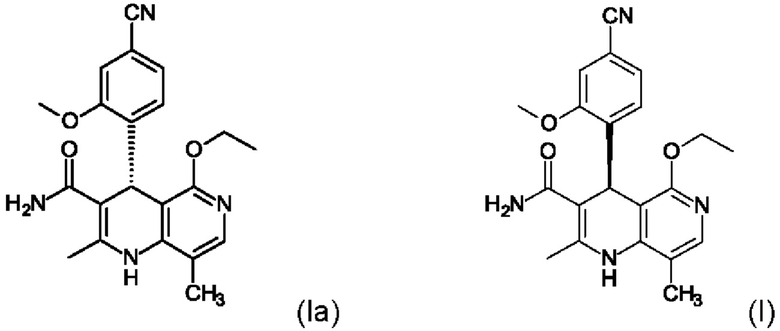
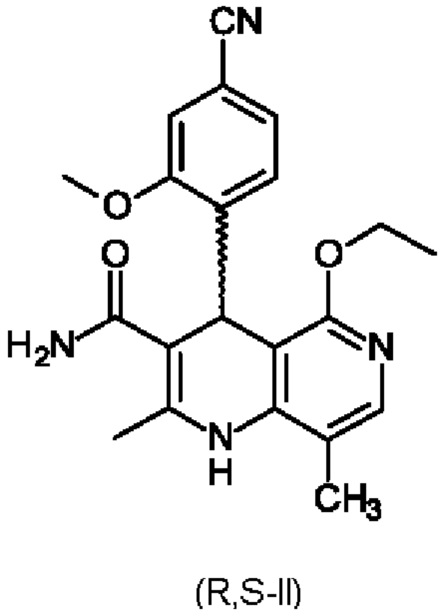
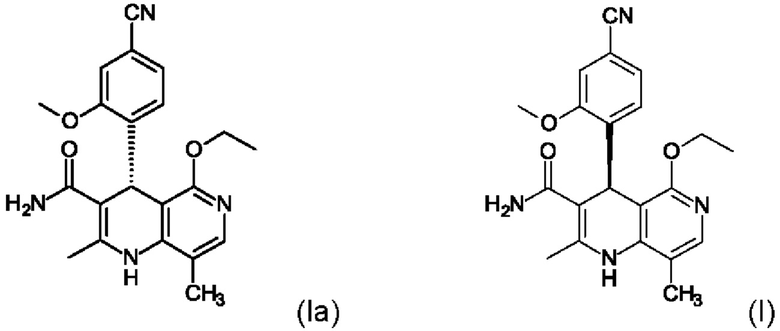
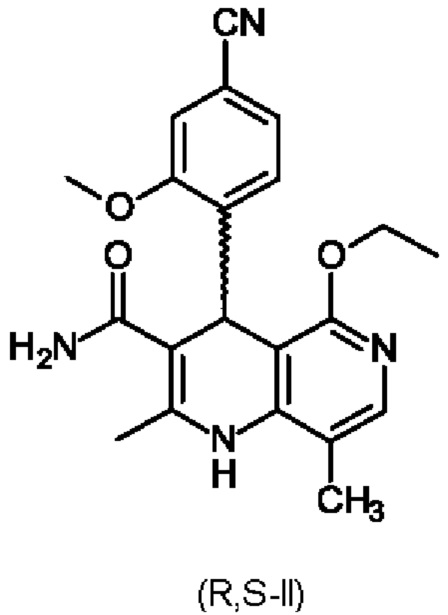
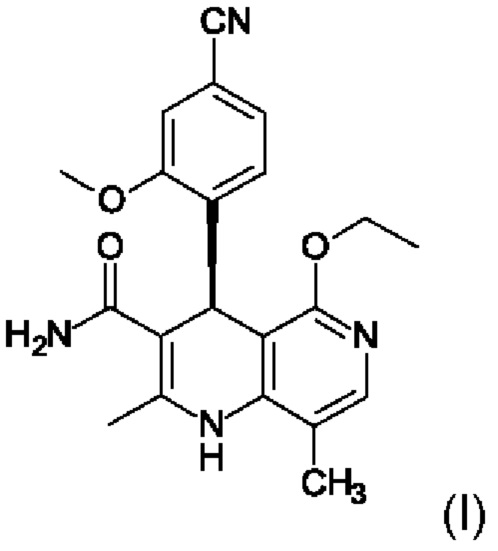
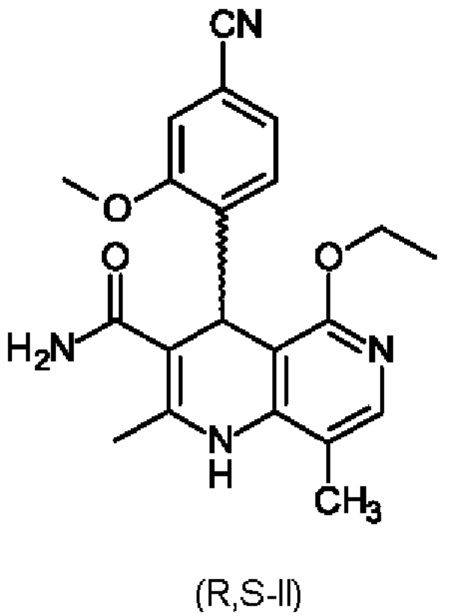
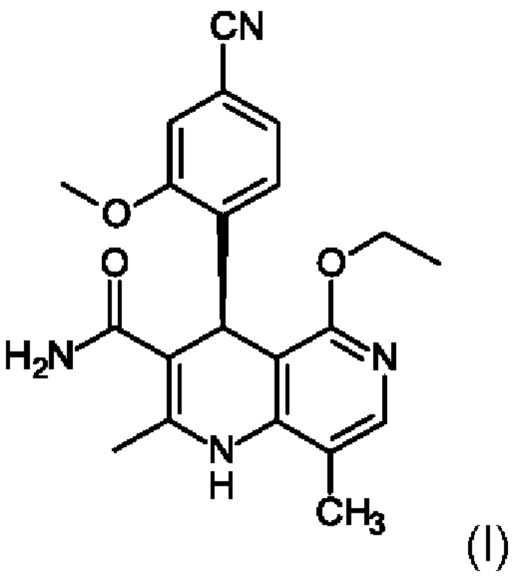
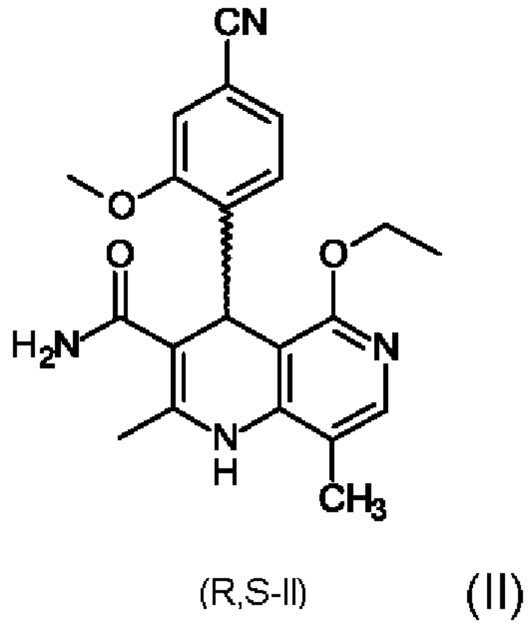
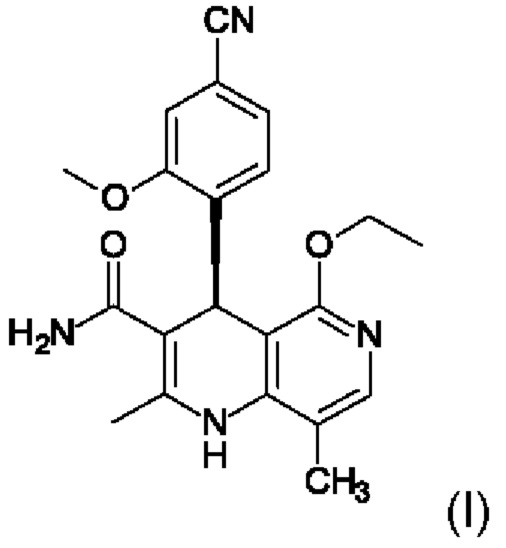
Настоящее изобретение относится к новому и улучшенному способу получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I)
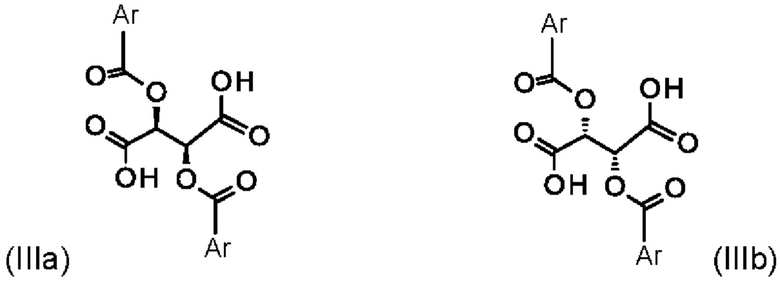
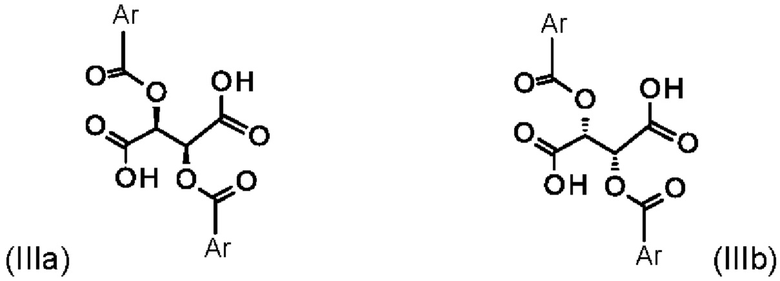
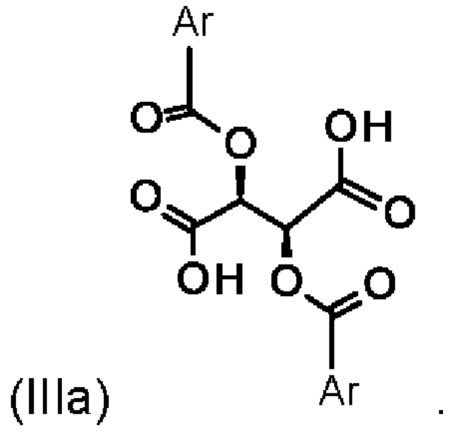
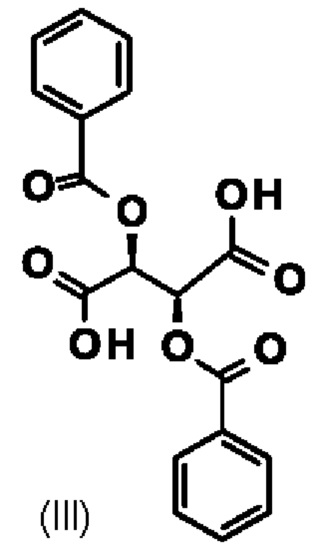
а также к получению энантиомера (Ia) путем разделения рацемата с хиральными замещенными сложными эфирами винной кислоты общих формул (Ша) и (IIIb)
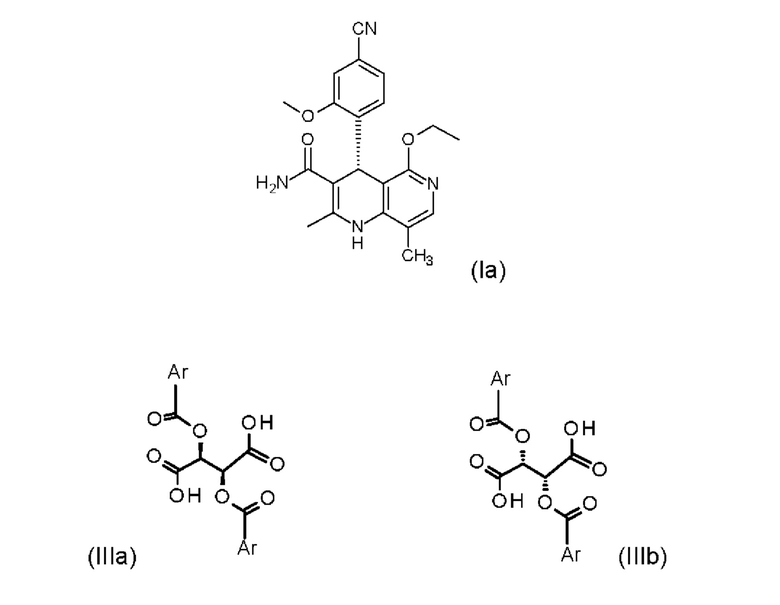
где Ar означает замещенные или незамещенные ароматические соединения или гетероароматические соединения.
Финеренон (I) обладает действием нестероидного антагониста минералокортикоидного рецептора и может находить применение в качестве средства для профилактики и/или лечения сердечно-сосудистых и почечных заболеваний, например, сердечной недостаточности и диабетической нефропатии.
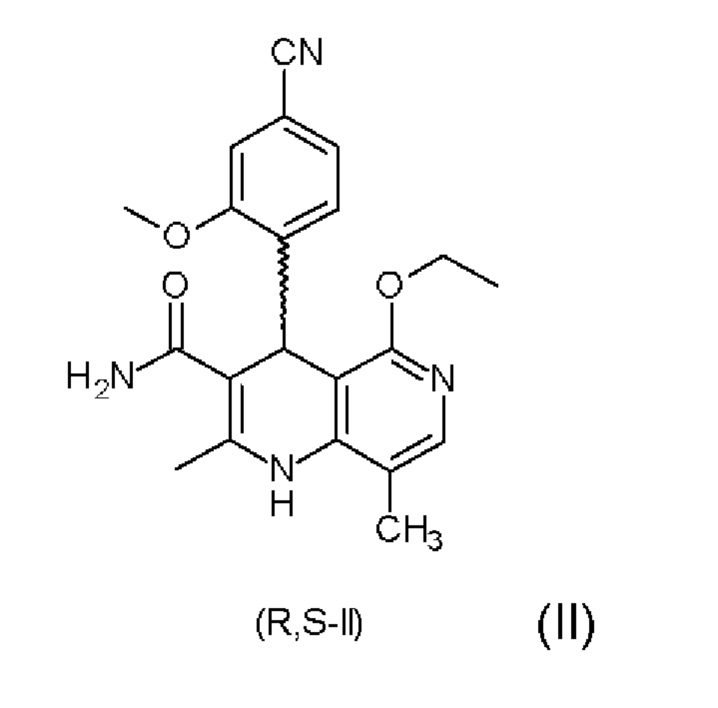
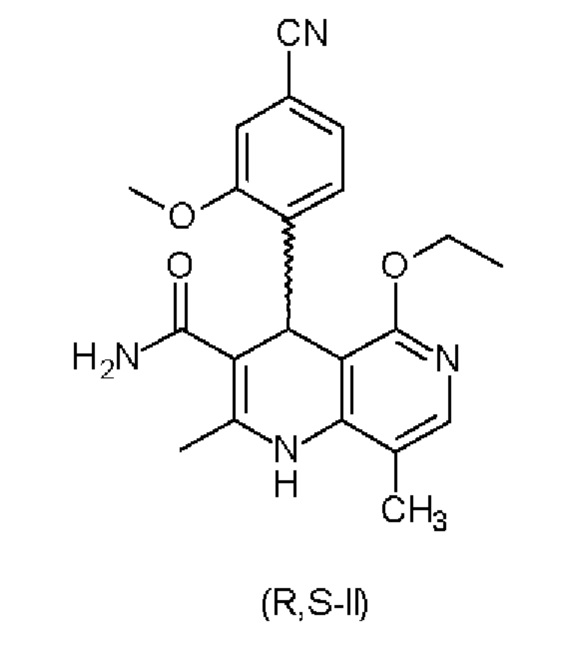
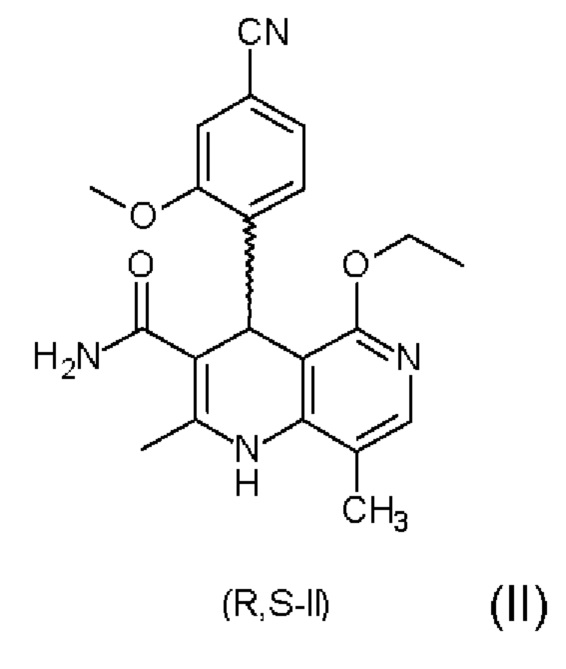
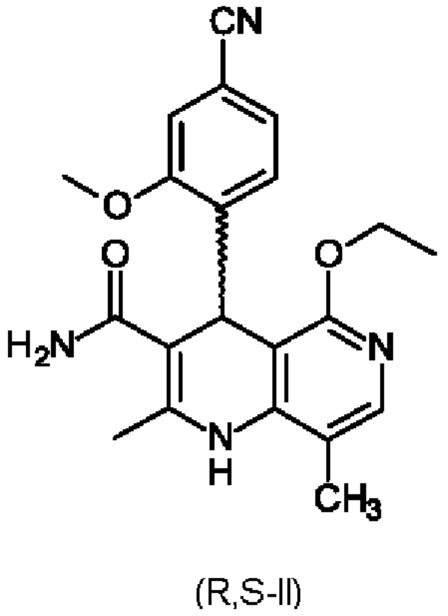
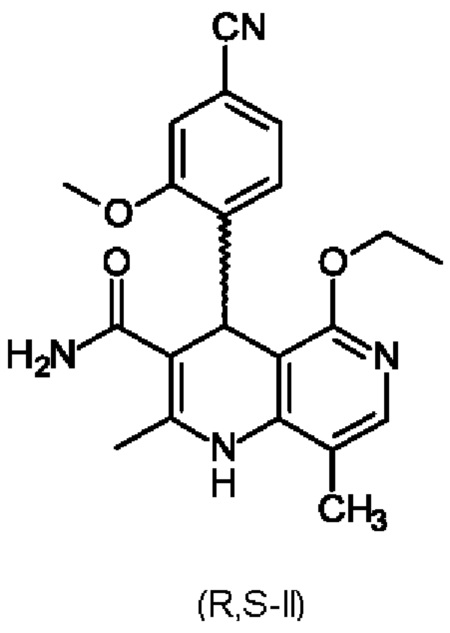
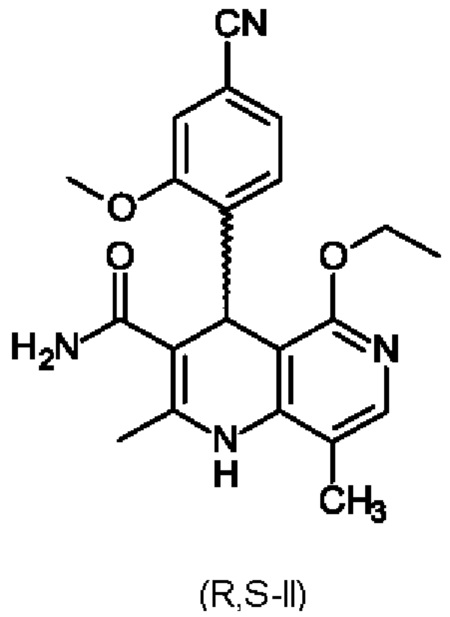
Соединение формулы (I) и способ его получения описаны в международной заявке на патент WO 2008/104306 и статье ChemMedChem 2012, 7, 1385, а также в международной заявке на патент WO 2016/016287 А1. Для получения соединения формулы (I) рацемическую смесь амидов (II)
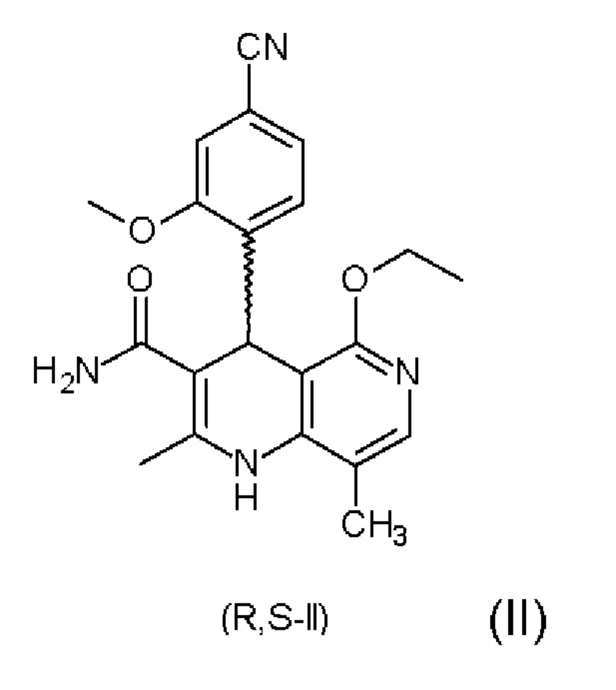
необходимо разделить на соответствующие антиподы, так как активен только антипод формулы (I).
В описанном выше научно-исследовательском синтезе (WO 2008/104306) для этой цели использовали специально синтезированную хиральную фазу (собственного производства), которая в качестве хирального селектора содержала поли-N-метакрилоил-D-лейцин-дициклопропилметиламид. Было обнаружено, что разделение также можно проводить на коммерчески легкодоступной фазе. При этом речь идет о фазе Chiralpak AS-V, 20 мкм. В качестве элюента была использована смесь метанол/ацетонитрил 60:40. В этом случае хроматографию можно проводить на коммерческой хроматографической колонке, но предпочтение отдается методам, известным специалисту в данной области, таким как SMB (моделируемый движущийся слой; G. Paredes, М. Mazotti, Journal of Chromatography A, 1142 (2007): 56-68) или Varicol (Computers and Chemical Engineering 27 (2003) 1883-1901).
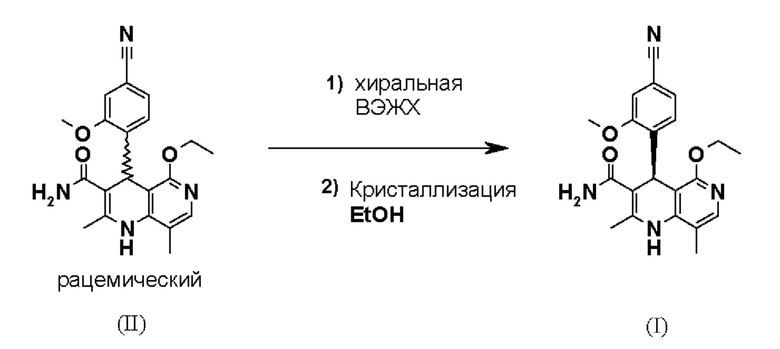
Хотя SMB-разделение обеспечивает очень хороший выход и оптическую чистоту, затраты на приобретение и эксплуатацию такой системы в условиях GMP являются серьезной проблемой и связаны с высокими затратами. Также хиральная фаза, используемая в каждом случае, является очень дорогой и имеет ограниченный срок службы, и ее необходимо раз за разом заменять в процессе производства. По производственно-техническим причинам это не оптимально, если отсутствует вторая система, чтобы гарантировать непрерывную работу, что связано с дополнительными затратами. Кроме того, особенно для продуктов, которые производятся в масштабе, измеряемом тоннами, регенерация растворителя является стадией, определяющей общую продолжительность, требует покупки огромных испарителей с падающей пленкой и сопряжена с потреблением огромного количества энергии.
Поэтому задача настоящего изобретения состояла в том, чтобы найти альтернативы для разделения смеси энантиомеров, которые были бы значительно более рентабельными и которые можно было бы осуществить с помощью обычного оборудования пилотной установки (резервуар с мешалкой / изолирующий аппарат). Такого рода установки традиционно входят в стандартную комплектацию фармацевтических производств и не требуют дополнительных инвестиций. Сертификация и валидация периодических процессов также намного проще, чем в случае хроматографических способов, что является дополнительным преимуществом.
Было предпринято множество попыток провести разделение с помощью обычных классических методов для разделения рацемата (вариации хиральной органической кислоты и растворителя), как показано в таблице 1:

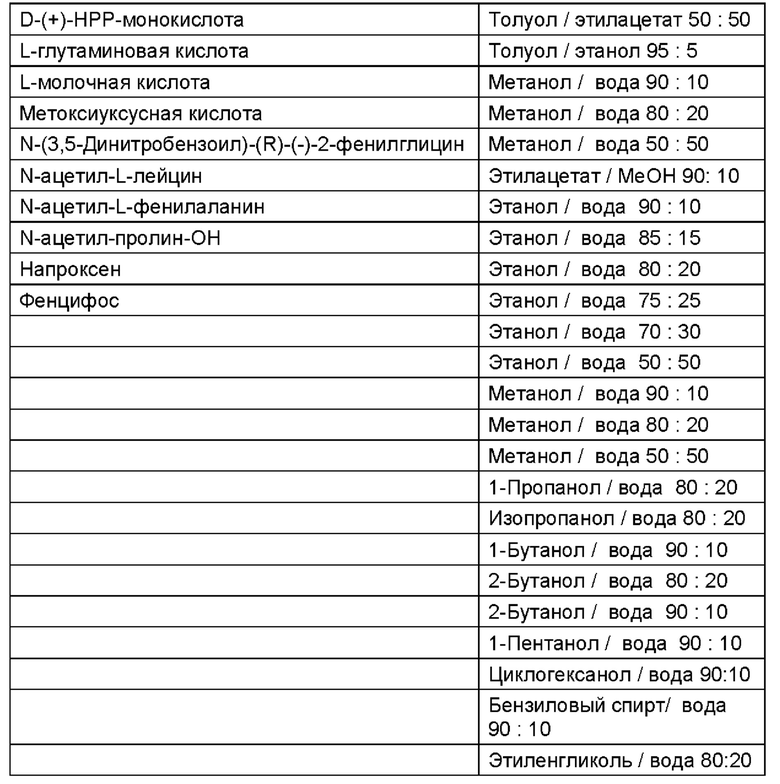
Среди прочего, проводили эксперименты с классическим разделительным реактивом (+)-винной кислотой.
Однако во всех случаях образования соли не наблюдалось; вместо этого из раствора всегда выпадал в осадок только рацемат в несолевой форме. Это в основном соответствует ожиданиям специалиста в данной области, которые можно было бы вывести из значения pKa молекулы (II), что классическое разделение рацемата посредством образования диастереомерной соли с органическими кислотами не должно быть возможным, поскольку измеренное значение pKa (для основания) составляет около 4,3 и, таким образом, почти исключает образование соли, так что предпочтительно возможность образования солей должна быть реализована только с использованием очень сильных неорганических или органических минеральных кислот, таких как хиральные сульфоновые кислоты или фосфоновые кислоты. Согласно литературе, например „Handbook of Pharmaceutical Salts - Properties, Selection and Use; P. Heinrich Stahl, Camille G. Wermuth (ред.); Wiley-VCH, стр. 166" разница значений pK должна составлять не менее 3 pK-единиц для обеспечения стабильного солеобразования. В действительности это наблюдается, если, например, используют сложный эфир циклической фосфорной кислоты хлоцифос, приведенный ниже:
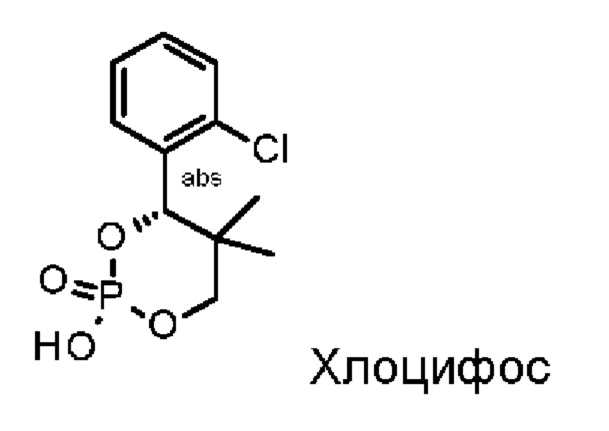
При взаимодействии 3 экв. указанного сложного эфира циклической фосфорной кислоты с рацематом (II) получают диастереомерную соль, в которой (I) присутствует с энантиомерным избытком лишь 44% ее.
Все усилия достичь избытка энантиомеров, близкого к >99% ее, были неэффективны; к тому же хлоцифос не был коммерчески доступен в больших количествах, поэтому были продолжены поиски дополнительных альтернатив.
Не наблюдалось образования солей и при взаимодействии с алкилзамещенными производными винной кислоты, такими как, например, (-)-O,O'-дипивалоил-L-винная кислота или (-)-O,O-диацетил-L-винная кислота.
При дальнейшей разработке темы было неожиданно обнаружено, что ароматически или соответственно гетероароматически замещенные производные винной кислоты отлично подходят для образования «диастереомерных солей» из рацемата (II).
Поэтому объектом настоящего изобретения является разделение рацемата (II)
на (Ia) и/или (I)
при помощи хиральных замещенных сложных эфиров винной кислоты общих формул (IIIa) или (IIIb)
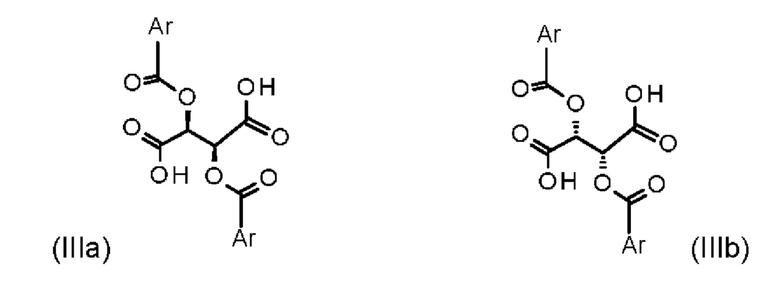
где Ar означает незамещенные или замещенные ароматические соединения или гетероароматические соединения.
Другой объект относится к способу получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I)
посредством разделения рацематов (II)
с хиральным замещенным сложным эфиром винной кислоты формулы (IIIa)
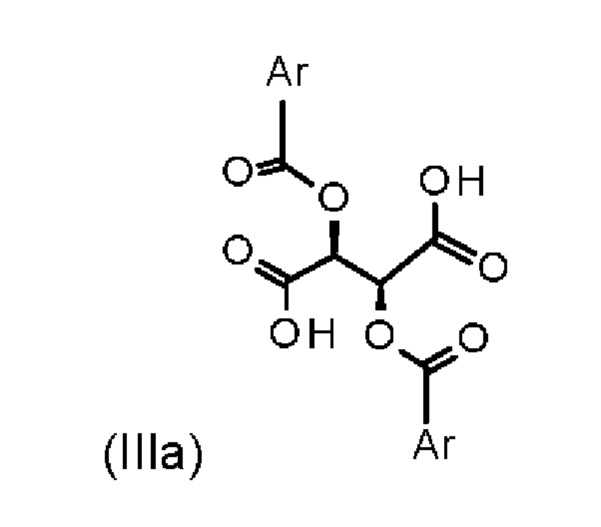
где Ar означает незамещенные или замещенные ароматические соединения или гетероароматические соединения.
Термин «замещенный» означает, что один или несколько атомов водорода у соответствующего атома или соответствующей группы заменен/заменены одним представителем из приведенной группы, при условии, что в данных обстоятельствах не превышается нормальная валентность соответствующего атома. Допускаются комбинации заместителей и/или переменных.
Термин «незамещенный» означает, что ни один атом водорода не был заменен.
Гетероарильная группа или соответственно гетероароматическая группа может быть 5-членной гетероарильной группой, такой как, например, тиенил, фуранил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, оксадиазолил, триазолил или тетразолил; или 6-членной гетероарильной группой, такой как, например, пиридинил, пиридазинил, пиримидинил, пиразинил или триазинил; или трициклической гетероарильной группой, такой как карбазолил, акридинил или феназинил; или 9-членной гетероарильной группой, такой как бензофуранил, бензотиенил, бензоксазолил, бензизоксазолил, бензимидазолил, бензотиазолил, бензотриазолил, индазолил, индолил, изоиндолил, индолизинил или пуринил; или 10-членной гетероарильной группой, такой как, например, хинолинил, хиназолинил, изохинолинил, циннолинил, фталазинил, хиноксалинил или птеридинил.
В частности, гетероарильная группа представляет собой пиридинильную, пиразинильную, пирролильную, пиразолильную или пиримидинильную группу.
В контексте настоящей заявки арильная группа представляет собой, в частности, фенильную группу.
В качестве заместителей в контексте настоящего изобретения пригодными являются галоген, алкил с 1-6 атомами углерода, алкокси с 1-6 атомами углерода, нитрил, нитрогруппа, цианогруппа, CF3-группа или амидная группа, такая как, например, NHCOR, в которой R может представлять собой метил, этил или фенил, или группа -NRCOR, в которой R имеет указанное выше значение, или CONHR, в которой R имеет указанное выше значение, или CONRR', в которой R' идентичен R, который был определен выше, или циклические амиды, такие как -СО-морфолиновый остаток, -СО-пиперидиновый остаток.
Термин "галоген" означает атом фтора, хлора, брома или йода, в частности атом фтора, хлора или брома.
Термин «алкил с 1-6 атомами углерода» означает линейную или разветвленную насыщенную одновалентную углеводородную группу с 1, 2, 3, 4, 5 или 6 атомами углерода, например, метильную, этильную, пропильную, изопропильную, бутильную, втор-бутильную, изобутильную, трет-бутильную, пентильную, изопентильную, 2-метилбутильную, 1-метилбутильную, 1-этилпропильную, 1,2-диметилпропильную, неопентильную, 1,1-диметилпропильную, гексильную, 1-метилпентильную, 2-метилпентильную, 3-метилпентильную, 4-метилпентильную, 1-этилбутильную, 2-этилбутильную, 1,1-диметилбутильную, 2,2-диметилбутильную, 3,3-диметилбутильную, 2,3-диметилбутильную, 1,2-диметилбутильную или 1,3-диметилбутильную группу или их изомер. В частности, группа имеет 1, 2, 3 или 4 атома углерода («алкил с 1-4 атомами углерода»), например метильная, этильная, пропильная, изопропильная, бутильная, втор-бутильная, изобутильная или трет-бутильная группа, в частности с 1, 2 или 3 атомами углерода («алкил с 1-3 атомами углерода»), например метильная, этильная, н-пропильная или изопропильная группа.
Термин «алкокси с 1-6 атомами углерода» означает линейную или разветвленную насыщенную одновалентную группу формулы (алкил с 1-6 атомами углерода)0-, в которой термин «алкил с 1-6 атомами углерода» имеет указанные выше значения, например метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, изобутокси, трет-бутокси, пентилокси, изопентилокси или н-гексилокси группу или их изомер.
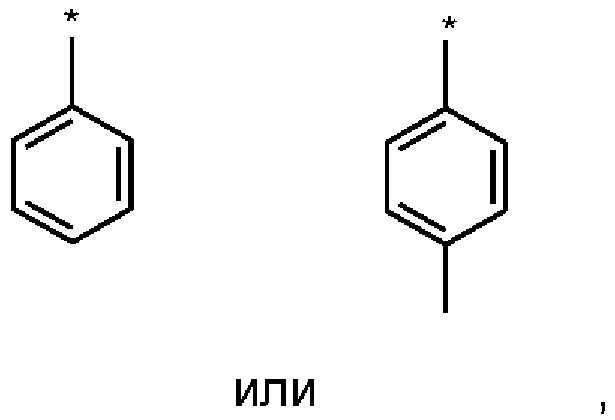
Предпочтительно Ar означает:
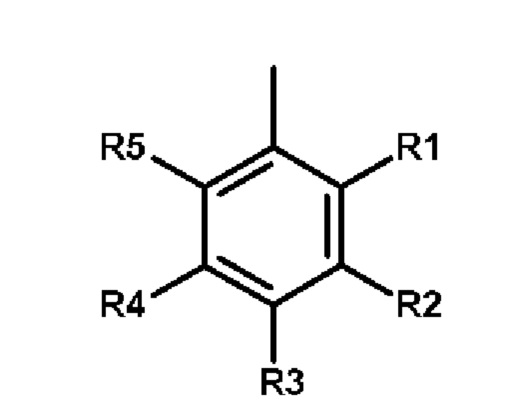
где R1, R2, R3, R4, R5 соответственно означают атом водорода или алкильный остаток, такой как, например, метил, этил, пропил, или атом галогена, такой как, например, фтор, хлор, бром или йод, или простую эфирную группу, такую как, например, О-метил, О-этил, О-фенил, или нитрогруппу, цианогруппу, CF3-группу или амидную группу, такую как, например, NHCOR, в которой R может представлять собой метил, этил или фенил, или группу -NRCOR, в которой R имеет указанное выше значение, или CONHR, в которой R имеет указанное выше значение, или CONRR', в которой R' идентичен R, который был определен выше, или циклические амиды, такие как -СО-морфолиновый остаток, -СО-пиперидиновый остаток. Схемы замещения могут быть очень разными, так теоретически может быть до 5 различных заместителей, но как правило предпочтительными являются монозамещенные Ar-остатки. Однако Ar также может быть замещенным гетероароматическим соединением, таким как предпочтительно пиридин или пиразин, пиридин или пиразин. Это также может быть полициклический ароматический углеводород, такой как, например, замещенный нафталин, антрацен или хинолин.
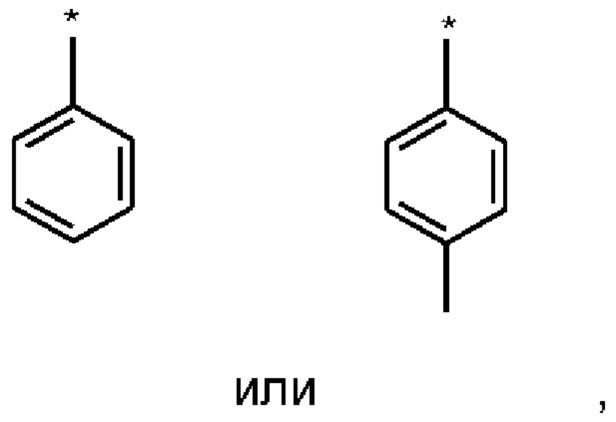
Особо предпочтительно в качестве Ar выступают:
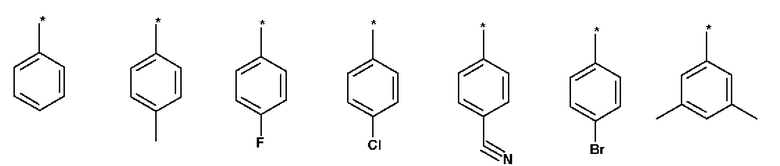
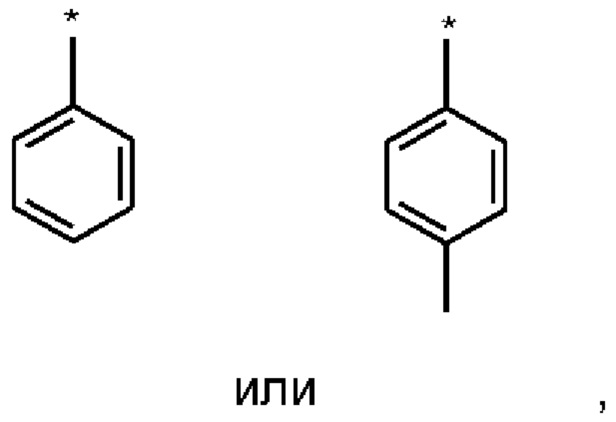
где * обозначает место присоединения.
Особенно предпочтительно Ar означает:
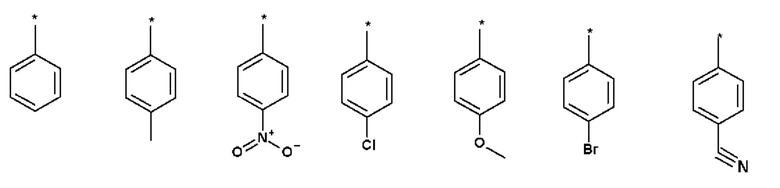
где * обозначает место присоединения.
Наиболее предпочтительными Ar-остатками являются:
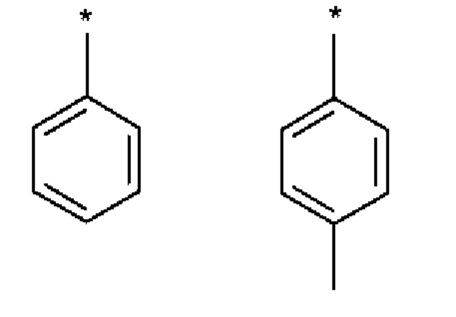
где * обозначает место присоединения.
Из указанных незамещенное кольцо (фенил) является особенно предпочтительным.
Получение сложных эфиров винной кислоты известно из литературы, например из Organic Synthesis, Coll. vol. 9, стр. 722 (1998); vol. 72, стр. 86 (1995), а также описано в Chirality 2011 (23), 3, стр. 228.
Еще один объект относится к диастереомерным солям в соответствии с формулами 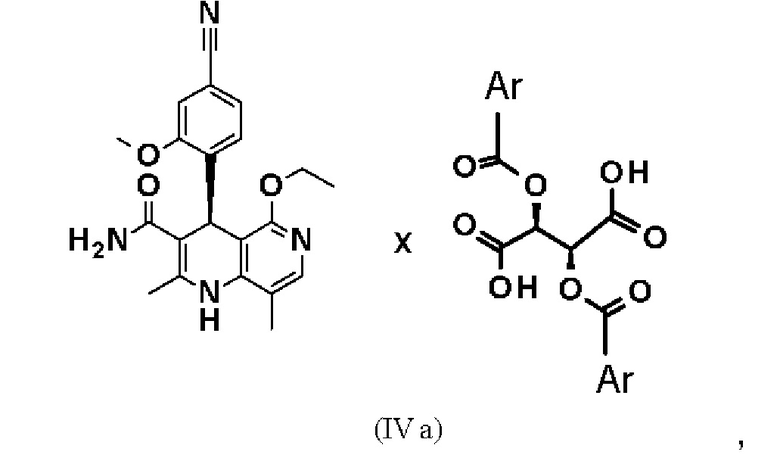
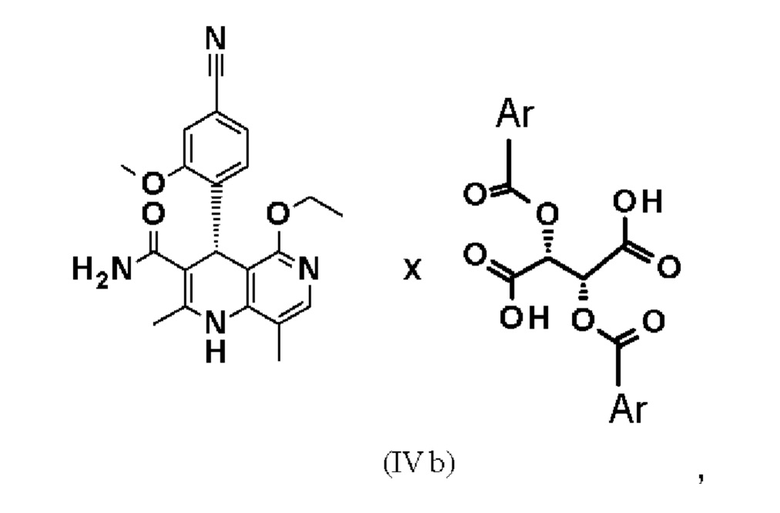
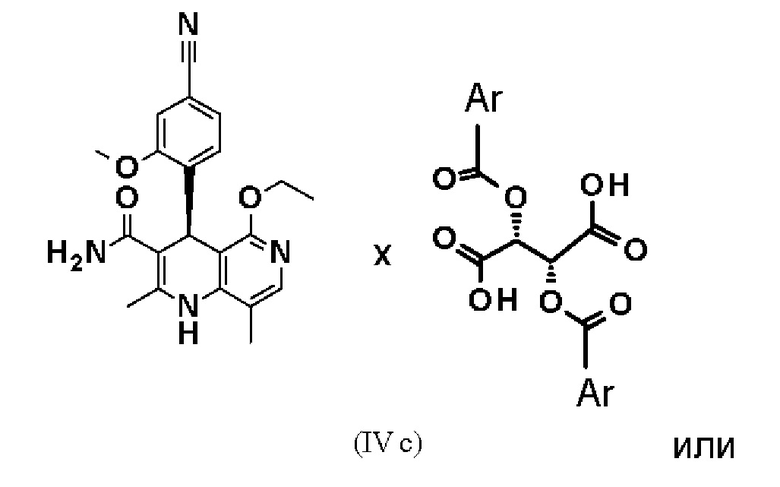
где Ar означает незамещенные или замещенные ароматические соединения или гетероароматические соединения и имеет вышеуказанные значения.
Особенно предпочтительными являются диастереомерные соли, в которых Ar означает фенил.
Невозможно однозначно предсказать, действительно ли в данном случае речь идет о классической диастереомерной соли или о стабилизированном водородной связью 1:1 молекулярном комплексе. Однако можно сказать наверняка, что указанные 1:1 молекулярные агрегаты являются очень стабильными и ведут себя и выделяются как классические диастереомерные соли, поэтому в дальнейшем мы будем использовать название диастереомерная соль. Для изображения диастереомерных солей используют производные винной кислоты общей формулы (IIIa) и (IIIb):
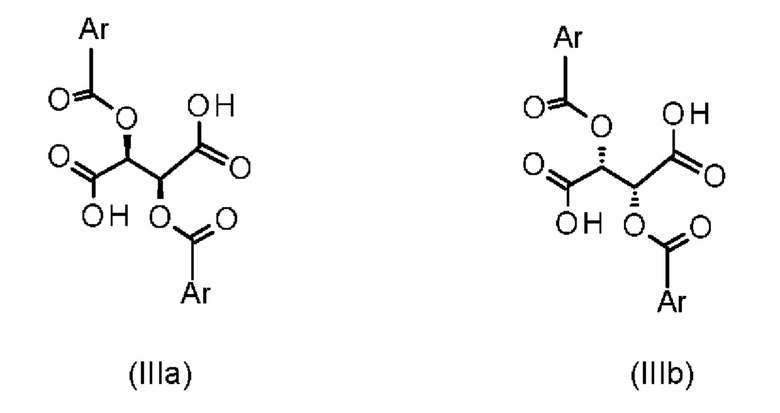
где Ar означает замещенные или незамещенные ароматические соединения или гетероароматические соединения.
Получение диастереомерных солей осуществляют следующим образом:
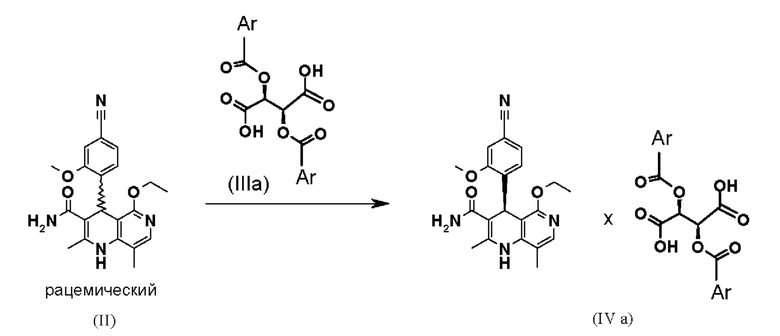
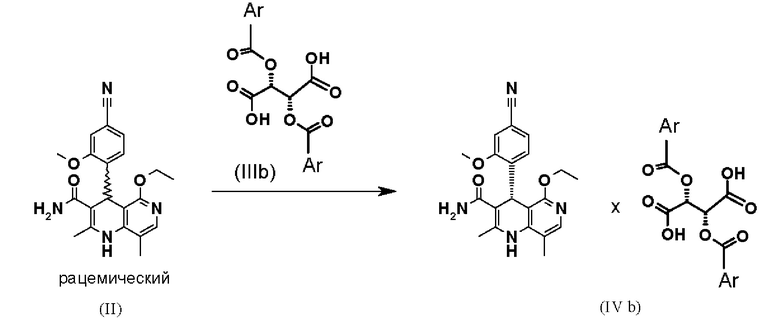
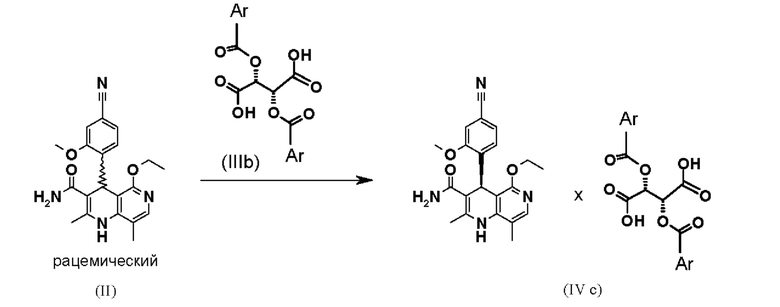
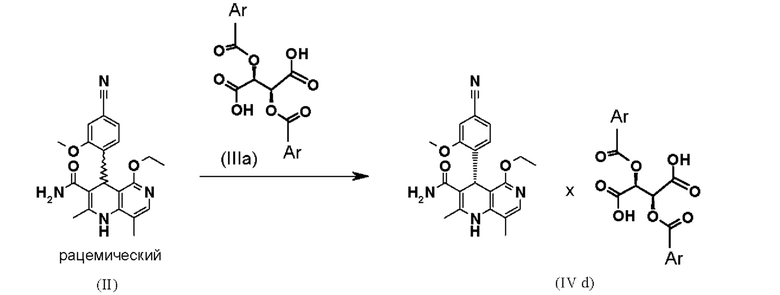
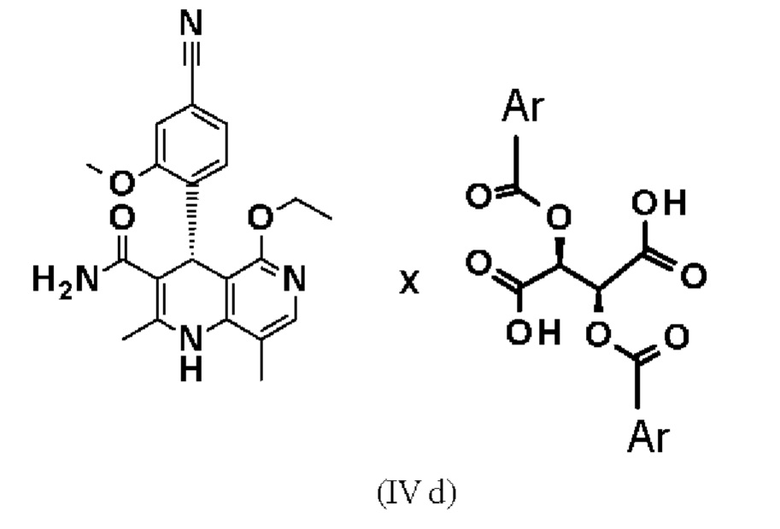
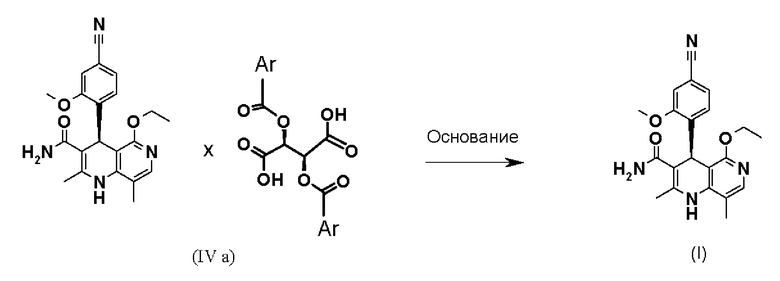
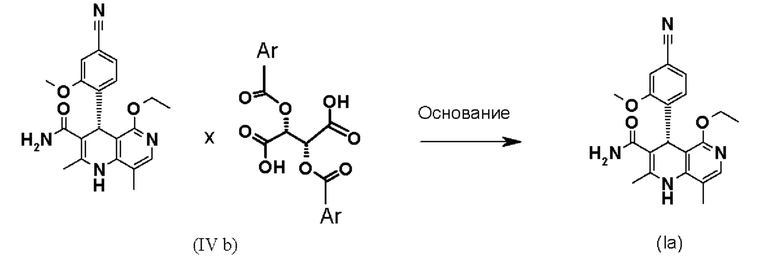
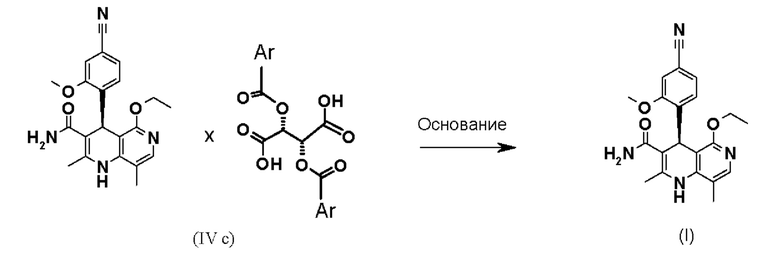
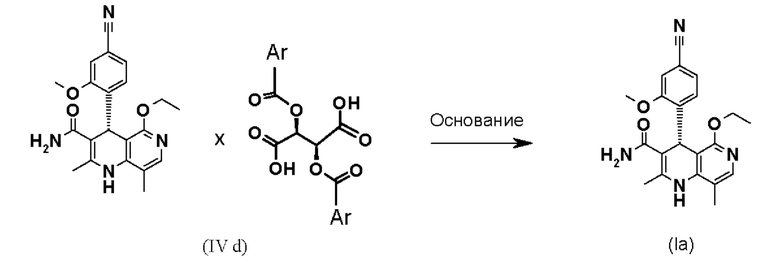
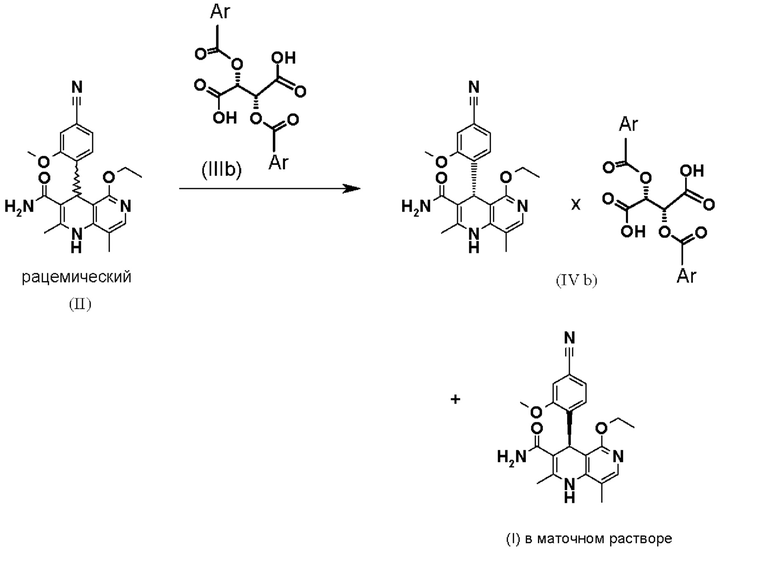
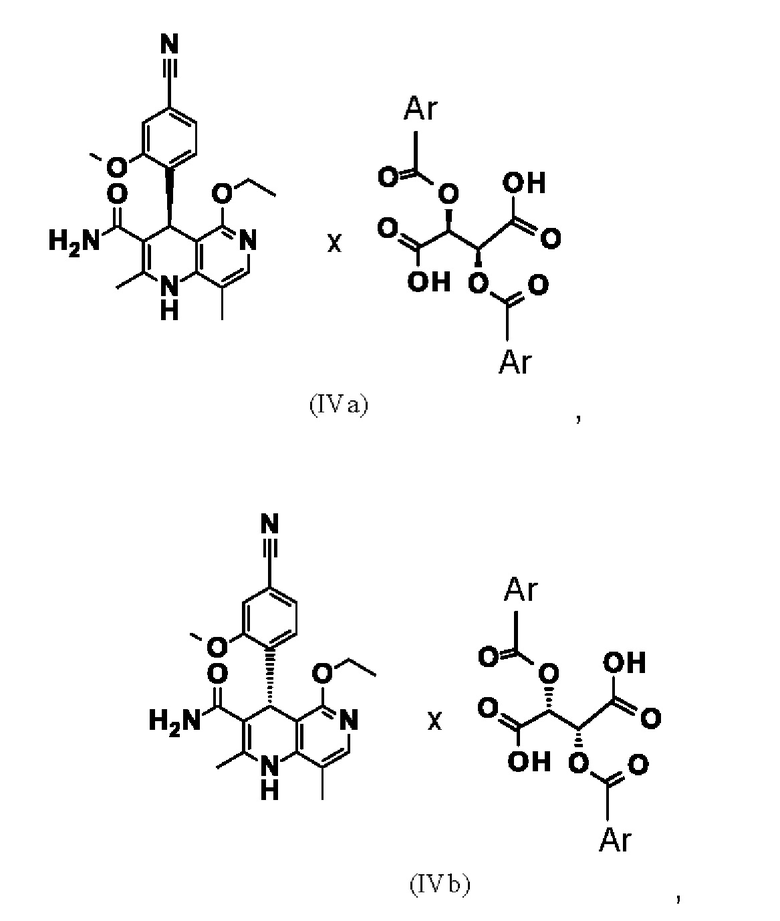
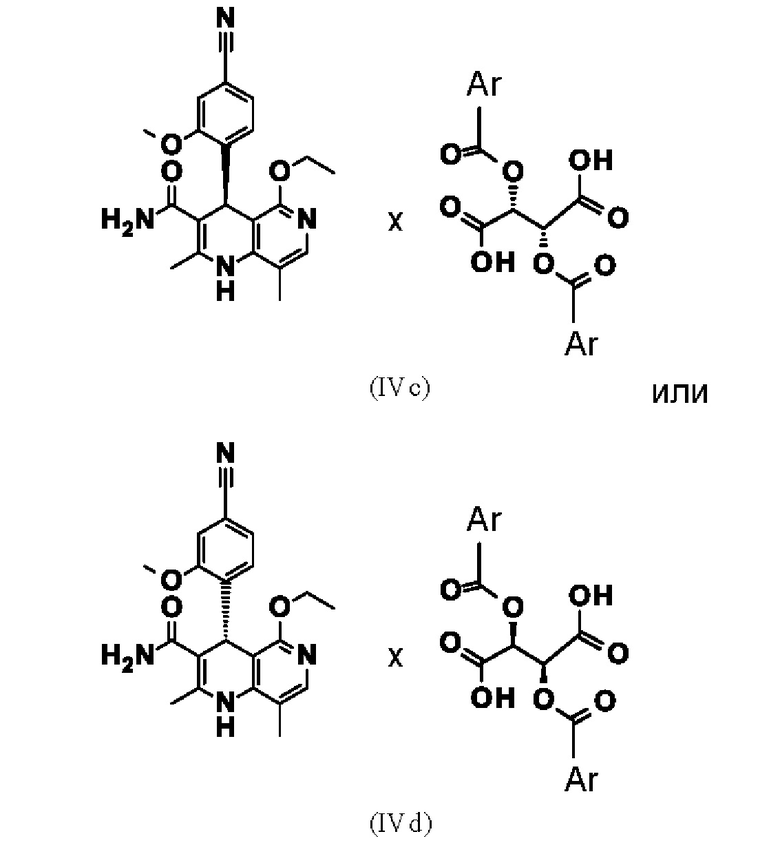
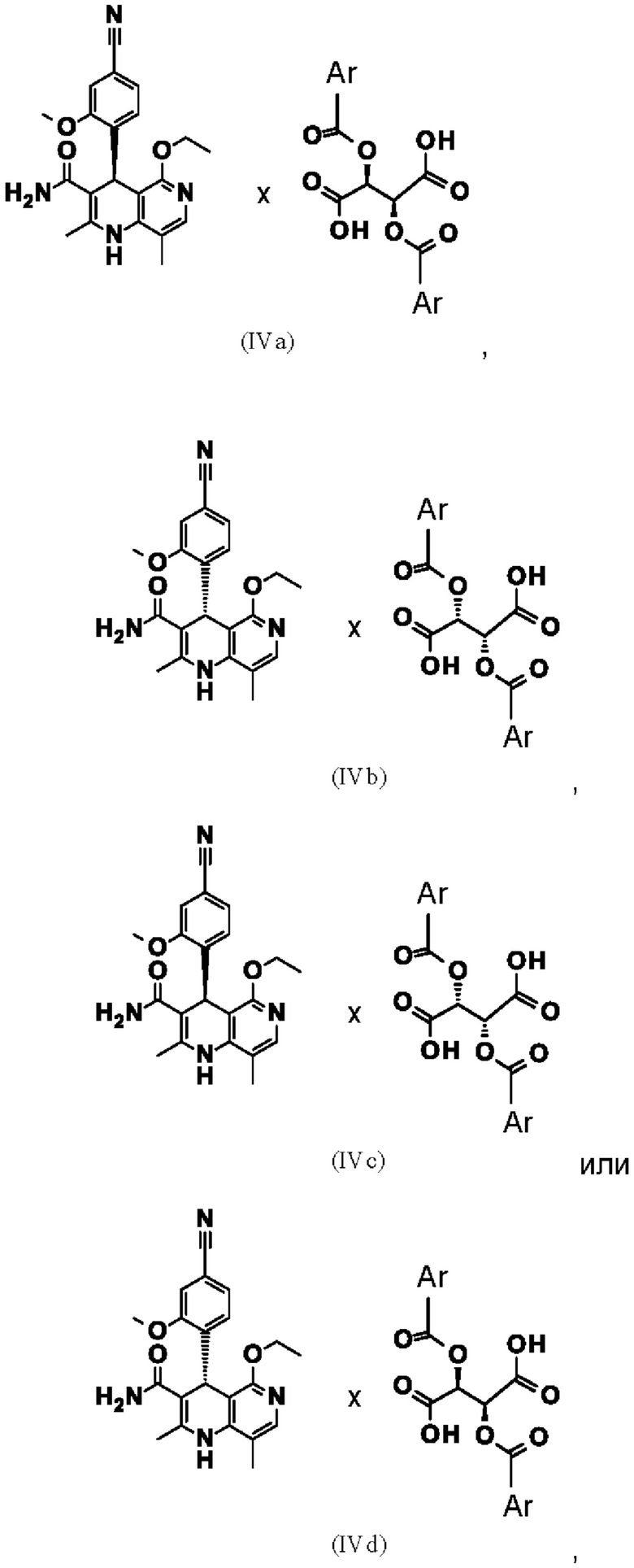
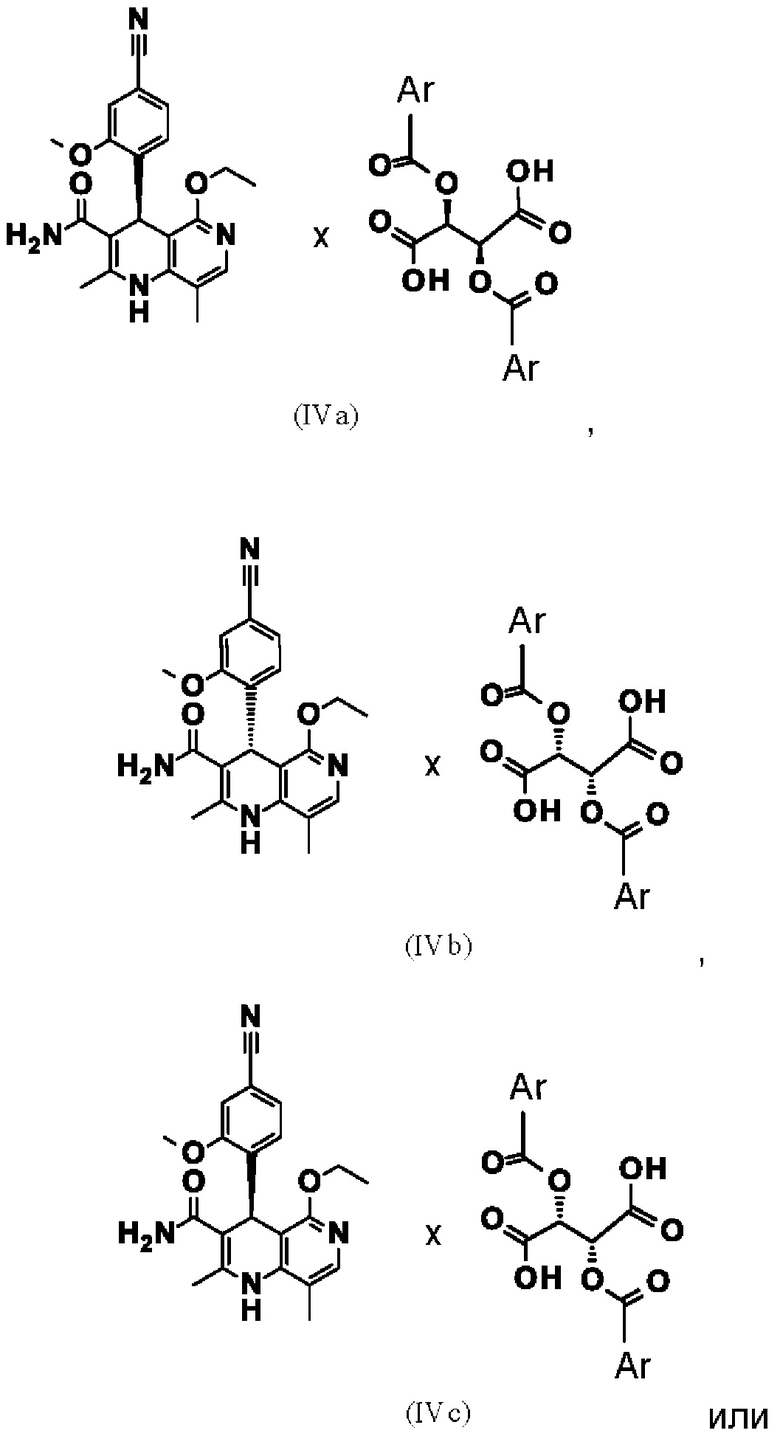
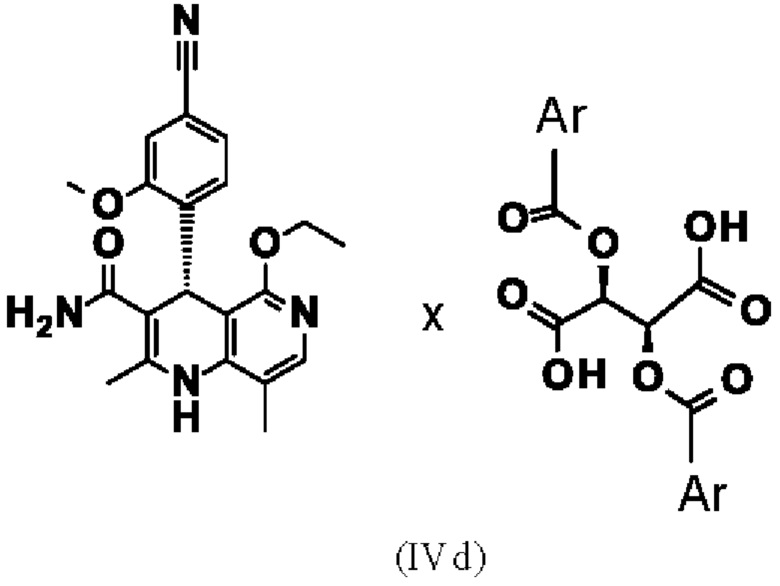
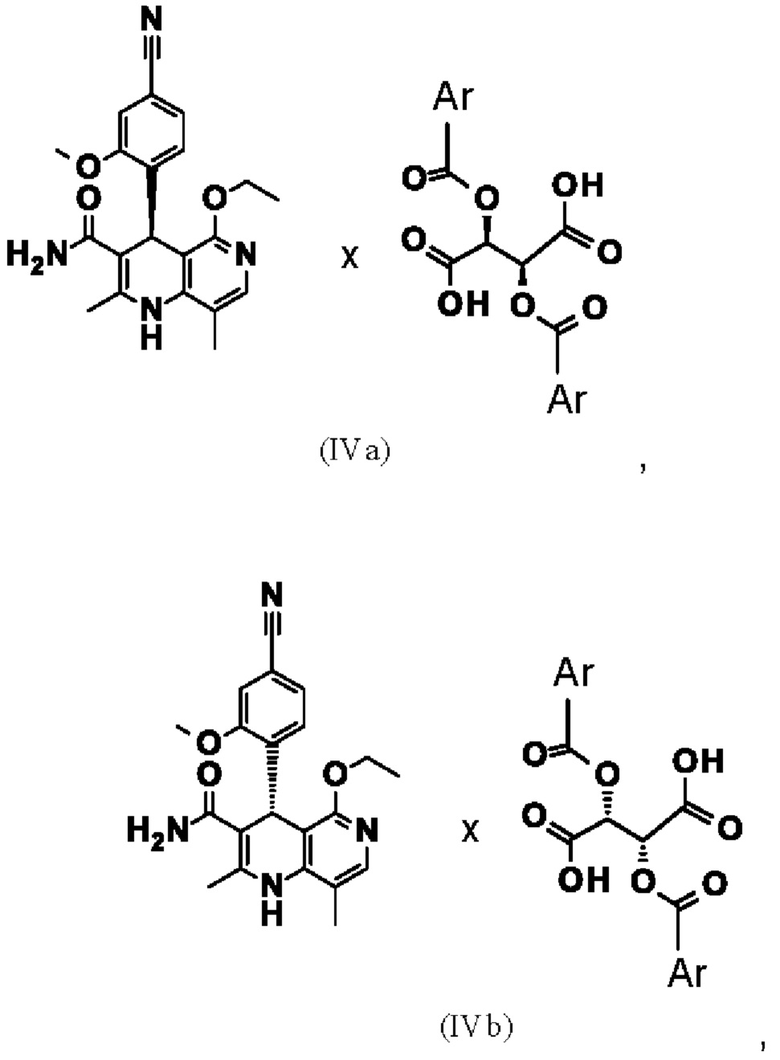
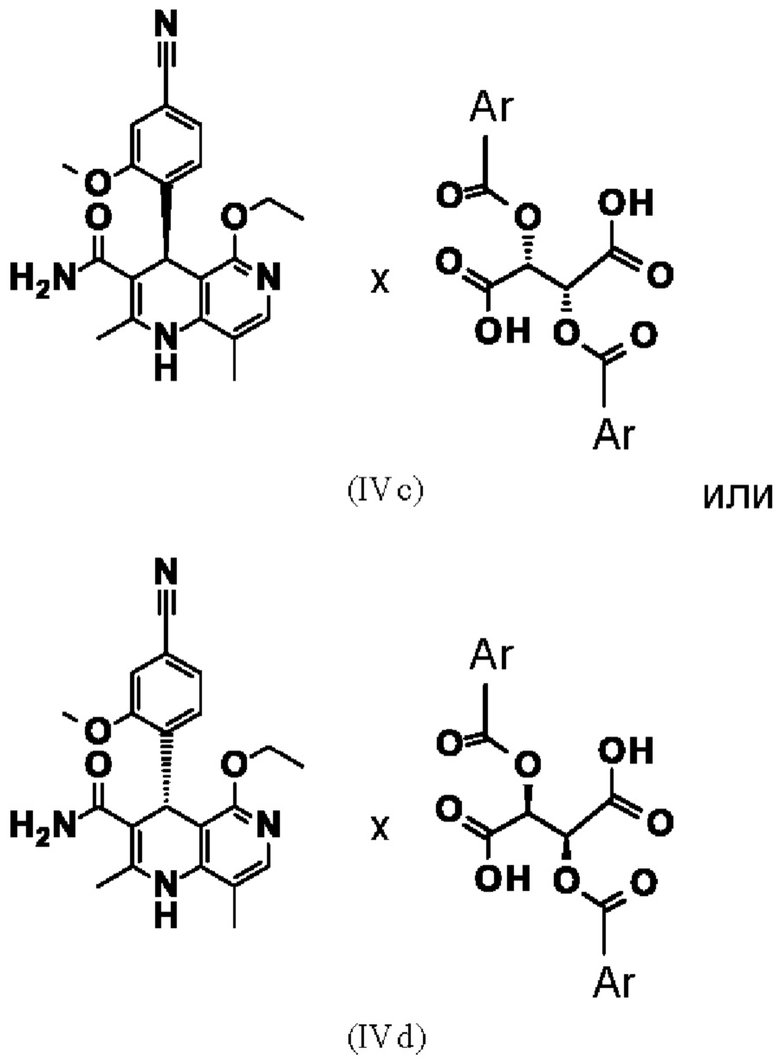
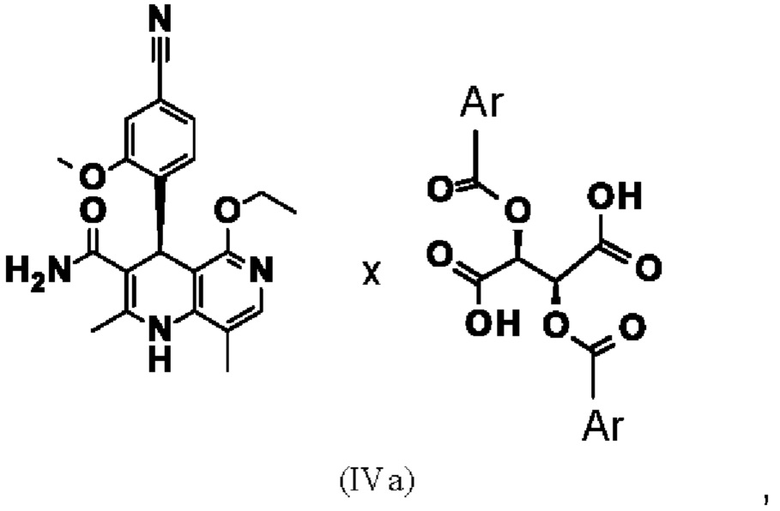
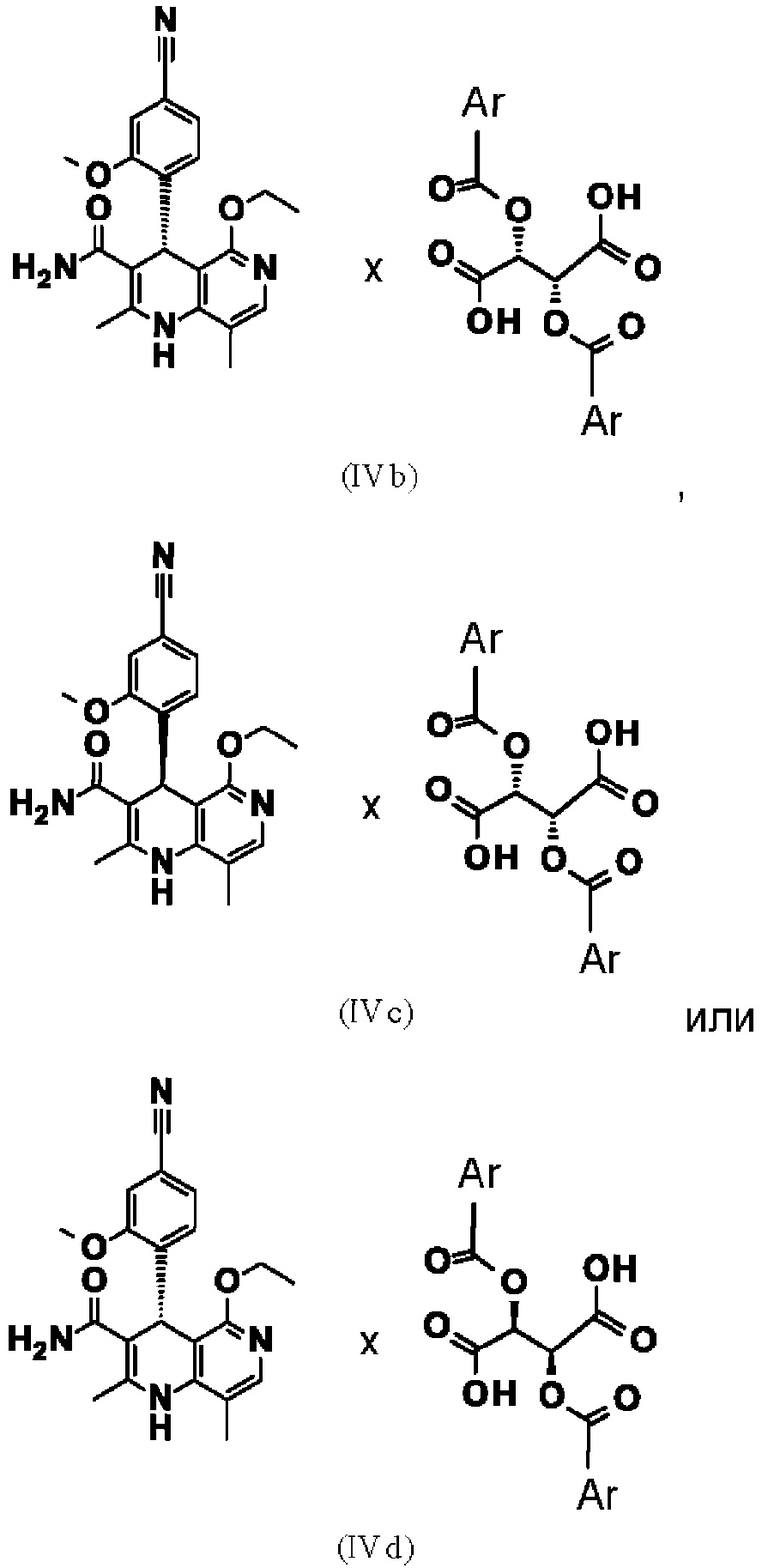
Взаимодействие рацемической смеси (II) с производным винной кислоты общей формулы (IIIa) или (IIIb) дает 4 возможности образования диастереомерной соли (IVa-d). Неожиданным образом наблюдается определенное предпочтение, если, например, рацемат-(II) взаимодействует с производным винной кислоты общей формулы (Ша), в большинстве случаев получается диастереомерная соль общей формулы (IVa), причем образует соль предпочтительно антипод S-конфигурации. Диастереомерная соль (IVa) почти количественно осаждается из раствора, из которого ее затем можно выделить, например, фильтрацией, при этом антипод с Реконфигурацией остается в растворе. Очень похожим образом получают зеркально-симметричную соль общей формулы (IVb), согласно которому подвергают взаимодействию рацемат-(II) с производным винной кислоты общей формулы (IIIb), причем образует соль предпочтительно антипод Реконфигурации. Осажденные диастереомерные соли можно отделить почти количественно, при этом S-антипод остается в растворе.
Посредством стехиометрического соотношения (II) к (IIIa) или соответственно (IIIb) и выбора растворителя можно оптимизировать выход и энантиомерную чистоту.
Поскольку финереноны (I) имеют S-конфигурацию, то предпочтительно для разделения рацематов используют производные винной кислоты с S,S-конфигурацией, так как в данном случае предпочтительно образуется диастереомерная соль S-антипода.
Для разделения рацематов используют от 0,4 до 1,2 экв. сложного эфира винной кислоты (IIIa) или (IIIb), предпочтительно однако от 0,4 до 0,7 экв., особенно предпочтительно однако от 0,45 до 0,6 экв., наиболее предпочтительно однако 0,50-0,55 экв.
Образование диастереомерной соли происходит в смесях растворителей, которые состоят из воды и смешивающихся с водой органических растворителей.
В качестве органических растворителей в контексте настоящей заявки подходящими являются, например, этанол, метанол, изопропанол, 1-пропанол, этилацетат, изобутанол, дихлорметан, 1-пентанол или ацетон, однако предпочтительно используют этанол. Растворители также можно использовать в коммерчески доступной денатурированной форме, содержащей используемые для этанола денатурирующие агенты, например, толуол, метилэтилкетон, тиофен, гексан, что имеет большие преимущества с точки зрения стоимости, поэтому для промышленного использования в частности подходит винный спирт, который в контексте настоящей заявки состоит из этанола, денатурированного при необходимости толуолом или метилэтилкетоном. Поэтому в контексте настоящей заявки, если в дальнейшем упоминается этанол в качестве растворителя, следует понимать, что он означает не только чистый этанол, но также, в частности, для промышленного использования винный спирт, в смысле указанного выше определения. Кроме того, также использовались следующие растворители: этилацетат / метанол 90:10; метанол / вода 80:20; этанол / вода 90:10; этанол / вода 85:15; этанол / вода 80:20; этанол / вода 75:25; этанол / вода 70:30; дихлорметан; 1-пропанол / вода 80:20; 1-пентанол; 1-пентанол / вода 90:10; изопропанол; изопропанол / вода 80:20; изобутанол / вода 90:10; изобутанол / вода 80:20; циклогексанол / вода 90:10; бензиловый спирт / вода 90:10; этиленгликоль; этиленгликоль / вода 80:20.
Предпочтительно разделение рацемата проводят в смеси этанол / вода, причем смеси имеют соотношения (об./об.) в диапазоне этанол: вода = от 1: 1 до 6: 1. Предпочтительно однако используют смесь этанол: вода = от 4: 1 до 3: 1. Особо предпочтительно однако используют смесь этанол: вода = 3: 1. Смесь можно приготовить заранее, или на месте, после того как все компоненты будут помещены в емкость. Смесь растворителей можно использовать в 10-40-кратном избытке в пересчете на рацемат (II), например на 1 кг рацемата используют от 10 л до 40 л смеси растворителей. Предпочтительным является 10-20-кратный избыток, в частности 13-16-кратный избыток, особенно предпочтительно 14-15-кратный избыток.
Обычно разделение рацематов проводят, загружая сначала все компоненты в смесь растворителей при комнатной температуре, затем нагревая до 60-80°С, предпочтительно однако до 75°С, дополнительно перемешивая при 75°С в течение от 2 до 10 часов, предпочтительно от 3 до 4 часов, а затем охлаждая в течение от 3 до 10 часов, предпочтительно от 4 до 5 часов, до комнатной температуры (приблизительно от 20 до 23°С) (посредством линейного изменения температуры). Затем смесь оставляют дополнительно перемешиваться от 2 до 24 часов, предпочтительно от 5 до 18 часов, наиболее предпочтительно от 12 до 16 часов при комнатной температуре. Разделение рацемата предпочтительно проводят при температуре 75°С.
По окончании выделяют осажденную диастереомерную соль (IVa), (IVb), (IVc) и/или (IVd).
Выделение проводят способами, известными специалисту в данной области, например, посредством фильтрация или с использованием центрифуги. Полученный таким образом осадок на фильтре может быть повторно промыт один или несколько раз растворителем или смесью растворителей. Затем осуществляют сушку в вакууме, предпочтительно <100 мбар, при повышенной температуре (от 50 до 80°С, предпочтительно 50°С). Применение газа-носителя в некоторых случаях оказалось выгодным.
Описанная выше процедура позволяет получить химически очень чистые диастереомерные соли. Энантиомерный избыток диастереомерных солей обычно составляет >97% ее (см. примеры).
Диастереомерные соли при необходимости должны подвергаться сушке, их также можно использовать влажными на следующей стадии процесса.
Стадии способа помимо обычного порядка выполнения, указанного выше, также могут быть скомбинированы или их последовательность может быть изменена, как представлено в следующей Таблице 2:
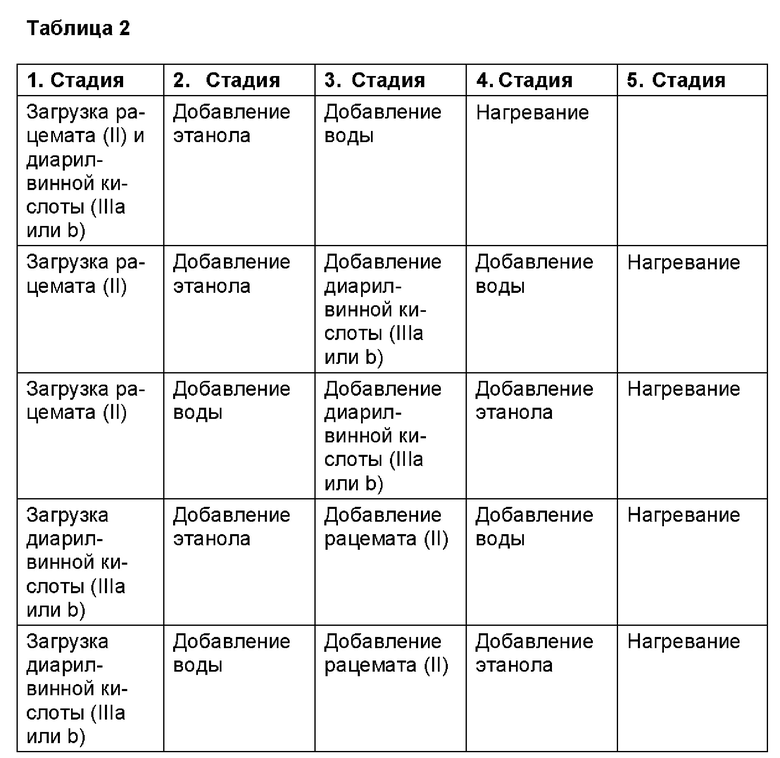
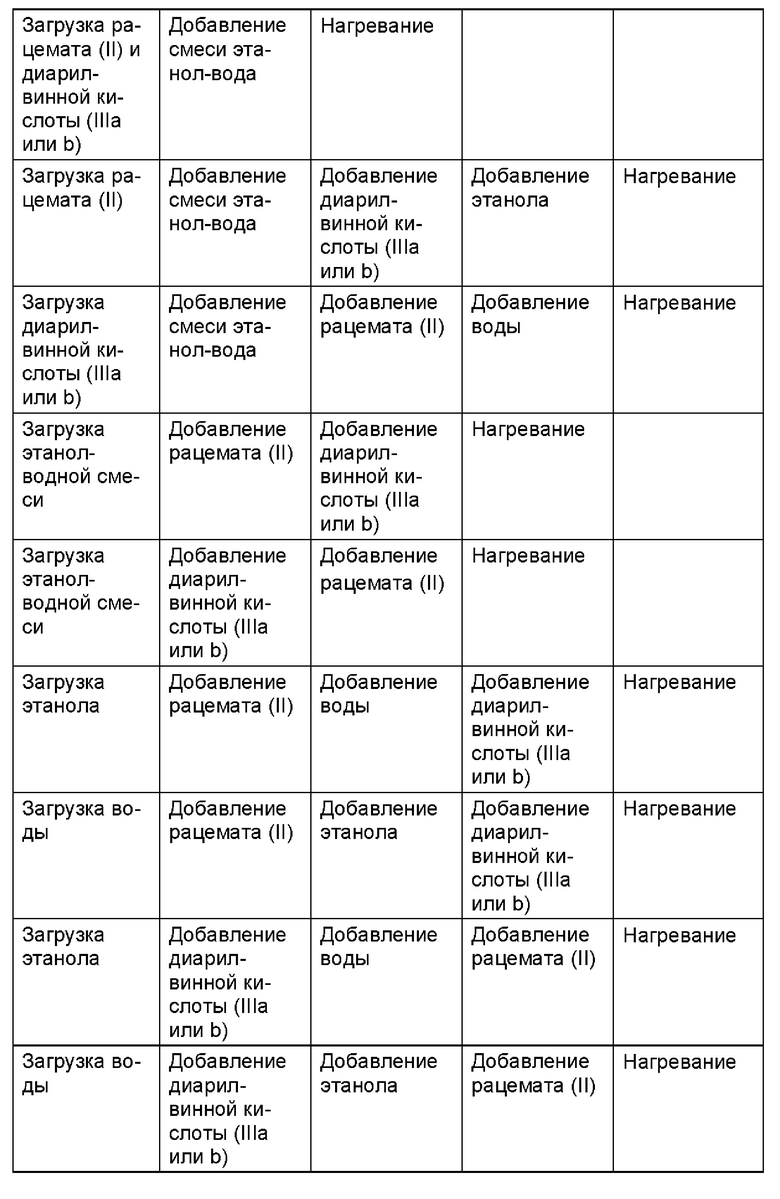
В зависимости от типа установки в пилотной системе или в производстве, может быть выгодным тот или иной вариант.
На следующей стадии диастереомерную соль обрабатывают основанием и удаляют растворитель. Растворитель удаляют известными специалисту в данной области методами, например, посредством дистилляции. Для получения хиральных соединений (I) и (Ia) диастереомерную соль общей формулы (IVa), (IVb), (IVc) или (IVd) необходимо обработать основанием, при этом целевая молекула (I) или (1а) выпадает в осадок после выпаривания органического растворителя из раствора, и ее выделяют - например, посредством фильтрования и промывания, а соответствующий сложный эфир винной кислоты формулы (IIIa) или (IIIb) остается в солевой форме в растворе.
В качестве оснований в контексте настоящего изобретения подходящими являются неорганические и органические основания. В случае неорганических оснований можно использовать аммиак, раствор гидроксида натрия, гидроксид лития, гидроксид калия, карбонат аммония, карбонат натрия, карбонат калия, карбонат лития, гидрокарбонат аммония, гидрокарбонат натрия, гидрокарбонат калия, фосфат натрия, фосфат калия, фосфат аммония. Однако предпочтительно используют гидроксид натрия, фосфат натрия или фосфат калия. Особо предпочтительно используют фосфат натрия или фосфат калия. Важно отметить, что неорганические основания могут быть использованы как в безводной форме, так и в форме их гидратов, так, например, можно успешно использовать фосфат натрия (безводный) и гидрат фосфата натрия. В качестве органического основания могут быть использованы алифатические или ароматические основания, такие как, например, триэтиламин, имидазол, N-метилимидазол, основание Хунига, пиридин, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU).
Высвобождение целевого соединения (I) или (Ia) происходит в смесях воды со смешивающимися с водой органическими растворителями, такими как этанол, изопропанол, 1,2-этандиол, метоксиэтанол, метанол или ацетон, но предпочтительно используют этанол. Растворители также можно использовать в коммерчески доступной денатурированной форме, содержащей используемые для этанола, денатурирующие агенты, например, толуол, метилэтилкетон, тиофен, гексан, предпочтительно используют винный спирт, который в контексте настоящей заявки состоит из этанола, который при необходимости может быть денатурирован толуолом или метилэтилкетоном, что имеет большие преимущества с точки зрения производственных затрат. Было доказано, что выгодно использовать смеси воды и этанола, причем смеси имеют соотношения (об./об.) в диапазоне этанол: вода = 1:6 до 1:3. Предпочтительно однако используют смесь этанол: вода = 1:4. Смесь можно приготовить заранее, или на месте, после того как все компоненты будут помещены в емкость. Указанную смесь можно использовать в 7-20-кратном количестве в пересчете на используемую диастереомерную соль (IVa или IVb или IVc или IVd), например 1 кг в 7-20 л указанной смеси. Предпочтительно указанную смесь применяют в 8-12-кратном количестве, особо предпочтительно указанную смесь применяют в 9-11-кратном количестве, наиболее предпочтительным является 10-кратное количество.
Высвобождение целевого соединения (I) или (1а) осуществляют посредством введения диастереомерной соли (IVa или IVb или IV с или IVd) в смесь растворителей при температуре от 0 до 80°С, предпочтительно от 20 до 50°С, устанавливая затем посредством добавления органического или неорганическое основание (либо в твердой форме, либо в виде раствора, предпочтительно в воде) значение рН от 6,9 до 9,5, предпочтительно от 7,0 до 8,0, особенно предпочтительно рН 7,5. По выбору основание можно добавлять очень быстро (в течение нескольких минут) или очень медленно (в течение нескольких часов), например, от 5 минут до 3 часов. В любом случае предпочтительным является более быстрое добавление, предпочтительно дозируют в течение от 5 минут до 1 часа. Для этой цели можно использовать рН-метр, встроенный в реактор, с помощью которого контролируют установление рН и медленно добавляют основание. Однако также с самого начала может быть добавлено фиксированное количество основания (твердого или растворенного в растворителе), которое гарантирует, на основании полученных опытным путем значений, что предпочтительно будет достигнут желаемый диапазон рН. Такая процедура особенно предпочтительна на производстве. Было доказано, что полезно, если после успешной установки рН смесь снова перемешивают при 10-80°С, предпочтительно 20-60°С, предпочтительно 40-60°С. Время ночного перемешивания составляет при этом от 1 до 10 часов, предпочтительно от 2 до 5 часов, особо предпочтительно от 3 до 4 часов. Затем оставляют охладиться до 15-25°С, например, с линейным изменением температуры, а затем можно снова перемешивать в течение от 1 до 64 часов (64 часа служат для демонстрации устойчивости способа, см. пример 3b), однако предпочтительно перемешивать от 3 до 24 часов, но, как правило, достаточно от 10 до 20 часов.
Выделение проводят способами, известными специалисту в данной области, например, посредством фильтрации или с использованием центрифуги. Полученный таким образом осадок на фильтре может быть повторно промыт один или несколько раз растворителем или смесью растворителей. Затем осуществляют сушку в вакууме, предпочтительно<100 мбар, при повышенной температуре (от 50 до 80°С, предпочтительно 50°С). Применение газа-носителя в некоторых случаях оказалось выгодным.
Однако также можно растворить полученный на фильтре материал в этаноле или смеси воды с этанолом, а затем, отрегулировав количество воды/этанола в элюате, сразу перейти к следующей стадии способа (см. ниже). Данная процедура особенно предпочтительна в промышленном масштабе, поскольку в результате этого позволяет избежать стадии сушки.
Описанная выше процедура позволяет получить химически очень чистые неочищенные продукты. Энантиомерный избыток неочищенных продуктов (I) и (Ia) как правило составляет >96,5% ее.
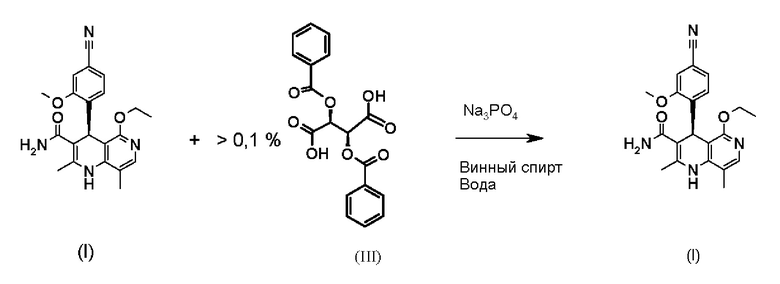
В рамках масштабирования было показано, что полное отделение сложных эфиров винной кислоты (IIIa) и (IIIb) от неочищенных продуктов (I) и (Ia) не всегда технически очень просто осуществить, и в случае содержания этих компонентов подходят очень близко к лимиту спецификации. Поскольку требования к спецификации конечного активного вещества очень высоки (<0,1% (IIIa) в конечном активном веществе), в некоторых случаях оказалось выгодным добавить дальнейшую стадию способа, которая гарантирует, что содержание сложного эфира винной кислоты IIIa) снижается до менее 0,15%, предпочтительно до менее 0,1% и, в частности, менее 0,05%, и конечное активное вещество воспроизводимо получают в соответствии со спецификацией. Сложный эфир винной кислоты (IIIa) практически, или почти совсем, не обедняется при окончательной кристаллизации до чистого продукта, так что такая дополнительная стадия способа гарантирует устойчивый режим выполнения всего способа и работу почти без потерь. Данная стадия способа, которая гарантирует, что соединение (IIIa) обедняется до менее 0,15%, предпочтительно менее 0,1% и, в частности, менее 0,05% в активном веществе, также является объектом настоящего изобретения. Способ снижения содержания (IIIa) осуществляют следующим образом. Для этого используют высушенный или предпочтительно еще влажный неочищенный продукт (I) или (Ia). Для этого опять применяют основание: Для удаления сложных эфиров винной кислоты формулы (IIIa) или (IIIb) можно применять неорганические и органические основания. В случае неорганических оснований можно использовать аммиак, раствор гидроксида натрия, гидроксид лития, гидроксид калия, карбонат аммония, карбонат натрия, карбонат калия, карбонат лития, гидрокарбонат аммония, гидрокарбонат натрия, гидрокарбонат калия, фосфат натрия, фосфат калия, фосфат аммония. Однако предпочтительно используют гидроксид натрия, фосфат натрия или фосфат калия. Особо предпочтительно используют фосфат натрия или фосфат калия. Важно отметить, что неорганические основания могут быть использованы как в безводной форме, так и в форме их гидратов, так, например, можно успешно использовать фосфат натрия (безводный) и гидрат фосфата натрия. В качестве органического основания могут быть использованы алифатические или ароматические основания, такие как, например, триэти-ламин, имидазол, N-метилимидазол, основание Хунига, пиридин, DBU.
Проведение обеднения (IIIa) или (IIIb) происходит в смесях воды со смешивающимися с водой органическими растворителями, такими как этанол, изопропанол, 1,2-этандиол, метоксиэтанол, метанол или ацетон, но предпочтительно используют этанол. Растворители также можно использовать в коммерчески доступной денатурированной форме, содержащей используемые для этанола денатурирующие агенты, например, толуол, метилэтилкетон, тиофен, гексан, что имеет большие преимущества с точки зрения производственных затрат. Было доказано, что выгодно использовать смеси воды и этанола, причем смеси имеют соотношения (об./об.) в диапазоне этанол: вода = 30:10 до 10:10. Предпочтительно однако используют смесь этанол: вода = 20: 10. Смесь можно приготовить заранее, или на месте, после того как все компоненты будут помещены в емкость. Указанную смесь растворителей можно использовать в 10-20-кратном избытке в пересчете на используемую диастереомерную соль IVa, IVb, IVc или IVd, например 1 кг в 10-20 л указанной смеси. Предпочтительно указанную смесь применяют в 10-14-кратном количестве, особо предпочтительно указанную смесь применяют в 11-12-кратном количестве.
Проведение обеднения (IIIa) или (IIIb) осуществляют таким образом, что вводят в смесь растворителей, описанную выше, при температуре от 40 до 80°С, предпочтительно от 50 до 70°С, устанавливая затем посредством добавления органического или неорганическое основание (либо в твердой форме, либо в виде раствора, предпочтительно в воде) значение рН от 6,9 до 9,5, предпочтительно от 7,5 до 9,0, особенно предпочтительно рН 8,5. По выбору основание можно добавлять очень быстро (в течение нескольких минут) или очень медленно (в течение нескольких часов), например, от 1 минуты до 3 часов. В любом случае предпочтительным является более быстрое добавление, предпочтительно дозируют в течение от 1 минуты до 1 часа. Для этой цели можно использовать рН-метр, встроенный в реактор, с помощью которого контролируют установление рН и медленно добавляют основание. Однако также с самого начала может быть добавлено фиксированное количество основания (твердого или растворенного в растворителе), которое гарантирует, на основании полученных опытным путем значений, что предпочтительно будет достигнут желаемый диапазон рН. Такая процедура наиболее предпочтительна на производстве. Было доказано, что полезно, если после успешной установки рН смесь снова перемешивают при 40-80°С, предпочтительно 50-75°С, предпочтительно 60-70°С. Время ночного перемешивания составляет при этом от 1 до 24 часов, предпочтительно от 2 до 10 часов, особо предпочтительно от 2 до 4 часов. Затем органический растворитель в значительной мере отгоняют; это можно выполнить при нормальном давлении или более осторожно при пониженном давлении. Как только органический растворитель в значительной мере отогнан, оставляют охладиться до 15-25°С, например, с линейным изменением температуры, а затем можно снова перемешивать в течение от 1 до 64 часов (64 часа служат для демонстрации устойчивости способа, см. пример 3b), однако предпочтительно перемешивать от 1 до 24 часов, но, как правило, достаточно от 1 до 5 часов.
Выделение проводят способами, известными специалисту в данной области, например, посредством фильтрации или с использованием центрифуги. Полученный таким образом осадок на фильтре может быть повторно промыт один или несколько раз растворителем или смесью растворителей. Затем осуществляют сушку в вакууме, предпочтительно <100 мбар, при повышенной температуре (от 50 до 80°С, предпочтительно 50°С). Применение газа-носителя в некоторых случаях оказалось выгодным.
Также можно перед проведением отгонки растворителя установить значение рН снова на 5-7 с помощью органической кислоты, такой как муравьиная кислота, лимонная кислота, уксусная кислота, или неорганической кислоты, такой как соляная кислота, серная кислота, фосфорная кислота, или кислотной соли, такой как дигидрофосфат натрия, гидросульфат калия или гидросульфат натрия, а затем продолжить, как описано выше.
Описанная выше процедура позволяет получить химически очень чистые неочищенные продукты. Энантиомерный избыток неочищенных продуктов (I) и (Ia), как правило, составляет >97% ее, а содержание (IIIa) или (IIIb) снижено до менее 0,15%, предпочтительно менее 0,1% и, в частности, менее 0,05%.
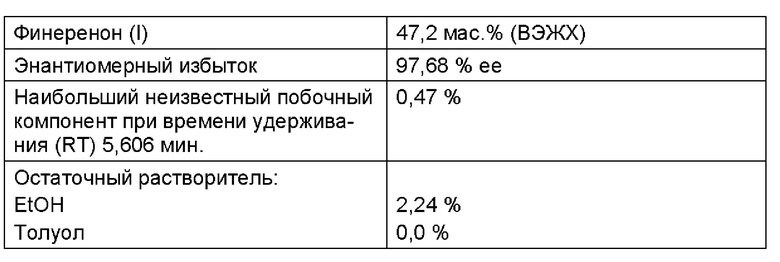
Было доказано, что полученный таким образом неочищенный продукт формулы (I) или (Ia) выгодно подвергнуть еще одной последующей кристаллизации для улучшения как химической, так и, прежде всего, оптической чистоты, поскольку энантиомерные избытки неочищенных продуктов, как правило, составляют >97% ее. Для этого был разработан способ окончательной кристаллизации: Для этой цели по GMP-техническим причинам неочищенный продукт (I) или (Ia) растворяют в этаноле (при необходимости посредством нагревания), а затем подвергают фильтрации частиц; предпочтительно используют винный спирт или этанол, денатурированный толуолом. В некоторых случаях, в зависимости от технического оборудования, кристаллизацию также можно проводить под давлением, при повышенной температуре (преимущество: меньше растворителя), но также можно перекристаллизовать из водных растворов этанола (под давлением или при нормальном давлении). Затем при нормальном или соответственно пониженном давлении объем уменьшают примерно в 3-6 раз, предпочтительно в 4-5 раз, при этом выкристаллизовывается продукт. В некоторых случаях, если, например, необходимо отгонять большие объемы в течение длительных периодов времени, оказалось выгодно отгонять при пониженном давлении, причем поддерживают низкую внутреннюю температуру, чтобы избежать разложения и образования побочных продуктов. После окончания дистилляции охлаждают до 0°С и затем кристаллы выделяют и сушат в вакууме при температуре от 40 до 50°С. Выходы, как правило, составляют >88% от теоретического. Достигаемая степень химической чистоты и содержание соответствуют критериям торгового продукта, принятым для оценки согласно рекомендациями ICH (Международной конференции по гармонизации технических требований к регистрации лекарственных средств для человека). Остаточный растворитель, в данном случае этанол, обычно составляет <0,05% в случае загрузок на пилотной установке. Оптическая чистота составляет >> 99% ее.
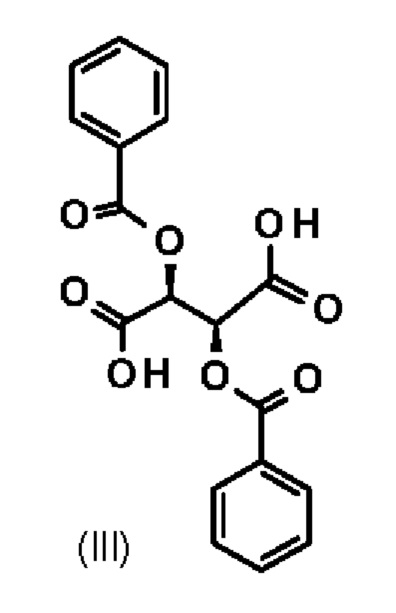
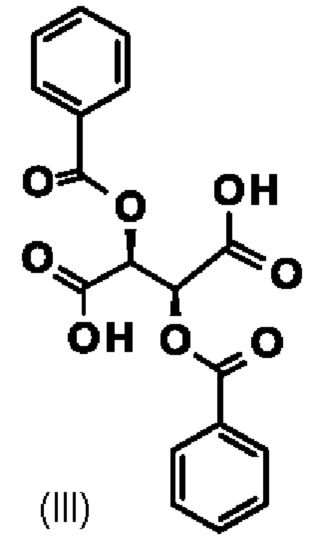
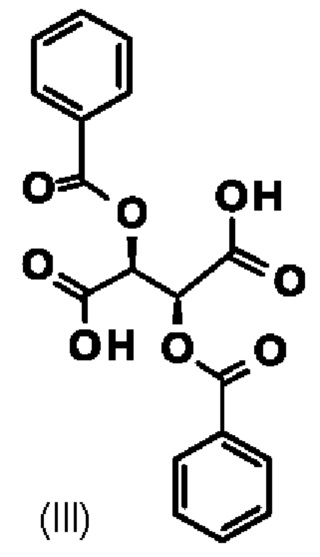
В качестве особенно предпочтительного способа, в частности для крупномасштабного производства, используют (+)-дибензоилвинную кислоту (III); может быть использована как безводная форма, так и гидрат:
Разделение рацемата предпочтительно проводят в смеси винного спирта и воды. Последующее высвобождение финеренона неочищенного
предпочтительно происходит в смеси винного спирта и воды с использованием фосфата натрия в качестве основания. В случаях, в которых необходима дополнительная обработка, если доля (+)-дибензоилвинной кислоты (III) составляет >0,15%
предпочтительно дополнительная обработка происходит в смеси винного спирта и воды с использованием фосфата натрия в качестве основания. Конечная кристаллизация до финеренона, чистого происходит предпочтительно в винном спирте в качестве растворителя. При этом достигается чистота 0,15%, предпочтительно 0,1%, особенно предпочтительно менее 0,05% примесей.
Также возможно выделить целевой энантиомер из маточного раствора. В таком случае сначала получают соответствующую диастереомерную соль (IVa), (IVb), (IVc) или (IVd) из (I) или (Ia), потом выделяют фильтрованием, а затем в маточном растворе, который в данном случае содержит соответствующий антипод, посредством добавления основания, например аммиака, раствора едкого натра, гидроксида лития, гидроксида калия, карбоната аммония, карбоната натрия, карбоната калия, карбоната лития, гидрокарбоната аммония, гидрокарбоната натрия, гидрокарбоната калия, фосфата натрия, фосфата калия, фосфата аммония, предпочтительно гидроксида натрия, фосфата натрия и фосфата калия, особенно предпочтительно фосфата натрия и фосфата калия, доводят значение рН до уровня >7, предпочтительным является рН 7,5. Затем органический растворитель, предпочтительно этанол, отгоняют либо при нормальном давлении, либо более осторожно при пониженном давлении, при этом соответствующий антипод выпадает в осадок. Продукт отфильтровывают, промывают водой или смесями вода/растворитель и сушат. Соответствующая окончательная кристаллизация из винного спирта, как например, описано в примере 1с, дает соединения (I) и (Ia) в соответствующей чистой форме.
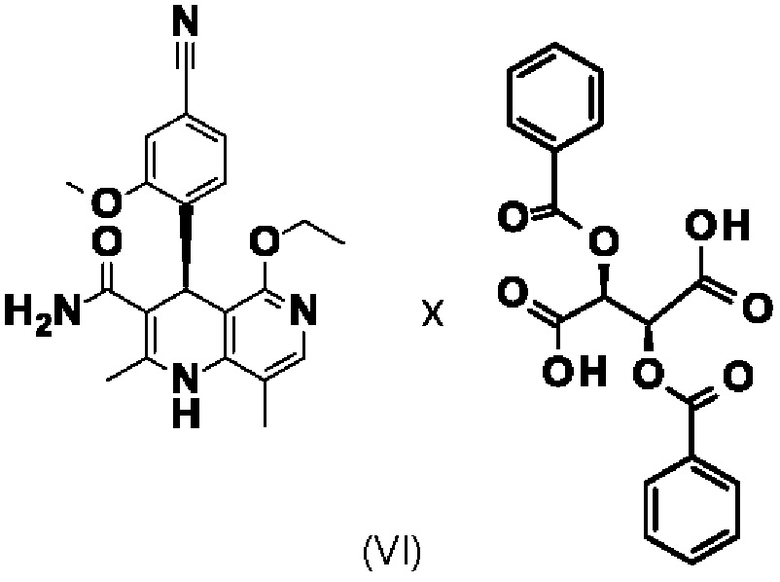
Поэтому другим объектом настоящей заявки также является финеренон (I) с содержанием дибензоилвинной кислоты ≤0,15%, полученный в результате того, что рацемат (II)
взаимодействует с дибензоилвинной кислотой формулы (III)
в смеси винного спирта и воды с получением диастереомерной соли (VI)
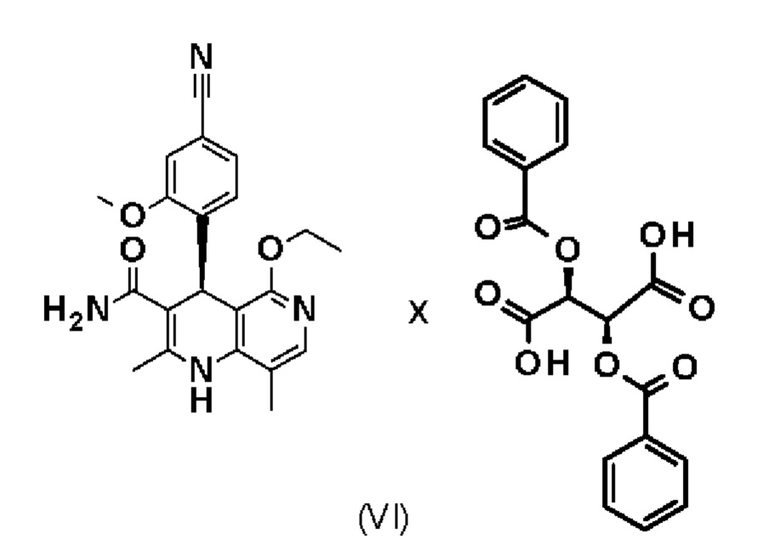
и затем финеренон (I)
высвобождается с использованием фосфата натрия также в смеси винного спирта и воды, а затем снова взаимодействует с фосфатом натрия в смеси винного спирта и воды, после чего кристаллизуется чистый финеренон с содержанием дибензоилвинной кислоты ≤0,15%; в особенно предпочтительном варианте осуществления финеренон получают с содержанием дибензоилвинной кислоты ≤0,1%, в частности предпочтительно содержание дибензоилвинной кислоты ниже 0,05%.
Экспериментальная часть
Сокращения и аббревиатуры:

Примеры
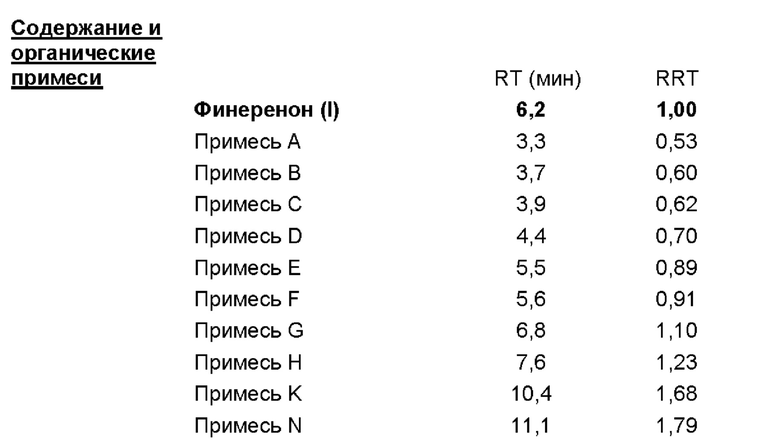
В далее следующей таблице 3 показаны структуры соединений, обнаруженных при ВЭЖХ. Сопоставление времен удерживания при ВЭЖХ приведено ниже.
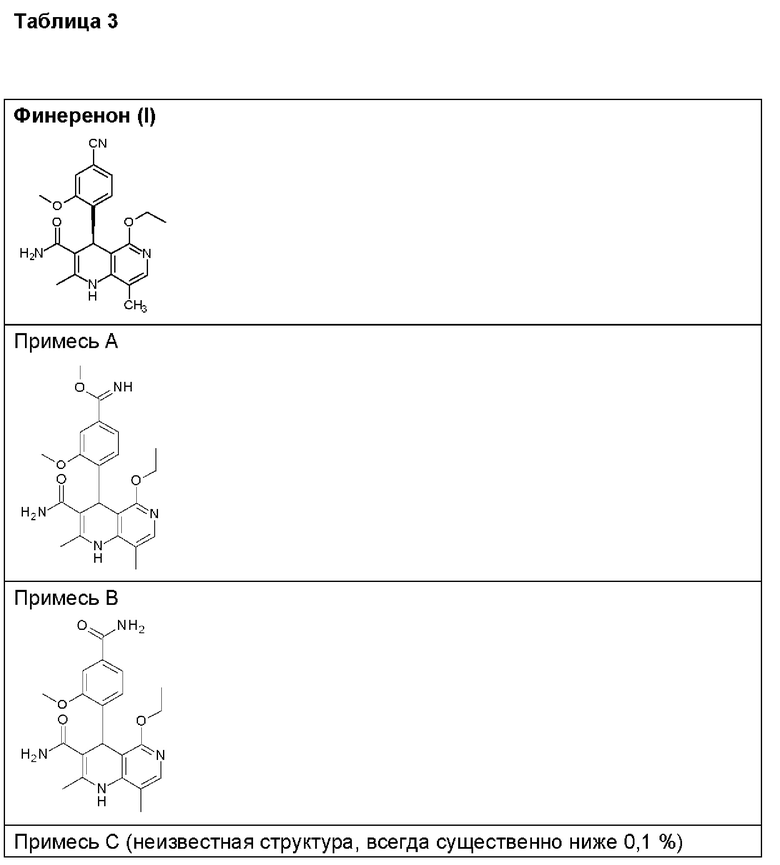
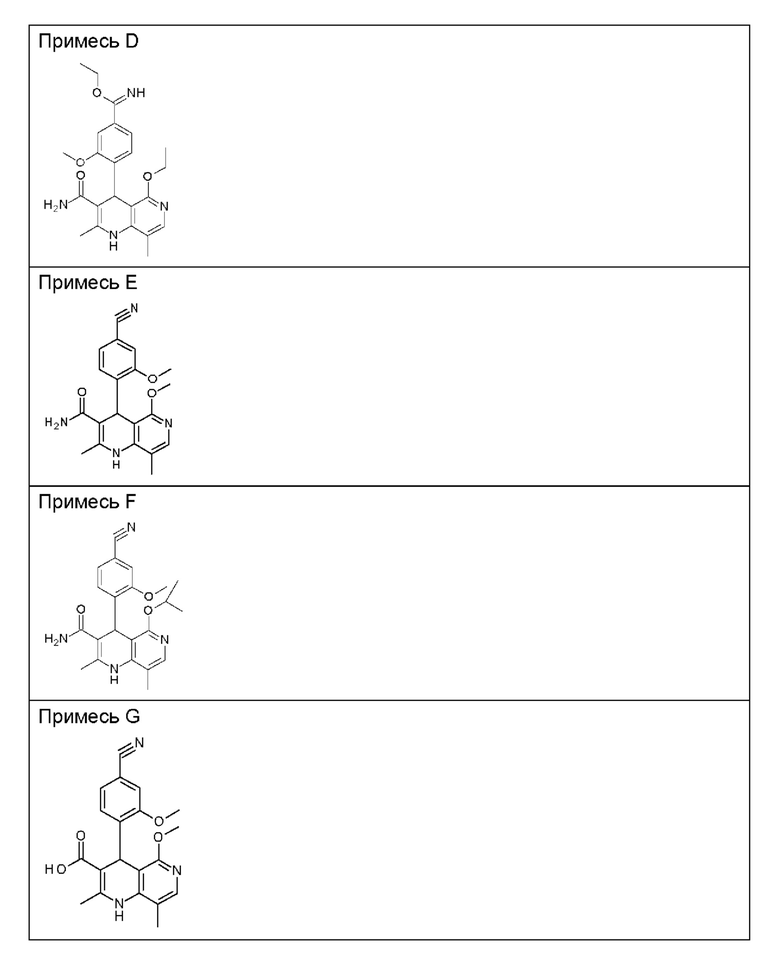
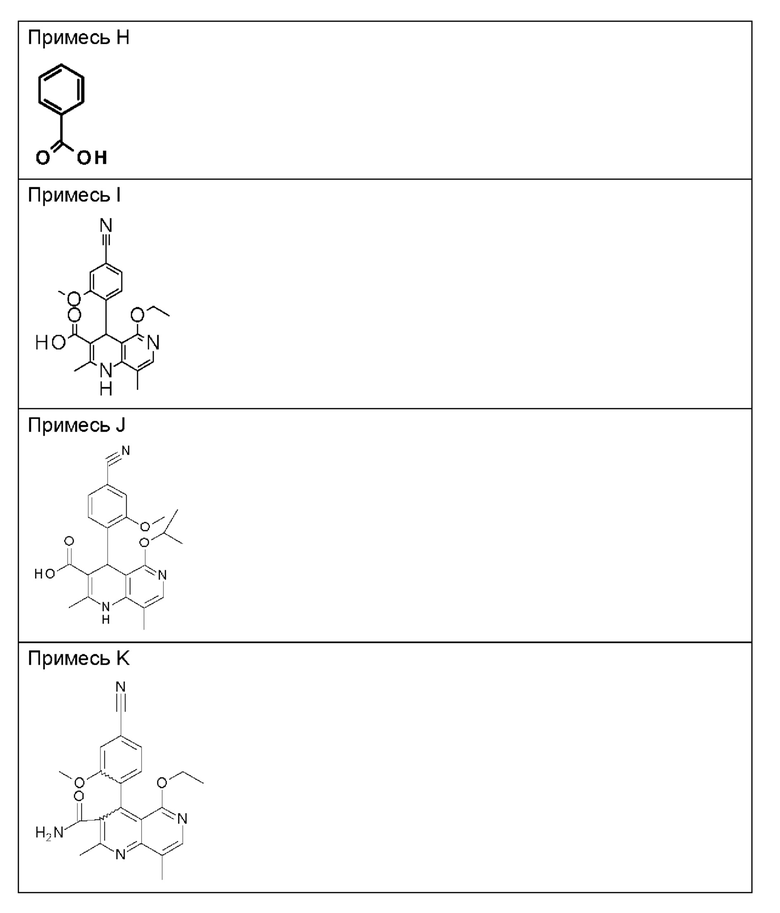

1) Аналитический метод проверки содержания примесей и энантиомерной чистоты на стадии дибензоилвинной кислоты.
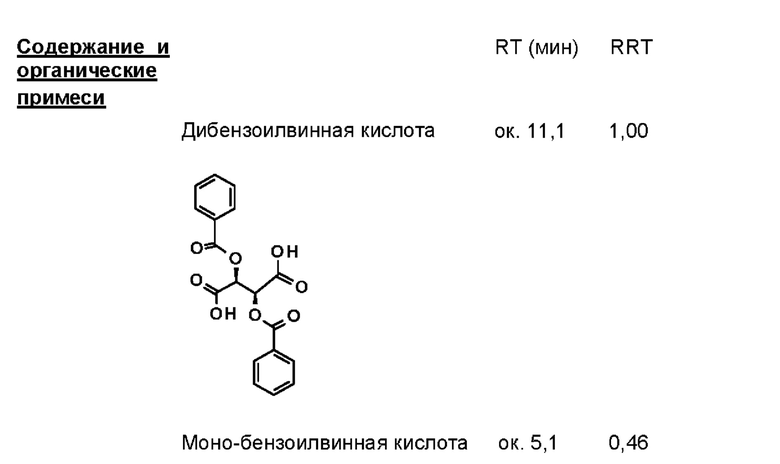
Прибор: Ультра-высокоэффективный жидкостный хроматограф (с диапазоном давления до 1200 бар с термостатируемой печью колонок и УФ-детектором)
Колонка: YMC Triart С8
длина: 100 мм, внутренний диаметр: 3,0 мм, размер зерна: 1,9 мкм
Максимальное давление: 1000 бар
Условия: 20°С; 0,50 мл/мин; 1,7 мкл(10°С); 240 нм/6 нм
Элюент: А: 0,1% TFA в воде; В: Ацетонитрил
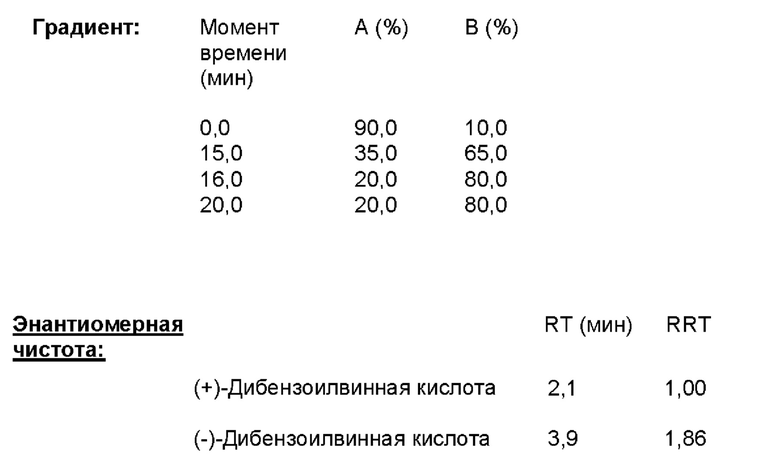
Прибор: Высокоэффективный жидкостный хроматограф с термостатируемой печью колонок и УФ-детектором
Колонка: Chiralpak IC
длина: 250 мм, внутренний диаметр: 4,6 мм, размер зерна: 5,0 мкм
Максимальное давление: 300 бар
Условия: 40°С; 2,0 мл/мин; 5 мкл; 234 нм/6 нм
Элюент: А: Гептан; В: 0,1% TFA в этаноле;
Изократически: А(%) 80: В (%) 20
2) Аналитический метод проверки содержания примесей и энантиомерной чистоты на стадии диастереомерной соли.

Прибор: Ультра-высокоэффективный жидкостный хроматограф (с диапазоном давления до 1200 бар с термостатируемой печью колонок и УФ-детектором)
Колонка: YMC Triart С8
длина: 100 мм, внутренний диаметр: 3,0 мм, размер зерна: 1,9 мкм
Максимальное давление: 1000 бар
Условия: 20°С; 0,50 мл/мин; 3,5 мкл(10°С); 242 нм/6 нм
Элюент: А: 0,1% TFA в воде; В: Ацетонитрил
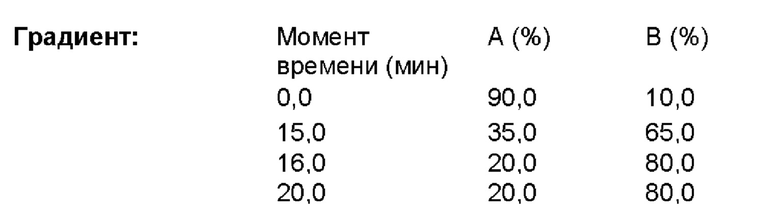
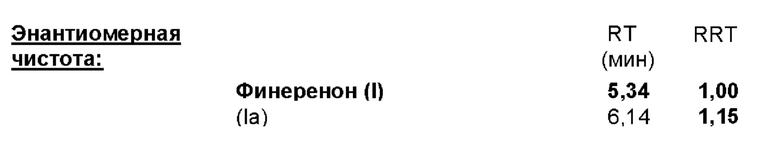
Прибор: Высокоэффективный жидкостный хроматограф с термостатируемой печью колонок и УФ-детектором
Колонка: Lux 3μm i-Cellulose-5
длина: 150 мм, внутренний диаметр: 4,6 мм, размер зерна: 3,0 мкм
Максимальное давление: 300 бар
Условия: 40 "С; 1,0 мл/мин; 10 мкл (20°С); 252 нм/6 нм
Элюент: А: 20 ммоль аммонийацетатный буффер рН 9,0 (при помощи 1,54 г ацетата аммония в 1 л воды качества Milli-Q и аммиака доводили до рН 9,0)
; В: Ацетонитрил
Изократически: А(%) 50: В (%) 50
3) Аналитический метод проверки содержания примесей и энан-тиомерной чистоты на стадии финеренона, неочищенного (I).
Прибор: Ультра-высокоэффективный жидкостный хроматограф
(с диапазоном давления до 1200 бар с термостатируемой печью колонок и УФ-детектором)
Колонка: YMC Triart С8
Длина: 100 мм, внутренний диаметр: 3,0 мм, размер зерна: 1,9 мкм
Максимальное давление: 1000 бар
Условия: 20°С; 0,50 мл/мин; 1,7 мкл (10°С); 252 нм/6 нм и 230 нм/6 нм для оценки ДБ-винной кислоты
Элюент: А: 0,1% TFA в воде; В: Ацетонитрил

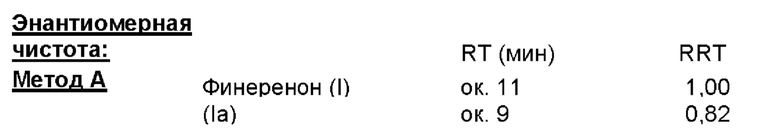
Прибор: Высокоэффективный жидкостный хроматограф с термостатируемой печью колонок и УФ-детектором
Колонка: Chiralpak IA
длина: 250 мм, внутренний диаметр: 4,6 мм, размер зерна: 5,0 мкм
Максимальное давление: 300 бар
Условия: 40 "С; 0,8 мл/мин; 5 мкл (20°С); 255 нм/6 нм
Элюент: А: Ацетонитрил; В: Метил-трет-бутиловый эфир (МТБЭ)
Изократически: А(%) 90: В (%) 10

Прибор/Детектор: Высокоэффективный жидкостный хроматограф с термостатируемой печью колонок, УФ-детектором и системой оценки данных
Измерительная длина волны: 252 нм
Температура печи: 40°С
Колонка: Chiralpak 1С
Длина: 150 мм, внутренний диаметр: 4,6 мм, размер зерна: 3 мкм
Подвижная фаза: А: 50% буффер 20 мМ Ацетата аммония рН 9 В: 50% ацетонитрил
Поток: 1 мл/мин
Время прохождения: 8 мин
Уравновешивание: не требуется, изократически
Растворитель образца: Элюент
Испытуемый раствор: примерно 0,5 мг/мл рацемата вещества, растворенного в растворителе образца
Сравнительный раствор: сравнительный раствор получают по аналогии с испытуемым раствором
Объем впрыска: 10 мкл
Все измеренные значения для определения энантиомеров, приведенные в следующих примерах, были определены в соответствии с методом В. Некоторые значения, прежде всего для партий, полученных на пилотной установке, были измерены для сравнения методом А и дали сопоставимые результаты.
Данные ВЭЖХ-анализа, приведенные в следующих примерах в отношении чистоты и содержания конечного продукта финеренона, чистого (I), относятся только к примесям, которые присутствуют в продукте в количеств е>0,05%. В основном это примесь Е. Все остальные примеси, указанные в таблице выше, как правило, составляют <0,05%. Структура таких примесей определялась путем выделения из обогащенных маточных растворов.
Пример 1
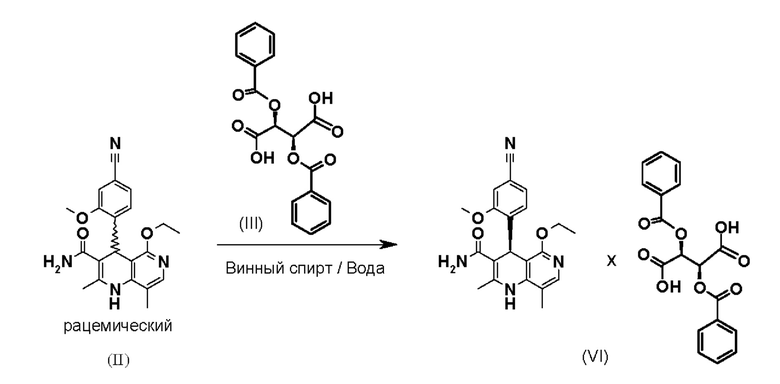
Лабораторная загрузка с использованием безводной (+)-O,O-дибензоил-D-винной кислоты (III)
Пример 1а
Получение тартратной соли (IV)
250 г (660,616 ммоль) рацемата финеренона (II) помещали в 3500 мл смеси, состоящей из этанола, денатурированного толуолом, и воды = 75:25 (об./об.) при комнатной температуре (приблизительно 23°С). 130,2 г (363,339 ммоль) (+)-O,O-дибензоил-D-винной кислоты (III) добавляли через воронку для твердых веществ, а затем промывали 250 мл смеси, состоящей из этанола, денатурированного толуолом, и воды = 75:25 (об./об.). Полученную таким образом суспензию нагревали до внутренней температуры 75°С в течение 0,75 часа, а затем дополнительно перемешивали при этой температуре в течение 3,0 часов. Затем смесь охлаждали с 23°С при линейной скорости охлаждения в течение 5,0 часов и дополнительно перемешивали при данной температуре в течение ночи (приблизительно 16 часов). Суспензию отфильтровывали на пористом стеклянном фильтре и один раз промывали 250 мл смеси, состоящей из этанола, денатурированного толуолом, и воды = 75:25 (об./об.). Выход влажного продукта: 334,7 г. Затем влажный продукт сушили в течение ночи (примерно 16 часов) при 50 0 С под вакуумом (<100 мбар). Выход: 250,2 г (100,08% от теор.) бесцветного кристаллического порошка.
Аналитические результаты:
МС (Elpos): m/z=379 [М+Н]+
1Н-ЯМР (500 МГц, диметилсульфоксид-d6): δ=1.05 (т, 3H), 2.12 (с, 3H), 2.18 (с, 3H), 3.82 (с, 3H), 3.99-4.07 (м, 2Н), 5.39 (с, 1Н), 5,89 (с, 2Н), 6.60-6.84 (м (уширенный сигнал), 2Н), 7.14 (д, 1Н), 7.28 (дд, 1Н), 7.37 (д, 1Н), 7.55 (с, 1Н), 7,61 (т, 4Н), 7.69 (с, 1Н), 7,75 (т, 2Н), 8,04 (д, 4Н), 12,50-15,40 (очень широкий сигнал, 2Н) и сигнал растворителя диметилсульфоксида и повышенный сигнал воды: δ=2,5-2,6, а также маленькие пики около δ=3,40-3,50 (кв) и δ=1,05-1,10 (т), наложенные сигналы от остаточного растворителя этанола.
Бесцветный кристаллический порошок (комплекс финеренон - (+)-O,O-дибензоил-D-винная кислота (VI)), который был получен согласно примеру 1а, исследовали с помощью порошковой рентгеновской дифракции (XRPD) при следующих условиях при 25°С:
Приготовление образца: Порошок готовится как тонкий слой между двумя пленками (например, из полиацетата).
Аппарат: Рентгеновский порошковый дифрактометр X'Pert Pro
Генератор: 40 кВ / 40 мА
Детектор: PIXcel
Излучение: CuKa-излучение
Длина волны: 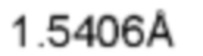
Техника: пропускание
Диапазон сканирования: 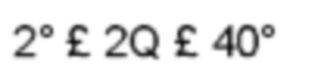
Ширина шага: 0.013° 2Q
Время измерения: 25 сек/шаг
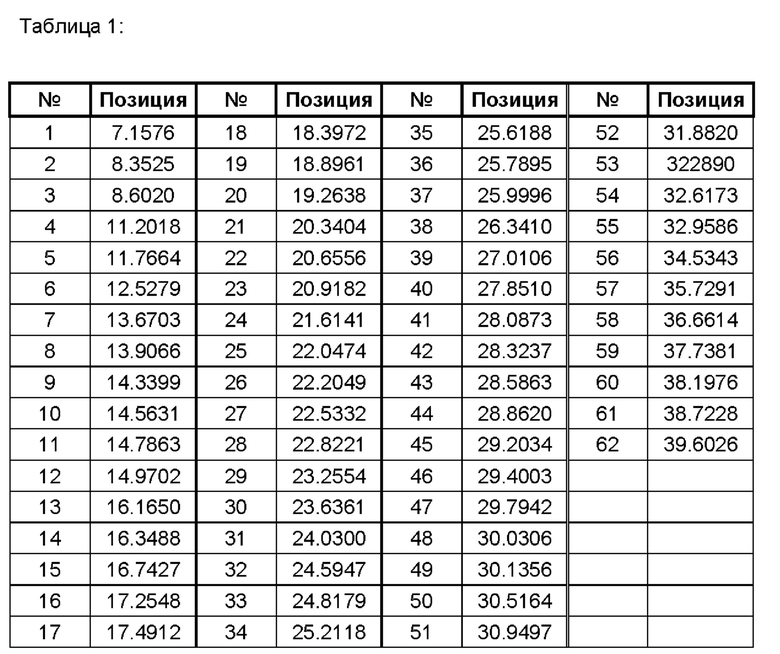
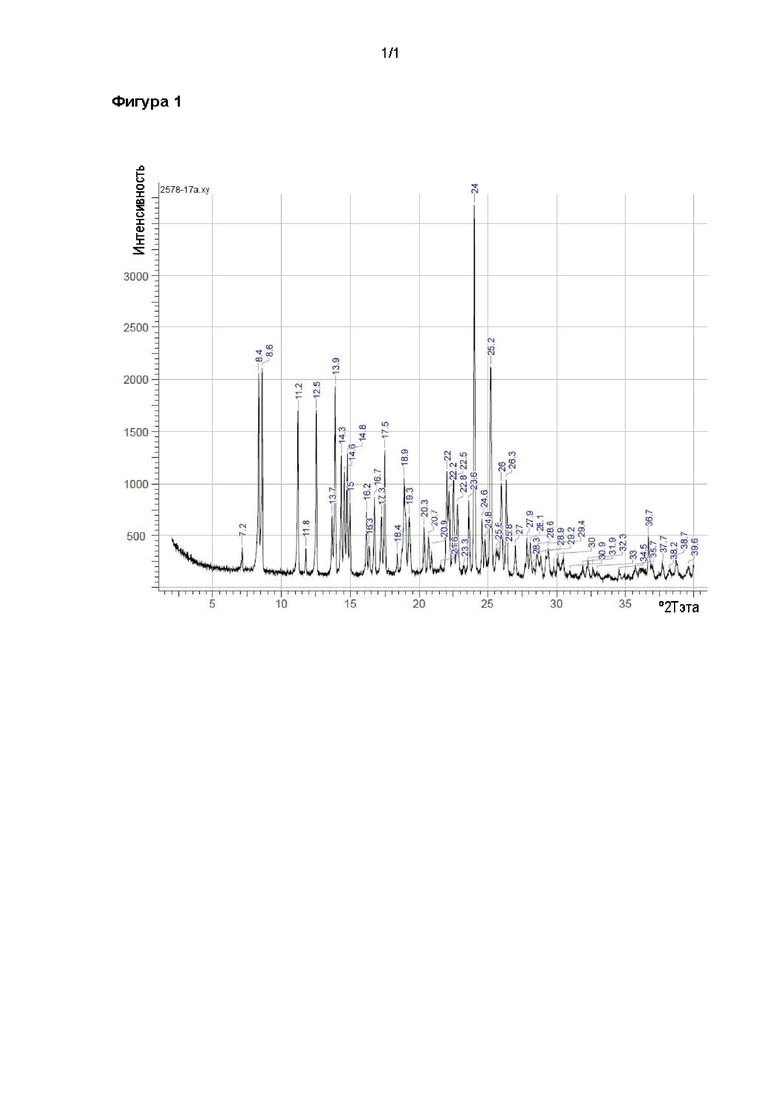
Результаты приведены на рисунке 1 и в таблице 1.
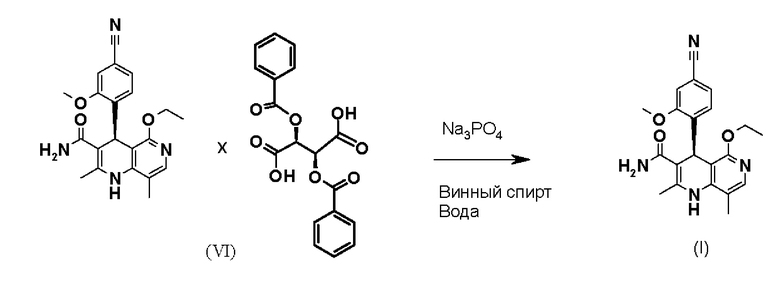
Пример 1b
Получение неочищенного продукта (I)
248 г полученного в Примере 1а соединения (IV) суспендировали при комнатной температуре в 2480 мл смеси, состоящей из этанола, денатурированного толуолом, и воды = 20:80 (об./об.) (измеренное значение рН составляло рН=4). Затем по каплям в течение 60 минут добавляли 819,6 г водного раствора фосфата натрия (100 г фосфата натрия, растворенного в 1000 мл воды) и доводили значение рН до рН=7,2. Смесь дополнительно перемешивали при 23°С в течение 50 минут (рН 7,1). Затем по каплям в течение 10 минут добавляли 98,3 г водного раствора фосфата натрия (100 г фосфата натрия, растворенного в 1000 мл воды) и доводили значение рН до рН=7,5. Нагревали в течение часа до внутренней температуры 50°С, и 3,0 часа дополнительно перемешивали при этой температуре. Охлаждали в течение часа до 22°С и дополнительно перемешивали в течение часа при этой температуре. Кристаллизат отфильтровывают на пористом стеклянном фильтре и промывают один раз 200 мл, и один раз 100 мл смеси, состоящей из этанола, денатурированного толуолом, и воды = 20:80 (об./об.) и два раза 200 г воды. Выход влажного продукта: 263,4 г. Затем влажный продукт сушили в течение выходных (>48 часов) при 50°С под вакуумом (<100 мбар). Выход: 116,9 г (93,52% от теор.) бесцветного кристаллического порошка.
Аналитические результаты:
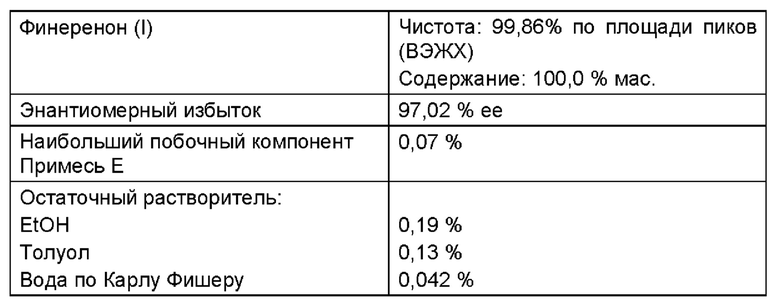
MC (Elpos): m/z=379 [M+H]+
1Н-ЯМР (500 МГц, диметилсульфоксид-d6): δ=1.05 (т, 3H), 2.12 (с, 3H), 2.18 (с, 3H), 3.82 (с, 3H), 3.99-4.07 (м, 2Н), 5.37 (с, 1Н), 6.60-6.84 (м (уширенный сигнал), 2Н), 7.14 (д, 1Н), 7.28 (дд, 1Н), 7.37 (д, 1Н), 7.55 (с, 1Н), 7.69 (с, 1Н) и сигнал растворителя диметилсульфоксида и значительно повышенный сигнал воды: δ=2,5-2,6, а также очень маленький пик около δ=3,38 (не классифицирован)
Пример 1с
Получение чистого продукта (I)
16,0 г полученного в примере 1b неочищенного продукта (I) суспендировали в 2330 мл этанола, денатурированного толуолом, и затем нагревали до кипячения с возвратом флегмы. При этом продукт перешел в раствор. Перемешивали в течение часа при этой температуре. Раствор отфильтровывали через нагретый (Т=75°С) фильтр, работающий под давлением, а затем фильтр, работающий под давлением, промывали 30 мл этанола, денатурированного толуолом. Затем отгоняли растворитель (было отогнано примерно 1920 мл) до достижения конечного, примерно 4-кратного объема (относительно используемого вещества: 116 г × 4 ~ 484 мл). Затем охлаждали до внутренней температуры 23°С (продолжительность приблизительно от 1,5 до 2 часов). Затем перемешивали два часа при внутренней температуре 3°С. Продукт отфильтровывали и дополнительно один раз промывали 100 мл этанола, денатурированного толуолом. Выход влажного продукта: 124 г. Затем влажный продукт сушили в течение выходных (>48 часов) при 50°С под вакуумом (<100 мбар). Выход: 112,6 г (97,07% от теор.) бесцветного кристаллического порошка, мелкие игольчатые кристаллы.
Аналитические результаты:
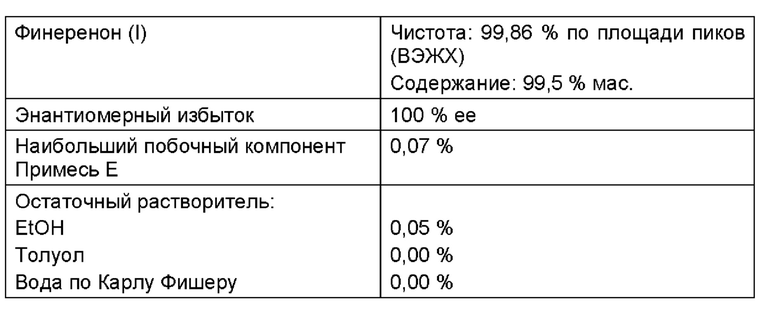
МС (Elpos): m/z=379 [М+Н]+
1Н-ЯМР (500 МГц, диметилсульфоксид-d6): δ=1.05 (т, 3H), 2.12 (с, 3H), 2.18 (с, 3H), 3.82 (с, 3H), 3.99-4.07 (м, 2Н), 5.37 (с, 1Н), 6.60-6.84 (м (уширенный сигнал), 2Н), 7.14 (д, 1Н), 7.28 (дд, 1Н), 7.37 (д, 1Н), 7.55 (с, 1Н), 7.69 (с, 1Н) и маленькие сигналы растворителя диметилсульфоксида и воды около δ=2,5-2,6, а также очень маленький пик около δ=3,38 (не классифицированный).
Модификация: Мод А (согласно определению в WO 2016/016287 А1, в котором абсолютная конфигурация была определена с помощью рентгеновского излучения)
Пример 2
Лабораторная загрузка с использованием гидрата (+)-O,O-дибензоил-D-винной кислоты (III)
Пример 2а
Получение тартрата
Сначала при комнатной температуре вводили 350,0 г рацемата (II), а затем добавляли 3112 г винного спирта. Затем добавляли 1200 г воды. 191,4 г моногидрата (+)-O,O-дибензоил-D-винной кислоты (III) добавляли через воронку для твердых веществ, а затем промывали 113 г воды. Суспензию в течение часа нагревали до внутренней температуры 75°С, а затем перемешивали в течение 3 часов при 75°С в течение 3 часов. Затем смесь охлаждали с 23°С при линейной скорости охлаждения в течение 5,0 часов и дополнительно перемешивали при данной температуре в течение ночи (приблизительно 18 часов). Суспензию отфильтровывали на пористом стеклянном фильтре и два раза промывали смесью из 332,3 г винного спирта и 140,2 г воды. Выход влажного продукта: 487,6 г. Затем влажный продукт сушили в течение выходных (>48 часов) при 50°С под вакуумом (<100 мбар). Выход: 351,0 г (100,29% от теор.) бесцветного кристаллического порошка.
Аналогичным образом, как описано в примерах 1b и 1с, может быть получен чистый финеренон (I) из данной тартратной соли.
Пример 3
Производство 3 партий чистого финеренона (I) в технической лаборатории, исходя из 2 кг рацемата (II).
Пример 3а
Получение тартрата (IV)
2,00 кг (5,285 моль) рацемического финеренона (II) помещали в 28,0 л смеси, состоящей из этанола, денатурированного толуолом, и воды = 75:25 (об./об.) при комнатной температуре (приблизительно 23°С). 1,042 кг (2,907 моль) (+)-O,O-дибензоил-D-винной кислоты (III) добавляли через воронку для твердых веществ, а затем промывали 2 л смеси, состоящей из этанола, денатурированного толуолом, и воды = 75:25 (об./об.). Полученную таким образом суспензию нагревали до внутренней температуры 75°С в течение 45 минут, а затем дополнительно перемешивали при этой температуре в течение 3,0 часов. Затем смесь охлаждали с 23°С при линейной скорости охлаждения в течение 5,0 часов и дополнительно перемешивали при данной температуре в течение ночи (приблизительно 16 часов). Суспензию отфильтровывали на пористом стеклянном фильтре и один раз промывали 2 л смеси, состоящей из этанола, денатурированного толуолом, и воды = 75:25 (об./об.) и 10 минут откачивали досуха. Выход влажного продукта: 2,69 кг. Затем влажный продукт сушили до постоянной массы при 50°С в вакууме (<100 мбар) (постоянная масса примерно через 17 часов). Выход: 2,009 кг (~100% от теор.) бесцветного кристаллического порошка.
Пример 3b
Получение неочищенного продукта (I)
2,006 кг полученного в Примере 3а соединения (IV) суспендировали при комнатной температуре (ок. 23°С) в 20,0 л смеси, состоящей из этанола, денатурированного толуолом, и воды = 1:4 (об./об.) (измеренное значение рН составляло рН=4). Затем по каплям добавляли 6,05 кг 9,09%-ного водного раствора фосфата натрия и доводили значение рН до рН=7,2. Смесь дополнительно перемешивали при 23°С в течение примерно 50 минут (рН 7,17). Затем по каплям добавляли 0,65 кг 9,09%-ного водного раствора фосфата натрия и доводили значение рН до рН=7,5. Нагревали в течение часа до внутренней температуры 50°С, и 3,0 часа дополнительно перемешивали при этой температуре. Охлаждали в течение часа до 22°С и дополнительно перемешивали в течение выходных (примерно 64 часа*) при этой температуре. Кристаллизат отфильтровывают на К800 пластине фильтра Зейтца и промывают один раз 1,60 л, и один раз 0,8 л смеси, состоящей из этанола, денатурированного толуолом, и воды = 20:80 (об./об.) и два раза соответственно 1,6 л воды. Выход влажного продукта: 1,82 кг. Затем влажный продукт сушили до постоянной массы при 50°С в вакууме (<100 мбар) (постоянная масса примерно через 17 часов). Выход: 0,978 кг (97,8% от теор.) бесцветного кристаллического порошка.
*) По техническим причинам перемешивание продолжали в течение выходных, обычно достаточно 14 часов дополнительного перемешивания.
Пример 3с
Получение чистого продукта (I)
250 г полученного в примере 3b неочищенного продукта (I) суспендировали в 5000 мл этанола, денатурированного толуолом, и затем нагревали до кипячения с возвратом флегмы. При этом продукт перходил в раствор. Перемешивали в течение часа при этой температуре. Раствор отфильтровывали через нагретый (Т=85°С) фильтр, работающий под давлением, а затем фильтр, работающий под давлением, промывали 200 мл этанола, денатурированного толуолом. Затем отгоняли растворитель (было отогнано примерно 4200 мл) до достижения конечного, примерно 4-кратного объема (относительно используемого вещества: 250 г × 4 ~ 1000 мл). Затем его охлаждали до внутренней температуры 4-5°С (линейное изменение: продолжительность примерно 4 часа). Затем дополнительно перемешивали в течение часа при внутренней температуре 4-5°С. Продукт отфильтровывали и дополнительно один раз промывали 220 мл этанола, денатурированного толуолом. Выход влажного продукта: 251,2 г. Затем влажный продукт сушили в течение выходных (>48 часов) при 50°С под вакуумом (<100 мбар). Выход: 223,0 г (89,2% от теор.) бесцветного кристаллического порошка.
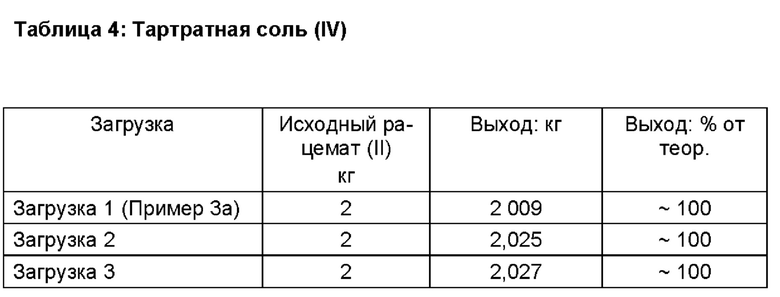
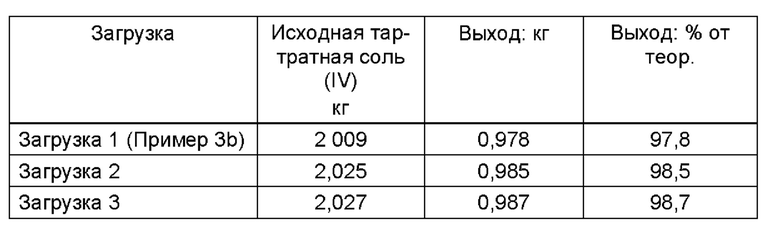
В следующей таблице 4 представлены результаты 3 производственных циклов, исходя из 2 кг рацемата финеренона (II):
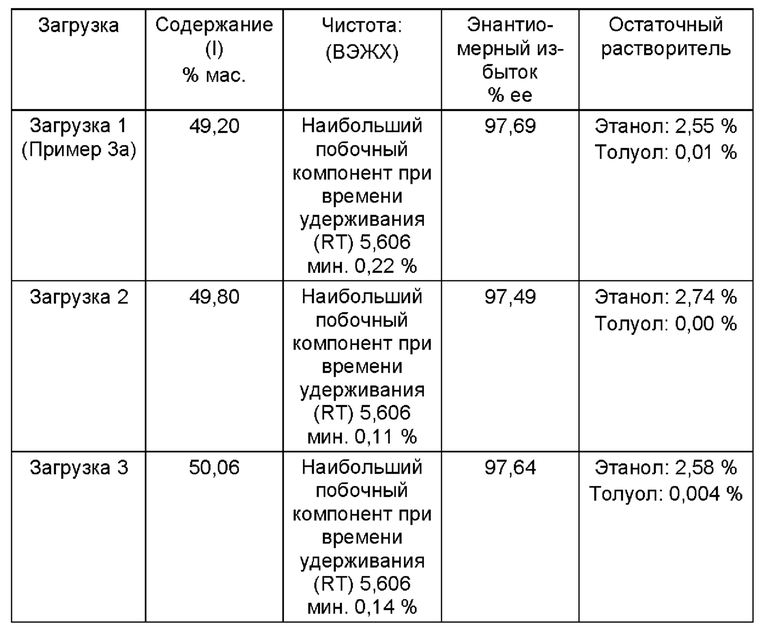
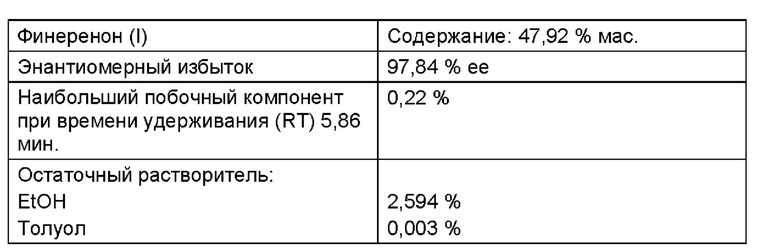
Неочищенный продукт (I)
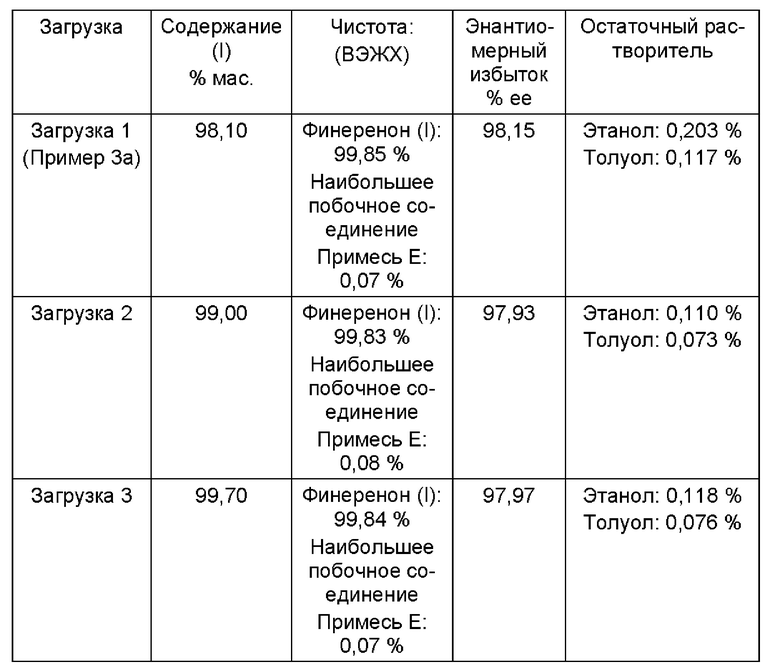
Во всех 3 загрузках содержание (+)-O,O-дибензоил-D-винной кислоты (III) было 0,00%
Чистый продукт (I)
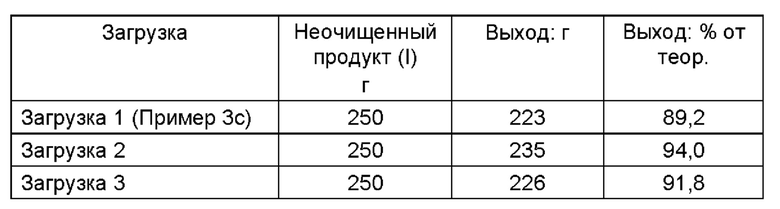
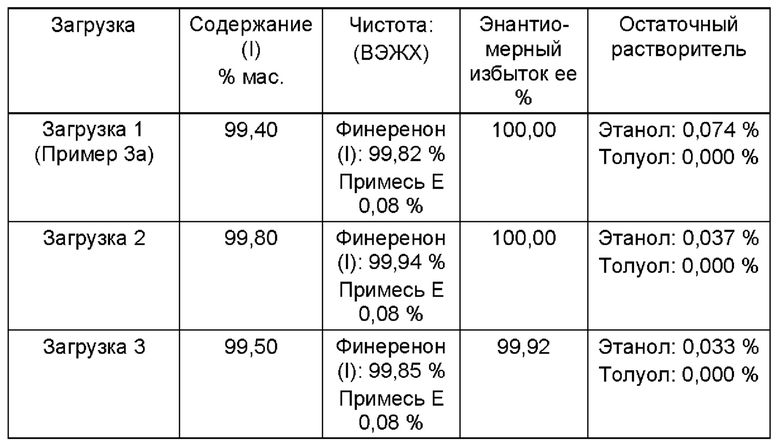
Во всех 3 загрузках содержание (+)-O,O-дибензоил-D-винной кислоты (III) было 0,00%
Пример 4
Промышленное исполнение способа. Следующие ниже примеры описывают реализацию способа в промышленном масштабе. Результаты, такие как выход и аналитические данные, 2х партий финеренона чистого (I), в заключение представлены в таблице:
Пример 4а
Получение тартратной соли (IV)
Размер загрузки соответствовал 80 кг рацемического финеренона (II).
• В перемешивающее устройство объемом 1600 л помещали 711,0 кг винного спирта при внутренней температуре (IT) 20°С, затем туда подавали 300 кг воды, потом 80,0 кг рацемата (II) и, наконец, 41,7 кг (+)-дибензоилвинной кислоты (III).
• Нагревали до IT 75°С и дополнительно перемешивали в течение 3 часов.
• Охлаждали в течение 5 часов (линейное изменение) до IT 23°С и дополнительно перемешивали в течение 16 часов.
• Суспензию переносили для центрифугирования в инвентированную центрифугу, центрифугировали и промывали смесью, состоящей из 10,7 кг винного спирта и 4,5 кг воды, затем центрифугировали досуха и выгружали.
• Сушку проводили в шаровой сушилке емкостью 300 л при температуре рубашки 50°С и 30 мбар. Процесс сушки завершали при внутренней температуре 45°С (продолжительность примерно 6 часов).
• Смесь охлаждали до 15°С, а затем выгружали продукт.
Пример 4b
Неочищенный продукт (I)
Размер загрузки соответствовал 89,3 кг тартратной соли (IV) для использования.
• Приготовление раствора фосфата натрия
• В перемешивающее устройство объемом 630 л помещали 360,0 кг деионизированной воды при 20°С.
• Добавляли и растворяли 36,0 кг фосфата натрия.
• В перемешивающее устройство объемом 1600 л помещали 141,1 кг винного спирта при 20°С.
• Потом добавляли 714,2 кг воды и 89,3 кг тартратной соли (IV).
• Нагревали до внутренней температуры 50°С.
• Затем дозировали 295,1 кг раствора фосфата натрия
• Через 1,5 часа посредством добавления 35,4 кг раствора фосфата натрия значение рН доводили до рН=7,5 (+/-0,1).
• Смесь оставляли перемешиваться и охлаждали посредством линейного изменения температуры (3 ч) до IT=22°С.
• Затем смесь дополнительно перемешивали при IT=22°С (18 ч).
• В отдельной емкости готовили смесь 129,6 кг воды и 25,6 кг винного спирта (для дополнительной промывки).
• Суспензию дозировали на инвентированную центрифугу (примерно 4 партии+оставшаяся партия) и промывали порциями один раз смесью 129,6 кг воды и 25,6 кг винного спирта и один раз 288,0 кг деионизированной воды.
• Продукт выгружали и сушили в шаровой сушилке (ТМ=50°С, 30 мбар, окончание при IT=45°С, примерно 6 часов).
• Охлаждали, а затем выгружали продукт при <30°С.
Пример 4с
Дополнительная обработка неочищенного продукта (I)
В некоторых случаях, когда предельное значение по спецификации для (+) - дибензоилвинной кислоты (III) в неочищенном продукте составляет >0,1%, оказалось целесообразным провести дополнительную обработку (см. Пример 6), которая гарантирует воспроизводимое достижение предельного значения <0,1%. Способ дополнительной обработки к тому же проходит почти без потерь и как правило дает неочищенный продукт с выходом >95%.
Дополнительно обработанный неочищенный продукт (I)
Размер загрузки соответствовал 80 кг неочищенного продукта (I).
• Приготовление раствора фосфата натрия: 2,3 кг фосфата натрия при комнатной температуре растворяли в 147,8 кг воды в 250-литровой отдельном емкости с мешалкой.
• 198,8 кг винного спирта помещали в перемешивающее устройство объемом 630 л при 20°С и добавляли 120 кг воды, затем вводили 30 кг неочищенного продукта (содержащего (+)-дибензоилвинную кислоту (III), как указано в таблице 5).
• Нагревали до внутренней температуры 75°С и дополнительно перемешивали в течение 30 минут.
• При 70°С добавляли 14,1 кг полученного ранее раствора фосфата натрия, устанавливали значение рН на рН=8,9 (+/-0,1) и затем перемешивали при 70°С в течение 20 часов.
• Смесь охлаждали до 44°С в течение 60 минут посредством линейного изменения температуры и добавляли 300 г затравочных кристаллов финеренона чистого (I).
• Затем при 200 мбар и максимальной внутренней температуре 60°С отгоняли растворитель и параллельно в перемешивающее устройство добавляли 369 кг воды.
• Наконец, в течение 1,5 часов охлаждали до 22°С, и продукт выделяли на нутч-фильтре, работающем под давлением, и в 3 порции промывали суммарно 180 л воды.
• Затем сушили при температуре рубашки 50°С под вакуумом (30 мбар) до постоянной массы.
• Позволяли охладиться до 15°С, а затем выгружали продукт.
Пример 4d
Чистый продукт (I)
Размер загрузки соответствовал 26 кг (2 раза по 13 кг) неочищенного продукта (I).
• В перемешивающем устройстве объемом 250 л помещали 13 кг дополнительно обработанного неочищенного продукта (I) в 184,9 кг винного спирта при 20°С, и перемешивающее устройство предварительно инертизировали.
• Раствор нагревали до температуры рубашки 80°С и перемешивали в течение 30 минут при 78°С (при этом продукт переходил в раствор).
• Раствор фильтровали через фильтровальную свечу 0,65 мкм (РР-свеча Sartorius) (GMP-фильтрация) и фильтрат переносили в перемешивающее устройство объемом 630 л.
• Дополнительно промыли 18,5 кг винного спирта.
• Повторяли для 2 порций (2 раза по 13 кг) и растворы объединяли.
• Растворитель отгоняли до конечного состояния примерно 130 л при нормальном давлении (двумя порциями, температура рубашки: 104°С, продолжительность примерно 6 часов).
• Раствор охлаждали до 0°С с линейным изменением температуры в течение 4 часов, а затем дополнительно перемешивали в течение одного часа.
• Суспензию продукта выделяли на нутч-фильтре, работающим под давлением, и промывали 45,9 кг винного спирта.
• Затем влажный продукт сушили в вакууме (30 мбар) при 50°С до постоянной массы (примерно 12 часов). Охлаждали до 15°С и выделяли продукт.
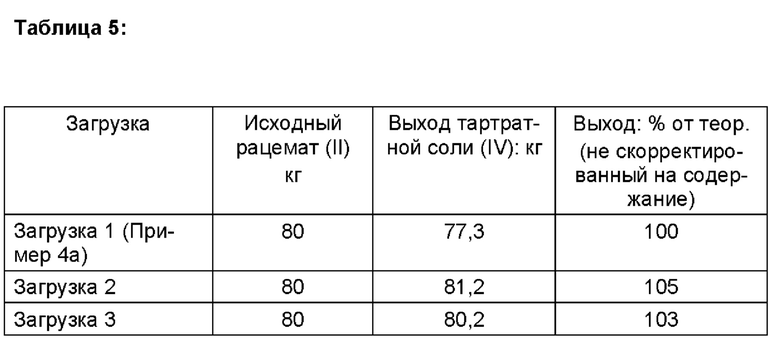
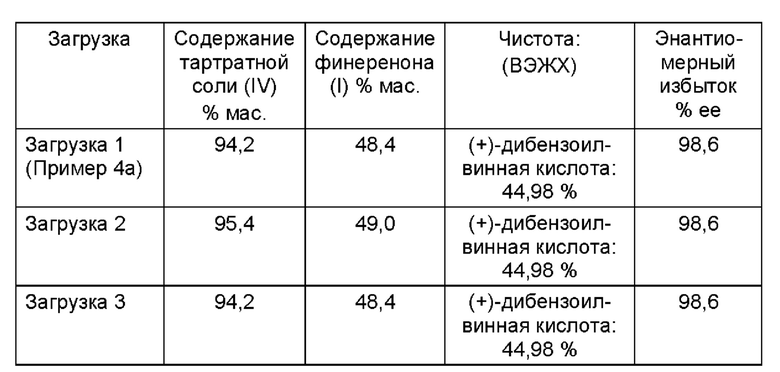
В таблице 5 представлен обзор выхода и аналитических результатов 3 промышленных загрузок для получения чистого финеренона (I).
Тартратная соль (IV)
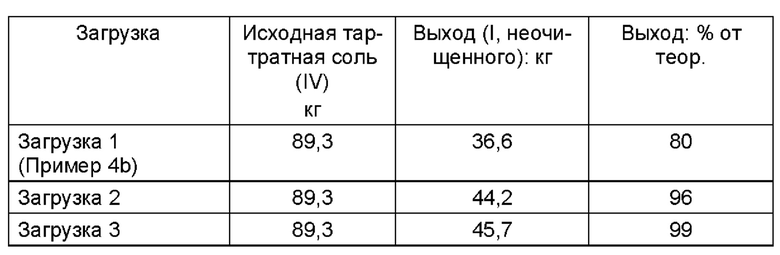
Неочищенный продукт (I)
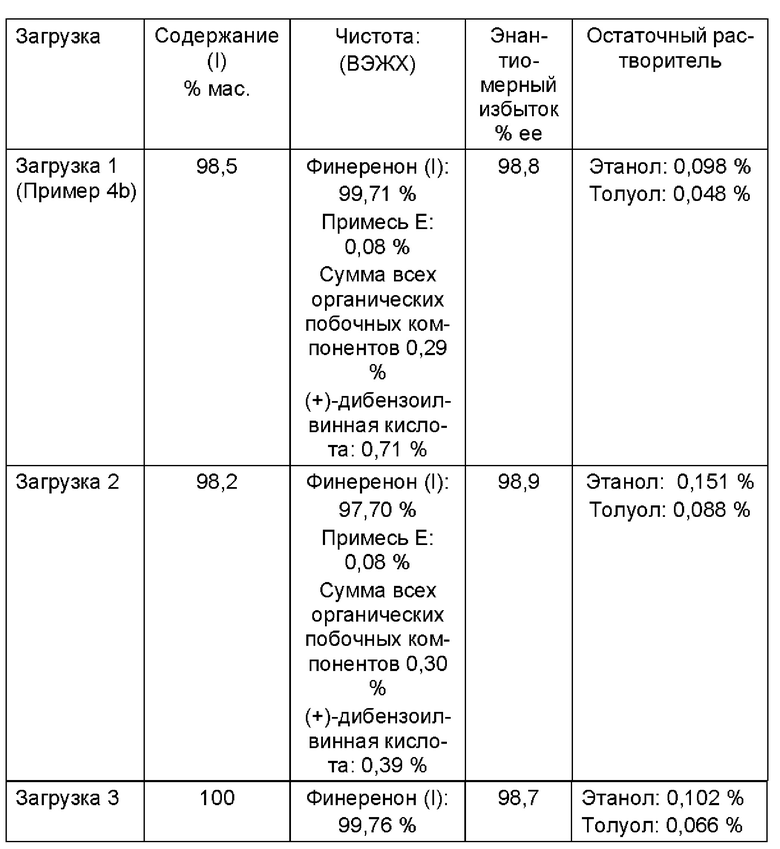

Неочищенный продукт (I) после дополнительной обработки
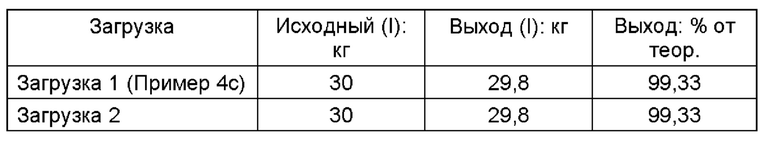
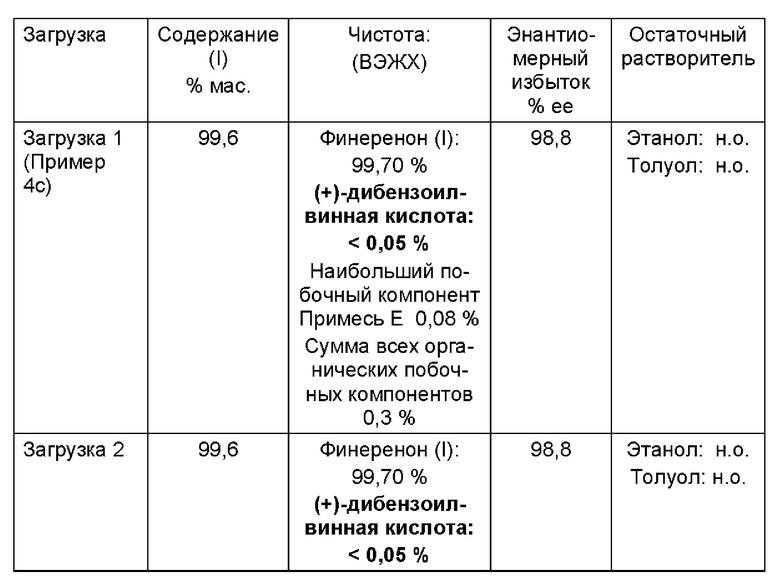

Чистый продукт, финеренон, чистый (I)
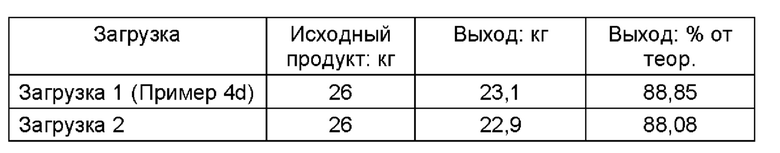
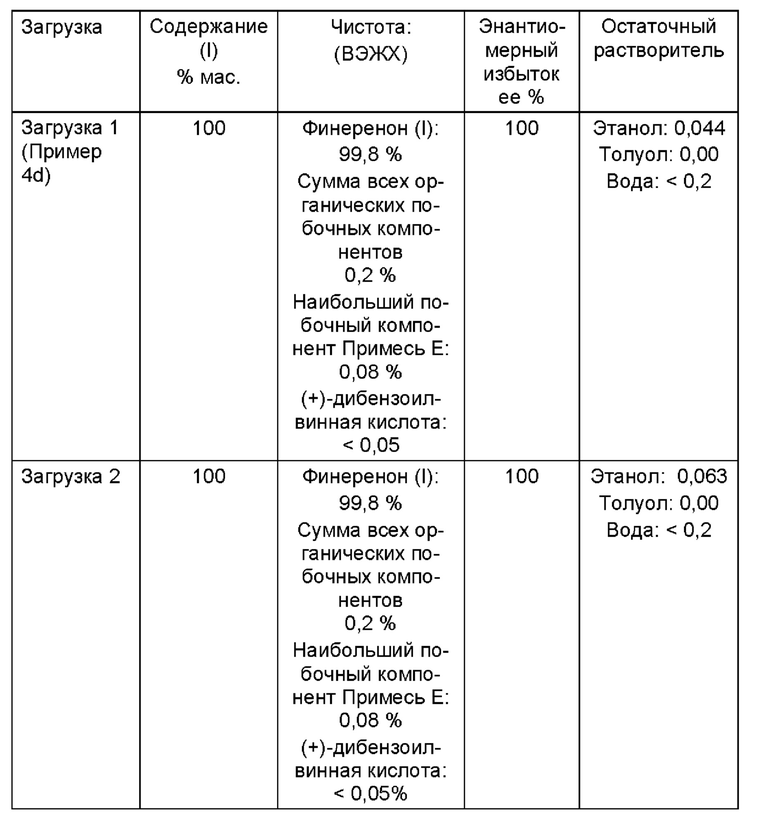
Пример 5
Для высвобождения неочищенного финеренона (I) подходят несколько вариантов, аналогичных процедуре из Примера 1b. Целью при этом было удержание как можно меньшей доли (+)-О,О-дибензоил-D-винной кислоты (III) в неочищенном продукте (0,15% и менее). При этом в частности изменяли конечное значение рН после дозирования раствора фосфата натрия, температуру, время дозирования раствора фосфата натрия и время последующего перемешивания и исследовали влияние на содержание (+)-O,O-дибензоил-D-винной кислоты в неочищенном продукте.
Размер загрузки: 100 г винной кислоты (IV) в 158,0 г винного спирта и 799,8 г воды
Аналитические данные исходной тартратной соли (IV):
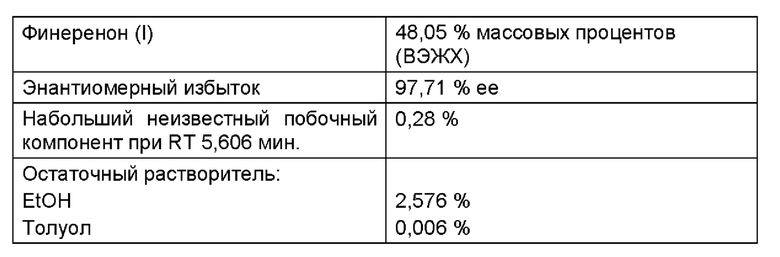
В далее следующей таблице 6 обобщены результаты:
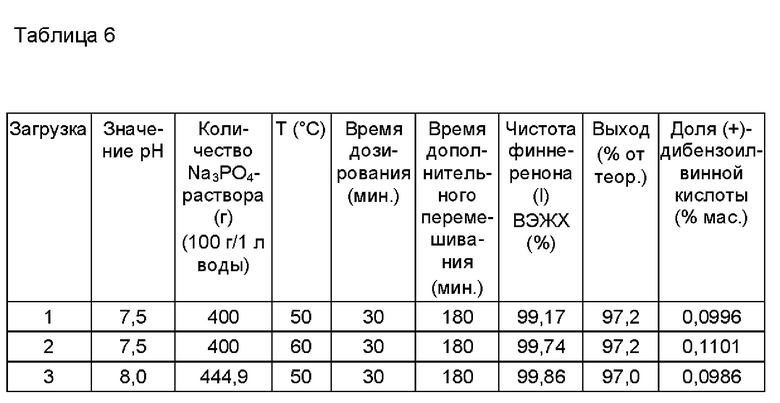
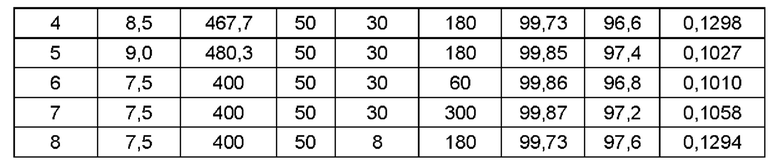
Пример 6
Способ дополнительной обработки
Пример 6а
Дополнительно обработанный неочищенный продукт: Снижение доли (+)-О.О-дибензоил-D-винной кислоты до <0.1%.
Данный эксперимент служил для демонстрации надежности процесса дополнительной обработки, в котором намеренно использовали партию с повышенным содержанием (+)-0,0-дибензоил-Р-винной кислоты (III).
Для дополнительной обработки использовали неочищенный продукт, который, согласно анализу, все еще содержал 0,77% (+) - О,О-дибензоил-D-винной кислоты (III).
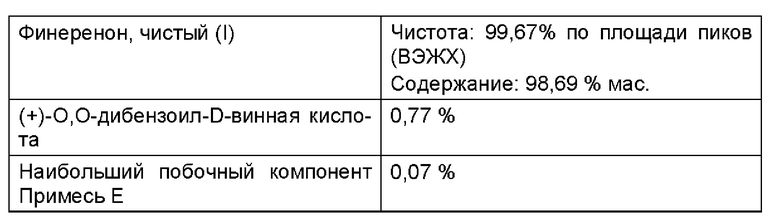
Дополнительная обработка неочищенного продукта: Снижение доли (+)-О.О-дибензоил-D-винной кислоты (III) до <0.1%.
Для дополнительной обработки использовали неочищенный продукт, который, согласно анализу, все еще содержал 0,77% (+) - О, О-дибензоил-D-винной кислоты (III). Процедура для этого была следующей:
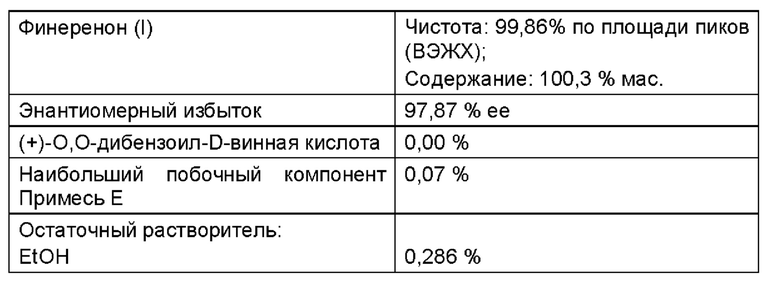
150 г неочищенного продукта (I) (содержащего 0,77% (+)-O,O-дибензоил-D-винной кислоты (III)) добавляли к 600 г воды и 953,9 г винного спирта и нагревали до 70°С. В течение одного часа все переходило в раствор. Затем дополнительно перемешивали при 70°С в течение 30 минут. Значение рН составляло рН=5,0. Значение рН посредством добавления 44,6 г водного раствора фосфата натрия (15 г фосфата натрия на 985 г воды) доводили до рН=8,5, и затем перемешивали при 70°С в течение 2 часов. В течение 30 минут охлаждали до 53°С и затравляли чистым материалом (затравочными кристаллами). Затем в течение 30 минут дополнительно перемешивали при 50-52°С. При давлении от 200 до 155 мбар и внутренней температуре почти полностью отгоняли этанол, и одновременно дозировали 1845 мл воды (уровень поддерживали постоянным: отогнанное количество = добавленное количество воды). Выключали нагревание и суспензию дополнительно перемешивали при 23°С в течение ночи. Кристаллизат отфильтровывали на пористом стеклянном фильтре и трижды промывали соответственно по 300 мл воды. Выход влажного продукта: 218,9 г. Затем влажный продукт сушили в течение выходных (>48 часов) при 50°С под вакуумом (<100 мбар). Выход: 146,7 г (97,8% от теор.)
Пример 6b
Демонстрация стабильности, перемешивание 20 ч при 70°С
Дополнительная обработка неочищенного продукта: Снижение доли (+)-О,О-дибензоил-D-винной кислоты (III) до <0,1%.
Для дополнительной обработки использовали неочищенный продукт, который, согласно анализу, все еще содержал 0,77% (+) - О,О-дибензоил-D-винной кислоты (III). Процедура для этого была следующей:
150 г неочищенного продукта (I) (содержащего 0,77% (+)-O,O-дибензоил-D-винной кислоты (III)) добавляли к 600 г воды и 953,9 г винного спирта и нагревали до 70°С. В течение одного часа все переходило в раствор. Затем дополнительно перемешивали при 70°С в течение 30 минут. Значение рН составляло рН=5,0. Значение рН посредством добавления 45,2 г водного раствора фосфата натрия (15 г фосфата натрия на 985 г воды) доводили до рН=8,5, и затем перемешивали при 70°С в течение 20 часов. В течение 30 минут охлаждали до 53°С и затравляли чистым материалом (затравочными кристаллами). Затем в течение 30 минут дополнительно перемешивали при 50-52°С. При давлении от 200 до 155 мбар и внутренней температуре почти полностью отгоняли этанол, и одновременно дозировали 1845 мл воды (уровень поддерживали постоянным: отогнанное количество = добавленное количество воды). Выключали нагревание и суспензию дополнительно перемешивали при 23°С в течение ночи. Кристаллизат отфильтровывали на пористом стеклянном фильтре и трижды промывали соответственно по 300 мл воды. Выход влажного продукта: 194,3 г. Затем влажный продукт сушили в течение ночи (ок. 16 часов) при 50°С под вакуумом (<100 мбар). Выход: 143,1 г (95,4% от теор.)
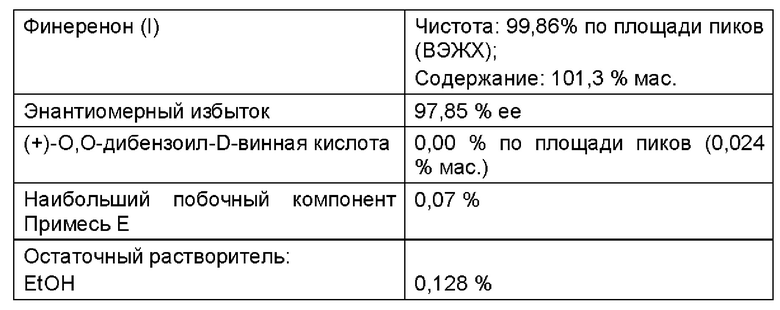
Пример 6с
Дополнительная обработка неочищенного продукта: Снижение доли (+)-О,О-дибензоил-D-винной кислоты (III) до <0.1%. Обратное титрование значения рН дигидрофосфатом натрия до рН 7,0.
Для дополнительной обработки использовали неочищенный продукт, который, согласно анализу, все еще содержал 0,77% (+) - О,О-дибензоил-D-винной кислоты (III). Процедура для этого была следующей:
375,0 г неочищенного продукта (I) (содержащего 0,77% (+)-O,O-дибензоил-D-винной кислоты (III)) добавляли к 1500 г воды и 2384,8 г винного спирта и нагревали до 70°С. В течение одного часа все переходило в раствор. Затем дополнительно перемешивали при 70°С в течение 30 минут. Значение рН составляло рН=4,9. Значение рН посредством добавления 101,4 г водного раствора фосфата натрия (15 г фосфата натрия на 985 г воды) доводили до рН=8,5, и затем перемешивали при 70°С в течение 20 часов. Затем значение рН с помощью раствора дигидрофосфата натрия (50 г дигидрофосфата натрия в 200 мл воды) доводили до рН=7. В течение 30 минут охлаждали до 53°С и затравляли чистым материалом (затравочными кристаллами). Затем в течение 30 минут дополнительно перемешивали при 50-52°С. При давлении от 200 до 155 мбар и внутренней температуре почти полностью отгоняли этанол, и одновременно дозировали 4610 мл воды (уровень поддерживали постоянным: отогнанное количество = добавленное количество воды). После этого в течение 90 минут охлаждали до 23°С. Затем в течение выходных (примерно 70 ч) дополнительно перемешивали при 22°С. Кристаллизат отфильтровывали на пористом стеклянном фильтре и трижды промывали соответственно по 750 мл воды. Выход влажного продукта: 402,3 г. Затем влажный продукт сушили в течение ночи (ок. 16 часов) при 50°С под вакуумом (<100 мбар). Выход: 336,6 г (89,76% от теор.)
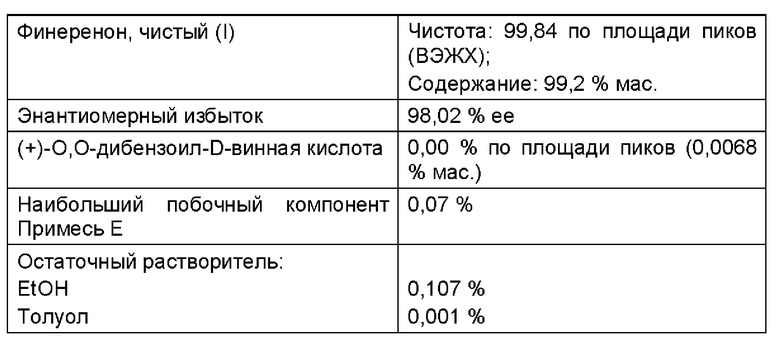
Пример 6d
Окончательная кристаллизация порций из Примера 6с
80,0 г полученного в примере 6 с неочищенного продукта (I) суспендировали в 1600 мл этанола, денатурированного толуолом, и затем нагревали до кипячения с возвратом флегмы. При этом продукт переходил в раствор. Перемешивали в течение часа при этой температуре. Раствор отфильтровывали через нагретый (Т=75°С) фильтр, работающий под давлением, а затем промывали 100 мл этанола, денатурированного толуолом. Затем отгоняли под вакуумом растворитель (температура рубашки 45-47°С) до достижения конечного, примерно 5-кратного объема (относительно используемого вещества: 80 г × 5 ~ 400 мл). Потом охлаждали в течение часа до внутренней температуры 2°С и перемешивали в течение примерно часа при этой температуре. Продукт отфильтровывали и дополнительно один раз промывали 80 мл этанола, денатурированного толуолом. Выход влажного продукта: 83,3 г. Затем влажный продукт сушили в течение ночи (ок. 16 часов) при 50°С под вакуумом (<100 мбар). Выход: 71,4 г (89,3% от теор.) бесцветного кристаллического порошка, мелкие игольчатые кристаллы.
Аналитические результаты:
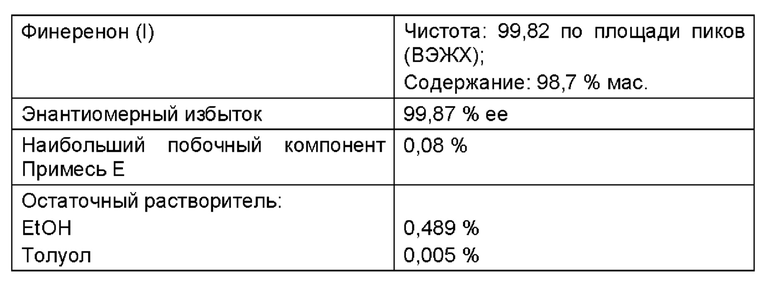
Пример 7
Выделение энантиомера финеренона (Ia)
Пример 7а
Получение соли (-)-О,О-дибензоил-L-винной кислоты
Сначала при комнатной температуре вводили 187,2 г рацемата (II), а затем добавляли 1662,5 г винного спирта. Затем добавляли 485,5 г воды. 97,5 г % (-)-O,O-дибензоил-L-винной кислоты (IIIa) добавляли через воронку для твердых веществ, а затем промывали 156,0 г воды. Суспензию в течение часа нагревали до внутренней температуры 75°С, а затем перемешивали в течение 3 часов при 75°С в течение 3 часов. Затем смесь охлаждали до 23°С при линейной скорости охлаждения в течение 5,0 часов и дополнительно перемешивали при данной температуре в течение ночи (приблизительно 18 часов). Суспензию отфильтровывали на пористом стеклянном фильтре и два раза промывали смесью из 178 г винного спирта и 75 г воды. Выход влажного продукта: 255,4 г. Затем влажный продукт сушили в течение ночи (примерно 16 часов) при 50°С под вакуумом (<100 мбар). Выход: 188,6 г (100,73% от теор.) бесцветного кристаллического порошка. Маточный раствор и промывочный раствор объединяли (приблизительно 3200 мл желтоватого раствора, рН=4,6), и из него выделяли финеренон, неочищенный (I) (Пример 7b).
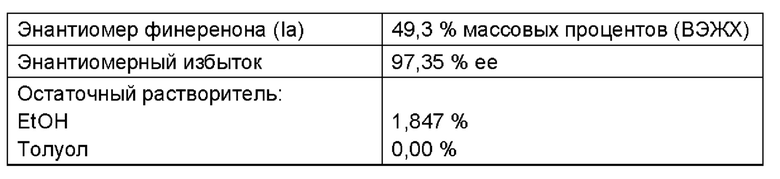
Аналогичным образом, как описано в примерах 1b и 1с, из данной тартратной соли может быть получен энантиомер финеренона (Ia).
Пример 7b
Выделение финеренона, неочищенного (I) из маточного раствора
Объединенные маточный раствор и промывочный раствор из Примера 7а (приблизительно 3200 мл желтоватого раствора, рН=4,6) при комнатной температуре посредством добавления 43,3 г водного раствора фосфата натрия (100 г растворенных в 1 л воды) доводили до рН=7,6. Затем при пониженном давлении (от 85 до 65 мбар, внутренняя температура от 38 до 20°С) в значительной степени отгоняли винный спирт и уменьшали до конечного объема примерно 0,8 л. Охлаждали до комнатной температуры и перемешивали выпадающую суспензию в течение 2 часов при 22°С. Суспензию отфильтровывали и 2 раза промывали соответственно по 150 мл воды. Выход влажного продукта: 159,1 г. Затем влажный продукт сушили в течение ночи (ок. 16 часов) при 50°С под вакуумом (<100 мбар). Выход: 86,3 г (92,2% от теор. в пересчете на использованный в примере 7а рацемат (II)).
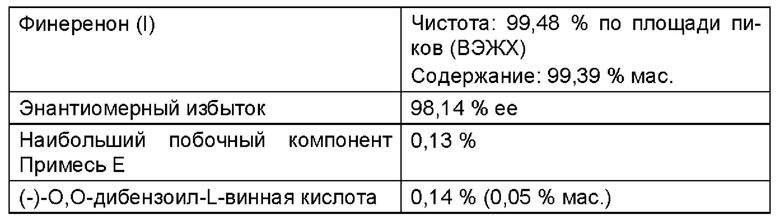
Аналогичным образом, как описано в примере 1с, из данного неочищенного продукта может быть получен финеренон (I) в чистой форме.
Пример 8
Пример 8а
Выделение нежелательного энантиомера (1а) из маточного раствора
Объединенные маточный раствор и промывочный раствор из Примера 1а (приблизительно 3750 мл желтоватого раствора, рН=4,5) при комнатной температуре посредством добавления 101,1 г водного раствора фосфата натрия (100 г растворенных в 1 л воды) доводили до рН=7,5. Затем при пониженном давлении (от 85 до 65 мбар, внутренняя температура от 38 до 20°С) в значительной степени отгоняли винный спирт и уменьшали до конечного объема примерно 0,85 л. Охлаждали до комнатной температуры и выпадающую суспензию перемешивали в течение выходных (>48 часов), а затем в течение дополнительных 2 часов перемешивали при 22°С. Суспензию отфильтровывали и 2 раза промывали соответственно по 200 мл воды. Выход влажного продукта: 139,1 г. Затем влажный продукт сушили в течение ночи (ок. 16 часов) при 50°С под вакуумом (<100 мбар). Выход: 103,1 г (82,48% от теор. в пересчете на использованный в примере 1а рацемат (II)).
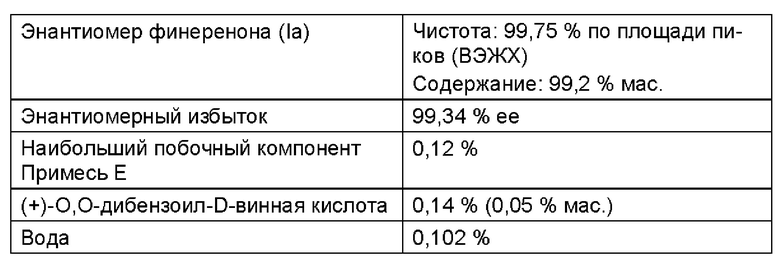
Пример 8b
Промышленная загрузка для выделения энантиомера финеренона (Ia)
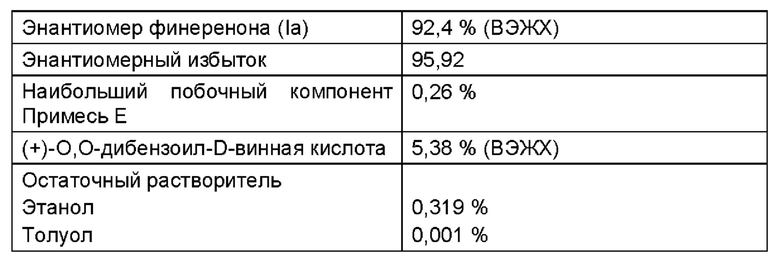
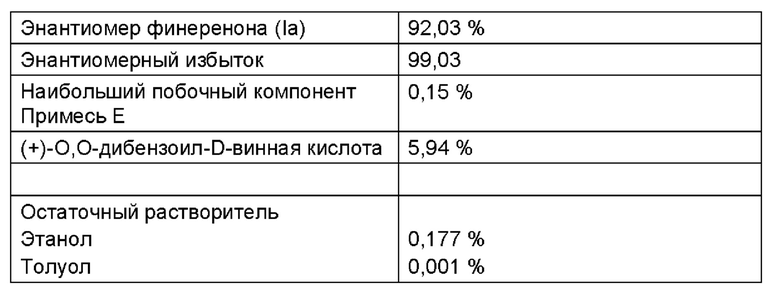
Объединенный маточный раствор и промывочный раствор из Примера 4 (получение тартрата в промышленных масштабах) обрабатывали следующим образом: Количество примерно 1165 кг раствора
• Приготовление раствора фосфата натрия: 7,3 кг фосфата натрия растворяли в 73,1 кг воды при 20°С.
• В перемешивающее устройство объемом 1600 л помещали при 20°С 1165 кг маточного раствора (включая промывочный раствор), и с помощью 28 кг приготовленного ранее раствора фосфата натрия, рН доводили до 7,5 (+/-0,1).
• Дополнительно перемешивали в течение 0,25 ч, и затем отгоняли при пониженном давлении (65 мбар, IT примерно 22°С) винный спирт до уровня примерно 310 л.
• Затем дополнительно перемешивали при 20°С в течение 2 часов и суспензию выделяли с помощью инвертированной центрифуги, причем в каждом случае промывали 20 кг воды.
• Влажный продукт сушили в шаровой сушилке при 50°С под вакуумом (30 мбар; примерно 6 ч).
• Охлаждали до 15°С и выделяли продукт.
Аналитические данные использованного маточного раствора (включая промывочный раствор) для загрузки 1
Аналитические данные использованного маточного раствора (включая промывочный раствор) для загрузки 2
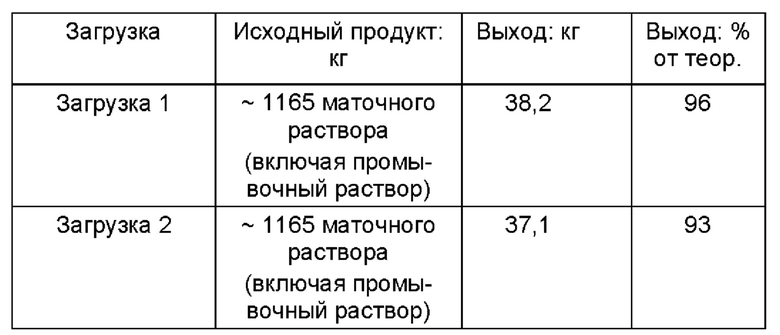
Аналитические данные загрузки 1
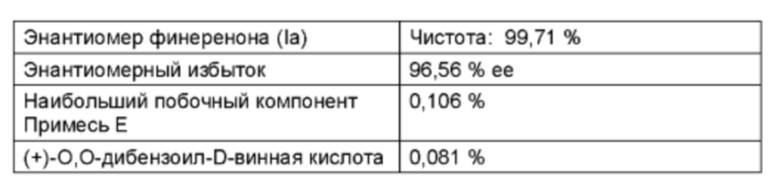
Аналитические данные загрузки 2
Пример 9
Примеры других производных винной кислоты и других растворителей
Пример 9а
(+)-O,O-ди-п-анизоил-D-винная кислота
100 мг рацемата растворяли с 111 мг (+)-O,O-ди-п-анизоил-D-винной кислоты в 5 мл дихлорметана и оставляли в покое. Через некоторое время выпадала диастереомерная соль. Ее отфильтровывали и измеряли энантиомерный избыток. Измерение показало энантиомерный избыток 68% ее с преобладанием Ia).
Пример 9b
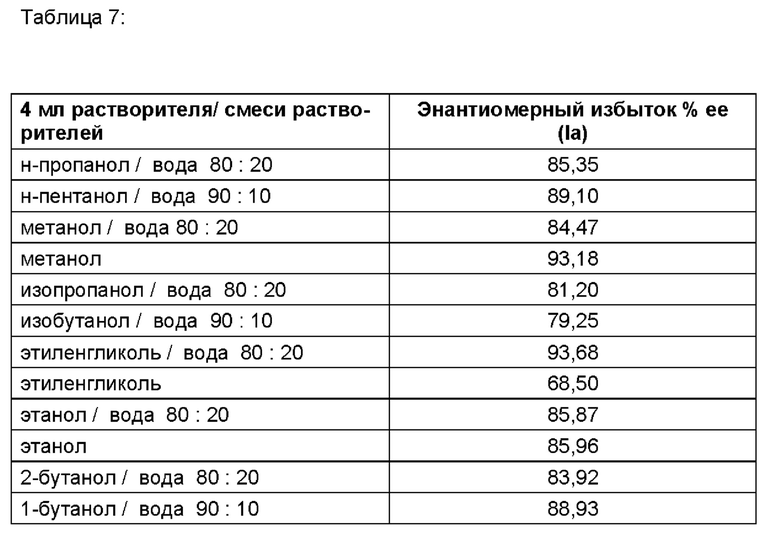
(-)-О,О-ди-п-толуил-L-винная кислота
100 мг рацемата растворяли с 0,55 экв. (-)-O,O-ди-п-толуил-L-винной кислоты в 4 мл растворителя и оставляли в покое. Через некоторое время выпадала диастереомерная соль. Ее отфильтровывали и измеряли энантиомерный избыток. Измерение показало энантиомерный избыток у % ее с преобладанием (Ia) В далее следующей таблице 7 обобщены результаты
Пример 9с
(+)-O,O'-дибензоил-D-винная кислота
100 мг рацемата растворяли с 0,55 экв. (+)-O,O-дибензоил-D-винной кислоты в 4 мл растворителя и оставляли в покое. Через некоторое время выпадала диастереомерная соль. Ее отфильтровывали и измеряли энантиомерный избыток. Измерение показало энантиомерный избыток у % ее с преобладанием (I). В далее следующей таблице 8 обобщены результаты
Описание чертежа
Фиг. 1: Порошковая рентгеновская дифракция (XRPD) соединения (VI), полученного в виде бесцветного кристаллического порошка согласно Примеру 1а.
Изобретение относится к способу разделения рацемата (II) на (Ia) и/или (I) при помощи хиральных замещенных сложных эфиров винной кислоты общих формул (IIIa) или (IIIb), где Ar означает
 где * обозначает место присоединения, при температуре в диапазоне от 60 до 80°С, с образованием диастереомерных солей (IVa), (IVb), (IVc) и/или (IVd), которое осуществляют в смесях растворителей, состоящих из воды и с водой смешивающихся органических растворителей. Технический результат – разработан новый способ разделения рацемата (II), который может найти свое применение для получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I) с высоким выходом и чистотой, обладающего действием нестероидного антагониста минералокортикоидного рецептора и может находить применение в медицине в качестве средства для профилактики и/или лечения сердечно-сосудистых и почечных заболеваний. 3 н. и 13 з.п. ф-лы, 1 ил., 8 табл., 9 пр.
где * обозначает место присоединения, при температуре в диапазоне от 60 до 80°С, с образованием диастереомерных солей (IVa), (IVb), (IVc) и/или (IVd), которое осуществляют в смесях растворителей, состоящих из воды и с водой смешивающихся органических растворителей. Технический результат – разработан новый способ разделения рацемата (II), который может найти свое применение для получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I) с высоким выходом и чистотой, обладающего действием нестероидного антагониста минералокортикоидного рецептора и может находить применение в медицине в качестве средства для профилактики и/или лечения сердечно-сосудистых и почечных заболеваний. 3 н. и 13 з.п. ф-лы, 1 ил., 8 табл., 9 пр.
1. Разделение рацемата (II)
на (Ia) и/или (I)
при помощи хиральных замещенных сложных эфиров винной кислоты общих формул (IIIa) или (IIIb)
где Ar означает
где * обозначает место присоединения,
отличающееся тем, что разделение рацемата осуществляют при температуре в диапазоне от 60 до 80°С,
образование диастереомерной соли осуществляют в смесях растворителей, состоящих из воды и с водой смешивающихся органических растворителей, и
с водой смешивающиеся органические растворители выбирают из этанола, изопропанола, 1,2-этандиола, метоксиэтанола, метанола, ацетона, винного спирта и их смесей.
2. Способ по п. 1, отличающийся тем, что разделение рацемата (II)
на (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид формулы (I)
осуществляют
при помощи хирального замещенного сложного эфира винной кислоты формулы (IIIa)
3. Способ по п. 1, отличающийся тем, что разделение рацемата осуществляют в смеси этанола и воды.
4. Способ по п. 1, отличающийся тем, что Ar означает фенил.
5. Способ по п. 1, отличающийся тем, что дибензоил-винную кислоту (III)
используют для разделения рацемата.
6. Способ по п. 1, отличающийся тем, что рацемат
взаимодействует с одним из хиральных замещенных сложных эфиров винной кислоты общих формул (IIIa) или (IIIb)
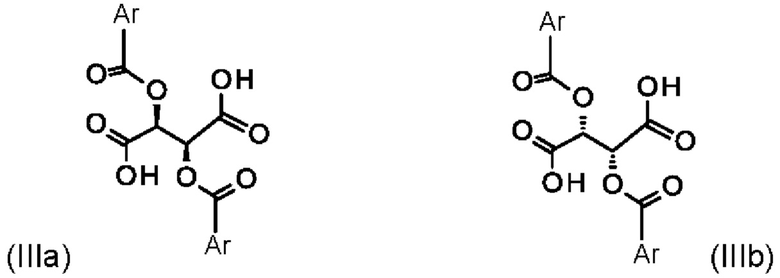
таким образом, что по меньшей мере получают одну из диастереомерных солей (IVa), (IVb), (IVc) и/или (IVd)
причем Ar означает
где * обозначает место присоединения.
7. Способ по п. 1, отличающийся тем, что по меньшей мере одну из диастереомерных солей (IVa), (IVb), (IVc) и/или (IVd)
осаждают, причем Ar означает
где * обозначает место присоединения.
8. Способ по п. 6, отличающийся тем, что осаждают диастереомерную соль (IVa), (IVb), (IVc) и/или (IVd)
и выделяют осажденную диастереомерную соль (IVa), (IVb), (IVc) и/или (IVd), причем Ar означает
где * обозначает место присоединения.
9. Способ по п. 8, отличающийся тем, что диастереомерную соль обрабатывают основанием и удаляют растворитель.
10. Способ по п. 9, отличающийся тем, что в качестве основания используют гидроксид калия, фосфат калия или фосфат натрия.
11. Способ по одному из пп. 1-10, причем рацемат
взаимодействует с дибензоил-винной кислотой формулы (III)
в смеси винного спирта и воды с получением диастереомерной соли (VI)
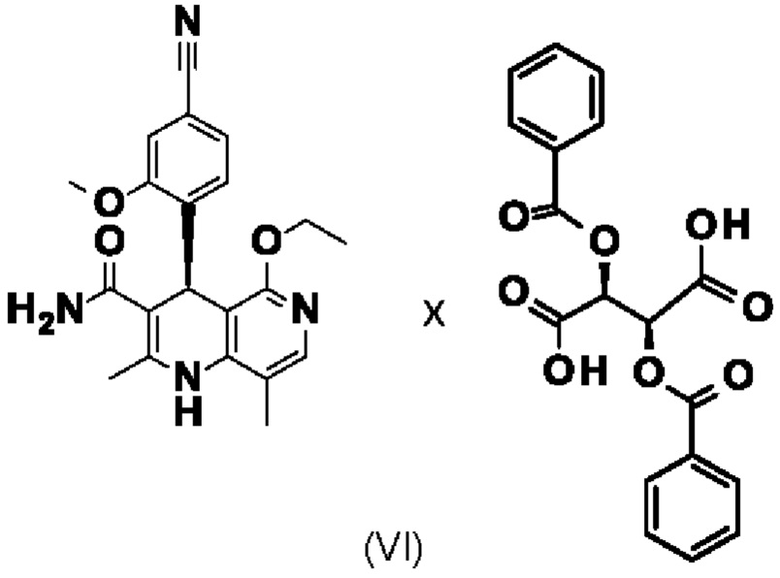
и затем финеренон (I)
высвобождают с использованием фосфата натрия также в смеси винного спирта с водой.
12. Применение разделения рацемата по одному из пп. 2-11 в способе получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I)
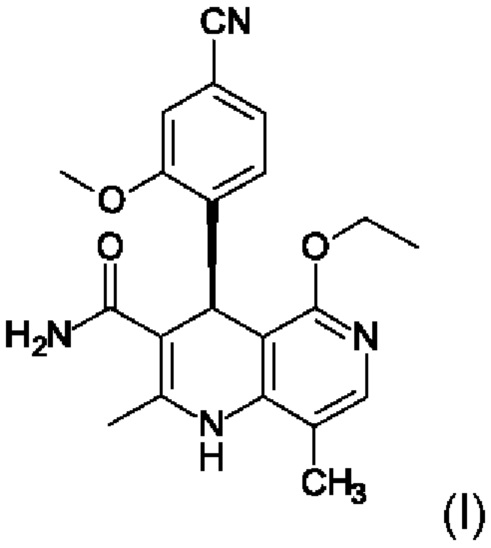
посредством разделения рацемата (II)
при помощи хирального замещенного сложного эфира винной кислоты формулы (IIIa)
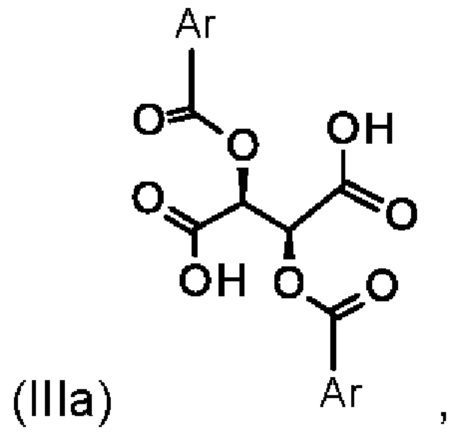
причем Ar означает
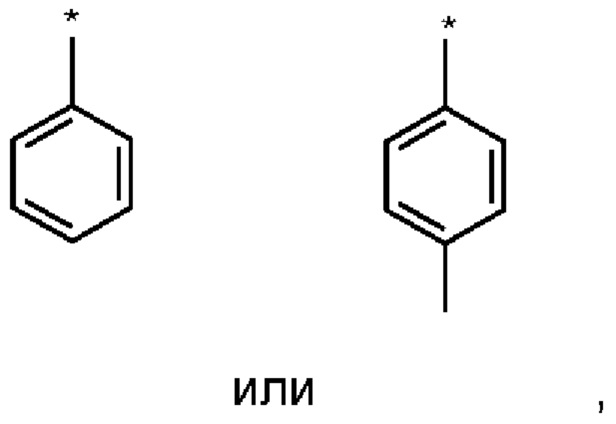
где * обозначает место присоединения.
13. Применение по п. 12, где финеренон (I) с содержанием дибензоил-винной кислоты ≤0,15% получают таким образом, что рацемат (II)
взаимодействует с дибензоилвинной кислотой формулы (III)
в смеси винного спирта и воды с получением диастереомерной соли (VI)
и затем финеренон (I)
высвобождается с использованием фосфата натрия также в смеси винного спирта и воды, а затем снова взаимодействует с фосфатом натрия в смеси винного спирта и воды, после чего кристаллизуется чистый финеренон с содержанием дибензоилвинной кислоты ≤0,15%.
14. Применение по п. 13, где финеренон (I) получают с содержанием дибензоилвинной кислоты ≤0,1%.
15. Диастереомерная соль согласно формулы
где Ar означает
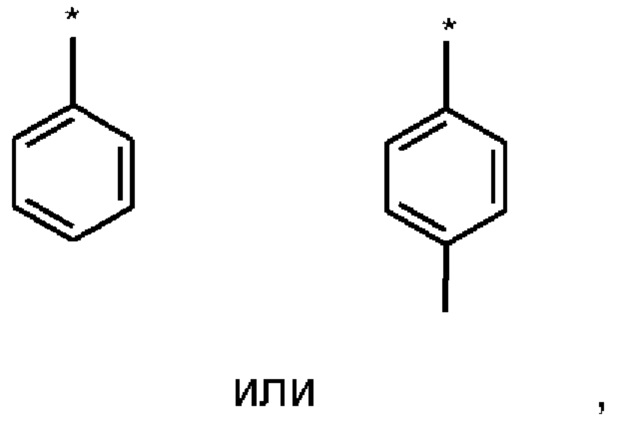
где * обозначает место присоединения.
16. Диастереомерная соль по п. 15, отличающаяся тем, что Ar означает фенил.
| WO 2017032673 A1, 02.03.2017 | |||
| WO 2017032673 A1, 02.03.2017 | |||
| LUDWIK SYNORADZKI et al., Organic Preparations and Procedures International: The Journal for Organic Synthesis, vol.40, no | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Деревянное стыковое устройство | 1920 |
|
SU163A1 |
| WO 2017032678 A1, 02.03.2017 | |||
| WO 2016016287 A1, 04.02.2016 | |||
| ЗАМЕЩЕННЫЕ 4-АРИЛ-1,4-ДИГИДРО-1,6-НАФТИРИДИНАМИДЫ И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2470932C9 |

