Изобретение относится к химической технологии, в частности к технологии получения ароматических нитросоединений.
Ароматические нитросоединения используются как взрывчатые вещества в качестве промежуточных продуктов в производстве фармацевтических препаратов, красителей, полиуретанов и других полимеров. Потребность в нитросоединениях измеряется миллионами тонн и постоянно растет. Кроме того, в последнее десятилетие резко возрос объем производства пенополиуретанов (производство в мире превышает 5 млн т в год), основным сырьем для которых являются нитробензол и нитротолуол.
Несмотря на стопятидесятилетнюю историю, химизм и технологическое оформление производства нитросоединений не подверглись значительным изменениям. Нитрование ароматических соединений проводится, как и прежде, смесью азотной и серной кислот. Основной недостаток этого способа - использование концентрированной серной кислоты. Ее присутствие, циркуляция и утилизация создают значительные инструментальные, технологические и экологические трудности. (Olah G.A., Malhotra R., Narang S.С., Nitration: Methods and Mechanism, VCH: New York, 1989.)
В последние 20 лет развиваются процессы гетерофазного нитрования ароматических соединений на твердых катализаторах оксидами азота или азотной кислотой. Такие процессы проводят при высоких температурах (100-300°С), характеризуются высокой взрывоопасностью, низкими выходами или требуют большого избытка азотной кислоты или использования дорогостоящих смесей, например уксусный ангидрид - азотная кислота (US 6791000, С04В 18/02, 2004; US 5004846, C07C 201/08, 1991; WO 96/36587, C07C 201/08, 1996).
Число публикаций, посвященных нитрованию ароматических соединений одной азотной кислотой в присутствии твердофазных катализаторов, мало. В большинстве опубликованных работ нитрование проводят в среде органического растворителя при использовании концентрированной (~100%) азотной кислоты (US 6376726, C07C 201/08, 2002). Недостатком данного метода является использование более дорогой дымящей азотной кислоты, необходимость отделения катализатора и растворителя от продуктов нитрования.
В качестве прототипа может быть использован патент РФ 2309142, C07C 201/08, 2007, в котором описан способ нитрования ароматических углеводородов на ячеистом высокопористом катализаторе 50-80% азотной кислотой при температуре 70-210°С в аппарате колонного типа, в котором катализаторный блок одновременно является реакционной и ректификационной зоной, в середину которой подается азотная кислота и нитруемый углеводород, а реакционная вода удаляется из верхней части блока в виде азеотропа с нитруемым соединением.
Недостатком этого способа является то, что удаление реакционной воды в виде азеотропа с нитруемым соединением возможно только на колонне с достаточно большим числом тарелок при большом потоке флегмы в ректификационной зоне, что увеличивает энергетические затраты на разделение. При этом азотная кислота концентрируется в более узкой зоне по высоте реактора, а локальный избыток кислоты приводит к увеличению выхода побочных продуктов.
Ограничены возможности регулирования диапазона температурного профиля по длине реактора, так как температура в каждой точке определяется только составом смеси.
Данное изобретение направлено на совершенствование способа нитрования ароматических соединений. Технический результат - уменьшение выхода побочных продуктов и улучшение изомерного состава продуктов в процессе нитрования.
Этот технический результат достигается тем, что в способе получения ароматических нитросоединений нитрованием ароматического соединения азотной кислотой в реакторе колонного типа, заполненном насадкой, являющейся одновременно реакционной и ректификационной зоной, в среднюю часть которой подают исходное ароматическое соединение и азотную кислоту, а реакционную воду отбирают из верхней части, нитрование проводят при давлении 0,4-2,0 атм, отбирая воду в виде смеси, содержащей воду, исходное ароматическое соединение и азотную кислоту, смесь конденсируют, органическую фазу, содержащую ароматическое соединение, отделяют от водной фазы и возвращают в реактор, а из водной фазы, содержащей воду и азотную кислоту, отгоняют воду, а укрепленную азотную кислоту возвращают в реактор, из кубового остатка реактора, содержащего исходное ароматическое соединение и нитросоединение, отгоняют ароматическое соединение и возвращают его реактор, а оставшееся нитросоединение подвергают очистке известными методами.
Может быть использована каталитически активная насадка в виде блоков ячеистого высокопористого носителя, содержащего сульфатированные диоксиды циркония или титана в количестве 5-20% от массы насадки или инертная насадка.
Нитрование проводят при давлении 0,4-2,0 атм. Температура в какой-либо точке реакционной зоны определяется составом жидкости и давлением и изменятся по высоте реактора. Изменяя давление в указанном диапазоне, можно варьировать температуру в конкретной точке в пределах 50-70°С.
Более высокую температуру в реакционной зоне поддерживают для менее реакционноспособных ароматических соединений, например для бензола, проводя синтез при атмосферном или повышенном давлении. При этом увеличение температуры в реакционной зоне приводит к увеличению скорости нитрования, а значит, и удельной производительности реактора.
Нитрование более реакционноспособных соединений, например толуола, следует проводить под вакуумом. Уменьшение температуры позволяет уменьшить скорость побочных реакций, а следовательно, увеличить селективность процесса.
Для увеличения конверсии исходного ароматического соединения необходимо удалять реакционную воду из зоны реакции. В прототипе воду удаляют из верхней части реактора в виде азеотропа исходного ароматического соединения с водой.
В предложенном способе реакционную воду отбирают из верхней части реактора в виде смеси с исходным ароматическим соединением и азотной кислотой. Такой способ позволяет более эффективно отводить воду из реакционной зоны, так как содержание воды в отгоняемой смеси больше, чем в азеотропе нитруемого соединения с водой. При этом азотная кислота более равномерно распределяется по реакционной зоне, что позволяет избежать локального избытка азотной кислоты по отношению к нитруемому соединению и таким образом уменьшить образование побочных продуктов нитрования. Кроме того, частичный отбор кислоты из верхней части колонны позволяет уменьшить время пребывания азотной кислоты в реакторе, а значит, уменьшить разложение азотной кислоты и соответственно увеличить селективность процесса по азотной кислоте.
Отбор реакционной воды в виде смеси с нитруемым соединением и азотной кислотой позволяет работать с меньшей флегмой, что приводит к уменьшению энергетических затрат.
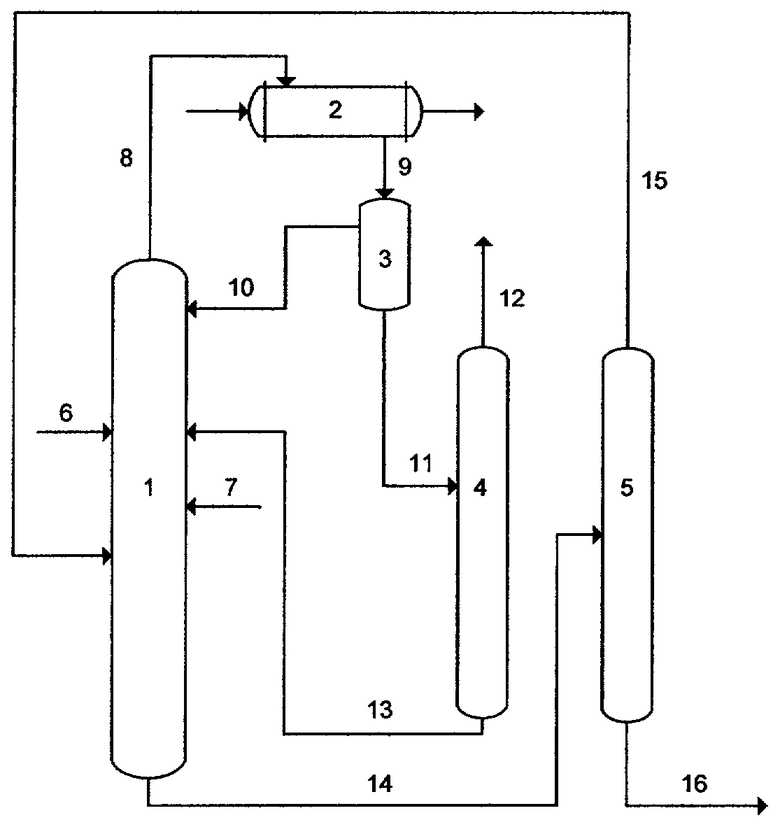
Схема установки получения ароматических нитросоединений показана на чертеже.
Установка состоит из реактора 1, дефлегматора 2, фазоразделителя 3, колонны для концентрирования азотной кислоты 4 и колонны для отделения продуктов нитрования от непрореагировавшего ароматического соединения 5, трубопроводов 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16.
Реактор представляет собой колонну с обогреваемым кубом, заполненную насадкой. Колонна является одновременно реакционной и ректификационной зоной. Колонна может быть заполнена либо инертной, либо каталитически активной насадкой. В качестве каталитически активной насадки используют сульфатированные диоксиды циркония или титана, нанесенные на блоки высокопористого ячеистого носителя, в количестве 5-20% от массы насадки. Носитель для каталитически активной насадки изготовлен в соответствии с патентом РФ №2233700 B01J 21/12, 2004. Изделия из высокопористых керамических материалов получаются воспроизведением структуры вспененного ретикулированного пенополиуретана путем нанесения керамического порошка, содержащего наполнитель и активный к спеканию порошок оксида алюминия с добавками связующего, с последующим выжиганием основы и нагревом оставшегося керамического каркаса до температуры 1450-1500°С. Общая пористость керамического изделия составляет 85-92% при открытой пористости перемычек между ячейками 20-30%, имеющих размер пор 1-2 мкм.
Свежее и возвращаемое после выделения из куба нитруемое ароматическое соединение по трубопроводам 6 и 15, а также свежую и возвратную азотную кислоту по трубопроводам 7 и 13 подают в среднюю часть реактора 1.
Смесь воды, нитруемого соединения и азотной кислоты, отгоняемая из верхней части реактора, подается по трубопроводу 8 в дефлегматор 2. После конденсации в дефлегматоре 2 смесь по трубопроводу 9 направляется в фазоразделитель 3, где расслаивается на две фазы. Органическая фаза, содержащая, в основном, нитруемое ароматическое соединение, возвращается по трубопроводу 10 в реактор. Водная фаза, содержащая, в основном, воду и азотную кислоту, по трубопроводу 11 направляется в колонну 4 для концентрирования азотной кислоты, в которой частично отгоняется вода по трубопроводу 12, а укрепленная азотная кислота по трубопроводу 13 возвращается в реактор.
Кубовая жидкость реактора, содержащая исходное ароматическое соединение и его нитропроизводное, подается по трубопроводу 14 в колонну 5 для отделения продуктов нитрования. Отогнанное в колонне 5 ароматическое соединение по трубопроводу 15 возвращают в реактор 1. Оставшееся нитропроизводное ароматического соединения (трубопровод 16) подвергают очистке известными методами.
Предложенный способ получения нитропроизводных ароматических соединений можно проиллюстрировать следующими примерами:
Пример 1.
Синтез нитробензола проводили в стеклянном реакторе непрерывного действия, который состоит из куба объемом 400 см3, реакционно-ректификационной зоны в виде колонны диаметром 26 мм и высотой 1,8 м, заполненной инертной стеклянной насадкой (кольца Фенске), и дефлегматора. Перед синтезом в куб реактора заливали 200 г возвратного бензола.
В колонну реактора подавали в течение 8 часов 177,8 г 80% азотной кислоты (полученной путем смешения 102,5 г свежей 100% кислоты и 75,4 г возвратной 53% азотной кислоты) и 636,0 г бензола (смешали 508,8 г возвратного и 127,2 г свежего бензола). Реактор работал при давлении 1 атм, температура в верхней части колонны 76-78°С, в кубе колонны 81-90°С. Из верхней части колонны выходит смесь воды, бензола и азотной кислоты, которая конденсируется в дефлегматоре 2 и в фазоразделителе 3 разделяется на две фазы: органическую фазу и водную. Органическую фазу возвращали в колонну реактора 1. Из водной фазы в количестве 104,8 г (38% азотная кислота с примесью бензола) отогнали на ректификационной колонне 28,8 г воды с примесью бензола, а остаток - 53% азотную кислоту (75,4 г) вернули в реактор. Из куба колонны отобрали смесь, содержащую 200,0 г нитробензола и 708,8 г/ч бензола с примесями воды и азотной кислоты (22% раствор нитробензола). Из кубовой жидкости отогнали на колонне ректификации 708,6 г бензола и получили 200,0 г нитробензола-сырца с массовой долей динитробензолов менее 0,001%.
Конверсия азотной кислоты - 72,2%. Селективность образования нитробензола по азотной кислоте - более 99,6%.
По сравнению с прототипом способ позволяет получить нитробензол с меньшим содержанием динитробензолов, а селективность образования нитробензола по азотной кислоте больше.
Пример 2.
Нитрование толуола проводили в реакторе непрерывного действия, который состоит из куба объемом 400 см3, реакционно-ректификационной зоны в виде колонны диаметром 26 мм и высотой 1,8 м, заполненной блоками ячеистого высокопористого катализатора (6,5% сульфатированного диоксида циркония от массы носителя) и дефлегматора. Перед синтезом в куб реактора заливали 200 г возвратного толуола.
В колонну реактора подавали в течение 12 часов 216 г 70% азотной кислоты, полученной смешением 83,1 г. свежей 100% азотной кислоты и 132,9 г 51,2% возвратной азотной кислоты, и 562,7 г толуола (474,0 г возвратного и 88,7 г свежего толуола). Реактор работал при давлении 1 атм, температура в верхней части колонны 106°С, в кубе колонны 110-120°С. Из верхней части колонны отобрали смесь, содержащую воду, толуол и азотную кислоту. Смесь сконденсировали в дефлегматоре, после разделения органическую фазу вернули в реактор. Из водной фазы в количестве 155,4 г (43,8% азотная кислота со следами толуола) отогнали на ректификационной колонне 22,5 г воды с примесями толуола, а остаток 132,8 г (51,2% азотная кислота) направили на смешение со свежей азотной кислотой. Из куба колонны отобрали массу, содержащую 147,9 г смеси изомеров мононитротолуола (18%), 674,0 г толуола, 6,1 г бензальдегида (0,7%) и следовые количества не идентифицированных продуктов конденсации. Из кубовой жидкости отогнали на колонне ректификации 674,0 г толуола и получили 154,0 г смеси продуктов реакции (массовое соотношение изомеров нитротолуола пара:орто:мета - 1:1,1:0,15).
Конверсия азотной кислоты - 54,5%. Селективность образования мононитропроизводных толуола - 82,5%. Селективность образования n-нитротолуола составила 36,8%.
Как видно из примера, способ позволяет получить наиболее востребованный изомер п-нитротолуол с большей селективностью, чем по классической технологии нитрования с использованием серной кислоты (примерное массовое соотношение изомеров нитротолуола пара:орто:мета - 1:1,7:0,1).
Пример 3.
Нитрование толуола проводили в реакторе, описанном в примере 2. Перед синтезом в куб реактора заливали 200 г возвратного толуола.
В колонну реактора подали в течение 12 часов 216,0 г 70% азотной кислоты, полученной смешением 80,6 г свежей 100% азотной кислоты и 135,4 г 52,2% возвратной азотной кислоты, и 517,4 г толуола (417,9 г возвратного и 99,4 г свежего толуола). Реактор работал при остаточном давлении 0,4 атм. Температура в верхней части колонны - 65°С, в кубе колонны - 85-88°С. Из верхней части колонны отбирали смесь, содержащую воду, толуол и азотную кислоту. Смесь конденсировали в дефлегматоре, после разделения в фазоразделителе органическую фазу непрерывно возвращали в колонну. Из водной фазы в количестве 158,3 г (44,6% азотная кислота со следами толуола) отогнали на ректификационной колонне 22,9 г воды с примесями толуола, а остаток 135,4 г (52,2% азотная кислота) направили на смешение со свежей азотной кислотой. Из куба колонны отобрали массу, содержащую 174,3 г смеси изомеров мононитротолуола (22%), 0,09 г бензальдегида (0,01%) и 617,9 г толуола. Из кубовой жидкости отогнали на колонне ректификации 617,9 г толуола и получили 174,4 г смеси нитропроизводных (молярное соотношение изомеров нитротолуола пара:орто:мета - 1:1,47:0,2).
Конверсия азотной кислоты - 53,2%. Селективность образования мононитропроизводных толуола - 99,6%. Селективность образования n-нитротолуола составила 37,4%.
Таким образом, проведение нитрования при пониженном давлении (0,4 атм) приводит к увеличению селективности образования мононитротолуолов с 82,5% (пример 2) до 99,6%.
В других примерах изменяли рабочее давление в реакторе синтеза. Увеличение давления более 2 атм не приводит к дальнейшему существенному увеличению скорости нитрования, так же как и уменьшение давления менее 0,4 атм нежелательно, так как скорость нитрования становится слишком низкой. При этом более высокое давление следует использовать для менее реакционноспособных соединений, а синтез под вакуумом - для таких реакционноспособных соединений, как толуол.
Кроме того, в примерах использовали каталитически активную насадку в виде сульфатированного диоксида титана, нанесенного на высокопористый ячеечный носитель. При этом результаты нитрования были аналогичны результатам, полученным при использовании диоксида циркония.
Содержание каталитически активного компонента меняли в диапазоне 5-20%. Увеличение содержания активного компонента в диапазоне от 0 до 5% не приводило к существенному увеличению скорости нитрования, а при увеличении содержания более 20% скорость увеличивается незначительно.
Предложенный способ получения ароматических нитросоединений позволяет уменьшить выход побочных продуктов и улучшить изомерный состав продуктов в процессе нитрования, а также уменьшить энергетические затраты на стадии синтеза.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КАТАЛИТИЧЕСКОГО НИТРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ АЗОТНОЙ КИСЛОТОЙ | 2010 |
|
RU2473536C2 |
| СПОСОБ КАТАЛИТИЧЕСКОГО НИТРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ НА ЯЧЕИСТОМ ВЫСОКОПОРИСТОМ КАТАЛИЗАТОРЕ | 2005 |
|
RU2309142C1 |
| СПОСОБ И УСТАНОВКА ДЛЯ РЕГЕНЕРАЦИИ ОТРАБОТАННОЙ СЕРНОЙ КИСЛОТЫ ИЗ ПРОЦЕССОВ НИТРОВАНИЯ | 2010 |
|
RU2511380C2 |
| СПОСОБ НЕПРЕРЫВНОГО ИЗОТЕРМИЧЕСКОГО ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛОВ В ПРИСУТСТВИИ ФОСФОРНОЙ КИСЛОТЫ | 2002 |
|
RU2293722C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛОВ | 2001 |
|
RU2274634C2 |
| СПОСОБ КАТАЛИТИЧЕСКОГО ЖИДКОФАЗНОГО НИТРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2002 |
|
RU2226187C1 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛА С ПОВЫШЕННЫМ СОДЕРЖАНИЕМ П-ИЗОМЕРА | 2007 |
|
RU2346930C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 1997 |
|
RU2116286C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5(6)-НИТРО-1-(4-НИТРОФЕНИЛ)-1,3,3-ТРИМЕТИЛИНДАНОВ | 2014 |
|
RU2551672C1 |
| СПОСОБ НИТРОВАНИЯ БЕНЗОЛА НА ЦЕОЛИТНЫХ КАТАЛИЗАТОРАХ | 1995 |
|
RU2095342C1 |
Настоящее изобретение относится к химической технологии, а именно к способу получения ароматических соединений нитрованием ароматического соединения азотной кислотой в реакторе колонного типа, заполненном насадкой, являющейся одновременно реакционной и ректификационной зоной, в среднюю часть которой подают исходное ароматическое соединение и азотную кислоту, а реакционную воду отбирают из верхней части, отличающийся тем, что нитрование проводят при давлении 0,4-2,0 атм, отбирая воду в виде смеси, содержащей воду, исходное ароматическое соединение и азотную кислоту, смесь конденсируют, органическую фазу, содержащую ароматическое соединение, отделяют от водной фазы и возвращают в реактор, из водной фазы, содержащей воду и азотную кислоту, отгоняют воду, а укрепленную азотную кислоту возвращают в реактор, из кубового остатка реактора, содержащего исходное ароматическое соединение и нитросоединение, отгоняют ароматическое соединение и возвращают его в реактор, оставшееся нитросоединение подвергают очистке известными методами. Технический результат: разработан новый способ получения ароматических нитросоединений, отличающийся уменьшенным выходом побочных продуктов и улучшенным изомерным составом продуктов. 2 з.п. ф-лы, 1 ил.
1. Способ получения ароматических нитросоединений нитрованием ароматического соединения азотной кислотой в реакторе колонного типа, заполненном насадкой, являющейся одновременно реакционной и ректификационной зоной, в среднюю часть которой подают исходное ароматическое соединение и азотную кислоту, а реакционную воду отбирают из верхней части, отличающийся тем, что нитрование проводят при давлении 0,4-2,0 атм, отбирая воду в виде смеси, содержащей воду, исходное ароматическое соединение и азотную кислоту, смесь конденсируют, органическую фазу, содержащую ароматическое соединение, отделяют от водной фазы и возвращают в реактор, из водной фазы, содержащей воду и азотную кислоту, отгоняют воду, а укрепленную азотную кислоту возвращают в реактор, из кубового остатка реактора, содержащего исходное ароматическое соединение и нитросоединение, отгоняют ароматическое соединение и возвращают его в реактор, оставшееся нитросоединение подвергают очистке известными методами.
2. Способ по п.1, отличающийся тем, что используют каталитически активную насадку в виде блоков ячеистого высокопористого носителя, содержащего сульфатированные диоксиды циркония или титана в количестве 5-20% от массы насадки.
3. Способ по п.1, отличающийся тем, что используют инертную насадку.
| СПОСОБ КАТАЛИТИЧЕСКОГО НИТРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ НА ЯЧЕИСТОМ ВЫСОКОПОРИСТОМ КАТАЛИЗАТОРЕ | 2005 |
|
RU2309142C1 |
| СПОСОБ КАТАЛИТИЧЕСКОГО ЖИДКОФАЗНОГО НИТРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2002 |
|
RU2226187C1 |
| US 6376726 В1, 23.04.2002. | |||

