Изобретение относится к способам получения соединений пиррольного ряда, конкретно 2-фенилпиррола.
Пиррол и его производные широко представлены в живой природе. Они являются структурными фрагментами многих биологически активных молекул, таких как гемоглобин, осуществляющий транспорт кислорода в живых организмах млекопитающих, хлорофилл, играющий ключевую роль в процессах фотосинтеза, пигменты желчи, витамин В12 и др. Соединения пиррольного ряда широко используются для синтеза фармацевтических препаратов, электропроводящих (таких как полипирролы) и нелинейно-оптических материалов. Производные 2-фенилпиррола могут использоваться в качестве прекурсоров лекарственных препаратов для лечения психических и сердечно-сосудистых заболеваний, лечения и профилактики заболеваний, вызванных ограничением функции головного мозга, а также для синтеза флуоресцирующих азаиндаценовых красителей семейства BODIPY и электропроводящих пигментных композиционных материалов. Производные 2-фенилпиррола применяются для изменения электропроводности полимеров, используемых в медицинских исследованиях.
2-Фенилпиррол получают различными методами.
Описаны методы, основанные на Pd-катализируемом кросс-сочетании производных пиррола с фенилгалогенидами. Например, сочетание бромбензола с 1-пирролилцинкхлоридом, получаемым in situ из 1-пирролилнатрия и хлорида цинка, в присутствии каталитической системы [PdCl2(PPh3)2]/PPh3 (N-метилпирролидон, 140°C, 2 ч) дает 2-фенилпиррол с 75%-ным выходом (Filippini L., Gusmeroli М., Riva R. Tetrahedron. Lett. 1992, 33, 1755). Однако в данном случае наряду с целевым продуктом (хроматографический выход 75%) образуется 5% его структурного изомера - 3-фенилпиррола - и 15% бифенила, что должно значительно затруднять выделение 2-фенилпиррола в чистом виде (способ выделения не описан). Кросс-сочетание пиррола с иодбензолом в присутствии каталитической системы Pd(OAc)2/PPh3/MgO (диоксан, 150°C, 12-15 ч) приводит к 2-фенилпирролу с выходом 86% (Sezen В., Sames D. J. Am. Chem. Soc. 2003, 125, 5274). 2-Фенилирование пиррола фенилиодиниевой солью [Ph2I]BF4 в присутствии 1,3-бис(2,4,6-триметилфенил)имидазол-2-илидендиацетата палладия (25°C, 15 ч) дает 2-фенилпиррол с выходом 69% (Deprez N.R., Kalyani D., Krause A., Sanford M. J. Am. Chem. Soc. 2006, 128, 4943). Несмотря на сравнительно высокие выходы 2-фенилпиррола, получаемого этими методами, использование дорогостоящих и трудносинтезируемых палладиевых катализаторов делает их пригодными только для его лабораторного получения. Кроме того, неселективность первого метода, использование горючего и токсичного диоксана и продолжительное нагревание во втором процессе создают дополнительные препятствия для технологического получения 2-фенилпиррола.
2-Фенилпиррол с выходом 55% получен внутримолекулярной циклизацией 1-амино-1-фенил-3-бутин-2-ола при нагревании в этаноле в присутствии 10 мольных процентов дирутенивого комплекса {[Cp*RuCl(µ2-SMe)2RuCp*(OH2)]OSO2CF3} (70°C, 24 ч). К недостаткам данного метода следует отнести умеренный выход продукта, применение труднодоступного катализатора и необходимость использования для синтеза исходного 1-амино-1-фенил-3-бутин-2-ола реактива Гриньяра (этилмагнийбромида) и, как следствие, пожаро- и взрывоопасных растворителей (Yukito Yada, Yoshihiro Miyake, Yoshiaki Nishibayashi Organometallics 2008, 27, 3614).
Описан способ синтеза 2-фенилпиррола с общим выходом, не превышающим 50%, из ацетофенона и O-(2-гидроксиэтил)гидроксиламина (уксусная кислота, пиридин/этанол, кипячение) с последующими иодированием метилтрифенилфосфонийиодидом (ацетонитрил, 20°C, 20 мин) и циклизацией промежуточных производных ацетофеноноксима в присутствии трет-бутилата калия в кипящем трет-бутиловом спирте (Dhanak D., Reese С.В., Romana S., Zappia G. J. Chem. Soc., Chem. Commun. 1986, 903). Многостадийность процесса, использование большого набора реактивов, в том числе пожароопасных трет-бутилата калия и трет-бутилового спирта, а также умеренный выход целевого продукта снижают перспективы использования этого метода в технологической практике для многотоннажного получения 2-фенилпиррола.
2-Фенилпиррол с выходом 45% получен ароматизацией 2-фенил-1-пирролина, синтезированного из 4-хлорбутиронитрила и фенилмагнийбромида, на активированном угле в кипящем толуоле (Carabineiro S.A., Bellabarba R.M., Gomes Р.Т., Fonseca I.M. Cat. Lett. 2006, 111, No 3-4, 221). Невысокий выход 2-фенилпиррола, необходимость использования реактива Гриньяра и абсолютированного и пожаро-взрывоопасного растворителя - диэтилового эфира - делают этот процесс труднореализуемым в производстве.
Описан способ получения 2-фенилпиррола (Авт. Свид. СССР №601282, 1976; Бюл. изобрет. №13, 1978) взаимодействием ацетофеноноксима с ацетиленом в присутствии гидроксида лития (10-30% от массы оксима) в среде диметилсульфоксида (ДМСО) под давлением ацетилена (12-15 атм) при 100°C (схема 1):
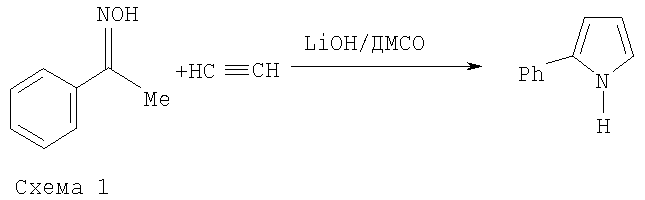
Для выделения 2-фенилпиррола реакционную смесь обрабатывают трехкратным (по объему) количеством воды, экстрагируют диэтиловым эфиром. После удаления эфира перегонка с паром дает продукт с выходом 56-64%, конверсия ацетофеноноксима не указана. Недостатками этого способа являются использование ацетилена под давлением (что, вследствие повышенной опасности, делает процесс труднореализуемым в производстве) и трудоемкая очистка целевого продукта с применением пожаро- и взрывоопасного диэтилового эфира.
Наиболее близким по технической сущности к предлагаемому изобретению является способ получения 2-фенилпиррола взаимодействием ацетофеноноксима с ацетиленом в системе гидроксид калия - ДМСО в присутствии 10% воды (от объема взятого растворителя) при атмосферном давлении ацетилена, мольном соотношении ацетофеноноксим : КОН=1:1.5-2.3 и продолжительности контакта реагентов 9-10 ч при 95-97°C (Авт. Свид. СССР №694504, 1978; Бюл. изобрет. №40, 1979). Как описано в авторском свидетельстве (пример 3), из 15 г (0.11 моль) ацетофеноноксима после обработки реакционной смеси водой и перекристаллизации отфильтрованного сырого продукта получают 4 г (72.5%) 2-фенилпиррола, при этом конверсия исходного оксима не указана. Теоретический выход 2-фенилпиррола из 15.0 г (0.11 моль) ацетофеноноксима составляет 15.75 г, т.о. 4.0 г 2-фенилпиррола составляют 25.4% на взятый оксим, следовательно, выход 72.5% рассчитан с учетом конверсии ацетофеноноксима, которая не превышает 34.5%. К недостаткам данного способа следует отнести низкий выход целевого продукта (24-25% на взятый ацетофеноноксим), низкую конверсию исходного кетоксима (34,5%), высокое содержание гидроокиси калия (1.5 и 2.3 моль на 1 моль оксима или 60 и 100% от массы оксима соответственно) и низкую концентрацию ацетофеноноксима (0.73 моль/л) в реакционной смеси, что приводит к необходимости использования повышенных количеств растворителя и, стало быть, повышению затрат на его регенерацию. Высокое содержание гидроксида калия в реакционной среде осложняет выделение целевого продукта и повышает количество отходов (водно-диметилсульфоксидные растворы КОН и ацетата калия).
Перечисленные недостатки препятствуют практическому использованию способа-прототипа.
Целью настоящего изобретения является разработка способа получения 2-фенилпиррола, лишенного недостатков способа-прототипа.
Фундаментальной причиной снижения выхода 2-фенилпиррола, получаемого по указанной реакции, является ее консекутивный характер, а именно последующее взаимодействие 2-фенилпиррола с ацетиленом, приводящее к 1-винил-2-фенилпирролу, в данном случае нежелательному побочному продукту. В результате при достаточно высокой конверсии исходного ацетофеноноксима образуется трудноразделяемая смесь 2-фенилпиррола и 1-винил-2-фенилпиррола. В общем случае это не позволяет достичь высокого выхода целевого продукта и хорошей селективности процесса при технологически приемлемых степенях превращения исходного оксима (см. схему 2).
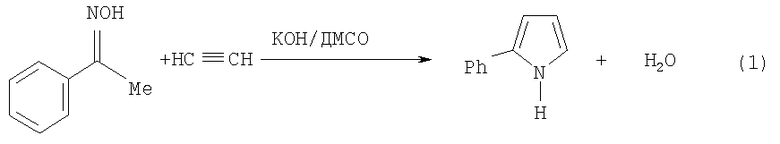
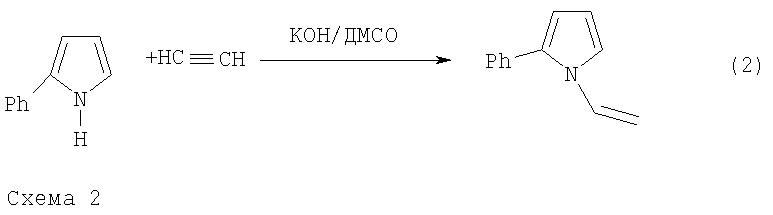
Для достижения поставленной цели мы учли различие механизмов этих двух последовательных реакций 1 и 2 (схема 2). Первая реакция представляет собой основно-каталитическое многостадийное построение пиррольного кольца из ацетофеноноксима и ацетилена, заканчивающееся выделением молекулы воды (Трофимов Б.А., Михалева А.И. N-Винилпирролы. Новосибирск: Наука, 1984, 264 с), тогда как вторая реакция является основно-каталитическим нуклеофильным присоединением 2-фенилпирррола к тройной связи. Из опубликованных работ (Трофимов Б.А., Михалева А.И. N-Винилпирролы. Новосибирск: Наука, 1984, 264 с.) следует, что присутствие воды незначительно влияет на реакцию построения пиррольного кольца. В то же время вода резко тормозит реакцию винилирования пирролов. Вместе с тем скорость последней реакции резко возрастает с повышением концентрации катализатора-основания (Трофимов Б.А., Михалева А.И. N-Винилпирролы. Новосибирск: Наука, 1984, 264 с). Важной особенностью изучаемого процесса является постепенное превращение щелочи под действием ацетилена и воды в каталитически неактивный (на стадии винилирования) ацетат калия по схеме 3 (Б.А.Трофимов. Гетероатомные производные ацетилена. Новые полифункциональные мономеры, реагенты и полупродукты. М.: Наука, 1981, с.14):
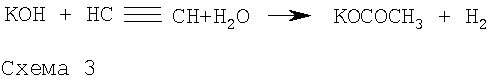
Таким образом, по мере накопления воды в реакционной смеси концентрация щелочи за счет этой реакции будет снижаться и, следовательно, снижаться скорость винилирования образующегося 2-фенилпиррола.
Используя эти особенности двух последовательных реакций 1 и 2 (схема 2), можно в принципе выйти на саморегулирующийся процесс, позволяющий существенно повысить выход 2-фенилпиррола и подавить нежелательную реакцию винилирования, поскольку вода, выделяющаяся в результате первой реакции, тормозит вторую реакцию, т.е. нежелательный синтез 1-винил-2-фенилпиррола. Скорость образования 1-винил-2-фенилпиррола будет также снижаться при уменьшении концентрации щелочного катализатора. Очевидно, что содержание 1-винил-2-фенилпиррола в реакционной смеси можно понизить также за счет сокращения продолжительности брутто-процесса. Учет перечисленных фундаментальных особенностей синтеза 2-фенилпиррола из ацетофеноноксима и ацетилена позволяет принципиально модифицировать процесс и достичь поставленной в изобретении цели - существенно (примерно в три раза) повысить выход и селективность брутто-реакции при полной конверсии исходного оксима.
В предлагаемом изобретении цель достигается:
1) значительным снижением концентрации гидроокиси калия в реакционной среде (с 60-100% в способе-прототипе до 8-16% в предлагаемом способе), что обеспечивает значительное снижение скорости винилирования образующегося пиррола и предотвращает связывание выделяющейся воды щелочью;
2) существенным повышением концентрации ацетофеноноксима в реакционной среде (от 0.73 моль/л в способе-прототипе до 1.32-1.85 моль/л в предлагаемом способе), что приводит к повышению скорости первой стадии и, следовательно, скорости образования воды, тормозящего вторую нежелательную стадию;
3) значительным повышением температуры реакции (от 95-97°C в способе-прототипе до 130-150°C в предлагаемом способе), что позволяет сократить продолжительность брутто-процесса в два раза, особенно его второй нежелательной стадии;
4) новым приемом выделения целевого продукта - отгон большей части (70-80%) ДМСО из реакционной среды с последующим разбавлением остатка водой и отделением целевого продукта фильтрованием.
Совокупность перечисленных принципиальных технологических модификаций позволяет довести выход высокочистого (чистота выше 99.9%) 2-фенилпиррола до 74% против 24-25% в способе-прототипе при полной конверсии ацетофеноноксима и высокой селективности процесса (90-100%).
Преимуществом предлагаемого способа является также меньший расход катализатора (КОН): в предлагаемом способе 8-16% от массы ацетофеноноксима против 60-100% в способе-прототипе.
К преимуществам предлагаемого способа следует отнести также сокращение времени процесса до 4-8 ч по сравнению с 9-10 ч.
Еще одним существенным преимуществом нового способа является то, что он позволяет работать с более концентрированными растворами, т.е. использовать меньшие количества активного растворителя (ДМСО): при реализации процесса по новому способу концентрация ацетофеноноксима в реакционной смеси составляет 1.32-1.85 моль/л против 0.73 моль/л в способе-прототипе, следовательно, используются в 1.8-2.5 раза более концентрированные реакционные смеси, что существенно повышает интенсивность процесса и снижает время и энергозатраты, необходимые на регенерацию ДМСО.
Очень важным технологическим и экономическим преимуществом предлагаемого способа является то, что в нем впервые применена прямая отгонка большей части ДМСО (70-80%) из реакционной смеси, что означает: 1 - почти полную регенерацию растворителя; 2 - многократное уменьшение объема водно-диметилсульфоксидного щелочного раствора на завершающей стадии выделения целевого продукта; 3 - существенное сокращение энергетических и временных затрат.
В общем виде предлагаемый способ получения 2-фенилпиррола осуществляется следующим образом: через нагретый до 130-150°C ДМСО, предпочтительно 140-150°C, содержащий ацетофеноноксим и КОН в количестве 8-16% от массы оксима, пропускается ацетилен со скоростью, обеспечивающей его конверсию 90-100%, предпочтительно 95%. Через 4-8 часов после достижения конверсии оксима, равной 81-95%, подача ацетилена прекращается, реакционная смесь выдерживается, охлаждается, отгоняется в вакууме 70-80% ДМСО, затем остаток, содержащий ДМСО, 2-фенилпиррол и 1-винил-2-фенилпиррол, разбавляется водой (1:1 по объему), выпавший 2-фенилпиррол-сырец отфильтровывается, промывается водой от ДМСО, сушится.
Таким образом, решается принципиальная для данной технологии проблема регенерации дорогостоящего растворителя ДМСО (нерешенная в способе-прототипе).
Снижение температуры реакции (до 120°C и ниже), а также количества KOH (меньше 8% от массы оксима) приводит к уменьшению конверсии ацетофеноноксима и уменьшению выхода 2-фенилпиррола. Проведение реакции при более высокой, чем заявляемая, температуре (выше 150°C) и количестве КОН, превышающем 16% от массы оксима, ускоряет реакцию винилирования целевого 2-фенилпиррола, тем самым снижая его выход.
Следующие неограничивающие примеры иллюстрируют изобретение.
Пример 1.
В стеклянный реактор емкостью 250 мл помещают 100 мл ДМСО, 1.63 г [0.025 моль, 8% от массы оксима товарного (88%-ного)] КОН, 17.79 г (0.132 моль) ацетофеноноксима (концентрация 1.32 моль/л), реакционную смесь нагревают (150°C) при перемешивании и барботируют ацетилен со скоростью ~30-40 мл/мин. Контроль за ходом реакции осуществляют методом ГЖХ путем отбора проб через 1 ч, который осуществляют следующим образом: 1 мл реакционной смеси разбавляют 2 мл воды, экстрагируют эфиром (3×1 мл), экстракты промывают водой (3×1 мл), сушат поташом и анализируют. Через 6 ч из реакционной смеси, содержащей 2.3% ацетофенона, 10.8% ацетофеноноксима, 4.7% 1-винил-2-фенилпиррола и 82.1% 2-фенилпиррола, в вакууме отгоняют часть (70 мл) ДМСО и остаток разбавляют водой (1:1 по объему). Выпавший осадок отфильтровывают, промывают водой, сушат. Получают 16.68 г 2-фенилпиррола-сырца (чистота 91.5%, примесь - 1-винил-2-фенилпиррол, 8.5%). После перекристаллизации из петролейного эфира (т.кип. 70-100°C) получают 13.73 г (72.5%) перламутровых светло-розовых кристаллов (чистота по данным ГЖХ [хроматограф Agilent Technologies 6890N] ~99.9%, на хроматограмме примеси отсутствуют), т.пл. 129°C. ИК (Bruker ISF 25, KBr, см-1): 3433, 3391 (v N-H), 3101 (валентные колебания =С-Н), 1947, 1867, 1808, 1734 (обертоны С-Н фенильного цикла), 1640, 1604, 1580, 1495, 1465 (валентные С=С пиррольного и фенильного колец), 1408, 1296, 1248, 1186, 1156, 1112, 1073, 1033 (деформационные С-С колебания остова), 916, 900, 880, 758, 719, 691 (неплоские деформационные колебания =С-Н), 535. 1Н ЯМР (Bruker-400DPX, 400.13 МГц, CDCl3, δ, м.д., внутренний стандарт ГМДС). Обозначения ядер см. структурную формулу, приведенную ниже:
8.40 (1H, уш. с, NH), 7.45 (2Н, м, o-Ph), 7.34 (2Н, м, м-Ph), 7.19 (1Н, м, р-Ph), 6.84 (1Н, м, Н-5), 6.51 (1Н, м, Н-3), 6.28 (1H, м, Н-4). 13С ЯМР (Bruker-400DPX, 100.6 МГц, ДМСО-d6, δ, м.д.): 132.7. 132.0, 128.8, 126.1, 123.8, 118.8 110.0, 105.9.
Пример 2.
В условиях примера 1 в том же реакторе, используя 25.00 г (1.85 моль/л) [вместо 17.79 г (1.32 моль/л) - в первом примере] ацетофеноноксима и 2.30 г КОН (8% от массы оксима, в пересчете на 88%-ный КОН, проводя реакцию при той же температуре (150°C) в течение того же времени, получают 21.35 г 2-фенилпиррола (чистота 98.0%, примесь - 1-винил-2-фенилпиррол, 2.0%). После обработки, описанной в 1 примере, выход 2-фенилпиррола-сырца 79.0%. После перекристаллизации из петролейного эфира (т.кип. 70-100°C) получают 19.22 г (72.6%) 2-фенилпиррола (чистота, по данным ГЖХ, ~ 99.9%, на хроматограмме примеси отсутствуют).
Пример 3.
В условиях примера 2 в том же реакторе, используя те же загрузки, проводя реакцию при той же температуре (150°C), но в течение 7 ч, получают (обрабатывают, как описано в примере 1) 23.96 г 2-фенилпиррола-сырца, чистота 89.0%, примесь 1-винил-2-фенилпиррол, 11.0%, по данным ГЖХ. После перекристаллизации из петролейного эфира (т.кип. 70-100°C) получают 19.19 г (72.4%) 2-фенилпиррола (чистота, по данным ГЖХ, ~99.9%, на хроматограмме примеси отсутствуют).
Пример 4.
В условиях примера 3 в том же реакторе, используя те же загрузки и то же время реакции, но при температуре 140°C, получают реакционную смесь, состоящую из ацетофенона (0.9%), ацетофеноноксима (32.5%), 1-винил-2-фенилпиррола (2.0%) и 2-фенилпиррола (64.6%). С целью увеличения конверсии ацетофеноноксима продолжают нагревание реакционной смеси еще 1 ч и после обработки, описанной в примере 1, получают 18.72 г смеси, содержащей 25.3% ацетофеноноксима (конверсия 81.0%), 1.5% 1-винил-2-фенилпиррола и 73.2% 2-фенилпиррола. Выход 2-фенилпиррола-сырца 51.7% на взятый ацетофеноноксим (63.7% с учетом конверсии оксима). После перекристаллизации из петролейного эфира (т.кип. 70-100°С) получают 12.16 г (45.9% на взятый оксим) 2-фенилпиррола (чистота, по данным ГЖХ, ~99.9%, на хроматограмме примеси отсутствуют).
Пример 5.
В условиях примера 4 в том же реакторе, используя те же загрузки, за исключением количества гидроксида калия (16% вместо 8% от массы оксима), при той же температуре (140°C), проводя реакцию в течение 4 ч, получают 26.12 г смеси, содержащей 2.1% ацетофенона, 4.5% ацетофеноноксима (конверсия 95.3%), 14.0% 1-винил-2-фенилпиррола и 79.4% 2-фенилпиррола. Выход 2-фенилпиррола-сырца 78.3%. После перекристаллизации из петролейного эфира (т.кип. 70-100°C) получают 18.25 г (68.9%) 2-фенилпиррола (чистота, по данным ГЖХ, - 99.9%, на хроматограмме примеси отсутствуют).
Пример 6.
В условиях примера 5 в том же реакторе, используя те же загрузки и то же время реакции, но при температуре 130°C, получают реакционную смесь, состоящую из ацетофенона (1.8%), ацетофеноноксима (9.4%), 1-винил-2-фенилпиррола (8.0%) и 2-фенилпиррола (80.7%). С целью увеличения конверсии ацетофеноноксима продолжают нагревание реакционной смеси еще 1 ч и после обработки, описанной в примере 1, получают 19.57 г 87.0%-ного 2-фенилпиррола-сырца (примесь - 1-винил-2-фенилпиррол, 13.0%). После перекристаллизации из петролейного эфира (т.кип. 70-100°C) получают 15.02 г (56.7%) 2-фенилпиррола (чистота, по данным ГЖХ, ~99.9%, на хроматограмме примеси отсутствуют).
Пример 7.
В стальной реактор емкостью 10 л с механической мешалкой, рубашкой для электроподогрева и змеевиком для внутреннего охлаждения загружают 5 л ДМСО, 1000 г ацетофеноноксима (концентрация 1.48 моль/л) и 91.5 г товарного КОН (8% от массы оксима в пересчете на 88%-ный КОН). Через реакционную смесь при нагревании (120-125°C) и перемешивании пропускают азот в течение 0.5 ч со скоростью 0.8-1 л/мин, а затем ацетилен со скоростью 2.1 л/мин в течение 5 ч. Температура в реакторе (136-143°C) регулируется скоростью подачи ацетилена и внутренним охлаждением (для снятия экзотермического эффекта в змеевик подается холодная вода), давление ацетилена в реакторе при этом не превышает 0.4-0.8 ати. После прекращения пропускания ацетилена реакционную смесь выдерживают еще 0.5 ч при 140-143°C до полного израсходования растворенного в ней ацетилена и обрабатывают, как описано в примере 1: отгоняют 80% (4 л) от взятого ДМСО, остаток разбавляют водой (2350 мл, 1:1 по объему), осадок отфильтровывают, промывают водой, сушат. Выделяют 972.35 г 2-фенилпиррола-сырца, чистота 91.1% (примесь - N-винил-2-фенилпиррол, 8.9%). После перекристаллизации из петролейного эфира (т.кип. 70-100°C) получают 784.69 г (74.1%) 2-фенилпиррола (чистота, по данным ГЖХ, - 99.9%, на хроматограмме примеси отсутствуют).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 1-ВИНИЛ-2-ФЕНИЛПИРРОЛА | 2009 |
|
RU2399615C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(2-ТИЕНИЛ)ПИРРОЛА | 2012 |
|
RU2514945C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-АЛКИЛ-2-(2-ТИЕНИЛ)ПИРРОЛОВ И ИХ N-ВИНИЛЬНЫХ ПРОИЗВОДНЫХ | 2011 |
|
RU2477725C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4,5,6,7-ТЕТРАГИДРОИНДОЛА | 2005 |
|
RU2297410C2 |
| СПОСОБ ПОЛУЧЕНИЯ 0-ВИНИЛОКСИМОВ | 1982 |
|
SU1095593A1 |
| Способ получения 2-или 2,3-замещенных пирролов | 1978 |
|
SU694504A1 |
| Способ получения винилового эфира циклогексанола | 1990 |
|
SU1796613A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛФУРФУРИЛОВЫХ ЭФИРОВ | 2008 |
|
RU2359965C1 |
| Способ получения виниловых эфиров аминофенолов | 2016 |
|
RU2640808C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАРИЛ (ГЕТАРИЛ) ЭТИНИЛКАРБИНОЛОВ | 2012 |
|
RU2479565C1 |
Изобретение относится к способу получения 2-фенилпиррола, который заключается в том, что ацетофеноноксим в среде ДМСО в присутствии гидроксида калия подвергают взаимодействию с ацетиленом при температуре 130-150°С, давлении ацетилена 0.4-0.8 ати в течение 4-8 ч, причем концентрация ацетофеноноксима в реакционной смеси составляет 1.32-1.85 моль/л, а гидроксид калия берется в количестве 8-16% от массы ацетофеноноксима. Способ характеризуется высокой селективностью процесса, полной конверсией исходных и высокой чистотой (99,9%) конечного продукта. Растворитель ДМСО регенерируют (до 70-80%) из реакционной смеси при помощи вакуумной дистилляции, а выделение 2-фенилпиррола осуществляют последующим разбавлением остатка водой, отделением целевого продукта фильтрованием и перекристаллизацией из петролейного эфира. 1 з.п. ф-лы.
1. Способ получения 2-фенилпиррола из ацетофеноноксима и ацетилена при давлении, близком к атмосферному, в среде ДМСО в присутствии гидроксида калия при нагревании, отличающийся высокой селективностью процесса, полной конверсией исходных и высокой чистотой (99,9%) конечного продукта, что достигается тем, что концентрация ацетофеноноксима в реакционной смеси составляет 1,32-1,85 моль/л, а гидроксид калия берется в количестве 8-16% от массы ацетофеноноксима, и процесс ведут при температуре 130-150°С, давлении ацетилена 0,4-0,8 ати в течение 4-8 ч.
2. Способ по п.1, отличающийся тем, что растворитель ДМСО регенерируют (до 70-80%) из реакционной смеси при помощи вакуумной дистилляции, а выделение 2-фенилпиррола осуществляют последующим разбавлением остатка водой, отделением целевого продукта фильтрованием и перекристаллизацией из петролейного эфира.
| Способ получения 2-или 2,3-замещенных пирролов | 1978 |
|
SU694504A1 |
| ХГС, 1975, 1425 | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| ТРОФИМОВ Б.А., МИХАЛЕВА А.И | |||
| N-Винилпирролы | |||
| - Новосибирск: Наука, 1984, с.7-13, 15-23, 39, 87-90. | |||

