Изобретение относится к усовершенствованному способу получения третичных пропаргиловых спиртов с ароматическими и гетероароматическими заместителями [алкиларил(гетарил)этинилкарбинолов] из кетонов жирноароматического и жирногетероароматического ряда. Соединения этого класса находят широкое применение в органическом синтезе, например при получении изопреноидов (включая промышленное производство изопрена) [Tedeschi R.J. in Encyclopedia of Physical Science and Technology, Vol.1, Acad. Press, Inc., San Diego, 1992, 27-65], каротиноидов [Tedeschi R.J. Acetylene-Based Chemicals from Coal and Other Natural Resources, Marcel Dekher, Inc., New York, 1982], витаминов А и E [Mercer C., Chabardes P.Pure & Appl. Chem. 1994, 66, 1509-1518], цветочных и ароматических композиций [Nowicki J. Molecules 2000, 5, 1033-1050], противоклещевых агентов, гербицидов, ингибиторов коррозии, неионогенных поверхностно-активных веществ (сурфинолов) [Tedeschi R.J. Acetylene-Based Chemicals from Coal and Other Natural Resources, Marcel Dekher, Inc., New York, 1982, Trofimov B.A. Curr. Org. Chem. 2002, 6, 1121-1162], замещенных инденов [Щукин А.О., Васильева А.В., Гриненко E.В. ЖОХ, 2007, 43, вып.5, 785-787; Щукин А.О., Васильева А.В., ЖОХ, 2010, 46, вып.1,81-97].
Известно несколько методов получения третичных ацетиленовых спиртов из алкиларил(гетарил)кетонов, в которых в качестве этинилирующего агента используют реактив Иоцича [Иоцич Ж.И. ЖРФХО, 1902, 34, 100], триметилэтинилалюминат натрия [Ju J.M., Ahn J.H., Yoon N.M. J. Org. Chem. 1996, 61, 4472-4475], ацетилениды щелочных металлов в среде жидкого аммиака [Gmitter Gr. Т., Benton F.L.J. Am. Chem. Soc., 1950, 72, 4586-4589] или тетрагидрофуране [Midland M. M. J. Org. Chem., 1975, 40, 2250-2252] при низких (-78°С) или умеренных (при использовании этилендиаминного комплекса ацетиленида лития) температурах [Beumel O.F., Harris R.F. J. Org. Chem., 1964, 29, 1872-1876]. Известен способ получения третичных ацетиленовых спиртов из кетонов и ацетилена в растворе диметилсульфоксида или этилендиамина в присутствии порошкообразного гидроксида калия [Blumental J.H. Патент США 2996552, 1961]. Однако в этом патенте приводится только один пример получения простейшего представителя алкиларилэтинилкарбинолов, а именно метилфенилэтинилкарбинола с конверсией всего 39%. Этот продукт был получен согласно описанию в растворе диметилсульфоксида в присутствии порошкового KOH 90%-ной чистоты со строго нормированной величиной зерен (100 меш), при этом целевой ацетиленовый спирт не был выделен в чистом виде, т.к. "сырой" продукт представлял собой трудно разделяемую смесь указанного спирта и побочного 2,5-дифенил-3-гексин-2,5-диола (20%). В этилендиамине указанный спирт был получен с конверсией 57% в присутствии порошкового KOH с тем же строго контролируемым размером зерен (100 меш). Очевидными недостатками этого способа в случае использования в качестве растворителя диметилсульфоксида являются его низкая эффективность (конверсия по спирту 39%), низкая селективность (20% побочного ацетиленового диола), а также необходимость использования КОН со строго контролируемым размером зерен. Вариант способа с использованием этилендиамина в качестве растворителя, не смотря на несколько большую конверсию, безусловно, не пригоден к широкому практическому применению из-за высокой токсичности этилендиамина.
До настоящего времени наиболее эффективным и наиболее близким к заявляемому изобретению остается способ (прототип) получения алкиларил(гетарил)этинилкарбинолов из ароматических кетонов и ацетилена под давлением (8-10 атм) в абсолютном диэтиловом эфире в присутствии гидроксида калия и этанола (1% от объема растворителя) при температуре 15-20°С. Выходы соответствующих этинилкарбинолов составляют ~90% [И.Н.Назаров, В.Ф.Рябченко. Изв. АН СССР, ОХН, 1956, 1370]. Принципиальными технологическими недостатками этого метода являются использование ацетилена под высоким (8-10 атм) давлением, необходимость непрерывной подачи ацетилена и кетона под давлением, проведение реакции в сухом диэтиловом эфире, что делает процесс взрыво- и пожароопасным, требует специального оборудования и особых защитных мер. Кроме того, для успешного проведения этот способ требует очень большого мольного избытка (4-6 кратного) гидроксида калия, что чрезвычайно затрудняет обработку реакционной смеси и приводит к образованию большого количества трудноутилизируемых высокощелочных отходов. При этом реакция протекает медленно: для ее завершения требуется 5-6 ч, а конверсия исходного кетона не всегда является полной. В качестве побочных продуктов в этом процессе всегда образуются ацетиленовые гликоли (до 5%).
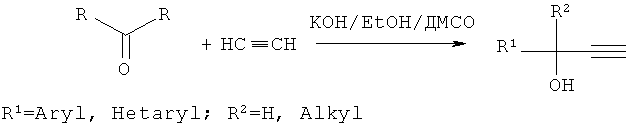
Нами предлагается новый способ получения алкиларил(гетарил)этинилкарбинолов, общей формулы:

где R1=Aryl, Hetaryl; R2=H, Alkyl, лишенный указанных недостатков.
Согласно предлагаемому способу кетоны жирноароматического или жирногетероароматического ряда реагируют с ацетиленом при атмосферном давлении в присутствии гидратированного гидроксида калия и этанола в среде диметилсульфоксида (ДМСО) при температуре 10-20°С, при этом гидроксид калия используется в количестве 0.50-2.50 моль на 1 моль кетона (предпочтительно 1 моль KOH на 1 моль кетона, что в среднем в 4-6 раз меньше по сравнению со способом-прототипом), а этанол в количестве 10% от объема растворителя (т.е. на порядок больше, чем в способе-прототипе). Процесс продолжается 1-2 ч (предпочтительно 1 ч) (в способе-прототипе 4-6 ч), выходы целевых продуктов составляют 79-93%.
Более конкретно способ реализуется следующим образом: смесь гидратированного гидроксида калия (KOH×0.5H2O), этанола и диметилсульфоксида в мольном соотношении 0.5-2.0:1.0-2.0:6.0-8.0 нагревают до 100-120°С (предпочтительно 110-115°С) при перемешивании в реакторе, представляющем собой круглодонную колбу, снабженную магнитной мешалкой и барботером, в течение 0.5-1.0 ч (предпочтительно 0.5 ч) до полной гомогенизации реакционной смеси. Затем смесь охлаждают до 10-20°С (предпочтительно 15°С) и насыщают ее ацетиленом при атмосферном давлении, пропуская последний через раствор в течение 15-60 мин (предпочтительно 30 мин). После этого в токе ацетилена прикалывают раствор кетона в ДМСО в течение 30-60 мин (предпочтительно 30 мин) и ведут реакцию в токе ацетилена еще 1-2 ч (предпочтительно 1 ч). По окончании реакции реакционную смесь разбавляют холодным (7-10°С) водным раствором хлорида аммония. Далее продукт выделяют экстракцией подходящим экстрагентом (например, дихлорметаном, диэтиловым эфиром и т.п.) и очищают известными методами (вакуумная перегонка, перекристаллизация, хроматография).
Существенная новизна предлагаемого способа по сравнению со способом-прототипом определяется следующим:
1. процесс проводится при атмосферном давлении ацетилена (вместо сжатого до 8-10 атм ацетилена в способе-прототипе);
2. отсутствием в реакционной смеси диэтилового эфира (легко воспламеняющегося и образующего с воздухом взрывчатые смеси);
3. использованием гидратированного гидроксида калия;
4. добавкой этанола к каталитической смеси в количестве 10% (вместо 1% в способе-прототипе);
5. многократным сокращением продолжительности процесса (1-2 ч по сравнению с 4-6 ч в способе-прототипе);
6. предварительной нейтрализацией реакционной смеси хлоридом аммония (в способе-прототипе применяется углекислый газ, причем нейтрализуют только эфирный экстракт).
Все эти новые отличительные признаки приводят к значительному положительному эффекту (повышению безопасности процесса, принципиальному упрощению аппаратурного оформления технологии, снижению количества агрессивных, трудно утилизируемых отходов) и в своей совокупности составляют предмет изобретения.
Возможность десятикратного снижения рабочего давления ацетилена (с 10 атм до атмосферного) в предлагаемом способе соответствует фактическому повышению скорости реакции на порядок, что однозначно связано с принципиальным изменением активности каталитической системы и физико-химических свойств среды. Это достигается за счет использования гидратированного гидроксида калия (KOH×0.5H2O) и повышенных (на порядок) концентраций этанола. В итоге в реакционной среде формируется сложная гомогенная (в отличие от гетерогенной в способе-прототипе) суперосновная каталитическая система, состоящая из этилата калия (вместо гидроксида калия в способе-прототипе), воды и диметилсульфоксида, согласно известному уравнению:
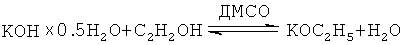
Таким образом, в данном процессе меняется не только концентрация катализатора (его полное растворение в реакционной среде), но и его природа. Тогда как в способе-прототипе следовые количества (1%) этанола и чрезвычайно высокое содержание KOH в реакционной среде, не говоря уже об отсутствии диметилсульфоксида, резко повышающего суперосновность каталитической системы (вплоть до суперосновности), не позволяют сформироваться указанной выше новой каталитической системе.
Следует особо подчеркнуть, что главным технологическим преимуществом предлагаемого способа по сравнению со способом-прототипом является использование ацетилена при атмосферном давлении. Это особенно важно при реализации данного метода в промышленных масштабах.
Дополнительным важным преимуществом заявляемого способа является замена сухого (т.е. требующего дополнительной осушки) пожаро- и взрывоопасного растворителя - диэтилового эфира - на высококипящий, пожаро- и взрывобезопасный и нетоксичный диметилсульфоксид, не требующий специальной осушки.
Заявляемый способ также позволяет существенно сократить количество используемого гидроксида калия: от 4.0-6.0 моль KOH на 1 моль кетона (в способе-прототипе) до 0.5-1.0 моль KOH на 1 моль кетона, что удешевляет процесс, упрощает стадию обработки и сокращает количество агрессивных (сильно щелочных) трудно утилизируемых отходов.
Еще одно важное преимущество заявляемого способа состоит в том, что в данном процессе практически не образуются диолы, как в прототипе (3-6%), так и в методе [Blumental J.H. Патент США 2996552, 1961] (20% и выше).
Следующие неограничивающие примеры иллюстрируют изобретение.
Пример 1
Смесь KOH×0.5H2O (10.0 г, 0.15 моль), 5 мл этанола и 50 мл ДМСО помещают в реактор, представляющий собой круглодонную колбу емкостью 100 мл, снабженную магнитной мешалкой и барботером для подачи ацетилена, и нагревают до 110°С при перемешивании в течение 30 мин до полной гомогенизации. Затем смесь охлаждают до 15°С и насыщают ацетиленом, пропуская его через реакционную смесь в течение 30 мин. После этого, не прекращая подачу ацетилена, прикалывают раствор ацетофенона (20.0 г, 0.16 моль) в 10 мл ДМСО в течение 1 ч. Далее ведут реакцию 1 ч в токе ацетилена. Реакционную смесь разбавляют холодным (7-10°С) раствором 16.5 г NH4Cl в 100 мл воды, экстрагируют диэтиловым эфиром (3×20 мл), эфирные экстракты промывают водой (3×10 мл) и сушат над MgSO4. После удаления эфира получают 24.65 г "сырого" продукта, из которого колоночной хроматографией (основный Al2O3, элюент - гексан) выделяют 20.52 г (выход 84%) 3-бутин-2-фенил-2-ола с чистотой 100%.
3-Бутин-2-фенил-2-ол. Бесцветные кристаллы. Тпл=43-47°С.ЯМР 1Н (400.13 МГц, CDCl3): δ 7.58 (м, 2Н, Но). 7.28 (м, 2Н, Hm), 7.22 (м, 1H, Нр), 2.74 (с, 1Н, ОН), 2.58 (с, 1Н, =СН), 1.70 (с, 3Н, Me). ЯМР 13С (100.61 МГц, CDCl3): δ 144.3 (Ci), 124.3 (Cm), 127.3 (Ср), 124.3 (Со), 86.7 (-С≡СН), 72.7 ((-С≡СН), 69.5 (С-ОН), 32.8 (Me). ИК (KBr): νmax 3288, 2983, 2929, 1489, 1448, 1366, 1225, 1151, 1091, 1054, 933, 766, 701, 660, 586 см-1. Элементный анализ (%): вычислено для С10Н10О: С 82.16, Н 6.89; найдено С 81.93, Н 6.56.
Пример 2
В условиях примера 1, в том же реакторе, используя такие же загрузки KOH×0.5H2O, этанола и ДМСО. Исключение составляет время насыщения ацетиленом (1 ч), и загрузка ацетофенона (10.0 г, 0.08 моль, в 10 мл ДМСО), и время реакции (1.5 ч). После описанной в примере 1 обработки получают 14.35 г "сырого" продукта, из которого перекристаллизацией из гексана выделяют 10.41 г 3-бутин-2-фенил-2-ола (выход 86%) с чистотой 100%.
Пример 3
В условиях примера 1, в том же реакторе смесь KOH×0.5H2O (2.5 г, 0.04 моль), 2.5 мл этанола и 25 мл ДМСО гомогенизируют при 115°С в течение 1 ч, насыщают ацетиленом при 15°С (0.5 ч), добавляют 5.0 г (0.33 моль) 3-метоксифенилацетофенона в 10 мл ДМСО в течение 0.5 ч. Далее ведут реакцию в токе ацетилена еще 1 ч. После вышеописанной обработки получают 5.72 г "сырого" продукта, из которого колоночной хроматографией (основной Al2O3, элюент - гексан) получают 5.26 г (выход 90%) 3-бутин-2-(3-метоксифенил)-2-ола с чистотой 100%.
3-Бутин-2-(3-метоксифенил)-2-ол. Желтое масло. ЯМР 1Н (400.13 МГц, CDCl3):δ 7.32-7.31 (м, 1Н, Hm), 7.25-7.23 (м, 1Н, Но), 7.04-7.02 (м, 1Н, Но), 6.66-6.64 (м, 1Н, Нр), 3.31 (с, 3Н, O-Ме), 2.61 (с, 1Н, ОН), 2.21 (с, 1Н, ≡СН), 1.65 (с, 3Н, Me). ЯМР 13С (100.61 МГц, CDCl3): δ 160.0 (C3aryl), 147.5 (C1aryl), 129.3 (C5aryl), 117.6 (C6aryl), 113.2 (C4aryl), 111.1 (C2aryl), 87.8 (-C≡CH), 72.7 (-С≡СН), 69.6 (С-ОН), 54.7 (O-Ме), 33.4 (Me). ИК (пленка): νmax 3419, 3290, 2987, 2936, 2837, 2113, 1675, 1600, 1486, 1452, 1363, 1289, 1152, 1078, 1043, 936, 879, 817, 786, 702, 651, 546 см-1. Элементный анализ (%): вычислено для С11Н12О (176.21): С 74.98, Н 6.86; найдено С 74.69, Н 6.70.
Пример 4
В условиях примера 4, в том же реакторе, используя такие же загрузки KOH, этанола и ДМСО, время реакции составляет 2.0 ч из 5.0 г (0.03 моль) 2-ацетилнафталина получают 5.0 г (выход 87%) 3-бутин-2-(2-нафтил)-2-ола с чистотой 100%.
3-Бутин-2-(2-нафтил)-2-ол. Желтое масло. ЯМР 1Н (400.13 МГц, CDCl3): δ 8.10 (с, 1Н, H1naphth), 7.66-7.64 (м, 1Н, H4naphth), 7.60-7.57 (м, 2Н, H5naphth, H8naphth), 7.55-7.54 (м, 1Н, H3naphth), 7.19-7.17 (м, 2Н, H6naphth, H7naphth), 2.48 (с, 1Н, ОН), 2.22 (с, 1Н, ≡СН), 1.69 (с, 3Н, Me). ЯМР 13С (100.61 МГц, CDCl3): δ 143.2 (C2naphth), 133.6-123.9 (9Cnaphth), 87.9 (-С≡СН), 73.2 (-C≡СН), 69.1 (С-ОН), 33.4 (С3-Ме). ИК (KBr): νmax 3537, 3394, 3293, 3057, 2986, 2930, 2113, 1923, 1670, 1629, 1600, 1570, 1445, 1368, 1272, 1220, 1187, 1128, 1080, 1051, 953, 934, 860, 820, 749, 669, 566, 478 cm-1. Элементный анализ (%): вычислено для С14Н12О (196.24): С 85.68, Н 6.16; найдено С 85.60, Н 6.19.
Пример 5
В условиях примера 4, в том же реакторе, используя такие же загрузки КОН, этанола и ДМСО, (время реакции составляет 1.5 ч) из 5.0 г (0.04 моль) 2-ацетилпиридина получают 5.63 г (выход 93%) 3-бутин-2-(4-пиридинил)-2-ола с чистотой 100%.
3-Бутин-2-(4-пиридинил)-2-ол. Белые кристаллы. Тпл=181-182°С. ЯМР 1Н (400.13 МГц, (CD3)2СО): δ 8.54 (д. J=5.6 Гц, 2Н, H2pyr H6pyr), 7.57 (д, J=5.6 Гц, 2Н, H3pyr, H5pyr), 5.45 (с, 1Н, ОН), 3.14 (с, 1Н, ≡СН), 1.69 (с, 3Н, Me). ЯМР 13С (100.61 МГц, (CD3)2SO): δ 155.4 (C4pyr), 150.1 (C2pyr, C6pyr). 120.5 (C3pyr, C5pyr), 88.0 (-С≡СН). 75.4 (-С≡СН), 67.7 (С-ОН), 33.3 (Me). ИК (пленка): νmax 3232, 3086, 2976, 2924, 2821, 2112, 1695, 1605, 1479, 1434, 1368, 1229, 1163, 1084, 1007, 942, 821, 709, 615, 587 см-1. Элементный анализ (%): вычислено для C9H9NO (147.17): С 73.45, Н 6.16, N 9.52; найдено С 73.91, Н 5.61, N 9.61.
Пример 6
В условиях примера 4, в том же реакторе, используя такие же загрузки катализатора и растворителя (время реакции составляет 1.0 ч), из 5.0 г (0.03 моль) пропилфенилкетона получают 5.00 г (выход 85%) 1-гексин-3-фенил-3-ола с чистотой 100%.
1-Гексин-3-фенил-3-ол. Желтое масло. ЯМР 1Н (400.13 МГц, CDCl3): δ 7.59-7.58 (м, 2Н, Но), 7.32-7.30 (м, 2Н, Hm), 7.26-7.25 (м, 1Н, Нр), 2.64 (с, 1Н, ≡СН), 2.53 (с, 1Н, ОН), 1.79-1.95 (м, 2Н, С(ОН)СН 2), 1.28-1.52 (м, 2Н, CH 2Me), 0.84-0.88 (м, 3Н, Me). ЯМР 13С (100.61 МГц, CDCl3): δ 144.2 (Ci), 128.1 (Cm), 127.7 (Cp), 125.4 (Co), 86.4 (-C≡CH), 74.0 (-С≡СН). 73.2 (С-ОН), 47.4 (C(OH)CH2), 17.9 (CH2Me), 13.9 (Me). ИК (пленка): νmax, 3410, 3303, 3062, 2961, 2873, 2113, 1954, 1888, 1812, 1680, 1600, 1489, 1449, 1379, 1317, 1203, 1139, 1110, 1030, 948, 609, 169, 700, 657, 584 см-1. Элементный анализ (%): вычислено для С12Н14О (174.239): С 85.72, Н 8.10; найдено С 82.53, Н 8.4.
Пример 7
В условиях примера 1, в том же реакторе, используя такие же загрузки катализатора и растворителя (время реакции составляет 2.0 ч), из 10.0 г (0.08 моль) 2-ацетилтиофена получают 10.02 г (выход 79%) 3-бутин-2-(2-тиенил)-2-ола (чистота 95%, примесь - исходный кетон, 5%).
3-Бутин-2-(2-тиенил)-2-ол. Желтое масло. ЯМР 1Н (400.13 МГц, C6D6): δ 7.15-7.14 (м, 1Н, H5thioph), 6.96-6.95 (м, 1Н, H4thioph), 6.76-6.75 (м, 1Н, H3thioph), 2.33 (с, 1Н, ОН), 2.27 (с, 1Н, ≡СН), 1.86 (м, 3Н, Me). ИК (пленка): νmax 3400, 3292, 2987, 2932, 1652, 1517, 1414, 1366, 1281, 1236, 1136, 1086, 1073, 1020, 972, 861, 841, 705, 659 см-1.
| название | год | авторы | номер документа |
|---|---|---|---|
| Атом-экономный безотходный способ получения 2-(4-фторфенил)-бут-3-ин-2-ола | 2016 |
|
RU2629024C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4,5,6,7-ТЕТРАГИДРОИНДОЛА | 2005 |
|
RU2297410C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ФЕНИЛПИРРОЛА | 2009 |
|
RU2397974C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛПРОПАРГИЛОВОГО СПИРТА | 2012 |
|
RU2515241C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-АЛКИЛ-2-(2-ТИЕНИЛ)ПИРРОЛОВ И ИХ N-ВИНИЛЬНЫХ ПРОИЗВОДНЫХ | 2011 |
|
RU2477725C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛСУЛЬФИДОВ | 2005 |
|
RU2284320C1 |
| ФЛУОРОФОР И СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА СОЛЕОТЛОЖЕНИЙ, СОДЕРЖАЩЕГО ФЛУОРОФОР В КАЧЕСТВЕ ФЛУОРЕСЦЕНТНОЙ МЕТКИ | 2016 |
|
RU2640339C2 |
| 3-ТРИАЗЕНОИНДОЛЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ПРОТИВ МИКОБАКТЕРИЙ | 2016 |
|
RU2724334C1 |
| Способ получения (2E,2'E)-2,2'-(1,2,4-тиадиазол-3,5-диил)бис[3-арил(гетарил)акрилонитрилов] | 2022 |
|
RU2802632C1 |
| Способ получения водорастворимых пропаргиловых эфиров полисахарида арабиногалактана | 2016 |
|
RU2650544C2 |
Изобретение относится к способу получения третичных ацетиленовых спиртов - алкиларил(гетарил)этинилкарбинолов общей формулы

где R1=Aryl, HetAryl; R2=H, Alkyl, которые используют при получении изопреноидов, каротиноидов, витаминов А и Е, цветочных и ароматических композиций, противоклещевых агентов, гербицидов, ингибиторов коррозии, неионогенных поверхностно-активных веществ (сурфинолов) и замещенных инденов. Способ заключается во взаимодействии алкиларил(гетарил)кетонов с ацетиленом при атмосферном давлении в присутствии сильного основания в растворе диметилсульфоксида. При этом процесс проводят в гомогенной каталитической системе, которую получают предварительным нагреванием гидратированного гидроксида калия, этанола и диметилсульфоксида при мольном соотношении KOH×0.5Н2О:EtOH:ДМСО, равном 0.5-2.0:1.0-2.0:6.0-8.0, при температуре 100-120°С в течение 0.5-1.0 ч, а основную реакцию проводят при температуре 10-20°С, в течение 1-2 ч. Предлагаемое изобретение позволяет повысить безопасность процесса, принципиально упростить аппаратурное оформление технологии, снизить количество агрессивных, трудно утилизируемых отходов. 1 з.п. ф-лы, 7 пр.
1. Способ получения третичных ацетиленовых спиртов алкиларил(гетарил)этинилкарбинолов общей формулы
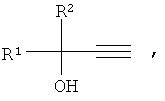
где R1 - Aryl, HetAryl; R2 - H, Alkyl, взаимодействием алкиларил(гетарил)кетонов с ацетиленом при атмосферном давлении в присутствии сильного основания в растворе диметилсульфоксида, отличающийся тем, что процесс проводят в гомогенной каталитической системе, которую получают предварительным нагреванием гидратированного гидроксида калия, этанола и диметилсульфоксида при мольном соотношении KOH·0,5Н2О:EtOH:ДМСО, равном 0,5-2,0:1,0-2,0:6,0-8,0, при температуре 100-120°С в течение 0,5-1,0 ч, а основную реакцию проводят при температуре 10-20°С в течение 1-2 ч.
2. Способ по п.1, отличающийся тем, что мольное соотношение кетон:KОН должно быть равно 1:0,5-2,0.
| Назаров Н.Н | |||
| и др | |||
| Производные ацетилена | |||
| Кулисный парораспределительный механизм | 1920 |
|
SU177A1 |
| Конденсация ароматических и жирноароматических кетонов с ацетиленом под давлением | |||
| III | |||
| Известия академии наук СССР, ОХН, 1956, №11, с.1370-1377 | |||
| Трофимов Б.А | |||
| и др | |||
| Синтез третичных ацетиленовых спиртов и их эфиров в системе КОН-ДМСО | |||
| ЖПХ, 1987, т | |||
| LX, №6, с.1366-1370 | |||
| WO 2000050370 А1, 07.11.1969 | |||
| RU 2059597 С1, 10.05.1996. | |||

