Настоящее изобретение относится к области биоорганической химии и касается фармацевтических композиций, содержащих N-ацильные производные аминокислот и их фармацевтически приемлемые соли, и их применения в медицине в качестве противоаллергических, противовоспалительных и антианафилактических средств.
Предшествующий уровень техники
Как известно в настоящее время аллергические заболевания и нарушения липидного обмена весьма распространены, вследствие плохой экологической обстановки, изменения структуры питания и образа жизни населения. Поэтому проблема создания лекарственных средств для борьбы с этими патологиями, а также с воспалительными процессами, как правило, сопровождающими аллергию, продолжает оставаться актуальной.
Наиболее распространенной группой противоаллергических препаратов являются блокаторы H1-гистаминовых рецепторов. В настоящее время выделяют 2 поколения антигистаминных препаратов (Машковский М.Д. Лекарственные средства / М.: Новая волна, 2005, с.285).
Антигистаминные препараты 1-го поколения проникают через гематоэнцефалический барьер и способны вызывать блокаду H1-рецепторов клеток центральной нервной системы, что обусловливает их нежелательный седативный эффект. Для достижения выраженного антигистаминного действия необходимы высокие концентрации этих препаратов в крови, что требует назначения их в больших дозах. Отрицательной характеристикой этих препаратов является довольно частое развитие тахифилаксии, влияние на ЦНС, проявляющееся в нарушении координации, головокружении, чувстве вялости, снижении способности концентрировать внимание. Несмотря на вышеуказанное, антигистаминные средства первого поколения по-прежнему применяются, особенно в тех ситуациях, когда необходим очень быстрый эффект от лечения, например при анафилаксии. К антигистаминным препаратам первого поколения относятся димедрол, пипольфен, перитол, супрастин, клемастин, тавегил, фенкарол.
Антигистаминные препараты 2-го поколения получили в последние годы широкое применение в аллергологической практике, поскольку не обладают побочными эффектами, присущими препаратам 1-го поколения. В частности, препараты 2-го поколения не проникают через гематоэнцефалический барьер, не имеют седативного и снотворного эффектов. Для них характерно быстрое и продолжительное антигистаминное действие. К антигистаминным средствам второго поколения относятся: кларитин (лоратадин), зиртек (цетиризин), кестин (эбастин). Однако проведенные клинические испытания выявили побочные действия и этих препаратов, обусловленные их взаимодействием с другими лекарственными средствами или нарушением их метаболизма цитохромом P 450. Таким образом, были выявлены потенциально седативные (цетиризин, лоратадин) и потенциально кардиотоксичные (терфенадин, астемизол, эбастин) эффекты у антигистаминных средств 2-го поколения.
В некоторых случаях, например, при бронхиальной астме применяют глюкокортикостероиды, оказывающие мощное антиаллергическое действие. Однако их применение сопровождается системными проявлениями в виде синдрома Иценко-Кушинга, гипертензии, гипергликемии, остеопороза и др. (Машковский М.Д. Лекарственные средства / М.: Медицина, 1993, т.1, с.565).
Особое значение в развитии аллергических заболеваний имеет патохимическая стадия аллергических реакций, которая в значительной степени определяется степенью активации клеток-мишеней аллергии 1-го порядка (базофилов и тучных клеток). Их важной особенностью является способность к накоплению и высвобождению под действием стимула (аллергена) биологически активных соединений, в первую очередь, гистамина. При IgE- и/или IgG-опосредованном ответе на антиген именно эти клетки определяют степень выраженности клинической картины немедленной аллергии (Паркер Ч.В./ Медиаторы: высвобождение и функции. // В книге: Иммунология. Под редакцией У. Пола., М.: Мир, 1989, т.3, с.170-247; Chakravarty N.K. // In: The mast cell: Its role in health and disease. ed. J Pepys, 1979, p. 38-46).
Существует группа препаратов (кромолин-натрия, кетотифен, оксатомид), применяемых при бронхиальной астме и бронхоспастических состояниях, в основе действия которых лежит способность тормозить дегрануляцию тучных клеток и задерживать высвобождение из них медиаторных веществ, способствующих развитию бронхоспазма, аллергии и воспаления (брадикинина, гистамина). В качестве побочных эффектов могут наблюдаться раздражение слизистых оболочек, головная боль, отек гортани, кашель, удушье (Машковский М.Д. Лекарственные средства / М.: Новая волна, 2005, с.297].
В связи с указанным выше, актуальным является поиск новых эффективных противоаллергических и антианафилактических средств, с альтернативными механизмами действия, способных проявлять активность в низких концентрациях и лишенных побочных эффектов. В этом отношении особый интерес представляют соединения, включающие остатки веществ природного происхождения, так как для них можно прогнозировать более низкую токсичность и частоту побочных эффектов.
В публикации международной заявки WO 99/01103 описано противоаллергическое и гиполипидемическое действие N-ацильных производных биогенных аминов, например, γ-глутамилгистамина, и его ближайшего аналога глутарилгистамина, которые наиболее близки по структуре и действию к заявляемым соединениям. В статье Кржечковская В.В., Желтухина Г.А., Небольсин В.Е. и др., Изучение антианафилактической активности и механизмов действия γ-L-глутамилгистамина // Патогенез, 2003, Т.1, №2, с. 60-64, показано, что γ-глутамилгистамин обладает выраженной антианафилактической активностью при использовании различных видов животных и способов введения. Полученные результаты свидетельствуют о том, что в тучных клетках животных под действием γ-глутамилгистамина достоверно снижается содержание гистамина и его антиген-стимулированная секреция. В тесте по исследованию влияния глутарилгистамина на выраженность антиген-индуцированного бронхоспазма было показано снижение величины бронхоспазма более чем на 50% по сравнению с контролем. Данный эффект проявлялся как при пероральном, так и интратрахеальном способе его введения в низкой дозе - 50 мкг/кг. Глутарилгистамин обладает способностью снижать проявления пассивной кожной анафилаксии от 34 до 42% и, таким образом, превосходит по эффективности супрастин, но уступает кларитину. В WO 99/01103 показано, что при введении животным глутарилгистамина в дозах 50 и 500 мкг/кг было продемонстрировано достоверное снижение интенсивности гиперчувствительности замедленного типа. Кроме того, глутарилгистамин в дозах 50 и 500 мкг/кг обладал также некоторой антихолестеринмимической активностью, снижая содержание общего холестерина по сравнению с животными с атерогенной нагрузкой на 5-7%.
Недостатком глутарилгистамина является его сравнительно высокая стоимость и малая доступность исходного сырья для его получения - гистамина. Кроме того, указанное вещество недостаточно эффективно в вышеперечисленных тестах.
С целью расширения арсенала технических средств и создания более эффективного и доступного противоаллергического, противовоспалительного и антианафилактического средства, авторами изобретения были выявлены некоторые специфические N-ацильные производные аминокислот общей формулы (I), раскрытой в публикации международной заявки WO 99/01103, но конкретно в ней не описанные, не полученные и не охарактеризованные.
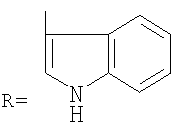
Так, под общую формулу (I) вышеуказанной международной заявки подпадают соединения настоящего изобретения Nα-сукцинил-L-триптофан (II), Nα-глутарил-L-триптофан (III), Nα-глутарил-L-гистидин (IV) и Nα-сукцинил-L-гистидин (V). Однако в указанной публикации не приведены ни конкретные структурные формулы данных соединений, ни какие-либо физико-химические характеристики, а также не описаны способы их получения. В публикации международной заявки WO 03/072124 приведены формулы Nα-глутарил-L-триптофана (II), Nα-глутарил-L-гистидина (IV) и Nα-сукцинил-L-гистидина (V), но не описан способ их синтеза и не приведены их физико-химические константы.
Одно из соединений глутарилгистидин упоминается только в виде метилового эфира по С-концу His [Glt-His(OMe)] в патенте США 3963691, в качестве промежуточного соединения в синтезе пептида Glt-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-poly-Lys.
Соединение сукцинилгистидин упоминается в публикации международной заявки WO 93/04690. В этой публикации указано, что добавление свободного имидазола или сукцинилгистидина ускоряет взаимодействие карнозина с дигидроксиацетоном. Методики синтеза сукцинилгистидина и его физико-химические константы не приведены.
Структурная формула сукцинилтриптофана упоминается в заявке США 2005079515. В указанной публикации ни методики синтеза сукцинилтриптофана, ни его физико-химические константы не приведены.
Получение Nα-глутарил-L-гистамина описано в публикации международной заявки WO 99/01103 и представляет собой N-ацилирование биогенного амина глутаровым ангидридом в среде безводного N,N-диметилформамида. Кроме того, в публикации Гершкович А.А., Киберев В.К.// Химический синтез пептидов/ Киев: Наукова думка, 1992, 360, описан способ ацилирования аминокислот в водно-органической, сильно щелочной среде.
В Sorm F., Pravda Z., Proteins and amino acids. X. Synthesis of two peptide analogs. // Chemicke Listy pro Vedu a Prumysl., 1951, V.45, p.423-425, описан способ синтеза сукцинилтирозина этилового эфира в смеси воды и этилацетата при соотношении (1:1), исходя из хлоргидрата этилового эфира тирозина и глутарового ангидрида в присутствии NaHCO3 для поддержания слабощелочного pH.
Однако ацилирование ангидридами дикарбоновых кислот свободного гистидина в литературе не описано.
Целью настоящего изобретения являются фармацевтические композиции, содержащие новые эффективные N-ацильные производные аминокислот, обладающие противоаллергическим и антианафилактическим действием в низких дозах и не проявляющие побочных эффектов.
Краткое описание изобретения
Настоящее изобретение относится к применению соединений общей формулы I
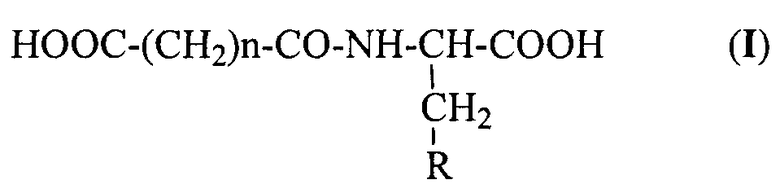
где n равно 2 или 3; и
R представляет
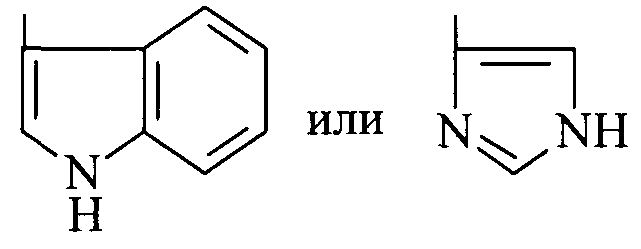
и их фармацевтически приемлемых солей в качестве противоаллергических, противовоспалительных и антианафилактических средств.
Далее, настоящее изобретение относится к фармацевтической композиции и средству, обладающим противоаллергическим, антианафилактическим и противовоспалительным действием, содержащим эффективное количество соединения общей формулы I или его фармацевтически приемлемой соли, а также, если требуется фармацевтически приемлемый носитель.
Еще одним объектом изобретения является способ лечения аллергических заболеваний, включающих бронхиальную астму, аллергический ринит, поллинозы, сезонный ринит, круглогодичный ринит, атопический дерматит, псориаз, крапивницу, аллергические (в том числе анафилактические) реакции на укусы насекомых и лекарственные препараты, холодовую аллергию, аллергический конъюктивит, хронических обструктивных заболеваний легких, а именно хронического обструктивного бронхита, эмфиземы, облитерирующего бронхита, муковисцидоза, включающий введение эффективного количества соединения общей формулы I или его фармацевтически приемлемой соли.
Детальное описание изобретения
Предпочтительные соединения общей формулы I представлены в таблице 1.
 (Ind)
(Ind) (Im)
(Im)
Синтез соединений общей формулы I может быть осуществлен двумя способами. Первый способ заключается в постепенном добавлении к водному раствору аминокислоты общей формулы
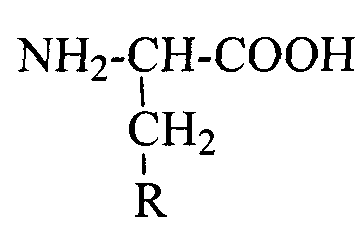
или ее соли, где
R представляет
глутарового или янтарного ангидрида в виде твердого вещества с последующим выделением целевого продукта ионообменной хроматографией, предпочтительно пропусканием реакционной смеси через колонку с катионитом и последующей кристаллизацией из водного раствора. Полученные кристаллы целевого продукта промывают подходящим растворителем, предпочтительно метанолом. Главное преимущество заявляемого способа состоит в отсутствии щелочи в водном растворе аминокислоты, что препятствует инактивации ангидрида дикарбоновой кислоты в результате гидролиза. Кроме того, остаток имидазола в составе молекулы аминокислоты может осуществлять кислотно-основный аутокатализ реакции ацилирования аминогруппы аминокислоты. Достаточно высокие выходы (55-60%) при использовании заявляемого способа достигаются, в частности, благодаря постепенному прибавлению ангидрида дикарбоновой кислоты, взятого в избытке, и интенсивному перемешиванию реакционной массы.
Соединения общей формулы I также могут быть получены альтернативным способом в двухфазной системе, включающим добавление ангидрида глутаровой или янтарной кислоты в несмешивающемся с водой органическом растворителе к водному раствору аминокислоты общей формулы:
или ее соли, где
R представляет
.
Данный способ позволяет использовать избыток ацилирующего агента, достичь полного ацилирования α-аминогруппы аминокислоты и выхода целевого продукта около 70%. Для поддержания необходимого pH вместо неорганической щелочи используют органическое основание - пиридин, который не гидролизует ангидрид, и, кроме того, как известно, является катализатором ацилирования. Использование пиридина позволяет избежать загрязнения конечного продукта неорганическими солями, которые вместе с продуктом реакции остаются в водном слое. Использованные подходы позволяют упростить отделение целевого продукта от непрореагировавших ангидрида и соответствующей аминокислоты и выделять целевой продукт простой кристаллизацией.
Предпочтительными несмешивающимися с водой органическими растворителями являются бутанол, этилацетат, хлороформ.
Предпочтительными растворителями, используемыми для кристаллизации целевого продукта, являются водноспиртовые смеси, в частности вода-этанол.
Соединения общей формулы I могут быть также получены в виде фармацевтически приемлемых солей с гидроксидом натрия, гидроксидом калия, карбонатом магния, гидроксидом лития, карбонатом кальция рутинными способами, широко описанными в литературе.
Соединения общей формулы I обладают противоаллергической, противовоспалительной и антианафилактической активностью и могут быть использованы для лечения аллергических, анафилактических, в том числе сопровождающихся воспалением заболеваний.
В частности, соединения настоящего изобретения могут быть использованы для лечения следующих аллергических заболеваний: бронхиальной астмы, аллергического ринита, поллинозов, сезонного ринита, круглогодичного ринита, атопического дерматита, псориаза, крапивницы, аллергических (в том числе анафилактических) реакций на укусы насекомых и лекарственные препараты, холодовой аллергии, аллергического конъюктивита, хронических обструктивных заболеваний легких, а именно хронического обструктивного бронхита, эмфиземы, облитерирующего бронхита, муковисцидоза.
Соединения настоящего изобретения вводятся в эффективном количестве, которое обеспечивает желаемый терапевтический результат.
Для лечения аллергических заболеваний, включающих бронхиальную астму, аллергический ринит, поллинозы, сезонный ринит, круглогодичный ринит, атопический дерматит, псориаз, крапивницу, аллергические (в том числе анафилактические) реакции на укусы насекомых и лекарственные препараты, холодовую аллергию, аллергический конъюктивит, хронические обструктивные заболевания легких, а именно хронический обструктивный бронхит, эмфизему, облитерирующий бронхит, муковисцидоз, соединения общей формулы I могут быть введены перорально, местно, парентерально, интраназально, ингаляционно и ректально в виде стандартных лекарственных форм, содержащих нетоксичные фармацевтически приемлемые носители. Используемый в настоящем описании термин «парентеральное введение» означает подкожные, внутривенные, внутримышечные или внутригрудные инъекции или вливания.
Соединения настоящего изобретения могут быть введены пациенту в дозах, составляющих от 0,01 до 10 мг/кг веса тела в день, предпочтительно в дозах от 0,05 до 5 мг/кг один или более раз в день.
При этом следует отметить, что конкретная доза для каждого конкретного пациента будет зависеть от многих факторов, включая активность данного используемого соединения, возраст, вес тела, пол, общее состояние здоровья, и режим питания пациента, время и способ введения лекарственного средства, скорость его выведения из организма, конкретно используемую комбинацию лекарственных средств, а также тяжесть заболевания у данного индивида, подвергаемого лечению.
Фармацевтические композиции по настоящему изобретению содержат соединение общей формулы (I) в количестве, эффективном для достижения желаемого результата, и могут быть введены в виде стандартных лекарственных форм (например, в твердой, полутвердой или жидкой формах), содержащих соединения настоящего изобретения в качестве активного ингредиента в смеси с носителем или наполнителем, пригодным для внутримышечного, внутривенного, перорального, сублингвального, ингаляционного, интраназального и интраректального введения. Активный ингредиент может быть включен в композицию вместе с обычно используемыми нетоксичными фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, драже, суппозиториев, эмульсий, суспензий, мазей, гелей и любых других лекарственных форм.
В качестве наполнителей могут быть использованы различные вещества, такие как сахариды, например глюкоза, лактоза или сахароза, манит или сорбит, производные целлюлозы и/или фосфаты кальция, например трикальций фосфат или кислый фосфат кальция, в качестве связующего компонента могут быть использованы такие, как крахмальная паста, например кукурузный, пшеничный, рисовый, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза и/или поливинилпирролидон. При необходимости могут быть использованы разрыхляющие агенты, такие как вышеупомянутые крахмалы и карбоксиметилкрахмал, поперечно сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия.
Могут быть использованы необязательные добавки, такие как агенты, регулирующие текучесть, и смазывающие агенты, такие как диоксид кремния, тальк, стеариновая кислота и ее соли, такие как стеарат магния или стеарат кальция, и/или пропиленгликоль.
Ядро драже обычно покрывают слоем, который устойчив к действию желудочного сока. Для этой цели могут быть использованы концентрированные растворы сахаридов, которые могут необязательно содержать аравийскую камедь, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, и подходящие органические растворители или их смеси.
В качестве добавок могут быть также использованы стабилизаторы, загустители, красители и отдушки.
В качестве мазевой основы могут быть использованы углеводородные мазевые основы, такие как вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции, такие как твердый парафин и воск; абсорбтивные мазевые основы, такие как гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens); мазевые основы, смываемые водой, такие как гидрофильная мазь (Unguentum hydrophylum); водорастворимые мазевые основы, такие как полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие.
В качестве основы для гелей могут быть использованы метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, оксипропилцеллюлоза, полиэтиленгликоль или полиэтиленоксид, карбопол.
В качестве основы для суппозитория могут быть использованы основы, нерастворимые в воде, такие как масло какао; основы, растворимые в воде или смешиваемые с водой, такие как желатиноглицериновые или полиэтиленоксидные; комбинированные основы - мыльно-глицериновые.
При приготовлении стандартной лекарственной формы количество активного ингредиента, используемого в комбинации с носителем, может варьироваться в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства.
Так, например, при использовании соединений настоящего изобретения в виде растворов для инъекций, содержание активного агента в них составляет 0,01-5%. В качестве разбавителей могут быть использованы 0,9% раствор хлорида натрия, дистиллированная вода, раствор новокаина для инъекций, раствор Рингера, раствор глюкозы, специфические добавки для растворения. При введении в организм соединений настоящего изобретения в виде таблеток и суппозиториев их количество составляет 5,0-500 мг на стандартную лекарственную форму.
Лекарственные формы настоящего изобретения получают по стандартным методикам, таким как, например, процессы смешивания, гранулирования, формирование драже, растворение и лиофилизация.
Следует отметить, что соединения настоящего изобретения проявляют биологическую активность в дозах на два-три порядка ниже по сравнению с известными препаратами, использованными для сравнения, при практически одинаковой эффективности, и для них не выявлено отрицательных побочных действий и не обнаружено противопоказаний к применению. При этом при исследовании токсичности соединений настоящего изобретения в дозе 3000 мг/кг, перорально, не зарегистрировали гибели экспериментальных животных.
Детальное описание соединений настоящего изобретения, их получения и исследования фармакологической активности представлено в нижеследующих примерах, предназначенных для иллюстрации предпочтительных вариантов изобретения и не ограничивающих его объем.
Примеры синтеза N-ацильных производных аминокислот общей формулы (I)
Индивидуальность полученных соединений проверялась методом ТСХ на пластинках “Kieselgel 60 F254” “Merck” (Германия) в системах: метанол (1), хлороформ-метанол-аммиак (4:3:1) (2).
Хроматограммы проявляли хлортолидиновым реактивом, нингидрином, йодом по свечению в УФ-свете.
Углы оптического вращения измеряли на поляриметре “Perkin Elmer 341” (Швеция).
1Н-ЯМР регистрировали на приборе “AMX-400 Bruker” (Германия).
Температуру плавления определяли на приборе “Boetius” (Германия).
Аналитическую ВЭЖХ поводили на приборе “System Gold” (“Beckman”, США): скорость элюции 0,25 мл/мин, детекция при 214 нм в условиях: колонка Ultrasphere ODS “Beckman”, 2×250 мм, 5 мкм, элюция 0,1%-ной TFA, скорость элюирования 0,25 мл/мин (1); скорость элюции 1 мл/мин, детекция при 220 нм, колонка Luna-5 “Phenomenex”, C18, 250×4,6 мм, элюция 25% ацетонитрила в 0,05 М фосфатном буфере (pH 3,0) (2).
Пример 1
N α -Глутарил-L-гистидин (IV)
Методика А
К раствору 103,4 г (0,67 моль) гистидина в 400 мл воды добавляют 83,7 г (0,73 моль) глутарового ангидрида. Суспензию перемешивают 1 час, образующийся раствор упаривают до объема 150 мл, оставляют в холодильнике на 16 часов. Выпавший осадок отфильтровывают, промывают 150 мл метанола и сушат. Очистку проводят ионообменной хроматографией на смоле Пьюролайт в Н+ форме, элюируя водой. Фракции, содержащие целевой продукт, объединяют, упаривают до начала выпадения осадка и оставляют на 16 часов при +4°C. Выпавший осадок отфильтровывают, промывают 200 мл метанола и сушат до постоянного веса. Выход 98,8 г (55%). Rf 0,55 (1), 0,37 (2). Тпл = 222-224°С. [α]D 20+15, 95° (C 0,53, вода). [М+Н]+ 270,1. 1H-ЯМР спектр (D2O), δ, м.д.: 1,60-1,80 (м, 2Н, β-CH2-Glt), 2,10-2,25 (м, 4Н, α,γ-CH2-Glt), 2,90-3,25 (м, 2Н, β-CH2-His), 4,40-4,50 (м, 1Н, α-CH-His), 7,15 (с, 1Н, CH-4-Im), 8,50 (с, 1Н, CH-2-Im). Найдено, %: C 49,18; H 5,91; N 15,42. C11H15N3O5. Вычислено, %: C 49,07; H 5,62; N 15,61.
Методика Б
К суспензии 0,3 г (1,93 ммоль) гистидина в 5 мл воды при интенсивном перемешивании добавляют 0,44 г (3,86 ммоль) глутарового ангидрида, растворенного в 2,5 мл этилацетата. Перемешивают 2 часа, пиридином доводят рН до 7 и перемешивают еще 1 час. Этилацетатный и водный слой разделяют. Водный слой дважды промывают эфиром, эфирный слой отбрасывают. Воду удаляют в вакууме, остаток растворяют в минимальном количестве воды и добавляют этанол до начала выпадения белого осадка, оставляют при +4°C на 20 ч. Осадок отделяют фильтрованием, сушат в вакууме. Выход 0,36 г (70%). Rf 0,56 (1), 0,35 (2). Тпл = 219-221°С. [α]D 20 = +15,71° (C 0,56, вода). [М+Н]+ 270,1. 1H-ЯМР спектр (D2O), δ, м.д.: 1,40-1,55 (м, 2Н, β-CH2-Glt), 1,90-2,0 (м, 4H, α, γ-CH2-Glt), 2,7-3,0 (м, 2H, β-CH2-His), 4,20-4,30 (м, 1H, α-CH-His), 6,95 (c, 1H, 4-CH-Im), 8,30 (c, 1H, 2-CH-Im). ВЭЖХ в условиях: (1) - индивидуальный пик, время удерживания 14,55 мин. Найдено, %: C 49,07; H 5,65; N 15,65. C11H15N3O5. Вычислено, %: C 49,07; H 5,62; N 15,61.
Пример 2
N α -Сукцинил-L-гистидин (V)
Синтез проводили в соответствии с методикой А, приведенной для соединения IV.
Выход 0,08 г (57%).
Rf 0,44 (1), 0,25 (2).
Тпл = 179-181°С.
[α]D 20 = +30,71° (C 0,56, вода).
[М]+ 255,2.
1H-ЯМР спектр (D2O), δ, м.д.: 2,15-2,30 (м, 4Н, (CH2)2-Suc), 2,75-2,95 (м, 2H, β-CH2-His), 4,25 (уш.с, 1H, α-CH-His), 6,95 (c, 1H, 4-CH-Jm), 8,25 (c, 1H, 2-CH-Jm).
Найдено %: C 47,09; H 5,04; N 16,40. C10H13N3O5. Вычислено %: C 47,06; H 5,13; N 16,46.
Синтез проводили в соответствии с методикой Б, приведенной для соединения IV.
Выход 0,101 г (67%).
Rf 0,45 (1), 0,27 (2).
Тпл = 178-180°С.
[α]D 20 = +30,8° (C 0,57, вода).
ВЭЖХ в условиях (1) - индивидуальный пик, время удерживания 7,54 мин.
Найдено %: C 47,15; H 5,2; N 16,50. C10H13N3O5. Вычислено %: C 47,06; H 5,13; N 16,46.
Пример 3
N α -Глутарилтриптофан (III)
К суспензии 1,0 г (4,9 ммоль) триптофана в 7 мл воды прибавляют по каплям раствор 1 N NaOH (4,9 ммоль). К полученному раствору добавляют раствор 0,56 г (4,9 мммоль) глутарового ангидрида в 3 мл этилацетата. Реакционную смесь перемешивают в течение 3 часов при комнатной температуре в атмосфере аргона в темноте, оставляют на 16 часов при +4°. Растворитель из реакционной смеси удаляют в вакууме. Полученный маслообразный остаток растворяют в 30 мл воды при перемешивании, охлаждают до 0°, добавляют раствор 1 N HCl до pH 4. Продукт экстрагируют этилацетатом (3×25 мл). Объединенный этилацетатный экстракт охлаждают до 0°, промывают водой (4×25 мл) до pH 7, раствором 5% HCl (5 мл), водой (4×25 мл) до p H7, сушат над безводным Na2SO4 в течение 1 часа. Осадок Na2SO4 отфильтровывают, промывают этилацетатом, растворитель удаляют в вакууме. Получают сероватый твердый остаток, который сушат в вакууме.
Выход 1,0 г (70%).
Rf 0,54 (1).
Тпл = 150-152°С.
[α]D 20 = +8,20° (C 0,5, метанол).
1H-ЯМР спектр (CD3OD), δ, м.д.: 1,75-1,84 (м, 2Н, β-CH2-Glt), 2,15-2,30 (м, 4Н, α,γ-CH2-Glt), 3,30-3,40 (м, 2Н, β-CH2-Trp), 3,80-3,90 (м, 1Н, α-CH-Trp), 6,97 (т, J=7 Гц, 1H, CH-6-Ind), 7,06 (т, J=7 Гц, 1Н, CH-7-Ind), 7,15 (д, J=7 Гц, 1H, CH-2-Ind), 7,33 (д, J=7 Гц, 1H, CH-5-Ind), 7,55 (д, J=7 Гц, 1Н, CH-8-Ind).
ВЭЖХ в условиях: (2) - индивидуальный пик, время удерживания 6,77 мин.
Найдено, %: С 60,07; H 5,65; N 8,75. C16H18N2O5 Вычислено, %: С 60,37; H 5,7; N 8,8.
Пример 4
N α -Сукцинил-L-триптофан (II)
Синтез проводили в соответствии с методикой, приведенной для соединения III.
Выход 100,5 мг (67%).
Rf 0,63 (1).
[α]D 20 = +21,05° (C 0,6, вода).
1H-ЯМР спектр (DMSO-d6), δ, м.д.: 2,33-2,41 (м, 4Н, α,β-CH2-Suc), 2,93-3,01 (м, 1Н, β-CH2-Trp), 3,10-3,16 (м, 1Н, β-CH2-Trp), 4,39-4,47 (м, 1Н, α-CH-Trp), 6,93-7,06 (м, 2H, CH-6,7-Ind), 7,11 (д, J 2,2 Гц, 1H, CH-2-Ind), 7,30-7,32 (м, 1H, CH-5-Ind), 7,44-7,47 (м, 1Н, CH-8-Ind). [М]+ 304,3.
ВЭЖХ в условиях: (2) - индивидуальный пик, время удерживания 6,35 мин.
Найдено, %: С 59,07; H 5,65; N 9,35. C15H16N2O5 Вычислено, %: С 59,21; H 5,3; N 9,21.
Пример 5
Мононатриевая соль N α -глутарил-L-гистидина (IV)
К раствору 1,0 г (3,7 ммоль) Nα-глутарил-L-гистидина в 15 мл воды при перемешивании и охлаждении до +5°С прибавляют раствор 0,15 г (3,7 ммоль) NaOH в 20 мл воды. Раствор перемешивают 30 мин, растворитель удаляют в вакууме. К маслообразному остатку порциями добавляют бензол, растворитель удаляют в вакууме. Твердый остаток сушат над гранулированной щелочью.
Выход 1,07 г (99,7%).
Тпл = 208-210°С.
[α]D 20 = +16,27° (C 0,58, вода).
Найдено, %: C 45,25; H 5,51; N 14,52. C11H15N3O5Na. Вычислено, %: C 45,21; H 5,17; N 14,38.
Пример 6
Мононатриевая соль N α -сукцинил-L-гистидина (V)
Синтез проводили в соответствии с методикой, приведенной для мононатриевой соли Nα-глутарил-L-гистидина (IV) (пример 5).
Выход 1,06 г (97,0%).
[α]D 20 = +40,21° (C 0,48, вода).
Найдено, %: C 43,25; H 4,51; N 15,52. C10H13N3O5Na. Вычислено, %: C 43,17; H 4,71; N 15,10.
Пример 7
Мононатриевая соль N α -сукцинил-L-триптофана (II)
Синтез проводили в соответствии с методикой, приведенной для мононатриевой соли Nα-глутарил-L-гистидина (IV) (пример 5).
Выход 0,21 г (98,0%).
Тпл = 147-150°С.
[α]D 20 = +22,02° (C 0,39, вода).
Найдено, %: C 55,25; H 4,51; N 8,32. C15H16N2O5Na. Вычислено, %: C 55,05; H 4,93; N 8,56.
ВЭЖХ в условиях: (2) - индивидуальный пик, время удерживания 6,56 мин.
Пример 8
Мононатриевая соль N α -глутарил-L-триптофана (III)
Синтез проводили в соответствии с методикой, приведенной для мононатриевой соли Nα-глутарил-L-гистидина (IV) (пример 5).
Выход 0,11 г (99,0 %).
Тпл = 128-130°С.
[α]D 20 = +22,06° (C 0,34, метанол).
Найдено, %: C 56,15; H 5,21; N 8,22. C16H18N2O5Na. Вычислено, %: C 56,30; H 5,32; N 8,21.
ВЭЖХ в условиях: (2) - индивидуальный пик, время удерживания 6,96 мин.
Пример 9
Динатриевая соль N α -глутарил-L-гистидина (IV)
К раствору 1,0 г (3,7 ммоль) Nα-глутарил-L-гистидина в 15 мл воды при перемешивании и охлаждении до +5°С прибавляют раствор 0,3 г (7,44 ммоль) NaOH в 15 мл воды. Раствор перемешивают 30 мин, растворитель удаляют в вакууме. К маслообразному остатку порциями добавляют бензол, растворитель удаляют в вакууме. Твердый остаток сушат над гранулированной щелочью.
Выход 1,15 г (99,0%).
[α]D 20 = +11,92° (C 0,57, вода).
Найдено, %: C 41,25; H 4,51; N 13,52. C11H15N3O5Na2. Вычислено, %: C 41,91; H 4,80; N 13,3.
Пример 10
Динатриевая соль N α -сукцинил-L-гистидина (V)
Синтез проводили в соответствии с методикой, приведенной для динатриевой соли Nα-глутарил-L-гистидина (IV) (пример 9).
Выход 1,16 г (99,0%).
Тпл = 124-128°С.
[α]D 20 = +20,06° (C 0,67, вода).
Найдено, %: C 39,55; H 4,31; N 13,52. C10H13N3O5Na2. Вычислено, %: C 39,88; H 4,35; N 13,95.
Пример 11
Динатриевая соль N α -сукцинил-L-триптофана (II)
Синтез проводили в соответствии с методикой, приведенной для динатриевой соли Nα-глутарил-L-гистидина (IV) (пример 9).
Выход 0,56 г (97,7%).
Найдено, %: C 51,35; H 4,31; N 8,22. C15H16N2O5Na2. Вычислено, %: C 51,43; H 4,60; N 8,0.
Пример 12
Динатриевая соль N α -глутарил-L-триптофана (III)
Синтез проводили в соответствии с методикой, приведенной для динатриевой соли Nα-глутарил-L-гистидина (IV) (пример 9).
Выход 0,56 г (98,5%).
Найдено, %: C 52,55; H 4,71; N 7,52. C16H18N2O5Na2. Вычислено, %: C 52,75; H 4,98; N 7,69.
Тесты на биологическую активность
Пример 13
Влияние соединений общей формулы I на аллергические реакции немедленного типа (тест индуцированной овальбумином (ОА) дегрануляции базофилов крови иммунизированной морской свинки in vitro)
Выделение лейкоцитов из крови морской свинки осуществляли по методу Фримеля (Иммунологические методы/Под ред. Г.Фримеля. М., Медицина,1987, стр.222 в нашей модификации).
Для постановки теста использовали морских свинок обоего пола массой 600-800 г. Животных иммунизировали однократно смесью овальбумина 10 мкг и 100 мг гидроокиси алюминия на животное по Andersson (Anderson P. Antigen-induced bronchial anaphulaxis in actively sensitized geinea-pigs. // Allergy, 1980, Vol.35, p. 63-71).
Под эфирным наркозом из сердца морской свинки отбирали 15 мл крови. Для выделения базофилов в составе лейкоцитарной взвеси использовали двойное осаждение клеток - посредством ЭДТА и с помощью цитратсодержащей осаждающей жидкости.
Кровь смешивали с 5% раствором ЭДТА·Na2 2H2O (“Sigma”) в соотношении 9:1 и через 30 мин мягко центрифугировали (12 мин при 80 g). Надосадочную жидкость собирали и центрифугировали 15 мин при 500 g.
К оставшимся клеткам крови добавляли цитратсодержащую осаждающую жидкость (3) в пропорции 3:10 (термостатируется при 37°С в течение 30 мин) Обогащенную лейкоцитами надосадочную фракцию центрифугировали 7 мин при 100 g. К осадку лейкоцитов добавляли 0,85% раствор NaCl и концентрацию клеток доводили до 30×103/мкл.
Постановка теста дегрануляции базофилов in vitro (Справочник по клиническим лабораторным методам исследования/Под ред. Е.А.Кост. М., Медицина, 1975, стр. 130).
Для постановки теста в центрифужную пробирку (используется по 3 пробирки на каждую пробу) помещали 300 мкл клеточной взвеси, затем добавляли 300 мкл солевого раствора исследуемого соединения (или солевого раствора в контроле спонтанной и максимальной дегрануляции) и преинкубировали при 37°С в течение 15 мин, затем добавляли по 300 мкл 1% солевого раствора ОА в каждую пробирку (в контроль спонтанной дегрануляции добавляли солевой раствор в таком же количестве) и еще раз преинкубировали при 37°С в течение 10 мин. Рабочая концентрация лейкоцитов составляет при этом 104/мкл. Из каждой пробирки отбирали пробы (100 мкл) в отдельные пробирки для оценки полной дегрануляции базофилов, а к оставшимся клеткам добавляли охлажденный солевой раствор (по 5 мл в каждую пробирку) для остановки реакции дегрануляции, затем центрифугировали 7 мин при 100 g, а из осадка готовили препараты для микроскопирования. Фиксацию и окраску препаратов проводили по методу Seder et al. (Seder R.A. et al. Mouse splenic and bone marrow cell populations that express high-affinity Fcε receptors and produce interleukin-4 are highly enriched in basophils. // Proc. Natl. Acad. USA, 1991, V.88, p.2835-2839).
Для выявления специфической зернистости базофилов использовали краситель 0,5% альциановый синий (рН 1,0), ядра докрашивали сафранином (0,1% раствор в 1% уксусной кислоте). Препараты использовали для оценки суммарного торможения дегрануляции.
Торможение суммарной дегрануляции (ТГ) (%) рассчитывали по формуле:
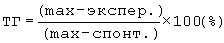 , где
, где
mах - % дегранулированных базофилов при максимальной дегрануляции (ОА),
спонт. - % дегранулированных базофилов при спонтанной дегрануляции (контроль),
экспер. - % дегранулированных базофилов после воздействия исследуемого соединения.
Оценка полной дегрануляции базофилов
Отобранные после постановки теста дегрануляции базофилов пробы (по 100 мкл) помещали в пробирки с красителем (0,5% альциановый синий, рН 1,0) в соотношении 1:1. Окрашивание проводили при комнатной температуре не менее 50 мин. Подсчет количества окрашенных базофилов проводили с использованием камеры Фукса-Розенталя. Торможение полной дегрануляции (ТПД) базофилов рассчитывали по формуле:
ТПД(%) = 1-[(М ср (к) - М ср (эксп.)]/[М ср (к) - М ср (ОА)] ×100, где
М ср (к) - среднее (по 3-м пробам) количество базофилов в тесте спонтанной дегрануляции;
М ср (ОА) - среднее (по 3-м пробам) количество базофилов в тесте максимальной антиген-индуцированной дегрануляции;
М ср (эксп) - среднее (по 3-м пробам) количество базофилов в тесте дегрануляции после инкубации с исследуемым соединением.
Торможение ОА-индуцированной дегрануляции базофилов крови иммунизированных морских свинок in vitro под воздействием соединений общей формулы I
(max дегрануляция)
(10-3 М)
(10-4 М)
(10-5 М)
(10-6 М)
(10-7 М)
(10-3 М)
(10-4 М)
(10-5 М)
(max дегрануляция)
(10-3 М)
(10-4 М)
(10-5 М)
(10-3 М)
(10-4 М)
(10-5 М)
Данные таблицы 2 показывают, что по сравнению с глутарилгистамином соединение (IV) оказывает выраженное антианафилактическое действие, проявляющееся практически 100% торможением дегрануляции в тесте полной ОА-индуцированной дегрануляции базофилов крови активно иммунизированных морских свинок (реакция анафилаксии in vitro в безкальциевой среде). Значительный антианафилактический эффект соединения IV проявляется и в уменьшении количества дегранулированных клеток, особенно выраженном в концентрации 10-5 М (отсутствие дегранулированных клеток).
Пример 14
Изучение влияния соединений общей формулы I на системную анафилаксию in vivo
Использовали модель бронхоспазма у ненаркотизированных активно сенсибилизированных морских свинок с аэрозольным воздействием овальбумина в качестве антигена [Ковалева В.Л. Методические указания по изучению бронхолитических, муколитических и противовоспалительных средств // Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ, М., 2000, с. 242-250).
Морских свинок сенсибилизировали овальбумином по методу Andersson (Anderson P. Antigen-induced bronchial anaphulaxis in actively sensitized geinea-pigs//Allergy, 1980, Vol.35, p.63-71) и через 1-2 месяца после сенсибилизации индуцировали бронхоспазм аэрозольным введением разрешающей дозы овальбумина (3 мг/кг в 1 мл физраствора).
В опытных группах морским свинкам в течение трех дней внутрижелудочно с помощью зонда вводили исследуемые соединения в дозах 10 мкг/кг, 50 мкг/кг и 150 мкг/кг. В другой серии экспериментов исследуемые соединения в дозе 50 мкг/кг (в 1 мл физраствора) вводили ингаляционно (с помощью небулайзера) также в течение трех дней 1 раз в сутки. Контрольной группе вводили физраствор. Через 1 час после последнего введения веществ ингалировали с помощью небулайзера овальбумин и оценивали длительность (в секундах) и интенсивность бронхоспастической реакции животных.
Торможение системной анафилактической реакции морских свинок при ингаляционном введении соединения IV в дозе 50 мкг/кг
(физраствор)
50 мкг/кг
Торможение системной анафилактической реакции морских свинок при внутрижелудочном введении соединения IV в дозах 10 и 150 мкг/кг (М±m)
(физраствор)
10 мкг/кг
150 мкг/кг
Результаты экспериментов, представленные в таблицах 3 и 4, показывают, что соединение IV при внутрижелудочном введении в дозах 10 и 150 мкг/кг и ингаляционном введении в дозе 50 мкг/кг проявило антианафилактическую активность. Ингаляционное введение вещества в дозе 50 мкг/кг блокировало развитие острой фазы бронхоконстрикторной реакции, которая, вызывая удушье, является причиной гибели животных. При внутрижелудочном введении соединения IV в дозах 10 мкг/кг и 150 мкг/кг, был выявлен значительный протективный эффект в отношении антиген-индуцированного бронхоспазма.
Таким образом, соединение IV проявляет значительный протективный эффект в отношении системной анафилактической реакции in vivo.
Пример 15
Противоаллергическое действие соединений общей формулы I на модели аллергического ринита у морских свинок
Использована модель аллергического ринита у морских свинок.
Морских свинок иммунизировали по определенной схеме в течение 1,5-2-х месяцев (Hutson P.A., Church M.K. et al., 1988): вначале животных иммунизировали внутрибрюшинным введением овальбумина в дозе 10 мг/кг с 7-дневным интервалом (дважды), затем свинкам ингалировали с помощью небулайзерной техники (Pari) раствор овальбумина в возрастающей концентрации, начиная с 0,1% до 1% с интервалом в 4 дня между ингаляциями. Последнюю дозу овальбумина вводили в назальные ходы с помощью микропипетки. Через 24 часа после последнего введения овальбумина производили забор назального смыва (через систему специальных трубочек), и изменения в слизистой носа оценивали с помощью комплекса методов: гистологических и цитологических. Исследуемые соединения (0,1% раствор) вводили ежедневно в виде ингаляций с помощью небулайзерной техники в течение 6 дней, на 6-й день введения вызывали провокацию антигеном (ОА). Назальный смыв получали через сутки после провокации.
Влияние соединения IV на цитоз (абсолютное количество клеток в 1 мкл) в назальном смыве
(0,1% р-р)
(° - Р< 0,05; * - Р < 0,01; ** - Р< 0,001)
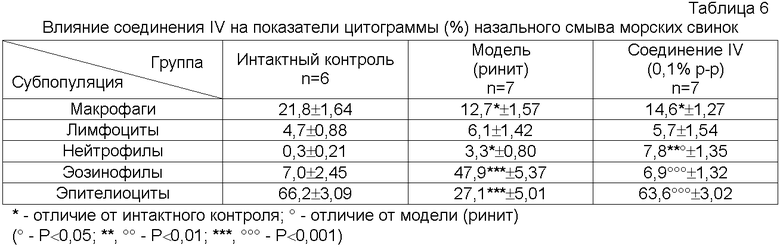

Из данных таблиц 5-7 следует, что в условиях моделирования аллергического ринита соединение IV значительно подавляет эозинофильное воспаление. Это подтверждается снижением до нормы абсолютного и относительного количества эозинофилов, а также существенным снижением цитоза в назальном смыве.
Пример 16
Противоаллергическая активность соединений общей формулы I на модели аллергического воспаления легких у морских свинок
Использована модель аллергического воспаления легких у морских свинок.
Иммунизация животных аналогична той, которая описана в примере 14.
Через 24 часа после последнего введения овальбумина проводили забор бронхоальвеолярного смыва (через канюлю, вставленную в трахею) и изменения в слизистой бронхов оценивали с помощью комплекса методов: гистологических и цитологических.
Исследуемые соединения (0,1% р-р) вводили ежедневно в виде ингаляций с помощью небулайзерной техники в течение 6 дней, на 6-й день введения вызывали провокацию антигеном (ОА).
Влияние соединения IV на цитоз (абсолютное количество клеток в 1 мкл) в бронхоальвеолярном смыве
(0,1% р-р)

Из данных таблиц 8 и 9 следует, что соединение IV в условиях модели аллергического воспаления легких существенно ингибирует воспалительный процесс, что проявляется уменьшением цитоза, снижением содержания ключевой клетки аллергического воспаления - эозинофилов, резким снижением нейтрофилов, а также уменьшением числа лимфоцитов.
Пример 17
Изучение противовоспалительного действия соединений общей формулы I на модели воспаления легких у крыс, индуцированного сефадексом
Модель сефадекс-индуцированного (6-дневного) воспаления легких у крыс
В опытах были использованы крысы-самцы породы Вистар с массой тела 270-300 г.
Воспаление в легких индуцировали однократным ингаляционным введением сефадекса А-25 (гидрофильного порошка с размерами частиц от 20 до 80 мкм) в дозе 5 мг/кг с помощью дозирующего устройства, являющегося лабораторным аналогом ингалятора "Циклохалера” (НИИ пульмонологии РФ).
Методика ингаляционного введения сефадекса и фармакологических веществ
Крысам с помощью оригинального дозирующего устройства для ингаляционного введения сухих порошков под эфирным наркозом вводили сефадекс А-25 в дозе 5 мг на 1 кг массы тела. Крысы Вистар после введения сефадекса А-25 быстро выходили из наркоза и внешне каких-либо особенностей в поведении, характере дыхания у них не отмечалось. Вещества в виде сухого порошка вводили ингаляционно в дозе 500 мкг/кг через 1 час после введения сефадекса, затем в течение 5 дней подряд ежедневно 1 раз в день в одни и те же утренние часы. Контроль представлен 2 группами: группой интактных животных, группой крыс, которым ингаляционно однократно ввели сефадекс.
Результаты терапевтического действия фармакологических веществ на развитие воспалительного процесса в легких оценивали через 6 суток после аэрозольного воздействия сефадекса с помощью морфологических и морфометрических показателей (объемная плотность эмфиземы и альвеолита).
Методы, использованные в работе
Гистологические
Проводили гистологическое исследование легких, окрашенных гематоксилином и эозином.
Морфометрические
Готовили гистологические срезы легких толщиной 4-5 мкм, в которых подсчитывали число нейтрофилов межальвеолярных перегородок, а также оценивали объемную плотность альвеолита и эмфиземы с помощью сетки Автандилова (Автандилов Г.Г. Введение в количественную патологическую морфологию // М.: Медицина, 1980, с. 203). Также проводили морфометрическое исследование лимфоидной ткани легких. С этой целью макропрепараты легких фиксировали по методу Веinenstock et al. (Bienenstock J., Johnson N., Perey D.Y.E. Bronchial lymphoid tissue 1. Morphologic characteristics. // Lab. Invest., 1973, v.28, p.693-698). Легкие с трахеей извлекали из грудной полости и макропрепарат помещали в 2% водный раствор уксусной кислоты. Через 18-24 часа трахею, главный и долевые бронхи рассекали; методом точечного счета под лупой (×7) проводили морфометрическую оценку объемной плотности лимфоидной ткани, ассоциированной с бронхами. Методом точечного счета определяли объемную плотность альвеолита и эмфиземы.
Цитологические
Бронхоальвеолярный смыв у крыс и морских свинок получали под гексеналовым наркозом путем двукратного промывания легких через трахею 10 мл физиологического раствора. Жизнеспособность клеток определяли в тесте с трипановым синим. В жидкости бронхоальвеолярного смыва (БАС) с помощью камеры Горяева определяли абсолютное число клеток в 1 мл (цитоз). В мазках из осадка жидкости БАС, полученного с помощью центрифугирования при 200 g в течение 10 минут, и затем окрашенных по Романовскому-Гимзе, подсчитывали эндопульмональную цитограмму (в процентах) (Авцын А.П., Лукомский Г.И. Романова Л.К. и соавт. Эндопульмональная цитограмма // Сов. мед., 1982, №7, с.8 14).
Результаты исследований обработаны методами вариационной статистики с использованием t-критерия Стьюдента.
Показатели цитоза бронхоальвеолярного смыва крыс Вистар после аэрозольного воздействия сефадекса А-25 и лечения фармакологическими aгентами (М±m)
Цитоз
Абсолютное количество клеток в 1 мкл БАЛ
n=5
(модель)
n=6
n=6
Показатели клеточного состава бронхоальвеолярного смыва крыс Вистар после аэрозольного воздействия сефадекса А-25 и лечения исследуемого соединения (М±m)
Цитоз
Абсолютное количество клеток различных субпопуляций в 1 мкл БАЛ
контроль
(сефадекс)
Гистологическое исследование легких
Соединение IV вызвало отчетливое противовоспалительное действие: распространенность альвеолита была достоверно ниже по сравнению с модельной группой животных; эмфизема практически не выявлялась; не отмечена инфильтрация нейтрофилами межальвеолярных перегородок. По уровню цитоза и количеству нейтрофилов в БАЛ воспалительный процесс также значительно менее выражен, чем у животных, получавших сефадекс в течение 5 дней.
Таким образом, весь комплекс использованных экспериментальных моделей свидетельствует о значительной противоаллергической, антианафилактической и противовоспалительной активности соединений общей формулы I, проявляемой как в тестах in vitro, так и при моделировании аллергической и воспалительной патологии in vivo.
Пример 18
Исследования противовоспалительной активности соединений общей формулы I (1%гель) на модели каррагенинового отека in vivo
Материалы и методы
Тесты проводят на нелинейных белых мышах массой 30-32 г. Используют модель каррагенин-индуцированного отека по методу, описанному в Winter et al. In: De Rosa M., Giroud J.P., Willoughby D.A. Studies of the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. // J. Phamacol., 1971, V.104, p.15-29. В правую лапу мыши субплантарно вводят 1% раствор каррагенина (SERVA) в объеме 0,05 мл. Вещества в виде 1% геля наносят на лапу 2 раза: первый раз - сразу после введения каррагенина; второй раз - через 1,5 часа. Измерение объема правой и левой (интактной) лап проводят с помощью штангенциркуля через 3 ч после введения каррагенина. Воспалительную реакцию и эффект терапевтического воздействия оценивают по формуле:
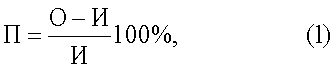
где
П - прирост отека,
О - величина объема лапы после введения флогогена,
И - величина объема лапы до введения флогогена.
Эффект терапевтического воздействия оценивают по степени угнетения воспалительной реакции по сравнению с контролем и рассчитывают по формуле:
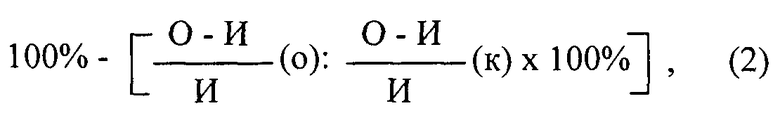 , (2)
, (2)
где
(о) - леченные животные,
(к) - нелеченые животные.
Влияние соединений общей формулы I (1% гель) на развитие каррагенинового отека лапы мышей
** - Р<0,01
Результаты экспериментов, представленные в таблице 12, демонстрируют выраженную противовоспалительную активность мононатриевой соли соединения II, сопоставимую с активностью препарата сравнения - вольтареном.
Cоединение V и его мононатриевая соль проявляют умеренную активность на данной модели каррагенинового отека лапы мышей, торможение отека составляет 43,8% и 26,2% соответственно.
Таким образом, весь комплекс использованных экспериментальных моделей свидетельствует о значительной противоаллергической, антианафилактической и противовоспалительной активности соединений общей формулы I, проявляемой как в тестах in vitro, так и при моделировании аллергической и воспалительной патологии in vivo. Показано, что описываемые соединения, соответствующие общей формуле I, проявляют значительный протективный эффект в отношении системной анафилактической реакции in vivo, в условиях моделирования аллергического ринита, кроме того, эти соединения подавляют эозинофильное воспаление и оказывают отчетливое противовоспалительное действие.
Примеры лекарственных форм
Пример 19
А. Таблетированная форма
Таблетированную форму получают, используя приведенные ниже ингредиенты:
Компоненты смешивают и прессуют для образования таблеток весом 300 мг каждая.
Б. Суппозитории
Пример состава суппозитория:
При необходимости возможно изготовление ректальных, вагинальных и уретральных суппозиториев с соответствующими наполнителями.
В. Мази
Пример состава мази:
Мази изготавливают по общеизвестной технологии.
Г. Гели
Пример состава геля:
Д. Сухой порошок для ингаляций
Пример состава порошка:
Порошок помещают в специальное устройство (контейнер) или в желатиновую капсулу.
Е. Назальный спрей
Пример состава спрея:
Е. Глазные капли
Пример состава капель:
Е. Раствор для инъекций
Пример состава раствора для инъекций:
| название | год | авторы | номер документа |
|---|---|---|---|
| N-АЦИЛЬНЫЕ ПРОИЗВОДНЫЕ АМИНОКИСЛОТ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕВЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГИПОЛИПИДЕМИЧЕСКИХ СРЕДСТВ | 2005 |
|
RU2335495C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ N-АЦИЛЬНЫХ ПРОИЗВОДНЫХ АМИНОКИСЛОТ (ВАРИАНТЫ) | 2008 |
|
RU2378284C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ГЛУТАРОВОЙ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ СРЕДСТВ | 2008 |
|
RU2373934C1 |
| ПРОТИВОАЛЛЕРГИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ МЕСТНОГО ГЛАЗНОГО ПРИМЕНЕНИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1994 |
|
RU2107497C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2141483C1 |
| НОВЫЕ АНТИДИАБЕТИЧЕСКИЕ АГЕНТЫ | 2000 |
|
RU2265012C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭКСТРАКТ ИЛИ ФРАКЦИЮ ИЗ РАСТЕНИЯ РОДА JUSTICIA | 2015 |
|
RU2671501C1 |
| ГЕМИНПЕПТИДЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМКОМПОЗИЦИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ | 2004 |
|
RU2280649C1 |
| АСПАРТИЛЬНЫЕ ПРОИЗВОДНЫЕ ГИСТАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ ФЕРМЕНТОВ АНТИОКСИДАНТНОЙ ЗАЩИТЫ | 2005 |
|
RU2287524C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕМИНА С АНТИБАКТЕРИАЛЬНОЙ И ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2011 |
|
RU2475498C1 |
Настоящее изобретение относится к применению N-ацильным производным аминокислот общей формулы (I), где n равно 2 или 3 и их фармацевтически приемлемых солей, в качестве противоаллергических, противовоспалительных и антианафилактических средств. Кроме того, объектами изобретения являются фармацевтическая композиция, содержащая указанные соединения в эффективном количестве, и способ лечения аллергических и воспалительных заболеваний, например, таких как бронхиальная астма, аллергический ринит, поллиноз, псориаз. 8 н. и 3 з.п. ф-лы, 12 табл.
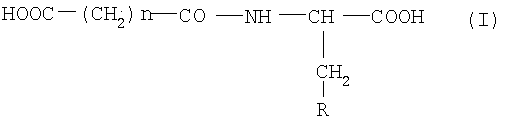 ,
,  или
или 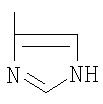
1. Фармацевтическая композиция, обладающая противоаллергической, антианафилактической и противовоспалительной активностью, включающая N-ацильные производные аминокислот общей формулы (I)
где n равно 2 или 3; и
R представляет
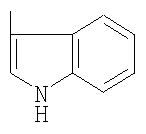 или
или
при условии, что соединение общей формулы (I) не является сукцинил-L-гистидином, сукцинил-L-триптофаном, сукцинил-D-триптофаном, сукцинил-D,L-триптофаном и его дикалиевой солью, или их фармацевтически приемлемые соли в эффективном количестве и фармацевтически приемлемые добавки.
2. Фармацевтическая композиция для регулирования содержания эозинофилов и снижения дегрануляции базофилов, включающая N-ацильные производные аминокислот общей формулы (I)
где n равно 2 или 3; и
R представляет
или
или их фармацевтически приемлемые соли в эффективном количестве и фармацевтически приемлемые добавки.
3. Применение N-ацильных производных аминокислот общей формулы (I)
где n равно 2 или 3; и
R представляет
или
при условии, что соединение общей формулы (I) не является сукцинил-L-гистидином, сукцинил-L-триптофаном, сукцинил-D-триптофаном, сукцинил-D,L-триптофаном и его дикалиевой солью или их фармацевтически приемлемых солей в качестве средств, обладающих противоаллергической, антианафилактической и противовоспалительной активностью.
4. Применение по п.3 N-ацильных производных аминокислот или их фармацевтически приемлемых солей в качестве средств, обладающих способностью снижать антигензависимую секрецию гистамина и регулировать содержание нейтрофилов и лимфоцитов.
5. Применение по п.3 N-ацильных производных аминокислот в качестве средств, способных устранять признаки бронхиальной астмы и псориаза.
6. Применение N-ацильных производных аминокислот общей формулы (I)
где n равно 2 или 3; и
R представляет
или
или их фармацевтически приемлемых солей в качестве средств, способных регулировать содержание эозинофилов и снижать дегрануляцию базофилов.
7. Применение N-ацильных производных аминокислот общей формулы (I)
где n равно 2 или 3; и
R представляет
или
или их фармацевтически приемлемых солей в качестве средств, способных устранять признаки аллергического ринита, поллинозов, сезонного и круглогодичного ринита, аллергического воспаления легких.
8. Средство, обладающее противоаллергической, антианафилактической и противовоспалительной активностью, которое представляет собой N-ацильные производные аминокислот общей формулы (I)
где n равно 2 или 3; и
R представляет
или
при условии, что соединение общей формулы (I) не является сукцинил-L-гистидином, сукцинил-L-триптофаном, сукцинил-D-триптофаном, сукцинил-D,L-триптофаном и его дикалиевой солью, или их фармацевтически приемлемые соли.
9. Способ лечения аллергических, анафилактических, в том числе заболеваний, сопровождающихся воспалением, включающий введение млекопитающему эффективного количества N-ацильных производных аминокислот общей формулы (I)
где n равно 2 или 3; и
R представляет
или
при условии, что соединение общей формулы (I) не является сукцинил-L-гистидином, сукцинил-L-триптофаном, сукцинил-D-триптофаном, сукцинил-D,L-триптофаном и его дикалиевой солью, или их фармацевтически приемлемых солей.
10. Способ по п.9 лечения бронхиальной астмы и псориаза.
11. Способ лечения аллергического ринита, поллинозов, сезонного и круглогодичного ринита, аллергического воспаления легких, включающий введение млекопитающему эффективного количества N-ацильных производных аминокислот общей формулы (I)
где n равно 2 или 3; и
R представляет
или
или их фармацевтически приемлемых солей.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Branco S., "Cyclodextrin assisted ensntiomeric recognition of benzo(de)isoquinoline-1,3-dione derived amino acids." Tetrahedron, 2005, 61(4), 919-926 | |||
| Галенко-Ярошевский П.А | |||
| Противоаритмическая активность бефола, суфана, мексидола и Т3-146 в сочетании с некоторыми антиаритмиками | |||
| БЮЛЛЕТЕНЬ ЭКСПЕРИМЕНТАЛЬНОЙ БИОЛОГИИ И МЕДИЦИНЫ, 1998, т.125, №5, с.544-547. | |||

