Область, к которой относится изобретение
Изобретение относится к химическим соединениям типа пери-замещенных бициклических арилсульфонамидов, которые могут быть использованы для лечения и профилактики окклюзионного заболевания артерий и родственных опосредуемых простагландином расстройств.
Предшествующий уровень техники
Атеросклероз представляет собой патологию, которая может приводить к развитию нескольких наиболее опасных смертельных заболеваний человека, таких как инфаркт миокарда и окклюзионное заболевание периферических артерий (ОЗПА). ОЗПА представляет собой атеросклероз крупных и средних артерий конечностей, а в частности, нижних конечностей, включая аорту и подвздошные артерии. Это заболевание часто сопровождается ишемической болезнью сердца и цереброваскулярным заболеванием. Индивидуумы, страдающие ОЗПА, подвержены повышенному риску возникновения других сосудистых заболеваний, таких как инфаркт миокарда или инсульт [Waters, RE, Terjung RL, Peters KG & Annex BH. J. Appl. Physiol. 2004; Ouriel K. Lancet, 2001, 258:1257-64; Kroger, K. Angiology, 2004, 55:135-138]. Такие серьезные с клинической точки зрения поражения могут приводить к постепенному сужению периферических артерий, вызывающему боли при ходьбе (хромоту), которые обычно стихают в состоянии покоя, ишемические язвы, гангрену, а иногда такие поражения могут привести к необходимости ампутации конечности. Лекарственная терапия обычно неэффективна, а операции по обходному шунтированию или замене пораженных артерий искусственными или венозными трансплантатами приводят к улучшению периферического кровообращения, по меньшей мере, только до тех пор, пока эти искусственные сосуды не станут снова подвергаться стенозу [Haustein, K.O., Int. J. Clin. Pharmacol. Ther., 35:266 (1997)]. Недавно, при проведении исследования по сцеплению генов у человека было обнаружено, что варианты ДНК гена PTGER3, который кодирует рецептор простагландина Е2 подтипа 3 (известный как ЕР3), вызывают увеличение риска развития у индивидуума ОЗПА (см. опубликованную заявку США 2003/0157599). Таким образом, антагонисты простагландина Е2 (PGE2), связывающиеся с рецептором ЕР3, могут обеспечивать эффективное лечение или профилактику ОЗПА.
Простагландины, в ответ на различные внешние раздражители, быстро генерируются из свободной арахидоновой кислоты под непрерывным действием циклооксигеназ и синтаз. Простагландины действуют в непосредственной близости от участка их синтеза. В настоящее время уже клонированы и охарактеризованы восемь простаноидных рецепторов. Эти рецепторы являются членами все растущего класса сопряженных с G-белком рецепторов. PGЕ2 связывается преимущественно с рецепторами EPl, EP2, EP3 и EP4; PGD2 связывается с рецепторами DP и FP; PGF2α связывается с рецепторами FP и EP3; PGI2 связывается с рецептором IP, а TXA2 связывается с рецептором TP. Было обнаружено, что PGЕ2, связывающийся с рецептором ЕР3, играет ключевую роль в регуляции транспорта ионов, в сокращении гладкой мышцы желудочно-кишечного тракта, в секреции кислоты, в сокращении матки во время оплодотворения и имплантации, в повышении температуры и в развитии гипералгезии. Рецептор ЕР3 был обнаружен во многих органах, таких как почки, желудочно-кишечный тракт, матка и головной мозг. В сердечно-сосудистой системе ЕР3 экспрессируется в васкулярном эндотелии и в гладкой мышце, и по меньшей мере, четыре изоформы ЕР3 экспрессируются на человеческих тромбоцитах [Paul, B.Z., B. Ashby, and S.B. Sheth, Distribution of prostaglandin IP and EP receptor subtypes and isoforms in platelets and human umbilical artery smooth muscle cells. British Journal of Haematology, 1998. 102(5): p. 1204-11].
Простаноиды, действующие посредством мембраноспецифических рецепторов, принадлежащих к суперсемейству сопряженных с G-белками рецепторов (GPCR), играют важную роль в гомеостазе сосудов, включая регуляцию функции тромбоцитов. Среди простаноидов, сильным стимулятором агрегации тромбоцитов является тромбоксан A2 (TxA2), а простагландин (PGI2) ингибирует их активацию. С другой стороны, сообщалось, что простагландин E2 (PGЕ2) обладает двухсторонним действием на ответ тромбоцитов, то есть при низких концентрациях он потенцирует их агрегацию, а при более высоких концентрациях ингибирует их агрегацию. Было показано, что стимулирующее действие PGЕ2 на агрегацию тромбоцитов происходит, главным образом, посредством рецептора ЕР3, т.е. одного из четырех подтипов рецепторов, активируемых PGЕ2.
Локальный синтез простагландинов в стенках артериальных сосудов может играть существенную роль в развитии атеросклероза. В стенках здоровых сосудов присутствует только COX-1, а в артериосклеротических бляшках присутствуют COX-1 и COX-2 [Schonbeck, U., et al., Augmented expression of cyclooxygenase-2 в human atherosclerotic lesions. Am J Pathol, 1999. 155(4): p. 1281-91; Cipollone, F., et al., Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of PGE2-dependent plaque instability. Circulation, 2001. 104(8): p. 921-7]. Их повышенная экспрессия, наряду с повышенной экспрессией простагландин-Е-синтазы, может приводить к увеличению продуцирования вышеуказанного PGЕ2. У генетически модифицированных мышей, у которых отсутствует рецептор липопротеина низкой плотности (LDL-R), образование атеросклеротических бляшек может быть уменьшено путем обработки рофекоксибом, селективным ингибитором COX-2, действующими посредством снижения уровня продуцирования PGЕ2 и других простагландинов [Burleigh ME, Babaev VR, Oates JA, Harris RC, Gautam S, Riendeau D, Marnett LJ, Morrow JD, Fazio S, Linton MF. Cyclooxygenase-2 promotes early atherosclerotic lesion formation в LDL receptor-deficient mice. Circulation. 2002 Apr 16; 105(15):1816-23].
Было показано, что в атеросклеротических бляшках, клетки сосудов гладких мышц экспрессируют рецепторы ЕР3, а PGЕ2 стимулирует их пролиферацию и миграцию, что является признаком образования атеросклеротических бляшек [Blindt R, Bosserhoff AK, vom Dahl J, Hanrath P, Schror K, Hohlfeld T, Meyer-Kirchrath J. Activation of JP and EP(3) receptors alters cAMP-dependent cell migration.Eur J Pharmacol. 2002 May 24;444(l-2):31-7]. Поэтому, вероятно, что в хронически воспаленных сосудах продуцируются PGЕ2 в количестве, достаточном для активации рецепторов EP3 на клетках сосудов гладких мышц (что способствует образованию атеросклеротических поражений) и на тромбоцитах (что способствует развитию тромбоза). Локально продуцируемые PGЕ2 (из самих тромбоцитов, из компонентов клеточных стенок и воспалительных клеток) потенцируют агрегацию тромбоцитов, осуществляемую под действием субоптимальных количеств протромбиновых тканевых факторов, которые сами по себе не вызывают агрегацию тромбоцитов, а действуют лишь посредством стимуляции протеинкиназы C. Внутриклеточные процессы, запускаемые активацией рецептора EP3, могут усиливать агрегацию тромбоцитов посредством предотвращения действия PGI2 и усиления действия основных факторов, усиливающих агрегацию, таких как коллаген. Поэтому, активация рецептора ЕР3 может стимулировать развитие атеросклероза и повышать риск возникновения тробмоза, наблюдаемого при таких патологических состояниях, как васкулиты и ОЗПА.
Современные методы лечения ОЗПА направлены либо на снижение повышенного риска развития сердечно-сосудистых заболеваний, таких как инфаркт миокарда и инсульт, либо на симптоматическое ослабление хромоты. Все эти методы лечения направлены на подавление функции тромбоцитов. Лечение, приводящее к снижению риска развития сердечно-сосудистых заболеваний, включает введение небольших доз аспирина (достаточных для снижения агрегации тромбоцитов, но недостаточных для прекращения продуцирования PGI2 в стенках сосудов) и ингибиторов тромбоцитарного аденозиндифосфатного рецептора (клопидогрель). Связывание аденозиндифосфата с тромбоцитарным аденозиндифосфатным рецептором приводит к снижению уровня тромбоцитарного cAMP с последующей активацией и агрегацией тромбоцитов. Лечение, способствующее симптоматическому ослаблению хромоты, включает введение ингибиторов тромбоцитарной фосфодиэстеразы типа 3, таких как цилостазол, который повышает внутриклеточные уровни cAMP. Ингибиторы тромбоцитарного аденозиндифосфатного рецептора или тромбоцитарной фосфодиэстеразы типа 3 оказывают прямое или опосредованное действие, повышающее содержание cAMP в тромбоцитах, ингибируя, тем самым, активацию тромбоцитов и последующую их агрегацию, приводящую к образованию тромбов. Связывание PGЕ2 с EP3 приводит к снижению уровней cAMP, а следовательно, антагониста PGЕ2, связывающегося с рецептором EP3, благодаря предотвращению PGЕ2-зависимого снижения уровня cAMP, необходимого для индуцирования активации тромбоцитов и последующей их агрегации, или благодаря предотвращению PGЕ2-зависимого снижения уровня cAMP в клетках сосудов гладких мышц, необходимого для стимуляции миграции тромбоцитов, а поэтому, это связывание может, вероятно, оказывать благоприятное терапевтическое действие при лечении ОЗПА. Такой антагонист может также ослаблять симптомы заболевания посредством ингибирования или снижения образования атеросклеротических бляшек.
Кроме того, простагландины участвуют в патогенезе ряда патологических состояний, включающих боль, повышение температуры или воспаление, ассоциированное с ревматической лихорадкой, гриппом или другими вирусными инфекциями; насморк, боли в пояснице и в области шеи, боли в позвоночнике, послеродовые боли, дисменорею, головную боль, мигрень, зубную боль, растяжение и деформацию суставов, миозит, невралгию, синовит, артрит, включая ревматоидный артрит, дегенеративные заболевания суставов (остеоартрит), подагру и анкилозирующий спондилит, бурсит, ожоги, включая ожоги, вызванные облучением и разъедающими химическими соединениями, солнечные ожоги, боли после хирургических операций и стоматологических процедур, иммунные и аутоиммунные заболевания; неопластическое перерождение клеток или метастатический рост опухолей; диабетическую ретинопатию, опухолевый ангиогенез; индуцированное простаноидами сокращение гладких мышц, ассоциированное с дисменореей, преждевременными родами, астмой или эозинофильными расстройствами; болезнь Альцгеймера; глаукому; резрежение кости; остеопороз; болезнь Пэджета; пептические язвы, гастрит, регионарный энтерит, язвенный колит, дивертикулез или другие поражения желудочно-кишечного тракта; кровотечения в желудочно-кишечном тракте; нарушения свертываемости крови, выбранные из гипопротромбинемии, гемофилии и других нарушений, связанных с повышенным кровотечением; и почечные заболевания.
Хотя уровни простаноидов в кровотоке у здоровых индивидуумов крайне низки [FitzGerald GA, Brash AR, Falardeau P & Oates JA. JCI 1981 68:12472-1275], однако, локальная концентрация PGЕ2 может резко возрастать при воспалительных заболеваниях. Так, например, было показано in vitro, что при окклюзионном заболевании аорты подвздошной кишки локальное продуцирование PGЕ2 возрастает более чем в 30 раз [Reilly J, Miralles M, Wester W & Sicard G. Surgery, 1999, 126:624-628]. Поэтому, вероятно, что в сосудах с хроническим воспалением PGЕ2 продуцируется в количестве, достаточном для активации рецепторов ЕР3 на тромбоцитах. В этом случае, внутриклеточные процессы, запускаемые активацией рецептора ЕР3, могут приводить к повышению агрегации тромбоцитов путем подавления действия PGI2 и усиления действия основных факторов, усиливающих агрегацию, таких как AДФ. Поэтому, активация рецептора ЕР3 может стимулировать развитие тромбоза, наблюдаемого при таких патологических состояниях, как васкулиты и атеросклероз. Окклюзионное заболевание периферических артерий (ОЗПА) представляет собой атеросклеротическое заболевание, которое развивается, главным образом, у пожилых людей вследствие окклюзии просвета периферических артерий, в основном, бедренных артерий, и это заболевание ассоциируется с повышенным риском развития сосудистых расстройств, таких как инфаркт миокарда или инсульт [Waters, RE, Terjung RL, Peters KG & Annex BH. J. Appl. Physiol. 2004; Ouriel K. Lancet, 2001, 258:1257-64; Kroger, K. Angiology, 2004, 55:135-138]. Различные клинические исследования показали, что лечение с использованием простагландинов способствует ослаблению симптомов ОЗПА [Reiter M, Bucek R, Stumpflen A & Minar E. Cochrane Database Syst. Rev. 2004, 1:CD000986; Bandiera G, Forletta M, Di Paola FM, Cirielli C. Int. Angiol. 2003, 22:58-63; Matsui K, Ikeda U, Murakami Y, Yoshioka T, Shimada K. Am. Heart J. 2003, 145:330-333], что подтверждает взаимосвязь ОЗПА с функцией простаноидного рецептора.
Орто-замещенные фенилацилсульфонамиды и их эффективность в лечении опосредуемых простагландином расстройств описаны в патенте США № 6242493 и в двух статьях Juteau et al. [BioOrg. Med. Chem. 9, 1977-1984 (2001)] и Gallant et al. [BioOrg. Med. Chem. Let. 12, 2583-2586 (2002)], которые вводятся в настоящее описание посредством ссылки.
Сущность изобретения
В одном из своих аспектов, настоящее изобретение относится к соединениям формулы I:
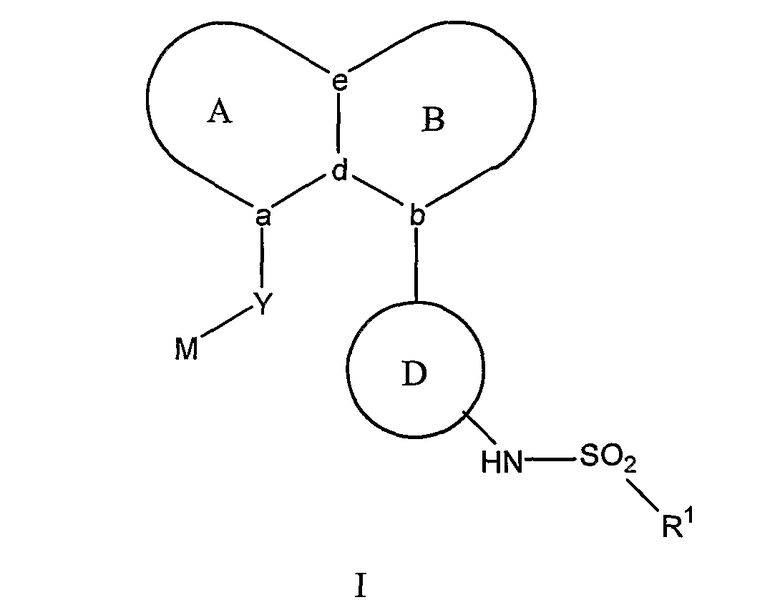
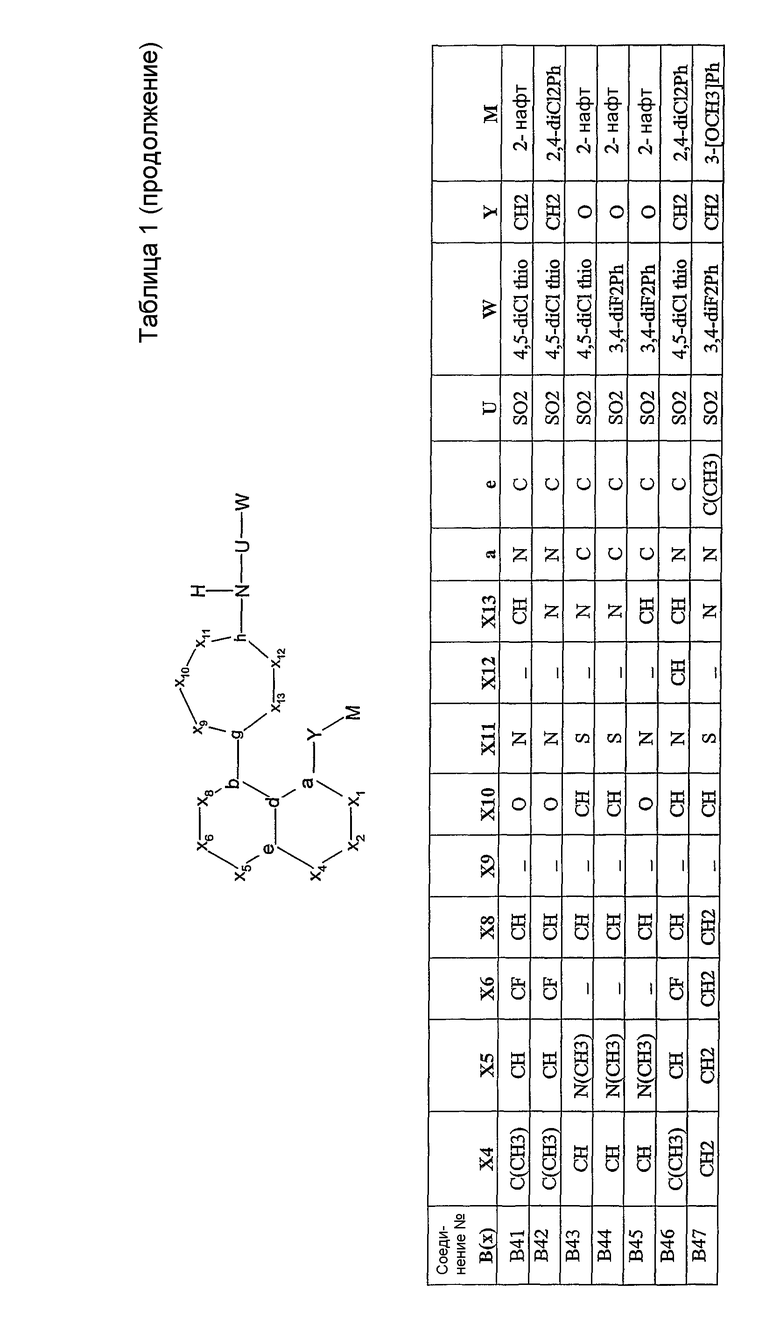
где А и В представляют собой пару конденсированных 5-, 6- или 7-членных колец. Конденсированная система колец A/B может содержать от 0 до 4 гетероатомов, выбранных из азота, кислорода и серы, и может быть дополнительно замещена 0-4 заместителями, независимо выбранными из галогена, -ОН, низшего алкила, -О-(низшего алкила), низшего фторалкила, -О-(низшего фторалкила), метилендиокси, этилендиокси, алкокси(низшего алкила), гидрокси(низшего алкила), оксо, оксида, -CN, нитро, -S-(низшего алкила), амино, низшего алкиламино, низшего диалкиламино, низшего диалкиламиноалкила, карбокси, карбоалкокси, ацила, карбоксамидо, низшего алкилсульфоксида, ациламино, фенила, бензила, спиро-тиазолидинила, фенокси и бензилокси. Узловые положения, обозначенные a и b, представляют собой положения присоединения остатков Y и D, соответственно, и эти положения a и b находятся в пери-положении по отношению друг к другу на конденсированной системе колец А/В. Узловые положения, обозначенные d и е, представляют собой конденсированные положения между кольцом А и кольцом В в конденсированной системе колец А/В. Каждое из узловых положений а, b, d и е может означать углерод или азот.
D представляет собой арильную или гетероарильную циклическую систему, которая может быть дополнительно замещена 0-4 заместителями. Такие заместители независимо выбраны из галогена, -ОН, низшего алкила, -О-(низшего алкила), низшего фторалкила, -О-(низшего фторалкила), метилендиокси, этилендиокси, алкокси(низшего алкила), гидрокси(низшего алкила), -CN, нитро, -S-(низшего алкила), амино, низшего алкиламино, низшего диалкиламино, низшего диалкиламиноалкила, карбокси, карбоалкокси, ацила, карбоксамидо, низшего алкилсульфоксида, ациламино, фенила, бензила, фенокси и бензилокси.
Y представляет собой линкер, содержащий от 0 до 8 атомов в цепи.
M выбран из арила, замещенного арила, гетероциклила, замещенного гетероциклила, С6-С20алкила и замещенного С6-С20алкила.
R1 выбран из арила, замещенного арила, гетероарила, замещенного гетероарила и СF3; и если Y представляет собой одноатомный линкер, то R1 дополнительно может представлять собой низший алкил.
В своем втором аспекте, настоящее изобретение относится к фармацевтическим композициям, содержащим фармацевтически приемлемый носитель и вышеописанное соединение или его сложный эфир, фармацевтически приемлемую соль или гидрат.
В своем третьем аспекте, настоящее изобретение относится к способам лечения или профилактики заболевания или состояния, опосредуемого простагландинами. Указанные способы включают введение млекопитающему терапевтически эффективного количества описанного здесь соединения. Такими заболеваниями или состояниями могут быть, например, повышенная температура или воспаление, ассоциированное с ревматической лихорадкой, гриппом или другими вирусными инфекциями; мигрень, насморк, дисменорея, растяжение и деформация суставов, миозит, невралгия, синовит, артрит, включая ревматоидный артрит, дегенеративные заболевания суставов (остеоартрит), подагра и анкилозирующий спондилит, бурсит, ожоги, включая ожоги, вызванные облучением и разъедающими химическими соединениями, солнечные ожоги, иммунные и аутоиммунные заболевания; и боли (например, боли в пояснице и в области шеи, боли в позвоночнике, послеродовые боли, головная боль, зубная боль, боли после хирургических операций и стоматологических процедур). Соединения-антагонисты ЕР3 согласно изобретению, которые проникают в ЦНС, являются особенно подходящими для устранения боли.
Соединения согласно изобретению, которые ингибируют агрегацию тромбоцитов и повышают региональное кровообращение, могут быть использованы для лечения первичной тромбоэмболии, тромбоза и окклюзионных сосудистых заболеваний. Эти соединения могут быть использованы преимущественно в комбинации с другими ингибиторами агрегации тромбоцитов и с ингибиторами биосинтеза или поглощения холестерина. Указанные соединения могут быть также преимущественно использованы в комбинации с ингибитором циклооксигеназы-2 для лечения воспалительных состояний.
Другими заболеваниями или состояниями, которые могут быть подвергнуты лечению, являются, например, неопластическое перерождение клеток или метастатический рост опухолей; диабетическая ретинопатия, опухолевый ангиогенез; индуцированное простаноидами сокращение гладких мышц, ассоциированное с дисменореей, преждевременными родами, астмой или эозинофильными расстройствами; болезнь Альцгеймера; глаукома; резрежение кости, остеопороз или болезнь Пэджета; пептические язвы, гастрит, регионарный энтерит, язвенный колит, дивертикулез или другие поражения желудочно-кишечного тракта; кровотечения в желудочно-кишечном тракте; нарушения свертываемости крови, выбранные из гипопротромбинемии, гемофилии и других нарушений кровообращения; и почечные заболевания. Этот аспект изобретения также включает способы стимуляции остеогенеза, цитопротекции и снижения количества бляшек при лечении атеросклероза.
В своем четвертом аспекте, настоящее изобретение относится к способам скрининга на селективные простаноидные рецепторы, а в частности, лиганды ЕР3.
Подробное описание изобретения
Соединениями определенного типа, представленными вышеуказанной формулой 1, являются антагонисты рецептора ЕР3. Такие соединения могут быть использованы для лечения и предупреждения опосредуемых простагландином состояний, описанных выше, а в частности, таких состояний, как окклюзионное сосудистое заболевание.
Композиции согласно изобретению включают эффективную дозу или фармацевтически эффективное количество или терапевтически эффективное количество соединения, описанного выше, и могут также включать другие терапевтические средства, такие как ингибиторы агрегации тромбоцитов (тирофибан, дипиридамол, клопидогрель, тиклопидин и т.п.), ингибиторы HMG-CoA-редуктазы (ловастатин, симвастатин, правастатин, розувастатин, мевастатин, аторвастатин, церивастатин, питавастатин, флувастатин и т.п.) и ингибиторы циклооксигеназы. Другие неограничивающие примеры гиполипидемических средств, которые могут быть использованы в комбинации с соединениями согласно изобретению, можно найти в колонках 5-6 патента США 6498156, описание которого вводится в настоящее изобретение посредством ссылки. Предпочтительными ингибиторами циклооксигеназы-2 являются ингибиторы, селективные по отношению к циклооксигеназе-2, но не к циклооксигеназе-1. Предпочтительными ингибиторами циклооксигеназы-2 являются рофекоксиб, мелоксикам, целекоксиб, эторикоксиб, лумиракоксиб, вальдекоксиб, парекоксиб, цимикоксиб, диклофенак, сулиндак, этодолак, кеторалак, кетопрофен и LAS-34475, хотя настоящее изобретение не ограничивается этими или другими известными ингибиторами циклооксигеназы-2.
Способы согласно изобретению включают использование как композиций, так и препаратов. Эти способы включают введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества пери-замещенного конденсированного циклического соединения A/B согласно изобретению. Настоящее изобретение также относится к способам скрининга на селективные агонисты или антагонисты простаноидных рецепторов. Простаноидными рецепторами являются рецепторы EP1, EP2, EP3, EP4, IP и FP. Наибольший интерес представляют селективные лиганды для EP3, способ применения которых включает контактирование меченого соединения согласно изобретению с клонированным человеческим рецептором EP3 и измерение вытеснения метки тест-соединением.
Соединениями согласно изобретению являются соединения формулы I:
где А и В представляют собой пару конденсированных 5-, 6- или 7-членных колец, а D представляет собой арильную или гетероарильную циклическую систему. В соединении одного из подвидов, D представляет собой фенил, который может быть замещенным или незамещенным. В соединении другого подвида, D представляет собой нафтил, который может быть замещенным или незамещенным. В соединении третьего подвида, D представляет собой моноциклический гетероарил, который может быть замещенным или незамещенным. В соединении четверного подвида, D представляет собой бициклический гетероарил, который может быть замещенным или незамещенным. В одном из вариантов изобретения, R1 выбран из фенила, замещенного фенила, 5-членного циклического гетероарила, замещенного 5-членного циклического гетероарила и СF3.
Каждый из А и В независимо представляет собой 5-, 6- или 7-членное кольцо. Конденсированная система колец A/B содержит от 0 до 4 гетероатомов, выбранных из азота, кислорода и серы, и эти кольца дополнительно замещены 0-4 заместителями. Подходящими заместителями являются галоген, -ОН, низший алкил, -О-(низший алкил), низший фторалкил, -О-(низший фторалкил), метилендиокси, этилендиокси, алкокси(низший алкил), гидрокси(низший алкил), оксо, оксид, -CN, нитро, -S-(низший алкил), амино, низший алкиламино, низший диалкиламино, низший диалкиламиноалкил, карбокси, карбоалкокси, ортоэфиры, ацил, карбоксамидо, низший алкилсульфоксид, ациламино, фенил, бензил, спиро-тиазолидинил, фенокси и бензилокси. Поскольку конденсированная система колец А/В может включать азот или серу, то заместителями могут быть оксиды, например, N→О и S→О.
В соединениях одного из подвидов, циклическая система A/B представляет собой пару конденсированных 5-членных колец:
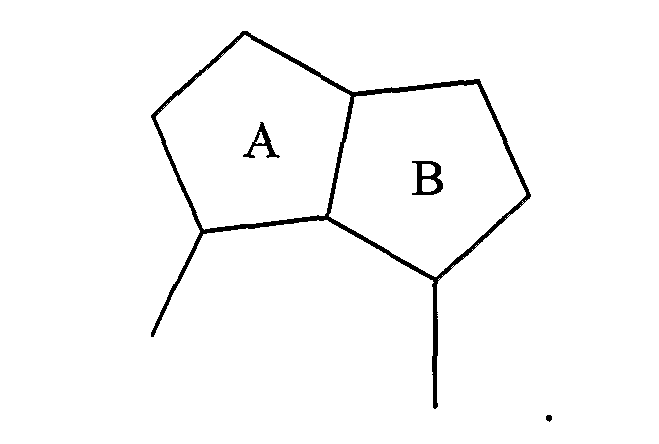
Примерами таких 5/5-циклических систем являются:
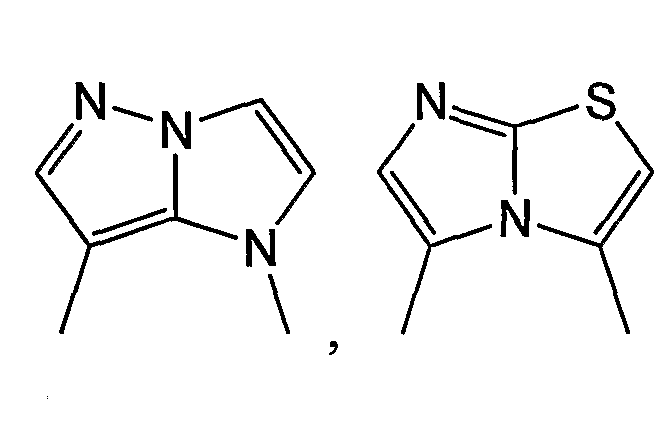 и
и 
В соединениях другого подвида, циклическая система A/В представляет собой пару 6-членных колец:
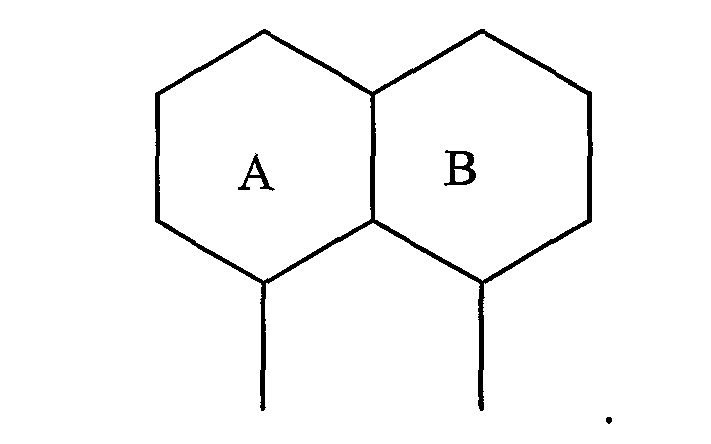
Примерами таких 6/6-циклических систем являются:
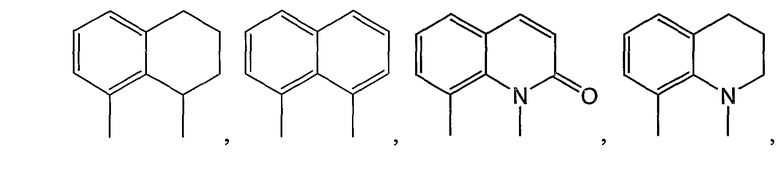
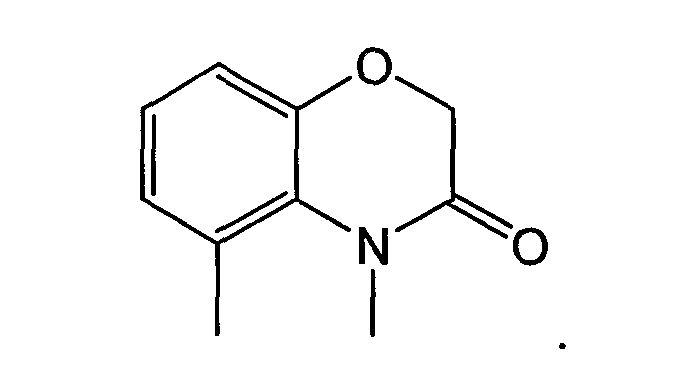
В соединениях другого подвида, циклическая система A/B представляет собой пару конденсированных 5- и 6-членных колец:
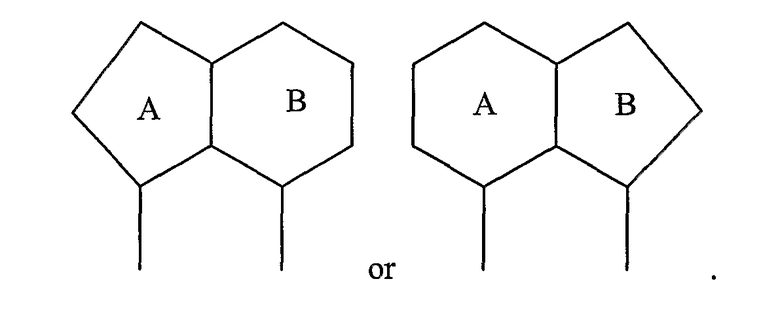 или
или
Примерами таких 5/6-циклических систем являются индолы, индолины, индолоны, изатины, бензимидазолы, бензоксазолиноны, бензофураны и индазолы:
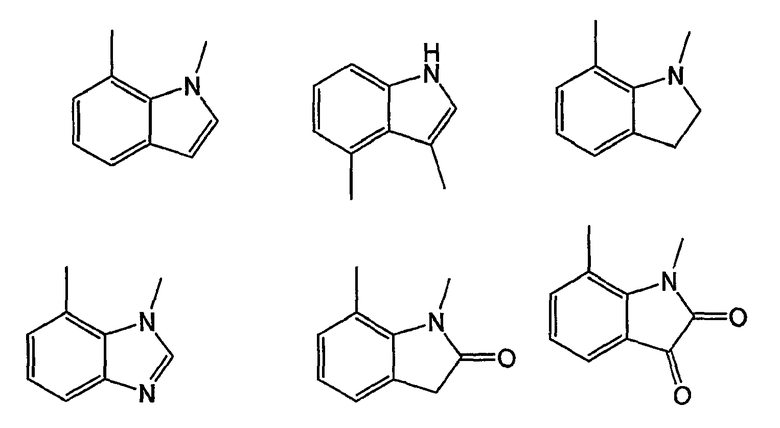
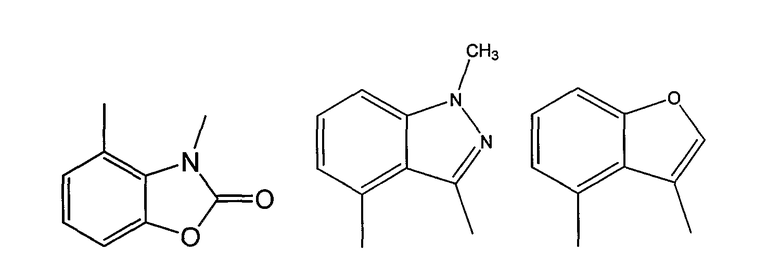
Как было показано ранее, циклические системы могут быть замещены, например:
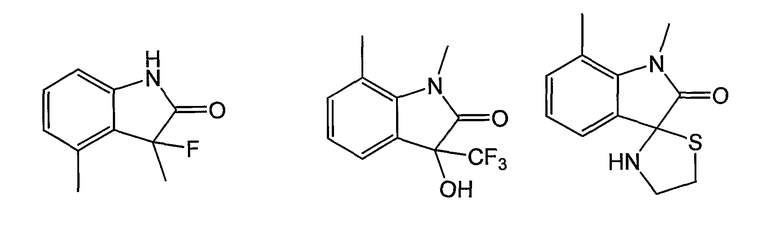
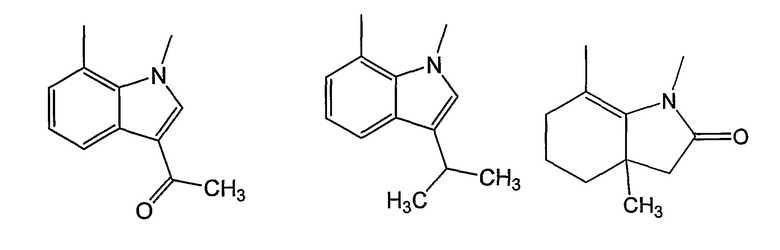
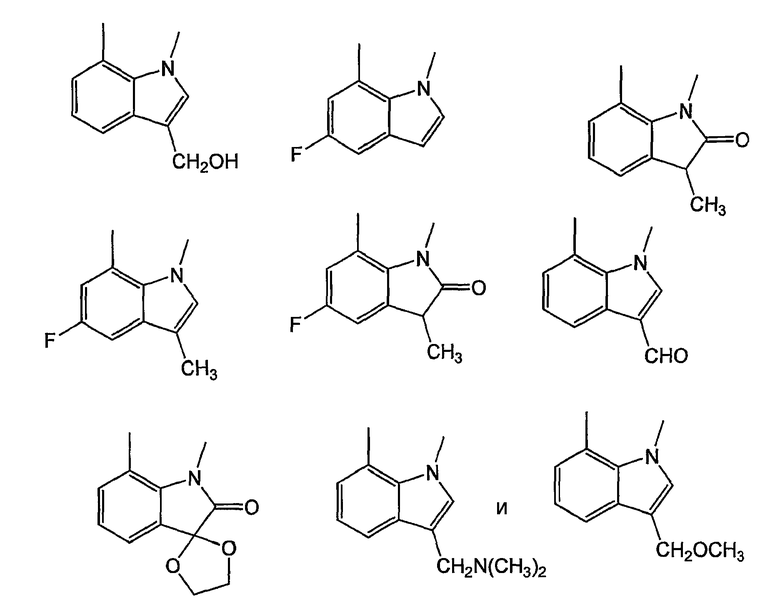
Y представляет собой линкер, содержащий от 0 до 8 атомов в цепи. Предпочтительно, Y собой С1-С8алкил, в котором один или два -СН2- могут быть заменены группами -O-, -C(=O)-, -CH=CH-, -CF2-, -S-, -SO-, -SO2-, -NH- или -N(алкил)-. Более предпочтительно, Y представляет собой двухатомную цепь, то есть С1- или С2алкил, в котором один или оба СН2 могут быть заменены группами, указанными выше. В одном из вариантов изобретения, Y выбран из -СН2-, -O-, -OCH2-, -S-, -SO- и -SO2-. Левосторонняя связь указывает на положение присоединения колец А или В.
M выбран из арила, замещенного арила, гетероциклила, замещенного гетероциклила, С6-С20алкила и замещенного С6-С20алкила. В одном из предпочтительных вариантов изобретения, M выбран из арила, замещенного арила, гетероциклила и замещенного гетероарила, а более предпочтительно, из фенила, замещенного фенила, нафтила, замещенного нафтила, гетероарила и замещенного гетероарила.
Указанные соединения могут быть получены в виде солей. Термин “фармацевтически приемлемая соль” означает соли, противоионы которых происходят от фармацевтически приемлемых нетоксичных кислот и оснований. Подходящими фармацевтически приемлемыми основно-аддитивными солями соединений согласно изобретению являются, но не ограничиваются ими, соли металлов, полученные из алюминия, кальция, лития, магния, калия, натрия и цинка, или органические соли, полученные из лизина, производных N,N-диалкиламинокислот (например, N,N-диметилглицина, пиперидин-1-уксусной кислоты и морфолин-1-уксусной кислоты), N,N'-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, меглумина (N-метилглюкамина) и прокаина. Если указанные соединения содержат основный остаток, то подходящими фармацевтически приемлемыми основно-аддитивными солями соединений согласно изобретению являются соли неорганических кислот и соли органических кислот. Примерами являются ацетат, бензолсульфонат (безилат), бензоат, бикарбонат, бисульфат, карбонат, камфорсульфонат, цитрат, этансульфонат, фумарат, глюконат, глутамат, бромид, хлорид, изетионат, лактат, малеат, малат, манделат, метансульфонат, мукат, нитрат, памоат, пантотенат, фосфат, сукцинат, сульфат, тартрат, п-толуолсульфонат и т.п.
Определения
В описании настоящей заявки приводятся определения используемых здесь терминов и заместителей.
“Алкил” означает прямые, разветвленные, и, если это не определено иначе, циклические углеводородные структуры и их комбинации. “Низший алкил” означает алкильные группы, имеющие от 1 до 6 атомов углерода. Примерами низших алкильных групп являются метил, этил, пропил, изопропил, бутил, втор- и трет-бутил и т.п. Предпочтительные алкильные и алкиленовые группы имеют 20 атомов углерода или менее. Циклоалкил представляет собой подгруппу алкилов и включает циклические углеводородные группы, имеющие от 3 до 8 атомов углерода. Примерами циклоалкильных групп являются циклопропил, циклобутил, циклопентил, норборнил, адамантил и т.п.
С1-С20-углеводородами являются алкил, циклоалкил, алкенил, алкинил, арил и их комбинации. Примерами являются бензил, фенетил, циклогексилметил, камфорил и нафтилэтил.
“Алкокси” или “алкоксил” означают группы, имеющие 1-8 атомов углерода в прямой, разветвленной и циклической структурах и их комбинациях, присоединенных к исходной структуре посредством атома кислорода. Примерами являются метокси, этокси, пропокси, изопропокси, циклопропилокси, циклогексилокси и т.п. “Низший алкокси” означает группы, содержащие от одного до четырех атомов углерода.
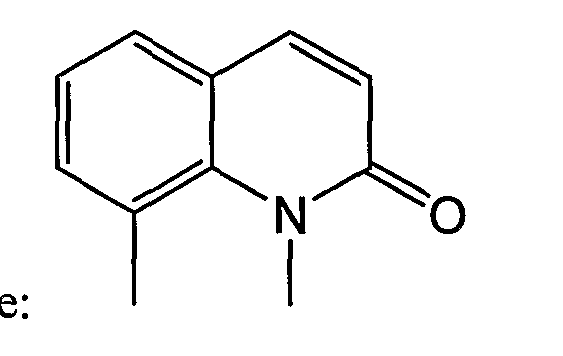
“Оксаалкил” означает алкильные остатки, в которых один или несколько атомов углерода (и связанные с ними атомы водорода) заменены атомом кислорода. Примерами являются метоксипропокси, 3,6,9-триоксадецил и т.п. Термин “оксаалкил” понятен специалистам [см. каталог Naming and Indexing of Chemical Substances for Chemical Abstracts, опубликованный Американским химическим обществом, см. §196, где 127(a) приводится без ограничений] и означает соединения, в которых атом кислорода связан со смежными атомами посредством простой связи (с образованием эфирных связей). Аналогичным образом, тиаалкил и азаалкил означают алкильные остатки, в которых одни или несколько атомов углерода заменены атомом серы или азота, соответственно. Примерами являются этиламиноэтил и метилтиопропил. Термин “оксо”, относящийся к заместителю, означает кислород, связанный двойными связями (карбонил). Так, например, 2-оксохинолин согласно изобретению имеет следующую формулу:
“Ацил” означает группы, имеющие от 1 до 8 атомов углерода в прямой цепи, разветвленной цепи и циклической структуре, в насыщенной, ненасыщенной и ароматической структуре и в их комбинациях, и присоединенные к исходной структуре посредством карбонильной функциональной группы. Один или несколько атомов углерода в ацильном остатке могут быть заменены азотом, кислородом или серой, при условии, что положение присоединения к исходной структуре находится у карбонила. Примерами являются формил, ацетил, пропионил, изобутирил, трет-бутоксикарбонил, бензоил, бензилоксикарбонил и т.п. Низший ацил означает группы, содержащие от одного до четырех атомов углерода.
“Арил” и “гетероарил” означают 5- или 6-членное ароматическое или гетероароматическое кольцо, содержащее 0-3 гетероатомов, выбранных из O, N или S; бициклическую 9- или 10-членную ароматическую или гетероароматическую циклическую систему, содержащую 0-3 гетероатомов, выбранных из O, N или S; или трициклическую 13- или 14-членную ароматическую или гетероароматическую циклическую систему, содержащую 0-3 гетероатомов, выбранных из O, N или S. Ароматическими 6-14-членными карбоциклическими кольцами являются, например, бензол, нафталин, индан, тетралин и флуорен, а 5-10-членными ароматическими гетероциклическими кольцами являются, например, имидазол, пиридин, индол, тиофен, бензопиранон, тиазол, фуран, бензимидазол, хинолин, изохинолин, хиноксалин, пиримидин, пиразин, тетразол и пиразол.
“Арилалкил” означает алкильный остаток, связанный с арильным кольцом. Примерами являются бензил, фенетил и т.п.
“Замещенные алкил, арил, циклоалкил, гетероциклил” и т.п. означают алкил, арил, циклоалкил или гетероциклил, где 1-3 атомов Н в каждом остатке заменены галогеном, низшим алкилом, галогеналкилом, гидрокси, низшим алкокси, карбокси, карбоалкокси (также называемым алкоксикарбонилом), карбоксамидо (также называемым алкиламинокарбонилом), циано, карбонилом, нитро, амино, алкиламино, диалкиламино, меркапто, алкилтио, сульфоксидом, сульфоном, ациламино, амидино, фенилом, бензилом, гетероарилом, фенокси, бензилокси или гетероарилокси. В нижеследующем описании настоящей заявки, метилендиокси и этилендиокси упомянуты как заместители. Метилендиокси связан с соседними атомами углерода на кольце, а этилендиокси может быть связан либо с соседними атомами углерода на кольце, либо с тем же самым атомом углерода, образуя, тем самым, спиродиоксол (кеталь), аналогично спиротиазолидинилу. Различными вариантами таких соединений являются соединения 114, 144 и 160.
Термин “галоген” означает фтор, хлор, бром или йод.
Термин “пролекарство” означает соединение, которое in vivo образует более активное соединение. Активация in vivo может происходить под действием химической реакции или посредством фермента. В активации in vivo может также участвовать микрофлора желудочно-кишечного тракта.
При определении символов было указано, что A и B представляют собой пару конденсированных 5-, 6- или 7-членных колец, и что конденсированная система колец A/B может содержать от нуля до четырех гетероатомов, выбранных из атомов азота, кислорода и серы. При этом подразумевается, что указанные кольца могут иметь различную степень насыщенности, то есть они могут быть полностью насыщенными или ароматическими. Причем, предпочтительными являются ароматические и частично ненасыщенные кольца.
При определении различных заместителей предусматривается, что указанные конденсированные кольца могут быть дополнительно замещены 0-4 заместителями, независимо выбранными из списка определяемых заместителей. Замещения такого типа отражены на проиллюстрированной ниже структуре. В этом примере, указанные конденсированные кольца замещены тремя заместителями: -СН3-, -ОН и оксо:
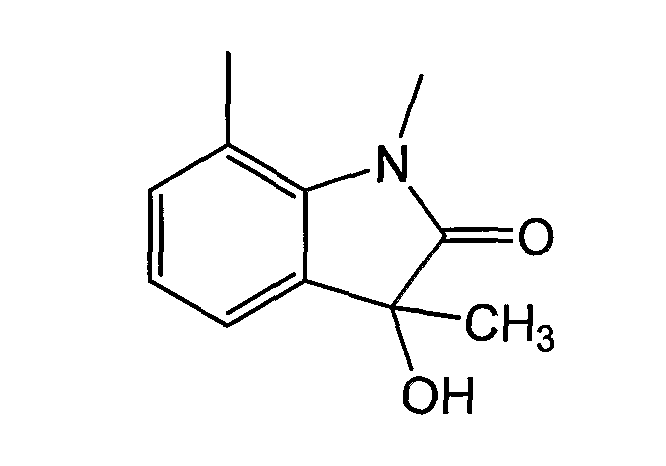
Следует отметить, что соединения согласно изобретению могут присутствовать в радиоактивно меченной форме, т.е. эти соединения могут содержать один или несколько атомов, имеющих атомную массу или массовое число, отличающееся от атомной массы или массового числа обычных элементов, встречающихся в природе. Радиоизотопами водорода, углерода, фосфора, фтора и хлора являются 2H, 3H, 13C, 14C, 15N, 35S, 18F и 36Cl, соответственно. Соединения, содержащие эти радиоизотопы и/или другие радиоизотопы других атомов, входят в объем настоящего изобретения. Для облегчения получения и детекции соединений особенно предпочтительными являются такие радиоизотопы, как тритий, т.е. 3H, и углерод-14, то есть 14C. Радиоактивно меченные соединения формулы Iа согласно изобретению и их пролекарства обычно получают методами, хорошо известными специалистам. Обычно, такие радиоактивно меченные соединения получают способами, описанными в разделе “Примеры” и “Схемы”, путем замены не меченного радиоизотопом реагента легко доступным радиоактивно меченным реагентом.
Используемый здесь и понятный специалистам термин “соединение” включает соли, сольваты и комплексы этого соединения, содержащие кристаллы и включения.
Термин “сольват" означает соединение формулы I в твердом состоянии, где молекулы подходящего растворителя включены в кристаллическую решетку. Растворитель, подходящий для терапевтического введения, хорошо переносится организмом во вводимых дозах. Примерами подходящих растворителей для терапевтического введения являются этанол и вода. Если растворителем является вода, то сольват называется гидратом. В общих чертах, сольваты получают путем растворения соединения в соответствующем растворителе и выделения полученного сольвата путем охлаждения или с использованием антирастворителя. Полученный сольват обычно осушают или подвергают азеотропной перегонке в условиях окружающей среды. Ко-кристаллы представляют собой комбинации двух или нескольких отличающихся молекул, расположенных так, что они образуют уникальную кристаллическую форму, физические свойства которой отличаются от свойств ее чистых компонентов. В последнее время, фармацевтические ко-кристаллы представляют все больший интерес с точки зрения повышения растворимости, облегчения приготовления и улучшения биологической доступности таких лекарственных средств, как итраконазол [см. Remenar et al. J.Am.Chem.Soc. 125, 8456-8457 (2003)] и флуоксетин. Комплексы включения описаны в руководстве Remington: The Science and Practice of Pharmacy 19th Ed. (1995) volume 1, page 176-177. Наиболее часто используемыми комплексами включения являются комплексы с циклодекстринами, и все указанные природные и синтетические циклодекстриновые комплексы, содержащие или не содержащие добавки и полимеры и описанные в патентах США №№ 5324718 и 5472954, входят в объем притязаний настоящего изобретения. Руководство Remington и патенты '718 и '954 вводятся в настоящее описание посредством ссылки.
Термин “способы лечения или предупреждения” означают уменьшение интенсивности, предупреждение или ослабление симптомов и/или эффектов заболеваний, ассоциированных с липидным обменом. Используемый здесь термин “предупреждение” означает предварительное введение лекарственного средства в целях предотвращения или ослабления острого приступа заболевания. Среднему специалисту-медику (на которого рассчитано описание способа, заявленного в настоящем изобретении) известно, что термин “предупреждение” не является абсолютным термином. В медицине, под этим термином подразумевается профилактическое введение лекарственного средства в целях значительного снижения вероятности возникновения или тяжести данного состояния, и в настоящей заявке указанный термин используется именно в этом смысле. Используемый здесь термин “лечение” пациента также включает и профилактику. В этой связи, в описании настоящей заявки имеются ссылки на различные публикации. Описания этих публикаций во всей своей полноте вводятся в настоящее изобретение посредством ссылки, если на это имеются указания.
Термин “млекопитающее” используется здесь в его обычном смысле, определяемом в словарях. Человек входит в группу млекопитающих и является предпочтительным млекопитающим, для лечения которого могут быть применены описанные способы.
Описанные здесь соединения могут содержать асимметрические центры и, тем самым, могут образовывать энантиомерные, диастереомерные и другие стереоизомерные формы. С точки зрения абсолютной стереохимической конфигурации, каждый хиральный центр может быть определен как (R)- или (S)-. Настоящее изобретение включает все такие возможные изомеры, а также их рацемические и оптически чистые формы. Оптические активные (R)- и (S)- или (D)- и (L)-изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов, либо эти реагенты могут быть разделены стандартными методами. Если описанные здесь соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если это не оговорено особо, то предусматривается, что указанные соединения включают геометрические Е- и Z-изомеры. Аналогичным образом, в настоящее изобретение также входят все таутомерные формы.
Графические представления используемых здесь рацемических, амбискалемных и скалемных или энантиомерно чистых соединений были взяты из работы Maehr J. Chem. Ed. 62, 114-120 (1985): заштрихованные и не заштрихованные клинообразные линии используются для обозначения абсолютной конфигурации хирального элемента; волнистые линии и одинарные тонкие линии указывают на то, что связь, которая может быть образована, не имеет какой-либо определенной стереохимии; сплошные и пунктирные жирные линии являются геометрическими дискрипторами, указывающими на относительную конфигурацию, иллюстрирующую и обозначающую рацемический характер; а клинообразные, пунктирные или прерывистые линии обозначают энантиомерно чистые соединения конкретно не определенной абсолютной конфигурации.
Представленные здесь конфигурации любой углерод-углеродной двойной связи выбраны лишь для иллюстрации и, если это не оговорено особо, не означают какую-либо конкретную конфигурацию. Так, например, вышеуказанная углерод-углеродная двойная связь, произвольно выбранная как E, может быть Z, E или смесью двух этих изомеров в любом соотношении.
Во всем описании настоящей заявки используются термины, относящиеся к “защите” функциональных групп, к “снятию защиты” с функциональных групп и к “защищенным” функциональным группам. Эти термины хорошо известны среднему специалисту в данной области и применяются при описании способов, которые включают последовательную обработку несколькими реагентами. В этом контексте, защитная группа означает группу, используемую для маскировки функциональной группы во время проведения стадии данного процесса, в которой данная группа подвергается той или иной реакции, но в которой эта реакция является нежелательной. Защитная группа предотвращает реакцию на этой стадии, но затем она может быть удалена для того, чтобы исходная функциональная группа стала снова доступной для реакции. Удаление защитной группы или “снятие защиты” осуществляют после завершения реакции или реакций, в которых участие данной функциональной группы является нежелательным. Так, например, если в способах согласно изобретению точно известна последовательность реакций, в которых участвуют данные реагенты, то средний специалист в этой области может легко определить группы, которые могут быть подходящими в качестве “защитных групп”. Группы, подходящие для использования в этих целях, обсуждаются в известных химических справочниках, таких как руководство Protective Groups в Organic Synthesis by T.W.Greene [John Wiley & Sons, New York, 1991], которое вводится в настоящее описание посредством ссылки. Особое внимание следует уделить главам: "Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols" (страницы 10-86).
Сокращения Me, Et, Ph, Tf, Ts и Ms означают метил, этил, фенил, трифторметансульфонил, толуолсульфонил и метансульфонил, соответственно. Исчерпывающий список сокращений, используемых химиками-органиками (то есть средними специалистами в данной области), приводится в первом издании каждого тома Journal of Organic Chemistry. Список, который обычно приводится в таблице, озаглавленной “Стандартный список сокращений” (“Stаndard List of Abbreviations"), вводится в настоящее описание посредством ссылки.
Хотя соединения формулы I могут быть введены в виде исходного химического соединения, однако, предпочтительно, чтобы эти соединения были введены в виде фармацевтической композиции. В соответствии с другим своим аспектом, настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль или сольват, в комбинации с одним или несколькими фармацевтическими носителями, и, необязательно, с одним или несколькими другими терапевтическими ингредиентами. Такие носители должны быть “приемлемыми” в смысле их совместимости с другими ингредиентами указанной композиции и не должны оказывать негативное воздействие на реципиента.
Указанными препаратами являются препараты, подходящие для перорального и парентерального введения (включая подкожное, чрескожное, внутримышечное, внутривенное и внутрисуставное введение), ректального и местного введения (включая кожное, трансбуккальное, подъязычное и внутриглазное введение). Наиболее подходящий способ введения может зависеть от состояния и расстройства данного реципиента. Обычно, указанные препараты могут быть получены в виде унифицированной лекарственной формы и могут быть приготовлены любым методом, хорошо известным фармацевтам. Все эти методы включают стадию смешивания соединения формулы I или его фармацевтически приемлемой соли или сольвата (“активного ингредиента”) с носителем, который включает одно или несколько вспомогательных ингредиентов. В общих чертах, указанные препараты получают путем тщательного и равномерного смешивания активного ингредиента с жидкими носителями или с тонкодисперсными твердыми носителями, или с теми и другими, а затем, если это необходимо, формования продукта с получением нужного препарата.
Препараты согласно изобретению, подходящие для перорального введения, могут быть приготовлены в виде дискретных дозированных единиц, таких как капсулы, саше или таблетки, каждая из которых содержит предварительно определенное количество активного ингредиента; порошки (включая порошки, состоящие из микрочастиц и наночастиц) или гранулы; растворы или суспензии в водной или в безводной жидкости; или жидкие эмульсии типа “масло в воде” или “вода в масле”. Активный ингредиент может быть приготовлен в виде болюса, лекарственной кашки или пасты.
Таблетка может быть приготовлена путем прессования или формования, необязательно, с одним или несколькими вспомогательными ингредиентами. Спрессованные таблетки могут быть получены путем прессования активного ингредиента в сыпучей форме, такой как порошок или гранулы, необязательно, в смеси со связующим агентом, замасливателем, инертным разбавителем, поверхностно-активным веществом или диспергирующим агентом, в подходящем аппарате. Сформованные таблетки могут быть изготовлены путем формования смеси порошкообразного соединения, увлажненного инертным жидким разбавителем, в подходящем аппарате. Такие таблетки могут, но необязательно, иметь покрытия или насечки и могут быть получены в виде лекарственных форм с замедленным, пролонгированным или регулируемым высвобождением описанного здесь активного ингредиента.
Фармацевтические композиции могут содержать “фармацевтически приемлемый инертный носитель”, и этот термин включает один или несколько инертных наполнителей, которыми являются крахмалы, полиолы, гранулирующие агенты, микрокристаллическая целлюлоза, разбавители, замасливатели, связующие вещества, дезинтеграторы и т.п. Если это необходимо, то на описанные здесь лекарственные композиции типа таблеток могут быть нанесены водные или безводные покрытия стандартными методами. Термин “фармацевтически приемлемый носитель” также включает средство с регулируемым высвобождением.
Композиции согласно изобретению могут также, но необязательно, включать и другие терапевтические ингредиенты, а именно вещества, предотвращающие спекание, консерванты, подсластители, окрашивающие средства, отдушки, эксикаторы, пластификаторы, красители и т.п. Совершенно очевидно, что для гарантии стабильности данного препарата, любой такой необязательный ингредиент должен быть совместим с соединением согласно изобретению.
Интервал доз для взрослого человека обычно составляет от 0,1 мкг до 10 г/день для перорального введения. Таблетки или другие формы, полученные в виде дискретных дозированных единиц, в основном, могут содержать соединение согласно изобретению в количестве, эффективном при такой дозе, либо, если эти препараты предназначены для многократного приема, то они могут содержать, например, от 0,1 мг до 500 мг, а обычно, примерно, от 5 мг до 200 мг данного соединения. Точное количество соединения, вводимого пациенту, должно быть назначено лечащим врачом. Однако применяемая доза будет зависеть от ряда факторов, включая возраст и пол пациента, конкретное расстройство, подвергаемое лечению, и его тяжесть. Частота введения будет зависеть от фармакодинамических свойств отдельного соединения и препарата данной лекарственной формы и может быть оптимизирована методами, хорошо известными специалистам (например, путем введения таблеток регулируемого или пролонгированного действия, таблеток с энтеросолюбильным покрытием и т.п.).
Комбинированная терапия может быть осуществлена путем введения двух или более средств, каждое из которых получают и вводят отдельно, либо путем введения двух или более средств, присутствующих в одном препарате. Комбинированная терапия может также включать и другие комбинации. Так, например, два лекарственных средства могут быть приготовлены вместе и введены в комбинации с отдельным препаратом, содержащим третье лекарственное средство. При комбинированной терапии, два или более средств могут быть, но необязательно, введены одновременно.
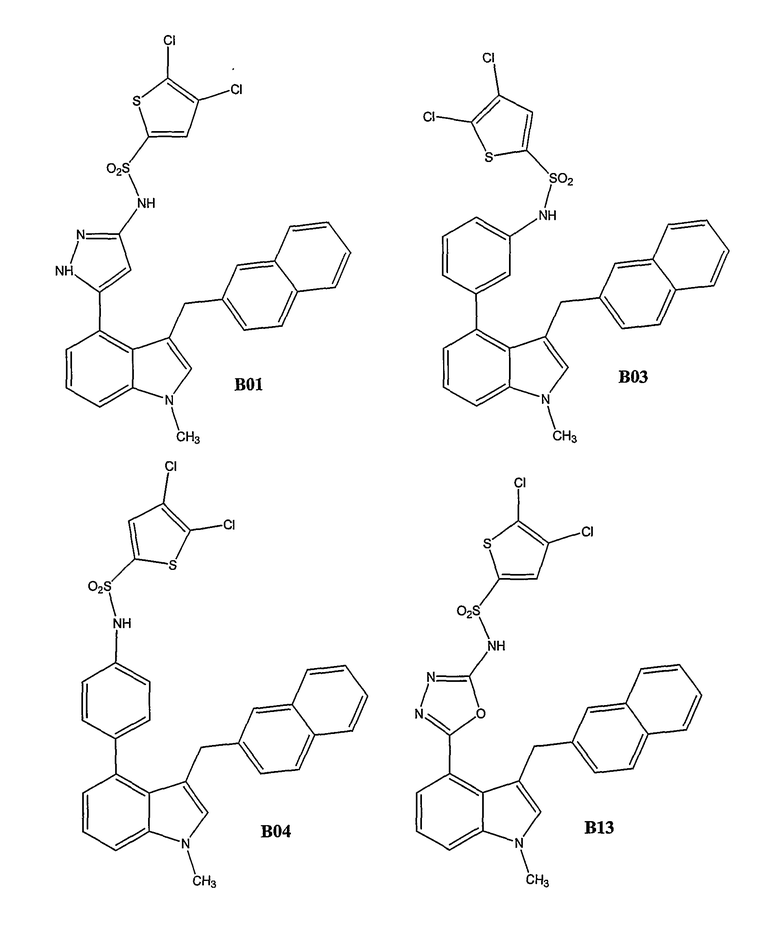
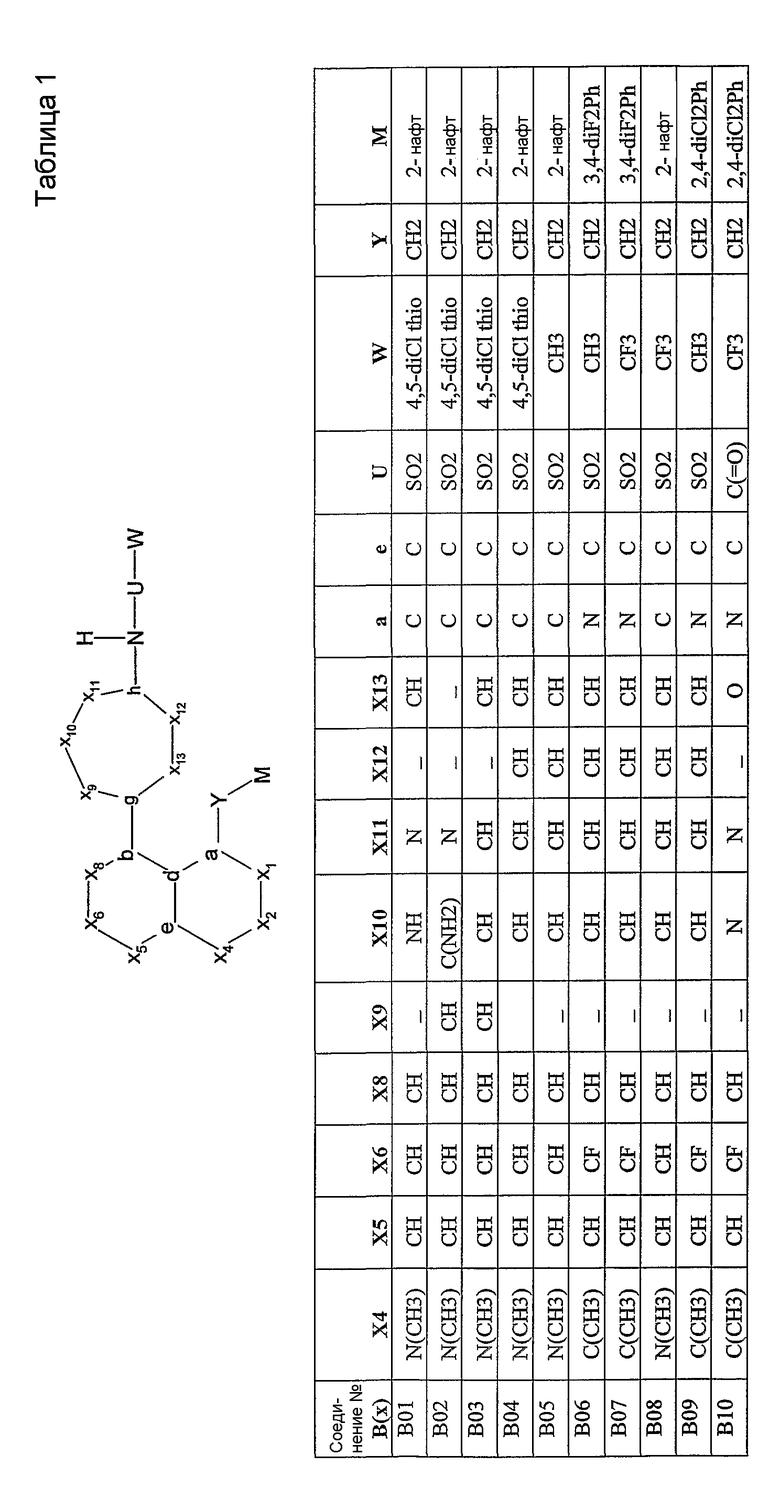
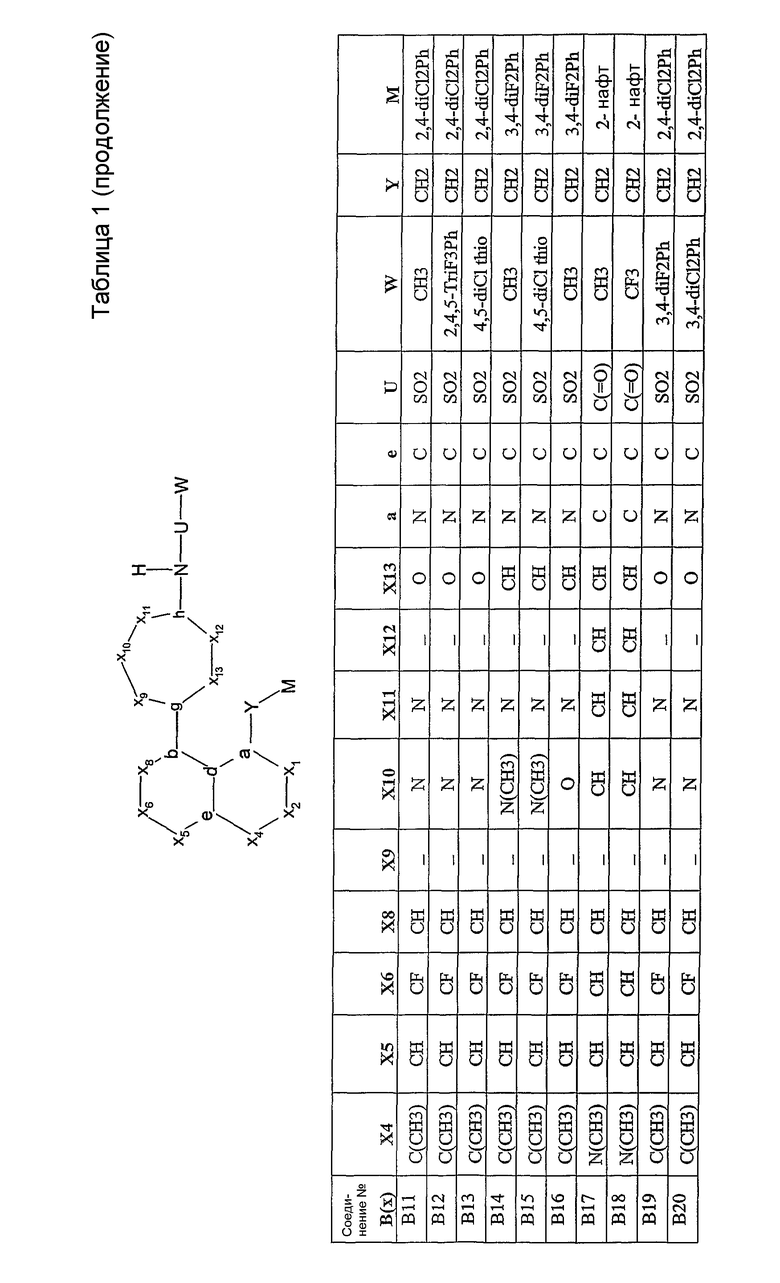
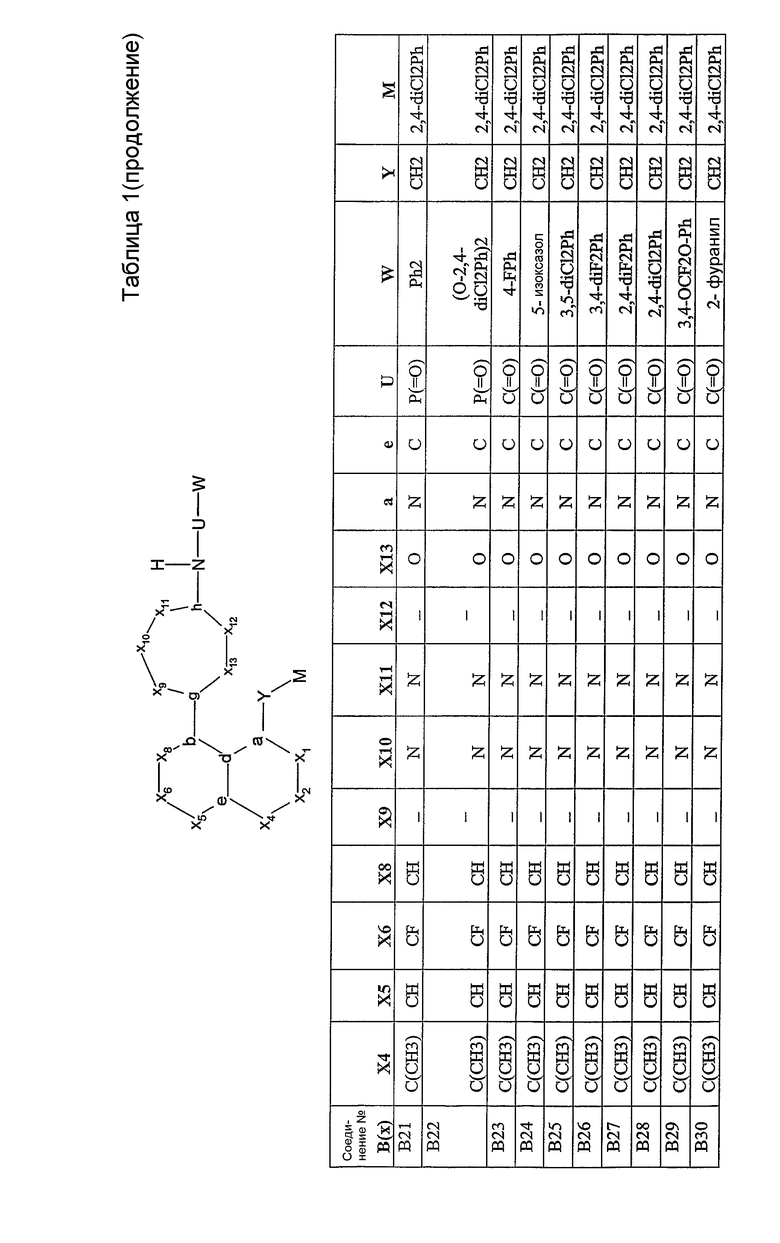
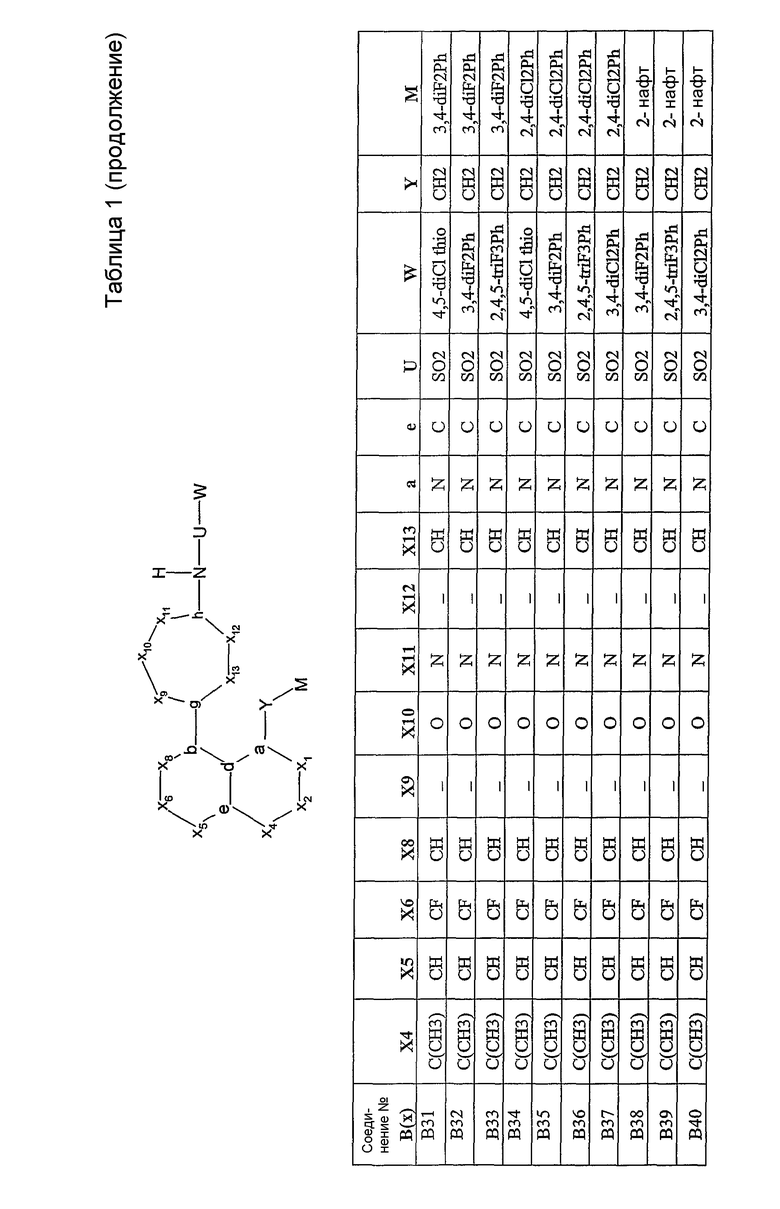
Было синтезировано приблизительно 300 репрезентативных соединений согласно общей концепции. Их структуры представлены в двух совместно рассматриваемых заявках, имеющих одну и ту же дату подачи, и озаглавленных "SULFONAMIDE PERI-SUBSTITUTED BICYCLICS FOR OCCLUSIVE ARTERY DISEASE" и "CARBOXYLIC ACID PERI-SUBSTITUTED BICYCLICS FOR OCCLUSIVE ARTERY DISEASE". Обе этих заявки вводятся в описание настоящего изобретения посредством ссылки. Примерами подвидов соединений, заявленных в настоящем изобретении, являются соединения В01, В03, В04 и В13.
Соединения согласно изобретению могут быть проанализированы на их связывание с простаноидными рецепторами ЕР3 методом, описанным Abramovitz et al. [Bioch. Biophys. Acta, 1473, 285-293 (2000)]. Все примеры соединений, приведенных в нижеследующих таблицах, были синтезированы, охарактеризованы и протестированы на связывание с рецептором ЕР3.
Соединения согласно изобретению могут быть также проанализированы на их влияние на агрегацию тромбоцитов in vitro. В экспериментах, проводимых с человеческими тромбоцитами, у доноров, которые не принимали пищу в течение ночи, брали цельную кровь. Каждый эксперимент проводили с использованием пробы крови, взятой у одного конкретного индивидуума. В экспериментах, проводимых с тромбоцитами грызунов, цельную кровь брали из сердца самок мышей или самцов крыс, анестезированных изофлураном (Abbott). В каждом эксперименте, проводимом на крысах и мышах, кровь брали у двух или десяти отдельных грызунов, соответственно. Во всех случаях, кровь собирали в пробирки с 3,8% цитратом натрия (Greiner Bio-one). Обогащенную тромбоцитами плазму (PRP) получали путем центрифугирования при 100·g в течение 15 мин при 25°С - для человека, при 150·g - для крыс или при 80·g в течение 10 мин - для мышей. Обедненную тромбоцитами плазму получали путем центрифугирования остальной части крови при 2400·g в течение 10 мин при 25°С. После подсчета числа тромбоцитов на счетчике Autocounter (Model 920 EO, Swelab), эти тромбоциты разводили, если это необходимо, до нужной концентрации (200000-300000 тромбоцитов/мкл) с использованием 0,9% изотонического раствора NaCl (Braun).
Агрегацию тромбоцитов определяли путем измерения оптической плотности с использованием агрегометра тромбоцитов, снабженного магнитной мешалкой непрерывного действия (Model 490, Chronolog Cop., Havertown, Pennsylvania, USA) в объеме 500 мкл на кювету. Во время проведения эксперимента, раствор тромбоцитов постоянно помешивали путем легкого встряхивания с горизонтальным сдвигом. В качестве ускорителей агрегации тромбоцитов использовали коллаген (Sigma) и PGE2 или сульпростон (Cayman Chemicals). Соединения, используемые в этом анализе, растворяли и хранили в 100% растворе ДМСО. После разведения, конечная концентрация ДМСО в этом анализе составляла менее 0,1% об./об. В этом анализе было определено, что такая концентрация ДМСО не ингибирует агрегацию тромбоцитов. Агенты, ускоряющие агрегацию тромбоцитов, и соединения, протестированные на связывание с ЕР3, разводили в изотоническом растворе в нужной концентрации. Для вычисления концентрации тест-соединения, необходимого для 50%-ного ингибирования агрегации тромбоцитов (IC50), применяли метод сигмоидальной нелинейной регрессии. Величины IC50 для тест-соединений вычисляли с помощью компьютерной программы GraphPad Prism 3.02 для Windows (GraphPad Software, San Diego California USA).
Анализ на тромбоэмболию легких: неанестезированным самкам мышей С57BL/6 перорально вводили указанные тест-соединения, и через 30 мин у этих животных индуцировали тромбоэмболию путем инъекции арахидоновой кислоты в хвостовую вену. Выживаемость оценивали через один час после введения арахидоновой кислоты, поскольку у мышей, которые выживали в течение указанного периода времени, обычно наблюдалось полное выздоровление. Инъекцию арахидоновой кислоты делали в боковую хвостовую вену мышей, тело которых быстро нагревали под нагревательной лампой (для расширения хвостовых вен, облегчающего введение инъекции). Для введения дозы использовали инсулиновый шприц емкостью 0,5 мл (от Becton Dickinson). Объем доз данного тест-соединения и арахидоновой кислоты корректировали в соответствии с массой мыши (объем перорально вводимых (p.o.) доз тест-соединений и внутривенно вводимых (i.v.) доз раствора арахидоновой кислоты составлял 10 мкл и 5 мкл на один грамм массы тела, соответственно). Затем оценивали степень выживаемости мышей с моделью тромбоэмболии, которые были обработаны тест-соединениями (100 мг/кг, перорально).
В общих чертах, соединения согласно изобретению могут быть получены методами, проиллюстрированными в общих схемах реакций, например, описанных ниже, или их различными модификациями, с применением доступных исходных веществ, реагентов и стандартных методов синтеза. В этих реакциях могут быть использованы варианты, которые, сами по себе, являются известными, но не упоминаются в описании настоящей заявки. Что касается исходных веществ, то такие вещества, в случае соответствующим образом замещенных конденсированных циклических соединений А/В, могут быть коммерчески доступными или могут быть получены методами, хорошо известными специалистам в данной области.
В основном, соединения формулы I могут быть получены из бициклических систем, соответствующим образом замещенных функциональными группами, как показано на схемах 1-16. В частности, если в узловом положении “a" присутствует атом азота, то после присоединения функциональной группы в этом положении с последующим проведением опосредуемой палладием реакции сочетания Судзуки получают ариламиновое производное G3, которое затем подвергают дериватизации с получением связанного с арилом амида, сульфонамида или фосфорамида G5 (схема 1). Альтернативно, N-функционализированное промежуточное соединение, посредством проведения опосредуемой палладием реакции сочетания Судзуки, превращают в производное арилового эфира G6, из которого, после гидролиза и реакции взаимодействия с Ph2P(O)N3 с продуцированием in situ ацилазида, образуется продукт реакции перегруппировки Курциуса - ариламин G8. Ацилазид может быть также получен из сложного эфира G6 с использованием гидразина с последующим проведением реакции взаимодействия с изоамилнитритом и с образованием промежуточного ацилазида. Затем амин G8 превращают в соединение G8, как показано на схеме 2. Кислота G7 может быть также подвергнута реакции взаимодействия, например, с сульфонамидом, с получением ацилсульфонамида G9. В нижепривиденных схемах, R1 представляет собой остаток, который в формуле изобретения представлен как М, а R2 представляет собой остаток, который в формуле изобретения представлен как R1.
Схема 1
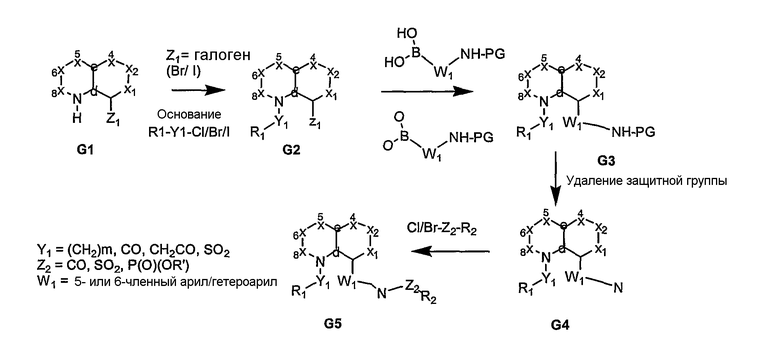
Схема 2
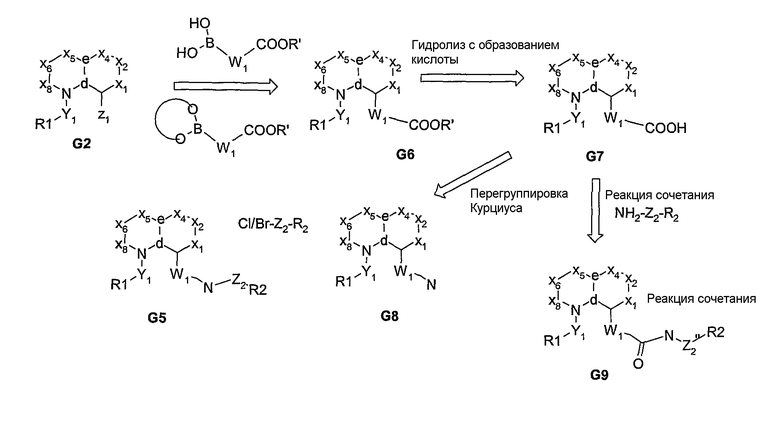
Если у углерода в узловом положении “b” присутствует сложноэфирная или нитрильная функциональная группа, то после реакции с анионом, образующимся in situ из ацетонитрила, образуется соответствующий β-гидроксиакрилонитрил G11 (схема 3) или β-аминоакрилонитрил G15 (схема 4), соответственно. Затем эти промежуточные соединения могут быть подвергнуты реакции циклизации с получением азотсодержащих 5- или 6-членных гетероциклических аминов (G12), которые превращают в дериватизированный амином продукт G13 (схемы 3 и 4). Альтернативно, посредством реакции Хека, из ароматической галогенидной бициклической системы может быть получен α,β-ненасыщенный нитрил, из которого, посредством реакции взаимодействия с гидразином или амидином, получают дигидрогетероциклы, которые после реакции окислительной ароматизации превращаются в гетероциклические амины G12, как показано на схеме 5.
Схема 3
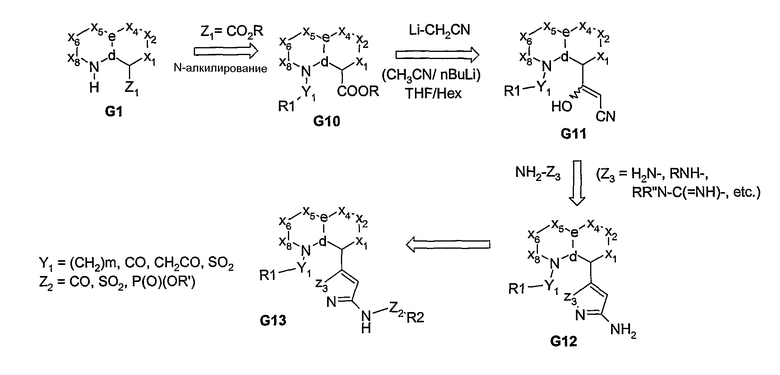
Схема 4
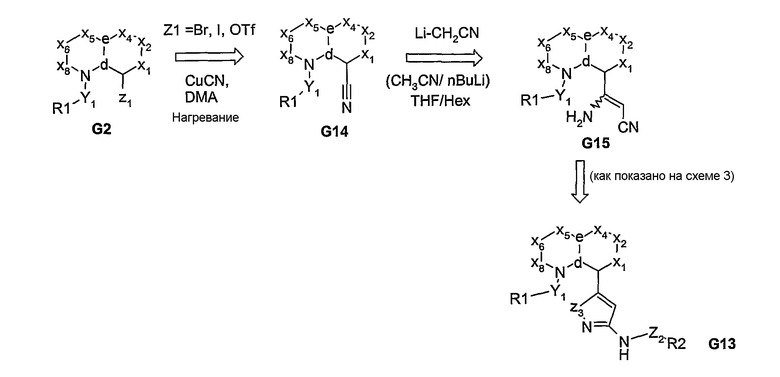
Схема 5
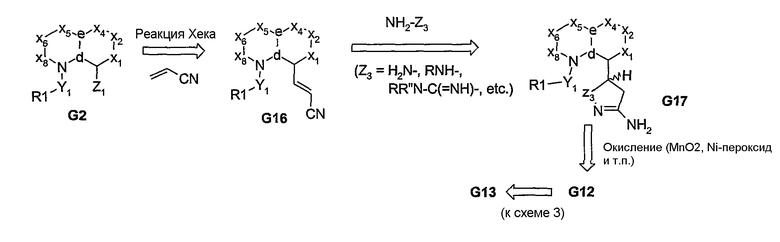
После реакции взаимодействия β-гидрокси- или β-аминоакрилонитриловых производных (G11 и G15, соответственно) с гидроксиламином получают аминоизоксазоловое производное G18, из которого затем получают продукт G19 с региоспецифичностью, показанной на схеме 6.
Схема 6
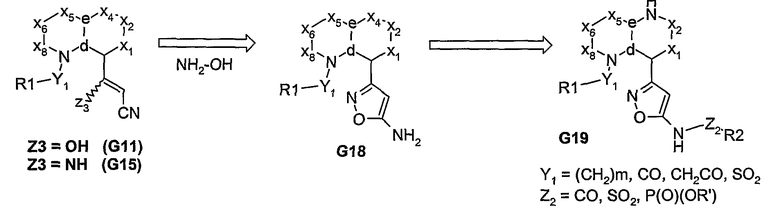
После гидролиза, из бициклических сложноэфирных систем получают соответствующие карбоновые кислоты. Благодаря разнообразию этих промежуточных соединений из них может быть получен широкий ряд 5-членных азоловых производных, как показано на схеме 7. Эта кислота, путем проведения реакции в одном сосуде, может быть превращена в аминотиадиазол (G22, где Z4=S). Соответствующий аминооксадиазол (G22, где Z4=О) может быть получен из соответствующего гидразида (G23) после его обработки бромцианом. Альтернативно, кислота G20 может быть подвергнута реакции взаимодействия с семикарбазидом с получением промежуточного соединения G21, которое может быть превращено в 5- или 6-членный гетероциклический амин, который может быть затем функционализирован с получением продуктов, представленных формулой I.
Схема 7
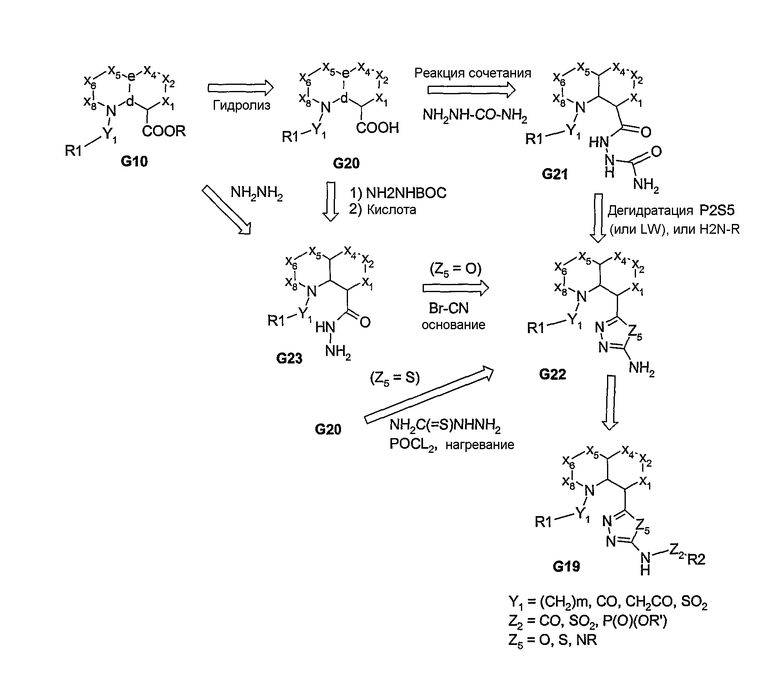
В вышеуказанных примерах, если в узловом положении “а” присутствует N, то это означает, что была введена одна из пери-замещенных линкерных групп. Если оба заместителя в бициклической системе связаны посредством атома углерода, то, как и в предыдущих примерах, может быть введен связанный с арилом амин и функционализированная аминовая часть. В случае бициклических систем, которые являются электрофильными по своей природе, могут быть введены вторые С-связанные пери-заместители, что приводит к образованию заместителей широкого ряда, у которых присоединение к узловому атому углерода осуществляется посредством гетероатома. Соединения, в которых присоединение к атому углерода осуществляется через атом серы, представлены на схеме 8. Благодаря высокой нуклеофильности тиолов использование таких систем, как G24, позволяет вводить вторые пери-заместители. Образование тиоэфирного линкера обеспечивает последующее получение сульфоксидных или сульфоновых продуктов, т.е. получение биарильных аналогов, несущих сульфид, сульфоксид или сульфоны в качестве линкеров. На схеме 9 представлены варианты, в которых аналоги химических соединений, описанных на схемах 3 и 4, придают вводимым реагентам и промежуточным соединениям гибкость структуры и, тем самым, обеспечивают получение большого разнообразия продуктов.
Пример реакции, которая позволяет вводить ацильный фрагмент (несущий группу R2) посредством электрофильной реакции, представлен на схеме 16. Такая реакция приводит к образованию аналогов, представленных G90 и G91. Бензилкарбонильная группа, присутствующая в G90 и G91, может быть затем дериватизирована, например, путем восстановления до спирта или СН2, путем образования оксима и т.п.
Схема 8
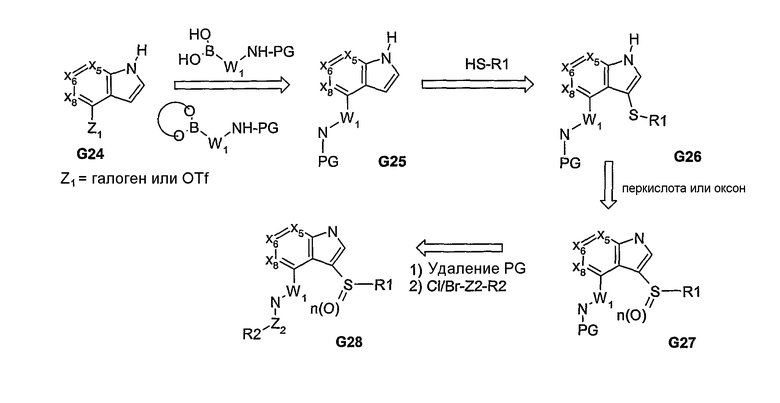
Схема 9
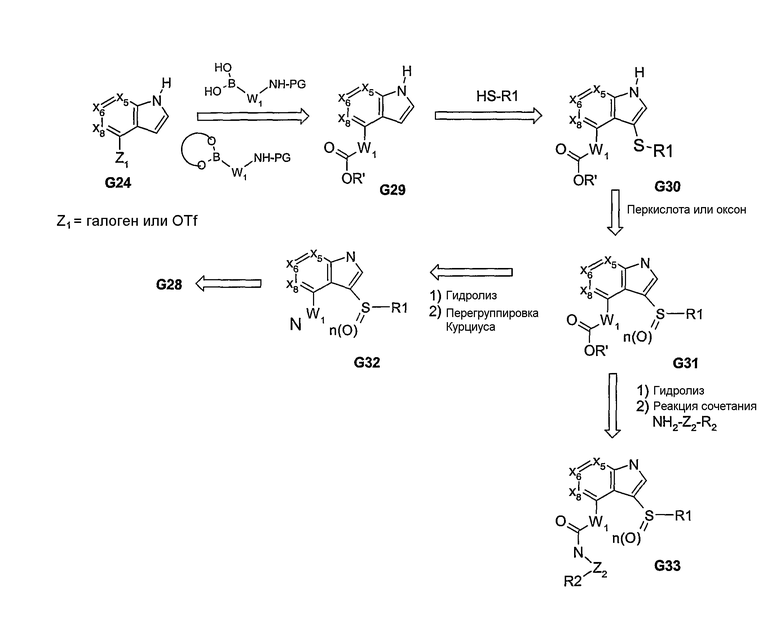
Для получения соответствующих аза- или окса-связанных арильных/гетероарильных/алкильных групп (R1) могут быть использованы реакционно-способные промежуточные соединения, родственные изатину, как показано в G37, и происходящие от G34 (схема 10). Как показано на схеме 10, из промежуточного соединения 37 получают различные аза-связанные соединения, каждое из которых образуется в результате присоединения связанной с углеродом группы к бициклической системе. Другие варианты происходящих от изатина промежуточных соединений представлены на схеме 11. Этот способ позволяет получать пери-замещенные арильные бициклические соединения, которые имеют функциональные группы, связанные через атомы углерода и азота с основной бициклической системой. Кроме того, доступность ключевого промежуточного соединения G48, содержащего реакционно-способный карбонил, расположенный на значительном расстоянии от пери-заместителей, заканчивающихся группой R1 и R2, позволяет специалисту в данной области проводить ряд химических реакций с получением продуктов, указанных на схеме 11. Такие химические реакции, например, образование кеталя, помимо карбонила, и реакция взаимодействия с DAST позволяют получать аналоги, содержащие различные функциональные группы, как показано в G47-G52. Из аналогов, представленных на схемах 10 и 11, могут быть также получены бициклические системы, содержащие одно или оба кольца, которые не являются ароматическими.
Схема 10
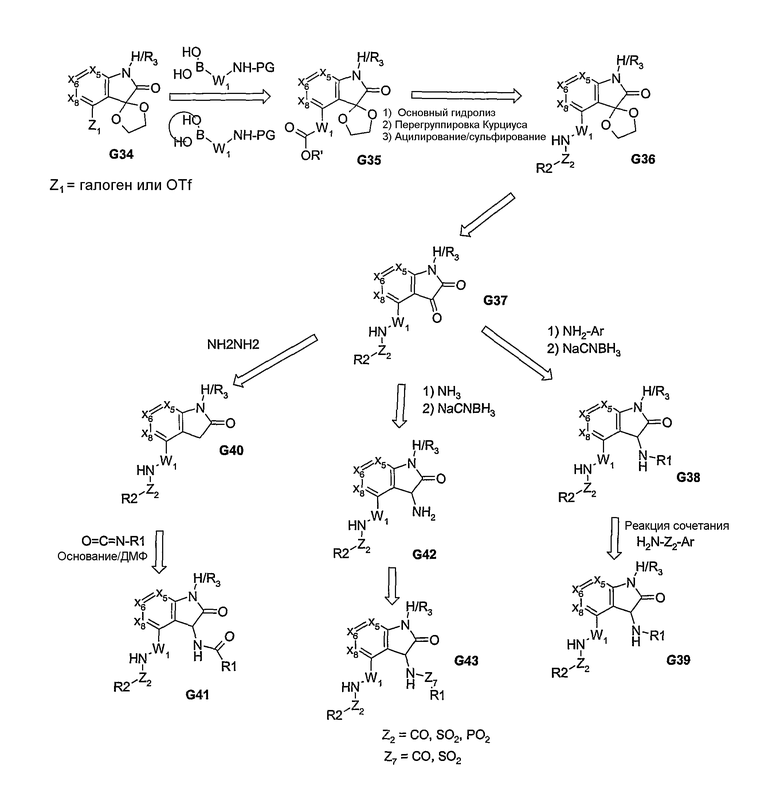
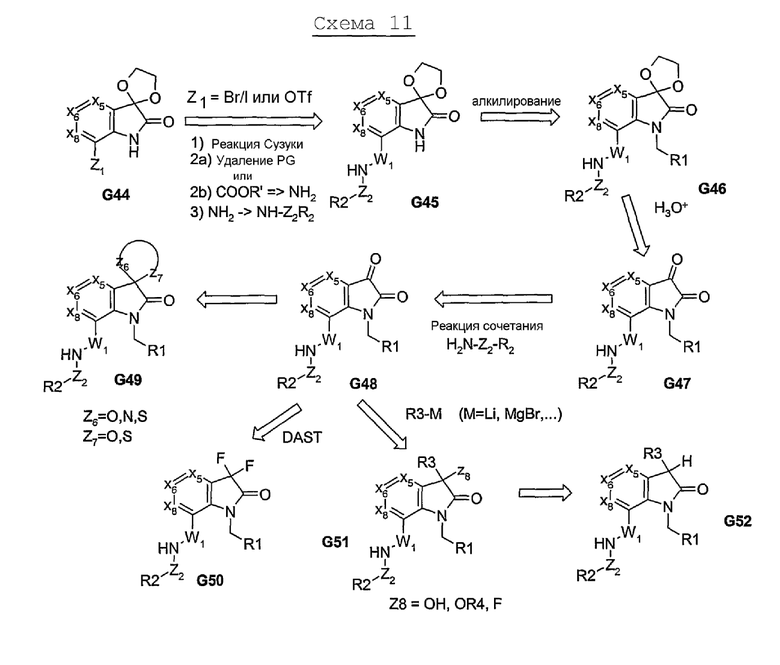
Пути синтеза, описанные выше, в основном, предусматривают использование бициклической системы, которую соответствующим образом дериватизируют с получением соединений, представленных формулой I. Нижеследующие химические реакции позволяют вводить, по меньшей мере, один из пери-фрагментов в процессе образования бициклической системы. Химические реакции на схеме 12 включают трехкомпонентную реакцию конденсации, посредством которой α,γ-дикетоэфир (G54), после его реакции взаимодействия с альдегидом и первичным амином, образует моноциклический продукт G63. Этот продукт G63, после его взаимодействия, например, с гидразином (или монозамещенным гидразином), образует пери-замещенную бициклическую систему (в данном случае, 5,5-циклическую систему, как показано в G64), из которой затем получают аналог G56. α,β-ненасыщенный сложный эфир может быть превращен в соответствующий α,β-ненасыщенный нитрил, из которого, посредством химических реакций, проиллюстрированных на схеме 5, получают 5- или 6-членную гетероциклическую систему, связанную с 5:5-бициклической системой, и тем самым, получают соединения, представленные в G58, как показано на формуле I.
Схема 12
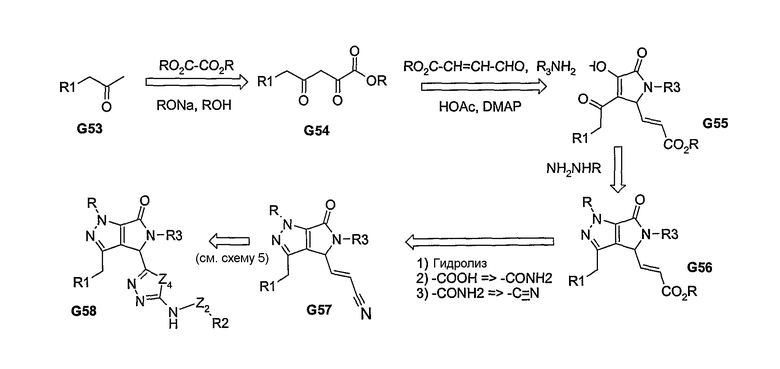
Другие примеры химических реакций, которые приводят к образованию бициклических систем, представлены на схемах 13 и 14. Эти примеры иллюстрируют синтез бензимидазоловых структур. Для получения пери-замещенной системы, группу Rl региоспецифически вводят на стадии получения соединений G61-G62. На схеме 14, нужное региоспецифическое введение группы Rl осуществляется посредством миграции ацила O→N с последующим восстановлением амида до вторичного амина. В этом случае, реакция замыкания кольца также приводит к введению нужных пери-заместителей, как показано в G70. Промежуточные соединения G62 и G70, после проведения последовательных стадий реакций, описанных на схеме 11, могут быть дериватизированы с получением нужных продуктов G64 и G71, соответственно.
Схема 13
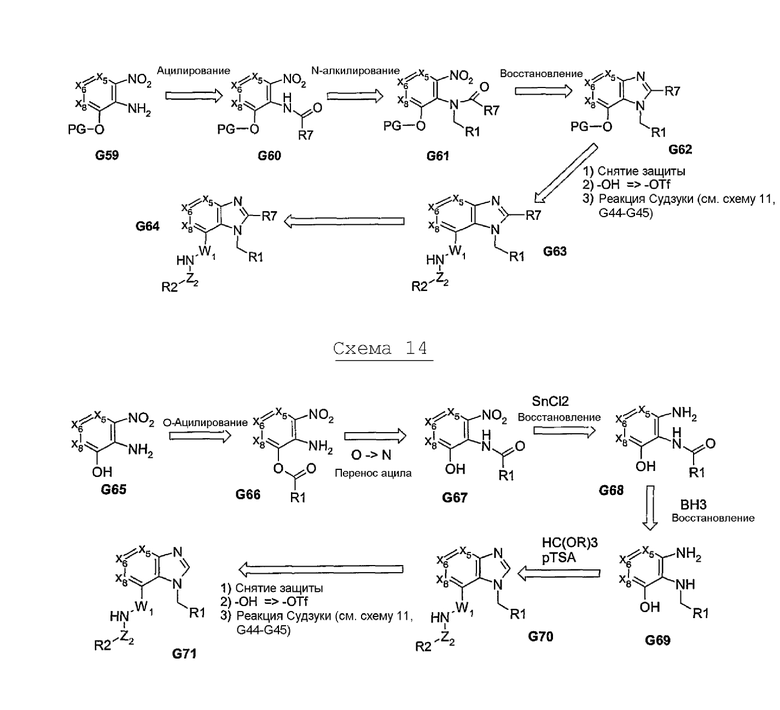
Другие примеры химических реакций, приводящих к образованию бициклических систем с нужными функциональными группами в пери-положении, проиллюстрированы на схеме 15. В данном случае, термоциклизация амина с циклической γ-кетокислотой G74 приводит к образованию нужного бициклического промежуточного соединения G75. Реакция бромирования позволяет получить ключевое промежуточное соединение, которое, путем проведения различных реакций, может быть превращено в различные нужные варианты соединений. Продукт G77 может быть получен путем проведения реакции Судзуки. Альтернативно, винилбромид может быть превращен в соответствующий тризамещенный ненасыщенный сложный эфир или нитрил, который может быть затем дериватизирован после проведения химических реакций, проиллюстрированных на схеме 3/4 [Jasbir: какой должна быть эта схема?] 6 и 7, с получением продуктов G80, G79 и G81, соответственно. Эти химические реакции позволяют осуществлять синтез, по существу, неароматических циклических систем, а также приводить к образованию бициклических систем, где кольцо (а) является 5-членным. Кольцо (a) образуется в процессе реакции циклизации, а размер кольца (b) регулируется с использованием циклического кетона в начальной стадии синтеза, что приводит к образованию бициклической системы “5-N". Помимо размера, гибкость структуре указанного циклического кетона придают также заместитель и гетероатомы. Природа третичной группы также может варьироваться, и такая группа может быть введена на стадии образования циклического кетона, что позволяет в значительной степени регулировать его региохимическую структуру. В положениях X5/X8 могут также присутствовать гетероатомы и/или другие заместители.
Схема 15
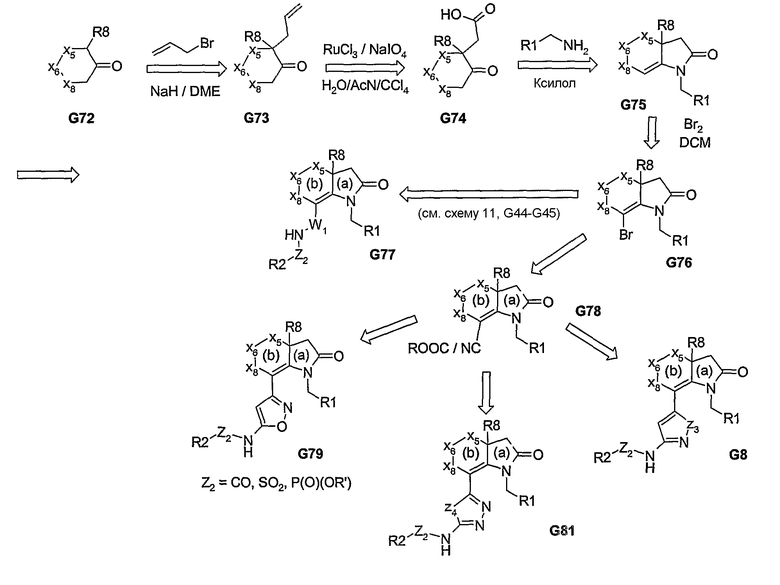
На схеме 16 проиллюстрирована альтернативная реакция замещения связанных с углеродом бициклических пери-заместителей по сравнению с реакцией, описанной выше на схемах 8 и 9. Проведение реакции бициклической системы индолового типа с циклическим кетоном, несущим соответствующим образом замещенный сложный эфир или защищенный амин, позволяет вводить заместители в положение С3. Может быть осуществлена функционализация или дериватизация указанного сложного эфира или амина с получением неарильных пери-заместителей (соединения G87 или соединения, полученного из G86 в виде ацилсульфонамида), либо сначала может быть проведена реакция ароматизации, а затем дериватизации аминового/кислотного заместителя с получением пери-замещенных бициклических арилсульфонамидов, амидов, фосфорамидов и т.п., представленных формулой I.
Схема 16
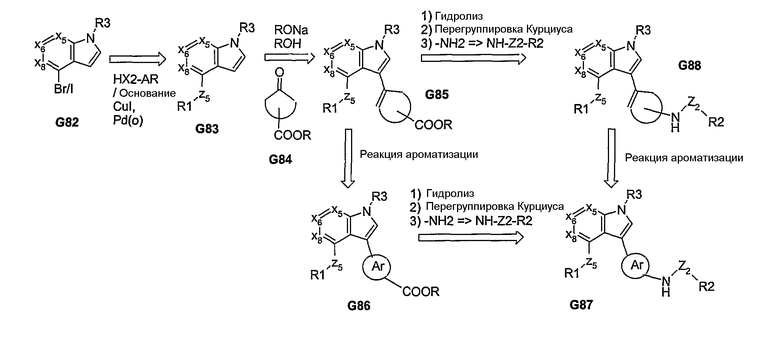
Из соединения G10, где R=алкил (например, Ме), после реакции взаимодействия с дианионом 2-бромуксусной кислоты, а затем реакции декарбоксилирования, получают α-бромкетон G89 (Х=Br). После реакции взаимодействия соединения G89 с тиомочевиной получают 2-аминотиазолы G90. Аминотиазол G90 может быть затем дериватизирован с получением соединения G91 методами, аналогичными методам, описанным выше. Эти серии реакций (реакции взаимодействия с дианионом бромуксусной кислоты и с тиомочевиной) могут быть также проведены с другими сложными эфирами, полученными из бициклосистемы, такими как G78 (схема 15), в результате чего могут быть получены соответствующие 2-аминотиазолы, которые подвергают последующей обработке.
Схема 17
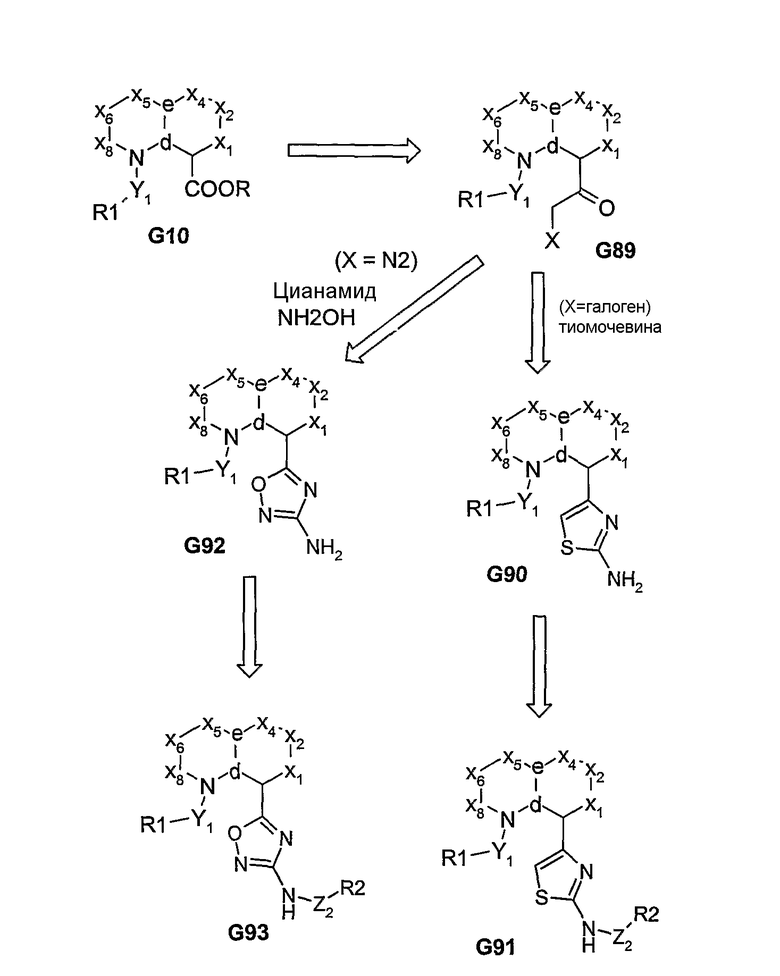
Соединение G10, где R=Н (карбоновая кислота), может быть превращено в соответствующий хлорангидрид, из которого, после проведения реакции взаимодействия с диазометаном, получают диазокетон G89 (Х=N2). Такое промежуточное соединение G89 может быть затем подвергнуто реакции взаимодействия с цианамидом, а затем с гидроксиламином, в результате чего могут быть получены 3-амино-1,2,4-оксадиазолы G92. После этого аминогруппу соединения G92 дериватизируют (например, сульфонилхлоридом) и получают соединение G93 (сульфонамид).
Функциональный α-бромкетон может быть также введен в положение С-3 индоловых систем, таких как G83, с использованием бромацетилхлорида. После реакции взаимодействия полученного α-бромкетона с тиомочевиной образуется 4-(2-аминотиазол)[аналог G91], присоединенный в положении С-3 индолового кольца. Функциональная аминогруппа полученных соединений может быть затем дериватизирована, как описано выше. Методы, описанные на схеме 17, служат дополнительными примерами получения различных аминогетероциклов в качестве ключевых производных, из которых могут быть получены соединения, относящиеся к типу соединений согласно изобретению.
И, наконец, несколько соответствующим образом функционализированных бициклических систем либо являются коммерчески доступными, либо их синтез описан в литературе, либо подразумевается, что они могут быть получены самим специалистом в данной области. Некоторые из этих систем описаны на конкретных примерах. Некоторые из этих систем систематизированы ниже.
Примерами бициклических систем, в которых одним из узловых положений является азот, могут служить легко доступные и широко используемые производные индола. 4-бром- и 4-гидроксииндолы являются коммерчески доступными. 7-замещенные индолы, например, 7-CO2R-, 7-алкокси-, 7-бензилокси- и т.п., могут быть получены посредством химической реакции Бачо-Лейнгрубера из соответствующим образом замещенного 2-нитротолуола (Org. Synthesis Co, Vol. 7). Этот метод также позволяет получить 7-Me-, 7-CHO-, 7-CN- и 7-OH-индолы путем модификаций с использованием функциональных групп. Альтернативно, 7-галогениндолы могут быть получены из 2-галогенанилинов посредством химической реакции Бартоли (Bartoli, G. et.al. Tett.Letters, 1989, 30, 2129-2132). Различные 7-замещенные индолы могут быть также получены путем селективной функционализации индола посредством прямого орто-металлирования в соответствии с процедурой Сникуса [Snieckus V. et.al. Org Letters 2003, 1899-1902]. Эти различные способы также позволяют получать другие замещенные индоловые производные. 8-гидрокситетрагидрохинолины, [6:6]-системы, могут быть получены из коммерчески доступного 8-гидроксихинолина посредством реакции восстановления. 8-OH-1H-хинолин-2-он, 8-OH-3,4-дигидро-1H-хинолин-2-он, 2,6-дигидроксианилины или родственные гетероциклы могут быть превращены в бициклические производные, 5-гидрокси-4H-бензо[l,4]оксазин-3-он, 5-гидрокси-4H-бензо[1,4]оксазин-2,3-дион, 4-гидрокси-3H-бензооксазол-2-он. В результате окисления индоловых 1,7-дизамещенных или 3,4-дизамещенных бицикло-аналогов образуются соответствующие оксииндоловые производные. Различные анилины могут быть превращены в аналоги изатина в соответствии с процедурами, описанными в литературе, и их примеры описаны ниже в разделе “Примеры”. Синтез ряда [5:5]-бицикло-систем (например, имидазотиазола и пирролопиразолона) описан в конкретных примерах. Другая группа [6:5]-бицикло-систем может быть также получена методами, аналогичными описанным в литературе методам синтеза таких систем, как имидазопиридин и имидазопиримидин [Katritzky A.R. et al. JOC 2003, 68, 4935-37], а также пирролопиримиды [Norman M. et al. JMC 2000, 43, 4288-4312]. Затем эти различные бицикло-структуры могут быть дериватизированы с получением аналогов формулы I.
В целом, различные химические реакции, описанные выше, позволяют получать эффективные антагонисты/агонисты простеноидов. Такие химические реакции позволяют модифицировать указанные структуры, а также вводить оптимальные функциональные группы для достижения гидрофобно-гидрофильного баланса; вводить донорную и акцепторную водородную связь с нужной топологией; регулировать нужные физические характеристики, подходящие для достижения нужных фармацевтических свойств и ADME-свойств (например, мембранной проницаемости, связывания с низкомолекулярным плазматическим белком, нужного метаболического профиля и т.п.). Возможность корректировать физические свойства позволяет получать подходящие препараты, удобные для перорального введения, что, в свою очередь, позволяет регулировать размер и частоту доз, вводимых млекопитающим для достижения желаемого фармакологического ответа. Возможность регулировать метаболический профиль позволяет минимизировать вероятные взаимодействия лекарственных средств друг с другом. Таким образом, объем настоящего изобретения включает не только получение эффективных антагонистов простеноидов с соответствующей изоферментной селективностью, которые являются ценным инструментом для проведения исследований, а также получение соединений, которые являются ценными терапевтическими средствами.
Настоящее изобретение проиллюстрировано на нижеследующих конкретных не ограничивающих примерах, представленных в таблице 1. В таблице 1 “Xl”=CH, за исключением B47, где Х1 представляет собой C(=O); “X2” отсутствует, за исключением B43, B44, B45, где Х2 представляет собой CH; “g”=C; “h”=C, за исключением B02, где h представляет собой N; “b” и “d” представляют собой =C.
Пример 1
Получение B0l
Синтез (4-бром-1H-индол-3-ил)нафталин-2-ил-метанона, I-1. К раствору 4-броминдола (5 г, 25,5 ммоль) в безводном метиленхлориде (100 мл) по каплям добавляли MeMgBr (3M раствор в эфире, 8,95 мл, 26,7 ммоль) при 20°С. При этом наблюдалась слабая экзотермическая реакция (максимальная температура составляла 28°С). Полученный оранжевый раствор перемешивали при комнатной температуре в течение 10 мин, а затем к этому раствору через капельную воронку добавляли ZnCl2 (1M раствор в эфире, 76,5 мл, 76,5 ммоль). Реакционную смесь перемешивали в течение 30 мин. Затем добавляли раствор нафтоилхлорида (5,1 г, 26,7 ммоль) в метиленхлориде (25 мл), и во время добавления цвет раствора менялся со светло-оранжевого на темно-красный. Полученную смесь перемешивали при комнатной температуре в течение ночи. ТСХ (EtOAc/гексан, 1:2) указывала на завершение реакции, после чего реакцию гасили насыщенным NH4Cl (100 мл). Полученную суспензию перемешивали в течение 15 мин. Полученные твердые вещества отфильтровывали и несколько раз промывали метиленхлоридом. Фильтрат промывали насыщенным NН4Cl, водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали в вакууме с получением неочищенного продукта (7 г). Полученное твердое вещество растворяли в 10% водном растворе HCl и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали с получением 500 мг неочищенного продукта. Объединенный неочищенный продукт (7,5 г) промывали MTBE (15 мл), растворитель декантировали, и твердые вещества суспендировали в смеси MTBE/гексан (1:1) (10 мл), а затем фильтровали, в результате чего получали 4,61 г чистого указанного в заголовке соединения. Фильтрат концентрировали, и остаток очищали колоночной хроматографией (SiO2), элюируя градиентом смеси этилацетат/гексан (1:3→1:1), в результате чего получали 2 г чистого указанного в заголовке соединения, I-1, всего 6,61 г (выход 74%). 1Н-ЯМР (400 МГц, CDC13) подтвердил указанную структуру.
Синтез (4-бром-1-метил-1H-индол-3-ил)нафталин-2-ил-метанона, I-2. Йодметан (4,55 г, 32 ммоль, 2 экв.) добавляли к перемешиваемому раствору соединения I-1 (5,55 г, 15,9 ммоль, 1 экв.) и K2CO3 (5,48 г, 39,6 ммоль, 2,5 экв.) в aцетоне (110 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем, реакционную смесь концентрировали, разбавляли водой (100 мл) и экстрагировали этилацетатом (3·100 мл). Объединенные органические слои промывали водой (50 мл), насыщенным раствором соли (50 мл), сушили над MgSO4, фильтровали и концентрировали, в результате чего получали 5,45 г (94%) указанного в заголовке соединения I-2 в виде коричневого масла. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 4-бром-1-метил-3-нафталин-2-илметил-1H-индола, I-3. 1 M раствор BH3·ТГФ (16,3 мл, 16,3 ммоль, 3,3 экв.) в ТГФ добавляли в течение 15 мин к перемешиваемому раствору I-2 (1,8 г, 4,9 ммоль, 1 экв.) в ТГФ (48 мл) при 0°С и медленно нагревали до комнатной температуры. Затем реакционную смесь перемешивали при комнатной температуре в течение ночи. После этого, по каплям в течение 5 мин добавляли MeOH (3 мл), а затем снова добавляли MeOH (50 мл). Растворитель выпаривали в вакууме, а затем снова добавляли MeOH (50 мл), и смесь упаривали в вакууме. Эту процедуру повторяли два раза и получали 2 г желтого масла. Полученное масло растворяли в смеси CH2Cl2/гексан, 1:4 (8 мл) при 40°С, а затем охлаждали до комнатной температуры и очищали хроматографией на SiО2 (27 г), элюируя градиентом смеси CH2Cl2/гексан (1:4→1:1), в результате чего получали соединение I-3 (1,04 г, 60%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 1-метил-3-нафталин-2-илметил-1H-индол-4-карбонитрила, I-4. Раствор соединения I-3 (200 мг, 0,571 ммоль, 1 экв.) и цианида меди(I) (153 мг, 1,713 ммоль, 3 экв.) в безводном диметилацетамиде (0,83 мл) дегазировали аргоном в течение 15 мин при комнатной температуре, а затем нагревали при 210°С в закрытом сосуде в течение 2 ч. Затем два раза добавляли воду и этилацетат (по 4 мл каждого), и полученную суспензию фильтровали через целит. Остаток два раза промывали этилацетатом (2 мл) и фильтровали. Органический слой отделяли, промывали водой (4·4 мл) и насыщенным раствором соли (4 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме, в результате чего получали соединение I-4 (167 мг, 99%) в виде коричневого масла, которое кристаллизовалось при отстаивании. Rf 0,42 (EtOAc/гексан, 1:3). МС (ESI-): 296 (M-1). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 3-амино-3-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)акрилонитрила, I-5. Раствор n-BuLi (1,6 M, 1,7 мл, 2,7 ммоль, 10 экв.) в гексане по каплям добавляли к раствору соединения I-4 (80 мг, 0,27 ммоль, 1 экв.) в безводном ацетонитриле (111 мг, 2,7 ммоль, 10 экв.) и ТГФ (2 мл) при -78°С. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 1,5 ч. Затем, реакцию гасили насыщенным NН4Cl, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и упаривали с получением неочищенного I-5 (186 мг) в виде темно-коричневого масла. Rf=0,52 (EtOAc/гексан, 1:1). МС (AP+): 338 (M+l). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 3-гидрокси-3-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)акрилонитрила, I-6. Раствор неочищенного соединения I-5 (186 мг) в СНCl3 (2 мл) перемешивали в 10% водной HCl (2 мл) при комнатной температуре в течение ночи. Органический слой отделяли, фильтровали через целит и промывали CHCl3 (2 мл). После концентрирования фильтрата получали неочищенное соединeние I-6 (106 мг, количественный выход) в виде темно-коричневого масла. Rf=0,73 (EtOAc/гексан, 1:1). МС (AP+): 338 (M+l). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 5-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)-1H-пиразол-3-иламина, I-7. Раствор соединения I-6 (46 мг, 0,136 ммоль, 1 экв.) и гидрата гидразина (68 мг, 1,36 ммоль, 10 экв.) в этаноле (0,3 мл) нагревали при 100°С в течение ночи, а затем при 120°С в течение 2 ч. Реакцию гасили насыщенным NН4Cl, и смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем упаривали в вакууме с получением 46 мг неочищенного продукта. Остаток хроматографировали на SiО2 (1 г), элюируя градиентом смеси этилацетат/гексан (1:4, 1:3, 1:1), а затем чистым этилацетатом, в результате чего получали соединение I-7 (10 мг, 46%) в виде желтого масла. Rf=0,19 (EtOAc). МС (AP+): 353 (M+l). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез [5-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)-1H-пиразол-3-ил]амида 4,5-дихлор-тиофен-2-сульфоновой кислоты, B0l, и l-(4,5-дихлор-тиофен-2-сульфонил)-5-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)-1H-пиразол-3-иламина, B02. Раствор соединения I-7 (12 мг, 0,034 ммоль, 1 экв.), 2,3-дихлортиофен-5-сульфонилхлорида (8,6 мг, 0,034 ммоль, 1 экв.) и DMAP (0,2 мг, 0,0017 ммоль, 0,05 экв.) в пиридине (0,2 мл) перемешивали при комнатной температуре в течение 1 ч. Затем реакцию гасили 10% водной HCl, и смесь экстрагировали этилацетатом. Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили над MgSO4. Раствор концентрировали в вакууме, и получали неочищенную смесь сульфонамидов (23 мг) в виде красного твердого вещества. Этот неочищенный продукт объединяли с неочищенным продуктом, полученным в предыдущей реакции (9 мг, полученных в результате реакции с 7 мг, 0,02 ммоль I-7). Объединенную неочищенную смесь хроматографировали на SiО2 (2 г), элюируя градиентом смеси этилацетат/гексан (1:4→1:1), в результате чего получали менее полярное соединение B02 (6,7 мг, 22%) в виде оранжевого твердого вещества; Rf= 0,26 (EtOAc/гексан, 1:3); ЖХ-МС (80%): ESI+, вычислено: 567 (M), найдено: 568,9 (M+l). 1Н-ЯМР (CDCl3) 3,72 (с, 3H), 4,05 (с, 2H), 4,70 (шир. с, 2H), 5,29 (с, 1Н), 6,65 (шир. с, 1Н), 7,04 (дд, J=8,8, 0,8 Гц, 1Н), 7,15 (дд, J=8,8, 2,0 Гц, 1Н), 7,21 (дд, J=8,0, 7,2 Гц, 1Н), 7,34 (дд, J=8,4, 1,2 Гц), 7,35-7,42 (м, 3H), 7,61 (с, 1Н), 7,66 (д, J=8,4 Гц, 1Н), 7,70-7,73 (м, 1Н), 7,75-7,78 (м, 1H), и BOl (8 мг, 26%) в виде красного твердого вещества; Rf=0,41 (EtOAc/гексан, 1:1); ЖХ-МС (92%): ESI+, вычислено: 566 (M), найдено: 567,3 (M+l). 1Н-ЯМР (CDCl3): 3,72 (с, 3H), 3,86 (с, 2H), 6,47 (с, 1Н), 6,65 (шир. с, 1Н), 7,05 (дд, J=7,2, 0,8 Гц, 1Н), 7,10 (дд, J=8,8, 2,0 Гц, 1Н), 7,22 (с, 1Н), 7,25 (д, J=8,0 Гц, 1Н), 7,27 (д, J=8,8 Гц, 1Н), 7,34 (шир. с, 1Н), 7,36-7,41 (м, 3H), 7,62-7,66 (м, 2H), 7,73 (дд, J=6,8, 2,8 Гц, 1Н).
Пример 2
Получение B03
Синтез 3-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)-фениламина, I-8. Смесь соединения I-3 (175 мг, 0,5 ммоль, 1 экв.), гидрата 3-аминобензолбороновой кислоты (103 мг, 0,75 ммоль, 1,5 экв.), гидроксида бария (103 мг, 0,75 ммоль, 1,5 экв.) и тетракистрифенилфосфинпалладия (58 мг, 0,05 ммоль, 0,1 экв.) в DME-H2O (1:1, 7,2 мл) нагревали при 110°С в течение 4 ч в закрытом сосуде. Затем добавляли тетракистрифенилфосфинпалладий (25 мг, 0,022 ммоль, 0,4 экв.) и карбонат цезия (160 мг, 0,5 ммоль, 1 экв.), и реакционную смесь снова нагревали при 110°С в течение 3 ч. Затем снова добавляли тетракистрифенилфосфинпалладий (58 мг, 0,05 ммоль, 0,1 экв.), реакционную смесь нагревали при 120°С в течение 3 ч. Реакционную смесь распределяли между водой и EtOAc (1:1), и водную фазу экстрагировали EtOAc. Органический слой фильтровали через небольшую колонку с SiO2-целитом и получали 0,32 г неочищенного продукта в виде масла. Неочищенный продукт очищали хроматографией на SiО2 (5 г), элюируя градиентом смеси СН2Cl2/гексан (1:3→2:3), в результате чего получали 113 мг (в виде желтого твердого вещества) неочищенного продукта, содержащего два пятна, на что указывала ТСХ (EtOAc/гексан, 1:3). Этот неочищенный продукт растворяли в MTBE (3 мл), а затем примесь осаждали путем добавления гексана (~6 мл). Смесь охлаждали при -20°С, и примесь отфильтровывали. Маточный раствор концентрировали, и получали соединение I-8 (64 мг, 35%) в виде желтых кристаллов. Rf=0,17 (EtOAc/гексан, 1:3); ЖХ-МС (ESI+): 364 (M+l) (95%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез [3-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)фенил]амида 4,5-дихлор-тиофен-2-сульфоновой кислоты, B03: Раствор соединения I-8 (20 мг, 0,055 ммоль, 1 экв.), 2,3-дихлортиофен-5-сульфонилхлорида (14 мг, 0,055 ммоль, 1 экв.) и DMAP (0,3 мг, 0,0028 ммоль, 0,05 экв.) в пиридине (0,2 мл) перемешивали при комнатной температуре в течение 2 ч. Реакцию гасили 10% водной HCl, и смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили над MgSO4. Раствор фильтровали и концентрировали в вакууме с получением неочищенного продукта (35 мг) в виде красного маслянистого твердого вещества. Неочищенный продукт хроматографировали на SiО2 (1 г), элюируя градиентом смеси этилацетат/гексан (3:17→1:1), в результате чего получали соединение B03 (13 мг, 41%) в виде белой пены. Rf=0,30 (EtOAc/гексан, 1:3). ЖХ-МС (92%): ESI-, вычислено: 576 (M), найдено: 577,3 (M-1). 1Н-ЯМР (400 МГц, CDCl3) 3,74 (с, 2H), 3,78 (с, 3H), 6,03 (шир. с, 1Н), 6,76 (с, 1Н), 6,78 (м, 1Н), 6,83 (дд, J=6,4, 1,2 Гц, 1Н), 7,01 (дд, J=8,4, 1,6 Гц, 1Н), 7,07 (с, 1Н), 7,15 (м, 1Н), 7,18 (м, 1Н), 7,20 (м, 1Н), 7,25 (м, 1Н), 7,34 (дд, J=6,4, 0,8 Гц, 1Н), 7,41-7,44 (м, 2H), 7,63 (м, 1Н), 7,65 (д, J=7,6, 1Н), 7,79 (м, 1Н).
Пример 3
Получение B04
Синтез 4-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)фениламина, I-9. Смесь соединения I-3 (175 мг, 0,5 ммоль, 1 экв.), 4-(4,4,5,5-тетраметил)-l,3,2-диоксаборолан-2-ил)анилина (164 мг, 0,75 ммоль, 1,5 экв.), тетракистрифенилфосфинпалладия (58 мг, 0,05 ммоль, 0,1 экв.) и карбоната цезия (244 мг, 0,75 ммоль, 1,5 экв.) в ДМЕ (3,8 мл) нагревали при 120°С в течение 3 ч в закрытом сосуде. Охлажденную реакционную смесь разбавляли этилацетатом и фильтровали через небольшую колонку с SiO2-целитом, в результате чего получали 0,34 г неочищенного продукта в виде масла. Этот неочищенный продукт очищали хроматографией на SiO2 (2 г), элюируя градиентом смеси СН2Cl2/гексан (1:3→1:1) и получали соединение I-9 (88 мг, 49%) в виде белого пенистого твердого вещества. Rf=0,22 (EtOAc/гексан, 1:3); ЖХ-МС (ESI+): 364 (M+l) (96%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез [4-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)фенил]амида 4,5-дихлор-тиофен-2-сульфоновой кислоты, B04. Раствор соединения I-9 (20 мг, 0,055 ммоль, 1 экв.), 2,3-дихлортиофен-5-сульфонилхлорида (14 мг, 0,055 ммоль, 1 экв.) и DMAP (0,3 мг, 0,0028 ммоль, 0,05 экв.) в пиридине (0,2 мл) перемешивали при комнатной температуре в течение 2 ч. Реакцию гасили 10% водной HCl, и смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили над MgSO4. Раствор фильтровали и концентрировали в вакууме с получением неочищенного продукта (39 мг) в виде розового масла. Неочищенный продукт очищали с помощью хроматографии на SiО2 (1 г), элюируя смесью этилацетат/гексан (1:8), в результате чего получали соединение B04 (8 мг, 25%) в виде не совсем белого пенистого твердого вещества. Rf=0,30 (EtOAc/гексан, 1:3). 1Н-ЯМР (400 МГц, CDCl3) 3,72 (с, 2H), 3,75 (с, 3H), 6,54 (шир. с, 1Н), 6,66 (с, 1Н), 6,91 (дд, J=7,2, 1,2 Гц, 1Н), 7,00-7,05 (м, 2H), 7,05 (с, 1Н), 7,23 (с, 1Н), 7,24-7,28 (м, 3H), 7,29 (м, 1Н), 7,33 (дд, J=7,2, 1,2 Гц, 1Н), 7,40 (м, 2H), 7,60 (д, J=8,4 Гц, 1Н), 7,61-7,7,64 (м, 1Н), 7,74-7,76 (м, 1Н). ЖХ-МС (89%): ESI-, вычислено: 576 (M), найдено: 577,3 (M-1).
Пример 4
Получение B17
Синтез N-[4-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)-фенил]ацетамида, B17. К раствору ариламина I-9 (0,06 ммоль) в ТГФ (0,2 мл) добавляли триэтиламин (2 экв.), а затем 2 экв. ангидрида уксусной кислоты при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Затем реакционную смесь концентрировали в вакууме, разбавляли этилацетатом и промывали 10% водной HCl. Органический слой отделяли, промывали водой и насыщенным раствором соли, а затем сушили и получали неочищенный продукт. Этот продукт очищали колоночной хроматографией и получали N-ацетиловый продукт B17 с выходом 73%. 1Н-ЯМР (CDCl3): 2,2 (с, 3H), 3,73 (с, 3H), 3,78 (с, 2H), 6,62 (с, 1Н), 6,91 (дд, J=6,8, 1,2 Гц, 1Н), 7,06 (дд, J=8,4, 1,6 Гц, 1Н), 7,13 (шир. с, 1Н), 7,21-7,31 (м, 4H), 7,29 (с, 1Н), 7,37-7,41 (м, 4H), 7,61 (д, J=8,4, 1Н), 7,64-7,65 (м, 1Н), 7,73-7,75 (м, 1Н). ЖХ-МС (APCI+): 405 (M+l), 94%.
Пример 5
Получение B18
Синтез 2,2,2-трифтор-N-[4-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)фенил]ацетамида, B18. К раствору ариламина I-9 (0,06 ммоль) в ТГФ (0,2 мл) добавляли триэтиламин (2 экв.) и 2 экв. ангидрида трифторуксусной кислоты при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Затем реакционную смесь концентрировали в вакууме, разбавляли этилацетатом и промывали 10% водной HCl. Органический слой отделяли, промывали водой и насыщенным раствором соли, а затем сушили, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Этот неочищенный продукт очищали колоночной хроматографией с получением N-трифторацетилового продукта с выходом 51%. 1Н-ЯМР (CDCl3): 1,25 (с, 3H), 3,77 (с, 3H), 3,79 (с, 2H), 6,71 (с, 1Н), 6,89 (дд, J=6,4, 0,8 Гц, 1Н), 7 (дд, J=8,4, 1,2 Гц, 1Н), 7,2 (шир. с, 1Н), 7,23-7,27 (м, 3H), 7,29 (с, 1Н), 7,33-7,39 (м, 4H), 7,57-7,6 (м, 2H), 7,73-7,76 (м, 2H). ЖХ-МС (APCI-): 457 (M-1), 100%.
Пример 6
Получение B05
Синтез N-[4-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)-фенил]метансульфонамида, B05. К раствору соединения I-9 (50 мг, 0,138 ммоль) в пиридине (0,25 мл), охлажденному до 0°С, добавляли метансульфонилхлорид (31,6 мг, 2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Затем реакционную смесь концентрировали в вакууме, добавляли 10% водную HCl, и водный слой экстрагировали этилацетатом (2·10 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали в вакууме с получением неочищенного продукта. Этот неочищенный продукт очищали колоночной хроматографией, элюируя смесью этилацетат/гексан (1:4), в результате чего получали неочищенный продукт B05 с выходом 93,7%. 1Н-ЯМР (CDCl3): 2,99 (с, 3H), 3,76 (с, 3H), 3,8 (с, 2H), 6,54 (шир. с, 1Н), 6,7 (с, 1Н), 6,9 (дд, J=7,2, 0,8 Гц, 1Н), 7,02 (дд, J=8,4, 1,6 Гц, 1Н), 7,08 (дд, J=8,4, 2 Гц, 2H), 7,21 (шир. с, 1Н), 7,25-7,27 (м, 1Н), 7,28 (дд, J=8,4, 2 Гц, 2H), 7,33 (дд, J=8,4, 1,2 Гц, 1Н), 7,37-7,4 (м, 2H), 7,6 (д, J=8,4 Гц, 1Н), 7,62-7,65 (м, 1Н), 7,73-7,75 (м, 1Н). ЖХ-МС (APCI-): 439 (M-1), 100%.
Пример 7
Получение B08
Синтез C,C,C-трифтор-N-[4-(1-метил-3-нафталин-2-илметил-1H-индол-4-ил)-фенил]метансульфонамида, B08. К раствору соединения I-9 (50 мг, 0,138 ммоль) и триэтиламина (14 мг, 2 экв.) в метиленхлориде (0,25 мл), охлажденном до -78°С, по каплям добавляли раствор ангидрида трифторметансульфоновой кислоты (58 мг, 1,5 экв.) в метиленхлориде (0,25 мл). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 4 ч. Реакцию гасили 10% водной HCl, и смесь экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией, элюируя смесью этилацетат/гексан (1:9), в результате чего получали 40 мг продукта B08 с выходом 58,8%. 1Н-ЯМР (CDCl3): 3,76 (с, 2H), 3,78 (с, 3H), 6,64 (шир. с, 1Н), 6,75 (с, 1Н), 6,88 (дд, J=6,8, 1,2 Гц, 1Н), 6,97 (дд, J=8,4, 1,6 Гц, 1Н), 7,1 (дд, J=6,4, 1,6 Гц, 1Н), 7,22 (шир. с, 1Н), 7,25-7,27 (м, 3H), 7,34 (дд, J=8,4, 1,2 Гц, 1Н), 7,36-7,4 (м, 3H), 7,59 (д, J=8, 1Н), 7,61-7,63 (м, 1Н), 7,73-7,76 (м, 1Н). ЖХ-МС (APCT-): 494 (M-1), 100%.
Пример 8
Получение B10
Синтез 7-бром-5-фтор-3-метил-1H-индола, I-10. Это соединение получали известным методом (Dobbs, A., J. Org. Chem., 66, 638-641 (2001).
Синтез 7-бром-l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индола, I-11. NaH (60% в минеральном масле, 526 мг, 13,15 ммоль, 1,5 экв.) добавляли к раствору соединения I-10 (2 г, 8,77 ммоль, 1 экв.) в ДМФ (30 мл) при -10°С. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 30 мин. Раствор 2,4-дихлорбензилхлорида (2,06 г, 10,52 ммоль, 1,2 экв.) в ДМФ (10 мл) добавляли в течение 2,5 мин при -10°С. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 1 ч. Эту реакционную смесь добавляли к перемешиваемой смеси 10% водная HCl/вода/эфир (1:1:2, 40 мл). Водный слой экстрагировали эфиром (2×10 мл). Объединенные органические слои промывали водой (3×75 мл) и насыщенным раствором соли (75 мл), а затем сушили над MgSO4, фильтровали и концентрировали в вакууме, в результате чего получали неочищенный продукт в виде коричневого твердого вещества. К этому неочищенному продукту добавляли эфир (4 мл), полученную суспензию охлаждали до -78°С и фильтровали, в результате чего получали соединение I-11 (2,49 г, 73%) в виде не совсем белого твердого вещества. Rf=0,70 (EtOAc/гексан, 1:5). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез этилового эфира l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-карбоновой кислоты, I-12. n-BuLi (1,6 M в гексане, 0,97 мл, 1,55 ммоль, 1,5 экв.) в течение 7 мин в атмосфере Ar добавляли к раствору соединения I-11 (400 мг, 1,03 ммоль, 1 экв.) в простом эфире (7 мл) при -78°С. Реакционную смесь перемешивали при -78°С еще 30 мин. Затем, к реакционной смеси медленно добавляли этилхлорформиат (0,2 мл, 2,07 ммоль, 2 экв.), и полученную смесь оставляли для нагревания до комнатной температуры (на водяной бане) и перемешивали при комнатной температуре в течение 30 мин. Реакцию гасили добавлением 10% водной HCl (5 мл). Органический слой промывали водой (2×10 мл) и насыщенным раствором соли (10 мл), а затем сушили над MgSO4, фильтровали и концентрировали в вакууме, в результате чего получали соединение I-12 (386 мг, 98%) в виде коричневого масла. Rf=0,45 (EtOAc/гексан, 1:19). МС (AP+): 380, 382 (M+l). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез гидразида l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-карбоновой кислоты, I-13. Раствор соединения I-12 (114 мг, 0,3 ммоль, 1 экв.) и гидразина (0,1 мл, 1,5 ммоль, 10 экв.) в этаноле (0,5 мл) нагревали при 120°С в закрытом сосуде в течение ночи. Реакцию гасили добавлением 10% водной HCl при 0°С, а затем смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта (100 мг). Этот неочищенный продукт растирали с MTBE и получали чистый I-13 (72 мг, 66%) в виде бежевого твердого вещества. Rf=0,52 (EtOAc/гексан, 1:1). МС (AP+): 366, 368 (M+l). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-иламина, I-14. Раствор бикарбоната натрия (16 мг, 0,188 ммоль, 1 экв.) в воде (0,45 мл) добавляли к раствору соединения I-13 (69 мг, 0,18 ммоль, 1 экв.) в диоксане (0,5 мл) при комнатной температуре и перемешивали в течение 5 мин с получением суспензии. Затем при комнатной температуре добавляли бромциан (20 мг, 0,184 ммоль, 1,02 экв.), и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После этого добавляли гексан (2 мл), и суспензию фильтровали, в результате чего получали соединение I-14 (54 мг, 73%) в виде бежевого твердого вещества. Rf=0,45 (EtOAc/гексан, 1:1). ЖХ-МС (ESI+): 391, 393 (M+l) (97%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}-2,2,2-трифторацетамида, B-10. Ангидрид трифторуксусной кислоты (13 мг, 0,061 ммоль, 1,5 экв.) добавляли к суспензии соединения I-14 (15 мг, 0,041 ммоль, 1 экв.) в триэтиламине (8 мг, 0,082 ммоль, 2 экв.) и метиленхлориде (0,2 мл) при -78°С. Реакционную смесь оставляли на 10 мин для нагревания до комнатной температуры. Затем реакцию гасили добавлением 10% водной HCl, и смесь экстрагировали метиленхлоридом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали в вакууме, в результате чего получали соединение B10 (17 мг, 91%) в виде желтого твердого вещества. Rf=0,17 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,24 (с, 3H), 5,60 (с, 2H), 6,04 (д, J=8,4 Гц, 1Н), 7,20 (дд, J=8,4, 2,4 Гц, 1Н), 7,34 (дд, J=8,8, 2,4 Гц, 1Н), 7,46 (шир. с, 1Н), 7,48 (д, J=2,0 Гц, 1Н), 7,74 (дд, J=8,8, 2,4 Гц, 1Н). ЖХ-МС (90%): ESI-, вычислено: 486 (M), найдено: 485,4 (M-1).
Пример 9
Получение B1l
Синтез N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}метансульфонамида, B1l. Метансульфонилхлорид (13 мг, 9 мл, 0,11 ммоль, 2 экв.) добавляли к раствору соединения I-14 (22 мг, 0,056 ммоль, 1 экв.) в пиридине (0,2 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем нагревали до 70°С в течение 2 ч. Реакцию гасили добавлением 10% водной HCl, и смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученное масло подвергали хроматографии на SiО2 (0,5 г), элюируя градиентом смеси этилацетат/гексан (1:3→1:1), а затем чистым этилацетатом, в результате чего получали B11 (6,8 мг, 26%) в виде оранжевого твердого вещества. Rf=0,24 (EtOAc). 1Н-ЯМР (400 МГц, CDCl3) 2,05 (с, 3H), 3,11 (с, 3H), 5,60 (с, 2H), 6,07 (д, J=8,4 Гц, 1Н), 7,00 (дд, J=8,4, 2,4 Гц, 1Н), 7,01 (с, 1Н), 7,27 (дд, J=8,4, 2,4 Гц, 1Н), 7,40 (д, J=2,0 Гц, 1Н), 7,50 (дд, J=8,4, 2,4 Гц, 1Н). ЖХ-МС (96%): ESI-, вычислено: 470 (M), найдено: 469,2 (M-1).
Пример 10
Получение B12
Синтез N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}-2,4,5-трифтор-бензолсульфонамида, B12. Раствор 2,4,5-трифторбензолсульфонилхлорида (92 мг, 0,4 ммоль, 4 экв.) в пиридине (0,3 мл) добавляли к смеси соединения I-14 (39 мг, 0,1 ммоль, 1 экв.) и DMAP (49 мг, 0,4 ммоль, 4 экв.) при комнатной температуре. Реакционную смесь нагревали до 90°С в течение 2 ч. Затем реакцию гасили добавлением 10% водной HCl, и смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученное масло подвергали хроматографии на SiО2 (0,5 г), элюируя СН2Cl2, в результате чего получали B12 (10 мг, 17%) в виде желтого масла. Rf=0,40 (EtOAc). 1Н-ЯМР (400 МГц, CDCl3) 2,34 (д, J=1,2 Гц, 3H), 5,64 (с, 2H), 6,05 (д, J=8,4 Гц, 1Н), 6,97 (дд, J=8,4, 2,0 Гц, 1Н), 7,03 (с, 1Н), 7,08 (м, 1Н), 7,29 (дд, J=9,6, 2,4 Гц, 1Н), 7,35 (д, J=2,0 Гц, 1Н), 7,51 (дд, J=8,4, 2,4 Гц, 1Н), 7,86 (м, 1Н). ЖХ-МС (95%): ESI-, вычислено: 586 (M), найдено: 585,1 (M-1).
Пример 11
Получение B13
Синтез {5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}амида 4,5-дихлор-тиофен-2-сульфоновой кислоты, B13. Раствор 2,3-дихлортиофен-5-сульфонилхлорида (75 мг, 0,3 ммоль, 3 экв.) в пиридине (0,20 мл) добавляли к раствору соединения I-14 (39 мг, 0,1 ммоль, 1 экв.) и DMAP (37 мг, 0,3 ммоль, 3 экв.) в пиридине (0,15 мл) при комнатной температуре. Реакционную смесь нагревали до 70°С в течение 2 ч. Затем реакцию гасили добавлением 10% водной HCl, и смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученное масло подвергали хроматографии на SiО2 (1 г), элюируя СН2Cl2, в результате чего получали B13 (17 мг, 27%) в виде белого твердого вещества. Rf=0,38 (EtOAc). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,30 (д, J=0,8 Гц, 3H), 5,59 (д, J=0,4 Гц, 2H), 5,92 (д, J=8,4 Гц, 1Н), 7,15 (дд, J=8,4, 2,0 Гц, 1Н), 7,24 (дд, J=9,6, 2,0 Гц, 1Н), 7,27 (м, 1Н), 7,45 (шир. с, 1Н), 7,70 (дд, J=8,4, 2,8 Гц, 1Н), 7,71 (с, 1Н). ЖХ-МС (96%): ESI-, вычислено: 606 (M), найдено: 605,4 (M-1).
Пример 12
Получение B06
Синтез 7-бром-1-(3,4-дифтор-бензил)-5-фтор-3-метил-1H-индола, I-15. К суспензии NaH (60% в минеральном масле, 263 мг, 10,5 ммоль, 1,5 экв.) в ДМФ (20 мл) добавляли 7-бром-5-фтор-3-метил-1H-индол, I-10 (1 г, 4,38 ммоль, 1 экв.) при -10°С. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 30 мин. 3,4-дифторбензилбромид (0,95 г, 4,6 ммоль, 1,05 экв.) добавляли в течение 2,5 мин при -10°С. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 1 ч. Затем реакционную смесь добавляли к перемешиваемому раствору смеси 10% водная HCl/вода/эфир (1:1:2, 40 мл). Слои разделяли, и водный слой экстрагировали эфиром (2·20 мл). Объединенные органические слои промывали водой (3·75 мл) и насыщенным раствором соли (25 мл), а затем сушили над MgSO4, фильтровали и концентрировали в вакууме, в результате чего получали неочищенный продукт в виде коричневого масла. Этот неочищенный продукт очищали колоночной хроматографией, элюируя смесью этилацетат/гексан (2,5%), в результате чего получали 1,4 г соединения I-15 с выходом 90%. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 4-[l-(3,4-дифтор-бензил)-5-фтор-3-метил-1H-индол-7-ил]-фениламина, I-16. Смесь соединения I-15 (345 мг, 0,974 ммоль), 4-(4,4,5,5-тетраметил)-l,3,2-диоксаборан-2-ил)анилина (320 мг, 1,46 ммоль), тетракистрифенилфосфинпалладия (60 мг, 0,048 ммоль) и карбоната цезия (476 мг, 1,46 ммоль) в ДМФ (4 мл) нагревали при 120°С в течение 3 ч в закрытом сосуде. Реакционную смесь олаждали до комнатной температуры и распределяли между водой и EtOAc. Водный слой экстрагировали EtOAc (2·20 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили (MgSO4) и концентрировали. Неочищенный продукт подвергали хроматографии на SiО2, элюируя смесью растворителей 10%-20% EtOAc/гексан, в результате чего получали соединение I-16 (180 мг, выход 50,6%) в виде белой пены. 1Н-ЯМР (400 МГц, CDCl3), 2,31 (с, 3H), 3,75 (шир. с, 2H), 4,86 (с, 2H), 6,19-6,22 (м, 1Н), 6,27-6,32 (м, 1Н), 6,59 (дд, J=8,4, 2 Гц, 2H), 6,71 (дд, J=8,8, 2,4 Гц, 1Н), 6,85 (с, 1Н), 6,88-6,91 (м, 1Н), 6,94 (дд, J=8,4, 2 Гц, 2H), 7,17 (дд, J=8,8, 2,4 Гц, 1Н). ЖХ-МС (ESI+): 367(M+1), 91%.
Синтез N-{4-[l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]фенил}метансульфониламида, B06. К раствору соединения I-16 (50 мг, 0,136 ммоль) в пиридине (0,25 мл) при 0°С добавляли метансульфонилхлорид (31,3 мг, 2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Затем реакционную смесь концентрировали в вакууме, и добавляли 10% водную HCl, после чего водный слой экстрагировали этилацетатом (2·10 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией, элюируя смесью этилацетат/гексан (1:4), в результате чего получали 57 мг B06 (выход 50%). 1Н-ЯМР (400 МГц, CDCl3), 2,32 (с, 3H), 3,1 (с, 3H), 4,83 (с, 2H), 6,1-6,13 (м, 1Н), 6,16-6,21 (м, 1Н), 6,5 (шир. с, 1Н), 6,69 (дд, J=9,6, 2,4 Гц, 1Н), 6,84-6,91 (м, 2H), 7,1-7,14 (м, перекрывающийся, 4H), 7,23 (дд, J=8,8, 2,4 Гц, 1Н). ЖХ-МС (ESI-): 443 (M-1), 97%.
Пример 13
Получение B07
Синтез N-{4-[l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]фенил}-C,C,C-трифторметансульфонамида, B07. К раствору соединения I-16 (63 мг, 0,172 ммоль) и триэтиламина (35 мг, 2 экв.) в метиленхлориде (0,7 мл) при -78°С по каплям добавляли раствор ангидрида трифторметансульфоновой кислоты (48,5 мг, 1,5 экв.) в метиленхлориде (0,25 мл). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 4 ч. Реакцию гасили добавлением 10% водной HCl, и смесь экстрагировали этилацетатом (2·10 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией, элюируя смесью этилацетат/гексан (1:9), в результате чего получали 40 мг продукта B07 с выходом 36,5%. 1Н-ЯМР (400 МГц, CDCl3), 2,34 (с, 3H), 4,8 (с, 2H), 6,05-6,06 (м, 1Н), 6,1-6,15 (м, 1Н), 6,69 (дд, J=9,2, 2,4 Гц, 1Н), 6,83-6,89 (м, 2H), 6,91 (с, 1Н), 7,12-7,2 (м, 4H), 7,25 (дд, J=9,2, 2,4 Гц, 1Н). ЖХ-МС (APCI-): 497 (М-1), 97%.
Пример 14
Получение B14
Синтез l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-карбонитрила, I-17. Раствор соединения I-15 (1,1 г, 3,106 ммоль, 1 экв.) и цианида меди(I) (834 мг, 9,32 ммоль, 3 экв.) в безводном диметилацетамиде (3,5 мл) дегазировали аргоном в течение 15 мин при комнатной температуре, а затем нагревали при 210°С в закрытом сосуде в течение 1,5 ч. Затем добавляли воду и EtOAc (каждого по 30 мл), и смесь фильтровали. Твердый остаток промывали этилацетатом. Органический слой отделяли, промывали водой (3·50 мл) и насыщенным раствором соли (30 мл), а затем сушили над MgSO4, фильтровали и концентрировали с получением соединения I-17 (903 мг, 97%) в виде твердого вещества. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез (Z)-3-амино-3-[l-(3,4-дифтор-бензил)-5-фтор-3-метил-1H-индол-7-ил]ацетонитрила, I-18. n-BuLi (1,6 M, 5,8 мл, 9,324 ммоль, 4 экв.) по каплям добавляли к раствору диизопропиламина (1,3 мл, 9,324 ммоль, 4 экв.) в безводном ТГФ (4 мл) при -78°С. Затем добавляли раствор соединения I-17 в безводном ацетонитриле (0,49 мл) и ТГФ (1,8 мл). Реакционную смесь оставляли на 1,5 ч для нагревания до комнатной температуры и перемешивали в течение 1,5 ч. Реакцию гасили добавлением насыщенного NН4Cl (20 мл), и смесь экстрагировали этилацетатом (20 мл). Органический слой промывали насыщенным раствором соли, сушили и концентрировали в вакууме, в результате чего получали неочищенный продукт I-18 (754 мг) в виде темно-коричневого масла, которое кристаллизовалось после отстаивания при комнатной температуре. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 5-[l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]-2-метил-2H-пиразол-3-иламина, I-19. К смеси соединения I-18 (150 мг, 0,438 ммоль) в изопропаноле (0,2 мл) и уксусной кислоте (0,2 мл) добавляли метилгидразин (100 мг, 0,115 мл, 2,19 ммоль, 5 экв.) при комнатной температуре. Реакционную смесь нагревали при температуре до 100°С в течение ночи. Затем реакционную смесь концентрировали в вакууме и распределяли между водой и этилацетатом. Органический слой промывали водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали в вакууме, в результате чего получали 100 мг неочищенного продукта. После очистки колоночной хроматографией на силикагеле с элюированием метиленхлоридом, получали 40 мг I-19 с выходом 25%. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез N-{5-[l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]-2-метил-2H-пиразол-3-ил}метансульфонамида, B14. К смеси соединения I-19 (18 мг, 0,048 ммоль) в пиридине (0,1 мл) добавляли метансульфонилхлорид (12 мг, 2 экв.) при 0°С. Смесь перемешивали при комнатной температуре в течение 2 ч, а затем нагревали при 60°С в течение 6 ч. Реакционную смесь концентрировали в вакууме и разбавляли этилацетатом (10 мл). Органический слой промывали 10% водной HCl (2 мл), водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали в вакууме с получением 20 мг неочищенного продукта. Этот неочищенный продукт растирали со смесью эфир/гексан (2:1) и фильтровали, в результате чего получали 14 мг соединения B14. 1Н-ЯМР (CDCl3) 2,32 (с, 3H), 2,99 (с, 3H), 3,92 (с, 3H), 5,29 (с, 2H), 6,07 (с, 1Н), 6,13 (шир. с, 1Н), 6,27-6,32 (м, 1Н), 6,32-6,36 (м, 1Н), 6,83 (дд, J=9,6, 2,4 Гц, 1Н), 6,9 (дд, J=8,4, 2 Гц, 1Н), 6,95 (с, 1Н), 7,25 (дд, J=8,8, 2,8 Гц, 1 H). ЖХ-МС (ESI-): 448 (М-1), 89%.
Пример 15
Получение B15
Синтез {5-[l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]-2-метил-2H-пиразол-3-ил}амида 4,5-дихлортиофен-2-сульфоновой кислоты, B15. Смесь соединения I-19 (15 мг, 0,04 ммоль) и 2,3-дихлортиофен-5-сульфонилхлорида (12,2 мг, 0,048 ммоль) в пиридине (0,1 мл) нагревали до 60°С в течение ночи. ТСХ-анализ указывал на то, что в данной реакции имело место лишь ~50% превращение. Затем добавляли DMAP (9,8 мг, 2 экв.), и смесь нагревали снова нагревали при 60°С в течение ночи. Реакционную смесь концентрировали в вакууме, разбавляли этилацетатом и промывали 10% водной HCl. Органический слой промывали водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали в вакууме, в результате чего получали 20 мг неочищенного продукта. Этот неочищенный продукт очищали препаративной ТСХ с использованием 1% MeOH/метиленхлорида и получали 10 мг B15. 1Н-ЯМР (CDCl3) 2,24 (с, 3H), 3,65 (с, 3H), 5,31 (с, 2H), 5,98 (шир. с, 1Н), 6,29-6,32 (м, 1Н), 6,46-6,51 (м, 1Н), 6,79 (дд, J=9,6, 2,4 Гц, 1Н), 7,05-7,14 (м, 2H), 7,29 (дд, J=9,2, 2,4 Гц, 1 H), 7,34 (с, 1Н), 7,50 (шир. с, 1Н). ЖХ-МС (ESI-): 585 (М-1), 91%.
Пример 16
Получение B16
Синтез этилового эфира 1-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-карбоновой кислоты, I-20. n-BuLi (1,6 M в гексане, 0,64 мл, 1,01 ммоль, 1,2 экв.) добавляли в течение 7 мин в атмосфере Ar к раствору 7-бром-l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индола, I-15 (300 мг, 0,847 ммоль, 1 экв.) в диэтиловом эфире (15 мл) при -78°С. Реакционную смесь перемешивали при -78°С еще 30 мин. Затем к реакционной смеси по каплям добавляли этилхлорформиат (0,09 мл, 1 ммоль, 1,2 экв.), и смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 30 мин. Реакцию гасили добавлением 10% водной HCl (5 мл), и смесь разбавляли простым эфиром (15 мл). Органический слой отделяли, промывали водой (2·10 мл) и насыщенным раствором соли (10 мл), а затем сушили над MgSO4, фильтровали и концентрировали в вакууме, в результате чего получали неочищенный сложный эфир в виде коричневого масла. Остаток очищали колоночной хроматографией, элюируя смесью этилацетат/гексан (1:19), в результате чего получали 260 мг I-20. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 3-[l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]-3-оксо-пропионитрила, I-21. Сухой ацетонирил (50 мкл, 1,1 экв.) добавляли к раствору n-BuLi (2,5 M в гексане, 0,375 мл, 0,93 ммоль, 1,25 экв.) в безводном ТГФ (1,5 мл) при -78°С. Полученную смесь перемешивали в течение 30 мин, а затем по каплям добавляли раствор соединения I-20 в ТГФ (1,5 мл). Реакционную смесь оставляли на 3 ч для нагревания до комнатной температуры. Реакцию гасили водой, а затем к смеси добавляли 10% водную HCl. Эту смесь перемешивали в течение 10 мин, а затем экстрагировали этилацетатом (3·20 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили, фильтровали и концентрировали в вакууме, в результате чего получали 280 мг неочищенного продукта I-27. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру. Продукт I-21 использовали в следующей стадии без дополнительной очистки.
Синтез 5-[l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]-изоксазол-3-иламина, I-22. К смеси соединения I-21 (160 мг, 0,46 ммоль) и гидрохлорида гидроксиламина (86 мг, 1,21 ммоль, 2,6 экв.) в этаноле (2,8 мл) добавляли раствор гидроксида натрия (48 мг, 1,21 ммоль, 2,6 экв.) в воде (0,6 мл). Полученную смесь кипятили с обратным холодильником в течение 1 ч. Затем реакционную смесь разбавляли водой (2 мл) и метиленхлоридом (5 мл), и pH доводили до 1 путем добавления 10% водной HCl. Органический слой отделяли, и рН водного слоя доводили до 8 добавлением твердого NaНСO3, и смесь экстрагировали этилацетатом (2·10 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем концентрировали в вакууме, в результате чего получали 80 мг неочищенного промежуточного соединения. Этот остаток смешивали с 2н. водной HCl (0,2 мл) и нагревали до 100°С в течение 3 ч. Смесь охлаждали до комнатной температуры, и pH доводили до 8 добавлением насыщенного NaНСO3. Водную смесь несколько раз экстрагировали метиленхлоридом, и объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили, фильтровали и концентрировали в вакууме с получением 100 мг неочищенной смеси изомеров 3-амино и 5-аминоизоксазола. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя метиленхлоридом, в результате чего получали 35 мг I-22. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез N-{5-[1-(3,4-дифтор-бензил)-5-фтор-3-метил-1H-индол-7-ил]-изоксазол-3-ил}метансульфонамида, B16. К раствору соединения I-22 (30 мг, 0,084 ммоль) в пиридине (0,2 мл) по каплям добавляли метансульфонилхлорид (19 мг, 0,168 ммоль, 2 экв.). Полученную смесь нагревали при 60°С в течение 6 ч. Затем смесь концентрировали в вакууме, разбавляли этилацетатoм и промывали 10% водной HCl. Органический слой промывали водой и насыщенным раствором соли, а затем сушили, фильтровали и концентрировали в вакууме, в результате чего получали 30 мг неочищенного продукта. Этот неочищенный продукт очищали препаративной ТСХ с использованием смеси 1% MeOH/метиленхлорид, в результате чего получали 10 мг B16. 1Н-ЯМР (400 МГц, CDCl3), 2,33 (с, 3H), 3,15 (с, 3H), 5,18 (с, 2H), 6,19 (с, 1Н), 6,39 (м, 1Н), 6,5 (м, 1Н), 6,93-6,99 (м, 2H), 7,0 (с, 1Н), 7,2 (шир. с, 1Н), 7,39 (дд, J=8,8, 2,4 Гц, 1Н). ЖХ-МС (ESI-): 435 (M-1), 88%.
Пример 17
Получение B19
Синтез (N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[1,3,4]оксадиазол-2-ил}-3,4-дифторбензолсульфонамида, B19. Раствор 3,4-дифторбензолсульфонилхлорида (159 мг, 0,75 ммоль, 2,5 экв.) в пиридине (0,5 мл) добавляли к раствору I-14 (117 мг, 0,3 ммоль, 1 экв.) и DMAP (92 мг, 0,75 ммоль, 2,5 экв.) в пиридине (0,8 мл) при комнатной температуре. Реакционную смесь перемешивали и нагревали при 80°С в течение 0,5 ч. Реакцию гасили добавлением 10% водной HCl (4 мл), и смесь экстрагировали EtOAc (4 мл). Органический слой промывали водой (3·4 мл) и насыщенным раствором соли (2 мл), а затем сушили над MgSO4, фильтровали и концентрировали. Полученное масло (154 мг) растирали с гексаном (4 мл) и фильтровали, в результате чего получали 145 мг твердого вещества. Это твердое вещество подвергали хроматографии на SiО2 (флеш-хроматографии, 2 г), элюируя СН2Cl2 (50 мл), смесью EtOAc/гексан, 1:3 (30 мл), EtOAc/гексан, 1:1 (30 мл), в результате чего получали коричневое масло. Это масло растирали с гексаном (2 мл), и получали указанное в заголовке соединение B-19 (33 мг, 19%) в виде коричневого твердого вещества. Rf=0,40 (EtOAc), 1Н-ЯМР (400 МГц, ДМСO-d6) 2,24 (с, 3H), 5,54 (шир. с, 2H), 5,86 (д, J=8,0 Гц, 1Н), 7,08 (дд, J=8,0, 2,4 Гц, 1Н), 7,15 (дд, J=10,4, 2,4 Гц, 1Н), 7,18 (д, J=2,0 Гц, 1Н), 7,38 (с, 1Н), 7,57-7,64 (м, 2H), 7,68 (шир. с, 1Н), 7,86 (м, 1Н). ЖХ-МС (85%): ESI-, вычислено: 566 (M), найдено: 565,3 (М-1).
Пример 18
Получение B20
Синтез (3,4-дихлор-N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}-бензолсульфонамида, B20. Раствор свежеприготовленного LDA (0,525 ммоль, 2,1 экв.) в ТГФ (0,5 мл) по каплям добавляли в течение 5 мин к раствору соединения I-14 (98 мг, 0,25 ммоль, 1 экв.) и HMPA (87 мг, 0,50 ммоль, 2,1 экв.) в ТГФ (0,5 мл) при -78°С. Реакционную смесь перемешивали в течение 15 мин при -78°С. Раствор 3,4-дихлорбензолсульфонилхлорида (153 мг, 0,625 мг, 2,5 экв.) в ТГФ (0,5 мл) по каплям добавляли в течение 3 мин, и реакционную смесь медленно в течение 1 ч нагревали до 0°С, а затем перемешивали в течение 1 ч при 0°С и медленно нагревали в течение 1 ч до комнатной температуры. Полученную реакционную смесь охлаждали до -78°С, после чего реакцию гасили добавлением 10% водной HCl (4 мл), и смесь экстрагировали EtOAc (2·4 мл). Объединенные органические фазы промывали водой (2·4 мл) и насыщенным раствором соли (4 мл), а затем сушили над MgSO4, фильтровали и концентрировали, в результате чего получали неочищенный продукт (140 мг) в виде оранжевого масла. После очистки хроматографией на SiО2 (флеш-хроматографией, 2 г) с использованием СН2Cl2 в качестве элюента получали неочищенный продукт (10 мг) в виде желтого масла. Это масло промывали гексаном, и получали указанное в заголовке соединение B20 (10 мг, 7%) в виде желтоватого твердого вещества. Rf=0,18 (EtOAc). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,34 (с, 3H), 5,57 (с, 2H), 6,00 (д, J=8,4 Гц, 1Н), 6,94 (дд, J=8,4, 2,0 Гц, 1Н), 7,01 (с, 1Н), 7,22 (дд, J=6,4, 2,0 Гц, 1Н), 7,50 (дд, J=8,4, 2,4 Гц, 1Н), 7,62 (д, J=8,8 Гц, 1Н), 7,78 (дд, J=8,4, 2,0 Гц, 1Н), 8,50 (д, J=2,0 Гц, 1Н). ЖХ-МС (91%): ESI+, вычислено: 598 (M), найдено: 599,1 (M+l).
Пример 19
Получение B21
Синтез B21. Раствор хлорангидрида дифенилфосфиновой кислоты (35 мг, 0,15 ммоль, 1,5 экв.) в пиридине (0,1 мл) добавляли к раствору соединения I-14 (39 мг, 0,1 ммоль, 1 экв.) и DMAP (1,2 мг, 0,01 ммоль, 0,1 экв.) в пиридине (0,3 мл) при 60°С. Реакционную смесь перемешивали и нагревали при 60°С в течение 16 ч. Затем реакцию гасили 10% водной HCl (2 мл), и смесь экстрагировали EtOAc (2·2 мл). Объединенные органические слои промывали водой (3·4 мл) и насыщенным раствором соли (4 мл), а затем сушили над MgSO4, фильтровали и концентрировали. Полученное масло (59 мг) растирали с гексаном (2·1 мл) и эфиром (1,5 мл), а затем фильтровали с получением соединения В21 (29 мг, 49%) в виде белого твердого вещества. Rf=0,37 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,29 (с, 3H), 5,60 (шир. с, 2H), 5,91 (д, J=8,4 Гц, 1Н), 7,00 (шир. с, 1Н), 7,16 (дд, J=8,4, 2,0 Гц, 1Н), 7,40 (шир. с, 1Н), 7,43 (с, 1Н), 7,46-7,58 (м, 7H), 7,66 (дд, J=8,8, 2,4 Гц, 1Н), 7,75-7,80 (м, 4H). ЖХ-МС (91%): ESI-; вычислено: 592 (M), найдено: 591,2 (M-1).
Пример 20
Получение B22
Синтез бис-2,4-дихлорфенилового эфира {5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}-фосфорамидиновой кислоты, B-22. Раствор бис(2,4-дихлорфенил)хлорфосфата (73 мг, 0,18 ммоль, 1,2 экв.) в пиридине (0,2 мл) добавляли к раствору соединения I-14 (59 мг, 0,15 ммоль, 1 экв.) и DMAP (1,8 мг, 0,015 ммоль, 0,1 экв.) в пиридине (0,2 мл) при комнатной температуре. Реакционную смесь перемешивали и нагревали при 60°С в течение 2 ч и при 70°С в течение 1 ч. Реакционную смесь охлаждали до -78°С, и реакцию гасили добавлением 10% водной HCl (4 мл), а затем смесь экстрагировали EtOAc (2·2 мл). Объединенные органические слои промывали водой (3·2 мл) и насыщенным раствором соли (2 мл), а затем сушили над MgSO4, фильтровали и концентрировали. Полученное масло (130 мг) растирали с гексаном (2 мл) и МТВЕ (1 мл), а затем фильтровали и получали указанное в заголовке соединение В-22 (29 мг, 25%) в виде белого твердого вещества. Rf=0,22 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,30 (д, J=0,8 Гц, 3H), 5,62 (с, 2H), 5,95 (д, J=8,4 Гц, 1Н), 7,16 (дд, J=8,4, 2,0 Гц, 1Н), 7,20 (дд, J=9,6, 1,6, 1Н), 7,30 (дд, J=8,8, 2,4, 1Н), 7,44 (д, J=2,4 Гц, 1Н), 7,45 (с, 1Н), 7,47 (д, J=2,8 Гц, 1Н), 7,51 (д, J=2,4 Гц, 1Н), 7,53 (д, J=1,2 Гц, 1Н), 7,55 (д, J=0,8 Гц, 1Н), 7,69 (дд, J=2,4, 0,8 Гц, 1Н), 7,73 (дд, J=8,8, 2,8 Гц, 1Н). ЖХ-МС (87%): ESI-, вычислено: 762 (M), найдено: 761,1 (M-1).
Пример 21
Получение B23
Общая процедура A-1. Раствор соответствующего ацилхлорида (0,30 ммоль, 1,2 экв.) в ТГФ (0,15 мл) в течение 1 мин добавляли к раствору соединения I-14 (98 мг, 0,25 ммоль, 1 экв.) и кат. DMAP (1,5 мг, 0,0125 ммоль, 0,05 экв.) в пиридине (0,6 мл) при комнатной температуре, и реакционную смесь перемешивали при комнатной температуре в течение 3-16 ч. Реакционную смесь охлаждали приблизительно до -70°С (на бане из сухого льда и ацетона), и добавляли 10% водную HCl (4 мл). Смесь экстрагировали EtOAc (2·2 мл). Объединенную органическую фазу промывали водой (3·4 мл) и насыщенным раствором соли (4 мл), а затем сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта в виде масла. Это масло кристаллизовали путем добавления гексана (2 мл). Полученное твердое вещество промывали смесью эфир/гексан, 1:1 (2 мл), и получали указанное в заголовке соединение.
Синтез (N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}-4-фторбензамида, B23. В соответствии с общей процедурой А-1 выделяли 70 мг (55%) соединения В23 в виде белого твердого вещества, Rf 0,15 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,32 (д, J=0,8 Гц, 3H), 5,66 (шир. с, 2H), 6,00 (д, J=8,4 Гц, 1Н), 7,18 (дд, J=6,4, 2,0 Гц, 1Н), 7,33 (дд, J=9,6, 2,4 Гц, 1Н), 7,41 (т, J=8,8 Гц, 2H), 7,45 (д, J=2,0 Гц, 1Н), 7,49 (шир. с, 1Н), 7,73 (дд, J=8,8, 2,4 Гц, 1Н), 8,06-8,09 (м, 2H). ЖХ-МС (92%): ESI-, вычислено: 514 (M), найдено: 513,3 (M-1).
Пример 22
Получение B24
Синтез {5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}амида изоксазол-5-карбоновой кислоты, B24. В соответствии с общей процедурой A-1 выделяли 41 мг (34%) соединения B24 в виде белого твердого вещества, Rf 0,17 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,32 (д, J = 0,8 Гц, 3H), 5,65 (шир. с, 2H), 6,01 (д, J=8,4 Гц, 1Н), 7,17 (дд, J=8,4, 2,4 Гц, 1Н), 7,34 (дд, J=9,6, 2,8 Гц, 1Н), 7,36-7,46 (м, 2H), 7,46 (д, J=2,0 Гц, 1Н), 7,49 (с, 1Н), 7,74 (дд, J=8,8, 2,4 Гц, 1Н), 8,86 (шир. с, 1Н). ЖХ-МС (90%): ESI-, вычислено: 485 (M), найдено: 484,3 (M-1).
Пример 23
Получение B25
Синтез (3,5-дихлор-N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}бензамида, B25. В соответствии с общей процедурой А-1 выделяли 52 мг (37%) соединения B25 в виде белого твердого вещества, Rf 0,31 (EtOAc/гексан, 1:2). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,32 (д, J=1,2 Гц, 3H), 5,64 (шир. с, 2H), 6,01 (д, J=8,4 Гц, 1Н), 7,18 (дд, J=8,0, 2,0 Гц, 1Н), 7,33 (дд, J=9,6, 2,0 Гц, 1Н), 7,44 (д, J=2,0 Гц, 1Н), 7,49 (шир. с, 1Н), 7,74 (дд, J=8,4, 2,4 Гц, 1Н), 7,86 (д, J = 2,0 Гц, 1Н), 7,95 (шир. с, 1Н), 8,0 (д, J=1,6 Гц, 2H). ЖХ-МС (78%): ESI-, вычислено: 564 (M), найдено: 563,1 (M-1).
Пример 24
Получение B26
Синтез (N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}-3,4-дифторбензамида, B26. В соответствии с общей процедурой А-1 выделяли 76 мг (57%) соединения B26 в виде белого твердого вещества, Rf 0,54 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,32 (д, J=0,8 Гц, 3H), 5,65 (шир. с, 2H), 6,01 (д, J=8,4 Гц, 1Н), 7,17 (дд, J=8,4, 2,0 Гц, 1Н), 7,33 (дд, J=9,2, 2,4 Гц, 1Н), 7,45 (д, J=2,0 Гц, 1Н), 7,49 (шир. с, 1Н), 7,66 (кв., J=8,4 Гц, 1Н), 7,23 (дд, J=8,8, 2,4 Гц, 1Н), 7,90 (шир. с, 1Н), 8,02-8,08 (м, 1Н), 12,28 (шир. с, 1Н). ЖХ-МС (92%): ESI-, вычислено: 532 (M), найдено: 531,1 (M-1).
Пример 25
Получение B27
Синтез (N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}-2,4-дифторбензамида, B27. В соответствии с общей процедурой A-1 выделяли 55 мг (41%) соединения B27 в виде белого твердого вещества, Rf 0,80 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСО-d6) 2,32 (д, J=0,8 Гц, 3H), 5,67 (с, 2H), 5,98 (д, J=8,4 Гц, 1Н), 7,17 (дд, J=8,4, 2,0 Гц, 1Н), 7,25 (дт, J=8,4, 2,0 Гц, 1Н), 7,31 (дд, J=8,4, 2,4 Гц, 1Н), 7,43-7,46 (м, 1Н), 7,49 (с, 1Н), 7,49 (д, J=2,4 Гц, 1Н), 7,73 (дд, J=8,8, 2,4 Гц, 1Н), 7,79 (кв., J=7,2 Гц, 1Н), 12,33 (шир. с, 1Н). ЖХ-МС (100%): APCI+, вычислено: 530 (M), найдено: 531,0 (M+l).
Пример 26
Получение B28
Синтез (2,4-дихлор-N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}бензамида, B28. В соответствии с общей процедурой A-1 выделяли 105 мг (74%) соединения B28 в виде белого твердого вещества, Rf 0,60 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСО-d6) 2,32 (д, J=0,8 Гц, 3H), 5,67 (с, 2H), 5,94 (д, J=8,4 Гц, 1Н), 7,17 (дд, J=8,4, 2,0 Гц, 1Н), 7,28 (шир. д, J=8,4 Гц, 1Н), 7,48 (шир. с, 1Н), 7,52 (д, J=1,6 Гц, 1Н), 7,60 (дд, J=8,0, 2,0 Гц, 1Н), 7,65 (д, J=8,0 Гц, 1Н), 7,73 (дд, J=8,8, 2,4 Гц, 1Н), 7,80 (д, J=2,0 Гц, 1Н), 12,52 (шир. с, 1Н). ЖХ-МС (100%): APCI+, вычислено: 563 (M), найдено: 564,0 (M+l).
Пример 27
Получение B29
Синтез {5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[1,3,4]оксадиазол-2-ил}амида 2,2-дифторбензо[1,3]диоксол-5-карбоновой кислоты, B29. В соответствии с общей процедурой А-1 выделяли 60 мг (35%) соединения B29 в виде желтоватого твердого вещества, Rf 0,27 (EtOAc/гексан, 1:2). 1Н-ЯМР (400 МГц, ДМСО-d6): 2,32 (д, J=0,8 Гц, 3H), 5,65 (шир. с, 2H), 6,00 (д, J=8,4 Гц, 1Н), 7,18 (дд, J=8,0, 2,0 Гц, 1Н), 7,36 (шир. кв, J=8,0 Гц, 2H), 7,45 (д, J=2,0 Гц, 1Н), 7,49 (шир. с, 1Н), 7,67 (шир. t, J=8,0 Гц, 2H), 7,74 (дд, J=8,8, 2,4 Гц, 1Н), 12,42 (шир. с, 1Н). ЖХ-МС (100%): APCI+, вычислено: 574 (M), найдено: 575,2 (M+l).
Пример 28
Получение B30
Синтез {5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,3,4]оксадиазол-2-ил}амида фуран-2-карбоновой кислоты, B30. В соответствии с общей процедурой А-1 выделяли 80 мг (55%) соединения B30 в виде желтоватого твердого вещества, Rf 0,23 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСО-d6) 2,32 (д, J = 0,8 Гц, 3H), 5,66 (шир. с, 2H), 5,99 (д, J =8,4 Гц, 1Н), 6,75 (дд, J = 3,6, 2,0 Гц, 1Н), 7,17 (дд, J = 8,4, 2,0 Гц, 1Н), 7,33 (дд, J =8,8, 2,8 Гц, 1Н), 7,44 (д, J = 2,0 Гц, 1Н), 7,48 (с, 1Н), 7,57 (д, J = 3,2 Гц, 1Н), 7,72 (дд, J = 8,8, 2,8 Гц, 1Н), 8,03 (д, J = 0,8 Гц, 1Н), 12,15 (шир. с, 1Н). ЖХ-МС (100%): APCI+, Вычислено: 484 (M), найдено: 485,2 (M+l).
Пример 29
Получение B31
Синтез {5-[l-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]изоксазол-3-ил}амида 4,5-дихлор-тиофен-2-сульфоновой кислоты, B31. К суспензии соединения I-22 (42 мг, 0,117 ммоль) в пиридине (0,2 мл) добавляли DMAP (28 мг, 0,23 ммоль, 2 экв.). Эту смесь нагревали при 70°С до образования раствора, а затем добавляли 2,3-дихлортиофен-5-сульфонилхлорид (58 мг, 0,23 ммоль, 2 экв.). Реакционную смесь перемешивали при этой температуре в течение 2 ч. Охлажденную реакционную смесь концентрировали с получением масла, и добавляли 10% водную HCl (1 мл). Смесь экстрагировали EtOAc (2·5 мл). Объединенные органические слои промывали 10% водной HCl (1 мл), водой (2·3 мл) и насыщенным раствором соли (2Ч3 мл), а затем сушили над MgSO4, фильтровали и концентрировали с получением 55 мг неочищенного продукта. После очистки колоночной хроматографией с использованием смеси 40%→10% гексана/метиленхлорида получали 12 мг соединения B31 (выход 18%). 1Н-ЯМР (400 МГц, CDCl3), 2,32 (с, 3H), 5,09 (с, 2H), 6,31 (с, 1Н), 6,41 (м, 1Н), 6,44-6,49 (м, 1Н), 6,92-6,99 (м, перекрывающийся, 2H), 6,96 (с, 1Н), 7,4 (дд, J=8,8, 2,4 Гц, 1Н), 7,47 (с, 1Н), 7,97 (шир. с, 1Н). ЖХ/МС (ESI-): 572 (M-1), 96%.
Пример 30
Получение B32
Общая процедура (A-2) сульфирования 3-аминоизоксазолов. В 5-миллилитровый сосуд добавляли соответствующий 3-аминоизоксазол (1 экв.), пиридин (1 мл/0,80 ммоль) и DMAP (2 экв.). Реакционную смесь нагревали до 75°С, и через 2-3 мин добавляли чистый сульфонилхлорид (2-3,5 экв.). Сразу после этого образовывалась суспензия, и реакционную смесь перемешивали и нагревали при 75°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры, и добавляли 10% водную HCl (10 мл/0,80 ммоль). Смесь экстрагировали EtOAc (10 мл). Органическую фазу промывали водой (2·10 мл), насыщенным раствором соли (10 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта в виде масла. Этот продукт очищали флеш-хроматографией на SiО2 (1 г на 0,05 ммоль исходного 3-аминоизоксазола с использованием СН2Cl2 в качестве элюента) с получением указанного в заголовке продукта в виде твердого вещества.
Синтез (N-{5-[l-(3,4-дифтор-бензил)-5-фтор-3-метил-1H-индол-7-ил]-изоксазол-3-ил}-3,4-дифторбензолсульфонамида, B32. В соответствии с общей процедурой А-2, из соединения I-22 (143 мг, 0,40 ммоль) и 3,4-дифторбензолсульфонилхлорида (212 мг, 1,00 ммоль) получали 93 мг (44%) указанного в заголовке соединения в виде желтого твердого вещества (гексан). Rf 0,18 (СН2Cl2-МеОН, 19:1). 1Н-ЯМР (400 МГц, CDCl3) 2,32 (д, J=1,2 Гц, 3H), 5,10 (с, 2H), 6,25 (с, 1Н), 6,34-6,39 (м, 1Н), 6,40-6,45 (м, 1Н), 6,86-6,95 (м, 2H), 6,97 (с, 1Н), 7,33 (м, 1Н), 7,39 (дд, J=8,4, 2,4 Гц, 1Н), 7,67-7,72 (м, 1Н), 7,74-7,78 (м, 1Н), 8,10 (шир. с, 1Н). ЖХ-МС (96%): ESI-, вычислено: 532,9 (M-1), найдено: 532,6.
Пример 31
Получение B33
Синтез (N-{5-[1-(3,4-дифторбензил)-5-фтор-3-метил-1H-индол-7-ил]-изоксазол-3-ил}-2,4,5-трифторбензолсульфонамида, B33. В соответствии с общей процедурой А-2, из соединения I-22 (143 мг, 0,40 ммоль) и 2,4,5-трифторбензолсульфонилхлорида (323 мг, 1,40 ммоль) получали 35 мг (16%) указанного в заголовке соединения в виде желтого твердого вещества (гексан). Rf 0,13 (СН2Cl2-МеОН, 19:1). 1Н-ЯМР (400 МГц, CDCl3) 2,32 (д, J=0,8 Гц, 3H), 5,10 (с, 2H), 6,23 (с, 1Н), 6,33-6,41 (м, 1Н), 6,41-6,46 (м, 1Н), 6,87-6,94 (м, 2H), 6,97 (с, 1Н), 7,12 (м, 1Н), 7,38 (дд, J=8,8, 1,6 Гц, 1Н), 7,80 (м, 1Н), 8,25 (шир. с, 1Н). ЖХ-МС (97%): ESI-, вычислено: 550,5, найдено: 550,7.
Пример 32
Получение B34
Синтез 3-[1-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-3-оксопропионитрила, I-23. К смеси n-BuLi (2,5 M в гексане, 13,7 мл, 2,25 экв.) в 90 мл безводного ТГФ при -78°С в течение 5 мин добавляли ацетонитрил (1,6 мл, 30,26 ммоль, 2 экв.). Суспензию перемешивали при этой температуре в течение 0,5 ч, a затем в течение 20 мин добавляли раствор соединения I-12 (5,75 г, 15,13 ммоль) в безводном ТГФ (40 мл). Смесь оставляли для нагревания до 10°С, и реакцию гасили медленным добавлением 10% водного HCl. Смесь экстрагировали EtOAc (2·100 мл). Объединенные органические слои промывали водой (2·50 мл) и насыщенным раствором соли (50 мл), а затем сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 5,9 г продукта I-23 в виде масла. Этот продукт использовали в следующей стадии без очистки. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]изоксазол-3-иламина, I-24. К раствору неочищенного I-23 (1 г, 2,66 ммоль) в смеси EtOH/вода (1:1, 54 мл) добавляли NaOH (124 мг, 3,06 ммоль) и сульфат гидроксиламина (486 мг, 2,93 ммоль). Эту смесь нагревали при 80°С в течение 22 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали до половины его первоначального объема и экстрагировали этилацетатом (2·50 мл). Объединенные органические слои промывали водой (2·20 мл), насыщенным раствором соли (20 мл), сушили (над MgSO4), фильтровали и концентрировали с получением 900 мг коричневого масла. После очистки этого остатка колоночной хроматографией с использованием 20-30% смеси EtOAc/гексан получали 290 мг продукта I-24 (выход 29%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез {5-[1-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]изоксазол-3-ил}амида 4,5-дихлор-тиофен-2-сульфоновой кислоты, B34. К суспензии соединения I-24 (180 мг, 0,447 ммоль) в пиридине (0,5 мл) добавляли DMAP (81 мг, 0,67 ммоль, 1,5 экв.). Эту смесь нагревали при 70°С до образования раствора, и добавляли 2,3-дихлортиофен-5-сульфонилхлорид (140 мг, 0,536 ммоль, 1,2 экв.). Реакционную смесь перемешивали при этой температуре в течение 3 ч. Охлажденную реакционную смесь концентрировали с образованием масла и разбавляли EtOAc (15 мл). Органический слой промывали 10% водной HCl (2·3 мл), водой (2·3 мл) и насыщенным раствором соли (2·3 мл), а затем сушили над MgSO4, фильтровали и концентрировали с получением 280 мг неочищенного остатка. После очистки этого остатка колоночной хроматографией с использованием 20-50% EtOAc/гексана получали 100 мг продукта В34 (выход 35%). 1Н-ЯМР (400 МГц, CDCl3), 2,32 (с, 3H), 5,05 (с, 2H), 6,24 (д, J=8 Гц, 1Н), 6,34 (с, 1Н), 6,9 (с, 1Н), 6,97 (дд, J=8,8, 2,8 Гц, 1Н), 7,03 (дд, J=8,8, 2 Гц, 1Н), 7,3 (д, J=2, 1Н), 7,41 (дд, J=8,8, 2,8 Гц, 1Н), 7,45 (с, 1Н), 7,55 (шир. с, 1Н). ЖХ/МС (ESI-) 604, 97%.
Пример 33
Получение B35
Синтез N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]изоксазол-3-ил}-3,4-дифторбензолсульфонамида, B35. К суспензии соединения I-24 (94 мг, 0,24 ммоль) в пиридине (0,3 мл) добавляли DMAP (44 мг, 0,36 ммоль, 1,5 экв.). Эту смесь нагревали при 70°С до получения раствора, и добавляли 3,4-дифторбензолсульфонилхлорид (64,4 мг, 0,028 ммоль, 1,2 экв.). Реакционную смесь перемешивали при этой температуре в течение 3 ч. Охлажденную реакционную смесь концентрировали с получением масла, и добавляли 10% водную HCl (2 мл). Смесь экстрагировали EtOAc (3·10 мл). Объединенные органические слои промывали водой (2·5 мл) и насыщенным раствором соли (5 мл), а затем сушили над MgSO4, фильтровали и концентрировали с получением 100 мг остатка. После очистки этого остатка колоночной хроматографией с использованием 20-50% EtOAc/гексан получали 20 мг продукта B35 (выход 15%). 1Н-ЯМР (400 МГц, CDCl3), 2,32 (с, 3H), 5,05 (с, 2H), 6,18 (д, J=8,4 Гц, 1Н), 6,29 (с, 1Н), 6,89 (с, 1Н), 6,93 (дд, J=9,2, 2,4 Гц, 1Н), 7,01 (дд, J=8,4, 2 Гц, 1Н), 7,27-7,31 (м, перекрывающийся, 1Н), 7,28 (д, J=2 Гц, 1Н), 7,4 (дд, J=8,8, 2,4 Гц, 1Н), 7,47 (шир. с, 1Н), 7,64-7,66 (м,1H), 7,7-7,74 (м, 1Н). ЖХ/МС (APCI-) 565, 91%.
Пример 34
Получение B36
Синтез (N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]изоксазол-3-ил}-2,4,5-трифторбензолсульфонамида, B36. В соответствии с общей процедурой А-2, из соединения I-24 (156 мг, 0,40 ммоль) и 2,4,5-трифторбензолсульфонилхлорида (185 мг, 0,80 ммоль) получали 64 мг (27%) указанного в заголовке соединения в виде желтого твердого вещества (гексан). Rf 0,15 (CH2Cl2-MeOH, 19:1). 1Н-ЯМР (400 МГц, CDCl3): 2,31 (д, J=1,2 Гц, 3H), 5,03 (с, 2H), 6,17 (д, J=8,4 Гц, 1Н), 6,26 (с, 1Н), 6,89 (с, 1Н), 6,92 (дд, J=8,8, 2,8 Гц, 1Н), 7,02 (дд, J=8,4, 2,4 Гц, 1Н), 7,11 (м, 1Н), 7,28 (д, J=2,4 Гц, 1Н), 7,39 (дд, J=8,4, 2,8 Гц, 1Н), 7,75 (м, 1Н), 8,00 (шир. с, 1Н). ЖХ-МС (96%): ESI-, вычислено: 585 (M), найдено: 584,1 (M-1).
Пример 35
Получение B37
Синтез (3,4-дихлор-N-{5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]изоксазол-3-ил}бензолсульфонамида, B37. В соответствии с общей процедурой A-2, из соединения I-24 (156 мг, 0,40 ммоль) и 3,4-дихлорбензолсульфонилхлорида (196 мг, 0,80 ммоль) получали 103 мг (43%) указанного в заголовке соединения в виде желтого твердого вещества (гексан). Rf=0,18 (СН2Cl2-MeOH, 19:1). 1Н-ЯМР (400 МГц, CDCl3) 2,32 (д, J=0,8 Гц, 3H), 5,02 (с, 2H), 6,19 (д, J=8,4 Гц, 1Н), 6,31 (с, 1Н), 6,90 (с, 1Н), 6,94 (дд, J=9,2, 2,4 Гц, 1Н), 7,02 (дд, J=8,0, 2,0 Гц, 1Н), 7,27 (д, J=2,0 Гц, 1Н), 7,40 (дд, J=8,8, 1,6 Гц, 1Н), 7,57 (д, J=8,4 Гц, 1Н), 7,67 (дд, J=8,4, 2,0 Гц, 1Н), 7,92 (шир. с, 1Н), 7,97 (д, J=2,0 Гц, 1Н). ЖХ-МС (98%): ESI-, вычислено: 599 (M), найдено: 598,3 (М-1).
Пример 36
Получение B38
Синтез 7-бром-5-фтор-3-метил-(l-нафталин-2-илметил)-1H-индола, I-25. Из соединения I-10 (4,8 г, 21,04 ммоль), NaH (1,26 г, 31,57 ммоль), 2-(бромметил)нафталина (5,58 г, 25,25 ммоль) и ДМФ (90 мл), в соответствии с процедурой, аналогичной процедуре превращения соединения I-10 в соединение I-11, получали 7,00 г (90%) соединения I-25 в виде светло-коричневого твердого вещества (гексан). Rf=0,33 (гексан/ацетон, 9:1). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез этилового эфира 5-фтор-3-метил-l-нафталин-2-илметил-1H-индол-7-карбоновой кислоты, I-26. Из соединения I-25 (7,00 г, 19,01 ммоль), 2,5н. BuLi (11,4 мл, 28,50 мл), этилхлорформиата (3,63 мл, 38,02 ммоль) и безводного эфира (120 мл), в соответствии с процедурой, аналогичной процедуре получения соединения I-12 из I-11, получали 7,09 г (количественный выход) соединения I-26 в виде коричневого масла. Rf=0,36 (гексан/ацетон, 9:1). 1Н-ЯМР (400 МГц, CDCl3) подтвердил указанную структуру.
Синтез 3-(5-фтор-3-метил-l-нафталин-2-илметил-1H-индол-7-ил)-3-оксо-пропионитрила, I-27. Соединение I-27 получали из соединения I-26 (7,06 г, 19,53 ммоль) в соответствии с процедурой, аналогичной описанной процедуре превращения соединения I-12 в I-23. Полученное неочищенное масло (7,16 г, колич.) растирали с гексаном (15 мл) и получали твердое вещество, которое фильтровали и промывали гексаном (2·5 мл) с получением соединения I-27 (5,56 г, 80%) в виде светло-коричневого твердого вещества. Rf 0,06 (гексан/ацетон, 9:1). 1Н-ЯМР (400 МГц, CDCl3) подтвердил указанную структуру.
Синтез 5-(5-фтор-3-метил-l-нафталин-2-илметил-1H-индол-7-ил)изоксазол-3-иламина, I-28. Из соединения I-27 (4,43 г, 12,43 ммоль), в соответствии процедурой, аналогичной описанной процедуре превращения соединения I-23 в I-24, получали соединение I-28 (1,69 г, 37%) в виде оранжевого твердого вещества. Rf=0,33 (СН2Cl2). 1Н-ЯМР (400 МГц, CDC13): 2,32 (д, J=0,8 Гц, 3H), 5,32 (с, 2H), 5,45 (с, 1Н), 6,89 (дд, J=9,6, 2,4 Гц, 1Н), 6,91 (дд, J=8,8, 1,6 Гц, 1Н), 7,01 (с, 1Н), 7,21 (шир. с, 1Н), 7,34 (дд, J=8,8, 2,4 Гц, 1Н), 7,39-7,44 (м, 2H), 7,65 (д, J=8,0 Гц, 1Н), 7,65-7,69 (м, 1Н), 7,73-7,77 (м, 1Н).
Синтез (3,4-дифтор-N-[5-(5-фтор-3-метил-l-нафталин-2-илметил-1H-индол-7-ил)изоксазол-3-ил]бензолсульфонамида, B38. В соответствии с общей процедурой А-2, из соединения I-28 (297 мг, 0,80 ммоль) и 3,4-дифторбензолсульфонилхлорида (340 мг, 1,60 ммоль) получали указанное в заголовке соединение B38 (106 мг, 24%) в виде оранжевого твердого вещества. Rf=0,14 (СН2Cl2). 1Н-ЯМР (400 МГц, CDC13) 2,31 (д, J=0,8 Гц, 3H), 5,22 (с, 2H), 6,19 (с, 1Н), 6,84 (дд, J=8,4, 1,6 Гц, 1Н), 6,91 (дд, J=9,2, 2,8 Гц, 1Н), 6,99 (с, 1Н), 7,07 (дкв., J=8,0, 1,6 Гц, 1Н), 7,14 (с, 1Н), 7,41 (дд, J=5,6, 2,4 Гц, 1Н), 7,42-7,45 (м, 2H), 7,51-7,55 (м, 1Н), 7,62-7,70 (м, 3H), 7,74-7,76 (м, 1Н), 7,92 (шир. с, 1Н). ЖХ-МС (98%): ESI-, вычислено: 547,56, найдено: 546,4 (M-1).
Пример 37
Получение B39
Синтез (2,4,5-трифтор-N-[5-(5-фтор-3-метил-l-нафталин-2-илметил-1H-индол-7-ил)изоксазол-3-ил]бензолсульфонамида, B39. В соответствии с общей процедурой А-2, из соединения I-28 (149 мг, 0,40 ммоль) и 2,4,5-трифторбензолсульфонилхлорида (185 мг, 0,80 ммоль) получали указанное в заголовке соединение B39 (42 мг, 19%) в виде не совсем белого твердого вещества. Rf 0,26 (СН2Cl2-MeOH, 19:1). 1Н-ЯМР (400 МГц, CDC13): 2,31 (д, J=0,8 Гц, 3H), 5,22 (с, 2H), 6,18 (с, 1Н), 6,85 (дд, J=8,8, 2,0 Гц, 1Н), 6,89 (дд, J=9,2, 2,4 Гц, 1Н), 6,95 (дд, J=9,2, 4,8 Гц, 1Н), 6,99 (с, 1Н), 7,14 (с, 1Н), 7,38 (дд, J=8,8, 2,4 Гц, 1Н), 7,43-7,46 (м, 2H), 7,63-7,77 (м, 4H), 8,09 (шир. с, 1Н). ЖХ-МС (94%): ESI- вычислено: 565,55, найдено: 564,6 (M-1).
Пример 38
Получение B40
Синтез (3,4-дихлор-N-[5-(5-фтор-3-метил-l-нафталин-2-илметил-1H-индол-7-ил)изоксазол-3-ил]бензолсульфонамида, B40. В соответствии с общей процедурой А-2, из соединения I-28 (149 мг, 0,40 ммоль) и 3,4-дихлорбензолсульфонилхлорида (196 мг, 0,80 ммоль) получали указанное в заголовке соединение B40 (76 мг, 33%) в виде не совсем белого твердого вещества. Rf=0,31 (СН2Cl2-MeOH, 19:1). 1Н-ЯМР (400 МГц, CDC13): 2,31 (д, J=0,8 Гц, 3H), 5,22 (с, 2H), 6,20 (с, 1Н), 6,84 (дд, J=8,4, 1,6 Гц, 1Н), 6,91 (дд, J=8,4, 1,6 Гц, 1Н), 6,99 (с, 1Н), 7,14 (с, 1Н), 7,35 (д, J=8,8 Гц, 1Н), 7,40 (дд, J=8,4, 2,4 Гц, 1Н), 7,42-7,45 (м, 2H), 7,56 (дд, J=8,4, 2,0 Гц, 1Н), 7,63 (д, J=7,6 Гц, 2H), 7,74-7,76 (м, 1Н), 7,95 (д, J=2,4 Гц, 1Н), 8,00 (шир. д, J=4,5 Гц, 1Н). ЖХ-МС (93%): ESI-, вычислено: 581 (M), найдено: 580,3 (M-1).
Пример 39
Получение B41
Синтез 5-(5-фтор-3-метил-l-нафталин-2-илметил-1H-индол-7-ил)изоксазол-3-ил]амида 4,5-дихлортиофен-2-сульфоновой кислоты, B41. В соответствии с общей процедурой А-2, из соединения I-28 (149 мг, 0,40 ммоль) и 2,3-дихлортиофен-5-сульфонилхлорида (201 мг, 0,80 ммоль) получали указанное в заголовке соединение B41 (132 мг, 33%) в виде не совсем белого твердого вещества. Rf=0,10 (СН2Cl2-MeOH, 19:1). 1Н-ЯМР (400 МГц, CDCl3): 2,32 (д, J=0,8 Гц, 3H), 5,26 (с, 2H), 6,23 (с, 1Н), 6,86 (дд, J=8,4, 2,0 Гц, 1Н), 6,95 (дд, J=9,2, 2,8 Гц, 1Н), 7,00 (с, 1Н), 7,19 (шир. с, 1Н), 7,39-7,43 (м, 3H), 7,63-7,66 (м, 2H), 7,74-7,76 (м, 1Н), 7,98 (с, 1Н). ЖХ-МС (99%): ESI-, вычислено: 587 (M), найдено: 586,2 (M-1).
Пример 40
Получение B42
Синтез l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-карбоновой кислоты, I-29. Раствор соединения I-11 (1,08 г, 2,84 ммоль, 1 экв.) в 2н. водном NaOH (7,1 мл, 14,20 ммоль, 5 экв.), метаноле (3 мл) и ТГФ (3 мл) перемешивали и нагревали в закрытом сосуде при 85°С в течение 1,5 ч. Реакционную смесь охлаждали до -70°С, и реакцию гасили добавлением 10% водной HCl (20 мл). Смесь экстрагировали EtOAc (50 мл), органический слой промывали водой (3·50 мл) и насыщенным раствором соли (50 мл), а затем сушили над MgSO4, фильтровали и концентрировали. Полученное твердое вещество фильтровали и промывали гексаном с получением соединения I-29 (694 мг, 69%) в виде не совсем белого твердого вещества. Rf=0,22 (EtOAc/гексан, 1:3). 1Н-ЯМР (400 МГц, CDCl3) подтвердил указанную структуру.
Синтез иминометиленамида l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-карбоновой кислоты, I-30. Оксалилхлорид (0,99 мл, 1,98 ммоль, 1,2 экв.) добавляли к раствору соединения I-29 (580 мг, 1,65 ммоль) в ТГФ (7 мл) при комнатной температуре в атмосфере Ar. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, а затем концентрировали с получением желтых кристаллов. К раствору цианамида (138 мг, 3,294 ммоль, 2 экв.) в ТГФ (7 мл) добавляли раствор 2н. водного NaOH (1,65 мл, 3,29 ммоль, 2 экв.), перемешивали при комнатной температуре в течение 20 мин, а затем полученный раствор добавляли в течение 2 мин к суспензии, полученной из соединения I-29 и оксалилхлорида в ТГФ (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь концентрировали и добавляли воду (4 мл), а затем добавляли 10% водную HCl (2 мл) и водную фазу экстрагировали EtOAc (8 мл). Органическую фазу промывали водой (2·6 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме с получением оранжевого масла (400 мг). Полученное масло промывали гексаном (4 мл, 2 мл) с получением указанного в заголовке соединения I-30 (325 мг, 52%) в виде желтоватого порошка. Rf=0,30 (EtOAc). МС: ESI-, вычислено: 375 (M), найдено: 374,3 (M-1). 1Н-ЯМР (400 МГц, CDCl3) подтвердил указанную структуру.
Синтез (5-[1-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,2,4]оксадиазол-3-иламина, I-31. Пиридин (0,5 мл) добавляли к смеси соединения I-30 (113 мг, 0,3 ммоль, 1 экв.) и гидроксиламина (21 мг, 1 экв.), и реакционную смесь перемешивали и нагревали при 45°С в течение 16 ч и при 60°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в смесь 10% водной HCl (4 мл) и EtOAc (4 мл). Органическую фазу промывали водой (3·6 мл) и насыщенным раствором соли (4 мл), а затем сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта (136 мг) в виде оранжевого масла. После очистки хроматографией на SiО2 (флеш-хроматографией, 2 г) с использованием смеси СН2Cl2/гексан, 1:1 (20 мл), СН2Cl2 (20 мл), получали неочищенный продукт (45 мг) в виде масла. Полученное масло растирали с гексаном, и получали указанное в заголовке соединение I-31 (30 мг, 26%) в виде белого порошка. Rf=0,78 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСO-d6): 2,31 (д, J=0,8 Гц, 3H), 5,70 (с, 2H), 5,89 (д, J=8,4 Гц, 1Н), 6,35 (с, 2H), 7,16 (дд, J=8,8, 2,0 Гц, 1Н), 7,32 (дд, J=9,6, 2,4 Гц, 1Н), 7,47 (с, 1Н), 7,54 (д, J=2,4 Гц, 1Н), 7,71 (дд, J=8,8, 2,4 Гц, 1Н). ЖХ-МС (99%): (ESI+), вычислено: 390 (M), найдено: 391,2 (M+l).
Синтез {5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]-[l,2,4]оксадиазол-3-ил}амида 5-дихлор-тиофен-2-сульфоновой кислоты, B42. Раствор свежеприготовленного LDA (0,537 ммоль, 2,1 экв.) в ТГФ (0,5 мл) по каплям в течение 2 мин добавляли к раствору соединения I-31 (100 мг, 0,256 ммоль, 1 экв.) и HMPA (96 мг, 0,537 ммоль, 2,1 экв.) в ТГФ (0,5 мл) при -78°С. Реакционную смесь перемешивали в течение 10 мин при -78°С. Затем в течение 2 мин по каплям добавляли раствор 2,3-дихлортиофен-5-сульфонилхлорида (161 мг, 0,639 мг, 2,5 экв.) в ТГФ (0,5 мл), и реакционную смесь медленно в течение 1 ч нагревали до -18°С, а затем перемешивали в течение 1 ч при -18°С и медленно нагревали в течение 1 ч до комнатной температуры. Затем реакционную смесь выливали в смесь 10% водной HCl (4 мл) и EtOAc (4 мл). Органическую фазу промывали водой (3·4 мл) и насыщенным раствором соли (4 мл), а затем сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта (134 мг) в виде оранжевого масла. После очистки полученного масла хроматографией на SiО2 (флеш-хроматографией, 5 г) с использованием смеси СН2Cl2/гексан, 1:2 (30 мл), СН2Cl2/гексан, 1:1 (10 мл), СН2Cl2 (10 мл), EtOAc (10 мл) получали неочищенный продукт (40 мг) в виде желтого масла. Полученное масло очищали хроматографией на SiО2 (флеш-хроматографией, 2 г) с использованием смеси СН2Cl2/гексан, 1:4 (30 мл) и получали частично очищенный продукт (35 мг) в виде желтого масла. Полученное масло перекристаллизовывали из смеси СН2Cl2-гексан, 2:1, и получали указанное в заголовке соединение B42 (15 мг, 9%) в виде белого твердого вещества. Rf=0,10 (EtOAc/гексан, 1:1). 1Н-ЯМР (400 МГц, ДМСO-d6) 2,31 (с, 3H), 5,59 (с, 2H), 5,89 (д, J=8,8 Гц, 1Н), 7,13 (дд, J=8,4, 2,4 Гц, 1Н), 7,26 (д, J=1,6 Гц, 1Н), 7,37 (дд, J=9,6, 2,4 Гц, 1Н), 7,46 (с, 1Н), 7,68 (с, 1Н), 7,74 (дд, J=8,8, 2,4 Гц, 1Н). ЖХ-МС (93%): ESI-, вычислено: 604 (M), найдено: 603,1 (М-1).
Пример 41
Получение B43
Синтез 4-бром-1-метил-1H-индола, I-32. К раствору NaH (60% в минеральном масле, 600 мг, 15 ммоль) в ДМФ (20 мл) добавляли 4-бром-1H-индол (1,96 г, 10 ммоль) при -10°С. Перемешиваемую смесь оставляли на 10 мин для нагревания до комнатной температуры, а затем снова охлаждали до -10°С, после чего при -10°С добавляли йодометан (6,7 г, 50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и разбавляли СН2Cl2 (-200 мл). Реакционную смесь промывали водой (3·200 мл), насыщенным раствором соли и сушили над сульфатом натрия. После фильтрации и удаления растворителя получали 3 г неочищенного продукта I-32. Полученное соединение непосредственно использовали в следующей стадии без дополнительной очистки.
1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 1-метил-4-(нафталин-2-илoкси)-1H-индол, I-33. Смесь соединения I-32 (2,4 г, 11,42 ммоль), CuI (217 мг, 1,142 ммоль), HCl-соли N,N-димeтилглицина (480 мг, 3,42 ммоль), 2-нафтола (2,47 г, 17,14 ммоль) и Cs2CO3 (7,42 г, 22,84 ммоль) в диоксане (22 мл) перемешивали в атмосфере Ar при 105°С в течение 2 дней. Реакционную смесь разбавляли этилацетатом, промывали водой и насыщенным раствором соли, а затем сушили над сульфатом натрия. После удаления растворителя остаток очищали колоночной хроматографией на силикагеле с использованием 2% этилацетата/гексана в качестве элюента, в результате чего получали 2,16 г 1-метил-4-(нафталин-2-илокси)-1H-индола, I-33 (выход 83%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 2-бром-l-[1-метил-4-(нафталин-2-илокси)-1H-индол-3-ил]этанона, I-34. К раствору соединения I-33 (500 мг, 1,83 ммоль) в безводном метиленхлориде (10 мл) при -70°С добавляли хлорид диэтилалюминия (1 M раствор в гексане, 2,74 мл, 2,74 ммоль) со скоростью, необходимой для поддержания температуры ниже -65°С. После добавления хлорида диэтилалюминия баню с сухим льдом-ацетоном заменяли на баню со смесью вода-соль-лед, и раствор нагревали до температуры -10°С. При этой температуре добавляли бромацетилхлорид (0,23 мл, 2,74 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 ч. ТСХ-анализ указывал на завершение реакции. Затем медленно, при перемешивании, добавляли воду (9 мл). Водную фазу экстрагировали метиленхлоридом (3·15 мл). Объединенные органические экстракты промывали водой и насыщенным раствором соли, а затем сушили и концентрировали, в результате чего получали 500 мг неочищенного продукта. После растирания с эфиром получали 450 мг соединения I-34 (выход 62%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 4-[1-метил-4-(нафталин-2-илокси)-1H-индол-3-ил]-тиазол-2-иламина, I-35. Суспензию соединения I-34 (220 мг, 0,558 ммоль) и тиомочевины (51 мг, 0,67 ммоль) в этаноле (5 мл) кипятили с обратным холодильником в течение 2 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой и подщелачивали насыщенным водным NaНСО3. Суспензию отфильтровывали, промывали водой и сушили. После растирания с эфиром получали 200 мг соединения I-35 в виде белого твердого вещества с выходом 96%. 1Н-ЯМР (400 МГц, CDCl3), 3,83 (с, 3H), 4,76 (шир. с, 2H), 6,7 (дд, J=7,2, 1,2 Гц, 1Н), 7,01 (с, 1Н), 7,12-7,25 (м, 2H), 7,28-7,3 (м, 2H), 7,34-7,43 (м, 2H), 7,6 (с, 1Н), 7,64 (д, J=8 Гц, 1Н), 7,78-7,81 (м, 2H). ЖХ/МС (ESI+) 372: 98%.
Общая процедура синтеза сульфонамида, (A-3).
К раствору соединения I-35 (0,1 ммоль) в безводном ТГФ (0,3 мл) добавляли NaH (2 экв., 60% дисперсия в масле). Реакционную смесь перемешивали при комнатной температуре в течение 15 мин, а затем добавляли соответствующий сульфонилхлорид (2 экв.). После завершения добавления, реакционную смесь подкисляли, разбавляли 10% водной HCl и экстрагировали EtOAc (2·5 мл). Объединенные органические слои промывали промывали водой и насыщенным раствором соли, а затем сушили и концентрировали с получением неочищенного продукта. После очистки препаративной ТСХ с использованием 5% MeOH/метиленхлорида получали целевой продукт.
Синтез {4-[1-метил-4-(нафталин-2-илокси)-1H-индол-3-ил]-тиазол-2-ил}амида 4,5-дихлортиофен-2-сульфоновой кислоты, B43. Соединение B43 синтезировали в соответствии с общей процедурой A-3. 1Н-ЯМР (400 МГц, CDCl3), 3,89 (с, 3H), 6,32 (с, 1Н), 6,81 (дд, J=7,6, 1,2 Гц, 1Н), 6,96 (м, 1Н), 7,19-7,38 (м, 4H), 7,38 (с, 1Н), 7,41-7,45 (м, 2H), 7,61 (д, J=7,6 Гц, 1Н), 7,73 (д, J=8,8 Гц, 1Н), 7,79-7,81 (м, 1Н), 10,6 (шир. с, 1Н). ЖХ/МС (ESI-) 586: 98%.
Пример 42
Получение B44
Синтез 3,4-дифтор-N-{4-[1-метил-4-(нафталин-2-илокси)-1H-индол-3-ил]тиазол-2-ил}бензолсульфонамида, B44. Соединение B43 синтезировали в соответствии с общей процедурой A-3. 1Н-ЯМР (400 МГц, CDCl3), 3,87 (с, 3H), 6,28 (с, 1Н), 6,79 (дд, J=7,6, 1,2 Гц, 1Н), 7,21-7,27 (м, 4H), 7,3 (д, J=2,4 Гц, 1Н), 7,38 (с, 1Н), 7,39-7,45 (м, 2H), 7,52-7,56 (м,1H), 7,59-7,61 (м, 1Н), 7,65-7,69 (м, 1Н), 7,71 (с, 1Н), 7,78-7,8 (м, 1Н), 10,57 (шир. с, 1Н). ЖХ/МС (AP+) 547: 98%.
Пример 43
Получение B45
Синтез 3-[1-метил-4-(нафталин-2-илокси)-1H-индол-3-ил]-3-оксо-пропионитрила, I-36. Смесь цианоуксусной кислоты (130 мг, 1,51 ммоль), ангидрида уксусной кислоты (1,5 г, 1,5 мл, 15,1 ммоль) и 1-33 (412 мг, 1,51 ммоль) нагревали при 50°С в течение 15 мин. ТСХ-анализ указывал на отсутствие исходного вещества. Смесь охлаждали до комнатной температуры, и твердое вещество осаждали. Затем смесь разбавляли простым эфиром (5 мл) и отфильтровывали. Твердое вещество растирали с эфиром (10 мл). После фильтрации и сушки на воздухе получали 346 мг (выход 67%) соединения 1-36 в виде светло-желтого вещества. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 5-[1-метил-4-(нафталин-2-илокси)-1H-индол-3-ил]изоксазол-3-иламина, 1-37. Суспензию соединения 1-36 (360 мг, 1,05 ммоль), сульфата гидроксиламина (104 мг, 1,15 ммоль) и гидроксида натрия (50,4 мг, 1,26 ммоль) в смеси этанола/воды (1:1, 5 мл) нагревали при 80°С в течение 24 ч. Реакция не была завершена, а поэтому добавляли еще 50 мг гидроксида натрия и 100 мг сульфата гидроксиламина. Смесь нагревали при 100°С в течение 24 ч. Реакционную смесь концентрировали до половины ее исходного объема и добавляли 36% HCl (0,25 мл). Затем реакционную смесь нагревали при 100°С в течение 3 ч. Смесь охлаждали до комнатной температуры, концентрировали до получения масла и разбавляли этилацетатом (10 мл). Раствор промывали 10% водным NaOH. Щелочную водную фазу экстрагировали этилацетатом (3·10 мл). Объединенные экстракты промывали водой и насыщенным раствором соли, а затем сушили над сульфатом магния, фильтровали и концентрировали, в результате чего получали коричневое твердое вещество (400 мг). Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием смеси 30% этилацетат/гексан, и получали 120 мг соединения 1-37 (выход 32%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 3,4-дифтор-N-{5-[1-метил-4-(нафталин-2-илокси)-1H-индол-3-ил]изоксазол-3-ил}бензолсульфонамида, B45. К раствору соединения I-37 (90 мг, 0,253 ммоль) в безводном ТГФ (0,8 мл) добавляли NaH (21 мг, 0,51 ммоль, 60% дисперсия в масле). Реакционную смесь перемешивали при комнатной температуре в течение 15 мин, а затем добавляли 3,4-дифторбензолсульфонилхлорид (83 мг, 0,38 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. После завершения перемешивания, смесь подкисляли 10% водной HCl и экстрагировали EtOAc. Объединенные органические экстракты промывали водой и насыщенным раствором соли, а затем сушили и концентрировали с получением неочищенного продукта. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием смеси 10, 15, 20% этилацетат/гексан, и получали 39 мг (выход 37%) B45. 1Н-ЯМР (400 МГц, CDCl3), 3,89 (с, 3H), 6,41-6,47 (м, 1Н), 6,79 (дд, J=7,6, 0,8 Гц, 1Н), 6,99 (с, 1Н), 7,02 (м, 1Н), 7,18 (д, J=8, 0,8 Гц, 1Н), 7,26 (т, J=8,4, 1Н), 7,38-7,48 (м, 5H), 7,68 (с,1H), 7,7 (д, J=8 Гц, 1Н), 7,84 (д, J=8, 1Н), 7,90 (д, J=8,8, 1Н), 8,39 (шир. с, 1Н). ЖХ/МС (APCI+) 532: 100%.
Пример 44
Получение B46
Синтез 5-бром-2-(2,5-диметил-пиррол-l-ил)пиридина, I-38. Смесь 5-бромпиридин-2-иламина (3,28 г, 19 ммоль), ацетонилацетона (2,17 г, 19 ммоль) и моногидрата п-толуолсульфоновой кислоты (0,95 г) в толуоле (20 мл) кипятили в течение ночи с обратным холодильником с использованием ловушки Дина-Старка. Реакционную смесь концентрировали в вакууме, разбавляли EtOAc (50 мл) и промывали водой (2·10 мл), 10% водным NaHCO3, водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали, в результате чего получали 4,2 г остатка. После очистки остатка колоночной хроматографией на силикагеле с использованием 2-4% EtOAc/гексан получали 3 г продукта I-38. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 2-(2,5-диметил-пиррол-1-ил)-5-(4,4,5,5-тетраметил-[l,3,2]диоксаборолан-2-ил)пиридина, I-39. К раствору соединения I-38 (220 мг, 0,876 ммоль) в безводном ТГФ (10 мл) при -78°С добавляли n-BuLi (2,5 M в гексане, 0,43 мл, 1,095 ммоль). Реакционную смесь перемешивали при этой температуре в течение 15 мин, а затем по каплям добавляли 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диoксаборолан (0,36 мл, 1,75 ммоль). Реакционную смесь перемешивали при -78°С в течение 1 ч, а затем баню с ацетоном и сухим льдом удаляли, и смесь оставляли для нагревания до 0°С, после чего реакцию гасили при этой температуре добавлением насыщенного водного NН4Cl. Смесь перемешивали при комнатной температуре в течение 15 мин, а затем экстрагировали EtOAc (2·10 мл). Объединенные органические экстракты промывали водой и насыщенным раствором соли, а затем сушили над Na2SO4, фильтровали и концентрировали, в результате чего получали 300 мг соединения I-39. Это соединение имело достаточную степень чистоты, а поэтому его использовали в следующей стадии. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 1-(2,4-дихлорбензил)-7-[6-(2,5-диметил-пиррол-1-ил)-пиридин-3-ил]-5-фтор-3-метил-1H-индола, I-40. К раствору соединения I-39 (300 мг, 1 ммоль) в DME (4 мл) добавляли соединение I-11 (258 мг, 0,66 ммоль), а затем карбонат цезия (326 мг, 1 ммоль). После дегазирования суспензии путем барботирования смеси аргоном в течение 5 мин добавляли катализатор Pd(Ph3P)4 (46 мг, 0,04 ммоль), и реакционную смесь перемешивали при 100°С в течение 3,5 ч. Затем реакционную смесь охлаждали до комнатной температуры и разбавляли водой. Полученную смесь экстрагировали EtOAc (2·15 мл). Объединенные органические экстракты промывали водой и насыщенным раствором соли, а затем сушили над Na2SO4, фильтровали и концентрировали, в результате чего получали 400 мг остатка. После очистки этого остатка колоночной хроматографией на силикагеле получали 100 мг соединения I-40. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 5-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]пиридин-2-иламина, I-41. Смесь соединения I-40 (95 мг, 0,198 ммоль), триэтиламина (110 мкл, 0,792 ммоль), гидрохлорида гидроксиламина (158 мг, 2,28 ммоль) в смеси растворителей: EtOH (1,2 мл), воды (0,4 мл) и хлороформа (0,2 мл) нагревали при 90°С в течение 24 ч в закрытом сосуде. ТСХ-анализ указывал на то, что реакция не была завершена. Поэтому добавляли еще 130 мг гидрохлорида гидроксиламина, и смесь нагревали при 100°С в течение 1 дня. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали, и добавляли 10% водную HCl до тех пор, пока pH не достигал 2, после чего смесь экстрагировали эфиром. Водный слой подщелачивали до pH 9 с использованием 6н. водного NaOH, и экстрагировали этилацетатом (3·10 мл). Объединенные экстракты промывали водой и насыщенным раствором соли, а затем сушили над Na2SO4, фильтровали и концентрировали, в результате чего получали 120 мг остатка. После очистки этого остатка колоночной хроматографией на силикагеле с использованием 10-50% этилацетата/гексана получали 50 мг соединения I-40 (исходного соединения) и 30 мг соединения I-41.
Синтез {5-[1-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]пиридин-2-ил}амида 4,5-дихлор-тиофен-2-сульфоновой кислоты, B46. К смеси соединения I-41 (12 мг, 0,03 ммоль) в пиридине (0,15 мл) добавляли при комнатной температуре 2,3-дихлортиофен-5-сульфонилхлорид (12 мг, 0,045 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 ч. ТСХ-анализ указывал на отсутствие образования продукта. Поэтому, на этой стадии добавляли DMAP (4 мг), и смесь перемешивали при комнатной температуре в течение 24 ч. Пиридин удаляли в вакууме, а затем добавляли 10% водную HCl (1 мл), и смесь экстрагировали этилацетатом (2·4 мл). Объединенные экстракты промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали с получением 20 мг остатка. После растирания этого остатка с метанолом (0,15 мл) и после фильтрации получали 8 мг соединения B46. 1Н-ЯМР (400 МГц, CDCl3), 2,34 (с, 3H), 4,94-5,03 (м, 2H), 5,97 (д, J=8,4, 1Н), 6,67 (дд, J=8,8, 2,4, 1Н), 6,93 (с, 1Н), 7,04 (дд, J=8, 2 Гц, 1Н), 7,1 (д, J=2, 1Н), 7,24-7,33 (м, 4H), 7,45 (с, 1H), 8,08 (шир. с, 1Н). ЖХ/МС (ESI-) 614: >80%.
Пример 45
Получение B47
Синтез 2-метил-2-аллилциклогексанона, I-42. К раствору гидрида натрия (1 экв.; 60% дисперсия в минеральном масле) в диметоксиэтиленгликоле при 5°С в атмосфере азота по каплям добавляли 2-метилциклогексанон (1 экв.). Раствор оставляли нагреваться до комнатной температуры, после чего его нагревали до 80°С в течение 1,5 ч. Полученный раствор охлаждали до комнатной температуры, а затем до 5°С. После этого по каплям добавляли аллилбромид (1 экв.), и реакционную смесь нагревали до 80°С в течение 1,5 ч. Затем реакционную смесь охлаждали до комнатной температуры и по каплям добавляли воду (~14 экв.). Водный слой два раза экстрагировали этиловым эфиром и сушили над сульфатом натрия. После концентрирования неочищенный продукт очищали хроматографией на силикагеле с использованием 2,5% этилового эфира в гексане, в результате чего получали соединение I-42 с выходом 35%. 1Н-ЯМР.
Синтез (1-метил-2-оксо-циклогексил)уксусной кислоты, I-43. К двухфазному раствору 1-метил-1-аллициклогексанона, I-42, в H2O/СН3CN/СCl4 в атмосфере азота добавляли NaIO4 (20 экв.), а затем RuCl3·H2O. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем по каплям добавляли 2-пропанол (~88 экв.), в результате чего реакционная смесь становилась черной. Полученную смесь разбавляли водой и этиловым эфиром, фильтровали через слой целита, и этот слой промывали этиловым эфиром. Водный слой экстрагировали дихлорметаном и этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме, в результате чего получали соединение I-43 с количественным выходом. 1Н-ЯМР подтвердил указанную структуру.
Общая процедура (A-4) получения гексагидро-индол-2-онов, I-44. Раствор (1-метил-2-оксо-циклогексил)уксусной кислоты, I-43 (1 экв.), и соответствующего бензиламина (1 экв.) в м-ксилоле кипятили с обратным холодильником в течение 3 ч при 145°С. Реакционную смесь концентрировали в вакууме, и остаток либо использовали в неочищенном виде, либо очищали хроматографией на силикагеле с использованием гексана в дихлорметане (10-20%) в качестве элюента, в результате чего получали нужный продукт, I-44. Структуру продукта подтверждали с помощью 1Н-ЯМР.
Общая процедура (A-5) бромирования гексагидро-индол-2-онов, I-45: К раствору соответствующего гексагидро-индол-2-она, I-44, в дихлорметане, при 0°С по каплям добавляли бром (1 экв.). Реакционную смесь перемешивали до тех пор, пока не исчезал цвет брома, а затем еще 5 мин. Затем одной порцией добавляли триэтиламин (3 экв.), и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Реакционную смесь промывали водой (3×) и сушили над сульфатом магния. Дихлорметановый раствор фильтровали и концентрировали в вакууме. Остаток использовали в следующей стадии либо в неочищенном виде, либо его очищали хроматографией на силикагеле с использованием дихлорметана в качестве элюента, в результате чего получали соответствующий винилбромид, I-45. Структуру продукта подтверждали с помощью 1Н-ЯМР.
Синтез l-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-она, I-44: В соответствии с общей процедурой A-4, (1-метил-2-оксо-циклогексил)уксусную кислоту (I-43) превращали в I-44. Структуру подтверждали с помощью 1Н-ЯМР.
Синтез 7-бром-l-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-она, I-45: В соответствии с общей процедурой A-5, 1-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-он I-44 превращали в соединение I-45. Структуру подтверждали с помощью 1Н-ЯМР.
Синтез 7-(l-этоксивинил)-l-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-она, I-46. К раствору бромида I-45 (350 мг, 1 ммоль) в сухом диoксане (5 мл) добавляли трибутил(1-этоксивинил)олово (390 мг, 1,05 ммоль) и дихлорбис(трифенилфосфин)палладий (36 мг, 0,05 ммоль). Реакционную смесь нагревали в закрытом сосуде при 100°С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, разбавляли метиленхлоридом (10 мл) и фильтровали через короткую пробку из целита. Эту пробку несколько раз промывали метиленхлоридом. Растворитель удаляли, и неочищенный остаток очищали колоночной хроматографией на силикагеле с использованием гексана и 2% этилацетата/гексана, в результате чего получали 224 мг соединения I-46 (выход 65,7%). 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 7-ацетил-l-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-она, I-47. К раствору соединения I-46 (220 мг, 0,645 ммоль) в ТГФ (5 мл) добавляли 2н. водную HCl (2 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь распределяли между водой и эфиром (20 мл, 1:1). Смесь переносили в делительную воронку, и органический слой отделяли. Водный слой экстрагировали эфиром (3×15 мл). Объединенные органические экстракты промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали с получением 203 мг соединения I-47. 1Н-ЯМР (500 МГц, CDCl3) подтвердил данную структуру.
Синтез 7-(2-бром-ацетил)-l-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-она, I-48. К раствору 7-ацетил-1-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-она, I-47 (160 мг, 0,511 ммоль), в смеси диоксан/хлороформ (1:1, 2 мл) через каждые 3 секунды добавляли одну каплю брома (81,8 мг, 26 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Смесь концентрировали, разбавляли этилацетатом (10 мл), промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали, в результате чего получали 208 мг соединения I-48. Этот продукт был достаточно чистым и был использован в следующей стадии. 1Н-ЯМР (500 МГц, CDCl3) подтвердил указанную структуру.
Синтез 7-(2-аминотиазол-4-ил)-1-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-oна, I-49. Смесь 7-(2-бромацетил)-1-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-oна, I-48 (200 мг, 0,51 ммоль), тиомочевины (34 мг, 0,51 ммоль) в этаноле (2 мл) нагревали при 80°С в течение 3 ч. Смесь концентрировали, разбавляли этилацетатом (15 мл) и промывали 10% раствором ацетата натрия (3 мл). Органический слой отделяли, промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали, в результате чего получали 150 мг неочищенного продукта. После растирания продукта с эфиром получали 75 мг соединения I-49. 1Н-ЯМР (500 МГц, CDCl3).
Синтез 3,4-дифтор-N-{4-[l-(3-метокси-бензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]тиазол-2-ил}-бензолсульфонамида, B47. К раствору 7-(2-амино-тиазол-4-ил)-l-(3-метокси-бензил)-3a-метил-l,3,3a,4,5,6-гексагидро-индол-2-она, I-49 (45 мг, 0,122 ммоль), в пиридине (0,2 мл) добавляли DMAP (30 мг, 0,24 ммоль). Эту смесь нагревали при 70°С, и добавляли 3,4-дифторбензолсульфонилхлорид (52 мг, 0,24 ммоль). Этот раствор превращался в суспензию, и через 10 мин реакция завершалась. Смесь охлаждали до комнатной температуры и концентрировали досуха. Остаток разбавляли этилацетатом (4 мл) и промывали 10% водной HCl. Водный слой еще раз экстрагировали этилацетатом. Объединенные экстракты промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали, в результате чего получали 70 мг неочищенного продукта. После очистки продукта с помощью препаративной TСХ на силикагеле с использованием смеси этилацетат/гексан (1:1) в качестве элюента получали 35 мг соединения B47 (выход 53%). 1Н-ЯМР (400 МГц, CDCl3), 1,21 (с, 3H), 1,61-1,67 (м, 2H), 1,77-1,83 (м, 2H), 2,18-2,27 (м, 2H), 2,24 (дд, J=16, 4,8 Гц, 2H), 3,73 (с, 1Н), 3,99 (д, J=16 Гц, 1Н), 5,03 (д, J=16 Гц, 1Н), 5,95 (с, 1Н), 6,36 (д, J=7,6 Гц, 1Н), 6,38 (с, 1Н), 6,68 (дд, J=8,4, 2 Гц, 1Н), 7 (т, J=8 Гц, 1Н), 7,23-7,29 (м, 2H), 7,66-7,76 (м, 2H). ЖХ/МС (ES1+) 546: 93%.
Пример 46
Получение B09
Синтез 4-(5-фтор-3-метил-1H-индол-7-ил)фениламина, I-50. Смесь соединения I-10 (220 мг, 0,96 ммоль), 4-(4,4,5,5-тетраметил)-l,3,2-диoксaборан-2-ил)анилина (316 мг, 1,44 ммоль), тетракистрифенилфосфинпалладия (56 мг, 0,048 ммоль) и карбоната цезия (470 мг, 1,44 ммоль) в ДМФ (4 мл) нагревали при 110°С в течение 2 ч в закрытом сосуде. Реакционную смесь охлаждали до комнатной температуры и распределяли между водой и EtOAc. Водный слой экстрагировали EtOAc (2·20 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили (MgSO4) и концентрировали с получением 250 мг неочищенного продукта. Неочищенный продукт подвергали хроматографии на SiО2 с использованием смеси растворителей 20% EtOAc/гексан, в результате чего получали соединение I-50 (120 мг, выход 52%) в виде белой пены. 1Н-ЯМР (400 МГц, CDCl3) подтвердил указанную структуру.
Синтез N-[4-(5-фтор-3-метил-1H-индол-7-ил)-фенил]метансульфонамида, I-51. К раствору 4-(5-фтор-3-метил-1H-индол-7-ил)фениламина, I-50 (120 мг, 0,5 ммоль), в пиридине (0,3 мл), охлажденному до 0°С, добавляли метансульфонилхлорид (114,55 мг, 2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь концентрировали, а затем к ней добавляли 10% водную HCl, и полученную водную смесь экстрагировали EtOAc (2·10 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали с получением остатка. Полученный остаток очищали колоночной хроматографией (SiО2) с использованием смеси растворителей 20% EtOAc/гексан, в результате чего получали 95,5 мг соединения I-51 (выход 60%). 1Н-ЯМР (400 МГц, CDCl3) подтвердил указанную структуру.
Синтез N-{4-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индол-7-ил]фенил}метансульфонамида, B09. К суспензии NaH (60% в минеральном масле, 24 мг, 0,59 ммоль, 2 экв.) в ДМФ (2 мл) добавляли N-[4-(5-фтор-3-метил-1H-индол-7-ил)фенил]метансульфонамид, I-51 (95 мг, 0,298 ммоль, 1 экв.) при -10°С. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь охлаждали до 0°С и постепенно добавляли 2,4-дихлорбензилхлорид (71 мг, 0,36 ммоль, 1,2 экв.). Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 4 ч. Реакцию гасили 10% водной HCl (10 мл), и смесь экстрагировали эфиром (3·20 мл). Объединенные органические экстракты промывали водой и насыщенным раствором соли, а затем сушили над MgSO4, фильтровали и концентрировали, в результате чего получали остаток. Полученный остаток очищали колоночной хроматографией с использованием смеси 7% EtOAc/гексан в качестве элюента, в результате чего получали 38 мг соединения B09 (выход 30%). 1Н-ЯМР (400 МГц, CDCl3): 2,34 (с, 3H), 3,07 (с, 3H), 4,83 (с, 2H), 5,99 (д, J=8 Гц, 1Н), 6,37 (шир. с, 1Н), 6,7 (дд, J=9,6, 2,4 Гц, 1Н), 6,86 (с, 1Н), 6,99 (дд, J=8,4, 2 Гц, 1Н), 7,03 (с, 4H), 7,2 (д, J=2 Гц, 1Н), 7,26 (дд, J=8,8, 2,4 Гц, 1Н). ЖХ-МС (ESI-): 447, 99%.
Соединения согласно изобретению были проанализированы на их связывание с простаноидными рецепторами EP3 методом, описанным в публикации Abramovitz et al. [Bioch. Biophys. Acta, 1473, 285-293 (2000)]. В главе 1, в столбце 2 указана активность. Соединения с IC50<1 мкМ отмечены +++; соединения с IC50=1-10 мкM отмечены ++; соединения с IC50>10 мкM отмечены +.
B(X)
| название | год | авторы | номер документа |
|---|---|---|---|
| СУЛЬФОНАМИДНЫЕ ПЕРИ-ЗАМЕЩЕННЫЕ БИЦИКЛЫ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ПОРАЖЕНИЯ АРТЕРИЙ | 2005 |
|
RU2403240C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ ДИГИДРОПИРАН-2-ОНА | 2007 |
|
RU2444519C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2382779C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ БЛОКИРОВАНИЯ ОБРАТНОГО ЗАХВАТА НОРЭПИНЕФРИНА, ДОПАМИНА И СЕРОТОНИНА | 2005 |
|
RU2388751C2 |
| ИНДОЛЫ, ОБЛАДАЮЩИЕ ПРОТИВОДИАБЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2003 |
|
RU2328483C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2528393C9 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
Изобретение относится к соединениям формулы:
где А и В представляют собой пару конденсированных насыщенных или ненасыщенных 5- или 6-членных колец, где указанная система конденсированных колец А/В содержит от 0 до 2 атомов азота, и указанные кольца дополнительно замещены 0-4 заместителями, независимо выбранными из галогена, низшего алкила или оксо; а и b представляют собой положения присоединения остатков Y и D, соответственно, и эти положения а и b находятся в пери-положении по отношению друг к другу на указанной конденсированной системе колец А/В; d и e представляют собой конденсированные положения между кольцом А и кольцом В в указанной конденсированной системе колец А/В; D представляет собой арильную или гетероарильную циклическую систему, которая означает 5- или 6-членное ароматическое кольцо, содержащее 0-3 гетероатомов, выбранных из О, N или S; которая может быть дополнительно замещена 0-4 заместителями, независимо выбранными из низшего алкила и амино; Y выбран из -СН2- и -O-; М выбран из арила, арила, замещенного галогеном или алкокси; R1 выбран из арила, арила, замещенного галогеном, гетероарила, гетероарила, замещенного галогеном, где гетероарил означает 5- или 6-членное ароматическое кольцо, содержащее 0-3 гетероатомов, выбранных из О, N или S, и CF3; и если Y представляет собой -СН3- или -O-, то R1 дополнительно представляет собой низший алкил. Изобретение также относится к применению соединений по п.1, к фармацевтической композиции, к способу скрининга на селективные лиганды простаноидных рецепторов, а также к соединениям формулы. Технический результат - получение новых биологически активных соединений, предназначенных для ингибирования связывания простагландина E2 с ЕР3 рецептором. 8 н. и 17 з.п. ф-лы, 1 табл.
1. Соединение формулы
где А и В представляют собой пару конденсированных насыщенных или ненасыщенных 5- или 6-членных колец, где указанная система конденсированных колец А/В содержит от 0 до 2 атомов азота, и указанные кольца дополнительно замещены 0-4 заместителями, независимо выбранными из галогена, низшего алкила или оксо;
а и b представляют собой положения присоединения остатков Y и D соответственно, и эти положения а и b находятся в пери-положении по отношению друг к другу на указанной конденсированной системе колец А/В;
d и e представляют собой конденсированные положения между кольцом А и кольцом В в указанной конденсированной системе колец А/В;
D представляет собой арильную или гетероарильную циклическую систему, которая означает 5- или 6-членное ароматическое кольцо, содержащее 0-3 гетероатомов, выбранных из О, N или S; которая может быть дополнительно замещена 0-4 заместителями, независимо выбранными из низшего алкила и амино;
Y выбран из -СН3- и -O-;
М выбран из арила, арила замещенного галогеном или алкокси;
R1 выбран из арила, арила замещенного галогеном, гетероарила, гетероарила замещенного галогеном, где гетероарил означает 5- или 6-членное ароматическое кольцо, содержащее 0-3 гетероатомов, выбранных из О, N или S, и CF3; и если Y представляет собой -СН2- или -O-, то R1 дополнительно представляет собой низший алкил.
2. Соединение по п.1, где D представляет собой фенил, замещенный 0-4 заместителями, выбранными из низшего алкила и амино.
3. Соединение по п.1, где D представляет собой нафтил, замещенный 0-4 заместителями, выбранными из низшего алкила и амино.
4. Соединение по п.1, где D представляет собой моноциклический гетероарил, замещенный 0-4 заместителями, выбранными из низшего алкила и амино.
5. Соединение по п.1, где R1 выбран из фенила, фенила, замещенного галогеном, 5-членного циклического гетероарила, 5-членного циклического гетероарила, замещенного галогеном, и CF3.
6. Соединение по п.1, где М выбран из арила, арила, замещенного галогеном или алкокси.
7. Соединение по п.6, где М выбран из фенила, фенила, замещенного галогеном или алкокси, нафтила, нафтила, замещенного галогеном или алкокси, гетероарила и гетероарила, замещенного галогеном или алкокси.
8. Соединение по п.1, где указанная система колец А/В представляет собой пару конденсированных 5-членных колец
9. Соединение по п.1, где указанная система колец А/В представляет собой пару конденсированных 6-членных колец
10. Соединение по п.1, где указанная система колец А/В представляет собой пару конденсированных 5- и 6-членных колец
 или
или 
11. Соединение по п.10, где указанная система колец А/В представляет собой индол.
12. Применение соединения или его сложного эфира по п.1 для получения фармацевтической композиции для лечения или профилактики опосредуемого простагландинами заболевания или состояния у млекопитающего.
13. Применение по п.12, где указанным заболеванием является окклюзионное сосудистое заболевание.
14. Применение соединения или его сложного эфира по п.1 для получения фармацевтической композиции для уменьшения количества бляшек при лечении атеросклероза у млекопитающего.
15. Применение соединения или его сложного эфира по п.1 для получения фармацевтической композиции для стимуляции остеогенеза или цитопротекции у млекопитающего.
16. Применение соединения или его сложного эфира по п.1 для получения фармацевтической композиции для лечения или профилактики боли, воспалений, атеросклероза, инфаркта миокарда, инсульта или окклюзионного сосудистого заболевания у млекопитающего.
17. Фармацевтическая композиция, предназначенная для ингибирования связывания простагландина Е2 с ЕР3 рецептором, содержащая фармацевтически приемлемый носитель и соединение по п.1.
18. Способ скрининга на селективные лиганды простаноидных рецепторов, включающий контактирование меченного соединения по п.1 с простаноидным рецептором и измерение вытеснения метки тест-соединением.
19. Способ по п.18, представляющий собой способ скрининга на селективные лиганды ЕР3, включающий контактирование указанного меченного соединения с клонированным рецептором человеческого ЕР3 и измерение вытеснения метки тест-соединением.
20. Соединение формулы
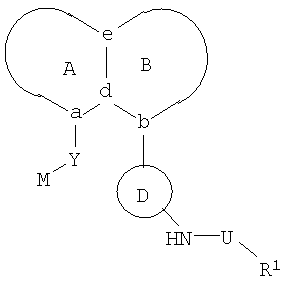
где А и В представляют собой пару конденсированных ненасыщенных 5-или 6-членных колец, и где указанная конденсированная система колец А/В содержит 1 гетероатом азота, и указанные кольца дополнительно замещены 0-4 заместителями, независимо выбранными из галогена и низшего алкила;
а и b представляют собой положения присоединения остатков Y и D соответственно, и эти положения а и b находятся в пери-положении по отношению друг к другу на указанной конденсированной системе колец А/В;
d и e представляют собой конденсированные положения между кольцом А и кольцом В в указанной конденсированной системе колец А/В;
U представляет собой С=O или Р=O;
D представляет собой арильную или гетероарильную циклическую систему, которая означает 5- или 6-членное ароматическое кольцо, содержащее 0-3 гетероатомов, выбранных из О, N или S, которая может быть дополнительно замещена 0-4 заместителями, независимо выбранными из галогена или низшего алкила;
Y представляет собой -CH2;
М выбран из арила, арила замещенного галогеном;
R1 выбран из арила, арила замещенного галогеном, гетероарила, который представляет собой 5- или 6-членное ароматическое кольцо, содержащее 0-3 гетероатомов, выбранных из О, N или S; CF3 и низшего алкила.
21. Соединение по п.20, где U представляет собой С=O.
22. Соединение по п.20, где U представляет собой Р=O.
23. Соединение по п.21 или 22, где указанная система колец А/В представляет собой индол.
24. Соединение по п.23, где D представляет собой фенил или оксадиазолил.
25. Соединение по п.24, где R1 выбран из фенила, фенила, замещенного галогеном, 5-членного циклического гетероарила, содержащего 0-3 гетероатомов, выбранных из О, N или S, СН3 и CF3.
| US 5981533 А, 09.11.1999 | |||
| Karpeiskii et al, BIOORGANISCHESKAYA KHIMIYA, vol.13, no.8, 1987, pages 1059-1065 | |||
| EP 1431267 A, 23.06.2004 | |||
| RU 95112848 A1, 20.03.1997. |

