Перекрестная ссылка на родственные заявки
Настоящая заявка заявляет приоритет к патентной заявке США, серийный № 12/635927, поданной 11 декабря 2009 года. Содержание этой заявки включено в настоящую заявку посредством ссылки во всей полноте.
Область изобретения
Настоящее изобретение относится к модуляторам АТФ-Связывающих Кассетных ("ABC") транспортеров или их фрагментам, включая регулятор трансмембранной проводимости кистозного фиброза ("CFTR"), их композициям и способам, в которых их используют. Настоящее изобретение также относится к способам лечения ABC транспортер-опосредованных заболеваний с использованием таких модуляторов.
Предпосылки изобретения
ABC транспортеры представляют собой семейство мембранных транспортных белков, которые регулируют транспорт множества различных фармакологических веществ, потенциально токсичных лекарственных средств и ксенобиотиков, а также анионов. ABC транспортеры представляют собой гомологичные мембранные белки, которые связывают и используют клеточный аденозинтрифосфат (АТФ) для их специфической активности. Было обнаружено, что некоторые из этих транспортеров являются полилекарственно-резистентными белками (например, MDR1-P гликопротеин или полилекарственно-резистентный белок MRP1), защищающими злокачественные раковые клетки против химиотерапевтических средств. К настоящему времени 48 ABC Транспортеров были идентифицированы и сгруппированы в 7 семейств на основании идентичности их последовательности и функции.
ABC транспортеры регулируют различные важные физиологические роли в организме и обеспечивают защиту против экологически вредных соединений. Поэтому они представляют собой важные потенциальные мишени для лекарственных средств для лечения заболеваний, связанных с дефектами транспортера, предотвращения транспорта лекарственного средства из клетки-мишени и вмешательства в другие заболевания, где модуляция активности ABC транспортера может быть полезной.
Одним членом семейства ABC транспортеров, обычно связанным с заболеванием, является cAMP/АТФ-опосредованный анионный канал, CFTR. CFTR экспрессируется в различных типах клеток, включая абсорбтивные и секреторные эпителиальные клетки, где он регулирует приток анионов через мембрану, а также активность других ионных каналов и белков. В эпителиальных клетках нормальное функционирование CFTR является критическим для поддержания транспорта электролитов в организме, включая респираторную и пищеварительную ткани. CFTR состоит приблизительно из 1480 аминокислот, которые кодируют белок, состоящий из тандемного повтора трансмембранных доменов, каждый из которых содержит шесть трансмембранных двойных спиралей и нуклеотидный связывающийся домен. Эти два трансмембраных домена связаны между собой большим полярным регуляторным (R)-доменом с множественными сайтами фосфорилирования, которые регулируют активность канала и клеточный траффик.
Ген, кодирующий CFTR, был идентифицирован и секвенирован (See Gregory, R. J. et al. (1990) Nature 347:382-386; Rich, D. P. et al. (1990) Nature 347:358-362), (Riordan, J. R. et al. (1989) Science 245:1066-1073). Дефект в этом гене вызывает мутации в CFTR, приводящие к кистозному фиброзу ("CF"), неиболее распространенному фатальному генетическому заболеванию у людей. Кистозный фиброз поражает приблизительно одного из каждых 2500 детей раннего возраста в США. Из населения США в целом, до 10 миллионов людей имеют одну копию дефективного гена без явно выраженных эффектов заболевания. В отличие от этого, субъекты с двумя копиями CF-ассоциированного гена страдают от изнурительных и фатальных эффектов CF, включая хроническое легочное заболевание.
У пациентов с кистозным фиброзом мутации в CFTR, эндогенно экспрессируемом в респираторном эпителии, приводят к уменьшенной апикальной анионной секреции, вызывая дисбаланс в транспорте ионов и жидкостей. Полученное в результате снижение анионного транспорта способствует повышенной аккумуляции слизи в легких с сопутствующими микробными инфекциями, которые, в конечном счете, приводят к смерти CF пациентов. Помимо респираторного заболевания, CF пациенты типично страдают от желудочно-кишечных проблем и недостаточной функции поджелудочной железы, что, если это не лечить, приводит к смерти. Кроме того, большинство мужчин с кистозным фиброзом являются бесплодными, и фертильность снижена у женщин с кистозным фиброзом. В отличие от тяжелых эффектов двух копий CF-ассоциированного гена, субъекты с одной копией CF-ассоциированного гена демонстрируют повышенную резистентность к холере и к обезвоживанию в результате диареи - возможно объясняющую относительно высокую распространенность CF гена среди населения.
Анализ последовательности CFTR гена CF хромосом выявил ряд различных заболеваний, вызывающих мутации (Cutting, G. R. et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61:863:870; and Kerem, B-S. et al. (1989) Science 245:1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). В настоящее время идентифицировано более чем 1000 заболеваний, вызывающих мутации в CF гене (http://www.genet.sickkids.on.ca/cftr/). Наиболее распространенной мутацией является делеция фенилаланина в положении 508 аминокислотной последовательности CFTR, и ее обычно указывают как ΔF508-CFTR. Эта мутация возникает приблизительно в 70% случаев кистозного фиброза, и она ассоциируется с тяжелым заболеванием.
Делеция остатка 508 в ΔF508-CFTR препятствует правильной укладке зарождающегося белка. Это приводит к неспособности мутантного белка выходить из ER и перемещаться к плазменной мембране. В результате, количество каналов, присутствующих в мембране, далеко от наблюдаемого в клетках, экспрессирующих CFTR дикого типа. Помимо нарушенного траффика, такая мутация приводит к дефекту воротного механизма канала. Все вместе, уменьшенное количество каналов в мембране и дефект воротного механизма, приводят к снижению анионного транспорта через эпителий, приводя к нарушенному транспорту ионов и жидкостей. (Quinton, P. M. (1990), FASEB J. 4: 2709-2727). Исследования, однако, показали, что уменьшенные количества ΔF508-CFTR в мембране являются функциональными, хотя меньше чем CFTR дикого типа. (Dalemans et al. (1991), Nature Lond. 354: 526-528; Denning et al., supra; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). Помимо ΔF508-CFTR, R117H-CFTR и G551D-CFTR другие вызывающие заболевание мутации в CFTR, которые приводят к нарушенному траффику, синтезу и/или воротному механизму канала, можно было бы регулировать либо путем активации, либо даун-регуляции для изменения анионной секреции и модификации прогрессирования и/или тяжести заболевания.
Хотя CFTR транспортирует различные молекулы, помимо анионов, ясно, что эта роль (транспорт анионов, хлорида и бикарбоната) представляет собой один элемент в важном механизме транспорта ионов и воды через эпителий. Другие элементы включают эпителиальный Na+ канал, ENaC, Na+/2C1-/K+ ко-транспортер, Na+-K+-АТФазный насос и базолатеральные мембранные K+ каналы, которые отвечают за поглощение хлора в клетке.
Эти элементы работают вместе для достижения направленного транспорта через эпителий через их селективную экспрессию и локализацию в клетке. Абсорбция хлора происходит в результате скоординированной активности ENaC и CFTR, присутствующих на апикальной мембране, и Na+-K+-АТФазного насоса и Cl ионных каналов, экспрессируемых на базолатеральной поверхности клетки. Вторичный активный транспорт хлора с люминальной стороны приводит к аккумуляции внутриклеточного хлора, который затем пассивно покидает клетку через Cl- каналы, приводя к векторному транспорту. Расположение Na+/2C1-/7K+ ко-транспортера, Na+-K+-АТФазного насоса и базолатеральных мембранных K+ каналов на базолатеральной поверхности и CFTR на люминальной стороне координирует секрецию хлора через CFTR на люминальной стороне. Поскольку вода, вероятно, никогда сама активно не транспортируется, ее протекание через эпителий зависит от очень малых трансэпителиальных осмотических градиентов, создаваемых объемным потоком натрия и хлора.
Предполагают, что дефект бикарбонатного транспорта из-за мутаций в CFTR вызывает дефекты некоторых секреторных функций. См., например, “Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis,” Paul M. Quinton, Lancet 2008; 372: 415-417.
Мутации в CFTR, которые связывают с умеренной CFTR дисфункцией, также очевидны у пациентов с состояниями, при которых некоторые проявления заболевания являются общими с CF, но не достигают диагностического критерия CF. Они включают врожденное двустороннее отсутствие семявыводящих протоков, идиопатический хронический панкреатит, хронический бронхит и хронический риносинусит. Другие заболевания, в которых, как считают, мутантный CFTR является фактором риска, вместе с модифицированными генами или экологическими факторами, включают первичный склерозирующий холангит, аллергический бронхолегочный аспергиллез и астму.
Также было продемонстрировано, что сигаретный дым, гипоксия и экологические факторы, которые индуцируют передачу гипоксического сигнала, нарушают CFTR функцию и могут способствовать некоторым формам респираторного заболевания, такого как хронический бронхит. Заболевания, которые могут возникать из-за нарушенной функции CFTR, но не достигают диагностического критерия CF, определяют как CFTR-связанные заболевания.
Помимо кистозного фиброза, модуляция CFTR активности может быть полезной для других заболеваний, не связанных непосредственно с мутациями в CFTR, таких как секреторные заболевания и другие связанные с укладкой белка заболевания, опосредованные CFTR. CFTR регулирует поток хлорида и бикарбоната через эпителий многих клеток для контроля движения жидкости, солюбилизации белка, вязкости слизи и ферментативной активности. Дефекты в CFTR могут вызывать блокаду дыхательных путей или протоков во многих органах, включая печень и поджелудочную железу. Потенциирующие средства представляют собой соединения, которые усиливают связанную с воротным механизмом активность CFTR, присутствующего в клеточной мембране. Любое заболевание, которое включает сгущение слизи, нарушение регуляции жидкости, нарушение слизистого клиренса или блокаду протоков, приводящую к воспалению и разрушению ткани, может быть кандидатом для потенцирующих средств.
Они включают, но не ограничиваются этим, хроническое обструктивное легочное заболевание (COPD), астму, индуцированное курением COPD, хронический бронхит, риносинусит, запор, болезнь сухих глаз и синдром Шегрена, гастро-эзофагеальный рефлюкс, желче-каменную болезнь, выпадение прямой кишки и воспалительное заболевание кишечника. COPD характеризуется ограничением воздушного потока, которое прогрессирует и не является полностью обратимым. Ограничение воздушного потока происходит из-за гиперсекреции слизи, эмфиземы и бронхиолита. Активаторы мутантного или дикого типа CFTR предлагают потенциальное лечение гиперсекреции слизи и нарушенного клиренса реснисчатого эпителия, что является обычным при COPD. В частности, повышение анионной секреции через CFTR может облегчать транспорт жидкости в поверхностную жидкость дыхательных путей для гидратирования слизи и оптимизирования вязкости перицилиарной жидкости. Это может привести к усиленному клиренсу реснисчатого эпителия и уменьшению симптомов, связанных с COPD. Кроме того, путем предотвращения развития инфекции и воспаления в результате улучшенного клиренса дыхательных путей модуляторы CFTR могут предотвращать или замедлять паренхимальную деструкцию дыхательных путей, которая характерна для эмфиземы, и уменьшать или обращать увеличение количества и размер клеток, секретирующих слизь, что является причиной гиперсекреции слизи при заболеваниях дыхательных путей. Болезнь сухих глаз характеризуется снижением продукции слезной жидкости и аномальными профилями слезной липидной пленки, белка и муцина. Существует множество причин болезни сухих глаз, некоторые из которых включают возраст, глазные операции Lasik, артрит, лекарственное лечение, химические/термальные ожоги, аллергии и заболевания, такие как кистозный фиброз и синдром Шегрена. Увеличение анионной секреции через CFTR может повысить транспорт жидкости из роговичных эндотелиальных клеток и секреторных желез, окружающих глаз, усиливая увлажнение роговицы. Это помогает облегчить симптомы, связанные с болезнью сухих глаз. Синдром Шегрена представляет собой аутоиммунное заболевание, при котором иммунная система атакует продуцирующие влагу железы по всему организму, включая глаз, рот, кожу, респираторную ткань, печень, влагалище и кишечник. Симптомы включают, сухость глаз, полости рта и влагалища, а также заболевание легких. Это заболевание также связано с ревматоидным артритом, системной волчанкой, системным склерозом и полимиозитом/дерматомиозитом. Считают, что нарушение белкового траффика вызывает это заболевание, возможности лечения которого ограничены. Модуляторы CFTR активности могут гидратировать различные органы, пораженные заболеванием, и могут способствовать облегчению связанных с этим симптомов. У субъектов с кистозным фиброзом повторяются эпизоды интестинальной обструкции и чаще наблюдаются случаи выпадения прямой кишки, желче-каменной болезни, гастро-эзофагеального рефлюкса, желудочно-кишечных опухолей и воспалительного заболевания кишечника, указывая на то, что функция CFTR может играть важную роль в предотвращении таких заболеваний.
Как обсуждалось выше, считается, что делеция остатка 508 в ΔF508-CFTR препятствует правильной укладке зарождающегося белка, что приводит к неспособности мутантного белка выходить из ER и перемещаться к плазменной мембране. В результате, на плазменной мембране присутствуют недостаточные количества зрелого белка, и транспорт хлора в эпителиальных тканях существенно снижен. Действительно, было показано, что этот клеточный феномен дефектного ER процессинга CFTR повредством ER механизма лежит в основе не только CF заболевания, но также широкого ряда других отдельных и наследственных заболеваний. Два пути, приводящие к неправильной работе ER механизма, включают либо потерю связи с ER экспортом белков, приводящую к деградации, либо ER аккумуляцию этих дефектных/имеющих неправильную укладку белков [Aridor M, et al., Nature Med., 5(7), pp 745-751 (1999); Shastry, B.S., et al., Neurochem. International, 43, pp 1-7 (2003); Rutishauser, J., et al., Swiss Med Wkly, 132, pp 211-222 (2002); Morello, JP et al., TIPS, 21, pp. 466-469 (2000); Bross P., et al., Human Mut., 14, pp. 186-198 (1999)]. Заболевания, связанные с первым классом неправильной функции ER, включают кистозный фиброз (из-за неправильной укладки ΔF508-CFTR, как обсуждается выше), наследственную эмфизему (из-за a1-антитрипсина; не Piz варианты), наследственный гемохроматоз, нарушения коагуляции-фибринолиза, такие как дефицит белка C, наследственную ангиоэдему типа 1, нарушения липидного процессинга, такие как семейная гиперхолестеринемия, хиломикронемию типа 1, абеталипопротеинемию, болезни лизосомального накопления, такие как I-клеточное заболевание/псевдо-Hurler, Мукополисахаридозы (из-за лизосомнных процессинговых ферментов), болезнь Сандхофа/Тэя-Сакса (из-за β-гексозаминидазы), синдром Криглера-Найяра типа II (из-за UDP-глюкуронил-сиалил-трансферазы), полиэндокринопатию/гиперинсулинемию, сахарный диабет (из-за инсулинового рецептора), карликовость Ларона (из-за рецептора гормона роста), дефицит милеопероксидазы, первичный гипопаратиреоидизм (из-за препропаратиреоидного гормона), меланому (из-за тирозиназы). Заболевания, связанные с последним классом неправильной функции ER, включают Гликаноз CDG типа 1, наследственную эмфизему (из-за α1-Антитрипсина (PiZ вариант), врожденный гипертиреоидизм, несовершенный остеогенез (из-за типа I, II, IV проколлагена), наследственную гипофибриногенемию (из-за фибриногена), дефицит ACT (из-за α1-антихимотрипсина), несахарный диабет (DI), нейрофизеальный DI (из-за гормона вазопрессина/V2-рецептора), нефрогенный DI (из-за аквапорина II), синдром Шарко-Мари-Тута (из-за периферийного миелинового белка 22), болезнь Perlizaeus-Merzbacher, нейродегенеративные заболевания, такие как болезнь Альцгеймера (из-за βAPP и пресенилинов), болезнь Паркинсона, амиотрофический боковой склероз, прогрессирующий супрануклеарный паралич, болезнь Пика, некоторые полиглутаминовые неврологические расстройства, такие как болезнь Гентингтона, спиноцеребуллярная атаксия типа I, спинальная и бульбарная мышечная атрофия, dentatorubal pallidoluysian и миотоническая дистрофия, а также спонгиформные энцефалопатии, такие как наследственная болезнь Крейцфельдта-Якоба (из-за дефекта процессинга прионового белка), болезнь Фабри (из-за лизосомной α-галактозидазы A), синдром Штросслера-Шейнкера (из-за дефекта процессинга Prp), бесплодие, панкреатит, панкреатическую недостаточность, остеопороз, остеопению, синдром Горэма, хлоридные каналопатии, врожденную миотонию (формы Томсона и Бекера), синдром Бартера типа III, болезнь Дента, гиперэкплексию, эпилепсию, гиперэкплексию, болезнь лизосомального накопления, синдром Ангельмана, первичную цилиарную дискинезию (PCD), PCD с situs inversus (также известна как синдром Картагенера), PCD без situs inversus и цилиарную аплазию и заболевание печени.
Другие заболевания, связанные с мутацией в CFTR, включают мужское бесплодие, вызванное врожденным двусторонним отсутствием семявыводящих протоков (CBAVD), слабую форму легочного заболевания, идиопатический панкреатит и аллергический бронхолегочный аспергиллез (ABPA). См. “CFTR-opathies: disease phenotypes associated with cystic fibrosis transmembrane regulator gene mutations,” Peader G. Noone и Michael R. Knowles, Respir. Res. 2001, 2: 328-332 (включен в настоящую заявку посредством ссылки).
Помимо положительной регуляции CFTR активности, снижение анионной секреции модуляторами CFTR может быть полезным для лечения секреторных диарей, при которых эпителиальный водный транспорт существенно увеличивается в результате действия усиливающего секрецию средства, активирующего хлоридный транспорт. Этот механизм включает повышение cAMP и стимуляцию CFTR.
Хотя существуют многочисленные причины диареи, основные последствия диарей, являющиеся результатом чрезмерного хлоридного транспорта, являются общими для всех, и они включают обезвоживание, ацидоз, задержку роста и смерть. Острые и хронические диареи представляют серьезную медицинскую проблему во многих регионах мира. Диарея является как серьезным фактором нарушения питания, так и основной причиной смертности (5000000 смертей/год) у детей возраста младше 5 лет.
Секреторные диареи также представляют собой опасное состояние у пациентов с синдромом приобретенного иммунодефицита (СПИД) и хроническим воспалительным заболеванием кишечника (IBD). У 16 миллионов путешественников, посещающих развивающиеся страны, из индустриально развитых стран каждый год развивается диарея, при этом тяжесть и количество случаев диареи варьирует в зависимости от посещаемой страны или области.
Диарея у домашнего скота и домашних животных, таких как коровы, свиньи и лошади, овцы, козы, кошки и собаки, также известная как дизентерия, является основной причиной гибели у этих животных. Диарея может возникать в результате какой-либо существенной перемены, такой как прекращение грудного кормления или физическое перемещение, а также в ответ на различные бактериальные или вирусные инфекции, и в основном возникает в течение первых нескольких часов жизни животного.
Бактерией, наиболее часто вызывающей диарею, является энтеротоксогенная E.coli (ETEC), содержащая K99 pilus антиген. Основными вирусами, вызывающими диарею, являются ротавирус и коронавирус. Другие инфекционные агенты включают, среди прочего, cryptosporidium, giardia lamblia и salmonella.
Симптомы ротавирусной инфекции включают экскрецию водных фекалий, обезвоживание и слабость. Коронавирус вызывает более тяжелое заболевание у новорожденных животных и имеет более высокий показатель смертности, чем ротавирусная инфекция. Однако часто молодое животное может быть инфицировано более чем одним вирусом или комбинацией вирусных и бактериальных микроорганизмов одновременно. Это существенно усиливает тяжесть заболевания.
Соответственно, существует потребность в модуляторах активности ABC транспортера и композициях на их основе, которые можно использовать для модуляции активности ABC транспортера в клеточной мембране млекопитающего.
Существует потребность в способах лечения ABC транспортер-опосредованных заболеваний с использованием таких модуляторов активности ABC транспортера.
Существует потребность в способах модуляции активности ABC транспортера в ex vivo клеточной мембране млекопитающего.
Существует потребность в модуляторах активности CFTR, которые можно использовать для модуляции активноси CFTR в клеточной мембране млекопитающего.
Также существует потребность в сильных и селективных CFTR-потенциирующих средствах, в том числе дикого типа и мутантных форм человеческого CFTR. Эти мутантные формы CFTR включают, но не ограничиваются этим, ΔF508del, G551D, R117H, 2789+5G->A.
Существует потребность в способах лечения CFTR-опосредованных заболеваний с использованием таких модуляторов активности CFTR.
Существует потребность в способах модуляции активности CFTR в ex vivo клеточной мембране млекопитающего.
Краткое описание изобретения
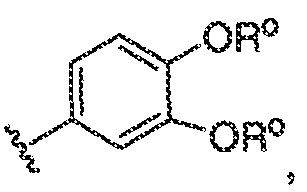
Было обнаружено, что соединения по настоящему изобретению и содержащие их фармацевтически приемлемые композиции являются полезными в качестве модуляторов активности ABC транспортера. Эти соединения имеют общую формулу I:
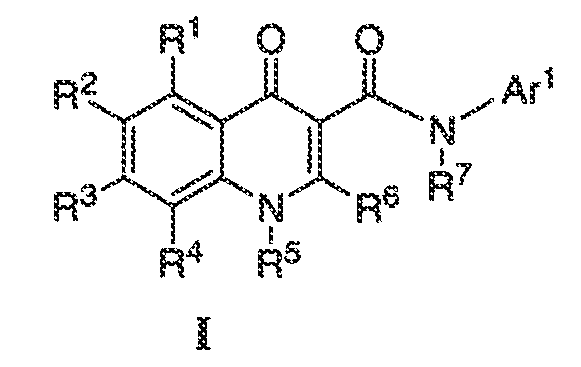
или фармацевтически приемлемая соль такого соединения, где R1, R2, R3, R4, R5, R6, R7 и Ar1 описаны в общем виде и в классах и подклассах ниже.
Эти соединения и фармацевтически приемлемые композиции являются полезными для лечения или ослабления тяжести различных заболеваний, расстройств или состояний, включая, но не ограничиваясь этим, кистозный фиброз, наследственную эмфизему, наследственный гемохроматоз, нарушения коагуляции-фибринолиза, такие как дефицит белка C, наследственная ангиоэдема типа 1, нарушения липидного процессинга, такие как семейная гиперхолестеринемия, хиломикронемия типа 1, абеталипопротеинемия, болезни лизосомального накопления, такие как I-клеточное заболевание/Псевдо-Hurler, мукополисахаридозы, болезнь Сандхофа/Тэя-Сакса, синдром Криглера-Найяра типа II, полиэндокринопатию/гиперинсулинемию, сахарный диабет, карликовость Ларона, дефицит милеопероксидазы, первичный гипопаратиреоидизм, меланому, гликаноз CDG типа 1, наследственную эмфизему, врожденный гипертиреоидизм, несовершенный остеогенез, наследственную гипофибриногенемию, дефицит ACT, несахарный диабет (DI), нейрофизеальный DI, нефрогенный DI, синдром Шарко-Мари-Тута, болезнь Perlizaeus-Merzbacher, нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз, прогрессирующий супрануклеарный паралич, болезнь Пика, некоторые полиглутаминовые неврологические расстройства, такие как болезнь Гентингтона, спиноцеребуллярная атаксия типа I, спинальная и бульбарная мышечная атрофия, Dentatorubal pallidoluysian и миотоническая дистрофия, а также спонгиформные энцефалопатии, такие как наследственная болезнь Крейцфельдта-Якоба, болезнь Фабри, синдром Штросслера-Шейнкера, COPD, болезнь сухих глаз и болезнь Шегрена.
Подробное описание изобретения
I. Общее описание соединений по настоящему изобретению:
Настоящее изобретение относится к соединениям формулы I, полезным в качестве модуляторов активности ABC транспортера:
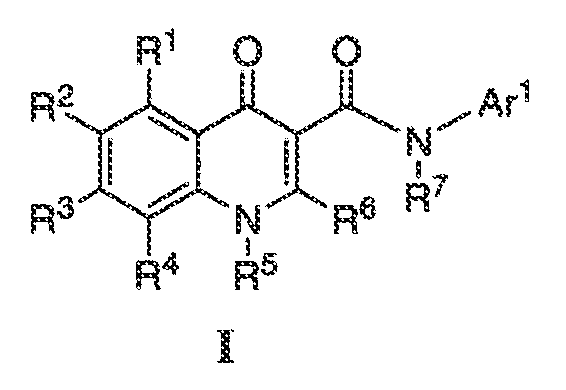
или фармацевтически приемлемой соли такого соединения, где:
Ar1 представляет собой 5-6 членное ароматическое моноциклическое кольцо, содержащее 0-4 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо необязательно является конденсированным с 5-12-членным моноциклическим или бициклическим ароматическим, частично ненасыщенным или насыщенным кольцом, где каждое кольцо содержит 0-4 гетероатома, независимо выбранных из азота, кислорода или серы, где Ar1 содержит m количество заместителей, каждый из которых независимо выбран из -WRW;
W представляет собой связь или представляет собой необязательно замещенную C1-C6 алкилиденовую цепь, где вплоть до двух метиленовых звеньев в W необязательно и независимо заменены -CO-, -CS-, -COCO-, -CONR'-, -CONR'NR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'NR', -NR'NR'CO-, -NR'CO-, -S-, -SO, -SO2-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-;
RW независимо представляет собой R', галоген, NO2, CN, CF3 или OCF3;
m имеет значение 0-5;
каждый из R1, R2, R3, R4 и R5 независимо представляет собой -X-RX;
X представляет собой связь или представляет собой необязательно замещенную C1-C6 алкилиденовую цепь, где вплоть до двух метиленовых звеньев в X необязательно и независимо заменены -CO-, -CS-, -COCO-, -CONR'-, -CONR'NR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'NR', -NR'NR'CO-, -NR'CO-, -S-, -SO, -SO2-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-;
RX независимо представляет собой R', галоген, NO2, CN, CF3 или OCF3;
R6 представляет собой водород, CF3, -OR', -SR' или необязательно замещенную C1-6 алифатическую группу;
R7 представляет собой водород или C1-6 алифатическую группу, необязательно замещенную группой -X-RX;
R' независимо выбран из водорода или необязательно замещенной группы, выбраной из C1-C8 алифатической группы, 3-8-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, или 8-12-членной насыщенной, частично ненасыщенной или полностью ненасыщенной бициклической кольцевой системы, содержащей 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы; или два присутствующих R' взяты вместе с атомом(атомами), с которым они связаны, с образованием необязательно замещенного 3-12-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического или бициклического кольца, содержащего 0-4 гетероатома, независимо выбранных из азота, кислорода или серы.
В некоторых других вариантах воплощения обеспечиваются соединения формулы I:
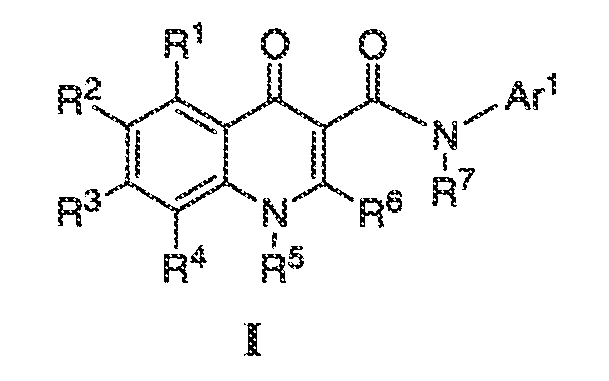
или фармацевтически приемлемая соль такого соединения, где:
Ar1 представляет собой 5-6 членное ароматическое моноциклическое кольцо, содержащее 0-4 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо необязательно является конденсированным с 5-12-членным моноциклическим или бициклическим ароматическим частично ненасыщенным или насыщенным кольцом, где каждое кольцо содержит 0-4 гетероатома, независимо выбранных из азота, кислорода или серы, где Ar1 содержит m количество заместителей, каждый из которых независимо выбран из -WRW;
W представляет собой связь или представляет собой необязательно замещенную C1-C6 алкилиденовую цепь, где вплоть до двух метиленовых звеньев в W необязательно и независимо заменены -CO-, -CS-, -COCO-, -CONR'-, -CONR'NR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'NR', -NR'NR'CO-, -NR'CO-, -S-, -SO, -SO2-, -NR'-, -SO2NR'-, NR'SO2-, -NR'SO2NR'-;
RW независимо представляет собой R', галоген, NO2, CN, CF3 или OCF3;
m имеет значение 0-5;
каждый из R1, R2, R3, R4 и R5 независимо представляет собой -X-RX;
X представляет собой связь или представляет собой необязательно замещенную C1-C6 алкилиденовую цепь, где вплоть до двух метиленовых звеньев в X необязательно и независимо заменены -CO-, -CS-, -COCO-, -CONR'-, -CONR'NR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'NR', -NR'NR'CO-, -NR'CO-, -S-, -SO, -SO2-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-;
RX независимо представляет собой R', галоген, NO2, CN, CF3 или OCF3;
R6 представляет собой водород, CF3, -OR', -SR' или необязательно замещенную C1-C8 алифатическую группу;
R7 представляет собой водород или C1-C6 алифатическую группу, необязательно замещенную группой -X-RX;
R' независимо выбран из водорода или необязательно замещенной группы, выбранной из C1-C8 алифатической группы, 3-8-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, или 8-12-членной насыщенной, частично ненасыщенной или полностью ненасыщенной бициклической кольцевой системы, содержащей 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы; или два присутствующих R' взяты вместе с атомом(атомами), с которым они связаны, с образованием необязательно замещенного 3-12-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического или бициклического кольца, содержащего 0-4 гетероатома, независимо выбранных из азота, кислорода или серы;
при условии, что:
i) когда R1, R2, R3, R4, R5, R6 и R7 представляют собой водород, тогда Ar1 не может представлять собой фенил, 2-метоксифенил, 4-метоксифенил, 2-метилфенил, 2,6-дихлорфенил, 2,4-дихлорфенил, 2-бромфенил, 4-бромфенил, 4-гидроксифенил, 2,4-динитрофенил, фенил 3,5-дикарбоновую кислоту, 2,4-диметилфенил, 2,6-диметилфенил, 2-этилфенил, 3-нитро-4-метилфенил, фенил 3-карбоновую кислоту, 2-фторфенил, 3-фторфенил, 3-трифторметилфенил, 3-этоксифенил, 4-хлорфенил, 3-метоксифенил, 4-диметиламинофенил, 3,4-диметилфенил, 2-этилфенил или 4-этоксикарбонилфенил;
ii) когда R1, R2, R3, R5, R6 и R7 представляют собой водород и R4 представляет собой метокси, тогда Ar1 не может представлять собой 2-фторфенил или 3-фторфенил;
iii) когда R1, R3, R4, R5, R6 и R7 представляют собой водород, R2 представляет собой 1,2,3,4-тетрагидроизохинолин-1-ил-сульфонил, тогда Ar1 не может представлять собой 3-трифторметилфенил;
iv) когда R1, R2, R3, R4, R5 и R7 представляют собой водород, R6 представляет собой метил, тогда Ar1 не может представлять собой фенил;
v) когда R1, R4, R5, R6 и R7 представляют собой водород, R2 и R3, взятые вместе, представляют собой метилендиокси, тогда Ar1 не может представлять собой 4-хлорфенил, 4-бромфенил, 4-нитрофенил, 4-карбоэтоксифенил, 6-этокси-бензотиазол-2-ил, 6-карбоэтокси-бензотиазол-2-ил, 6-галоген-бензотиазол-2-ил, 6-нитро-бензотиазол-2-ил или 6-тиоциано-бензотиазол-2-ил.
vi) когда R1, R4, R5, R6 и R7 представляют собой водород, R2 и R3, взятые вместе, представляют собой метилендиокси, тогда Ar1 не может представлять собой 4-замещенный фенил, где указанный заместитель представляет собой -SO2NHRxx, где Rxx представляет собой 2-пиридинил, 4-метил-2-пиримидинил, 3,4-диметил-5-изоксазолил;
vii) когда R1, R2, R3, R4, R5, R6 и R7 представляют собой водород, тогда Ar1 не может представлять собой тиазол-2-ил, 1H-1,2,4-триазол-3-ил или 1H-1,3,4-триазол-2-ил;
viii) когда R1, R2, R3, R5, R6 и R7 представляют собой водород, и R4 представляет собой CF3, OMe, хлор, SCF3 или OCF3, тогда Ar1 не может представлять собой 5-метил-1,2-оксазол-3-ил, тиазол-2-ил, 4-фторфенил, пиримидин-2-ил, 1-метил-1,2-(1H)-пиразол-5-ил, пиридин-2-ил, фенил, N-метил-имидазол-2-ил, имидазол-2-ил, 5-метил-имидазол-2-ил, 1,3-оксазол-2-ил или 1,3,5-(1H)-триазол-2-ил;
ix) когда R1, R2, R3, R4, R5, R6 и R7, каждый, представляет собой водород, тогда Ar1 не может представлять собой пиримидин-2-ил, 4,6-диметил-пиримидин-2-ил, 4-метокси-6-метил-1,3,5-триазин-2-ил; 5-бром-пиридин-2-ил, пиридин-2-ил или 3,5-дихлор-пиридин-2-ил;
x) когда R1, R2, R3, R4, R5 и R7, каждый, представляет собой водород, R6 представляет собой гидрокси, тогда Ar1 не может представлять собой 2,6-дихлор-4-аминосульфонил-фенил;
xi) когда R2 или R3 представляет собой необязательно замещенный н-пиперазил, н-пиперидил или N-морфолинил, тогда Ar1 не может представлять собой необязательно замещенное кольцо, выбранное из тиазол-2-ила, пиридила, фенила, тиадиазолила, бензотиазол-2-ила или индазолила;
xii) когда R2 представляет собой необязательно замещенный циклогексиламино, тогда Ar1 не может представлять собой необязательно замещенный фенил, пиридил или тиадиазолил;
xiii) Ar1 не может представлять собой необязательно замещенный тетразолил;
xiv) когда R2, R4, R5, R6 и R7, каждый, представляет собой водород, и R1 и R3 оба одновременно представляют собой CF3, хлор, метил или метокси, тогда Ar1 не может представлять собой 4,5-дигидро-1,3-тиазол-2-ил, тиазол-2-ил или [3,5-бис(трифторметил)-1H-пиразол-1-ил]фенил;
xv) когда R1, R4, R5, R6 и R7, каждый, представляет собой водород, и Ar1 представляет собой тиазол-2-ил, тогда ни R2 ни R3 не может представлять собой изопропил, хлор или CF3;
xvi) когда Ar1 представляет собой 4-метоксифенил, 4-трифторметилфенил, 2-фторфенил, фенил или 3-хлорфенил, тогда:
a) когда R1, R2, R4, R5, R6 и R7, каждый, представляет собой водород, тогда R3 не может представлять собой метокси; или
b) когда R1, R3, R4, R5, R6 и R7, каждый, представляет собой водород, тогда R2 не может представлять собой хлор; или
c) когда R1, R2, R3, R5, R6 и R7, каждый, представляет собой водород, тогда R4 не может представлять собой метокси; или
d) когда R1, R3, R4, R6 и R7, каждый, представляет собой водород, и R5 представляет собой этил, тогда R2 не может представлять собой хлор;
e) когда R1, R2, R4, R5, R6 и R7, каждый, представляет собой водород, тогда R3 не может представлять собой хлор;
xvi) когда R1, R3, R4, R5, R6 и R7, каждый, представляет собой водород, и R2 представляет собой CF3 или OCF3, тогда Ar1 не может представлять собой [3,5-бис(трифторметил)-1H-пиразол-1-ил]фенил;
xvii) когда R1, R2, R4, R5, R6 и R7, каждый, представляет собой водород, и R3 представляет собой водород или CF3, тогда Ar1 не может представлять собой фенил, замещенный -OCH2CH2Ph, -OCH2CH2(2-трифторметил-фенил), -OCH2CH2-(6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ил) или замещенным 1H-пиразол-3-илом; и
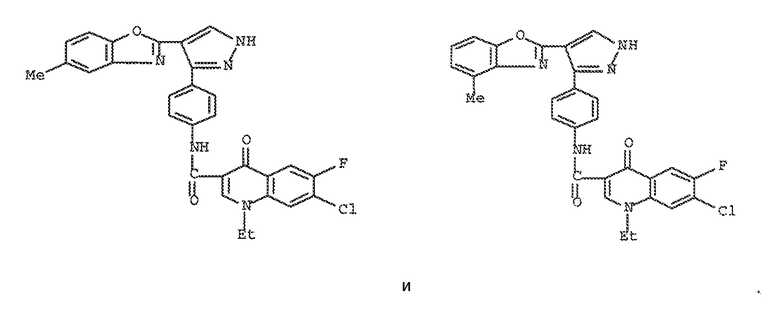
xviii) следующие два соединения исключаются:
2. Соединения и определения:
Соединения по настоящему изобретению включают соединения, описанные в общем виде выше и далее проиллюстрированные на примере классов, подклассов и видов, раскрытых в настоящей заявке. Как это используется в настоящей заявке, применимы следующие определения, если не указано иное.
Термин "ABC-транспортер", как он используется в настоящей заявке, означает ABC-транспортерный белок или его фрагмент, содержащий, по меньшей мере, один связывающий домен, где указанный белок или его фрагмент присутствует in vivo или in vitro. Термин "связывающий домен", как он используется в настоящей заявке, означает домен на ABC-транспортере, который может связываться с модулятором. Смотри, например, Hwang, T. C. et al., J. Gen. Physiol. (1998): 111(3), 477-90.
Термин "CFTR", как он используется в настоящей заявке, означает регулятор трансмембранной проводимости кистозного фиброза или его мутацию, обладающую способностью регулятора активности, включая, но не ограничиваясь этим, ΔF508 CFTR и G551D CFTR (смотри, например, http://www.genet.sickkids.on.ca/cftr/, для CFTR мутаций).
Термин "модулирующий", как он используется в настоящей заявке, означает увеличение или уменьшение на количество, которое можно измерить.
Для целей настоящего изобретения, химические элементы обозначаются в соответствии с Периодической Таблицей Элементов, CAS version, Handbook of Chemistry and Physics, 75th Ed. Кроме того, общие принципы органической химии описаны в "Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito: 1999 и в "March's Advanced Organic Chemistry", 5th Ed., Ed.: Smith, M. B. and March, J., John Wiley & Sons, New York: 2001, полное содержание которых включено в настоящую заявку посредством ссылки.
Как описано в настоящей заявке, соединения по настоящему изобретению необязательно могут быть замещены одним или несколькими заместителями, такими как в общем виде проиллюстрированные выше или представленные на примере конкретных классов, подклассов и видов по настоящему изобретению. Должно быть понятно, что фраза "необязательно замещенный" используется взаимозаменяемо с фразой "замещенный или незамещенный". Как правило, термин "замещенный", независимо от того, стоит перед ним слово "необязательно" или нет, относится к замещению водородных радикалов в данной структуре радикалом указанного заместителя. Если не указано иное, необязательно замещенная группа может содержать заместитель в каждом замещаемом положении этой группы, и когда более чем одно положение в какой-либо определенной структуре может быть замещено более чем одним заместителем, выбранным из указанной группы, такие заместители могут быть либо одинаковыми, либо отличными друг от друга в каждом положении. Комбинации заместителей, предусматриваемые настоящим изобретением, предпочтительно представляют собой такие, которые приводят к образованию стабильных или химически достижимых соединений. Термин "стабильный", как он используется в настоящей заявке, относится к соединениям, которые, по существу, не изменяются, будучи подвержены условиям, которые делают возможным их получение, детекцию и, предпочтительно, их выделение, очистку и использование по одному или нескольким назначениям, раскрытым в настоящей заявке. В некоторых вариантах воплощения стабильное соединение или химически достижимое соединение представляет собой такое, которое, по существу, не изменяется при выдерживании при температуре 40ºC или ниже, в отсутствие влаги или в других химически реакционных условиях, в течение, по меньшей мере, недели.
Термин "алифатический" или "алифатическая группа", как он используется в настоящей заявке, означает линейную (т.е. неразветвленную) или разветвленную, замещенную или незамещенную углеводородную цепь, которая является полностью насыщенной или которая содержит одну или несколько единиц ненасыщенности, или моноциклический углеводород или бициклический углеводород, который является полностью насыщенным или который содержит одну или несколько единиц ненасыщенности, но который не является ароматическим (также указанный в настоящей заявке как "карбоцикл", "циклоалифатический" или "циклоалкил"), который имеет одну точку присоединения к остальной части молекулы. Если не указано иное, алифатические группы содержат 1-20 алифатических атомов углерода. В некоторых вариантах воплощения алифатические группы содержат 1-10 алифатических атомов углерода. В других вариантах воплощения, алифатические группы содержат 1-8 алифатических атомов углерода. В следующих вариантах воплощения, алифатические группы содержат 1-6 алифатических атомов углерода, и еще в некоторых вариантах воплощения алифатические группы содержат 1-4 алифатических атома углерода. В некоторых вариантах воплощения "циклоалифатический" (или "карбоцикл" или "циклоалкил") относится к моноциклическому C3-8 углеводороду или бициклическому или трициклическому C8-14 углеводороду, который является полностью насыщенным или который содержит одну или несколько единиц ненасыщенности, но который не является ароматическим, который имеет одну точку присоединения к остальной части молекулы, где любое отдельное кольцо в указанной бициклической кольцевой системе содержит 3-7 членов. Подходящие алифатические группы включают, но не ограничиваются этим, линейные или разветвленные, замещенные или незамещенные алкильные, алкенильные, алкинильные группы и их гибриды, такие как (циклоалкил)алкил, (циклоалкенил)алкил или (циклоалкил)алкенил. Подходящие циклоалифатические группы включают циклоалкил, бициклический циклоалкил (например, декалин), связанный мостиковой связью бициклоалкил, такой как норборнил или [2,2,2]бицикло-октил или связанный мостиковой связью трициклическую группу, такую как адамантил.
Термин "гетероалифатический", как он используется в настоящей заявке, означает алифатические группы, где один или два атома углерода независимо заменены одним или несколькими атомами, выбранными из кислорода, серы, азота, фосфора или кремния. Гетероалифатические группы могут быть замещенными или незамещенными, разветвленными или неразветвленными, циклическими или нециклическими и включают "гетероцикл", "гетероциклил", "гетероциклоалифатические" или "гетероциклические" группы.
Термин "гетероцикл", "гетероциклил", "гетероциклоалифатический" или "гетероциклический", как он используется в настоящей заявке, означает неароматические, моноциклические, бициклические или трициклические кольцевые системы, в которых один или несколько кольцевых членов представляет собой независимо выбранный гетероатом. В некоторых вариантах воплощения "гетероцикл", "гетероциклил", "гетероциклоалифатическая" или "гетероциклическая" группа содержит от трех до четырнадцати кольцевых членов, где один или несколько кольцевых членов представляют собой гетероатом, независимо выбранный из группы, включающей кислород, серу, азот или фосфор и каждое кольцо в системе содержит от 3 до 7 кольцевых членов.
Термин "гетероатом" означает один или несколько атомов, выбранных из кислорода, серы, азота, фосфора или кремния (включая, любую окисленную форму азота, серы, фосфора или кремния; кватернизированную форму любого основного азота или; замещаемый азот гетероциклического кольца, например, N (как в 3,4-дигидро-2H-пирролиле), NH (как в пирролидиниле) или NR+ (как в N-замещенном пирролидиниле)).
Термин "ненасыщенный", как он используется в настоящей заявке, означает, что группа содержит одну или несколько единиц ненасыщенности.
Термин "алкокси" или "тиоалкил", используемый в настоящей заявке, относится к алкильной группе, определенной выше, присоединенной к основной углеродной цепи через атом кислорода ("алкокси") или серы ("тиоалкил").
Термины "галогеналифатический" и "галогеналкокси" означает алифатический или алкокси, в зависимости от конкретного случая, замещенный одним или несколькими атомами галогена. Термин "галоген" или "гало" означает F, Cl, Br или I. Примеры галогеналифатической группы включают -CHF2, -CH2F, -CF3, -CF2 - или пергалогеналкил, такой как, -CF2CF3.
Термин "арил", используемый отдельно или как часть более крупной группы, как в "аралкиле", "аралкокси" или "арилоксиалкиле", относится к моноциклической, бициклической и трициклической кольцой системе, содержащей в целом от пяти до четырнадцати кольцевых членов, где, по меньшей мере, одно кольцо в системе является ароматическим, и где каждое кольцо в системе содержит от 3 до 7 кольцевых членов. Термин "арил" можно использовать взаимозаменяемо с термином "арильное кольцо". Термин "арил" также относится к гетероарильным кольцевым системам, определенным в настоящей заявке ниже.
Термин "гетероарил", используемый отдельно или или как часть более крупной группы, как в "гетероаралкиле" или "гетероарилалкокси", относится к моноциклическим, бициклическим и трициклическим кольцевым системам, содержащим в целом от пяти до четырнадцати кольцевых членов, где по меньшей мере одно кольцо в системе является ароматическим, по меньшей мере одно кольцо в системе содержит один или несколько гетероатомов, и где каждое кольцо в системе содержит от 3 до 7 кольцевых членов. Термин "гетероарил" можно использовать взаимозаменяемо с термином "гетероарильное кольцо" или термином "гетероароматический".
Арильная (включая аралкил, аралкокси, арилоксиалкил и подобные) или гетероарильная (включая гетероаралкил и гетероарилалкокси и подобные) группа может содержать один или несколько заместителей. Подходящие заместители по ненасыщенному атому углерода арильной или гетероарильной группы выбраны из галогена; -RO; -ORO; -SRO; 1,2-метилен-диокси; 1,2-этилендиокси; фенила (Ph), необязательно замещенного группой RO; -O(Ph), необязательно замещенного группой RO; -(CH2)1-2(Ph), необязательно замещенного группой RO; -CH=CH(Ph), необязательно замещенного группой RO; -NO2; -CN; -N(RO)2; -NROC(O)RO; -NROC(O)N(RO)2; -NROCO2RO; -NRONROC(O)RO; -NRONROC(O)N(RO)2; -NRONROCO2RO; -C(O)C(O)RO; -C(O)CH2C(O)RO; -CO2RO; -C(O)RO; -C(O)N(RO)2; -OC(O)N(RO)2; -S(O)2RO; -SO2N(RO)2; -S(O)RO; -NROSO2N(RO)2; -NROSO2RO; -C(=S)N(RO)2; -C(=NH)-N(RO)2; или -(CH2)0-2NHC(O)RO, где в каждом независимом случае RO выбран из водорода, необязательно замещенной C1-6 алифатической группы, незамещенного 5-6-членного гетероарильного или гетероциклического кольца, фенила, -O(Ph) или -CH2(Ph), или, несмотря на определения выше, две независимо присутствующие группы RO, на одном и том же заместителе или на разных заместителях, взятые вместе с атомом(ами), с которым связана каждая RO группа, образуют 3-8-членное циклоалкильное, гетероциклильное, арильное или гетероарильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из группы, включающей азот, кислород или серу. Необязательные заместители в алифатической группе RO выбраны из NH2, NH(C1-4алифатическая группа), N(C1-4алифатическая группа)2, галогена, C1-4 алифатическая группа, OH, О(C1-4алифатическая группа), NO2, CN, CO2H, CO2(C1-4 алифатическая группа), O(галоген C1-4алифатическая группа) или галоген C1-4 алифатической группы, где каждая из перечисленных выше C1-4алифатических групп RO является незамещенной.
Алифатическая или гетероалифатическая группа или неароматическое гетероциклическое кольцо может содержать один или несколько заместителей. Подходящие заместители по насыщенному углероду алифатической или гетероалифатической группы или неароматического гетероциклического кольца выбраны из перечисленных выше для ненасыщенного углерода арильной или гетероарильной группы и дополнительно включают следующие: =О, =S, =NNHR*, =NN(R*)2, =NNHC(O)R*, =NNHCO2(алкил), =NNHSO2(алкил) или =NR*, где каждый R* независимо выбран из группы, включающей водород или необязательно замещенную C1-6 алифатическую группу. Необязательные заместители в алифатической группе R* выбраны из NH2, NH(C1-4 алифатическая группа), N(C1-4 алифатическая группа)2, галогена, C1-4 алифатической группы, OH, 0(C1-4 алифатическая группа), NO2, CN, CO2H, CO2(C1-4 алифатическая группа), O(галоген C1-4 алифатическая группа) или галоген(C1-4 алифатическая группа), где каждая из перечисленных выше C1-4 алифатических групп R* является незамещенной.
Необязательные заместители по азоту неароматического гетероциклического кольца выбраны из -R+, -N(R+)2, -C(O)R+, -CO2R+, -C(O)C(O)R+, -C(O)CH2C(O)R+, -SO2R+, -SO2N(R+)2, -C(=S)N(R+)2, -C(=NH)-N(R+)2 или -NR+SO2R+; где R+ представляет собой водород, необязательно замещенную C1-6 алифатическую группу, необязательно замещенный фенил, необязательно замещенный -O(Ph), необязательно замещенный -CH2(Ph), необязательно замещенный -(CH2)1-2(Ph); необязательно замещенный -CH=CH(Ph); или незамещенное 5-6-членное гетероарильое или гетероциклическое кольцо, содержащее от одного до четырех гетероатомов, независимо выбранных из группы, включающей кислород, азот или серу, или, несмотря на определение выше, две независимо присутствующие группы R+, на одном и том же заместителе или на разных заместителях, взятые вместе с атомом(ами), с которым связана каждая R+ группа, образуют 3-8-членное циклоалкильное, гетероциклильное, арильное или гетероарильное кольцо, содержащее 0-3 гетероатома, независимо выбранных из группы, включающей азот, кислород или серу. Необязательные заместители в алифатической группе или фенильном кольце R+ выбраны из NH2, NH(C1-4 алифатическая группа), N(C1-4 алифатическая группа)2, галогена, C1-4 алифатической группы, OH, О(C1-4 алифатическая группа), NO2, CN, CO2H, CO2(C1-4 алифатическая группа), O(галоген C1-4 алифатическая группа) или галоген(C1-4 алифатическая группа), где каждая из перечисленных выше C1-4 алифатических групп R+ является незамещенной.
Термин "алкилиденовая цепь" относится к прямой или разветвленной углеродной цепи, которая может быть полностью насыщенной или может содержать одну или несколько единиц ненасыщенности и содержит две точки присоединения к остальной части молекулы. Термин "спироциклоалкилиден" относится к карбоциклическому кольцу, которое может быть полностью насыщенным или может содержать одну или несколько единиц ненасыщенности и содержит две точки присоединения от одного и того же кольцевого атома углерода к остальной части молекулы.
Как объясняется выше, в некоторых вариантах воплощения, два независимо присутствующих Ro (или R+ или любая другая переменная, подобным образом определенная в настоящей заявке), взяты вместе с атомом(атомами), с которым каждая переменная связана, с образованием 3-8-членного циклоалкильного, гетероциклического, арильного или гетероарильного кольца, содержащего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы. Примеры колец, которые образуются, когда два независимо присутствующих Ro (или R+ или любая другая переменная, подобным образом определенная в настоящей заявке) взяты вместе с атомом(атомами), с которым каждая переменная связана, включают, но не ограничиваются этим, следующие: a) два независимо присутствующих Ro (или R+ или любая другая переменная, подобным образом определенная в настоящей заявке), которые связаны с одним и тем же атомом и взяты вместе этим атомом с образованием кольца, например, N(Ro)2, где оба присутствующих Ro взяты вместе с атомом азота с образованием пиперидин-1-ильной, пиперазин-1-ильной или морфолин-4-ильной группы; и b) два независимо присутствующих Ro (или R+ или любая другая переменная, подобным образом определенная в настоящей заявке), которые связаны с разными атомами и взяты вместе с обоими этими атомами с образованием кольца, например где фенильная группа замещена двумя присутствующими ORO
 эти два присутствующих Ro взяты вместе с атомами кислорода, с которыми они связаны, с образованием конденсированного 6-членного кислород-содержащего кольца:
эти два присутствующих Ro взяты вместе с атомами кислорода, с которыми они связаны, с образованием конденсированного 6-членного кислород-содержащего кольца:  Должно быть понятно, что множество других колец могут быть образованы, когда два независимо присутствующих Ro (или R+ или любая другая переменная, подобным образом определенная в настоящей заявке) взяты вместе с атомом(атомами), с которым каждая переменная связана, и что примеры, приведенные выше, не предназначены для ограничения.
Должно быть понятно, что множество других колец могут быть образованы, когда два независимо присутствующих Ro (или R+ или любая другая переменная, подобным образом определенная в настоящей заявке) взяты вместе с атомом(атомами), с которым каждая переменная связана, и что примеры, приведенные выше, не предназначены для ограничения.
Заместитель, представляющий собой связь, например, в бициклической кольцевой системе, как показано ниже, означает, что заместитель может быть присоединен к любому кольцевому атому, который может быть замещен, в любом кольце бициклической кольцевой системы:

Если не указано иное, предполагается, что структуры, представленные в настоящей заявке, также включают все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационные)) формы структур; например, R и S конфигурации для каждого асимметрического центра, (Z) и (E) изомеры по двойной связи и (Z) и (E) конформационные изомеры. Поэтому отдельные стереохимические изомеры, а также энантиомерные, диастереомерные и геометрические (или конформационные) смеси соединений по настоящему изобретению включены в объем настоящего изобретения. Если не указано иное, все таутомерные формы соединений по настоящему изобретению включены в объем настоящего изобретения. Например, когда R5 в соединениях формулы I представляет собой водород, соединения формулы I могут существовать в виде таутомеров:

Кроме того, если не указано иное, структуры, представленные в настоящей заявке, также включают соединения, которые отличаются только присутствием одного или нескольких изотопно обогащенных атомов. Например, соединения, имеющие представленные структуры, за исключением замещения водорода дейтерием или тритием, или замещения углерода углеродом 13C или 14C, включены в объем настоящего изобретения. Такие соединения являются полезными, например, в качестве аналитических инструментов или зондов в биологических анализах.
3. Описание иллюстративных соединений:
В некоторых вариантах воплощения настоящего изобретения, Ar1 выбран из:
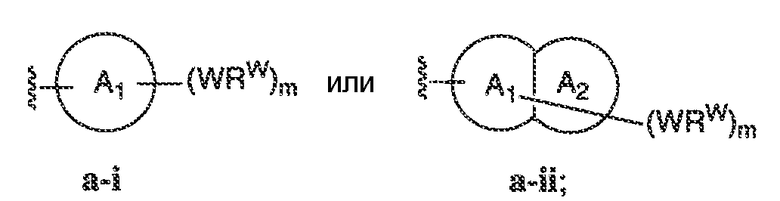
где кольцо A1 представляет собой 5-6-членное ароматическое моноциклическое кольцо, содержащее 0-4 гетероатома, независимо выбранных из азота, кислорода или серы; или
A1 и A2, вместе представляют собой 8-14 ароматическое бициклическое или трициклическое арильное кольцо, где каждое кольцо содержит 0-4 гетероатома, независимо выбранных из азота, кислорода или серы.
В некоторых вариантах воплощения, A1 представляет собой необязательно замещенное 6-членное ароматическое кольцо, содержащее 0-4 гетероатома, где указанный гетероатом представляет собой азот. В некоторых вариантах воплощения, A1 представляет собой необязательно замещенный фенил. Или A1 представляет собой необязательно замещенный пиридил, пиримидинил, пиразинил или триазинил. Или A1 представляет собой необязательно замещенный пиразинил или триазинил. Или A1 представляет собой необязательно замещенный пиридил.
В некоторых вариантах воплощения, A1 представляет собой необязательно замещенное 5-членное ароматическое кольцо, содержащее 0-3 гетероатома, где указанный гетероатом представляет собой азот, кислород или серу. В некоторых вариантах воплощения, A1 представляет собой необязательно замещенное 5-членное ароматическое кольцо, содержащее 1-2 атома азота. В одном варианте воплощения, A1 представляет собой необязательно замещенное 5-членное ароматическое кольцо, отличное от тиазолила.
В некоторых вариантах воплощения, A2 представляет собой необязательно замещенное 6-членное ароматическое кольцо, содержащее 0-4 гетероатома, где указанный гетероатом представляет собой азот. В некоторых вариантах воплощения, A2 представляет собой необязательно замещенный фенил. Или A2 представляет собой необязательно замещенный пиридил, пиримидинил, пиразинил или триазинил.
В некоторых вариантах воплощения, A2 представляет собой необязательно замещенное 5-членное ароматическое кольцо, содержащее 0-3 гетероатома, где указанный гетероатом представляет собой азот, кислород или серу. В некоторых вариантах воплощения, A2 представляет собой необязательно замещенное 5-членное ароматическое кольцо, содержащее 1-2 атома азота. В некоторых вариантах воплощения, A2 представляет собой необязательно замещенный пирролил.
В некоторых вариантах воплощения, A2 представляет собой необязательно замещенное 5-7-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1-3 гетероатома, независимо выбранных из азота, серы или кислорода. Примеры таких колец включают пиперидил, пиперазил, морфолинил, тиоморфолинил, пирролидинил, тетрагидрофуранил и т.п.
В некоторых вариантах воплощения, A2 представляет собой необязательно замещенное 5-10-членное насыщенное или ненасыщенное карбоциклическое кольцо. В одном варианте воплощения, A2 представляет собой необязательно замещенное 5-10-членное насыщенное карбоциклическое кольцо. Примеры таких колец включают циклогексил, циклопентил и т.п.
В некоторых вариантах воплощения, кольцо A2 выбрано из:
где кольцо A2 конденсировано с кольцом A1 через два смежных кольцевых атома.
В других вариантах воплощения, W представляет собой связь или представляет собой необязательно замещенную C1-6 алкилиденовую цепь, где одно или два метиленовых звена необязательно и независимо заменены заместителем O, NR', S, SO, SO2 или COO, CO, SO2NR', NR'SO2, C(O)NR', NR'C(O), OC(O), OC(O)NR', и RW представляет собой R' или галоген. В следующих вариантах воплощения, каждый присутствующий WRW независимо представляет собой -C1-C3 алкил, C1-C3 пергалогеналкил, -O(C1-C3алкил), -CF3, -OCF3, -SCF3, -F, -Cl, -Br или -COOR', -COR', -O(CH2)2N(R')(R'), -O(CH2)N(R')(R'), -CON(R')(R'), -(CH2)2OR', -(CH2)OR', необязательно замещенное моноциклическое или бициклическое ароматическое кольцо, необязательно замещенный арилсульфон, необязательно замещенное 5-членное гетероарильное кольцо, -N(R')(R'), -(CH2)2N(R')(R') или -(CH2)N(R')(R').
В некоторых вариантах воплощения, m имеет значение 0. Или m имеет значение 1. Или m имеет значение 2. В некоторых вариантах воплощения, m имеет значение 3. В других вариантах воплощения, m имеет значение 4.
В одном варианте воплощения, R5 представляет собой X-RX. В некоторых вариантах воплощения R5 представляет собой водород. Или R5 представляет собой необязательно замещенную C1-8 алифатическую группу. В некоторых вариантах воплощения, R5 представляет собой необязательно замещенную C1-4 алифатическую группу. Или R5 представляет собой бензил.
В некоторых вариантах воплощения R6 представляет собой водород. Или R6 представляет собой необязательно замещенную C1-8 алифатическую группу. В некоторых вариантах воплощения, R6 представляет собой необязательно замещенную C1-4 алифатическую группу. В некоторых других вариантах воплощения, R6 представляет собой -(O-C1-4 алифатическая группа) или -(S-C1-4 алифатическая группа). Предпочтительно, R6 представляет собой -OMe или -SMe. В некоторых других вариантах воплощения, R6 представляет собой CF3.
В одном варианте воплощения настоящего изобретения, R1, R2, R3 и R4 одновременно представляют собой водород. В другом варианте воплощения, R6 и R7 оба одновременно представляют собой водород.
В другом варианте воплощения настоящего изобретения, R1, R2, R3, R4 и R5 одновременно представляют собой водород. В другом варианте воплощения настоящего изобретения, R1, R2, R3, R4, R5 и R6 одновременно представляют собой водород.
В другом варианте воплощения настоящего изобретения, R2 представляет собой X-RX, где X представляет собой -SO2NR'- и RX представляет собой R'; т.е. R2 представляет собой -SO2N(R')2. В одном варианте воплощения, два R', взятые вместе, образуют необязательно замещенное 5-7-членное кольцо с 0-3 дополнительными гетероатомами, выбранными из азота, кислорода или серы. Или R1, R3, R4, R5 и R6 одновременно представляют собой водород и R2 представляет собой SO2N(R')2.
В некоторых вариантах воплощения, X представляет собой связь или представляет собой необязательно замещенную C1-6 алкилиденовую цепь, где один или два несмежных метиленовых звена необязательно и независимо заменены O, NR', S, SO2 или COO, CO, и RX представляет собой R' или галоген. В следующих вариантах воплощения, каждый присутствующий XRX независимо представляет собой -C1-3алкил, -O(C1-3алкил), -CF3, -OCF3, -SCF3, -F, -Cl, -Br, OH, -COOR', -COR', -O(CH2)2N(R')(R'), -O(CH2)N(R')(R'), -CON(R')(R'), -(CH2)2OR', -(CH2)OR', необязательно замещенный фенил, -N(R')(R'), -(CH2)2N(R')(R') или -(CH2)N(R')(R').
В некоторых вариантах воплощения, R7 представляет собой водород. В некоторых других вариантах воплощения, R7 представляет собой C1-4 линейную или разветвленную алифатическую группу.
В некоторых вариантах воплощения, RW выбран из галогена, циано, CF3, CHF2, OCHF2, Me, Et, CH(Me)2, CHMeEt, н-пропила, трет-бутила, OMe, OEt, OPh, O-фторфенила, O-дифторфенила, O-метоксифенила, O-толила, O-бензила, SMe, SCF3, SCHF2, SEt, CH2CN, NH2, NHMe, N(Me)2, NHEt, N(Et)2, C(O)CH3, C(O)Ph, C(O)NH2, SPh, SO2-(амино-пиридил), SO2NH2, SO2Ph, SO2NHPh, SO2-N-морфолино, SO2-N-пирролидила, н-пирролила, N-морфолино, 1-пиперидила, фенила, бензила, (циклогексил-метиламино)метила, 4-Метил-2,4-дигидро-пиразол-3-он-2-ила, бензимидазол-2-ила, фуран-2-ила, 4-метил-4H-[1,2,4]триазол-3-ила, 3-(4'-хлорфенил)-[1,2,4]оксадиазол-5-ила, NHC(O)Me, NHC(O)Et, NHC(O)Ph, NHSO2Me, 2-индолила, 5-индолила, -CH2CH2OH, -OCF3, O-(2,3-диметилфенил), 5-метилфурила, -SO2-N-пиперидила, 2-толила, 3-толила, 4-толила, O-бутила, NHCO2C(Me)3, CO2C(Me)3, изопропенила, н-бутила, O-(2,4-дихлорфенил), NHSO2PhMe, O-(3-хлор-5-трифторметил-2-пиридил), фенилгидроксиметила, 2,5-диметилпирролила, NHCOCH2C(Me)3, O-(2-трет-бутил)фенила, 2,3-диметилфенила, 3,4-диметилфенила, 4-гидроксиметилфенила, 4-диметиламинофенила, 2-трифторметилфенила, 3-трифторметилфенила, 4-трифторметилфенила, 4-цианометилфенила, 4-изобутилфенила, 3-пиридила, 4-пиридила, 4-изопропилфенила, 3-изопропилфенила, 2-метоксифенила, 3-метоксифенила, 4-метоксифенила, 3,4-метилендиоксифенила, 2-этоксифенила, 3-этоксифенила, 4-этоксифенила, 2-метилтиофенила, 4-метилтиофенила, 2,4-диметоксифенила, 2,5-диметоксифенила, 2,6-диметоксифенила, 3,4-диметоксифенила, 5-хлор-2-метоксифенила, 2-OCF3-фенила, 3-трифторметокси-фенила, 4-трифторметоксифенила, 2-феноксифенила, 4-феноксифенила, 2-фтор-3-метокси-фенила, 2,4-диметокси-5-пиримидила, 5-изопропил-2-метоксифенила, 2-фторфенила, 3-фторфенила, 4-фторфенила, 3-цианофенила, 3-хлорфенила, 4-хлорфенила, 2,3-дифторфенила, 2,4-дифторфенила, 2,5-дифторфенила, 3,4-дифторфенила, 3,5-дифторфенила, 3-хлор-4-фтор-фенила, 3,5-дихлорфенила, 2,5-дихлорфенила, 2,3-дихлорфенила, 3,4-дихлорфенила, 2,4-дихлорфенила, 3-метоксикарбонилфенила, 4-метоксикарбонилфенила, 3-изопропилоксикарбонилфенила, 3-ацетамидофенила, 4-фтор-3-метилфенила, 4-метансульфинил-фенила, 4-метансульфонил-фенила, 4-N-(2-N,N-диметиламиноэтил)карбамоилфенила, 5-ацетил-2-тиенила, 2-бензотиенила, 3-бензотиенила, фуран-3-ила, 4-метил-2-тиенила, 5-циано-2-тиенила, N'-фенилкарбонил-N-пиперазинила, -NHCO2Et, -NHCO2Me, н-пирролидинила, -NHSO2(CH2)2 н-пиперидина, -NHSO2(CH2)2 N-морфолина, -NHSO2(CH2)2N(Me)2, COCH2N(Me)COCH2NHMe, -CO2Et, O-пропила, -CH2CH2NHCO2C(Me)3, гидрокси, аминометила, пентила, адамантила, циклопентила, этоксиэтила, C(Me)2CH2OH, C(Me)2CO2Et, -CHOHMe, CH2CO2Et, -C(Me)2CH2NHCO2C(Me)3, O(CH2)2OEt, O(CH2)2OH, CO2Me, гидроксиметила, 1-метил-1-циклогексила, 1-метил-1-циклооктила, 1-метил-1-циклогептила, C(Et)2C(Me)3, C(Et)3, CONHCH2CH(Me)2, 2-аминометил-фенила, этенила, 1-пиперидинилкарбонила, этинила, циклогексила, 4-метилпиперидинила, -OCO2Me, -C(Me)2CH2NHCO2CH2CH(Me)2, -C(Me)2CH2NHCO2CH2CH2CH3, -C(Me)2CH2NHCO2Et, -C(Me)2CH2NHCO2Me, -C(Me)2CH2NHCO2CH2C(Me)3, -CH2NHCOCF3, -CH2NHCO2C(Me)3, -C(Me)2CH2NHCO2(CH2)3CH3, C(Me)2CH2NHCO2(CH2)2OMe, C(OH)(CF3)2, -C(Me)2CH2NHCO2CH2-тетрагидрофуран-3-ила, C(Me)2CH2O(CH2)2OMe или 3-этил-2,6-диоксопиперидин-3-ила.
В одном варианте воплощения, R' представляет собой водород.
В одном варианте воплощения, R' представляет собой C1-C8 алифатическую группу, необязательно имеющую вплоть до 3 заместителей, выбранных из галогена, CN, CF3, CHF2, OCF3 или OCHF2, где вплоть до двух метиленовых звеньев в указанной C1-C8 алифатической группе необязательно замещены -CO-, -CONH(C1-C4 алкил)-, -CO2-, -OCO-, -N(C1-C4 алкил)CO2-, -O-, -N(C1-C4 алкил)CON(C1-C4 алкил)-, -OCON(C1-C4 алкил)-, -N(C1-C4 алкил)CO-, -S-, -N(C1-C4 алкил)-, -SO2N(C1-C4 алкил)-, N(C1-C4 алкил)SO2- или -N(C1-C4 алкил)SO2N(C1-C4 алкил)-.
В одном варианте воплощения, R' представляет собой 3-8-членное насыщенное, частично ненасыщенное или полностью ненасыщенное моноциклическое кольцо, содержащее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где R' необязательно имеет вплоть до 3 заместителей, выбранных из галогена, CN, CF3, CHF2, OCF3, OCHF2 или C1-C6 алкила, где вплоть до двух метиленовых звеньев в указанном C1-C6 алкиле необязательно замещены -CO-, -CONH(C1-C4 алкил)-, -CO2-, -OCO-, -N(C1-C4 алкил)CO2-, -O-, -N(C1-C4 алкил)CON(C1-C4 алкил)-, -OCON(C1-C4 алкил)-, -N(C1-C4 алкил)CO-, -S-, -N(C1-C4 алкил)-, -SO2N(C1-C4 алкил)-, N(C1-C4 алкил)SO2- или -N(C1-C4 алкил)SO2N(C1-C4 алкил)-.
В одном варианте воплощения, R' представляет собой 8-12-членную насыщенную, частично ненасыщенную или полностью ненасыщенную бициклическую кольцевую систему, содержащую 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы; где R' необязательно имеет вплоть до 3 заместителей, выбранных из галогена, CN, CF3, CHF2, OCF3, OCHF2 или C1-C6 алкила, где вплоть до двух метиленовых звеньев в указанном C1-C6 алкиле необязательно заменены -CO-, -CONH(C1-C4 алкил)-, -CO2-, -OCO-, -N(C1-C4 алкил)CO2-, -O-, -N(C1-C4 алкил)CON(C1-C4 алкил)-, -OCON(C1-C4 алкил)-, -N(C1-C4 алкил)CO-, -S-, -N(C1-C4 алкил)-, -SO2N(C1-C4 алкил)-, N(C1-C4 алкил)SO2- или -N(C1-C4 алкил)SO2N(C1-C4 алкил)-.
В одном варианте воплощения, два присутствующих R' взяты вместе с атомом(атомами), с которым они связаны, с образованием необязательно замещенного 3-12-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического или бициклического кольца, содержащего 0-4 гетероатома, независимо выбранных из азота, кислорода или серы, где R' необязательно имеет вплоть до 3 заместителей, выбранных из галогена, CN, CF3, CHF2, OCF3, OCHF2 или C1-C6 алкила, где вплоть до двух метиленовых звеньев в указанном C1-C6 алкиле необязательно замещены -CO-, -CONH(C1-C4 алкил)-, -CO2-, -OCO-, -N(C1-C4 алкил)CO2-, -O-, -N(C1-C4 алкил)CON(C1-C4 алкил)-, -OCON(C1-C4 алкил)-, -N(C1-C4 алкил)CO-, -S-, -N(C1-C4 алкил)-, -SO2N(C1-C4 алкил)-, N(C1-C4 алкил)SO2- или -N(C1-C4 алкил)SO2N(C1-C4 алкил)-.
В соответствии с одним вариантом воплощения, настоящее изобретение обеспечивает соединения формулы IIA или формулы IIB:
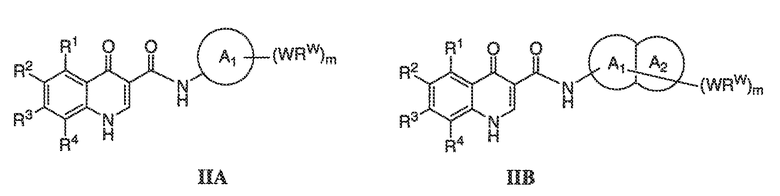
В соответствии с другим вариантом воплощения, настоящее изобретение обеспечивает соединения формулы IIIA, формулы IIIB, формулы IIIC, формулы IIID или формулы IIIE:
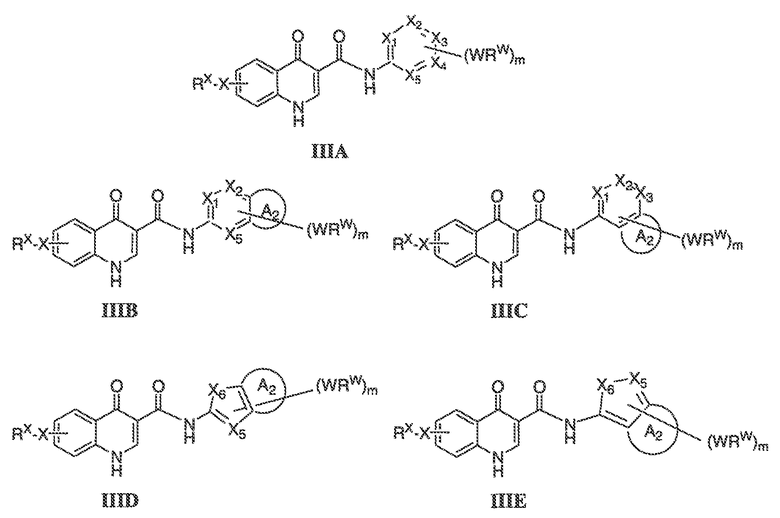
где каждый из X1, X2, X3, X4 и X5 независимо выбран из CH или N; и
X6 представляет собой O, S или NR'.
В одном варианте воплощения, соединения формулы IIIA, формулы IIIB, формулы IIIC, формулы IIID или формулы IIIE имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. Или y имеет значение 1. Или y имеет значение 2.
В некоторых вариантах воплощения формулы IIIA, X1, X2, X3, X4 и X5, взятые вместе с WRW и m, представляют собой необязательно замещенный фенил.
В некоторых вариантах воплощения формулы IIIA, X1, X2, X3, X4 и X5, взятые вместе, представляют собой необязательно замещенное кольцо, выбранное из:
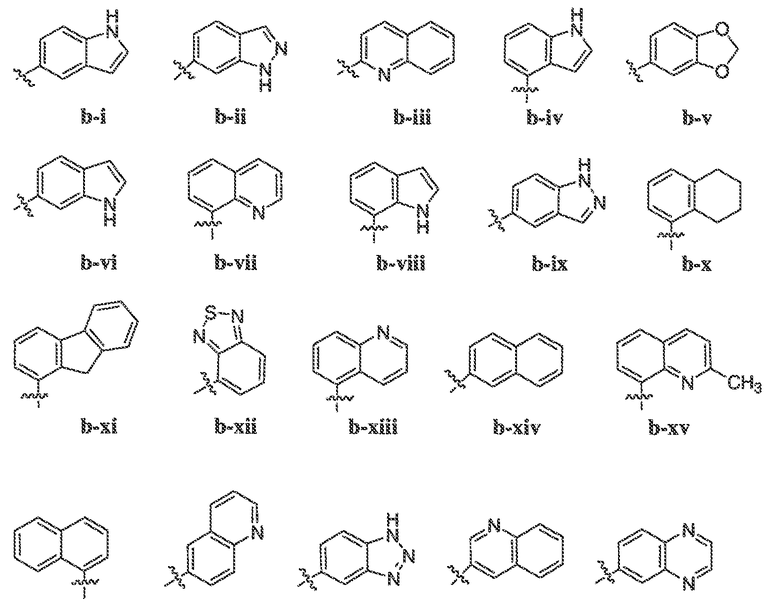
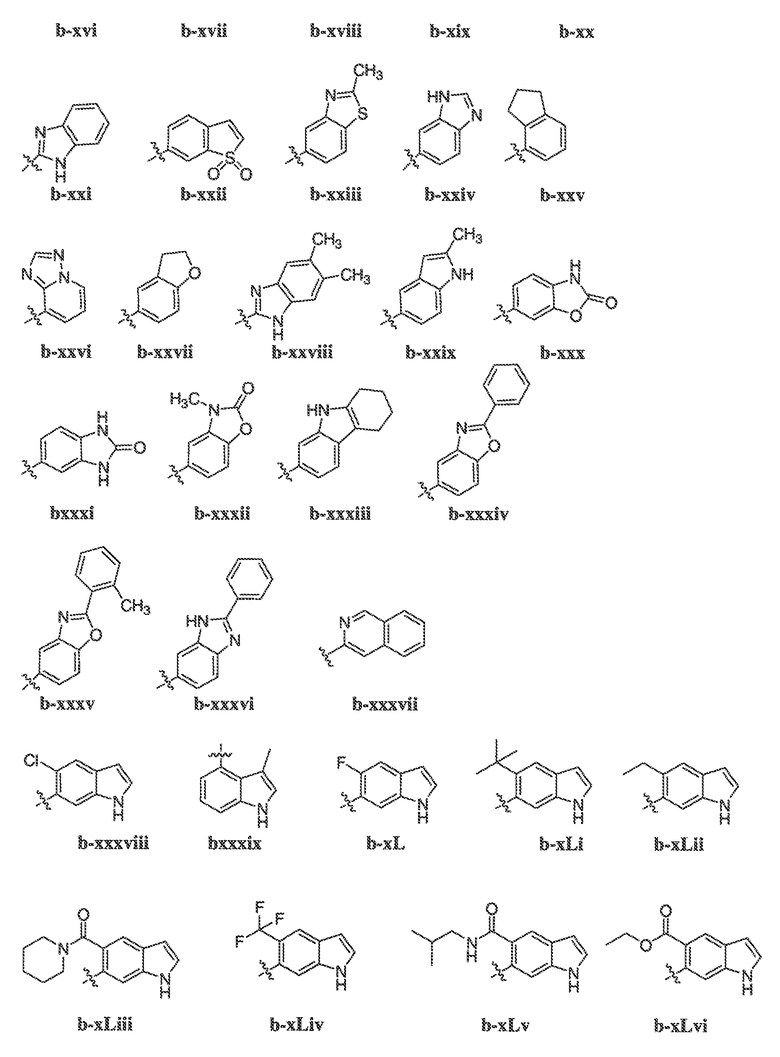
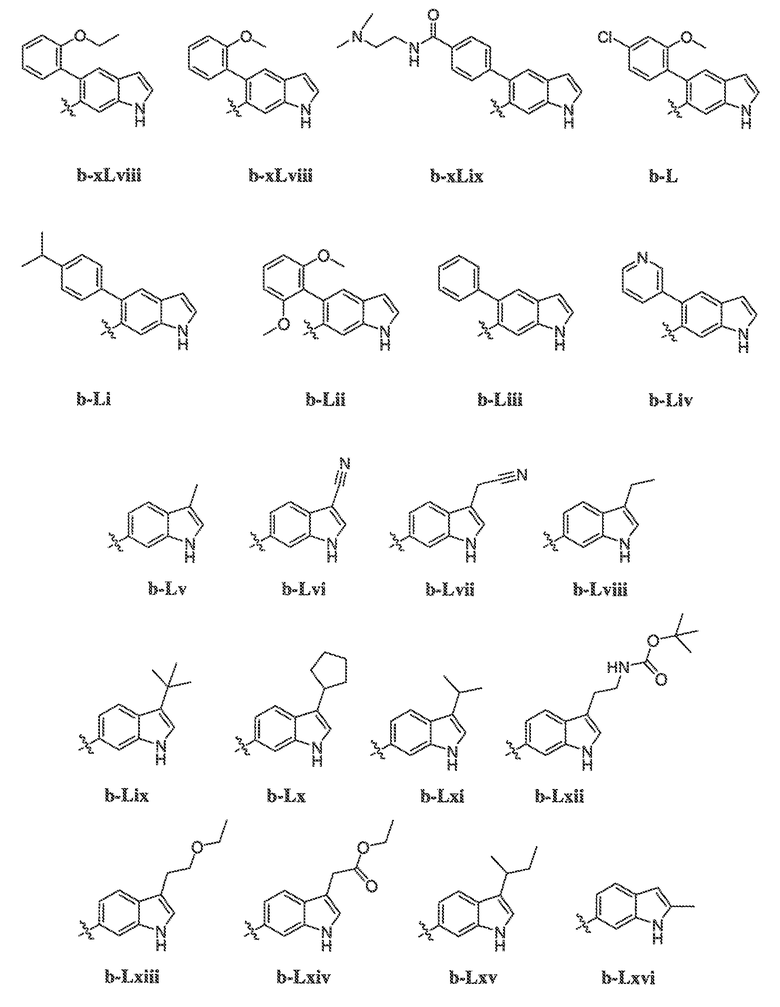
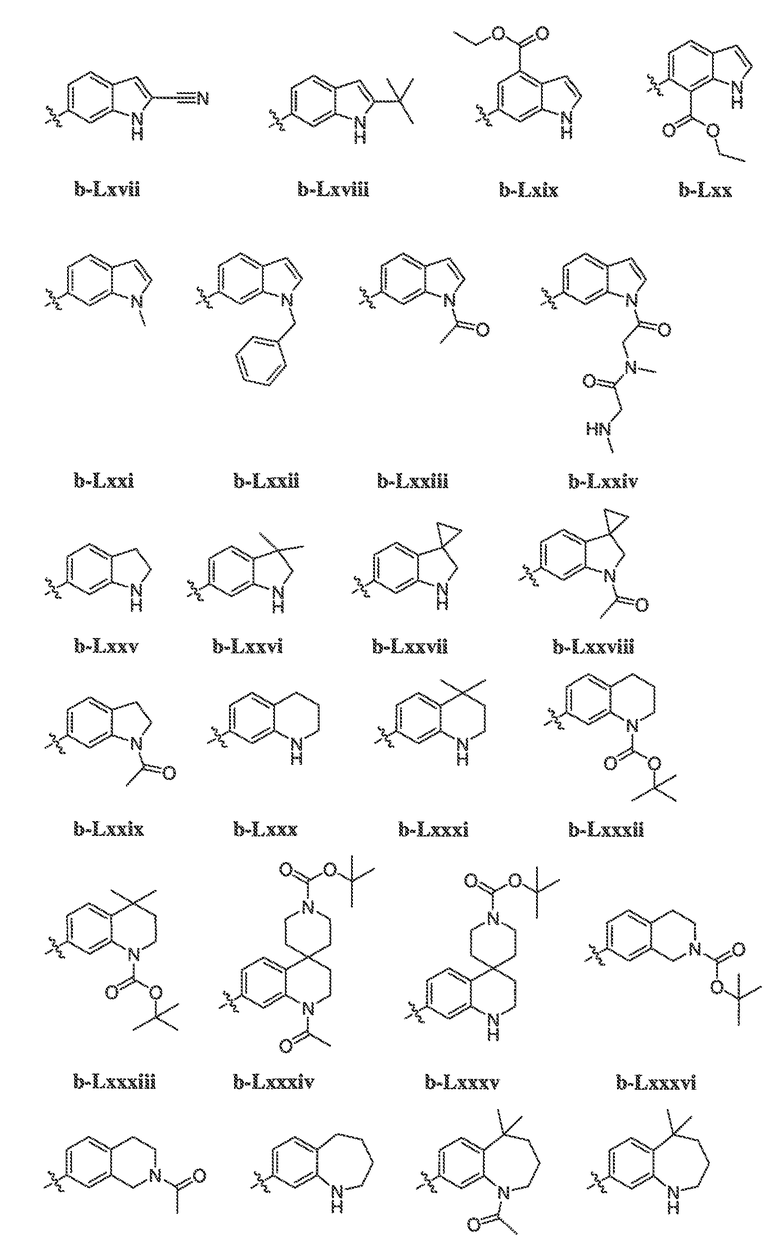
В некоторых вариантах воплощения формулы IIIB, формулы IIIC, формулы IIID или формулы IIIE, X1, X2, X3, X4, X5 или X6, взятые вместе с кольцом A2, представляют собой необязательно замещенное кольцо, выбранное из:
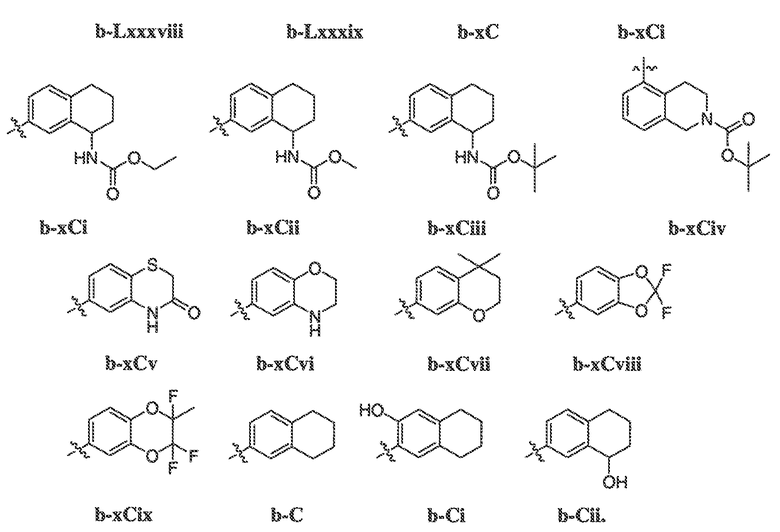
В некоторых вариантах воплощения, RW выбран из галогена, циано, CF3, CHF2, OCHF2, Me, Et, CH(Me)2, CHMeEt, н-пропила, трет-бутила, OMe, OEt, OPh, O-фторфенила, O-дифторфенила, O-метоксифенила, O-толила, O-бензила, SMe, SCF3, SCHF2, SEt, CH2CN, NH2, NHMe, N(Me)2, NHEt, N(Et)2, C(O)CH3, C(O)Ph, C(O)NH2, SPh, SO2-(амино-пиридил), SO2NH2, SO2Ph, SO2NHPh, SO2-N-морфолино, SO2-N-пирролидила, н-пирролила, N-морфолино, 1-пиперидила, фенила, бензила, (циклогексил-метиламино)метила, 4-Метил-2,4-дигидро-пиразол-3-он-2-ила, бензимидазол-2-ила, фуран-2-ила, 4-метил-4H-[1,2,4]триазол-3-ила, 3-(4'-хлорфенил)-[1,2,4]оксадиазол-5-ила, NHC(O)Me, NHC(O)Et, NHC(O)Ph или NHSO2Me.
В некоторых вариантах воплощения, X и RX, взятые вместе, представляют собой Me, Et, галоген, CN, CF3, OH, OMe, OEt, SO2N(Me)(фторфенил), SO2-(4-метил-пиперидин-1-ил или SO2-N-пирролидинил.
В соответствии с другим вариантом воплощения, настоящее изобретение обеспечивает соединения формулы IVA, формулы IVB или формулы IVC:
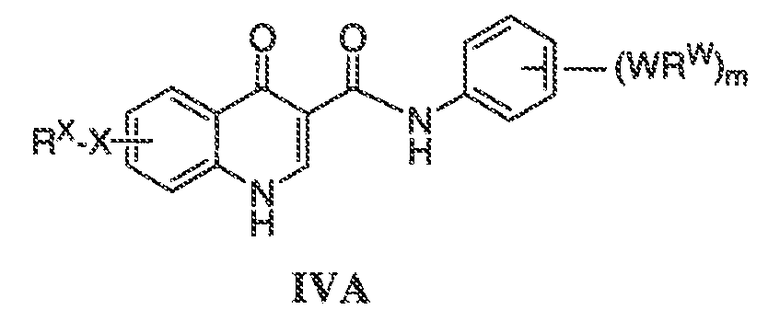
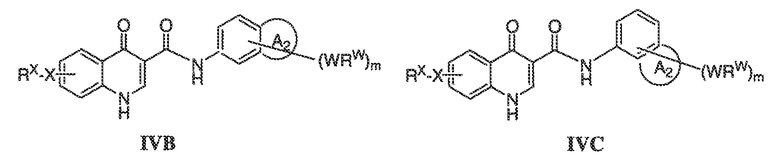
В одном варианте воплощения соединения формулы IVA, формулы IVB и формулы IVC имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы IVA, формулы IVB и формулы IVC, где X представляет собой связь и RX представляет собой водород.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы формулы IVB и формулы IVC, где кольцо A2 представляет собой необязательно замещенное насыщенное, ненасыщенное или ароматическое семи-членное кольцо с 0-3 гетероатомами, выбранными из O, S или N. Примеры колец включают азепанил, 5,5-диметил азепанил и т.п.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы IVB и IVC, где кольцо A2 представляет собой необязательно замещенное насыщенное, ненасыщенное или ароматическое шести-членное кольцо с 0-3 гетероатомами, выбранными из O, S или N. Примеры колец включают пиперидинил, 4,4-диметилпиперидинил и т.п.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы IVB и IVC, где кольцо A2 представляет собой необязательно замещенное насыщенное, ненасыщенное или ароматическое пяти-членное кольцо с 0-3 гетероатомами, выбранными из O, S или N.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы IVB и IVC, где кольцо A2 представляет собой необязательно замещенное пяти-членное кольцо с одним атомом азота, например, пирролил или пирролидинил.
В соответствии с одним вариантом воплощения формулы IVA, обеспечивается следующее соединение формулы VA-1:
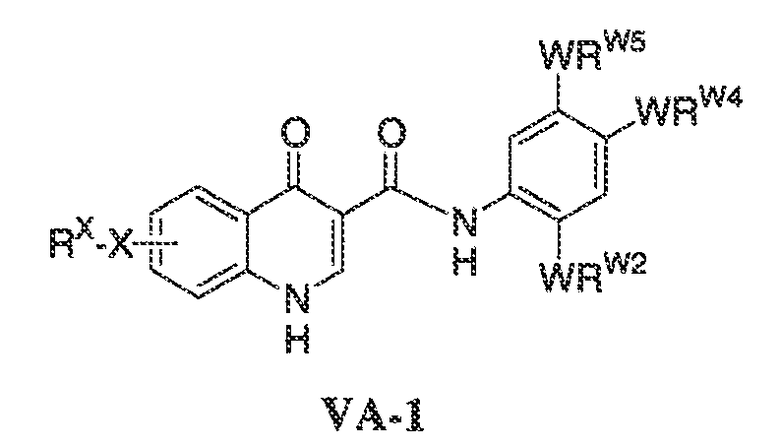
где каждый из WRW2 и WRW4 независимо выбран из водорода, CN, CF3, галогена, C1-C6 линейного или разветвленного алкила, 3-12-членной циклоалифатической группы, фенила, C5-C10 гетероарила или C3-C7 гетероциклической группы, где указанный гетероарил или гетероциклическая группа содержит вплоть до 3 гетероатомов, выбранных из O, S или N, где указанный WRW2 и WRW4 независимо и необязательно имеет вплоть до трех заместителей, выбранных из -OR', -CF3, -OCF3, SR', S(O)R', SO2R', -SCF3, галоген, CN, -COOR', -COR', -O(CH2)2N(R')(R'), -O(CH2)N(R')(R'), -CON(R')(R'), -(CH2)2OR', -(CH2)OR', CH2CN, необязательно замещенного фенила или фенокси, -N(R')(R'), -NR'C(O)OR', -NR'C(O)R', -(CH2)2N(R')(R') или -(CH2)N(R')(R'); и
WRW5 выбран из водорода, -OH, NH2, CN, CHF2, NHR', N(R')2, -NHC(O)R', -NHC(O)OR', NHSO2R', -OR', CH2OH, CH2N(R')2, C(O)OR', SO2NHR', SO2N(R')2 или CH2NHC(O)OR'. Или WRW4 и WRW5 взятые вместе, образуют 5-7-членное кольцо, содержащее 0-3 гетероатома, выбранных из N, O или S, где указанное кольцо необязательно имеет вплоть до трех WRW заместителей.
В одном варианте воплощения, соединения формулы VA-1 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы VA-1, где X представляет собой связь и RX представляет собой водород.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы VA-1, где:
каждый из WRW2 и WRW4 независимо выбран из водорода, CN, CF3, галогена, C1-C6 линейного или разветвленного алкила, 3-12-членной циклоалифатической группы или фенила, где указанный WRW2 и WRW4 независимо и необязательно имеет вплоть до трех заместителей, выбранных из -OR', -CF3, -OCF3, -SCF3, галогена, -COOR', -COR', -O(CH2)2N(R')(R'), -O(CH2)N(R')(R'), -CON(R')(R'), -(CH2)2OR', -(CH2)OR', необязательно замещенного фенила, -N(R')(R'), -NC(O)OR', -NC(O)R', -(CH2)2N(R')(R') или -(CH2)N(R')(R'); и
WRW5 выбран из водорода, -OH, NH2, CN, NHR', N(R')2, -NHC(O)R', -NHC(O)OR', NHSO2R', -OR', CH2OH, C(O)OR', SO2NHR' или CH2NHC(O)O-(R').
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы VA-1, где:
WRW2 представляет собой фенильное кольцо, необязательно содержащее вплоть до трех заместителей, выбранных из -OR', -CF3, -OCF3, SR', S(O)R', SO2R', -SCF3, галогена, CN, -COOR', -COR', -O(CH2)2N(R')(R'), -O(CH2)N(R')(R'), -CON(R')(R'), -(CH2)2OR', -(CH2)OR', CH2CN, необязательно замещенного фенила или фенокси, -N(R')(R'), -NR'C(O)OR', -NR'C(O)R', -(CH2)2N(R')(R') или -(CH2)N(R')(R');
WRW4 представляет собой C1-C6 линейный или разветвленный алкил; и
WRW5 представляет собой OH.
В одном варианте воплощения, каждый из WRW2 и WRW4 независимо выбран из CF3 или галогена. В одном варианте воплощения, каждый из WRW2 и WRW4 независимо выбран из необязательно замещенного водорода, C1-C6 линейного или разветвленного алкила. В некоторых вариантах воплощения, каждый из WRW2 и WRW4 независимо выбран из необязательно замещенного н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила, 1,1-диметил-2-гидроксиэтила, 1,1-диметил-2-(этоксикарбонил)-этила, 1,1-диметил-3-(трет-бутоксикарбонил-амино) пропила или н-пентила.
В одном варианте воплощения, каждый из WRW2 и WRW4 независимо выбран из необязательно замещенной 3-12-членной циклоалифатической группы. Примеры вариантов воплощения такой циклоалифатической группы включают циклопентил, циклогексил, циклогептил, норборнил, адамантил, [2.2.2.]бицикло-октил, [2.3.1.]бицикло-октил или [3.3.1]бицикло-нонил.
В некоторых вариантах воплощения WRW2 представляет собой водород, и WRW4 представляет собой C1-C6 линейный или разветвленный алкил. В некоторых вариантах воплощения, WRW4 выбран из метила, этила, пропила, н-бутила, втор-бутила или трет-бутила.
В некоторых вариантах воплощения WRW4 представляет собой водород, и WRW2 представляет собой C1-C6 линейный или разветвленный алкил. В некоторых вариантах воплощения, WRW2 выбран из метила, этила, пропила, н-бутила, втор-бутила, трет-бутила или н-пентила.
В некоторых вариантах воплощения каждый из WRW2 и WRW4 представляет собой C1-C6 линейный или разветвленный алкил. В некоторых вариантах воплощения, каждый из WRW2 и WRW4 выбран из метила, этила, пропила, н-бутила, втор-бутила, трет-бутила или пентила.
В одном варианте воплощения, WRW5 выбран из водорода, CHF2, NH2, CN, NHR', N(R')2, CH2N(R')2, -NHC(O)R', -NHC(O)OR', -OR', C(O)OR' или SO2NHR'. Или WRW5 представляет собой -OR',например, OH.
В некоторых вариантах воплощения, WRW5 выбран из водорода, NH2, CN, CHF2, NH(C1-C6 алкил), N(C1-C6 алкил)2, -NHC(O)(C1-C6 алкил), -CH2NHC(O)O(C1-C6 алкил), -NHC(O)O(C1-C6 алкил), -OH, -O(C1-C6 алкил), C(O)O(C1-C6 алкил), CH2O(C1-C6 алкил) или SO2NH2. В другом варианте воплощения, WRW5 выбран из -OH, OMe, NH2, -NHMe, -N(Me)2, -CH2NH2, CH2OH, NHC(O)OMe, NHC(O)OEt, CN, CHF2, -CH2NHC(O)O(трет-бутил), -O-(этоксиэтил), -O-(гидроксиэтил), -C(O)OMe или -SO2NH2.
В одном варианте воплощения, соединение формулы VA-1 имеет один, предпочтительно более одного, или более предпочтительно все из следующих характерных признаков:
i) WRW2 представляет собой водород;
ii) WRW4 представляет собой C1-C6 линейный или разветвленный алкил или моноциклическую или бициклическую алифатическую группу; и
iii) WRW5 выбран из водорода, CN, CHF2, NH2, NH(C1-C6 алкил), N(C1-C6 алкил)2, -NHC(O)(C1-C6 алкил), -NHC(O)O(C1-C6 алкил), -CH2C(O)O(C1-C6 алкил), -OH, -O(C1-C6 алкил), C(O)O(C1-C6 алкил) или SO2NH2.
В одном варианте воплощения, соединени формулы VA-1 имеет один, предпочтительно более одного, или более предпочтительно все из следующих характерных признаков:
i) WRW2 представляет собой галоген, C1-C6 алкил, CF3, CN или фенил, который необязательно имеет вплоть до 3 заместителей, выбранных из C1-C4 алкила, -O(C1-C4 алкил) или галогена;
ii) WRW4 представляет собой CF3, галоген, C1-C6 алкил или C6-C10 циклоалифатическую группу; и
iii) WRW5 представляет собой OH, NH2, NH(C1-C6 алкил) или N(C1-C6 алкил).
В одном варианте воплощения, X-RX находится в положении 6 хинолинилового кольца. В некоторых вариантах воплощения, X-RX, взятые вместе, представляют собой C1-C6 алкил, -O-(C1-C6 алкил) или галоген.
В одном варианте воплощения, X-RX находится в положении 5 хинолинилового кольца. В некоторых вариантах воплощения, X-RX, взятые вместе, представляют собой -OH.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы VA-1, где WRW4 и WRW5 взятые вместе, образуют 5-7-членное кольцо, содержащее 0-3 гетероатома, выбранных из N, O или S, где указанное кольцо необязательно имеет вплоть до трех WRW заместителей.
В некоторых вариантах воплощения, WRW4 и WRW5, взятые вместе, образуют необязательно замещенное 5-7-членное насыщенное, ненасыщенное или ароматическое кольцо, содержащее 0 гетероатомов. В других вариантах воплощения, WRW4 и WRW5, взятые вместе, образуют необязательно замещенное 5-7-членное кольцо, содержащее 1-3 гетероатома, выбранных из N, O или S. В некоторых других вариантах воплощения, WRW4 и WRW5, взятые вместе, образуют необязательно замещенное насыщенное, ненасыщенное или ароматическое 5-7-членное кольцо, содержащее 1 гетероатом азота. В некоторых других вариантах воплощения, WRW4 и WRW5, взятые вместе, образуют необязательно замещенное 5-7-членное кольцо, содержащее 1 гетероатом кислорода.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-A-2:
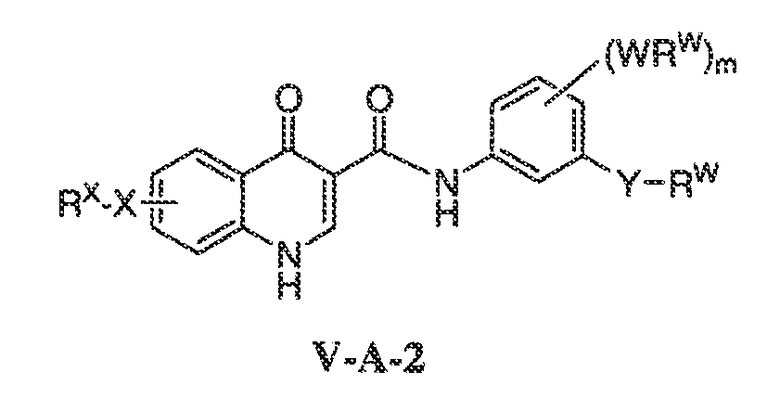
где:
Y представляет собой CH2, C(O)O, C(O) или S(O)2;
m имеет значение 0-4; и
X, RX, W и RW имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы VA-2 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, Y представляет собой C(O). В другом варианте воплощения, Y представляет собой C(O)O. Или Y представляет собой S(O)2. Или Y представляет собой CH2.
В одном варианте воплощения, m имеет значение 1 или 2. Или m имеет значение 1. Или m имеет значение 0.
В одном варианте воплощения, W представляет собой связь.
В другом варианте воплощения, RW представляет собой C1-C6 алифатическую группу, галоген, CF3 или фенил, необязательно замещенный C1-C6 алкилом, галогеном, циано или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно замещены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
Примеры вариантов воплощения WRW включают метил, этил, пропил, трет-бутил или 2-этоксифенил.
В другом варианте воплощения, RW в Y-RW представляет собой C1-C6 алифатическую группу, необязательно замещенную N(R”)2, где R” представляет собой водород, C1-C6 алкил, или два R”, взятые вместе, образуют 5-7-членное гетероциклическое кольцо, содержащее вплоть до 2 дополнительных гетероатомов, выбранных из O, S или NR'. Примеры таких гетероциклических колец включают пирролидинил, пиперидил, морфолинил или тиоморфолинил.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-A-3:
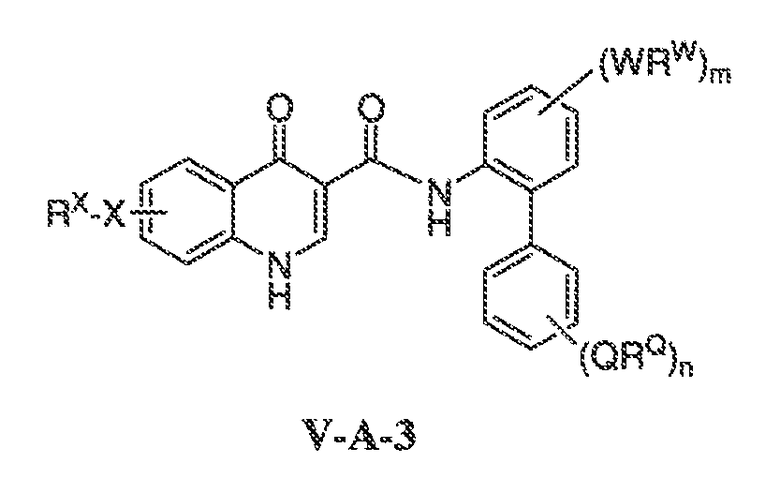
где:
Q представляет собой W;
RQ представляет собой RW;
m имеет значение 0-4;
n имеет значение 0-4; и
X, RX, W и RW имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы VA-3 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, n имеет значение 0-2.
В другом варианте воплощения, m имеет значение 0-2. В одном варианте воплощения, m имеет значение 0. В одном варианте воплощения, m имеет значение 1. Или m имеет значение 2.
В одном варианте воплощения, QRQ, взятые вместе, представляют собой галоген, CF3, OCF3, CN, C1-C6 алифатическую группу, O-C1-C6 алифатическую группу, O-фенил, NH(C1-C6 алифатическая группа) или N(C1-C6 алифатическая группа)2, где указанные алифатическая группа и фенил необязательно имеют вплоть до трех заместителей, выбранных из C1-C6 алкила, O-C1-C6 алкила, галогена, циано, OH или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно замещены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, SOR', SO2R', -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
Примеры QRQ включают метил, изопропил, втор-бутил, гидроксиметил, CF3, NMe2, CN, CH2CN, фтор, хлор, OEt, OMe, SMe, OCF3, OPh, C(O)OMe, C(O)O-iPr, S(O)Me, NHC(O)Me или S(O)2Me.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-A-4:
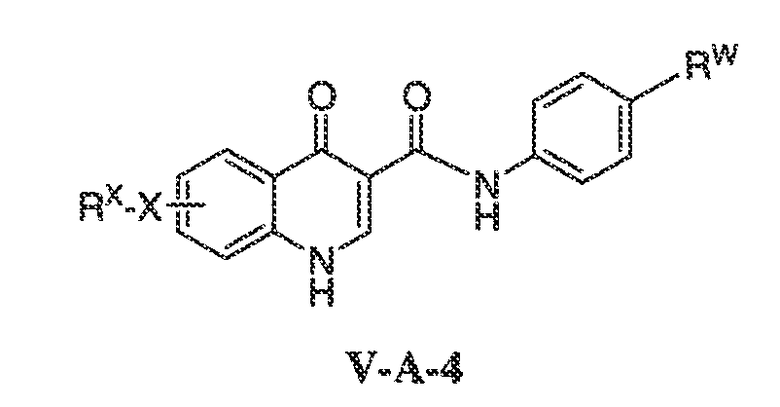
где X, RX и RW имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы VA-4 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, RW представляет собой C1-C12 алифатическую группу, C5-C10 циклоалифатическую группу или C5-C7 гетероциклическое кольцо, где указанная алифатическая группа, циклоалифатическая группа или гетероциклическое кольцо необязательно имеет вплоть до трех заместителей, выбранных из C1-C6 алкила, галогена, циано, оксо, OH или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно замещены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
Примеры RW включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, винил, цианометил, гидроксиметил, гидроксиэтил, гидроксибутил, циклогексил, адамантил или -C(CH3)2-NHC(O)O-T, где T представляет собой C1-C4 алкил, метоксиэтил или тетрагидрофуранилметил.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-A-5:
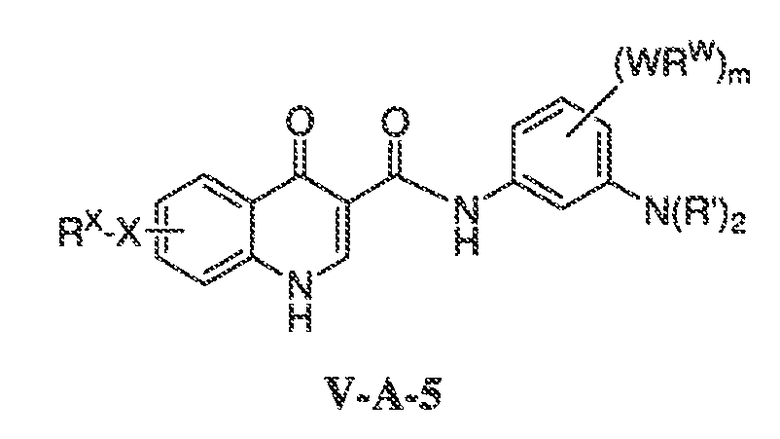
где:
m имеет значение 0-4; и
X, RX, W, RW и R' имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы VA-5 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, m имеет значение 0-2. Или m имеет значение 1. Или m имеет значение 2.
В другом варианте воплощения, оба R' представляют собой водород. Или один R' представляет собой водород, а другой R' представляет собой C1-C4 алкил, например, метил. Или оба R' представляют собой C1-C4 алкил, например, метил.
В другом варианте воплощения, m имеет значение 1 или 2, и RW представляет собой галоген, CF3, CN, C1-C6 алифатическую группу, O-C1-C6 алифатическую группу или фенил, где указанная алифатическая группа и фенил необязательно имеют вплоть до трех заместителей, выбранных из C1-C6 алкила, O-C1-C6 алкила, галогена, циано, OH или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно замещены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, R' выше представляет собой C1-C4 алкил.
Примеры вариантов воплощения RW включают хлор, CF3, OCF3, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, метокси, этокси, пропилокси или 2-этоксифенил.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-A-6:
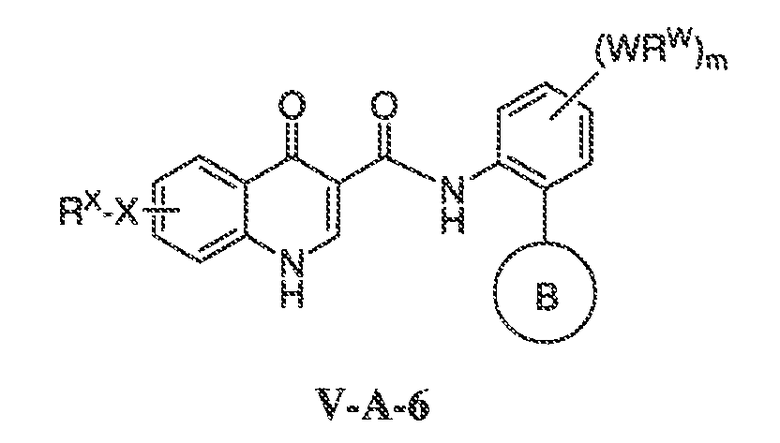
где:
кольцо B представляет собой 5-7-членное моноциклическое или бициклическое, гетероциклическое или гетероарильное кольцо, которое необязательно имеет вплоть до n присутствующих заместителей -Q-RQ, где n имеет значение 0-4, и Q и RQ имеют значения, определенные выше; и
Q, RQ, X, RX, W и RW имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы VA-6 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, m имеет значение 0-2. Или m имеет значение 0. Или m имеет значение 1.
В одном варианте воплощения, n имеет значение 0-2. Или n имеет значение 0. Или n имеет значение 1.
В другом варианте воплощения, кольцо B представляет собой 5-7-членное моноциклическое, гетероциклическое кольцо, содержащее до 2 гетероатомов, выбранных из O, S или N, которое необязательно имеет вплоть до n присутствующих заместителей -Q-RQ. Примеры гетероциклических колец включают N-морфолинил, N-пиперидинил, 4-бензоил-пиперазин-1-ил, пирролидин-1-ил или 4-метил-пиперидин-1-ил.
В другом варианте воплощения, кольцо B представляет собой 5-6-членное моноциклическое, гетероарильное кольцо, содержащее до 2 гетероатомов, выбранных из O, S или N, которое необязательно имеет вплоть до n присутствующих заместителей -Q-RQ. Примеры таких колец включают бензимидазол-2-ил, 5-метил-фуран-2-ил, 2,5-диметил-пиррол-1-ил, пиридин-4-ил, индол-5-ил, индол-2-ил, 2,4-диметокси-пиримидин-5-ил, фуран-2-ил, фуран-3-ил, 2-ацил-тиен-2-ил, бензотиофен-2-ил, 4-метил-тиен-2-ил, 5-циано-тиен-2-ил, 3-хлор-5-трифторметил-пиридин-2-ил.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-B-1:
где:
один из Q1 и Q3 представляет собой N(WRW), а другой из Q1 и Q3 выбран из O, S или N(WRW);
Q2 представляет собой C(O), CH2-C(O), C(O)-CH2, CH2, CH2-CH2, CF2 или CF2-CF2;
m имеет значение 0-3; и
X, W, RX и RW имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы V-B-1 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, Q3 представляет собой N(WRW); примеры WRW включают водород, C1-C6 алифатическую группу, C(O)C1-C6 алифатическую группу или C(O)OC1-C6 алифатическую группу.
В другом варианте воплощения, Q3 представляет собой N(WRW), Q2 представляет собой C(O), CH2, CH2-CH2, и Q1 представляет собой O.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-B-2:
где:
RW1 представляет собой водород или C1-C6 алифатическую группу;
каждый из RW3 представляет собой водород или C1-C6 алифатическую группу; или
оба RW3 взятые вместе, образуют C3-C6 циклоалкил или гетероциклическое кольцо, содержащее до двух гетероатомов, выбранных из O, S или NR', где указанное кольцо необязательно имеет вплоть до двух WRW заместителей;
m имеет значение 0-4; и
X, RX, W и RW имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы V-B-2 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, WRW1 представляет собой водород, C1-C6 алифатическую группу, C(O)C1-C6 алифатическую группу или C(O)OC1-C6 алифатическую группу.
В другом варианте воплощения, каждый RW3 представляет собой водород, C1-C4 алкил. Или оба RW3, взятые вместе, образуют C3-C6 циклоалифатическое кольцо или 5-7-членное гетероциклическое кольцо, содержащее до двух гетероатомов, выбранных из O, S или N, где указанное циклоалифатическое или гетероциклическое кольцо необязательно имеет вплоть до трех заместителей, выбранных из WRW1. Примеры таких колец включают циклопропил, циклопентил, необязательно замещенный пиперидил и т.п.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-B-3:
где:
Q4 представляет собой связь, C(O), C(O)O или S(O)2;
RW1 представляет собой водород или C1-C6 алифатическую группу;
m имеет значение 0-4; и
X, W, RW и RX имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы V-B-3 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0.
В одном варианте воплощения, Q4 представляет собой C(O). Или Q4 представляет собой C(O)O. В другом варианте воплощения, RW1 представляет собой C1-C6 алкил. Примеры RW1 включают метил, этил или трет-бутил.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-B-4:

где:
m имеет значение 0-4; и
X, RX, W и RW имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы V-B-4 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, m имеет значение 0-2. Или m имеет значение 0. Или m имеет значение 1.
В другом варианте воплощения, указанное циклоалифатическое кольцо представляет собой 5-членное кольцо. Или указанное кольцо представляет собой шестичленное кольцо.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-B-5:
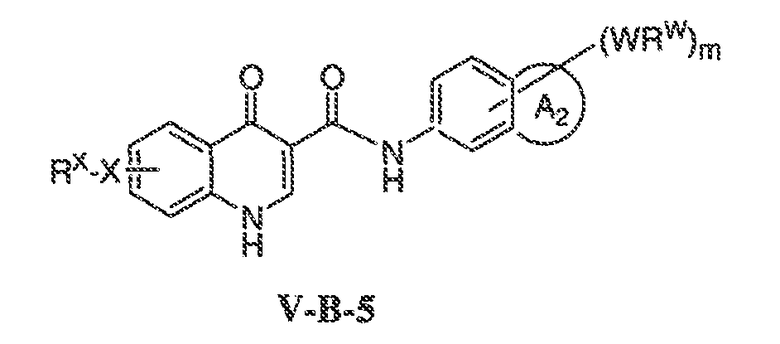
где:
кольцо A2 представляет собой фенил или 5-6-членное гетероарильное кольцо, где кольцо A2 и фенильное кольцо, которые являются конденсированными, вместе содержат до 4 заместителей, независимо выбранных из WRW;
m имеет значение 0-4; и
X, W, RW и RX имеют значения, определенные выше.
В одном варианте воплощения, соединения формулы V-B-5 имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, кольцо A2 представляет собой необязательно замещенное 5-членное кольцо, выбранное из пирролила, фуранила, тиенила, пиразолила, имидазолила, тиазолила, оксазолила, тиадиазолила, оксадиазолила или триазолила.
В одном варианте воплощения, кольцо A2 представляет собой необязательно замещенное 5-членное кольцо, выбранное из пирролила, пиразолила, тиадиазолила, имидазолила, оксазолила или триазолила. Примеры таких колец включают:
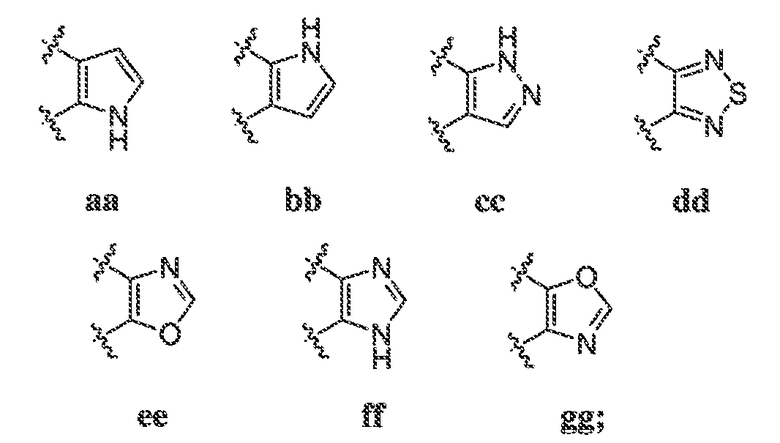
где указанное кольцо является необязательно замещенным, как описано выше.
В другом варианте воплощения, кольцо A2 представляет собой необязательно замещенное 6-членное кольцо. Примеры таких колец включают пиридил, пиразинил или триазинил. В другом варианте воплощения, указанное кольцо представляет собой необязательнный пиридил.
В одном варианте воплощения, кольцо A2 представляет собой фенил.
В другом варианте воплощения, кольцо A2 представляет собой пирролил, пиразолил, пиридил или тиадиазолил.
Примеры W в формуле V-B-5 включают связь, C(O), C(O)O или C1-C6 алкилен.
Примеры RW в формуле V-B-5 включают циано, галоген, C1-C6 алифатическую группу, C3-C6 циклоалифатическую группу, арил, 5-7-членное гетероциклическое кольцо, содержащее до двух гетероатомов, выбранных из O, S или N, где указанная алифатическая группа, фенил и гетероциклическая группа независимо и необязательно имеют вплоть до трех заместителей, выбранных из C1-C6 алкила, O-C1-C6 алкила, галогена, циано, OH или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно заменены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы V-B-5-a:
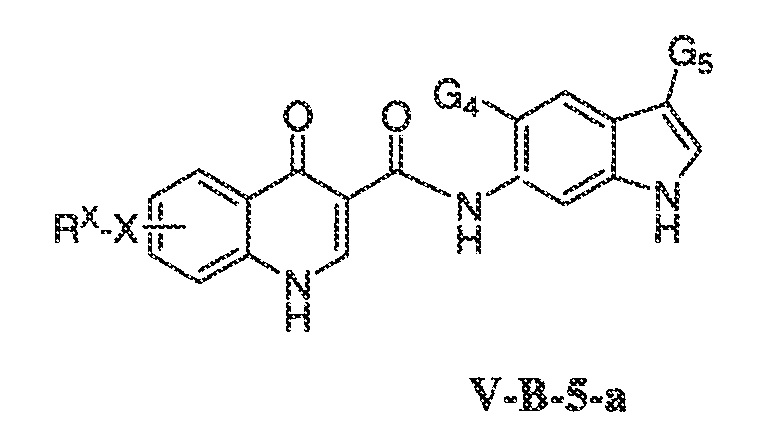
где:
G4 представляет собой водород, галоген, CN, CF3, CHF2, CH2F, необязательно замещенную C1-C6 алифатическую группу, арил-C1-C6 алкил или фенил, где G4 необязательно имеет вплоть до 4 заместителей WRW; где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно заменены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. ;
G5 представляет собой водород или необязательно замещенную C1-C6 алифатическую группу;
где указанная индольная кольцевая система необязательно имеет вплоть до 3 дополнительных заместителей, независимо выбранных из WRW.
В одном варианте воплощения, соединения формулы V-B-5-a имеют y количество присутствующих заместителей X-RX, где y имеет значение 0-4. В одном варианте воплощения, y имеет значение 0. Или y имеет значение 1. Или y имеет значение 2.
В одном варианте воплощения, G4 представляет собой водород. Или G5 представляет собой водород.
В другом варианте воплощения, G4 представляет собой водород, и G5 представляет собой C1-C6 алифатическую группу, где указанная алифатическая группа необязательно замещена C1-C6 алкилом, галогеном, циано или CF3 и где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно заменены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
В другом варианте воплощения, G4 представляет собой водород, и G5 представляет собой циано, метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, цианометил, метоксиэтил, CH2C(O)OMe, (CH2)2-NHC(O)O-трет-бутил или циклопентил.
В другом варианте воплощения, G5 представляет собой водород, и G4 представляет собой галоген, C1-C6 алифатическую группу или фенил, где указанная алифатическая группа или фенил необязательно замещены C1-C6 алкилом, галогеном, циано или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно заменены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
В другом варианте воплощения, G5 представляет собой водород, и G4 представляет собой галоген, CF3, этоксикарбонил, трет-бутил, 2-метоксифенил, 2-этоксифенил, (4-C(O)NH(CH2)2-NMe2)-фенил, 2-метокси-4-хлор-фенил, пиридин-3-ил, 4-изопропилфенил, 2,6-диметоксифенил, втор-бутиламинокарбонил, этил, трет-бутил или пиперидин-1-илкарбонил.
В другом варианте воплощения, G4 и G5 оба представляют собой водород, и кольцевой атом азота в указанном индольном кольце замещен C1-C6 алифатической группой, C(O)(C1-C6 алифатическая группа) или бензилом, где указанная алифатическая группа или бензил необязательно замещены C1-C6 алкилом, галогеном, циано или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно заменены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
В другом варианте воплощения, G4 и G5 обы представляют собой водород, и кольцевой атом азота в указанном индольном кольце замещен ацилом, бензилом, C(O)CH2N(Me)C(O)CH2NHMe или этоксикарбонилом.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы I':
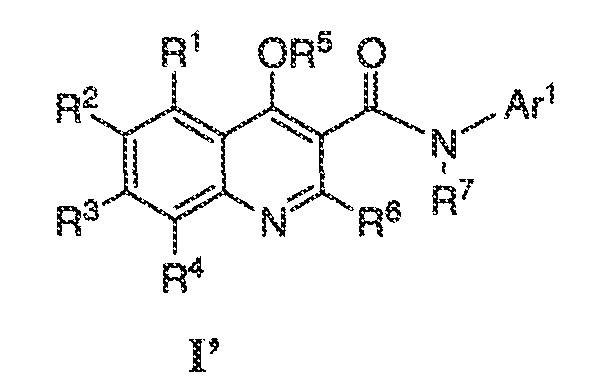
или их фармацевтически приемлемые соли,
где R1, R2, R3, R4, R5, R6, R7 и Ar1 имеют значения, определенные выше для соединений формулы I'.
В одном варианте воплощения, каждый из R1, R2, R3, R4, R5, R6, R7 и Ar1 в соединениях формулы I' независимо имеет значение, определенное выше для любого из вариантов воплощения соединений формулы I.
Репрезентативные соединения по настоящему изобретению представлены в Таблице 1 в графической части.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения, полезные в качестве промежуточных соединений в синтезе соединений формулы I. В одном варианте воплощения, такие соединения имеют формулу A-I:
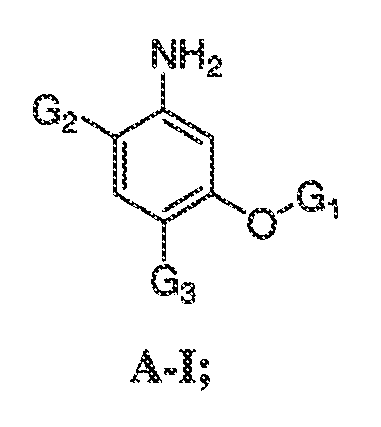
или соль такого соединения;
где:
G1 представляет собой водород, R', C(O)R', C(S)R', S(O)R', S(O)2R', Si(CH3)2R', P(O)(OR')3, P(S)(OR')3, или B(OR')2;
G2 представляет собой галоген, CN, CF3, изопропил или фенил, где указанный изопропил или фенил необязательно имеет вплоть до 3 заместителей, независимо выбранных из WRW, где W и RW имеют значения, определенные выше для формулы I и ее вариантов воплощения;
G3 представляет собой изопропил или C3-C10 циклоалифатическое кольцо, где указанный G3 необязательно имеет вплоть до 3 заместителей, независимо выбранных из WRW, где W и RW имеют значения, определенные выше для формулы I и ее вариантов воплощения;
при условии, что когда G1 представляет собой метокси, G3 представляет собой трет-бутил, тогда G2 не может представлять собой 2-амино-4-метокси-5-трет-бутил-фенил.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы A-I, при условии, что когда G2 и G3, каждый, представляет собой трет-бутил, тогда G1 не может представлять собой водород.
В другом варианте воплощения:
G1 представляет собой водород;
G2 представляет собой галоген или изопропил, где указанный изопропил необязательно имеет вплоть до 3 заместителей, независимо выбранных из R'; и
G3 представляет собой изопропил или C3-C10 циклоалифатическое кольцо, где указанный G3 необязательно имеет вплоть до 3 заместителей, независимо выбранных из R'.
В другом варианте воплощения:
G1 представляет собой водород;
G2 представляет собой галоген, предпочтительно фтор; и
G3 представляет собой C3-C10 циклоалифатическое кольцо, где указанный G3 необязательно имеет вплоть до 3 заместителей, независимо выбранных из метила, этила, пропила или бутила.
В другом варианте воплощения:
G1 представляет собой водород;
G2 представляет собой CN, галоген или CF3; и
G3 представляет собой изопропил или C3-C10 циклоалифатическое кольцо, где указанный G3 необязательно имеет вплоть до 3 заместителей, независимо выбранных из R'.
В другом варианте воплощения:
G1 представляет собой водород;
G2 представляет собой фенил, который необязательно имеет вплоть до 3 заместителей, независимо выбранных из -OC1-C4 алкила, CF3, галогена или CN; и
G3 представляет собой изопропил или C3-C10 циклоалифатическое кольцо, где указанный G3 необязательно имеет вплоть до 3 заместителей, независимо выбранных из R'.
Примеры G3 включают необязательно замещенный циклопентил, циклогексил, циклогептил или адамантил. Или G3 представляет собой C3-C8 разветвленную алифатическую цепь. Примеры G3 включают изопропил, трет-бутил, 3,3-диэтил-проп-3-ил или 3,3-диэтил-2,2-диметил-проп-3-ил.
В другом варианте воплощения:
G1 представляет собой водород;
G2 представляет собой трет-бутил; и
G3 представляет собой трет-бутил.
В другом варианте воплощения, настоящее изобретение обеспечивает соединение формулы A-II:
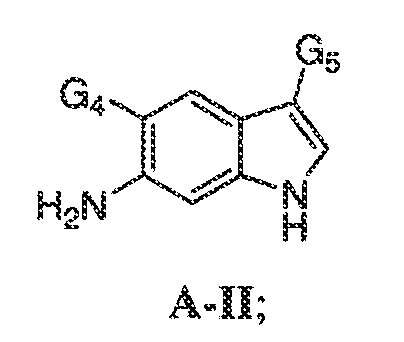
или его соль, где:
G4 представляет собой водород, галоген, CN, CF3, CHF2, CH2F, необязательно замещенную C1-C6 алифатическую группу, аралкил или фенильное кольцо, которое необязательно имеет вплоть до 4 заместителей WRW;
G5 представляет собой водород или необязательно замещенную C1-C6 алифатическую группу;
при условии, что оба, G4 и G5, одновременно не могут представлять собой водород;
где указанная индольная кольцевая система, кроме того, необязательно имеет вплоть до 3 дополнительных заместителей, независимо выбранных из WRW.
В одном варианте воплощения, G4 представляет собой водород. Или G5 представляет собой водород.
В другом варианте воплощения, G4 представляет собой водород, и G5 представляет собой C1-C6 алифатическую группу, где указанная алифатическая группа необязательно замещена C1-C6 алкилом, галогеном, циано или CF3, и где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно заменены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
В другом варианте воплощения, G4 представляет собой водород, и G5 представляет собой циано, метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, цианометил, метоксиэтил, CH2C(O)OMe, (CH2)2-NHC(O)O-трет-But или циклопентил.
В другом варианте воплощения, G5 представляет собой водород, и G4 представляет собой галоген, C1-C6 алифатическую группу или фенил, где указанная алифатическая группа или фенил необязательно замещены C1-C6 алкилом, галогеном, циано или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно заменены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
В другом варианте воплощения, G5 представляет собой водород, и G4 представляет собой галоген, этоксикарбонил, трет-бутил, 2-метоксифенил, 2-этоксифенил, 4-C(O)NH(CH2)2-NMe2, 2-метокси-4-хлор-фенил, пиридин-3-ил, 4-изопропилфенил, 2,6-диметоксифенил, втор-бутиламинокарбонил, этил, трет-бутил или пиперидин-1-илкарбонил.
В родственных вариантах воплощения формулы A-II, кольцевой атом азота в указанном индольном кольце замещен C1-C6 алифатической группой, C(O)(C1-C6 алифатическая группа) или бензилом, где указанная алифатическая группа или бензил необязательно замещенны C1-C6 алкилом, галогеном, циано или CF3, где вплоть до двух метиленовых звеньев в указанной C1-C6 алифатической группе или C1-C6 алкиле необязательно заменены -CO-, -CONR'-, -CO2-, -OCO-, -NR'CO2-, -O-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO2NR'-, NR'SO2- или -NR'SO2NR'-. В другом варианте воплощения, указанный выше R' представляет собой C1-C4 алкил.
В другом варианте воплощения кольцевой атом азота в указанном индольном кольце замещен ацилом, бензилом, C(O)CH2N(Me)C(O)CH2NHMe или этоксикарбонилом.
4. Общие схемы синтеза
Соединения по настоящему изобретению легко получают способами, известными из уровня техники. Ниже проиллюстрированы примеры способов получения соединений по настоящему изобретению.
Представленнная ниже схема иллюстрирует синтез кислотных предшественников соединений по настоящему изобретению.
Синтез кислотных предшественников P-IV-A, P-IV-B или P-IV-C:
a) (CO2Et)2CH2; b) (CO2Et)2CH=CH(OEt); c) CF3CO2H, PPh3, CCl4, Et3N; d) MeI; e) PPA или дифениловый эфир; f) NaOH.
Синтез кислотных предшественников P-IV-A, P-IV-B или P-IV-C:
a) AcONH4; b) EtOCHC(CO2Et)2, 130ºC; c) Ph2O, ∆T; d) I2, EtOH; e) NaOH.
Синтез кислотных предшественников P-IV-A, P-IV-B или P-IV-C
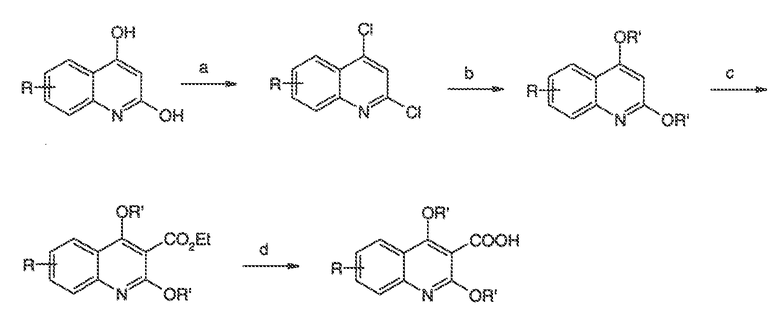
a) POCl3; b) R'ONa; c) n-BuLi, ClCO2Et; d) NaOH
Синтез аминового предшественника P-III-A:
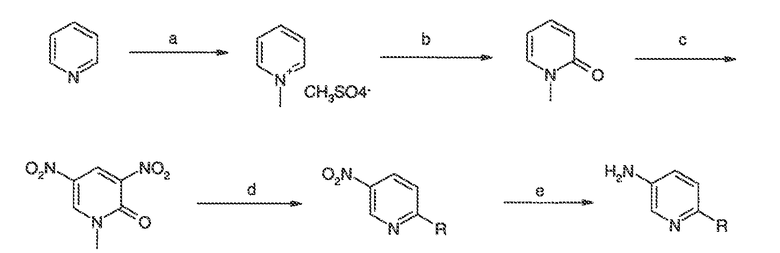
a) (CH3)2SO4; b) K3Fe(CN)6, NaOH, H2O; c) HNO3, H2SO4; d) RCOCH3, MeOH, NH3; e) H2, Ni Ренея
Синтез аминового предшественника P-IV-A:
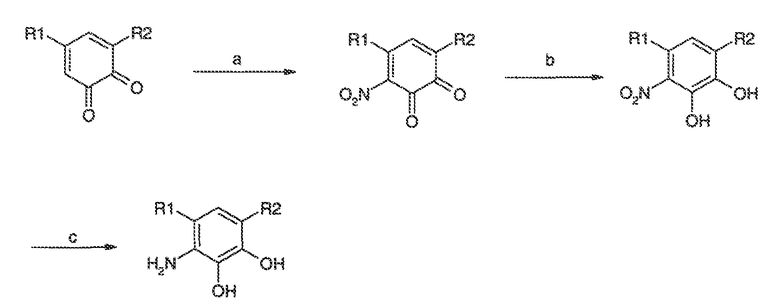
а) HNO3, HOAc; b) Na2S2O4, THF/H2O; c) H2, Pd/C.
Синтез аминового предшественника P-V-A-1:
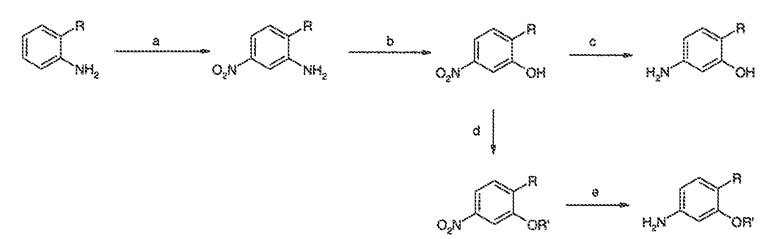
a) KNO3, H2SO4; b) NaNO2, H2SO4-H2O; c) NH4CO2H, Pd-C; d) R'X; e) NH4CO2H, Pd-C
Синтез аминового предшественника P-V-A-1:

a) SO2Cl2, R2=Cl; b) R2OH, R2=алкил; c) NBS, R1=Br; d) ClCO2R, TEA; e) HNO3, H2SO4; f) основание; g) ArB(OH)2, R1=Br; h) [H]; I) R'X, R1= Br; j) ClCF2CO2Me; k) [H]; l) [H].
Синтез аминового предшественника P-V-A-1:
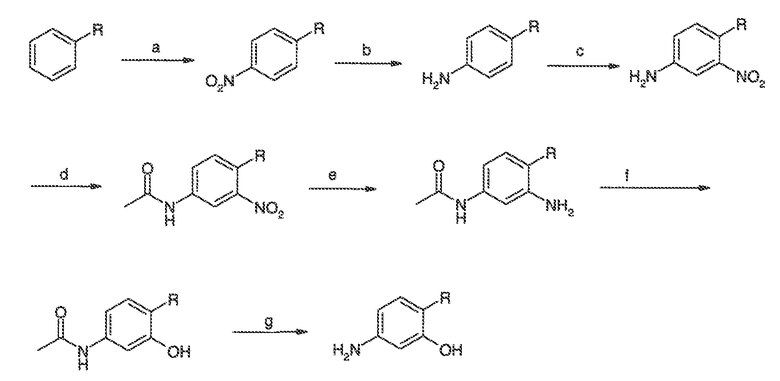
a) KNO3; b) [H]; c) KNO3; d) AcCl; e) [H]; f) i) NaNO2; ii) H2O; г) HCl
Синтез аминового предшественника P-V-A-1:
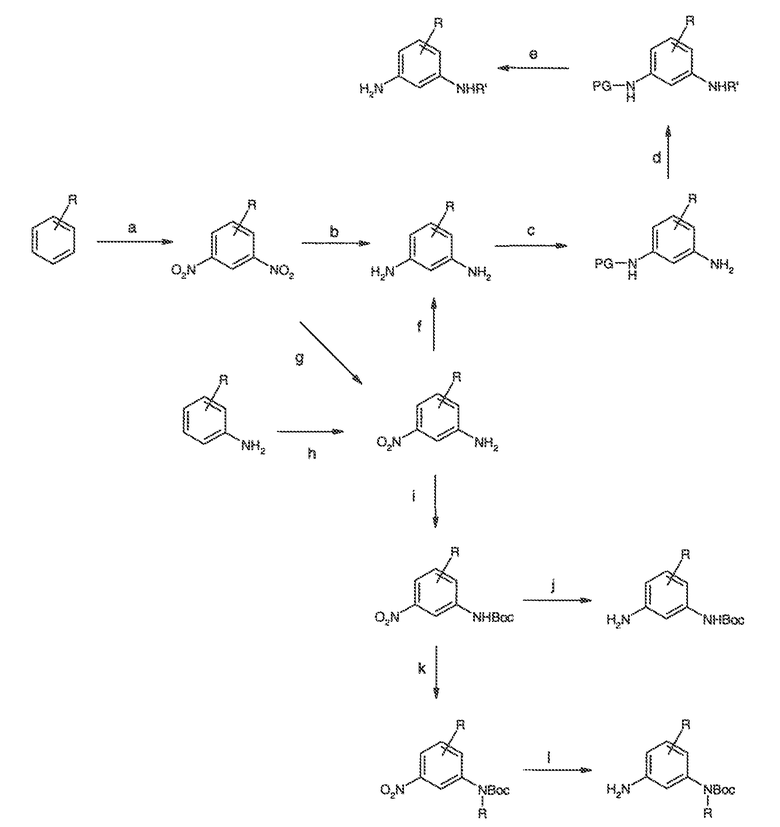
a) HNO3, H2SO4; b) [H]; c) защита; d) R'CHO; e) удаление защиты; f) [H]; g) Na2S, S, H2O; h) нитрование; i) (BOC)2O; j) [H]; k) RX; l) [H]; PG=защитная группа
Синтез аминового предшественника P-V-A-1 или P-V-A-2:

a) Br2; b) Zn(CN)2, Pd(PPh)3; c) [H]; d) BH3; e) (BOC)2O; f) [H]; g) H2SO4, H2O; h) R'X; i) [H]; j) LiAlH4
Синтез аминового предшественника P-V-A-1 или P-V-A-2:
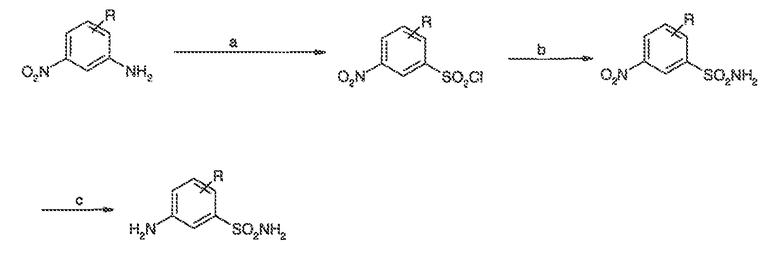
(i) NaNO2, HCl; ii) Na2SO3, CuSO4, HCl; b) NH4Cl; c) [H]
Синтез аминовых предшественников P-V-A-1:
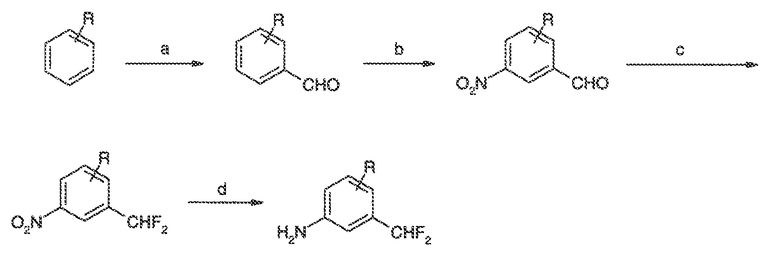
a) CHCl2OMe; b) KNO3, H2SO4; c) Деоксо-Фтор; d) Fe
Синтез аминовых предшественников P-V-A-3:
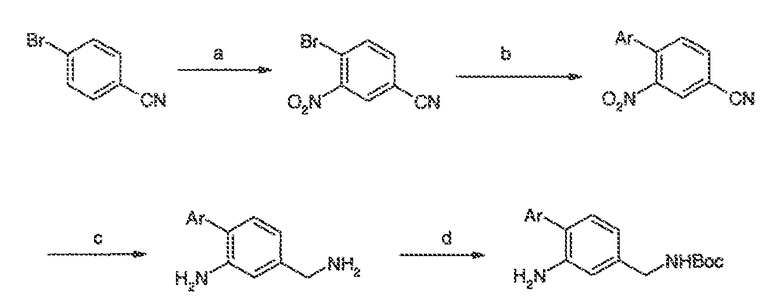
Ar = Арил или Гетероарил
a) Нитрование; b) ArB(OH)2, Pd; c) BH3; d) (BOC)2O
Синтез аминовых предшественников P-V-B-1:
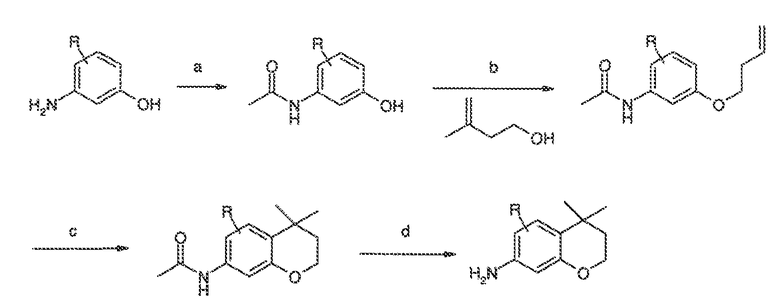
a) AcCl; b) DEAD; c) AlCl3; d) NaOH
Синтез аминовых предшественников P-V-B-1:

a) ClCH2COCl; b) [H]; c) защита; d) [H]
PG=защитная группа
Синтез аминовых предшественников P-V-B-1:
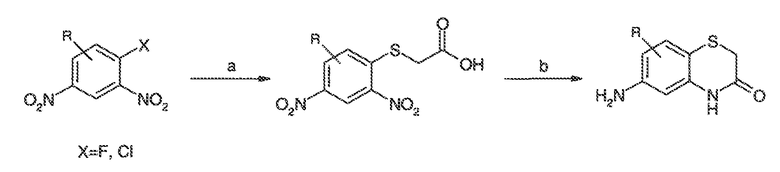
a) HSCH2CO2H; b) [H]
Синтез аминовых предшественников P-V-B-2:
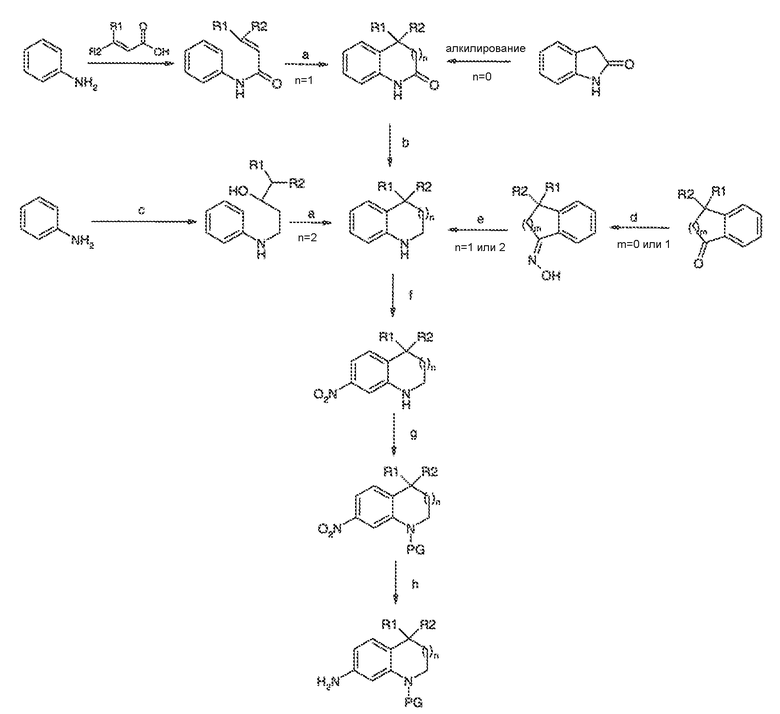
a) AlCl3; b) [H]; c) i) R1R2CHCOCH2CH2Cl; ii) NaBH4; d) NH2OH; e) DIBAL-H; f) нитрование; г) защита; h) [H]
PG=защитная группа
Синтез аминовых предшественников P-V-B-3:
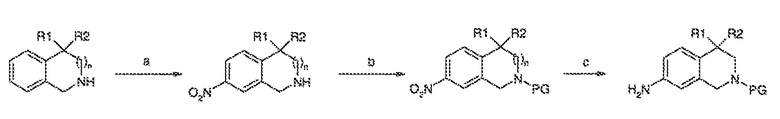
А) Нитрование; b) Защита; c) [H]
PG=защитная группа
Синтез аминовых предшественников P-V-B-5:
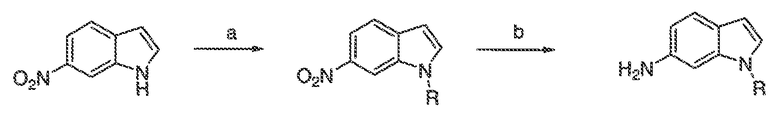
a) когда X=Cl, Br, I: RX, K2CO3, DMF или CH3CN; когда X=OH: RX, TFFH, DIEA, ТГФ b) H2, Pd-C, EtOH или SnCl2.2H2O, EtOH или SnCl2.2H2O, DIEA, EtOH.
Синтез аминовых предшественников P-V-B-5:
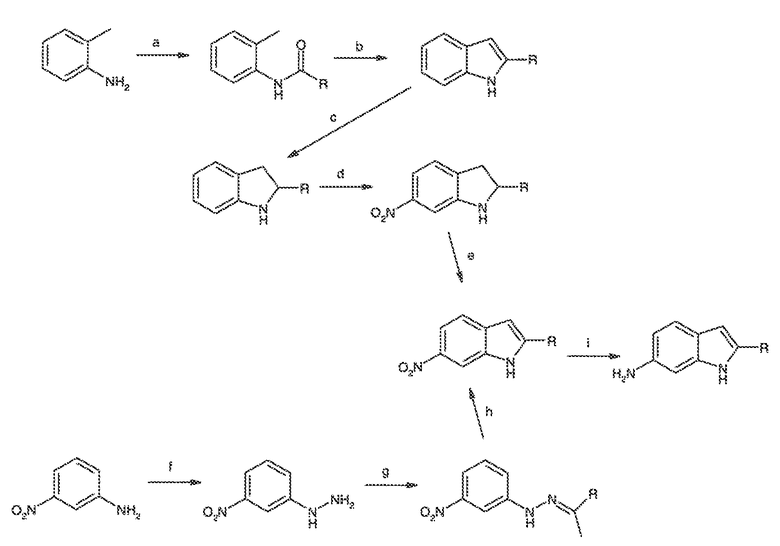
a) RCOCl, Et3N, CH2Cl2; b) n-BuLi, ТГФ; c) NaBH4, AcOH; d) KNO3, H2SO4; e) DDQ, 1,4-диоксан; f) NaNO2, HCl, SnCl2.2H2O, H2O; г) MeCOR, EtOH; h) PPA; i) LiAlH4, ТГФ или H2, Ni Ренея, EtOH или MeOH
Синтез аминовых предшественников V-B-5:
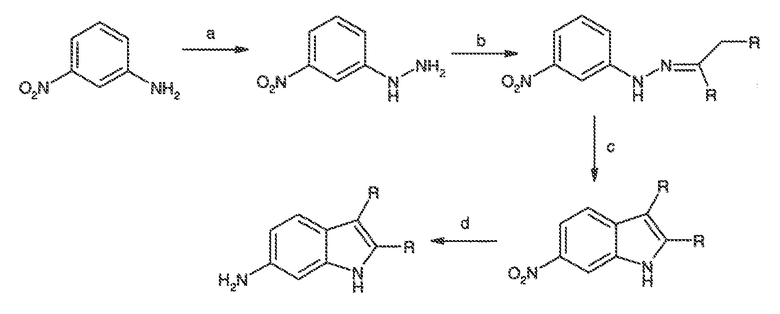
a) NaNO2, HCl, SnCl2.2H2O, H2O; b) RCH2COR, AcOH, EtOH; c) H3PO4, толуол; d) H2, Pd-C, EtOH
Синтез аминовых предшественников P-V-B-5:
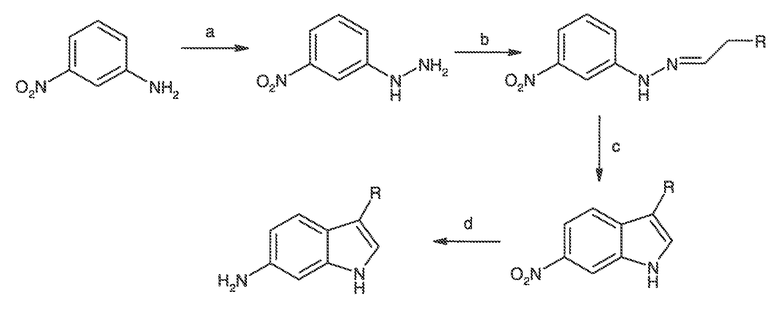
a) NaNO2, HCl, SnCl2.2H2O, H2O; b) RCH2COH, AcOH, EtOH; c) H3PO4, толуол; d) H2, Pd-C, EtOH
Синтез аминовых предшественников P-V-B-5:
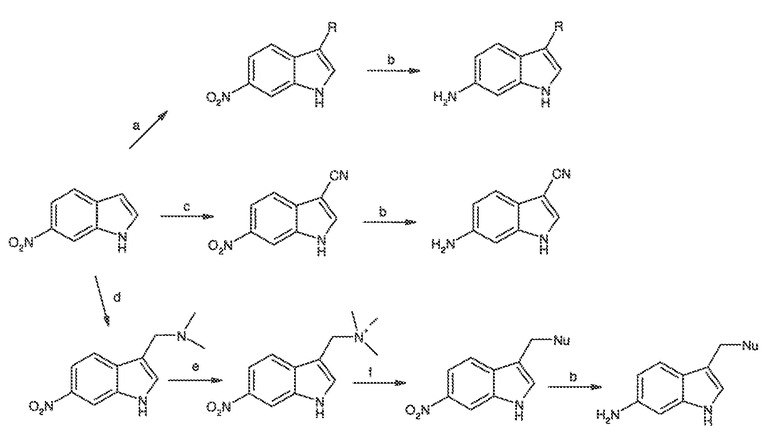
a) RX (X=Br, I), трифлат цинка, TBAI, DIEA, толуол; b) H2, Ni Ренея, EtOH или H2, Pd-C, EtOH или SnCl2.2H2O, EtOH; c) ClSO2NCO, DMF, CH3CN; d) Me2NH, H2CO, AcOH; e) MeI, DMF, ТГФ, H2O; f) MNu (M= Na, K, Li; Nu= нуклеофил)
Синтез аминовых предшественников P-V-B-5:
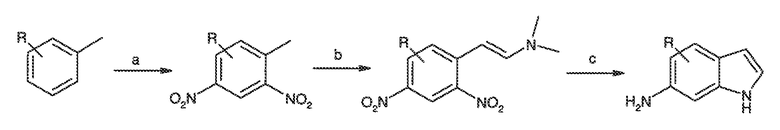
a) HNO3, H2SO4; b) Me2NCH(OMe)2, DMF; c) H2, Ni Ренея, EtOH
Синтез аминовых предшественников P-V-B-5:
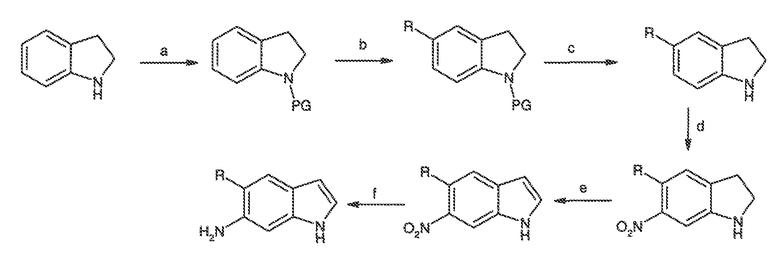
a) Когда PG= SO2Ph: PhSO2Cl, Et3N, DMAP, CH2Cl2; Когда PG= Ac: AcCl, NaHCO3, CH2Cl2; b) Когда R= RCO: (RCO)2O, AlCl3, CH2Cl2; Когда R=Br: Br2, AcOH; c) HBr или HCl; d) KNO3, H2SO4; e) MnO2, CH2Cl2 или DDQ, 1,4-диоксан; f) H2, Ni Ренея, EtOH.
Синтез аминовых предшественников P-V-B-5:
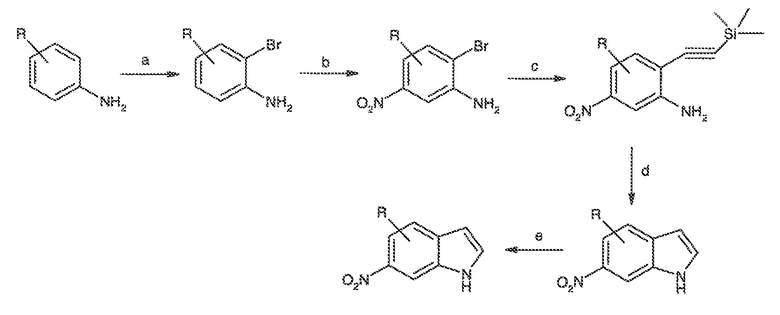
a) NBS, DMF; b) KNO3, H2SO4; c) HC≡CSiMe3, Pd(PPh3)2Cl2, CuI, Et3N, Толуол, H2O; d) CuI, DMF; e) H2, Ni Ренея, MeOH
Синтез аминовых предшественников P-V-A-3 и P-V-A-6:
Ar= Арил или гетероарил
a) ArB(OH)2, Pd(PPh3)4, K2CO3, H2O, ТГФ или ArB(OH)2, Pd2(dba)3, P(tBu)3, KF, ТГФ
Синтез аминовых предшественников P-V-A-4:
R= CN, CO2Et; a) MeI, NaOtBu, DMF; b) HCO2K, Pd-C, EtOH или HCO2NH4, Pd-C, EtOH
Синтез аминовых предшественников P-V-A-4:
a) ArBr, Pd(OAc)2, PS-PPh3, K2CO3, DMF
Синтез аминовых предшественников P-V-B-4:

H2, Pd-C, MeOH
Синтез аминовых предшественников P-V-B-4:
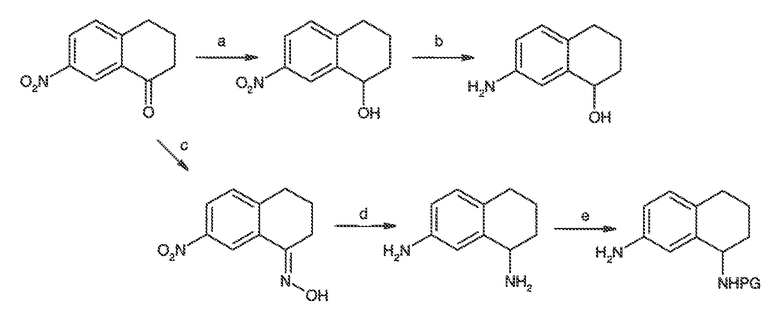
a) NaBH4, MeOH; b) H2, Pd-C, MeOH; c) NH2OH, Пиридин; d) H2, Pd-C, MeOH; e) Boc2O, Et3N, MeOH
Синтез соединений формулы I:
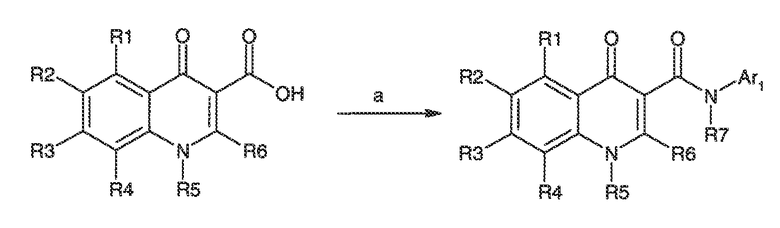
a) Ar1R7NH, связывающий реагент, основание, растворитель. Примеры используемых условий:
HATU, DIEA; BOP, DIEA, DMF; HBTU, Et3N, CH2Cl2; PFPTFA, пиридин.
Синтез соединений формулы I':
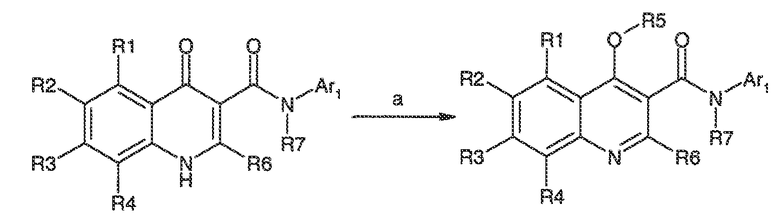
R5 = алифатическая группа:
a) R5X (X= Br, I), Cs2CO3, DMF
Синтез соединений формулы V-B-5:
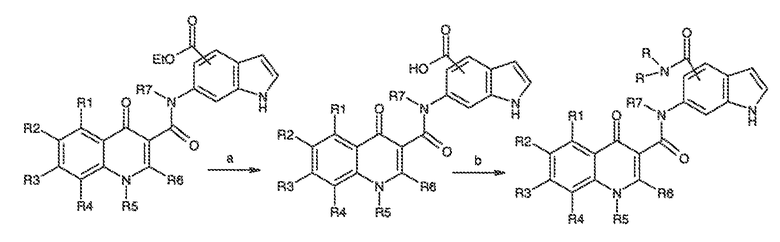
а) NaOH, ТГФ; b) HNR2, HATU, DIEA, DMF
Синтез соединений формулы V-B-5:
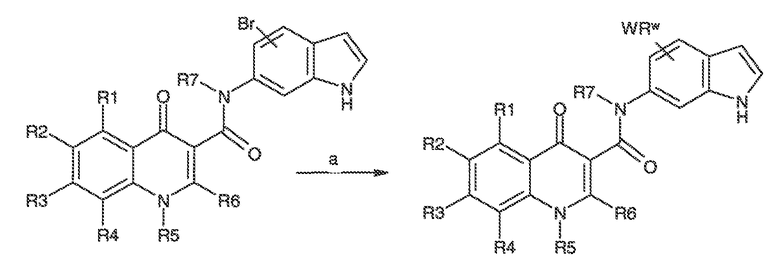
WRw = арил или гетероарил: a) ArB(OH)2, (dppf)PdCl2, K2CO3, DMF
Синтез соединений формулы V-A-2 и V-A-5:
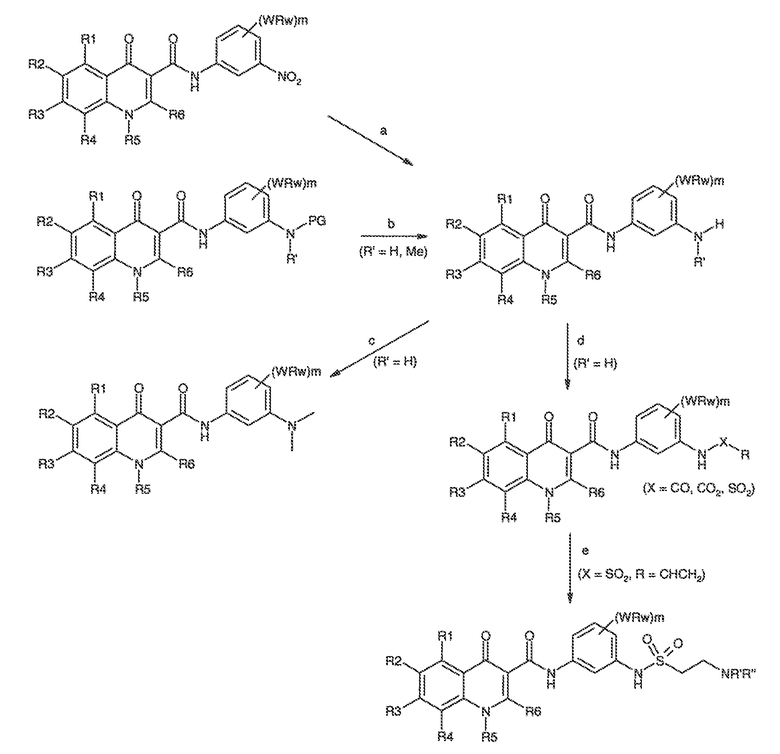
a) SnCl2.2H2O, EtOH; b) PG= BOC: TFA, CH2Cl2; c) CH2O, NaBH3CN, CH2Cl2, MeOH; d) RXCl, DIEA, ТГФ или RXCl, NMM, 1,4-диоксан или RXCl, CH2Cl2, DMF; e) R'R''NH, LiClO4, CH2Cl2, iPrOH
Синтез соединений формулы V-B-2:
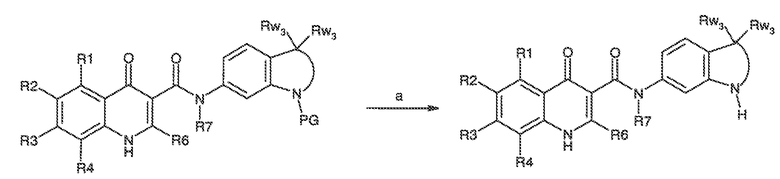
a) Когда PG = BOC: TFA, CH2Cl2; Когда PG = Ac: NaOH или HCl, EtOH или ТГФ
Синтез соединений формулы V-A-2:
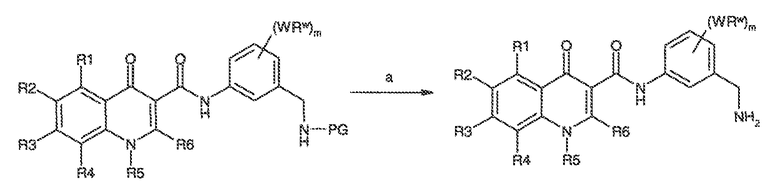
a) Когда PG = BOC: TFA, CH2Cl2
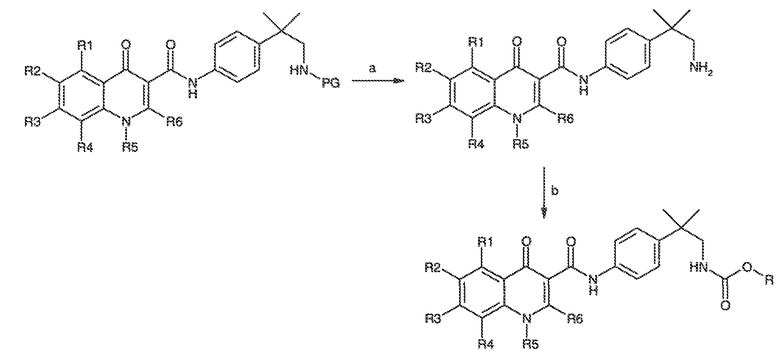
a) Когда PG = BOC: TFA, CH2Cl2; b) ROCOCl, Et3N, DMF
Синтез соединений формулы V-A-4:
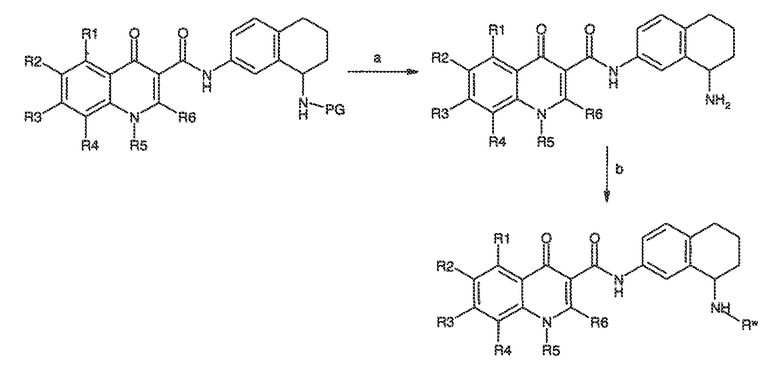
a) Когда PG = BOC: TFA, CH2Cl2; b) Когда Rw = CO2R: ROCOCl, DIEA, MeOH
На представленных выше схемах, указанный радикал R представляет собой заместитель, например, RW имеет значение, определенное выше. Специалистам в данной области должно быть понятно, что пути синтеза, подходящие для различных заместителей по настоящему изобретению, являются такими, чтобы испоьзуемые условия реакции и стадии не модифицировали предполагаемые заместители.
5. Применения, формулирование и введение
Фармацевтически приемлемые композиции
Как обсуждается выше, настоящее изобретение обеспечивает соединения, которые являются полезными в качестве модуляторов ABC транспортеров и, таким образом, являются полезными в лечении заболеваний, расстройств или состояний, таких как кистозный фиброз, наследственная эмфизема, наследственный гемохроматоз, нарушения коагуляции-фибринолиза, такие как дефицит белка C, наследственная ангиоэдема типа 1, нарушения липидного процессинга, такие как семейная гиперхолестеринемия, хиломикронемия типа 1, абеталипопротеинемия, болезни лизосомального накопления, такие как I-клеточная болезнь/псевдо-Hurler, мукополисахаридозы, болезнь Сандхофа/Тэя-Сакса, синдром Криглера-Найяра типа II, полиэндокринопатия/гиперинсулинемия, сахарный диабет, карликовость Ларона, дефицит милеопероксидазы, первичный гипопаратиреоидизм, меланома, гликаноз CDG типа 1, врожденный гипертиреоидизм, несовершенный остеогенез, наследственная гипофибриногенемия, дефицит ACT, несахарный диабет (DI), нейрофизеальный DI, нефрогенный DI, синдром Шарко-Мари-Тута, болезнь Perlizaeus-Merzbacher, нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз, прогрессирующий супрануклеарный паралич, болезнь Пика, некоторые полиглутаминовые неврологические расстройства, такие как болезнь Гентингтона, спиноцеребуллярная атаксия типа I, спинальная и бульбарная мышечная атрофия, dentatorubal pallidoluysian и миотоническая дистрофия, а также спонгиформные энцефалопатии, такие как наследственная болезнь Крейцфельдта-Якоба (из-за дефекта процессинга прионового белка), болезнь Фабри, синдром Штросслера-Шейнкера, COPD, болезнь сухих глаз или болезнь Шегрена.
Соответственно, в другом аспекте настоящего изобретения обеспечиваются фармацевтически приемлемые композиции, при этом такие композиции включают любое из соединений, описанных в настоящей заявке, и необязательно включают фармацевтически приемлемый носитель, адъювант или наполнитель. В некоторых вариантах воплощения, эти композиции необязательно дополнительно включают одно или несколько дополнительных терапевтических средств.
Также должно быть понятно, что некоторые соединения по настоящему изобретению могут существовать в свободной форме для лечения или, если это является подходящим, в виде фармацевтически приемлемого производного или его пролекарства. В соответствии с настоящим изобретением, фармацевтически приемлемое производное или пролекарство включает, но не ограничивается этим, фармацевтически приемлемые соли, сложные эфиры, соли таких сложных эфиров или любой другой продукт присоединения или производное, которое при введении пациенту, нуждающемуся в этом, способно обеспечивать, непосредственно или опосредованно, соединение, которое описано в настоящей заявке, или его метаболит или остаток.
Используемый в настоящей заявке термин "фармацевтически приемлемая соль" относится к тем солям, которые, в соответствии со взвешенной медицинской оценкой, являются подходящими для использования в контакте с тканями человека и низших животных, без чрезмерной токсичности, раздражения, аллергических реакций и т.п., и соизмеримы с разумным соотношением польза/риск. "Фармацевтически приемлемая соль" означает любую нетоксичную соль или соль сложного эфира соединения настоящего изобретения, которая при введении реципиенту способна обеспечивать, либо непосредственно или опосредованно, соединение настоящего изобретения или обладающий ингибиторной активностью метаболит или остаток такого соединения.
Фармацевтически приемлемые соли хорошо известны из уровня техники. Например, S. M. Berge, et al. подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 1977, 66, 1-19, который включен в настоящую заявку посредством ссылки. Фармацевтически приемлемые соли соединений настоящего изобретения включают соли, полученные из подходящих неорганических и органических кислот и оснований. Примеры фармацевтически приемлемых нетоксичных кислотно-аддитивных солей включают соли аминогруппы, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с использованием других способов, используемых в данной области техники, таких как ионообмен. Другие фармацевтически приемлемые соли включают адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидроиодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканоат, валерат и подобные. Соли, полученные из подходящих оснований, включают соли щелочных металлов, щелочно-земельных металлов, аммония и N+(C1-4алкила)4. Настоящее изобретение также предусматривает кватернизацию любой основной азот-содержащей группы соединений, раскрытых в настоящей заявке. Водо- или масло-растворимые или диспергируемые продукты можно получить при помощи такой кватернизации. Репрезентативные соли щелочных или щелочно-земельных металлов включают соли натрия, лития, калия, кальция, магния и подобные. Кроме того, фармацевтически приемлемые соли включают, если это является подходящим, нетоксичные катионы аммония, четвертичного аммония и амина, образованные с использованием противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, низший алкилсульфонат и арилсульфонат.
Как описано выше, фармацевтически приемлемые композиции по настоящему изобретению дополнительно включают фармацевтически приемлемый носитель, адъювант или наполнитель, который, как это используется в настоящей заявке, включает любой и все растворители, разбавители или другие жидкие носители, вещества, способствующие диспергированию или суспендированию, поверхностно-активные вещества, изотонические агенты, загустители или эмульгаторы, консерванты, твердые связующие, смазывающие вещества и подобные, как это является подходящим для конкретной лекарственной формы, которая является желательной. Remington's Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) раскрывает различные носители, используемые для формулирования фармацевтически приемлемых композиций, и известные способы их получения. За исключением только тех случаев, когда какая-либо традиционная среда, ипользуемая в качестве носителя, несовместима с соединениями по настоящему изобретению, например, вызывает какой-либо нежелательный биологический эффект или иначе взаимодействует неблагоприятным образом с любым другим компонентом(компонентами) фармацевтически приемлемой композиции, ее использование предусматривается как охватываемое объемом настоящего изобретения. Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничиваются этим, ионообменники, оксид алюминия, стеарат алюминия, лецитин, белки сыворотки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота или сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, вторичный кислый фосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, полиэтилен-полиоксипропиленовые блок-полимеры, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, масло семян хлопчатника; саффлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли; такие пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатно-буферные растворы, а также другие нетоксичные совместимые смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, вещества, способствующие высвобождению из формы, вещества покрытий, подсластители, отдушки и ароматизаторы, консерванты и антиоксиданты также могут присутствовать в композици, в соответствии с тем, как это сочтет нужным специалист, занимающийся формулированием композиции.
Применения соединений и фармацевтически приемлемые композиции
Еще в одном аспекте настоящее изобретение обеспечивает способ лечения или ослабления тяжести состояния, заболевания или расстройства, связанного с CFTR мутацией. В некоторых вариантах воплощения, настоящее изобретение обеспечивает способ лечения состояния, заболевания или расстройства, связанного с дефицитом CFTR активности, при этом способ включает введение композиции, включающей соединение формулы 1, субъекту, предпочтительно млекопитающему, нуждающемуся в этом.
В некоторых вариантах воплощения, настоящее изобретение обеспечивает способ лечения заболеваний, связанных с пониженной CFTR функцией из-за мутаций в гене, кодирующем CFTR, или из-за экологических факторов (например, курение). Эти заболевания включают, кистозный фиброз, хронический бронхит, рецидивирующий бронхит, острый бронхит, мужское бесплодие, вызванное врожденным двусторонним отсутствием семявыводящих протоков (CBAVD), женское бесплодие, вызванное врожденным отсутствием матки или влагалища (CAUV), идиопатический хронический панкреатит (ICP), идиопатический рецидивирующий панкреатит, идиопатический острый панкреатит, хронический риносинусит, первичный склерозирующий холангит, аллергический бронхолегочный аспергиллез, диабет, синдром сухих глаз, запор, аллергический бронхолегочный аспергиллез (ABPA), заболевания кости (например, остеопороз) и астму.
В некоторых вариантах воплощения, настоящее изобретение обеспечивает способ лечения заболеваний, связанных с нормальной CFTR функцией. Эти заболевания включают, хроническое обструктивное легочное заболевание (COPD), хронический бронхит, рецидивирующий бронхит, острый бронхит, риносинусит, запор, панкреатит, включая хронический панкреатит, рецидивирующий панкреатит и острый панкреатит, панкреатическую недостаточность, мужское бесплодие, вызванное врожденным двусторонним отсутствием семявыводящих протоков (CBAVD), слабую форму легочного заболевания, идиопатический панкреатит, заболевание печени, наследственную эмфизему, желче-каменную болезнь, гастро-эзофагеальный рефлюкс, желудочно-кишечные опухоли, воспалительное заболевание кишечника, запор, диабет, артрит, остеопороз и остеопению.
В некоторых вариантах воплощения, настоящее изобретение обеспечивает способ лечения заболеваний, связанных с нормальной CFTR функцией, включающих наследственный гемохроматоз, нарушения коагуляции-фибринолиза, такие как дефицит белка C, наследственная ангиоэдема типа 1, нарушения липидного процессинга, такие как семейная гиперхолестеринемия, хиломикронемия типа 1, абеталипопротеинемия, болезни лизосомального накопления, такие как I-клеточная болезнь/псевдо-Hurler, мукополисахаридозы, болезнь Сандхофа/Тэя-Сакса, синдром Криглера-Найяра типа II, полиэндокринопатию/гиперинсулинемию, сахарный диабет, карликовость Ларона, дефицит милеопероксидазы, первичный гипопаратиреоидизм, меланому, гликаноз CDG типа 1, врожденный гипертиреоидизм, несовершенный остеогенез, наследственную гипофибриногенемию, дефицит ACT, несахарный диабет (DI), нейрофизеальный DI, нефрогенный DI, синдром Шарко-Мари-Тута, болезнь Perlizaeus-Merzbacher, нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз, прогрессирующий супрануклеарный паралич, болезнь Пика, некоторые полиглутаминовые неврологические расстройства, такие как болезнь Гентингтона, спиноцеребуллярная атаксия типа I, спинальная и бульбарная мышечная атрофия, dentatorubal pallidoluysian и миотоническая дистрофия, а также спонгиформные энцефалопатии, такие как наследственная болезнь Крейцфельдта-Якоба (из-за дефекта процессинга прионового белка), болезнь Фабри, синдром Штросслера-Шейнкера, синдром Горэма, хлоридные каналопатии, врожденную миотонию (формы Томсона и Бекера), синдром Бартера типа III, болезнь Дента, гиперэкплексию, эпилепсию, гиперэкплексию, болезнь лизосомального накопления, синдром Ангельмана, первичную цилиарную дискинезию (PCD), PCD с situs inversus (также известный как синдром Картагенера), PCD без situs inversus и цилиарную аплазию или болезнь Шегрена, включающий стадию введения указанному млекопитающему эффективного количества композиции, включающей соединение по настоящему изобретению.
В соответствии с альтернативным предпочтительным вариантом воплощения, настоящее изобретение обеспечивает способ лечения кистозного фиброза, включающий стадию введения указанному млекопитающему эффективного количества композиции, включающей соединение по настоящему изобретению.
В соответствии с настоящим изобретением, “эффективное количество” соединения или фармацевтически приемлемой композиции представляет собой такое количество, которое является эффективным для лечения или ослабления тяжести одного или нескольких из заболеваний, расстройств или состояний, указанных выше.
Соединения и композиции, в соответствии со способом по настоящему изобретению, можно вводить с использованием любого количества и любого пути введения, эффективного для лечения или ослабления тяжести одного или нескольких из заболеваний, расстройств или состояний, указанных выше.
В некоторых вариантах воплощения, соединения и композиции по настоящему изобретению являются полезными для лечения или ослабления тяжести кистозного фиброза у пациентов, которые демонстрируют остаточную CFTR активность в апикальной мембране респираторного и нереспираторного эпителия. Присутствие остаточной CFTR активности на поверхности эпителия можно легко определить при помощи способов, известных из уровня техники, например, стандартными электрофизиологическими, биохимическими или гистохимическими методами. Такими способами определяют CFTR активность с использованием in vivo или ex vivo электрофизиологических методов, измерения Cl- концентраций в выделениях потных или слюнных желез или с использованием ex vivo биохимических или гистохимических методов для контроля плотности на клеточной поверхности. С использованием таких способов можно легко определить остаточную CFTR активность у пациентов гомозиготных или гетерозиготных для наиболее распространенной мутации, ΔF508.
Еще в одном варианте воплощения, соединения и композиции по настоящему изобретению являются полезными для лечения или ослабления тяжести кистозного фиброза у пациентов, которые имеют остаточную CFTR активность, индуцируемую или усиливаемую с использованием фармакологических способов или генной терапии. Такие способы повышают количество CFTR, присутствующего на клеточной поверхности, индуцируя, таким образом, отсутствующую до этого CFTR активность у пациента или повышая существующий уровень остаточной CFTR активности у пациента.
В одном варианте воплощения, соединения и композиции по настоящему изобретению являются полезными для лечения или ослабления тяжести кистозного фиброза у пациентов в рамках определенных генотипов, демонстрирующих остаточную CFTR активность, например, мутации класса III (нарушение регуляции или воротного механизма), мутации класса IV (изменение проводимости) или мутации класса V (уменьшение синтеза) (Lee R. Choo-Kang, Pamela L., Zeitlin, Type I, II, III, IV and V cystic fibrosis Transmembrane Conductance Regulator Defects and Opportunities of Therapy; Current Opinion in Pulmonary Medicine 6:521-529, 2000). Другие генотипы пациентов, которые демонстрируют остаточную CFTR активность, включают пациентов гомозиготных для одного из этих классов или гетерозиготных с любым другим классом мутаций, включая мутации класса I, мутации класса II или мутации, которые не классифицированы.
В одном варианте воплощения, соединения и композиции по настоящему изобретению являются полезными для лечения или ослабления тяжести кистозного фиброза у пациентов в пределах определенных клинических фенотипов, например, от средней тяжести до легкой формы клинического фенотипа, который типично соотносится с количеством остаточной CFTR активности в апикальной мембране эпителия. Такие фенотипы включают пациентов, демонстрирующих недостаточность функции поджелудочной железы, или пациентов, у которых диагностирован идиопатический панкреатит и врожденное двустороннее отсутствие семявыносящих протоков или легкая форма легочного заболевания.
Точное количество, которое необходимо, будет разным для разных субъектов, в зависимости от конкретного вида, возраста и общего состояния субъекта, тяжести инфекции, конкретного средства, способа его введения и т.п. Соединения по настоящему изобретению предпочтительно формулируют в лекарственную форму, содержащую стандартные единицы дозирования, для простоты введения и равномерного дозирования. Выражение "стандартные единицы дозирования", используемое в настоящей заявке, относится к физически дискретной единице средства, подходящего для пациента, подлежащего лечению. Однако должно быть понятно, что общий суточный прием соединений и композиций по настоящему изобретению определяет лечащий врач в соответствии с взвешенной медицинской оценкой. Конкретный уровень эффективной дозы для любого конкретного пациента или организма будет зависеть от различных факторов, включая расстройство, подлежащее лечению, и тяжесть этого расстройства; активность конкретного используемого соединения; конкретную используемую композицию; возраст, массу тела, общее состояние здоровья, пол и режим питания пациента; время введения, пути введения и скорость выведения из организма конкретного используемого соединения; продолжительность лечения; лекарственные средства, используемые в сочетании или случайно с конкретным используемым соединением, и подобные факторы, хорошо известные в медицине. Термин "пациент", как он используется в настоящей заявке, означает животное, предпочтительно - млекопитающее, и наиболее предпочтительно - человека.
Фармацевтически приемлемые композиции по настоящему изобретению можно вводить человеку и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонеально, местным путем (например, в виде порошков, мазей, капель или пластыря), буккально, в виде перорального или назального спрея или т.п., в зависимости от тяжести инфекции, подлежащей лечению. В некоторых вариантах воплощения, соединения по настоящему изобретению можно вводить перорально или парентерально при дозах на уровне от около 0,01 мг/кг до около 50 мг/кг, и предпочтительно от около 1 мг/кг до около 25 мг/кг массы тела субъекта в день, один или несколько раз в день, для получения желаемого терапевтического эффекта.
Жидкие лекарственные формы для перорального введения включают, но не ограничиваются этим, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активного соединения, жидкие лекарственные формы могут содержать инертные разбавители, традиционно используемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие вещества и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, масло семян хлопчатника, арахисовое масло, кукурузное масло, масло из проростков семян, оливковое масло, касторовое масло и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот сорбитана, и их смеси. Помимо инертных разбавителей, пероральные композиции также могут включать адъюванты, такие как смачивающие вещества, эмульгаторы и суспендирующие вещества, подсластители, отдушки и ароматизаторы.
Препараты для инъекций, например, стерильные водные или масляные суспензии для инъекций, можно сформулировать в соответствии с известными из уровня техники способами с использованием подходящих диспергирующих или смачивающих веществ и суспендирующих веществ. Стерильный препарат для инъекций также может представлять собой стерильный раствор, суспензию или эмульсию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Из приемлемых наполнителей и растворителей, которые можно использовать, можно указать воду, раствор Рингера U.S.P. и изотонический раствор хлорида натрия. Кроме того, стерильные, нелетучие масла традиционно используют в качестве растворителя или среды для суспендирования. Для этих целей можно использовать любое светлое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, в препаратах для инъекций используют жирные кислоты, такие как олеиновая кислота.
Композиции для инъекций можно стерилизовать, например, путем фильтрования через удерживающий бактерии фильтр или путем включения стерилизующих веществ в форме стерильных твердых композиций, которые можно растворить или диспергировать в стерильной воде или другой стерильной среде для инъекций перед использованием.
Для пролонгирования эффекта соединения по настоящему изобретению, часто желательно замедлить абсорбцию соединения из подкожной или внутримышечной инъекции. Это можно осуществить с использованием жидкой суспензии кристаллического или аморфного вещества с плохой водо-растворимостью. Скорость абсорбции соединения в этом случае зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристалла и кристаллической формы. Альтернативно, замедленную абсорбцию парентерально вводимой формы соединения получают путем растворения или суспендирования соединения в масляном наполнителе. Депо формы для инъекций получают путем образования матриц для микроинкапсулирования соединения в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения соединения и полимера и природы конкретного используемого полимера, можно контролировать скорость высвобождения соединения. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Композиции депо препаратов для инъекций также получают путем заключения соединения в липосомы или микроэмульсии, которые совместимы с тканями организма.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые можно получить путем смешивания соединения по настоящему изобретению с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но являются жидкими при температуре тела, и поэтому плавятся в прямой кишке или вагинальной полости и высвобождают активное соединение.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешано с, по меньшей мере, одним инертным, фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или вторичный кислый фосфат кальция и/или a) наполнителями или объемными веществами, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремневая кислота, b) связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и аравийская камедь, c) увлажнителями, такими как глицерин, d) разрыхлителями, такими как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия, e) замедляющими растворение веществами, такими как парафин, f) ускорителями абсорбции, такими как четвертичные аммониевые соединения, g) смачивающими веществами, такими как, например, цетиловый спирт и глицеринмоностеарат, h) абсорбентами, такими как каолиновая и бентонитовая глина, и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и смесями таких веществ. В случае капсул, таблеток и пилюль, лекарственная форма также может включать буферные вещества.
Твердые композиции подобного типа также можно использовать в качестве наполнителей в мягких и твердых желатиновых капсулах, с использованием таких эксципиентов, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и подобные. Твердые лекарственные формы в виде таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области фармацевтического формулирования. Они необязательно содержат светонепроницаемые агенты, и также могут иметь такую композицию, которая делает возможным высвобождение активного ингредиента(ингредиентов), только или предпочтительно, в определенной части пищеварительного тракта, необязательно замедленным образом. Примеры композиций инкапсулирующих веществ, которые можно использовать, включают полимерные вещества и воски. Твердые композиции подобного типа также можно использовать в качестве наполнителей, заключенных в мягкие и твердые желатиновые капсулы, с использованием таких эксципиентов, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и подобные.
Активные соединения также могут быть в микроинкапсулированной форме с одним или несколькими эксципиентами, как указано выше. Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, контролирующие высвобождение покрытия и другие покрытия, хорошо известные в области фармацевтического формулирования. В таких твердых лекарственных формах активное соединение может быть смешано с, по меньшей мере, одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы также могут включать, как это имеет место в обычной практике, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие вещества для таблетирования и другие вспомогательные вещества для таблетирования, такие стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, такие лекарственные формы также могут включать буферные вещества. Они необязательно содержат светонепроницаемые агенты, и также могут иметь такую композицию, которая делает возможным высвобождение активного ингредиента(ингредиентов), только или предпочтительно, в определенной части пищеварительного тракта, необязательно замедленным образом. Примеры композиций инкапсулирующих веществ, которые можно использовать, включают полимерные вещества и воски.
Лекарственные формы для введения соединения по настоящему изобретению местным или чрескожным путем включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, препараты для ингаляций или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, которые могут для этого потребоваться. Глазные препараты, ушные капли и глазные капли также предусматриваются как охватываемые объемом настоящего изобретения. Кроме того, настоящее изобретение предусматривает использование чрескожных пластырей, которые имеют дополнительное преимущество, обеспечивая контролируемую доставку соединения в организм. Такие лекарственные формы получают путем растворения или диспергирования соединения в подходящей среде. Также можно использовать усилители абсорбции, используемые для усиления проникновения соединения через кожу. Скорость можно контролировать либо путем обеспечения контролирующей скорость мембраны, либо путем диспергирования соединения в полимерной матрице или геле.
Как описано в общем виде выше, соединения по настоящему изобретению являются полезными в качестве модуляторов ABC транспортеров. Таким образом, не желая быть связанным с какой-либо конкретной теорией, соединения и композиции являются особенно полезными для лечения или ослабления тяжести заболевания, состояния или расстройства, где такое заболевание, состояние или расстройство связано с гиперактивностью или отсутствием активности ABC транспортеров. Когда конкретное заболевание, состояние или расстройство связано с гиперактивностью или отсутствием активности ABC транспортера, такое заболевание, состояние или расстройство также может быть указано как “ABC транспортер-опосредованное заболевание, состояние или расстройство”. Соответственно, в другом аспекте настоящее изобретение обеспечивает способ лечения или ослабления тяжести заболевания, состояния или расстройства, где в состояние такого заболевания вовлечена гиперактивность или отсутствие активности ABC транспортера.
Анализ активности соединения, используемого в настоящем изобретении в качестве модулятора ABC транспортера, можно осуществить в соответствии со способами, в общем описанными в известном уровне техники и в Примерах, представленных в настоящей заявке.
Также должно быть понятно, что соединения и фармацевтически приемлемые композиции по настоящему изобретению можно использовать в комбинированной терапии, то есть соединения и фармацевтически приемлемые композиции можно вводить одновременно с введением, до или после введения одного или нескольких других желательных терапевтических средств или медицинских процедур. Конкретные комбинированные терапии (терапевтические средства или процедуры) для использования в комбинированной схеме лечения должны учитывать совместимость желаемых терапевтических средств и/или процедур и желаемого терапевтического эффекта, достижение которого необходимо. Также должно быть понятно, что используемые терапии могут достигать желаемого эффекта при одном и том же расстройстве (например, соединение по настоящему изобретению можно вводить одновременно с другим средством, используемым для лечения этого же расстройства), или они могут быть предназначены для достижения разных эффектов (например, контроль каких-либо побочных эффектов). Как это используется в настоящей заявке, дополнительные терапевтические средства, которые обычно вводят для лечения или профилактики определенного заболевания или состояния, известны как "подходящие для заболевания или состояния, подлежащего лечению".
В одном варианте воплощения, дополнительное средство выбрано из муколитического средства, бронходилататора, антибиотика, противоинфекционного средства, противоивоспалительного средства, модулятора CFTR, отличного от соединения по настоящему изобретению, или питательного вещества. В другом варианте воплощения, дополнительное средство представляет собой модулятор CFTR, отличный от соединения по настоящему изобретению.
В одном варианте воплощения, дополнительное средство представляет собой антибиотик. Примеры антибиотиков, полезных в настоящем изобретении, включают тобрамицин, включая тобрамицин в виде порошка для ингаляции (TIP), азитромицин, азтреонам, включая аэрозольную форму азтреонама, амикацин, включая его липосомальную композицию, ципрофлоксацин, включая его композицию, подходящую для введения путем ингаляции, левофлаксацин, включая его аэрозольные композиции, и сочетания двух антибиотиков, например, фосфомицина и тобрамицина.
Еще в одном варианте воплощения, дополнительное средство представляет собой муколитическое средство. Примеры муколитических средств, полезных в настоящем изобретении, включают Pulmozyme®.
Еще в одном варианте воплощения, дополнительное средство представляет собой бронходилататор. Пример бронходилататоров включают албутерол, метапротенерол сульфат, пирбутерол ацетат, салметерол или тетрабулин сульфат.
Еще в одном варианте воплощения, дополнительное средство является эффективным для восстановления поверхностной жидкости в дыхательных путях в легких. Такие средства улучшают перемещение соли в клетки и из клеток, делая слизь в дыхательных путях в легких более гидратированной, поэтому они более легко выводятся. Примеры таких средств включают гипертонический солевой раствор, денуфозол тетранатрий ([[(3S,5R)-5-(4-амино-2-оксопиримидин-1-ил)-3-гидроксиоксолан-2-ил]метокси-гидроксифосфорил][[[(2R,3S,4R,5R)-5-(2,4-диоксопиримидин-1-ил)-3,4-дигидроксиоксолан-2-ил]метокси-гидроксифосфорил]окси-гидроксифосфорил]гидрофосфат) или бронхитол (препарат для ингаляций с использованием маннита).
Еще в одном варианте воплощения, дополнительное средство представляет собой противоивоспалительное средство, т.е. средство, которое может уменьшать воспаление в легких. Примеры таких средств, полезных в настоящем изобретении, включают ибупрофен, докозагексановую кислоту (DHA), силденафил, глутатион для ингаляций, пиоглитазон, гидроксихлорохин или симавастатин.
В другом варианте воплощения, дополнительное средство снижает активность блокатора эпителиальных натриевых каналов (ENaC) либо непосредственно путем блокирования канала, либо опосредованно путем модуляции протеаз, что приводит к повышению активности ENaC (например, сериновых протеаз, канал-активирующих протеаз). Примеры таких средств включают камостат (ингибитор трипсин-подобной протеазы), QAU145, 552-02, GS-9411, INO-4995, Aerolytic и амилорид. Дополнительные средства, которые снижают активность блокатора эпителиальных натриевых каналов (ENaC), можно найти, например в Публикации PCT № WO2009/074575, полное содержание которой включено в настоящую заявку во всей полноте.
Среди других заболеваний, описанных в настоящей заявке, комбинации модуляторов CFTR, таких как соединения формулы I, и средств, которые снижают активность ENaC, являются полезными для лечения синдрома Лиддла, воспалительного или аллергического состояния, включая кистозный фиброз, первичную цилиарную дискинезию, хронический бронхит, хроническое обструктивное легочное заболевание, астму, инфекции респираторного тракта, карциному легкого, ксеростомия и кератоконъюнктивит sire, инфекции респираторного тракта (острые и хронические; вирусные и бактериальные) и карциному легкого.
Комбинации модуляторов CFTR, такие как соединения формулы I и средства, которые снижают активность ENaC, также являются полезными для лечения заболеваний, опосредованных блокадой эпителиальных натриевых каналов, также включающих заболевания, отличные от респираторных заболеваний, которые связаны с аномальной регуляцией жидкости через эпителий, возможно связанных с аномальной физиологией защитных поверхностных жидкостей на их поверхности, например, ксеростомия (сухость во рту) или кератоконъюнктивит sire (синдром сухих глаз). Кроме того, блокаду эпителиальных натриевых каналов в почке можно использовать для поддержания диуреза индуцируя, таким образом, гипотензивный эффект.
Астма включает как наследственную (не-аллергическую) астму и приобретенную (аллергическую) астму, слабовыраженную астму, умеренную форму астмы, тяжелую форму астмы, бронхитную астму, индуцированную физическими нагрузками астму, профессиональную астму и астму, являющуюся результатом бактериальной инфекции. Лечение астмы также следует рассматривать, как лечение субъектов, например, возраста менее 4 или 5 лет, у которых имеются симптомы одышки и поставлен диагноз, или может быть поставлен диагноз, "дети младшего возраста, страдающие одышкой ", которые представляют категорию пациентов, вызывающую серьезную медицинскую озабоченность, и которых часто определяют как инципиент или астматики в ранней фазе. (Для удобства это конкретное астматическое состояние указано как "синдром детей с одышкой"). Доказательством профилактической эффективности в лечении астмы может быть пониженная частота или тяжесть симптоматических приступов, например, острого астматического или бронхоконстрикторного приступа, улучшение функции легких или улучшение состояния гиперреактивности дыхательных путей. Кроме того, это может быть подтверждено снижением необходимости в другом симптоматическом лечении, т.е. лечении симптоматического приступа, когда он возникает, или которое предназначено для ограничения или предотвращения такого приступа, например, с использованием противовоспалительных средств (например, кортикостероидов) или бронходилататоров. Профилактическая польза при астме, в частности, может быть очевидна у субъектов, склонных к "утренним приступам". "Утренний приступ" представляет собой признанный астматический синдром, характерный для существенного процента астматиков и характеризующийся астматическим приступом, например, в период между 4-6 часов утра, т.е. во время, которое обычно достаточно отдалено от какого-либо предшествующего приема симптомтического лечения астмы.
Хроническое обструктивное легочное заболевание включает хронический бронхит или связанную с этим одышку, эмфизему, а также усиление гиперреактивности дыхательных путей в результате приема другого лекарственного средства, в частности, другого лекарственного препарата для ингаляции. В некоторых вариантах воплощения, комбинации модуляторов CFTR, таких как соединения формулы I, и средств, которые снижают активность ENaC, являются полезными для лечения бронхита любого типа или генезиса, включая, например, острый, арахидальный, катаральный, крупозный, хронический или гнойный туберкулезный бронхит.
Еще в одном варианте воплощения, дополнительное средство представляет собой модулятор CFTR, отличный от соединения 1, т.е. средство, которое обладает эффектом модулирования CFTR активности. Примеры таких средств включают аталурен ("PTC 124®"; 3-[5-(2-фторфенил)-1,2,4-оксадиазол-3-ил]бензойная кислота), синапултит, ланковутид, депелестат (человеческий рекомбинантный ингибитор эластазы нейтрофилов), кобипростон (7-{(2R,4aR,5R,7aR)-2-[(3S)-1,1-дифтор-3-метилпентил]-2-гидрокси-6-оксооктагидроциклопента[b]пиран-5-ил}гептановая кислота) или (3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойная кислота. Еще в одном варианте воплощения, дополнительное средство представляет собой (3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту.
Еще в одном варианте воплощения, дополнительное средство представляет собой питательное вещество. Примеры таких веществ включают панкрелипазу (заместитель панкреатических ферментов), включая Pancrease®, Pancreacarb®, Ultrase® или Creon®, Liprotomase® (прежнее название Trizytek®), Aquadeks®, или глутатион для ингаляций. В одном варианте воплощения, дополнительное питательное вещество представляет собой панкрелипазу.
Количество дополнительного терапевтического средства, присутствующего в композиции по настоящему изобретению, не должно превышать количество, являющееся нормально возможным для введения в композиции, включающей такое терапевтическое средство в качестве единственного активного вещества. Предпочтительно, количество дополнительного терапевтического средства в композициях, раскрытых в настоящем изобретении, должно находиться в пределах от около 50% до 100% от количества, обычно присутствующего в композиции, включающей такое терапевтическое средство в качестве единственного активного вещества.
Соединения по настоящему изобретению или содержащие их фармацевтически приемлемые композиции также могут быть включены в композиции для покрытия имплантируемых медицинских устройств, таких как протезы, искусственные клапаны, сосудистые трансплантаты, стенты и катетеры. Соответственно, настоящее изобретение, еще в одном аспекте, включает композицию для покрытия имплантируемого устройства, включающую соединение по настоящему изобретению, описанное в целом выше и в классах и подклассах, описанных в настоящей заявке, и носитель, подходящий для покрытия указанного имплантируемого устройства. Еще в одном аспекте настоящее изобретение включает имплантируемое устройство, имеющее покрытие с композицией, включающей соединение по настоящему изобретению, описанное в целом выше и в классах и подклассах, описанных в настоящей заявке, и носитель, подходящий для покрытия указанного имплантируемого устройства. Подходящие покрытия и общее описание получения имеющих покрытия имплантируемых устройств можно найти в патентах США №№ 6099562; 5886026; и 5304121. Покрытия типично представляют собой биосовместимые полимерные вещества, такие как полимерные гидрогели, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленвинилацетат и их смеси. Покрытия, необязательно, могут быть дополнительно покрыты подходящим верхним слоем из фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или представляющим собой комбинацию таких веществ для придания композиции характеристик контролируемого высвобождения.
Другой аспект настоящего изобретения относится к модулированиию активности ABC транспортера в биологическом образце или у пациента (например, in vitro или in vivo), при этом способ включает введение пациенту или контактирование указанного биологического образца с соединением формулы 1 или композицией, включающей указанное соединение. Термин "биологический образец", используемый в настоящей заявке, включает, без ограничения, клеточные культуры или их экстракты; материал биопсии, полученный от млекопитающего, или его экстракты; и кровь, слюну, мочу, кал, семенную жидкость, слезы или другие жидкости организма, или их экстракты.
Модуляция активности ABC транспортера, т.е. CFTR, в биологическом образце является полезной для различных целей, которые известны специалистам в данной области. Примеры таких целей включают, но не ограничиваются этим, исследование ABC транспортеров в биологических и патологических явлениях; и сравнительную оценку новых модуляторов ABC транспортеров.
В следующем варианте воплощения, обеспечивается способ модулирования активности анионного канала in vitro или in vivo, включающий стадию контактирования указанного канала с соединением формулы I. В предпочтительных вариантах воплощения, анионный канал представляет собой хлоридный канал или бикарбонатный канал. В других предпочтительных вариантах воплощения, анионный канал представляет собой хлоридный канал.
В соответствии с альтернативным вариантом воплощения, настоящее изобретение обеспечивает способ увеличения количества функциональных ABC транспортеров в мембране клетки, включающий стадию контактирования указанной клетки с соединением формулы I. Термин “функциональный ABC транспортер”, как он используется в настоящей заявке, означает ABC транспортер, который может обладать транспортирующей активностью. В предпочтительных вариантах воплощения, ABC транспортер представляет собой CFTR.
В соответствии с другим вариантом воплощения, активность ABC транспортера определяют путем измерения трансмембраного потенциала. Средства для измерения потенциала через мембрану в биологическом образце могут включать любой из способов, известных из уровня техники, такой как оптический анализ мембранного потенциала или другие электрофизиологические методы.
Оптический анализ мембранного потенциала включает использование потенциал-чувствительных FRET сенсоров, описанных Gonzalez и Tsien (см. Gonzalez, J. E. and R. Y. Tsien (1995) "Voltage sensing by fluorescence resonance energy transfer in single cells." Biophys J 69(4): 1272-80 ; Gonzalez, J. E. and R. Y. Tsien (1997); "Improved indicators of cell membrane potential that use fluorescence resonance energy transfer" Chem Biol 4(4): 269-77), в сочетании со средствами измерения изменений флуоресценции, такими как Voltage/Ion Probe Reader (VIPR) (см. Gonzalez, J. E., K. Oades, et al. (1999) "Cell-based assays and instrumentation for screening ion-channel targets" Drug Discov Today 4(9): 431-439).
Эти потенциал-чувствительные анализы основаны на изменении передачи резонансной энергии флуоресценции (FRET) между мембрано-растворимым потенциал-чувствительным красителем, DiSBAC2(3) и флуоресцентным фосфолипидом, CC2-DMPE, который присоединен к внешней створке плазменной мембраны и действует в качестве FRET донора. Изменения мембранного потенциала (Vm) вызывают редистрибуцию отрицательно заряженного DiSBAC2(3) через плазменную мембрану, и количество передаваемой энергии от CC2-DMPE соответственно изменяется. Изменения эмиссии флуоресценции можно отслеживать с использованием VIPR™ II, который представляет собой интегрированное устройство, включающее подачу жидкости и детектор флуоресценции, предназначенное для осуществления клеточных скрининговых анализов в 96- или 384-луночных микротитровальных планшетах.
В другом аспекте настоящее изобретение обеспечивает набор для использования в измерении активности ABC транспортера или его фрагмента в биологическом образце in vitro или in vivo, включающий (i) композицию, включающую соединение формулы I или любой из описанных выше вариантов воплощения; и (ii) инструкции для a) контактирования композиции с биологическим образцом, и b) измерения активности указанного ABC транспортера или его фрагмента. В одном варианте воплощения, набор дополнительно включает инструкции для a) контактирования дополнительной композиции с биологическим образцом; b) измерение активности указанного ABC транспортера или его фрагмента в присутствии указанного дополнительного соединения, и c) сравнение активности ABC транспортера в присутствии дополнительного соединения с плотностью ABC транспортера в присутствии композиции формулы I. В предпочтительных вариантах воплощения, набор используют для измерения плотности CFTR.
Для того, чтобы лучше понять изобретение, описанное в настоящей заявке, далее представлены примеры. Должно быть понятно, что эти примеры представлены только в иллюстративных целях и не должны рассматриваться как каким-либо образом ограничивающие настоящее изобретение.
ПРИМЕРЫ
Пример 1:
Общая схема получения кислотных групп:
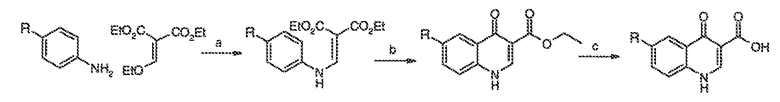
a) 140-150ºC; b) PPA, POCl3, 70ºC или дифениловый эфир, 220ºC; c) i) 2н NaOH ii) 2н HCl
Специальный пример: диэтиловый эфир 2-фениламинометилен-малоновой кислоты
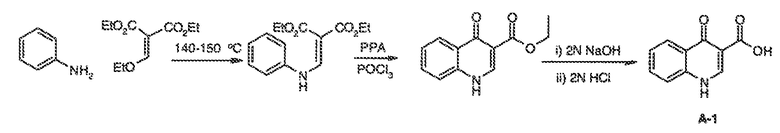
Смесь анилина (25,6 г, 0,28 моль) и диэтил 2-(этоксиметилен)малонат (62,4 г, 0,29 моль) нагревали при 140-150ºC в течение 2 часов. Смесь охлаждали до комнатной температуры и сушили при пониженном давлении с получением диэтилового эфира 2-фениламинометилен-малоновой кислоты в виде твердого вещества, которое использовали на следующей стадии без дополнительной очистки. 1H ЯМР (d-DMSO) δ 11,00 (д, 1H), 8,54 (д, J=13,6 Гц, 1H), 7,36-7,39 (м, 2H), 7,13-7,17 (м, 3H), 4,17-4,33 (м, 4H), 1,18-1,40 (м, 6H).
Этиловый эфир 4-гидроксихинолин-3-карбоновой кислоты
В 1-л трехгорлую колбу, снабженную механической мешалкой, загружали диэтиловый эфир 2-фениламинометилен-малоновой кислоты (26,3 г, 0,1 моль), полифосфорную кислоту (270 г) и фосфорилхлорид (750 г). Смесь нагревали до около 70ºC и перемешивали в течение 4 часов. Смесь охлаждали до комнатной температуры и фильтровали. Остаток обрабатывали водным раствором Na2CO3, фильтровали, промывали водой и сушили. Этиловый эфир 4-гидроксихинолин-3-карбоновой кислоты получали в виде бледно-коричневого твердого вещества (15,2 г, 70%). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
A-1; 4-Оксо-1,4-дигидрохинолин-3-карбоновая кислота
Этиловый эфир 4-гидроксихинолин-3-карбоновой кислоты (15 г, 69 ммоль) суспендировали в растворе гидроксида натрия (2н, 150 мл) и перемешивали в течение 2 часов при температуре кипения с обратным холодильником. После охлаждения смесь фильтровали и фильтрат подкисляли до pH 4 2н раствором HCl. Образовавшийся осадок собирали фильтрованием, промывали водой и сушили в вакууме с получением 4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (A-1) в виде бледно-белого твердого вещества (10,5 г, 92%). 1H ЯМР (d-DMSO) δ 15,34 (с, 1H), 13,42 (с, 1H), 8,89 (с, 1H), 8,28 (д, J=8,0 Гц, 1H), 7,88 (м, 1H), 7,81 (д, J=8,4 Гц, 1H), 7,60 (м, 1H).
Специальный пример: A-2; 6-Фтор-4-гидрокси-хинолин-3-карбоновая кислота
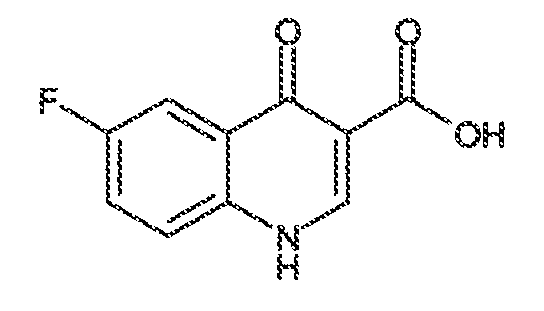
6-Фтор-4-гидрокси-хинолин-3-карбоновую кислоту (A-2) синтезировали в соответствии с общей схемой, представленной выше, исходя из 4-фтор-фениламина. Общий выход (53%). 1H ЯМР (DMSO-d6) δ 15,2 (шир.с, 1H), 8,89 (с, 1H), 7,93-7,85 (м, 2H), 7,80-7,74 (м, 1H); ESI-MS 207,9 m/z (MH+).
Пример 2:
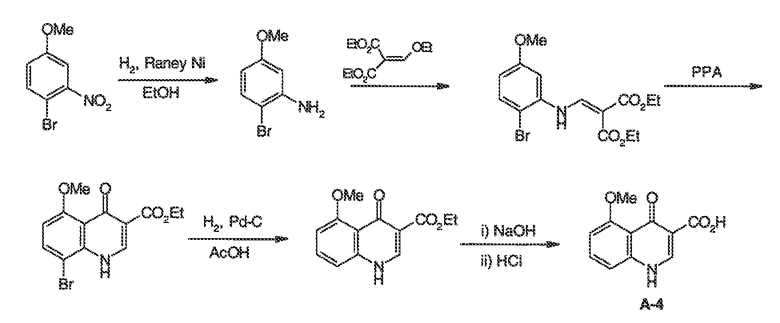
2-Бром-5-метокси-фениламин
Смесь 1-бром-4-метокси-2-нитро-бензола (10 г, 43 ммоль) и Ni Ренея (5 г) в этаноле (100 мл) перемешивали в атмосфере H2 (1 атм) в течение 4 часов при комнатной температуре. Ni Ренея отфильтровывали и фильтрат концентрировали при пониженном давлении. Полученное твердое вещество очищали колоночной хроматографией с получением 2-бром-5-метокси-фениламина (7,5 г, 86%).
Диэтиловый эфир 2-[(2-бром-5-метокси-фениламино)-метилен]-малоновой кислоты
Смесь 2-бром-5-метокси-фениламина (540 мг, 2,64 ммоль) и диэтил 2-(этоксиметилен)малоната (600 мг, 2,7 ммоль) перемешивали при 100ºC в течение 2 часов. После охлаждения реакционную смесь перекристаллизовывали из метанола (10 мл) с получением диэтилового эфира 2-[(2-бром-5-метокси-фениламино)-метилен]-малоновой кислоты в виде желтого твердого вещества (0,8 г, 81%).
Этиловый эфир 8-бром-5-метокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты
Диэтиловый эфир 2-[(2-бром-5-метокси-фениламино)-метилен]-малоновой кислоты (9 г, 24,2 ммоль) медленно добавляли к полифосфорной кислоте (30 г) при 120ºC. Смесь перемешивали при этой температуре еще в течение 30 минут и затем охлаждали до комнатной температуры. Добавляли абсолютный этанол (30 мл) и полученную смесь кипятили с обратным холодильником в течение 30 минут. Смесь подщелачивали водным раствором бикарбоната натрия при 25ºC и экстрагировали при помощи EtOAc (4×100 мл). Органические слои объединяли, сушили и растворитель выпаривали с получением этилового эфира 8-бром-5-метокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (2,3 г, 30%).
Этиловый эфир 5-метокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты
Смесь этилового эфира 8-бром-5-метокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (2,3 г, 7,1 ммоль), ацетата натрия (580 мг, 7,1 ммоль) и 10% Pd/C (100 мг) в ледяной уксусной кислоте (50 мл) перемешивали в атмосфере H2 (2,5 атм) в течение ночи. Катализатор удаляли фильтрованием и реакционную смесь концентрировали при пониженном давлении. Полученное масло растворяли в CH2Cl2 (100 мл) и промывали водным раствором бикарбоната натрия и водой. Органический слой сушили, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией с получением этилового эфира 5-метокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты в виде желтого твердого вещества (1 г, 57%).
A-4; 5-Метокси-4-оксо-1,4-дигидро-хинолин-3-карбоновая кислота
Смесь этилового эфира 5-метокси-4-оксо-1, 4-дигидро-хинолин-3-карбоновой кислоты (1 г, 7,1 ммоль) в 10% растворе NaOH (50 мл) нагревали до температуры кипения с обратным холодильником в течение ночи и затем охлаждали до комнатной температуры. Смесь экстрагировали простым эфиром. Водную фазу отделяли и подкисляли концентрированным раствором HCl до pH 1-2. Образовавшийся осадок собирали фильтрованием с получением 5-метокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (A-4) (530 мг, 52%). 1H ЯМР (DMSO) δ: 15,9 (с, 1H), 13,2 (шир., 1H), 8,71 (с, 1H), 7,71 (т, J=8,1 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 6,82 (д, J=8,4 Гц, 1H), 3,86 (с, 3H); ESI-MS 219,9 m/z (MH+).
Пример 3:
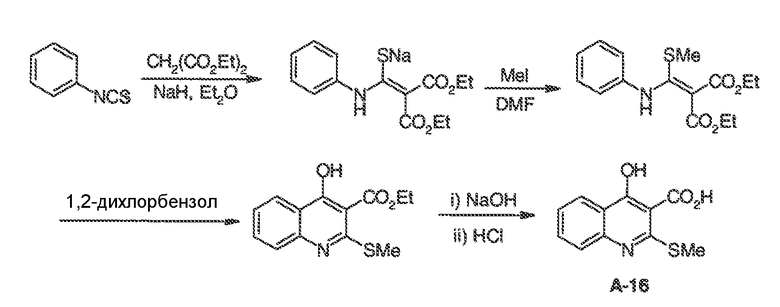
Диэтиловый эфир натрий 2-(меркапто-фениламино-метилен)-малоновой кислоты
К суспензии NaH (60% в минеральном масле, 6 г, 0,15 моль) в Et2O при комнатной температуре добавляли по каплям в течение 30 минут этилмалонат (24 г, 0,15 моль). Затем добавляли по каплям фенилизотиоцианат (20,3 г, 0,15 моль) при перемешивании в течение 30 минут. Смесь кипятили с обратным холодильником в течение 1 часа и затем перемешивали в течение ночи при комнатной температуре. Твердое вещество отделяли, промывали безводным эфиром (200 мл) и сушили в вакууме с получением диэтилового эфира натрий 2-(меркапто-фениламино-метилен)-малоновой кислоты в виде бледно-желтого порошка (46 г, 97%).
Диэтиловый эфир 2-(метилсульфанил-фениламино-метилен)-малоновой кислоты
В течение 30-минутного периода, метилиодид (17,7 г, 125 ммоль) добавляли по каплям к раствору диэтилового эфира натрий 2-(меркапто-фениламино-метилен)-малоновой кислоты (33 г, 104 ммоль) в DMF (100 мл), охлажденному на ледяной бане. Смесь перемешивали при комнатной температуре в течение 1 часа и затем выливали в ледяную воду (300 мл). Полученное твердое вещество собирали фильтрованием, промывали водой и сушили с получением диэтилового эфира 2-(метилсульфанил-фениламино-метилен)-малоновой кислоты в виде бледно-желтого твердого вещества (27 г, 84%).
Этиловый эфир 4-гидрокси-2-метилсульфанил-хинолин-3-карбоновой кислоты
Смесь диэтилового эфира 2-(метилсульфанил-фениламино-метилен)-малоновой кислоты (27 г, 87 ммоль) в 1,2-дихлорбензоле (100 мл) нагревали до температуры кипения с обратным холодильником в течение 1,5 часов. Растворитель удаляли при пониженном давлении и маслянистый остаток растирали в порошок с гексаном с получением бледно-желтого твердого вещества, которое очищали препаративной ВЭЖХ с получением этилового эфира 4-гидрокси-2-метилсульфанил-хинолин-3-карбоновой кислоты (8 г, 35%).
A-16; 2-Метилсульфанил-4-оксо-1,4-дигидро-хинолин-3-карбоновая кислота
Этиловый эфир 4-гидрокси-2-метилсульфанил-хинолин-3-карбоновой кислоты (8 г, 30 ммоль) нагревали при температуре кипения с обратным холодильником в растворе NaOH (10%, 100 мл) в течение 1,5 часов. После охлаждения смесь подкисляли концентрированной HCl до pH 4. Полученное твердое вещество собирали фильтрованием, промывали водой (100 мл) и MeOH (100 мл) с получением 2-метилсульфанил-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (A-16) в виде белого твердого вещества (6 г, 85%). 1H ЯМР (CDCl3) δ 16,4 (шир.с, 1H), 11,1 (шир.с, 1H), 8,19 (д, J=8 Гц, 1H), 8,05 (д, J=8 Гц, 1H), 7,84 (т, J=8, 8 Гц, 1H), 7,52 (т, J=8 Гц, 1H), 2,74 (с, 3H); ESI-MS 235,9 m/z (MH+).
Пример 4:

a) PPh3, Et3N, CCl4, CF3CO2H; b) диэтилмалонат; c) T~200ºC; d) 10% NaOH
2,2,2-Трифтор-N-фенил-ацетимидоилхлорид
Смесь Ph3P (138,0 г, 526 ммоль), Et3N (21,3 г, 211 ммоль), CCl4 (170 мл) и TFA (20 г, 175 ммоль) перемешивали в течение 10 минут на ледяной бане. Анилин (19,6 г, 211 ммоль) растворяли в CCl4 (20 мл) и добавляли. Смесь перемешивали при температуре кипения с обратным холодильником в течение 3 часов. Растворитель удаляли в вакууме и добавляли гексан. Осажденные вещества (Ph3PO и Ph3P) отфильтровывали и промывали гексаном. Фильтрат подвергали перегонке при пониженном давлении с получением 2,2,2-трифтор-N-фенил-ацетимидоилхлорида (19 г), которое использовали на следующей стадии без дополнительной очистки.
Диэтиловый эфир 2-(2,2,2-трифтор-1-фенилимино-этил)-малоновой кислоты
К суспензии NaH (3,47 г, 145 ммоль, 60% в минеральном масле) в ТГФ (200 мл) добавляли диэтилмалонат (18,5 г, 116 ммоль) при 0ºC. Смесь перемешивали в течение 30 минут при этой температуре и добавляли 2,2,2-трифтор-N-фенил-ацетимидоилхлорид (19 г, 92 ммоль) при 0ºC. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Смесь разбавляли при помощи CH2Cl2, промывали насыщенным раствором бикарбоната натрия и насыщенным солевым раствором. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением диэтилового эфира 2-(2,2,2-трифтор-1-фенилимино-этил)-малоновой кислоты, который использовали непосредственно на следующей стадии без дополнительной очистки.
Этиловый эфир 4-гидрокси-2-трифторметил-хинолин-3-карбоновой кислоты
Диэтиловый эфир 2-(2,2,2-трифтор-1-фенилимино-этил)-малоновой кислоты нагревали при 210ºC в течение 1 часа при постоянном перемешивании. Смесь очищали колоночной хроматографией (петролейный эфир) с получением этилового эфира 4-гидрокси-2-трифторметил-хинолин-3-карбоновой кислоты (12 г, 24% за 3 стадии).
A-15; 4-Гидрокси-2-трифторметил-хинолин-3-карбоновая кислота
Суспензию этилового эфира 4-гидрокси-2-трифторметил-хинолин-3-карбоновой кислоты (5 г, 17,5 ммоль) в 10% водном растворе NaOH нагревали при температуре кипения с обратным холодильником в течение 2 часов. После охлаждения добавляли дихлорметан и водную фазу отделяли и подкисляли концентрированной HCl до pH 4. Образовавшийся осадок собирали фильтрованием, промывали водой и Et2O с получением 4-гидрокси-2-трифторметил-хинолин-3-карбоновой кислоты (A-15) (3,6 г, 80%). 1H ЯМР (DMSO-d6) δ 8,18-8,21 (д, J=7,8 Гц, 1H), 7,92-7,94 (д, J=8,4 Гц, 1H), 7,79-7,83 (т, J=14,4 Гц, 1H), 7,50-7,53 (т, J=15 Гц, 1H); ESI-MS 257,0 m/z (MH+).
Пример 5:

a) CH3C(O)ONH4, толуол; b) EtOCHC(CO2Et)2, 130ºC; c) Ph2O; d) I2, EtOH; e) NaOH
3-Амино-циклогекс-2-енон
Смесь циклогексан-1,3-дион (56,1 г, 0,5 моль) и AcONH4 (38,5 г, 0,5 моль) в толуоле нагревали при температуре кипения с обратным холодильником в течение 5 часов с использованием ловушки Дина-Старка. Полученный маслянистый слой отделяли и концентрировали при пониженном давлении с получением 3-амино-циклогекс-2-енона (49,9 г, 90%), который использовали непосредственно на следующей стадии без дополнительной очистки.
Диэтиловый эфир 2-[(3-оксо-циклогекс-1-ениламино)-метилен]-малоновой кислоты
Смесь 3-амино-циклогекс-2-енона (3,3 г, 29,7 ммоль) и диэтил 2-(этоксиметилен)малоната (6,7 г, 31,2 ммоль) перемешивали при 130ºC в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении и полученное масло очищали колоночной хроматографией (силикагель, этилацетат) с получением диэтилового эфира 2-[(3-оксо-циклогекс-1-ениламино)-метилен]-малоновой кислоты (7,5 г, 90%).
Этиловый эфир 4,5-диоксо-1,4,5,6,7,8-гексагидро-хинолин-3-карбоновой кислоты
Смесь диэтилового эфира 2-[(3-оксо-циклогекс-1-ениламино)-метилен]-малоновой кислоты (2,8 г, 1 ммоль) и дифенилового эфира (20 мл) кипятили с обратным холодильником в течение 15 минут. После охлаждения добавляли н-гексан (80 мл). Полученное твердое вещество выделяли фильтрованием и перекристаллизовывали из метанола с получением этилового эфира 4,5-диоксо-1,4,5,6,7,8-гексагидро-хинолин-3-карбоновой кислоты (1,7 г, 72%).
Этиловый эфир 5-гидрокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты
К раствору этилового эфира 4,5-диоксо-1,4,5,6,7,8-гексагидро-хинолин-3-карбоновой кислоты (1,6 г, 6,8 ммоль) в этаноле (100 мл) добавляли иод (4,8 г, 19 ммоль). Смесь кипятили с обратным холодильником в течение 19 часов и затем концентрировали при пониженном давлении. Полученное твердое вещество промывали этилацетатом, водой и ацетоном и затем перекристаллизовывали из DMF с получением этилового эфира 5-гидрокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (700 мг, 43%).
A-3; 5-Гидрокси-4-оксо-1, 4-дигидро-хинолин-3-карбоновая кислота
Смесь этилового эфира 5-гидрокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (700 мг, 3 ммоль) в 10% NaOH (20 мл) нагревали при температуре кипения с обратным холодильником в течение ночи. После охлаждения смесь экстрагировали простым эфиром. Водную фазу отделяли и подкисляли при помощи концентрированной HCl до pH 1-2. Образовавшийся осадок собирали фильтрованием с получением 5-гидрокси-4-оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (A-3) (540 мг, 87%). 1H ЯМР (DMSO-d6) δ 13,7 (шир., 1H), 13,5 (шир., 1H), 12,6 (с, 1H), 8,82 (с, 1H), 7,68 (т, J=8,1 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 6,82 (д, J=8,4 Гц, 1H); ESI-MS 205,9 m/z (MH+).
Пример 6:
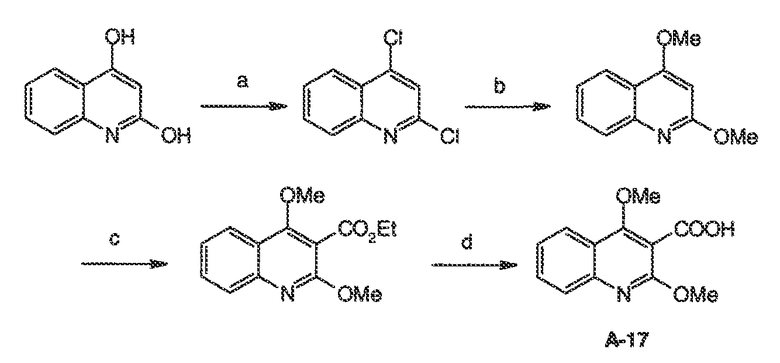
a) POCl3; b) MeONa; c) n-BuLi, ClCO2Et; d) NaOH
2,4-Дихлорхинолин
Суспензию хинолин-2,4-диола (15 г, 92,6 ммоль) в POCl3 нагревали при температуре кипения с обратным холодильником в течение 2 часов. После охлаждения растворитель удаляли при пониженном давлении с получением 2,4-дихлорхинолина, который использовали без дополнительной очистки.
2,4-Диметоксихинолин
К суспензии 2,4-дихлорхинолина в MeOH (100 мл) добавляли метоксид натрия (50 г). Смесь нагревали при температуре кипения с обратным холодильником в течение 2 дней. После охлаждения смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением остатка, который растворяли в воде и экстрагировали при помощи CH2Cl2. Объединенные органические слои сушили над Na2SO4 и концентрировали с получением 2,4-диметоксихинолина в виде белого твердого вещества (13 г, 74% за 2 стадии).
Этил 2,4-диметоксихинолин-3-карбоксилат
К раствору 2,4-диметоксихинолина (11,5 г, 60,8 ммоль) в безводном ТГФ добавляли по каплям n-BuLi (2,5 M в гексане, 48,6 мл, 122 ммоль) при 0ºC. После перемешивания в течение 1,5 часов при 0ºC смесь добавляли к раствору этил хлорформиата в безводном ТГФ и перемешивали при 0ºC еще в течение 30 минут и затем при комнатной температуре в течение ночи. Реакционную смесь выливали в воду и экстрагировали при помощи CH2Cl2. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией (петролейный эфир/EtOAc = 50/1) с получением этил 2,4-диметоксихинолин-3-карбоксилата (9,6 г, 60%).
A-17; 2,4-Диметоксихинолин-3-карбоновая кислота
Этил 2,4-диметоксихинолин-3-карбоксилат (1,5 г, 5,7 ммоль) нагревали при температуре кипения с обратным холодильником в растворе NaOH (10%, 100 мл) в течение 1 часа. После охлаждения смесь подкисляли концентрированной HCl до pH 4. Образовавшийся осадок собирали фильтрованием и промывали водой и простым эфиром с получением 2,4-диметоксихинолин-3-карбоновой кислот (A-17) в виде белого твердого вещества (670 мг, 50%). 1H ЯМР (CDCl3) δ 8,01-8,04 (д, J=12 Гц, 1H), 7,66-7,76 (м, 2H), 7,42-7,47 (т, J=22 Гц, 2H), 4,09 (с, 3H), 3,97 (с, 3H); ESI-MS 234,1 m/z (MH+).
Коммерчески доступные кислоты
Группы амина
N-1 Замещенные 6-аминоиндолы
Пример 1:
Общая Схема:
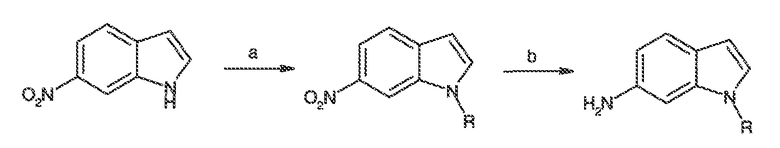
a) RX (X = Cl, Br, I), K2CO3, DMF или CH3CN; b) H2, Pd-C, EtOH или SnCl2.2H2O, EtOH.
Специальный пример:
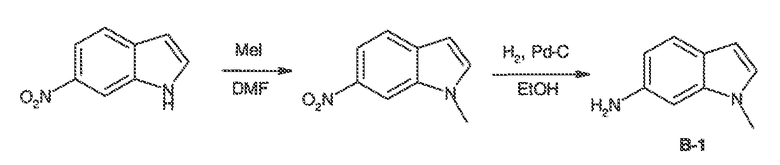
1-Метил-6-нитро-1H-индол
К раствору 6-нитроиндола (4,05 г, 25 ммоль) в DMF (50 мл) добавляли K2CO3 (8,63 г, 62,5 ммоль) и MeI (5,33 г, 37,5 ммоль). После перемешивания при комнатной температуре в течение ночи смесь выливали в воду и экстрагировали этилацетатом. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением продукта 1-метил-6-нитро-1H-индола (4,3 г, 98%).
B-1; 1-Метил-1H-индол-6-иламин
Суспензию 1-метил-6-нитро-1H-индола (4,3 г, 24,4 ммоль) и 10% Pd-C (0,43 г) в EtOH (50 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение ночи. После фильтрования фильтрат концентрировали и подкисляли при помощи HCl-MeOH (4 моль/л) с получением гидрохлоридной соли 1-метил-1H-индол-6-иламина (B-1) (1,74 г, 49%) в виде серого порошка. 1H ЯМР (DMSO-d6): δ 9,10 (с, 2H), 7,49 (д, J=8,4 Гц, 1H), 7,28 (д, J=2,0 Гц, 1H), 7,15(с, 1H), 6,84 (д, J=8,4 Гц, 1H), 6,38 (д, J=2,8 Гц, 1H), 3,72 (с, 3H); ESI-MS 146,08 m/z (MH+).
Другие примеры:
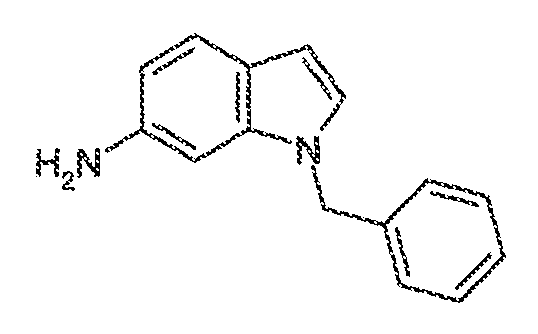
B-2; 1-Бензил-1H-индол-6-иламин
1-Бензил-1H-индол-6-иламин (B-2) синтезировали в соответствии с общей схемой, представленной выше, исходя из 6-нитроиндола и бензилбромида. Общий выход (~40%). ВЭЖХ время удерживания 2,19 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 223,3 m/z (MH+).
B-3; 1-(6-Амино-индол-1-ил)-этанон
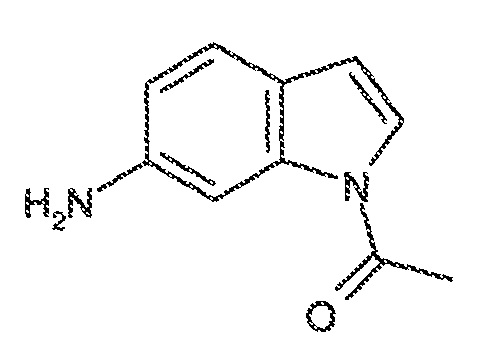
1-(6-Амино-индол-1-ил)-этанон (B-3) синтезировали в соответствии с общей схемой, представленной выше, исходя из 6-нитроиндола и ацетилхлорида. Общий выход (~40%). ВЭЖХ время удерживания 0,54 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 175,1 m/z (MH+).
Пример 2:
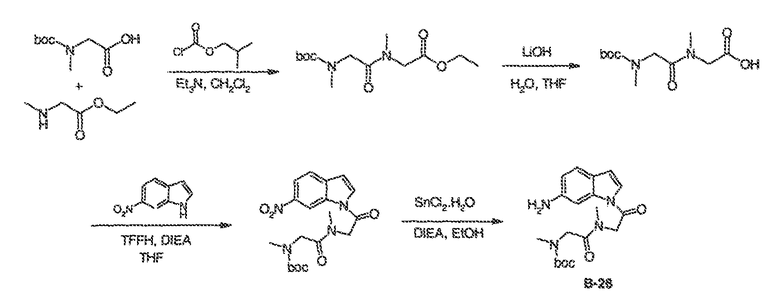
Этиловый эфир {[2-(трет-бутоксикарбонил-метил-амино)-ацетил]-метил-амино}-уксусной кислоты
К перемешиваемому раствору (трет-бутоксикарбонил-метил-амино)-уксусной кислоты (37 г, 0,2 моль) и Et3N (60,6 г, 0,6 моль) в CH2Cl2 (300 мл) добавляли изобутилхлорформиат (27,3 г, 0,2 ммоль) по каплям при -20ºC в атмосфере аргона. После перемешивания в течение 0,5 часов добавляли по каплям этиловый эфир метиламино-уксусной кислоты гидрохлорид (30,5 г, 129 ммоль) при -20ºC. Смеси давали нагреться до комнатной температуры (~1 час) и гасили водой (500 мл). Органический слой отделяли, промывали 10% раствором лимонной кислоты, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир/EtOAc 1:1) с получением этилового эфира {[2-(трет-бутоксикарбонил-метил-амино)-ацетил]-метил-амино}-уксусной кислоты (12,5 г, 22%).
{[2-(трет-Бутоксикарбонил-метил-амино)-ацетил]-метил-амино}-уксусная кислота
Суспензию этилового эфира {[2-(трет-бутоксикарбонил-метил-амино)-ацетил]-метил-амино}-уксусной кислоты (12,3 г, 42,7 ммоль) и LiOH (8,9 г, 214 ммоль) в H2O (20 мл) и ТГФ (100 мл) перемешивали в течение ночи. Летучий растворитель удаляли в вакууме и остаток экстрагировали простым эфиром (2×100 мл). Водную фазу подкисляли до pH 3 разбавленным раствором HCl и затем экстрагировали при помощи CH2Cl2 (2×300 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали в вакууме с получением {[2-(трет-бутоксикарбонил-метил-амино)-ацетил]-метил-амино}-уксусной кислоты в виде бесцветного масла (10 г, 90%). 1H ЯМР (CDCl3) δ 7,17 (шир.с, 1H), 4,14-4,04 (м, 4H), 3,04-2,88 (м, 6H), 1,45-1,41 (м, 9H); ESI-MS 282,9 m/z (M+Na+).
трет-Бутиловый эфир метил-({метил-[2-(6-нитро-индол-1-ил)-2-оксо-этил]-карбамоил}-метил)-карбаминовой кислот
К смеси {[2-(трет-бутоксикарбонил-метил-амино)-ацетил]-метил-амино}-уксусной кислоты (13,8 г, 53 ммоль) и TFFH (21,0 г, 79,5 ммоль) в безводном ТГФ (125 мл) добавляли DIEA (27,7 мл, 159 ммоль) при комнатной температуре в атмосфере азота. Раствор перемешивали при комнатной температуре в течение 20 минут. Добавляли раствор 6-нитроиндола (8,6 г, 53 ммоль) в ТГФ (75 мл) и реакционную смесь нагревали при 60ºC в течение 18 часов. Растворитель выпаривали и неочищенную смесь снова распределяли между EtOAc и водой. Органический слой отделяли, промывали водой (×3), сушили над Na2SO4 и концентрировали. Добавляли диэтиловый эфир, затем EtOAc. Полученное твердое вещество собирали фильтрованием, промывали диэтиловым эфиром и сушили на воздухе с получением трет-бутилового эфира метил-({метил-[2-(6-нитро-индол-1-ил)-2-оксо-этил]-карбамоил}-метил)-карбаминовой кислоты (6,42 г, 30%). 1H ЯМР (400 МГц, DMSO-d6) δ 1,37 (м, 9H), 2,78 (м, 3H), 2,95 (д, J=1,5 Гц, 1H), 3,12 (д, J=2,1 Гц, 2H), 4,01 (д, J=13,8 Гц, 0,6H), 4,18 (д, J=12,0 Гц, 1,4H), 4,92 (д, J=3,4 Гц, 1,4H), 5,08 (д, J=11,4 Гц, 0,6H), 7,03 (м, 1H), 7,90 (м, 1H), 8,21 (м, 1H), 8,35 (д, J=3,8 Гц, 1H), 9,18 (м, 1H); ВЭЖХ время удерживания 3,12 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 405,5 m/z (MH+).
B-26; трет-бутиловый эфир ({[2-(6-амино-индол-1-ил)-2-оксо-этил]-метил-карбамоил}-метил)-метил-карбаминовой кислоты
Смесь трет-бутилового эфира метил-({метил-[2-(6-нитро-индол-1-ил)-2-оксо-этил]-карбамоил}-метил)-карбаминовой кислоты (12,4 г, 30,6 ммоль), SnCl2.2H2O (34,5 г, 153,2 ммоль) и DIEA (74,8 мл, 429 ммоль) в этаноле (112 мл) нагревали до 70ºC в течение 3 часов. Добавляли воду и EtOAc и смесь фильтровали через короткую пробку из Целита. Органический слой отделяли, сушили над Na2SO4 и концентрировали с получением трет-бутилового эфира ({[2-(6-амино-индол-1-ил)-2-оксо-этил]-метил-карбамоил}-метил)-метил-карбаминовой кислоты (B-26) (11,4 г, количественный). ВЭЖХ время удерживания 2,11 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 375,3 m/z (MH+).
2-Замещенные 6-аминоиндолы
Пример 1:
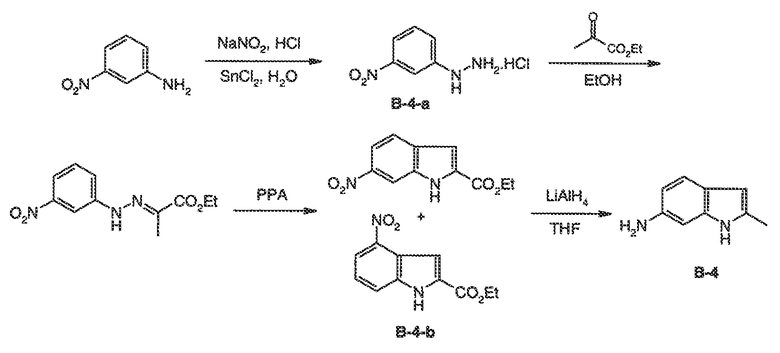
B-4-a; (3-Нитро-фенил)-гидразин, гидрохлоридная соль
3-Нитро-фениламин (27,6 г, 0,2 моль) растворяли в смеси H2O (40 мл) и 37% HCl (40 мл). Добавляли раствор NaNO2 (13,8 г, 0,2 моль) в H2O (60 мл) при 0ºC с последующим добавлением SnCl2.H2O (135,5 г, 0,6 моль) в 37% HCl (100 мл) при этой температуре. После перемешивания при 0ºC в течение 0,5 часов твердое вещество выделяли фильтрованием и промывали водой с получением (3-нитро-фенил)-гидразина в виде гидрохлоридной соли (B-4-a) (27,6 г, 73%).
Этиловый эфир 2-[(3-нитро-фенил)-гидразоно]-пропионовой кислоты
Гидрохлоридную соль (3-нитро-фенил)-гидразина (B-4-a) (30,2 г, 0,16 моль) и этиловый эфир 2-оксо-пропионовой кислоты (22,3 г, 0,19 моль) растворяли в этаноле (300 мл). Смесь перемешивали при комнатной температуре в течение 4 часов. Растворитель выпаривали при пониженном давлении с получением этилового эфира 2-[(3-нитро-фенил)-гидразоно]-пропионовой кислоты, который использовали непосредственно на следующей стадии.
B-4-b; этиловый эфир 4-нитро-1H-индол-2-карбоновой кислоты и этиловый эфир 6-Нитро-1H-индол -2-карбоновой кислоты
Этиловый эфир 2-[(3-нитро-фенил)-гидразоно]-пропионовой кислоты с предыдущей стадии растворяли в толуоле (300 мл). Добавляли PPA (30 г). Смесь нагревали при температуре кипения с обратным холодильником в течение ночи и затем охлаждали до комнатной температуры. Растворитель удаляли с получением смеси этилового эфира 4-нитро-1H-индол-2-карбоновой кислоты и этилового эфира 6-нитро-1H-индол-2-карбоновой кислоты (B-4-b) (15 г, 40%).
B-4; 2-Метил-1H-индол-6-иламин
К суспензии LiAlH4 (7,8 г, 0,21 моль) в ТГФ (300 мл) добавляли по каплям смесь этилового эфира 4-нитро-1H-индол-2-карбоновой кислоты и этилового эфира 6-нитро-1H-индол-2-карбоновой кислоты (B-4-b) (6 г, 25,7 ммоль) в ТГФ (50 мл) при 0ºC в атмосфере N2. Смесь нагревали при температуре кипения с обратным холодильником в течение ночи и затем охлаждали до 0ºC. К смеси добавляли H2O (7,8 мл) и 10% NaOH (7,8 мл) при 0ºC. Нерастворимое твердое вещество удаляли фильтрованием. Фильтр сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией с получением 2-метил-1H-индол-6-иламина (B-4) (0,3 г, 8%). 1H ЯМР (CDCl3) δ 7,57 (шир.с, 1H), 7,27 (д, J=8,8 Гц, 1H), 6,62 (с, 1H), 6,51-6,53 (м, 1H), 6,07 (с, 1H), 3,59-3,25 (шир.с, 2H), 2,37 (с, 3H); ESI-MS 147,2 m/z (MH+).
Пример 2:
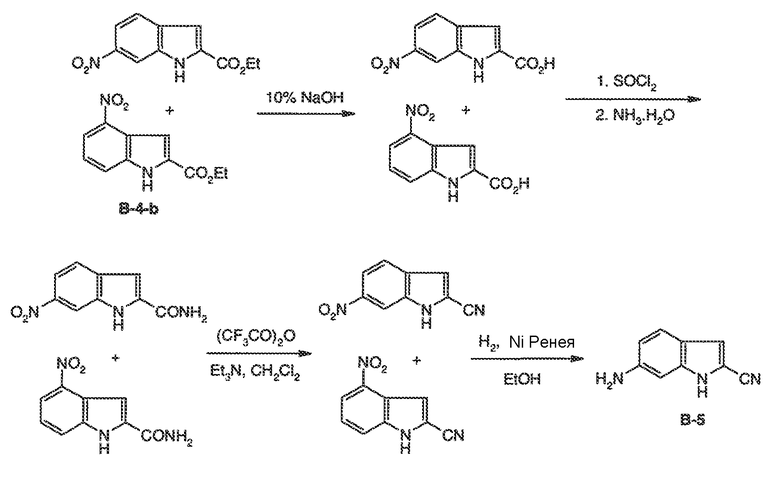
6-Нитро-1H-индол-2-карбоновая кислота и 4-Нитро-1H-индол-2-карбоновая кислота
Смесь этилового эфира 4-нитро-1H-индол-2-карбоновой кислоты и этилового эфира 6-нитро-1H-индол-2-карбоновой кислоты (B-4-b) (0,5 г, 2,13 ммоль) в 10% NaOH (20 мл) нагревали при температуре кипения с обратным холодильником в течение ночи и затем охлаждали до комнатной температуры. Смесь экстрагировали простым эфиром. Водную фазу отделяли и подкисляли при помощи HCl до pH 1-2. Полученное твердое вещество выделяли фильтрованием с получением смеси 6-нитро-1H-индол-2-карбоновой кислоты и 4-нитро-1H-индол-2-карбоновой кислоты (0,3 г, 68%).
Амид 6-нитро-1H-индол-2-карбоновой кислоты и амид 4-нитро-1H-индол-2-карбоновой кислоты
Смесь 6-нитро-1H-индол-2-карбоновой кислоты и 4-нитро-1H-индол-2-карбоновой кислоты (12 г, 58 ммоль) и SOCl2 (50 мл, 64 ммоль) в бензоле (150 мл) кипятили с обратным холодильником в течение 2 часов. Бензол и избыточное количество SOCl2 удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (250 мл). NH4OH (21,76 г, 0,32 моль) добавляли по каплям при 0ºC. Смесь перемешивали при комнатной температуре в течение 1 часа. Полученное твердое вещество выделяли фильтрованием с получением неочищенной смеси амида 6-нитро-1H-индол-2-карбоновой кислоты и амида 4-нитро-1H-индол-2-карбоновой кислоты (9 г, 68%), которую использовали непосредственно на следующей стадии.
6-Нитро-1H-индол-2-карбонитрил и 4-Нитро-1H-индол-2-карбонитрил
Смесь амида 6-нитро-1H-индол-2-карбоновой кислот и амида 4-нитро-1H-индол-2-карбоновой кислоты (5 г, 24 ммоль) растворяли в CH2Cl2 (200 мл). Добавляли Et3N (24,24 г, 0,24 моль) с последующим добавлением (CF3CO)2O (51,24 г, 0,24 моль) при комнатной температуре. Смесь перемешивали в течение 1 часа и выливали в воду (100 мл). Органический слой отделяли. Водный слой экстрагировали при помощи EtOAc (100 мл ×3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией с получением смеси 6-нитро-1H-индол-2-карбонитрила и 4-нитро-1H-индол-2-карбонитрила (2,5 г, 55%).
B-5; 6-Амино-1H-индол-2-карбонитрил
Смесь 6-нитро-1H-индол-2-карбонитрила и 4-нитро-1H-индол-2-карбонитрила (2,5 г, 13,4 ммоль) и Ni Ренея (500 мг) в EtOH (50 мл) перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 1 часа. Ni Ренея отфильтровывали. Фильтрат упаривали при пониженном давлении и очищали колоночной хроматографией с получением 6-амино-1H-индол-2-карбонитрила (B-5) (1 г, 49%). 1H ЯМР (DMSO-d6) δ 12,75 (шир.с, 1H), 7,82 (д, J=8 Гц, 1H), 7,57 (с, 1H), 7,42 (с, 1H), 7,15 (д, J=8 Гц, 1H); ESI-MS 158,2 m/z (MH+).
Пример 3:
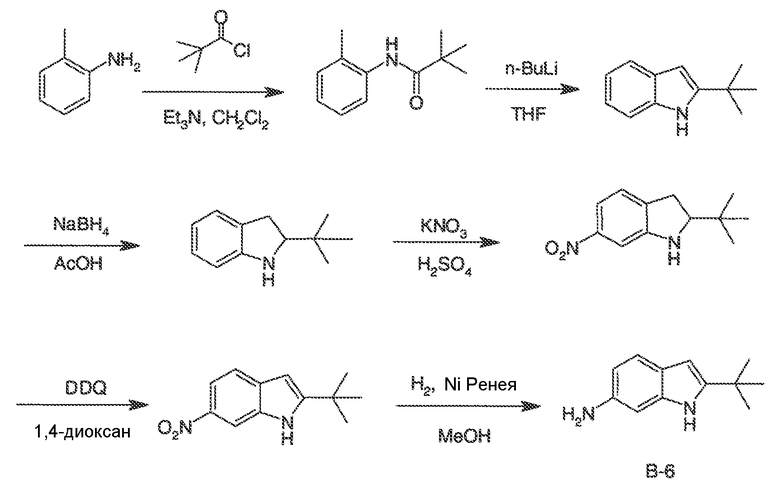
2,2-Диметил-N-o-толил-пропионамид
К раствору o-толиламина (21,4 г, 0,20 моль) и Et3N (22,3 г, 0,22 моль) в CH2Cl2 добавляли 2,2-диметил-пропионилхлорид (25,3 г, 0,21 моль) при 10ºC. Смесь перемешивали в течение ночи при комнатной температуре, промывали водным раствором HCl (5%, 80 мл), насыщенным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали в вакууме с получением 2,2-диметил-N-o-толил-пропионамида (35,0 г, 92%).
2-трет-бутил-1H-индол
К раствору 2,2-диметил-N-o-толил-пропионамида (30,0 г, 159 ммоль) в безводном ТГФ (100 мл) добавляли по каплям n-BuLi (2,5 M, в гексане, 190 мл) при 15ºC. Смесь перемешивали в течение ночи при 15ºC, охлаждали на бане лед-вода и обрабатывали насыщенным раствором NH4Cl. Органический слой отделяли и водный слой экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией с получением 2-трет-бутил-1H-индола (23,8 г, 88%).
2-трет-бутил-2,3-дигидро-1H-индол
К раствору 2-трет-бутил-1H-индола (5,0 г, 29 ммоль) в AcOH (20 мл) добавляли NaBH4 при 10ºC. Смесь перемешивали в течение 20 минут при 10ºC, обрабатывали по каплям при помощи H2O при охлаждении льдом и экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением смеси исходного вещества и 2-трет-бутил-2,3-дигидро-1H-индола (4,9 г), которую использовали непосредственно на следующей стадии.
2-трет-бутил-6-нитро-2,3-дигидро-1H-индол
К раствору смеси 2-трет-бутил-2,3-дигидро-1H-индола и 2-трет-бутил-1H-индола (9,7 г) в H2SO4 (98%, 80 мл) медленно добавляли KNO3 (5,6 г, 55,7 ммоль) при 0ºC. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, осторожно выливали на колотый лед, подщелачивали при помощи Na2CO3 до pH~8 и экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией с получением 2-трет-бутил-6-нитро-2,3-дигидро-1H-индола (4,0 г, 32% за 2 стадии).
2-трет-бутил-6-нитро-1H-индол
К раствору 2-трет-бутил-6-нитро-2,3-дигидро-1H-индола (2,0 г, 9,1 ммоль) в 1,4-диоксане (20 мл) добавляли DDQ при комнатной температуре. После кипячения с обратным холодильником в течение 2,5 часов смесь фильтровали и фильтрат концентрировали в вакууме. Остаток очищали колоночной хроматографией с получением 2-трет-бутил-6-нитро-1H-индола (1,6 г, 80%).
B-6; 2-трет-бутил-1H-индол-6-иламин
К раствору 2-трет-бутил-6-нитро-1H-индола (1,3 г, 6,0 ммоль) в MeOH (10 мл) добавляли Ni Ренея (0,2 г). Смесь перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 3 часов. Реакционную смесь фильтровали и фильтрат концентрировали. Остаток промывали петролейным эфиром с получением 2-трет-бутил-1H-индол-6-иламина (B-6) (1,0 г, 89%). 1H ЯМР (DMSO-d6) δ 10,19 (с, 1H), 6,99 (д, J=8,1 Гц, 1H), 6,46 (с, 1H), 6,25 (дд, J=1,8, 8,1 Гц, 1H), 5,79 (д, J=1,8 Гц, 1H), 4,52 (с, 2H), 1,24 (с, 9H); ESI-MS 189,1 m/z (MH+).
3-Замещенные 6-аминоиндолы
Пример 1:
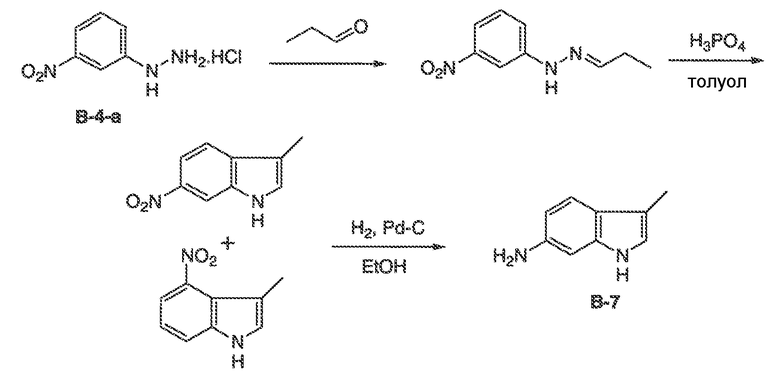
N-(3-Нитро-фенил)-N'-пропилиден-гидразин
Раствор гидроксида натрия (10%, 15 мл) добавляли медленно к перемешиваемой суспензии гидрохлоридной соли (3-нитро-фенил)-гидразина (B-4-a) (1,89 г, 10 ммоль) в этаноле (20 мл) до достижения pH 6. К смеси добавляли уксусную кислоту (5 мл) с последующим добавлением пропиональдегида (0,7 г, 12 ммоль). После перемешивания в течение 3 часов при комнатной температуре смесь выливали в ледяную воду и образовавшийся осадок выделяли фильтрованием, промывали водой и сушили на воздухе с получением N-(3-нитро-фенил)-N'-пропилиден-гидразина, который использовали непосредственно на следующей стадии.
3-Метил-4-нитро-1H-индол и 3-Метил-6-нитро-1H-индол
Смесь N-(3-нитро-фенил)-N'-пропилиден-гидразина растворяли в 85% H3PO4 (20 мл) и толуоле (20 мл) и нагревали при 90-100ºC в течение 2 часов. После охлаждения толуол удаляли при пониженном давлении. Полученное масло подщелачивали 10% раствором NaOH до pH 8. Водный слой экстрагировали при помощи EtOAc (100 мл ×3). Объединенные органические слои сушили, фильтровали и концентрировали при пониженном давлении с получением смеси 3-метил-4-нитро-1H-индола и 3-метил-6-нитро-1H-индола (1,5 г, 86% за две стадии), которую использовали непосредственно на следующей стадии.
B-7; 3-Метил-1H-индол-6-иламин
Смесь 3-метил-4-нитро-1H-индола и 3-метил-6-нитро-1H-индола (3 г, 17 моль) и 10% Pd-C (0,5 г) в этаноле (30 мл) перемешивали в течение ночи в атмосфере H2 (1 атм) при комнатной температуре. Pd-C отфильтровывали и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией с получением 3-метил-1H-индол-6-иламина (B-7) (0,6 г, 24%). 1H ЯМР (CDCl3) δ 7,59 (шир.с, 1H), 7,34 (д, J=8,0 Гц, 1H), 6,77 (с, 1H), 6,64 (с, 1H), 6,57 (м, 1H), 3,57 (шир.с, 2H), 2,28 (с, 3H); ESI-MS 147,2 m/z (MH+).
Пример 2:
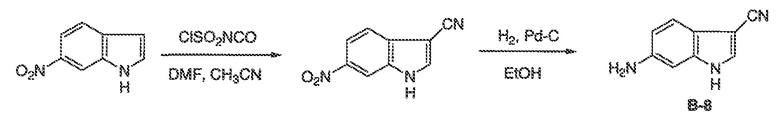
6-Нитро-1H-индол-3-карбонитрил
К раствору 6-нитроиндола (4,86 г, 30 ммоль) в DMF (24,3 мл) и CH3CN (243 мл) добавляли по каплям раствор ClSO2NCO (5 мл, 57 ммоль) в CH3CN (39 мл) при 0ºC . После добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 2 часов. Смесь выливали в ледяную воду, подщелачивали насыщенным раствором NaHCO3 до pH 7-8 и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением 6-нитро-1H-индол-3-карбонитрила (4,6 г, 82%).
B-8; 6-Амино-1H-индол-3-карбонитрил
Суспензию 6-нитро-1H-индол-3-карбонитрила (4,6 г, 24,6 ммоль) и 10% Pd-C (0,46 г) в EtOH (50 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение ночи. После фильтрования фильтрат концентрировали и остаток очищали колоночной хроматографией (петролейный эфир/EtOAc = 3/1) с получением 6-амино-1H-индол-3-карбонитрила (B-8) (1 г, 99%) в виде розового порошка. 1H ЯМР (DMSO-d6) δ 11,51 (с, 1H), 7,84 (д, J=2,4 Гц, 1H), 7,22 (д, J=8,4 Гц, 1H), 6,62 (с, 1H), 6,56 (д, J=8,4 Гц, 1H), 5,0 (с, 2H); ESI-MS 157,1 m/z (MH+).
Пример 3:

Диметил-(6-нитро-1H-индол-3-илметил)-амин
Раствор диметиламина (25 г, 0,17 моль) и формальдегида (14,4 мл, 0,15 моль) в уксусной кислоте (100 мл) перемешивали при 0ºC в течение 30 минут. К этому раствору добавляли 6-нитро-1H-индол (20 г, 0,12 моль). После перемешивания в течение 3 дней при комнатной температуре смесь выливали в 15% водный раствор NaOH (500 мл) при 0ºC. Осадок собирали фильтрованием и промывали водой с получением диметил-(6-нитро-1H-индол-3-илметил)-амина (23 г, 87%).
B-9-a; (6-Нитро-1H-индол-3-ил)-ацетонитрил
К смеси DMF (35 мл) и MeI (74,6 г, 0,53 моль) в воде (35 мл) и ТГФ (400 мл) добавляли диметил-(6-нитро-1H-индол-3-илметил)-амин (23 г, 0,105 моль). Реакционную смесь кипятили с обратным холодильником в течение 10 минут, затем добавляли цианид калия (54,6 г, 0,84 моль) и продолжали кипячение смеси с обратным холодильником в течение ночи. Смесь затем охлаждали до комнатной температуры и фильтровали. Фильтрат промывали насыщенным солевым раствором (300 мл ×3), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией с получением (6-нитро-1H-индол-3-ил)-ацетонитрила (B-9-a) (7,5 г, 36%).
B-9; (6-Амино-1H-индол-3-ил)-ацетонитрил
Смесь (6-нитро-1H-индол-3-ил)-ацетонитрила (B-9-a) (1,5 г, 74,5 мл) и 10% Pd-C (300 мг) в EtOH (50 мл) перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 5 часов. Pd-C удаляли фильтрованием и фильтрат упаривали с получением (6-амино-1H-индол-3-ил)-ацетонитрила (B-9) (1,1 г, 90%). 1H ЯМР (DMSO-d6) δ 10,4 (шир.с, 1H), 7,18 (д, J=8,4 Гц, 1H), 6,94 (с, 1H), 6,52 (с, 1H), 6,42 (дд, J=8,4, 1,8 Гц, 1H), 4,76(с, 2H), 3,88 (с, 2H); ESI-MS 172,1 m/z (MH+).
Пример 4:
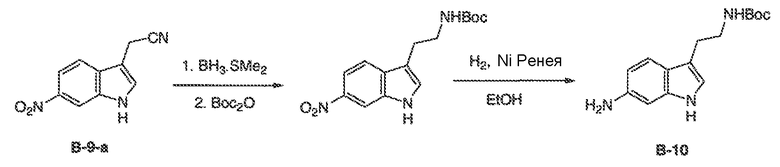
трет-бутиловый эфир [2-(6-нитро-1H-индол-3-ил)-этил]-карбаминовой кислоты
К раствору (6-нитро-1H-индол-3-ил)-ацетонитрила (B-9-a) (8,6 г, 42,8 ммоль) в безводном ТГФ (200 мл) добавляли 2 M раствор комплекса боран-диметилсульфид в ТГФ (214 мл. 0,43 моль) при 0ºC. Смесь нагревали при температуре кипения с обратным холодильником в течение ночи в атмосфере азота. Смесь затем охлаждали до комнатной температуры и добавляли раствор (Boc)2O (14 г, 64,2 ммоль) и Et3N (89,0 мл, 0,64 моль) в ТГФ. Реакционную смесь продолжали перемешивать в течение ночи и затем выливали в ледяную воду. Органический слой отделяли и водную фазу экстрагировали при помощи EtOAc (200×3 мл). Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением трет-бутилового эфира [2-(6-нитро-1H-индол-3-ил)-этил]-карбаминовой кислоты (5 г, 38%).
B-10; трет-бутиловый эфир [2-(6-амино-1H-индол-3-ил)-этил]-карбаминовой кислоты
Смесь трет-бутилового эфира [2-(6-нитро-1H-индол-3-ил)-этил]-карбаминовой кислоты (5 г, 16,4 ммоль) и Ni Ренея (1 г) в EtOH (100 мл) перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 5 часов. Ni Ренея отфильтровывали и фильтрат упаривали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией с получением трет-бутилового эфира [2-(6-амино-1H-индол-3-ил)-этил]-карбаминовой кислоты (B-10) (3 г, 67%). 1H ЯМР (DMSO-d6) δ 10,1 (шир.с, 1H), 7,11 (д, J=8,4 Гц, 1H), 6,77-6,73 (м, 2H), 6,46 (д, J=1,5 Гц, 1H), 6,32 (дд, J=8,4, 2,1 Гц, 1H), 4,62 (с, 2H), 3,14-3,08 (м, 2H), 2,67-2,62 (м, 2H), 1,35 (с, 9H); ESI-MS 275,8 m/z (MH+).
Пример 5:
Общая схема:
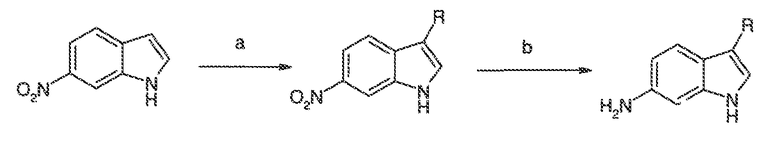
a) RX (X=Br,I), трифлат цинка, TBAI, DIEA, толуол; b) H2, Ni Ренея, EtOH или SnCl2.2H2O, EtOH.
Специальный пример:
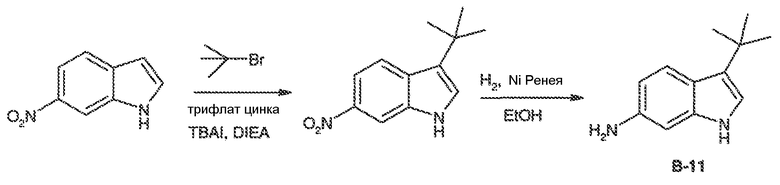
3-трет-бутил-6-нитро-1H-индол
К смеси 6-нитроиндола (1 г, 6,2 ммоль), трифлата цинка (2,06 г, 5,7 ммоль) и TBAI (1,7 г, 5,16 ммоль) в безводном толуоле (11 мл) добавляли DIEA (1,47 г, 11,4 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали в течение 10 минут при 120ºC, с последующим добавлением трет-бутилбромида (0,707 г, 5,16 ммоль). Полученную смесь перемешивали в течение 45 минут при 120ºC. Твердое вещество отфильтровывали и фильтрат концентрировали досуха и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc 20:1) с получением 3-трет-бутил-6-нитро-1H-индола в виде желтого твердого вещества (0,25 г, 19%). 1H ЯМР (CDCl3) δ 8,32 (д, J=2,1 Гц, 1H), 8,00 (дд, J=2,1, 14,4 Гц, 1H), 7,85 (д, J=8,7 Гц, 1H), 7,25 (с, 1H), 1,46 (с, 9H).
B-11; 3-трет-бутил-1H-индол-6-иламин
Суспензию 3-трет-бутил-6-нитро-1H-индола (3,0 г, 13,7 ммоль) и Ni Ренея (0,5 г) в этаноле перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 3 часов. Катализатор отфильтровывали и фильтрат концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc 4:1) с получением 3-трет-бутил-1H-индол-6-иламина (B-11) (2,0 г, 77,3%) в виде серого твердого вещества. 1H ЯМР (CDCl3): δ 7,58 (м, 2H), 6,73 (д, J=1,2 Гц, 1H), 6,66 (с, 1H), 6,57(дд, J=0,8, 8,6 Гц, 1H), 3,60 (шир.с, 2H), 1,42 (с, 9H).
Другие примеры:
B-12; 3-Этил-1H-индол-6-иламин
3-Этил-1H-индол-6-иламин (B-12) синтезировали в соответствии с общей схемой, представленной выше, исходя из 6-нитроиндола и этилбромида. Общий выход (42%). ВЭЖХ время удерживания 1,95 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 161,3 m/z (MH+).
B-13; 3-Изопропил-1H-индол-6-иламин
3-Изопропил-1H-индол-6-иламин (B-13) синтезировали в соответствии с общей схемой, представленной выше, исходя из 6-нитроиндола и изопропилиодида. Общий выход (17%). ВЭЖХ время удерживания 2,06 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 175,2 m/z (MH+).
B-14; 3-втор-Бутил-1H-индол-6-иламин
3-втор-Бутил-1H-индол-6-иламин (B-14) синтезировали в соответствии с общей схемой, представленной выше, исходя из 6-нитроиндола и 2-бромбутана. Общий выход (20%). ВЭЖХ время удерживания 2,32 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 189,5 m/z (MH+).
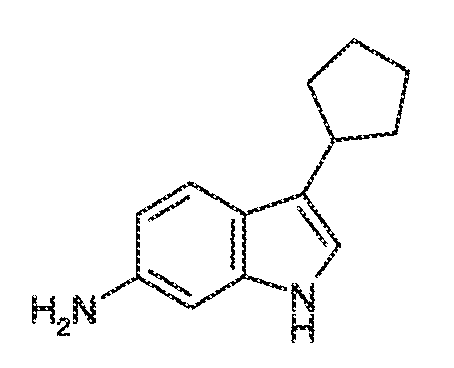
B-15; 3-Циклопентил-1H-индол-6-иламин
3-Циклопентил-1H-индол-6-иламин (B-15) синтезировали в соответствии с общей схемой, представленной выше, исходя из 6-нитроиндола и иод-циклопентана. Общий выход (16%). ВЭЖХ время удерживания 2,39 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 201,5 m/z (MH+).
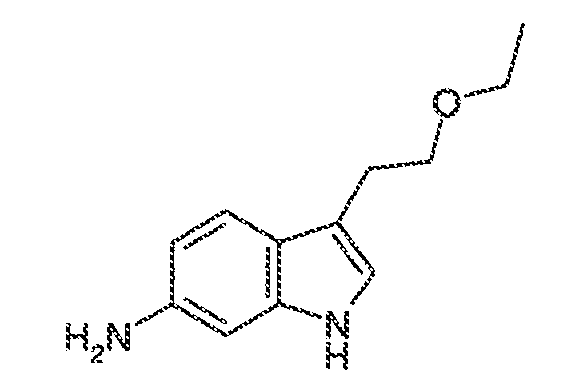
B-16; 3-(2-Этокси-этил)-1H-индол-6-иламин
3-(2-Этокси-этил)-1H-индол-6-иламин (B-16) синтезировали в соответствии с общей схемой, представленной выше, исходя из 6-нитроиндола и 1-бром-2-этокси-этана. Общий выход (15%). ВЭЖХ время удерживания 1,56 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 205,1 m/z (MH+).
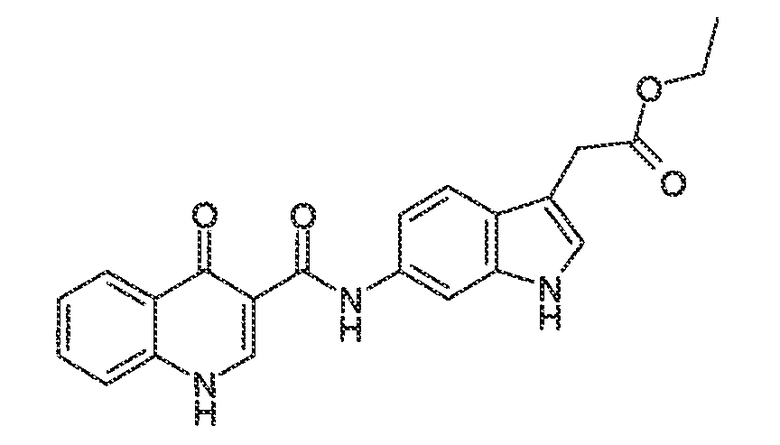
B-17; этиловый эфир (6-амино-1H-индол-3-ил)-уксусной кислоты
Этиловый эфир (6-Амино-1H-индол-3-ил)-уксусной кислоты (B-17) синтезировали в соответствии с общей схемой, представленной выше, исходя из 6-нитроиндола и этилового эфира иод-уксусной кислоты. Общий выход (24%). ВЭЖХ время удерживания 0,95 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 219,2 m/z (MH+).
4-Замещенный 6-аминоиндол
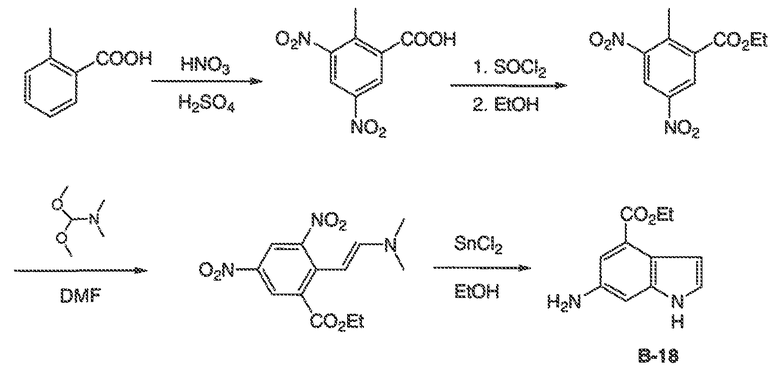
2-Метил-3,5-динитро-бензойная кислота
К смеси HNO3 (95%, 80 мл) и H2SO4 (98%, 80 мл) медленно добавляли 2-метилбензойную кислоту (50 г, 0,37 моль) при 0ºC. После добавления реакционную смесь перемешивали в течение 1,5 часов, поддерживая при этом температуру ниже 30ºC, выливали в ледяную воду и перемешивали в течение 15 минут. Образовавшийся осадок собирали фильтрованием и промывали водой с получением 2-метил-3,5-динитро-бензойной кислоты (70 г, 84%).
Этиловый эфир 2-метил-3,5-динитро-бензойной кислоты
Смесь 2-метил-3,5-динитро-бензойной кислоты (50 г, 0,22 моль) в SOCl2 (80 мл) нагревали при температуре кипения с обратным холодильником в течение 4 часов и затем концентрировали досуха. Добавляли CH2Cl2 (50 мл) и EtOH (80 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, выливали в ледяную воду и экстрагировали при помощи EtOAc (3×100 мл). Объединенные экстракты промывали насыщенным раствором Na2CO3 (80 мл), водой (2×100 мл) и насыщенным солевым раствором (100 мл), сушили над Na2SO4 и концентрировали досуха с получением этилового эфира 2-метил-3,5-динитро-бензойной кислоты (50 г, 88%).
Этиловый эфир 2-(2-диметиламино-винил)-3,5-динитро-бензойной кислоты
Смесь этилового эфира 2-метил-3,5-динитро-бензойной кислоты (35 г, 0,14 моль) и диметоксиметил-диметил-амина (32 г, 0,27 моль) в DMF (200 мл) нагревали при 100ºC в течение 5 часов. Смесь выливали в ледяную воду. Осадок собирали фильтрованием и промывали водой с получением этилового эфира 2-(2-диметиламино-винил)-3,5-динитро-бензойной кислоты (11,3 г, 48%).
B-18; этиловый эфир 6-амино-1H-индол-4-карбоновой кислоты
Смесь этилового эфира 2-(2-диметиламино-винил)-3,5-динитро-бензойной кислоты (11,3 г, 0,037 моль) и SnCl2 (83 г, 0,37 моль) в этаноле нагревали при температуре кипения с обратным холодильником в течение 4 часов. Смесь концентрировали досуха и остаток выливали в воду и подщелачивали насыщенным раствором Na2CO3 до pH 8. Осадок отфильтровывали и фильтрат экстрагировали этилацетатом (3×100 мл). Объединенные экстракты промывали водой (2×100 мл) и насыщенным солевым раствором (150 мл), сушили над Na2SO4 и концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле с получением этилового эфира 6-амино-1H-индол-4-карбоновой кислоты (B-18) (3 г, 40%). 1H ЯМР (DMSO-d6) δ 10,76 (шир.с, 1H), 7,11-7,14 (м, 2H), 6,81-6,82 (м, 1H), 6,67-6,68 (м, 1H), 4,94 (шир.с, 2H), 4,32-4,25 (кв., J=7,2 Гц, 2H), 1,35-1,31 (т, J=7,2, 3H), ESI-MS 205,0 m/z (MH+).
5-Замещенные 6-аминоиндолы
Пример 1:
Общая схема:
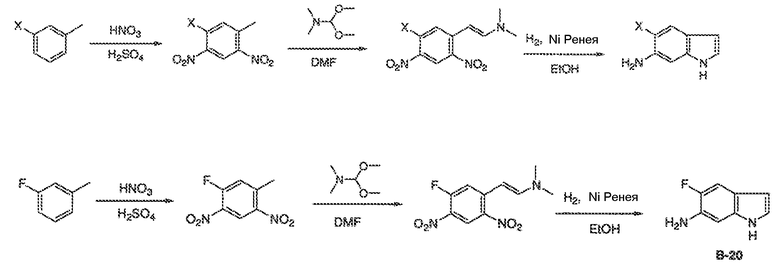
Специальный пример:
1-Фтор-5-метил-2,4-динитро-бензол
К перемешиваемому раствору HNO3 (60 мл) и H2SO4 (80 мл), охлажденному на ледяной бане, добавляли 1-фтор-3-метил-бензол (27,5 г, 25 ммоль) при такой скорости, чтобы температура не повышалась выше 35ºC. Смесь оставляли для перемешивания в течение 30 минут при комнатной температуре и выливали в ледяную воду (500 мл). Образовавшийся осадок (смесь желаемого продукта и 1-фтор-3-метил-2,4-динитро-бензола, приблизительно 7:3) собирали фильтрованием и очищали перекристаллизацией из 50 мл изопропилового эфира с получением 1-фтор-5-метил-2,4-динитро-бензола в виде белого твердого вещества (18 г, 36%).
[2-(5-Фтор-2,4-динитро-фенил)-винил]-диметил-амин
Смесь 1-фтор-5-метил-2,4-динитро-бензола (10 г, 50 ммоль), диметоксиметил-диметиламина (11,9 г, 100 ммоль) и DMF (50 мл) нагревали при 100ºC в течение 4 часов. Раствор охлаждали и выливали в воду. Красный осадок собирали фильтрованием, промывали водой в достаточном количестве и сушили с получением [2-(5-фтор-2,4-динитро-фенил)-винил]-диметил-амина (8 г, 63%).
B-20; 5-Фтор-1H-индол-6-иламин
Суспензию [2-(5-фтор-2,4-динитро-фенил)-винил]-диметил-амина (8 г, 31,4 ммоль) и Ni Ренея (8 г) в EtOH (80 мл) перемешивали в атмосфере H2 (40 ф/дюйм (2,812 кг/см2)) при комнатной температуре в течение 1 часа. После фильтрования фильтрат концентрировали и остаток очищали хроматографией (петролейный эфир/EtOAc = 5/1) с получением 5-фтор-1H-индол-6-иламина (B-20) в виде коричневого твердого вещества (1 г, 16%). 1H ЯМР (DMSO-d6) δ 10,56 (шир.с, 1H), 7,07 (д, J=12 Гц, 1H), 7,02 (м, 1H), 6,71 (д, J=8 Гц, 1H), 6,17 (с, 1H), 3,91 (шир.с, 2H); ESI-MS 150,1 m/z (MH+).
Другие примеры:
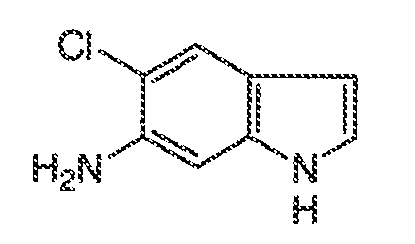
B-21; 5-Хлор-1H-индол-6-иламин
5-Хлор-1H-индол-6-иламин (B-21) синтезировали в соответствии с общей схемой, представленной выше, исходя из 1-хлор-3-метил-бензола. Общий выход (7%). 1H ЯМР (CDCl3) δ 7,85 (шир.с, 1H), 7,52 (с, 1H), 7,03 (с, 1H), 6,79 (с, 1H), 6,34 (с, 1H), 3,91 (шир.с, 2H); ESI-MS 166,0 m/z (MH+).
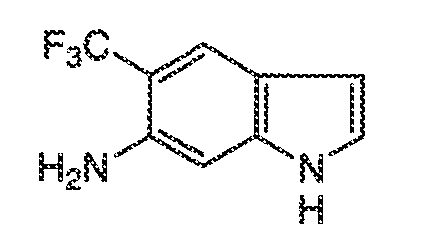
B-22; 5-Трифторметил-1H-индол-6-иламин
5-Трифторметил-1H-индол-6-иламин (B-22) синтезировали в соответствии с общей схемой, представленной выше, исходя из 1-метил-3-трифторметил-бензола. Общий выход (2%). 1H ЯМР (DMSO-d6) 10,79 (шир.с, 1H), 7,55 (с, 1H), 7,12 (с, 1H), 6,78 (с, 1H), 6,27(с, 1H), 4,92 (с, 2H); ESI-MS 200,8 m/z (MH+).
Пример 2:
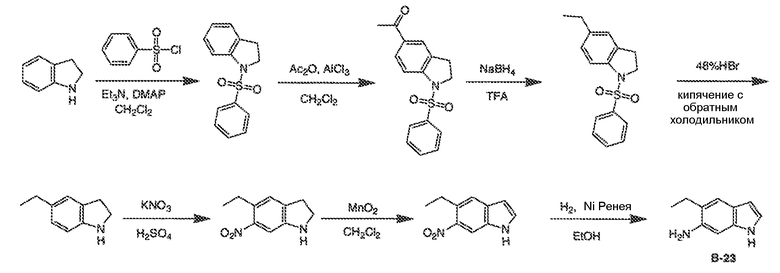
1-Бензолсульфонил-2,3-дигидро-1H-индол
К смеси DMAP (1,5 г), бензолсульфонилхлорида (24 г, 136 ммоль) и 2,3-дигидро-1H-индола (14,7 г, 124 ммоль) в CH2Cl2 (200 мл) добавляли по каплям Et3N (19 г, 186 ммоль) на бане лед-вода. После добавления смесь перемешивали при комнатной температуре в течение ночи, промывали водой, сушили над Na2SO4 и концентрировали досуха при пониженном давлении с получением 1-бензолсульфонил-2,3-дигидро-1H-индола (30,9 г, 96%).
1-(1-Бензолсульфонил-2,3-дигидро-1H-индол-5-ил)-этанон
К перемешиваемой суспензии AlCl3 (144 г, 1,08 моль) в CH2Cl2 (1070 мл) добавляли уксусный ангидрид (54 мл). Смесь перемешивали в течение 15 минут. Добавляли по каплям раствор 1-бензолсульфонил-2,3-дигидро-1H-индола (46,9 г, 0,18 моль) в CH2Cl2 (1070 мл). Смесь перемешивали в течение 5 часов и гасили путем медленного добавления колотого льда. Органический слой отделяли и водный слой экстрагировали при помощи CH2Cl2. Объединенные органические слои промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали в вакууме с получением 1-(1-бензолсульфонил-2,3-дигидро-1H-индол-5-ил)-этанона (42,6 г, 79%).
1-Бензолсульфонил-5-этил-2,3-дигидро-1H-индол
К перемешиваемому при помощи магнитной мешалки TFA (1600 мл) добавляли при 0ºC борогидрид натрия (64 г, 1,69 моль) в течение 1 часа. К этой смеси добавляли по каплям раствор 1-(1-бензолсульфонил-2,3-дигидро-1H-индол-5-ил)-этанона (40 г, 0,13 моль) в TFA (700 мл) в течение 1 часа. Смесь перемешивали в течение ночи при 25ºC, разбавляли при помощи H2O (1600 мл) и подщелачивали гидроксидом натрия в виде таблеток при 0ºC. Органический слой отделяли и водный слой экстрагировали при помощи CH2Cl2. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 1-бензолсульфонил-5-этил-2,3-дигидро-1H-индола (16,2 г, 43%).
5-Этил-2,3-дигидро-1H-индол
Смесь 1-бензолсульфонил-5-этил-2,3-дигидро-1H-индола (15 г, 0,05 моль) в HBr (48%, 162 мл) нагревали при температуре кипения с обратным холодильником в течение 6 часов. Смесь подщелачивали насыщенным раствором NaOH до pH 9 и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 5-этил-2,3-дигидро-1H-индола (2,5 г, 32%).
5-Этил-6-нитро-2,3-дигидро-1H-индол
К раствору 5-этил-2,3-дигидро-1H-индола (2,5 г, 17 ммоль) в H2SO4 (98%, 20 мл) медленно добавляли KNO3 (1,7 г, 17 ммоль) при 0ºC. После добавления смесь перемешивали при 0-10ºC в течение 10 минут, осторожно выливали на лед, подщелачивали при помощи раствора NaOH до pH 9 и экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле с получением 5-этил-6-нитро-2,3-дигидро-1H-индола (1,9 г, 58%).
5-Этил-6-нитро-1H-индол
К раствору 5-этил-6-нитро-2,3-дигидро-1H-индола (1,9 г, 9,9 ммоль) в CH2Cl2 (30 мл) добавляли MnO2 (4 г, 46 ммоль). Смесь перемешивали при комнатной температуре в течение 8 часов. Твердое вещество отфильтровывали и фильтрат концентрировали досуха с получением неочищенного 5-этил-6-нитро-1H-индола (1,9 г, количественный).
B-23; 5-Этил-1H-индол-6-иламин
Суспензию 5-этил-6-нитро-1H-индола (1,9 г, 10 ммоль) и Ni Ренея (1 г) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 2 часов. Катализатор отфильтровывали и фильтрат концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле с получением 5-этил-1H-индол-6-иламина (B-23) (760 мг, 48%). 1H ЯМР (CDCl3) δ 7,90 (шир.с, 1H), 7,41 (с, 1H), 7,00 (с, 1H), 6,78 (с, 2H), 6,39 (с, 1H), 3,39 (шир.с, 2H), 2,63 (кв., J=7,2 Гц, 2H), 1,29 (т, J=6,9 Гц, 3H); ESI-MS 161,1 m/z (MH+).
Пример 3:
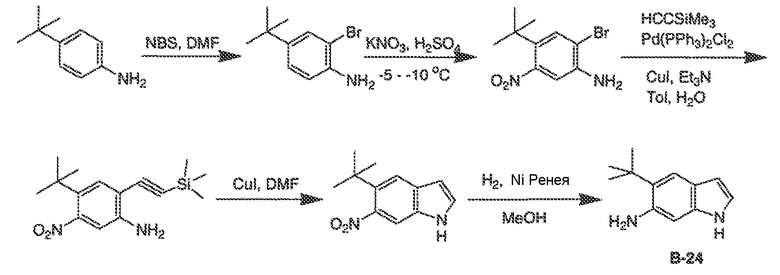
2-Бром-4-трет-бутил-фениламин
К раствору 4-трет-бутил-фениламина (447 г, 3 моль) в DMF (500 мл) добавляли по каплям NBS (531 г, 3 моль) в DMF (500 мл) при комнатной температуре. После завершения добавления реакционную смесь разбавляли водой и экстрагировали при помощи EtOAc. Органический слой промывали водой, насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный продукт непосредственно использовали на следующей стадии без дополнительной очистки.
2-Бром-4-трет-бутил-5-нитро-фениламин
2-Бром-4-трет-бутил-фениламин (162 г, 0,71 моль) добавляли по каплям к H2SO4 (410 мл) при комнатной температуре с получением прозрачного раствора. Этот прозрачный раствор затем охлаждали до темпеературы -5 до -10ºC. Добавляли по каплям раствор KNO3 (82,5 г, 0,82 моль) в H2SO4 (410 мл), поддерживая при этом температуру в пределах от -5 до -10ºC. После завершения добавления реакционную смесь выливали на лед/воду и экстрагировали при помощи EtOAc. Объединенные органические слои промывали 5% раствором Na2CO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (EtOAc/петролейный эфир 1/10) с получением 2-бром-4-трет-бутил-5-нитро-фениламина в виде желтого твердого вещества (152 г, 78%).
4-трет-бутил-5-нитро-2-триметилсиланилэтинил-фениламин
К смеси 2-бром-4-трет-бутил-5-нитро-фениламина (27,3 г, 100 ммоль) в толуоле (200 мл) и воде (100 мл) добавляли Et3N (27,9 мл, 200 ммоль), Pd(PPh3)2Cl2 (2,11 г, 3 ммоль), CuI (950 мг, 0,5 ммоль) и триметилсилилацетилен (21,2 мл, 150 ммоль) в атмосфере азота. Реакционную смесь нагревали при 70ºC в герметично закрытой колбе высокого давления в течение 2,5 часов, охлаждали до комнатной температуры и фильтровали через короткую пробку из Целита. Фильтровальную лепешку промывали при помощи EtOAc. Объединенный фильтрат промывали 5% раствором NH4OH и водой, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали колоночной хроматографией (0-10% EtOAc/петролейный эфир) с получением 4-трет-бутил-5-нитро-2-триметилсиланилэтинил-фениламина в виде коричневой вязкой жидкости (25 г, 81%).
5-трет-бутил-6-нитро-1H-индол
К раствору 4-трет-бутил-5-нитро-2-триметилсиланилэтинил-фениламина (25 г, 86 ммоль) в DMF (100 мл) добавляли CuI (8,2 г, 43 ммоль) в атмосфере азота. Смесь нагревали при 135ºC в герметично закрытой колбе высокого давления в течение ночи, охлаждали до комнатной температуры и фильтровали через короткую пробку из Целита. Фильтровальную лепешку промывали при помощи EtOAc. Объединенный фильтрат промывали водой, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали колоночной хроматографией (10-20% EtOAc/Гексан) с получением 5-трет-бутил-6-нитро-1H-индола в виде желтого твердого вещества (12,9 г, 69%).
B-24; 5-трет-бутил-1H-индол-6-иламин
Ni Ренея (3 г) добавляли к 5-трет-бутил-6-нитро-1H-индолу (14,7 г, 67 ммоль) в метаноле (100 мл). Смесь перемешивали в атмосфере водорода (1 атм) при 30ºC в течение 3 часов. Катализатор отфильтровывали. Фильтрат сушили над Na2SO4 и концентрировали. Неочищенное темно-коричневое вязкое масло очищали колоночной хроматографией (10-20% EtOAc/петролейный эфир) с получением 5-трет-бутил-1H-индол-6-иламина (B-24) в виде серого твердого вещества (11 г, 87%). 1H ЯМР (300 МГц, DMSO-d6) δ 10,3 (шир.с, 1H), 7,2 (с, 1H), 6,9 (м, 1H), 6,6 (с, 1H), 6,1 (м, 1H), 4,4 (шир.с, 2H), 1,3 (с, 9H).
Пример 4:
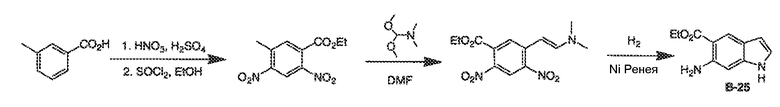
5-Метил-2,4-динитро-бензойная кислота
К смеси HNO3 (95%, 80 мл) и H2SO4 (98%, 80 мл) медленно добавляли 3-метилбензойную кислоту (50 г, 0,37 моль) при 0ºC. После добавления смесь перемешивали в течение 1,5 часов, поддерживая при этом температуру ниже 30ºC. Смесь выливали в ледяную воду и перемешивали в течение 15 минут. Осадок собирали фильтрованием и промывали водой с получением смеси 3-метил-2,6-динитро-бензойной кислоты и 5-метил-2,4-динитро-бензойной кислоты (70 г, 84%). К раствору этой смеси в EtOH (150 мл) добавляли по каплям SOCl2 (53,5 г, 0,45 моль). Смесь нагревали при температуре кипения с обратным холодильником в течение 2 часов и концентрировали досуха при пониженном давлении. Остаток растворяли в EtOAc (100 мл) и экстрагировали 10% раствором Na2CO3 (120 мл). Было обнаружено, что органический слой содержал этиловый эфир 5-метил-2,4-динитро-бензойной кислоты, тогда как водный слой содержал 3-метил-2,6-динитро-бензойную кислоту. Органический слой промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4 и концентрировали досуха с получением этилового эфира 5-метил-2,4-динитро-бензойной кислоты (20 г, 20%).
Этиловый эфир 5-(2-диметиламино-винил)-2,4-динитро-бензойной кислоты
Смесь этилового эфира 5-метил-2,4-динитро-бензойной кислоты (39 г, 0,15 моль) и диметоксиметил-диметиламина (32 г, 0,27 моль) в DMF (200 мл) нагревали при 100ºC в течение 5 часов. Смесь выливали в ледяную воду. Осадок собирали фильтрованием и промывали водой с получением этилового эфира 5-(2-диметиламино-винил)-2,4-динитро-бензойной кислоты (15 г, 28%).
B-25; Этиловый эфир 6-амино-1H-индол-5-карбоновой кислоты
Смесь этилового эфира 5-(2-диметиламино-винил)-2,4-динитро-бензойной кислоты (15 г, 0,05 моль) и Ni Ренея (5 г) в EtOH (500 мл) перемешивали в атмосфере H2 (50 ф/дюйм2 (3,515 кг/см2)) при комнатной температуре в течение 2 часов. Катализатор отфильтровывали и фильтрат концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле с получением этилового эфира 6-амино-1H-индол-5-карбоновой кислоты (B-25) (3 г, 30%). 1H ЯМР (DMSO-d6) δ 10,68 (с, 1H), 7,99 (с, 1H), 7,01-7,06 (м, 1H), 6,62 (с, 1H), 6,27-6,28 (м, 1H), 6,16 (с, 2H), 4,22 (кв., J=7,2 Гц, 2H), 1,32-1,27 (т, J=7,2 Гц, 3H).
Пример 5:
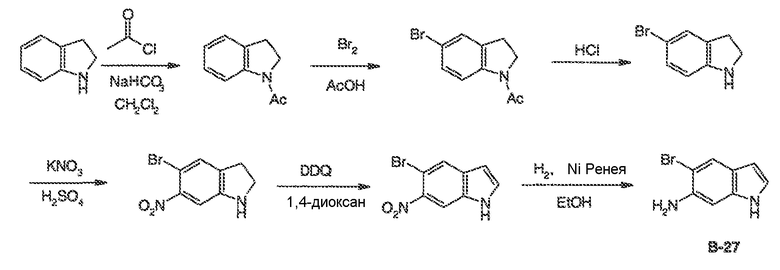
1-(2,3-Дигидро-индол-1-ил)-этанон
К суспензии NaHCO3 (504 г, 6,0 моль) и 2,3-дигидро-1H-индола (60 г, 0,5 моль) в CH2Cl2 (600 мл), охлажденной на бане лед-вода, добавляли по каплям ацетилхлорид (78,5 г, 1,0 моль). Смесь перемешивали при комнатной температуре в течение 2 часов. Твердое вещество отфильтровывали и фильтрат концентрировали с получением 1-(2,3-дигидро-индол-1-ил)-этанона (82 г, 100%).
1-(5-Бром-2,3-дигидро-индол-1-ил)-этанон
К раствору 1-(2,3-дигидро-индол-1-ил)-этанона (58,0 г, 0,36 моль) в уксусной кислоте (3000 мл) добавляли Br2 (87,0 г, 0,54 моль) при 10ºC. Смесь перемешивали при комнатной температуре в течение 4 часов. Осадок собирали фильтрованием с получением неочищенного 1-(5-бром-2,3-дигидро-индол-1-ил)-этанона (100 г, 96%), который использовали непосредственно на следующей стадии.
5-Бром-2,3-дигидро-1H-индол
Смесь неочищенного 1-(5-бром-2,3-дигидро-индол-1-ил)-этанона (100 г, 0,34 моль) в HCl (20%, 1200 мл) нагревали при температуре кипения с обратным холодильником в течение 6 часов. Смесь подщелачивали при помощи Na2CO3 до pH 8,5-10 и затем экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением 5-бром-2,3-дигидро-1H-индола (37 г, 55%).
5-Бром-6-нитро-2,3-дигидро-1H-индол
К раствору 5-бром-2,3-дигидро-1H-индола (45 г, 0,227 моль) в H2SO4 (98%, 200 мл) медленно добавляли KNO3 (23,5 г, 0,23 моль) при 0ºC. После добавления смесь перемешивали при 0-10ºC в течение 4 часов, осторожно выливали на лед, подщелачивали при помощи Na2CO3 до pH 8 и экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле с получением 5-бром-6-нитро-2,3-дигидро-1H-индола (42 г, 76%).
5-Бром-6-нитро-1H-индол
К раствору 5-бром-6-нитро-2,3-дигидро-1H-индола (20 г, 82,3 ммоль) в 1,4-диоксане (400 мл) добавляли DDQ (30 г, 0,13 моль). Смесь перемешивали при 80ºC в течение 2 часов. Твердое вещество отфильтровывали и фильтрат концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле с получением 5-бром-6-нитро-1H-индола (7,5 г, 38%).
B-27; 5-Бром-1H-индол-6-иламин
Смесь 5-бром-6-нитро-1H-индола (7,5 г, 31,1 ммоль) и Ni Ренея (1 г) в этаноле перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 2 часов. Катализатор отфильтровывали и фильтрат концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле с получением 5-бром-1H-индол-6-иламина (B-27) (2 г, 30%). 1H ЯМР (DMSO-d6) δ 10,6 (с, 1H), 7,49 (с, 1H), 6,79-7,02 (м, 1H), 6,79 (с, 1H), 6,14-6,16 (м, 1H), 4,81 (с, 2H).
7-Замещенный 6-аминоиндол
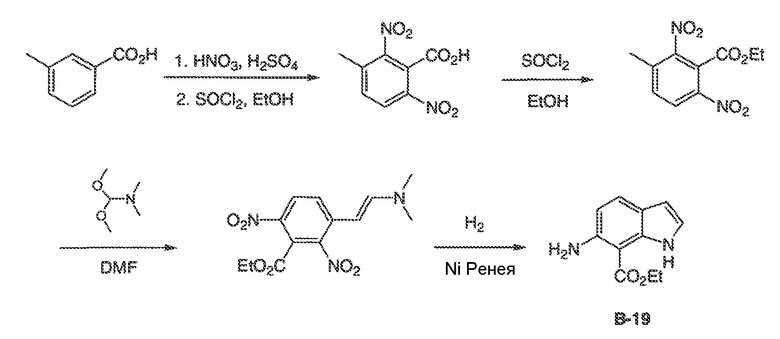
3-Метил-2,6-динитро-бензойная кислота
К смеси HNO3 (95%, 80 мл) и H2SO4 (98%, 80 мл) медленно добавляли 3-метилбензойную кислоту (50 г, 0,37 моль) при 0ºC. После добавления смесь перемешивали в течение 1,5 часов, поддерживая при этом температуру ниже 30ºC. Смесь выливали в ледяную воду и перемешивали в течение 15 минут. Осадок собирали фильтрованием и промывали водой с получением смеси 3-метил-2,6-динитро-бензойной кислоты и 5-метил-2,4-динитро-бензойной кислоты (70 г, 84%). К раствору этой смеси в EtOH (150 мл) добавляли по каплям SOCl2 (53,5 г, 0,45 моль). Смесь нагревали до температуры кипения с обратным холодильником в течение 2 часов и концентрировали досуха при пониженном давлении. Остаток растворяли в EtOAc (100 мл) и экстрагировали 10% раствором Na2CO3 (120 мл). Было обнаружено, что органический слой содержал этиловый эфир 5-метил-2,4-динитро-бензойной кислоты. Водный слой подкисляли при помощи HCl до pH 2~3 и образовавшийся осадок собирали фильтрованием, промывали водой и сушили на воздухе с получением 3-метил-2,6-динитро-бензойной кислоты (39 г, 47%).
Этиловый эфир 3-метил-2,6-динитро-бензойной кислоты
Смесь 3-метил-2,6-динитро-бензойной кислоты (39 г, 0,15 моль) и SOCl2 (80 мл) нагревали при температуре кипения с обратным холодильником в течение 4 часов. Избыток SOCl2 удаляли при пониженном давлении и остаток добавляли по каплям к раствору EtOH (100 мл) и Et3N (50 мл). Смесь перемешивали при 20ºC в течение 1 часа и концентрировали досуха. Остаток растворяли в EtOAc (100 мл), промывали раствором Na2CO3 (10%, 40 мл ×2), водой (50 мл ×2) и насыщенным солевым раствором (50 мл), сушили над Na2SO4 и концентрировали с получением этилового эфира 3-метил-2,6-динитро-бензойной кислоты (20 г, 53%).
Этиловый эфир 3-(2-диметиламино-винил)-2,6-динитро-бензойной кислоты
Смесь этилового эфира 3-метил-2,6-динитро-бензойной кислоты (35 г, 0,14 моль) и диметоксиметил-диметиламина (32 г, 0,27 моль) в DMF (200 мл) нагревали при 100ºC в течение 5 часов. Смесь выливали в ледяную воду и осадок собирали фильтрованием и промывали водой с получением этилового эфира 3-(2-диметиламино-винил)-2,6-динитро-бензойной кислоты (25 г, 58%).
B-19; этиловый эфир 6-амино-1H-индол-7-карбоновой кислоты
Смесь этилового эфира 3-(2-диметиламино-винил)-2,6-динитро-бензойной кислоты (30 г, 0,097 моль) и Ni Ренея (10 г) в EtOH (1000 мл) перемешивали в атмосфере H2 (50 ф/дюйм2 (3,515 кг/см2)) в течение 2 часов. Катализатор отфильтровывали и фильтрат концентрировали досуха. Остаток очищали колоночной хроматографией на силикагеле с получением этилового эфира 6-амино-1H-индол-7-карбоновой кислоты (B-19) в виде не совсем белого твердого вещества (3,2 г, 16%). 1H ЯМР (DMSO-d6) δ 10,38 (с, 1H), 7,44-7,41 (д, J=8,7 Гц, 1H), 6,98 (т, 1H), 6,65 (с, 2H), 6,50-6,46 (м, 1H), 6,27-6,26 (м, 1H), 4,43-4,36 (кв., J=7,2 Гц, 2H), 1,35 (т, J=7,2 Гц, 3H).
Фенолы
Пример 1:
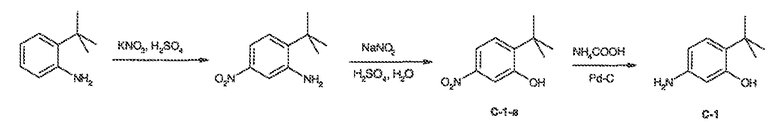
2-трет-бутил-5-нитроанилин
К охлажденному раствору серной кислоты (90%, 50 мл) добавляли по каплям 2-трет-бутил-фениламин (4,5 г, 30 ммоль) при 0ºC. Добавляли нитрат калия (4,5 г, 45 ммоль) по порциям при 0ºC. Реакционную смесь перемешивали при 0-5ºC в течение 5 минут, выливали в ледяную воду и затем экстрагировали при помощи EtOAc три раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. После удаления растворителя остаток очищали перекристаллизацией с использованием 70% EtOH-H2O с получением 2-трет-бутил-5-нитроанилина (3,7 г, 64%). 1H ЯМР (400 МГц, CDCl3) δ 7,56 (дд, J=8,7, 2,4 Гц, 1H), 7,48 (д, J=2,4 Гц, 1H), 7,36 (д, J=8,7 Гц, 1H), 4,17 (с, 2H), 1,46 (с, 9H); ВЭЖХ время удерживания 3,27 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 195,3 m/z (MH+).
C-1-a; 2-трет-бутил-5-нитрофенол
К смеси 2-трет-бутил-5-нитроанилина (1,94 г, 10 ммоль) в 40 мл 15% раствора H2SO4 добавляли по каплям раствор NaNO2 (763 мг, 11,0 ммоль) в воде (3 мл) при 0ºC. Полученную смесь перемешивали при 0-5ºC в течение 5 минут. Избыток NaNO2 нейтрализовали при помощи мочевины, затем добавляли 5 мл H2SO4-H2O (об/об 1:2) и смесь кипятили с обратным холодильником в течение 5 минут. Добавляли три дополнительные 5 мл аликвоты H2SO4-H2O (об/об 1:2) при нагревании при температуре кипения с обратным холодильником. Реакционную смесь охлаждали до комнатной температуры и экстрагировали при помощи EtOAc два раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После удаления растворителя остаток очищали колоночной хроматографией (0-10% EtOAc-гексан) с получением 2-трет-бутил-5-нитрофенола (C-1-a) (1,2 г, 62%). 1H ЯМР (400 МГц, CDCl3) δ 7,76 (дд, J=8,6, 2,2 Гц, 1H), 7,58 (д, J=2,1 Гц, 1H), 7,43 (д, J=8,6 Гц, 1H), 5,41 (с, 1H), 1,45 (с, 9H); ВЭЖХ время удерживания 3,46 минут, 10-99% CH3CN, 5 мин цикл.
C-1; 2-трет-бутил-5-аминофенол
К нагреваемому при температуре кипения с обратным холодильником раствору 2-трет-бутил-5-нитрофенола (C-1-a) (196 мг, 1,0 ммоль) в EtOH (10 мл) добавляли формиат аммония (200 мг, 3,1 ммоль) с последующим добавлением 140 мг 10% Pd-C. Реакционную смесь кипятили с обратным холодильником еще в течение 30 минут, охлаждали до комнатной температуры и фильтровали через слой Целита. Фильтрат концентрировали досуха и очищали колоночной хроматографией (20-30% EtOAc-гексан) с получением 2-трет-бутил-5-аминофенола (C-1) (144 мг, 87%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,76 (с, 1H), 6,74 (д, J=8,3 Гц, 1H), 6,04 (д, J=2,3 Гц, 1H), 5,93 (дд, J=8,2, 2,3 Гц, 1H), 4,67 (с, 2H), 1,26 (с, 9H); ВЭЖХ время удерживания 2,26 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 166,1 m/z (MH+).
Пример 2:
Общая схема:
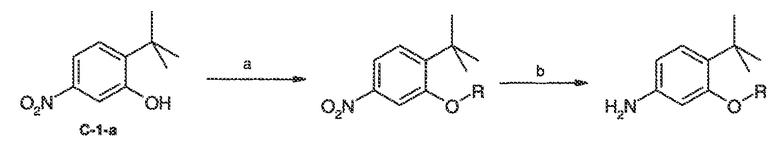
a) RX (X = Br, I), K2CO3 или Cs2CO3, DMF; b) HCO2NH4 или HCO2K, Pd-C, EtOH
Специальный пример:

1-трет-бутил-2-метокси-4-нитробензол
К смеси 2-трет-бутил-5-нитрофенола (C-1-a) (100 мг, 0,52 ммоль) и K2CO3 (86 мг, 0,62 ммоль) в DMF (2 мл) добавляли CH3I (40 мкл, 0,62 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, разбавляли водой и экстрагировали при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После фильтрования фильтрат упаривали досуха с получением 1-трет-бутил-2-метокси-4-нитробензола (82 мг, 76%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,77 (т, J=4,3 Гц, 1H), 7,70 (д, J=2,3 Гц, 1H), 7,40 (д, J=8,6 Гц, 1H), 3,94 (с, 3H), 1,39 (с, 9H).
C-2; 4-трет-бутил-3-метоксианилин
К нагреваемому при температуре кипения с обратным холодильником раствору 1-трет-бутил-2-метокси-4-нитробензола (82 мг, 0,4 ммоль) в EtOH (2 мл) добавляли формиат калия (300 мг, 3,6 ммоль) в воде (1 мл), с последующим добавлением 10% Pd-C (15 мг). Реакционную смесь кипятили с обратным холодильником еще в течение 60 минут, охлаждали до комнатной температуры и фильтровали через Целит. Фильтрат концентрировали досуха с получением 4-трет-бутил-3-метоксианилина (C-2) (52 мг, 72%), который использовали без дополнительной очистки. ВЭЖХ время удерживания 2,29 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 180,0 m/z (MH+).
Другие примеры:
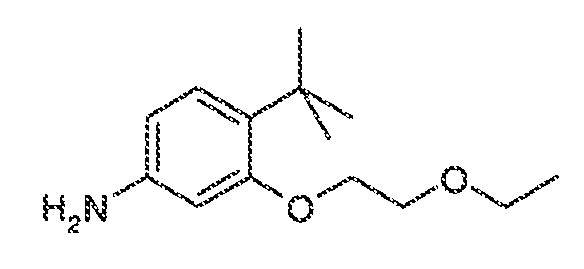
C-3; 3-(2-Этоксиэтокси)-4-трет-бутилбензоламин
3-(2-Этоксиэтокси)-4-трет-бутилбензоламин (C-3) синтезировали в соответствии с общей схемой, представленной выше, исходя из 2-трет-бутил-5-нитрофенола (C-1-a) и 1-бром-2-этоксиэтана. 1H ЯМР (400 МГц, CDCl3) δ 6,97 (д, J=7,9 Гц, 1H), 6,17 (с, 1H), 6,14 (д, J=2,3 Гц, 1H), 4,00 (т, J=5,2 Гц, 2H), 3,76 (т, J=5,2 Гц, 2H), 3,53 (кв., J=7,0 Гц, 2H), 1,27 (с, 9H), 1,16 (т, J=7,0 Гц, 3H); ВЭЖХ время удерживания 2,55 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 238,3 m/z (MH+).
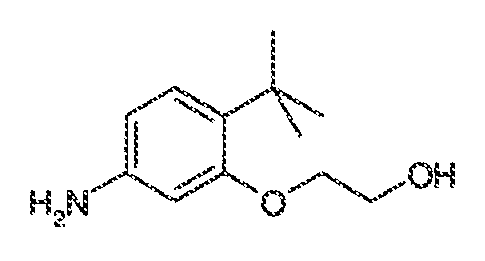
C-4; 2-(2-трет-бутил-5-аминофенокси)этанол
2-(2-трет-бутил-5-аминофенокси)этанол (C-4) синтезировали в соответствии с общей схемой, представленной выше, исходя из 2-трет-бутил-5-нитрофенола (C-1-a) и 2-бромэтанола. ВЭЖХ время удерживания 2,08 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 210,3 m/z (MH+).
Пример 3:
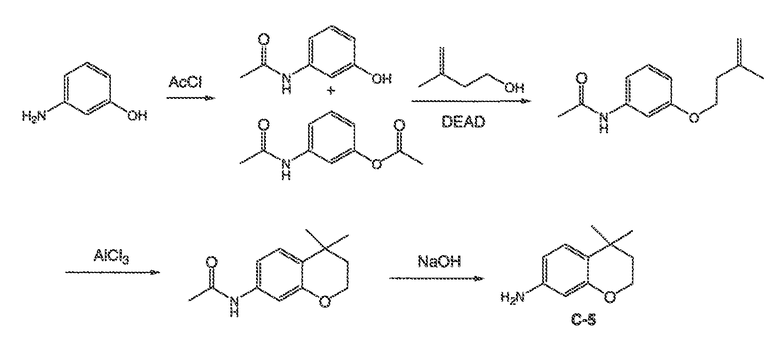
N-(3-гидрокси-фенил)-ацетамид и 3-формиламино-фениловый эфир уксусной кислоты
К тщательно перемешиваемой суспензии 3-амино-фенола (50 г, 0,46 моль) и NaHCO3 (193,2 г, 2,3 моль) в хлороформе (1 л) добавляли по каплям хлорацетилхлорид (46,9 г, 0,6 моль) в течение 30 минут при 0ºC. После завершения добавления реакционную смесь кипятили с обратным холодильником в течение ночи и затем охлаждали до комнатной температуры. Избыток NaHCO3 удаляли фильтрованием. Фильтрат выливали в воду и экстрагировали при помощи EtOAc (300×3 мл). Объединенные органические слои промывали насыщенным солевым раствором (500 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением смеси N-(3-гидрокси-фенил)-ацетамида и 3-формиламино-фенилового эфира уксусной кислоты (35 г, 4:1 по данным анализа ЯМР). Смесь использовали непосредственно на следующей стадии.
N-[3-(3-Метил-бут-3-енилокси)-фенил]-ацетамид
Суспензию смеси N-(3-гидрокси-фенил)-ацетамида и 3-формиламино-фенилового эфира уксусной кислоты (18,12 г, 0,12 моль), 3-метил-бут-3-ен-1-ола (8,6 г, 0,1 моль), DEAD (87 г, 0,2 моль) и Ph3P (31,44 г, 0,12 моль) в бензоле (250 мл) нагревали при температуре кипения с обратным холодильником в течение ночи и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду и органический слой отделяли. Водную фазу экстрагировали при помощи EtOAc (300×3 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией с получением N-[3-(3-метил-бут-3-енилокси)-фенил]-ацетамида (11 г, 52%).
N-(4,4-Диметил-хроман-7-ил)-ацетамид
Смесь N-[3-(3-метил-бут-3-енилокси)-фенил]-ацетамида (2,5 г, 11,4 ммоль) и AlCl3 (4,52 г, 34,3 ммоль) в фтор-бензоле (50 мл) нагревали при температуре кипения с обратным холодильником в течение ночи. После охлаждения реакционную смесь выливали в воду. Органический слой отделяли и водную фазу экстрагировали при помощи EtOAc (40×3 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией с получением N-(4,4-диметил-хроман-7-ил)-ацетамида (1,35 г, 54%).
C-5; 3,4-Дигидро-4,4-диметил-2H-хромен-7-амин
Смесь N-(4,4-диметил-хроман-7-ил)-ацетамида (1,35 г, 6,2 ммоль) в 20% растворем HCl (30 мл) нагревали при температуре кипения с обратным холодильником в течение 3 часов и затем охлаждали до комнатной температуры. Реакционную смесь подщелачивали 10% водным раствором NaOH до pH 8 и экстрагировали при помощи EtOAc (30×3 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением 3,4-дигидро-4,4-диметил-2H-хромен-7-амина (C-5) (1 г, 92%). 1H ЯМР (DMSO-d6) δ 6,87 (д, J=8,4 Гц, 1H), 6,07 (дд, J=8,4, 2,4 Гц, 1H), 5,87 (д, J=2,4 Гц, 1H), 4,75 (с, 2H), 3,99 (т, J=5,4 Гц, 2H), 1,64 (т, J=5,1 Гц, 2H), 1,15 (с, 6H); ESI-MS 178,1 m/z (MH+).
Пример 4:
Общая схема:
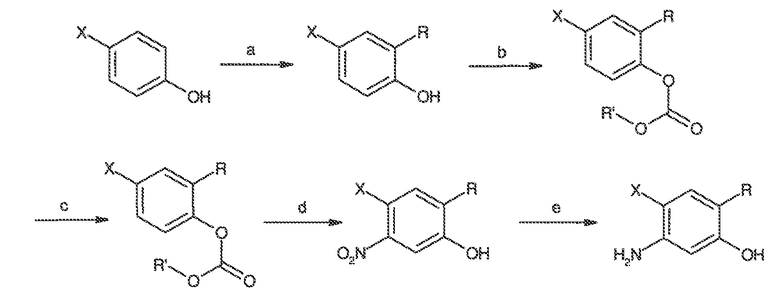
X = F, Cl; a) ROH, H2SO4 или MeSO3H, CH2Cl2; b) R'CO2Cl, Et3N, 1,4-диоксан или CHCl3; c) HNO3, H2SO4 или KNO3, H2SO4 или HNO3, AcOH; d) пиперидин, CH2Cl2; e) HCO2NH4, Pd-C, EtOH или SnCl2·2H2O, EtOH или H2, Pd-C, MeOH.
Специальный пример

2-трет-бутил-4-фторфенол
4-Фторфенол (5 г, 45 ммоль) и трет-бутанол (5,9 мл, 63 ммоль) растворяли в CH2Cl2 (80 мл) и обрабатывали концентрированной серной кислотой (98%, 3 мл). Смесь перемешивали при комнатной температуре в течение ночи. Органический слой промывали водой, нейтрализовали при помощи NaHCO3, сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией (5-15% EtOAc-гексан)с получением 2-трет-бутил-4-фторфенола (3,12 г, 42%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,32 (с, 1H), 6,89 (дд, J=11,1, 3,1 Гц, 1H), 6,84-6,79 (м, 1H), 6,74 (дд, J=8,7, 5,3 Гц, 1H), 1,33 (с, 9H).
2-трет-бутил-4-фторфенилметилкарбонат
К раствору 2-трет-бутил-4-фторфенола (2,63 г, 15,7 ммоль) и NEt3 (3,13 мл, 22,5 ммоль) в диоксане (45 мл) добавляли метилхлорформиат (1,27 мл, 16,5 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Осадок удаляли фильтрованием. Фильтрат затем разбавляли водой и экстрагировали простым эфиром. Эфирный экстракт промывали водой и сушили над MgSO4. После удаления растворителя остаток очищали колоночной хроматографией с получением 2-трет-бутил-4-фторфенилметилкарбоната (2,08 г, 59%). 1H ЯМР (400 МГц, DMSO-d6) δ 7,24 (дд, J=8,8, 5,4 Гц, 1H), 7,17-7,10 (м, 2H), 3,86 (с, 3H), 1,29 (с, 9H).
2-трет-бутил-4-фтор-5-нитрофенилметилкарбонат (C-7-a) и 2-трет-бутил-4-фтор-6-нитрофенилметилкарбонат (C-6-a)
К раствору 2-трет-бутил-4-фторфенилметилкарбоната (1,81 г, 8 ммоль) в H2SO4 (98%, 1 мл) добавляли медленно охлажденную смесь H2SO4 (1 мл) и HNO3 (1 мл) при 0ºC. Смесь перемешивали в течение 2 часов, нагревая при этом до комнатной температуры, выливали на лед и экстрагировали диэтиловым эфиром. Эфирный экстракт промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией (0-10% EtOAc-гексан) с получением 2-трет-бутил-4-фтор-5-нитрофенилметилкарбоната (C-7-a) (1,2 г, 55%) и 2-трет-бутил-4-фтор-6-нитрофенилметилкарбоната (C-6-a) (270 мг, 12%). 2-трет-бутил-4-фтор-5-нитрофенилметилкарбонат (C-7-a): 1H ЯМР (400 МГц, DMSO-d6) δ 8,04 (дд, J=7,6, 3,1 Гц, 1H), 7,69 (дд, J=10,1, 3,1 Гц, 1H), 3,91 (с, 3H), 1,35 (с, 9H). 2-трет-бутил-4-фтор-6-нитрофенилметилкарбонат (C-6-a): 1H ЯМР (400 МГц, DMSO-d6) δ 8,04 (дд, J=7,6, 3,1 Гц, 1H), 7,69 (дд, J=10,1, 3,1 Гц, 1H)3,91 (с, 3H), 1,35 (с, 9H).
2-трет-бутил-4-фтор-5-нитрофенол
К раствору 2-трет-бутил-4-фтор-5-нитрофенилметилкарбоната (C-7-a) (1,08 г, 4 ммоль) в CH2Cl2 (40 мл) добавляли пиперидин (3,94 мл, 10 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и экстрагировали 1н NaOH (3×). Водный слой подкисляли 1н раствором HCl и экстрагировали диэтиловым эфиром. Эфирный экстракт промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали с получением 2-трет-бутил-4-фтор-5-нитрофенола (530 мг, 62%). 1H ЯМР (400 МГц, DMSO-d6) δ 10,40 (с, 1H), 7,49 (д, J=6,8 Гц, 1H), 7,25 (д, J=13,7 Гц, 1H), 1,36 (с, 9H).
C-7; 2-трет-бутил-5-амино-4-фторфенол
К нагреваемому при температуре кипения с обратным холодильником раствору 2-трет-бутил-4-фтор-5-нитрофенола (400 мг, 1,88 ммоль) и формиата аммония (400 мг, 6,1 ммоль) в EtOH (20 мл) добавляли 5% Pd-C (260 мг). Смесь кипятили с обратным холодильником еще в течение 1 часа, охлаждали и фильтровали через Целит. Растворитель удаляли выпариванием с получением 2-трет-бутил-5-амино-4-фторфенола (C-7) (550 мг, 83%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,83 (шир.с, 1H), 6,66 (д, J=13,7 Гц, 1H), 6,22 (д, J=8,5 Гц, 1H), 4,74 (шир.с, 2H), 1,26 (с, 9H); ВЭЖХ время удерживания 2,58 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 184,0 m/z (MH+).
Другие примеры:
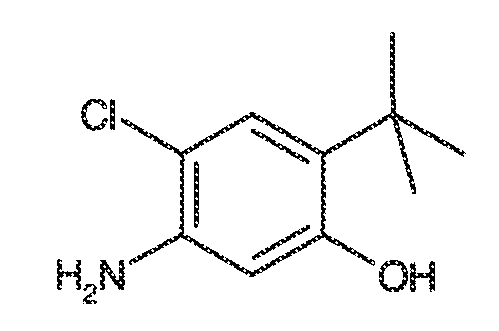
C-10; 2-трет-бутил-5-амино-4-хлорфенол
2-трет-бутил-5-амино-4-хлорфенол (C-10) синтезировали в соответствии с общей схемой, представленной выше, исходя из 4-хлорфенола и трет-бутанола. Общий выход (6%). ВЭЖХ время удерживания 3,07 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 200,2 m/z (MH+).
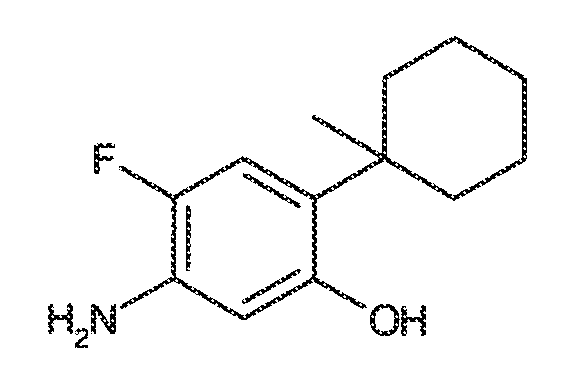
C-13; 5-Амино-4-фтор-2-(1-метилциклогексил)фенол
5-Амино-4-фтор-2-(1-метилциклогексил)фенол (C-13) синтезировали в соответствии с общей схемой, представленной выше, исходя из 4-фторфенола и 1-метилциклогексанола. Общий выход (3%). ВЭЖХ время удерживания 3,00 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 224,2 m/z (MH+).
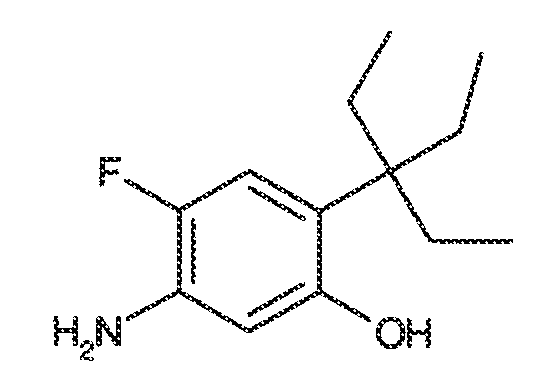
C-19; 5-Амино-2-(3-этилпентан-3-ил)-4-фтор-фенол
5-Амино-2-(3-этилпентан-3-ил)-4-фтор-фенол (C-19) синтезировали в соответствии с общей схемой, представленной выше, исходя из 4-фторфенола и 3-этил-3-пентанола. Общий выход (1%).
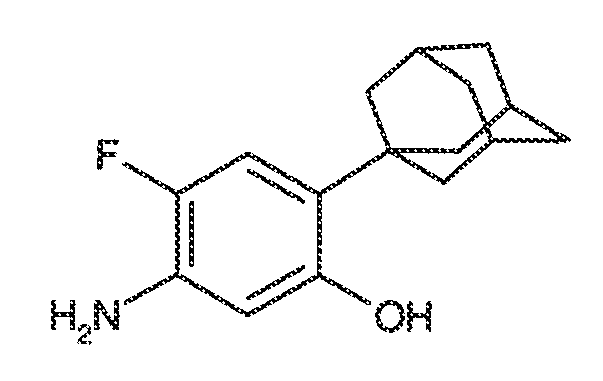
C-20; 2-Адамантил-5-амино-4-фтор-фенол
2-Адамантил-5-амино-4-фтор-фенол (C-20) синтезировали в соответствии с общей схемой, представленной выше, исходя из 4-фторфенола и адамантан-1-ола.
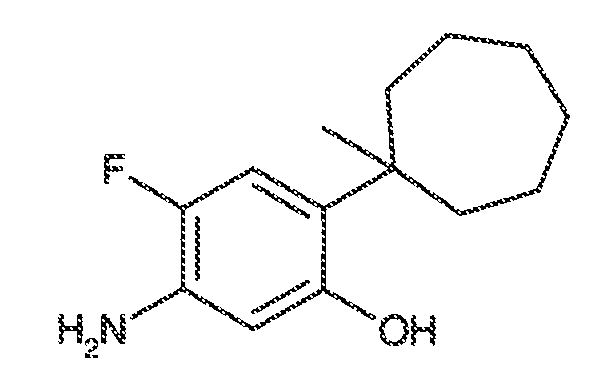
C-21; 5-Амино-4-фтор-2-(1-метилциклогептил)фенол
5-Амино-4-фтор-2-(1-метилциклогептил)фенол (C-21) синтезировали в соответствии с общей схемой, представленной выше, исходя из 4-фторфенола и 1-метил-циклогептанола.
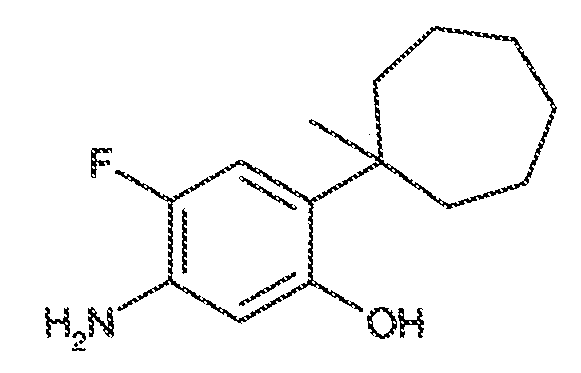
C-22; 5-Амино-4-фтор-2-(1-метилциклооктил)фенол
5-Амино-4-фтор-2-(1-метилциклооктил)фенол (C-22) синтезировали в соответствии с общей схемой, представленной выше, исходя из 4-фторфенола и 1-метил-циклооктанола.
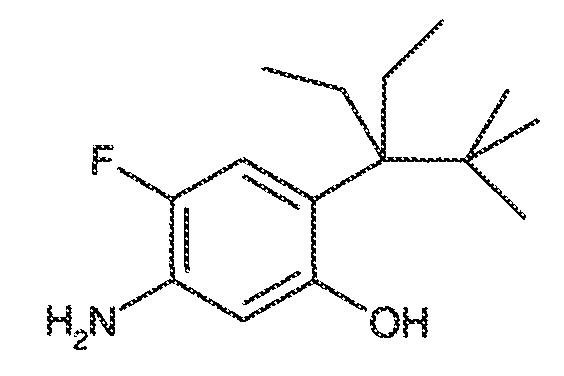
C-23; 5-Амино-2-(3-этил-2,2-диметилпентан-3-ил)-4-фтор-фенол
5-Амино-2-(3-этил-2,2-диметилпентан-3-ил)-4-фтор-фенол (C-23) синтезировали в соответствии с общей схемой, представленной выше, исходя из 4-фторфенола и 3-этил-2,2-диметил-пентан-3-ола.
Пример 5:
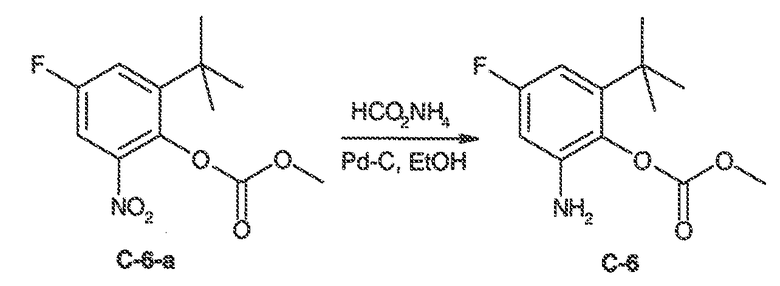
C-6; 2-трет-бутил-4-фтор-6-аминофенилметилкарбонат
К нагреваемому при температуре кипения с обратным холодильником раствору 2-трет-бутил-4-фтор-6-нитрофенилметилкарбоната (250 мг, 0,92 ммоль) и формиата аммония (250 мг, 4 ммоль) в EtOH (10 мл) добавляли 5% Pd-C (170 мг). Смесь кипятили с обратным холодильником еще в течение 1 часа, охлаждали и фильтровали через Целит. Растворитель удаляли выпариванием и остаток очищали колоночной хроматографией (0-15%, EtOAc-гексан) с получением 2-трет-бутил-4-фтор-6-аминофенилметилкарбоната (C-6) (60 мг, 27%). ВЭЖХ время удерживания 3,35 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 242,0 m/z (MH+).
Пример 6:
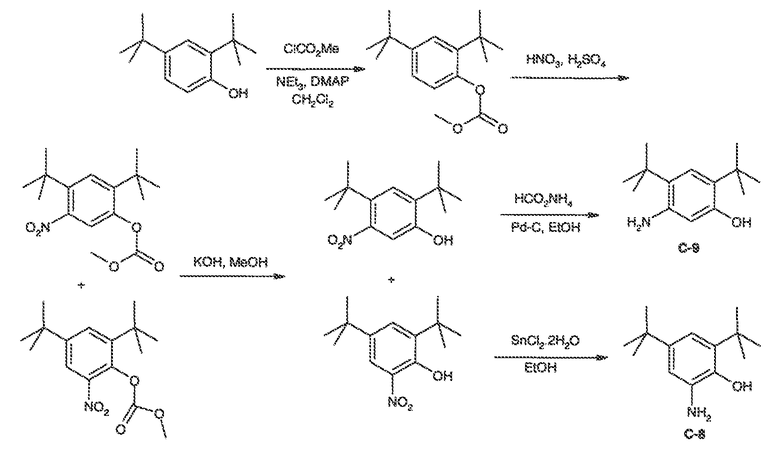
2,4-ди-трет-бутил-фениловый эфир метиловый эфир угольной кислоты
Метилхлорформиат (58 мл, 750 ммоль) добавляли по каплям к раствору 2,4-ди-трет-бутил-фенола (103,2 г, 500 ммоль), Et3N (139 мл, 1000 ммоль) и DMAP (3,05 г, 25 ммоль) в дихлорметане (400 мл), охлажденному на бане лед-вода до 0ºC. Смеси давали нагреться до комнатной температуры при перемешивании в течение ночи, затем фильтровали через силикагель (приблизительно 1 л) с использованием смеси 10% этилацетата-гексан (~4 л) в качестве элюента. Объединенные фильтраты концентрировали с получением 2,4-ди-трет-бутил-фенилового эфира метилового эфира угольной кислоты в виде желтого масла (132 г, количественный). 1H ЯМР (400 МГц, DMSO-d6) δ 7,35 (д, J=2,4 Гц, 1H), 7,29 (дд, J=8,5, 2,4 Гц, 1H), 7,06 (д, J=8,4 Гц, 1H), 3,85 (с, 3H), 1,30 (с, 9H), 1,29 (с, 9H).
2,4-ди-трет-бутил-5-нитро-фениловый эфир метиловый эфир угольной кислоты и 2,4-ди-трет-бутил-6-нитро-фениловый эфир метиловый эфир угольной кислоты
К перемешиваемой смеси 2,4-ди-трет-бутил-фенилового эфира метилового эфира угольной кислоты (4,76 г, 18 ммоль) в концентрированной серной кислоте (2 мл), охлажденной на бане лед-вода, добавляли охлажденную смесь серной кислоты (2 мл) и азотной кислоты (2 мл). Добавление осуществляли медленно, чтобы температура реакции не превышала 50ºC. Реакционную смесь оставляли для перемешивания в течение 2 часов, нагревая при этом до комнатной температуры. Реакционную смесь затем добавляли в ледяную воду и экстрагировали в диэтиловый эфир. Эфирный слой сушили (MgSO4), концентрировали и очищали колоночной хроматографией (0-10% этилацетат-гексан) с получением смеси 2,4-ди-трет-бутил-5-нитро-фенилового эфира метилового эфира угольной кислоты и 2,4-ди-трет-бутил-6-нитро-фенилового эфира метилового эфира угольной кислоты в виде бледно-желтого твердого вещества (4,28 г), которое использовали непосредственно на следующей стадии.
2,4-Ди-трет-бутил-5-нитро-фенол и 2,4-Ди-трет-бутил-6-нитро-фенол
Смесь угольной кислот 2,4-ди-трет-бутил-5-нитро-фенилового эфира метилового эфира угольной кислоты и 2,4-ди-трет-бутил-6-нитро-фенилового эфира метилового эфира угольной кислоты (4,2 г, 12,9 ммоль) растворяли в MeOH (65 мл) и добавляли KOH (2,0 г, 36 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь затем подкисляли (pH 2-3) путем добавления концентрированной HCl и распределяли между водой и диэтиловым эфиром. Эфирный слой сушили (MgSO4), концентрировали и очищали колоночной хроматографией (0-5% этилацетата-гексан) с получением 2,4-ди-трет-бутил-5-нитро-фенола (1,31 г, 29% за 2 стадии) и 2,4-ди-трет-бутил-6-нитро-фенола. 2,4-Ди-трет-бутил-5-нитро-фенол: 1H ЯМР (400 МГц, DMSO-d6) δ 10,14 (с, 1H, OH), 7,34 (с, 1H), 6,83 (с, 1H), 1,36 (с, 9H), 1,30 (с, 9H). 2,4-Ди-трет-бутил-5-нитро-фенол: 1H ЯМР (400 МГц, CDCl3) δ 11,48 (с, 1H), 7,98 (д, J=2,5 Гц, 1H), 7,66 (д, J=2,4 Гц, 1H), 1,47 (с, 9H), 1,34 (с, 9H).
C-9; 5-Амино-2,4-ди-трет-бутил-фенол
При температуре кипения с обратным холодильником к раствору 2,4-ди-трет-бутил-5-нитро-фенола (1,86 г, 7,4 ммоль) и формиата аммония (1,86 г) в этаноле (75 мл) добавляли Pd-5% масс. на активированном угле (900 мг). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение 2 часов, охлаждали до комнатной температуры и фильтровали через Целит. Целит промывали метанолом и объединенные фильтраты концентрировали с получением 5-амино-2,4-ди-трет-бутил-фенола в виде серого твердого вещества (1,66 г, количественный). 1H ЯМР (400 МГц, DMSO-d6) δ 8,64 (с, 1H, OH), 6,84 (с, 1H), 6,08 (с, 1H), 4,39 (с, 2H, NH2), 1,27 (м, 18H); ВЭЖХ время удерживания 2,72 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 222,4 m/z (MH+).
C-8; 6-Амино-2,4-ди-трет-бутил-фенол
Раствор 2,4-ди-трет-бутил-6-нитро-фенола (27 мг, 0,11 ммоль) и SnCl2·2H2O (121 мг, 0,54 ммоль) в EtOH (1,0 мл) нагревали в микроволновой печи при 100ºC в течение 30 минут. Смесь разбавляли EtOAc и водой, подщелачивали насыщенным раствором NaHCO3 и фильтровали через Целит. Органический слой отделяли и сушили над Na2SO4. Растворитель удаляли выпариванием с получением 6-амино-2,4-ди-трет-бутил-фенола (C-8), который использовали без дополнительной очистки. ВЭЖХ время удерживания 2,74 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 222,5 m/z (MH+).
Пример 7:
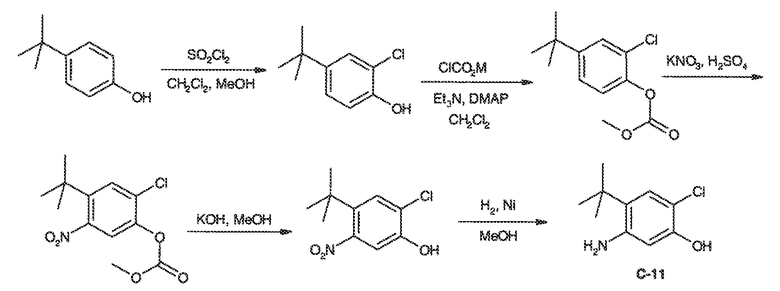
4-трет-бутил-2-хлор-фенол
К раствору 4-трет-бутил-фенола (40,0 г, 0,27 моль) и SO2Cl2 (37,5 г, 0,28 моль) в CH2Cl2 добавляли MeOH (9,0 г, 0,28 моль) при 0ºC. После завершения добавления смесь перемешивали в течение ночи при комнатной температуре и затем добавляли воду (200 мл). Полученный раствор экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией (петролейный эфир/EtOAc, 50:1) с получением 4-трет-бутил-2-хлор-фенола (47,0 г, 95%).
4-трет-бутил-2-хлорфенилметилкарбонат
К раствору 4-трет-бутил-2-хлорфенола (47,0 г, 0,25 моль) в дихлорметане (200 мл) добавляли Et3N (50,5 г, 0,50 моль), DMAP (1 г) и метилхлорформиат (35,4 г, 0,38 моль) при 0ºC. Реакционной смеси давали нагреться до комнатной температуры и перемешивали еще в течение 30 минут. Реакционную смесь промывали при помощи H2O и органический слой сушили над Na2SO4 и концентрировали с получением 4-трет-бутил-2-хлорфенилметилкарбоната (56,6 г, 92%), который использовали непосредственно на следующей стадии.
4-трет-бутил-2-хлор-5-нитрофенилметилкарбонат
4-трет-бутил-2-хлорфенилметилкарбонат (36,0 г, 0,15 моль) растворяли в концентрированной H2SO4 (100 мл) при 0ºC. KNO3 (0,53 г, 5,2 ммоль) добавляли по порциям в течение 25 минут. Реакционную смесь перемешивали в течение 1,5 часов и выливали на лед (200 г). Водный слой экстрагировали дихлорметаном. Объединенные органические слои промывали водным раствором NaHCO3, сушили над Na2SO4 и концентрировали в вакууме с получением 4-трет-бутил-2-хлор-5-нитрофенилметилкарбоната (41,0 г), который использовали без дополнительной очистки.
4-трет-бутил-2-хлор-5-нитро-фенол
Гидроксид калия (10,1 г, 181 ммоль) добавляли к 4-трет-бутил-2-хлор-5-нитрофенилметилкарбонату (40,0 г, 139 ммоль) в MeOH (100 мл). Через 30 минут реакционную смесь подкисляли 1н раствором HCl и экстрагировали дихлорметаном. Объединенные органические слои объединяли, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный остаток очищали колоночной хроматографией (петролейный эфир/EtOAc, 30:1) с получением 4-трет-бутил-2-хлор-5-нитро-фенола (23,0 г, 68% за 2 стадии).
C-11; 4-трет-бутил-2-хлор-5-амино-фенол
К раствору 4-трет-бутил-2-хлор-5-нитро-фенола (12,6 г, 54,9 ммоль) в MeOH (50 мл) добавляли Ni (1,2 г). Реакционную смесь встряхивали в атмосфере H2 (1 атм) в течение 4 часов. Реакционную смесь фильтровали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир/EtOAc, 20:1) с получением 4-трет-бутил-2-хлор-5-амино-фенола (C-11) (8,5 г, 78%). 1H ЯМР (DMSO-d6) δ 9,33 (с, 1H), 6,80 (с, 1H), 6,22 (с, 1H), 4,76 (с, 1H), 1,23 (с, 9H); ESI-MS 200,1 m/z (MH+).
Пример 8:
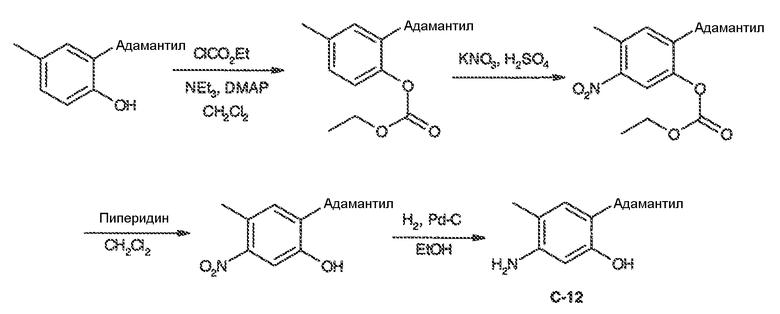
2-Адамантил-4-метил-фенилэтилкарбонат
Этилхлорформиат (0,64 мл, 6,7 ммоль) добавляли по каплям к раствору 2-адамантил-4-метилфенола (1,09 г, 4,5 ммоль), Et3N (1,25 мл, 9 ммоль) и DMAP (каталитическое количество) в дихлорметане (8 мл), охлажденному на бане лед-вода до 0ºC. Смеси давали нагреться до комнатной температуры при перемешивании в течение ночи, затем фильтровали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (10-20% этилацетата-гексан) с получением 2-адамантил-4-метил-фенилэтилкарбоната в виде желтого масла (1,32 г, 94%).
2-Адамантил-4-метил-5-нитрофенилэтилкарбонат
К охлажденному раствору 2-адамантил-4-метил-фенилэтилкарбоната (1,32 г, 4,2 ммоль) в H2SO4 (98%, 10 мл) добавляли KNO3 (510 мг, 5,0 ммоль) небольшими порциями при 0ºC. Смесь перемешивали в течение 3 часов, нагревая при этом до комнатной температуры, выливали на лед и затем экстрагировали дихлорметаном. Объединенные органические слои промывали NaHCO3 и насыщенным солевым раствором, сушили над MgSO4 и концентрировали досуха. Остаток очищали колоночной хроматографией (0-10% EtOAc-гексан) с получением 2-адамантил-4-метил-5-нитрофенилэтилкарбоната (378 мг, 25%).
2-Адамантил-4-метил-5-нитрофенол
К раствору 2-адамантил-4-метил-5-нитрофенилэтилкарбоната (378 мг, 1,05 ммоль) в CH2Cl2 (5 мл) добавляли пиперидин (1,0 мл). Раствор перемешивали при комнатной температуре в течение 1 часа, адсорбировали на силикагель при пониженном давлении и очищали флэш-хроматографией на силикагеле (0-15%, EtOAc-гексан) с получением 2-адамантил-4-метил-5-нитрофенола (231 мг, 77%).
C-12; 2-Адамантил-4-метил-5-аминофенол
К раствору 2-адамантил-4-метил-5-нитрофенола (231 мг, 1,6 ммоль) в EtOH (2 мл) добавляли Pd-5% масс. на углероде (10 мг). Смесь перемешивали в атмосфере H2 (1 атм) в течение ночи и затем фильтровали через Целит. Фильтрат упаривали досуха с получением 2-адамантил-4-метил-5-аминофенола (C-12), который использовали без дополнительной очистки. ВЭЖХ время удерживания 2,52 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 258,3 m/z (MH+).
Пример 9:
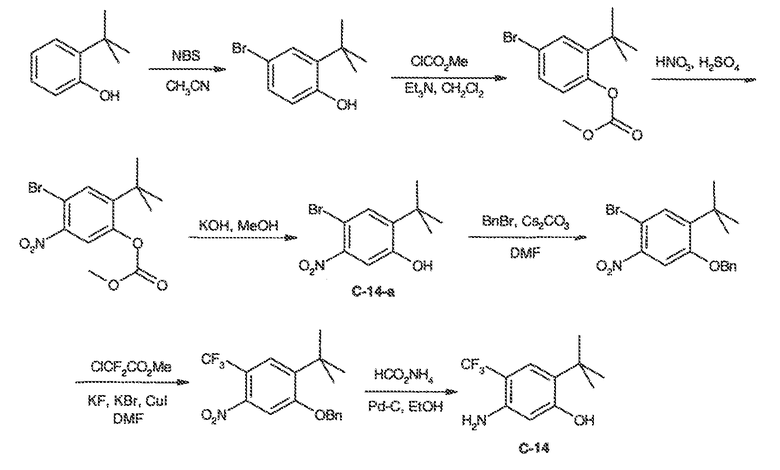
2-трет-бутил-4-бромфенол
К раствору 2-трет-бутилфенола (250 г, 1,67 моль) в CH3CN (1500 мл) добавляли NBS (300 г, 1,67 моль) при комнатной температуре. После добавления смесь перемешивали при комнатной температуре в течение ночи и затем растворитель удаляли. Добавляли петролейный эфир (1000 мл) и образовавшийся белый осадок отфильтровывали. Фильтрат концентрировали при пониженном давлении с получением неочищенного 2-трет-бутил-4-бромфенола (380 г), который использовали без дополнительной очистки.
Метил (2-трет-бутил-4-бромфенил) карбонат
К раствору 2-трет-бутил-4-бромфенола (380 г, 1,67 моль) в дихлорметане (1000 мл) добавляли Et3N (202 г, 2 моль) при комнатной температуре. К этому раствору добавляли по каплям метилхлорформиат (155 мл) при 0ºC. После добавления смесь перемешивали при 0ºC в течение 2 часов, гасили насыщенным раствором хлорида аммония и разбавляли водой. Органический слой отделяли и промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного метил (2-трет-бутил-4-бромфенил)карбоната (470 г), который использовали без дополнительной очистки.
Метил (2-трет-бутил-4-бром-5-нитрофенил)карбонат
Метил (2-трет-бутил-4-бромфенил)карбонат (470 г, 1,67 моль) растворяли в концентрированной H2SO4 (1000 мл) при 0ºC. Добавляли по порциям KNO3 (253 г, 2,5 моль) в течение 90 минут. Реакционную смесь перемешивали при 0ºC в течение 2 часов и выливали в ледяную воду (20 л). Образовавшийся осадок собирали фильтрованием и тщательно промывали водой, сушили и перекристаллизовывали из простого эфира с получением метил (2-трет-бутил-4-бром-5-нитрофенил)карбоната (332 г, 60% за 3 стадии).
C-14-a; 2-трет-бутил-4-бром-5-нитро-фенол
К раствору метил (2-трет-бутил-4-бром-5-нитрофенил)карбоната (121,5 г, 0,366 моль) в метаноле (1000 мл) добавляли гидроксид калия (30,75 г, 0,549 моль) по порциям. После добавления смесь перемешивали при комнатной температуре в течение 3 часов и подкисляли 1н раствором HCl до pH 7. Метанол удаляли и добавляли воду. Смесь экстрагировали этилацетатом и органический слой отделяли, сушили над Na2SO4 и концентрировали с получением 2-трет-бутил-4-бром-5-нитро-фенола (C-14-a) (100 г, 99%).
1-трет-бутил-2-(бензилокси)-5-бром-4-нитробензол
К смеси 2-трет-бутил-4-бром-5-нитрофенола (C-14-a) (1,1 г, 4 ммоль) и Cs2CO3 (1,56 г, 4,8 ммоль) в DMF (8 мл) добавляли бензилбромид (500 мкл, 4,2 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов, разбавляли при помощи H2O и экстрагировали два раза при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После удаления растворителя остаток очищали колоночной хроматографией (0-5% EtOAc-гексан) с получением 1-трет-бутил-2-(бензилокси)-5-бром-4-нитробензола (1,37 г, 94%). 1H ЯМР (400 МГц, CDCl3) 7,62 (с, 1H), 7,53 (с, 1H), 7,43 (м, 5H), 5,22 (с, 2H), 1,42 (с, 9H).
1-трет-бутил-2-(бензилокси)-5-(трифторметил)-4-нитробензол
Смесь 1-трет-бутил-2-(бензилокси)-5-бром-4-нитробензола (913 мг, 2,5 ммоль), KF (291 мг, 5 ммоль), KBr (595 мг, 5 ммоль), CuI (570 мг, 3 ммоль), метилхлордифторацетата (1,6 мл, 15 ммоль) и DMF (5 мл) перемешивали при 125ºC в герметично закрытой пробирке в течение ночи, охлаждали до комнатной температуры, разбавляли водой и экстрагировали три раза при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным MgSO4. После удаления растворителя остаток очищали колоночной хроматографией (0-5% EtOAc-гексан) с получением 1-трет-бутил-2-(бензилокси)-5-(трифторметил)-4-нитробензола (591 мг, 67%). 1H ЯМР (400 МГц, CDCl3) 7,66 (с, 1H), 7,37 (м, 5H), 7,19 (с, 1H), 5,21 (с, 2H), 1,32 (с, 9H).
C-14; 5-Амино-2-трет-бутил-4-трифторметил-фенол
К нагреваемому при температуре кипения с обратным холодильником раствору 1-трет-бутил-2-(бензилокси)-5-(трифторметил)-4-нитробензола (353 мг, 1,0 ммоль) и формиата аммония (350 мг, 5,4 ммоль) в EtOH (10 мл) добавляли 10% Pd-C (245 мг). Смесь кипятили с обратным холодильником еще в течение 2 часов, охлаждали до комнатной температуры и фильтровали через Целит. После удаления растворителя остаток очищали колоночной хроматографией с получением 5-амино-2-трет-бутил-4-трифторметил-фенола (C-14) (120 мг, 52%). 1H ЯМР (400 МГц, CDCl3) δ 7,21 (с, 1H), 6,05 (с, 1H), 1,28 (с, 9H); HPLC время удерживания 3,46 мин, 10-99 % CH3CN, 5 мин. цикл; ESI-MS 234,1 m/z (MH+).
Пример 10:
Общая схема:
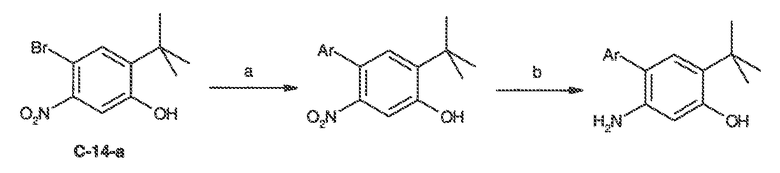
a) ArB(OH)2, K2CO3, Pd(PPh3)4, H2O, DMF или ArB(OH)2, (dppf)PdCl2, K2CO3, EtOH; b) H2, Ni Ренея, MeOH или HCO2NH4, Pd-C, EtOH или SnCl2.2H2O.
Специальный пример:
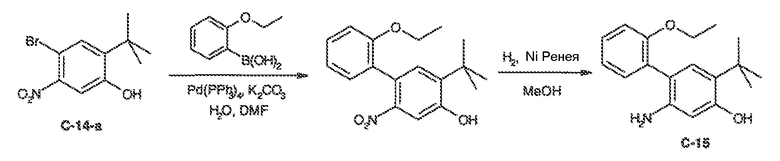
2-трет-бутил-4-(2-этоксифенил)-5-нитрофенол
К раствору 2-трет-бутил-4-бром-5-нитрофенола (C-14-a) (8,22 г, 30 ммоль) в DMF (90 мл) добавляли 2-этоксифенилбороновую кислоту (5,48 г, 33 ммоль), карбонат калия (4,56 г, 33 ммоль), воду (10 мл) и Pd(PPh3)4 (1,73 г, 1,5 ммоль). Смесь нагревали при 90ºC в течение 3 часов в атмосфере азота. Растворитель удаляли при пониженном давлении. Остаток распределяли между водой и этилацетатом. Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили и очищали колоночной хроматографией (петролейный эфир-этилацетат, 10:1) с получением 2-трет-бутил-4-(2-этоксифенил)-5-нитрофенола (9,2 г, 92%). 1H ЯМР (DMSO-d6) δ 10,38 (с, 1H), 7,36 (с, 1H), 7,28 (м, 2H), 7,08 (с, 1H), 6,99 (т, 1H, J=7,35 Гц), 6,92 (д, 1H, J=8,1 Гц), 3,84 (кв., 2H, J=6,6 Гц), 1,35 (с, 9H), 1,09 (т, 3H, J=6,6 Гц); ESI-MS 314,3 m/z (MH+).
C-15; 2-трет-бутил-4-(2-этоксифенил)-5-аминофенол
К раствору 2-трет-бутил-4-(2-этоксифенил)-5-нитрофенола (3,0 г, 9,5 ммоль) в метаноле (30 мл) добавляли Ni Ренея (300 мг). Смесь перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 2 часов. Катализатор отфильтровывали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир-этилацетат, 6:1) с получением 2-трет-бутил-4-(2-этоксифенил)-5-аминофенола (C-15) (2,35 г, 92%). 1H ЯМР (DMSO-d6) δ 8,89 (с, 1H), 7,19 (т, 1H, J=4,2 Гц), 7,10 (д, 1H, J=1,8 Гц), 7,08 (д, 1H, J=1,8 Гц), 6,94 (т, 1H, J=3,6 Гц), 6,67 (с, 1H), 6,16 (с, 1H), 4,25 (с, 1H), 4,00 (кв., 2H, J=6,9 Гц), 1,26 (с, 9H), 1,21 (т, 3H, J=6,9 Гц); ESI-MS 286,0 m/z (MH+).
Другие примеры:
C-16; 2-трет-бутил-4-(3-этоксифенил)-5-аминофенол
2-трет-бутил-4-(3-этоксифенил)-5-аминофенол (C-16) синтезировали в соответствии с общей схемой, представленной выше, исходя из 2-трет-бутил-4-бром-5-нитрофенола (C-14-a) и 3-этоксифенилбороновой кислоты. ВЭЖХ время удерживания 2,77 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 286,1 m/z (MH+).
C-17; 2-трет-бутил-4-(3-метоксикарбонилфенил)-5-аминофенол (C-17)
2-трет-бутил-4-(3-метоксикарбонилфенил)-5-аминофенол (C-17) синтезировали в соответствии с общей схемой, представленной выше, исходя из 2-трет-бутил-4-бром-5-нитрофенола (C-14-a) и 3-(метоксикарбонил)фенилбороновой кислоты. ВЭЖХ время удерживания 2,70 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 300,5 m/z (MH+).
Пример 11;
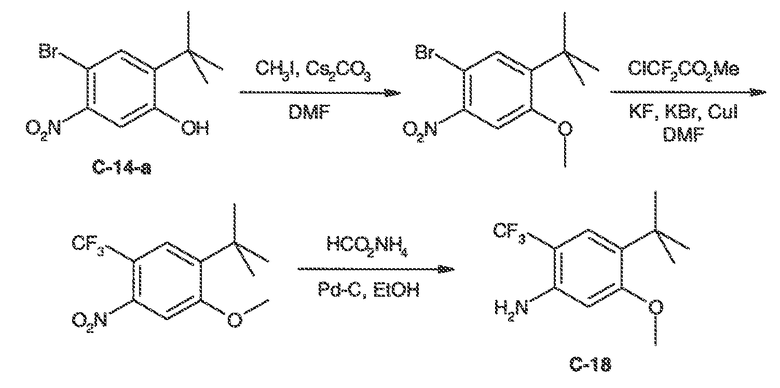
1-трет-бутил-2-метокси-5-бром-4-нитробензол
К смеси 2-трет-бутил-4-бром-5-нитрофенола (C-14-a) (1,5 г, 5,5 ммоль) и Cs2CO3 (2,2 г, 6,6 ммоль) в DMF (6 мл) добавляли метилиодид (5150 мкл, 8,3 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов, разбавляли при помощи H2O и экстрагировали два раза при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После удаления растворителя остаток промывали гексаном с получением 1-трет-бутил-2-метокси-5-бром-4-нитробензола (1,1 г, 69%). 1H ЯМР (400 МГц, CDCl3) δ 7,58 (с, 1H), 7,44 (с, 1H), 3,92 (с, 3H), 1,39 (с, 9H).
1-трет-бутил-2-метокси-5-(трифторметил)-4-нитробензол
Смесь 1-трет-бутил-2-метокси-5-бром-4-нитробензола (867 мг, 3,0 ммоль), KF (348 мг, 6 ммоль), KBr (714 мг, 6 ммоль), CuI (684 мг, 3,6 ммоль), метилхлордифторацетата (2,2 мл, 21,0 ммоль) в DMF (5 мл) перемешивали при 125ºC в герметично закрытой пробирке в течение ночи, охлаждали до комнатной температуры, разбавляли водой и экстрагировали три раза при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным MgSO4. После удаления растворителя остаток очищали колоночной хроматографией (0-5% EtOAc-гексан) с получением 1-трет-бутил-2-метокси-5-(трифторметил)-4-нитробензола (512 мг, 61%). 1H ЯМР (400 МГц, CDCl3) δ 7,60 (с, 1H), 7,29 (с, 1H), 3,90 (с, 3H), 1,33 (с, 9H).
C-18; 1-трет-бутил-2-метокси-5-(трифторметил)-4-аминобензол
К нагреваемому при температуре кипения с обратным холодильником раствору 1-трет-бутил-2-метокси-5-(трифторметил)-4-нитробензола (473 мг, 1,7 ммоль) и формиата аммония (473 мг, 7,3 ммоль) в EtOH (10 мл) добавляли 10% Pd-C (200 мг). Смесь кипятили с обратным холодильником в течение 1 часа, охлаждали и фильтровали через Целит. Растворитель удаляли выпариванием с получением 1-трет-бутил-2-метокси-5-(трифторметил)-4-аминобензола (C-18) (403 мг, 95%). 1H ЯМР (400 МГц, CDCl3) δ 7,19 (с, 1H), 6,14 (с, 1H), 4,02 (шир.с, 2H), 3,74 (с, 3H), 1,24 (с, 9H).
Пример 12:
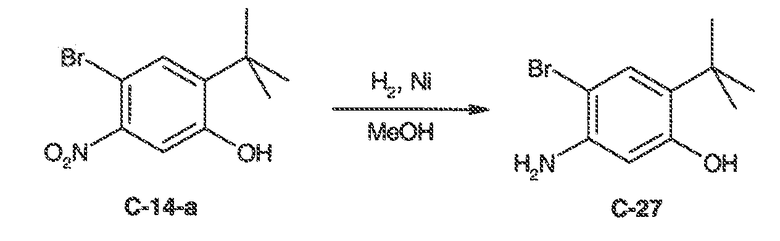
C-27; 2-трет-бутил-4-бром-5-амино-фенол
К раствору 2-трет-бутил-4-бром-5-нитрофенола (C-14-a) (12 г, 43,8 ммоль) в MeOH (90 мл) добавляли Ni (2,4 г). Реакционную смесь перемешивали в атмосфере H2 (1 атм) в течение 4 часов. Смесь фильтровали и фильтрат концентрировали. Неочищенный продукт перекристаллизовывали из этилацетата и петролейного эфира с получением 2-трет-бутил-4-бром-5-амино-фенола (C-27) (7,2 г, 70%). 1H ЯМР (DMSO-d6) δ 9,15 (с, 1H), 6,91 (с, 1H), 6,24 (с, 1H), 4,90 (шир.с, 2H), 1,22 (с, 9H); ESI-MS 244,0 m/z (MH+).
Пример 13:
C-24; 2,4-Ди-трет-бутил-6-(N-метиламино)фенол
Смесь 2,4-ди-трет-бутил-6-амино-фенола (C-9) (5,08 г, 23 ммоль), NaBH3CN (4,41 г, 70 ммоль) и параформальдегида (2,1 г, 70 ммоль) в метаноле (50 мл) перемешивали при температуре кипения с обратным холодильником в течение 3 часов. После удаления растворителя остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 30:1) с получением 2,4-ди-трет-бутил-6-(N-метиламино)фенола (C-24) (800 мг, 15%). 1H ЯМР (DMSO-d6) δ 8,67 (с, 1H), 6,84 (с, 1H), 5,99 (с, 1H), 4,36 (кв., J=4,8 Гц, 1H), 2,65 (д, J=4,8 Гц, 3H), 1,23 (с, 18H); ESI-MS 236,2 m/z (MH+).
Пример 14
2-Метил-2-фенил-пропан-1-ол
К раствору 2-метил-2-фенил-пропионовой кислоты (82 г, 0,5 моль) в ТГФ (200 мл) добавляли по каплям боран-диметилсульфид (2M, 100 мл) при 0-5ºC. Смесь перемешивали при этой температуре в течение 30 минут и затем нагревали при температуре кипения с обратным холодильником в течение 1 часа. После охлаждения добавляли метанол (150 мл) и воду (50 мл). Смесь экстрагировали при помощи EtOAc (100 мл ×3) и объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением 2-метил-2-фенил-пропан-1-ола в виде масла (70 г, 77%).
2-(2-Метокси-этокси)-1,1-диметил-этил]-бензол
К суспензии NaH (29 г, 0,75 моль) в ТГФ (200 мл) добавляли по каплям раствор 2-метил-2-фенил-пропан-1-ола (75 г, 0,5 моль) в ТГФ (50 мл) при 0ºC. Смесь перемешивали при 20ºC в течение 30 минут и затем добавляли по каплям раствор 1-бром-2-метокси-этана (104 г, 0,75 моль) в ТГФ (100 мл) при 0ºC. Смесь перемешивали при 20ºC в течение ночи, выливали в воду (200 мл) и экстрагировали при помощи EtOAc (100 мл ×3). Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, петролейный эфир) с получением 2-(2-Метокси-этокси)-1,1-диметил-этил]-бензола в виде масла (28 г, 27%).
1-[2-(2-Метокси-этокси)-1,1-диметил-этил]-4-нитро-бензол
К раствору 2-(2-метокси-этокси)-1,1-диметил-этил]-бензола (52 г, 0,25 моль) в CHCl3 (200 мл) добавляли KNO3 (50,5 г, 0,5 моль) и TMSCl (54 г, 0,5 моль). Смесь перемешивали при 20ºC в течение 30 минут и затем добавляли AlCl3 (95 г, 0,7 моль). Реакционную смесь перемешивали при 20ºC в течение 1 часа и выливали в ледяную воду. Органический слой отделяли и водный слой экстрагировали при помощи CHCl3 (50 мл ×3). Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, петролейный эфир) с получением 1-[2-(2-метокси-этокси)-1,1-диметил-этил]-4-нитро-бензола (6 г, 10%).
4-[2-(2-Метокси-этокси)-1,1-диметил-этил]-фениламин
Суспензию 1-[2-(2-метокси-этокси)-1,1-диметил-этил]-4-нитро-бензола (8,1 г, 32 ммоль) и Ni Ренея (1 г) в MeOH (50 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 1 часа. Катализатор отфильтровывали и фильтрат концентрировали с получением 4-[2-(2-метокси-этокси)-1,1-диметил-этил]-фениламина (5,5 г, 77%).
4-[2-(2-Метокси-этокси)-1,1-диметил-этил]-3-нитро-фениламин
К раствору 4-[2-(2-метокси-этокси)-1,1-диметил-этил]-фениламина (5,8 г, 26 ммоль) в H2SO4 (20 мл) добавляли KNO3 (2,63 г, 26 ммоль) при 0ºC. После завершения добавления смесь перемешивали при этой температуре в течение 20 минут и затем выливали в ледяную воду. Смесь экстрагировали при помощи EtOAc (50 мл ×3). Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 100:1) с получением 4-[2-(2-метокси-этокси)-1,1-диметил-этил]-3-нитро-фениламина (5 г, 71%).
N-{4-[2-(2-Метокси-этокси)-1,1-диметил-этил]-3-нитро-фенил}-ацетамид
К суспензии NaHCO3 (10 г, 0,1 моль) в дихлорметане (50 мл) добавляли 4-[2-(2-метокси-этокси)-1,1-диметил-этил]-3-нитро-фениламин (5 г, 30 ммоль) и ацетилхлорид (3 мл, 20 ммоль) при 0-5ºC. Смесь перемешивали в течение ночи при 15ºC и затем выливали в воду (200 мл). Органический слой отделяли и водный слой экстрагировали дихлорметаном (50 мл ×2). Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали досуха с получением N-{4-[2-(2-метокси-этокси)-1,1-диметил-этил]-3-нитро-фенил}-ацетамида (5,0 г, 87%).
N-{3-Амино-4-[2-(2-метокси-этокси)-1,1-диметил-этил]-фенил}-ацетамид
Смесь N-{4-[2-(2-метокси-этокси)-1,1-диметил-этил]-3-нитро-фенил}-ацетамида (5 г, 16 ммоль) и Ni Ренея (1 г) в MeOH (50 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 1 часа. Катализатор отфильтровывали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 100:1) с получением N-{3-амино-4-[2-(2-метокси-этокси)-1,1-диметил-этил]-фенил}-ацетамида (1,6 г, 35%).
N-{3-Гидрокси-4-[2-(2-метокси-этокси)-1,1-диметил-этил]-фенил}-ацетамид
К раствору N-{3-амино-4-[2-(2-метокси-этокси)-1,1-диметил-этил]-фенил}-ацетамида (1,6 г, 5,7 ммоль) в H2SO4 (15%, 6 мл) добавляли NaNO2 при 0-5ºC. Смесь перемешивали при этой температуре в течение 20 минут и затем выливали в ледяную воду. Смесь экстрагировали при помощи EtOAc (30 мл ×3). Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 100:1) с получением N-{3-гидрокси-4-[2-(2-метокси-этокси)-1,1-диметил-этил]-фенил}-ацетамида (0,7 г, 38%).
C-25; 2-(1-(2-Метоксиэтокси)-2-метилпропан-2-ил)-5-аминофенол
Смесь N-{3-гидрокси-4-[2-(2-метокси-этокси)-1,1-диметил-этил]-фенил}-ацетамида (1 г, 3,5 ммоль) и HCl (5 мл) нагревали при температуре кипения с обратным холодильником в течение 1 часа. Смесь подщелачивали раствором Na2CO3 до pH 9 и затем экстрагировали при помощи EtOAc (20 мл ×3). Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали досуха. Остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 100:1) с получением 2-(1-(2-метоксиэтокси)-2-метилпропан-2-ил)-5-аминофенола (C-25) (61 мг, 6%). 1H ЯМР (CDCl3) δ 9,11 (шир.с, 1H), 6,96-6,98 (д, J=8 Гц, 1H), 6,26-6,27 (д, J=4 Гц, 1H), 6,17-6,19 (м, 1H), 3,68-3,69 (м, 2H), 3,56-3,59 (м, 4H), 3,39 (с, 3H), 1,37 (с, 6H); ESI-MS 239,9 m/z (MH+).
Пример 15:
4,6-ди-трет-бутил-3-нитроциклогекса-3,5-диен-1,2-дион
К раствору 3,5-ди-трет-бутилциклогекса-3,5-диен-1,2-диона (4,20 г, 19,1 ммоль) в уксусной кислоте (115 мл) медленно добавляли HNO3 (15 мл). Смесь нагревали при 60ºC в течение 40 минут, затем выливали в H2O (50 мл). Смеси давали выстояться при комнатной температуре в течение 2 часов, затем помещали на ледяную баню на 1 час. Твердое вещество собирали и промывали водой с получением 4,6-ди-трет-бутил-3-нитроциклогекса-3,5-диен-1,2-диона (1,2 г, 24%). 1H ЯМР (400 МГц, DMSO-d6) δ 6,89 (с, 1H), 1,27 (с, 9H), 1,24 (с, 9H).
4,6-Ди-трет-бутил-3-нитробензол-1,2-диол
В делительную воронку помещали ТГФ/H2O (1:1, 400 мл), 4,6-ди-трет-бутил-3-нитроциклогекса-3,5-диен-1,2-дион (4,59 г, 17,3 ммоль) и Na2S2O4 (3 г, 17,3 ммоль). Делительную воронку закупоривали и встряхивали в течение 2 минут. Смесь разбавляли при помощи EtOAc (20 мл). Слои разделяли и органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали с получением 4,6-ди-трет-бутил-3-нитробензол-1,2-диола (3,4 г, 74%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ 9,24 (с, 1H), 8,76 (с, 1H), 6,87 (с, 1H), 1,35 (с, 9H), 1,25 (с, 9H).
C-26; 4,6-Ди-трет-бутил-3-аминобензол-1,2-диол
К раствору 4,6-ди-трет-бутил-3-нитробензол-1,2-диола (1,92 г, 7,2 ммоль) в EtOH (70 мл) добавляли Pd-5% масс. на углероде (200 мг). Смесь перемешивали в атмосфере H2 (1 атм) в течение 2 часов. В реакционную смесь снова загружали Pd-5% масс. на углероде (200 мг) и перемешивали в атмосфере H2 (1 атм) еще в течение 2 часов. Смесь фильтровали через Целит и фильтрат концентрировали и очищали колоночной хроматографией (10-40% этилацетата-гексан) с получением 4,6-ди-трет-бутил-3-аминобензол-1,2-диола (C-26) (560 мг, 33%). 1H ЯМР (400 МГц, CDCl3) δ 7,28 (с, 1H), 1,42 (с, 9H), 1,38 (с, 9H).
Анилины
Пример 1:
Общая схема

Специальный пример:
D-1; 4-Хлор-бензол-1,3-диамин
Смесь 1-хлор-2,4-динитро-бензола (100 мг, 0,5 ммоль) и SnCl2.2H2O (1,12 г, 5 ммоль) в этаноле (2,5 мл) перемешивали при комнатной температуре в течение ночи. Добавляли воду и затем смесь подщелачивали до pH 7-8 насыщенным раствором NaHCO3. Раствор экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением 4-хлор-бензол-1,3-диамина (D-1) (79 мг, количественный). ВЭЖХ время удерживания 0,38 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 143,1 m/z (MH+)
Другие примеры:
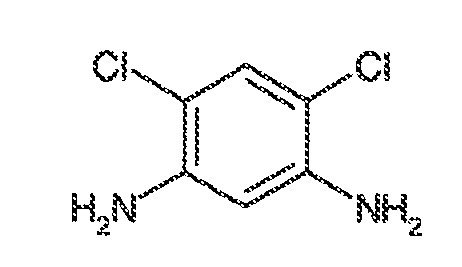
D-2; 4,6-Дихлор-бензол-1,3-диамин
4,6-Дихлор-бензол-1,3-диамин (D-2) синтезировали в соответствии с общей схемой, представленной выше, исходя из 1,5-дихлор-2,4-динитро-бензола. Выход (95%). ВЭЖХ время удерживания 1,88 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 177,1 m/z (MH+).
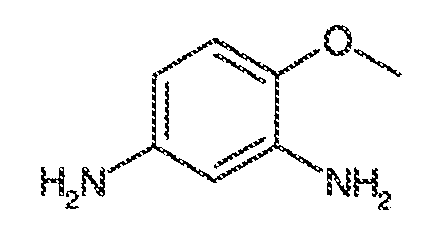
D-3; 4-Метокси-бензол-1,3-диамин
4-Метокси-бензол-1,3-диамин (D-3) синтезировали в соответствии с общей схемой, представленной выше, исходя из 1-метокси-2,4-динитро-бензола. Выход (количественный). ВЭЖХ время удерживания 0,31 минут, 10-99% CH3CN, 5 мин цикл.
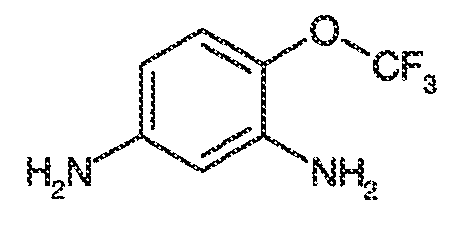
D-4; 4-Трифторметокси-бензол-1,3-диамин
4-Трифторметокси-бензол-1,3-диамин (D-4) синтезировали в соответствии с общей схемой, представленной выше, исходя из 2,4-динитро-1-трифторметокси-бензола. Выход (89%). ВЭЖХ время удерживания 0,91 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 193,3 m/z (MH+).
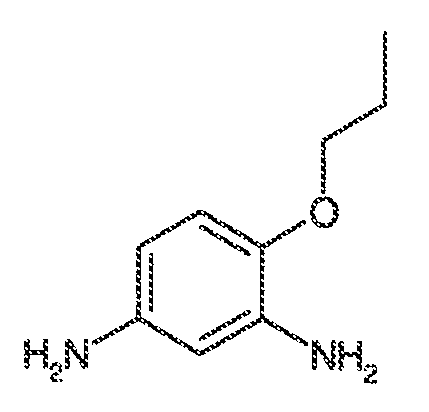
D-5; 4-Пропоксибензол-1,3-диамин
4-Пропоксибензол-1,3-диамин (D-5) синтезировали в соответствии с общей схемой, представленной выше, исходя из 5-нитро-2-пропокси-фениламина. Выход (79%). ВЭЖХ время удерживания 0,54 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 167,5 m/z (MH+).
Пример 2:
Общая схема

a) HNO3, H2SO4; b) SnCl2.2H2O, EtOH или H2, Pd-C, MeOH
Специальный пример:
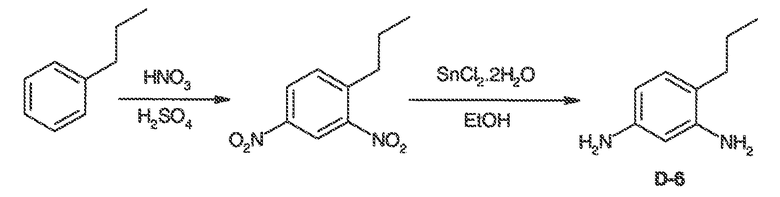
2,4-Динитро-пропилбензол
Раствор пропилбензола (10 г, 83 ммоль) в концентрированной H2SO4 (50 мл) охлаждали при 0ºC в течение 30 минут и добавляли по порциям раствор концентрированной H2SO4 (50 мл) и дымящей HNO3 (25 мл), предварительно охлажденный до 0ºC, в течение 15 минут. Смесь перемешивали при 0ºC еще в течение 30 минут и затем давали нагреться до комнатной температуры. Смесь выливали в лед (200 г) - воду (100 мл) и экстрагировали простым эфиром (2×100 мл). Объединенные экстракты промывали H2O (100 мл) и насыщенным солевым раствором (100 мл), сушили над MgSO4, фильтровали и концентрировали с получением 2,4-динитро-пропилбензола (15,6 г, 89%). 1H ЯМР (CDCl3, 300 МГц) δ 8,73 (д, J=2,2 Гц, 1H), 8,38 (дд, J=8,3, J=2,2, 1H), 7,6 (д, J=8,5 Гц, 1H), 2,96 (дд, 2H), 1,73 (м, 2H), 1,06 (т, J=7,4 Гц, 3H).
D-6; 4-Пропил-бензол-1,3-диамин
К раствору 2,4-динитро-пропилбензола (2,02 г, 9,6 ммоль) в этаноле (100 мл) добавляли SnCl2 (9,9 г, 52 ммоль) с последующим добавлением концентрированной HCl (10 мл). Смесь кипятили с обратным холодильником в течение 2 часов, выливали в ледяную воду (100 мл) и нейтрализовали при помощи твердого бикарбоната натрия. Раствор дополнительно подщелачивали 10% раствором NaOH до pH~10 и экстрагировали простым эфиром (2×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (100 мл), сушили над MgSO4, фильтровали и концентрировали с получением 4-пропил-бензол-1,3-диамина (D-6) (1,2 г, 83%). Не требовалось никакой дополнительной очистки для использования на следующей стадии; однако продукт не был стабильным в течение продолжительного периода времени. 1H ЯМР (CDCl3, 300 МГц) δ 6,82 (д, J=7,9 Гц, 1H), 6,11 (дд, J=7,5, J=2,2 Гц, 1H), 6,06 (д, J=2,2 Гц, 1H), 3,49 (шир.с, 4H, NH2), 2,38 (т, J=7,4 Гц, 2H), 1,58 (м, 2H), 0,98 (т, J=7,2 Гц, 3H); ESI-MS 151,5 m/z (MH+).
Другие примеры:
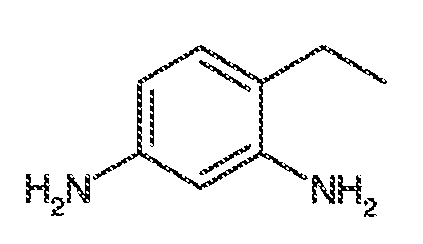
D-7; 4-Этилбензол-1,3-диамин
4-Этилбензол-1,3-диамин (D-7) синтезировали в соответствии с общей схемой, представленной выше, исходя из этилбензола. Общий выход (76%).
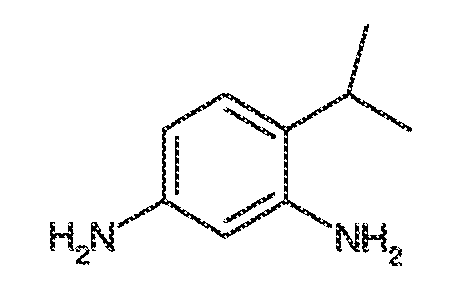
D-8; 4-Изопропилбензол-1,3-диамин
4-Изопропилбензол-1,3-диамин (D-8) синтезировали в соответствии с общей схемой, представленной выше, исходя из изопропилбензола. Общий выход (78%).
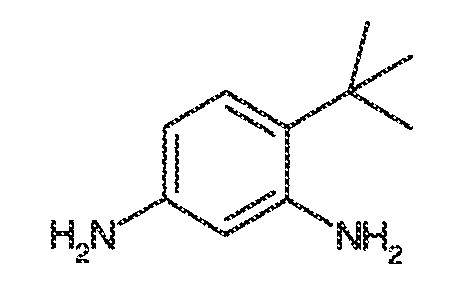
D-9; 4-трет-бутилбензол-1,3-диамин
4-трет-бутилбензол-1,3-диамин (D-9) синтезировали в соответствии с общей схемой, представленной выше, исходя из трет-бутилбензола. Общий выход (48%). 1H ЯМР (400 МГц, CDCl3) δ 7,01 (д, J=8,3 Гц, 1H), 6,10 (дд, J=2,4, 8,3 Гц, 1H), 6,01 (д, J=2,4 Гц, 1H), 3,59 (шир., 4H), 1,37 (с, 9H); 13C ЯМР (100 МГц, CDCl3) δ 145,5, 145,3, 127,6, 124,9, 105,9, 104,5, 33,6, 30,1; ESI-MS 164,9 m/z (MH+).
Пример 3:
Общая схема
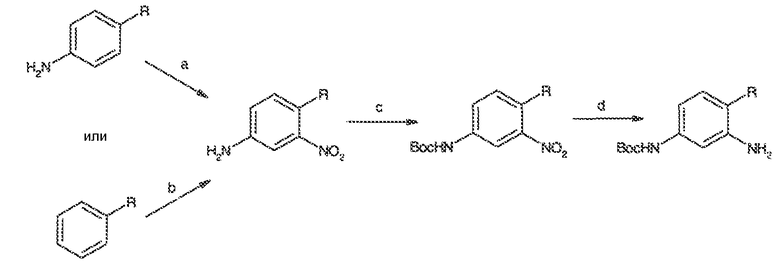
a) KNO3, H2SO4; b) (i) HNO3, H2SO4; (ii) Na2S, S, H2O; c) Boc2O, NaOH, ТГФ; d) H2, Pd-C, MeOH
Специальный пример:
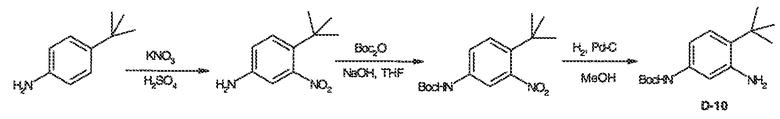
4-трет-бутил-3-нитро-фениламин
К смеси 4-трет-бутил-фениламина (10,0 г, 67,01 ммоль), растворенного в H2SO4 (98%, 60 мл), медленно добавляли KNO3 (8,1 г, 80,41 ммоль) при 0ºC. После добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Смесь затем выливали в ледяную воду и подщелачивали насыщенным раствором NaHCO3 до pH 8. Смесь экстрагировали несколько раз при помощи CH2Cl2. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 10:1) с получением 4-трет-бутил-3-нитро-фениламина (10 г, 77%).
трет-бутиловый эфир (4-трет-бутил-3-нитро-фенил)-карбаминовой кислоты
Смесь 4-трет-бутил-3-нитро-фениламина (4,0 г, 20,6 ммоль) и Boc2O (4,72 г, 21,6 ммоль) в NaOH (2N, 20 мл) и ТГФ (20 мл) перемешивали при комнатной температуре в течение ночи. ТГФ удаляли при пониженном давлении. Остаток растворяли в воде и экстрагировали при помощи CH2Cl2. Органический слой промывали NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением трет-бутилового эфира (4-трет-бутил-3-нитро-фенил)-карбаминовой кислоты (4,5 г, 74%).
D-10; трет-бутиловый эфир (3-амино-4-трет-бутил-фенил)-карбаминовой кислоты
Суспензию трет-бутилового эфира (4-трет-бутил-3-нитро-фенил)-карбаминовой кислоты (3,0 г, 10,19 моль) и 10% Pd-C (1 г) в MeOH (40 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение ночи. После фильтрования фильтрат концентрировали и остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 5:1) с получением трет-бутилового эфира (3-амино-4-трет-бутил-фенил)-карбаминовой кислоты (D-10) в виде коричневого масла (2,5 г, 93%). 1H ЯМР (CDCl3) δ 7,10 (д, J=8,4 Гц, 1H), 6,92 (с, 1H), 6,50-6,53 (м, 1H), 6,36 (с, 1H), 3,62 (шир.с, 2H), 1,50 (с, 9H), 1,38 (с, 9H); ESI-MS 528,9 m/z (2M+H+).
Другие примеры:
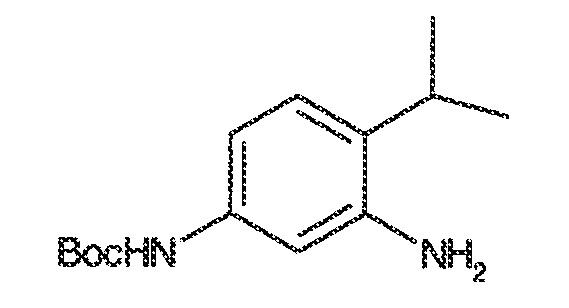
D-11; трет-бутиловый эфир (3-амино-4-изопропил-фенил)-карбаминовой кислоты
трет-Бутиловый эфир (3-амино-4-изопропил-фенил)-карбаминовой кислоты (D-11) синтезировали в соответствии с общей схемой, представленной выше, исходя из изопропилбензола. Общий выход (56%).
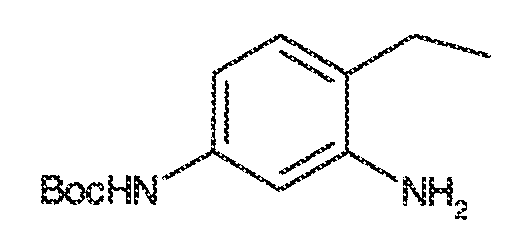
D-12; трет-бутиловый эфир (3-амино-4-этил-фенил)-карбаминовой кислоты
трет-Бутиловый эфир (3-амино-4-этил-фенил)-карбаминовой кислоты (D-12) синтезировали в соответствии с общей схемой, представленной выше, исходя из этилбензола. Общий выход (64%). 1H ЯМР (CD3OD, 300 МГц) δ 6,87 (д, J=8,0 Гц, 1H), 6,81 (д, J=2,2 Гц, 1H), 6,63 (дд, J=8,1, J=2,2, 1H), 2,47 (кв., J=7,4 Гц, 2H), 1,50 (с, 9H), 1,19 (т, J=7,4 Гц, 3H); ESI-MS 237,1 m/z (MH+).
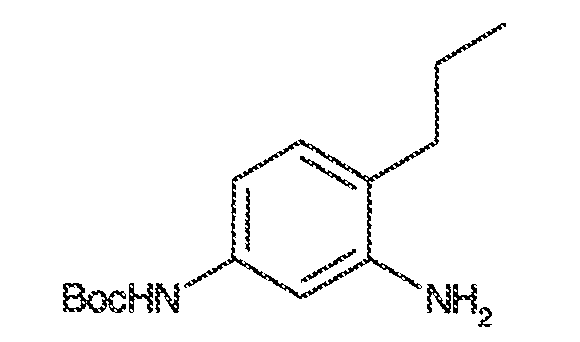
D-13; трет-бутиловый эфир (3-амино-4-пропил-фенил)-карбаминовой кислоты
трет-Бутиловый эфир (3-амино-4-пропил-фенил)-карбаминовой кислоты (D-13) синтезировали в соответствии с общей схемой, представленной выше, исходя из пропилбензола. Общий выход (48%).
Пример 4:
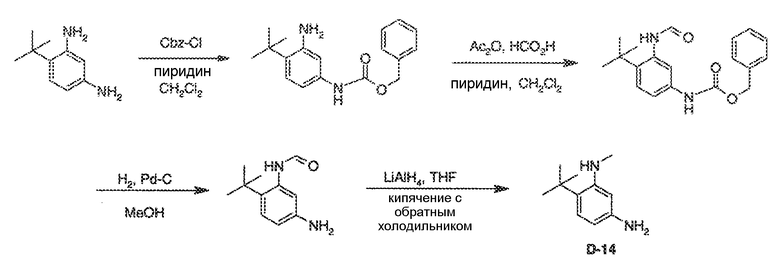
Бензиловый эфир (3-амино-4-трет-бутил-фенил)-карбаминовой кислоты
Раствор 4-трет-бутилбензол-1,3-диамина (D-9) (657 мг, 4 ммоль) и пиридина (0,39 мл, 4,8 ммоль) в CH2Cl2/MeOH (12/1, 8 мл) охлаждали до 0ºC и добавляли по каплям раствор бензилхлорформиата (0,51 мл, 3,6 ммоль) в CH2Cl2 (8 мл) в течение 10 минут. Смесь перемешивали при 0ºC в течение 15 минут, затем нагревали до комнатной температуры. Через 1 час смесь промывали раствором 1M лимонной кислоты (2×20 мл), насыщенным водным раствором бикарбоната натрия (20 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного бензилового эфира (3-амино-4-трет-бутил-фенил)-карбаминовой кислоты в виде коричневого вязкой смолы (0,97 г), которую использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,41-7,32 (м, 6H), 7,12 (д, J=8,5 Гц, 1H), 6,89 (шир.с, 1H), 6,57 (дд, J=2,3, 8,5 Гц, 1H), 5,17 (с, 2H), 3,85 (шир.с, 2H), 1,38 (с, 9H); 13C ЯМР (100 МГц, CDCl3, ротамерный) δ 153,3 (шир.), 145,3, 136,56, 136,18, 129,2, 128,73, 128,59, 128,29, 128,25, 127,14, 108,63 (шир.), 107,61 (шир.), 66,86, 33,9, 29,7; ESI-MS 299,1 m/z (MH+).
Бензиловый эфир (4-трет-бутил-3-формиламино-фенил)-карбаминовой кислоты
Раствор бензилового эфира (3-амино-4-трет-бутил-фенил)-карбаминовой кислоты (0,97 г, 3,25 ммоль) и пиридина (0,43 мл, 5,25 ммоль) в CH2Cl2 (7,5 мл) охлаждали до 0ºC и добавляли по каплям раствор муравьиного-уксусного ангидрида (3,5 ммоль, полученного путем смешивания муравьиной кислоты (158 мкл, 4,2 ммоль, 1,3 экв.) и уксусного ангидрида (0,32 мл, 3,5 ммоль, 1,1 экв.) в чистом виде и старения в течение 1 часа) в CH2Cl2 (2,5 мл) в течение 2 минут. После завершения добавления смеси давали нагреться до комнатной температуры, при этом происходило образование осадка, и полученную суспензию перемешивали в течение ночи. Смесь промывали раствором 1 M лимонной кислоты (2×20 мл), насыщенным водным раствором бикарбоната натрия (20 мл), сушили (Na2SO4) и фильтровали. Из мутной смеси осаждался тонкий слой твердого вещества над осушителем, ВЭЖХ анализ показал, что это желаемый формамид. Фильтрат концентрировали до приблизительно 5 мл и разбавляли гексаном (15 мл) для осаждения дополнительного количества формамида. Осушитель (Na2SO4) суспендировали с метанолом (50 мл), фильтровали и фильтрат объединяли с веществом из перекристаллизации из CH2Cl2/гексана. Полученную смесь концентрировали с получением бензилового эфира (4-трет-бутил-3-формиламино-фенил)-карбаминовой кислоты в виде не совсем белого твердого вещества (650 мг, 50% за 2 стадии). 1H и 13C ЯМР (CD3OD) показали продукт в виде ротамерной смеси. 1H ЯМР (400 МГц, CD3OD, ротамерный) δ 8,27 (с, 1H-a), 8,17 (с, 1H-b), 7,42-7,26 (м, 8H), 5,17 (с, 1H-a), 5,15 (с, 1H-b), 4,86 (с, 2H), 1,37 (с, 9H-a), 1,36 (с, 9H-b); 13C ЯМР (100 МГц, CD3OD, ротамерный) δ 1636,9, 163,5, 155,8, 141,40, 141,32, 139,37, 138,88, 138,22, 138,14, 136,4, 135,3, 129,68, 129,65, 129,31, 129,24, 129,19, 129,13, 128,94, 128,50, 121,4 (шир.), 118,7 (шир.), 67,80, 67,67, 35,78, 35,52, 31,65, 31,34; ESI-MS 327,5 m/z (MH+).
N-(5-Амино-2-трет-бутил-фенил)-формамид
В 100-мл колбу загружали бензиловый эфир (4-трет-бутил-3-формиламино-фенил)-карбаминовой кислоты (650 мг, 1,99 ммоль), метанол (30 мл) и 10% Pd-C (50 мг) и перемешивали в атмосфере H2 (1 атм) в течение 20 часов. Добавляли CH2Cl2 (5 мл) для гашения катализатора и смесь затем фильтровали через Целит и концентрировали с получением N-(5-амино-2-трет-бутил-фенил)-формамида в виде не совсем белого твердого вещества (366 мг, 96%). Ротамерный по данным 1H и 13C ЯМР (DMSO-d6). 1H ЯМР (400 МГц, DMSO-d6, ротамерный) δ 9,24 (д, J=10,4 Гц, 1H), 9,15 (с, 1H), 8,23 (д, J=1,5 Гц, 1H), 8,06 (д, J=10,4 Гц, 1H), 7,06 (д, J=8,5 Гц, 1H), 7,02 (д, J=8,5 Гц, 1H), 6,51 (д, J=2,5 Гц, 1H), 6,46 (дд, J=2,5, 8,5 Гц, 1H), 6,39 (дд, J=2,5, 8,5 Гц, 1H), 6,29 (д, J=2,5Гц, 1H), 5,05 (с, 2H), 4,93 (с, 2H), 1,27 (с, 9H); 13C ЯМР (100 МГц, DMSO-d6, ротамер) δ 164,0, 160,4, 147,37, 146,74, 135,38, 135,72, 132,48, 131,59, 127,31, 126,69, 115,15, 115,01, 112,43, 112,00, 33,92, 33,57, 31,33, 30,92; ESI-MS 193,1 m/z (MH+).
D-14; 4-трет-бутил-N 3 -метил-бензол-1,3-диамин
В 100-мл колбу загружали N-(5-амино-2-трет-бутил-фенил)-формамид (340 мг, 1,77 ммоль) и продували азотом. Добавляли ТГФ (10 мл) и раствор охлаждали до 0ºC. Добавляли раствор литийалюминийгидрида в ТГФ (4,4 мл, 1M раствор) в течение 2 минут. Смеси затем давали нагреться до комнатной температуры. После кипячения с обратным холодильником в течение 15 часов желтую суспензию охлаждали до 0ºC, гасили водой (170 мкл), 15% водным раствором NaOH (170 мкл) и водой (510 мкл), которые добавляли последовательно, и перемешивали при комнатной температуре в течение 30 минут. Смесь фильтровали через Целит и фильтровальную лепешку промывали метанолом (50 мл). Объединенные фильтраты концентрировали в вакууме с получением серо-коричневого твердого вещества, которое распределяли между хлороформ (75 мл) и водой (50 мл). Органический слой отделяли, промывали водой (50 мл), сушили (Na2SO4), фильтровали и концентрировали с получением 4-трет-бутил-N3-метил-бензол-1,3-диамина (D-14) в виде коричневого масла, которое отверждалось при выстаивании (313 мг, 98%). 1H ЯМР (400 МГц, CDCl3) δ 7,01 (д, J=8,1 Гц, 1H), 6,05 (дд, J=2,4, 8,1 Гц, 1H), 6,03 (д, J=2,4 Гц, 1H), 3,91 (шир.с, 1H), 3,52 (шир.с, 2H), 2,86 (с, 3H), 1,36 (с, 9H); 13C ЯМР (100 МГц, CDCl3) δ 148,4, 145,7, 127,0, 124,3, 103,6, 98,9, 33,5, 31,15, 30,31; ESI-MS 179,1 m/z (MH+).
Пример 5:
Общая схема:
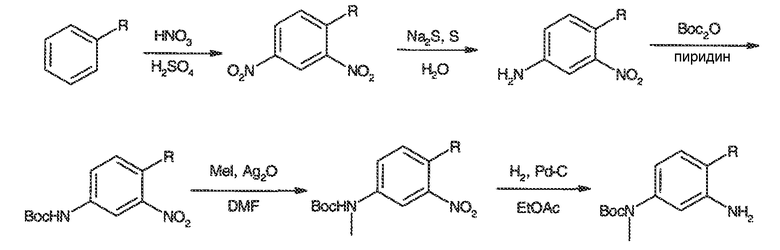
Специальный пример:
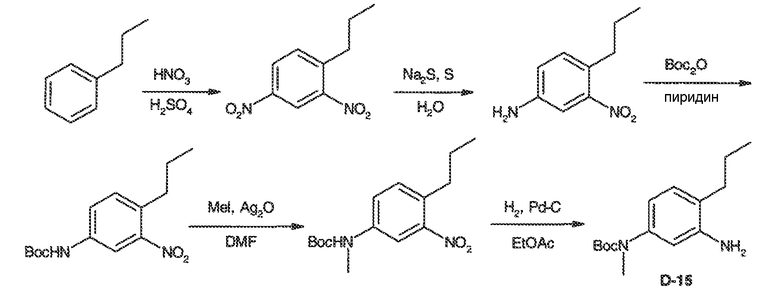
2,4-Динитро-пропилбензол
Раствор пропилбензола (10 г, 83 ммоль) в концентрированной H2SO4 (50 мл) охлаждали при 0ºC в течение 30 минут и добавляли по порциям раствор концентрированной H2SO4 (50 мл) и дымящей HNO3 (25 мл), предварительно охлажденный до 0ºC, в течение 15 минут. Смесь перемешивали при 0ºC еще в течение 30 минут и затем давали нагреться до комнатной температуры. Смесь выливали в лед (200 г) - воду (100 мл) и экстрагировали простым эфиром (2×100 мл). Объединенные экстракты промывали H2O (100 мл) и насыщенным солевым раствором (100 мл), сушили над MgSO4, фильтровали и концентрировали с получением 2,4-динитро-пропилбензола (15,6 г, 89%). 1H ЯМР (CDCl3, 300 МГц) δ 8,73 (д, J=2,2 Гц, 1H), 8,38 (дд, J=8,3, 2,2 Гц, 1H), 7,6 (д, J=8,5 Гц, 1H), 2,96 (м, 2H), 1,73 (м, 2H), 1,06 (т, J=7,4 Гц, 3H).
4-Пропил-3-нитроанилин
Суспензию 2,4-динитро-пропилбензола (2 г, 9,5 ммоль) в H2O (100 мл) нагревали при температуре около температуры кипения с обратным холодильником и интенсивно перемешивали. Добавляли по каплям прозрачный оранжево-красный раствор полисульфида (300 мл (10 экв.), предварительно полученного путем нагревания сульфида натрия наногидрата (10,0 г), порошка серы (2,60 г) и H2O (400 мл), в течение 45 минут. Красно-коричневый раствор нагревали при температуре кипения с обратным холодильником в течение 1,5 часов. Смесь охлаждали до 0ºC и затем экстрагировали простым эфиром (2×200 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 4-пропил-3-нитроанилина (1,6 г, 93%), который использовали без дополнительной очистки.
трет-бутиловый эфир (3-нитро-4-пропил-фенил)-карбаминовой кислота
4-Пропил-3-нитроанилин (1,69 г, 9,4 ммоль) растворяли в пиридине (30 мл) при перемешивании. Добавляли Boc ангидрид (2,05 г, 9,4 ммоль). Смесь перемешивали и нагревали при температуре кипения с обратным холодильником в течение 1 часа, затем растворитель удаляли в вакууме. Полученное масло снова растворяли в CH2Cl2 (300 мл) и промывали водой (300 мл) и насыщенным солевым раствором (300 мл), сушили над Na2SO4, фильтровали и концентрировали. Неочищенное масло, которое содержало как моно-, так и бис-ацилированные нитропродукты, очищали колоночной хроматографией (0-10% CH2Cl2-MeOH) с получением трет-бутилового эфира (3-нитро-4-пропил-фенил)-карбаминовой кислоты (2,3 г, 87%).
трет-бутиловый эфир метил-(3-нитро-4-пропил-фенил)-карбаминовой кислоты
К раствору трет-бутилового эфира (3-нитро-4-пропил-фенил)-карбаминовой кислоты (200 мг, 0,71 ммоль) в DMF (5 мл) добавляли Ag2O (1,0 г, 6,0 ммоль) с последующим добавлением метилиодида (0,20 мл, 3,2 ммоль). Полученную суспензию перемешивали при комнатной температуре в течение 18 часов и фильтровали через слой Целита. Фильтровальную лепешку промывали при помощи CH2Cl2 (10 мл). Фильтрат концентрировали в вакууме. Неочищенное масло очищали колоночной хроматографией (0-10% CH2Cl2-MeOH) с получением трет-бутилового эфира метил-(3-нитро-4-пропил-фенил)-карбаминовой кислоты в виде желтого масла (110 мг, 52%). 1H ЯМР (CDCl3, 300 МГц) δ 7,78 (д, J=2,2 Гц, 1H), 7,42 (дд, J=8,2, 2,2 Гц, 1H), 7,26 (д, J=8,2 Гц, 1H), 3,27 (с, 3H), 2,81 (т, J=7,7 Гц, 2H), 1,66 (м, 2H), 1,61 (с, 9H), 0,97 (т, J=7,4 Гц, 3H).
D-15; трет-бутиловый эфир (3-амино-4-пропил-фенил)-метил-карбаминовой кислоты
К раствору трет-бутилового эфира метил-(3-нитро-4-пропил-фенил)-карбаминовой кислоты (110 мг, 0,37 ммоль) в EtOAc (10 мл) добавляли 10% Pd-C (100 мг). Полученную суспензию перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 2 дней. Развие реакции отслеживали при помощи ТСХ. После завершения добавления реакционную смесь фильтровали через слой Целита. Фильтрат концентрировали в вакууме с получением трет-бутилового эфира (3-амино-4-пропил-фенил)-метил-карбаминовой кислоты (D-15) в виде бесцветного кристаллического соединения (80 мг, 81%). ESI-MS 265,3 m/z (MH+).
Другие примеры:
D-16; трет-бутиловый эфир (3-амино-4-этил-фенил)-метил-карбаминовой кислоты
трет-Бутиловый эфир (3-амино-4-этил-фенил)-метил-карбаминовой кислоты (D-16) синтезировали в соответствии с общей схемой, представленной выше, исходя из этилбензола. Общий выход (57%).
D-17; трет-бутиловый эфир (3-амино-4-изопропил-фенил)-метил-карбаминовой кислоты
трет-Бутиловый эфир (3-амино-4-изопропил-фенил)-метил-карбаминовой кислоты (D-17) синтезировали в соответствии с общей схемой, представленной выше, исходя из изопропилбензола. Общий выход (38%).
Пример 6:
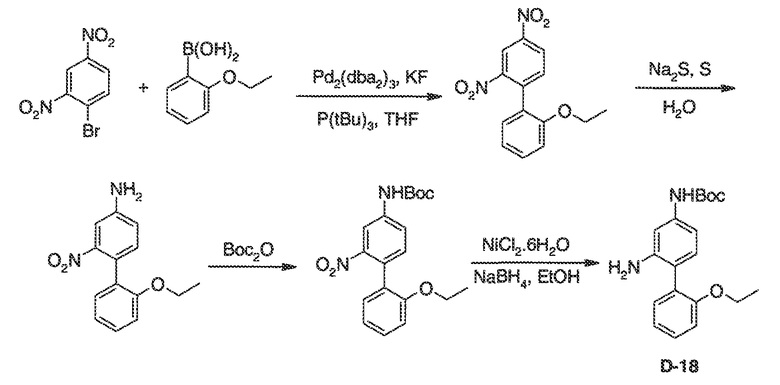
2'-Этокси-2,4-динитро-бифенил
В колбу высокого давления загружали 2-этоксифенилбороновую кислоту (0,66 г, 4,0 ммоль), KF (0,77 г, 13 ммоль), Pd2(dba)3 (16 мг, 0,02 ммоль) и 2,4-динитро-бромбензол (0,99 г, 4,0 ммоль) в ТГФ (5 мл). Сосуд продували аргоном в течение 1 минуты с последующим добавлением три-трет-бутилфосфина (0,15 мл, 0,48 ммоль, 10% раствор в гексане). Реакционный сосуд продували аргоном еще в течение 1 минуты, герметично закрывали и нагревали при 80ºC в течение ночи. После охлаждения до комнатной температуры раствор фильтровали через слой Целита. Фильтровальную лепешку промывали при помощи CH2Cl2 (10 мл) и объединенные органические экстракты концентрировали при пониженном давлении с получением неочищенного продукта 2'-этокси-2,4-динитро-бифенила (0,95 г, 82%). Не осуществляли никакой дополнительной очистки. 1H ЯМР (300 МГц, CDCl3) δ 8,75 (с, 1H), 8,43 (д, J=8,7 Гц, 1H), 7,60 (д, J=8,4 Гц, 1H), 7,40 (т, J=7,8 Гц, 1H), 7,31 (д, J=7,5 Гц, 1H), 7,08 (т, J=7,5 Гц, 1H), 6,88 (д, J=8,4 Гц, 1H), 3,44 (кв., J=6,6 Гц, 2H), 1,24 (т, J=6,6 Гц, 3H); ВЭЖХ время удерживания 3,14 минут, 10-100% CH3CN, 5 мин градиент.
2'-Этокси-2-нитробифенил-4-иламин
Прозрачный оранжево-красный раствор полисульфида (120 мл, 7,5 экв.), предварительно полученный путем нагревания сульфида натрия моногидрата (10 г), серы (1,04 г) и воды (160 мл), добавляли по каплям при 90ºC в течение 45 минут к суспензии 2'-этокси-2,4-динитро-бифенила (1,2 г, 4,0 ммоль) в воде (40 мл). Красно-коричневый раствор нагревали при температуре кипения с обратным холодильником в течение 1,5 часов. Смесь охлаждали до комнатной температуры и добавляли твердый NaCl (5 г). Раствор экстрагировали при помощи CH2Cl2 (3×50 мл) и объединенные органические экстракты концентрировали с получением 2'-этокси-2-нитробифенил-4-иламина (0,98 г, 95%), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (300 МГц, CDCl3) δ 7,26 (м, 2H), 7,17 (д, J=2,7 Гц, 1H), 7,11 (д, J=7,8 Гц, 1H), 7,00 (т, J=6,9 Гц, 1H), 6,83 (м, 2H), 3,91 (кв., J=6,9 Гц, 2H), 1,23 (т, J=7,2 Гц, 3H); ВЭЖХ время удерживания 2,81 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 259,1 m/z (MH+).
трет-бутиловый эфир (2'-этокси-2-нитробифенил-4-ил)-карбаминовой кислоты
Смесь 2'-этокси-2-нитробифенил-4-иламина (0,98 г, 4,0 ммоль) и Boc2O (2,6 г, 12 ммоль) нагревали при помощи фена. После того, как исходное вещество было израсходовано, как показал анализ ТСХ, неочищенную смесь очищали флэш-хроматографией (силикагель, CH2Cl2) с получением трет-бутилового эфира (2'-этокси-2-нитробифенил-4-ил)-карбаминовой кислоты (1,5 г, 83%). 1H ЯМР (300 МГц, CDCl3) δ 7,99 (с, 1H), 7,55 (д, J=8,4 Гц, 1H), 7,25 (м, 3H), 6,99 (т, J=7,5 Гц, 1H), 6,82 (м, 2H), 3,88 (кв., J=6,9 Гц, 2H), 1,50 (с, 9H), 1,18 (т, J=6,9 Гц, 3H); ВЭЖХ время удерживания 3,30 минут, 10-100% CH3CN, 5 мин градиент.
D-18; трет-бутиловый эфир (2'-этокси-2-аминобифенил-4-ил)-карбаминовой кислоты
К раствору NiCl2.6H2O (0,26 г, 1,1 ммоль) в EtOH (5 мл) добавляли NaBH4 (40 мг, 1,1 ммоль) при -10ºC. Наблюдали выделение газа, и происходило образование черного осадка. После перемешивания в течение 5 минут добавляли раствор трет-бутилового эфира 2'-этокси-2-нитробифенил-4-ил)карбаминовой кислоты (0,50 г, 1,1 ммоль) в EtOH (2 мл). Добавляли дополнительное количество NaBH4 (80 мг, 60 ммоль) 3 порциями в течение 20 минут. Реакционную смесь перемешивали при 0ºC в течение 20 минут с последующим добавлением NH4OH (4 мл, 25% водный раствор). Полученный раствор перемешивали в течение 20 минут. Неочищенную смесь фильтровали через короткую пробку из диоксида кремния. Лепешку из диоксида кремния промывали 5% раствором MeOH в CH2Cl2 (10 мл) и объединенные органические экстракты концентрировали при пониженном давлении с получением трет-бутилового эфира (2'-этокси-2-аминобифенил-4-ил)-карбаминовой кислоты (D-18) (0,36 г, количественный), который использовали без дополнительной очистки. ВЭЖХ время удерживания 2,41 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 329,3 m/z (MH+).
Пример 7:
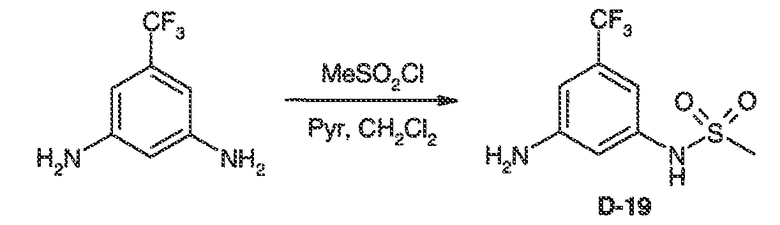
D-19; N-(3-Амино-5-трифторметил-фенил)-метансульфонамид
Раствор 5-трифторметил-бензол-1,3-диамина (250 мг, 1,42 ммоль) в пиридине (0,52 мл) и CH2Cl2 (6,5 мл) охлаждали до 0ºC. Медленно добавляли метансульфонилхлорид (171 мг, 1,49 ммоль) при такой скорости, чтобы температура раствора оставалась ниже 10ºC. Смесь перемешивали при ~8ºC и затем через 30 минут давали нагреться до комнатной температуры. После перемешивания при комнатной температуре в течение 4 часов реакция практически завершалась, как показал анализ LCMS. Реакцию гасили насыщенным водным раствором NH4Cl (10 мл), экстрагировали при помощи CH2Cl2 (4×10 мл), сушили над Na2SO4, фильтровали и концентрировали с получением N-(3-амино-5-трифторметил-фенил)-метансульфонамида (D-19) в виде красноватого полутвердого вещества (0,35 г, 97%), которое использовали без дополнительной очистки. 1H-ЯМР (CDCl3, 300 МГц) δ 6,76 (м, 1H), 6,70 (м, 1H), 6,66 (с, 1H), 3,02 (с, 3H); ESI-MS 255,3 m/z (MH+).
Циклические амины
Пример 1:
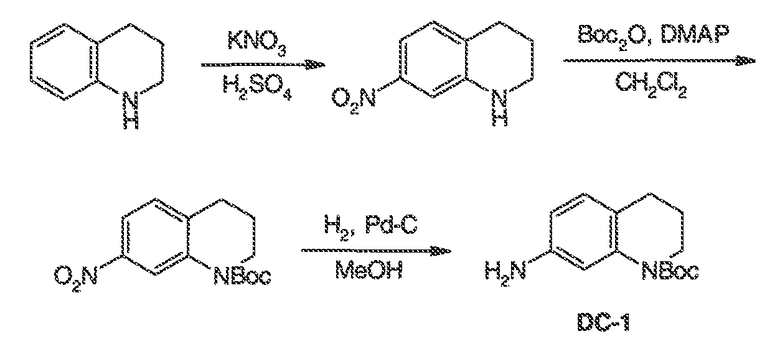
7-Нитро-1,2,3,4-тетрагидро-хинолин
К смеси 1,2,3,4-тетрагидро-хинолина (20,0 г, 0,15 моль), растворенного в H2SO4 (98%, 150 мл), медленно добавляли KNO3 (18,2 г, 0,18 моль) при 0ºC. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Смесь затем выливали в ледяную воду и подщелачивали насыщенным раствором NaHCO3 до pH 8. После экстрагирования при помощи CH2Cl2 объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 10:1) с получением 7-нитро-1,2,3,4-тетрагидро-хинолина (6,6 г, 25%).
трет-бутиловый эфир 7-нитро-3,4-дигидро-2H-хинолин-1-карбоновой кислоты
Смесь 7-нитро-1,2,3,4-тетрагидро-хинолина (4,0 г, 5,61 ммоль), Boc2O (1,29 г, 5,89 ммоль) и DMAP (0,4 г) в CH2Cl2 перемешивали при комнатной температуре в течение ночи. После разбавления водой смесь экстрагировали при помощи CH2Cl2. Объединенные органические слои промывали NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного трет-бутилового эфира 7-нитро-3,4-дигидро-2H-хинолин-1-карбоновой кислоты, который использовали на следующей стадии без дополнительной очистки.
DC-1; трет-бутил 7-амино-3,4-дигидрохинолин-1(2H)-карбоксилат
Суспензию неочищенного трет-бутилового эфира 7-нитро-3,4-дигидро-2H-хинолин-1-карбоновой кислоты (4,5 г, 16,2 моль) и 10% Pd-C (0,45 г) в MeOH (40 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение ночи. После фильтрования фильтрат концентрировали и остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 5:1) с получением трет-бутил 7-амино-3,4-дигидрохинолин-1(2H)-карбоксилата (DC-1) в виде коричневого твердого вещества (1,2 г, 22% за 2 стадии). 1H ЯМР (CDCl3) δ 7,15 (д, J=2 Гц, 1H), 6,84 (д, J=8 Гц, 1H), 6,36-6,38 (м, 1H), 3,65-3,68 (м, 2H), 3,10 (шир.с, 2H), 2,66 (т, J=6,4 Гц, 2H), 1,84-1,90 (м, 2H), 1,52 (с, 9H); ESI-MS 496,8 m/z (2M+H+).
Пример 2:
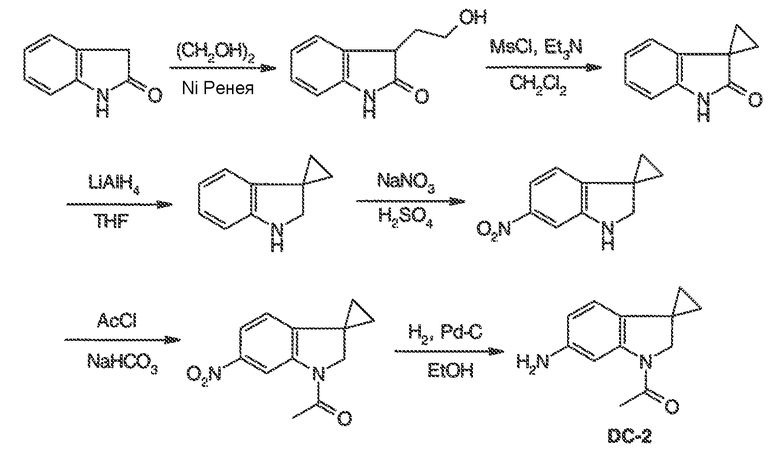
3-(2-Гидрокси-этил)-1,3-дигидро-индол-2-он
Перемешиваемую смесь оксиндола (5,7 г, 43 ммоль) и никеля Ренея (10 г) в этан-1,2-диоле (100 мл) нагревали в автоклаве. После завершения реакции смесь фильтровали и избыток диола удаляли в вакууме. Остаточное масло растирали в порошок с гексаном с получением 3-(2-гидрокси-этил)-1,3-дигидро-индол-2-она в виде бесцветного кристаллического твердого вещества (4,6 г, 70%).
1,2-Дигидро-3-спиро-1'-циклопропил-1H-индол-2-он
К раствору 3-(2-гидрокси-этил)-1,3-дигидро-индол-2-она (4,6 г, 26 ммоль) и триэтиламина (10 мл) в CH2Cl2 (100 мл) добавляли MsCl (3,4 г, 30 ммоль) по каплям при -20ºC. Смеси затем давали нагреться до комнатной температуры и перемешивали в течение ночи. Смесь фильтровали и фильтрат концентрировали в вакууме. Остаток очищали колоночной хроматографией с получением неочищенного 1,2-дигидро-3-спиро-1'-циклопропил-1H-индол-2-она в виде желтого твердого вещества (2,5 г), которое использовали непосредственно на следующей стадии.
1,2-Дигидро-3-спиро-1'-циклопропил-1H-индол
К раствору 1,2-дигидро-3-спиро-1'-циклопропил-1H-индол-2-она (2,5 г, неочищенный) в ТГФ (50 мл) добавляли LiAlH4 (2 г, 52 ммоль) по порциям. После нагревания смеси до температуры кипения с обратным холодильником смесь выливали в колотый лед, подщелачивали водным раствором аммиака до pH 8 и экстрагировали при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного 1,2-дигидро-3-спиро-1'-циклопропил-1H-индола в виде желтого твердого вещества (около 2 г), который использовали непосредственно на следующей стадии.
6-Нитро-1,2-дигидро-3-спиро-1'-циклопропил-1H-индол
К охлажденному раствору (-5ºC до -10ºC) NaNO3 (1,3 г, 15,3 ммоль) в H2SO4 (98%, 30 мл) добавляли 1,2-дигидро-3-спиро-1'-циклопропил-1H-индол (2 г, неочищенн) по каплям в течение 20 минут. После добавления реакционную смесь перемешивали еще в течение 40 минут и выливали в колотый лед (20 г). Охлажденную смесь затем подщелачивали при помощи NH4OH и экстрагировали при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении с получением 6-нитро-1,2-дигидро-3-спиро-1'-циклопропил-1H-индола в виде темно-серого твердого вещества (1,3 г)
1-Ацетил-6-нитро-1,2-дигидро-3-спиро-1'-циклопропил-1H-индол
NaHCO3 (5 г) суспендировали в растворе 6-нитро-1,2-дигидро-3-спиро-1'-циклопропил-1H-индола (1,3 г, неочищенный) в CH2Cl2 (50 мл). При интенсивном перемешивании добавляли по каплям ацетилхлорид (720 мг). Смесь перемешивали в течение 1 часа и фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле с получением 1-ацетил-6-нитро-1,2-дигидро-3-спиро-1'-циклопропил-1H-индола (0,9 г, 15% за 4 стадии).
DC-2; 1-Ацетил-6-амино-1,2-дигидро-3-спиро-1'-циклопропил-1H-индол
Смесь 1-ацетил-6-нитро-1,2-дигидро-3-спиро-1'-циклопропил- 1H-индола (383 мг, 2 ммоль) и Pd-C (10%, 100 мг) в EtOH (50 мл) перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 1,5 часов. Катализатор отфильтровывали и фильтрат концентрировали при пониженном давлении. Остаток обрабатывали при помощи HCl/MeOH с получением 1-ацетил-6-амино-1,2-дигидро-3-спиро-1'-циклопропил-1H-индола (DC-2) (300 мг, 90%) в виде гидрохлоридной соли.
Пример 3:
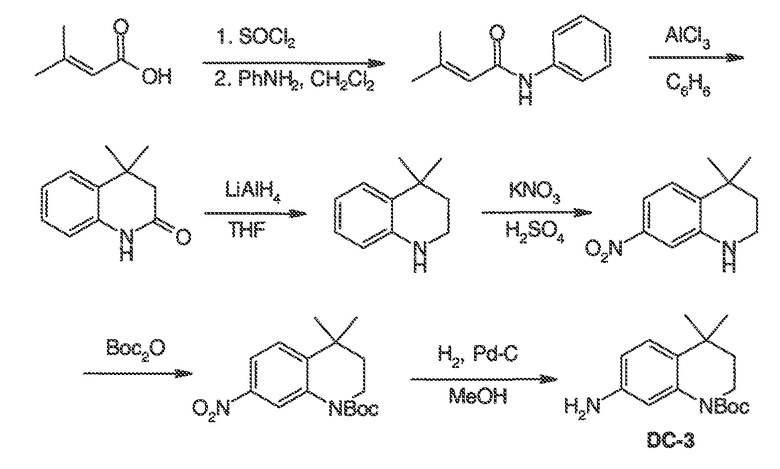
Фениламид 3-метил-бут-2-еновой кислоты
Смесь 3-метил-бут-2-еновой кислоты (100 г, 1 моль) и SOCl2 (119 г, 1 моль) нагревали при температуре кипения с обратным холодильником в течение 3 часов. Избыток SOCl2 удаляли при пониженном давлении. Добавляли CH2Cl2 (200 мл) с последующим добавлением анилина (93 г, 1,0 моль) в Et3N (101 г, 1 моль) при 0ºC. Смесь перемешивали при комнатной температуре в течение 1 часа и гасили при помощи HCl (5%, 150 мл). Водный слой отделяли и экстрагировали при помощи CH2Cl2. Объединенные органические слои промывали водой (2×100 мл) и насыщенным солевым раствором (100 мл), сушили над Na2SO4 и концентрировали с получением фениламида 3-метил-бут-2-еновой кислоты (120 г, 80%).
4,4-Диметил-3,4-дигидро-1H-хинолин-2-он
AlCl3 (500 г, 3,8 моль) осторожно добавляли к суспензии 3-фениламида метил-бут-2-еновой кислоты (105 г, 0,6 моль) в бензоле (1000 мл). Реакционную смесь перемешивали при 80ºC в течение ночи и выливали в ледяную воду. Органический слой отделяли и водный слой экстрагировали этилацетатом (250 мл ×3). Объединенные органические слои промывали водой (200 мл ×2) и насыщенным солевым раствором (200 мл), сушили над Na2SO4 и концентрировали с получением 4,4-диметил-3,4-дигидро-1H-хинолин-2-она (90 г, 86%).
4,4-Диметил-1,2,3,4-тетрагидро-хинолин
Раствор 4,4-диметил-3,4-дигидро-1H-хинолин-2-она (35 г, 0,2 моль) в ТГФ (100 мл) добавляли по каплям к суспензии LiAlH4 (18 г, 0,47 моль) в ТГФ (200 мл) при 0ºC. После добавления смесь перемешивали при комнатной температуре в течение 30 минут и затем медленно нагревали до температуры кипения с обратным холодильником в течение 1 часа. Смесь затем охлаждали до 0ºC. Осторожно добавляли воду (18 мл) и раствор NaOH (10%, 100 мл) для гашения реакции. Твердое вещество отфильтровывали и фильтрат концентрировали с получением 4,4-диметил-1,2,3,4-тетрагидро-хинолина.
4,4-Диметил-7-нитро-1,2,3,4-тетрагидро-хинолин
К смеси 4,4-диметил-1,2,3,4-тетрагидро-хинолина (33 г, 0,2 моль) в H2SO4 (120 мл) медленно добавляли KNO3 (20,7 г, 0,2 моль) при 0ºC. После добавления смесь перемешивали при комнатной температуре в течение 2 часов, осторожно выливали в ледяную воду и подщелачивали при помощи Na2CO3 до pH 8. Смесь экстрагировали этилацетатом (3×200 мл). Объединенные экстракты промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением 4,4-диметил-7-нитро-1,2,3,4-тетрагидро-хинолина (21 г, 50%).
трет-бутиловый эфир 4,4-диметил-7-нитро-3,4-дигидро-2H-хинолин-1-карбоновой кислоты
Смесь 4,4-диметил-7-нитро-1,2,3,4-тетрагидро-хинолина (25 г, 0,12 моль) и Boc2O (55 г, 0,25 моль) перемешивали при 80ºC в течение 2 дней. Смесь очищали хроматографией на силикагеле с получением трет-бутилового эфира 4,4-диметил-7-нитро-3,4-дигидро-2H-хинолин-1-карбоновой кислота (8 г, 22%).
DC-3; трет-бутил 7-амино-3,4-дигидро-4,4-диметилхинолин-1(2H)-карбоксилат
Смесь трет-бутилового эфира 4,4-диметил-7-нитро-3,4-дигидро-2H-хинолин-1 карбоновой кислоты (8,3 г, 0,03 моль) и Pd-C (0,5 г) в метаноле (100 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение ночи. Катализатор отфильтровывали и фильтрат концентрировали. Остаток промывали петролейным эфиром с получением трет-бутил 7-амино-3,4-дигидро-4,4-диметилхинолин-1(2H)-карбоксилата (DC-3) (7,2 г, 95%). 1H ЯМР (CDCl3) δ 7,11-7,04 (м, 2H), 6,45-6,38 (м, 1H), 3,71-3,67 (м, 2H), 3,50-3,28 (м, 2H), 1,71-1,67 (м, 2H), 1,51 (с, 9H), 1,24 (с, 6H).
Пример 4:
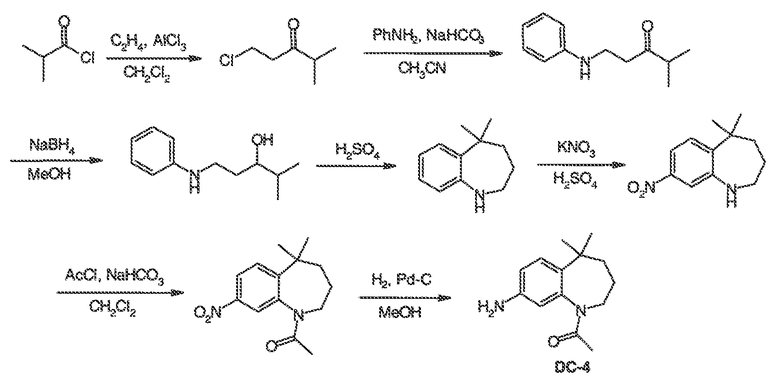
1-Хлор-4-метилпентан-3-он
Этилен пропускали через раствор изобутирилхлорида (50 г, 0,5 моль) и AlCl3 (68,8 г, 0,52 моль) в безводном CH2Cl2 (700 мл) при 5ºC. Через 4 часа абсорбция этилена прекращалась, и смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в холодный разбавленный раствор HCl и экстрагировали при помощи CH2Cl2. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного 1-хлор-4-метилпентан-3-она, который использовали непосредственно на следующей стадии без дополнительной очистки.
4-Метил-1-(фениламино)-пентан-3-он
Суспензию неочищенного 1-хлор-4-метилпентан-3-она (около 60 г), анилина (69,8 г, 0,75 моль) и NaHCO3 (210 г, 2,5 моль) в CH3CN (1000 мл) нагревали при температуре кипения с обратным холодильником в течение ночи. После охлаждения нерастворимую соль отфильтровывали и фильтрат концентрировали. Остаток разбавляли при помощи CH2Cl2, промывали 10% раствором HCl (100 мл) и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного 4-метил-1-(фениламино)-пентан-3-она.
4-Метил-1-(фениламино)-пентан-3-ол
При -10ºC, NaBH4 (56,7 г, 1,5 моль) постепенно добавляли к смеси неочищенного 4-метил-1-(фениламино)-пентан-3-она (около 80 г) в MeOH (500 мл). После добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 20 минут. Растворитель удаляли и остаток снова распределяли между водой и CH2Cl2. Органическую фазу отделяли, промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Полученную смолу растирали в порошок с простым эфиром с получением 4-метил-1-(фениламино)-пентан-3-ола в виде белого твердого вещества (22 г, 23%).
5,5-Диметил-2,3,4,5-тетрагидро-1H-бензо[b]азепин
Смесь 4-метил-1-(фениламино)-пентан-3-ола (22 г, 0,11 моль) в 98% H2SO4 (250 мл) перемешивали при 50ºC в течение 30 минут. Реакционную смесь выливали в ледяную воду, подщелачивали насыщенным раствором NaOH до pH 8 и экстрагировали при помощи CH2Cl2. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир) с получением 5,5-диметил-2,3,4,5-тетрагидро-1H-бензо[b]азепина в виде коричневого масла (1,5 г, 8%).
5,5-Диметил-8-нитро-2,3,4,5-тетрагидро-1H-бензо[b]азепин
При 0ºC, KNO3 (0,76 г, 7,54 ммоль) добавляли по порциям к раствору 5,5-диметил-2,3,4,5-тетрагидро-1H-бензо[b]азепина (1,1 г, 6,28 ммоль) в H2SO4 (15 мл). После перемешивания в течение 15 минут при этой температуре смесь выливали в ледяную воду, подщелачивали насыщенным раствором NaHCO3 до pH 8 и экстрагировали при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного 5,5-диметил-8-нитро-2,3,4,5-тетрагидро-1H-бензо[b]азепина (1,2 г), который использовали непосредственно на следующей стадии без дополнительной очистки.
1-(5,5-диметил-8-нитро-2,3,4,5-тетрагидробензо[b]азепин-1-ил)этанон
Ацетилхлорид (0,77 мл, 11 ммоль) добавляли к суспензии неочищенного 5,5-диметил-8-нитро-2,3,4,5-тетрагидро-1H-бензо[b]азепина (1,2 г, 5,45 ммоль) и NaHCO3 (1,37 г, 16,3 ммоль) в CH2Cl2 (20 мл). Смесь нагревали при температуре кипения с обратным холодильником в течение 1 часа. После охлаждения смесь выливали в воду и экстрагировали при помощи CH2Cl2. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией с получением 1-(5,5-диметил-8-нитро-2,3,4,5-тетрагидробензо[b]азепин-1-ил)этанона (1,05 г, 64% за две стадии).
DC-4; 1-(8-Амино-2,3,4,5-тетрагидро-5,5-диметилбензо[b]азепин-1-ил)этанон
Суспензию 1-(5,5-диметил-8-нитро-2,3,4,5-тетрагидробензо[b]азепин-1-ил)этанона (1,05 г, 40 ммоль) и 10% Pd-C (0,2 г) в MeOH (20 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 4 часов. После фильтрования фильтрат концентрировали с получением 1-(8-амино-2,3,4,5-тетрагидро-5,5-диметилбензо[b]азепин-1-ил)этанона в виде белого твердого вещества (DC-4) (880 мг, 94%). 1H ЯМР (CDCl3) δ 7,06 (д, J=8,0 Гц, 1H), 6,59 (дд, J=8,4, 2,4 Гц, 1H), 6,50 (шир.с, 1H), 4,18-4,05 (м, 1H), 3,46-3,36 (м, 1H), 2,23 (с, 3H), 1,92-1,85 (м, 1H), 1,61-1,51 (м, 3H), 1,21 (с, 3H), 0,73 (т, J=7,2 Гц, 3H); ESI-MS 233,0 m/z (MH+).
Пример 5:

Спиро[1H-инден-1,4'-пиперидин]-3(2H)-он, 1'-бензил
Смесь спиро[1H-инден-1,4'-пиперидин]-1'-карбоновой кислоты, 2,3-дигидро-3-оксо-, 1,1-диметилэтилового эфира (9,50 г, 31,50 ммоль) в насыщенном растворе HCl/MeOH (50 мл) перемешивали при 25ºC в течение ночи. Растворитель удаляли при пониженном давлении с получением не совсем белого твердого вещества (7,50 г). К раствору этого твердого вещества в безводном CH3CN (30 мл) добавляли безводный K2CO3 (7,85 г, 56,80 ммоль). Суспензию перемешивали в течение 5 минут и добавляли по каплям бензилбромид (5,93 г, 34,65 ммоль) при комнатной температуре. Смесь перемешивали в течение 2 часов, выливали на колотый лед и экстрагировали при помощи CH2Cl2. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением неочищенного спиро[1H-инден-1,4'-пиперидин]-3(2H)-она, 1'-бензила (7,93 г, 87%), который использовали без дополнительной очистки.
Спиро[1H-инден-1,4'-пиперидин]-3(2H)-он, 1'-бензил, оксим
К раствору спиро[1H-инден-1,4'-пиперидин]-3(2H)-она, 1'-бензила (7,93 г, 27,25 ммоль) в EtOH (50 мл) добавляли гидроксиламин гидрохлорид (3,79 г, 54,50 ммоль) и безводный ацетат натрия (4,02 г, 49,01 ммоль) одной порцией. Смесь кипятили с обратным холодильником в течение 1 часа и затем охлаждали до комнатной температуры. Растворитель удаляли при пониженном давлении и добавляли 200 мл воды. Смесь экстрагировали при помощи CH2Cl2. Объединенные органические слои сушили над Na2SO4 и концентрировали с получением спиро[1H-инден-1,4'-пиперидин]-3(2H)-он, 1'-бензил, оксима (7,57 г, 91%), который использовали без дополнительной очистки.
1,2,3,4-Тетрагидрохинолин-4-спиро-4'-(N'-бензил-пиперидин)
К раствору спиро[1H-инден-1,4'-пиперидин]-3(2H)-он, 1'-бензил, оксима (7,57 г, 24,74 ммоль) в безводном CH2Cl2 (150 мл) добавляли по каплям DIBAL-H (135,7 мл, 1M в толуоле) при 0ºC. Смесь перемешивали при 0ºC в течение 3 часов, разбавляли при помощи CH2Cl2 (100 мл) и гасили при помощи NaF (20,78 г, 495 ммоль) и воды (6,7 г, 372 ммоль). Полученную суспензию интенсивно перемешивали при 0ºC в течение 30 минут. После фильтрования остаток промывали при помощи CH2Cl2. Объединенные фильтраты концентрировали в вакууме с получением коричневатого масла, которое очищали колоночной хроматографией на силикагеле (CH2Cl2-MeOH, 30:1) с получением 1,2,3,4-тетрагидрохинолин-4-спиро-4'-(N'-бензил-пиперидин) (2,72 г, 38%).
1,2,3,4-Тетрагидрохинолин-4-спиро-4'-пиперидин
Суспензию 1,2,3,4-Тетрагидрохинолин-4-спиро-4'-(N'-бензил-пиперидин) (300 мг, 1,03 ммоль) и Pd(OH)2-C (30 мг) в MeOH (3 мл) перемешивали в атмосфере H2 (55 ф/дюйм2 (3,867 кг/см2)) при 50ºC в течение ночи. После охлаждения катализатор отфильтровывали и промывали при помощи MeOH. Объединенные фильтраты концентрировали при пониженном давлении с получением 1,2,3,4-тетрагидрохинолин-4-спиро-4'-пиперидина в виде белого твердого вещества (176 мг, 85%), которое использовали без дополнительной очистки.
трет-бутиловый эфир 7'-Нитро-спиро[пиперидин-4,4'(1'H)-хинолин], 2',3'-дигидро-карбоновой кислоты
KNO3 (69,97 мг, 0,69 ммоль) добавляли по порциям к суспензии 1,2,3,4-тетрагидрохинолин-4-спиро-4'-пиперидина (133 мг, 0,66 ммоль) в 98% H2SO4 (2 мл) при 0ºC. После завершения добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали еще в течение 2 часов. Смесь затем выливали на колотый лед и подщелачивали 10% раствором NaOH до pH~8. Добавляли по каплям Boc2O (172 мг, 0,79 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь затем экстрагировали при помощи EtOAc и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутилового эфира 7'-нитро-спиро[пиперидин-4,4'(1'H)-хинолин], 2',3'-дигидро-карбоновой кислоты (230 мг), который использовали на следующей стадии без дополнительной очистки.
трет-бутиловый эфир 7'-нитро-спиро[пиперидин-4,4'(1'H)-1-ацетил-хинолин], 2',3'-дигидро-карбоновой кислоты
Ацетилхлорид (260 мг, 3,30 ммоль) добавляли по каплям к суспензии трет-бутилового эфира 7'-нитро-спиро[пиперидин-4,4'(1'H)-хинолин], 2',3'-дигидро-карбоновой кислоты (230 мг) и NaHCO3 (1,11 г, 13,17 ммоль) в MeCN (5 мл) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 4 часов. После охлаждения суспензию фильтровали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир-EtOAc, 10:1) с получением трет-бутилового эфира 7'-нитро-спиро[пиперидин-4,4'(1'H)-1-ацетил-хинолин], 2',3'-дигидро-карбоновой кислоты (150 мг, 58% за 2 стадии).
DC-5; трет-бутиловый эфир 7'-Амино-спиро[пиперидин-4,4'(1'H)-1-ацетил-хинолин], 2',3'-дигидро-карбоновой кислоты
Суспензию трет-бутилового эфира 7'-нитро-спиро[пиперидин-4,4'(1'H)-1-ацетил-хинолин], 2',3'-дигидро-карбоновой кислоты (150 мг, 0,39 ммоль) и Ni Ренея (15 мг) в MeOH (2 мл) перемешивали в атмосфере H2 (1 атм) при 25ºC в течение ночи. Катализатор удаляли фильтрованием и промывали при помощи MeOH. Объединенные фильтраты сушили над Na2SO4, фильтровали и концентрировали с получением трет-бутилового эфира 7'-амино-спиро[пиперидин-4,4'(1'H)-1-ацетил-хинолин], 2',3'-дигидро-карбоновой кислоты (DC-5) (133 мг, 96%).
Пример 7:
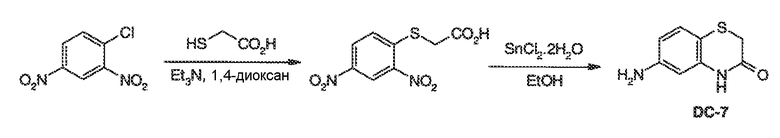
2-(2,4-Динитрофенилтио)-уксусная кислота
Et3N (1,5 г, 15 ммоль) и меркапто-уксусную кислоту (1 г, 11 ммоль) добавляли к раствору 1-хлор-2,4-динитробензола (2,26 г, 10 ммоль) в 1,4-диоксане (50 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение 5 часов добавляли H2O (100 мл). Полученную суспензию экстрагировали этилацетатом (100 мл ×3). Этилацетатный экстракт промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением 2-(2,4-динитрофенилтио)-уксусной кислоты (2,3 г, 74%), которую использовали без дополнительной очистки.
DC-7; 6-Амино-2H-бензо[b][1,4]тиазин-3(4H)-он
Раствор 2-(2,4-динитрофенилтио)-уксусной кислоты (2,3 г, 9 ммоль) и хлорида олова(II) дигидрата (22,6 г, 0,1 моль) в этаноле (30 мл) кипятили с обратным холодильником в течение ночи. После удаления растворителя при пониженном давлении оставшуюся суспензию разбавляли водой (100 мл) и подщелачивали 10% раствором Na2CO3 до pH 8. Полученную суспензию экстрагировали этилацетатом (3×100 мл). Этилацетатный экстракт промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток промывали при помощи CH2Cl2 с получением 6-амино-2H-бензо[b][1,4]тиазин-3(4H)-она (DC-7) в виде желтого порошка (1 г, 52%) 1H ЯМР (DMSO-d6) δ 10,24 (с, 1H), 6,88 (д, 1H, J=6 Гц), 6,19-6,21 (м, 2H), 5,15 (с, 2H), 3,28 (с, 2H); ESI-MS 181,1 m/z (MH+).
Пример 7:
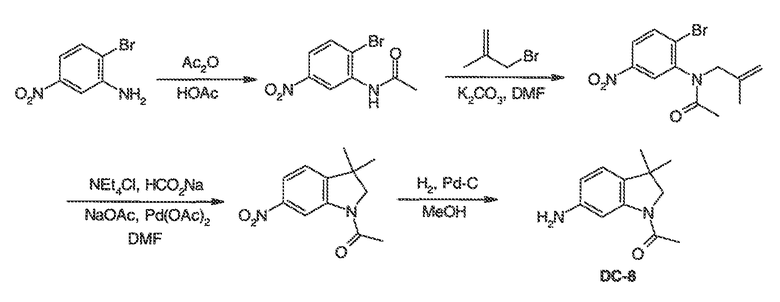
N-(2-Бром-5-нитрофенил)ацетамид
Уксусный ангидрид (1,4 мл, 13,8 ммоль) добавляли по каплям к перемешиваемому раствору 2-бром-5-нитроанилина (3 г, 13,8 ммоль) в ледяной уксусной кислоте (30 мл) при 25ºC. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем выливали в воду. Осадок собирали фильтрованием, промывали водой и сушили в вакууме с получением N-(2-бром-5-нитрофенил)ацетамида в виде не совсем белого твердого вещества (3,6 г, 90%).
N-(2-Бром-5-нитрофенил)-N-(2-метилпроп-2-енил)ацетамид
При 25ºC, раствор 3-бром-2-метилпропена (3,4 г, 55,6 ммоль) в безводном DMF (30 мл) добавляли по каплям к раствору N-(2-бром-5-нитрофенил)ацетамида (3,6 г, 13,9 ммоль) и карбоната калия (3,9 г, 27,8 ммоль) в безводном DMF (50 мл). Реакционную смесь перемешивали при 25ºC в течение ночи. Реакционную смесь затем фильтровали и фильтрат обрабатывали насыщенным раствором Na2CO3. Органический слой отделяли и водный слой экстрагировали при помощи EtOAc. Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением N-(2-бром-5-нитрофенил)-N-(2-метилпроп-2-енил)ацетамида в виде золотистого твердого вещества (3,1 г, 85%). ESI-MS 313 m/z (MH+).
1-(3,3-Диметил-6-нитроиндолин-1-ил)этанон
Раствор N-(2-бром-5-нитрофенил)-N-(2-метилпроп-2-енил)ацетамида (3,1 г, 10,2 ммоль), гидрата тетраэтиламмонийхлорида (2,4 г, 149 ммоль), формиата натрия (1,08 г, 18 ммоль), ацетата натрия (2,76 г, 34,2 ммоль) и ацетата палладия (0,32 г, 13,2 ммоль) в безводном DMF (50 мл) перемешивали при 80ºC в течение 15 часов в атмосфере N2. После охлаждения смесь фильтровали через Целит. Целит промывали при помощи EtOAc и объединенные фильтраты промывали насыщенным раствором NaHCO3. Отделенный органический слой промывали водой и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 1-(3,3-диметил-6-нитроиндолин-1-ил)этанона в виде коричневого твердого вещества (2,1 г, 88%).
DC-8; 1-(6-Амино-3,3-диметил-2,3-дигидро-индол-1-ил)-этанон
10% Pd-C (0,2 г) добавляли к суспензии 1-(3,3-диметил-6-нитроиндолин-1-ил)этанона (2,1 г, 9 ммоль) в MeOH (20 мл). Реакционную смесь перемешивали в атмосфере H2 (40 ф/дюйм2 (2,812 кг/см2)) при комнатной температуре в течение ночи. Pd-C отфильтровывали и фильтрат концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией с получением 1-(6-амино-3,3-диметил-2,3-дигидро-индол-1-ил)-этанона (DC-8) (1,3 г, 61%).
Пример 8:
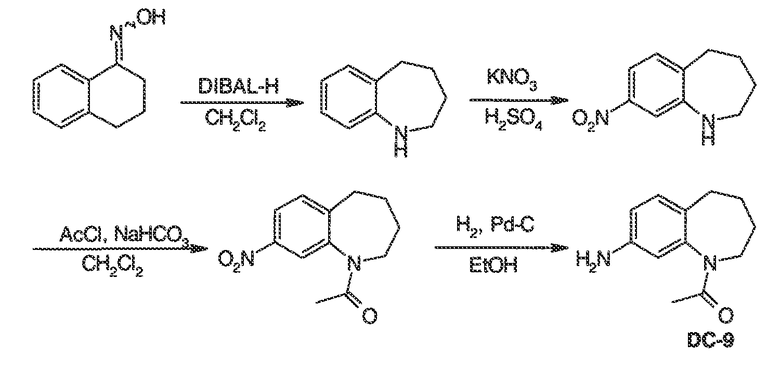
2,3,4,5-Тетрагидро-1H-бензо[b]азепин
DIBAL (90 мл, 90 ммоль) добавляли по каплям к раствору 4-дигидро-2H-нафталин-1-он оксима (3 г, 18 ммоль) в дихлорметане (50 мл) при 0ºC. Смесь перемешивали при этой температуре в течение 2 часов. Реакционную смесь гасили дихлорметаном (30 мл) с последующей обработкой при помощи NaF (2 г, 0,36 моль) и H2O (5 мл, 0,27 моль). Интенсивное перемешивание полученной суспензии продолжали при 0ºC в течение 30 минут. После фильтрования фильтрат концентрировали. Остаток очищали колоночной флэш-хроматографией с получением 2,3,4,5-тетрагидро-1H-бензо[b]азепина в виде бесцветного масла (1,9 г, 70%).
8-Нитро-2,3,4,5-тетрагидро-1H-бензо[b]азепин
При -10ºC, 2,3,4,5-тетрагидро-1H-бензо[b]азепин (1,9 г, 13 ммоль) добавляли по каплям к раствору KNO3 (3 г, 30 ммоль) в H2SO4 (50 мл). Смесь перемешивали в течение 40 минут, выливали на колотый лед, подщелачивали водным раствором аммиака до pH 13 и экстрагировали при помощи EtOAc. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением 8-нитро-2,3,4,5-тетрагидро-1H-бензо[b]азепина в виде черного твердого вещества (1,3 г, 51%), которое использовали без дополнительной очистки.
1-(8-Нитро-2,3,4,5-тетрагидро-бензо[b]азепин-1-ил)-этанон
Ацетилхлорид (1 г, 13 ммоль) добавляли по каплям к смеси 8-нитро-2,3,4,5-тетрагидро-1H-бензо[b]азепина (1,3 г, 6,8 ммоль) и NaHCO3 (1 г, 12 ммоль) в CH2Cl2 (50 мл). После перемешивания в течение 1 часа смесь фильтровали и фильтрат концентрировали. Остаток растворяли в CH2Cl2, промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией с получением 1-(8-нитро-2,3,4,5-тетрагидро-бензо[b]азепин-1-ил)-этанона в виде желтого твердого вещества (1,3 г, 80%).
DC-9; 1-(8-Амино-2,3,4,5-тетрагидро-бензо[b]азепин-1-ил)-этанон
Смесь 1-(8-нитро-2,3,4,5-тетрагидро-бензо[b]азепин-1-ил)-этанона (1,3 г, 5,4 ммоль) и Pd-C (10%, 100 мг) в EtOH (200 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 1,5 часов. Смесь фильтровали через слой Целита и фильтрат концентрировали с получением 1-(8-амино-2,3,4,5-тетрагидро-бензо[b]азепин-1-ил)-этанона (DC-9) в виде белого твердого вещества (1 г, 90%). 1H ЯМР (CDCl3) δ 7,01 (д, J=6,0 Гц, 1H), 6,56 (дд, J=6,0, 1,8 Гц, 1H), 6,50 (д, J=1,8 Гц, 1H), 4,66-4,61 (м, 1H), 3,50 (шир.с, 2H), 2,64-2,55 (м, 3H), 1,94-1,91 (м, 5H), 1,77-1,72 (м, 1H), 1,32-1,30 (м, 1H); ESI-MS 204,1 m/z (MH+).
Пример 9:
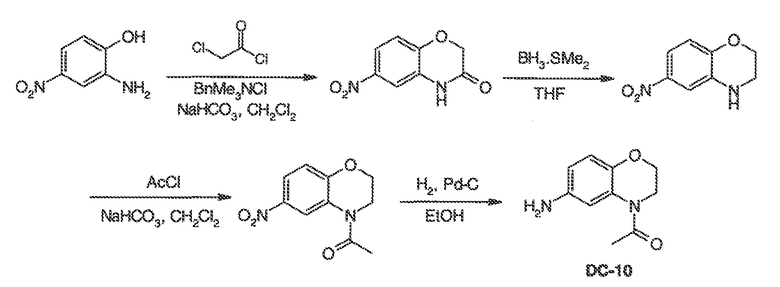
6-Нитро-4H-бензо[1,4]оксазин-3-он
При 0ºC, хлорацетилхлорид (8,75 мл, 0,11 моль) добавляли по каплям к смеси 4-нитро-2-аминофенола (15,4 г, 0,1 моль), бензилтриметиламмонийхлорида (18,6 г, 0,1 моль) и NaHCO3 (42 г, 0,5 моль) в хлороформе (350 мл) в течение 30 минут. После добавления реакционную смесь перемешивали при 0ºC в течение 1 часа, затем при 50ºC в течение ночи. Растворитель удаляли при пониженном давлении и остаток обрабатывали водой (50 мл). Твердое вещество собирали фильтрованием, промывали водой и перекристаллизовывали из этанола с получением 6-нитро-4H-бензо[1,4]оксазин-3-она в виде бледно-желтого твердого вещества (8 г, 41%).
6-Нитро-3,4-дигидро-2H-бензо[1,4]оксазин
Раствор BH3.Me2S в ТГФ (2 M, 7,75 мл, 15,5 ммоль) добавляли по каплям к суспензии 6-нитро-4H-бензо[1,4]оксазин-3-она (0,6 г, 3,1 ммоль) в ТГФ (10 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили при помощи MeOH (5 мл) при 0ºC и затем добавляли воду (20 мл). Смесь экстрагировали при помощи Et2O и объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением 6-нитро-3,4-дигидро-2H-бензо[1,4]оксазина в виде красного твердого вещества (0,5 г, 89%), которое использовали без дополнительной очистки.
4-Ацетил-6-нитро-3,4-дигидро-2H-бензо[1,4]оксазин
При интенсивном перемешивании при комнатной температуре, ацетилхлорид (1,02 г, 13 ммоль) добавляли по каплям к смеси 6-нитро-3,4-дигидро-2H-бензо[1,4]оксазина (1,8 г, 10 ммоль) и NaHCO3 (7,14 г, 85 ммоль) в CH2Cl2 (50 мл). После добавления реакционную смесь перемешивали в течение 1 часа при этой температуре. Смесь фильтровали и фильтрат концентрировали в вакууме. Остаток обрабатывали при помощи Et2O:гексан (1:2, 50 мл) при перемешивании в течение 30 минут и затем фильтровали с получением 4-ацетил-6-нитро-3,4-дигидро-2H-бензо[1,4]оксазина в виде бледно-желтого твердого вещества (2 г, 90%).
DC-10; 4-Ацетил-6-амино-3,4-дигидро-2H-бензо[1,4]оксазин
Смесь 4-ацетил-6-нитро-3,4-дигидро-2H-бензо[1,4]оксазина (1,5 г, 67,6 ммоль) и Pd-C (10%, 100 мг) в EtOH (30 мл) перемешивали в атмосфере H2 (1 атм) в течение ночи. Катализатор отфильтровывали и фильтрат концентрировали. Остаток обрабатывали при помощи HCl/MeOH с получением 4-ацетил-6-амино-3,4-дигидро-2H-бензо[1,4]оксазин гидрохлорида (DC-10) в виде не совсем белого твердого вещества (1,1 г, 85%). 1H ЯМР (DMSO-d6) δ 10,12 (шир.с, 2H), 8,08 (шир.с, 1H), 6,90-7,03 (м, 2H), 4,24 (т, J=4,8 Гц, 2H), 3,83 (т, J=4,8 Гц, 2H), 2,23 (с, 3H); ESI-MS 192,1 m/z (MH+).
Пример 10:
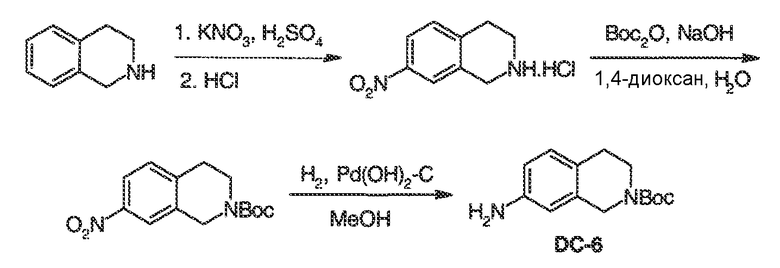
1,2,3,4-Тетрагидро-7-нитроизохинолин гидрохлорид
1,2,3,4-Тетрагидроизохинолин (6,3 мл, 50,0 ммоль) добавляли по каплям к перемешиваемому охлажденному льдом раствору концентрированной H2SO4 (25 мл). Добавляли по порциям KNO3 (5,6 г, 55,0 ммоль), поддерживая при этом температуру ниже 5ºC. Смесь перемешивали при комнатной температуре в течение ночи, осторожно выливали в охлажденный льдом раствор концентрированноого NH4OH и затем экстрагировали три раза при помощи CHCl3. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Полученное темно-коричневое масло брали для поглощения EtOH, охлаждали на ледяной бане и обрабатывали концентрированной HCl. Желтый осадок собирали фильтрованием и перекристаллизовывали из метанола с получением 1,2,3,4-тетрагидро-7-нитроизохинолин гидрохлорида в виде желтого твердого вещества (2,5 г, 23%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,86 (с, 2H), 8,22 (д, J=1,6 Гц, 1H), 8,11 (дд, J=8,5, 2,2 Гц, 1H), 7,53 (д, J=8,5 Гц,1H), 4,38 (с, 2H), 3,38 (с, 2H), 3,17-3,14 (м, 2H); ВЭЖХ время удерживания 0,51 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 179,0 m/z (MH+).
трет-Бутил 3,4-дигидро-7-нитроизохинолин-2(1H)-карбоксилат
Смесь 1,2,3,4-тетрагидро-7-нитроизохинолина (2,5 г, 11,6 ммоль), 1,4-диоксана (24 мл), H2O (12 мл) и 1н NaOH (12 мл) охлаждали на ледяной бане и добавляли Boc2O (2,8 г, 12,8 ммоль). Смесь перемешивали при комнатной температуре в течение 2,5 часов, подкисляли 5% раствором KHSO4 до pH 2-3 и затем экстрагировали при помощи EtOAc. Органический слой сушили над MgSO4 и концентрировали с получением трет-бутил 3,4-дигидро-7-нитроизохинолин-2(1H)-карбоксилата (3,3 г, количественный), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ 8,13 (д, J=2,3 Гц, 1H), 8,03 (дд, J=8,4, 2,5 Гц, 1H), 7,45 (д, J=8,5 Гц, 1H), 4,63 (с, 2H), 3,60-3,57 (м, 2H), 2,90 (т, J=5,9 Гц, 2H), 1,44 (с, 9H); ВЭЖХ время удерживания 3,51 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 279,2 m/z (MH+).
DC-6; трет-бутил 7-амино-3,4-дигидроизохинолин-2(1H)-карбоксилат
Pd(OH)2 (330,0 мг) добавляли к перемешиваемому раствору трет-бутил 3,4-дигидро-7-нитроизохинолин-2(1H)-карбоксилата (3,3 г, 12,0 ммоль) в MeOH (56 мл) в атмосфере N2. Реакционную смесь перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 72 часов. Твердое вещество удаляли фильтрованием через Целит. Фильтрат концентрировали и очищали колоночной хроматографией (15-35% EtOAc-гексан) с получением трет-бутил 7-амино-3,4-дигидроизохинолин-2(1H)-карбоксилата (DC-6) в виде розового масла (2,0 г, 69%). 1H ЯМР (400 МГц, DMSO-d6) δ 6,79 (д, J=8,1 Гц, 1H), 6,40 (дд, J=8,1, 2,3 Гц, 1H), 6,31 (с, 1H), 4,88 (с, 2H), 4,33 (с, 2H), 3,48 (т, J=5,9 Гц, 2H), 2,58 (т, J=5,9 Гц, 2H), 1,42 (с, 9H); ВЭЖХ время удерживания 2,13 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 249,0 m/z (MH+).
Другие амины
Пример 1:
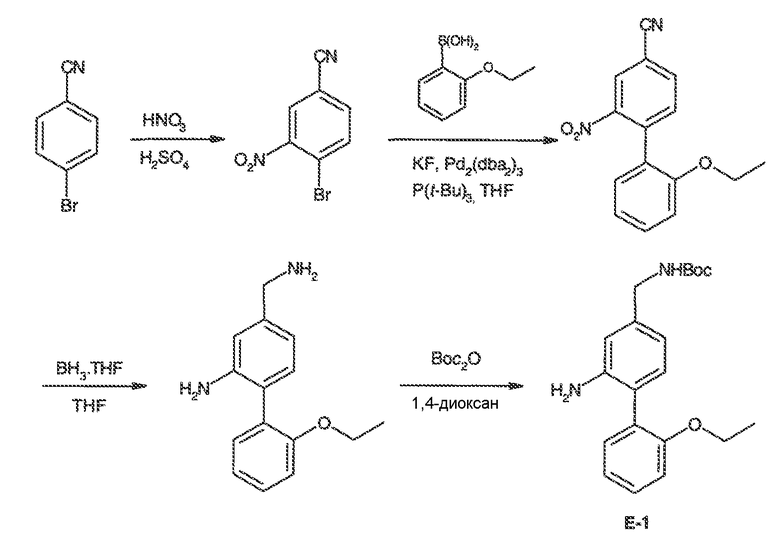
4-Бром-3-нитробензонитрил
К раствору 4-бромбензонитрила (4,0 г, 22 ммоль) в концентрированной H2SO4 (10 мл) добавляли по каплям при 0ºC азотную кислоту (6 мл). Реакционную смесь перемешивали при 0ºC в течение 30 минут и затем при комнатной температуре в течение 2,5 часов. Полученный раствор выливали в ледяную воду. Белый осадок собирали фильтрованием и промывали водой до тех пор, пока промывки не становились нейтральными. Твердое вещество перекристаллизовывали из смеси этанол/вода (1:1, 20 мл) два раза с получением 4-бром-3-нитробензонитрила в виде белого кристаллического твердого вещества (2,8 г, 56%). 1H ЯМР (300 МГц, DMSO-d6) δ 8,54 (с, 1H), 8,06 (д, J=8,4 Гц, 1H), 7,99 (д, J=8,4 Гц, 1H); 13C ЯМР (75 МГц, DMSO-d6) δ 150,4, 137,4, 136,6, 129,6, 119,6, 117,0, 112,6; ВЭЖХ время удерживания 1,96 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 227,1 m/z (MH+).
2'-Этокси-2-нитробифенил-4-карбонитрил
В 50-мл круглодонную колбу загружали 4-бром-3-нитробензонитрил (1,0 г, 4,4 ммоль), 2-этоксифенилбороновую кислоту (731 мг, 4,4 ммоль), Pd2(dba)3 (18 мг, 0,022 ммоль) и фторид калия (786 мг, 13,5 ммоль). В реакционном сосуде создавали вакуум и заполняли аргоном. Добавляли безводный ТГФ (300 мл) с последующим добавлением P(t-Bu)3 (0,11 мл, 10% масс. в гексане). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и затем нагревали при 80ºC в течение 16 часов. После охлаждения до комнатной температуры полученную смесь фильтровали через слой Целита и концентрировали. 2'-Этокси-2-нитробифенил-4-карбонитрил выделяли в виде желтого твердого вещества (1,12 г, 95%). 1H ЯМР (300 МГц, DMSO-d6) δ 8,51 (с, 1H), 8,20 (д, J=8,1 Гц, 1H), 7,68 (д, J=8,4 Гц, 1H), 7,41 (т, J=8,4 Гц, 1H), 7,37 (д, J=7,5 Гц, 1H), 7,08 (т, J=7,5 Гц, 1H), 7,03 (д, J=8,1 Гц, 1H), 3,91 (кв., J=7,2 Гц, 2H), 1,12 (т, J=7,2 Гц, 3H); 13C ЯМР (75 МГц, DMSO-d6) δ 154,9, 149,7, 137,3, 137,2, 134,4, 131,5, 130,4, 128,4, 125,4, 121,8, 117,6, 112,3, 111,9, 64,1, 14,7; ВЭЖХ время удерживания 2,43 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 269,3 m/z (MH+).
4-Аминометил-2'-этокси-бифенил-2-иламин
К раствору 2'-этокси-2-нитробифенил-4-карбонитрила (500 мг, 1,86 ммоль) в ТГФ (80 мл) добавляли раствор BH3.ТГФ (5,6 мл, 10% масс. в ТГФ, 5,6 ммоль) при 0ºC в течение 30 минут. Реакционную смесь перемешивали при 0ºC в течение 3 часов и затем при комнатной температуре в течение 15 часов. Реакционный раствор охлаждали до 0ºC и добавляли смесь H2O/ТГФ (3 мл). После перемешивания при комнатной температуре в течение 6 часов летучие вещества удаляли при пониженном давлении. Остаток растворяли в EtOAc (100 мл) и экстрагировали 1н раствором HCl (2×100 мл). Водную фазу подщелачивали 1н раствором NaOH до pH 1 и экстрагировали при помощи EtOAc (3×50 мл). Объединенные органические слои промывали водой (50 мл), сушили над Na2SO4, фильтровали и упаривали. После сушки в вакууме 4-аминометил-2'-этокси-бифенил-2-иламин выделяли в виде коричневого масла (370 мг, 82%). 1H ЯМР (300 МГц, DMSO-d6) δ 7,28 (дт, J=7,2 Гц, J=1,8 Гц, 1H), 7,09 (дд, J=7,2 Гц, J=1,8 Гц, 1H), 7,05 (д, J=7,5 Гц, 1H), 6,96 (дт, J=7,2 Гц, J=0,9 Гц, 1H), 6,83 (д, J=7,5 Гц, 1H), 6,66 (д, J=1,2 Гц, 1H), 6,57 (дд, J=7,5 Гц, J=1,5 Гц, 1H), 4,29 (с, 2H), 4,02 (кв., J=6,9 Гц, 2H), 3,60 (с, 2H), 1,21 (т, J=6,9 Гц, 3H); ВЭЖХ время удерживания 1,54 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 243,3 m/z (MH+).
E-1; трет-бутиловый эфир (2-Амино-2'-этокси-бифенил-4-илметил)карбаминовой кислоты
Раствор Boc2O (123 мг, 0,565 ммоль) в 1,4-диоксане (10 мл) добавляли в течение 30 минут к раствору 4-аминометил-2'-этокси-бифенил-2-иламина (274 мг, 1,13 ммоль) в 1,4-диоксане (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Летучие вещества удаляли на роторном испарителе. Остаток очищали флэш-хроматографией (силикагель, EtOAc-CH2Cl2, 1:4) с получением трет-бутилового эфира (2-Амино-2'-этокси-бифенил-4-илметил)карбаминовой кислоты (E-1) в виде бледно-желтого масла (119 мг, 31%). 1H ЯМР (300 МГц, DMSO-d6) δ 7,27 (м, 2H), 7,07 (дд, J=7,2 Гц, J=1,8 Гц, 1H), 7,03 (д, J=7,8 Гц, 1H), 6,95 (дт, J=7,2 Гц, J=0,9 Гц, 1H), 6,81 (д, J=7,5 Гц, 1H), 6,55 (с, 1H), 6,45 (дд, J=7,8 Гц, J=1,5 Гц, 1H), 4,47 (с, 2H), 4,00 (кв., J=7,2 Гц, 2H), 1,38 (с, 9H), 1,20 (т, J=7,2 Гц, 3H); ВЭЖХ время удерживания 2,34 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 343,1 m/z (MH+).
Пример 2:
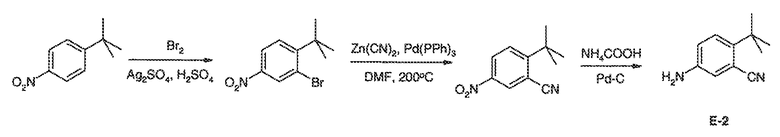
2-Бром-1-трет-бутил-4-нитробензол
К раствору 1-трет-бутил-4-нитробензола (8,95 г, 50 ммоль) и сульфата серебра (10 г, 32 ммоль) в 50 мл 90% серной кислоты добавляли по каплям бром (7,95 г, 50 ммоль). Перемешивание продолжали при комнатной температуре в течение ночи и затем смесь выливали в разбавленный раствор гидросульфита натрия и экстрагировали при помощи EtOAc три раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После фильтрования фильтрат концентрировали с получением 2-бром-1-трет-бутил-4-нитробензола (12,7 г, 98%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,47 (д, J=2,5 Гц, 1H), 8,11 (дд, J=8,8, 2,5 Гц, 1H), 7,63 (д, J=8,8 Гц, 1H), 1,57 (с, 9H); ВЭЖХ время удерживания 4,05 минут, 10-100% CH3CN, 5 мин градиент.
2-трет-бутил-5-нитробензонитрил
К раствору 2-бром-1-трет-бутил-4-нитробензола (2,13 г, 8,2 ммоль) и Zn(CN)2 (770 мг, 6,56 ммоль) в DMF (10 мл) добавляли Pd(PPh3)4 (474 мг, 0,41 ммоль) в атмосфере азота. Смесь нагревали в герметично закрытом сосуде при 205ºC в течение 5 часов. После охлаждения до комнатной температуры смесь разбавляли водой и экстрагировали при помощи EtOAc два раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После удаления растворителя остаток очищали колоночной хроматографией (0-10% EtOAc-гексан) с получением 2-трет-бутил-5-нитробензонитрила (1,33 г, 80%). 1H ЯМР (400 МГц, CDCl3) δ 8,55 (д, J=2,3 Гц, 1H), 8,36 (дд, J=8,8, 2,2 Гц, 1H), 7,73 (д, J=8,9 Гц, 1H), 1,60 (с, 9H); ВЭЖХ время удерживания 3,42 минут, 10-100% CH3CN, 5 мин градиент.
E-2; 2-трет-бутил-5-аминобензонитрил
К нагреваемому при температуре кипения с обратным холодильником раствору 2-трет-бутил-5-нитробензонитрила (816 мг, 4,0 ммоль) в EtOH (20 мл) добавляли формиат аммония (816 мг, 12,6 ммоль), с последующим добавлением 10% Pd-C (570 мг). Реакционную смесь кипятили с обратным холодильником еще в течение 90 минут, охлаждали до комнатной температуры и фильтровали через Целит. Фильтрат концентрировали с получением 2-трет-бутил-5-аминобензонитрила (E-2) (630 мг, 91%), который использовали без дополнительной очистки. ВЭЖХ время удерживания 2,66 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 175,2 m/z (MH+).
Пример 3:
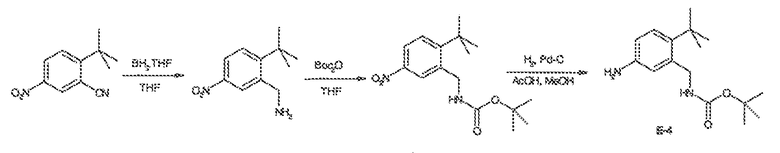
(2-трет-бутил-5-нитрофенил)метанамин
К раствору 2-трет-бутил-5-нитробензонитрила (612 мг, 3,0 ммоль) в ТГФ (10 мл) добавляли раствор BH3.ТГФ (12 мл, 1M в ТГФ, 12,0 ммоль) в атмосфере азота. Реакционную смесь перемешивали при 70ºC в течение ночи и охлаждали до 0ºC. Добавляли метанол (2 мл) с последующим добавлением 1н раствора HCl (2 мл). После кипячения с обратным холодильником в течение 30 минут раствор разбавляли водой и экстрагировали при помощи EtOAc. Водный слой подщелачивали 1н раствором NaOH и экстрагировали при помощи EtOAc два раза. Объединенные органические слои промывали насыщенным солевым раствором и сушили над Mg2SO4. После удаления растворителя остаток очищали колоночной хроматографией (0-10% MeOH-CH2Cl2) с получением (2-трет-бутил-5-нитрофенил)метанамина (268 мг, 43%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,54 (д, J=2,7 Гц, 1H), 7,99 (дд, J=8,8, 2,8 Гц, 1H), 7,58 (д, J=8,8 Гц, 1H), 4,03 (с, 2H), 2,00 (т, J=2,1 Гц, 2H), 1,40 (с, 9H); ВЭЖХ время удерживания 2,05 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 209,3 m/z (MH+).
трет-Бутил 2-трет-бутил-5-нитробензилкарбамат
Раствор (2-трет-бутил-5-нитрофенил)метанамина (208 мг, 1 ммоль) и Boc2O (229 мг, 1,05 ммоль) в ТГФ (5 мл) кипятили с обратным холодильником в течение 30 минут. После охлаждения до комнатной температуры раствор разбавляли водой и экстрагировали при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После фильтрования фильтрат концентрировали с получением трет-бутил 2-трет-бутил-5-нитробензилкарбамата (240 мг, 78%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ 8,26 (д, J=2,3 Гц, 1H), 8,09 (дд, J=8,8, 2,5 Гц, 1H), 7,79 (т, J=5,9 Гц, 1H), 7,68 (д, J=8,8 Гц, 1H), 4,52 (д, J=6,0 Гц, 2H), 1,48 (с, 18H); ВЭЖХ время удерживания 3,72 минут, 10-100% CH3CN, 5 мин градиент.
E-4; трет-бутил 2-трет-бутил-5-аминобензилкарбамат
К раствору трет-бутил 2-трет-бутил-5-нитробензилкарбамата (20 мг, 0,065 ммоль) в 5% AcOH-MeOH (1 мл) добавляли 10% Pd-C (14 мг) в атмосфере азота. Смесь перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 1 часа. Катализатор удаляли фильтрованием через Целит и фильтрат концентрировали с получением трет-бутил 2-трет-бутил-5-аминобензилкарбамата (E-4), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,09 (д, J=8,5 Гц, 1H), 6,62 (д, J=2,6 Гц, 1H), 6,47 (дд, J=8,5, 2,6 Гц, 1H), 4,61 (шир.с, 1H), 4,40 (д, J=5,1 Гц, 2H), 4,15 (шир.с, 2H), 1,39 (с, 9H), 1,29 (с, 9H); ВЭЖХ время удерживания 2,47 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 279,3 m/z (MH+).
Пример 4:
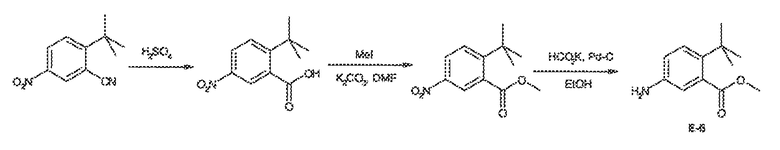
2-трет-бутил-5-нитробензойная кислота
Раствор 2-трет-бутил-5-нитробензонитрила (204 мг, 1 ммоль) в 5 мл 75% H2SO4 нагревали в микроволновой печи при 200ºC в течение 30 минут. Реакционную смесь выливали на лед, экстрагировали при помощи EtOAc, промывали насыщенным солевым раствором и сушили над MgSO4. После фильтрования фильтрат концентрировали с получением 2-трет-бутил-5-нитробензойной кислоты (200 мг, 90%), которую использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,36 (д, J=2,6 Гц, 1H), 8,24 (дд, J=8,9, 2,6 Гц, 1H), 7,72 (д, J=8,9 Гц, 1H) 1,51 (с, 9H); ВЭЖХ время удерживания 2,97 минут, 10-100% CH3CN, 5 мин градиент.
Метил 2-трет-бутил-5-нитробензоат
К смеси 2-трет-бутил-5-нитробензойной кислоты (120 мг, 0,53 ммоль) и K2CO3 (147 мг, 1,1 ммоль) в DMF (5,0 мл) добавляли CH3I (40 мкл, 0,64 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут, разбавляли водой и экстрагировали при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После фильтрования фильтрат концентрировали с получением метил 2-трет-бутил-5-нитробензоата, который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,20 (д, J=2,6 Гц, 1H), 8,17 (т, J=1,8 Гц, 1H), 7,66 (д, J=8,6 Гц, 1H), 4,11 (с, 3H), 1,43 (с, 9H).
E-6; Метил 2-трет-бутил-5-аминобензоат
К нагреваемому при температуре кипения с обратным холодильником раствору 2-трет-бутил-5-нитробензоата (90 мг, 0,38 ммоль) в EtOH (2,0 мл) добавляли формиат калия (400 мг, 4,76 ммоль) в воде (1 мл), с последующим добавлением 20 мг 10% Pd-C. Реакционную смесь кипятили с обратным холодильником еще в течение 40 минут, охлаждали до комнатной температуры и фильтровали через Целит. Фильтрат концентрировали с получением метил 2-трет-бутил-5-аминобензоата (E-6) (76 мг, 95%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,24 (д, J=8,6 Гц, 1H), 6,67 (дд, J=8,6, 2,7 Гц, 1H), 6,60 (д, J=2,7 Гц, 1H), 3,86 (с, 3H), 1,34 (с, 9H); ВЭЖХ время удерживания 2,19 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 208,2 m/z (MH+).
Пример 5:
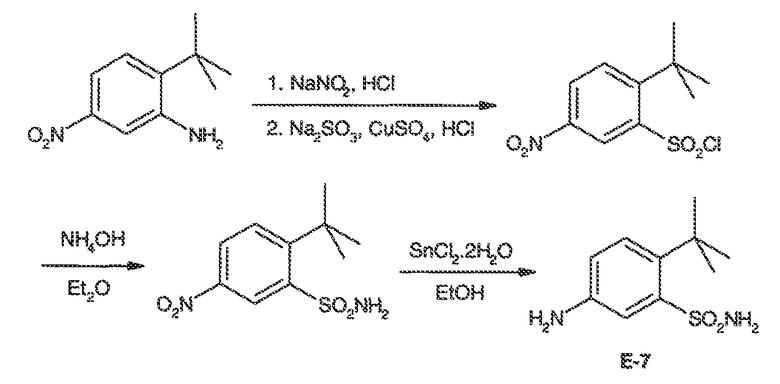
2-трет-бутил-5-нитробензол-1-сульфонилхлорид
Суспензию 2-трет-бутил-5-нитробензоламина (0,971 г, 5 ммоль) в концентрированной HCl (5 мл) охлаждали до 5-10ºC и добавляли по каплям раствор NaNO2 (0,433 г, 6,3 ммоль) в H2O (0,83 мл). Перемешивание продолжали в течение 0,5 часов, затем смесь подергали вакуум-фильтрации. Фильтрат добавляли, одновременно с раствором Na2SO3 (1,57 г, 12,4 ммоль) в H2O (2,7 мл), к перемешиваемому раствору CuSO4 (0,190 г, 0,76 ммоль) и Na2SO3 (1,57 г, 12,4 ммоль) в HCl (11,7 мл) и H2O (2,7 мл) при 3-5ºC. Перемешивание продолжали в течение 0,5 часов и образовавшийся осадок отфильтровывали, промывали водой и сушили с получением 2-трет-бутил-5-нитробензол-1-сульфонилхлорида (0,235 г, 17%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,13 (д, J=2,5 Гц, 1H), 8,36 (дд, J=8,9, 2,5 Гц, 1H), 7,88 (д, J=8,9 Гц, 1H), 1,59 (с, 9H).
2-трет-бутил-5-нитробензол-1-сульфонамид
К раствору 2-трет-бутил-5-нитробензол-1-сульфонилхлорида (100 мг, 0,36 ммоль) в простом эфире (2 мл) добавляли водный раствор NH4OH (128 мкл, 3,6 ммоль) при 0ºC. Смесь перемешивали при комнатной температуре в течение ночи, разбавляли водой и экстрагировали простым эфиром. Объединенные эфирные экстракты промывали насыщенным солевым раствором и сушили над Na2SO4. После удаления растворителя остаток очищали колоночной хроматографией (0-50% EtOAc-гексан) с получением 2-трет-бутил-5-нитробензол-1-сульфонамида (31,6 мг, 34%).
E-7; 2-трет-бутил-5-аминобензол-1-сульфонамид
Раствор 2-трет-бутил-5-нитробензол-1-сульфонамида (32 мг, 0,12 ммоль) и SnCl2.2H2O (138 мг, 0,61 ммоль) в EtOH (1,5 мл) нагревали в микроволновой печи при 100ºC в течение 30 минут. Смесь разбавляли EtOAc и водой, подщелачивали насыщенным раствором NaHCO3 и фильтровали через Целит. Органический слой отделяли от воды и сушили над Na2SO4. Растворитель удаляли выпариванием с получением 2-трет-бутил-5-аминобензол-1-сульфонамида (E-7) (28 мг, 100%), который использовали без дополнительной очистки. ВЭЖХ время удерживания 1,99 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 229,3 m/z (MH+).
Пример 6:
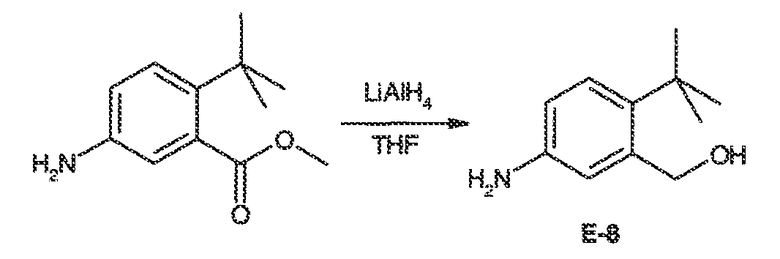
E-8; (2-трет-бутил-5-аминофенил)метанол
К раствору метил 2-трет-бутил-5-аминобензоата (159 мг, 0,72 ммоль) в ТГФ (5 мл) добавляли по каплям LiAlH4 (1,4 мл, 1M в ТГФ, 1,4 ммоль) при 0ºC. Реакционную смесь кипятили с обратным холодильником в течение 2 часов, разбавляли при помощи H2O и экстрагировали при помощи EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. После фильтрования фильтрат концентрировали с получением (2-трет-бутил-5-аминофенил)метанола (E-8) (25 мг, 20%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,17 (д, J=8,5 Гц, 1H), 6,87 (д, J=2,6 Гц, 1H), 6,56 (дд, J=8,4, 2,7 Гц, 1H), 4,83 (с, 2H), 1,36 (с, 9H).
Пример 7:
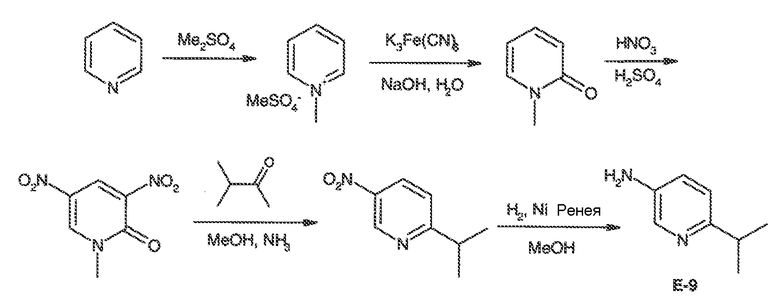
1-Метил-пиридиний, соль монометилсерной кислоты
Метилсульфат (30 мл, 39,8 г, 0,315 моль) добавляли по каплям к безводному пиридину (25,0 г, 0,316 моль). Смесь перемешивали при комнатной температуре в течение 10 минут, затем при 100ºC в течение 2 часов. Смесь охлаждали до комнатной температуры с получением неочищенного 1-метил-пиридиния, соли монометилсерной кислоты (64,7 г, количественный), который использовали без дополнительной очистки.
1-Метил-2-пиридон
Раствор соли монометилсерной кислоты 1-метил-пиридиния (50 г, 0,243 моль) в воде (54 мл) охлаждали до 0ºC. Получали отдельные растворы феррицианида калия (160 г, 0,486 моль) в воде (320 мл) и гидроксида натрия (40 г, 1,000 моль) в воде (67 мл) и добавляли по каплям из делительных воронок к тщательно перемешиваемому раствору соли монометилсерной кислоты 1-метил-пиридиния при такой скорости, чтобы температура реакционной смеси не повышалась выше 10ºC. Скорость добавления этих двух растворов регулировали так, чтобы весь раствор гидроксида натрия был введен в реакционную смесь, когда была добавлена половина раствора феррицианида калия. После завершения добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Добавляли безводный карбонат натрия (91,6 г) и смесь перемешивали в течение 10 минут. Органический слой отделяли и водный слой экстрагировали при помощи CH2Cl2 (100 мл ×3). Объединенные органические слои сушили и концентрировали с получением 1-метил-2-пиридона (25,0 г, 94%), который использовали без дополнительной очистки.
1-Метил-3,5-динитро-2-пиридон
1-Метил-2-пиридон (25,0 г, 0,229 моль) добавляли к серной кислоте (500 мл) при 0ºC. После перемешивания в течение 5 минут добавляли по каплям азотную кислоту (200 мл) при 0ºC. После добавления температуру реакции медленно повышали до 100ºC и затем поддерживали в течение 5 часов. Реакционную смесь выливали на лед, подщелачивали при помощи карбоната калия до pH 8 и экстрагировали при помощи CH2Cl2 (100 мл ×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением 1-метил-3,5-динитро-2-пиридона (12,5 г, 28%), который использовали без дополнительной очистки.
2-Изопропил-5-нитро-пиридин
К раствору 1-метил-3,5-динитро-2-пиридона (8,0 г, 40 ммоль) в метиловом спирте (20 мл) добавляли по каплям 3-метил-2-бутанон (5,1 мл, 48 ммоль), с последующим добавлением раствора аммиака в метиловом спирте (10,0 г, 17%, 100 ммоль). Реакционную смесь нагревали при 70ºC в течение 2,5 часов при атмосферном давлении. Растворитель удаляли в вакууме и остаточное масло растворяли в CH2Cl2 и затем фильтровали. Фильтрат сушили над Na2SO4 и концентрировали с получением 2-изопропил-5-нитро-пиридина (1,88 г, 28%).
E-9; 2-Изопропил-5-амино-пиридин
2-Изопропил-5-нитро-пиридин (1,30 г, 7,82 ммоль) растворяли в метиловом спирте (20 мл) и добавляли Ni Ренея (0,25 г). Смесь перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение 2 часов. Катализатор отфильтровывали и фильтрат концентрировали в вакууме с получением 2-изопропил-5-амино-пиридина (E-9) (0,55 г, 52%). 1H ЯМР (CDCl3) δ 8,05 (с, 1H), 6,93-6,99 (м, 2H), 3,47 (шир.с, 2H), 2,92-3,02 (м, 1H), 1,24-1,26 (м, 6H), ESI-MS 137,2 m/z (MH+).
Пример 8:
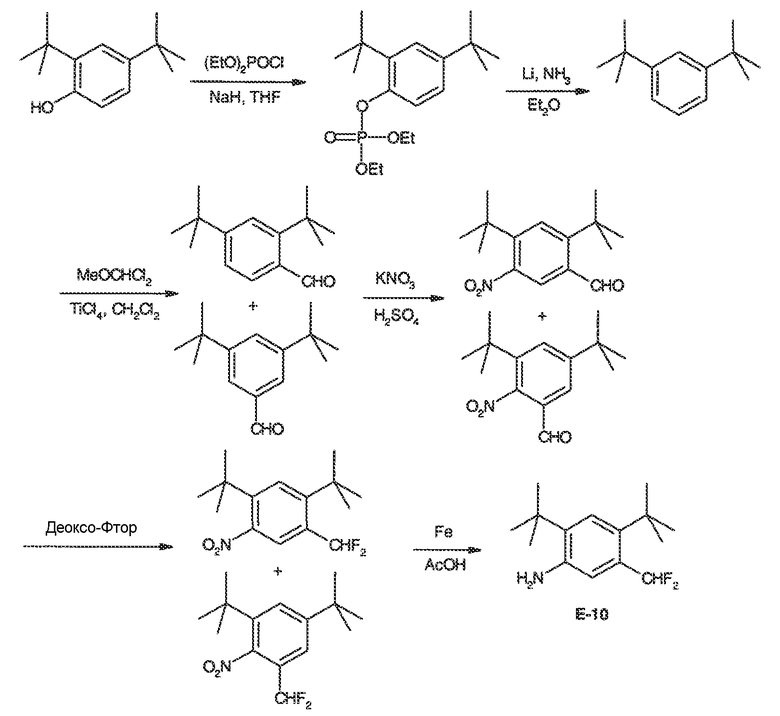
2,4-ди-трет-бутил-фениловый эфир диэтиловый эфир фосфорной кислоты
К суспензии NaH (60% в минеральном масле, 6,99 г, 174,7 ммоль) в ТГФ (350 мл) добавляли по каплям раствор 2,4-ди-трет-бутилфенола (35 г, 169,6 ммоль) в ТГФ (150 мл) при 0ºC. Смесь перемешивали при 0ºC в течение 15 минут и затем добавляли по каплям диэтиловый эфир фосфорохлоридной кислоты (30,15 г, 174,7 ммоль) при 0ºC. После добавления смесь перемешивали при этой температуре в течение 15 минут. Реакционную смесь гасили насыщенным раствором NH4Cl (300 мл). Органический слой отделяли и водную фазу экстрагировали при помощи Et2O (350 мл ×2). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного 2,4-ди-трет-бутил-фенилового эфира диэтилового эфира фосфорной кислоты в виде желтого масла (51 г, с некоторой примесью минерального масла), который использовали непосредственно на следующей стадии.
1,3-Ди-трет-бутил-бензол
К NH3 (жидкий, 250 мл) добавляли раствор 2,4-ди-трет-бутил-фенилового эфира диэтилового эфира фосфорной кислоты (51 г, неочищенный с последней стадии, около 0,2 моль) в Et2O (безводный, 150 мл) при -78ºC в атмосфере N2. К раствору добавляли металлический литий небольшими кусками до получения стойкого синего цвета. Реакционную смесь перемешивали при -78ºC в течение 15 минут и затем гасили насыщенным раствором NH4Cl, пока смесь не становилась бесцветной. Жидкий NH3 выпаривали и остаток растворяли в воде, экстрагировали при помощи Et2O (300 мл ×2). Объединенные органические фазы сушили над Na2SO4 и концентрировали с получением неочищенного 1,3-ди-трет-бутил-бензоле в виде желтого масла (30,4 г, 94% за 2 стадии, с некоторой примесью минерального масла), который использовали непосредственно на следующей стадии.
2,4-Ди-трет-бутил-бензальдегид и 3,5-ди-трет-бутил-бензальдегид
К перемешиваемому раствору 1,3-ди-трет-бутил-бензола (30 г, 157,6 ммоль) в безводном CH2Cl2 (700 мл) добавляли TiCl4 (37,5 г, 197 ммоль) при 0ºC, с последующим добавлением по каплям MeOCHCl2 (27,3 г, 236,4 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 1 часа. Смесь выливали в ледяную воду и экстрагировали при помощи CH2Cl2. Объединенные органические фазы промывали NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир) с получением смеси 2,4-ди-трет-бутил-бензальдегида и 3,5-ди-трет-бутил-бензальдегида (21 г, 61%).
2,4-Ди-трет-бутил-5-нитро-бензальдегид и 3,5-ди-трет-бутил-2-нитро-бензальдегид
К смеси 2,4-ди-трет-бутил-бензальдегида и 3,5-ди-трет-бутил-бензальдегида в H2SO4 (250 мл) добавляли KNO3 (7,64 г, 75,6 ммоль) по порциям при 0ºC. Реакционную смесь перемешивали при этой температуре в течение 20 минут и затем выливали в колотый лед. Смесь подщелачивали раствором NaOH до pH 8 и экстрагировали при помощи Et2O (10 мл ×3). Объединенные органические слои промывали водой и насыщенным солевым раствором и концентрировали. Остаток очищали колоночной хроматографией (петролейный эфир) с получением смеси 2,4-ди-трет-бутил-5-нитро-бензальдегида и 3,5-ди-трет-бутил-2-нитро-бензальдегида (2:1 по данным ЯМР) в виде желтого твердого вещества (14,7 г, 82%). После дополнительной очистки колоночной хроматографией (петролейный эфир) выделяли 2,4-ди-трет-бутил-5-нитро-бензальдегид (2,5 г, содержит 10% 3,5-ди-трет-бутил-2-нитро-бензальдегида).
1,5-Ди-трет-бутил-2-дифторметил-4-нитро-бензол и 1,5-Ди-трет-бутил-3-дифторметил-2-нитро-бензол
2,4-Ди-трет-бутил-5-нитро-бензальдегид (2,4 г, 9,11 ммоль, с примесью 10% раствора 3,5-ди-трет-бутил-2-нитро-бензальдегид) в растворе чистого деоксофтора перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь выливали в охлажденный насыщенный раствор NaHCO3 и экстрагировали дихлорметаном. Объединяли органические слои сушили над Na2SO4, концентрировали и очищали колоночной хроматографией (петролейный эфир) с получением 1,5-ди-трет-бутил-2-дифторметил-4-нитро-бензола (1,5 г) и смеси 1,5-ди-трет-бутил-2-дифторметил-4-нитро-бензола и 1,5-ди-трет-бутил-3-дифторметил-2-нитро-бензола (0,75 г, содержит 28% 1,5-ди-трет-бутил-3-дифторметил-2-нитро-бензола).
E-10; 1,5-Ди-трет-бутил-2-дифторметил-4-амино-бензол
К суспензии порошка железа (5,1 г, 91,1 ммоль) в 50% уксусной кислоте (25 мл) добавляли 1,5-ди-трет-бутил-2-дифторметил-4-нитро-бензол (1,3 г, 4,56 ммоль). Реакционную смесь нагревали при 115ºC в течение 15 минут. Твердое вещество отфильтровывали, промывали уксусной кислотой и CH2Cl2. Объединенный фильтрат концентрировали и обрабатывали при помощи HCl/MeOH. Осадок собирали фильтрованием, промывали при помощи MeOH и сушили с получением 1,5-ди-трет-бутил-2-дифторметил-4-амино-бензола в форме HCl соли (E-10) в виде белого твердого вещества (1,20 г, 90%). 1H ЯМР (DMSO-d6) δ 7,35-7,70 (т, J=53,7 Гц, 1H), 7,56 (с, 1H), 7,41 (с, 1H), 1,33-1,36 (д, J=8,1 Гц, 1H); ESI-MS 256,3 m/z (MH+).
Пример 9
Общая схема:
A) Pd(PPh3)4, K2CO3, H2O, ТГФ; B) Pd2(dba)3, P(tBu)3, KF, ТГФ
Способ A
В сосуде вместимостью 2 драхмы, 2-броманилин (100 мг, 0,58 ммоль) и соответствующую арилбороновую кислоту (0,82 ммоль) растворяли в ТГФ (1 мл). Добавляли H2O (500 мкл) с последующим добавлением K2CO3 (200 мг, 1,0 ммоль) и Pd(PPh3)4 (100 мг, 0,1 ммоль). Сосуд продували аргоном и герметично закрывали. Сосуд затем нагревали при 75ºC в течение 18 часов. Неочищенный образец разбавляли в EtOAc и фильтровали через пробку силикагеля. Органические слои концентрировали через Savant Speed-vac. Неочищенный амин использовали без дополнительной очистки.
Способ B
В сосуд вместимостью 2 драхмы добавляли соответствующую арилбороновую кислоту (0,58 ммоль) с последующим добавлением KF (110 мг, 1,9 ммоль). Твердые вещества суспендировали в ТГФ (2 мл) и затем добавляли 2-броманилин (70 мкл, 0,58 ммоль). Сосуд продували аргоном в течение 1 минуты. Добавляли P(tBu)3 (100 мкл, 10% раствор в гексане) с последующим добавлением Pd2(dba)3 (900 мкл, 0,005 M в ТГФ). Сосуд снова продували аргоном и герметично закрывали. Сосуд встряхивали на орбитальном встряхивающем устройстве при комнатной температуре в течение 30 минут и нагревали в нагревательном блоке при 80ºC в течение 16 часов. Сосуд затем охлаждали до 20ºC и суспензию пропускали через слой Целита. Слой промывали при помощи EtOAc (5 мл). Органические слои объединяли и концентрировали в вакууме с получением неочищенного амина, который использовали без дополнительной очистки.
Таблица, представленная ниже, включает амины, полученные в соответствии с общей схемой, представленной выше.
Пример 10:
Этил 2-(4-нитрофенил)-2-метилпропаноат
трет-Бутоксид натрия (466 мг, 4,85 ммоль) добавляли к DMF (20 мл) при 0ºC. Мутный раствор снова охлаждали до 5ºC. Добавляли этил 4-нитрофенилацетат (1,0 г, 4,78 ммоль). Фиолетовую суспензию охлаждали до 5ºC и добавляли метилиодид (0,688 мл, 4,85 ммоль) в течение 40 минут. Смесь перемешивали при 5-10ºC в течение 20 минут и затем снова загружали трет-бутоксид натрия (466 мг, 4,85 ммоль) и метилиодид (0,699 мл, 4,85 ммоль). Смесь перемешивали при 5-10ºC в течение 20 минут и добавляли третью загрузку трет-бутоксида натрия (47 мг, 0,48 ммоль) с последующим добавлением метилиодида (0,057 мл, 0,9 ммоль). Добавляли этилацетат (100 мл) и HCl (0,1 н, 50 мл). Органический слой отделяли, промывали насыщенным солевым раствором и сушили над Na2SO4. После фильтрования фильтрат концентрировали с получением этил 2-(4-нитрофенил)-2-метилпропаноата (900 мг, 80%), который использовали без дополнительной очистки.
G-1; Этил 2-(4-аминофенил)-2-метилпропаноат
Раствор этил 2-(4-нитрофенил)-2-метилпропаноата (900 мг, 3,8 ммоль) в EtOH (10 мл) обрабатывали 10% раствором Pd-C (80 мг) и нагревали до 45ºC. Добавляли раствор формиата калия (4,10 г, 48,8 ммоль) в H2O (11 мл) в течение 15 минут. Реакционную смесь перемешивали при 65ºC в течение 2 часов и затем обрабатывали дополнительными 300 мг Pd/C. Реакционную смесь перемешивали в течение 1,5 часов и затем фильтровали через Целит. Объем растворителя уменьшали приблизительно на 50% при пониженном давлении и экстрагировали при помощи EtOAc. Органические слои сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением этил 2-(4-аминофенил)-2-метилпропаноата (G-1) (670 мг, 85%). 1H ЯМР (400 МГц, CDCl3) δ 7,14 (д, J=8,5 Гц, 2H), 6,65 (д, J=8,6 Гц, 2H), 4,10 (кв., J=7,1 Гц, 2H), 1,53 (с, 6H), 1,18 (т, J=7,1 Гц, 3H).
Пример 11:
G-2; 2-(4-Аминофенил)-2-метилпропан-1-ол
Раствор этил 2-(4-аминофенил)-2-метилпропаноата (30 мг, 0,145 ммоль) в ТГФ (1 мл) обрабатывали при помощи LiAlH4 (1M раствор в ТГФ, 0,226 мл, 0,226 ммоль) при 0ºC и перемешивали в течение 15 минут. Реакционную смесь обрабатывали 0,1н раствором NaOH, экстрагировали при помощи EtOAc и органические слои сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением 2-(4-аминофенил)-2-метилпропан-1-ола (G-2), который использовали без дополнительной очистки: 1H ЯМР (400 МГц, CDCl3) δ 7,17 (д, J=8,5 Гц, 2H), 6,67 (д, J=8,5 Гц, 2H), 3,53 (с, 2H), 1,28 (с, 6H).
Пример 12:
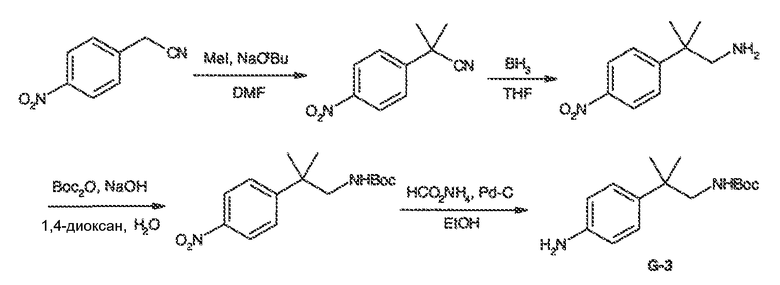
2-метил-2-(4-нитрофенил)пропаннитрил
Суспензию трет-бутоксида натрия (662 мг, 6,47 ммоль) в DMF (20 мл) при 0ºC обрабатывали 4-нитрофенилацетонитрилом (1000 мг, 6,18 ммоль) и перемешивали в течение 10 минут. Добавляли по каплям метилиодид (400 мкл, 6,47 ммоль) в течение 15 минут. Раствор перемешивали при 0-10ºC в течение 15 минут и затем при комнатной температуре еще в течение 15 минут. К этому фиолетовому раствору добавляли трет-бутоксид натрия (662 мг, 6,47 ммоль) и раствор перемешивали в течение 15 минут. Добавляли по каплям метилиодид (400 мкл, 6,47 ммоль) в течение 15 минут и раствор перемешивали в течение ночи. Добавляли трет-бутоксид натрия (192 мг, 1,94 ммоль) и реакционную смесь перемешивали при 0ºC в течение 10 минут. Добавляли метилиодид (186 мкл, 2,98 ммоль) и реакционную смесь перемешивали в течение 1 часа. Реакционную смесь затем распределяли между 1н раствором HCl (50 мл) и EtOAc (75 мл). Органический слой промывали 1 н раствором HCl и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением 2-метил-2-(4-нитрофенил)пропаннитрила в виде зеленого воскообразного твердого вещества (1,25 г, 99%). 1H ЯМР (400 МГц, CDCl3) δ 8,24 (д, J=8,9 Гц, 2H), 7,66 (д, J=8,9 Гц, 2H), 1,77 (с, 6H).
2-Метил-2-(4-нитрофенил)пропан-1-амин
К охлажденному раствору 2-метил-2-(4-нитрофенил)пропаннитрила (670 мг, 3,5 ммоль) в ТГФ (15 мл) добавляли BH3 (1M в ТГФ, 14 мл, 14 ммоль) по каплям при 0ºC. Смесь нагревали до комнатной температуры и нагревали при 70ºC в течение 2 часов. Добавляли 1н раствор HCl (2 мл) с последующим добавлением NaOH до достижения pH >7. Смесь экстрагировали простым эфиром и эфирный экстракт концентрировали с получением 2-метил-2-(4-нитрофенил)пропан-1-амина (610 мг, 90%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,20 (д, J=9,0 Гц, 2H), 7,54 (д, J=9,0 Гц, 2H), 2,89 (с, 2H), 1,38 (с, 6H).
трет-Бутил 2-метил-2-(4-нитрофенил)пропилкарбамат
К охлажденному раствору 2-метил-2-(4-нитрофенил)пропан-1-амина (600 мг, 3,1 ммоль) и 1н NaOH (3 мл, 3 ммоль) в 1,4-диоксане (6 мл) и воде (3 мл) добавляли Boc2O (742 мг, 3,4 ммоль) при 0ºC. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь подкисляли 5% раствором KHSO4 и затем экстрагировали этилацетатом. Органический слой сушили над MgSO4 и концентрировали с получением трет-бутил 2-метил-2-(4-нитрофенил)пропилкарбамата (725 мг, 80%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,11 (д, J=8,9 Гц, 2H), 7,46 (д, J=8,8 Гц, 2H), 3,63 (с, 2H), 1,31-1,29 (м, 15H).
G-3; трет-бутил 2-метил-2-(4-аминофенил)пропилкарбамат
К нагреваемому при температуре кипения с обратным холодильником раствору трет-бутил 2-метил-2-(4-нитрофенил)пропилкарбамата (725 мг, 2,5 ммоль) и формиата аммония (700 мг, 10,9 ммоль) в EtOH (25 мл) добавляли Pd-5% масс. на углероде (400 мг). Смесь кипятили с обратным холодильником в течение 1 часа, охлаждали и фильтровали через Целит. Фильтрат концентрировали с получением трет-бутил 2-метил-2-(4-аминофенил)пропилкарбамата (G-3) (550 мг, 83%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ 6,99 (д, J=8,5 Гц, 2H), 6,49 (д, J=8,6 Гц, 2H), 4,85 (с, 2H), 3,01 (д, J=6,3 Гц, 2H), 1,36 (с, 9H), 1,12 (с, 6H); ВЭЖХ время удерживания 2,02 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 265,2 m/z (MH+).
Пример 13:
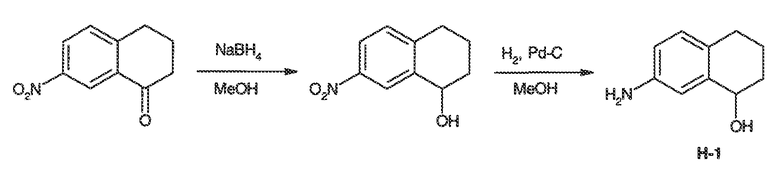
7-Нитро-1,2,3,4-тетрагидро-нафталин-1-ол
7-Нитро-3,4-дигидро-2H-нафталин-1-он (200 мг, 1,05 ммоль) растворяли в метаноле (5 мл) и добавляли по порциям NaBH4 ((78 мг, 2,05 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 минут и затем концентрировали и очищали колоночной хроматографией (10-50% этилацетат-гексан) с получением 7-нитро-1,2,3,4-тетрагидро-нафталин-1-ола (163 мг, 80%). 1H ЯМР (400 МГц, CD3CN) δ 8,30 (д, J=2,3 Гц, 1H), 8,02 (дд, J=8,5, 2,5 Гц, 1H), 7,33 (д, J=8,5 Гц, 1H), 4,76 (т, J=5,5 Гц, 1H), 2,96-2,80 (м, 2H), 2,10-1,99 (м, 2H), 1,86-1,77 (м, 2H); ВЭЖХ время удерживания 2,32 минут, 10-99% CH3CN, 5 мин цикл.
H-1; 7-Амино-1,2,3,4-тетрагидро-нафталин-1-ол
7-нитро-1,2,3,4-тетрагидро-нафталин-1-ол (142 мг, 0,73 ммоль) растворяли в метаноле (10 мл) и колбу промывали при помощи N2 (газ). Добавляли 10% Pd-C (10 мг) и реакционную смесь перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали с получением 7-амино-1,2,3,4-тетрагидро-нафталин-1-ола (H-1) (113 мг, 95%). ВЭЖХ время удерживания 0,58 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 164,5 m/z (MH+).
Пример 14:

7-Нитро-3,4-дигидро-2H-нафталин-1-он оксим
К раствору 7-нитро-3,4-дигидро-2H-нафталин-1-она (500 мг, 2,62 ммоль) в пиридине (2 мл) добавляли раствор гидроксиламина (1 мл, ~50% раствор в воде). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали и очищали колоночной хроматографией (10-50% этилацетат-гексан) с получением 7-нитро-3,4-дигидро-2H-нафталин-1-он оксима (471 мг, 88%). ВЭЖХ время удерживания 2,67 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 207,1 m/z (MH+).
1,2,3,4-Тетрагидро-нафталин-1,7-диамин
7-Нитро-3,4-дигидро-2H-нафталин-1-он оксим (274 мг, 1,33 ммоль) растворяли в метаноле (10 мл) и колбу промывали при помощи N2 (газ). Добавляли 10% Pd-C (50 мг) и реакционную смесь перемешивали в атмосфере H2 (1 атм) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали с получением 1,2,3,4-тетрагидро-нафталин-1,7-диамина (207 мг, 96%). 1H ЯМР (400 МГц, DMSO-d6) δ 6,61-6,57 (м, 2H), 6,28 (дд, J=8,0, 2,4 Гц, 1H), 4,62 (с, 2H), 3,58 (м, 1H), 2,48-2,44 (м, 2H), 1,78-1,70 (м, 2H), 1,53-1,37 (м, 2H).
H-2; трет-бутиловый эфир (7-Амино-1,2,3,4-тетрагидро-нафталин-1-ил)-карбаминовой кислоты
К раствору 1,2,3,4-тетрагидро-нафталин-1,7-диамина (154 мг, 0,95 ммоль) и триэтиламина (139 мкл, 1,0 ммоль) в метаноле (2 мл), охлажденному до 0ºC, добавляли ди-трет-бутилдикарбонат (207 мг, 0,95 ммоль). Реакционную смесь перемешивали при 0ºC и затем концентрировали и очищали колоночной хроматографией (5-50% метанола-дихлорметан) с получением трет-бутилового эфира (7-амино-1,2,3,4-тетрагидро-нафталин-1-ил)-карбаминовой кислоты (H-2) (327 мг, количественный). ВЭЖХ время удерживания 1,95 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 263,1 m/z (MH+).
Пример 15:
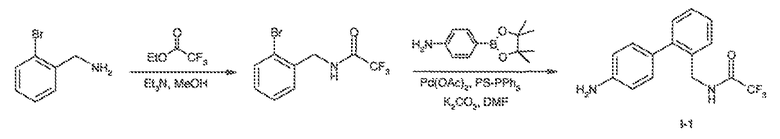
N-(2-Бром-бензил)-2,2,2-трифтор-ацетамид
К раствору 2-бромбензиламина (1,3 мл, 10,8 ммоль) в метаноле (5 мл) добавляли этилтрифторацетат (1,54 мл, 21,6 ммоль) и триэтиламин (1,4 мл, 10,8 ммоль) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь затем концентрировали в вакууме с получением N-(2-бром-бензил)-2,2,2-трифтор-ацетамида (3,15 г, количественный). ВЭЖХ время удерживания 2,86 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 283,9 m/z (MH+).
I-1; N-(4'-Амино-бифенил-2-илметил)-2,2,2-трифтор-ацетамид
Смесь N-(2-бром-бензил)-2,2,2-трифтор-ацетамида (282 мг, 1,0 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (284 мг, 1,3 ммоль), Pd(OAc)2 (20 мг, 0,09 ммоль) и PS-PPh3 (40 мг, 3 ммоль/г, 0,12 ммоль) растворяли в DMF (5 мл) и добавляли 4M K2CO3 раствор (0,5 мл). Реакционную смесь нагревали при 80ºC в течение ночи. Смесь фильтровали, концентрировали и очищали колоночной хроматографией (0-50% этилацетата-гексан) с получением N-(4'-амино-бифенил-2-илметил)-2,2,2-трифтор-ацетамида (I-1) (143 мг, 49%). ВЭЖХ время удерживания 1,90 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 295,5 m/z (MH+).
Коммерчески доступные амины
Амиды (Соединения формулы I)
Общая схема:
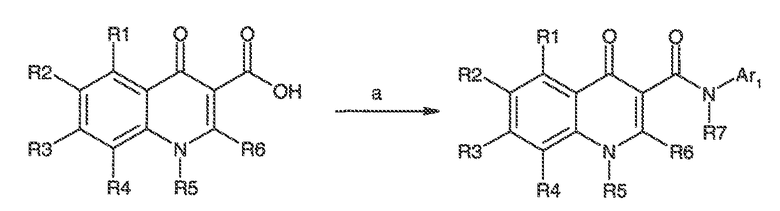
a) Ar1R7NH, связывающий реагент, основание, растворитель. Примеры используемых условий: HATU, DIEA, DMF; BOP, DIEA, DMF; HBTU, Et3N, CH2Cl2; PFP-TFA, пиридин
Специальный пример:
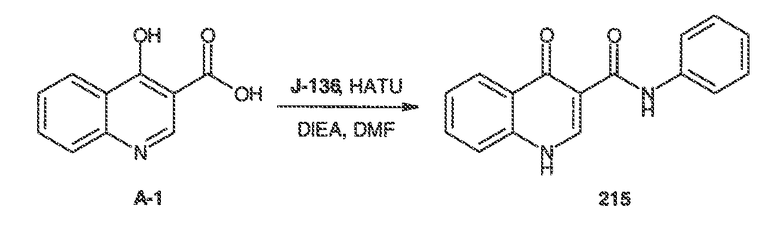
215; 4-Оксо-N-фенил-1H-хинолин-3-карбоксамид
К раствору 4-гидрокси-хинолин-3-карбоновой кислоты (A-1) (19 мг, 0,1 ммоль), HATU (38 мг, 0,1 ммоль) и DIEA (34,9 мкл, 0,2 ммоль) в DMF (1 мл) добавляли анилин (18,2 мкл, 0,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Полученный раствор фильтровали и очищали при помощи ВЭЖХ (10-99% CH3CN/H2O) с получением 4-оксо-N-фенил-1H-хинолин-3-карбоксамида (215) (12 мг, 45%). 1H ЯМР (400 МГц, DMSO-d6) δ 12,97 (с, 1H), 12,50 (с, 1H), 8,89 (с, 1H), 8,34 (дд, J=8,1, 1,1 Гц, 1H), 7,83 (т, J=8,3 Гц, 1H), 7,75 (м, 3H), 7,55 (т, J=8,1 Гц, 1H), 7,37 (т, J=7,9 Гц, 2H), 7,10 (т, J=6,8 Гц, 1H); ВЭЖХ время удерживания 3,02 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 265,1 m/z (MH+).
В представленной ниже Таблице перечислены другие примеры, синтезированные в соответствии с Общей схемой, представленной выше.
Индолы
Пример 1:
Общая схема:
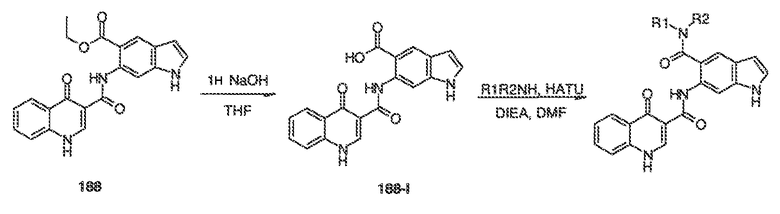
Специальный пример:
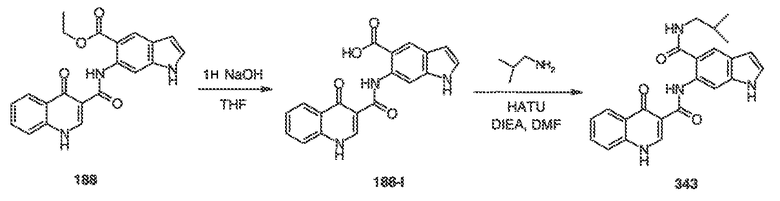
188-I; 6-[(4-Оксо-1H-хинолин-3-ил)карбониламино]-1H-индол-5-карбоновая кислота
Смесь этилового эфира 6-[(4-оксо-1H-хинолин-3-ил)карбониламино]-1H-индол-5-карбоновой кислоты (188) (450 мг, 1,2 ммоль) и 1н раствора NaOH (5 мл) в ТГФ (10 мл) нагревали при 85ºC в течение ночи. Реакционную смесь распределяли между EtOAc и водой. Водный слой подкисляли 1н раствором HCl до pH 5 и осадок фильтровали, промывали водой и сушили на воздухе с получением 6-[(4-оксо-1H-хинолин-3-ил)карбониламино]-1H-индол-5-карбоновой кислоты (188-I) (386 мг, 93%). 1H-ЯМР (400 МГц, DMSO-d6) δ 12,92-12,75 (м, 2H), 11,33 (с, 1H), 8,84 (с, 1H), 8,71 (с, 1H), 8,30 (дд, J=8,1, 0,9 Гц, 1H), 8,22 (с, 1H), 7,80-7,72 (м, 2H), 7,49 (т, J=8,0 Гц, 1H), 7,41 (т, J=2,7 Гц, 1H), 6,51 (м, 1H); ВЭЖХ время удерживания 2,95 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 376,2 m/z (MH+).
343; N-[5-(Изобутилкарбамоил)-1H-индол-6-ил]-4-оксо-1H-хинолин-3-карбоксамид
К раствору 6-[(4-оксо-1H-хинолин-3-ил)карбониламино]-1H-индол-5-карбоновой кислоты (188-I) (26 мг, 0,08 ммоль), HATU (38 мг, 0,1 ммоль) и DIEA (35 мкл, 0,2 ммоль) в DMF (1 мл) добавляли изобутиламин (7 мг, 0,1 ммоль) и реакционную смесь перемешивали при 65ºC в течение ночи. Полученный раствор фильтровали и очищали при помощи ВЭЖХ (10-99% CH3CN/H2O) с получением продукта, N-[5-(изобутилкарбамоил)-1H-индол-6-ил]-4-оксо-1H-хинолин-3-карбоксамида (343) (20 мг, 66%). 1H-ЯМР (400 МГц, DMSO-d6) δ 12,66 (д, J=7,4 Гц, 1H), 12,42 (с, 1H), 11,21 (с, 1H), 8,81 (д, J=6,6 Гц, 1H), 8,47 (с, 1H), 8,36 (т, J=5,6 Гц, 1H), 8,30 (д, J=8,4 Гц, 1H), 7,79 (т, J=7,9 Гц, 1H), 7,72-7,71 (м, 2H), 7,51 (т, J=7,2 Гц, 1H), 7,38 (м, 1H), 6,48 (м, 1H), 3,10 (т, J=6,2 Гц, 2H), 1,88 (м, 1H), 0,92 (д, J=6,7 Гц, 6H); ВЭЖХ время удерживания 2,73 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 403,3 m/z (MH+).
Другой пример:
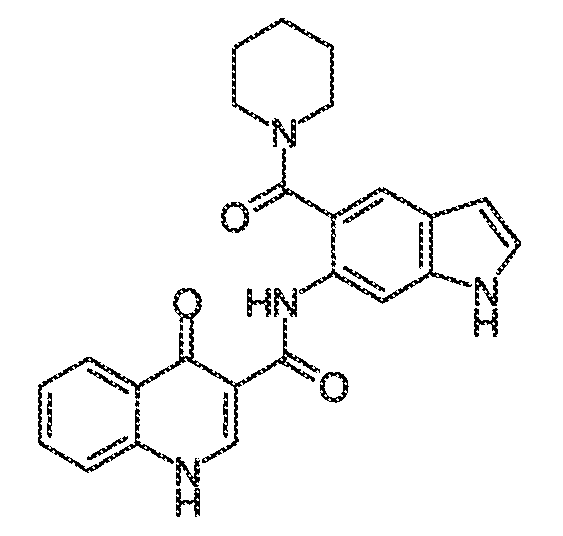
148; 4-Оксо-N-[5-(1-пиперидилкарбонил)-1H-индол-6-ил]-1H-хинолин-3-карбоксамид
4-Оксо-N-[5-(1-пиперидилкарбонил)-1H-индол-6-ил]-1H-хинолин-3-карбоксамид (148) синтезировали в соответствии с общей схемой, представленной выше, связывая кислоту (188-I) с пиперидином. Общий выход (12%). ВЭЖХ время удерживания 2,79 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 415,5 m/z (MH+).
Пример 2:
Общая схема:
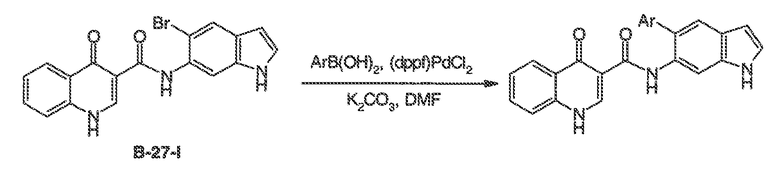
Специальный пример:
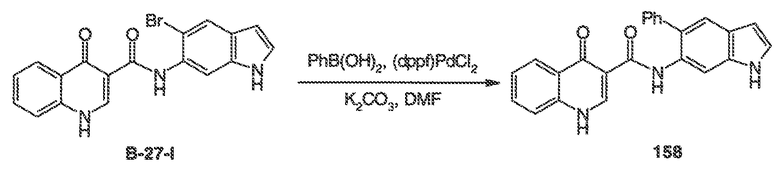
158; 4-Оксо-N-(5-фенил-1H-индол-6-ил)-1H-хинолин-3-карбоксамид
Смесь N-(5-бром-1H-индол-6-ил)-4-оксо-1H-хинолин-3-карбоксамида (B-27-I) (38 мг, 0,1 моль), фенилбороновой кислоты (18 мг, 0,15 ммоль), (dppf)PdCl2 (каталитическое количество) и K2CO3 (100 мкл, 2M раствор) в DMF (1 мл) нагревали в микроволновой печи при 180ºC в течение 10 минут. Реакционную смесь фильтровали и очищали при помощи ВЭЖХ (10-99% CH3CN/H2O) с получением продукта, 4-оксо-N-(5-фенил-1H-индол-6-ил)-1H-хинолин-3-карбоксамида (158) (5 мг, 13%). ВЭЖХ время удерживания 3,05 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 380,2 m/z (MH+).
В Таблице ниже перечислены другие примеры, синтезированные в соответствии с общей схемой, представленной выше.
Пример 3:
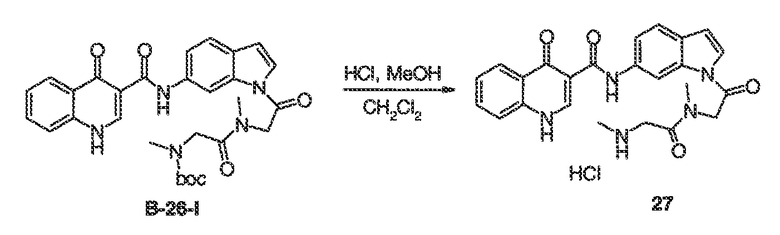
27; N-[1-[2-[Метил-(2-метиламиноацетил)-амино]ацетил]-1H-индол-6-ил]-4-оксо-1H-хинолин-3-карбоксамид
К раствору трет-бутилового эфира метил-{[метил-(2-оксо-2-{6-[(4-оксо-1,4-дигидро-хинолин-3-карбонил)-амино]-индол-1-ил}-этил)-карбамоил]-метил}-карбаминовой кислоты (B-26-I) (2,0 г, 3,7 ммоль), растворенного в смеси CH2Cl2 (50 мл) и метанола (15 мл), добавляли раствор HCl (60 мл, 1,25 M в метаноле). Реакционную смесь перемешивали при комнатной температуре в течение 64 часов. Осажденный продукт собирали фильтрованием, промывали диэтиловым эфиром и сушили в условиях высокого вакуума с получением HCl соли продукта, N-[1-[2-[метил-(2-метиламиноацетил)-амино]ацетил]-1H-индол-6-ил]-4-оксо-1H-хинолин-3-карбоксамида (27), в виде серовато-белого твердого вещества (1,25 г, 70%). 1H-ЯМР (400 МГц, DMSO-d6) δ 13,20 (д, J=6,7 Гц, 1H), 12,68 (с, 1H), 8,96-8,85 (м, 1H), 8,35 (д, J=7,9 Гц, 1H), 7,91-7,77 (м, 3H), 7,64-7,54 (м, 3H), 6,82 (м, 1H), 5,05 (с, 0,7H), 4,96 (с, 1,3H), 4,25 (т, J=5,6 Гц, 1,3H), 4,00 (т, J=5,7 Гц, 0,7H), 3,14 (с, 2H), 3,02 (с, 1H), 2,62 (т, J=5,2 Гц, 2H), 2,54 (т, J=5,4 Гц, 1H); ВЭЖХ время удерживания 2,36 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 446,5 m/z (MH+).
Фенолы
Пример 1:
Общая схема:
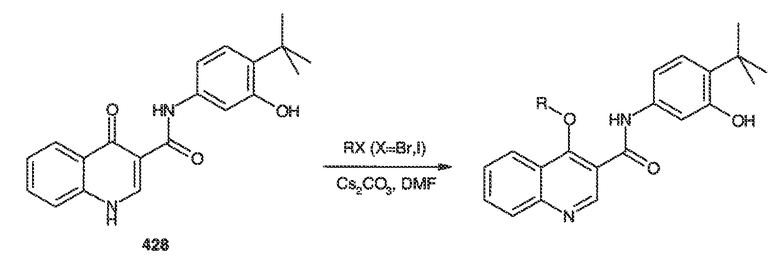
Специальный пример:
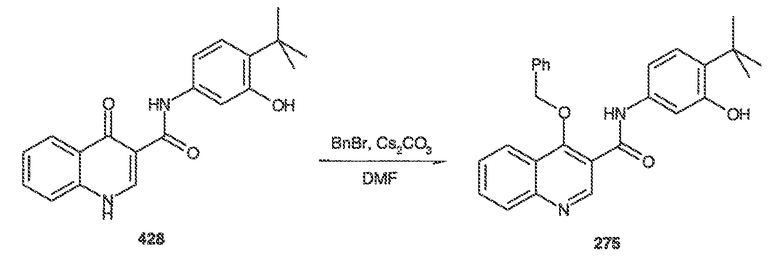
275; 4-Бензилокси-N-(3-гидрокси-4-трет-бутил-фенил)-хинолин-3-карбоксамид
К смеси N-(3-гидрокси-4-трет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (428) (6,7 мг, 0,02 ммоль) и Cs2CO3 (13 мг, 0,04 ммоль) в DMF (0,2 мл) добавляли BnBr (10 мкл, 0,08 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь фильтровали и очищали с использованием ВЭЖХ с получением 4-бензилокси-N-(3-гидрокси-4-трет-бутил-фенил)-хинолин-3-карбоксамида (275). 1H ЯМР (400 МГц, DMSO-d6) δ 12,23 (с, 1H), 9,47 (с, 1H), 9,20 (с, 1H), 8,43 (д, J=7,9 Гц, 1H), 7,79 (т, J=2,0 Гц, 2H), 7,56 (м, 1H), 7,38-7,26 (м, 6H), 7,11 (д, J=8,4 Гц, 1H), 6,99 (дд, J=8,4, 2,1 Гц, 1H), 5,85 (с, 2H), 1,35 (с, 9H). ВЭЖХ время удерживания 3,93 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 427,1 m/z (MH+).
Другой пример:
415; N-(3-Гидрокси-4-трет-бутил-фенил)-4-метокси-хинолин-3-карбоксамид
N-(3-Гидрокси-4-трет-бутил-фенил)-4-метокси-хинолин-3-карбоксамид (415) синтезировали в соответствии с общей схемой, представленной выше, осуществляя взаимодействие N-(3-гидрокси-4-трет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (428) с метилиодидом. 1H ЯМР (400 МГц, DMSO-d6) δ 12,26 (с, 1H), 9,46 (с, 1H), 8,99 (с, 1H), 8,42 (т, J=4,2 Гц, 1H), 7,95-7,88 (м, 2H), 7,61-7,69 (м, 1H), 7,38 (д, J=2,1 Гц, 1H), 7,10 (д, J=8,4 Гц, 1H), 6,96 (дд, J=8,4, 2,1 Гц, 1H), 4,08 (с, 3H), 1,35 (с, 9H); ВЭЖХ время удерживания 3,46 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 351,5 m/z (MH+).
Пример 2:
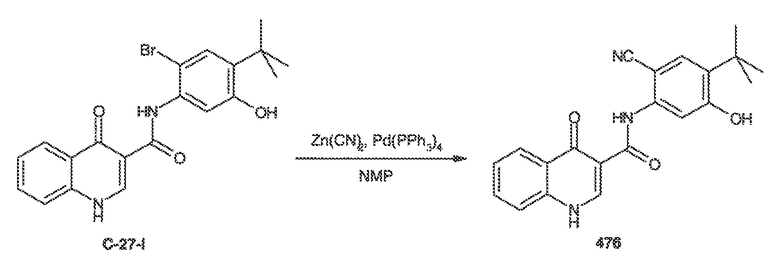
476; N-(4-трет-бутил-2-циано-5-гидроксифенил)-1,4-дигидро-4-оксохинолин-3-карбоксамид
К суспензии N-(4-трет-бутил-2-бром-5-гидроксифенил)-1,4-дигидро-4-оксохинолин-3-карбоксамида (C-27-I) (84 мг, 0,2 ммоль), Zn(CN)2 (14 мг, 0,12 ммоль) в NMP (1 мл) добавляли Pd(PPh3)4 (16 мг, 0,014 ммоль) в атмосфере азота. Смесь нагревали в микроволновой печи при 200ºC в течение 1 часа, фильтровали и очищали с использованием препаративной ВЭЖХ с получением N-(4-трет-бутил-2-циано-5-гидроксифенил)-1,4-дигидро-4-оксохинолин-3-карбоксамида (476). 1H ЯМР (400 МГц, DMSO-d6) δ 13,00 (д, J=6,4 Гц, 1H), 12,91 (с, 1H), 10,72 (с, 1H), 8,89 (д, J=6,8Гц, 1H), 8,34 (д, J=8,2Гц, 1H), 8,16 (с, 1H), 7,85-7,75 (м, 2H), 7,56-7,54 (м, 1H), 7,44 (с, 1H), 1,35 (с, 9H); ВЭЖХ время удерживания 3,42 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 362,1 m/z (MH+).
Анилины
Пример 1:
Общая схема:
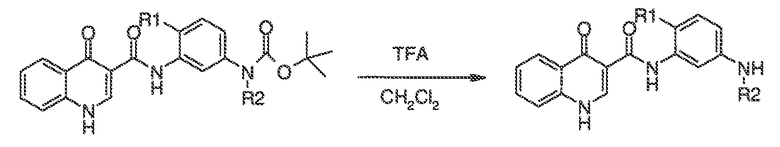
Специальный пример:
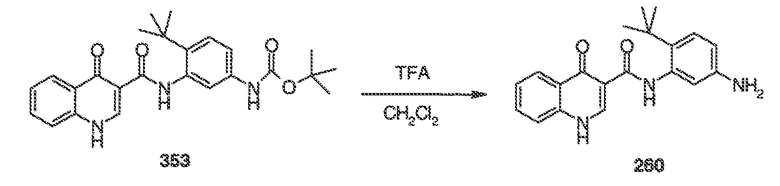
260; N-(5-Амино-2-трет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамид
Смесь трет-бутилового эфира [3-[(4-оксо-1H-хинолин-3-ил)карбониламино]-4-трет-бутил-фенил]аминомуравьиной кислоты (353) (33 мг, 0,08 ммоль), TFA (1 мл) и CH2Cl2 (1 мл) перемешивали при комнатной температуре в течение ночи. Раствор концентрировали и остаток растворяли в DMSO (1 мл) и очищали при помощи ВЭЖХ (10-99% CH3CN/H2O) с получением продукта, N-(5-амино-2-трет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (260) (15 мг, 56%). 1H ЯМР (400 МГц, DMSO-d6) δ 13,23 (д, J=6,6 Гц, 1H), 12,20 (с, 1H), 10,22 (шир.с, 2H), 8,88 (д, J=6,8 Гц, 1H), 8,34 (д, J=7,8 Гц, 1H), 7,86-7,80 (м, 3H), 7,56-7,52 (м, 2H), 7,15 (дд, J=8,5, 2,4 Гц, 1H), 1,46 (с, 9H); ВЭЖХ время удерживания 2,33 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 336,3 m/z (MH+).
В Таблице ниже перечислены другие примеры, синтезированные в соответствии с общей схемой, представленной выше.
Пример 2:
Общая схема:
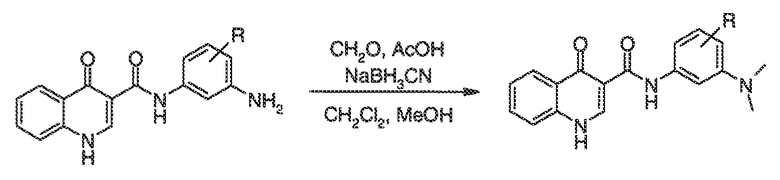
Специальный пример:
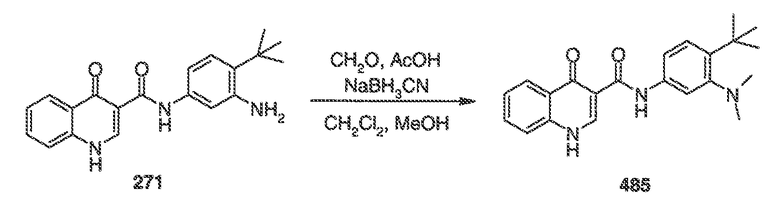
485; N-(3-Диметиламино-4-трет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамид
К суспензии N-(3-амино-4-трет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (271) (600 мг, 1,8 ммоль) в CH2Cl2 (15 мл) и метаноле (5 мл) добавляли уксусную кислоту (250 мкл) и формальдегид (268 мкл, 3,6 ммоль, 37% масс. в воде). Через 10 минут добавляли цианоборогидрид натрия (407 мг, 6,5 ммоль) одной порцией. Добавляли дополнительное количество формальдегида (135 мкл, 1,8 ммоль, 37% масс. в воде) через 1,5 и 4,2 часа. Через 4,7 часа смесь разбавляли простым эфиром (40 мл), промывали водой (25 мл) и насыщенным солевым раствором (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Полученное красно-коричневое пенистое вещество очищали препаративной ВЭЖХ с получением N-(3-диметиламино-4-трет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (485) (108 мг, 17%). 1H ЯМР (300 МГц, CDCl3) δ 13,13 (шир.с, 1H), 12,78 (с, 1H), 8,91 (шир.с, 1H), 8,42 (шир.с, 1H), 8,37 (д, J=8,1 Гц, 1H), 7,72-7,58 (м, 2H), 7,47-7,31 (м, 3H), 3,34 (с, 6H), 1,46 (с, 9H); ВЭЖХ время удерживания 2,15 минут, 10-100% CH3CN, 5 мин цикл; ESI-MS 364,3 m/z (MH+).
В Таблице ниже перечислены другие примеры, синтезированные в соответствии с общей схемой, представленной выше.
Пример 3:
Общая схема:

Специальный пример:
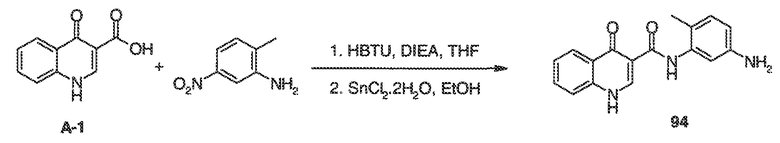
94; N-(5-Амино-2-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамид
К раствору 4-гидрокси-хинолин-3-карбоновой кислоты (A-1) (50 мг, 0,26 ммоль), HBTU (99 мг, 0,26 ммоль) и DIEA (138 мкл, 0,79 ммоль) в ТГФ (2,6 мл) добавляли 2-метил-5-нитро-фениламин (40 мг, 0,26 ммоль). Смесь нагревали при 150ºC в микроволновой печи в течение 20 минут и полученный раствор концентрировали. Остаток растворяли в EtOH (2 мл) и добавляли SnCl2.2H2O (293 мг, 1,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь подщелачивали насыщенным раствором NaHCO3 до pH 7-8 и экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток растворяли в DMSO и очищали при помощи ВЭЖХ (10-99% CH3CN/H2O) с получением продукта, N-(5-амино-2-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (94) (6 мг, 8%). ВЭЖХ время удерживания 2,06 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 294,2 m/z (MH+).
Другой пример:
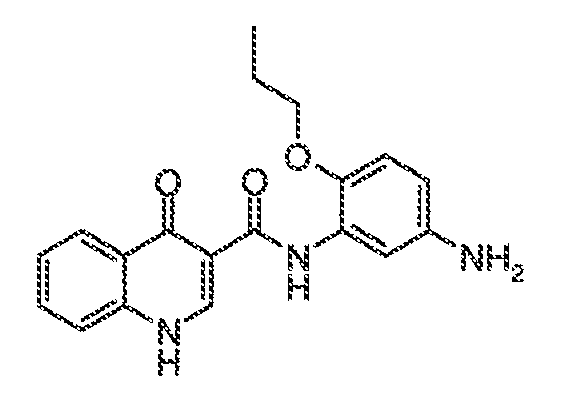
17; N-(5-Амино-2-пропокси-фенил)-4-оксо-1H-хинолин-3-карбоксамид
N-(5-Амино-2-пропокси-фенил)-4-оксо-1H-хинолин-3-карбоксамид (17) получали в соответствии с общей схемой, представленной выше, исходя из 4-гидрокси-хинолин-3-карбоновой кислоты (A-1) и 5-нитро-2-пропокси-фениламина. Выход (9%). ВЭЖХ время удерживания 3,74 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 338,3 m/z (MH+).
Пример 4:
Общая схема:
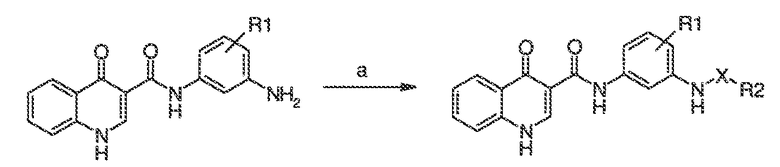
X= CO, CO2, SO2: a) R2XCl, DIEA, ТГФ или R2XCl, NMM, 1,4-диоксан или R2XCl, Et3N, CH2Cl2, DMF.
Специальный пример:
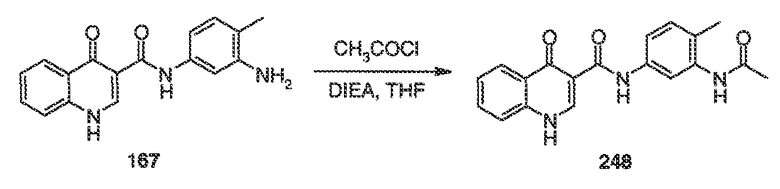
248; N-(3-Ацетиламино-4-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамид
К раствору N-(3-амино-4-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (167) (33 мг, 0,11 ммоль) и DIEA (49 мкл, 0,28 ммоль) в ТГФ (1 мл) добавляли ацетилхлорид (16 мкл, 0,22 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. LCMS анализ показал, что произошло диацилирование. Добавляли раствор пиперидина (81 мкл, 0,82 ммоль) в CH2Cl2 (2 мл) и реакционную смесь перемешивали еще в течение 30 минут, после этого только желаемый продукт определялся анализом LCMS. Реакционный раствор концентрировали и остаток растворяли в DMSO и очищали при помощи ВЭЖХ (10-99% CH3CN/H2O) с получением продукта, N-(3-ацетиламино-4-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (248) (4 мг, 11%). 1H ЯМР (400 МГц, DMSO-d6) δ 12,95 (д, J=6,6 Гц, 1H), 12,42 (с, 1H), 9,30 (с, 1H), 8,86 (д, J=6,8 Гц, 1H), 8,33 (дд, J=8,1, 1,3 Гц, 1H), 7,85-7,81 (м, 2H), 7,76 (д, J=7,8 Гц, 1H), 7,55 (т, J=8,1 Гц, 1H), 7,49 (дд, J=8,2, 2,2 Гц, 1H), 7,18 (д, J=8,3 Гц, 1H), 2,18 (с, 3H), 2,08 (с, 3H); ВЭЖХ время удерживания 2,46 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 336,3 m/z (MH+).
В Таблице ниже перечислены другие примеры, синтезированные в соответствии с общей схемой, представленной выше.
Пример 5:
Общая схема:

Специальный пример:
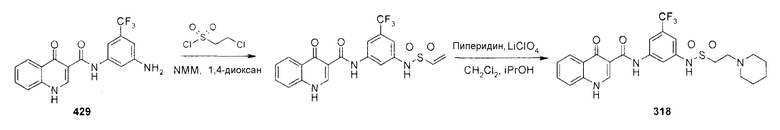
4-Оксо-N-[3-(трифторметил)-5-(винилсульфонамидо)фенил]-1,4-дигидрохинолин-3-карбоксамид
К суспензии N-[3-амино-5-(трифторметил)фенил]-4-оксо-1H-хинолин-3-карбоксамида (429) (500 мг 1,4 ммоль) в 1,4-диоксане (4 мл) добавляли NMM (0,4 мл, 3,6 ммоль). Добавляли β-хлорэтилсульфонилхлорид (0,16 мл, 1,51 ммоль) в атмосфере аргона. Смесь перемешивали при комнатной температуре в течение 6 ½ часов, после чего анализ ТСХ (CH2Cl2-EtOAc, 8:2) показал новое пятно с очень схожим Rf с исходным веществом. Добавляли еще 0,5 экв. NMM и смесь перемешивали при комнатной температуре в течение ночи. LCMS анализ неочищенной смеси показал >85% конверсию в желаемый продукт. Смесь концентрировали, обрабатывали раствором 1M HCl (5 мл) и экстрагировали при помощи EtOAc (3×10 мл) и CH2Cl2 (3×10 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали с получением 4-оксо-N-[3-(трифторметил)-5-(винилсульфонамидо)фенил]-1,4-дигидрохинолин-3-карбоксамида в виде оранжевого пенообразного вещества (0,495 г, 79%), которое использовали на следующей стадии без дополнительной очистки. 1H-ЯМР (d6-Ацетон, 300 МГц) δ 8,92 (с, 1H), 8,41-8,38 (м, 1H), 7,94 (м, 2H), 7,78 (шир.с, 2H), 7,53-7,47 (м, 1H), 7,30 (с, 1H), 6,87-6,79 (дд, J=9,9 Гц, 1H), 6,28 (д, J=16,5 Гц, 1H), 6,09 (д, J=9,9 Гц, 1H); ESI-MS 436,4 m/z (MH-).
318; 4-Оксо-N-[3-[2-(1-пиперидил)этилсульфониламино]-5-(трифторметил)фенил]-1H-хинолин-3-карбоксамид
Смесь 4-оксо-N-[3-(трифторметил)-5-(винилсульфонамидо)фенил]-1,4-дигидрохинолин-3-карбоксамида (50 мг, 0,11 ммоль), пиперидина (18 мкл, 1,6 экв.) и LiClO4 (20 мг, 1,7 экв.) суспендировали в 1:1 растворе CH2Cl2:изопропанол (1,5 мл). Смесь кипятили с обратным холодильником при 75ºC в течение 18 часов. После этого LCMS анализ показал >95% конверсию в желаемый продукт. Неочищенную смесь очищали обращенно-фазовой ВЭЖХ с получением 4-оксо-N-[3-[2-(1-пиперидил)этилсульфониламино]-5-(трифторметил)фенил]-1H-хинолин-3-карбоксамида (318) в виде желтоватого твердого вещества (15 мг, 25%). 1H-ЯМР (d6-Ацетон, 300 МГц) δ 8,92 (шир.с, 1H), 8,4 (д, J=8,1 Гц, 1H), 8,05 (шир.с, 1H), 7,94 (шир.с, 1H), 7,78 (шир.с, 2H), 7,53-751 (м, 1H), 7,36 (шир.с, 1H), 3,97 (т, J=7,2 Гц, 2H), 3,66 (т, J=8 Гц, 2H), 3,31-3,24 (м, 6H), 1,36-1,31 (м, 4H); ESI-MS 489,1 m/z (MH+).
В Таблице ниже перечислены другие примеры, синтезированные в соответствии с общей схемой, представленной выше.
Пример 6:
Общая схема:
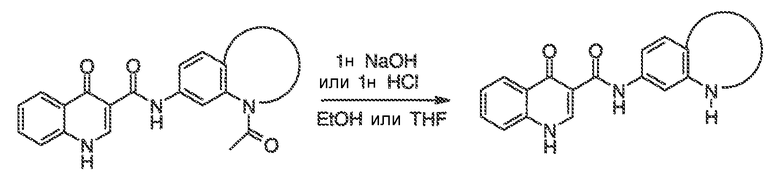
Специальный пример:
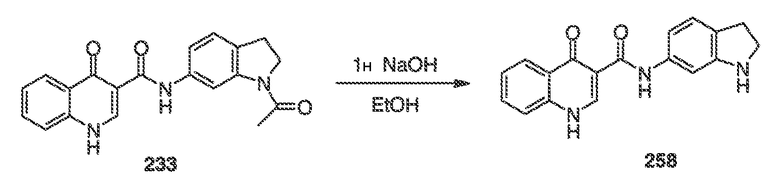
258; N-Индолин-6-ил-4-оксо-1H-хинолин-3-карбоксамид
Смесь N-(1-ацетилиндолин-6-ил)-4-оксо-1H-хинолин-3-карбоксамида (233) (43 мг, 0,12 ммоль), 1н раствора NaOH (0,5 мл) и этанола (0,5 мл) нагревали до температуры кипения с обратным холодильником в течение 48 часов. Раствор концентрировали и остаток растворяли в DMSO (1 мл) и очищали при помощи ВЭЖХ (10-99% CH3CN-H2O) с получением продукта, N-индолин-6-ил-4-оксо-1H-хинолин-3-карбоксамида (258) (10 мг, 20%). ВЭЖХ время удерживания 2,05 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 306,3 m/z (MH+).
В Таблице ниже перечислены другие примеры, синтезированные в соответствии с общей схемой, представленной выше.
Пример 2:
Общая схема:
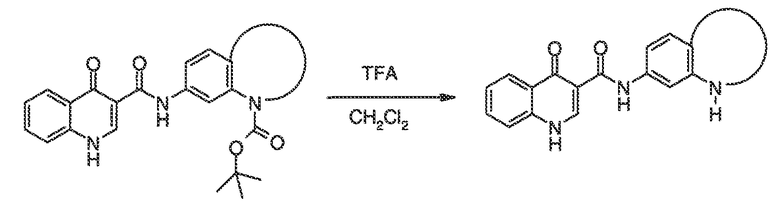
Специальный пример:
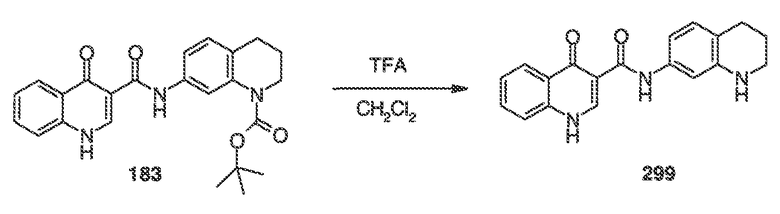
299; 4-Оксо-N-(1,2,3,4-тетрагидрохинолин-7-ил)-1H-хинолин-3-карбоксамид
Смесь трет-бутилового эфира 7-[(4-оксо-1H-хинолин-3-ил)карбониламино]-1,2,3,4-тетрагидрохинолин-1-карбоновой кислоты (183) (23 мг, 0,05 ммоль), TFA (1 мл) и CH2Cl2 (1 мл) перемешивали при комнатной температуре в течение ночи. Раствор концентрировали и остаток растворяли в DMSO (1 мл) и очищали при помощи ВЭЖХ (10-99% CH3CN-H2O) с получением продукта, 4-оксо-N-(1,2,3,4-тетрагидрохинолин-7-ил)-1H-хинолин-3-карбоксамида (299) (7 мг, 32%). ВЭЖХ время удерживания 2,18 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 320,3 m/z (MH+).
Другой пример:
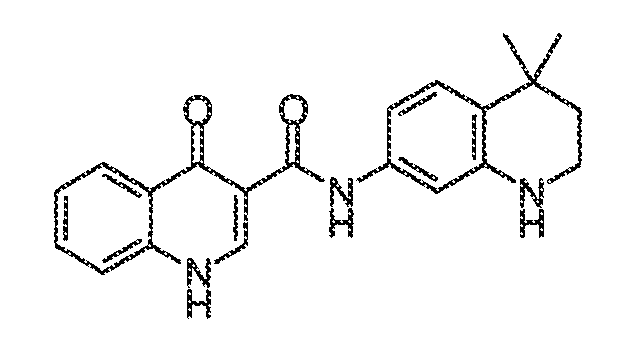
300; N-(4,4-Диметил-1,2,3,4-тетрагидрохинолин-7-ил)-4-оксо-1H-хинолин-3-карбоксамид
N-(4,4-Диметил-1,2,3,4-тетрагидрохинолин-7-ил)-4-оксо-1H-хинолин-3-карбоксамид (300) синтезировали в соответствии с общей схемой, представленной выше, исходя из трет-бутилового эфира 4,4-диметил-7-[(4-оксо-1H-хинолин-3-ил)карбониламино]-1,2,3,4-тетрагидрохинолин-1-карбоновой кислоты (108). Выход (33%). 1H ЯМР (400 МГц, DMSO-d6) δ 13,23 (д, J=6,6 Гц, 1H), 12,59 (с, 1H), 8,87 (д, J=6,8 Гц, 1H), 8,33 (д, J=7,7 Гц, 1H), 7,86-7,79 (м, 3H), 7,58-7,42 (м, 3H), 3,38 (м, 2H), 1,88 (м, 2H), 1,30 (с, 6H); ВЭЖХ время удерживания 2,40 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 348,2 m/z (MH+).
Другие
Пример 1:
Общая схема:
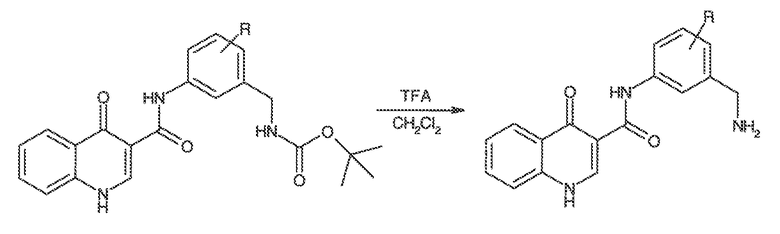
Специальный пример:
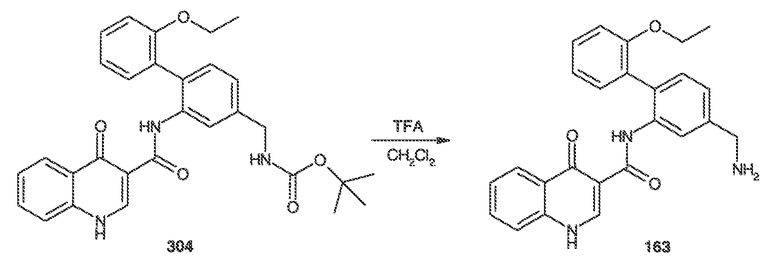
163; 4-Оксо-1,4-дигидро-хинолин-3-карбоновой кислоты (4-аминометил-2'-этокси-бифенил-2-ил)-амид
трет-Бутиловый эфир {2'-этокси-2-[(4-оксо-1,4-дигидрохинолин-3-карбонил)-амино]-бифенил-4-илметил}-карбаминовой кислоты (304) (40 мг, 0,078 ммоль) перемешивали в CH2Cl2/TFA смеси (3:1, 20 мл) при комнатной температуре в течение 1 часа. Летучие вещества удаляли на роторном испарителе. Неочищенный продукт очищали препаративной ВЭЖХ с получением (4-аминометил-2'-этоксибифенил-2-ил)амина 4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (163) в виде желтовато-коричневого твердого вещества (14 мг. 43%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,87 (д, J=6,3 Гц, 1H), 11,83 (с, 1H), 8,76 (д, J=6,3 Гц, 1H), 8,40 (с, 1H), 8,26 (шир.с, 2H), 8,01 (дд, J=8,4 Гц, J=1,5 Гц, 1H), 7,75 (дт, J=8,1 Гц, J=1,2 Гц, 1H), 7,67 (д, J=7,8 Гц, 1H), 7,47-7,37 (м, 2H), 7,24 (с, 2H), 7,15 (дд, J=7,5 Гц, J=1,8 Гц, 1H), 7,10 (д, J=8,1 Гц, 1H), 7,02 (дт, J=7,5 Гц, J=0,9 Гц, 1H), 4,09 (м, 2H), 4,04 (кв., J=6,9 Гц, 2H), 1,09 (т, J=6,9 Гц, 3H); ВЭЖХ время удерживания 1,71 минут, 10-100% CH3CN, 5 мин градиент; ESI-MS 414,1 m/z (MH+).
Другой пример:
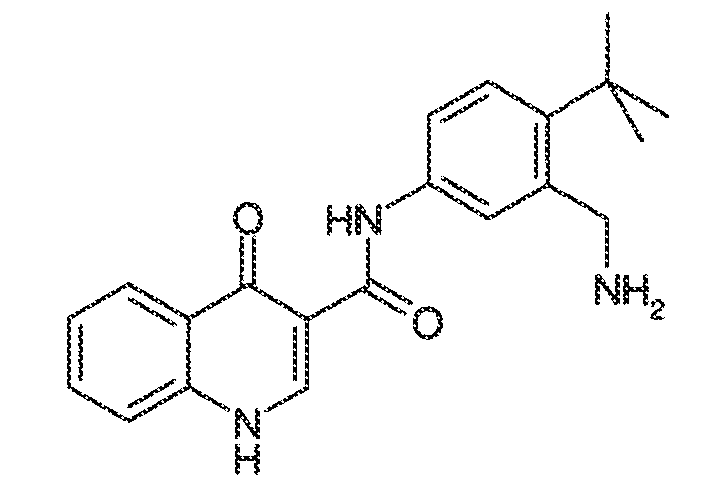
390; N-[3-(Аминометил)-4-трет-бутил-фенил]-4-оксо-1H-хинолин-3-карбоксамид
N-[3-(Аминометил)-4-трет-бутил-фенил]-4-оксо-1H-хинолин-3-карбоксамид (390) синтезировали в соответствии с общей схемой, представленной выше, исходя из трет-бутилового эфира [5-[(4-оксо-1H-хинолин-3-ил)карбониламино]-2-трет-бутил-фенил]метиламиномуравьиной кислоты (465). ВЭЖХ время удерживания 2,44 минут, 10-99% CH3CN, 5 мин градиент; ESI-MS m/z 350,3 (M + H)+.
Пример 2:
Общая схема:
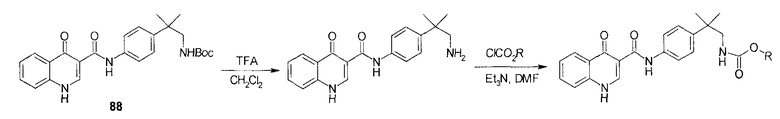
Специальный пример:
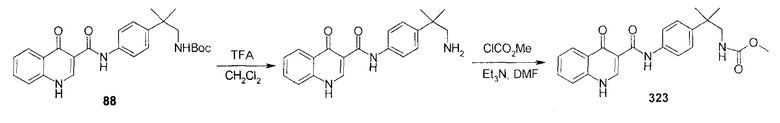
3-(2-(4-(1-Амино-2-метилпропан-2-ил)фенил)ацетил)хинолин-4(1H)-он
трет-Бутиловый эфир (2-метил-2-{4-[2-оксо-2-(4-оксо-1,4-дигидро-хинолин-3-ил)-этил]-фенил}-пропил)-карбаминовой кислоты (88) (0,50 г, 1,15 ммоль), TFA (5 мл) и CH2Cl2 (5 мл) объединяли и перемешивали при комнатной температурев течение ночи. Реакционную смесь затем нейтрализовали 1н раствором NaOH. Осадок собирали фильтрованием с получением продукта 3-(2-(4-(1-амино-2-метилпропан-2-ил)фенил)ацетил)хинолин-4(1H)-она в виде коричневого твердого вещества (651 мг, 91%). ВЭЖХ время удерживания 2,26 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 336,5 m/z (MH+).
323; метиловый эфир [2-метил-2-[4-[(4-оксо-1H-хинолин-3-ил)карбониламино]фенил]-пропил]аминомуравьиной кислоты
Метилхлорформиат (0,012 г, 0,150 ммоль) добавляли к раствору 3-(2-(4-(1-амино-2-метилпропан-2-ил)фенил)ацетил)хинолин-4(1H)-она (0,025 г, 0,075 ммоль), TEA (0,150 ммоль, 0,021 мл) и DMF (1 мл) и перемешивали при комнатной температуре в течение 1 часа. Затем добавляли пиперидин (0,074 мл, 0,750 ммоль) и реакционную смесь перемешивали еще в течение 30 минут. Реакционную смесь фильтровали и очищали препаративной ВЭЖХ (10-99% CH3CN-H2O) с получением продукта метилового эфира [2-метил-2-[4-[(4-оксо-1H-хинолин-3-ил)карбониламино]фенил]-пропил]аминомуравьиной кислоты (323). 1H ЯМР (400 МГц, DMSO-d6) δ 12,94 (шир.с, 1H), 12,44 (с, 1H), 8,89 (с, 1H), 8,33 (дд, J=8,2, 1,1 Гц, 1H), 7,82 (т, J=8,3 Гц, 1H), 7,76 (д, J=7,7 Гц, 1H), 7,67 (д, J=8,8 Гц, 2H), 7,54 (т, J=8,1 Гц, 1H), 7,35 (д, J=8,7 Гц, 2H), 7,02 (т, J=6,3 Гц, 1H), 3,50 (с, 3H), 3,17 (д, J=6,2 Гц, 2H), 1,23 (с, 6H); ВЭЖХ время удерживания 2,93 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 394,0 m/z (MH+).
В Таблице ниже перечислены другие примеры, синтезированные в соответствии с общей схемой, представленной выше.
Пример 3:
Общая схема:

Специальный пример:
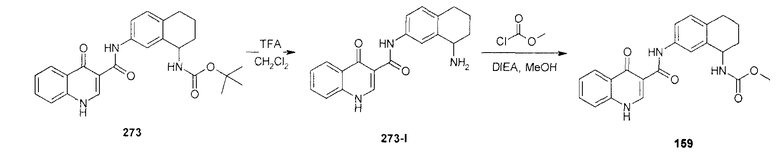
273-I; N-(1-Аминотетралин-7-ил)-4-оксо-1H-хинолин-3-карбоксамид
К раствору трет-бутилового эфира [7-[(4-оксо-1H-хинолин-3-ил)карбониламино]тетралин-1-ил]аминомуравьиной кислоты (273) (250 мг, 0,6 ммоль) в дихлорметане (2 мл) добавляли TFA (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси снова добавляли дихлорметан (10 мл) и раствор промывали насыщенным раствором NaHCO3 (5 мл). Начиналось образование осадка в органическом слое, поэтому объединенные органические слои концентрировали с получением N-(1-аминотетралин-7-ил)-4-оксо-1H-хинолин-3-карбоксамида (273-I) (185 мг, 93%). ВЭЖХ время удерживания 1,94 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 334,5 m/z (MH+).
159; метиловый эфир [7-[(4-оксо-1H-хинолин-3-ил)карбониламино]тетралин-1-ил]аминомуравьиной кислоты
К раствору N-(1-аминотетралин-7-ил)-4-оксо-1H-хинолин-3-карбоксамида (273-I) (65 мг, 0,20 ммоль) и DIEA (52 мкл, 0,29 ммоль) в метаноле (1 мл) добавляли метилхлорформиат (22 мкл, 0,29 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. LCMS анализ реакционной смеси показал пики, соответствующие обоим продуктам единственного и бис присоединения. Добавляли пиперидин (2 мл) и реакционную смесь перемешивали в течение ночи, после чего наблюдали только продукт единственного присоединения. Полученный раствор фильтровали и очищали при помощи ВЭЖХ (10-99% CH3CN-H2O) с получением продукта, метилового эфира [7-[(4-оксо-1H-хинолин-3-ил)карбониламино]тетралин-1-ил]аминомуравьиной кислоты (159) (27 мг, 35%). ВЭЖХ время удерживания 2,68 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 392,3 m/z (MH+).
Другой пример:
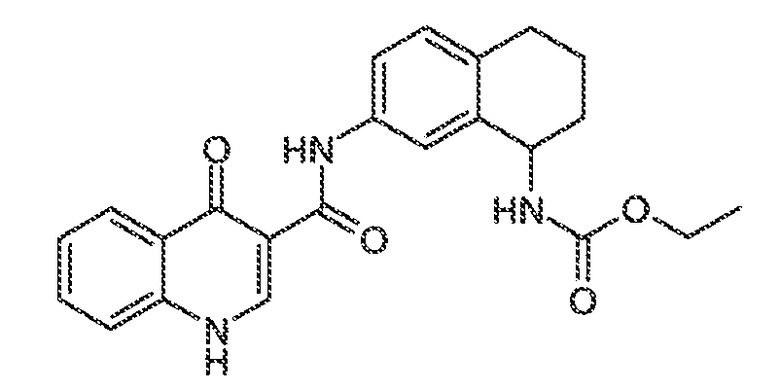
482; этиловый эфир [7-[(4-оксо-1H-хинолин-3-ил)карбониламино]тетралин-1-ил]аминомуравьиной кислоты
Этиловый эфир [7-[(4-оксо-1H-хинолин-3-ил)карбониламино]тетралин-1-ил]аминомуравьиной кислоты (482) синтезировали в соответствии с общей схемой, представленной выше, исходя из амина (273-I) и этилхлорформиата. Общий выход (18%). ВЭЖХ время удерживания 2,84 минут, 10-99% CH3CN, 5 мин цикл; ESI-MS 406,5 m/z (MH+).
Дополнительные примеры
Кислотное промежуточное соединение Пример 1: Синтез 7-фтор-6-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
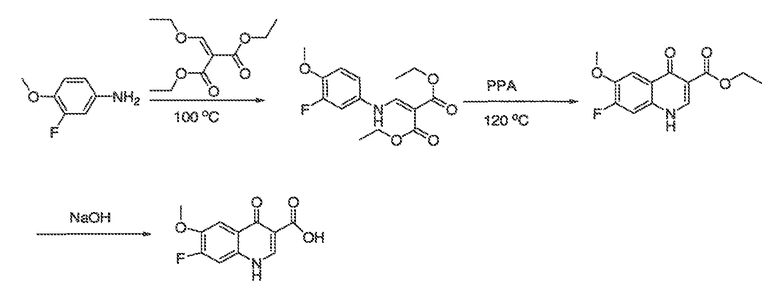
Смесь 3-фтор-4-метокси-анилина (0,7 г, 4,96 ммоль) и диэтил 2-(этоксиметилен)пропандиоата (1,1 г, 4,96 ммоль) нагревали при 100ºC в течение 4 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении и затем очищали колоночной хроматографией на силикагеле с использованием 1 до 60% EtOAc в гексане с получением диэтил 2-((3-фтор-4-метоксифениламино)метилен)малоната (1,2 г, 78%). LC/MS: m/z 312,3 (M+H)+ при 1,69 минут (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Колбу, содержащую диэтил 2-((3-фтор-4-метоксифениламино)метилен)малонат (1,2 г, 3,86 ммоль) и полифосфорную кислоту (4,8 г), нагревали при 120ºC в течение 4 часов. Реакционную смесь затем охлаждали до комнатной температуры и фильтровали. Остаток обрабатывали водным раствором NaHCO3, фильтровали, промывали водой и сушили. Твердое вещество затем очищали колоночной хроматографией на силикагеле с использованием 20 до 70% EtOAc в гексане с получением этил 7-фтор-6-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксилата (410 мг, 40%). LC/MS: m/z 266,3 (M+H)+ к 0,91 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Этил 7-фтор-6-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксилат (409 мг, 1,54 ммоль) суспендировали в растворе NaOH (3,55 мл 4% масс/об, 3,55 ммоль) и реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение 2 часов. После охлаждения реакционную смесь подкисляли при помощи концентрированной HCl. Образовавшийся осадок собирали фильтрованием, промывали водой и сушили с получением 7-фтор-6-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (50 мг, 14%). LC/MS: m/z 238,3 (M+H)+ при 0,95 минут (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
6-фтор-7-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
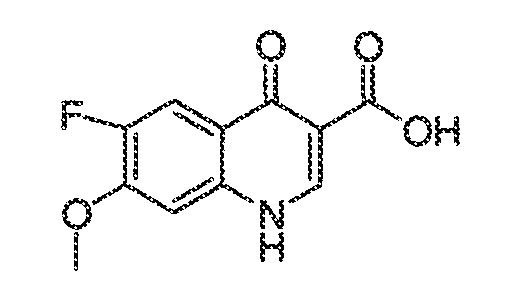
6-фтор-7-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 4-фтор-3-метоксианилина
Кислотное промежуточное соединение Пример 2: Синтез 5-метил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
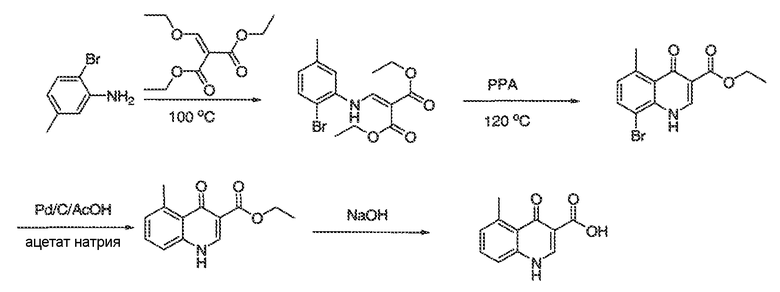
Смесь 2-бром-5-метил-анилина (1,0 г, 5,34 ммоль) и диэтил 2-(этоксиметилен)пропандиоата (1,23 г, 5,90 ммоль) нагревали при 100ºC в течение 4 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток затем очищали колоночной хроматографией на силикагеле с использованием 1 до 60% EtOAc в гексане с получением диэтил 2-[[(2-бром-5-метил-фенил)амино]метилен]пропандиоата (1,6 г, 84%). 1H ЯМР (300 МГц, CDCl3) δ 11,19 (д, J=13,2 Гц, 1H), 8,48 (д, J=13,2 Гц, 1H), 7,43 (д, J=8,1 Гц, 1H), 7,08 (с, 1H), 6,81 (д, J=8,1 Гц, 1H), 4,37-4,23 (м, 4H), 2,35 (с, 3H), 1,42-1,25 (м, 6H).
Колбу, содержащую диэтил 2-[[(2-бром-5-метил-фенил)амино]метилен]пропандиоат (1,6 г, 4,49 ммоль) и полифосфорную кислоту (5,6 г), нагревали при 120ºC в течение 4 часов. Реакционную смесь затем охлаждали до комнатной температуры и фильтровали. Остаток обрабатывали водным раствором NaHCO3, фильтровали, промывали водой и сушили. Остаток затем очищали колоночной хроматографией на силикагеле с использованием 20 до 70% EtOAc в гексане с получением этил 8-бром-5-метил-4-оксо-1H-хинолин-3-карбоксилата (895 мг, 64%). 1H ЯМР (300 МГц, DMSO-d6) δ 11,22 (с, 1H), 8,32 (с, 1H), 7,82 (д, J=7,8 Гц, 1H), 7,04 (д, J=7,8 Гц, 1H), 4,19 (кв., J=7,2, 14,4 Гц, 2H), 2,72 (с, 3H), 1,23 (т, J=10,2 Гц, 3H).
Колбу, содержащую этил 8-бром-5-метил-4-оксо-1H-хинолин-3-карбоксилат (895 мг, 2,89 ммоль), ацетат натрия (237 мг, 2,89 ммоль) и Pd/C (180 мг, 1,69 ммоль), промывали в атмосфере N2 с последующим удалением газа в вакууме. Добавляли уксусную кислоту (21 мл) в инертной атмосфере с последующим удалением газа в вакууме. Реакционную смесь затем перемешивали в течение 4 часов в атмосфере H2. Реакционную смесь фильтровали для удаления Pd катализатора и растворитель выпаривали с получением этил 5-метил-4-оксо-1H-хинолин-3-карбоксилата (145 мг, 22%).
Этил 5-метил-4-оксо-1H-хинолин-3-карбоксилат (142 мг, 0,61 ммоль) суспендировали в водном растворе NaOH (1,54 мл 4% масс/об, 1,54 ммоль) и реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение 2 часов. После охлаждения реакционную смесь подкисляли при помощи концентрированной HCl. Образовавшийся осадок собирали фильтрованием, промывали водой и сушили с получением 5-метил-4-оксо-1H-хинолин-3-карбоновой кислоты (42 мг, 34%). 1H ЯМР (400 МГц, DMSO-d6) δ 15,69 (шир.с, 1H), 13,81 (шир.с, 1H), 8,68 (д, J=6,4 Гц, 1H), 7,70-7,64 (м, 2H), 7,27(д, J=6,4 Гц, 1H), 2,84 (с, 3H), MS (ESI) m/z: 202,2 [M-H]-.
7-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
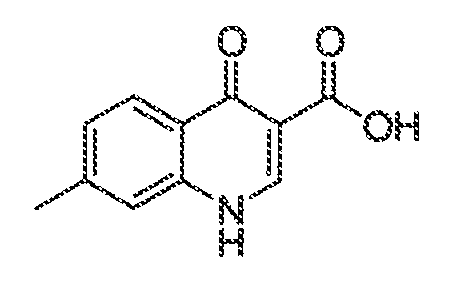
7-метил-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 2-бром-3-метиланилина. Общий выход (16%). 1H ЯМР (400 МГц, DMSO-d6) δ 15,24 (с, 1H), 12,30 (с, 1H), 8,63 (с, 1H), 8,07 (д, J=8,0 Гц, 2H), 7,28 (д, J=8,0 Гц, 1H), 2,85 (с, 3H).
8-бром-5-нитро-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
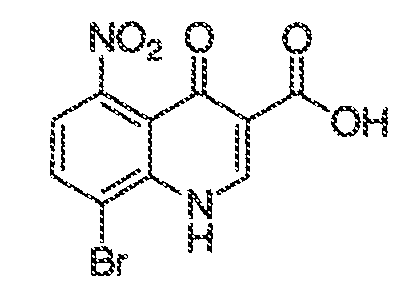
8-бром-5-нитро-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 2-бром-5-нитроанилина. LC/MS: m/z 314,9 (M+H)+ через 1,0 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Кислотное промежуточное соединение Пример 3: Синтез 4-оксо-5-(трифторметил)-1,4-дигидрохинолин-3-карбоновой кислоты
2-хлор-5-(трифторметил)анилин (52 г, 260 ммоль) и диэтил 2-(этоксиметилен)пропандиоат (85 г, 389 ммоль) объединяли в 250-мл колбе и колбу снабжали ловушкой Дина-Старка. Смесь нагревали до 110ºC в течение 4 часов. Реакционную смесь охлаждали до ~80ºC и медленно добавляли гексан (~150 мл). Образовавшийся осадок перемешивали до достижения комнатной температуры, затем фильтровали с получением белого кристаллического твердого вещества. Твердое вещество промывали гексаном и сушили на воздухе (93 г, 94%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,28 (д, J=13,0 Гц, 1H), 8,63 (д, J=13,0 Гц, 1H), 8,10 (с, 1H), 7,80 (д, J=8,3 Гц, 1H), 7,50 (дд, J=8,4, 1,5 Гц, 1H), 4,21 (д.кв., J=28,3, 7,1 Гц, 4H), 1,27 (тд, J=7,1, 2,9 Гц, 6H).
6 мл Dowtherm добавляли в длинногорлую 25 мл колбу, снабженную обратным холодильником. Dowtherm нагревали до 200ºC и дегазировали в течение 20 минут. Затем добавляли диэтил 2-((2-хлор-5-(трифторметил)фениламино)метилен)малонат (1 г, 2,73 ммоль) и раствор затем нагревали до температуры кипения с обратным холодильником в течение 2,5 часов в атмосфере N2. Реакционную смесь охлаждали через 2,5 часа, затем разбавляли гексаном (12 мл) с получением тонкодисперсного светло-коричневого осадка. Реакционную смесь фильтровали и осадок промывали гексаном до исчезновения цвета. Продукт сушили на воздухе с получением этил 8-хлор-4-оксо-5-(трифторметил)-1,4-дигидрохинолин-3-карбоксилата в виде светло-коричневого твердого вещества (0,57 г, 65%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,91 (с, 1H), 8,39 (с, 1H), 8,06 (д, J=8,3 Гц, 1H), 7,81 (д, J=8,4 Гц, 1H), 4,24 (кв., J=7,1 Гц, 2H), 1,29 (т, J=7,1 Гц, 3H).
Этил 8-хлор-4-оксо-5-(трифторметил)-1,4-дигидрохинолин-3-карбоксилат (500 мг, 1,56 ммоль) растворяли в NaOH (16 мл 2,0 M, 31 ммоль) и этаноле (3 мл) и нагревали до 100ºC в течение 2 часов. Прозрачный светло-желтый раствор охлаждали до 50ºC, реакционную смесь дегазировали при помощи N2 и затем обрабатывали 10% раствором Pd/C (65 мг, 0,03 ммоль). Реакционную смесь нагревали при 70ºC в течение 3 часов в атмосфере H2. Реакционную смесь охлаждали и затем фильтровали, подкисляли при помощи концентрированной HCl до образования белого осадка, затем оставляли для перемешивания в течение ночи. Реакционную смесь фильтровали, промывали водой и сушили при помощи CH3CN с получением белого порошка (350 мг, 87%). 1H ЯМР (400 МГц, DMSO-d6) δ 15,26 (с, 1H), 13,68 (с, 1H), 8,98 (с, 1H), 8,16-8,09 (м, 1H), 8,08-7,97 (м, 2H).
Коммерчески доступные кислоты и сложные эфиры
Аминовое промежуточное соединение Пример 1: Синтез 5-амино-2-(трифторметил)фенола
Смесь 1-бром-2-метокси-4-нитро-бензола (20 г, 86,2 ммоль), метил 2,2-дифтор-2-фторсульфонил-ацетата (100 г, 520,5 ммоль) и CuI (65 г, 341,3 ммоль) в безводном DMF (200 мл) перемешивали при 75ºC в атмосфере N2 в течение ночи. Растворитель выпаривали при пониженном давлении. К остатку добавляли EtOAc и твердое вещество удаляли фильтрованием. Фильтрат промывали водой (100 мл ×2), насыщенным солевым раствором (100 мл), сушили над безводным Na2SO4 и очищали колоночной хроматографией на силикагеле (петролейный эфир в качестве элюента) с получением смеси 1-бром-2-метокси-4-нитро-бензола и 2-метокси-4-нитро-1-(трифторметил)бензола (16 г). 1H ЯМР (300 МГц, CDCl3) δ 7,90-7,86 (м, 1H), 7,85 (с, 1H), 7,77-7,69 (м, 1H), 4,02 (с, 3H).
Смесь 2-метокси-4-нитро-1-(трифторметил)бензола и 1-бром-2-метокси-4-нитро-бензола (16 г, 72 ммоль) и пиридингидрохлорида (100 г, 865,3 ммоль) перемешивали при 210ºC в течение 40 минут. Затем реакционную смесь выливали в ледяную воду и экстрагировали при помощи EtOAc (80 мл ×3). Объединенные органические слои промывали водой (100 мл ×2) и насыщенным солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали, концентрировали и очищали колоночной хроматографией на силикагеле (5% EtOAc в петролейном эфире в качестве элюента) с получением смеси 5-нитро-2-(трифторметил)фенол и 2-бром-5-нитро-фенола (10 г). 1H ЯМР (400 МГц, CDCl3) δ 9,16 (с, 1H), 7,00 (д, J=8,4 Гц, 1H), 6,06 (с, 1H), 5,95 (д, J=8,4 Гц, 1H), 4,07 (шир.с, 1H).
К раствору 5-нитро-2-(трифторметил)фенола и 2-бром-5-нитро-фенола (10 г, 48,28 ммоль) в метаноле (60 мл) добавляли никель Ренея (2,83 г, 318 мкл, 48,28 ммоль) в атмосфере азота. Реакционную смесь затем перемешивали в течение 4 часов при комнатной температуре в атмосфере водорода (1 атм). Катализатор удаляли фильтрованием через слой Целита и фильтрат упаривали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ с получением 5-амино-2-(трифторметил)фенола (1,7 г, 20%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,79 (с, 1H), 7,04 (д, J=8,8 Гц, 1H), 6,10 (с, 1H), 6,01 (д, J=8,4 Гц, 1H), 5,58 (шир.с, 2H).
Аминовое промежуточное соединение Пример 2: Синтез 5-амино-2-(трифторметил)фенола
2-Изопропиланилин (13,5 г, 99,85 ммоль) добавляли по порциям к концентрированной H2SO4 (100 мл) для получения желтого гомогенного раствора. Раствор затем охлаждали до 0ºC и добавляли по порциям KNO3 (15,2 г, 150,3 ммоль), поддерживая внутреннюю температуру ниже 5ºC. Реакционную смесь перемешивали в течение 2 часов и затем выливали на ледяную воду, затем подщелачивали 10% раствором NaOH. Водный слой экстрагировали при помощи EtOAc, сушили над MgSO4, фильтровали и концентрировали с получением 2-изопропил-5-нитроанилина (14,9 г, 83%). 1H ЯМР (400 МГц, CDCl3) δ 7,60 (дд, J=8,4, 2,2 Гц, 1H), 7,50 (д, J=2,4 Гц, 1H), 7,27-7,21 (м, 1H), 3,96 (с, 2H), 2,98-2,79 (м, 1H), 1,29 (д, J=6,8 Гц, 6H).
2-Изопропил-5-нитро-анилин (1,89 г, 10,49 ммоль) добавляли по каплям к смеси концентрированной H2SO4 (9 мл) и H2O (50 мл). Эту реакционную смесь охлаждали до 0ºC и добавляли раствор NaNO2 (763 мг, 11,06 ммоль) в H2O (2 мл). Реакционную смесь перемешивали в течение 10 минут и затем добавляли 1 г мочевины добавляли для разложения избытка NaNO2, с последующим добавлением 10 мл 1:2 концентрированной H2SO4:H2O. Реакционную смесь затем кипятили с обратным холодильником в течение 10 минут, охлаждали до комнатной температуры и экстрагировали при помощи EtOAc, промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Остаток затем очищали колоночной хроматографией на силикагеле с использованием 10 до 20% EtOAc в гексане с получением 2-изопропил-5-метил-фенола (1,41 г, 89%). 1H ЯМР (400 МГц, CDCl3) δ 7,79 (дд, J=8,5, 2,3 Гц, 1H), 7,63 (д, J=2,3 Гц, 1H), 7,33 (д, J=8,5 Гц, 1H), 7,26 (с, 1H), 3,39-3,18 (м, 1H), 1,29-1,26 (м, 6H).
К нагреваемому при температуре кипения с обратным холодильником раствору 2-изопропил-5-метил-фенола (1,3 г, 8,65 ммоль) и формиата аммония (1,3 г, 20,62 ммоль) в этаноле (50 мл) добавляли 10% Pd/C (887 мг, 8,33 ммоль). Смесь кипятили с обратным холодильником еще в течение 5 минут, охлаждали и фильтровали через слой Целита. Растворитель удаляли выпариванием с получением 5-амино-2-изопропилфенола, который использовали без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ 8,70 (с, 1H), 6,71 (д, J=8,1 Гц, 1H), 6,04 (д, J=2,2 Гц, 1H), 5,97 (дд, J=8,1, 2,2 Гц, 1H), 4,65 (с, 2H), 3,08-2,94 (м, 1H), 1,07 (д, J=6,9 Гц, 6H).
3-амино-4-трет-бутилфенол
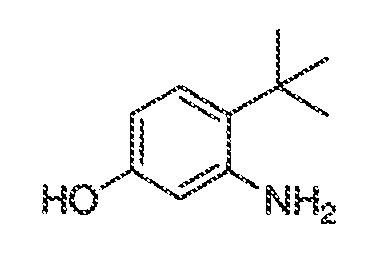
3-амино-4-трет-бутилфенол можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 4-трет-бутиланилина.
Аминовое промежуточное соединение Пример 3: Синтез 5-амино-2-циклогексилфенола
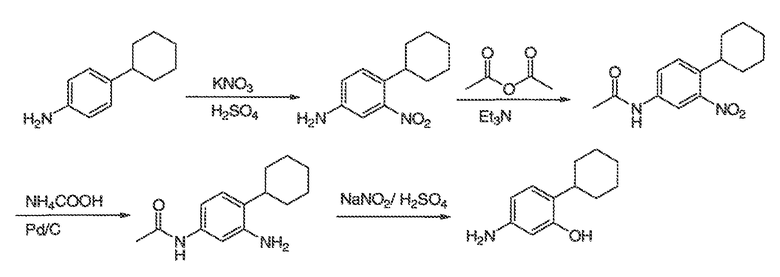
4-циклогексиланилин (15 г, 85,58 ммоль) добавляли по порциям к концентрированной серной кислоте (85 мл, 1,6 моль) для получения гомогенного раствора. Раствор затем охлаждали до 0ºC и добавляли по порциям KNO3 (13 г, 128,6 ммоль), поддерживая внутреннюю температуру ниже 5ºC. Реакционную смесь перемешивали в течение 5 минут при -10ºC и затем выливали на ледяную воду, подщелачивали 6н раствором NaOH и водный слой экстрагировали при помощи EtOAc, сушили над MgSO4, фильтровали и концентрировали с получением 4-циклогексил-3-нитроанилина (17 г, 921%). 1H ЯМР (400 МГц, CDCl3) δ 7,20 (д, J=8,5 Гц, 1H), 6,97 (д, J=2,5 Гц, 1H), 6,82 (дд, J=8,4, 2,5 Гц, 1H), 2,85 (ддд, J=11,4, 8,3, 3,2 Гц, 1H), 1,89-1,69 (м, 6H), 1,49-1,29 (м, 4H).
К раствору 4-циклогексил-3-нитро-анилина (1,54 г, 6,99 ммоль) в DCM (15 мл) добавляли Et3N (1,9 мл, 13,63 ммоль), с последующим добавлением уксусного ангидрида (3,3 мл, 34,98 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, гасили водой, слои разделяли и органический слой промывали 0,1н раствором HCl, с последующей промывкой при помощи H2O. Органический слой сушили над MgSO4, фильтровали и концентрировали с получением N-(4-циклогексил-3-нитрофенил)ацетамида (1,7 г, 93%). 1H ЯМР (400 МГц, CDCl3) δ 7,91 (д, J=2,2 Гц, 1H), 7,69 (дд, J=8,6, 2,2 Гц, 1H), 7,52 (с, 1H), 7,40 (д, J=8,6 Гц, 1H), 3,01-2,87 (м, 1H), 2,22 (с, 3H), 1,95-1,78 (м, 5H), 1,42 (т, J=10,4 Гц, 4H), 1,32-1,21 (м, 1H).
К нагреваемому при температуре кипения с обратным холодильником раствору N-(4-циклогексил-3-нитро-фенил)ацетамида (1,8 г, 6,86 ммоль) и формиата аммония (1,8 г, 28,55 ммоль) в этаноле (50 мл) добавляли 10% Pd/C (1,3 г, 12,22 ммоль). Смесь кипятили с обратным холодильником еще в течение 5 минут, охлаждали и фильтровали через слой Целита. Растворитель удаляли выпариванием с получением N-(3-амино-4-циклогексилфенил)ацетамида (1,4 г, 90%), который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,15 (д, J=2,1 Гц, 2H), 7,03 (д, J=8,3 Гц, 1H), 6,67 (дд, J=8,2, 2,1 Гц, 1H), 3,72 (д, J=9,8 Гц, 2H), 2,42 (д, J=8,5 Гц, 1H), 2,15 (с, 3H), 1,88 (д, J=9,5 Гц, 4H), 1,78 (д, J=12,1 Гц, 1H), 1,51-1,33 (м, 5H).
N-(3-амино-4-циклогексил-фенил)ацетамид (696 мг, 3,0 ммоль) добавляли по каплям к смеси концентрированной H2SO4 (3 мл, 56,28 ммоль) и H2O (17 мл). Эту реакционную смесь охлаждали до 0ºC и добавляли раствор NaNO2 (229 мг, 3,32 ммоль) в H2O (2 мл). Реакционную смесь перемешивали в течение 5 минут при 0ºC и затем добавляли 1 г мочевины с последующим добавлением 10 мл 1:2 H2SO4:H2O. Реакционную смесь затем кипятили с обратным холодильником в течение 1 часа, охлаждали до комнатной температуры и экстрагировали при помощи EtOAc. Водный слой подщелачивали твердым NaOH и экстрагировали при помощи EtOAc. Объединенный органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали с получением 5-амино-2-циклогексилфенола (426 мг, 74%), который использовали без дополнительной очистки.
Аминовое промежуточное соединение Пример 4: Синтез 5-амино-2-трет-бутил-4-этилфенола
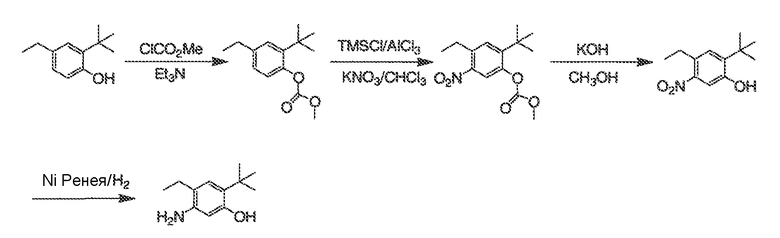
К раствору 2-трет-бутил-4-этил-фенола (10 г, 56,1 ммоль) в DCM (50 мл) добавляли Et3N (17 г, 168,0 ммоль) и метилхлорформиат (11 г, 116,4 ммоль) при 0ºC. Смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду для гашения реакции и смесь экстрагировали при помощи EtOAc (200 млЧ3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали в вакууме с получением 2-трет-бутил-4-этилфенилметилкарбоната (12 г, 91%). 1H ЯМР (300 МГц, CDCl3), 7,06-7,02 (м, 2H), 7,02 (с, 1H), 3,92 (с, 3H), 2,64 (кв., J=7,5 Гц, 2H), 1,37 (с, 9H), 1,54 (т, J=7,5 Гц, 3H).
К раствору KNO3 (3,9 г, 38,57 ммоль) в DCM (50 мл) добавляли TMSCl (5,5 г, 50,62 ммоль) и 2-трет-бутил-4-этилфенилметилкарбонат (6 г, 25,39 ммоль) при 0ºC. После перемешивания в течение 15 минут добавляли AlCl3 (10 г, 0,08 моль) и затем реакционную смесь перемешивали в течение 2 часов. Реакционную смесь выливали на ледяную воду и экстрагировали при помощи EtOAc (50 млЧ3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали в вакууме с получением неочищенного соединения, которое очищали колоночной хроматографией на силикагеле (10-15% EtOAc в петролейном эфире в качестве элюента) с получением 2-трет-бутил-4-этил-5-нитрофенилметилкарбоната (5 г, 70%). 1H ЯМР (400 МГц, CDCl3) 7,69 (с, 1H), 7,25 (с, 1H), 3,82 (с, 3H), 2,54 (кв., J=7,6 Гц, 2H), 1,26 (с, 9H), 1,15 (д, J=7,6 Гц, 3H).
К раствору (2-трет-бутил-4-этил-5-нитро-фенил)метилкарбоната (3,9 г, 13,86 ммоль) в метаноле (100 мл) добавляли KOH (1,8 г, 32,08 ммоль) при комнатной температуре. Смесь перемешивали в течение ночи. Добавляли воду и реакционную смесь экстрагировали при помощи EtOAc (50 млЧ3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали в вакууме с получением 2-трет-бутил-4-этил-5-нитрофенола (2,8 г, 91%), который использовали на следующей стадии без дополнительной очистки.
К раствору 2-трет-бутил-4-этил-5-нитро-фенола (3,2 г, 14,3 ммоль) в метаноле (20 мл) добавляли никель Ренея (200 мг, 3,4 ммоль) в атмосфере азота. Реакционную смесь затем перемешивали в течение ночи при комнатной температуре в атмосфере водорода (1 атм). Катализатор удаляли фильтрованием через слой Целита и фильтрат упаривали в вакууме с получением 5-амино-2-трет-бутил-4-этил-фенола (1,2 г, 43%). 1H ЯМР (400 МГц, CDCl3) δ 6,92 (с, 1H), 6,06 (с, 1H), 2,45 (кв., J=7,6 Гц, 2H), 1,37 (с, 9H), 1,21 (т, J=7,6 Гц, 3H), MS (ESI) m/e (M+H+) 194,2.
5-амино-2-трет-бутил-4-метилфенол
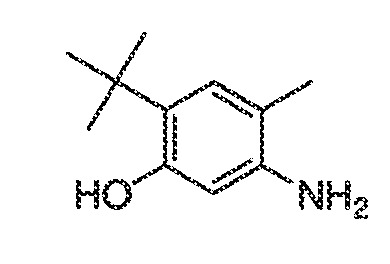
5-амино-2-трет-бутил-4-метилфенол можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 2-трет-бутил-4-метилфенола. 1H ЯМР (400 МГц, CDCl3) δ 6,91 (с, 1H), 6,05 (с, 1H), 4,73 (шир.с, 1H), 3,44 (шир.с, 2H), 2,09 (с, 3H), 1,37 (с, 9H), MS (ESI) m/z (M+H+) 179,3.
Аминовое промежуточное соединение Пример 5: Синтез 4-трет-бутил-3-фторанилин
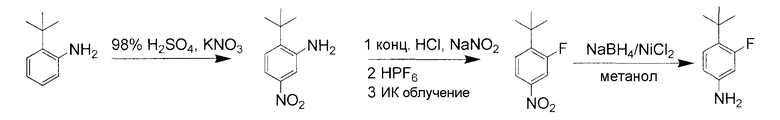
KNO3 (7,5 г, 74,18 ммоль) в концентрированной H2SO4 (50 мл) медленно добавляли к смеси 2-трет-бутиланилина (11 г, 73,71 ммоль) в концентрированной H2SO4 (50 мл) при -10ºC. Смесь перемешивали при -10ºC в течение 1 часа и выливали в ледяную воду. Смесь экстрагировали при помощи EtOAc (150 мл ×3). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и очищали колоночной хроматографией на силикагеле с получением 2-трет-бутил-5-нитро-анилина (9 г, 63%). 1H ЯМР (400 МГц, CDCl3) δ 7,53 (дд, J=2,8, 8,8 Гц, 1H), 7,46 (д, J=2,8 Гц, 1H), 7,34 (д, J=8,8 Гц, 1H), 4,12 (шир.с, 2H), 1,44 (с, 9H).
К перемешиваемому раствору 2-трет-бутил-5-нитро-анилина (5,0 г, 25,74 ммоль) в H2O (20 мл) добавляли концентрированную HCl (10 мл). После растворения смесь охлаждали до 0ºC с последующим медленным добавлением NaNO2 (1,8 г, 819 мкл, 25,74 ммоль) в H2O (10 мл). Реакционную смесь перемешивали при 0ºC еще в течение 0,5 часов. Затем добавляли раствор HPF6 (2×20 мл) партиями. Образовавшийся осадок выделяли фильтрованием, затем нагревали в условиях инфракрасного излучения при приблизительно 130-150ºC. Серое твердое вещество медленно превращалось в темное вязкое масло, которое очищали колоночной хроматографией на силикагеле с получением 1-трет-бутил-2-фтор-4-нитробензола (0,6 г, 12%). 1H ЯМР (400 МГц, CDCl3) δ 7,96 (дд, J=2,4, 8,8 Гц, 1H), 7,87 (дд, J=2,4, 12,0 Гц, 1H), 7,48 (т, J=8,0 Гц, 1H), 1,43 (с, 9H).
NaBH4 (144 мг, 152 мкл, 3,8 ммоль) добавляли к раствору 1-трет-бутил-2-фтор-4-нитро-бензола (750 мг, 3,8 ммоль) и NiCl2.6H2O (2,6 г, 11 ммоль) в метаноле (15 мл) при -15ºC. После добавления смесь перемешивали в течение 2 минут и добавляли воду для гашения реакции. Реакционную смесь экстрагировали этилацетатом (50 мл ×3). Объединенные органические слои сушили над безводным Na2SO4 и упаривали в вакууме с получением 4-трет-бутил-3-фтор-анилина (470 мг, 74%), который использовали без дополнительной очистки. 1H ЯМР (300 МГц, CDCl3) δ 7,08-7,02 (м, 1H), 6,42-6,34 (м, 2H), 1,32 (с, 9H), MS (ESI) m/z: 168,2 [M+H].
Аминовое промежуточное соединение Пример 6: Синтез 4-трет-бутил-3-фторанилина
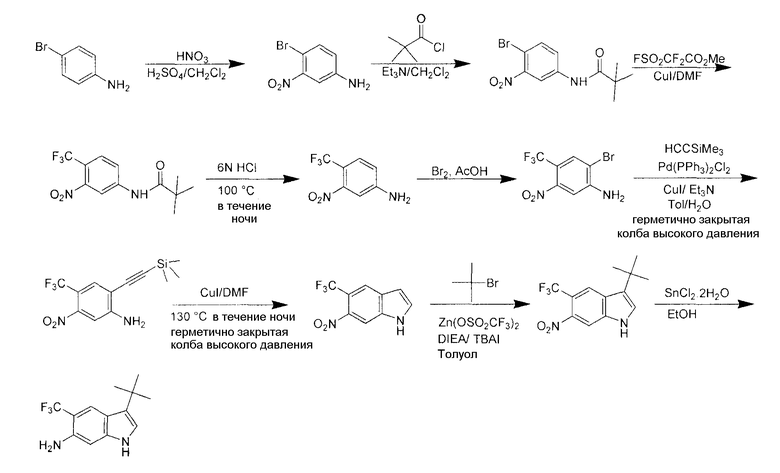
Концентрированную азотную кислоту (15,8 мл, 376,9 ммоль) добавляли к раствору 4-броманилина (40 г, 232,5 ммоль) в H2SO4/CH2Cl2 (400 мл, 1:1, об/об) по каплям при 0ºC в атмосфере N2. Охлаждающую баню удаляли и смесь перемешивали при 20ºC до тех пор, пока исходное вещество не было израсходовано (около 1 часа). Реакционную смесь выливали на воду и нейтрализовали раствором NaOH до pH=9 и экстрагировали при помощи DCM. Органический слой сушили, фильтровали и упаривали в вакууме с получением неочищенного продукта 4-бром-3-нитро-анилина, который использовали непосредственно на следующей стадии.
2,2-диметилпропаноилхлорид (28 г, 232,2 ммоль) добавляли к перемешиваемому раствору 4-бром-3-нитро-анилина (42 г, 193,5 ммоль) и Et3N (51 мл, 366 ммоль) в безводном DCM (200 мл) при 0ºC в атмосфере N2. Охлаждающую баню удаляли и перемешивание продолжали при комнатной температуре в течение 2 часов. Реакционную смесь выливали на лед (500 г) и экстрагировали при помощи CH2Cl2. Объединенный органический слой сушили над Na2SO4 и упаривали в вакууме с получением неочищенного продукта N-(4-бром-3-нитрофенил)пиваламида (55 г, 94%), который использовали непосредственно на следующей стадии. 1H ЯМР (300 МГц, CDCl3) δ: 8,18-8,17 (м, 1H), 7,64-7,63 (м, 2H), 7,50 (шир.с, 1H), 1,32 (с, 9H).
CuI (69,8 г, 366,5 ммоль) и метил 2,2-дифтор-2-фторсульфонил-ацетат (70,4 г, 366,4 ммоль) добавляли к перемешиваемому раствору N-(4-бром-3-нитро-фенил)-2,2-диметил-пропанамида (55,0 г, 182,6 ммоль) в безводном DMF (300 мл) при комнатной температуре. Реакционную смесь перемешивали при 100ºC до тех пор, пока исходное вещество не было израсходовано (около 12 часов). Растворитель выпаривали в вакууме с получением неочищенного продукта N-(3-нитро-4-(трифторметил)фенил)пиваламида (45,0 г, 85%)), который использовали непосредственно на следующей стадии без очистки. 1H ЯМР (400 МГц, CDCl3) δ: 8,21 (д, J=2,0 Гц, 1H), 7,87 (дд, J=2,0, 8,8 Гц, 1H), 7,74 (д, J=8,8 Гц, 1H), 7,64 (шир.с, 1H), 1,34 (с, 9H), 19F ЯМР (282,4 МГц, CDCl3): -60,62 (S).
2,2-диметил-N-[3-нитро-4-(трифторметил)фенил]пропанамид (45,0 г, 155,0 ммоль) в 6н HCl (200 мл) перемешивали при 100ºC в течение ночи. Реакционную смесь охлаждали до комнатной температуры и осторожно нейтрализовали при помощи твердого NaHCO3 до pH=9. Реакционную смесь экстрагировали при помощи CH2Cl2 и сушили над Na2SO4. Растворитель выпаривали в вакууме с получением неочищенного продукта 3-нитро-4-(трифторметил)анилина (31,0 г, 97%), который использовали непосредственно на следующей стадии.
К раствору 3-нитро-4-(трифторметил)анилина (31,0 г, 150,4 ммоль) в HOAc (200 мл) добавляли бром (9,3 мл, 180,5 ммоль) при 0ºC в атмосфере N2. Охлаждающую баню удаляли и смесь перемешивали при 20ºC в течение 1 часа. Растворитель удаляли в вакууме с получением неочищенного продукта 2-бром-5-нитро-4-(трифторметил)анилина (40,0 г, 93%), который использовали непосредственно на следующей стадии.
К раствору 2-бром-5-нитро-4-(трифторметил)анилина (10,0 г, 35,1 ммоль) в толуоле/H2O (100 мл, 1:1, об/об) добавляли CuI (0,4 г, 2,10 ммоль), Et3N (9,5 мл, 68,16 ммоль), Pd(PPh3)2Cl2 (5,0 г, 7,12 ммоль) и этинилтриметилсилан (5,2 г, 53 ммоль) последовательно в атмосфере азота при комнатной температуре. Реакционную смесь переносили в герметично закрываемую колбу высокого давления и нагревали при 70ºC в течение 10 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали, упаривали в вакууме и очищали колоночной хроматографией на силикагеле (1 до 20% EtOAc в петролейном эфире в качестве элюента) с получением 5-нитро-4-(трифторметил)-2-((триметилсилил)этинил)анилина (6,0 г, 57%) в виде желтого твердого вещества. 1H ЯМР (300 МГц, CDCl3): δ 7,67 (с, 1H), 7,13 (с, 1H), 4,86 (шир.с, 2H), 0,29 (с, 9H).
К раствору 5-нитро-4-(трифторметил)-2-((триметилсилил)этинил)анилина (6,0 г, 19,85 ммоль) в DMF (30 мл) добавляли CuI (1,9 г, 9,976 ммоль) в атмосфере азота. Реакционную смесь нагревали при 135ºC в герметично закрытой колбе высокого давления в течение ночи. Реакционную смесь затем фильтровали и фильтрат промывали водой, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (5 до 20% EtOAc в петролейном эфире в качестве элюента) с получением 6-нитро-5-(трифторметил)-1H-индола (1,4 г, 31%). 1H ЯМР (400 МГц, CDCl3): δ 8,83 (шир.с, 1H), 8,15 (с, 1H), 8,11 (с, 1H), 7,58-7,57 (м, 1H), 6,80-6,79 (м, 1H); 19F ЯМР (282,4 МГц, CDCl3) : δ -57,82; MS (ESI): m/z [M-H]-229.
В микроволновой сосуд загружали 6-нитро-5-(трифторметил)-1H-индол (100 мг, 0,43 ммоль), трет-бутилбромид (30 мг, 0,22 ммоль), трифлат цинка (95 мг, 0,26 ммоль), TBAI (80 мг, 0,22 ммоль), DIEA (63 мг, 0,49 ммоль) и толуол (1 мл), герметично закрывали и нагревали в микроволновой печи в течение 10 минут при 120ºC. Реакционную смесь гасили водой, слои разделяли и водный слой экстрагировали при помощи DCM. Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали. Смесь затем очищали колоночной хроматографией на силикагеле с использованием 0 до 20% EtOAc в гексане с получением 3-трет-бутил-6-нитро-5-(трифторметил)-1H-индола. 1H ЯМР (400,0 МГц, CDCl3) δ 8,25 (с, 1H), 8,10 (с, 1H), 7,33 (д, J=2,5 Гц, 1H), 7,28 (с, 1H) и 1,50 (с, 9H) м.д.
В микроволновой сосуд загружали 3-трет-бутил-6-нитро-5-(трифторметил)-1H-индол (95 мг, 0,3319 ммоль), SnCl2.2H2O (375 мг, 1,66 ммоль) и этанол (1 мл), герметично закрывали и нагревали при 62ºC в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли при помощи EtOAc и гасили насыщенным раствором NaHCO3 до достижения pH 7. Реакционную смесь затем фильтровали через слой Целита. Слои разделяли и органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием 0 до 40% EtOAc в гексане с получением 3-трет-бутил-5-(трифторметил)-1H-индол-6-амина. LC/MS: m/z 257,3 (M+H)+ при 1,54 минут (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Общая схема: Получение мета-замещенных анилинов
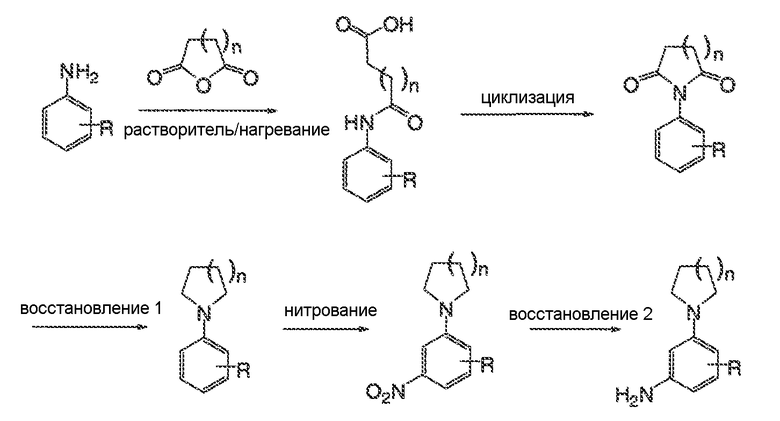
Подходящие растворители включают: бензол, толуол, DMSO; подходящие условия циклизации включают: NaOAc, Ac2O или тионилхлорид, NaOAc; подходящие условия 1 восстановления включают: BH3 или LiALH4 в Et2O или ТГФ; подходящие условия нитрования включают: HNO3, H2SO4 или KNO3, H2SO4; подходящие условия 2 восстановления включают: Pd/C, H2 или Zn, AcOH или Fe, AcOH
3-(пирролидин-1-ил)-5-(трифторметил)анилин
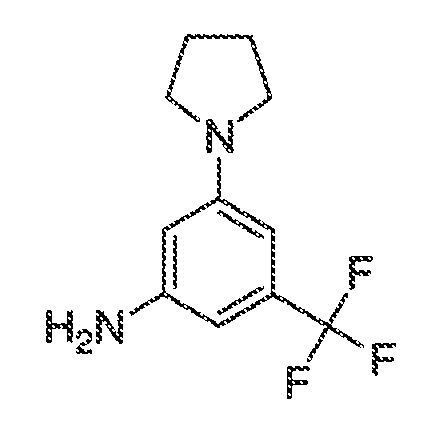
3-(пирролидин-1-ил)-5-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 3-нитро-5-(трифторметил)анилина и тетрагидрофуран-2,5-диона. LC/MS: m/z 230,9 (M+H)+ при 1,22 минут (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA))
3-(пиперидин-1-ил)-5-(трифторметил)анилин

3-(пиперидин-1-ил)-5-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 4-нитро-3-(трифторметил)анилина и дигидро-2H-пиран-2,6(3H)-диона. LC/MS: m/z 245,1 (M+H)+ к 0,77 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
3-(пиперидин-1-ил)-4-(трифторметокси)анилин

3-(пиперидин-1-ил)-4-(трифторметокси)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 4-(трифторметокси)бензол-1,3-диамина и дигидро-2H-пиран-2,6(3H)-диона. LC/MS: m/z 361,3 (M+H)+ при 1,15 минут (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
3-(пирролидин-1-ил)-4-(трифторметокси)анилин
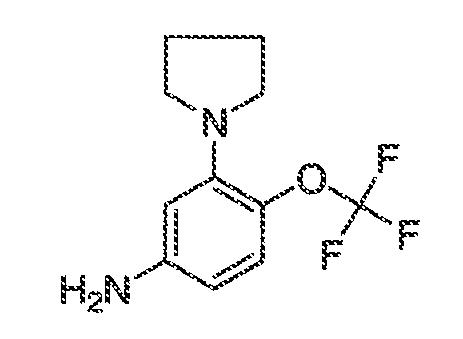
3-(пирролидин-1-ил)-4-(трифторметокси)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 4-(трифторметокси)бензол-1,3-диамина и тетрагидрофуран-2,5-диона. LC/MS: m/z 247,1 (M+H)+ при 1,13 минут (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Аминовое промежуточное соединение Пример 7: Синтез 4-трет-бутил-3-(пирролидин-1-ил)анилина
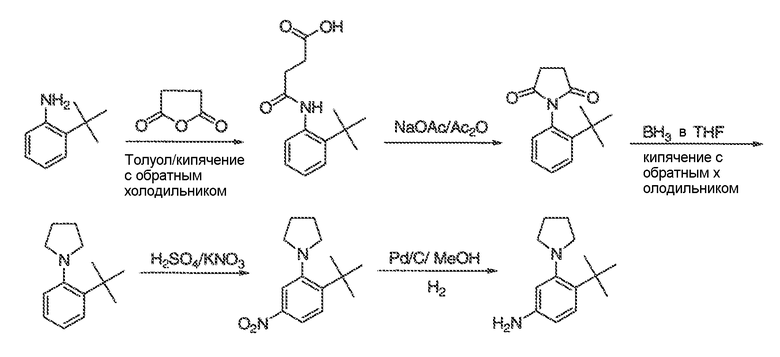
К раствору 2-трет-бутиланилина (1,0 г, 6,7 ммоль) в толуоле (15 мл) добавляли тетрагидрофуран-2,5-дион (0,81 г, 8,1 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали и фильтровали с получением 4-(2-трет-бутилфениламино)-4-оксобутановой кислоты, которую растворяли в уксусной кислоте (20 мл) и ацетате натрия (3,02 г, 36,86 ммоль) и перемешивали при 80ºC в течение ночи. Реакционную смесь гасили водой, слои разделяли и водный слой экстрагировали при помощи DCM. Объединенный органический слой сушили над MgSO4, фильтровали и концентрировали. Полученное твердое вещество перекристаллизовывали из этанола с получением чистого 1-(2-трет-бутилфенил)пирролидин-2,5-диона (930 мг, 60%). LC/MS: m/z 232,3 (M+H)+ при 1,264 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
К раствору 1-(2-трет-бутилфенил)пирролидин-2,5-диона (500 мг, 2,16 ммоль) в ТГФ (10 мл) добавляли BH3 в толуоле (350 мг, 1,72 ммоль) по каплям и реакционную смесь нагревали до температуры кипения с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры и гасили метанолом (до прекращения выделения H2). Растворитель выпаривали с получением 1-(2-трет-бутилфенил)пирролидина (350 мг, 80%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 7,30 (с, 1H), 7,28 (с, 1H), 7,15 (д, J=0,9 Гц, 1H), 7,07-7,01 (м, 1H), 2,90 (с, 4H), 1,88-1,78 (м, 4H), 1,35 (с, 9H). LC/MS: m/z 204,1 (M+H)+ при 0,88 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
1-(2-трет-бутилфенил)пирролидин (350 мг, 1,72 ммоль) добавляли по порциям к концентрированной H2SO4 (1 мл) для получения желтого гомогенного раствора. Раствор затем охлаждали до 0ºC и добавляли по порциям KNO3 (191 мг, 1,9 ммоль), поддерживая внутреннюю температур ниже 5ºC. Реакционную смесь перемешивали в течение 2 часов и затем выливали на ледяную воду и экстрагировали при помощи DCM, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с получением 1-(2-трет-бутил-5-нитрофенил)пирролидина (325 мг, 76%). LC/MS: m/z 248,9 (M+H)+ при 2,39 минут (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Колбу, содержащую 1-(2-трет-бутил-5-нитрофенил)пирролидин (150 мг, 0,60 ммоль) и Pd/C (15 мг, 0,14 ммоль), промывали в атмосфере N2 с последующим удалением газа в вакууме. Добавляли метанол (2 мл) в инертной атмосфере с последующим удалением газа в вакууме. Реакционную смесь перемешивали в течение ночи в атмосфере H2. Реакционную смесь фильтровали и растворитель выпаривали с получением 4-трет-бутил-3-(пирролидин-1-ил)анилина (119 мг, 90%). LC/MS: m/z 218,5 (M+H)+ при 2 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-трет-бутил-3-(пиперидин-1-ил)анилин
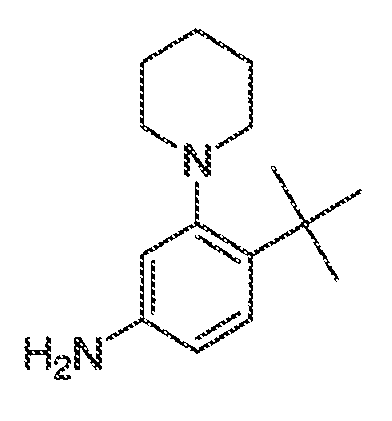
4-трет-бутил-3-(пиперидин-1-ил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 2-трет-бутиланилина и дигидро-2H-пиран-2,6(3H)-диона. LC/MS: m/z 232,9 (M+H)+ при 1,23 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Получение пара-замещенных анилинов
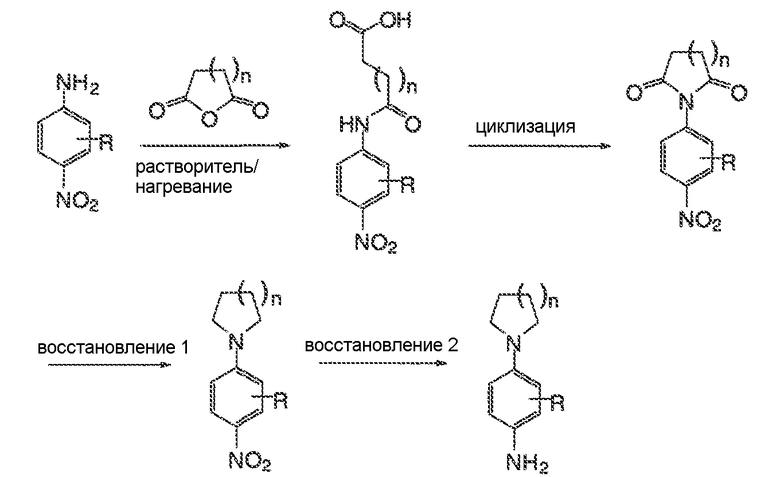
Подходящие растворители включают: бензол, толуол, DMSO; подходящие условия циклизации включают: NaOAc, Ac2O или тионилхлорид, NaOAc; подходящие условия 1 восстановления включают: BH3 или LiALH4 в Et2O или ТГФ; подходящие условия нитрования включают: HNO3, H2SO4 или KNO3, H2SO4; подходящие условия 2 восстановления включают: Pd/C, H2 или Zn, AcOH или Fe, AcOH.
Получение пара-замещенных анилинов через SNAr химию
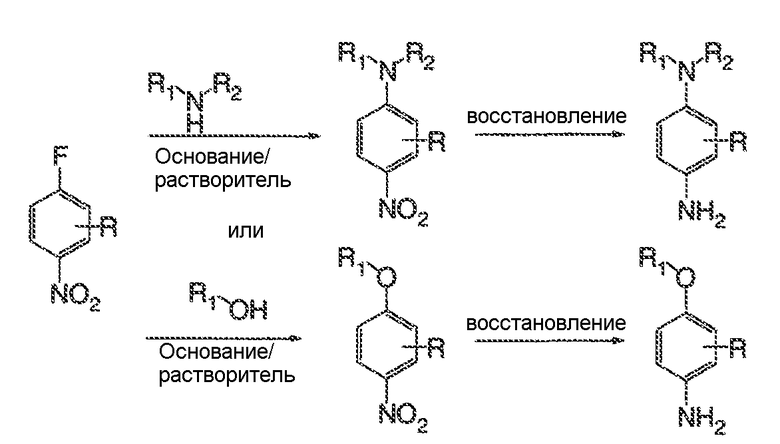
Подходящие основания включают: триэтиламин, калия трет-бутоксид, диизопропилэтиламин, карбонат калия; подходящие растворители включают: DMSO, DMF, CH3CN, ТГФ; подходящие условия восстановления включают: Pd/C, H2; Zn, AcOH, Fe, AcOH.
Аминовое промежуточное соединение Пример 8: Синтез 4-(пирролидин-1-ил)-2-(трифторметил)анилина
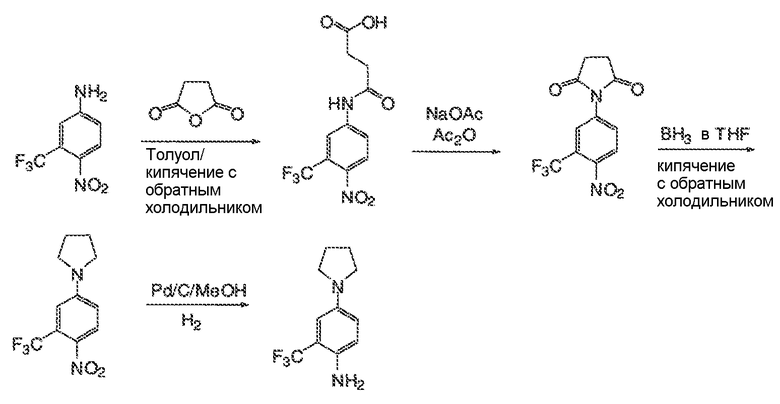
К раствору 4-нитро-3-(трифтор)анилина (2,0 г, 9,7 ммоль) в толуоле (30 мл) добавляли дигидрофуран-2,5-дион (1,16 г, 11,6 ммоль) и реакционную смесь нагревали при температуре кипения с обратным холодильником в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Осадок промывали простым эфиром и сушили с получением 4-(4-нитро-3-(трифторметил)фениламино)-4-оксобутановой кислоты (1,1 г, 37%). LC/MS: m/z 307,3 (M+H)+.
Раствор 4-(4-нитро-3-(трифторметил)фениламино)-4-оксобутановой кислоты (1,1 г, 3,6 ммоль) и NaOAc (1,62 г, 19,75 ммоль) в уксусном ангидриде (15 мл) перемешивали в течение ночи при 80ºC. Реакционную смесь разбавляли водой и экстрагировали два раза дихлорметаном. Органические слои объединяли и промывали при помощи NaOH (1 н) до достижения pH 9. Органический слой отделяли, сушили при помощи MgSO4, фильтровали и концентрировали в вакууме с получением 1-(4-нитро-3-(трифторметил)фенил)пирролидин-2,5-диона (0,4 г, 39%). LC/MS: m/z 288,9 (M+H)+.
К раствору 1-(4-нитро-3-(трифторметил)фенил)пирролидин-2,5-диона (400 мг, 1,38 ммоль) в ТГФ (10 мл) добавляли раствор 1 M BH3 в ТГФ (11,10 мл) по каплям в течение 5 минут. Реакционную смесь кипятили с обратным холодильником в течение 16 часов в инертной атмосфере, охлаждали, затем гасили при помощи MeOH и концентрировали в вакууме. Неочищенный продукт использовали на следующей стадии без очистки. LC/MS: m/z 261,1 (M+H)+.
Раствор 1-(4-нитро-3-(трифторметил)фенил)пирролидин (350 мг, 1,34 ммоль) в метаноле (2 мл) и Pd/C (30 мг, 0,3 ммоль) перемешивали в течение 16 часов в атмосфере H2. Реакционную смесь фильтровали и растворитель выпаривали в вакууме с получением 4-(пирролидин-1-ил)-2-(трифторметил)анилина (0,3 г, 97%). LC/MS: m/z 231,3 (M+H)+. 1H ЯМР (400 МГц, DMSO-d6) δ 6,84 (д, J=8,8 Гц, 1H), 6,72 (дд, J=8,8, 2,5 Гц, 1H), 6,53 (д, J=2,7 Гц, 1H), 4,74 (с, 2H), 3,21-3,11 (м, 4H), 2,00-1,93 (м, 4H).
4-(пиперидин-1-ил)-2-(трифторметил)анилин

4-(пиперидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 4-нитро-3-(трифторметил)анилина и дигидро-2H-пиран-2,6(3H)-диона. 1H ЯМР (400,0 МГц, DMSO-d6) δ 7,08 (дд, J=2,7, 8,8 Гц, 1H), 6,88 (д, J=2,8 Гц, 1H), 6,83 (д, J=8,9 Гц, 1H), 5,09 (с, 2H), 2,96 (т, J=5,4 Гц, 4H), 1,67 (кв., J=5,5 Гц, 4H) и 1,55-1,42 (м, 2H) м.д. LC/MS: m/z 244,9 (M+H)+ при 0,67 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
2-метил-4-(пирролидин-1-ил)анилин
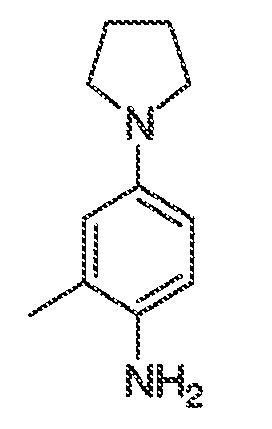
2-метил-4-(пирролидин-1-ил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 3-метил-4-нитроанилина и тетрагидрофуран-2,5-диона. 1H ЯМР (400,0 МГц, DMSO-d6) δ 6,56 (д, J=8,4 Гц, 1H), 6,33 (д, J=2,6 Гц, 1H), 6,27 (дд, J=2,7, 8,4 Гц, 1H), 4,10 (с, 2H), 3,13 (т, J=6,5 Гц, 4H), 2,09-2,04 (м, 3H) и 1,99-1,90 (м, 4H) м.д. LC/MS: m/z 177,3 (M+H)+ при 0,35 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Аминовое промежуточное соединение Пример 8: Синтез 1,4,4-триметил-1,2,3,4-тетрагидрохинолин-7-амина
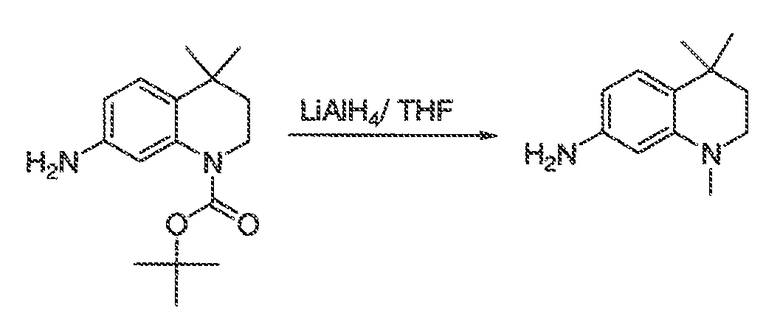
К раствору трет-бутил 7-амино-4,4-диметил-2,3-дигидрохинолин-1-карбоксилата (100 мг, 0,36 ммоль) в ТГФ (2,2 мл) в инертной атмосфере добавляли LiAlH4 (1,8 мл раствора 1 M, 1,8 ммоль) по каплям. Реакционную смесь нагревали до температуры кипения с обратным холодильником и перемешивали в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и гасили раствором 0,4 M NaOH (0,6 мл), водный слой экстрагировали при помощи DCM, сушили над Na2SO4, фильтровали и концентрировали с получением оранжевого масла. Остаток очищали обращенно-фазовой ВЭЖХ. LC/MS: m/z 191,5 (M+H)+ при 0,92 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Аминовое промежуточное соединение Пример 9: Синтез 5-(бензилокси)-4-циклогексил-2-(трифторметил)анилина
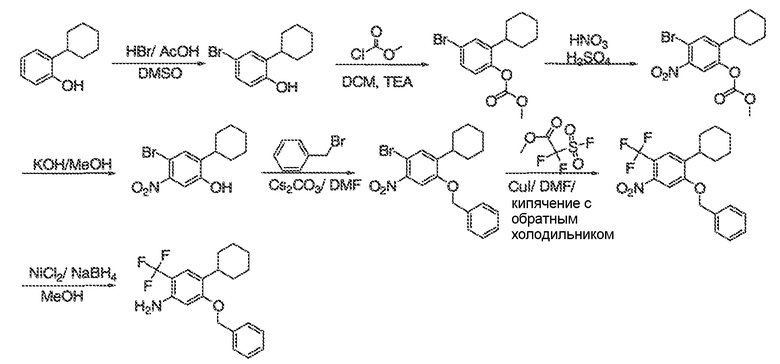
К перемешиваемому раствору 2-циклогексилфенола (26,0 г, 146,8 ммоль) в ледяной уксусной кислоте (100 мл) добавляли HBr в уксусной кислоте (150 мл 33% масс/масс) и H2O (52 мл), с последующим добавлением по каплям DMSO (100 мл) в течение 10 минут. Реакционную смесь затем осторожно гасили насыщенным раствором NaHCO3 и концентрировали в вакууме. Остаток обрабатывали простым эфиром(400 мл), промывали водой (2×100 мл) и насыщенным солевым раствором (1×100 мл) и сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла, которое очищали колоночной хроматографией на силикагеле с использованием градиента 15-30% EtOAc/гексан с получением 4-бром-2-циклогексил-фенола (35,0 г, 93%)
4-Бром-2-циклогексил-фенол (35,0 г, 137,2 ммоль) растворяли в DCM (200 мл) и Et3N (38 мл, 272,6 ммоль), охлаждали до 0ºC, затем обрабатывали метилхлорформиатом (15,0 г, 153,4 ммоль) и давали нагреться до комнатной температуры в течение 16 часов. Реакционную смесь гасили 30 мл насыщенного раствора NaHCO3, промывали 50% насыщенным раствором NaHCO3 (1×100 мл) и насыщенным солевым раствором (1×100 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла, которое очищали колоночной хроматографией на силикагеле, с использованием градиента 20% EtOAc/гексан с получением (4-бром-2-циклогексил-фенил)метилкарбоната (38,0 г, 88%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,41 (д, J=2,3 Гц, 1H), 7,32-7,29 (м, 1H), 6,98 (д, J=8,6 Гц, 1H), 3,92 (с, 3H), 2,71-2,64 (м, 1H), 1,85-1,74 (м, 5H), 1,39-1,20 (м, 5H).
(4-Бром-2-циклогексил-фенил) метилкарбонат (5,0 г, 15,96 ммоль) добавляли по порциям к концентрированной H2SO4 (15 мл) для получения бесцветного гомогенного раствора. Этот раствор затем охлаждали до 0ºC и добавляли по порциям KNO3 (1,77 г, 17,51 ммоль), поддерживая внутреннюю температуру ниже 5ºC. Реакционную смесь перемешивали в течение 2 часов и затем выливали на ледяную воду и водный слой экстрагировали при помощи DCM (3×10 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток затем очищали колоночной хроматографией на силикагеле с использованием градиента 10% EtOAc/гексан с получением 4-бром-2-циклогексил-5-нитро-фенилметилкарбоната (3,25 г, 58%). 1H ЯМР (400 МГц, CDCl3) δ 7,80 (с, 1H), 7,69 (с, 1H), 3,98 (с, 3H), 2,81-2,75 (м, 1H), 1,90-1,79 (м, 5H), 1,44-1,26 (м, 5H).
К раствору (4-бром-2-циклогексил-5-нитро-фенил)-метилкарбоната (1,5 г, 4,2 ммоль) в метаноле (15 мл) добавляли KOH (353 мг, 6,3 ммоль) по порциям при 0ºC. После добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь подкисляли 1н раствором HCl. Растворитель выпаривали и добавляли воду. Водный слой экстрагировали при помощи DCM (3×10 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали с получением 4-бром-2-циклогексил-5-нитрофенола (1,2 г, 95%). 1H ЯМР (400 МГц, CDCl3) δ 7,50 (с, 1H), 7,39 (с, 1H), 5,10 (с, 1H), 2,90-2,83 (м, 1H), 1,91-1,89 (м, 4H), 1,83-1,80 (м, 1H), 1,51-1,24 (м, 5H).
К раствору 4-бром-2-циклогексил-5-нитро-фенола (1,19 г, 4,0 ммоль) и Cs2CO3 (1,54 г, 4,72 ммоль) в DMF (10 мл) добавляли бензилбромид (1,02 г, 707 мкл, 6,0 ммоль) по каплям. Реакционную смесь перемешивали при комнатной температуре в инертной атмосфере в течение 2 часов. Реакционную смесь гасили водой и водный слой экстрагировали при помощи EtOAc (3×10 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием градиента 5% EtOAc/гексан с получением 1-(бензилокси)-4-бром-2-циклогексил-5-нитробензола (1,35 г, 87%). 1H ЯМР (400 МГц, CDCl3) δ 7,42 (с, 1H), 7,39 (с, 1H), 7,36-7,25 (м, 5H), 5,06 (с, 2H), 2,99-2,93 (м, 1H), 1,79 (д, J=11,5 Гц, 4H), 1,70 (д, J=13,4 Гц, 1H), 1,39-1,14 (м, 5H).
К раствору 1-бензилокси-4-бром-2-циклогексил-5-нитро-бензола (500 мг, 1,28 ммоль) и CuI (487,0 мг, 2,56 ммоль) в DMF (5 мл) при комнатной температуре добавляли метил 2,2-дифтор-2-фторсульфонил-ацетат (492 мг, 326,0 мл, 2,56 ммоль) по каплям в инертной атмосфере. Реакционную смесь затем нагревали при 105ºC в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, гасили при помощи NaHCO3 и фильтровали через слой Целита (для удаления Cu солей). Водный слой экстрагировали этилацетатом, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием градиента 10% EtOAc/гексан с получением 1-(бензилокси)-2-циклогексил-5-нитро-4-(трифторметил)бензола (435 мг, 90%). 1H ЯМР (400 МГц, CDCl3) δ 7,77 (с, 1H), 7,62 (с, 1H), 7,42-7,28 (м, 5H), 5,27 (с, 2H), 2,98-2,92 (м, 1H), 1,75-1,62 (м, 5H), 1,47-1,13 (м, 5H).
К раствору 1-(бензилокси)-2-циклогексил-5-нитро-4-(трифторметил)бензола (250 мг, 0,66 ммоль) и NiCl2 (170 мг, 1,31 ммоль) в метаноле (2,5 мл) добавляли NaBH4 (50 мг, 1,32 ммоль) по порциям при 0ºC. Реакционная смесь становилась черной через 5 минут. Реакционную смесь гасили при помощи NaHCO3 и разбавляли при помощи EtOAc. Реакционную смесь фильтровали через слой Целита, слои разделяли и водный слой экстрагировали при помощи EtOAc. Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток затем очищали колоночной хроматографией на силикагеле с использованием градиента 10% EtOAc/гексан с получением 5-бензилокси-4-циклогексил-2-(трифторметил)анилина (150 мг, 65%). LC/MS: m/z 350,3 (M+H)+ при 2,40 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
5-(бензилокси)-4-изопропил-2-(трифторметил)анилин
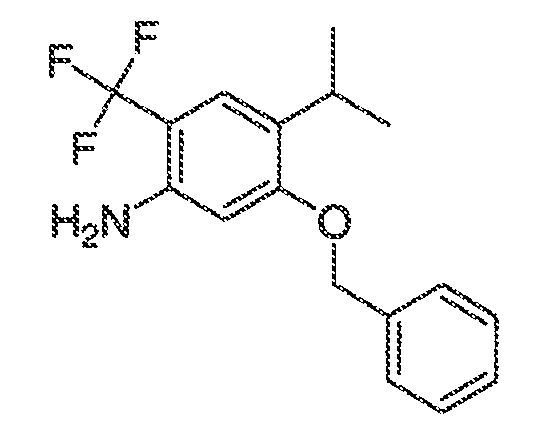
5-(бензилокси)-4-изопропил-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 2-изопропилфенола. LC/MS: m/z 310,3 (M+H)+ при 2,21 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-бром-2-циклопентил-5-нитрофенилметилкарбонат
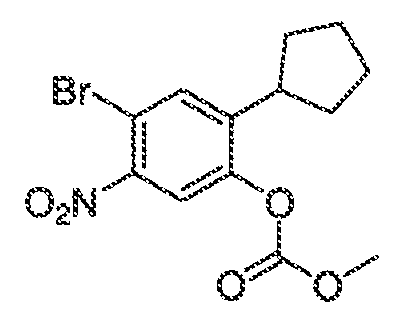
4-бром-2-циклопентил-5-нитрофенилметилкарбонат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 2-циклопентилфенола). 1H ЯМР (400,0 МГц, DMSO-d6) δ 8,12 (с, 1H), 7,88 (с, 1H), 3,88 (д, J=5,7 Гц, 3H), 3,13 (дд, J=9,4, 17,2 Гц, 1H), 1,96-1,92 (м, 2H), 1,80-1,75 (м, 2H), 1,68-1,54 (м, 4H).
Аминовое промежуточное соединение Пример 10: Синтез 5-(бензилокси)-2-фтор-4-(трифторметил)анилина
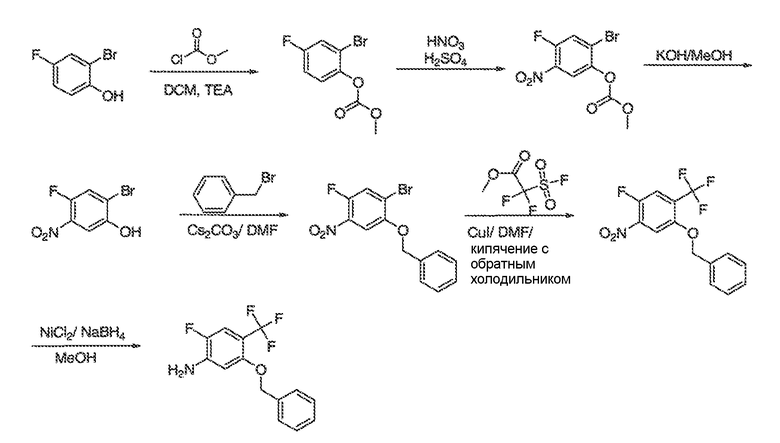
Раствор 2-бром-4-фтор-фенола (7,0 г, 36,65 ммоль) и DMAP (224 мг, 1,83 ммоль) в DCM (15 мл) и Et3N (7,42 г, 10 мл, 73,30 ммоль) охлаждали до 0ºC, затем обрабатывали метилхлорформиатом (14,5 г, 153,4 ммоль) по каплям и давали нагреться до комнатной температуры в течение 16 часов. Реакционную смесь гасили при помощи 30 мл насыщенного раствора NaHCO3, промывали 50% насыщенным раствором NaHCO3 (1×100 мл) и насыщенным солевым раствором (1×100 мл). Органический слой затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного масла, которое очищали колоночной хроматографией на силикагеле, с использованием градиента 15% EtOAc/гексан с получением (2-бром-4-фтор-фенил)метилкарбоната (8,25 г, 90%). 1H ЯМР (400 МГц, CDCl3) δ 7,38 (дд, J=7,7, 2,9 Гц, 1H), 7,23 (дд, J=9,0, 5,0 Гц, 1H), 7,08 (ддд, J=9,0, 7,6, 2,9 Гц, 1H), 3,97 (с, 3H). 19F ЯМР (376 МГц, CDCl3) δ -113,78 (тд, J=7,9, 5,2 Гц, 1H).
(2-Бром-4-фтор-фенил)метилкарбонат (8,25 г, 33,13 ммоль) добавляли по порциям к концентрированной H2SO4 (45 мл) для получения бесцветного гомогенного раствора. Этот раствор затем охлаждали до 0ºC и добавляли по порциям KNO3 (3,7 г, 36,44 ммоль), поддерживая внутреннюю температуру ниже 5ºC. Реакционную смесь перемешивали в течение 2 часов и затем выливали на ледяную воду и водный слой экстрагировали при помощи DCM (3×10 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток затем очищали колоночной хроматографией на силикагеле с использованием градиента 15% EtOAc/гексан с получением 2-бром-4-фтор-5-нитрофенилметилкарбоната (8,93 г, 92%). 1H ЯМР (400 МГц, CDCl3) δ 8,05 (д, J=6,7 Гц, 1H), 7,65 (д, J=9,6 Гц, 1H), 4,01 (с, 3H).
К раствору 2-бром-4-фтор-5-нитрофенилметилкарбоната (8,90 г, 30,27 ммоль) в метаноле (100 мл) добавляли KOH (4,25 г, 75,68 ммоль) по порциям при 0ºC. После завершения добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 1,5 часов. Реакционную смесь подкисляли 1н раствором HCl. Растворитель выпаривали и добавляли воду. Водный слой экстрагировали при помощи DCM (3×10 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали с получением 2-бром-4-фтор-5-нитро-фенола (7,05 г, 99%). 1H ЯМР (400 МГц, CDCl3) δ 7,75 (д, J=6,6 Гц, 1H), 7,51 (д, J=9,6 Гц, 1H), 5,62 (с, 1H).
К раствору 2-бром-4-фтор-5-нитро-фенола (3,5 г, 14,83 ммоль) и Cs2CO3 (5,80 г, 17,80 ммоль) в DMF (26 мл) добавляли бензилбромид (2,80 г, 1,94 мл, 16,31 ммоль) по каплям. Реакционную смесь перемешивали при комнатной температуре в инертной атмосфере в течение 2 часов. Реакционную смесь гасили водой и водный слой экстрагировали при помощи EtOAc (3×10 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием градиента 40% DCM/гексан с получением 1-(бензилокси)-2-бром-4-фтор-5-нитробензола. 1H ЯМР (400 МГц, CDCl3) δ 7,65 (д, J=6,4 Гц, 1H), 7,59 (д, J=9,9 Гц, 1H), 7,52-7,34 (м, 5H), 5,23 (с, 2H).
К раствору 1-бензилокси-2-бром-4-фтор-5-нитро-бензола (500 мг, 1,53 ммоль) и CuI (584 мг, 3,07 ммоль) в DMF (5 мл) при комнатной температуре добавляли метил 2,2-дифтор-2-фторсульфонил-ацетат (589,0 мг, 3,07 ммоль) по каплям в инертной атмосфере. Реакционную смесь затем нагревали при 105ºC в течение 3 часов, затем охлаждали до комнатной температуры, гасили при помощи NaHCO3 и фильтровали через слой Целита (для удаления Cu солей). Водный слой экстрагировали этилацетатом, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием градиента 10% EtOAc/гексан с получением 1-(бензилокси)-4-фтор-5-нитро-2-(трифторметил)бензола (400 мг, 83%). 1H ЯМР (400 МГц, CDCl3) δ 7,71 (д, J=5,7 Гц, 1H), 7,62-7,57 (м, 1H), 7,48-7,37 (м, 5H), 5,27 (с, 2H). 19F ЯМР (376 МГц, CDCl3) δ -63,28 (с, 3H), -126,02 (дд, J=10,2, 5,7 Гц, 1H).
К раствору 1-(бензилокси)-4-фтор-5-нитро-2-(трифторметил)бензола (382 мг, 1,21 ммоль) и NiCl2 (314 мг, 2,42 ммоль) в метаноле (40 мл) добавляли NaBH4 (50 мг, 1,32 ммоль) по порциям при 0ºC. Реакционную смесь становилась черной через 20 минут. Реакционную смесь гасили при помощи NaHCO3 и разбавляли при помощи EtOAc. Реакционную смесь затем фильтровали через слой Целита, слои разделяли и водный слой экстрагировали при помощи EtOAc. Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток затем очищали колоночной хроматографией на силикагеле с использованием градиента 15% EtOAc/гексан с получением 5-(бензилокси)-2-фтор-4-(трифторметил)анилина (150 мг, 43%). LC/MS: m/z 286,1 (M+H)+ при 1,89 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Аминовое промежуточное соединение Пример 11: Синтез (1S,4R)-2-азабицикло[2,2,1]гептана
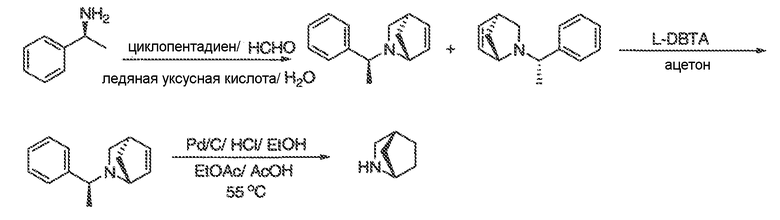
Охлажденный раствор (1S)-1-фенилэтанамина (19,0 г, 156,8 ммоль) в H2O (60 мл) при 0ºC обрабатывали раствором ледяной уксусной кислоты (9 мл) в воде (20 мл), с последующим добавлением свежеперегнанного циклопентадиена (20,73 г, 26,01 мл, 313,6 ммоль) и формальдегида (7,06 г, 6,5 мл, 235,2 ммоль). Полученную реакционную смесь перемешивали в течение 48 часов при 5ºC. Реакционную смесь затем выливали на ледяную воду (100 мл) и 50% этилацетата в гексане (100 мл) и затем подщелачивали таблетками NaOH (pH ≈10) при 0ºC. Слои разделяли и водный слой экстрагировали при помощи 50% этилацетата/гексан (2×100 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением смеси (1R,4R)-2-((S)-1-фенилэтил)-2-азабицикло[2,2,1]гепт-5-ена и (1S,4S)-2-((S)-1-фенилэтил)-2-азабицикло[2,2,1]гепт-5-ена в виде масла (33,0 г). Продукт имеет тенденцию вступать в реакцию ретро-Дильса-Альдера, поэтому его непосредственно использовали на следующей стадии. LC/MS: m/z 199,8 (M+H)+ при 0,38 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
К раствору (2R,3R)-2,3-дибензоилоксибутандиовой кислоты (55,75 г, 155,6 ммоль) в ацетоне (500 мл) добавляли раствор (1R,4R)-5-[(1S)-1-фенилэтил]-5-азабицикло[2,2,1]гепт-2-ена и (1S,4S)-5-[(1S)-1-фенилэтил]-5-азабицикло[2,2,1]гепт-2-ена (31,01 г, 155,6 ммоль) в ацетоне (200 мл) по каплям. Реакционную смесь перемешивали в течение 15 часов при комнатной температуре. Образовавшийся осадок собирали фильтрованием и промывали ацетоном (2×75 мл) и сушили при пониженном давлении. Остаток медленно добавляли к охлажденному (0ºC) 10% раствору NaOH (350 мл) и этилацетата (300 мл) и перемешивали в течение 15 мин (pH 10). Слои разделяли и водный слой экстрагировали этилацетатом (3×150 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением (1R,4R)-2-((S)-1-фенилэтил)-2-азабицикло[2,2,1]гепт-5-ена, который использовали непосредственно использовали на следующей стадии. LC/MS: m/z 199,8 (M+H)+ при 0,38 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Колбу, содержащую (1R,4R)-5-[(1S)-1-фенилэтил]-5-азабицикло[2,2,1]гепт-2-ен (19 г, 95,34 ммоль) и палладий на углероде (4 г, 133,0 ммоль), продували при помощи N2 с последующим удалением газа в вакууме. EtOAc (50 мл) и EtOH (200 мл) добавляли в инертной атмосфере с последующим удалением газа в вакууме. Реакционную смесь затем перемешивали в течение ночи в атмосфере H2. Добавляли AcOH (60 мл) и затем реакционную смесь перемешивали в течение ночи при 55ºC в атмосфере H2. Реакционную смесь охлаждали до комнатной температуры и фильтровали через слой Целита для удаления палладиевого катализатора, промывали этилацетатом, концентрировали в вакууме и остаток обрабатывали 1н раствором HCl в эфире. Растворители выпаривали при пониженном давлении с получением (1S,4R)-2-азабицикло[2,2,1]гептана.
Аминовое промежуточное соединение Пример 12: Синтез (S)-4-(2-метилпирролидин-1-ил)-2-(трифторметил)анилина
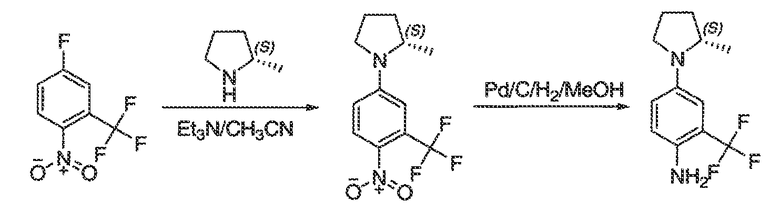
К раствору 5-фтор-2-нитробензотрифторида (2,0 г, 9,56 ммоль) в CH3CN (20 мл) добавляли Et3N (2,41 г, 23,90 ммоль) с последующим добавлением (S)-2-метилпирролидинтозилата (3,18 г, 12,43 ммоль). Реакционную смесь перемешивали при 80ºC в течение ночи. Реакционную смесь затем гасили водой и водный слой экстрагировали при помощи DCM. Объединенный органические слои промывали 1н раствором HCl, сушили над MgSO4, фильтровали и концентрировали с получением (S)-2-метил-1-(4-нитро-3-(трифторметил)фенил)пирролидина (2,45 г, 94% выход). 1H ЯМР (400 МГц, DMSO-d6) δ 8,11 (д, J=9,1 Гц, 1H), 6,88 (с, 1H), 6,85 (д, J=2,6 Гц, 1H), 4,17-4,13 (м, 1H), 3,59-3,54 (м, 1H), 3,33-3,28 (м, 1H), 2,14-2,00 (м, 3H), 1,80-1,71 (м, 1H), 1,14 (д, J=6,3 Гц, 3H). LC/MS: m/z 275,3 (M+H)+ при 1,98 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Колбу, содержащую (S)-2-метил-1-(4-нитро-3-(трифторметил)фенил)пирролидин (2,45 г, 8,9 ммоль) и палладий на углероде (245 мг, 10% масс.), продували при помощи N2 с последующим удалением газа в вакууме. Добавляли метанол (15 мл) в инертной атмосфере с последующим удалением газа в вакууме. Реакционную смесь перемешивали в течение ночи в атмосфере H2. Палладиевый катализатор удаляли фильтрованием и растворитель удаляли при пониженном давлении с получением (S)-4-(2-метилпирролидин-1-ил)-2-(трифторметил)анилина с количественным выходом. 1H ЯМР (400 МГц, DMSO-d6) δ 6,78 (д, J=8,8 Гц, 1H), 6,70-6,67 (м, 1H), 6,47 (д, J=2,7 Гц, 1H), 4,68 (с, 2H), 3,76-3,69 (м, 1H), 3,32-3,27 (м, 1H), 3,02-2,96 (м, 1H), 2,05-1,94 (м, 2H), 1,93-1,84 (м, 1H), 1,64-1,58 (м, 1H), 1,05 (д, J=6,1 Гц, 3H). LC/MS: m/z 245,1 (M+H)+ при 0,48 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Следующие соединения можно получить в соответствии с общей схемой, представленной выше.
(R)-4-(2-Метилпирролидин-1-ил)-2-(трифторметил)анилин
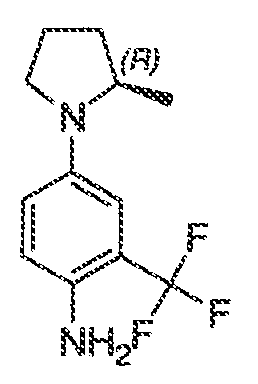
(R)-4-(2-Метилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (R)-2-метилпирролидина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 6,79 (д, J=8,8 Гц, 1H), 6,68 (дд, J=2,3, 8,7 Гц, 1H), 6,47 (д, J=2,5 Гц, 1H), 4,69 (с, 2H), 3,73 (т, J=5,6 Гц, 1H), 3,33-3,27 (м, 1H), 2,99 (кв., J=8,0 Гц, 1H), 2,03-1,87 (м, 3H), 1,64-1,60 (м, 1H) и 1,05 (д, J=6,1 Гц, 3H). LC/MS: m/z 245,3 (M+H)+ при 0,57 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-Морфолино-2-(трифторметил)анилин
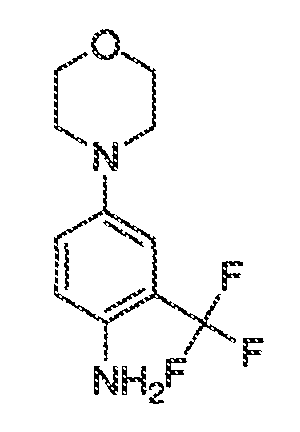
4-Морфолино-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и морфолина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 7,04 (дд, J=2,6, 8,9 Гц, 1H), 6,84-6,79 (м, 2H), 5,07 (с, 2H), 3,71 (т, J=4,7 Гц, 4H) и 2,93 (т, J=4,7 Гц, 4H). LC/MS: m/z 247,1 (M+H)+ при 0,43 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-(3,3-Диметилпирролидин-1-ил)-2-(трифторметил)анилин
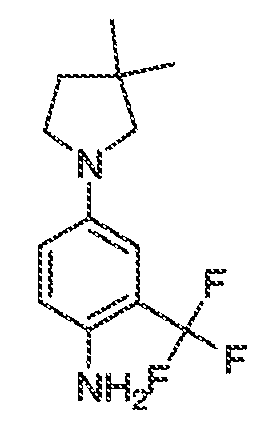
4-(3,3-Диметилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и 3,3-диметилпирролидина. LC/MS: m/z 259,1 (M+H)+ при 1,18 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-(3-Метилпирролидин-1-ил)-2-(трифторметил)анилин
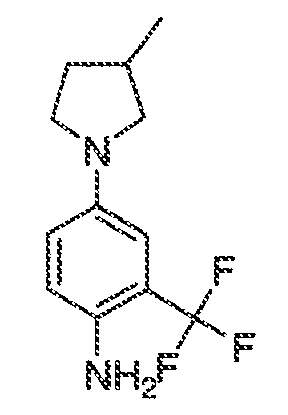
4-(3-Метилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и 3-метилпирролидина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 6,78 (д, J=8,8 Гц, 1H), 6,64 (дд, J=2,5, 8,8 Гц, 1H), 6,44 (д, J=2,7 Гц, 1H), 4,67 (с, 2H), 3,31 (т, J=7,6 Гц, 1H), 3,20-3,15 (м, 2H), 2,73-2,69 (м, 1H), 2,33 (дд, J=7,0, 15,1 Гц, 1H), 2,10-2,04 (м, 1H), 1,54 (тд, J=8,2, 4,0 Гц, 1H) и 1,06 (д, J=6,7 Гц, 3H) м.д. LC/MS: m/z 245,1 (M+H)+ при 0,94 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-(Азетидин-1-ил)-2-(трифторметил)анилин
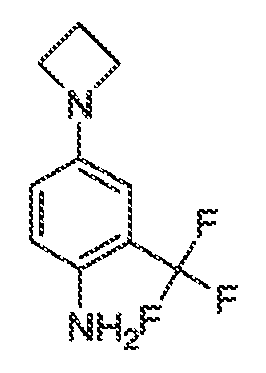
4-(Азетидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и азетидина. LC/MS: m/z 217,3 (M+H)+ при 0,34 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(R)-1-(4-Амино-3-(трифторметил)фенил)-N,N-диметилпирролидин-3-амин
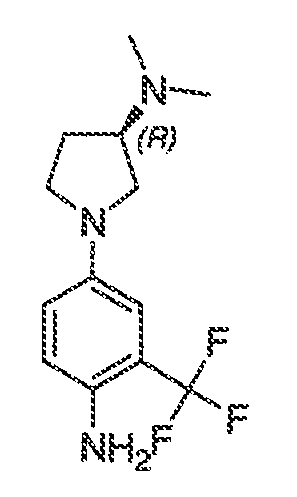
(R)-1-(4-Амино-3-(трифторметил)фенил)-N,N-диметилпирролидин-3-амин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (R)-N,N-диметилпирролидин-3-амина. LC/MS: m/z 274,5 (M+H)+ при 1,32 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(R)-трет-бутил 1-(4-амино-3-(трифторметил)фенил)пирролидин-2-карбоксилат
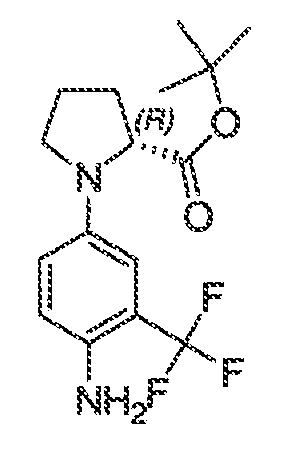
(R)-трет-бутил 1-(4-амино-3-(трифторметил)фенил)пирролидин-2-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (R)-трет-бутил пирролидин-2-карбоксилата. LC/MS: m/z 331,5 (M+H)+ при 1,70 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-трет-бутил 1-(4-амино-3-(трифторметил)фенил)пирролидин-2-карбоксилат
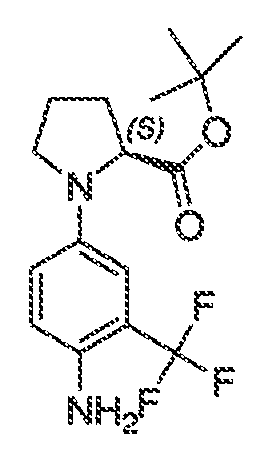
(S)-трет-бутил 1-(4-амино-3-(трифторметил)фенил)пирролидин-2-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-трет-бутил пирролидин-2-карбоксилата. LC/MS: m/z 331,5 (M+H)+ при 1,70 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-(4-Изопропилпиперазин-1-ил)-2-(трифторметил)анилин
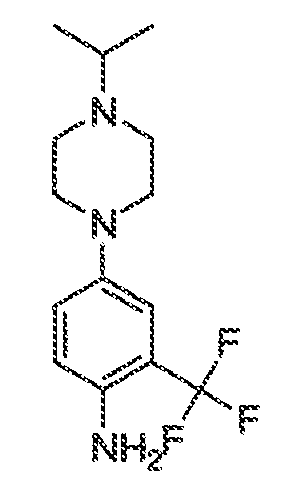
4-(4-Изопропилпиперазин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и 1-изопропилпиперазина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 7,02 (дд, J=2,6, 8,8 Гц, 1H), 6,82-6,77 (м, 2H), 5,03 (с, 2H), 2,95-2,92 (м, 4H), 2,65 (т, J=6,5 Гц, 1H), 2,56-2,50 (м, 4H) и 0,99 (д, J=6,5 Гц, 6H) м.д. LC/MS: m/z 288,3 (M+H)+ при 0,97 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(R)-(1-(4-амино-3-(трифторметил)фенил)пирролидин-2-ил)метанол
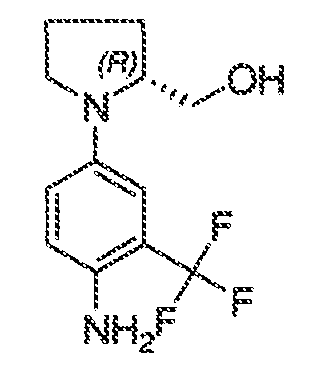
(R)-(1-(4-амино-3-(трифторметил)фенил)пирролидин-2-ил)метанол можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (R)-пирролидин-2-илметанола. LC/MS: m/z 261,1 (M+H)+ при 0,90 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-(1-(4-Амино-3-(трифторметил)фенил)пирролидин-2-ил)метанол
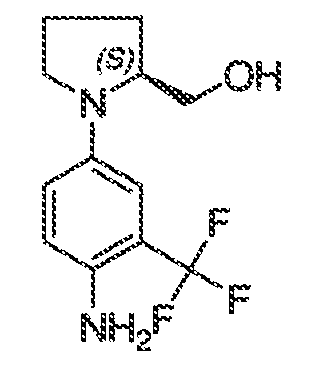
(S)-(1-(4-Амино-3-(трифторметил)фенил)пирролидин-2-ил)метанол можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-пирролидин-2-илметанола. LC/MS: m/z 261,1 (M+H)+ при 0,91 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(1-(4-Амино-3-(трифторметил)фенил)пиперидин-4-ил)метанол
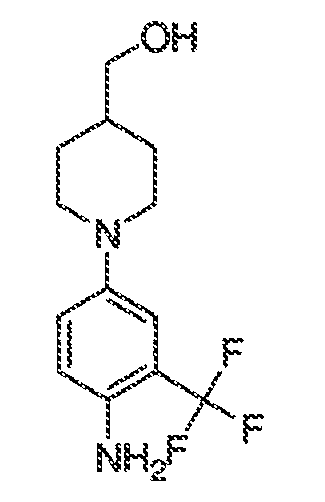
(1-(4-Амино-3-(трифторметил)фенил)пиперидин-4-ил)метанол можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и пиперидин-4-илметанола. LC/MS: m/z 275,3 (M+H)+ при 0,91 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-1-(4-Амино-3-(трифторметил)фенил)-N,N-диметилпирролидин-3-амин

(S)-1-(4-Амино-3-(трифторметил)фенил)-N,N-диметилпирролидин-3-амин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-N,N-диметилпирролидин-3-амина. LC/MS: m/z 274,1 (M+H)+ при 0,78 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-1-(4-Амино-3-(трифторметил)фенил)пирролидин-3-ол
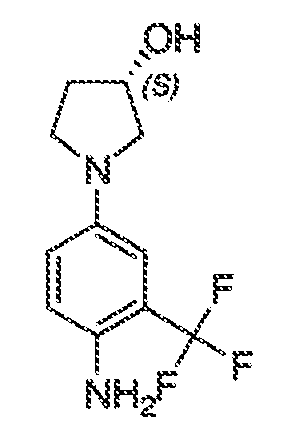
(S)-1-(4-Амино-3-(трифторметил)фенил)пирролидин-3-ол можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-пирролидин-3-ола. LC/MS: m/z 277,1 (M+H)+ при 1,28 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(R)-1-(4-Амино-3-(трифторметил)фенил)пирролидин-3-ол
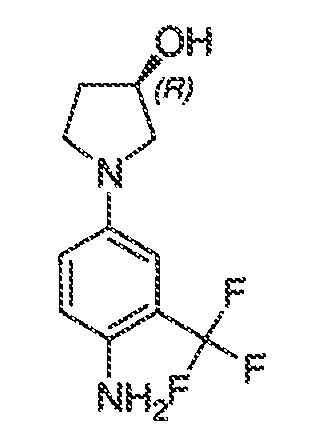
(R)-1-(4-Амино-3-(трифторметил)фенил)пирролидин-3-ол можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (R)-пирролидин-3-ола. LC/MS: m/z 277,1 (M+H)+ при 1,28 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-4-(2-Метилпиперидин-1-ил)-2-(трифторметил)анилин
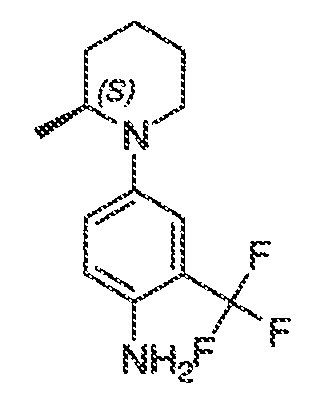
(S)-4-(2-Метилпиперидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-2-метилпиперидина. LC/MS: m/z 259,1 (M+H)+ при 0,66 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-Этил 1-(4-амино-3-(трифторметил)фенил)пиперидин-3-карбоксилат
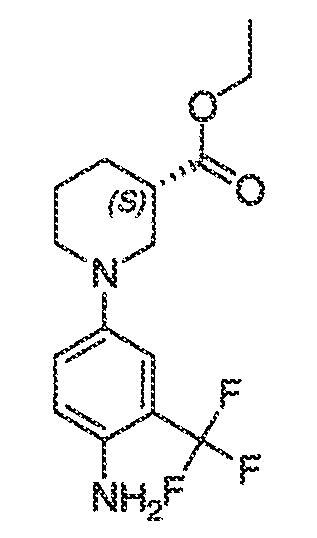
(S)-Этил 1-(4-амино-3-(трифторметил)фенил)пиперидин-3-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-этил пиперидин-3-карбоксилата. LC/MS: m/z 317,1 (M+H)+ при 1,01 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-(3,3-Диметилпиперидин-1-ил)-2-(трифторметил)анилин
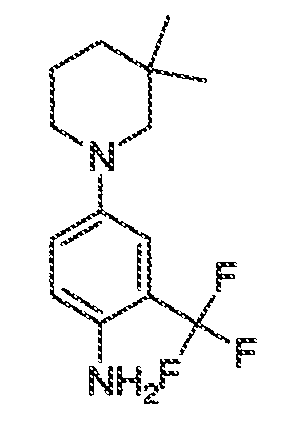
4-(3,3-Диметилпиперидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и 3,3-диметилпиперидина. LC/MS: m/z 273,1 (M+H)+ при 0,96 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(R)-Метил 1-(4-амино-3-(трифторметил)фенил)пиперидин-2-карбоксилат
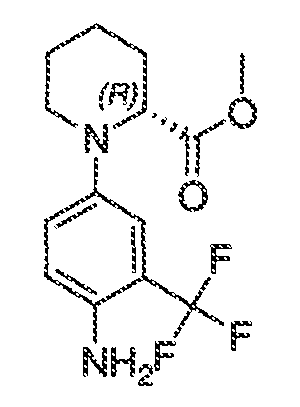
(R)-Метил 1-(4-амино-3-(трифторметил)фенил)пиперидин-2-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (R)-метил пиперидин-2-карбоксилата. LC/MS: m/z 303,3 (M+H)+ при 1,03 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-Метил 1-(4-амино-3-(трифторметил)фенил)пиперидин-2-карбоксилат
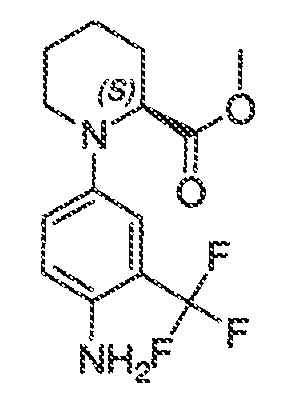
(S)-Метил 1-(4-амино-3-(трифторметил)фенил)пиперидин-2-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-метил пиперидин-2-карбоксилата. LC/MS: m/z 303,3 (M+H)+ при 1,03 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-Метил 1-(4-амино-3-(трифторметил)фенил)-2-метилпирролидин-2-карбоксилат
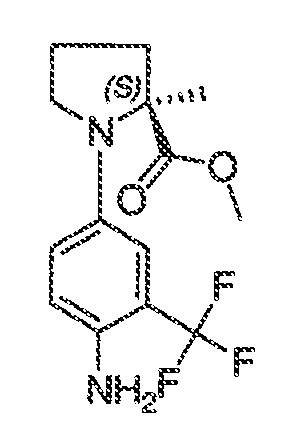
(S)-Метил 1-(4-амино-3-(трифторметил)фенил)-2-метилпирролидин-2-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-метил 2-метилпирролидин-2-карбоксилата. LC/MS: m/z 303,1 (M+H)+ при 1,27 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(2S,3S)-Метил 1-(4-амино-3-(трифторметил)фенил)-3-метилпирролидин-2-карбоксилат
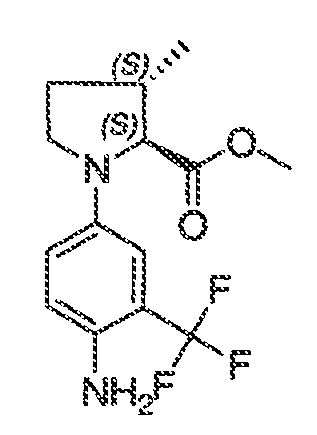
(2S,3S)-Метил 1-(4-амино-3-(трифторметил)фенил)-3-метилпирролидин-2-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (2S,3S)-метил 3-метилпирролидин-2-карбоксилата. LC/MS: m/z 303,3 (M+H)+ при 1,30 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(1R,2R,5S)-Метил 3-(4-амино-3-(трифторметил)фенил)-3-азабицикло[3,1,0]гексан-2-карбоксилат
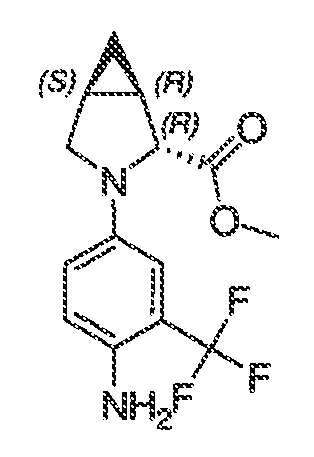
(1R,2R,5S)-Метил 3-(4-амино-3-(трифторметил)фенил)-3-азабицикло[3,1,0]гексан-2-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (1R,2R,5S)-метил 3-азабицикло[3,1,0]гексан-2-карбоксилата. LC/MS: m/z 301,5 (M+H)+ при 1,31 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(2S,4R)-Метил 1-(4-амино-3-(трифторметил)фенил)-4-трет-бутоксипирролидин-2-карбоксилат
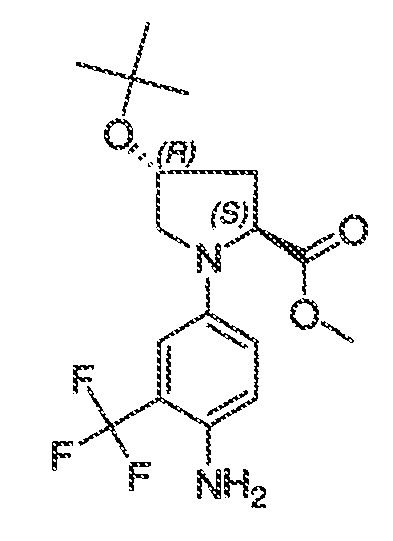
(2S,4R)-Метил 1-(4-амино-3-(трифторметил)фенил)-4-трет-бутоксипирролидин-2-карбоксилат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (2S,4R)-метил 4-трет-бутоксипирролидин-2-карбоксилата. LC/MS: m/z 301,5 (M+H)+ при 1,31 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
N-(4-Амино-3-(трифторметил)фенил)пиваламид
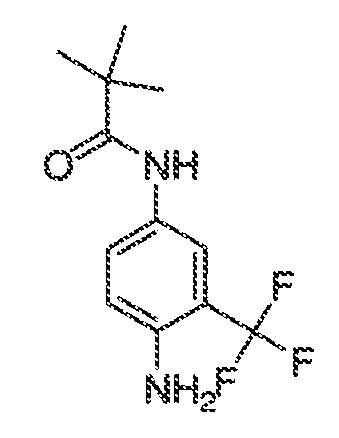
N-(4-Амино-3-(трифторметил)фенил)пиваламид можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и пиваламида. LC/MS: m/z 261,1 (M+H)+ при 1,28 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
трет-бутил 4-амино-3-(трифторметил)фенилкарбамат
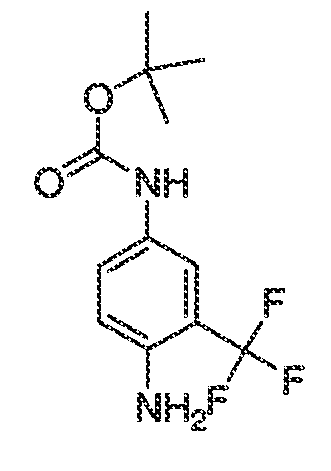
трет-бутил 4-амино-3-(трифторметил)фенилкарбамат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и трет-бутил карбамата. LC/MS: m/z 277,3 (M+H)+ при 1,56 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(R)-4-(3-Фторпирролидин-1-ил)-2-(трифторметил)анилин
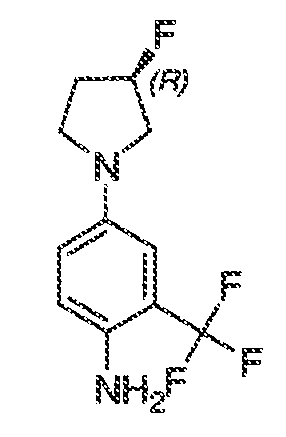
(R)-4-(3-Фторпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (R)-3-фторпирролидина. LC/MS: m/z 249,3 (M+H)+ при 0,92 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-4-(3-Фторпирролидин-1-ил)-2-(трифторметил)анилин

(S)-4-(3-Фторпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-3-фторпирролидина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 6,81 (д, J=8,8 Гц, 1H), 6,72 (дд, J=2,6, 8,8 Гц, 1H), 6,51 (д, J=2,8 Гц, 1H), 5,48 (д, J=3,1 Гц, 1H), 4,76 (с, 2H), 3,50 (дд, J=3,9, 11,8 Гц, 1H), 3,43-3,38 (м, 1H), 3,33-3,24 (м, 2H), 2,24-2,21 (м, 1H) и 2,15 (дд, J=3,9, 7,9 Гц, 1H) м.д. LC/MS: m/z 249,2 (M+H)+ при 0,92 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(R)-4-(3-Метилпирролидин-1-ил)-2-(трифторметил)анилин
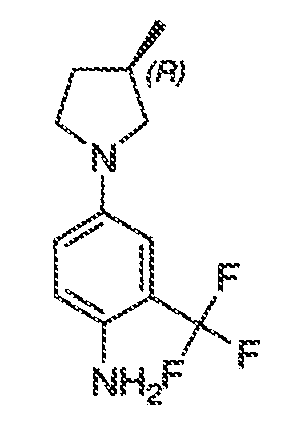
(R)-4-(3-Метилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (R)-3-метилпирролидина. LC/MS: m/z 245,1 (M+H)+ при 0,92 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-4-(3-Метилпирролидин-1-ил)-2-(трифторметил)анилин
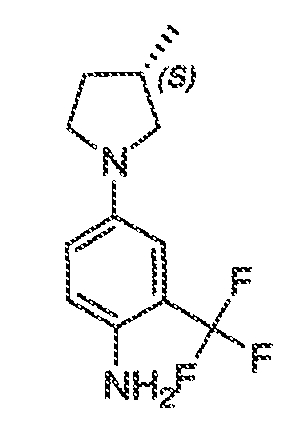
(S)-4-(3-Метилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (S)-3-метилпирролидина. LC/MS: m/z 245,1 (M+H)+ при 0,93 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-((2R,5R)-2,5-Диметилпирролидин-1-ил)-2-(трифторметил)анилин
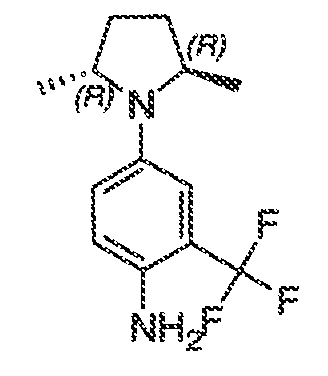
4-((2R,5R)-2,5-Диметилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (2R,5R)-2,5-диметилпирролидина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 6,77 (д, J=8,8 Гц, 1H), 6,70 (д, J=9,0 Гц, 1H), 6,49 (с, 1H), 4,70 (с, 2H), 3,88 (т, J=5,9 Гц, 2H), 2,18-2,14 (м, 2H), 1,56 (с, 2H) и 0,96 (д, J=6,1 Гц, 6H) м.д. LC/MS: m/z 259,3 (M+H)+ при 0,66 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-(3,3-Дифторпирролидин-1-ил)-2-(трифторметил)анилин
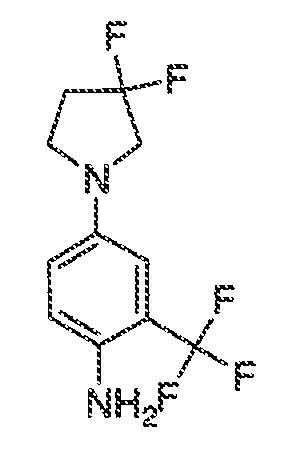
4-(3,3-Дифторпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и 3,3-дифторпирролидина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 6,83-6,76 (м, 2H), 6,58 (д, J=2,6 Гц, 1H), 4,89 (с, 2H), 3,59 (т, J=13,5 Гц, 2H), 3,36 (кв., J=7,0 Гц, 3H) и 1,18 (т, J=7,3 Гц, 1H) м.д. LC/MS: m/z 267,2 (M+H)+ при 1,35 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-((2S,5S)-2,5-Диметилпирролидин-1-ил)-2-(трифторметил)анилин
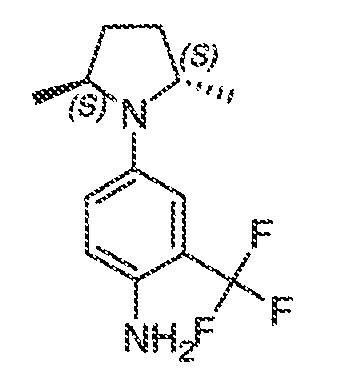
4-((2S,5S)-2,5-Диметилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (2S,5S)-2,5-диметилпирролидина. LC/MS: m/z 259,1 (M+H)+ при 0,79 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-((2S,5R)-2,5-Диметилпирролидин-1-ил)-2-(трифторметил)анилин
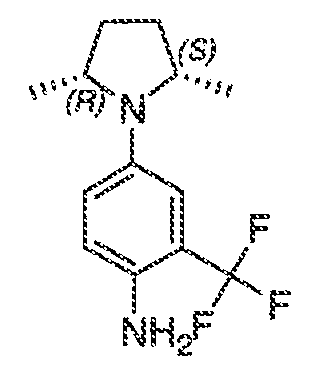
4-((2S,5R)-2,5-Диметилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (2S,5R)-2,5-диметилпирролидина. LC/MS: m/z 259,1 (M+H)+ при 0,79 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-((2S,5R)-2,5-Диметилпирролидин-1-ил)-2-(трифторметил)анилин
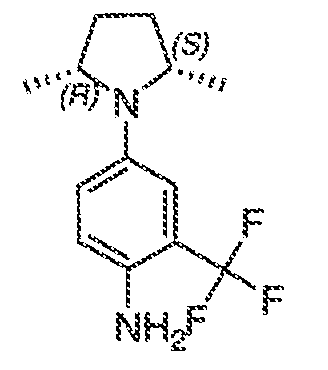
4-((2S,5R)-2,5-Диметилпирролидин-1-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (2S,5R)-2,5-диметилпирролидина. LC/MS: m/z 259,1 (M+H)+ при 0,79 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-(Циклопентилокси)-2-(трифторметил)анилин

4-(Циклопентилокси)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и циклопентанола. 1H ЯМР (400,0 МГц, CDCl3) δ 6,96 (д, J=2,8 Гц, 1H), 6,88 (дд, J=2,8, 8,7 Гц, 1H), 6,69-6,67 (м, 1H), 4,68-4,63 (м, 1H), 4,12 (д, J=7,1 Гц, 2H) и 1,90-1,56 (м, 8H) м.д.
4-Изопропокси-2-(трифторметил)анилин
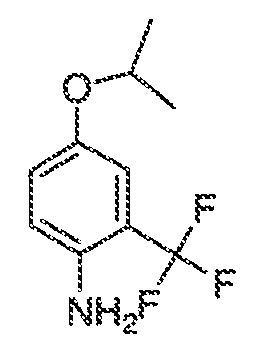
4-Изопропокси-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и пропан-2-ола. 1H ЯМР (400,0 МГц, CDCl3) δ 6,99 (д, J=2,8 Гц, 1H), 6,90 (дд, J=2,7, 8,7 Гц, 1H), 6,68 (д, J=8,7 Гц, 1H), 4,39 (т, J=6,1 Гц, 1H), 3,88 (с, 2H) и 1,30-1,26 (м, 6H) м.д.
2-Амино-5-(пирролидин-1-ил)бензонитрил
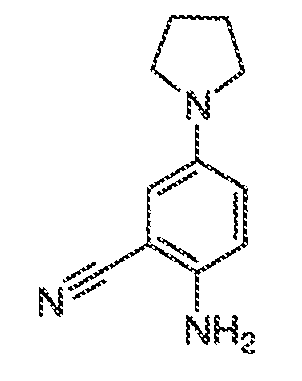
2-Амино-5-(пирролидин-1-ил)бензонитрил можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 2-амино-5-фторбензонитрила и пирролидина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 6,74 (д, J=1,1 Гц, 2H), 6,48 (с, 1H), 5,16 (с, 2H), 3,10 (м, 4H), 1,90 (м, 4H). LC/MS: m/z 188,5 (M+H)+ при 0,44 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
2-Метокси-4-(пирролидин-1-ил)анилин
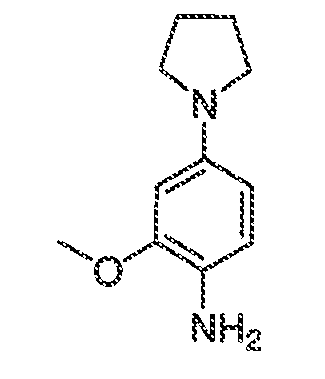
2-Метокси-4-(пирролидин-1-ил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 4-фтор-2-метоксианилина и пирролидина. 1H ЯМР (400,0 МГц, DMSO-d6) δ 6,51 (д, J=8,3 Гц, 1H), 6,14 (д, J=2,4 Гц, 1H), 5,95 (дд, J=2,4, 8,3 Гц, 1H), 3,91 (с, 2H), 3,74 (с, 3H), 3,11 (м, 4H), 1,90 (м, 4H). LC/MS: m/z 193,5 (M+H)+ при 1,06 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-(3-Метилпиперидин-1-ил)-2-(трифторметил)анилин

2-Метокси-4-(пирролидин-1-ил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и 3-метилпиперидина. 1H ЯМР (400 МГц, CDCl3) δ 7,05-6,93 (м, 2H), 6,69 (д, J=8,7 Гц, 1H), 3,85 (с, 2H), 3,42-3,29 (м, 2H), 2,52 (тд, J=11,5, 3,1 Гц, 1H), 2,25-2,15 (м, 1H), 1,86-1,62 (м, 5H), 0,94 (д, J=6,4 Гц, 3H). LC/MS: m/z 259,0 (M+H)+ при 0,79 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
4-((1S,4R)-2-Азабицикло[2,2,1]гептан--2-ил)-2-(трифторметил)анилин
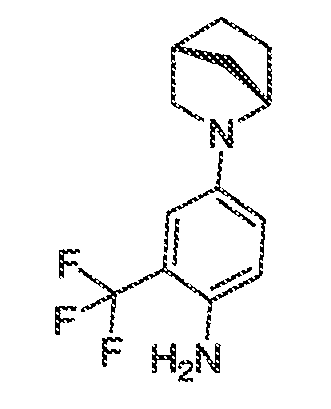
4-((1S,4R)-2-Азабицикло[2,2,1]гептан--2-ил)-2-(трифторметил)анилин можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и (1S,4R)-2-азабицикло[2,2,1]гептана. 1H ЯМР (400 МГц, CDCl3) δ 6,68 (д, J=8,5 Гц, 1H), 6,58 (дт, J=8,7, 2,5 Гц, 2H), 4,02 (с, 1H), 3,65 (с, 2H), 3,49 (дт, J=5,9, 3,2 Гц, 1H), 2,64 (д, J=8,0 Гц, 1H), 2,56 (с, 1H), 1,83-1,53 (м, 5H), 1,48 (д, J=9,3 Гц, 1H), 1,37-1,22 (м, 2H).
2-(4-(4-Амино-3-(трифторметил)фенил)пиперазин-1-ил)этанол
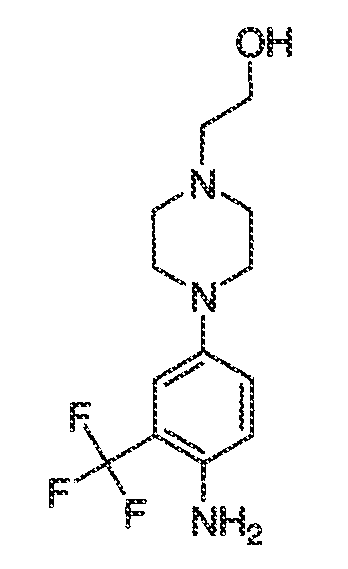
2-(4-(4-Амино-3-(трифторметил)фенил)пиперазин-1-ил)этанол можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 5-фтор-2-нитробензотрифторида и 2-(пиперазин-1-ил)этанола.
Аминовое промежуточное соединение Пример 13: Синтез 5-амино-2-изопропоксифенола
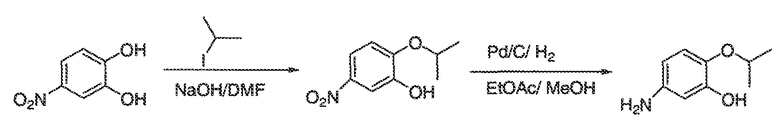
К перемешиваемому раствору гидроксида натрия (387 мг, 9,67 ммоль) в безводном DMSO добавляли 4-нитробензол-1,2-диол (500 мг, 3,22 ммоль) при 0ºC. После перемешивания в течение 5 минут добавляли по каплям 2-иод-пропан (603 мг, 3,55 ммоль). Полученную темную смесь перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь гасили водой и затем подкисляли при помощи 1M HCl (pH=4) и затем водный слой экстрагировали этилацетатом (3×20 мл). Объединенный органический слой промывали насыщенным раствором NaCl и затем сушили над MgSO4, фильтровали и концентрировали при пониженном давлении досуха. Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием градиента 0-20% EtOAc в гексане с получением 2-изопропокси-5-нитро-фенола (163 мг, 51%). 1H ЯМР (400,0 МГц, CDCl3) δ 7,86 (дд, J=2,5, 8,8 Гц, 1H), 7,76 (д, J=2,5 Гц, 1H), 6,99 (д, J=8,8 Гц, 1H), 4,73 (квин., J=6,1 Гц, 1H) и 1,43 (д, J=6,1 Гц, 6H).
Колбу, содержащую 2-изопропокси-5-нитро-фенол (100 мг, 0,47 ммоль) и палладий на углероде (50 мг, 0,47 ммоль), продували при помощи N2 с последующим удалением газа в вакууме. Добавляли EtOAc (2 мл) и метанол (2 мл) в инертной атмосфере с последующим удалением газа в вакууме. Реакционную смесь затем перемешивали в течение ночи в атмосфере водорода. Палладиевый катализатор удаляли фильтрованием и растворитель удаляли при пониженном давлении с получением 5-амино-2-изопропоксифенола (65 мг, 76%). 1H ЯМР (400,0 МГц, CDCl3) δ 6,73 (д, J=8,3 Гц, 1H), 6,31 (д, J=2,5 Гц, 1H), 6,22 (дд, J=2,5, 8,3 Гц, 1H), 5,22 (с, 1H), 4,50 (квин., J=6,1 Гц, 1H), 3,37 (с, 2H) и 1,37-1,26 (м, 6H) м.д.
Аминовое промежуточное соединение Пример 14: Синтез 6-метокси-5-(трифторметил)пиридин-2-амина
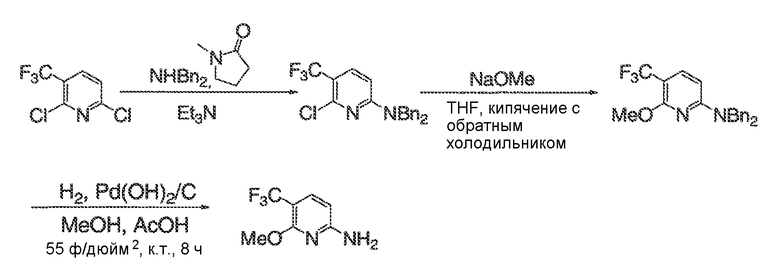
Смесь 2,6-дихлор-3-(трифторметил)пиридина (1,0 г, 4,63 ммоль), дибензиламина (913 мг, 890 мкл, 4,63 ммоль), триэтиламина (1,2 мл, 8,61 ммоль) и 1-метил-2-пирролидона (6 мл, 62,22 ммоль) нагревали при 120ºC в течение 9 часов. Реакционную смесь охлаждали до комнатной температуры и выливали в ледяную воду, экстрагировали при помощи EtOAc (20 мл ×3), объединенные органические слои сушили над безводным Na2SO4, фильтровали, концентрировали и очищали колоночной хроматографией на силикагеле (2%-10% EtOAc в петролейном эфире в качестве элюента) с получением N,N-дибензил-6-хлор-5-(трифторметил)пиридин-2-амина (1,5 г, 86%) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 7,59 (д, J=8,7 Гц, 1H), 7,37-7,21 (м, 10H), 6,32 (д, J=8,7 Гц, 1H), 4,80 (с, 4H).
К перемешиваемому раствору N,N-дибензил-6-хлор-5-(трифторметил)пиридин-2-амина (100 мг, 0,27 ммоль) в ТГФ (5 мл) добавляли свежеполученный CH3ONa (42 мг, 0,78 ммоль) при комнатной температуре. Смесь нагревали до температуры кипения с обратным холодильником в течение 10 часов и затем охлаждали до комнатной температуры, разбавляли водой (5 мл) и экстрагировали при помощи CH2Cl2 (6 мл ×3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с использованием градиента 2%-10% EtOAc в петролейном эфире в качестве элюента с получением N,N-дибензил-6-метокси-5-(трифторметил)пиридин-2-амина (90 мг, 91%). 1H ЯМР (400 МГц, CDCl3) δ 7,53 (д, J=8,4 Гц, 1H), 7,35-7,22 (м, 10H), 6,02 (д, J=8,4 Гц, 1H), 4,79 (с, 4H), 3,90 (с, 3H).
К раствору N,N-дибензил-6-метокси-5-(трифторметил)пиридин-2-амина (3,0 г, 8,056 ммоль) в смеси метанола (30 мл) и AcOH (3 мл) добавляли гидроксид палладия (0,3 г, 0,43 ммоль) в атмосфере азота. Смесь перемешивали в атмосфере водорода (55 ф/дюйм2 (3,867 кг/см2), 25 ºC) в течение ночи. Катализатор удаляли фильтрованием через слой Целита и фильтрат нейтрализовали насыщенным раствором Na2CO3. Водный слой экстрагировали при помощи CH2Cl2 (30 мл ×3) и объединенные органические слои концентрировали в вакууме с получением 6-метокси-5-(трифторметил)пиридин-2-амина (1,4 г, 90%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,56 (д, J=8,4 Гц, 1H), 6,03 (д, J=8,0 Гц, 1H), 4,64 (шир.с, 2H), 3,93 (с, 3H), MS (ESI) m/e (M+H+) 193, 13C ЯМР (100 МГц, CDCl3) 159,4, 138,0 (д, J=5,9 Гц), 125,9, 122,3, 101,9 (д, J=44,4 Гц), 98,3, 53,6, 19F ЯМР (282,4 МГц, CDCl3) -62,6.
Аминовое промежуточное соединение Пример 15: Синтез (R)-4-(4,4-дифтор-2-метилпирролидин-1-ил)-2-(трифторметил)анилина
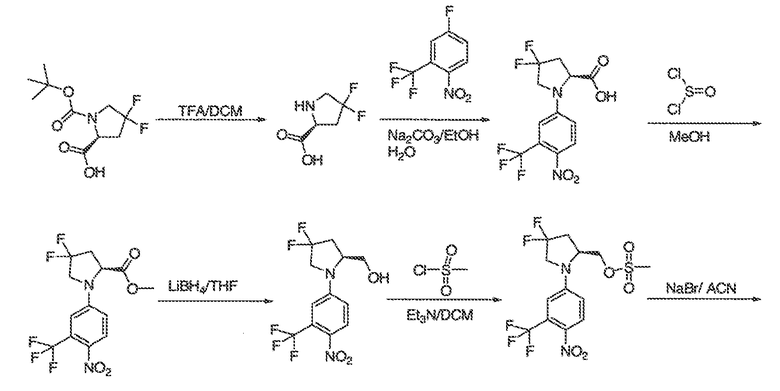
(S)-2-(бромметил)-4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин
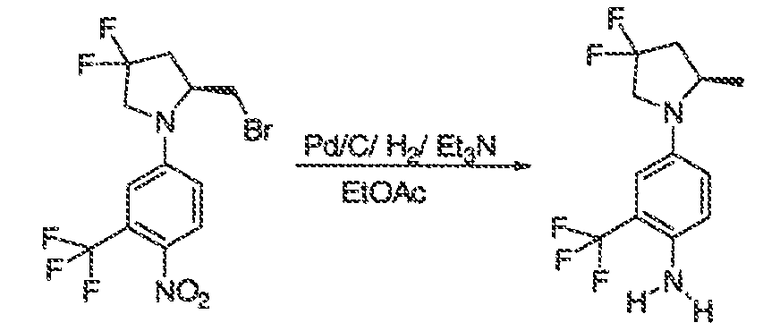
(R)-4-(4,4-дифтор-2-метилпирролидин-1-ил)-2-(трифторметил)анилин
(S)-1-(трет-бутоксикарбонил)-4,4-дифторпирролидин-2-карбоновую кислоту (500 мг, 2,0 ммоль) растворяли в дихлорметане и обрабатывали по каплям при помощи TFA (227 мг, 153 мкл, 2 ммоль). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре, растворители выпаривали с получением (S)-4,4-дифторпирролидин-2-карбоновой кислоты, которую использовали на следующей стадии без дополнительной очистки. LC/MS: m/z 152,2 (M+H)+ при 0,53 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
К 4-фтор-1-нитро-2-(трифторметил)бензолу (416 мг, 2 ммоль), (S)-4,4-дифторпирролидин-2-карбоновой кислоте (301 мг, 2 ммоль) и карбонату натрия (633 мг, 574 мкл, 6,0 ммоль) добавляли 1:1 воду/этанол (8 мл) и реакционную смесь нагревали при 90ºC в течение 72 часов. Реакционную смесь разбавляли водой (50 мл) затем подкисляли 1 н раствором HCl и продукт экстрагировали этилацетатом. Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением (S)-4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин-2-карбоновой кислоты в виде коричневого масла (600 мг, 89%), которое использовали непосредственно в следующей реакции. LC/MS: m/z 341,00 (M+H)+ при 1,49 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
(S)-4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин-2-карбоновую кислоту (600 мг, 1,76 ммоль) растворяли в метаноле (10 мл), охлаждали до 0ºC и обрабатывали по каплям тионилхлоридом (840 мг, 515 мкл, 7,08 ммоль). Реакционную смесь нагревали при 50ºC в течение 48 часов. Реакционную смесь упаривали в вакууме и остаток очищали колоночной хроматографией на силикагеле (0-60% этилацетата в гексане) с получением (S)-метил 4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин-2-карбоксилата (300 мг, 48% выход). 1H ЯМР (400 МГц, DMSO-d6) δ 8,12 (д, J=9,2 Гц, 1H), 7,04 (д, J=2,6 Гц, 1H), 6,97 (дд, J=9,2, 2,7 Гц, 1H), 5,19 (дд, J=9,8, 1,9 Гц, 1H), 4,14-3,99 (м, 2H), 3,69 (с, 3H), 3,11-2,95 (м, 1H), 2,78 (т, J=14,6 Гц, 1H).
К суспензии LiBH4 (55 мг, 2,54 ммоль) в безводном ТГФ (3 мл) добавляли раствор (S)-метил 4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин-2-карбоксилата (300 мг, 0,85 ммоль) в безводном ТГФ при 0ºC. Реакционную смесь перемешивали при 0ºC в течение 20 минут, затем давали нагреться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли этилацетатом (100 мл), промывали водой (50 мл) и затем насыщенным солевым раствором. Слои разделяли и органический слой сушили над Na2SO4, фильтровали и концентрировали с получением (S)-(4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин-2-ил)метанола в виде желтого вязкого масла (275 мг, 100%). LC/MS: m/z 327,2 (M+H)+ при 1,54 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
К раствору (S)-(4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин-2-ил)метанола (275 мг, 0,84 ммоль) в безводном DCM добавляли триэтиламин (171 мг, 235 мкл, 1,7 ммоль) с последующим добавлением метансульфонилхлорида (106 мг, 72 мкл, 0,93 ммоль) при 0ºC. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь разбавляли при помощи DCM и гасили при помощи 3 мл насыщенного раствора NaHCO3. Органический слой отделяли и промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением (S)-(4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин-2-ил)метилметансульфоната в виде желтого твердого вещества (330 мг, 96%). LC/MS: m/z 405 (M+H)+ при 1,71 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
NaBr (420 мг, 131 мкл, 4,08 ммоль) добавляли к раствору (S)-(4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин-2-ил)метилметансульфоната (330 мг, 0,82 ммоль) в ацетонитриле (2,0 мл) и нагревали при 80ºC в течение 16 часов. Реакционную смесь разбавляли этилацетатом (10 мл) и водой (5 мл). Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (15-50% этилацетат/гексан) с получением (S)-2-(бромметил)-4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидина (230 мг, 72%) в виде желтого масла. 1H ЯМР (400 МГц, DMSO-d6) δ 8,14 (д, J=8,7 Гц, 1H), 7,09-7,07 (м, 2H), 4,76-4,71 (м, 1H), 4,16-3,99 (м, 2H), 3,74 (дд, J=10,6, 3,0 Гц, 1H), 3,60 (т, J=9,7 Гц, 1H), 2,94-2,80 (м, 1H), 2,66-2,55 (м, 1H).
(S)-2-(бромметил)-4,4-дифтор-1-(4-нитро-3-(трифторметил)фенил)пирролидин (230 мг, 0,59 ммоль) растворяли в этилацетате (5 мл) и TEA (90 мг, 124 мкл, 0,89 ммоль) и колбу продували азотом. Добавляли 10% Pd/C (58 мг, 0,05 ммоль) катализатор и реакционную смесь перемешивали в течение ночи в атмосфере водорода. Реакционную смесь фильтровали, обрабатывали 50% насыщенным раствором NaHCO3 и экстрагировали этилацетатом. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением (R)-4-(4,4-дифтор-2-метилпирролидин-1-ил)-2-(трифторметил)анилина в виде фиолетового масла (164 мг, 99% выход). 1H ЯМР (400 МГц, DMSO-d6) δ 6,81 (с, 2H), 6,58 (с, 1H), 4,92 (с, 2H), 4,07-4,01 (м, 1H), 3,74-3,63 (м, 1H), 3,60-3,50 (м, 1H), 2,75-2,60 (м, 1H), 2,22-2,11 (м, 1H), 1,10 (д, J=6,2 Гц, 3H).
Аминовое промежуточное соединение Пример 16: Синтез 1-(2-метоксиэтил)-5-(трифторметил)-1H-индол-6-амина
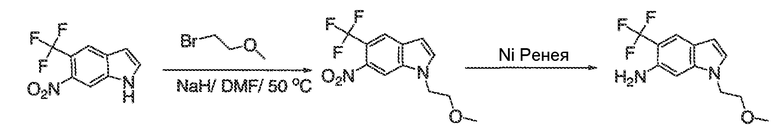
6-нитро-5-(трифторметил)-1H-индол (50 мг, 0,22 ммоль) растворяли в DMF (2 мл) и обрабатывали гидридом натрия (35 мг, 0,87 ммоль) с образованием темно-красного непрозрачного раствора. Раствор перемешивали в течение 20 минут и затем добавляли по каплям к раствору 2-бромэтилметилового эфира (121 мг, 82 мкл, 0,87 ммоль) в 1 мл DMF. Реакционную смесь нагревали при 50ºC в течение 30 минут. Реакционную смесь разбавляли этилацетатом (50 мл) и промывали 50% насыщенным раствором NaHCO3 (2×10 мл) и насыщенным солевым раствором. Органический слой сушили над Na2SO4, фильтровали и сушили с получением 1-(2-метоксиэтил)-6-нитро-5-(трифторметил)-1H-индола, который использовали на следующей стадии без дополнительной очистки.
1-(2-метоксиэтил)-6-нитро-5-(трифторметил)-1H-индол растворяли в 10 мл EtOH и гидрировали с использованием Ni Ренея в качестве катализатора (H-cube:1,2 мл/мин 30ºC) с получением 1-(2-метоксиэтил)-5-(трифторметил)-1H-индол-6-амина с количественным выходом. LC/MS: m/z 259,0 (M+H)+ при 1,12 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Аминовое промежуточное соединение Пример 17: Синтез N-(4-амино-3-(трифторметил)фенил)пиваламида
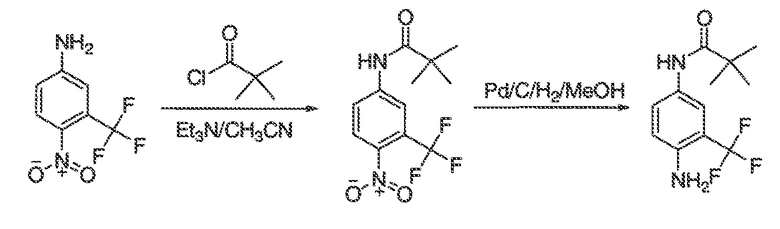
К раствору 4-нитро-3-(трифторметил)анилина (1,0 г, 4,85 ммоль) в CH2Cl2 (15 мл) и TEA (2,95 г, 4,1 мл, 29,11 ммоль) в круглодонной колбе при 0ºC добавляли триметилацетилхлорид (1,75 г, 1,8 мл, 14,55 ммоль) по каплям. Реакционную смесь нагревали до комнатной температуры и гасили, выливая в воду. Слои разделяли и водный слой экстрагировали при помощи DCM. Объединенный органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Остаток затем очищали колоночной хроматографией на силикагеле с использованием градиента (0-100% EtOAc/Гекс.) с получением N-(4-нитро-3-(трифторметил)фенил)пиваламида (1,64 г, 81%). LC/MS: m/z 291,1 (M+H)+ при 1,83 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
К раствору 2,2-диметил-н-(4-нитро-3-трифторметил-фенил)-пропионамида (1,64 г, 5,65 ммоль) в MeOH (50 мл) добавляли Pd/C (150 мг, 0,14 ммоль) в атмосфере азота. Смесь перемешивали в атмосфере водорода в течение 4 часов. Катализатор удаляли фильтрованием через слой Целита и растворитель выпаривали в вакууме с получением N-(4-амино-3-(трифторметил)фенил)пиваламида (1,2 г, 78%). LC/MS: m/z 261,5 (M+H)+ при 1,83 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
трет-бутил 4-амино-3-(трифторметил)фенилкарбамат
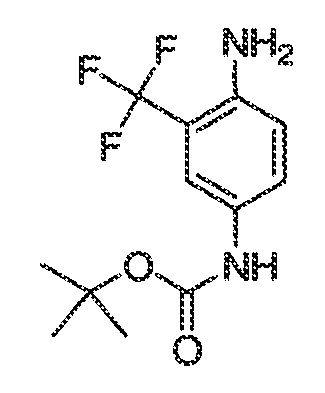
трет-бутил 4-амино-3-(трифторметил)фенилкарбамат можно синтезировать в соответствии с общей схемой, представленной выше, исходя из 4-нитро-3-(трифторметил)анилина и ди-трет-бутилдикарбоната. LC/MS: m/z 277,35 (M+H)+ при 1,56 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Аминовое промежуточное соединение Пример 18: Синтез (S)-6-(2-метилпирролидин-1-ил)-2-(трифторметил)пиридин-3-амина.
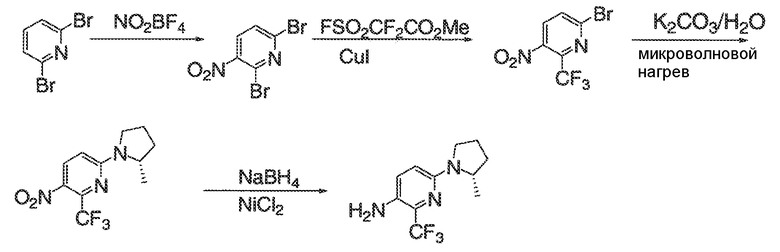
К раствору 2,6-дибромпиридина (10,0 г, 42,6 ммоль) в безводном CH3CN (100 мл) медленно добавляли NO2 +BF4 (11,3 г, 85,2 ммоль). Реакционную смесь нагревали до 80ºC в атмосфере азота в течение 24 часов. Смесь затем упаривали в вакууме с получением неочищенного продукта. Остаток очищали колоночной хроматографией на силикагеле с получением 2,6-дибром-3-нитропиридина (5,7 г, 48%). 1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, J=8,4 Гц, 1H), 7,65 (д, J=8,4 Гц, 1H).
К раствору 2,6-дибром-3-нитропиридина (5,7 г, 20,4 ммоль) в DMF (40 мл) добавляли CuI (3,9 г, 20,4 ммоль) и FSO2CF2CO2Me (4,7 г, 24,5 ммоль). Реакционную смесь перемешивали при 80ºC в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь выливали в воду (100 мл) и экстрагировали при помощи EtOAc (100 мл ×3). Объединенный органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и очищали колоночной хроматографией на силикагеле с получением 6-бром-3-нитро-2-(трифторметил)пиридина (3,0 г, 53%, чистота 70%). 1H ЯМР (400 МГц, CDCl3) δ 8,12 (д, J=8,4 Гц, 1H), 7,94 (д, J=8,4 Гц, 1H).
Суспензию 6-бром-3-нитро-2-(трифторметил)пиридина (400 мг, 1,5 ммоль), тозилатной соли (S)-2-метилпирролидина (381 мг, 1,5 ммоль) и K2CO3 (620 мг, 4,5 ммоль) в H2O (4 мл) нагревали при 140ºC в условиях микроволнового облучения в течение 30 минут. Раствор экстрагировали при помощи EtOAc (50 мл ×3) и объединенный органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и очищали колоночной хроматографией на силикагеле с получением (S)-6-(2-метилпирролидин-1-ил)-3-нитро-2-(трифторметил)пиридина (350 мг, 88% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,14 (д, J=9,2 Гц, 1H), 6,44 (д, J=8,4 Гц, 1H), 4,55-4,42 (м, 1H), 4,02-3,30 (м, 2H), 2,30-2,00 (м, 3H), 1,81 (шир.с, 1H), 1,27 (д, J=6,4 Гц, 3H).
К раствору (S)-6-(2-метилпирролидин-1-ил)-3-нитро-2-(трифторметил)пиридина (300 мг, 1,1 ммоль) в 5 мл метанола добавляли NiCl2∙6H2O (772 мг, 3,3 ммоль). После перемешивания в течение 5 минут добавляли по каплям NaBH4 (84 мг, 2,2 ммоль) тремя порциями при 0ºC. Реакционную смесь перемешивали в течение 5 минут, гасили водой (10 мл) и экстрагировали при помощи EtOAc (15 мл ×3). Объединенный органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4, концентрировали и очищали хроматографией на силикагеле с получением (S)-6-(2-метилпирролидин-1-ил)-2-(трифторметил)пиридин-3-амина (260 мг, 92,6% выход).
Аминовое промежуточное соединение Пример 19: Синтез 3-амино-2,6-ди-трет-бутилфенола
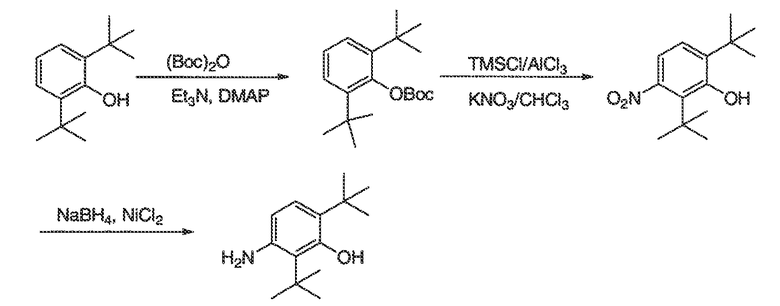
DMAP (885 мг, 7,24 ммоль) добавляли к раствору 2,6-ди-трет-бутилфенола (30 г, 145,4 ммоль) и ди-трет-бутилдикарбоната (38 г, 174,1 ммоль) в Et3N (29,43 г, 40,5 мл, 290,8 ммоль) и гексане (700 мл). Реакционную смесь перемешивали в течение ночи и гасили водой и экстрагировали при помощи EtOAc. Органический слой промывали водным раствором NaHCO3, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир в качестве элюента) с получением трет-бутил (2,6-ди-трет-бутилфенил)карбоната (30 г, 67%). 1H ЯМР (400 МГц, CDCl3) δ: 7,31 (д, J=8,0 Гц, 2H), 7,12 (т, J=8,0 Гц, 1H), 1,54 (с, 9H), 1,38 (с, 8H).
TMSCl (8,5 г, 78,24 ммоль) и трет-бутил (2,6-ди-трет-бутилфенил)карбонат (12,0 г, 39,16 ммоль) добавляли к суспензии KNO3 (5,9 г, 58,36 ммоль) в CHCl3 (100 мл) последовательно при 0ºC. Реакционную смесь перемешивали в течение 0,5 часов и добавляли AlCl3 (15,5 г, 116,20 ммоль). Перемешивание продолжали в течение 2 часов и полученную смесь выливали в ледяную воду и экстрагировали при помощи CH2Cl2. Объединенные органические слои промывали насыщенным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир) с получением 2,6-ди-трет-бутил-3-нитрофенола (2,5 г, 25%). 1H ЯМР (300 МГц, CDCl3) δ 7,18 (д, J=8,4 Гц, 1H), 6,79 (д, J=8,4 Гц, 1H), 5,57 (с, 1H), 1,51 (с, 9H), 1,44 (с, 9H).
NaBH4 (433 мг, 11,45 ммоль) добавляли к раствору 2,6-ди-трет-бутил-3-нитрофенола (950 мг, 3,780 ммоль) и NiCl2 (1,24 г, 9,568 ммоль) в метаноле (15 мл) при -15ºC. После завершения добавления реакционную смесь перемешивали в течение 20 секунд и сразу добавляли воду и реакционную смесь экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4 и упаривали в вакууме с получением 3-амино-2,6-ди-трет-бутил-фенола (650 мг, 78%). 1H ЯМР (300 МГц, CDCl3) δ 6,92 (д, J=8,4 Гц, 1H), 6,24 (д, J=8,4 Гц, 1H), 5,39 (с, 1H), 1,62 (с, 9H), 1,39 (с, 9H); MS (ESI) m/z: 222,2 [M+H+].
Аминовое промежуточное соединение Пример 20: Синтез 3-(трифторметил)-1H-индол-6-амина
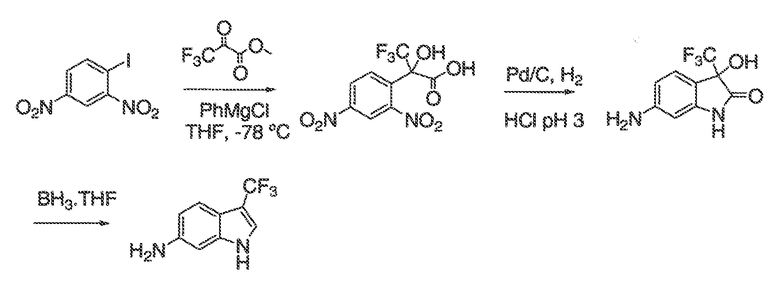
К раствору 1,4-динитроиодбензола (2,12 г, 7,21 ммоль) в тетрагидрофуране (11 мл) при -78ºC в атмосфере N2 добавляли фенилмагнийхлорид (2M в ТГФ) (4 мл, 8,0 ммоль, 1,1 экв.) по каплям. Темно-красный раствор перемешивали в течение 30 минут при -78ºC, затем добавляли по каплям метилтрифторпируват (0,75 мл, 8,65 ммоль). Реакционную смесь перемешивали в течение 30 минут при -78ºC и затем в течение 2 часов при комнатной температуре. Реакционную смесь охлаждали до -10ºC и гасили путем добавления 1 M HCl (6 мл). Реакционную смесь разбавляли водой (10 мл) и дихлорметаном (30 мл). Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3×30 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (0,5-30% этилацетата/гексан) с получением метил 2-(2,4-динитрофенил)-3,3,3-трифтор-2-гидроксипропаноата (1,34 г, 60%)
К раствору метил 2-(2,4-динитрофенил)-3,3,3-трифтор-2-гидроксипропаноата (1,3 г, 4,01 ммоль) в этилацетате (18 мл) добавляли (pH3) HCl (5,2 мл) с последующим добавлением 10% Pd/C (350 мг) в этилацетате (3 мл). Реакционную смесь перемешивали в течение ночи в атмосфере H2. Реакционную смесь фильтровали через слой Целита и фильтрат концентрировали в вакууме. Полученный неочищенный остаток распределяли между дихлорметаном (25 мл) и насыщенным водным раствором NaHCO3 (15 мл). Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (2×25 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (50-100% этилацетат/гексан) с получением 6-амино-3-гидрокси-3-(трифторметил)индолин-2-она (921 мг, 99%)
К раствору 6-амино-3-гидрокси-3-(трифторметил)индолин-2-она (58 мг, 0,25 ммоль) в ТГФ (0,5 мл) при 0ºC добавляли по каплям комплекс BH3 .ТГФ (1 M в ТГФ, 1 мл, 0,95 ммоль). Реакционную смесь перемешивали в течение 5 минут при 0ºC, затем в течение 3 часов при комнатной температуре. Реакционную смесь гасили путем добавления, очень осторожно, 6M HCl (3,5 мл) до тех пор, пока не прекращалось выделение газа. Реакционную смесь затем перемешивали при 80ºC в течение 2 часов. Растворитель удаляли при пониженном давлении и полученный твердый остаток растворяли в DMF (3 мл), фильтровали и очищали при помощи обращенно-фазовой ВЭЖХ (10-99% CH3CN/H2O) с получением 3-(трифторметил)-1H-индол-6-амина (30 мг, 54%, TFA соль).
Аминовое промежуточное соединение Пример 21: Синтез 2-(трифторметил)-1H-индол-6-амина

К раствору 4-метилбензол-1,3-диамина (500 мг, 4,1 ммоль) в безводном пиридине (25 мл) при 0ºC в атмосфере N2 добавляли трифторуксусный ангидрид (1,2 мл) по каплям. Охлаждающую баню удаляли и реакционную смесь перемешивали при комнатной температуре до достижения полной конверсии исходного вещества в желаемый продукт. Реакционную смесь концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (10-45% AcOEt в гексане) с получением N,N'-(4-метил-1,3-фенилен)бис(2,2,2-трифторацетамид) (807 мг, 63%). LC/MS: m/z 315,3 (M+H)+ при 1,39 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Смесь N,N'-(4-метил-1,3-фенилен)бис(2,2,2-трифторацетамид) (1,02 г, 3,25 ммоль), NBS (0,80 г, 4,5 ммоль) в CCl4 (6 мл) перемешивали в течение ночи при комнатной температуре в условиях облучения 300 Вт лампой. Образовавшийся осадок собирали фильтрованием и промывали при помощи CCl4. Неочищенный остаток брали для поглощения в безводный толуол (9,7 мл) в присутствии PPh3 (1,3 г, 4,96 ммоль) и перемешивали при 60oC в течение 16 часов. Фосфониевый осадок собирали фильтрованием, затем растворяли в безводном DMF (10 мл) и перемешивали при 165ºC до достижения полной конверсии в продукт (6,5 часов). Растворитель удаляли в вакууме и полученный остаток очищали колоночной хроматографией на силикагеле (5-25% AcOEt в гексане) с получением 2,2,2-трифтор-N-(2-(трифторметил)-1H-индол-6-ил)ацетамида (243 мг, 25%). LC/MS: m/z 297,3 (M+H)+ при 1,68 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
2,2,2-трифтор-N-(2-(трифторметил)-1H-индол-6-ил)ацетамид (28 мг, 0,09 ммоль) растворяли в MeOH (0,9 мл) и воде (0,4 мл) в присутстви K2CO3 (90 мг, 0,65 ммоль) и перемешивали в течение ночи при комнатной температуре. Очистка (10 до 99% ACN в воде) при помощи LC-MS давала 3-(трифторметил)-1H-индол-6-амин 2,2,2-трифторацетат (30 мг, количественный выход). LC/MS: m/z 201,1 (M+H)+ при 0,90 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Аминовое промежуточное соединение Пример 22: Синтез 4-(3-метилоксетан-3-ил)анилина
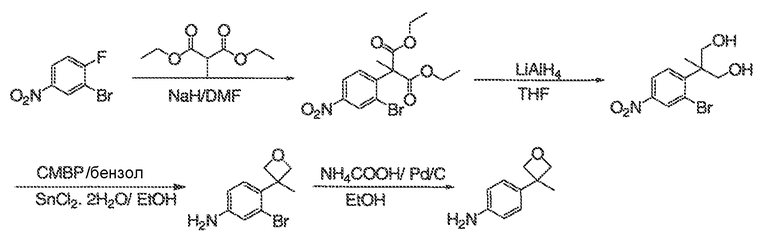
К раствору диэтил 2-метилпропандиоата (21,8 г, 125,0 ммоль) в безводном DMF (125 мл) медленно добавляли NaH (5,2 г, 130 ммоль) при 0ºC в атмосфере азота. Полученную реакционную смесь оставляли для перемешивания в течение 10 минут при 0ºC и затем при комнатной температуре в течение 10 минут. Быстро добавляли 2-бром-1-фтор-4-нитро-бензол (25,0 г, 113,6 ммоль) и реакционная смесь становилась ярко-красной. После перемешивания в течение 10 минут при комнатной температуре неочищенную смесь упаривали досуха и затем распределяли между дихлорметаном и насыщенным солевым раствором. Слои разделяли и объединенные органические слои сушили над безводным Na2SO4 и упаривали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc 10:1) с получением диэтил 2-(2-бром-4-нитрофенил)-2-метилмалоната (33,0 г, 78%)
К раствору диэтил 2-(2-бром-4-нитрофенил)-2-метилмалоната (7,5 г, 20,0 ммоль) в безводном тетрагидрофуране (80 мл) медленно добавляли раствор литийалюминийгидрида (22 мл, 22,0 ммоль, 1,0 M в ТГФ) при 0ºC в атмосфере азота. После перемешивания в течение 10 минут реакция завершалась. Реакцию гасили путем медленного добавления метанола при 0ºC. Реакционную смесь затем распределяли между дихлорметаном и 1 н раствором хлористоводородной кислоты. Слои разделяли и водный слой экстрагировали три раза дихлорметаном. Объединенные органические слои сушили над безводным Na2SO4 и упаривали в вакууме с получением неочищенного продукты, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc 2:1) с получением 2-(2-бром-4-нитрофенил)-2-метилпропан-1,3-диола (1,4 г, 24%) в виде красного твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 8,31 (д, J=2,4 Гц, 1H), 8,13 (дд, J=2,4, 8,8 Гц, 1H), 7,74 (д, J=8,8 Гц, 1H), 4,74 (т, J=5,2 Гц, 2H), 3,93 (кв., J=5,2 Гц, 2H), 3,79 (м, кв., J=5,2 Гц, 2H), 1,38 (с, 3H).
К раствору 2-(2-бром-4-нитрофенил)-2-метилпропан-1,3-диола (7,2 г, 24,82 ммоль) в безводном бензоле (75 мл) добавляли цианометилентрибутилфосфоран (9,0 г, 37,29 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 72 часов, затем упаривали досуха и снова растворяли в этаноле (100 мл). Затем добавляли хлорид олова(II) дигидрат (28 г, 94,42 ммоль) и полученный раствор нагревали до 70ºC в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и затем гасили насыщенным водным раствором бикарбоната натрия. Реакционную смесь затем экстрагировали три раза этилацетатом. Объединенные органические слои сушили над безводным Na2SO4 и упаривали в вакууме с получением неочищенного продукта, который очищали при помощи обращенно-фазовой ВЭЖХ с получением 3-бром-4-(3-метилоксетан-3-ил)анилина (1,5 г, 18%) TFA соль. 1H ЯМР (400 МГц, CD3CN) δ 6,93 (д, J=2,4 Гц, 1H), 6,78 (д, J=8,4 Гц, 1H), 6,71 (дд, J=2,4, 8,4 Гц, 1H), 4,94 (д, J=5,6 Гц, 2H), 4,46 (д, J=6,0 Гц, 2H), 1,70 (с, 3H).
К нагреваемому при температуре кипения с обратным холодильником раствору 3-(2-бром-4-нитрофенил)-3-метилоксетана (68 мг, 0,2499 ммоль) в этаноле (5 мл) добавляли формиат аммония (68 мг, 1,078 ммоль) с последующим добавлением Pd/C (32 мг, 0,3007 ммоль). Реакционную смесь кипятили с обратным холодильником еще в течение 5 минут, охлаждали до комнатной температуры и фильтровали через слой Целита. Растворитель выпаривали с получением 4-(3-метилоксетан-3-ил)анилина. 1H ЯМР (400 МГц, DMSO-d6) δ 6,93-6,90 (м, 2H), 6,56 (д, J=8,5 Гц, 2H), 4,70 (д, J=5,4 Гц, 2H), 4,45 (д, J=5,6 Гц, 2H), 1,55 (с, 3H).
Аминовое промежуточное соединение Пример 23: Синтез 2-(4-аминофенил)-2-метилпропан-1,3-диола

К нагреваемому при температуре кипения с обратным холодильником раствору 2-(2-бром-4-нитрофенил)-2-метилпропан-1,3-диола (211 мг, 0,73 ммоль) в этаноле (15,5 мл) добавляли формиат аммония (211 мг, 3,35 ммоль) с последующим добавлением Pd/C (140 мг, 1,32 ммоль). Реакционную смесь кипятили с обратным холодильником еще в течение 10 минут, охлаждали до комнатной температуры и фильтровали через слой Целита. Растворитель выпаривали при пониженном давлении с получением 2-(4-аминофенил)-2-метилпропан-1,3-диола (112 мг, 85%).
Специальные примеры
Синтез 4-оксо-N-(3-трет-бутилфенил)-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 603)

Колбу, содержащую 4-оксо-1H-хинолин-3-карбоновую кислоту (38 мг, 0,20 ммоль), HBTU (76 мг, 0,20 ммоль), Et3N (61 мг, 84 мкл, 0,60 ммоль) и DMF (2 мл), нагревали при 60ºC в течение 15 минут. К реакционной смеси затем добавляли 3-трет-бутиланилин (29,98 мг, 0,2009 ммоль) и реакционную смесь перемешивали при 60ºC еще в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры, фильтровали и очищали обращенно-фазовой ВЭЖХ с использованием 10 до 99% CH3CN в H2O с получением N-(3-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамида. 1H ЯМР (400 МГц, DMSO-d6) δ 12,94 (с, 1H), 12,45 (с, 1H), 8,89 (с, 1H), 8,34 (дд, J=1,1, 8,2 Гц, 1H), 7,84-7,80 (м, 1H), 7,76-7,71 (м, 2H), 7,62-7,53 (м, 2H), 7,29 (т, J=7,9 Гц, 1H), 7,15-7,12 (м, 1H), 1,31 (с, 9H). LC/MS: m/z 321,5 (M+H)+ при 1,8 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез N-[4-циклопентил-5-гидрокси-2-(3-гидроксипроп-1-инил)фенил]-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 512)
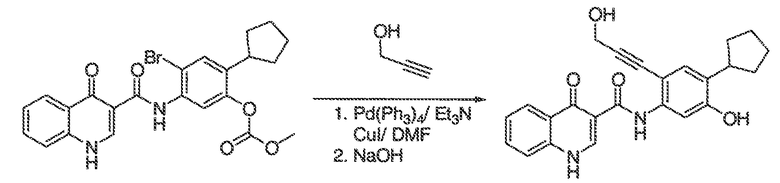
4-бром-2-циклопентил-5-(4-оксо-1,4-дигидрохинолин-3-карбоксамидо)фенилметилкарбонат (50 мг, 0,10 ммоль), Pd(PPh3)2Cl2 (4 мг, 0,005 ммоль) и иодид меди (1 мг, 0,10 мкл, 0,003 ммоль) добавляли в микроволновой сосуд, который продували при помощи N2 и закрывали крышкой. Добавляли дегазированный раствор DMF (1 мл), триэтиламин (2 мл) и проп-2-ин-1-ол (57,75 мг, 59,97 мкл, 1,030 ммоль) и реакционную смесь нагревали при 80ºC в течение 16 часов. Реакционную смесь фильтровали и очищали при помощи ВЭЖХ (20-99% CH3CN/0,05% TFA) с получением 2-циклопентил-4-(3-гидроксипроп-1-инил)-5-(4-оксо-1,4-дигидрохинолин-3-карбоксамидо)фенилметилкарбоната/N-(4-циклопентил-5-гидрокси-2-(3-гидроксипроп-1-инил)фенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида. Эти содержащие продукт фракции объединяли, концентрировали для удаления ацетонитрила и обрабатывали 5н раствором NaOH (2 мл). pH раствора доводили до 7 и реакционную смесь экстрагировали этилацетатом. Органический слой промывали 50% насыщенным раствором бикарбоната натрия (2×20 мл) и насыщенным солевым раствором. Раствор сушили над безводным Na2SO4, фильтровали и сушили с получением N-(4-циклопентил-5-гидрокси-2-(3-гидроксипроп-1-инил)фенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида. LC/MS: m/z 403,2 (M+H)+ при 0,92 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез N-(4-циклогексил-2-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 648)

К N-(4-бром-2-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамиду (50 мг, 0,14 ммоль), циклогексен-1-илбороновой кислоте (35 мг, 0,28) и Pd(dppf)Cl2-DCM (11 мг, 0,01 ммоль) добавляли Na2CO3 (980 мкл 2 M раствора, 1,96 ммоль) и ацетонитрил (2 мл). Реакционную смесь нагревали в условиях микроволнового облучения в течение 10 минут при 150ºC в атмосфере N2. Реакционную смесь разбавляли этилацетатом, промывали 50% насыщенным раствором бикарбоната натрия (2×20 мл), водой и насыщенным солевым раствором. Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (30-100% этилацетата/гексан) с получением N-(4-циклогексен-1-ил-2-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамида в виде белого твердого вещества (35 мг, 70%). N-(4-циклогексен-1-ил-2-метил-фенил)-4-оксо-1H-хинолин-3-карбоксамид (35 мг, 0,10 ммоль) интенсивно перемешивали с 10% раствором Pd/C (мокрый) (30 мг, 0,01 ммоль) в атмосфере H2 в течение 30 минут при 50ºC. Реакционную смесь фильтровали, концентрировали в вакууме и очищали при помощи ВЭЖХ (30-95% CH3CN/5 мМ HCl) с получением N-(4-циклогексил-2-метилфенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида (20 мг, 57% выход). LC/MS m/z 361,4 [M+H]+. 1H ЯМР (400,0 МГц, DMSO-d6) δ 12,94 (д, J=6,0 Гц, 1H), 12,24 (с, 1H), 8,89 (д, J=6,5 Гц, 1H), 8,35 (д, J=7,7 Гц, 1H), 8,22 (д, J=8,3 Гц, 1H), 7,82 (т, J=7,6 Гц, 1H), 7,76 (д, J=7,8 Гц, 1H), 7,55-7,51 (м, J=7,6 Гц, 1H), 7,11 (с, 1H), 7,05 (д, J=8,4 Гц, 1H), 2,45 (м, 1H), 2,38 (с, 3H), 1,80-1,69 (м, 5H), 1,42-1,21 (м, 5H).
Синтез N-(4-гидрокси-2-нафтил)-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 552)
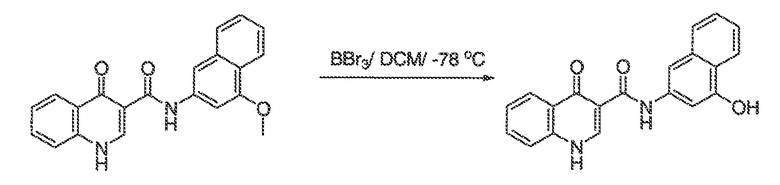
К раствору N-(4-метокси-2-нафтил)-4-оксо-1H-хинолин-3-карбоксамида (73 мг, 0,21 ммоль) в DCM (4 мл) добавляли BBr3 (1,1 мл, 11,64 ммоль) по каплям при -78ºC. После завершения добавления охлаждающую баню удаляли и полученную реакционную смесь нагревали до комнатной температуры и затем нагревали до 50ºC в течение 2 часов, охлаждали до -10ºC и гасили насыщенным раствором NaHCO3. Водный слой экстрагировали при помощи DCM и объединяли органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали обращенно-фазовой ВЭЖХ с получением N-(4-гидрокси-2-нафтил)-4-оксо-1H-хинолин-3-карбоксамида. LC/MS: m/z 331 (M+H)+ при 1,47 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез N-[5-гидрокси-4-изопропил-2-(трифторметил)фенил]-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 497)
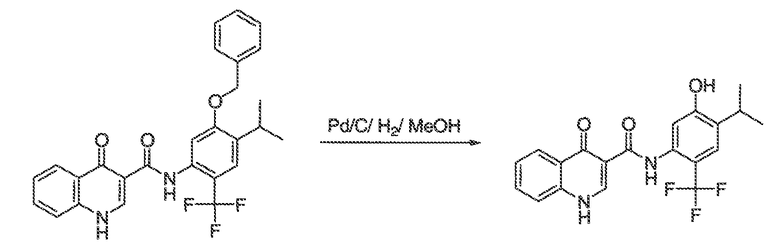
Колбу, содержащую N-[5-бензилокси-4-изопропил-2-(трифторметил)фенил]-4-оксо-1H-хинолин-3-карбоксамид (118 мг, 0,25 ммоль) и Pd/C (12 мг, 0,11 ммоль), разрежали в вакууме с последующей продувкой при помощи N2. Добавляли метанол (2 мл) в инертной атмосфере, с последующим удалением газа в вакууме. Реакционную смесь перемешивали в течение ночи в атмосфере водорода, фильтровали через слой Целита и концентрировали с получением N-[5-гидрокси-4-изопропил-2-(трифторметил)фенил]-4-оксо-1H-хинолин-3-карбоксамида. 1H ЯМР (400,0 МГц, DMSO-d6) δ 12,93 (с, 1H), 12,59 (с, 1H), 10,29 (с, 1H), 8,87 (с, 1H), 8,32 (дд, J=1,0, 8,1 Гц, 1H), 7,95 (с, 1H), 7,83-7,74 (м, 2H), 7,52 (т, J=8,0 Гц, 1H), 7,36 (с, 1H), 3,20 (квин., J=6,9 Гц, 1H) и 1,24-1,19 (м, 6H) м.д. LC/MS: m/z 391,36 (M+H)+ при 1,77 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез N-[4-циклогексил-5-гидрокси-2-(трифторметил)фенил]-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 620)
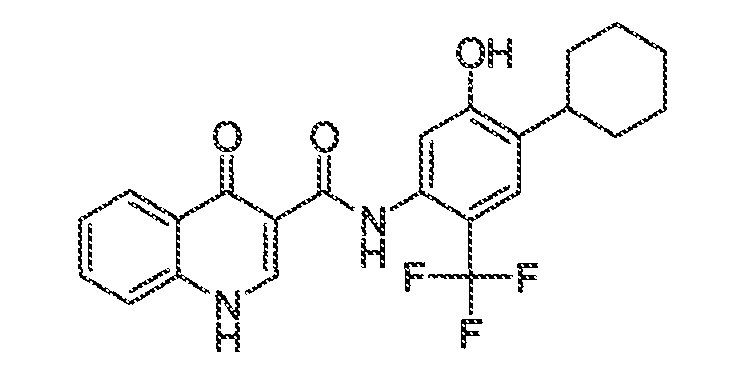
(N-(4-циклогексил-5-гидрокси-2-(трифторметил)фенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамид можно синтезировать в соответствии с общей схемой, представленной выше, исходя из N-(5-(бензилокси)-4-циклогексил-2-(трифторметил)фенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида. 1H ЯМР (400 МГц, DMSO-d6) δ 12,93 (д, J=6,6 Гц, 1H), 12,54 (с, 1H), 10,28 (с, 1H), 8,87 (д, J=6,8 Гц, 1H), 8,32 (д, J=7,3 Гц, 1H), 7,95 (с, 1H), 7,81 (ддд, J=23,1, 15,0, 4,7 Гц, 2H), 7,53 (дд, J=11,5, 4,6 Гц, 1H), 7,34 (с, 1H), 2,84 (с, 1H), 1,85-1,69 (м, 5H), 1,38 (т, J=10,3 Гц, 5H). LC/MS: m/z 431,5 (M+H)+ при 2,01 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез N-[2-фтор-5-гидрокси-4-(трифторметил)фенил]-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 501)
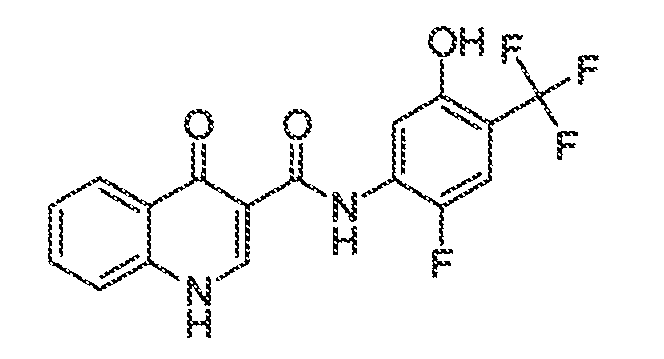
N-(2-фтор-5-гидрокси-4-(трифторметил)фенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамид можно синтезировать в соответствии с общей схемой, представленной выше, исходя из N-(5-(бензилокси)-2-фтор-4-(трифторметил)фенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида. LC/MS: m/z 367,10 (M+H)+ при 1,65 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез 6-[(4-оксо-1H-хинолин-3-ил)карбониламино]-1H-индол-4-карбоновой кислоты (Таблица 1, Соединение 712)
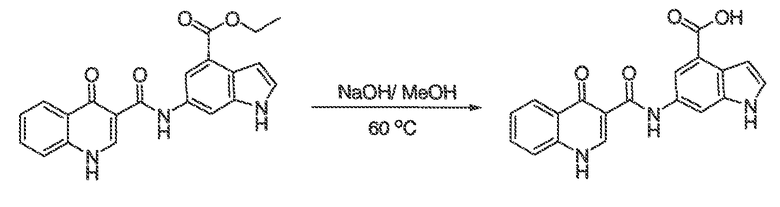
Этил 6-(4-оксо-1,4-дигидрохинолин-3-карбоксамидо)-1H-индол-4-карбоксилат (6 мг, 0,02 ммоль) суспендировали в 1 M NaOH (400 мкл, 0,40 ммоль) и нагревали до 50ºC в течение 30 минут. Прозрачный коричневый раствор разбавляли водой (1 мл) и подкисляли 1н раствором HCl (450 мкл). Раствор промывали водой (3×1 мл) и очищали обращенно-фазовой ВЭЖХ (75% ацетонитрила/вода) с получением 6-(4-оксо-1,4-дигидрохинолин-3-карбоксамидо)-1H-индол-4-карбоновой кислоты. LC/MS: m/z 347,8 (M+H)+ при 1,07 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA))
Синтез 5-амино-N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединени 586)
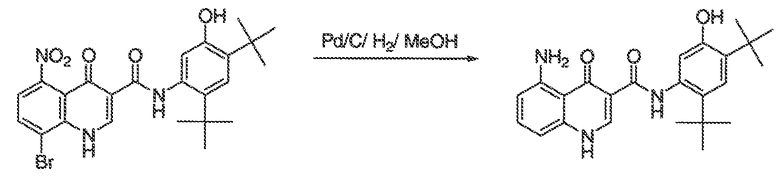
Колбу, содержащую 8-бром-N-(2,4-ди-трет-бутил-5-гидроксифенил)-5-нитро-4-оксо-1,4-дигидрохинолин-3-карбоксамид (430 мг, 0,83 ммоль) и Pd/C (60 мг, 0,56 ммоль), разрежали в вакууме, с последующей продувкой при помощи N2. Добавляли EtOAc (4 мл) и HCl (1 мл раствора 1 M, 1,000 ммоль) с последующим удалением газа в вакууме. Реакционную смесь перемешивали в течение ночи в атмосфере водорода, фильтровали через слой Целита и концентрировали с получением 5-амино-N-(2,4-дитрет-бутил-5-гидрокси-фенил)-4-оксо-1H-хинолин-3-карбоксамида (164 мг, 48%). LC/MS: m/z 408,5 (M+H)+ при 1,98 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез N-(5-гидрокси-2,4-дитрет-бутил-фенил)-5-метиламино-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединени 543)
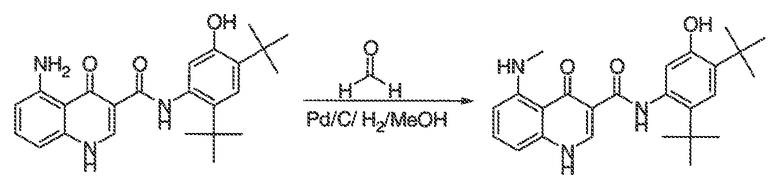
Колбу, содержащую 5-амино-N-(2,4-дитрет-бутил-5-гидрокси-фенил)-4-оксо-1H-хинолин-3-карбоксамид (23 мг, 0,06 ммоль), Pd/C (5 мг, 0,05 ммоль) и формальдегид (5 мкл 38% масс/об, 0,06 ммоль), разрежали в вакууме, с последующей продувкой при помощи N2. Добавляли метанол (1 мл) с последующим удалением газа в вакууме. Реакционную смесь перемешивали в течение ночи в атмосфере водорода, фильтровали через слой Целита и концентрировали с получением N-(2,4-дитрет-бутил-5-гидрокси-фенил)-5-метиламино-4-оксо-1H-хинолин-3-карбоксамида. LC/MS: m/z 422,5 (M+H)+ при 2,24 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез N-(2,4-ди-трет-бутил-5-гидрокси-фенил)-7-гидрокси-4-оксо-1H-хинолин-3-карбоксамид (Таблица 1, Соединение 653)

К раствору N-(2,4-ди-трет-бутил-5-гидрокси-фенил)-7-метокси-4-оксо-1H-хинолин-3-карбоксамида (120 мг, 0,28 ммоль) в DCM (1,5 мл) добавляли BBr3 (1,5 мл, 15,23 ммоль) по каплям при -78ºC. После завершения добавления охлаждающую баню удаляли и полученную реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакцию гасили насыщенным раствором NaHCO3. Водный слой экстрагировали при помощи DCM и объединенный органический слой сушили над MgSO4, фильтровали и концентрировали. Остаток очищали обращенно-фазовой ВЭЖХ с получением N-(2,4-дитрет-бутил-5-гидрокси-фенил)-7-гидрокси-4-оксо-1H-хинолин-3-карбоксамида . LC/MS: m/z 409,5 (M+H)+ при 1,83 мин (10%-99% CH 3 CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез 6-гидрокси-4-оксо-N-(5-трет-бутил-1H-индол-6-ил)-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 680)
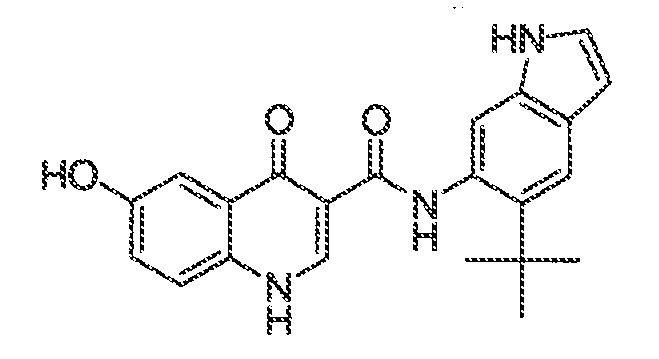
6-гидрокси-4-оксо-N-(5-трет-бутил-1H-индол-6-ил)-1H-хинолин-3-карбоксамид можно синтезировать в соответствии с общей схемой, представленной выше, исходя из N-(5-трет-бутил-1H-индол-6-ил)-6-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксамида. LC/MS: m/z 378,00 (M+H)+ при 1,38 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез 6-гидрокси-N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (Таблица 1, Соединение 703)
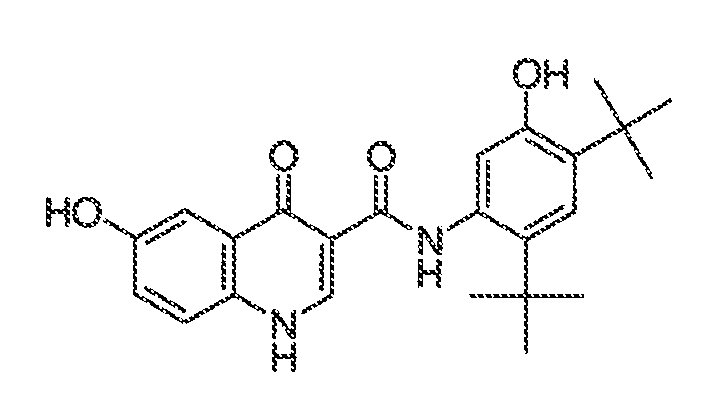
6-гидрокси-N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамид можно синтезировать в соответствии с общей схемой, представленной выше, исходя из N-(2,4-ди-трет-бутил-5-гидроксифенил)-6-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксамида. LC/MS: m/z 409,00 (M+H)+ при 1,73 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез (R)-1-[4-[(4-оксо-1H-хинолин-3-ил)карбониламино]-3-(трифторметил)фенил]пирролидин-2-карбоновой кислоты (Таблица 1, Соединение 699)
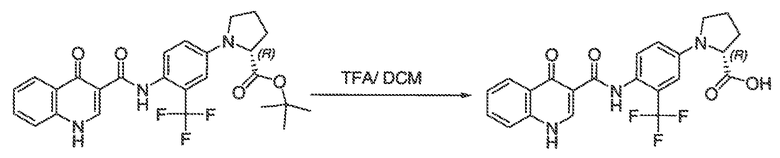
К раствору трет-бутил (2R)-1-[4-[(4-оксо-1H-хинолин-3-карбонил)амино]-3-(трифторметил)фенил]пирролидин-2-карбоксилата (35 мг, 0,06979 ммоль) в DCM (500 мкл) добавляли TFA (1 мл) при комнатной температуре и реакционную смесь перемешивали в течение 3 часов. Растворители выпаривали при пониженном давлении и остаток очищали обращенно-фазовой ВЭЖХ с получением (2R)-1-[4-[(4-оксо-1H-хинолин-3-карбонил)амино]-3-(трифторметил)фенил]пирролидин-2-карбоновой кислоты. LC/MS: m/z 466,3 (M+H)+ при 1,46 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
Синтез (S)-1-[4-[(4-оксо-1H-хинолин-3-ил)карбониламино]-3-(трифторметил)фенил]пирролидин-2-карбоновой кислоты (Таблица 1, Соединение 568)
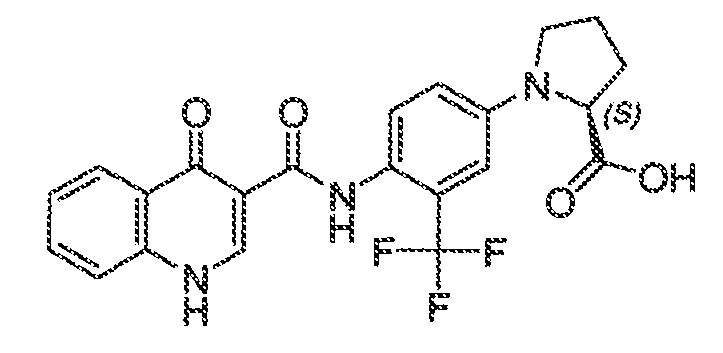
(S)-1-(4-(4-оксо-1,4-дигидрохинолин-3-карбоксамидо)-3-(трифторметил)фенил)пирролидин-2-карбоновую кислоту можно синтезировать в соответствии с общей схемой, представленной выше, исходя из трет-бутил (2S)-1-[4-[(4-оксо-1H-хинолин-3-карбонил)амино]-3-(трифторметил)фенил]пирролидин-2-карбоксилата. LC/MS: m/z 466,3 (M+H)+ при 1,47 мин (10%-99% CH3CN (0,035% TFA)/H2O (0,05% TFA)).
В таблице 2, приведенной в графической части, представлены данные, характеризующие соединения по настоящему изобретению, полученные в соответствии с приведенными выше Примерами .
Данные ЯМР для избранных соединений представлены в Таблице 2-А, приведенной в графической части.
B) Анализы для определения и измерения корректирующих свойств соединений в отношении ΔF508-CFTR
I) Оптические методы определения мембранного потенциала для анализа свойств соединений, направленных на модуляцию ΔF508-CFTR
Оптический анализ мембранного потенциала включает использование потенциал-чувствительных FRET сенсоров, описанных Gonzalez и Tsien (см. Gonzalez, J. E. and R. Y. Tsien (1995) "Voltage sensing by fluorescence resonance energy transfer in single cells." Biophys J 69(4): 1272-80 ; Gonzalez, J. E. and R. Y. Tsien (1997); "Improved indicators of cell membrane potential that use fluorescence resonance energy transfer" Chem Biol 4(4): 269-77), в сочетании со средствами измерения изменений флуоресценции, такими как Voltage/Ion Probe Reader (VIPR) (см. Gonzalez, J. E., K. Oades, et al. (1999) "Cell-based assays and instrumentation for screening ion-channel targets" Drug Discov Today 4(9): 431-439).
Эти потенциал-чувствительные анализы основаны на изменении передачи резонансной энергии флуоресценции (FRET) между мембрано-растворимым потенциал-чувствительным красителем, DiSBAC2(3) и флуоресцентным фосфолипидом, CC2-DMPE, который присоединен к внешней створке плазменной мембраны и действует в качестве FRET донора. Изменения мембранного потенциала (Vm) вызывают редистрибуцию отрицательно заряженного DiSBAC2(3) через плазменную мембрану, и количество передаваемой энергии от CC2-DMPE соответственно изменяется. Изменения эмиссии флуоресценции можно отслеживать с использованием VIPR™ II, который представляет собой интегрированное устройство, включающее подачу жидкости и детектор флуоресценции, предназначенное для осуществления клеточных скрининговых анализов в 96- или 384-луночных микротитровальных планшетах.
Идентификация корректирующих соединений
Для определения малых молекул, которые корректируют дефект траффика, связанный с ΔF508-CFTR, был разработан анализ в формате одного добавления HTS. Клетки инкубировали в бессывороточной среде в течение 16 часов при 37ºC в присутстви или в отсутствие (отрицательный контроль) испытываемого соединения. В качестве положительного контроля, клетки высевали в 384-луночные планшеты и инкубировали в течение 16 часов при 27ºC для “температурной коррекции” ΔF508-CFTR. Клетки затем промывали 3× раствором Кребса-Рингера и нагружали потенциал-чувствительными красителями. Для активации ΔF508-CFTR в каждую лунку добавляли 10 мкМ форсколина и CFTR-потенциирующего средства генистеина (20 мкМ) вместе с не содержащей Cl- средой. Добавление не содержащей Cl- среды промотировало отток Cl- в ответ на активацию ΔF508-CFTR, происходящую в результате этого деполяризацию мембраны оптически отслеживали с использованием потенциал-чувствительных красителей на основе FRET.
Идентификация потенциирующующих соединений
Для определения потенциаторов ΔF508-CFTR, был разработан анализ в формате двойного добавления HTS. При первом добавлении не содержащую Cl- среду, с или без испытываемого соединения, добавляли в каждую лунку. Через 22 сек осуществляли второе добавление не содержащей Cl- среды, которая содержала 2-10 мкМ форсколина, для активации ΔF508-CFTR. Внеклеточная Cl- концентрация после обоих добавлений была 28 мМ, что способствовало оттоку Cl- в ответ на активацию ΔF508-CFTR, и происходящую в результате этого деполяризацию мембраны оптически отслеживали с использованием потенциал-чувствительных красителей на основе FRET.
Растворы
ванночке
#1: (в мМ)
ванночке
без хлоридов:
Клеточная культура
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, использовали для оптических измерений мембранного потенциала. Клетки поддерживали при 37ºC в 5% CO2 и 90% влажности в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1× NEAA, β-ME, 1× пенициллина/стрептомицина и 25 мМ HEPES в 175 см2 колбах для культивирования. Для всех оптических анализов клетки высевали при плотности 30000/лунка в 384-луночных матригель-покрытых планшетах и культивировали в течение 2 часов при 37ºC, затем культивировали при 27ºC в течение 24 часов для анализа потенциирующего средства. Для корректирующих анализов, клетки культивировали при 27ºC или 37ºC с и без соединения в течение 16-24 часов.
Электрофизиологические анализы для определения ΔF508-CFTR-модулирующих свойств соединений.
1. Анализ с использованием камеры Ussing
Эксперименты с использованием камеры Ussing осуществляли на поляризованных эпителиальных клетках дыхательных путей, экспрессирующих ΔF508-CFTR, для дальнейшей характеризации модуляторов ΔF508-CFTR, идентифицированных в оптических анализах. Эпителиальные клетки FRT?F5O8-CFTR выращенные на включениях клеточной культуры Costar Snapwell, помещали в камеру Ussing (Physiologic Instruments, Inc., San Diego, CA) и постоянно осуществляли короткое замыкание в монослоях с использованием системы фиксации потенциала (Department of Bioengineering, University of Iowa, IA, и Physiologic Instruments, Inc., San Diego, CA). Трансэпителиальное сопротивление измеряли путем приложения 2-мВ импульса. В этих условиях FRT эпителии показали сопротивление 4КΩ/см2 или больше. Растворы поддерживали при 27ОС и продували воздухом. Напряжение смещения на электроде и сопротивление жидкости были скорректированы с использованием бесклеточного включения. В этих условиях ток отражает поток Cl- через ΔF508-CFTR, экспрессируемые в апикальной мембране. Данные ISC получали в цифровом формате с использованием МР100А-СЕ интерфейса и программы AcqKnowledge (ν3.2.6; BIOPAC Systems, Santa Barbara, CA).
Идентификация корректирующих соединений
Типичный протокол включал использование градиента концентраци Cl- от базолатеральной к апикальной мембране. Для установления этого градиента, на базолатеральной мембране использовали нормальные растворы Рингера, тогда как на апикальной NaCl заменяли эквимолярным количеством глюконата натрия (титровали до pH 7,4 при помощи NaOH) с получением выского градиента концентрации Cl- через эпителий. Все эксперименты осуществляли с интактным монослоем. Добавляли полностью активированный ΔF508-CFTR, форсколин (10 мкМ) и ингибитор PDE, IBMX (100 мкМ), с последующим добавлением потенциатора CFTR-генистеина (50 мкМ).
Как наблюдали в других типах клеток, инкубация при низких температурах FRT клеток, стабильно экспрессирующих ΔF508-CFTR, увеличивает функциональную плотность CFTR в плазменной мембране. Для определения активности корректирующих соединений клетки инкубировали с 10 мкМ испытываемого соединения в течение 24 часов при 37ºC и затем промывали 3× до осуществления регистрации. cAMP- и генистеин-опосредованный ISC в обработанных соединением клетках нормализовали к 27ºC и 37ºC контролям и выражали в виде процента активности. Предварительная инкубация клеток с корректирующим соединением существенно увеличивала cAMP- и генистеин-опосредованный ISC по сравнению с 37ºC контролями.
Идентификация потенцирующих соединений
Типичный протокол включал использование градиента концентраци Cl- от базолатеральной к апикальной мембране. Для установления этого градиента, на базолатеральной мембране использовали нормальный раствор Рингера и для проницаемости мембраны использовании нистатин (360 мкг/мл), тогда как на апикальной NaCl заменяли эквимолярным количеством глюконата натрия (титровали до pH 7,4 при помощи NaOH) с получением выского градиента концентрации Cl- через эпителий. Все эксперименты осуществляли через 30 минут после обработки нистатином для проницаемости мембраны. Форсколин (10 мкМ) и все испытываемые соединения добавляли на обеих сторонах включений клеточной культуры. Эффективность предполагаемых средств, потенцирующих ΔF508-CFTR, сравнивали с известным потенциирующим средством генистеином.
Растворы
Клеточная культура
Эпителиальные клетки крыс Fisher (FRT), экспрессирующие ΔF508-CFTR (FRT?F5O8-CFTR), использовали для экспериментов с использованием камеры для предполагаемых модуляторов ΔF508-CFTR, определенных в оптических анализах. Клетки культивировали на включениях клеточной культуры Costar Snapwell и культивировали в течение пяти дней при 37ºC и 5% CO2 в модифицированной Куном среде Хэма F-12, среде дополненной 5% фетальной телячьей сыворотки, 100 Ед./мл пенициллина и 100 мкг/мл стрептомицина. Перед их использованием для характеристики потенциирующей активности соединений, клетки инкубировали при 27ºC в течение 16-48 часов для коррекции на ΔF508-CFTR. Для определения активности корректирующих соединений, клетки инкубировали при 27ºC или 37ºC с и без соединений в течение 24 часов.
2. Регистрация в формате “целая клетка”
Макроскопический ΔF508-CFTR ток (IΔF508) в температура- и испытываемое соединение-скорректированных NIH3T3 клетках, стабильно экспрессирующих ΔF508-CFTR, отслеживали с использованием перфорированный-пэтч метода в формате “целая клетка”. Вкратце, вольт-кламп регистрации IΔF508 осуществляли при комнатной температуре с использованием Axopatch 200B пэтч-кламп усилителя (Axon Instruments Inc., Foster City, CA). Все регистрации осуществляли при частоте 10 кГц и с использованием низкочастотного фильтра при 1 кГц. Пипетки имели сопротивление 5-6 MΩ, когда были заполнены внутриклеточным раствором. В этих условиях регистрации, рассчитанный потенциал реверсии для Cl- (ECl) при комнатной температуре был -28 мВ. Все регистрации имели изолирующее сопротивление >20GΩ и последовательное сопротивление <15 GΩ. Генерацию импульсов, сбор данных и анализ осуществляли с использованием персонального компьютера, снабженного Digidata 1320 A/D интерфейсом вместе с Clampex 8 (Axon Instruments Inc.). Ванночка содержала <250 мкл физиологического солевого раствора, и осуществляли непрерывное перфундирование при скорости 2 мл/мин с использованием гравитационно-управляемой перфузионной системы.
Идентификация корректирующих соединений
Для определения активности корректирующих соединений для увеличения плотности ΔF508-CFTR в плазменной мембране, использовали описанные выше медоты перфорированный-пэтч-регистрации для измерения плотности тока после 24-часовой обработки корректирующими соединениями. Полностью активированный ΔF508-CFTR, 10 мкМ форсколина и 20 мкМ генистеина добавляли к клеткам. В этих условиях регистрации, плотность тока после 24-часовой инкубации при 27ºC была выше, чем плотность тока, наблюдаемая после 24-часовой инкубации при 37ºC. Эти результаты согласуются с известными эффектами низкотемпературной инкубации на плотность ΔF508-CFTR в плазменной мембране. Для определения эффектов корректирующих соединений на CFTR плотность тока, клетки инкубировали с 10 мкМ испытываемого соединения в течение 24 часов при 37ºC и плотность тока сравнивали с 27ºC и 37ºC контролями (% активности). Перед осуществлением регистрации клетки промывали 3× внеклеточной средой для регистрации для удаления остатков испытываемого соединения. Предварительная инкубация с 10 мкМ корректирующих соединений существенно повышала cAMP- и генистеин-зависимый ток по сравнению с 37ºC контролями.
Идентификация потенциирующих соединений
Способность ΔF508-CFTR-потенциирующих соединений повышать макроскопический ΔF508-CFTR ток (IΔF508) в NIH3T3 клетках, стабильно экспрессирующих ΔF508-CFTR, также исследовали с использованием перфорированный-пэтч метода. Потенциирующие средства, идентифицированные в оптических анализах, вызывали дозо-зависимое увеличение IΔF508 с такой же активностью и эффективностью, котрые наблюдали в оптических анализах. Во всех исследуемых клетках, потенциал реверсии до и в процессе применения потенциирующего средства был около -30 мВ, что представляет собой рассчитанное значение ECl (-28 мВ).
Растворы
Клеточная культура
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, использовали для регистрации в формате “целая клетка”. Клетки поддерживали при 37ºC в 5% CO2 и 90% влажности в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1× NEAA, β-ME, 1× пенициллина/стрептомицина и 25 мМ HEPES в 175 см2 колбах для культивирования. Для регистрации в формате “целая клетка” 2500-5000 клеток высевали на покрытые поли-L-лизином покровные стекла и культивировали в течение 24-48 часов при 27ºC перед использованием для испытания активности потенциирующих средств; и инкубировали с или без корректирующего соединения при 37ºC для измерения активности корректирующих соединений.
3. Регистрация одиночных каналов
Активности одиночных каналов температура-скорректированного ΔF508-CFTR, экспрессируемого в NIH3T3 клетках, наблюдали с использованием изолированных участков(пэтчи) мембран в конфигурации “внутренняя сторона наружу”. Вкратце, вольт-кламп регистрации активности одиночных каналов осуществляли при комнатной температуре с использованием Axopatch 200B пэтч-кламп усилителя (Axon Instruments Inc.). Все регистрации осуществляли при частоте 10 кГц и с использованием низкочастотного фильтра при 400 Гц. Пэтч-пипетки были изготовлены из стекла Corning Kovar Sealing #7052 (World Precision Instruments, Inc., Sarasota, FL) и имели сопротивление 5-8 МΩ, когда были заполнены внеклеточным раствором. ΔF508-CFTR был активирован после эксцизии, путем добавления 1 мМ Mg-АТФ и 75 нМ cAMP-зависимой протеинкиназы, каталитической субъединицы (PKA; Promega Corp. Madison, WI). После стабилизации активности канала, пэтч перфундировали с использованием гравитационно-управляемой микроперфузионной системы. Поступающий раствор был в непосредственной близости к пэтчу, что обеспечивало полный обмен растворов в течение 1-2 секунд. Для поддержания ΔF508-CFTR активности в процессе быстрой перфузии, неспецифический ингибитор фосфатазы F- (10 мМ NaF) добавляли к раствору в ванночке. В этих условиях регистрации, активность канала оставалась постоянной на протяжении всей пэтч-регистрации (до 60 мин). Токи, образуемые положительным зарядом, перемещающимся от внутри- к внеклеточным растворам (анионы перемещаются в обратном направлении) представлены как положительные токи. Потенциал на пипетке (Vp) поддерживали при 80 мВ.
Активность каналов анализировали из мембранных пэтчей, содержащих ≤2 активных канала. Максимальное количество одновременных открытий определяло количество активных каналов в процессе эксперимента. Для определения амплитуды тока одиночного канала, данные, полученные из 120 сек активности ΔF508-CFTR, фильтровали “off-line” при 100 Гц и затем использовали для построения гистограмм амплитуды с использованием всех точек, которые подгоняли с использованием мультигауссовых функций и с использованием программы Bio-Patch Analysis (Bio-Logic Comp. France). Общий микроскопический ток и возможность открытия (Po) определяли из 120 сек активности канала. Po определяли с использованием программы Bio-Patch или из отношения Po = I/i(N), где I = средний ток, i = амплитуда тока одиночного канала и N = количество активных каналов в пэтче.
Растворы
Клеточная культура
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, использовали для пэтч-кламп регистрации с использованием изолированных участков. Клетки поддерживали при 37ºC в 5% CO2 и 90% влажности в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1× NEAA, β-ME, 1× пенициллина/стрептомицина и 25 мМ HEPES в 175 см2 колбах для культивирования. Для регистрации одиночных каналов 2500-5000 клеток высевали на покрытые поли-L-лизином покровные стекла и культивировали в течение 24-48 часов при 27ºC перед использованием.
Соединения по настоящему изобретению являются полезными в качестве модуляторов АТФ-связывающих кассетных транспортеров. Таблица 3 ниже иллюстрирует EC50 и относительную эффективность некоторых вариантов воплощения, представленных в Таблице 1.
В Таблице 3, приведенной в графической части, используемые символы имеют следующие значения:
EC50: “+++” означает <10 мкМ; “++” означает от 10 мкМ до 25 мкМ; “+” означает от 25 мкМ до 60 мкМ.
% Эффективности: “+” означает <25%; “++” означает от 25% до 100%; “+++” означает >100%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2382779C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| МОДУЛЯТОРЫ ЯДЕРНЫХ РЕЦЕПТОРОВ | 2016 |
|
RU2715897C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА МИНЕРАЛОКОРТИКОИДОВ | 2012 |
|
RU2598842C2 |
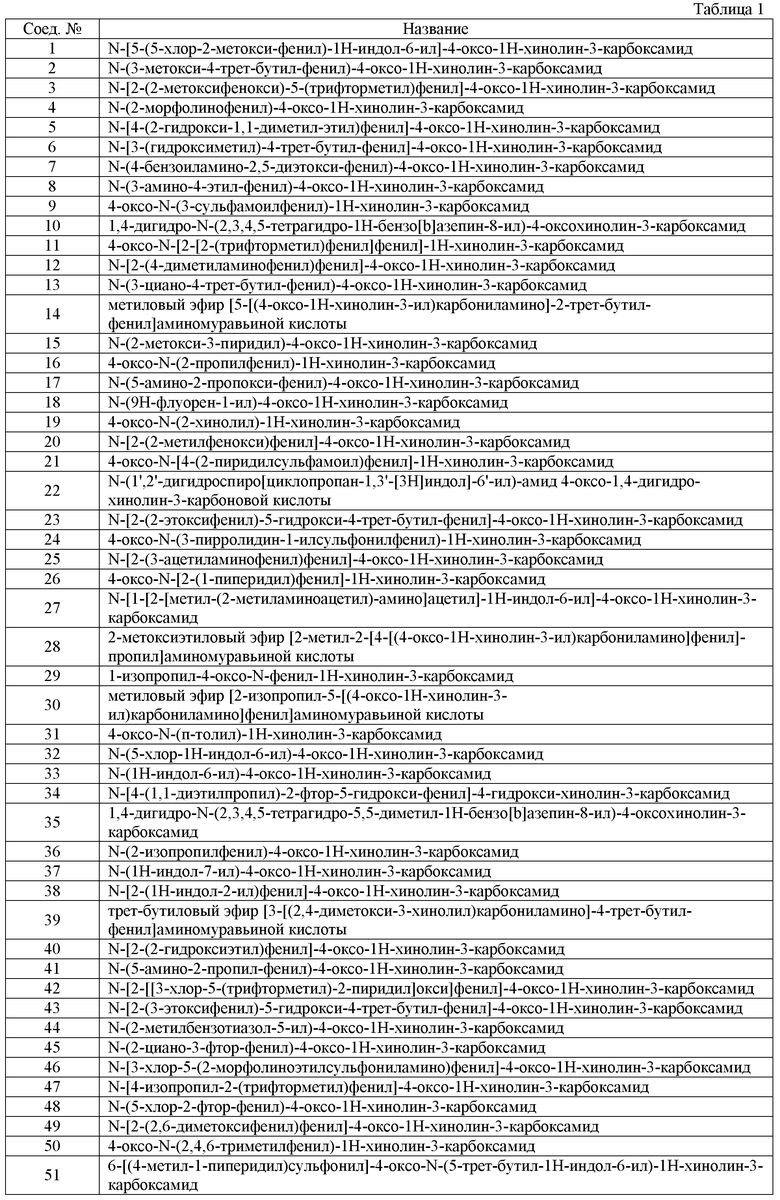




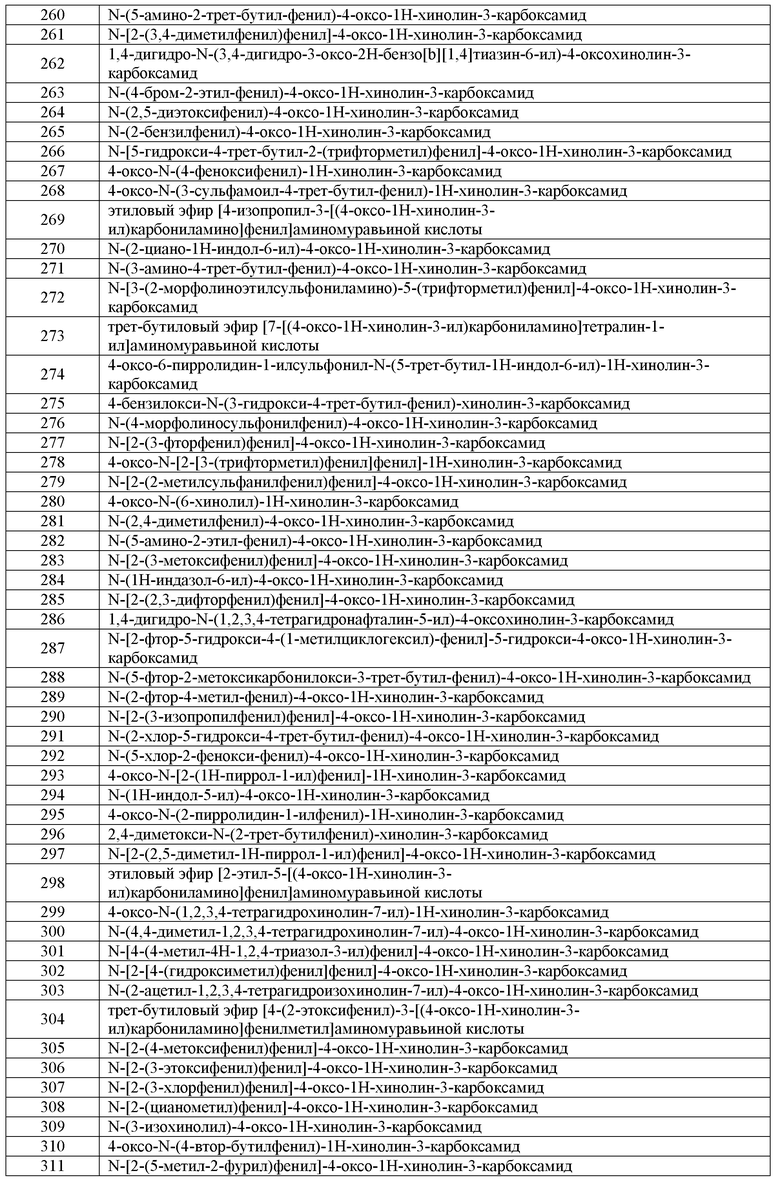
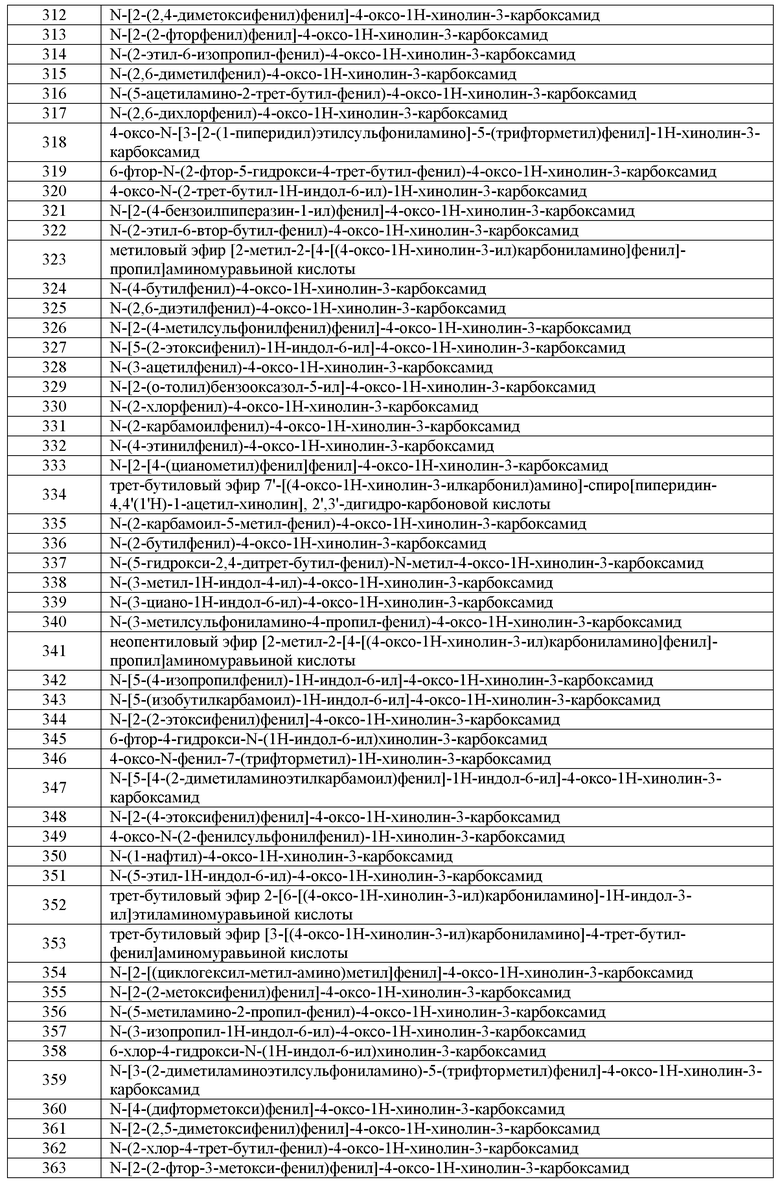
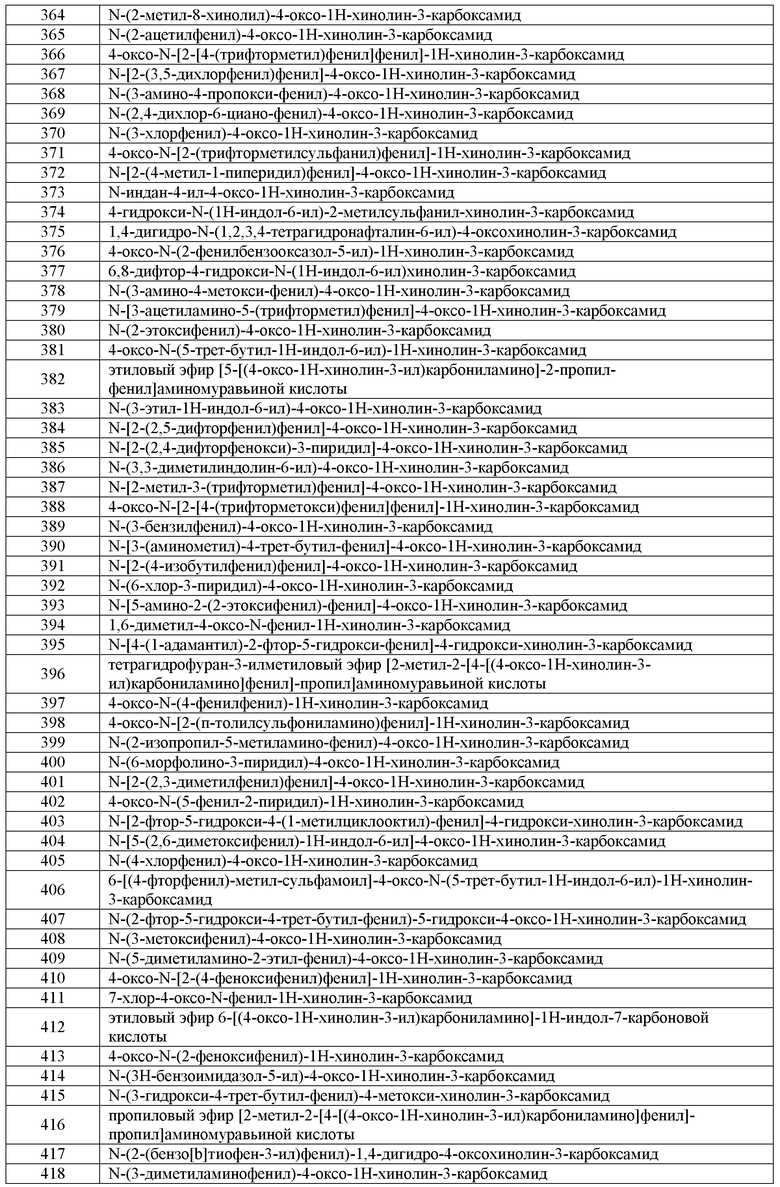

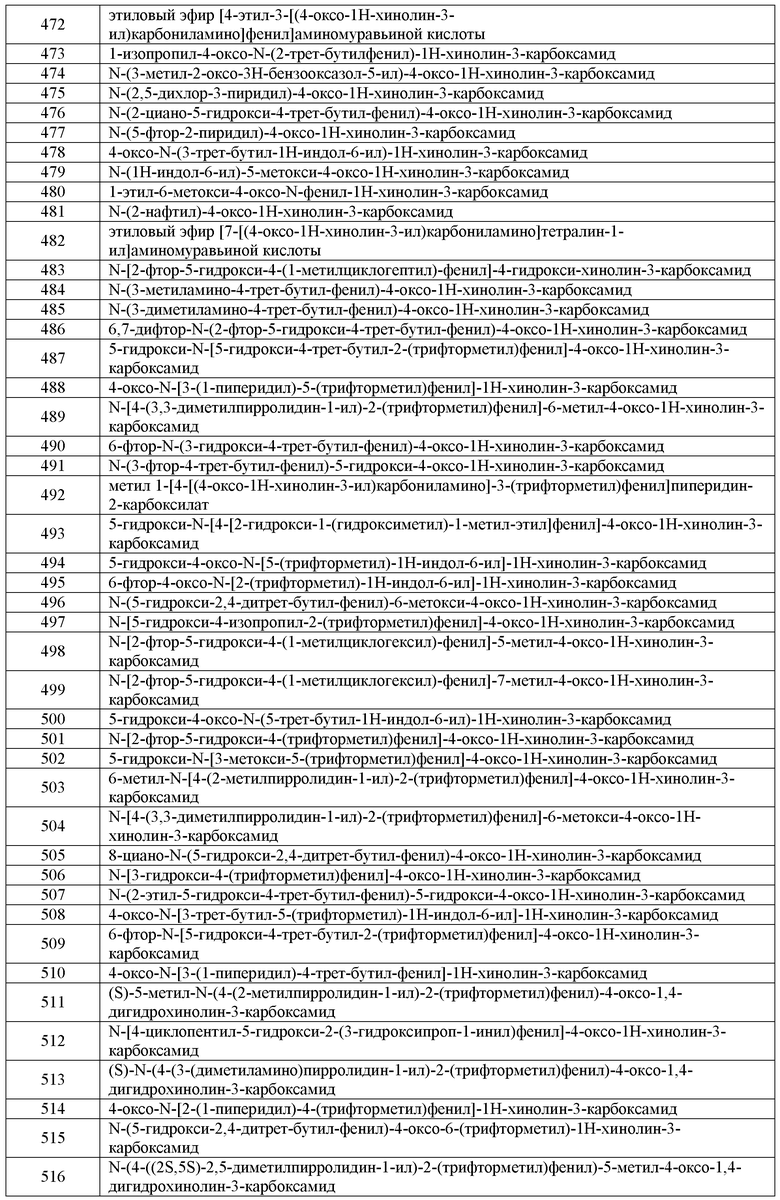





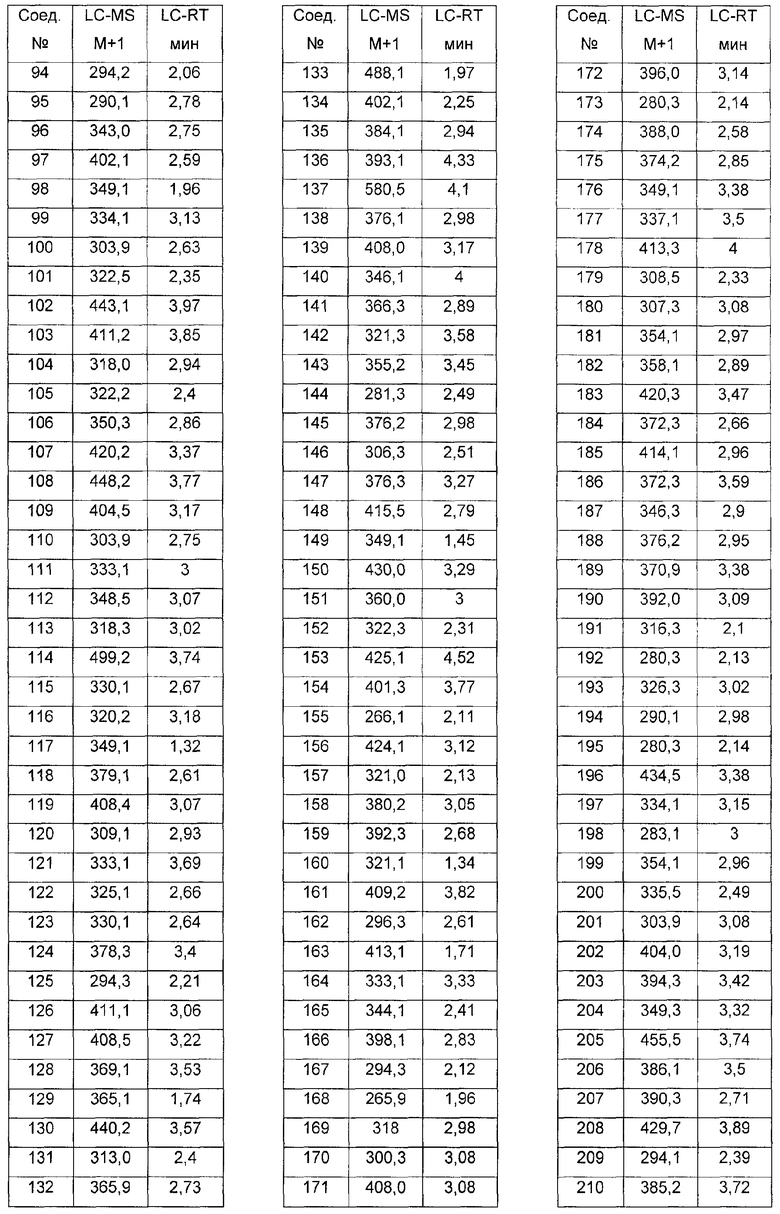
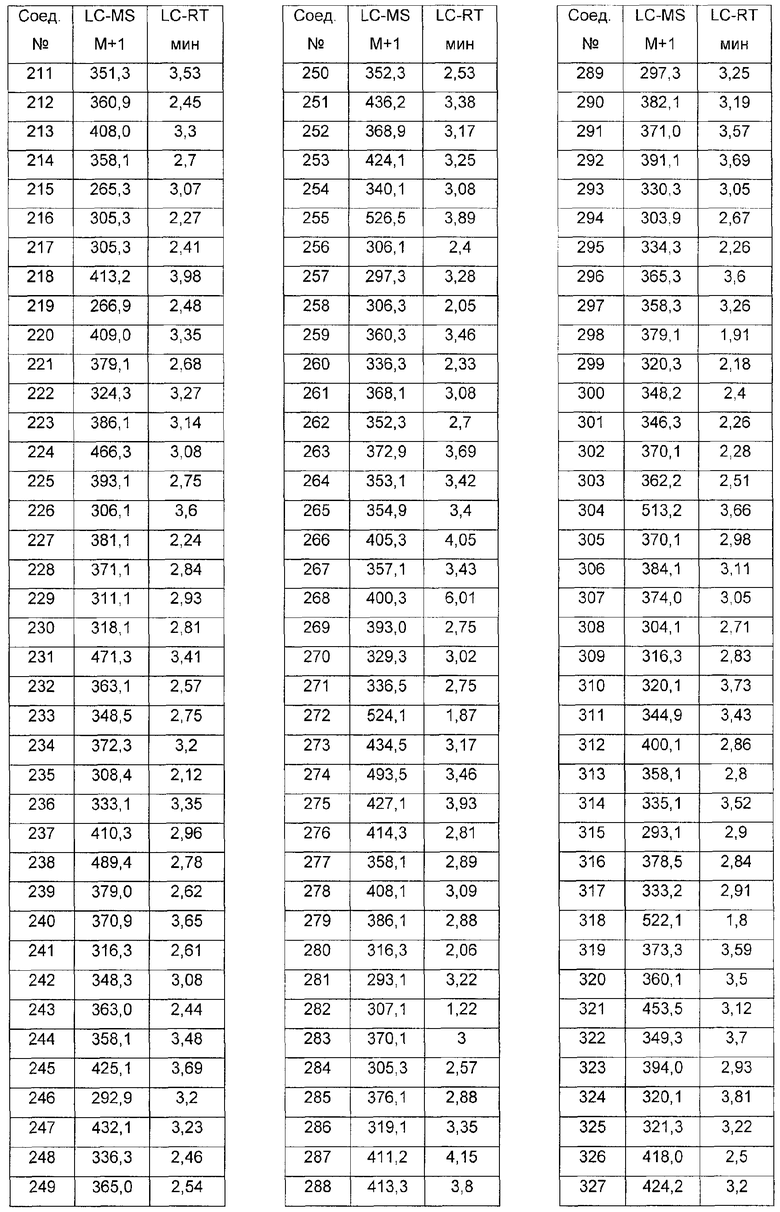
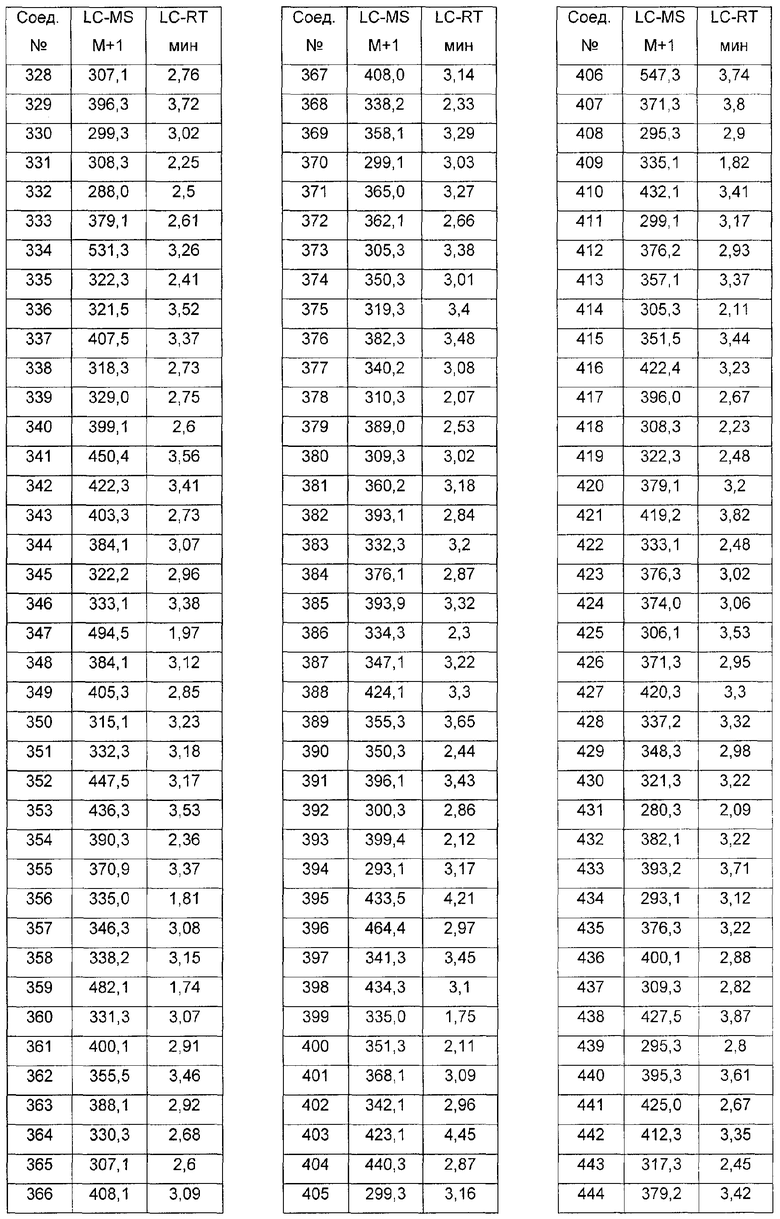
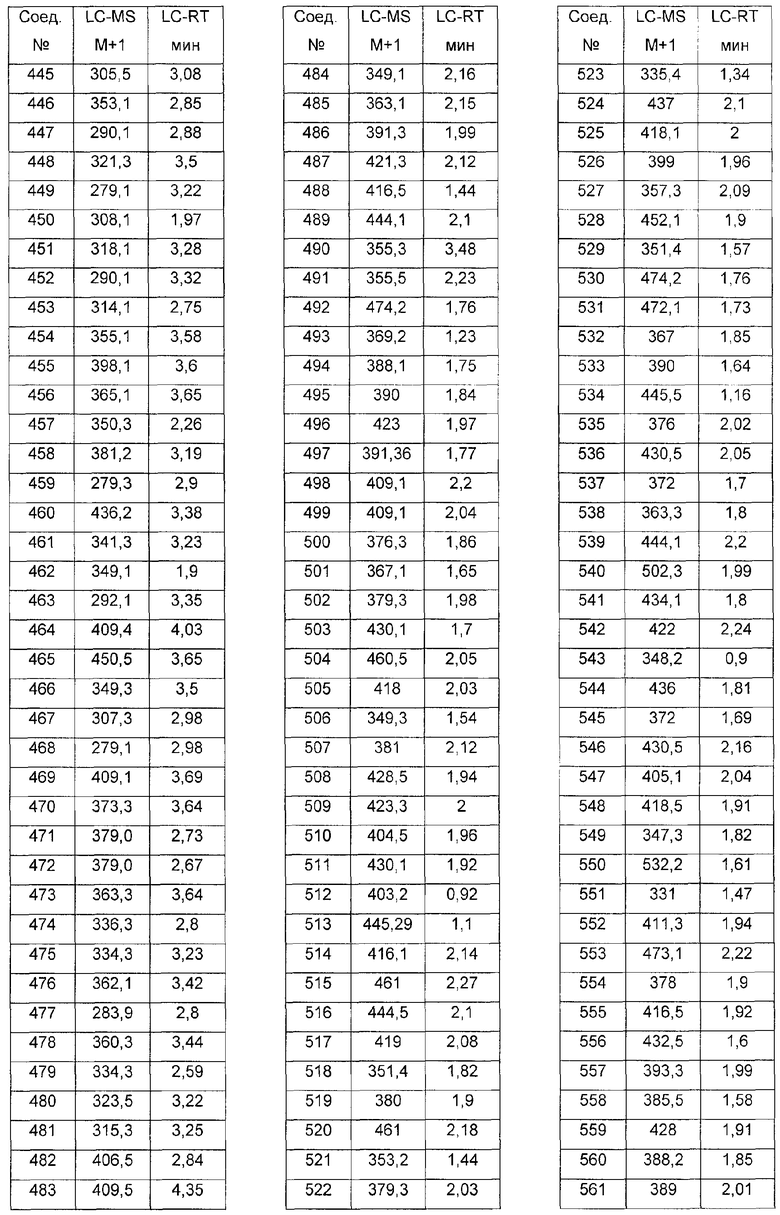
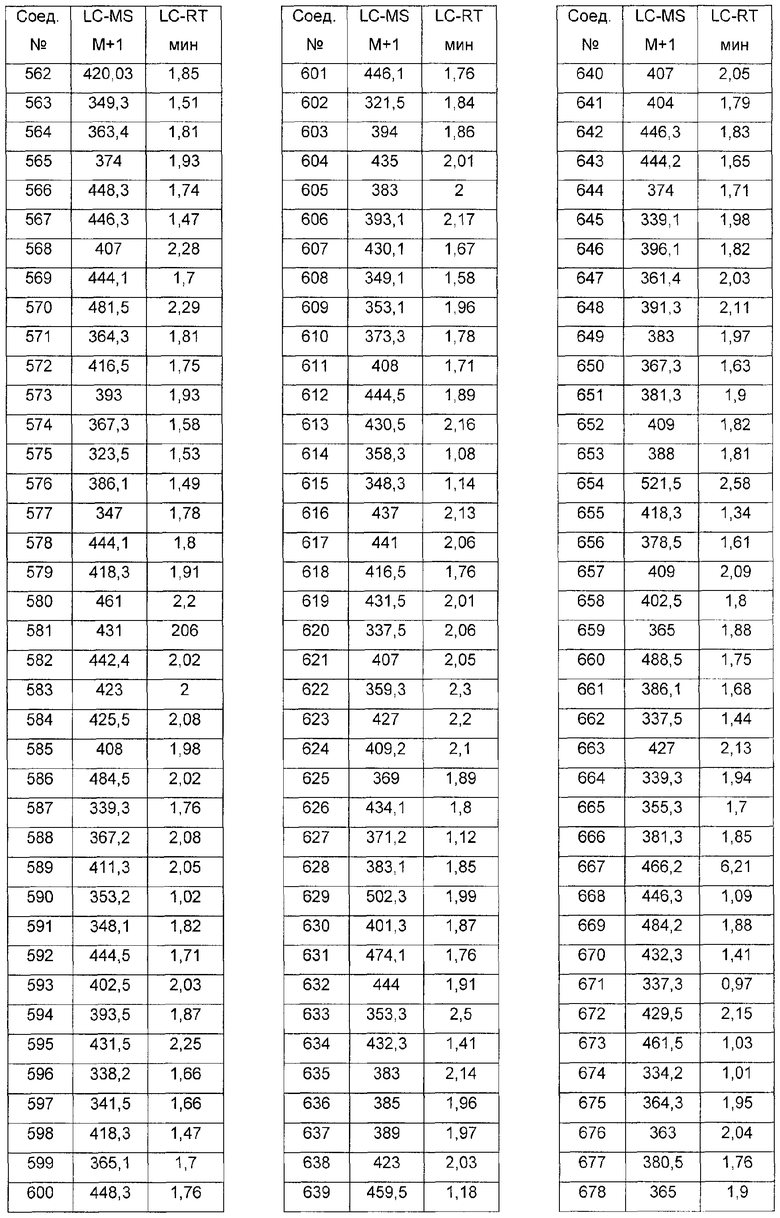
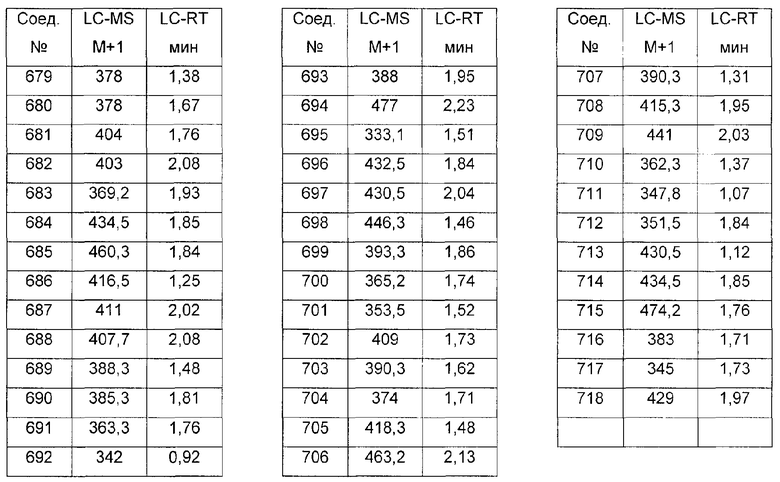
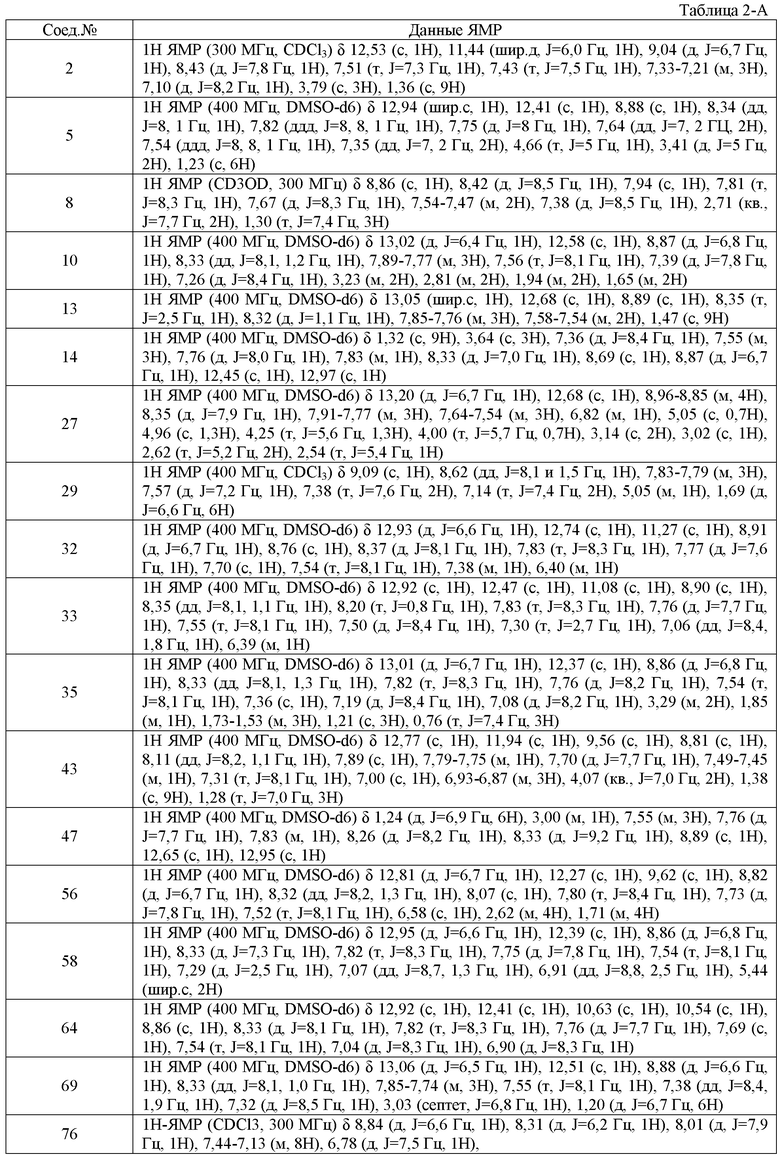
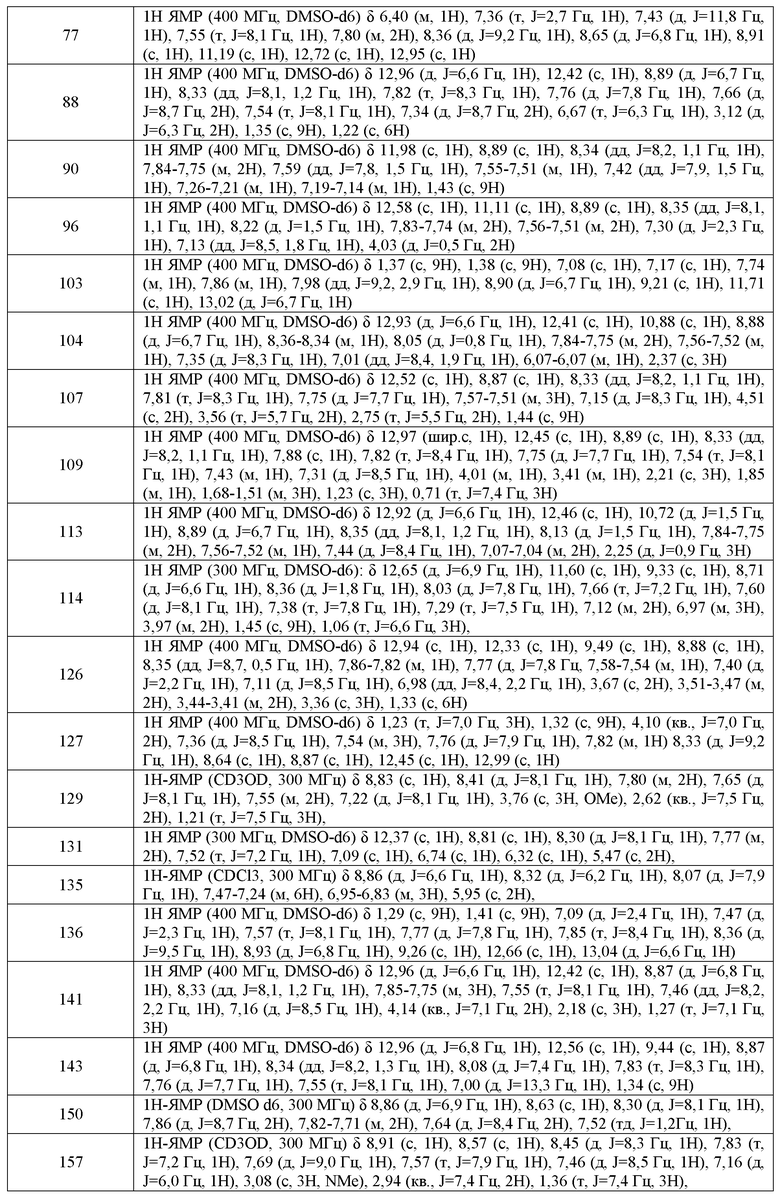
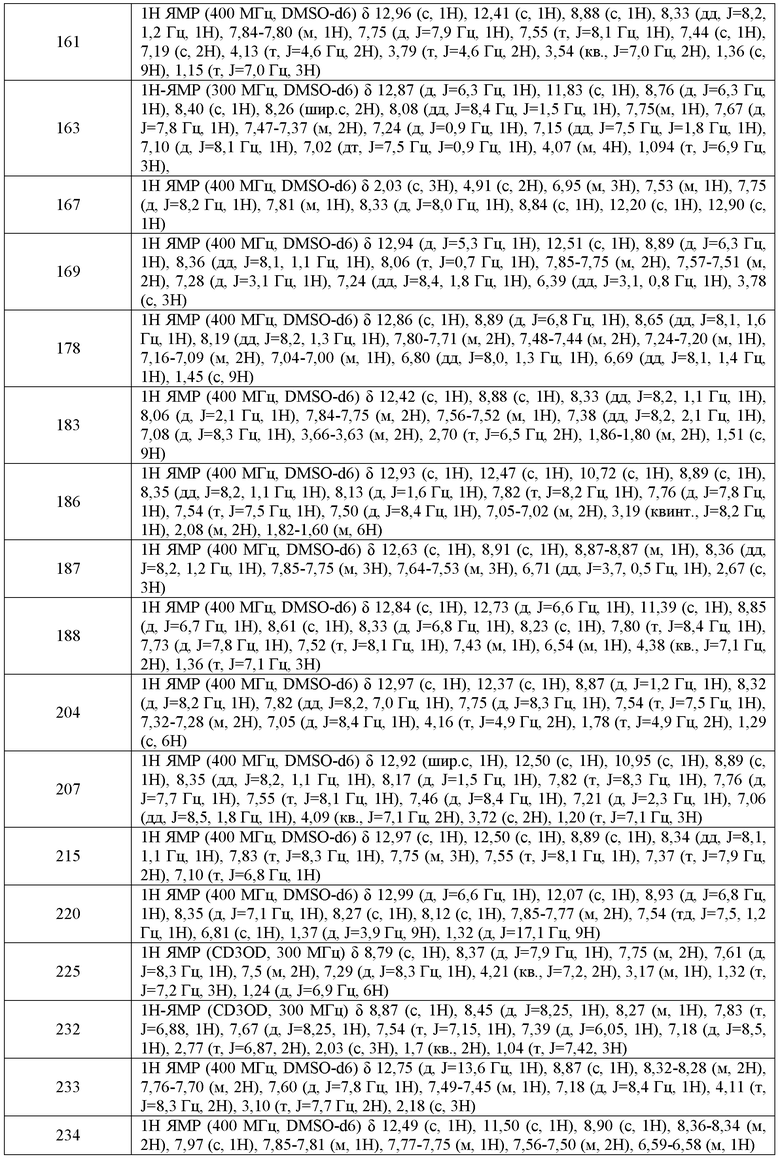
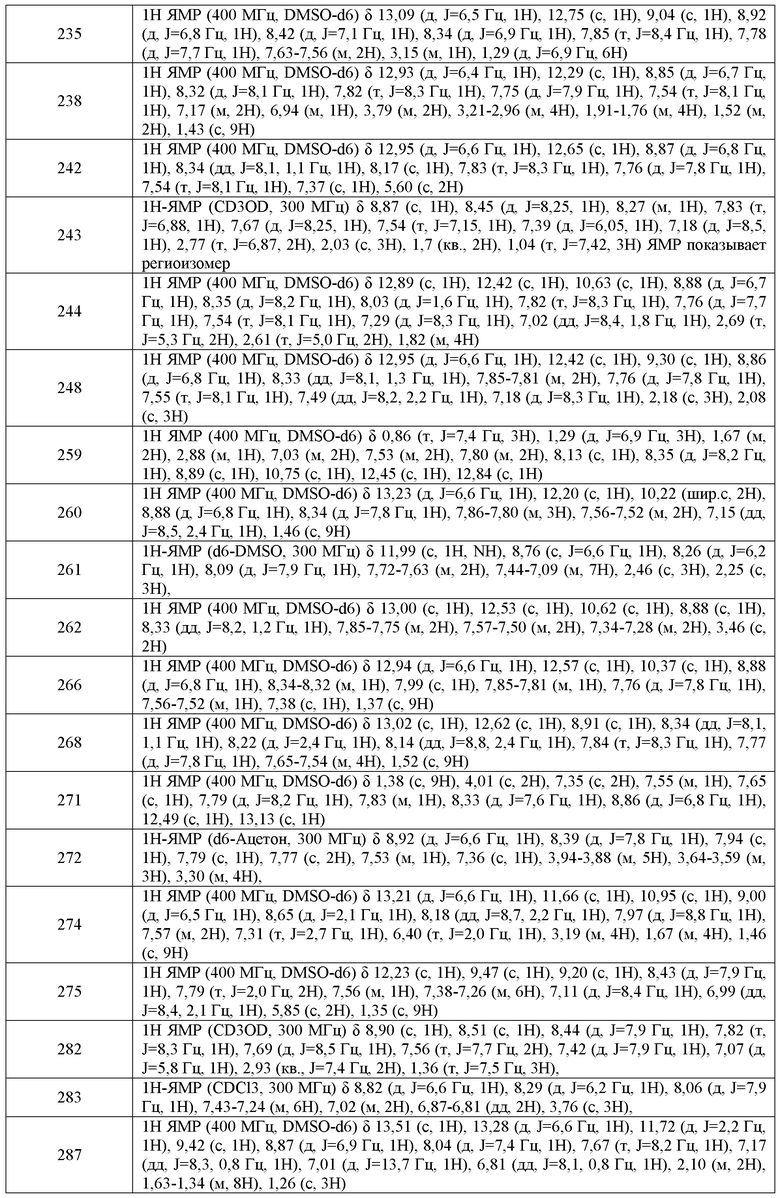
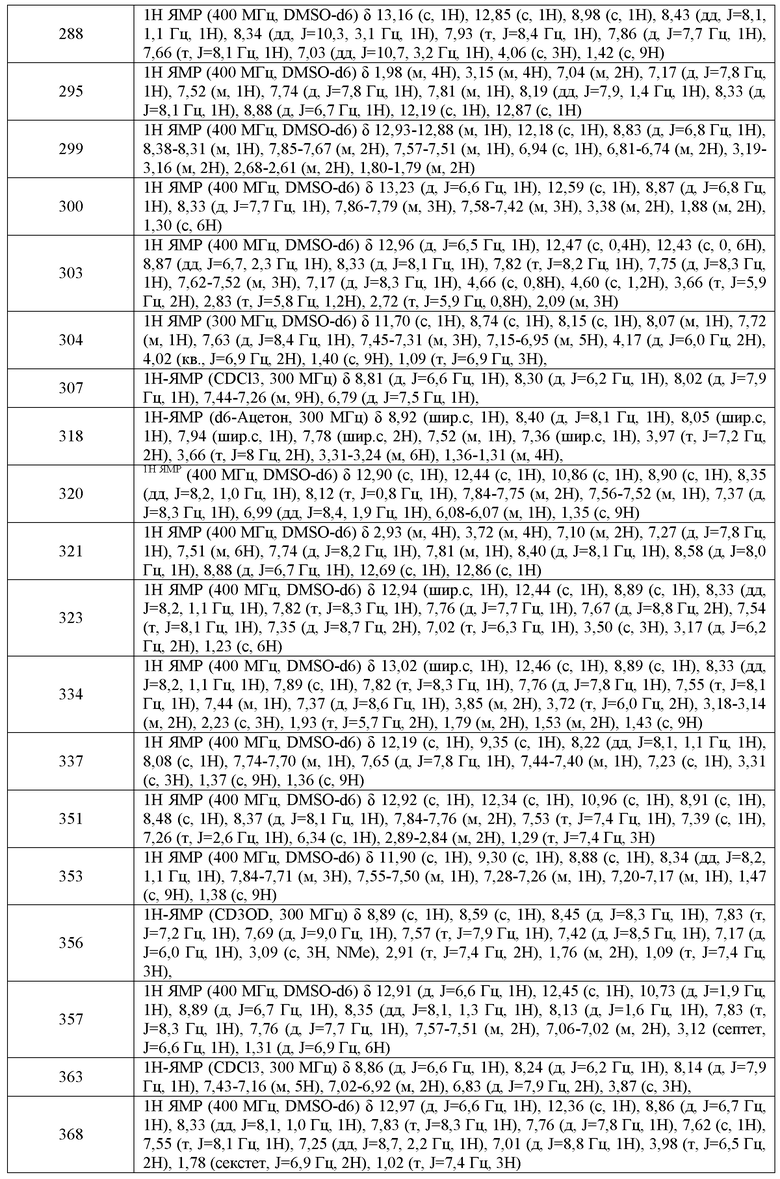

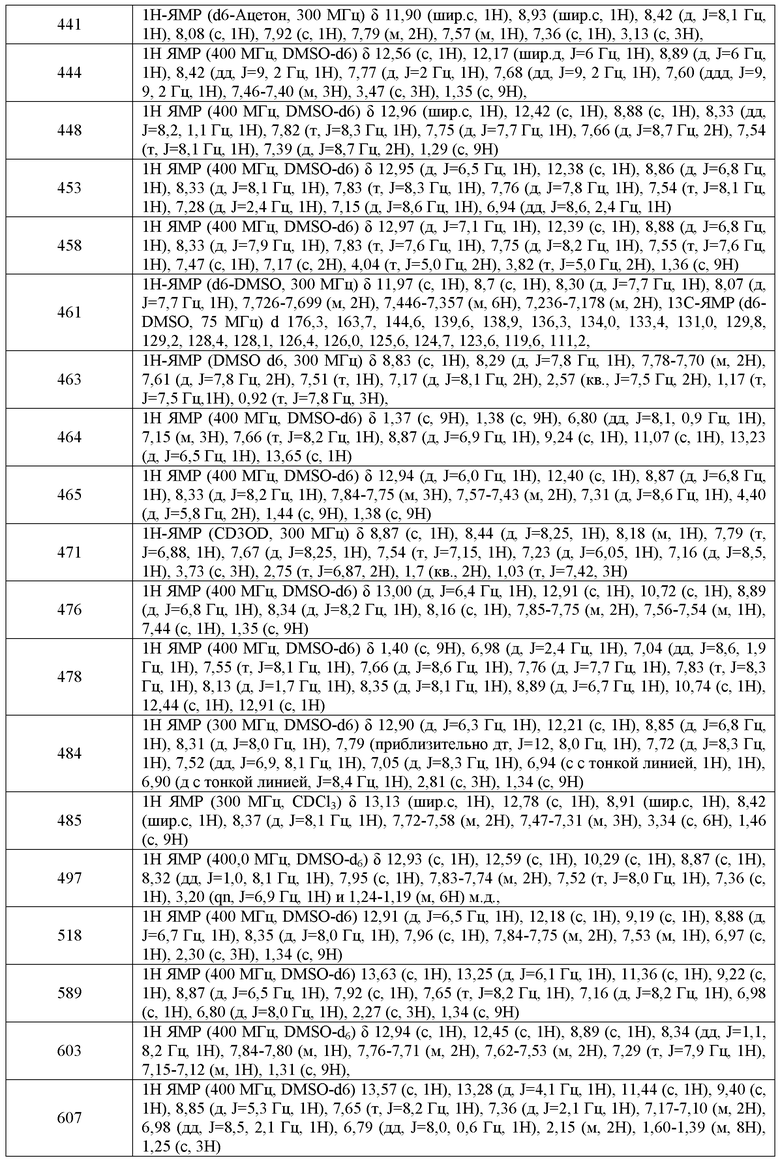

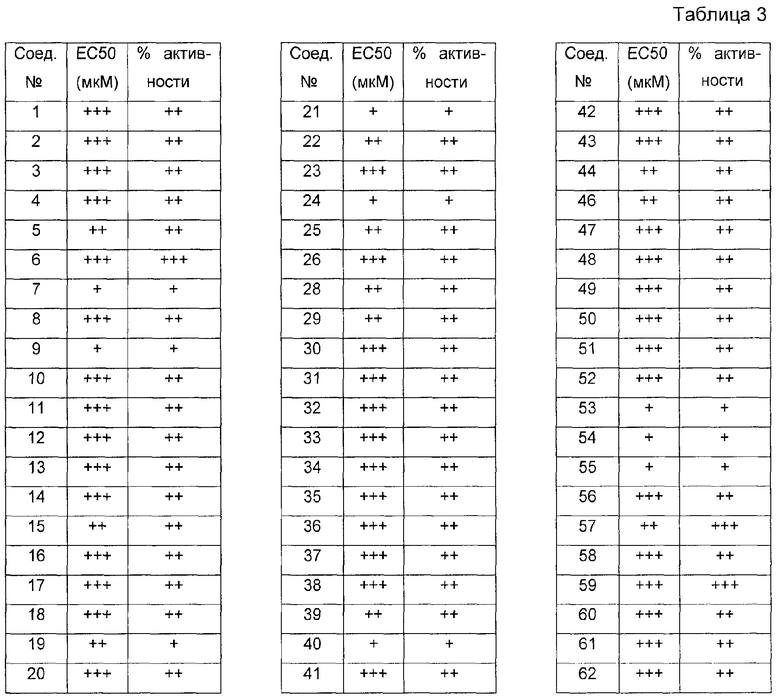
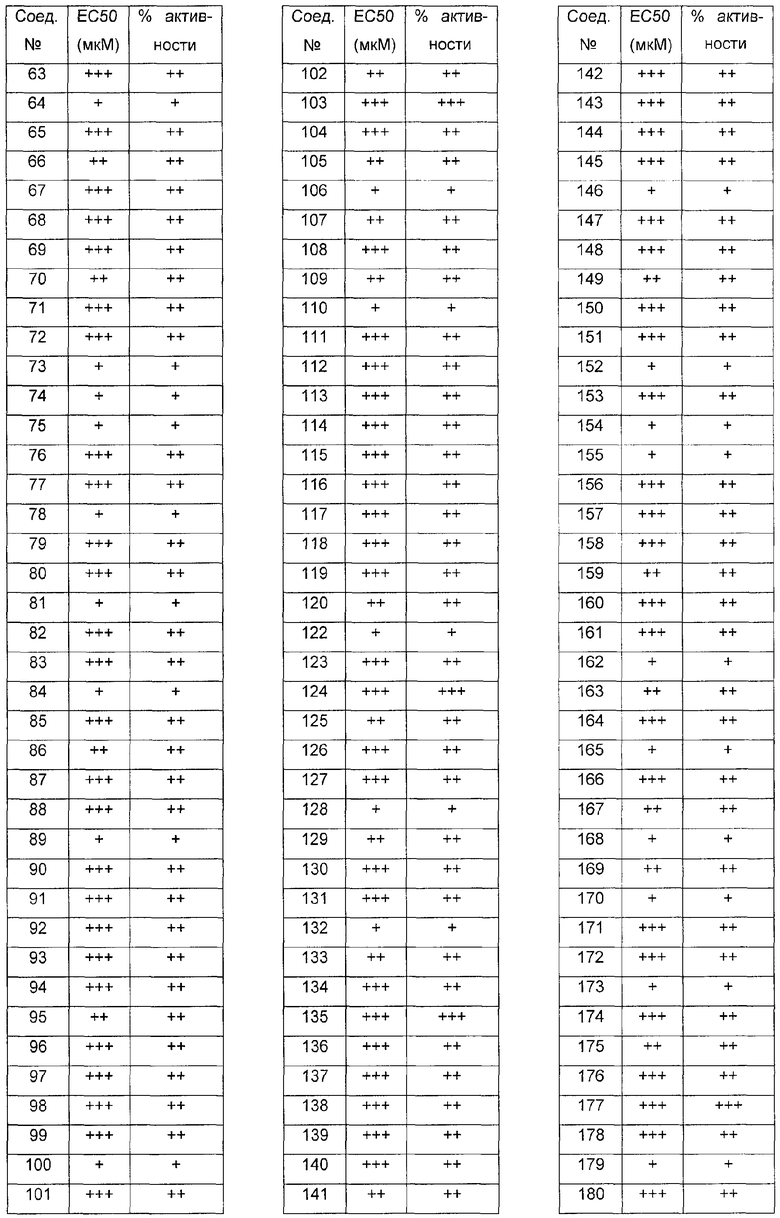
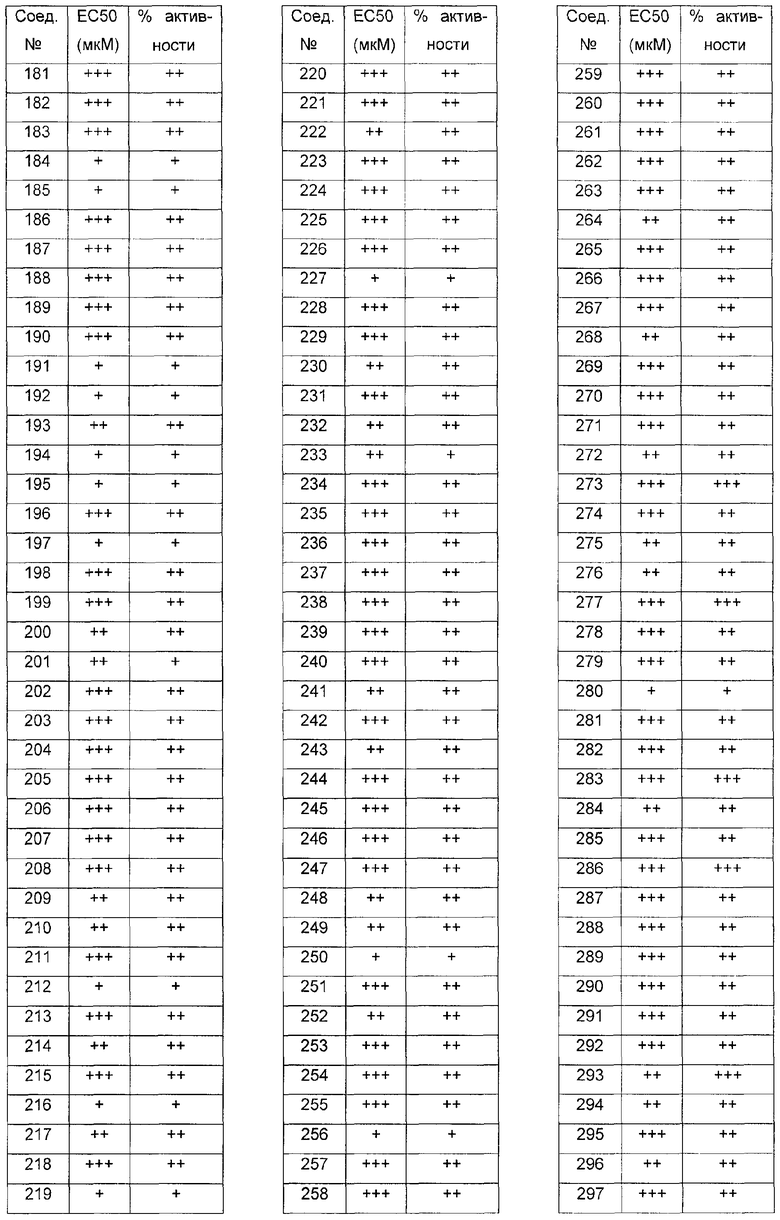
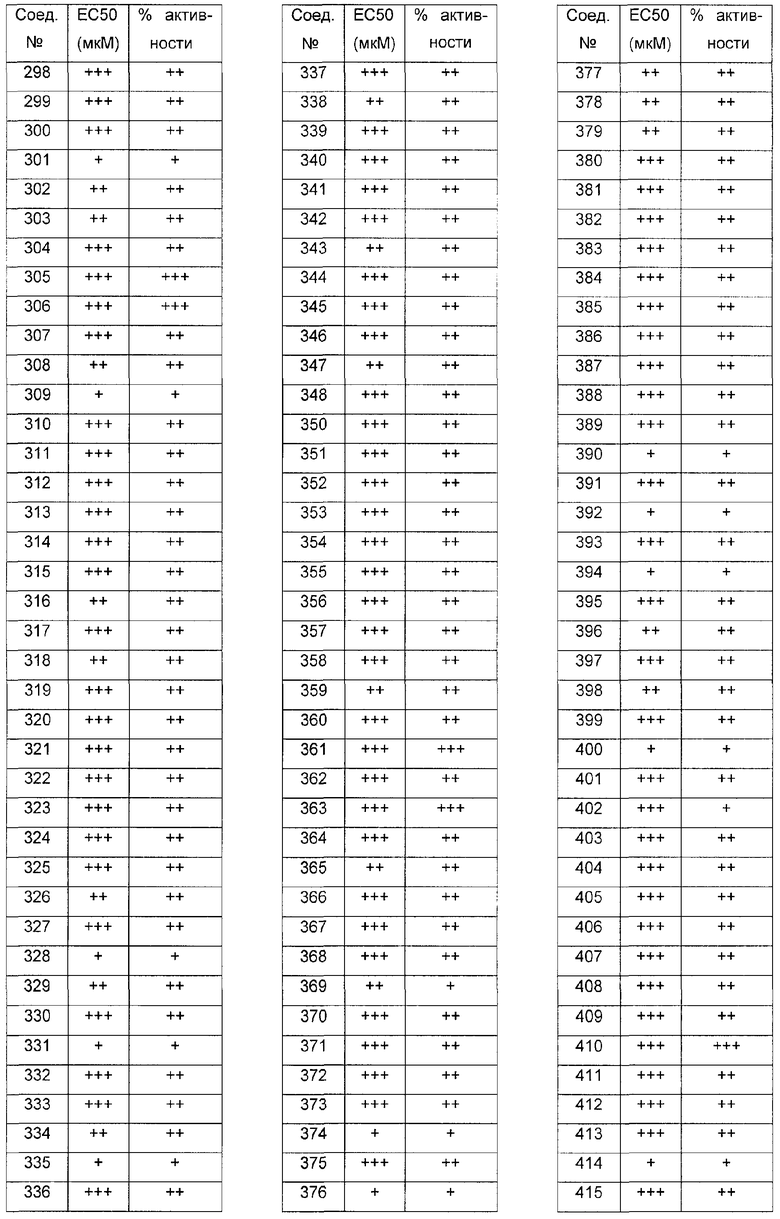
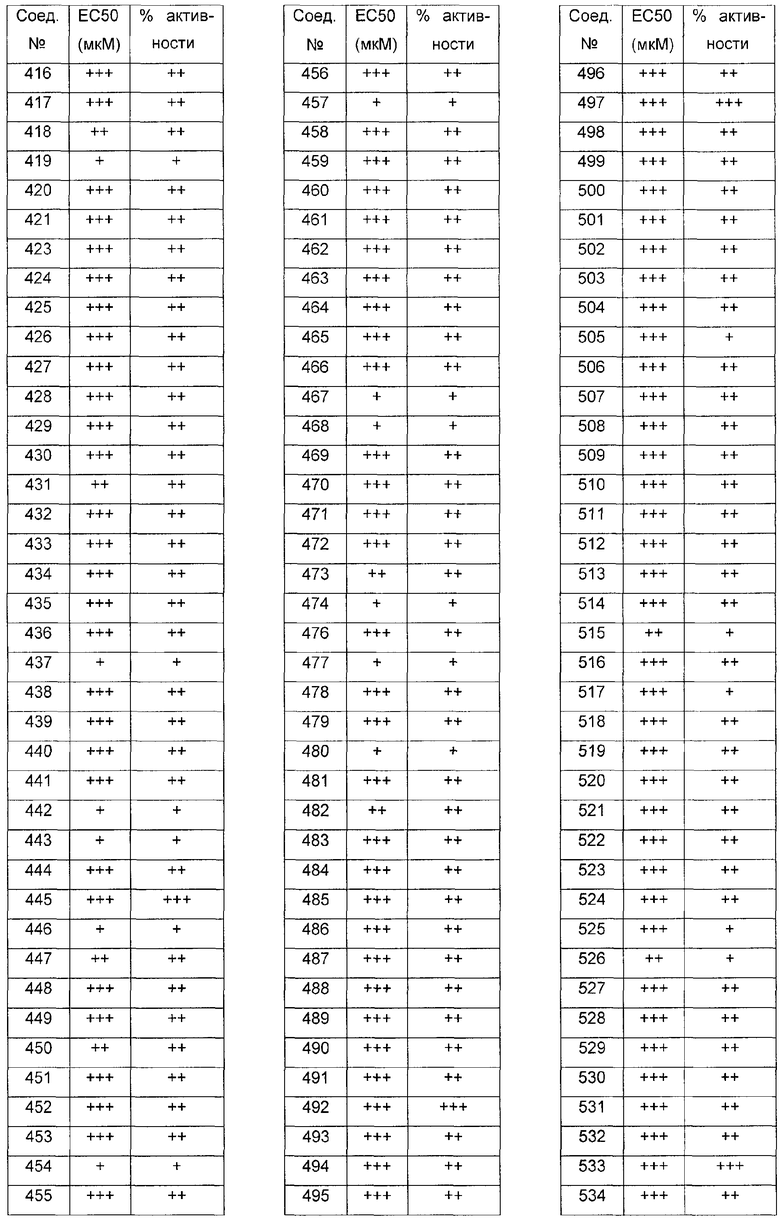
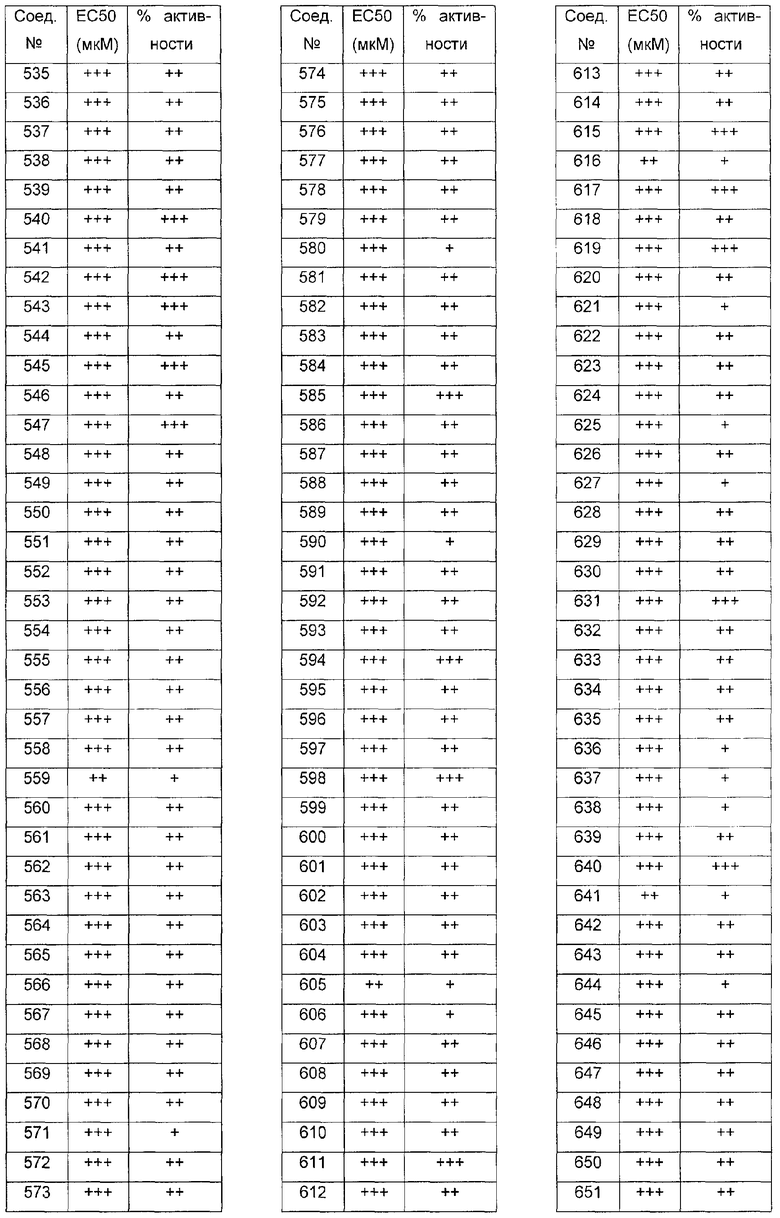
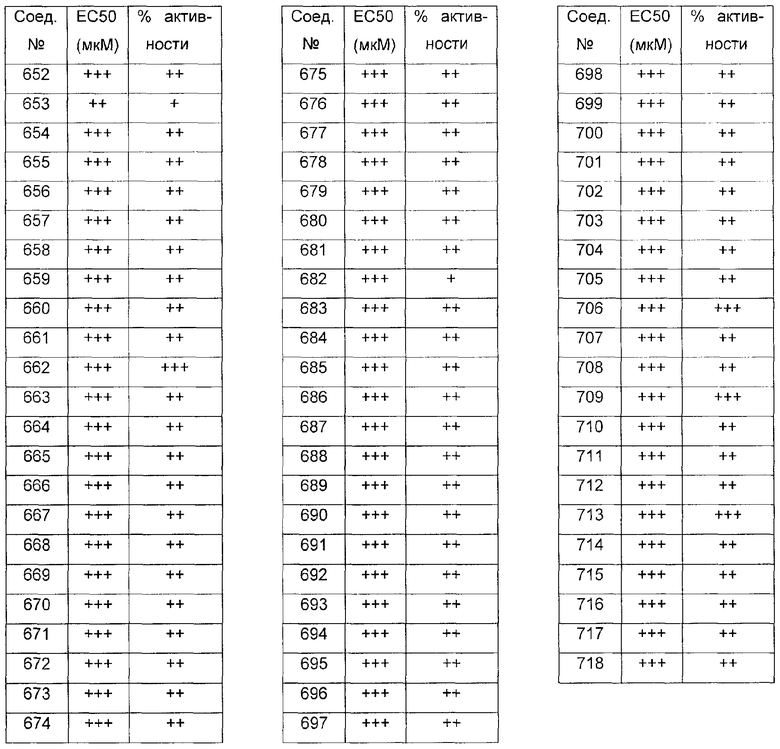
Изобретение относится к способу лечения способу лечения или ослабления тяжести кистозного фиброза у пациента, где у пациента имеется трансмембранный рецептор кистозного фиброза (CFTR) с R117H мутацией, включающий стадию введения указанному пациенту эффективного количества N-(5-гидрокси-2,4-дитрет-бутил-фенил)-N-метил-4-оксо-1H-хинолин-3-карбоксамида. Технический результат: разработан способ лечения кистозного фиброза, основанный на использовании N-(5-гидрокси-2,4-дитрет-бутил-фенил)-N-метил-4-оксо-1H-хинолин-3-карбоксамида. 2 з.п. ф-лы, 4 табл., 30 пр.
1. Способ лечения или ослабления тяжести кистозного фиброза у пациента, где у пациента имеется трансмембранный рецептор кистозного фиброза (CFTR) с R117H мутацией, включающий стадию введения указанному пациенту эффективного количества N-(5-гидрокси-2,4-дитрет-бутил-фенил)-N-метил-4-оксо-1H-хинолин-3-карбоксамида.
2. Способ по п. 1, где пациент является гетерозиготным для R117H мутации.
3. Способ по п. 1, где пациент является гомозиготным для R117H мутации.
| WO 2006002421 A2, 05.01.2006 | |||
| ПРИСПОСОБЛЕНИЕ К ТАНКУ ТЕЙЛОР ДЛЯ ПЕРЕДВИЖЕНИЯ ЕГО ПО ЖЕЛЕЗНОДОРОЖНЫМ ПУТЯМ | 1924 |
|
SU4043A1 |
| Способ получения нафтиридинхинолин-или бензоксазинкарбоновых кислот или их фармацевтически допустимых солей присоединения кислоты | 1983 |
|
SU1360584A3 |
| Способ получения производных 3-хинолинкарбоновой кислоты или их солей | 1977 |
|
SU867299A3 |

