Настоящее изобретение относится к комбинациям фармацевтически активных веществ для применения в лечении респираторных заболеваний, особенно хронического обструктивного заболевания легких (COPD) и астмы.
Предшествующий уровень техники
Для осуществления основной функции легких требуется хрупкая структура, которая испытывает огромное воздействие окружающей среды, включая загрязняющие вещества, микробы, аллергены и канцерогены. Факторы организма-хозяина, являющиеся результатом взаимодействия выбора образа жизни и генетической композиции, влияют на ответ на такое воздействие. Повреждение или инфекция легких может привести к развитию широкого ряда заболеваний респираторной системы (или респираторных заболеваний). Некоторые из этих заболеваний имеют большое значение в здравоохранении. Респираторные заболевания включают острое повреждение легких, острый респираторный дистресс-синдром (ARDS), профессиональное заболевание легких, рак легких, туберкулез, фиброз, пневмокониоз, пневмонию, эмфизему, хроническое обструктивное заболевание легких (COPD) и астму.
Астма является одним из наиболее распространенных респираторных заболеваний. Как правило, астму определяют как воспалительное расстройство дыхательных путей с клиническими симптомами, являющимися следствием периодической обструкции дыхательных путей. Клинически она характеризуется приступами стерторозного дыхания, одышкой и кашлем. Астма является хроническим расстройством, ограничивающим трудоспособность, распространенность и интенсивность которого, как представляется, увеличиваются. По оценкам 15% детей и 5% взрослых среди населения развитых стран страдают от астмы. Поэтому терапия должна быть направлена на контролирование симптомов, чтобы сделать возможным нормальный образ жизни и в то же время обеспечить основу для лечения лежащего в основе воспаления.
COPD является термином, который относится к большой группе легочных заболеваний, которые могут мешать нормальному дыханию. Действующие клинические руководства определяют COPD как болезненное состояние, характеризующееся ограничением дыхания, которое не является полностью обратимым. Ограничение дыхания обычно является и прогрессирующим и ассоциированным с аномальным воспалительным ответом легких на вредные частицы и газы. Наиболее важным, вносящим свой вклад источником таких частиц и газов, по меньшей мере в западном мире, является табачный дым. У пациентов с COPD наблюдается ряд симптомов, включая кашель, нехватку воздуха и избыточное продуцирование мокроты; подобные симптомы являются следствием дисфункции некоторых клеточных компартментов, включая нейтрофилы, макрофаги и эпителиальные клетки. Двумя наиболее важными состояниями, которые охватывает COPD, являются хронический бронхит и эмфизема.
Хронический бронхит представляет собой продолжительное воспаление бронхов, которое вызывает увеличенное продуцирование слизи и другие изменения. У пациента наблюдаются такие симптомы, как кашель и отхаркивание мокроты. Хронический бронхит может привести к более частым и тяжелым респираторным инфекциям, сужению и забиванию бронхов, затрудненному дыханию и нетрудоспособности.
Эмфизема представляет собой хроническое легочное заболевание, которое поражает альвеолы и/или концы самых мелких бронхов. Легкое теряет свою эластичность и, следовательно, эти области легких увеличиваются. Эти увеличенные области захватывают несвежий воздух и не обменивают его эффективно на свежий воздух. Это приводит к затрудненному дыханию и может привести к недостатку поступления кислорода в кровь. Основным симптомом у пациентов с эмфиземой является нехватка воздуха.
Терапевтические агенты, используемые в лечении респираторных заболеваний, включают глюкокортикостероиды. Глюкокортикостероиды (также известные как кортикостероиды или глюкокортикоиды) представляют собой мощные противовоспалительные агенты. В то время как точный механизм их действия не ясен, конечным результатом лечения глюкокортикостероидами является уменьшение числа, активности и перемещений воспалительных клеток в бронхиальной подслизистой ткани, что приводит к пониженной восприимчивости дыхательных путей. Глюкокортикоиды также могут являться причиной пониженного слущивания выстилки бронхиального эпителия, пониженной проницаемости сосудов и секреции слизи.
Хотя лечение глюкокортикоидами и может обеспечивать важные преимущества, эффективность этих агентов часто является далеко не удовлетворительной, особенно при COPD. Кроме того, хотя применение стероидов и может привести к терапевтическим эффектам, желательно иметь возможность использовать стероиды в низких дозах для минимизации частоты и тяжести нежелательных побочных эффектов, которые могут быть связаны с их регулярным введением. Недавние исследования также выдвинули на первый план проблему приобретения устойчивости к стероидам среди пациентов, страдающих от респираторных заболеваний. Например, обнаружено, что курящие субъекты с астмой являются нечувствительными к кратковременной ингаляционной терапии кортикостероидами, но различие в реакции между курящими и некурящими, как оказалось, снижается при высокой дозе ингалируемого кортикостероида (Tomlinson et al., Thorax 2005; 60:282-287). Следовательно имеется настоятельная медицинская необходимость в новых терапевтических средствах против респираторных заболеваний, таких как COPD и астма, в частности в терапевтических средствах с потенциальной возможностью модификации заболевания.
В WO 01/98273 и WO 03/051839 описаны соединения, обладающие активностью в качестве фармацевтических средств, в частности в качестве модуляторов хемокиновых рецепторов (особенно рецептора хемокина МIР-1α (макрофагального воспалительного протеина 1α)), их соли и фармацевтические композиции и их возможное применение в лечении различных заболеваний.
Рецептор хемокина МIР-1α CCR1 (хемокиновый рецептор 1) интенсивно экспрессируется в тканях, пораженных при различных аутоиммунных, воспалительных, пролиферативных, гиперпролиферативных и иммунологически опосредованных заболеваниях, например астме и хроническом обструктивном заболевании легких. Кроме того, воспалительные клетки (например, нейтрофилы и моноциты/макрофаги) вносят вклад в патогенез респираторных заболеваний, таких как COPD, путем секреции протеолитических ферментов, окислителей и фармакологических медиаторов. Рекрутмент и активация этих клеток в тканях легкого зависят от функции CCR1.
Теперь неожиданно было обнаружено, что может наблюдаться неожиданно благоприятный терапевтический эффект в лечении респираторных заболеваний, если антагонист рецептора CCR1 применяют в комбинации с глюкокортикостероидом. Например, комбинация по данному изобретению считается особенно эффективной в снижении притока воспалительных клеток в легкое. Такой благоприятный эффект может наблюдаться, когда два активных вещества вводят одновременно (либо в одном фармацевтическом препарате, либо посредством отдельных препаратов), либо последовательно или раздельно посредством отдельных фармацевтических препаратов.
Таким образом, согласно настоящему изобретению предложен фармацевтический продукт, содержащий в комбинации:
(а) первый активный ингредиент, представляющий собой соединение общей формулы
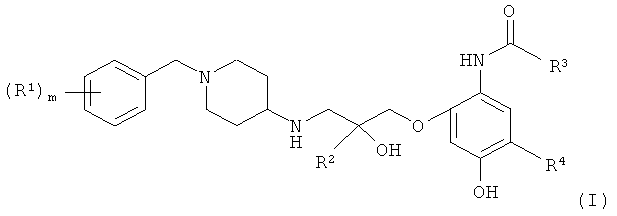
где m равен 0, 1 или 2;
каждый R1 независимо представляет собой галоген или циано;
R2 представляет собой атом водорода или метил;
R3 представляет собой группу С1-С4алкил
и
R4 представляет собой водород или галоген,
или его фармацевтически приемлемую соль и
(б) второй активный ингредиент, который представляет собой глюкокортикостероид.
Фармацевтический продукт по настоящему изобретению может, например, представлять собой фармацевтическую композицию, содержащую первый и второй активные ингредиенты в смеси. Альтернативно фармацевтический продукт может, например, представлять собой набор, содержащий препарат первого активного ингредиента и препарат второго активного ингредиента и, возможно, инструкции для одновременного, последовательного или раздельного введения этих препаратов пациенту, нуждающемуся в этом.
В контексте настоящего описания изобретения алкильная группа заместителя или алкильная группировка в группе заместителя может быть линейной или разветвленной.
Целое число m предпочтительно равно 1 или 2.
Каждый R1 независимо представляет собой галоген (например, хлор, фтор, бром или иод) или циано.
В одном воплощении изобретения m равно 1 и R1 представляет собой атом галогена, в частности атом хлора.
В другом воплощении m равно 1 и R1 представляет собой атом галогена (например, хлор) в положении 4 бензольного кольца относительно атома углерода, к которому присоединена связующая группа СН2.
R2 представляет собой атом водорода или метил. В одном воплощении настоящего изобретения R2 представляет собой метил.
R3 представляет собой группу С1-С4алкил (например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил). Обычно R3 представляет собой метил или этил, в частности метил.
R4 представляет собой водород или галоген (например, фтор, хлор, бром или иод). В одном воплощении настоящего изобретения R4 представляет собой водород или хлор.
Соединения формулы (I) могут существовать в стереоизомерных формах. Следует понимать, что изобретение включает применение всех геометрических и оптических изомеров соединений формулы (I) и их смесей, включая рацематы. Применение таутомеров и их смесей также составляет аспект настоящего изобретения. Предпочтительными оптическими изомерами являются (S)-энантиомеры (т.е. соединения с S-конфигурацией в стереоцентре с присоединенными R2 и ОН).
Соединения формулы (I) согласно настоящему изобретению можно синтезировать, используя методики, представленные в WO 01/98273 и WO 03/051839.
Соединения формул (I) могут применяться в форме фармацевтически приемлемой соли, предпочтительно соли присоединения кислоты, такой как гидрохлорид, гидробромид, фосфат, сульфат, ацетат, аскорбат, бензоат, фумарат, гемифумарат, фуроат, сукцинат, малеат, тартрат, цитрат, оксалат, ксинафоат, метансульфонат или пара-толуолсульфонат. Фармацевтически приемлемая соль также включает формы внутренних солей (цвиттер-ионные). Любая ссылка на соединения формулы (I) или их соли также включает сольваты таких соединений и сольваты таких солей (например, гидраты).
Следует учесть, что соединения формулы (I) и их соли могут существовать в виде цвиттер-ионов. Таким образом, хотя соединения изображены и упоминаются в гидроксильной форме, они также могут существовать в форме внутренней соли (цвиттер-ионной). Изображенная формула (I) и примеры по настоящему изобретению охватывают как гидроксильную, так и цвиттер-ионную формы и их смеси в любых пропорциях.
В другом воплощении настоящего изобретения соединение формулы (I) выбирают из
N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида,
N-{5-хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида,
N-{5-хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}ацетамида,
N-{5-хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}пропанамида или
N-{5-хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}пропанамида
или их фармацевтически приемлемой соли.
В другом воплощении настоящего изобретения соединение формулы (I) представляет собой соль N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида, например гидрохлорид, гидробромид, фосфат, сульфат, ацетат, аскорбат, бензоат, фумарат, гемифумарат, фуроат, сукцинат, малеат, тартрат, цитрат, оксалат, ксинафоат, метансульфонат или пара-толуолсульфонат. Солями с особенно хорошими свойствами (например, подходящей кристалличностью и другими физическими свойства, подходящими, например, для приготовления в виде сухого порошкового препарата для легочного введения) являются бензоат, фумарат или гемифумарат N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида, включая любые формы солей, упоминаемые в примерах.
В одном воплощении изобретения соединение формулы (I) или его соль имеет кристаллические свойства и является, например, по меньшей мере на 50% кристаллическим, по меньшей мере на 60% кристаллическим, по меньшей мере на 70% кристаллическим или по меньшей мере на 80% кристаллическим. Степень кристалличности может быть оценена посредством традиционных методик рентгеновской дифрактометрии.
В другом воплощении изобретения соединение формулы (I) или его соль является кристаллической в степени от 50%, 60%, 70%, 80% или 90% до 95%, 96%, 97%, 98%, 99% или 100%.
Следует заметить, что где приведены пики дифракции рентгеновских лучей на порошке (в градусах 2θ), предел погрешности соответствует общей главе Фармакопеи Соединенных Штатов по дифракции рентгеновских лучей (USP941), смотри Конвенцию о Фармакопее Соединенных Штатов. Дифракция рентгеновских лучей, Общий анализ <941>. United States Pharmacopeia, 25th ed. Rockville, MD: United States Pharmacopeial Convention; 2002:2088-2089).
В одном воплощении изобретения соединение формулы (I) представляет собой гемифумаратную соль N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида, который демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (выраженные в градусах 2θ):
(1) 6,2, 10,7 и 12,5 или
(2) 6,2, 10,7 и 18,8 или
(3) 6,2, 10,7 и 18,0 или
(4) 6,2, 10,7, 12,5, 18,0 и 18,8 или
(5) 6,2, 10,7, 12,5, 18,0, 18,8, 19,7 и 19,8.
В другом воплощении изобретения соединение формулы (I) представляет собой фуроатную соль N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида, который демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (выражены в градусах 2θ):
(1) 6,3, 11,0 и 12,7 или
(2) 6,3, 10,7 и 12,7 или
(3) 6,3, 11,0, 12,7 и 15,9 или
(4) 6,3, 10,7, 11,0, 12,7, 13,9, 14,2 и 15,9 или
(5) 6,3, 10,7, 11,0, 12,7, 15,9, 17,7, 19,1, 19,7 и 25,5 или
(6) 6,3, 10,7, 11,0, 12,7, 13,9, 14,2, 15,9, 17,7, 19,1, 19,7, 19,9, 21,6 и 25,5.
В другом воплощении изобретения соединение формулы (I) представляет собой фуроатную соль N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида, которая демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (выраженные в градусах 2θ):
(1) 6,7, 11,0 и 13,4 или
(2) 6,7, 10,4, 11,0 и 13,4 или
(3) 6,7, 10,4, 12,4, 13,4 и 13,7 или
(4) 6,7, 10,4, 13,4 и 20,9 или
(5) 6,7, 10,4, 11,0, 12,4, 13,4, 13,7, 15,6, 16,0 и 17,6 или
(6) 6,7, 10,4, 11,0, 12,4, 13,4, 13,7, 15,6, 16,0, 16,1, 17,6, 18,0, 18,6, 18,9, 20,1, 20,9 и 23,4.
В другом воплощении изобретения соединение формулы (I) представляет собой бензоатную соль N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида, которая демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (выраженные в градусах 2θ):
(1) 6,1, 10,7 и 19,3 или
(2) 6,1, 12,2 и 14,1 или
(3) 6,1, 10,7, 12,2, 14,1, 18,1 и 19,3 или
(4) 6,1, 10,7, 12,2, 14,1, 15,7, 18,1 и 19,3 или
(5) 6,1, 10,7, 12,2, 14,1, 15,1 и 19,3 или
(6) 6,1, 10,7, 12,2, 14,1, 15,1, 15,7, 18,1 и 19,3 или
(7) 6,1, 10,7, 12,2, 14,1, 15,1, 15,7, 18,1, 19,3, 21,2 и 24,6.
В другом воплощении изобретения соединение формулы (I) представляет собой бензоатную соль N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида, которая демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (выраженные в градусах 2θ):
(1) 6,5, 9,3 и 10,5 или
(2) 6,5, 9,3, 17,6 и 17,8 или
(3) 6,5, 9,3, 10,5, 12,0 и 12,4 или
(4) 6,5, 9,3, 10,5, 12,0, 12,4, 13,0, 13,6, 15,5, 17,6 и 17,8 или
(5) 6,5, 13,0 и 20,2 или
(6) 6,5, 9,3, 10,5, 12,0, 12,4, 13,0, 13,6, 15,5, 17,6, 17,8 и 19,2 или
(7) 6,5, 9,3, 10,5, 12,0, 12,4, 13,0, 13,6, 15,5, 17,6, 17,8, 19,2, 20,2, 22,8 и 26,0 или
(8) 6,5, 9,3, 10,5, 12,0, 12,4, 13,0, 13,6, 15,5, 17,6, 17,8, 19,2, 20,2, 22,8, 24,2, 26,0 и 30,7.
В одном воплощении изобретения соединение формулы (I) представляет собой фуроатную или бензоатную соль N-{5-хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида.
Вторым активным ингредиентом в комбинации по настоящему изобретению является глюкокортикостероид. Глюкокортикостероид по настоящему изобретению может быть любым синтетическим или встречающимся в природе глюкокортикостероидом. Примеры глюкокортикостероида, который можно использовать согласно настоящему изобретению, включают будесонид, флутиказон (например, в виде пропионатного эфира), мометазон (например, в виде фуроатного эфира), беклометазон (например, в виде 17-пропионатного или 17,21-дипропионатного эфира), циклесонид, лотепреднол (например, в виде этабоната), этипреднол (например, в виде диклоацетата), триамцинолон (например, в виде ацетонида), флунизолид, зотиказон, флумоксонид, рофлепонид, бутиксокорт (например, в виде пропионатного эфира), преднизолон, преднизон, типредан, стероидные эфиры согласно WO 2002/12265, WO 2002/12266 и WO 2002/88167, например 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботионовой кислоты S-фторметиловый эфир, 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-пропионилоксиандроста-1,4-диен-17β-карботионовой кислоты S-(2-оксотетрагидрофуран-3S-ил)овый эфир и 6α,9α-дифтор-11β-гидрокси-16α-метил-17α-[(4-метил-1,3-тиазол-5-карбонил)окси]-3-оксоандроста-1,4-диен-17β-карботионовой кислоты S-фторметиловый эфир, эфиры стероидов, такие как эфиры, описанные в DE 4129535, стероиды, например GSK 870086, GSK 685698, GSK 799943 и стероиды согласно WO 2002/00679, WO 2005/041980 и тому подобное.
В контексте настоящего описания изобретения, если не указано иное, любая ссылка на глюкокортикостероид включает все активные соли, сольваты или производные, которые могут быть образованы из указанного глюкокортикостероида. Примеры возможных солей или производных глюкокортикоидов включают натриевые соли, сульфобензоаты, фосфаты, изоникотинаты, ацетаты, пропионаты, дигидрофосфаты, пальмитаты, пивалаты, фумараты и фармацевтически приемлемые эфиры (например, C1-С6алкиловые эфиры). Глюкокортикостероиды и их активные соли или производные также могут находиться в форме их сольватов, например гидратов.
В одном воплощении настоящего изобретения глюкокортикостероид представляет собой будесонид. Химическое название будесонида 16,17-[бутилиденбис(окси)]-11,21-дигидроксипрегна-1,4-диен-3,20-дион. Будесонид и его получение описаны, например, в Arzneimittel-Forschung (1979), 29 (11), 1687-1690, DE 2323215 и US 3929768. Имеющиеся в настоящее время препараты будесонида поступают на рынок под торговым наименованием "Энтокорт".
Считается, что применение соединений формулы (I) демонстрирует особенно любопытные эффекты при использовании в комбинации с глюкокортикостероидами и особенно в комбинации с будесонидом. Например, эксперименты на животных in vivo показывают, что комбинация глюкокортикостероида и соединения формулы (I) с уровнями доз, при которых любой компонент по отдельности не оказывает значительного влияния на легочное воспаление, в комбинации обеспечивает значительное уменьшение притока воспалительных клеток. Уменьшение притока клеток для комбинации было больше, чем ожидаемое от аддитивного эффекта этих двух ингредиентов. Данный синергический эффект, наблюдающийся при комбинировании ингредиентов, можно было бы использовать, например, для снижения терапевтической дозы стероида или, при той же самой дозе, для достижения повышенной эффективности в отношении воспаления по сравнению с применением одного стероида. Данный синергический эффект может быть особенно полезным, когда желательными являются более низкие дозы стероида, например у индивидуумов, которые приобрели устойчивость к таким стероидам.
Фармацевтический продукт по настоящему изобретению возможно может дополнительно содержать в качестве третьего активного ингредиента бронходилататор. Бронходилататор представляет собой лекарственное средство, которое расслабляет и расширяет бронхиальные каналы и улучшает поступление воздуха в легкие. Примеры бронходилататора, который можно использовать в настоящем изобретении, включают β2-агонист или антихолинергическое соединение.
β2-Агонисты (также известные как агонисты бета2 (β2)-адренорецептора) могут быть использованы для облегчения симптомов респираторных заболеваний путем расслабления бронхиальных гладких мышц, снижения обструкции дыхательных путей, снижения чрезмерного расширения легких и снижения одышки, β2-Агонист по настоящему изобретения может представлять собой любое соединение или вещество, способное стимулировать β2-рецептор и действовать в качестве бронходилататора. Примеры β2-агонистов, которые могут применяться в настоящем изобретении, включают бамбутерол, битолтерол, карбутерол, индакатерол, кленбутерол, фенотерол, формотерол, гексопреналин, ибутерол, пирбутерол, прокатерол, репротерол, салметерол, сульфонтерол, тербуталин, толубутерол, ТА 2005 (химически идентифицируемый как 2(1Н)-хинолон, 8-гидрокси-5-[1-гидрокси-2-[[2-(4-метоксифенил)-1-метилэтил]амино]этил]моногидрохлорид, [R-(R*,R*)], также идентифицируемый по регистрационному номеру Chemical Abstract Service 137888-11-0 и раскрытый в патенте США 4579854 (=CHF-4226), GSK159797, производные форманилида, например 3-(4-{[6-({(2R)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфамид, как раскрыто в WO 2002/76933, производные бензолсульфамида, например 3-(4-{[6-({(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этил}амино)гексил]окси}бутил)бензолсульфамид, как раскрыто в WO 2002/88167, ариланилиновые агонисты рецепторов, такие как раскрыты в WO 2003/042164 и WO 2005/025555, и производные индола, такие как раскрыты в WO 2004/032921.
В одном аспекте β2-агонист изобретения является β2-агонистом длительного действия, т.е. β2-агонистом, активность которого сохраняется в течение более 12 часов. Примеры β2-агонистов длительного действия включают формотерол, бамбутерол и салметерол.
В контексте настоящего описания изобретения, если не указано иное, любая ссылка на бронходилататор (включая β2-агонисты, β2-агонисты длительного действия и антихолинергические соединения) включает активные соли, сольваты или производные, которые могут образоваться из указанного бронходилататора, и любые энантиомеры и их смеси, включая рацематы. Примерами возможных солей или производных являются соли присоединения кислоты, такие как соли соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, уксусной кислоты, фумаровой кислоты, янтарной кислоты, молочной кислоты, лимонной кислоты, винной кислоты, 1-гидрокси-2-нафталинкарбоновой кислоты, малеиновой кислоты и фармацевтически приемлемые эфиры (например, C1-С6алкиловые эфиры). Бронходилататор (включая соли и его производные) также может находиться в форме сольвата, например гидрата.
Примеры антихолинергического соединения включают ипратропий (например, в виде бромида), тиотропий (например в виде бромида), окситропий (например, в виде бромида), толтеродин, AD-237 (Arakis) и производные хинуклидина, как раскрыто в US 2003/0055080.
В одном воплощении настоящего изобретения бронходилататор представляет собой формотерол. Химическим названием формотерола является N-[2-гидрокси-5-[(1)-1-гидрокси-2-[[(1)-2-(4-метоксифенил)-1-метилэтил]амино]этил]фенил]формамид. Получение формотерола описано, например, в WO 92/05147. Как очевидно из приведенного выше, подразумевается, что термин формотерол включает все его фармацевтически приемлемые соли. В одном аспекте этого воплощения бронходилататором является формотерола фумарат, например формотерола фумарата дигидрат.
Как акцентируется выше, следует понимать, что изобретение охватывает применение всех оптических изомеров формотерола и их смесей, включая рацематы. Таким образом, например, термин формотерол включает N-[2-гидрокси-5-[(1R)-1-гидрокси-2-[[(1R)-2-(4-метоксифенил)-1-метилэтил]амино]этил]фенил]формамид, N-[2-гидрокси-5-[(1S)-1-гидрокси-2-[[(1S)-2-(4-метоксифенил)-1-метилэтил]амино]этил]фенил]формамид или смесь таких энантиомеров, включая рацемат.
В еще одном воплощении настоящего изобретения бронходилататор представляет собой индакатерол. Как очевидно из указанного выше, предполагается, что термин индакатерол включает все его фармацевтически приемлемые соли, включая, например, малеат индакатерола и гидрохлорид индакатерола.
Для лечения респираторных заболеваний соединение формулы (I) или его фармацевтически приемлемую соль (первый активный ингредиент), глюкокортикостероид (второй активный ингредиент) и возможно бронходилататор (третий активный ингредиент) по настоящему изобретению можно вводить одновременно, последовательно или раздельно для лечения респираторных заболеваний. Последовательное введение означает, что активные ингредиенты вводят в любом порядке непосредственно один за другим. Они также оказывают желаемый эффект при раздельном введении, но при введении таким способом их обычно вводят с промежутком времени менее 4 часов, более удобно с промежутком времени менее двух часов, более удобно с промежутком времени менее 30 минут и наиболее удобно с промежутком времени менее 10 минут.
Активные ингредиенты по настоящему изобретению можно вводить посредством перорального или парентерального (например, внутривенного, подкожного, внутримышечного или внутрисуставного) введения, используя традиционные системные лекарственные формы, такие как таблетки, капсулы, пилюли, порошки, водные или масляные растворы или суспензии, эмульсии и стерильные инъекционные водные или масляные растворы или суспензии. Активные ингредиенты можно также вводить местно (в легкие и/или дыхательные пути) в форме растворов, суспензий, аэрозолей и сухих порошковых препаратов. Эти лекарственные формы обычно будут включать один или более чем один фармацевтически приемлемый ингредиент, который может быть выбран, например, из адъювантов, носителей, связующих агентов, смазывающих веществ, разбавителей, стабилизаторов, буферных агентов, эмульгаторов, агентов, регулирующих вязкость, поверхностно-активных веществ, консервантов, корригентов и красителей. Как очевидно специалистам в данной области, наиболее подходящий способ введения активных ингредиентов зависит от целого ряда факторов.
В одном воплощении настоящего изобретения активные ингредиенты вводят посредством отдельных фармацевтических препаратов.
Таким образом, в одном аспекте настоящего изобретения предложен набор, содержащий препарат первого активного ингредиента, представляющего собой соединение формулы (I) или его фармацевтически приемлемую соль, и препарат второго активного ингредиента, представляющего собой глюкокортикостероид, и возможно инструкции по одновременному, последовательному или раздельному введению препаратов нуждающемуся в них пациенту. Данный набор возможно может дополнительно содержать препарат третьего активного ингредиента, представляющего собой бронходилататор.
В другом воплощении активные ингредиенты могут быть введены посредством одной фармацевтической композиции. Таким образом, в настоящем изобретении также предлагается фармацевтическая композиция, содержащая в смеси первый активный ингредиент, представляющий собой соединение формулы (I) или его фармацевтически приемлемую соль, и второй активный ингредиент, представляющий собой глюкокортикостероид. Фармацевтическая композиция может дополнительно содержать третий активный ингредиент, представляющий собой бронходилататор. В настоящем изобретении также предложен способ получения фармацевтической композиции, который включает смешивание первого активного ингредиента со вторым активным ингредиентом и возможно с третьим активным ингредиентом.
Фармацевтические композиции по настоящему изобретению можно получить путем смешивания первого активного ингредиента и второго активного ингредиента с фармацевтически приемлемым адъювантом, разбавителем или носителем и возможно с третьим активным ингредиентом.
Следовательно, в еще одном аспекте настоящего изобретения предложен способ получения фармацевтической композиции, который включает смешивание соединения формулы (I) или его фармацевтически приемлемой соли с глюкокортикостероидом и фармацевтически приемлемым адъювантом, разбавителем или носителем и, возможно, с бронходилататором.
Следует понимать, что терапевтическая доза каждого активного ингредиента, вводимого в соответствии с настоящим изобретением, будет меняться в зависимости от конкретного применяемого активного ингредиента, способа, посредством которого предполагается вводить активный ингредиент, и состояния или расстройства, подлежащего лечению.
В одном аспекте настоящего изобретения каждый из первого, второго (и, если присутствует, третьего) активного ингредиента по настоящему изобретению вводят путем ингаляции. В данном аспекте ингаляцию активных ингредиентов осуществляют одновременно, последовательно или раздельно.
Везде в данном описании изобретения количество применяемых активных ингредиентов относится к стандартным дозам для ингаляции, если ясно не указано иное.
При введении путем ингаляции доза первого активного ингредиента (соединения формулы (I) или его фармацевтически приемлемой соли или сольвата) будет обычно находиться в диапазоне от 0,1 мкг до 10000 мкг, от 0,1 до 5000 мкг, от 0,1 до 1000 мкг, от 0,1 до 500 мкг, от 0,1 до 200 мкг, от 0,1 до 100 мкг, от 0,1 до 50 мкг, от 5 мкг до 5000 мкг, от 5 до 1000 мкг, от 5 до 500 мкг, от 5 до 200 мкг, от 5 до 100 мкг, от 5 до 50 мкг, от 10 до 5000 мкг, от 10 до 1000 мкг, от 10 до 500 мкг, от 10 до 200 мкг, от 10 до 100 мкг, от 10 до 50 мкг, от 20 до 5000 мкг, от 20 до 1000 мкг, от 20 до 500 мкг, от 20 до 200 мкг, от 20 до 100 мкг, от 20 до 50 мкг, от 50 до 5000 мкг, от 50 до 1000 мкг, от 50 до 500 мкг, от 50 до 200 мкг, от 50 до 100 мкг, от 100 до 5000 мкг, от 100 до 1000 мкг или от 100 до 500 мкг.
Первый активный ингредиент (соединение формулы (I) или его фармацевтически приемлемую соль) также можно вводить перорально. Пероральное введение антагониста рецептора CCR1, например, можно использовать в фармацевтическом продукте или наборе, причем другой(ие) активный(ые) ингредиент(ы) вводят посредством ингаляции.
При введении посредством ингаляции доза второго активного ингредиента (глюкокортикостероида) обычно будет находиться в диапазоне от 0,1 микрограмма (мкг) до 1000 мкг, от 0,1 до 500 мкг, от 0,1 до 200 мкг, от 0,1 до 100 мкг, от 0,1 до 50 мкг, от 0,1 до 5 мкг, от 5 до 1000 мкг, от 5 до 500 мкг, от 5 до 200 мкг, от 5 до 50 мкг, от 5 до 10 мкг, от 10 до 1000 мкг, от 10 до 500 мкг, от 10 до 200 мкг, от 10 до 100 мкг, от 10 до 50 мкг, от 20 до 1000 мкг, от 20 до 500 мкг, от 20 до 200 мкг, от 20 до 100 мкг, от 20 до 50 мкг, от 50 до 1000 мкг, от 50 до 500 мкг, от 50 до 200 мкг, от 50 до 100 мкг, от 100 до 1000 мкг или от 100 до 500 мкг.
В одном воплощении количество первого используемого активного агента находится в диапазоне от 1 мкг до 200 мкг и количество второго агента находится в диапазоне от 1 до 200 мкг.
Молярное отношение второго активного ингредиента к первому активному ингредиенту в дозе типично может находиться в диапазоне от 1:10 до 10:1. Предпочтительно данное отношение находится в диапазоне от 1:1 до 10:1 и еще более предпочтительно в диапазоне от 5:1 до 20:1.
Третий активный ингредиент (бронходилататор), когда он присутствует, удобно может быть введен посредством ингаляции в дозе, обычно находящейся в диапазоне от 0,1 до 100 мкг, от 0,1 до 50 мкг, от 0,1 до 40 мкг, 0,1 до 30 мкг, от 0,1 до 20 мкг, от 0,1 до 10 мкг, от 5 до 100 мкг, от 5 до 50 мкг, от 5 до 40 мкг, от 5 до 30 мкг, от 5 до 20 мкг, от 5 до 10 мкг, от 10 до 100 мкг, от 10 до 50 мкг, от 10 до 40 мкг, от 10 до 30 мкг, от 10 до 20 мкг. В одном воплощении настоящего изобретения доза третьего активного ингредиента находится в диапазоне от 1 до 30 мкг.
Дозы первого и второго (и, когда он присутствует, третьего) активного ингредиентов обычно будут вводиться от 1 до 4 раз в сутки, удобно один или два раза в сутки и наиболее удобно один раз в сутки.
В одном воплощении настоящего изобретения предлагается фармацевтический продукт, содержащий, в комбинации, первый активный ингредиент, представляющий собой соединение формулы (I) или его фармацевтически приемлемую соль, и второй активный ингредиент, представляющий собой глюкокортикостероид, и возможно третий активный ингредиент, который представляет собой бронходилататор, где каждый активный ингредиент приготовлен в виде препарата для введения путем ингаляции.
Активные ингредиенты удобно вводить путем ингаляции (например, местно в легкие и/или дыхательные пути) в форме растворов, суспензий, аэрозолей или сухих порошковых препаратов. Введение можно осуществлять посредством ингаляции перорально или интраназально. Активные ингредиенты предпочтительно адаптированы для введения либо совместно, либо по отдельности из ингалятора для сухих порошков, дозирующего ингалятора под давлением или небулайзера.
Активные ингредиенты могут применяться в смеси с одним или более фармацевтически приемлемыми добавками, разбавителями или носителями. Примеры подходящих разбавителей или носителей включают лактозу (например, моногидрат), декстран, маннит или глюкозу.
Устройства для ингаляции отмеренной дозы можно применять для введения активных ингредиентов, диспергированных в подходящем пропелленте, и с дополнительными эксципиентами, такими как этанол, поверхностно-активные вещества, смазывающее вещество, антиоксидант или стабилизирующий агент, или без них. Подходящие пропелленты включают углеводородные, хлорфторуглеродные и гидрофторалкановые (например, гептафторалкановые) пропелленты или смеси любых таких пропеллентов. Предпочтительными пропеллентами являются Р134а и Р227, каждый из которых может применяться один или в комбинации с другими пропеллентами и/или поверхностно-активными веществами и/или другими эксципиентами. Также можно применять распыленные водные суспензии или предпочтительно растворы, с регулировкой подходящего рН и/или тоничности или без нее, в виде препаратов с однократной дозой либо многократными дозами.
Ингаляторы для сухих порошков можно использовать для введения активных ингредиентов, по отдельности или в комбинации с фармацевтически приемлемым носителем, в последнем случае либо в качестве тонкодиспергированного порошка, либо в качестве упорядоченной смеси. Ингалятор для сухого порошка может быть предназначен для единичной дозы или для многократных доз и в нем может быть использован сухой порошок или капсула, содержащая порошок.
Когда активные ингредиенты адаптированы для введения либо совместно, либо по отдельности, посредством небулайзера, они могут находиться в форме распыленной водной суспензии или раствора с регулировкой подходящего рН или тоничности или без нее, в виде устройства для введения единичной дозы или многократных доз.
Дозированный ингалятор, небулайзер и устройства для ингаляции сухих порошков общеизвестны и имеется множество таких устройств.
В одном воплощении настоящего изобретения соединение формулы (I) или его фармацевтически приемлемая соль могут быть введены перорально, а другой(ие) активный(ые) ингредиент(ы) - путем ингаляции.
В настоящем изобретении также предлагается фармацевтический продукт, набор или фармацевтическая композиция согласно изобретению для одновременного, последовательного или раздельного применения в терапии.
В настоящем изобретении также предлагается применение фармацевтического продукта, набора или фармацевтической композиции согласно изобретению в изготовлении лекарственного средства для лечения респираторного заболевания, в частности хронического обструктивного заболевания легких или астмы.
В настоящем изобретении, кроме того, предлагается способ лечения респираторного заболевания, который включает одновременное, последовательное или раздельное введение:
(а) (терапевтически эффективной) дозы первого активного ингредиента, представляющего собой соединение формулы (I) или его фармацевтически приемлемую соль, и
(б) (терапевтически эффективной) дозы второго активного ингредиента, представляющего собой глюкокортикостероид, и возможно
(в) (терапевтически эффективной) дозы третьего активного ингредиента, представляющего собой бронходилататор;
пациенту, нуждающемуся в этом.
В контексте настоящего описания изобретения термин «терапия» также включает «профилактику», если нет конкретных указаний на противоположное. Термины «терапевтический» и «терапевтически» следует интерпретировать соответственно. Ожидается, что профилактика будет особенно важна для лечения субъектов, которые в прошлом претерпевали эпизод рассматриваемого состояния или расстройства, или иным образом имеют повышенный риск их развития. Субъекты с повышенным риском развития определенного состояния или расстройства обычно включают тех, у кого это состояние или расстройство имеется в семейном анамнезе, или тех, кого по результатам генетического тестирования или скрининга определили, как особенно склонных к развитию данного состоянии или расстройства.
Настоящее изобретение ниже будет дополнительно пояснено посредством ссылки на следующие иллюстративные примеры.
На чертеже показаны результаты эксперимента по притоку клеток у крыс, подвергнутых контрольной провокации LPS (липополисахаридами), с использованием комбинации по настоящему изобретению.
Общие способы
Спектры 1H ЯМР (ядерно-магнитного резонанса) были записаны на Varian Unity Inova 400 или Varian Mercury VX 300 и данные приведены в виде значений дельта, выраженные в миллионных долях (м.д.) по отношению к тетраметилсилану (ТМС), используемому в качестве внутреннего стандарта.
Центральный пик растворителей: хлороформа-d (δH 7,27 м.д.), ацетона-d6 (δH 2,05 м.д.) или DMSO-d6 (диметилсульфоксид) (δH 2,50 м.д.) использовали в качестве внутреннего стандарта.
Масс-спектры низкого разрешения и точное определение массы получали в системе Agilent MSD 1100 LC-MS, оборудованной ионизационными камерами APCI (химическая ионизация при атмосферном давлении)/ESI (ионизация электрораспылением).
Все растворители и имеющиеся в продаже реагенты имели лабораторную чистоту и применялись в том виде, в котором были получены.
Используемые аббревиатуры:
Анализы дифракции рентгеновских лучей на порошке
Анализы дифракции рентгеновских лучей на порошке (XRPD) проводили на образцах, полученных согласно стандартным способам (смотрите, например, Giacovazzo et al., eds., Fundamentals of Crystallography, Oxford University Press (1992); Jenkins & Snyder, eds., Introduction to X-Ray Powder Diffractometry, John Wiley & Sons, New York (1996); Bunn, ed., Chemical Crystallography, Clarendon Press, London (1948), и Klug & Alexander eds., X-ray Diffraction Procedures, John Wiley & Sons, New York (1974)).
Картину дифракции рентгеновских лучей на порошке гемифумаратной соли, описанной выше в примере 1 (в безводной форме), получали, как описано ниже.
Для анализов использовали парафокусирующий дифрактометр рентгеновских лучей на порошке Брега-Брентано, использующий монохроматическое CuKα излучение (45 кВ и 40 мА). Первичная оптика включала щели Соллера и автоматическую щель дивергенции. Плоские образцы готовили на пластинах с нулевым фоном, которые вращались во время измерений. Вторичная оптика включала щели Соллера, автоматическую щель против рассеивания, принимающую щель и монохроматор. Дифрагированный сигнал определяли, используя пропорциональный детектор, заполненный ксеноном. Картины дифракции получали в диапазоне 2° ≤ 2θ (тета) ≤ 40° в режиме непрерывного сканирования с величиной шага 0,016° 2θ со скоростью 4° 2θ в минуту. Необработанные данные сохраняли в электронном виде. Оценку проводили на необработанных и сглаженных дифракционных картинах.
Для вышеупомянутых измерений использовали дифрактометр Panalytical X'pert PRO MPD θ-θ в режиме отражения. Квалифицированный специалист в данной области может установить инструментальные параметры для дифрактометра рентгеновских лучей на порошке так, чтобы можно было получить данные по дифракции, сравнимые с представленными данными.
Получение антагонистов рецептора хемокина MIP-1α
Пример 1(а)
N-{2-[((2S)-3-{[1-(4-Хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида гемифумарат (соль 2:1).
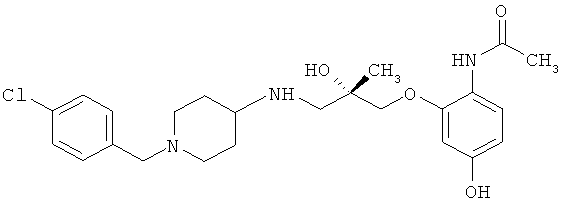
К перемешиваемому раствору неочищенного N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (24,0 г, 36,5 ммоль; полученному посредством экстракции при рН 9 из соответствующей соли с трифторуксусной кислотой, как описано в примере 1 WO 03/051839) в метаноле (240 мл) добавляли раствор фумаровой кислоты (2,13 г, 18,3 ммоль) в метаноле (55 мл) в течение 15 минут. Наблюдали, что осадок начинал формироваться, когда было добавлено около двух третей раствора фумаровой кислоты. Когда был добавлен весь раствор фумаровой кислоты, перемешивание прекращали и реакционную смесь оставили на ночь при температуре окружающей среды (20°С) в закрытом сосуде. Осадок выделяли с помощью фильтровальной воронки, промывали метанолом (3×50 мл) и сушили в вакууме при 60°С в течение ночи с получением указанной в заголовке соли в виде не совсем белого твердого вещества (14,0 г, 73%).
1H ЯМР (399,99 МГц, DMSO) δ 8,91 (s, 1H), 7,48 (d, J=8,6 Гц, 1Н), 7,38 (d, J=8,5 Гц, 2H), 7,31 (d, J=8,4 Гц, 2H), 6,50 (s, 1H), 6,42 (d, J=2,5 Гц, 1Н), 6,31 (dd, J=8,6, 2,5 Гц, 1Н), 3,79 (s, сильно связанная АВ-система, 2H), 3,44 (s, 2H), 2,88 (d, J=12,2 Гц, 1Н), 2,82-2,72 (m, 3H), 2,64-2,55 (m, 1Н), 2,02 (s, 3H), 2,00-1,92 (m, 2H), 1,91-1,83 (m, 2H), 1,47-1,35 (m, 2H), 1,23 (s, 3H).
APCI-MS: m/z 462 [MH+].
Стехиометрия, отношение основания к кислоте 2:1, была подтверждена посредством ЯМР.
Гемифумаратная соль демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (XRPD) (выраженные в градусах 2θ) (предел погрешности соответствует общей главе Фармакопеи Соединенных Штатов по дифракции рентгеновских лучей (USP941), смотри Конвенцию Фармакопеи Соединенных Штатов. X-ray Diffraction, General Test <941>. United States Pharmacopeia, 25th ed. Rockville, MD: United States Pharmacopeial Convention; 2002:2088-2089):
(1) 6,2, 10,7 и 12,5 или
(2) 6,2, 10,7 и 18,8 или
(3) 6,2, 10,7 и 18,0 или
(4) 6,2, 10,7, 12,5, 18,0 и 18,8 или
(5) 6,2, 10,7, 12,5, 18,0, 18,8, 19,7 и 19,8.
Пример 1(б)
Получение бензоата N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (соль 1:1), Форма А.
(а) Смешивали горячие растворы N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (который может быть получен способами, описанными в WO 03/051839; 462 мг, 1,0 ммоль) в этилацетате (10 мл) и бензойной кислоты (244 мг, 2,0 ммоль) в этилацетате (10 мл). Полученную смесь оставляли охлаждаться до температуры окружающей среды (20°С) в закрытом сосуде. Без помутнения образовывался белый осадок. После выстаивания при температуре окружающей среды в течение ночи полученный осадок промывали этилацетатом (3×10 мл) и сушили в вакууме при температуре 60°С в течение ночи с получением указанной в заголовке соли в виде не совсем белого твердого вещества (506 мг, 86%). Соль содержала следы этилацетата.
1H ЯМР (399,99 МГц, ацетон-d6) δ 8,77 (s, 1H), 8,07-8,04 (m, 2H), 7,83 (d, J=8,7 Гц, 1H), 7,55-7,50 (m, 1H), 7,46-7,41 (m, 2H), 7,36-7,31 (m, 4H), 6,52 (d, J=2,6 Гц, 1H), 6,40 (dd, J=8,7, 2,6 Гц, 1H), 3,97 (d, J=9,3 Гц, 1Н), 3,89 (d, J=9,3 Гц, 1H), 3,48 (s, 2H), 3,29 (d, J=12,1 Гц, 1H), 2,94 (d, J=12,2 Гц, 1Н), 2,91-2,77 (m, 3H), 2,09-2,00 (m, 4H), 1,98 (s, 3H), 1,72-1,59 (m, 2H), 1,30 (s, 3H).
APCI-MS: m/z 462 [MH+].
Стехиометрия, отношение основания к кислоте 1:1, была подтверждена посредством ЯМР.
Дополнительные количества указанной в заголовке соли получали следующим способом.
(б) Смешивали горячие растворы N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (4,0 г, 8,65 ммоль) в этилацетате (75 мл) и бензойной кислоты (1,16 г, 9,5 ммоль) в этилацетате (75 мл). Когда полученную смесь охлаждали до температуры окружающей среды (20°С), в нее вносили в качестве затравки частицу указанной в заголовке соли, полученной описанным выше способом (а), и оставляли на ночь в закрытом флаконе. Полученный осадок промывали этилацетатом (3×50 мл) и сушили в вакууме при температуре 60°С в течение ночи с получением указанной в заголовке соли в виде не совсем белого твердого вещества (4,41 г, 87%). Соль содержала следы этилацетата.
Форма А бензоатной соли демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (XRPD) (выраженные в градусах 2θ) (предел погрешности соответствует общей главе Фармакопеи Соединенных Штатов по дифракции рентгеновских лучей (USP941), смотри Конвенцию Фармакопеи Соединенных Штатов. Дифракция рентгеновских лучей, Общий анализ <941>. United States Pharmacopeial, 25th ed. Rockville, MD: United States Pharmacopeial Convention; 2002:2088-2089):
(1) 6,1, 10,7 и 19,3 или
(2) 6,1, 12,2 и 14,1 или
(3) 6,1, 10,7, 12,2, 14,1, 18,1 и 19,3 или
(4) 6,1, 10,7, 12,2, 14,1, 15,7, 18,1 и 19,3 или
(5) 6,1, 10,7, 12,2, 14,1, 15,1 и 19,3 или
(7) 6,1, 10,7, 12,2, 14,1, 15,1, 15,7, 18,1 и 19,3 или
(8) 6,1, 10,7, 12,2, 14,1, 15,1, 15,7, 18,1, 19,3, 21,2 и 24,6.
Получение бензоата N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (соль 1:1), Форма Б
(а) Бензоатную соль, полученную методом примера 1(б) (Форма А, от 10 до 15 мг), помещали в кювету для дифференциальной сканирующей калориметрии (с гофрированной крышкой) и нагревали до достижения температуры 155°С, используя скорость нагрева 5 К/мин. После плавления соли (в используемых условиях была зафиксирована температура начала плавления 146,5°С) расплавленную соль охлаждали со скоростью 5 К/мин до температуры окружающей среды (20°С). Затем ту же кювету снова нагревали со скоростью нагрева 5 К/мин до достижения температуры 151°С и при сканировании регистрировали изотерму при температуре 148°С в течение 10 минут. Затем кювету быстро охлаждали до температуры окружающей среды, что приводило к образованию кристаллов, которые представляли собой новую физическую форму бензоата N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (Форму Б). Некоторое количество аморфной бензоатной соли может быть образовано в качестве побочного продукта способа.
(б) Соль Формы Б, описанную выше в (а), также получали путем растворения в сосуде 20 мас.% образца бензоатной соли, полученной способом, описанным в примере 1(б) (Форма А), в растворителе, таком как метанол (>20 мг/мл), этанол (>20 мг/мл), н-пропанол (>20 мг/мл), изопропанол (8,5 мг/мл) или ацетон (9,6 мг/мл). Цифры в скобках указывают ожидаемую растворимость соли в этих растворителях. Затем сосуд герметично закрывали и суспензию гомогенизировали при температуре окружающей среды (20°С), используя магнит. Перемешивание и температуру поддерживали в течение по меньшей мере 7 суток, после чего образец полученного материала сушили и тестировали с помощью XRPD. XRPD подтвердила полную трансформацию Формы А в Форму Б.
(в) Соль Формы Б, описанную выше в (а), также получали путем растворения бензоатной соли, полученной способом, описанным в Примере 1(6) (Форма А) (22,0 г, 37,7 ммоль) и бензойной кислоты (0,46 г, 3,8 ммоль) в горячем 2-пропаноле (190 мл) во флаконе с круглым дном с получением красноватого раствора. Флакон вращали с помощью устройства Rotavapor на водяной бане при 40°С до охлаждения раствора до 40°С, после чего в него вносили затравку из нескольких кристаллов соли Формы Б. Водяную баню оставляли медленно остывать до температуры окружающей среды в течение ночи, при этом флакон продолжали вращать, и в смесь периодически вносили затравку нескольких кристаллов соли Формы Б. Образовывавшийся розовый осадок отделяли отсасыванием, промывали 2-пропанолом (2×50 мл) и сушили в вакууме при 100°С в течение 20 часов с получением указанной в заголовке соли (что было подтверждено XRPD) в виде бледно-розового твердого вещества (18,5 г, 84%). Соль содержала следы 2-пропанола.
1H ЯМР (299,95 МГц, DMSO-d6) δ 8,87 (s, 1H), 7,96-7,91 (m, 2H), 7,59-7,52 (m, 1H), 7,49-7,47 (m, 1H), 7,46-7,42 (m, 2H), 7,36 (d, J=8,6 Гц, 2H), 7,29 (d, J=8,6 Гц, 2H), 6,39 (d, J=2,5 Гц, 1H), 6,29 (dd, J=8,5, 2,5 Гц, 1H), 3,78-3,72 (m, 2H), 3,41 (s, 2H), 2,79-2,66 (m, 4H), 1,98 (s, 3H), 1,97-1,88 (m, 2H), 1,85-1,76 (m, 2H), 1,41-1,25 (m, 2H), 1,19 (s, 3H).
APCI-MS: m/z 462 [MH+].
Стехиометрия, отношение основания к кислоте 1:1, была подтверждена посредством ЯМР.
Бензоатная соль Формы Б демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (XRPD) (выраженные в градусах 2θ) (предел погрешности соответствует общей главе Фармакопеи Соединенных Штатов по дифракции рентгеновских лучей (USP941), смотри Конвенцию Фармакопеи Соединенных Штатов. Дифракция рентгеновских лучей, Общий анализ<941>. United States Pharmacopeia, 25th ed. Rockville, MD: United States Pharmacopeial Convention; 2002:2088-2089):
(1) 6,5, 9,3 и 10,5 или
(2) 6,5, 9,3, 17,6 и 17,8 или
(3) 6,5, 9,3, 10,5, 12,0 и 12,4 или
(4) 6,5, 9,3, 10,5, 12,0, 12,4, 13,0, 13,6, 15,5, 17,6 и 17,8 или
(5) 6,5, 13,0 и 20,2 или
(6) 6,5, 9,3, 10,5, 12,0, 12,4, 13,0, 13,6, 15,5, 17,6, 17,8 и 19,2 или
(7) 6,5, 9,3, 10,5, 12,0, 12,4, 13,0, 13,6, 15,5, 17,6, 17,8, 19,2, 20,2, 22,8 и 26,0 или
(8) 6,5, 9,3, 10,5, 12,0, 12,4, 13,0, 13,6, 15,5, 17,6, 17,8, 19,2, 20,2, 22,8, 24,2, 26,0 и 30,7.
Пример 1(в)
Получение фуроата N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (соль 1:1), Форма А.
(а) К перемешиваемому раствору N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (который может быть получен способами, описанными в WO 03/051839; 46 мг, 0,1 ммоль) и фуранкарбоновой кислоты (23 мг, 0,2 ммоль) в метаноле (0,2 мл), находящемуся в сосуде, добавляли диэтиловый эфир (5 мл) и сосуд закрывали. Полученную смесь перемешивали в течение 3 суток, образовавшийся осадок выделяли, промывали диэтиловым эфиром и сушили в вакууме с получением не совсем белого твердого вещества (38 мг). Это твердое вещество содержало указанную в заголовке соль в виде кристаллического вещества вместе с некоторым количеством аморфной соли. Указанная в заголовке соль содержала следовые количества диэтилового эфира.
1H ЯМР (299,946 МГц, DMSO-d6) δ 8,92 (s, 1H), 7,75-7,73 (m, 1H), 7,46 (d, J=8,6 Гц, 1H), 7,37 (d, J=4,4 Гц, 2Н), 7,29 (d, J=4,4 Гц, 2Н), 6,97-6,94 (m, 1H), 6,54 (dd, J=3,4, 1,7 Гц, 1H), 6,40 (d, J=2,4 Гц, 1H), 6,29 (dd, J=8,6, 2,4 Гц, 1H), 3,78 (s, 2Н), 3,43 (s, 2Н), 2,93 (d, J=12,1 Гц, 1H), 2,84-2,71 (m, 3H), 2,70-2,58 (m, 1H), 1,99 (s, 3H), 1,96-1,83 (m, 4H), 1,51-1,34 (m, 2H), 1,22 (s, 3H).
APCI-MS: m/z 462 [MH+].
Стехиометрия, отношение основания к кислоте 1:1, была подтверждена посредством ЯМР.
Дополнительные количества указанной в заголовке соли получали следующим способом.
(б) К раствору N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (230 мг, 0,5 ммоль) в метаноле (0,5 мл), находящемуся в сосуде, добавляли фуранкарбоновую кислоту (62 мг, 0,55 ммоль) в виде твердого вещества. Смесь встряхивали до образования раствора. Раствор разбавляли этилацетатом (6 мл), вносили затравку из частиц указанной в заголовке соли, полученной, как описано выше в (а), и оставляли на ночь в закрытом сосуде. Полученный осадок промывали этилацетатом и сушили в вакууме при 60°С в течение ночи с получением указанной в заголовке соли в виде не совсем белого твердого вещества (200 мг, 70%). Указанная в заголовке соль содержала следовые количества этилацетата.
1H ЯМР (299,946 МГц, DMSO-d6) δ 8,94 (s, 1H), 7,73-7,71 (m, 1H), 7,47 (d, J=8,6 Гц, 1Н), 7,37 (d, J=8,4 Гц, 2Н), 7,30 (d, J=8,4 Гц, 2Н), 6,94-6,91 (m, 1H), 6,52 (dd, J=3,3, 1,8 Гц, 1Н), 6,40 (d, J=2,2 Гц, 1Н), 6,30 (dd, J=8,6, 2,2 Гц, 1Н), 3,78 (s, 2Н), 3,43 (s, 2Н), 2,97 (d, J=11,9 Гц, 1H), 2,87-2,61 (m, 4H), 1,98 (s, 3H), 1,96-1,85 (m, 4H), 1,53-1,38 (m, 2Н), 1,23 (s, 3H).
APCI-MS: m/z 462 [MH+].
Стехиометрия, отношение основания к кислоте 1:1, была подтверждена посредством ЯМ Р.
Фуроатная соль Формы А демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (XRPD) (выраженные в градусах 2θ) (предел погрешности соответствует общей главе Фармакопеи Соединенных Штатов по дифракции рентгеновских лучей (USP941), смотри Конвенцию Фармакопеи Соединенных Штатов. Дифракция рентгеновских лучей, Общий анализ <941>. United States Pharmacopeia, 25th ed. Rockville, MD: United States Pharmacopeial Convention; 2002:2088-2089):
(1) 6,3, 11,0 и 12,7 или
(2) 6,3, 10,7 и 12,7 или
(3) 6,3, 11,0, 12,7 и 15,9 или
(4) 6,3, 10,7, 11,0, 12,7, 13,9, 14,2 и 15,9 или
(5) 6,3, 10,7, 11,0, 12,7, 15,9, 17,7, 19,1, 19,7 и 25,5 или
(6) 6,3, 10,7, 11,0, 12,7, 13,9, 14,2, 15,9, 17,7, 19,1, 19,7, 19,9, 21,6 и 25,5.
Получение фуроата N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (1:1 соль), Форма Б
(а) Форму Б получают путем растворения в сосуде 20 мас.% образца фуроатной соли, полученной методом, описанным в примере 1(б) (Форма А), в таком растворителе, как этанол (16 мг/мл) или 2-бутанол (8 мг/мл). Цифры в скобках указывают ожидаемую растворимость соли в этих растворителях. Затем сосуд запечатывали и суспензию гомогенизировали при температуре окружающей среды (20°С), используя магнит. Перемешивание и температуру поддерживали в течение по меньшей мере 7 суток, после чего образец полученного материала сушили и тестировали с помощью XRPD. XRPD подтвердила полную трансформацию Формы А в Форму Б.
Дополнительные количества указанной в заголовке соли получали с помощью следующего метода.
(б) Растворы N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида (46 мг, 0,10 ммоль) в 2-бутаноле (0,5 мл) и фуранкарбоновой кислоты (12,5 мг, 0,11 ммоль) в 2-бутаноле (0,5 мл) смешивали и вносили затравку нескольких кристаллов Формы Б. Смесь оставляли в закрытом сосуде при температуре окружающей среды в течение 3 суток. Полученный осадок промывали 2-бутанолом и сушили в вакууме при температуре 60°С в течение ночи с получением указанной в заголовке соли в виде не совсем белого твердого вещества. Соль содержала следы 2-бутанола.
Подлинность и стехиометрию, отношение основания к кислоте 1:1, подтверждали посредством ЯМР.
Фуроатная соль Формы Б демонстрирует по меньшей мере следующие характеристические пики дифракции рентгеновских лучей на порошке (XRPD) (выраженные в градусах 2θ) (предел погрешности соответствует общей главе Фармакопеи Соединенных Штатов по дифракции рентгеновских лучей (USP941), смотри Конвенцию Фармакопеи Соединенных Штатов. Дифракция рентгеновских лучей, Общий анализ<941>. United States Pharmacopeia, 25th ed. Rockville, MD: United States Pharmacopeial Convention; 2002:2088-2089):
(1) 6,7, 11,0 и 13,4 или
(2) 6,7, 10,4, 11,0 и13,4 или
(3) 6,7, 10,4, 12,4, 13,4 и 13,7 или
(4) 6,7, 10,4, 13,4 и 20,9 или
(5) 6,7, 10,4, 11,0, 12,4, 13,4, 13,7, 15,6, 16,0 и 17,6 или
(6) 6,7, 10,4, 11,0, 12,4, 13,4, 13,7, 15,6, 16,0, 16,1, 17,6, 18,0, 18,6, 18,9, 20,1, 20,9 и 23,4.
Пример 2
Ди-трифторацетат N-{5-хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}пропанамида.

(1) N-(2-Гидрокси-4-метоксифенил)пропанамид
К охлажденному на льду раствору гидрохлорида 2-гидрокси-4-метоксианилина (600 мг, 3,4 ммоль) и триэтиламина (3 экв.) в дихлорметане (25 мл) добавляли по каплям пропионовый ангидрид (1,1 экв.). Раствор оставляли при температуре окружающей среды в течение 5 часов. Реакцию гасили водой, слои разделяли и органическую фазу экстрагировали 1 н. NaOH (водн.) (3×25 мл). рН водной фазы доводили концентрированной HCl до 5 и экстрагировали дихлорметаном (3×25 мл). Органическую фазу сушили с помощью безводного сульфата натрия, фильтровали и удаляли в вакууме, получая указанное в подзаголовке соединение в виде коричневого твердого вещества (555 мг, 83%).
1H ЯМР (300 МГц, CDCl3-d6) δ 7,04 (b), 6,83 (d, J=8,4, 1 Н), 6,58 (d, J=2,8, 1H), 6,43 (dd, J1=8,4, J2=2,8, 1H), 3,77 (s, 3H), 2,49 (q, J=7,6, 2H), 1,29 (t, J=7,5, 3H). APCI-MS: m/z 196 [MH+].
(2) N-(5-Хлор-2-гидрокси-4-метоксифенил)пропанамид
К охлажденному на льду раствору N-(2-гидрокси-4-метоксифенил)пропанамида (500 мг, 2,6 ммоль) и гидрохлорида диметилформамида (1 экв.) в DMF (5 мл) добавляли небольшими порциями МСРВА (мета-хлорпербензойную кислоту) (70%, 1 экв.). Реакционную смесь перемешивали в течение еще 5 минут, после чего ее гасили 1 М NaHCO3 (водн.) (50 мл). Водную фазу промывали этилацетатом (50 мл). Органическую фазу промывали водой (3×25 мл), сушили и удаляли в вакууме, получая указанное в заголовке соединение в виде твердого вещества пурпурного цвета (408 мг, 71%).
1H ЯМР (300 МГц, ацетон-d6) δ 9,68 (b, 1H), 9,12 (b, 1H), 7,37 (s, 1H), 6,62 (s, 1H), 3,83 (s, 3H), 2,49 (q, J=7,7, 2H), 1,18 (t, J=7,5, 3H). APCI-MS: m/z 229 [M+].
(3) N-(5-Хлор-4-метокси-2-{[(2S)-метилоксиран-2-ил]метокси}фенил)пропанамид
Суспензию N-(5-хлор-2-гидрокси-4-метоксифенил)пропанамида (202 мг, 0,88 ммоль), [(2S)-2-метилоксиран-2-ил]метил-3-нитробензолсульфоната (1 экв.) и карбоната цезия (1,2 экв.) в DMF (4 мл) перемешивали при комнатной температуре в течение 4 часов. Смесь разделяли между водой (50 мл) и этилацетатом (50 мл). Органическую фазу промывали водой (2×30 мл), сушили и удаляли в вакууме, получая указанное в заголовке соединение в виде не совсем белого твердого вещества (249 мг, 95%).
1H ЯМР (300 МГц, CDCl3) δ 8,43 (s, 1Н), 7,80 (b, 1Н), 6,61 (s, 1H), 4,14 (m, 1H), 3,98 (m, 1Н), 3,85 (s, 3H), 2,94 (m, 1Н), 2,79 (m, 1Н), 2,42 (q, J=7,6, 2H), 1,47 (s, 3H), 1,25 (t, J=7,5, 3H).
APCI-MS: m/z 299 [MH+].
(4) N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}пропанамида ди-трифторацетат
К раствору 1-(4-хлорбензил)пиперидин-4-иламина (50 мг, 0,2 ммоль) и N-(5-хлор-4-метокси-2-{[(2S)-метилоксиран-2-ил]метокси}фенил)пропанамида (1 экв.) в ацетонитриле (5 мл) добавляли перхлорат лития (10 экв.). Реакционную смесь нагревали с обратным холодильником в течение 18 часов. Реакционную смесь наносили на колонку MEGA BE-SCX (Bond Elut®, 5 г, 20 мл). Колонку сначала промывали метанолом (3×10 мл) и затем смесью аммиак/метанол (1/20, 3×10 мл). Основные слои объединяли и растворитель удаляли в вакууме, получая N-{5-хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-метоксифенил}пропанамид в виде светло-коричневого масла (100 мг, 86%), который повторно растворяли в дихлорметане (4 мл). Раствор охлаждали до 0°С и добавляли по каплям 1 М BBr3 в дихлорметане (1 мл). Реакционную смесь перемешивали в течение 18 часов, после чего ее гасили метанолом. Растворитель удаляли в вакууме и осадок очищали с помощью обращенно-фазовой препаративной ВЭЖХ, используя ацетонитрил и воду, содержащую 0,1% TFA, в градиенте в качестве мобильной фазы. Объединенные фракции были лиофильно высушены с получением указанного в заголовке продукта в виде аморфного белого твердого вещества (38 мг, 39%).
1H ЯМР (300 МГц, ацетон-d6) δ 8,66 (уширенный), 8,09 (3, 1H), 7,60 (d, J=8,4, 4H), 7,47 (d, J=8,4, 4H), 6,78 (s, 1H), 4,41 (s, 2H), 4,10-3,93 (m, 2H), 3,70-3,65 (m, 4H), 3,44-2,39 (m, 1H), 3,20 (m, 2H), 2,52-2,37 (m, 6H), 1,38 (s, 3H), 1,10 (t, J=7,5, 3H). APCI-MS: m/z 510 [MH+].
Пример 3
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида ди-трифторацетат.
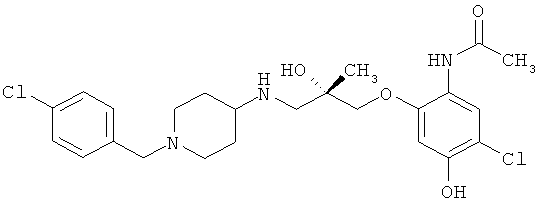
Синтез аналогичен описанному в примере 2, но где гидрохлорид 2-гидрокси-4-метоксианилина взаимодействует с уксусным ангидридом (1,1 экв.).
1H ЯМР (300 МГц, ацетон-d6) δ 8,77 (s, 1H), 8,06 (s, 1H), 7,61 (d, J=8,2 Гц, 2H), 7,47 (d, J=8,6 Гц, 2H), 6,79 (s, 1H), 4,43 (s, 2H), 4,08 (d, J=9,9 Гц, 1H), 3,94 (d, J=9,9 Гц, 1H), 3,79-3,61 (m, 3H), 3,68 (d, J=12.5 Гц, 1H), 3,42 (d, J=12,7 Гц, 1H), 3,32-3,13 (m, 2H), 2,63-2,48 (m, 2H), 2,49-2,29 (m, 2H), 2,08 (s, 3H), 1,38 (s, 3H). APCI-MS: m/z 496 [MH+].
Пример 4
N-{5-хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}ацетамида ди-трифторацетат.
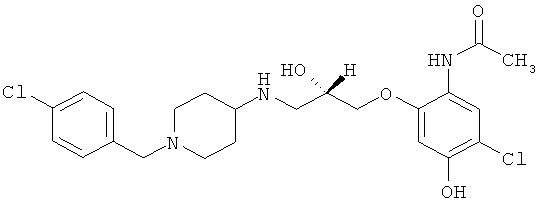
Синтез аналогичен описанному в примере 3, но N-(5-хлор-2-гидрокси-4-метоксифенил)ацетамид взаимодействует с S-(+)-глицидилнозилатом (1 экв.).
1H ЯМР (300 МГц, ацетон-d6) δ 8,64 (уширенный, NH), 8,21 (s, 1H), 7,59 (d, J=9,0 Гц, 2H), 7,47 (d, J=9,0 Гц, 2H), 6,74 (s, 1H), 4,41-4,35 (m, 3H), 4,13-4,01 (m, 2H), 3,69-3,40 (m, 5H), 3,14 (m, 2H), 2,55-2,47 (m, 2H), 2,31 (m, 2H), 2,09 (s, 3H). APCI-MS: m/z 482 [MH+].
Пример 5
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}пропанамида ди-трифторацетат.
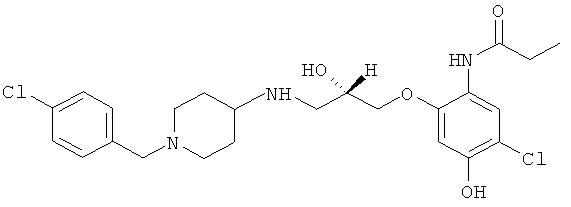
Синтез аналогичен описанному в примере 2, но N-(5-хлор-2-гидрокси-4-метоксифенил)ацетамид взаимодействует с S-(+)-глицидилнозилатом (1 экв.).
1H ЯМР (300 МГц, DMSO-d6) δ 10,05 (уширенный), 9,78 (уширенный), 9,79 (уширенный), 9,00 (уширенный), 8,88 (уширенный), 7,79 (m, 1H), 7,62-7,50 (m, 4H), 6,63 (s, 1H), 5,98 (уширенный), 4,29 (m, 2H), 4,16 (m, 1H), 3,95-3,88 (m, 2H), 3,41-2,97 (m, 7H), 2,35-2,22 (m, 4H), 1,82-1,75 (m, 2H), 1,07 (m, 3H). APCI-MS: m/z 496 [MH+].
Анализ связывания CCR1 человека
Мембраны
Для получения клеточных мембран, содержащих CCR1, использовали клетки HEK293 из ЕСАСС (Европейская коллекция культур клеток животных), стабильно экспрессирующие рекомбинантные CCR1 человека (HEK-CCR1). Мембраны хранили при -70°С. Концентрацию мембран в каждой партии доводили до 10% специфического связывания 33 пкМ [125I] MIP-1α.
Анализ связывания
100 мкл мембран HEK-CCR1 разбавляли в буфере для анализа с рН 7,4 (137 мМ NaCl (Merck, каталожный №1.06404), 5,7 мМ глюкозы (Sigma, каталожный № G5400), 2,7 мМ KCl (Sigma, каталожный №P-9333), 0,36 мМ NaH2PO4×H2O (Merck, каталожный №1.06346), 10 мМ HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота) (Sigma, каталожный №Н3375), 0,1% (мас./мас.) желатина (Sigma, каталожный №G2625) с добавлением 17500 единиц/л бацитрацина (Sigma, каталожный №В1025) добавляли в каждую лунку 96-луночного фильтровального планшета (0,45 мкм непрозрачный Millipore, каталожный номер MHVB N4550). 12 мкл соединения в буфере для анализа, содержащем 10% DMSO, добавляли с получением конечных концентраций соединения 1×10-5,5-1×10-9,5 M. В определенные лунки (без соединения) в качестве контроля неспецифического связывания (NBS) вносили по 12 мкл холодного рекомбинантного человеческого М1Р-1α (270-LD-050, R&D Systems, Oxford, UK), конечная концентрация в буфере для анализа с добавлением 10% DMSO составляла 10 нМ. В некоторые лунки (без соединения) добавляли по 12 мкл буфера для анализа с 10% DMSO для определения максимального связывания (ВО).
В лунки добавляли по 12 мкл [125I] MIP-1α, разведенного в буфере для анализа до конечной концентрации 33 пкМ. Планшеты с крышкой затем инкубировали в течение 1,5 часов при комнатной температуре. После инкубации лунки опустошали с помощью вакуумной фильтрации (Multiscreen Resist Vacuum Manifold system, Millipore) и промывали один раз 200 мкл буфера для анализа. После промывания во все лунки добавляли по 50 мкл сцинтилляционной жидкости (OptiPhase "Supermix", Wallac Oy, Turko, Финляндия). Связывание [125I] MIP-1α измеряли с помощью счетчика Wallac Trilux 1450 MicroBeta. Параметры окна: низкое 5, высокое 1020, 1-минутное считывание на лунку.
Вычисление процента замещения и IC50
Для вычисления процента замещения использовали следующее уравнение:
Процент замещения = 1 - ((импульс/мин тест - импульс/мин NSB) / (импульс/мин ВО - импульс/мин NSB)),
где импульс/мин тест = среднее значение импульс/мин в двух лунках с мембранами и соединением и [125I] MIP-1α, импульс/мин;
NSB = среднее значение импульс/мин в лунках с мембранами и MIP-1α и [125I] MIP-1α (неспецифическое связывание), импульс/мин;
ВО = среднее значение импульс/мин в лунках с мембранами и буфером для анализа и [125I] MIP-1α (максимальное связывание).
Молярную концентрацию соединения, приводящую к 50% замещению (IC50), получали, используя программу, основанную на Excel XLift (версия 2.0.9) для экстраполяции данных к 4-параметрической логарифмической функции.
Все соединения из примеров от 1 до 5 имели значения IC50 менее 20 нМ.
Эксперимент по притоку воспалительных клеток у крыс, подвергнутых контрольной провокации LPS
Эффект антагониста рецептора CCR1 (N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида гемифумарата (соль 2:1), упоминаемого здесь как Соединение А, и будесонида, и их комбинации на приток воспалительных клеток анализировали путем отслеживания эффекта на общее число клеток в жидкости бронхоальвеолярного лаважа (BAL) крыс, подвергнутых внутритрахеальной (в.т.) контрольной провокации липополисахаридом (LPS) [N=10 крыс на группу обработки].
Методология
Инстилляция LPS: Крыс анестезировали эфраном и размещали в положении супинации вверх головой на подставке под наклоном 30°. LPS (липополисахарид В E. coli 026:В6) (2,5 мкг/мл), растворенный в физиологическом растворе (0,9% NaCl), или один физиологический раствор (негативный контроль) в объеме 200 мкл вводили в.т., используя модифицированную металлическую канюлю. Крысы оставались в этом положении, пока к ним не возвращалось сознание.
Получение растворов. Гомогенизированный будесонид растворяли в этаноле и затем смешивали с 0,9%-ным раствором NaCl с получением конечной концентрации 0,002 мг будесонида/мл. Соединение А растворяли в 0,9%-ном растворе NaCl до конечной концентрации 0,034 мг/мл соединения А.
Смешанные препараты будесонид/соединение А получали путем растворения соединения А в суспензиях будесонида с получением конечной концентрации 0,034 мг соединения А/мл и 0,002 мг будесонида/мл.
Обработки. Животным внутритрахеально закапывали растворы (1 мл/кг) будесонида/соединения А (0,002/0,034 мг/кг), или одного будесонида (0,002 мг/кг), или одного соединения А (0,34 мг/кг), или физиологический раствор (негативные и позитивные контрольные животные). Данные обработки проводили под легкой анестезией (эфран), чтобы обеспечить достижение раствором легких. Лекарственные средства вводили за 30 мин до капельного введения LPS.
Завершение. Через 4 часа после контрольной провокации LPS крысам интраперитонеально инъецировали смесь (0,3 мл) пентобарбитала (60 мг/мл, Apoteksbolaget, Швеция) и PBS (1:1) в течение 1-2 мин.
Бронхоальвеолярный лаваж. После завершения дважды получали BAL с PBS. Жидкость BAL центрифугировали и клеточный осадок ресуспендировали в PBS. Общее число клеток BAL подсчитывали в счетчике клеток SYSMEX.
Результаты данного эксперимента показаны на чертеже, где крысы «физиологический раствор/физиологический раствор» представляют собой крыс негативного контроля, обработанных физиологическим раствором и подвергнутых контрольной провокации физиологическим раствором. Животные «физиологический раствор/LPS» представляют собой крыс позитивного контроля, обработанных физиологическим раствором и подвергнутых контрольной провокации LPS. Все остальные три группы обрабатывали указанными лекарственными средствами и подвергали контрольной провокации LPS.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВАЯ СОЛЬ I | 2006 |
|
RU2417220C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2298550C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2419608C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| ПРОИЗВОДНЫЕ ПИРАЗИНОНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЛЕГОЧНЫХ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2479580C2 |
| НОВЫЕ ТРИЦИКЛИЧЕСКИЕ СПИРОПИПЕРИДИНЫ ИЛИ СПИРОПИРРОЛИДИНЫ | 2003 |
|
RU2320664C2 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА, ИНГИБИРУЮЩИЕ Р38 КИНАЗУ | 2009 |
|
RU2512318C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ АНАБОЛИЧЕСКИХ АГЕНТОВ СКОТА | 2007 |
|
RU2419621C2 |
| АЗАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ ОПОСРЕДОВАННЫХ СЕРОТОНИНОМ ЗАБОЛЕВАНИЙ | 2001 |
|
RU2398765C1 |
| АГОНИСТЫ TLR7 | 2019 |
|
RU2817014C2 |

Изобретение относится к фармацевтическому продукту для лечения респираторного заболевания, содержащему в комбинации
(а) первый активный ингредиент, представляющий собой соединение общей формулы
где m равен 0, 1 или 2;
каждый R1 независимо представляет собой галоген или циано;
R2 представляет собой атом водорода или метил;
R3 представляет собой группу С1-С4алкил
и
R4 представляет собой водород или галоген,
или его фармацевтически приемлемую соль и
(б) второй активный ингредиент, который представляет собой глюкокортикостероид.
Изобретение также относится к применению продукта по любому из пп.1-15, к способу лечения респираторного заболевания, к набору, а также к фармацевтической композиции.
Технический результат - получение нового фармацевтического продукта для лечения респираторного заболевания. 5 н. и 30 з.п. ф-лы, 1 ил.
1. Фармацевтический продукт для лечения респираторного заболевания, содержащий в комбинации
(а) первый активный ингредиент, представляющий собой соединение общей формулы:

где m равен 0, 1 или 2;
каждый R1 независимо представляет собой галоген или циано;
R2 представляет собой атом водорода или метил;
R3 представляет собой группу С1-С4алкил; и
R4 представляет собой водород или галоген;
или его фармацевтически приемлемую соль; и
(б) второй активный ингредиент, который представляет собой
глюкокортикостероид.
2. Продукт по п.1, где R1 представляет собой галоген.
3. Продукт по п.1, где R4 представляет собой водород или хлор.
4. Продукт по п.1, где R4 представляет собой водород.
5. Продукт по п.1, где R3 представляет собой метил или этил.
6. Продукт по п.1, где первый активный ингредиент выбран из:
N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}пропанамида или
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}пропанамида,
или фармацевтически приемлемой соли любого из них.
7. Продукт по п.1, где первый активный ингредиент представляет собой соль, выбранную из бензоатной, фуроатной или гемифумаратной соли
N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида.
8. Продукт по п.1, где глюкокортикостероид представляет собой будесонид.
9. Продукт по п.1, который дополнительно содержит третий активный ингредиент, представляющий собой бронходилататор.
10. Продукт по п.9, где третий активный ингредиент представляет собой β2-агонист.
11. Продукт по п.10, где третий активный ингредиент выбран из формотерола или индакатерола.
12. Продукт по п.11, где третий активный ингредиент представляет собой формотерол.
13. Продукт по п.12, где третий активный ингредиент представляет собой формотерола фумарата дигидрат.
14. Продукт по п.1, который находится в форме, подходящей для введения посредством ингаляции.
15. Продукт по любому из пп.1-14 для применения в терапии респираторного заболевания.
16. Применение продукта по любому из пп.1-15 в изготовлении лекарственного средства для лечения респираторного заболевания.
17. Применение по п.16, где респираторное заболевание представляет собой хроническую обструктивную болезнь легких.
18. Применение по п.16, где респираторное заболевание представляет собой астму.
19. Способ лечения респираторного заболевания, включающий одновременное, последовательное или раздельное введение:
(а) (терапевтически эффективной) дозы первого активного ингредиента, представляющего собой соединение формулы (I), как оно определено в любом из пп.1-7, или его фармацевтически приемлемой соли;
(б) (терапевтически эффективной) дозы второго активного ингредиента, представляющей собой глюкокортикостероид; и возможно
(в) (терапевтически эффективной) дозы третьего активного ингредиента, представляющей собой бронходилататор;
пациенту, нуждающемуся в этом.
20. Набор для лечения респираторного заболевания, содержащий препарат первого активного ингредиента, представляющего собой соединение формулы (I), как оно определено в любом из пп.1-7, или его фармацевтически приемлемую соль, и препарат второго активного ингредиента, представляющего собой глюкокортикостероид, и возможно инструкции для одновременного, последовательного или раздельного введения данных препаратов пациенту, нуждающемуся в этом.
21. Набор по п.20, где первый активный ингредиент выбран из:
N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}пропанамида или
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}пропанамида,
или фармацевтически приемлемой соли любого из них.
22. Набор по п.20 или 21, где глюкокортикостероид представляет собой будесонид.
23. Набор по любому из пп.20-21, который дополнительно содержит препарат третьего активного ингредиента, представляющего собой бронходилататор.
24. Набор по п.23, где третий активный ингредиент представляет собой β2-агонист.
25. Набор по п.24, где третий активный ингредиент представляет собой формотерол или индакатерол.
26. Набор по п.25, где третий активный ингредиент представляет собой формотерол.
27. Набор по п.26, где третий активный ингредиент представляет собой формотерола фумарата дигидрат.
28. Фармацевтическая композиция для лечения респираторного заболевания, содержащая в смеси первый активный ингредиент, представляющий собой соединение формулы (I), как оно определено в любом из пп.1-7, или его фармацевтически приемлемую соль, и второй активный ингредиент, представляющий собой глюкокортикостероид.
29. Композиция по п.28, где первый активный ингредиент выбран из:
N-{2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}ацетамида,
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидроксипропил)окси]-4-гидроксифенил}пропанамида, или
N-{5-Хлор-2-[((2S)-3-{[1-(4-хлорбензил)пиперидин-4-ил]амино}-2-гидрокси-2-метилпропил)окси]-4-гидроксифенил}пропанамида,
или фармацевтически приемлемой соли любого из них.
30. Композиция по п.28, где глюкокортикостероид представляет собой будесонид.
31. Композиция по любому из пп.29-30, которая дополнительно содержит третий активный ингредиент, представляющий собой бронходилататор.
32. Композиция по п.31, где третий активный ингредиент представляет собой β2-агонист.
33. Композиция по п.31, где третий активный ингредиент представляет собой формотерол или индакатерол.
34. Композиция по п.32, где третий активный ингредиент представляет собой формотерол.
35. Композиция по п.34, где третий активный ингредиент представляет собой формотерола фумарата дигидрат.
| WO 03051839 A1, 26.06.2003 | |||
| US 3929768 A, 30.12.1975 | |||
| US 3994974 A, 30.11.1976 | |||
| WO 03082292 A1, 09.10.2003 | |||
| СПОСОБ ЛЕЧЕНИЯ ДЕФОРМИРУЮЩЕГО ГОНАРТРОЗА ПРИ ПОСЛЕДСТВИЯХ НЕПРАВИЛЬНО СРОСШЕГОСЯ ПЕРЕЛОМА ВНУТРЕННЕГО МЫЩЕЛКА БОЛЬШЕБЕРЦОВОЙ КОСТИ | 1994 |
|
RU2088167C1 |
| СПОСОБ РАСЧЕТА И НАСТИЛАНИЯ ТКАНИ | 0 |
|
SU245703A1 |
| Г ИДРОИМПУЛ ЬСАТОР | 0 |
|
SU198270A1 |
| RU 2004117887 C2, 20.04.2005. | |||

