ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Область изобретения
Данное изобретение относится к ряду новых гетероциклических соединений, полезных в лечении гиперпролиферативных заболеваний, таких как рак и воспаление, у млекопитающих. Данное изобретение также относится к способу применения таких соединений в лечении гиперпролиферативных заболеваний у млекопитающих, в частности у людей, и к фармацевтическим композициям, содержащим такие соединения.
Описание состояния уровня техники
Передача сигналов в клетках через рецепторы факторов роста и протеинкиназы является важным регулятором роста, пролиферации и дифференцировки клеток. При нормальном росте клеток факторы роста посредством активации рецепторов (то есть PDGF (фактор роста тромбоцитов) или EGF (эпидермальный фактор роста) и другие) активируют МАР-киназные пути (MAP - активируемый митогеном протеин). Одним из наиболее важных и наиболее хорошо изученных МАР-киназных путей, вовлеченных в нормальный и неконтролируемый клеточный рост, является Ras/Raf-киназный путь. Активный GТР (гуанозинтрифосфат)-связанный Ras приводит к активации и непрямому фосфорилированию Raf-киназы. Затем Raf фосфорилирует МЕК 1 и 2 (киназы митоген-активируемых ERK (киназ, регулируемых внеклеточными сигналами)) по двум остаткам серина (S218 и S222 для МЕК1 и S222 и S226 для МЕК2) (Ahn et al., Methods in Enzymology, 2001, 332, 417-431). Затем активированная МЕК фосфорилирует единственные известные для нее субстраты, МАР-киназы, ERK1 и 2. Фосфорилирование ERK посредством МЕК происходит по Y204 и Т202 для ERK1 и по Y185 и Т183 для ERK2 (Ahn et al., Methods in Enzymology, 2001, 332, 417-431). Фосфорилированная ERK димеризуется и затем транслоцируется в ядро, где происходит ее накопление (Khokhlatchev et al., Сеll, 1998, 93, 605-615). В ядре ERK вовлечена в ряд важных клеточных функций, включая, без ограничения, ядерный транспорт, передачу сигнала, репарацию ДНК, сборку и транслокацию нуклеосом, процессинг и трансляцию мРНК (Аhn et al., Molecular Cell, 2000, 6, 1343-1354). В целом, воздействие факторов роста на клетки приводит к активации ERK1 и 2, что вызывает пролиферацию и в некоторых случаях, дифференцировку (Lewis et al., Adv. Cancer Res., 1998, 74, 49-139).
При пролиферативных заболеваниях генетические мутации и/или сверхэкспрессия рецепторов факторов роста, расположенных ниже сигнальных белков или протеинкиназ, вовлеченных в ERK-киназный путь, приводит к неконтролируемой клеточной пролиферации и в итоге к образованию опухоли. Например, при некоторых злокачественных новообразованиях имеются мутации, вызывающие постоянную активацию этого пути вследствие непрерывного продуцирования факторов роста. Другие мутации могут приводить к дефектам в дезактивации активированного комплекса GTP-связанного Ras, снова приводя к активации МАР-киназного пути. Мутантные онкогенные формы Ras обнаружены в 50% случаев рака толстой кишки и более 90% случаев рака поджелудочной железы, а также при многих других видах рака (Kohl et al., Science, 1993, 260, 1834-1837). Недавно мутации bRaf были обнаружены более чем в 60% случаев злокачественной меланомы (Davies, H. et al., Nature, 2002, 417, 949-954). Эти мутации в bRaf приводят к конститутивно активному МАР-киназному каскаду. Исследования образцов первичных опухолей и клеточных линий также показали конститутивную или сверхактивацию МАР-киназного пути при раке поджелудочной железы, раке толстой кишки, раке легкого, раке яичника и раке почки (Hoshino, R. et al., Oncogene, 1999, 18, 813-822). Следовательно, существует строгая корреляция между раковыми заболеваниями и сверхактивным МАР-киназным путем в результате генетических мутаций.
Поскольку как конститутивная или сверхактивация МАР-киназного каскада играет решающую роль в клеточной пролиферации, дифференцировке, ингибирование этого каскада считается благоприятным при гиперпролиферативных заболеваниях. МЕК играет ключевую роль в этом пути, поскольку она расположена ниже Ras и Raf. К тому же она является привлекательной терапевтической мишенью, поскольку известными субстратами для МЕК-фосфорилирования являются только МАР-киназы, ERK1 и 2. В нескольких исследованиях было показано, что ингибирование МЕК имеет потенциальную терапевтическую пользу. Например, было показано, что ингибиторы МЕК, имеющие небольшие молекулы, ингибируют рост опухолей человека в ксенотрансплантатах "голых" мышей (Sebolt-Leopold et al., Nature-Medicine 1999, 5 (7), 810-816; Trachet et al., AACR April 6-10, 2002 Poster №5426; Tecle, H. IBC 2nd International Conference of Protein Kinases, September 9-10, 2002), блокируют статическую аллодинию у животных (WO 01/05390) и ингибируют рост клеток при остром миелоидном лейкозе (Milella et al., J. Clin. Invest. 2001, 108 (6), 851-859).
Ингибиторы МЕК, имеющие небольшие молекулы, были раскрыты, в том числе в публикациях патентов США №№2003/0232869, 2004/0116710 и 2003/0216460 и в заявках на патент США №№10/654580 и 10/929295, каждая из которых включена в данное описание посредством ссылки. За последние несколько лет появилось по меньшей мере пятнадцать дополнительных патентных заявок (см., например, патент США 5525625; WO 98/43960; WO 99/01421; WO 99/01426; WO 00/41505; WO 00/42002; WO 00/42003; WO 00/41994; WO 00/42022; WO 00/42029; WO 00/68201; WO 01/68619; WO 02/06213; WO 03/077914; и WO 03/077855.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение предусматривает новые гетероциклические соединения и их фармацевтически приемлемые соли и пролекарства, которые полезны в лечении гиперпролиферативных заболеваний. Обнаружено, что 6-оксо-1,6-дигидропиридазиновые и 6-оксо-1,6-дигидропиридиновые соединения, имеющие конкретные заместители, которые указаны в данном описании, являются сильнодействующими ингибиторами фермента МЕК.
Более конкретно в одном аспекте настоящего изобретения предложены соединения, включая таутомеры, метаболиты, разделенные энантиомеры, диастереомеры, сольваты и их фармацевтически приемлемые соли, имеющие формулу I:
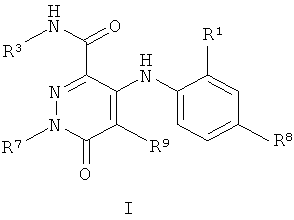
где R1 представляет собой Cl или F;
R3 представляет собой Н, Me, Et, ОН, МеО-, ЕtO-, НОСН2СН2O-, НОСН2С(Ме)2O-, (S)-MeCH(OH)CH2O-, (R)-HOCH2CH(OH)CH2O-, циклопропил-CH2O-, НОСН2СН2-,
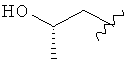 ,
, 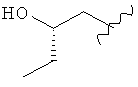 ,
, 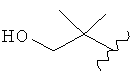 ,
, 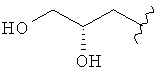 ,
,  или
или 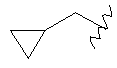 ;
;
R7 представляет собой циклопропил-СН2- или С1-С4алкил, где указанный алкил возможно замещен одним или более чем одним F;
R8 представляет собой Вr, I или SMe; и
R9 представляет собой СН3, CH2F, CHF2, СF3, F или Cl.
В еще одном аспекте этого изобретения предложены соединения, включая таутомеры, метаболиты, разделенные энантиомеры, диастереомеры, сольваты и их фармацевтически приемлемые соли, имеющие формулу IV:
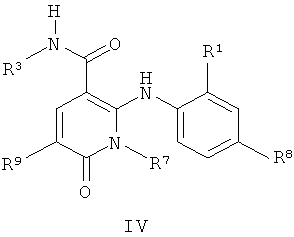
где R1 представляет собой Cl или F;
R3 представляет собой Н, Me, Et, ОН, МеО-, ЕtO-, НОСH2СН2О-, НОСН2С(Ме)2O-, (S)-MeCH(OH)CH2O-, (R)-HOCH2CH(OH)CH2O-, циклопропил-СН2О-, НОСН2СН2-,
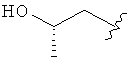 ,
, 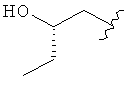 ,
, 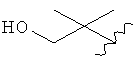 ,
, 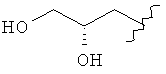 ,
,  или
или 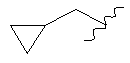 ;
;
R7 представляет собой метил или этил, где указанные метил и этил возможно замещены одним или более чем одним F;
R8 представляет собой Вr, I или SMe; и
R9 представляет собой Н, С1-C4алкил, Cl или CN, где указанный алкил возможно замещен одной или более группами, независимо выбранными из F или CN, при условии, что:
а) когда R1 представляет собой F, R8 представляет собой Br, R9 представляет собой Н, и R3 представляет собой НОСН2СН2O, тогда R7 не может представлять собой Me или Et;
б) когда R1 представляет собой F, R8 представляет собой I, R9 представляет собой Н, и R3 представляет собой МеО, тогда R7 не может представлять собой Me;
в) когда R1 представляет собой F, R8 представляет собой Me, R9 представляет собой Н, и R3 представляет собой НОСН2СН2O, тогда R7 не может представлять собой Me; и
г) когда R1 представляет собой F, R8 представляет собой Br, R9 представляет собой Н, и R3 представляет собой циклопропил-СH2О, тогда R7 не может представлять собой Me.
В другом аспекте настоящего изобретения предложены две кристаллические формы соединения формулы XI:
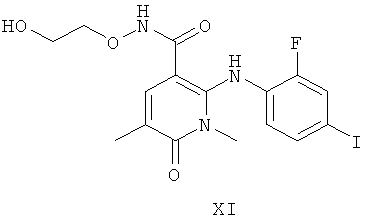
которые обозначены как Форма 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида и Форма 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
Предложены также способы получения Формы 1 и Формы 2 соединения формулы XI.
В другом аспекте настоящего изобретения предложены композиции, которые ингибируют МЕК, содержащие одно или более соединений по настоящему изобретению.
Согласно изобретению предложены также способы получения соединений по настоящему изобретению.
В еще одном аспекте настоящего изобретения предложен способ применения соединений по данному изобретению в качестве лекарственного средства для лечения заболеваний или медицинских состояний, опосредованных МЕК. Например, согласно данному изобретению предложено соединение по данному изобретению в качестве лекарственного средства для лечения гиперпролиферативного расстройства или воспалительного состояния у млекопитающего, включающего введение указанному млекопитающему одного или более соединений по настоящему изобретению или их фармацевтически приемлемых солей или пролекарств в количестве, эффективном для лечения указанного гиперпролиферативного расстройства. В другом аспекте данного изобретения предложено соединение по данному изобретению в изготовлении лекарственного средства для лечения гиперпролиферативного расстройства или воспалительного состояния.
В другом аспекте настоящего изобретения предложен способ продуцирования эффекта ингибирования МЕК у теплокровного животного, например человека, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения по данному изобретению.
В другом аспекте настоящего изобретения предложено лечение или предупреждение состояния, опосредованного МЕК, включающее введение человеку или животному, нуждающемуся в этом, фармацевтической композиции, содержащей соединение по настоящему изобретению или его фармацевтически приемлемую соль или in vivo расщепляемое пролекарство в количестве, эффективном для лечения или предупреждения указанного состояния, опосредованного МЕК.
Соединения по изобретению могут быть также предпочтительно использованы в комбинации с другими известными терапевтическими агентами.
Изобретение также относится к фармацевтическим композициям, которые ингибируют МЕК, содержащим эффективное количество соединения, выбранного из соединений по настоящему изобретению или их фармацевтически приемлемых пролекарств, фармацевтически активных метаболитов или фармацевтически приемлемых солей.
В дополнительном аспекте изобретения предложено применение соединения по настоящему изобретению в изготовлении лекарственного средства для лечения или предупреждения заболевания или медицинского состояния, опосредованного МЕК, у теплокровного животного, предпочтительно млекопитающего, более предпочтительно человека, страдающего таким расстройством. Более конкретно изобретение включает в себя применение соединения по изобретению в изготовлении лекарственного средства для лечения или предупреждения гиперпролиферативного расстройства или воспалительного состояния у млекопитающего.
Дополнительные преимущества и новые признаки данного изобретения частично изложены в нижеследующем описании и будут очевидны специалистам в данной области техники по мере изучения нижеследующего описания или могут быть изучены при осуществлении изобретения на практике. Преимущества изобретения могут быть реализованы и достигнуты с помощью средств, комбинаций, композиций и способов, в частности, указанных в прилагаемой формуле изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Сопровождающие графические материалы, которые включены в данное описание и составляют его часть, иллюстрируют неограничивающие воплощения настоящего изобретения и вместе с описанием служат для объяснения принципов изобретения.
В графических материалах:
На Фиг.1 представлена реакционная схема синтеза соединения 96.
На Фиг.2 представлена реакционная схема синтеза соединений 96, 100, 101 и 102.
На Фиг.3 представлена реакционная схема синтеза соединений 109, 110 и 111.
На Фиг.4 представлена альтернативная реакционная схема синтеза соединений 109, 110 и 111.
На Фиг.5 представлена реакционная схема синтеза соединений 119, 120 и 121.
На Фиг.6 представлена реакционная схема синтеза соединений 124 и 125.
На Фиг.7 представлена реакционная схема синтеза соединений 128, 129 и 130.
На Фиг.8 представлена реакционная схема синтеза соединений 145 и 146.
На Фиг.9 представлена альтернативная реакционная схема синтеза соединения 145.
На Фиг.10 представлена картина дифракции рентгеновских лучей на порошке для Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, полученной согласно Примеру 16А, Стадия 3.
На Фиг.11 представлена картина дифракции рентгеновских лучей на порошке для Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, полученной согласно Примеру 16А, Стадия 4.
На Фиг.12 представлена картина дифракции рентгеновских лучей на порошке для Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, полученной согласно Примеру 16Б.
На Фиг.13 представлена картина дифракции рентгеновских лучей на порошке для Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, полученной согласно Примеру 16Г.
На Фиг.14 представлена DSC-термограмма (DSC - дифференциальная сканирующая калориметрия) для Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
На Фиг.15 представлена DSC-термограмма для Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения по настоящему изобретению и таутомеры, метаболиты, разделенные энантиомеры, диастереомеры, сольваты и их фармацевтически приемлемые соли и пролекарства полезны в лечении гиперпролиферативных заболеваний. В общем, один аспект настоящего изобретения относится к соединениям по настоящему изобретению, которые действуют в качестве ингибиторов МЕК.
Более конкретно согласно одному аспекту настоящего изобретения предложены соединения, включая таутомеры, метаболиты, разделенные энантиомеры, диастереомеры, сольваты и их фармацевтически приемлемые соли, имеющие формулу I:
где R1 представляет собой Cl или F;
R3 представляет собой Н, Me, Et, ОН, МеО-, ЕtO-, HOCH2CH2O-, HOCH2C(Me)2O-, (S)-MeCH(OH)CH2O-, (R)-HOCH2CH(OH)CH2O-, циклопропил-CH2O-, НОСН2СН2-,
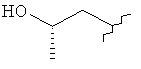 ,
, 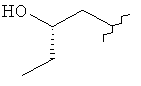 ,
, 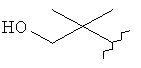 ,
,  ,
,  или
или 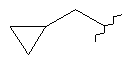 ;
;
R7 представляет собой циклопропил-СН2- или С1-С4алкил, где указанный алкил возможно замещен одним или более чем одним F;
R8 представляет собой Вr, I или SMe; и
R9 представляет собой СН3, CH2F, CHF2, СF3, F или Cl.
В одном из воплощений изобретения предложены соединения, включая таутомеры, метаболиты, разделенные энантиомеры, диастереомеры, сольваты, и их фармацевтически приемлемые соли, имеющие формулу IA:
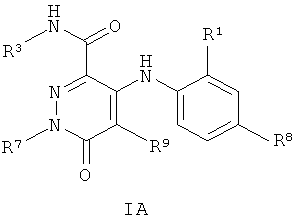
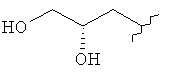
где R1 представляет собой Cl или F;
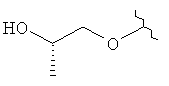
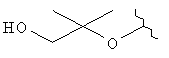
R3 представляет собой Н, Me, ОН, МеО, ЕtO, HOCH2CH2O, MeOCH2CH2O, HOCH2CH2CH2, 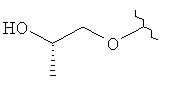 ,
, 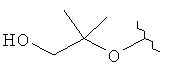 ,
, 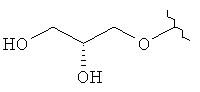 ,
, 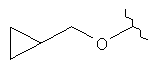 ,
, 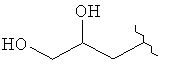 ,
, 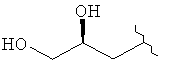 или
или 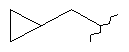 ;
;
R7 представляет собой циклопропил-СН2- или С1-С4алкил, где указанный алкил возможно замещен одним или более чем одним F;
R8 представляет собой Вr, I или SMe; и
R9 представляет собой СН3, CH2F, CHF2, СF3, F или Cl.
В одном из воплощений в соединениях формулы I или IА R7 представляет собой циклопропил-СН2- или Me. В другом воплощении R9 представляет собой СН3, F или Cl.
В другом воплощении предложено соединение формулы II
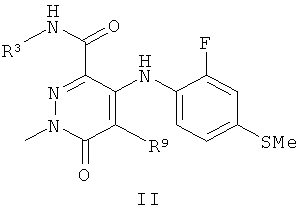
или его фармацевтически приемлемая соль,
где R3 представляет собой Н, МеО, НОСН2СН2O, МеОСН2СН2O, НОСН2СН2СН2, 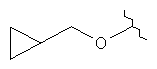 ,
,  или ; и
или ; и
R9 представляет собой Н, СН3, F или Cl.
Соединения формулы II, имеющие метильный заместитель в положении N1 и конкретные группы R3 и R9, являются сильнодействующими ингибиторами МЕК.
Конкретные новые соединения по изобретению включают любое из следующих соединений:
4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
N-(циклопропилметокси)-4-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-(метилтио)фениламино)-N-(2-метоксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-(метилтио)фениламино)-N-метокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(S)-4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
5-фтор-4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(S)-5-фтор-4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
5-хлор-4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(S)-5-хлор-4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-(метилтио)фениламино)-N-(3-гидроксипропил)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид; и
(S)-N-(2,3-дигидроксипропил)-4-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид.
В другом воплощении предложено соединение формулы III:
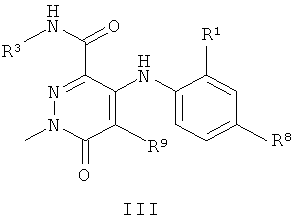
или его фармацевтически приемлемая соль,
где R1 представляет собой Cl или F;
R3 представляет собой Н, Me, MeO, HOCH2CH2O, HOCH2CH2CH2, НОСН2СН2,
,  , , , , или
, , , , или 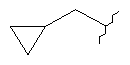 ;
;
R8 представляет собой Вr или I; и
R9 представляет собой СН3, F, Cl или Вr.
Соединения формулы III, имеющие метильный заместитель в положении N1 и конкретные группы R1, R3 R8 и R9, являются сильнодействующими ингибиторами МЕК.
Конкретные новые соединения по изобретению включают любое из следующих соединений:
5-бром-4-(4-бром-2-фторфениламино)-N-(циклопропилметокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(R)-N-(2,3-дигидроксипропокси)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-йодфениламино)-N-метокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
N-(циклопропилметокси)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(S)-4-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-хлор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(S)-4-(2-хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(4-бром-2-хлорфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(S)-4-(4-бром-2-хлорфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(R)-4-(4-бром-2-фторфениламино)-N-(2,3-дигидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(4-бром-2-фторфениламино)-N-(1-гидрокси-2-метилпропан-2-илокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(4-бром-2-фторфениламино)-5-фтор-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-йодфениламино)-N,1,5-триметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
N-(циклопропилметил)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-йодфениламино)-N-(3-гидроксипропил)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
5-фтор-4-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
4-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтил)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
N-(2,3-дигидроксипропил)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
5-хлор-4-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(S)-5-хлор-4-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
5-хлор-4-(2-фтор-4-йодфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
5-хлор-N-(2,3-дигидроксипропил)-4-(2-фтор-4-йодфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид;
(S)-N-(2,3-дигидроксипропил)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид; и
(S)-5-хлор-N-(2,3-дигидроксипропил)-4-(2-фтор-4-йодфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид.
Конкретные новые соединения по изобретению также включают следующие соединения:
4-(4-бром-2-фторфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид; и
(S)-4-(4-бром-2-фторфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид.
В еще одном аспекте данного изобретения предложены соединения, включая таутомеры, метаболиты, разделенные энантиомеры, диастереомеры, сольваты, и их фармацевтически приемлемые соли, имеющие формулу IV:
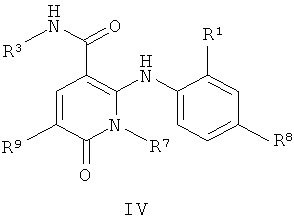
где R1 представляет собой Cl или F;
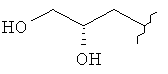
R3 представляет собой Н, Me, Et, ОН, МеО-, ЕtO-, НОСН2СН2O-, HOCH2C(Me)2O-, (S)-MeCH(OH)CH2O-, (R)-HOCH2CH(OH)CH2O-, циклопропил-СН2O-, НОСН2СН2-,  ,
,  ,
,  ,
,  ,
, 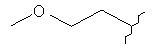 или
или 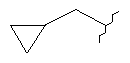 ;
;
R7 представляет собой метил или этил, которые возможно замещены одним или более чем одним F;
R8 представляет собой Вr, I или SMe; и
R9 представляет собой Н, С1-С4алкил, Cl или CN, где указанный алкил возможно замещен одной или более группами, независимо выбранными из F или CN, при условии, что когда
а) R1 представляет собой F, R8 представляет собой Вr, R9 представляет собой Н, и R7 представляет собой либо Me, либо Et, тогда R3 не может представлять собой НОСН2СН2O;
б) R1 представляет собой F, R8 представляет собой I, R9 представляет собой Н, и R3 представляет собой МеО, тогда R7 не может представлять собой Me;
в) R1 представляет собой F, R8 представляет собой Me, R9 представляет собой Н, и R3 представляет собой HOCH2CH2O, тогда R7 не может представлять собой Me; и
г) R1 представляет собой F, R8 представляет собой Вr, R9 представляет собой Н, и R3 представляет собой циклопропил-СН2О, тогда R7 не может представлять собой Me.
В одном из воплощений в соединениях формулы IV R9 представляет собой Н, Me, Et, Cl или CN.
В одном из воплощений соединения по изобретению или их фармацевтически приемлемые соли имеют формулу V:
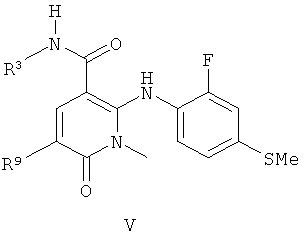
где R3 представляет собой НОСН2СН2O или (S)-MeCH(OH)CH2O; и
R9 представляет собой Н, СН3, F или Cl при условии, что когда R1 представляет собой F, R8 представляет собой SMe, R9 представляет собой Cl, и R7 представляет собой Me, тогда R3 не может представлять собой HOCH2CH2O.
Соединения формулы V, где R3 представляет собой HOCH2CH2O или (S)-MeCH(OH)CH2O, являются сильнодействующими ингибиторами МЕК.
Конкретные новые соединения по изобретению включают любое из следующих соединений:
2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид; и
(S)-5-хлор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид.
В одном воплощении соединения по изобретению или их фармацевтически приемлемые соли имеют формулу VI:
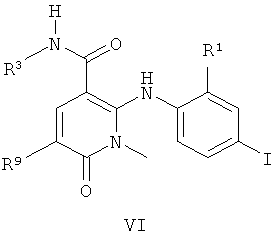
где R1 представляет собой Cl или F;
R3 представляет собой Н, HOCH2CH2O или (S)-МеСН(ОН)СН2O; и
R9 представляет собой Н, Me, F или Cl.
Соединения формулы VI, где R1 представляет собой Cl, R3 представляет собой НОСН2СН2O или (S)-МеСН(ОН)СН2O, и R9 представляет собой Н, являются сильнодействующими ингибиторами МЕК.
Соединения формулы VI, где R1 представляет собой F, R3 представляет собой Н, и R9 представляет собой Me, являются сильнодействующими ингибиторами МЕК.
Соединения формулы VI, где R3 представляет собой HOCH2CH2O или (S)-MeCH(OH)CH2O, являются сильнодействующими ингибиторами МЕК.
Соединение формулы VI, где R1 представляет собой F, R3 представляет собой НОСH2СН2О, и R9 представляет собой Me, является сильнодействующим ингибитором МЕК, а также имеет хорошую растворимость. Используемый здесь термин «хорошая растворимость» относится к соединению, которое имеет растворимость более 50 мкг/мл, например растворимость приблизительно 50-270 мкг/мл, которая определена способом из Примера В.
Конкретные новые соединения формулы VI по изобретению включают любое из следующих соединений:
2-(2-хлор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
2-(2-хлор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
5-хлор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-5-хлор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
5-фтор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид; и
(S)-5-фтор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид.
В другом воплощении предложено соединение формулы VI, где R1 представляет собой F, R3 представляет собой HOCH2CH2O, и R9 представляет собой метил, или его фармацевтически приемлемая соль.
Было обнаружено, что соединение формулы XI:
может существовать в двух кристаллических формах, далее обозначенных как Форма 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида и Форма 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, и что Форма 2 может превращаться в Форму 1.
Образцы конкретных кристаллических форм соединения формулы XI анализировали, используя комбинацию анализа дифракции рентгеновских лучей на порошке (XRPD) и дифференциальной сканирующей калориметрии, как описано в Примерах 16Д и 16Е.
Если установлено, что настоящее изобретение относится к кристаллической форме соединения формулы XI, обычно степень кристалличности по данным дифракции рентгеновских лучей на порошке составляет более приблизительно 60%, предпочтительно более приблизительно 80%, предпочтительно более приблизительно 90% и еще более предпочтительно более приблизительно 95%.
Согласно другому аспекту изобретения предложена кристаллическая форма соединения формулы XI по существу в виде Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
Согласно другому аспекту изобретения предложена кристаллическая форма соединения формулы XI по существу в виде Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
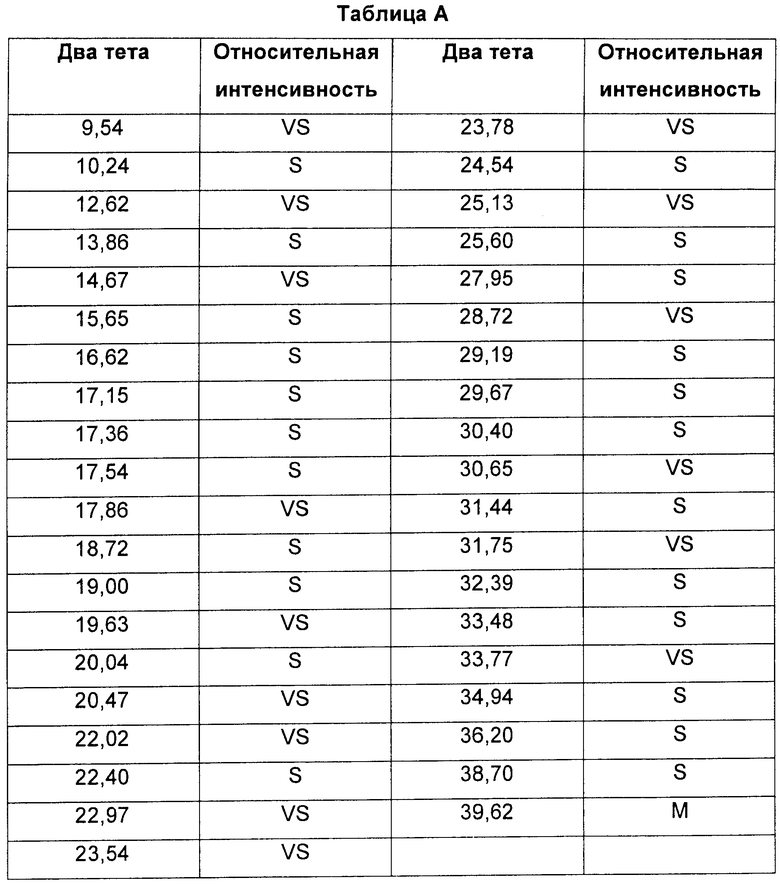
Соединение формулы XI в виде Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида имеет картину дифракции рентгеновских лучей с характеристическими пиками по шкале 2 тета (θ) при примерно 9,5 и 12,6. Согласно другому аспекту изобретения предложено соединение формулы XI в виде Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, которое имеет картину дифракции рентгеновских лучей с характеристическими пиками по шкале 2 тета (θ) при примерно 9,5; 12,6; 14,7 и 19,6.
Форма 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида имеет картину дифракции рентгеновских лучей по существу такую, как показано на Фиг.10, с характеристическими пиками [по шкале 2 тета (θ)] примерно в положениях, указанных в Таблице А.
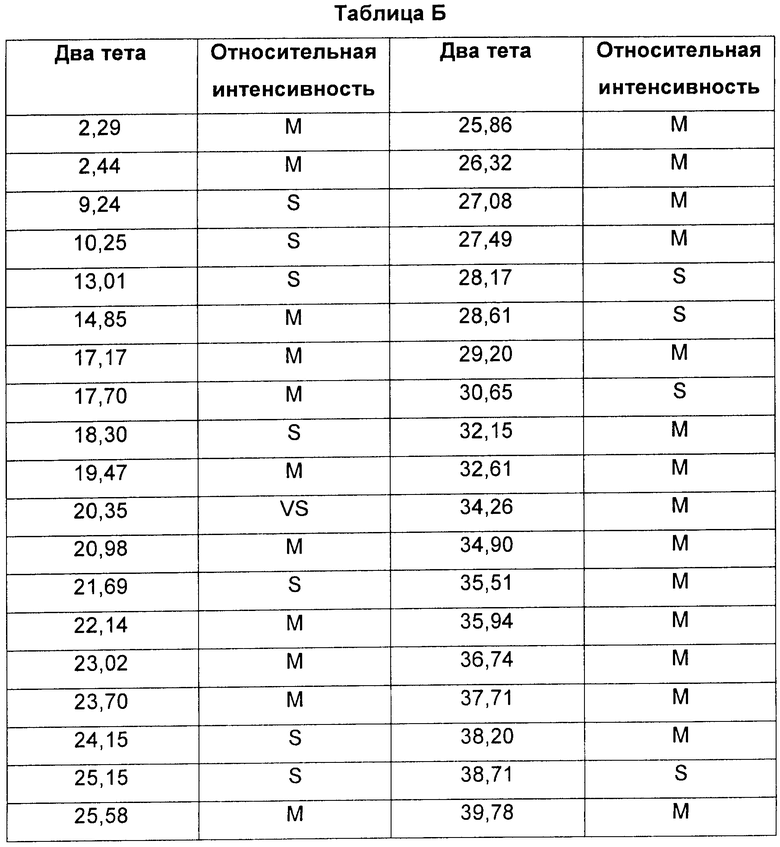
Соединение формулы XI в виде Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида имеет картину дифракции рентгеновских лучей с характеристическими пиками по шкале 2 тета (θ) при примерно 9,2 и 13,0. Согласно другому аспекту изобретения предложено соединение формулы XI в виде Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, которое имеет картину дифракции рентгеновских лучей с характеристическими пиками по шкале 2 тета (θ) при примерно 9,2; 13,0; 18,3; 21,0 и 21,7.
Форма 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида имеет картину дифракции рентгеновских лучей по существу такую, как показано на Фиг.11 или 12, с характеристическими пиками [по шкале 2 тета (θ)] примерно в положениях, указанных в Таблице Б.
Как упомянуто выше, интенсивности пиков на XRPD-дифрактограмме могут демонстрировать некоторую вариабельность в зависимости от использованных условий измерений. Соответственно в Таблицах А и Б числовые значения относительных интенсивностей не приведены. Точнее использованы следующие определения для интенсивности:
где относительные интенсивности получены из картин дифракции рентгеновских лучей, измеренных с использованием вариабельных щелей.
На Фиг.13 представлена картина дифракции рентгеновских лучей на порошке для Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, полученной согласно Примеру 16Г.
Видно, что некоторые из более минорных пиков, присутствующих на картине дифракции рентгеновских лучей на Фиг.10-13, не указаны в Таблицах А и Б.
Соединение формулы XI в виде Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида имеет картину дифракции рентгеновских лучей, по существу такую же, как показано на Фиг.10.
Соединение формулы XI в виде Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида имеет картину дифракции рентгеновских лучей, по существу такую же, как показано на Фиг.11 или 12.
В предыдущих фрагментах текста, где определены пики рентгеновской порошковой дифракции для кристаллических форм 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, термин «при примерно» использован в выражении «…по шкале 2 тета (θ) при примерно…», чтобы указать, что точное положение пиков (то есть приведенные значения угла 2-тета) не следует истолковывать как абсолютные величины, поскольку, как очевидно специалистам в данной области, точное положение пиков может незначительно варьировать от прибора к прибору, от образца к образцу или как результат небольших вариаций в использованных условиях измерения. Кроме того, в предыдущих абзацах установлено, что кристаллические формы 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида дают картины дифракции рентгеновских лучей на порошке «по существу» такие же, как картины дифракции рентгеновских лучей на порошке, представленные на Фиг.10-13, и имеют по существу самые отчетливые пики (значения углов 2-тета), показанные в Таблицах А и Б соответственно. Ясно, что использованный в этом контексте термин «по существу» также предназначен для указания того, что значения углов 2-тета на картинах дифракции рентгеновских лучей на порошке могут слегка варьировать от прибора к прибору, от образца к образцу или как результат незначительных вариаций в использованных условиях измерений, поэтому положения пиков, показанные на Фигурах или указанные в Таблицах А и Б, не следует истолковывать как абсолютные величины.
В данном описании раскрыты способы получения соединения формулы XI либо в Форме 1 либо в Форме 2.
В одном аспекте предложен способ получения соединения формулы XI по существу в виде Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, включающий:
а) приведение (2-винилоксиэтокси)-амида 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты в контакт с кислотной смесью в течение времени, достаточного для превращения этого соединения в 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
б) выдержку вещества со стадии (а) до кристаллизации из органического растворителя, содержащего затравку из Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида; и
в) выделение Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
В одном аспекте кислотная смесь на стадии (а) может представлять собой неорганическую или органическую кислоту. В другом аспекте стадия (а) может быть проведена в двухфазной системе растворителей водная кислота - этилацетат. В одном аспекте органическим растворителем на стадии (б) является этилацетат.
В другом аспекте предложен способ получения соединения формулы XI по существу в виде Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, включающий:
а) перемешивание Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида с небольшим количеством Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида в органическом растворителе; и
б) выделение Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
В одном аспекте количество вещества Формы 1, использованное на стадии (а), равно примерно 5% мас./мас.
В другом аспекте стадию (а) проводят в этилацетате при температуре слегка выше температуры окружающей среды, например примерно 50-60°С.
В другом аспекте изобретения предложен способ получения соединения формулы XI по существу в виде Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида по п.1 формулы изобретения, включающий:
а) приведение в контакт (2-винилоксиэтокси)-амида 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты с кислотной смесью в течение времени, достаточного для превращения соединения в 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
б) выдержку вещества со стадии (а) до кристаллизации из органического растворителя; и
в) выделение Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
В одном аспекте органический растворитель на стадии (б) содержит затравку из Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида. Кислотная смесь на стадии (а) может представлять собой неорганическую или органическую кислоту, и стадия (а) может быть проведена в органическом растворителе, таком как THF (тетрагидрофуран). В одном аспекте органический растворитель на стадии (б) может быть выбран из этилацетата и метилизобутилкетона, оба возможно в присутствии изогексана.
Некоторые соединения по данному изобретению могут существовать в виде двух или более таутомерных форм. «Таутомер» представляет собой один из двух или более структурных изомеров, которые существуют в равновесии и легко превращаются из одной изомерной формы в другую, например структуры, образованные в результате перемещения водорода из одного места в другое в пределах одной и той же молекулы. Другие таутомерные формы соединений могут взаимно обмениваться, например, посредством енолизации/деенолизации и тому подобного. Соответственно настоящее изобретение охватывает получение всех таутомерных форм соединений по данному изобретению.
Соединения по данному изобретению могут иметь один или более асимметрических центров, поэтому такие соединения могут быть получены в виде индивидуальных (R)- или (S)-стереоизомеров или в виде их смесей. Если не указано иное, описание или название конкретного соединения в описании изобретения и в формуле изобретения включает оба его индивидуальных энантиомера, его диастереомерные смеси, рацемические или иные. Соответственно изобретение также охватывает все такие изомеры, в том числе диастереомерные смеси и разделенные энантиомеры соединений по данному изобретению. Диастереомерные смеси могут быть разделены на их индивидуальные диастереомеры на основе их физико-химических различий способами, известными специалистам в данной области техники, например хроматографией или фракционной кристаллизацией. Энантиомеры могут быть разделены путем превращения энантиомерной смеси в диастереомерную смесь в результате взаимодействия с подходящим оптически активным соединением (например спиртом), разделения диастереомеров и превращения (например гидролизом) индивидуальных диастереомеров в соответствующие чистые энантиомеры. Методы определения стереохимии и разделения стереоизомеров общеизвестны в данной области (см. обсуждение в Главе 4 "Advanced Organic Chemistry", 4th edition, J. March, John Wiley and Sons, New York, 1992).
Данное изобретение также охватывает фармацевтические композиции, содержащие соединение по настоящему изобретению, и способы лечения пролиферативных расстройств или аномального роста клеток путем введения соединения по настоящему изобретению. Соединения по настоящему изобретению, имеющие свободные амино, амидо, гидрокси или карбоксильные группы, могут быть превращены в фармацевтически приемлемые пролекарства.
«Пролекарство» представляет собой соединение, которое в физиологических условиях или в результате сольволиза может превращаться в конкретное соединение или в фармацевтически приемлемую соль такого соединения. Пролекарства включают соединения, где аминокислотный остаток или полипептидная цепь из двух или более (например двух, трех или четырех) аминокислотных остатков ковалентно присоединены посредством амидной или эфирной связи к свободной амино, гидрокси или карбоксильной группе соединений по настоящему изобретению. Аминокислотные остатки включают 20 существующих в природе аминокислот, обычно обозначаемых трехбуквенными символами, а также включают 4-гидроксипролин, гидроксилизин, демозин, изодемозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитрулин, гомоцистеин, гомосерин, орнитин и метионинсульфон, но ими не ограничиваются. Одно предпочтительное пролекарство по данному изобретению представляет собой соединение по настоящему изобретению, ковалентно соединенное с остатком валина.
Также охвачены дополнительные типы пролекарств. Например, могут быть получены производные свободных карбоксильных групп в виде амидов или алкиловых сложных эфиров. В качестве другого примера в виде пролекарств производные соединений по данному изобретению, содержащие свободные гидроксильные группы, могут быть получены путем превращения гидроксильной группы в фосфатный сложный эфир, гемисукцинаты, диметиламиноацетат или фосфорилоксиметилоксикарбонил, как описано в Advanced Drug Delivery Reviews, 1996, 19, 115. Также охвачены карбаматные пролекарства гидрокси и аминогрупп, как представляющие собой карбонатные пролекарства, сульфонатные сложные эфиры и сульфатные сложные эфиры гидроксильных групп. Также охвачено получение производных гидроксильных групп в виде (ацилокси)метиловых и (ацилокси)этиловых простых эфиров, где ацильная группа может быть алкиловым сложным эфиром, возможно замещенным группами, включающими, без ограничения, эфирные, аминные или карбоновокислотные функциональные группы, или где ацильная группа является сложным эфиром аминокислоты, как описано выше. Пролекарства этого типа описаны в J. Med. Chem., 1996, 39, 10. Более конкретные примеры включают замену атома водорода спиртовой группы, такой группой, как (С1-С6)алканоилоксиметил, 1-((С1-С6)алканоилокси)этил, 1-метил-1-((С1-С6)алканоилокси)этил, (С1-С6)алкоксикарбонилоксиметил, N-(С1-С6)алкоксикарбониламинометил, сукциноил, (С1-С6)алканоил, α-амино(С1-С4)алканоил, арилацил и α-аминоацил или α-аминоацил-α-аминоацил, где каждая α-аминоацильная группа независимо выбрана из существующих в природе L-аминокислот, групп Р(O)(ОН)2, -Р(O)(O(С1-С6)алкил)2 или гликозила (радикала, образующегося в результате удаления гидроксильной группы полуацетальной формы углевода).
Также могут быть получены производные свободных аминов в виде амидов, сульфонамидов или фосфонамидов. Например, пролекарство может быть образовано при замене атома водорода в аминной группе такой группой, как R-карбонил, RO-карбонил, NRR'-карбонил, где R и R' каждый независимо представляет собой (С1-С10)алкил, (С3-С7)циклоалкил, бензил, или R-карбонил представляет собой природный α-аминоацил или (природный α-аминоацил)-(природный α-аминоацил), -C(OH)C(O)OY, где Y представляет собой Н, (С1-С6)алкил или бензил, -C(OY0)Y1, где Y0 представляет собой (С1-С4)алкил, и Y1 представляет собой (С1-С6)алкил, карбокси(С1-С6)алкил, амино(С1-С4)алкил или моно-N- либо ди-N,N-(С1-С6)алкиламиноалкил, -С(Y2)Y3, где Y2 представляет собой Н или метил, и Y3 представляет собой моно-N- или ди-N,N-(С1-С6)алкиламино, морфолино, пиперидин-1-ил или пирролидин-1-ил.
Все эти пролекарственные группировки могут инкорпорировать группы, включая эфирные, аминные или карбоновокислотные функциональные группы, но ими не ограничиваются.
Пролекарства соединения по настоящему изобретению могут быть идентифицированы с использованием рутинных методик, известных в данной области. В данной области известны различные формы пролекарств. Примеры таких пролекарственных производных см., например, в a) Design of Prodrugs, edited by Н.Bundgaard (Elsevier, 1985) и Methods in Enzymology, Vol.42, p.309-396, edited by K.Widder, et al. (Academic Press, 1985); б) A Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H.Bundgaard, Chapter 5 "Design and Application of Prodrugs" by H.Bundgaard, p.113-191 (1991); в) H.Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992); г) H.Bundgaard, et al., Journal of Pharmaceutical Sciences, 77: 285 (1988); и д) N.Kakeya, et al., Chem. Pharm. Bull., 32: 692 (1984), которые все включены в данное описание посредством ссылки.
Изобретение также охватывает сольваты, метаболиты и фармацевтически приемлемые соли соединений по настоящему изобретению.
Термин «сольват» относится к агрегату молекулы с одной или более молекулами растворителя.
«Метаболит» представляет собой фармакологически активный продукт, образующийся в процессе in vivo метаболизма конкретного соединения или его соли в организме. Такие продукты могут образовываться, например, в результате окисления, восстановления, гидролиза, амидирования, деамидирования, этерификации, деэтерификации, ферментативного расщепления и т.п. введенного соединения. Соответственно изобретение охватывает метаболиты соединений по настоящему изобретению, в том числе соединения, полученные способом, включающим приведение соединения по данному изобретению в контакт с млекопитающим в течение периода времени, достаточного для получения его метаболического продукта.
Обычно метаболиты идентифицируют путем получения меченного радиоактивным изотопом (например, 14С или 3H) соединения по изобретению, введения его парентерально в детектируемой дозе (например, более примерно 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна или человек, выдержки его в течение времени, достаточного для осуществления метаболизма (обычно от примерно 30 секунд до 30 часов) и выделения продуктов его превращения из мочи, крови или других биологических образцов. Эти продукты выделяют без труда, поскольку они меченые (другие выделяют путем использования антител, способных связывать эпитопы, оставшиеся в метаболите). Структуры метаболитов определяют общепринятыми методами, например MS (масс-спектрометрия), LC/MS (жидкостная хроматография/масс-спектрометрия) или ЯМР (ядерный магнитный резонанс). Как правило, анализ метаболитов выполняют так же, как и традиционные исследования метаболизма лекарственных средств, хорошо известные специалистам в данной области. Метаболиты, если только иным способом они не обнаруживаются in vivo, полезны в диагностических тестах по терапевтическому дозированию соединений по изобретению.
Используемый здесь термин «фармацевтически приемлемая соль», если не указано иное, охватывает соли, которые сохраняют биологическую эффективность свободных кислот и оснований конкретного соединения и которые не являются биологически или по-иному нежелательными. Соединение по изобретению может иметь достаточно кислотные, достаточно основные или и те и другие функциональные группы и соответственно могут взаимодействовать с любым из целого ряда неорганических или органических оснований и неорганических и органических кислот с образованием фармацевтически приемлемой соли. Примеры фармацевтически приемлемых солей включают такие соли, полученные в результате взаимодействия соединений по настоящему изобретению с минеральной или органической кислотой или неорганическим основанием, причем такие соли включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себакаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксиленсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, γ-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты и манделаты. Поскольку одно соединение по настоящему изобретению может содержать более чем одну кислотную или основную группировку, соединения по настоящему изобретению могут включать моно-, ди- или три-соли в одном соединении.
Если соединение по изобретению представляет собой основание, то желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, известным в данной области техники, например путем обработки свободного основания кислотным соединением, в частности неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, или органической кислотой, такой как уксусная кислота, яблочная кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-оксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота, или тому подобное.
Если соединение по изобретению представляет собой кислоту, желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, например путем обработки свободной кислоты неорганическим или органическим основанием. Предпочтительными неорганическими солями являются соли, образованные со щелочными и щелочноземельными металлами, такими как литий, натрий, калий, барий и кальций. Предпочтительные соли органических оснований включают, например, соли аммония, дибензиламмония, бензиламмония, 2-гидроксиэтиламмония, бис(2-гидроксиэтил)аммония, фенилэтилбензиламина, дибензилэтилендиамина и тому подобные соли. Другие соли кислотных группировок могут включать, например, соли, образованные с прокаином, хинином и N-метилглюкозамином, плюс соли, образованные с основными аминокислотами, такими как глицин, орнитин, гистидин, фенилглицин, лизин и аргинин.
Способы получения соединений по настоящему изобретению предложены в качестве дополнительных признаков изобретения. Соединения по изобретению могут быть получены согласно реакционным путям и схемам синтеза, как описано ниже, с использованием методик, известных в данной области, с использованием исходных веществ, которые доступны или могут быть синтезированы способами, известными в данной области.
Иллюстрации получения соединений по настоящему изобретению представлены на Фиг.1-7.
Получение соединения 96 изображено на Фиг.1. Замещенный гидразин 28 может быть превращен в гидразонопропаноат 29 по двухстадийной методике. На первой стадии гидразин 28 подвергают конденсации с этилпируватом в стандартных условиях дегидратирования, например в присутствии MgSO4 в подходящем органическом растворителе, таком как хлороформ или метиленхлорид, при температуре в диапазоне от 0°С до температуры окружающей среды. На второй стадии ацилирование достигается в результате обработки основанием при пониженной температуре в подходящем органическом растворителе, таком как THF, DMF (диметилформамид), диоксан или МеСN, с последующим добавлением метилмалонилхлорида. В одном воплощении гидразон обрабатывают LiH в THF при 0°С, после чего добавляют метилмалонилхлорид и нагревают до комнатной температуры. Гидроксипиридазинон 31 получают из гидразонопропаноата 29 путем циклизации в сильно основных условиях с последующим декарбоксилированием. Циклизация может быть осуществлена путем обработки гидразонопропаноата 29 сильным основанием, таким как DBU (1,8-диазабицикло[5.4.0]ундец-7-ен), LDA (диизопропиламид лития) или NaH, в подходящем органическом растворителе, таком как THF или МеСN, при комнатной температуре. В одном воплощении циклизация достигается с использованием DBU в MeCN при комнатной температуре. Декарбоксилирование с образованием гидроксипиридазинона 31 может быть достигнуто путем нагревания группировки сложного метилового эфира пиразинона в подходящем органическом растворителе, таком как диоксан или декалин или смесь диоксан/декалин, до повышенных температур в присутствии концентрированной HCl. Карбоновая кислота 94 может быть получена из гидроксипиридазинона 31 в двухстадийном процессе, т.е. хлорированием с последующим окислением. Стадия хлорирования может быть осуществлена путем обработки РОСl3, тионилхлоридом, оксалилхлоридом или PCl5. В одном воплощении это превращение достигается с использованием чистого РОСl3 при повышенной температуре (приблизительно 85°С). После стадии хлорирования карбоновая кислота 94 может быть получена путем окисления в стандартных условиях, включая, без ограничений, КМnO4 в воде, SeO2 в органическом растворителе, таком как диоксан, ксилол или пиридин, NaOCl/RuCl3, СrО3 в водной Н2SO4, K2Cr2O7 и Na2Cr2O7 в воде. В одном воплощении это превращение достигается с использованием К2Сr2O7-Н2SO4. Карбоновая кислота 94 может быть превращена в сложный эфир пиридазинона 95 двухстадийным способом, включающим этерификацию пиридазиноновой кислоты 94 с последующей опосредованной палладием реакцией поперечного связывания. Этерификация может быть осуществлена в стандартных условиях, включая, без ограничений, концентрированную НСl в МеОН, TMSCl в МеОН или TMSCHN2 в подходящих органических растворителях, таких как смесь эфир/МеОН, THF/MeOH или РhМе/МеОН. Опосредованная палладием реакция поперечного связывания может быть осуществлена стандартными способами, включая, без ограничений, обработку эфира хлорпиридазинона анилином, палладиевым катализатором, таким как Pd(OAc)2, PdCl2(dppf), Pd(Ph3P)4 или Pd2dba3, фосфиновым лигандом и основанием в подходящем органическом растворителе, таком как THF, DMF, PhMe, DME (диметиловый эфир) или MeCN при повышенной температуре. В одном воплощении реакция поперечного связывания включает в себя обработку сложного эфира 94 Pd(OAc)2, рацемическим 2,2-бис(дифенилфосфино)-1,1'-бинафтилом и Сs2СО3 в толуоле при 70-100°С. В воплощениях соединения 95, где желательно, чтобы R9 представлял собой Вr, заместитель бром может быть введен после реакции поперечного связывания. Бромирование пиридазинона может быть осуществлено с использованием NBS (N-бромсукцинимид) в подходящем органическом растворителе, таком как DMF, MeCN или смешанные системы растворителей, при комнатной температуре. В одном воплощении бромирование проводят в DMF. Гидроксамат 96 может быть получен путем обработки эфира пиридазинона 95 соответствующим гидроксиламином и амидным основанием, таким как LDA, LiHMDS (гексаметилдисилазид лития) или NaHMDS (гексаметилдисилазид натрия), в подходящем органическом растворителе, таком как THF, при пониженной температуре. В одном воплощении раствор LiHMDS добавляют к раствору эфира пиридазинона 95 и гидроксиламина в THF при 0°С. Реакционную смесь затем нагревают до комнатной температуры с получением желаемого гидроксамата 96. В некоторых случаях гидроксиламин, используемый в реакции сочетания, содержит стандартную защитную группу. В таких случаях защитная группа может быть удалена в стандартных условиях, известных в данной области техники.
На Фиг.2 представлен синтез соединений 96, 100, 101 и 102. Замещенный гидразин 28 может быть превращен в гидразономалонат 97 по одной из двух методик. В одном воплощении конденсация замещенного гидразина 28 с последующим ацилированием особенно подходит для аналогов, где R9 представляет собой алкил или галоген. В этом воплощении гидразин 28 может быть подвергнут конденсации с диэтил-2-оксомалонатом в стандартных условиях дегидратирования с использованием ловушки Дина-Старка в подходящем органическом растворителе, таком как бензол или толуол, при температуре в диапазоне от 80 до 120°С. Ацилирование реагентом, который доставляет ацильную группу, с получением гидразономалоната 97 достигается путем обработки основанием при соответствующей температуре в подходящем органическом растворителе, таком как THF, DMF, диоксан или MeCN, с последующим добавлением ацилирующего реагента. Примеры ацилирующих реагентов общеизвестны специалистам в данной области и включают, без ограничений, хлорангидриды кислот, ангидриды карбоновых кислот и активированные сложные эфиры. В одном воплощении гидразон обрабатывают LiH в THF при 0°С, после чего добавляют хлорангидрид кислоты и перемешивают при 25-60°С с получением соединения 97. Альтернативный способ синтеза соединения 97, где R9 не является галогеном, включает ацилирование гидразина 28 реагентом, который доставляет ацильную группу, затем конденсацию с диэтил-2-оксомалонатом с получением гидразономалоната 97. В соответствии с этим способом замещенный гидразин 28 может быть превращен в гидразид стандартными способами ацилирования. В одном воплощении это превращение достигается с использованием соответствующего хлорангидрида кислоты в метиленхлориде при температуре от 0°С до температуры окружающей среды. Полученный гидразид подвергают конденсации с диэтилкетомалонатом в стандартных условиях дегидратирования, используя ловушку Дина-Старка, в подходящем органическом растворителе, таком как бензол или толуол, при температуре от 80 до 130°С. Пиридазинон 99 получают из гидразономалоната 97 путем циклизации в щелочных условиях с получением промежуточной кислоты или промежуточного сложного эфира 98, затем хлорированием с получением пиридазинона 99. Циклизация может быть осуществлена путем обработки гидразономалоната 97 амидным основанием, таким как LiHMDS, NaHMDS, KHMDS или LDA, в подходящем органическом растворителе, таком как THF или эфир, при пониженной температуре. В одном воплощении циклизация достигается с использованием LiHMDS в THF при пониженной температуре (от -78 до -40°С) с последующей обработкой концентрированной НСl с получением сложноэфирного производного 98 (R=Et). В другом воплощении кислотное производное 98 (R=Н) получают путем омыления in situ сложноэфирного производного пиридазинона 98. По завершении циклизации реакционную смесь гасят водой при низкой температуре (от -78 до -40°С), затем нагревают до температуры окружающей среды при перемешивании, после чего подкисляют. Пиридазинон 99 затем получают из кислотного или сложноэфирного производного пиридазинона 98 путем обработки РОСl3, тионилхлоридом, оксалилхлоридом или PCl5. В одном воплощении это превращение достигается с использованием чистого РОСl3 при повышенной температуре (приблизительно 85°С). Когда R9 не представляет собой F, кислотное производное пиридазинона 99 (когда R=Н) далее может быть превращено в пиридазинон 101. Введение анилиновой группировки достигается в результате SNАr реакции в подходящем органическом растворителе, таком как THF, с использованием амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при подходящей температуре (от -78°С до комнатной температуры). В одном воплощении анилин добавляют к LDA или LiHMDS в THF при низкой температуре (от -20 до -80°С). Затем добавляют кислотное производное пиридазинона 99 (R=Н), и реакционную смесь нагревают до комнатной температуры до образования карбоновой кислоты 101. Гидроксаматы 96 и амиды 102 далее могут быть получены из кислоты 101 с использованием стандартного реагента сочетания, такого как, без ограничений, 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), 1-гидроксибензотриазол-6-сульфонамидометил-гидрохлорид (HOBt) или бензотриазол-1-ил-окситрипирролидинофосфонийгексафторфосфат (РуВОР), и соответствующего амина или гидроксиламина в подходящем органическом растворителе, таком как DMF, THF или метиленхлорид. В некоторых случаях амин или гидроксиламин содержит стандартную защитную группу. В таких случаях защитная группа может быть удалена в стандартных условиях, известных в данной области техники. Альтернативно, эфирное производное пиридазинона 99 (R=Et) может быть превращено в гидроксамат 96 через эфирное производное пиридазинона 100 стандартными способами, представленными на Фиг.1. Когда требуется, чтобы R8 представлял собой Вr или I, требуемый галоген может быть введен с использованием NBS или NIS (N-йодсукцинимид) в подходящем органическом растворителе или смешанной системе растворителей, таких как DMF, THF-MeOH или AcOH-THF, в присутствии подходящего кислотного катализатора.
На Фиг.3 представлен синтез соединений 109, 110 и 111, где в качестве исходного вещества используют 2,6-дихлорникотиновую кислоту. Никотиновую кислоту 103 превращают в монохлорсодержащую кислоту 104 путем кипячения с обратным холодильником в 2 н. водном NaOH по методике, описанной в патенте США №3682932. Алкилирование соединения 104 с получением соединения 105 может быть достигнуто в стандартных условиях основного алкилирования, включающих использование алкилгалогенидов, с использованием двух эквивалентов соответствующего алкилгалогенида и основания с получением смеси сложноэфирного производного N-алкилпиридона 105 и сложноэфирного производного региоизомерного O-алкилпиридина, которые легко разделяются колоночной хроматографией. Эти условия включают, без ограничений, К2СО3 в ацетоне или DMF при комнатной или повышенной температуре или NaH в THF при температуре окружающей среды или повышенной температуре и последующее добавление алкилгалогенида. В некоторых воплощениях это алкилирование достигается с использованием LiH в DMF при 0°С с последующим добавлением алкилбромида или алкилйодида и нагреванием до комнатной температуры. Бромирование сложноэфирного производного пиридона 105 может быть осуществлено с использованием либо Вr2 и уксусной кислоты либо NBS в подходящем органическом растворителе, таком как DMF. В некоторых воплощениях NBS добавляют к раствору сложноэфирного производного пиридона 105 в DMF с получением соединения 106. Превращение бромида 106 в соединение 107 может быть достигнуто с использованием условий Pd-опосредованного поперечного связывания. Когда R9 представляет собой алкенил или алкинил, их далее можно восстановить, используя подходящий восстанавливающий агент, с получением алкильных заместителей по R9. Как правило, эта химическая реакция может быть осуществлена с использованием различных Pd катализаторов и лигандов, с добавлением основания или без добавления основания, в подходящем органическом растворителе, таком как DMF, PhMe, DME, THF, СН3СN, при повышенной температуре. Партнер сочетания будет зависеть от природы R9. Например, если желательно, чтобы R9 представлял собой CN, партнером сочетания является Zn(CN)2. Эта реакция может быть проведена с использованием Pd2dba3 и dppf в NMP (N-метилпирролидон) при 120°С. Такие реакции палладий-опосредованного поперечного связывания хорошо задокументированы в литературе и хорошо известны специалисту в данной области техники. Введение соответствующим образом замещенной анилиновой группировки с получением соединения 108 достигается SNAR реакцией. Она может быть проведена в подходящем органическом растворителе, таком как THF, с использованием амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при подходящей температуре (от -78°С до комнатной температуры). В некоторых воплощениях анилин добавляют к LDA или LiHMDS в THF при пониженной температуре (от -20 до -80°С). Затем добавляют пиридон 105, и эту смесь перемешивают при пониженной температуре до образования сложного эфира 108. Затем может быть получена карбоновая кислота 109 в стандартных условиях омыления, таких как LiOH или NaOH в стандартных смешанных системах водных/органических растворителей. Гидроксамат 110 и амид 111 могут быть получены по стандартным методикам сочетания, включая, без ограничений, EDCI, HOBt или РуВОР и соответствующий амин или гидроксиламин в подходящих органических растворителях, таких как DMF, THF или метиленхлорид. В некоторых воплощениях сочетание осуществляют с использованием HOBt и EDCI в DMF. В некоторых случаях амин или гидроксиламин, используемый в реакции сочетания, содержит стандартную защитную группу. В таких случаях защитная группа может быть удалена в стандартных условиях, известных в данной области техники.
На Фиг.4 представлена альтернативная реакционная схема синтеза соединений 109, 110 и 111. Этот путь особенно подходит для аналогов, где R7 не представляет собой Me или Et. Никотиновая кислота 103 может быть превращена в метиловый эфир N-алкилпиридона 114 после проведения семистадийной процедуры, где сначала 2,6-дихлор-никотиновую кислоту 103 превращают в метоксипиридиновую кислоту, которую этерифицируют до образования метилового эфира, а затем удаляют защиту с получением монохлорсодержащего эфира 112. В некоторых воплощениях превращение в метоксипиридиновую кислоту осуществляют путем добавления трет-бутоксида калия к раствору кислоты 103 в МеОН, и эту смесь затем нагревают до температуры дефлегмации в течение нескольких суток. Этерификация с получением метилового эфира может быть проведена в стандартных условиях, включающих, без ограничений, этерификацию по Фишеру (МеОН, H2SO4), TMSCI в МеОН или TMSCHN2 в подходящих органических растворителях, таких как РhМе/МеОН. Затем может быть осуществлено деметилирование метоксипиридина в стандартных условиях, включающих, без ограничений, HCl при повышенной температуре, п-TsOH (п-толуолсульфоновая кислота) в уксусной кислоте при повышенной температуре и водный НВr в МеОН при повышенной температуре. Предпочтительно деметилирование с получением пиридона 112 достигается путем обработки метоксипиридина водным НВr в уксусной кислоте при повышенной температуре (80-120°С). Алкилирование соединения 112 может быть достигнуто в стандартных условиях основного алкилирования, включающего использование алкилгалогенидов, с использованием одного эквивалента соответствующего алкилгалогенида и основания с получением смеси эфирного производного N-алкилпиридона 113 и эфирного производного региоизомерного O-алкилпиридина, которые легко разделяются колоночной хроматографией. Эти условия включают, без ограничений, К2СО3 в ацетоне или DMF при комнатной или повышенной температуре или NaH в THF при температуре окружающей среды или повышенной температуре и последующее добавление алкилгалогенида. В некоторых воплощениях это алкилирование достигается с использованием LiH в DMF при 0°С с последующим добавлением алкилбромида или алкилйодида и нагреванием до комнатной температуры. Бромирование сложноэфирного производного пиридона 113 может быть выполнено с использованием либо Br2 и уксусной кислоты либо NBS в подходящем органическом растворителе, таком как DMF. В некоторых воплощениях NBS добавляют к раствору сложноэфирного производного пиридона 113 в DMF с получением соединения 114. Превращение бромида 114 в соединение 115 может быть достигнуто с использованием условий палладий-опосредованного поперечного связывания. Когда R9 представляет собой алкенил или алкинил, их далее можно восстановить, используя подходящий восстанавливающий агент, с получением алкильных заместителей по R9. Как правило, эта химическая реакция может быть осуществлена с использованием различных Pd катализаторов и лигандов, с добавлением основания или без добавления основания, в подходящем органическом растворителе, таком как DMF, РhМе, DME, THF, СН3СN, при повышенной температуре. Партнер сочетания будет зависеть от природы R9. Такие реакции Pd-опосредованного поперечного связывания хорошо задокументированы в литературе и хорошо известны специалисту в данной области техники. Введение соответствующим образом замещенной анилиновой группировки с получением соединения 116 достигается SNAR реакцией. Она может быть проведена в подходящем органическом растворителе, таком как THF, с использованием амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при подходящей температуре (от -78°С до комнатной температуры). В некоторых воплощениях анилин добавляют к LDA или LiHMDS в THF при пониженной температуре (от -20 до -80°С). Затем добавляют пиридон 115, и эту смесь перемешивают при низкой температуре до образования сложного эфира 116. Превращение соединения 116 в карбоновую кислоту 109, а также в гидроксамат 110 и амид 111 может быть осуществлено так, как описано для Фиг.3. Альтернативно гидроксамат 110 может быть получен прямо из метилового эфира 116 в подходящем органическом растворителе, таком как THF, с использованием соответствующего гидроксиламина и амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при подходящей температуре (от -78°С до комнатной температуры). В некоторых воплощениях раствор LiHMDS добавляют к раствору сложного эфира 116 и гидроксиламина в THF при 0°С. Реакционную смесь затем нагревают до комнатной температуры с получением желаемого гидроксамата 110. В некоторых случаях гидроксиламин, используемый в реакции сочетания, содержит стандартную защитную группу. В таких случаях защитная группа может быть удалена в стандартных условиях, известных в данной области техники.
На Фиг.5 представлена реакционная схема синтеза соединений 119, 120 и 121, где в качестве исходного вещества используют метиловый эфир N-алкилпиридона 112. Образование соединения 117 может быть осуществлено в результате введения соответствующим образом замещенной анилиновой группировки SNAR реакцией. Она может быть проведена в подходящем органическом растворителе, таком как THF, с использованием амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при подходящей температуре (от -78°С до комнатной температуры). В некоторых воплощениях анилин добавляют к LDA или LiHMDS в THF при низкой температуре (от -20 до -80°С). Затем добавляют пиридон 112, и эту смесь перемешивают при низкой температуре с получением сложного эфира 117. Это может быть выполнено в подходящем органическом растворителе, таком как THF, с использованием амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при соответствующих температурах (от -78°С до комнатной температуры). Хлорирование пиридона 117 с получением пиридона 118 может быть осуществлено с использованием стандартных условий, таких как NCS (N-хлорсукцинимид) в подходящем органическом растворителе, таком как DMF. Превращение соединения 118 в карбоновую кислоту 119, а также в гидроксамат 120 и амид 121 может быть осуществлено так, как описано для Фиг.3 и 4.
На Фиг.6 представлена реакционная схема синтеза соединений 124 и 125. 4-Фторпиридазинон 123 может быть получен из 4-хлорпиридазинона 122 путем обработки KF или HF в присутствии или в отсутствие основания, такого как Et3N или Me3N, в подходящих органических растворителях, таких как CH3CN, THF, DMF, NMP или DMSO (диметилсульфоксид). В одном воплощении это превращение достигается с использованием KF в DMSO при повышенной температуре (например, 160°С). Сложноэфирное производное пиридазинона 123 (когда R=Et) может быть превращено в пиридазинон 124, причем введение анилиновой группировки достигается в результате проведения SNAR реакции. Она может быть проведена в подходящем органическом растворителе, таком как DMF, EtOH, iPrOH, CH3CN или THF, с использованием основания, такого как Сs2СО3, NаНСО3, К2СО3 или Nа2СО3, при температуре от 80 до 160°С. В одном воплощении анилин и Сs2СО3 добавляют к раствору пиридазинона 123 в DMF, и эту реакционную смесь нагревают до 80°С. Альтернативно кислотное производное пиридазинона 123 (R=Н) может быть превращено в пиридазинон 125 стандартными способами, например, как представлено на Фиг.2. Пиридазинон 124 или 125 может быть превращен в гидроксаматы или амиды, как представлено на Фиг.1 или 2.
На Фиг.7 представлена реакционная схема синтеза соединений 128, 129 и 130, где в качестве исходного вещества используют метилэфирное производное пиридона 117. Бромирование эфирного производного пиридона 117 может быть осуществлено с использованием либо Br2 и уксусной кислоты либо NBS в подходящем органическом растворителе, таком как DMF. Предпочтительно NBS добавляют к раствору эфирного производного пиридона 117 в DMF с получением 126. Превращение бромида 126 в соединение 127, где R9 представляет собой циано, может быть достигнуто с использованием условий Pd-опосредованного поперечного связывания. Как правило, эта химическая реакция может быть осуществлена с использованием различных Pd катализаторов и лигандов, с добавлением основания или без добавления основания, в подходящем органическом растворителе, таком как DMF, РhМе, DME, THF, CH3CN или NMP, при повышенной температуре. Предпочтительно эту реакцию проводят с использованием Zn(CN)2 и Рd2dba3 и dppf в DMF при 120°С. Превращение соединения 127 в карбоновую кислоту 128, а также в гидроксамат 129 и амид 130 может быть осуществлено так, как описано для Фиг.3 и 4.
На Фиг.8 представлен синтез соединений формулы V, где R9 представляет собой Н или F, где в качестве исходного вещества используют 2,6-дихлор-никотиновую кислоту или 2,6-дихлор-6-фтор-никотиновую кислоту. Этот путь особенно подходит для аналогов, где R7 представляет собой Me. Никотиновую кислоту 140 превращают в монохлорсодержащую кислоту 141 путем кипячения с обратным холодильником в 2 н. водном NaOH по методике, описанной в патенте США 3682932 (1972). Алкилирование соединения 141 может быть достигнуто в стандартных условиях основного алкилирования, включающего использование алкилгалогенидов, с использованием двух эквивалентов соответствующего алкилгалогенида и основания с получением смеси сложноэфирного производного N-алкилпиридона и сложноэфирного производного региоизомерного O-алкилпиридина, которые легко разделяются колоночной хроматографией. Эти условия включают, без ограничений, К2СО3 в ацетоне или DMF при комнатной или повышенной температуре либо NaH в THF при температуре окружающей среды или повышенной температуре и последующее добавление алкилгалогенида. Предпочтительно это алкилирование достигается с использованием LiH в DMF при 0°С с последующим добавлением алкилбромида или алкилйодида и нагреванием до комнатной температуры. Введение соответствующим образом замещенной анилиновой группировки с получением соединения 143 достигается в результате SNAR реакции. Она может быть проведена в подходящем органическом растворителе, таком как THF, с использованием амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при подходящей температуре (от -78°С до комнатной температуры). Предпочтительно анилин добавляют к LDA или LiHMDS в THF при низкой температуре (от -20 до -80°С). Затем добавляют пиридон, и смесь перемешивают при низкой температуре с получением сложного эфира 143. Карбоновая кислота 144 затем может быть получена в стандартных условиях омыления, таких как LiOH или NaOH в стандартных смешанных системах водных/органических растворителей. Гидроксамат 145 и амид 146 могут быть получены с использованием стандартных методик сочетания, включая, без ограничений, EDCI, HOBt или РуВОР и соответствующий амин или гидроксиламин в подходящих органических растворителях, таких как DMF, THF или метиленхлорид. Предпочтительно сочетание осуществляют с использованием HOBt и EDCI в DMF. В некоторых случаях амин или гидроксиламин, используемый в реакции сочетания, содержит стандартную защитную группу. В таких случаях защитная группа может быть удалена в стандартных условиях, известных в данной области техники.
На Фиг.9 представлен альтернативный синтез соединений формулы V, где R9 представляет собой Н или F, где в качестве исходного вещества используют 2,6-дихлор-никотиновую кислоту или 2,6-дихлор-5-фторникотиновую кислоту. Никотиновая кислота 140 может быть превращена в метилэфирное производное N-алкилпиридона 149 после проведения пятистадийной процедуры, где сначала 2,6-дихлорникотиновую кислоту 140 превращают в метоксипиридиновую кислоту, которую этерифицируют с получением метилового эфира, а затем удаляют защиту с получением монохлорсодержащего эфира 147. Превращение в метоксипиридиновую кислоту предпочтительно осуществляют путем добавления трет-бутоксида калия к раствору кислоты 140 в МеОН, и эту смесь затем нагревают до температуры дефлегмации в течение нескольких суток. Этерификация с получением метилового эфира может быть проведена в стандартных условиях, включающих, без ограничений, этерификацию по Фишеру (МеОН, H2SO4), TMSCI в МеОН или TMSCHN2 в подходящих органических растворителях, таких как РhМе/МеОН. Затем может быть проведено деметилирование метоксипиридина в стандартных условиях, включающих, без ограничений, HCl при повышенной температуре, п-TsOH в уксусной кислоте при повышенной температуре и водный НВr в МеОН при повышенной температуре. Предпочтительно деметилирование с получением пиридона 147 достигается путем обработки метоксипиридина водным НВr в уксусной кислоте при повышенной температуре (80-120°С). Алкилирование соединения 147 с получением соединения 148 может быть достигнуто в стандартных условиях основного алкилирования, включающего использование алкилгалогенидов, с использованием одного эквивалента соответствующего алкилгалогенида и основания с получением смеси сложноэфирного производного N-алкилпиридона и сложноэфирного производного региоизомерного O-алкилпиридина, которые легко разделяются колоночной хроматографией. Эти условия включают, без ограничений, К2СО3 в ацетоне или DMF при комнатной или повышенной температуре или NaH в THF при температуре окружающей среды или повышенной температуре и последующее добавление алкилгалогенида. Предпочтительно это алкилирование достигается с использованием LiH в DMF при 0°С с последующим добавлением алкилбромида или алкилйодида и нагреванием до комнатной температуры. Введение соответствующим образом замещенной анилиновой группировки достигается в результате SNAR реакции. Она может быть проведена в подходящем органическом растворителе, таком как THF, с использованием амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при подходящей температуре (от -78°С до комнатной температуры). Предпочтительно анилин добавляют к LDA или LiHMDS в THF при низкой температуре (от -20 до -80°С). Затем добавляют пиридон, и эту смесь перемешивают при низкой температуре с получением сложного эфира 149. Гидроксамат 145 может быть получен прямо из метилового эфира 149 в подходящем органическом растворителе, таком как THF, с использованием соответствующего гидроксиламина и амидного основания, такого как LDA, LiHMDS, NaHMDS или KHMDS, при подходящей температуре (от -78°С до комнатной температуры). Предпочтительно раствор LiHMDS добавляют к раствору метилового эфира 149 и гидроксиламина в THF при 0°С.Реакционную смесь затем нагревают до комнатной температуры с получением желаемого гидроксамата 145. В некоторых случаях гидроксиламин, используемый в реакции сочетания, содержит стандартную защитную группу. В таких случаях защитная группа может быть удалена в стандартных условиях, известных в данной области техники.
В еще одном аспекте данного изобретения предложен способ получения соединения формулы IA, включающий:
взаимодействие соединения формулы 100 или 101
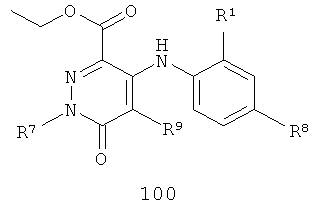
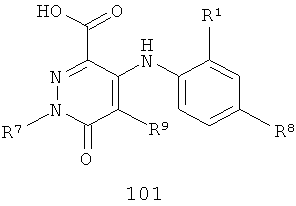
с R3NH2 в присутствии либо (1) реагента сочетания, когда R3 такой, как определено в формуле IA, либо (2) амидного основания, когда R3 такой, как определено в формуле IA, за исключением того, что R3 не представляет собой Н или Me, с получением указанного соединения формулы IA.
В еще одном аспекте данного изобретения предложен способ получения соединения формулы IV, включающий:
взаимодействие соединения формулы 108 или 109
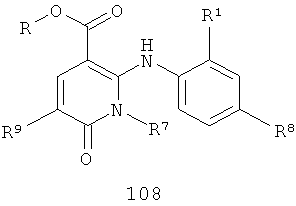
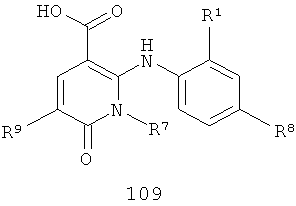
с R3NН2, где R3 такой, как определено в формуле IV, в присутствии либо (1) реагента сочетания, либо (2) амидного основания, когда R3 такой, как определено в формуле IV, за исключением того, что R3 не представляет собой Н или Me.
В еще одном аспекте этого изобретения предложен способ получения соединения формулы VI, включающий:
(а) бромирование соединения, имеющего формулу 105,
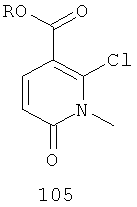
где R представляет собой алкил, с получением соединения 106
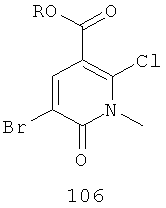 ;
;
(б) взаимодействие соединения 106 с Zn(Me)2 в присутствии палладиевого катализатора и лиганда и возможно в присутствии основания с получением соединения 107
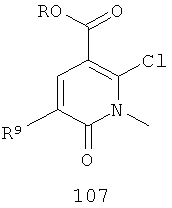 ;
;
(в) взаимодействие соединения 107 с анилином, имеющим формулу
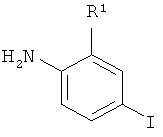 ,
,
в присутствии палладиевого катализатора, фосфинового лиганда и амидного основания с получением соединения 108
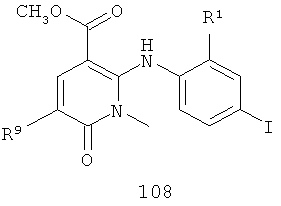 ;
;
(г) возможно гидролиз соединения 108 в щелочных условиях с получением соединения 109
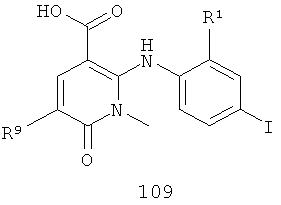 ; и
; и
(д) взаимодействие либо соединения 108 либо соединения 109 с R3NН2, либо в присутствии (1) реагента сочетания, когда R3 такой, как определено в формуле VI, либо (2) амидного основания, когда R3 такой, как определено в формуле VI, за исключением того, что R3 не представляет собой Н, с получением указанного соединения формулы VI.
В одном из воплощений соединение 105 получают способом, включающим:
(а) взаимодействие соединения 103
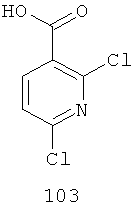
с водным гидроксидом натрия с получением соединения 104
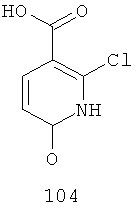 ; и
; и
(б) взаимодействие соединения 104 с RX, где R представляет собой Me, и Х представляет собой галогенид, в присутствии основания с получением соединения 105.
В еще одном аспекте данного изобретения предложен способ получения соединения формулы II, включающий:
(а) взаимодействие гидразина, имеющего формулу Me-NH-NH2, с:
(1) диэтил-2-оксомалонатом с последующей обработкой ацилирующим реагентом, который доставляет ацильную группу, имеющую формулу C(=O)CH2R9, где R9 такой, как определено в формуле II, или
(2) ацилирующим реагентом, который доставляет ацильную группу, имеющую формулу C(=O)CH2R9, где R9 такой, как определено в формуле II, с последующей обработкой диэтилкетомалонатом с получением соединения 97
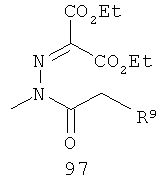 ;
;
(б) обработку соединения 97 амидным основанием при температуре ниже -40°С с последующей обработкой концентрированной HCl с получением соединения формулы 98
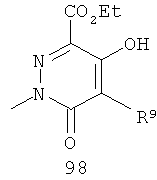 ;
;
(в) хлорирование соединения 98 с получением соединения 99
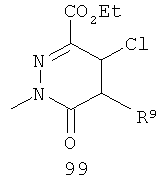 ;
;
(г) взаимодействие соединения 99 с анилином, имеющим формулу
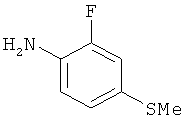 ,
,
в присутствии палладиевого катализатора, лиганда и амидного основания с получением соединения 100
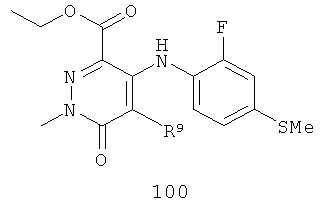 ; и
; и
(д) взаимодействие соединения формулы 100 с R3NH2 либо в присутствии (1) реагента сочетания, когда R3 такой, как определено в формуле II, либо (2) амидного основания, когда R3 такой, как определено в формуле II, за исключением того, что R3 не представляет собой Н, с получением указанного соединения формулы II.
В еще одном аспекте данного изобретения предложен способ получения соединения формулы II, где R9 представляет собой Н, Me или Cl, включающий:
(а) взаимодействие гидразина, имеющего формулу Me-NH-NH2, с:
(1) диэтил-2-оксомалонатом с последующей обработкой ацилирующим реагентом, который доставляет ацильную группу, имеющую формулу C(=O)CH2R9, где R9 представляет собой Н, Me или Cl; или
(2) ацилирующим реагентом, который доставляет ацильную группу, имеющую формулу C(=O)CH2R9, где R9 представляет собой Н, Me или Cl, с последующей обработкой диэтилкетомалонатом с получением соединения 97
;
(б) обработку соединения 97 амидным основанием при температуре ниже -40°С с получением соединения формулы 98
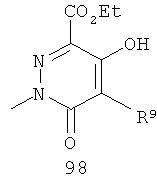 ;
;
(в) хлорирование соединения 98 с получением соединения 99
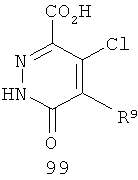 ;
;
(г) взаимодействие соединения 99 с анилином, имеющим формулу
,
в присутствии амидного основания с получением соединения 101
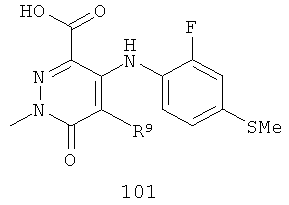 ; и
; и
(д) взаимодействие соединения 101 с R3NH2 либо в присутствии (1) реагента сочетания, когда R3 такой, как определено в формуле II, либо (2) амидного основания, когда R3 такой, как определено в формуле II, за исключением того, что R3 не представляет собой Н, с получением указанного соединения формулы II.
В еще одном аспекте данного изобретения предложен способ получения соединения формулы V, где R9 представляет собой Me, включающий:
(а) бромирование соединения, имеющего формулу 105,
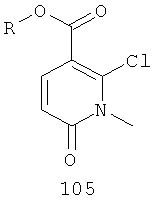
где R представляет собой алкил, с получением соединения 106
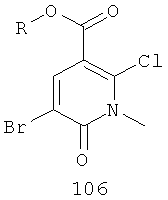 ;
;
(б) взаимодействие соединения 106 с Zn(Me)2 в присутствии палладиевого катализатора и лиганда и возможно в присутствии основания с получением соединения 107
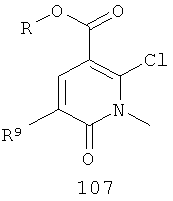 ;
;
(в) взаимодействие соединения 107 с анилином, имеющим формулу F
,
в присутствии амидного основания с получением соединения 108
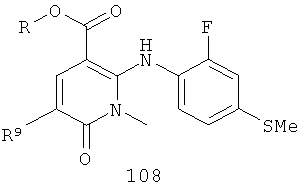 ;
;
(г) возможно гидролиз соединения 108 в щелочных условиях с получением соединения 109
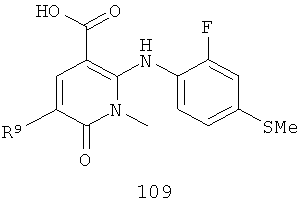 ; и
; и
(д) взаимодействие либо соединения 108 либо соединения 109 с R3NH2, где R3 такой, как определено в формуле V, в присутствии реагента сочетания или амидного основания с получением указанного соединения формулы V.
В еще одном аспекте этого изобретения предложен способ получения соединения формулы V, где R9 представляет собой Cl, включающий:
(а) взаимодействие соединения формулы 112
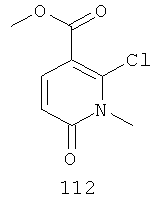
с анилином, имеющим формулу
,
в присутствии амидного основания с получением соединения 117
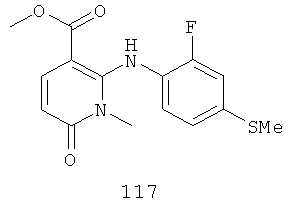 ;
;
(б) хлорирование соединения 117 с получением соединения 118
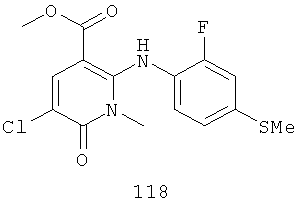 ; и
; и
(в) возможно гидролиз соединения 118 с получением соединения 118А
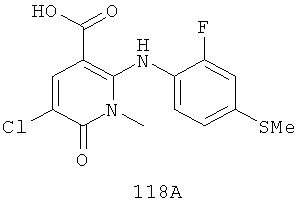 ;
;
(г) взаимодействие либо соединения 118 либо соединения 118А с (S)-MeCH(OH)CH2ONH2 или HOCH2CH2ONH2 в присутствии реагента сочетания или амидного основания с получением указанного соединения формулы V.
В еще одном аспекте данного изобретения предложен способ получения соединения формулы V, где R9 представляет собой Н или F, включающий:
(а) обработку соединения формулы 140,
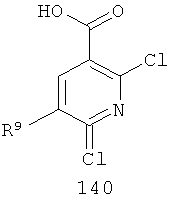
где R9 представляет собой Н или F, водным NaOH с получением соединения 141
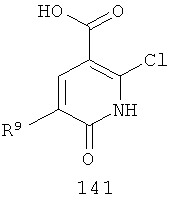 ;
;
(б) взаимодействие соединения 141 с СН3Х, где Х представляет собой галогенид, в присутствии основания с получением соединения 142
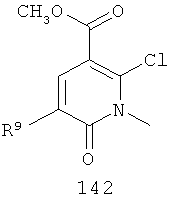 ;
;
(в) взаимодействие соединения 142 с анилином, имеющим формулу
,
в присутствии амидного основания с получением соединения 143
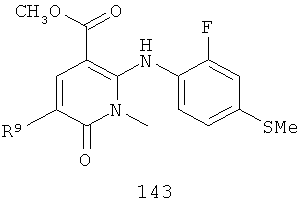 ;
;
(г) возможно гидролиз соединения 143 с получением соединения 144
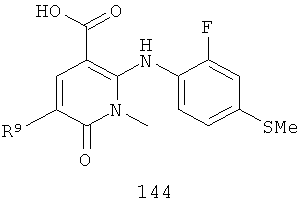 ; и
; и
(д) взаимодействие либо соединения 143 либо соединения 144 с R3NH2, где R3 такой, как определено в формуле V, в присутствии реагента сочетания или амидного основания с получением указанного соединения формулы V.
В одном воплощении приведенных выше способов агентом сочетания является 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид, 1-гидроксибензотриазол-6-сульфонамидометилгидрохлорид или бензотриазол-1-ил-окситрипирролидинофосфонийгексафторфосфат.
В другом аспекте настоящего изобретения предложен способ применения соединений по данному изобретению в качестве лекарственного средства для лечения заболеваний или медицинских состояний, опосредованных МЕК. Например, согласно данному изобретению предложено соединение по данному изобретению в качестве лекарственного средства для лечения гиперпролиферативного расстройства или воспалительного состояния у млекопитающего, включающего введение указанному млекопитающему одного или более соединений по настоящему изобретению или их фармацевтически приемлемых солей или пролекарств в количестве, эффективном для лечения указанного гиперпролиферативного расстройства. В другом аспекте данного изобретения предложено соединение по данному изобретению в изготовлении лекарственного средства для лечения гиперпролиферативного расстройства или воспалительного состояния.
В другом аспекте настоящего изобретения предложен способ продуцирования ингибирующего МЕК эффекта у теплокровного животного, например человека, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения по данному изобретению.
В другом аспекте настоящего изобретения предложено лечение или предупреждение состояния, опосредованного МЕК, включающее введение человеку или животному, нуждающемуся в этом, фармацевтической композиции, содержащей соединение по настоящему изобретению или его фармацевтически приемлемую соль или in vivo расщепляемое пролекарство в количестве, эффективном для лечения или предупреждения указанного состояния, опосредованного МЕК.
Изобретение также относится к фармацевтическим композициям, которые ингибируют МЕК, содержащим эффективное количество соединения, выбранного из соединений по настоящему изобретению или их фармацевтически приемлемых пролекарств, фармацевтически активных метаболитов или фармацевтически приемлемых солей.
Изобретение также относится к фармацевтической композиции для лечения гиперпролиферативного расстройства у млекопитающего, ссодержащей терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтически приемлемый носитель. В одном воплощении указанная фармацевтическая композиция предназначена для лечения рака, такого как рак головного мозга, рак легкого, плоскоклеточный рак, рак мочевого пузыря, рак желудка, рак поджелудочной железы, рак молочной железы, рак в области головы, шеи, рак почки, рак яичника, рак предстательной железы, рак толстой и прямой кишки, рак пищевода, рак яичка, гинекологический рак или рак щитовидной железы. В другом воплощении указанная фармацевтическая композиция предназначена для лечения гиперпролиферативного расстройства нераковой природы, такого как доброкачественная гиперплазия кожи (например, псориаз), рестеноз или расстройство предстательной железы (например, доброкачественная гипертрофия предстательной железы (ВРН)).
Изобретение также относится к фармацевтической композиции для лечения панкреатита или заболевания почек (включая пролиферативный гломерулонефрит и индуцированное диабетом почечное заболевание) или лечения боли у млекопитающего, содержащей терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтически приемлемый носитель.
Изобретение также относится к фармацевтической композиции для предупреждения имплантации бластоцитов у млекопитающего, содержащей терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтически приемлемый носитель.
Изобретение также относится к фармацевтической композиции для лечения заболевания, связанного с васкулогенезом или ангиогенезом, у млекопитающего, содержащей терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтически приемлемый носитель. В одном воплощении указанная фармацевтическая композиция предназначена для лечения заболевания, выбранного из группы, состоящей из опухолевого ангиогенеза, хронического воспалительного заболевания или другого воспалительного состояния, такого как ревматоидный артрит, атеросклероз, воспалительное заболевание кишечника, кожные заболевания, такие как псориаз, экзема и склеродерма, диабет, диабетическая ретинопатия, ретролентальная фиброплазия, возрастная дегенерация желтого пятна, гемангиома, глиома, меланома, саркома Капоши и рак яичника, рак молочной железы, рак легкого, рак поджелудочной железы, рак предстательной железы, рак толстой кишки и эпидермоидный рак.
Изобретение также относится к способу лечения гиперпролиферативного расстройства у млекопитающего, включающему введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата. В одном воплощении указанный способ относится к лечению рака, такого как рак головного мозга, рак легкого, плоскоклеточный рак, рак мочевого пузыря, рак желудка, рак поджелудочной железы, рак молочной железы, рак в области головы, шеи, рак почки, рак яичника, рак предстательной железы, рак толстой и прямой кишки, рак пищевода, рак яичка, гинекологический рак или рак щитовидной железы. В другом воплощении указанный способ относится к лечению гиперпролиферативного расстройства нераковой природы, такого как доброкачественная гиперплазия кожи (например псориаз), рестеноз или расстройство предстательной железы (например, доброкачественная гипертрофия предстательной железы (ВРН)).
Изобретение также относится к способу лечения гиперпролиферативного расстройства у млекопитающего, включающему введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата в комбинации с противоопухолевым агентом, выбранным из группы, состоящей из ингибиторов митоза, алкилирующих агентов, антиметаболитов, интеркалирующих антибиотиков, ингибиторов факторов роста, ингибиторов клеточного цикла, ферментных ингибиторов, ингибиторов топоизомеразы, модификаторов биологического ответа, антигормонов, ингибиторов ангиогенеза и антиандрогенов.
Изобретение также относится к способу лечения панкреатита или заболевания почек у млекопитающего, включающему введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата.
Изобретение также относится к способу предупреждения имплантации бластоцитов у млекопитающего, включающему введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата.
Изобретение также относится к способу лечения заболеваний, связанных с васкулогенезом или ангиогенезом, у млекопитающего, включающему введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата. В одном воплощении указанный способ предназначен для лечения заболевания, выбранного из группы, состоящей из опухолевого ангиогенеза, хронического воспалительного заболевания, такого как ревматоидный артрит, атеросклероза, воспалительного заболевания кишечника, кожных заболеваний, таких как псориаз, экзема и склеродерма, диабета, диабетической ретинопатии, ретролентальной фиброплазии, возрастной дегенерации желтого пятна, гемангиомы, глиомы, меланомы, саркомы Калоши и рака яичника, рака молочной железы, рака легкого, рака поджелудочной железы, рака предстательной железы, рака толстой кишки и эпидермоидного рака.
Изобретение также относится к фармацевтической композиции для лечения заболевания или состояния, связанного с воспалительным заболеванием, аутоиммунным заболеванием, деструктивными костными нарушениями, пролиферативными расстройствами, инфекционным заболеванием, вирусным заболеванием, фиброзным заболеванием или нейродегенеративным заболеванием у млекопитающего, содержащей терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтически приемлемый носитель. Примеры упомянутых выше заболеваний и/или состояний включают, но ими ограничиваются, ревматоидный артрит, атеросклероз, воспалительное заболевание кишечника, кожные заболевания, такие как псориаз, экзема и склеродерма, диабет и осложнения диабета, диабетическую ретинопатию, ретролентальную фиброплазию, возрастную дегенерацию желтого пятна, гемангиому, хроническую обструктивную болезнь легких, идиопатический легочный фиброз, аллергические реакции, включая астматический аллергический ринит и атопический дерматит, почечное заболевание и почечную недостаточность, поликистозное почечное заболевание, острый коронарный синдром, застойную сердечную недостаточность, остеоартрит, нейрофиброматоз, отторжение органных трансплантатов, кахексию и боль.
Кроме того, соединение по настоящему изобретению предложено для применения в качестве лекарственного средства в лечении описанных выше заболеваний и состояний у теплокровного животного, предпочтительно млекопитающего, более предпочтительно человека, страдающего таким расстройством. Также предложено применение соединения по настоящему изобретению в изготовлении лекарственного средства для лечения описанных выше заболеваний и состояний у теплокровного животного, предпочтительно млекопитающего, более предпочтительно человека, страдающего таким расстройством.
Пациенты, которые могут быть подвергнуты лечению соединениями по настоящему изобретению или фармацевтически приемлемыми солями, пролекарствами и гидратами указанных соединений в соответствии со способами по данному изобретению, включают, например, пациентов с диагнозом псориаз, рестеноз, атеросклероз, ВРН, рак легкого, рак кости, хронический миеломоноцитарный лейкоз (CMML), рак поджелудочной железы, рак кожи, рак в области головы и шеи, кожная или внутриглазная меланома, рак матки, рак яичника, рак прямой кишки, рак в анальной области, рак желудка, рак толстой кишки, рак молочной железы, опухоли яичек, гинекологические опухоли (например, саркомы матки, карцинома фаллопиевых труб, карцинома эндометрия, карцинома шейки, карцинома влагалища или карцинома вульвы), болезнь Ходжкина, рак пищевода, рак тонкого кишечника, рак эндокринной системы (например, рак щитовидной, паращитовидной желез или надпочечников), саркомы мягких тканей, рак уретры, рак пениса, рак предстательной железы, хронический или острый лейкоз, солидные опухоли у детей, лимфоцитарная лимфома, рак мочевого пузыря, рак почки или мочеточника (например, почечно-клеточный рак, карцинома почечной лоханки) или опухоли в центральной нервной системе (например, первичная лимфома ЦНС, опухоли позвоночного столба, глиомы ствола мозга или аденомы гипофиза).
Изобретение также относится к фармацевтической композиции для ингибирования аномального клеточного роста у млекопитающего, содержащей определенное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата или пролекарства в комбинации с некоторым количеством химиотерапевтического средства, причем количества соединения, соли, сольвата или пролекарства и химиотерапевтического средства вместе эффективны в ингибировании аномального клеточного роста. В настоящее время в данной области техники известно множество химиотерапевтических средств. В одном воплощении химиотерапевтическое средство выбрано из группы, состоящей из ингибиторов митоза, алкилирующих агентов, антиметаболитов, интеркалирующих антибиотиков, ингибиторов факторов роста, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомеразы, модификаторов биологического ответа, антигормонов, ингибиторов ангиогенеза и антиандрогенов.
В другом аспекте настоящего изобретения предложены фармацевтические композиции, которые ингибируют МЕК, содержащие одно или более соединений по настоящему изобретению.
Кроме того, изобретение относится к способу ингибирования аномального клеточного роста у млекопитающего или лечения пролиферативного расстройства, включающему введение млекопитающему некоторого количества соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата или пролекарства в комбинации с лучевой терапией, где количества соединения, соли, сольвата или пролекарства в комбинации с лучевой терапией эффективны в ингибировании аномального клеточного роста или лечении пролиферативного расстройства у млекопитающего. Методики применения лучевой терапии известны в данной области техники, и эти методики могут быть использованы в описанной здесь комбинированной терапии. Введение соединения по изобретению в такой комбинированной терапии может быть определено, как описано в данном описании.
Предполагается, что соединения по настоящему изобретению могут делать аномальные клетки более чувствительными к лечению облучением в целях клеточного лизиса и/или ингибирования роста таких клеток. Соответственно данное изобретение также относится к способу сенсибилизации аномальных клеток у млекопитающего к лечению облучением, включающему введение данному млекопитающему соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата или пролекарства в количестве, которое эффективно в сенсибилизации аномальных клеток к лечению облучением. Количество соединения, соли или сольвата в этом способе может быть определено согласно методикам выявления эффективных количеств таких соединений, описанным в данном описании.
Соединения по изобретению преимущественно могут быть использованы также в комбинации с другими известными терапевтическими агентами. Например, изобретение также относится к способу и к фармацевтической композиции для ингибирования аномального клеточного роста у млекопитающего, которая содержит некоторое количество соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата, его пролекарства или его меченного изотопом производного и некоторое количество одного или более веществ, выбранных из антиангиогенезных агентов, ингибиторов сигнальной трансдукции и антипролиферативных агентов.
Антиангиогенезные агенты, такие как ингибиторы ММР-2 (матриксная металлопротеиназа 2), ингибиторы ММР-9 (матриксная металлопротеиназа 9) и ингибиторы СОХ-11 (циклооксигеназа II), могут быть использованы вместе с соединением по настоящему изобретению и фармацевтическими композициями, изложенными в данном описании. Примеры полезных ингибиторов СОХ-II включают CELEBREX™ (алекоксиб), валдекоксиб и рофекоксиб. Примеры полезных ингибиторов матриксных металлопротеиназ описаны в WO 96/33172, WO 96/27583, ЕР 818442, ЕР 1004578, WO 98/07697, WO 98/03516, WO 98/34918, WO 98/34915, WO 98/33768, WO 98/30566, ЕР 606046, ЕР 931788, WO 90/05719, WO 99/52910, WO 99/52889, WO 99/29667, WO 99/07675, ЕР 945864, патенте США №5863949, патенте США №5861510 и ЕР 780386, которые во всей своей полноте включены в данное описание посредством ссылки. Предпочтительными ингибиторами ММР-2 и ММР-9 являются ингибиторы, обладающие незначительной активностью ингибирования ММР-1 или не имеющие такую активность. Более предпочтительными являются ингибиторы, селективно ингибирующие ММР-2 и/или ММР-9 по сравнению с другими матриксными металлопротеиназами (т.е. ММР-1, ММР-3, ММР-4, ММР-5, ММР-6, ММР-7, ММР-8, ММР-10, ММР-11, ММР-12 и ММР-13).
Термины «аномальный клеточный рост» и «гиперпролиферативное расстройство» используются в данной заявке как взаимозаменяемые.
«Аномальный клеточный рост», как он использован в данном описании и если не указано иное, относится к клеточному росту, который не зависит от нормальных регуляторных механизмов (например, утрата контактного торможения). Он включает в себя, например, аномальный рост: (1) опухолевых клеток (опухолей), которые пролиферируют в результате экспрессирования мутантной тирозинкиназы или сверхэкспрессии рецепторной тирозинкиназы; (2) доброкачественных или злокачественных клеток при других пролиферативных заболеваниях, при которых происходит аберрантная активация тирозинкиназы; (3) любых опухолей, которые пролиферируют под действием рецепторных тирозинкиназ; (4) любых опухолей, которые пролиферируют в результате аберрантной активации серин/треонинкиназ; и (5) доброкачественных и злокачественных клеток при других пролиферативных заболеваниях, при которых наблюдается аберрантная активация серин/треонинкиназ.
Термин «лечение», как он использован в данном описании и если не указано иное, означает реверсирование, облегчение, торможение развития или предупреждение расстройства или состояния, к которым применяется такой термин, или одного или более симптомов такого расстройства или состояния. Термин «лечение», как он использован в данном описании и если не указано иное, также относится к акту лечения как "лечения", которое определено непосредственно выше.
Количество данного агента, которое будет соответствовать такому количеству, будет варьировать в зависимости от таких факторов, как конкретное соединение, болезненное состояние и его тяжесть, личность (например, масса) млекопитающего, нуждающегося в лечении, но тем не менее может быть рутинно определено специалистом в данной области техники. Предполагается, что «лечение» означает по меньшей мере облегчение болезненного состояния у млекопитающего, например человека, которое подвергается воздействию, по меньшей мере частично, активности МЕК, и оно включает, без ограничений, предупреждение возникновения болезненного состояния у млекопитающего, в частности когда обнаружено, что млекопитающее имеет предрасположенность к данному болезненному состоянию, но диагноз, что он его имеет, еще не поставлен; модулирование и/или ингибирование болезненного состояния; и/или ослабление болезненного состояния.
Чтобы использовать соединение по настоящему изобретению или его фармацевтически приемлемую соль или пролекарство для терапевтического лечения (включая профилактическое лечение) млекопитающих, включая людей, его обычно вводят в состав фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Согласно этому аспекту изобретения предложена фармацевтическая композиция, которая содержит соединение по настоящему изобретению или его фармацевтически приемлемую соль или пролекарство, как они определены выше, вместе с фармацевтически приемлемым разбавителем или носителем.
Для приготовления фармацевтических композиций согласно одному воплощению данного изобретения терапевтически или профилактически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, сольвата, метаболита или пролекарства (одного или вместе с дополнительным терапевтическим агентом) сначала смешивают с фармацевтически приемлемым носителем в соответствии с традиционными фармацевтическими технологиями смешения для получения дозы. Носитель может иметь различные формы в зависимости от формы препарата, желательного для введения, например перорального или парентерального. Примеры подходящих носителей включают любые и все растворители, дисперсионные среды, адъюванты, покрытия, антибактериальные и противогрибковые агенты, изотонические агенты и агенты, замедляющие всасывание, подсластители, стабилизаторы (для обеспечения длительного хранения), эмульгаторы, связывающие агенты, загустители, соли, консерванты, растворители, корригенты и смешанные вещества, такие как буферы и абсорбенты, которые могут потребоваться для приготовления конкретной терапевтической композиции. Использование таких сред и агентов с фармацевтически активными веществами общеизвестно в данной области. Их использование в терапевтических композициях и препаратах предполагается, за исключением случаев, когда какие-либо традиционные среды или агенты несовместимы с соединением по настоящему изобретению. В композиции и препараты, рассмотренные в данном описании, также могут быть включены дополнительные активные ингредиенты.
Композиции по изобретению могут быть в форме, подходящей для перорального применения (например, в виде таблеток, пастилок, твердых и мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения ингаляцией (например в виде тонкоизмельченного порошка или жидкого аэрозоля), для введения инсуффляцией (например, в виде тонкоизмельченного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного или внутримышечного введения или в виде суппозитория для ректального введения). Например, композиции, предназначенные для перорального применения, могут содержать, например, один или более красителей, подсластителей, корригентов и/или консервантов.
Подходящие фармацевтически приемлемые эксципиенты для таблеточного препарата включают, например, инертные разбавители, такие как лактоза, карбонат натрия, фосфат кальция или карбонат кальция, гранулирующие агенты и разрыхлители, такие как кукурузный крахмал или альгиновая кислота; связывающие агенты, такие как крахмал; смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк; консерванты, такие как этил- или пропил-п-гидроксибензоат; и антиоксиданты, такие как аскорбиновая кислота. Таблеточные препараты могут быть не покрытыми оболочкой или могут быть покрыты оболочкой либо для модификации их распадаемости и последующего всасывания активного ингредиента в желудочно-кишечном тракте, либо для улучшения их стабильности и/или внешнего вида, в обоих случаях с использованием традиционных для нанесения покрытий агентов и методик, хорошо известных в данной области.
Композиции для перорального применения могут быть в форме твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или маслом, таким как арахисовое масло, вазелиновое масло или оливковое масло.
Как правило, водные суспензии содержат активный ингредиент в тонкоизмельченной порошковой форме вместе с одним или более суспендирующими агентами, такими как натриевая соль карбоксиметилцеллюлозы, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующими или смачивающими агентами, такими как лецитин или продукты конденсации алкиленоксида с жирными кислотами (например, полиоксиэтиленстеарат), или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, производными жирных кислот и гексита, такие как полиоксиэтиленсорбитолмоноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, производными жирных кислот и ангидридов гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или более консервантов (таких как этил- или пропил-п-гидроксибензоат), антиоксидантов (таких как аскорбиновая кислота), красителей, корригентов и/или подсластителей (таких как сахароза, сахарин или аспартам).
Масляные суспензии могут быть приготовлены путем суспендирования активного ингредиента в растительном масле (таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло) или в минеральном масле (таком как вазелиновое масло). Масляные суспензии также могут содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Для придания приятного вкуса пероральной композиции могут быть добавлены подсластители, такие как подсластители, упомянутые выше, и корригенты. Для сохранения этих композиций может быть добавлен такой антиоксидант, как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии путем добавления воды, обычно содержат активный ингредиент вместе с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или более консервантами. Примеры подходящих диспергирующих или смачивающих агентов и суспендирующих агентов приведены выше. Также могут присутствовать дополнительные эксципиенты, такие как подсластители, корригенты и красители.
Фармацевтические композиции по изобретению также могут быть в форме эмульсий типа масло-в-воде. Масляная фаза может представлять собой растительное масло, такое как оливковое масло или арахисовое масло, или минеральное масло, такое как, например, вазелиновое масло, или смесь любых из них. Подходящими эмульгирующими агентами могут быть, например, камеди природного происхождения, такие как аравийская камедь или трагакантовая камедь, фосфатиды природного происхождения, например из соевых бобов, лецитин, сложные эфиры или неполные сложные эфиры, производные жирных кислот и ангидридов гексита (например, сорбитанмоноолеат), и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, такие как полиоксиэтиленсорбитанмоноолеат. Эмульсии также могут содержать подсластители, корригенты и консерванты.
Сиропы и эликсиры могут быть приготовлены с подсластителями, такими как глицерин, пропиленгликоль, сорбит, аспартам или сахароза, а также могут содержать смягчающее средство, консервант, корригент и/или краситель.
Фармацевтические композиции также могут быть в форме стерильной инъекционной водной или масляной суспензии, которая может быть приготовлена по известным методикам с использованием одного или более соответствующих диспергирующих или смачивающих агентов и суспендирующих агентов, которые упомянуты выше. Стерильная инъекционная композиция также может представлять собой стерильный(ую) инъекционный(ую) раствор или суспензию в нетоксичном, пригодном для парентерального введения разбавителе или растворителе, например раствор в 1,3-бутандиоле.
Суппозиторные композиции могут быть приготовлены путем смешивания активного ингредиента с подходящим, не вызывающим раздражения эксципиентом, который является твердым при обычных температурах, но жидким при ректальной температуре, и поэтому будет плавиться в прямой кишке с высвобождением лекарственного средства. Подходящие эксципиенты включают, например, масло какао и полиэтиленгликоли.
Композиции для местного применения, такие как кремы, мази, гели и водные или масляные растворы или суспензии, в общем случае могут быть получены путем объединения активного ингредиента с традиционным, пригодным для местного применения наполнителем или разбавителем с использованием стандартных методик, общеизвестных в данной области.
Композиции для введения инсуффляцией могут быть в форме тонкоизмельченного порошка, содержащего частицы среднего диаметра, например 30 мкм или намного меньше, при этом сам порошок содержит активный ингредиент либо один либо разбавленный одним или более физиологически приемлемыми носителями, такими как лактоза. Удобно затем порошок для инсуффляции поместить в капсулу, содержащую, например, 1-50 мг активного ингредиента, для использования в турбоингаляторном устройстве, таком как устройство, которое используют для инсуффляции известного агента кромогликата натрия.
Композиции для введения ингаляцией могут быть в форме традиционного аэрозоля под давлением, приспособленного для дозирования активного ингредиента либо в виде аэрозоля, содержащего тонкодисперсное твердое вещество, либо в виде капелек жидкости. Могут быть использованы традиционные аэрозольные пропелленты, такие как летучие фторированные углеводороды или углеводороды, и удобно, если аэрозольное устройство приспособлено для дозирования отмеренного количества активного ингредиента.
Дополнительную информацию по технологиям приготовления композиций смотри в главе 25.2 в томе 5 Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press, 1990, которая включена в данное описание посредством ссылки.
Количество соединения по данному изобретению, которое объединяют с одним или более эксципиентами для изготовления разовой лекарственной формы, обязательно будет варьировать в зависимости от подвергаемого лечению субъекта, тяжести расстройства или состояния, скорости введения, характера соединения и выбора лечащего врача. Тем не менее, эффективная дозировка находится в диапазоне от приблизительно 0,001 до приблизительно 100 мг на кг массы тела в сутки, предпочтительно от приблизительно 1 до приблизительно 35 мг/кг/сутки, в разовой или разделенных дозах. Для человека массой 70 кг это количество будет составлять от приблизительно 0,07 до 2,45 г/сутки, предпочтительно от приблизительно 0,05 до приблизительно 1,0 г/сутки. В некоторых случаях уровни дозировок меньше низшего предела вышеуказанного диапазона могут быть более чем достаточны, а в других случаях могут быть использованы более высокие дозы, не вызывающие никакого вредного побочного эффекта, при условии, что такие более высокие дозы сначала делят на несколько небольших доз для введения в течение суток. Дополнительную информацию по способам введения и режимам дозирования смотри главу 25.3 в томе 5 Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press, 1990, которая включена в данное описание посредством ссылки.
Величина дозы соединения по настоящему изобретению в терапевтических или профилактических целях естественно будет варьировать в зависимости от природы и тяжести состояний, возраста и пола животного или пациента и пути введения, согласно общеизвестным принципам медицины.
Соединения по данному изобретению можно использовать одни или в комбинации с другими используемыми в лечении болезненных состояний лекарственными средствами и терапиями, при которых полезно ингибирование МЕК. Такое лечение может включать, в дополнение к соединениям по изобретению, традиционную хирургию, или радиотерапию, или химиотерапию. Такая химиотерапия может включать одну или более из следующих категорий противоопухолевых агентов:
(1) антипролиферативные/антинеопластические лекарственные средства и их комбинации, которые используются в медицинской онкологии, такие как алкилирующие агенты (например, цисплатин, оксалиплатин, карбоплатин, циклофосфамид, азотистый аналог иприта, мелфалан, хлорамбуцил, бусульфан, темозоламид и нитрозомочевины); антиметаболиты (например, гемцитабин, антифолаты, такие как фторпиримидины, подобные 5-фторурацилу и тегафуру, ралтитрексед, метотрексат, цитозинарабинозид, гидроксимочевина или один из предпочтительных антиметаболитов, раскрытых в европейской патентной заявке №239362, такой как N-(5-[N-(3,4-дигидро-2-метил-4-оксохиназолин-6-илметил)-N-метиламино]-2-теноил)-L-глутаминовая кислота); противоопухолевые антибиотики (например, антрациклины, подобные адриамицину, блеомицину, доксорубицину, дауномицину, эпирубицину, идарубицину, митомицину-С, дактиномицину и митрамицину); антимитотические агенты (например, алкалоиды барвинка, подобные винкристину, винбластину, виндезину и винорелбину, и таксоиды, подобные таксолу и таксотеру, и ингибиторы polo-киназы); и ингибиторы топоизомеразы (например, эпиподофиллотоксины, подобные эптопозиду и тенипозиду, амсакрин, топотекан и камптотецин);
(2) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен и йодоксифен), понижающие регуляторы эстрогеновых рецепторов (например, фулвестрант), антиандрогены (например, бикалутамид, флутамид, нилутамид, ципроксерона ацетат и Casodex™ (4'-циано-3-(4-фторфенилсульфонил)-2-гидрокси-2-метил-3'-(трифторметил)пропионанилид)), антагонисты LHRH (рилизинг-фактор лютеинизирующего гормона) или агонисты LHRH (например, гозерелин, лейпорелин и бусерелин), прогестогены (например, мегестрола ацетат), ингибиторы ароматазы (например, азанастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы, такие как финастерид;
(3) антиинвазивные агенты (например, ингибиторы c-Src-киназного семейства и ингибиторы металлопротеиназ, подобные маримастату, и ингибиторы функционирования рецептора плазминогенного активатора урокиназы или антитела к гепараназе);
(4) ингибиторы функционирования факторов роста, подобные антителам к факторам роста, антителам к рецепторам факторов роста (например, анти-erbВ2-антитело трастумузаб [Herceptin™], анти-ЕGFR(рецептор эпидермального фактора роста)-антитело панитумумаб и анти-erbВ1-антитело цетуксимаб [Erbitux C225]) и любым антителам к факторам роста или к рецепторам факторов роста, раскрытым в Stern et al. Critical Reviews in Oncology/Haematology, 2005, vol.54, pp.11-29): такие ингибиторы включают ингибиторы тирозинкиназ (например, ингибиторы тирозинкиназ семейства эпидермальных факторов роста, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, AZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)-хиназолин-4-амин (CI 1033)), ингибиторы erbВ2-тирозинкиназ, такие как лапатиниб, ингибиторы семейства факторов роста гепатоцитов, ингибиторы семейства факторов роста из тромбоцитов, такие как иматиниб, ингибиторы серин/треонинкиназ (например, ингибиторы Ras/Raf-сигналинга, такие как ингибиторы фарнезилтрансферазы, например сорафениб (BAY 43-9006)), ингибиторы клеточного сигналинга через киназы МЕК и/или АКТ (протеинкиназа В), ингибиторы семейства факторов роста гепатоцитов, ингибиторы из с-набора, ингибиторы киназы аbl, ингибиторы рецепторной киназы IGF (инсулиноподобный фактор роста); ингибиторы киназ семейства Aurora (например, AZD1152, РН739358, VX-680, MLN8054, R763, МР235, МР529, VX-528 и АХ39459) и ингибиторы циклинзависимых киназ (CDK), такие как ингибиторы CDK2 и/или CDK4;
(5) антиангиогенные агенты, такие как агенты, которые ингибируют эффекты васкулярного эндотелиального фактора роста (VEGF) (например, антитело против фактора роста васкулярных эндотелиальных клеток бевацизумаб [Avastin™] и ингибиторы рецепторных тирозинкиназ к VEGF, такие как 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (ZD6474; Пример 2 в WO 01/32651), 4-(4-фтор-2-метилиндол-5-илокси)-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолин (AZD2171; Пример 240 в WO 00/47212), ваталаниб (РТК787; WO 98/35985) и SU11248 (сунитиниб; WO 01/60814), соединения, как, например, раскрытые в публикациях международных заявок №№ WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354), и соединения, действующие по другим механизмам (например, линомид, ингибиторы функции αγβ3-интегринов, ингибиторы ММР, ингибиторы СОХ-2 и ангиостатин);
(6) агенты, повреждающие сосуды, такие как комбретастатин А4 и соединения, раскрытые в публикациях международных заявок №№ WO 99/02166, WO 0/40529, WO 00/41669, WO 01/92224, WO 02/04434 и WO 02/08213;
(7) антисмысловые терапии (например, терапии, которые направлены на мишени, перечисленные выше, такие как ISIS 2503, и анти-ras антисмысловые);
(8) генные терапевтические подходы, включая, например, GVAX™, подходы для замены аберрантных генов, таких как аберрантный р53 или аберрантный BRCA1 или BRCA2, подходы GDEPT (ген-направленная ферментативная пролекарственная терапия), такие как подходы с использованием цитозиндезаминазы, тимидинкиназы или бактериального фермента нитроредуктаза, и подходы для улучшения переносимости пациентами химиотерапии или радиотерапии, такие как генная терапия мультилекарственной устойчивости;
(9) интерферон; и
(10) иммунотерапевтические подходы, включая, например, ex vivo и in vivo подходы для повышения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, например интерлейкином-2, интерлейкином-4 или гранулоцитарно-макрофагальным колониестимулирующим фактором, подходы для снижения Т-клеточной анергии, подходы с использованием трансфицированных иммунных клеток, например трансфицированных цитокинами дендритных клеток, подходы с использованием трансфицированных цитокинами опухолевых клеточных линий и подходы с использованием антиидиотипических антител.
Такого сочетанного лечения можно достичь путем одновременного, последовательного или раздельного введения индивидуальных компонентов лечения. В таких комбинированных продуктах используют соединения по данному изобретению в пределах диапазона доз, описанного выше, и другой фармацевтически активный агент в пределах его установленного диапазона доз.
Согласно этому аспекту изобретения предложен фармацевтический продукт, содержащий соединение по настоящему изобретению, как оно определено в данном описании, и дополнительный противоопухолевый агент, как определено выше, для сочетанного лечения рака.
Несмотря на то, что соединения по настоящему изобретению главным образом полезны в качестве терапевтических агентов для применения у теплокровных животных (включая человека), они также полезны везде, где необходимо ингибировать эффекты МЕК. Так, они полезны в качестве фармакологических стандартов для применения в разработке новых биологических тестов и при поиске новых фармакологических агентов.
Репрезентативные соединения по настоящему изобретению, которые охвачены настоящим изобретением, включают, без ограничений, соединения, примеры которых приведены, и их фармацевтически приемлемые соли присоединения кислоты или основания или их пролекарства. Представленные ниже примеры предназначены для иллюстрации конкретных воплощений изобретения и никоим образом не предназначены для ограничения объема изобретения.
Описания всех процитированных здесь публикаций, включая патенты, включены в данное описание посредством ссылки.
В другом воплощении изобретения предложен продукт производства или «набор», содержащий вещества, полезные для лечения расстройств, описанных выше. В одном воплощении набор включает в себя контейнер, содержащий соединение по настоящему изобретению или содержащую его композицию. Набор также может включать в себя этикетку на упаковке или листок-вкладыш в упаковке. Термин «листок-вкладыш» используется для ссылки на инструкции, обычно вкладываемые в промышленные упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предостережениях, касающихся применения таких терапевтических продуктов. Подходящие контейнеры включают, например, бутыли, флаконы, шприцы, блистерную упаковку и т.д. Контейнер может быть сформован из различных материалов, таких как стекло или пластик. В контейнере содержится соединение по настоящему изобретению или содержащая его композиция, которые эффективны для лечения состояния, и может иметь стерильное входное отверстие (например, контейнер может представлять собой мешок для внутривенных растворов или флакон, имеющий пробку, которую можно проткнуть иглой для подкожных инъекций). Этикетка или листок-вкладыш может содержать указание на то, что композицию используют для лечения состояния выбора, такого как рак. В одном воплощении этикетки или листки-вкладыши содержат указания на то, что соединение по настоящему изобретению или содержащая его композиция могут быть использованы для лечения заболевания или медицинского состояния, опосредованного МЕК. В дополнение к этому этикетка или листок-вкладыш может содержать указания на то, что пациентом, которого следует лечить, является пациент с таким заболеванием или медицинским состоянием, опосредованным МЕК, как гиперпролиферативное расстройство или воспалительное состояние. Этикетка или листовка-вкладыш также может содержать указания на то, что композиция может быть использована для лечения других расстройств. Альтернативно или в дополнение к этому продукт производства может дополнительно включать в себя второй контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Он может дополнительно включать в себя другие вещества, которые с точки зрения продавца и потребителя желательны, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
Согласно другому воплощению набор может включать в себя (а) первый контейнер с помещенным в него соединением по настоящему изобретению или содержащей его композицией и возможно (б) второй контейнер с помещенной в него второй фармацевтической композицией, причем вторая фармацевтическая композиция содержит второе соединение с антипролиферативной или противовоспалительной активностью. Альтернативно или в дополнение к этому продукт производства может дополнительно включать в себя третий контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Он может дополнительно включать в себя другие вещества, которые желательны с точки зрения продавца и потребителя, включая другие буферы, разбавители, фильтры, иглы и шприцы.
Набор может дополнительно включать в себя инструкции по введению соединения по настоящему изобретению или содержащей его композиции и, если она присутствует, второй фармацевтической композиции. Например, если набор включает в себя соединение по настоящему изобретению или содержащую его композицию («первую композицию») и вторую фармацевтическую композицию, набор может дополнительно включать в себя инструкции по одновременному, последовательному или раздельному ведению первой и второй фармацевтических композиций нуждающемуся в этом пациенту.
В другом воплощении наборы подходят для доставки твердых пероральных форм соединения по настоящему изобретению, таких как таблетки или капсулы. Такой набор предпочтительно включает в себя много стандартных дозировок. Такие наборы могут включать в себя карточку с дозировками, ориентированными в порядке их намеченного применения. Примером такого набора является «блистерная упаковка». Блистерные упаковки хорошо известны в упаковочной промышленности и широко используются для упаковки фармацевтических стандартных лекарственных форм. Если это желательно, она может быть снабжена памяткой, например, в виде чисел, букв или других знаков, или календарем-вкладышем, где указаны дни в схеме лечения, в которые могут быть введены дозы.
В некоторых других воплощениях, где набор включает в себя соединение по настоящему изобретению или содержащую его композицию и второй терапевтический агент, набор может включать в себя контейнер для помещения в него отдельных компонентов, такой как секционный флакон или секционный пакет из фольги, однако отдельные композиции также могут находиться в едином, неразделенном контейнере. Обычно набор включает в себя инструкции по введению отдельных компонентов. Форма набора особенно полезна в тех случаях, когда отдельные компоненты предпочтительно вводят в разных лекарственных формах (например, пероральной и парентеральной), вводят с различными интервалами дозирования, или в тех случаях, когда по предписанию врача желательно титрование индивидуальных компонентов комбинации.
Биологические анализы
Для измерения эффектов соединений по настоящему изобретению в качестве ингибиторов МЕК могут быть использованы следующие анализы.
Пример А
Анализ фермента МЕК (тест 1а)
Активность соединений по настоящему изобретению может быть определена по следующей методике. 6His-меченую с N-конца конститутивно активную МЕК-1 (2-393) экспрессируют в E.Соli, и белок очищают общепринятыми методами (Ahn et al., Science, 1994, 265, 966-970). Активность МЕК1 оценивают путем измерения включения γ-33P-фосфата из γ-33Р-АТФ в His-меченую с N-конца ERK2, которая экспрессирована в E.Соli и очищена общепринятыми методами, в присутствии МЕК-1. Анализ проводят в 96-луночном полипропиленовом планшете. Инкубационная смесь (100 мкл) состоит из 25 мМ Hepes, pH 7,4, 10 мМ MgCl2, 5 мМ β-глицерофосфата, 100 мкМ Na-ортованадата, 5 мМ DTT (дитиотреит), 5 нМ МЕК1 и 1 мкМ ERK2. Ингибиторы суспендируют в DMSO, и все реакционные смеси, включая контроли, готовят в конечной концентрации 1% DMSO. Реакции инициируют добавлением 10 мкМ АТФ (с 0,5 мкКи γ-33Р-АТФ/лунка), и реакционные смеси инкубируют при температуре окружающей среды в течение 45 минут. Для остановки реакции и осаждения белков добавляют равный объем 25%-ной ТСА (трихлоруксусная кислота). Выпавшие в осадок белки улавливают на фильтровальных пластинах В из стекловолокна, и избыток меченого АТФ отмывают, используя харвестер Tomtec MACH III. Планшеты оставляют сушиться на воздухе, после чего добавляют Packard Microscint 20 из расчета 30 мкл/лунка, и планшеты считывают, используя Packard TopCount.
Пример Б
Клеточный анализ фосфорилирования ERK 1/2 (тест 1б)
Свойства соединений по изобретению в отношении ингибирования МЕК 1/2 могут быть определены следующим клеточным анализом in vitro. Ингибирование фонового фосфорилирования ERK1/2 определяли путем инкубирования клеток с соединением в течение 1 часа и количественного измерения флуоресцентного сигнала pERK на фиксированных клетках и приведения к сигналу общей ERK.
Материалы и методы: клетки Malme-3М получили из АТСС (Американская коллекция типовых культур) и выращивали на RPMI-1640, дополненной 10% сыворотки плода коровы. Клетки высевали в 96-луночные планшеты по 15000 клеток/лунка и давали им возможность прикрепиться в течение 1-2 часов. Затем добавляли разведенные соединения в конечной концентрации 1% DMSO. Через 1 час клетки промывали PBS и фиксировали в 3,7% формальдегиде в PBS в течение 15 минут. После этого промывали в смеси PBS/0,2% Тритон X-100 и подвергают пермеабилизации в 100% МеОН в течение 15 минут. Клетки блокировали в блокирующем буфере Odyssey (LI-COR Biosciences) в течение по меньшей мере 1 часа. К клеткам добавляли антитела против фосфорилированной ERK 1/2 (Cell Signaling, №9106, моноклональные) и общей ERK 12 (Santa Cruz Biotechnology, № sc-94, поликлональные) и инкубировали в течение по меньшей мере 1 часа. После промывки смесью PBS/0,2% Тритон Х-100 клетки инкубировали с меченными флуоресцентной меткой вторичными антителами (антикроличьими IgG-IRDye800 козы, Rockland, и анти-мышиными IgG-Alexa Fluor 680 козы, Molecular Probes) в течение еще одного часа. Затем клетки промывали и анализировали на флуоресценцию на обеих длинах волн, используя систему визуализации в инфракрасной области Odyssey (LI-COR Biosciences). Сигнал фосфорилированной ERK приводили к сигналу общей ERK.
Пример В
Анализ растворимости в воде
Термодинамическую растворимость в воде соединений измеряли, используя модифицированный способ встряхивания в колбе. Кристалличность каждого соединения подтверждали с использованием поляризационного оптического микроскопа (Olympus BX51). Для каждого тестируемого соединения во флакон отвешивали приблизительно по 0,5 мг сухого соединения для использования для приготовления стандартных растворов. Также приблизительно по 0,5 мг отвешивали в несколько флаконов для измерения водных искомых параметров, один флакон для каждого рН, который тестируют.
Для измерения каждого водного искомого параметра к 0,5 мг сухого соединения добавляли по 0,5 мл водного буфера (10 мМ фосфат калия) при желаемом рН (для рН 1,2 использовали 0,1 н. HCl). Таким образом, верхний предел концентрации для этого анализа составлял 1 мг/мл. Каждый образец для измерения водного искомого параметра затем центрифугировали при 350 об/мин при комнатной температуре в течение 24 часов для уравновешивания. После центрифугирования окончательное значение рН каждого образца проверяли и подтверждали. После этого отбирали аликвоты и фильтровали их во флаконы для HPLC-анализа (HPLC - высокоэффективная жидкостная хроматография).
Исходный раствор для каждого соединения готовили путем растворения 0,5 мг соединения в общем объеме 1 мл метанола для исходной концентрации 500 мкг/мл. Исходный раствор затем последовательно разводили для построения калибровочной кривой от 5 до 250 мкг/мл.
Образцы и стандарты немедленно анализировали методом LC/UV (жидкостная хроматография с ультрафиолетовым детектированием). Для каждого из водных образцов по два разных объема впрыскивали в трех повторах. Для каждого из стандартов по два разных объема впрыскивали однократно. Образцы, дающие пики за пределами калибровочного диапазона, последовательно разводили и анализировали повторно.
Система HPLC/PDA (высокоэффективная жидкостная хроматография/детектор на диодной матрице) состояла из разделительной системы Alliance 2795 (Waters) или разделительной системы Acquity UPLC (Waters), объединенной с фотодиодным матричным детектором 2996 Photodiode Array Detector (Waters). На системе Alliance хроматографического разделения аналита достигали, используя колонку YMC ODS-Aq C18 (3,0×50 мм, размер частиц 3 мкм, 120Ǻ, Waters) в сочетании с градиентными условиями с использованием подвижных фаз А (водная, 0,01% гептафтормасляной кислоты (HFBA), 1% изопропилового спирта) и Б (0,01% HFBA и 1% изопропилового спирта в ацетонитриле). Общее время выполнения, включая время на повторное уравновешивание, для однократного впрыскивания составляло 5 минут. Характеристики аналитов измеряли путем мониторинга поглощения при 220 нм и 254 нм. Предел детектирования для большинства соединений на этой системе составлял приблизительно 1 мкг/мл.
На системе Acquity хроматографического разделения аналита достигали, используя колонку Acquity UPLC ВЕН C18 (2,1×50 мм, размер частиц 1,7 мкм, Waters) в сочетании с градиентными условиями с использованием подвижных фаз А (водная, 0,1% муравьиной кислоты (FA), 1% изопропилового спирта) и Б (0,1% FA и 1% изопропилового спирта в ацетонитриле). Общее время выполнения, включая время на повторное уравновешивание, для однократного впрыскивания составляло 3 минуты. Характеристики аналитов измеряли путем мониторинга поглощения при 220 нм и 254 нм. Предел детектирования для большинства соединений на этой системе составлял приблизительно 1 мкг/мл. На выходе из системы Acquity находится отдельный квадрупольный масс-спектрометр ZQ-2000 (Waters). Для идентификации массы родительского соединения использовали положительную ESI (электрораспылительная ионизация с регистрацией положительных ионов).
Данные получали и обрабатывали с использованием программного обеспечения Waters Empower. Калибровку осуществляли, строя график зависимости площади пика аналита от номинальной концентрации стандартных образцов. Калибровочную модель создавали в результате линейной регрессии калибровочной кривой. Эту модель использовали для расчета концентраций во всех водных образцах.
Несмотря на то, что фармакологические свойства соединений формул I-V варьируют в зависимости от структурного изменения, как ожидалось, в общем случае активность и/или растворимость, которые имеют соединения, может быть продемонстрирована в следующих концентрациях или дозах:
Соединения формулы II
Тест 1а (ферментный анализ): IC50≤250 нМ, например ≤100 нМ, в качестве еще одного примера ≤30 нМ.
Тест 1б: (клеточный анализ): IС50≤180 нМ, например ≤80 нМ, в качестве еще одного примера ≤10 нМ.
Соединения формулы III
Тест 1а (ферментный анализ): IС50≤250 нМ, например ≤50 нМ, в качестве еще одного примера <20 нМ; и
Тест 1б: (клеточный анализ): IC50≤600 нМ, например ≤30 нМ, в качестве еще одного примера ≤10 нМ.
Соединения формулы V
Тест 1а (ферментный анализ): IC50≤40 нМ, например ≤20 нМ; и
Тест 1б: (клеточный анализ): IC50≤10 нМ.
Соединения формулы VI
Тест 1а (ферментный анализ): IC50≤35 нМ, в качестве еще одного примера ≤15 нМ, и в качестве еще одного примера ≤10 нМ.
Тест 1б: (клеточный анализ): IC50≤5 нМ, в качестве еще одного примера ≤1 нМ.
ПРИМЕРЫ
Для иллюстрации изобретения приводятся следующие далее примеры. Однако следует иметь в виду, что эти примеры не ограничивают изобретение и предназначены только для того, чтобы предложить способ практического применения изобретения. Специалистам в данной области техники известно, что описанные химические реакции легко могут быть адаптированы для получения ряда других ингибиторов МЕК по изобретению, и считается, что альтернативные способы получения соединений по данному изобретению входят в объем данного изобретения. Например, синтез не проиллюстрированных примерами соединений по изобретению может быть с успехом осуществлен с использованием модификаций, очевидных специалистам в данной области техники, например путем соответствующей защиты вмешивающихся групп, путем использования других подходящих реагентов, известных в данной области, отличающихся от описанных, и/или путем выполнения рутинных модификаций реакционных условий. Альтернативно будут известны другие реакции, раскрытые в данном описании или известные в данной области, как находящие применение для получения других соединений по изобретению.
В описанных ниже примерах, если не указано иное, все температуры приведены в градусах Цельсия. Реагенты были приобретены у коммерческих поставщиков, таких как Aldrich Chemical Company, Lancaster, TCI или Maybridge, и использованы без дополнительной очистки, если не указано иное. Тетрагидрофуран (THF), N,N-диметилформамид (DMF), дихлорметан, толуол, диоксан и 1,2-дифторэтан были приобретены у Aldrich в герметично закрытых бутылях и использованы при получении.
Приведенные ниже реакции обычно проводили при положительном давлении азота или аргона или с использованием осушительной трубки (если не указано иное) в безводных растворителях, а реакционные колбы обычно закрывали подогнанными по размеру резиновыми мембранами для введения веществ и реагентов посредством шприца. Стеклянную посуду сушили в сушильном шкафу и/или сушили нагреванием.
Колоночную хроматографию выполняли на системе Biotage (производитель: Dyax Corporation) с силикагелевой колонкой или на картридже с диоксидом кремния SepPak (Waters).
Спектры 1H-ЯМР записывали на приборе Varian, работающем при 400 МГц. Спектры 1H-ЯМР получали для растворов в CDCl3 (выраженные в млн-1), используя хлороформ в качестве стандарта сравнения (7,25 млн-1). При необходимости использовали другие растворители для ЯМР. Если приведены множественные пики, то использованы следующие сокращения: s (синглет), d (дублет), t (триплет), m (мультиплет), br (уширенный), dd (дублет дублетов), dt (дублет триплетов). Константы связывания, если они приведены, приведены в герцах (Гц).
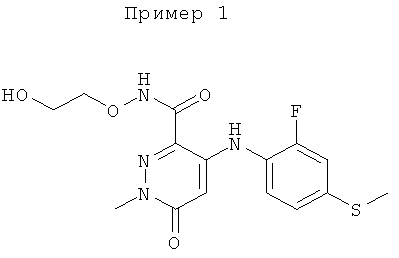
4-(2-Фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
Стадия А: Получение этил-2-(2-метилгидразоно)пропаноата
К суспензии этилпирувата (37,8 мл; 338 ммоль) и MgSO4 (40,8 г; 339 ммоль) в СНСl3 (500 мл) добавляли раствор метилгидразина (18,0 мл; 332 ммоль) в СНСl3 (100 мл) при 0°С. Реакционную смесь нагревали до комнатной температуры. После перемешивания в течение 24 часов при комнатной температуре реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением 44 г (94%) целевого продукта, который использовали непосредственно без дополнительной очистки.
Стадия Б: Получение метил-3-(2-(1-этокси-1-оксопропан-2-илиден)-1-метилгидразинил)-3-оксопропаноата
К раствору этил-2-(2-метилгидразоно)пропаноата (25,0 мл; 186 ммоль) в THF (500 мл) при 0°С добавляли LiH (2,02 г; 241 ммоль). Полученную смесь перемешивали в течение 10 минут при 0°С, нагревали до комнатной температуры и перемешивали в течение 6 часов. Добавляли метилмалонилхлорид (26,7 мл; 242 ммоль) в THF (20 мл) при 0°С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Реакцию осторожно гасили 1 н. водной HCl при 0°С, концентрировали при пониженном давлении и разбавляли ЕtOАс. Органический слой сушили над МgSO4, фильтровали и концентрировали при пониженном давлении с получением 46 г (99%) целевого продукта, который использовали непосредственно без дополнительной очистки.
Стадия В: Получение метил-5-гидрокси-2,6-диметил-3-оксо-2,3-дигидропиридазин-4-карбоксилата
К раствору этил-2-(2-метил-2-(метил-3-оксопропаноил)гидразоно)-пропаноата (1,02 г; 4,09 ммоль) в MeCN (10 мл) при 0°С добавляли DBU (2,0 мл; 13 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и разбавляли EtOAc. Органический слой промывали 10% водной HCl, сушили над МgSO4, фильтровали и концентрировали при пониженном давлении с получением 0,39 г (48%) неочищенного продукта, который использовали непосредственно без дополнительной очистки.
Стадия Г: Получение 5-гидрокси-2,6-диметилпиридазин-3(2Н)-она
Смесь метил-5-гидрокси-2,6-диметил-3-оксо-2,3-дигидропиридазин-4-карбоксилата (3,00 г; 15,1 ммоль) и 6 н. водной HCl (25 мл; 150 ммоль) в диоксане (25 мл) кипятили с обратным холодильником в течение 48 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением неочищенного вещества, которое разбавляли EtOAc. Органический слой промывали водой и рассолом, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 0,74 г (35%) целевого продукта. Водный слой концентрировали при пониженном давлении. Полученное твердое вещество разбавляли водой и EtOAc-THF. Органический слой отделяли. Водный слой экстрагировали EtOAc (2×). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 0,80 г (37%) дополнительного целевого продукта. Суммарно получили 1,54 г (72%) целевого продукта, который использовали непосредственно без дополнительной очистки.
Стадия Д: Получение 5-хлор-2,6-диметилпиридазин-3(2Н)-она
Смесь 5-гидрокси-2,6-диметилпиридазин-3(2Н)-она (736 мг; 5,25 ммоль) и РОСl3 (4,5 мл) перемешивали при 85°С в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного вещества, которое гасили насыщенным водным Nа2СО3. Полученную смесь перемешивали в течение 2 часов и экстрагировали EtOAc (3×). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 587 мг (70%) целевого продукта, который использовали непосредственно без дополнительной очистки.
Стадия Е: Получение 4-хлор-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты
К раствору 5-хлор-2,6-диметилпиридазин-3(2Н)-она (780 мг; 4,67 ммоль) в дымящей H2SO4 (25 мл) при 0°С медленно добавляли K2Cr2O7 (3,33 г; 11,2 ммоль) при перемешивании. После добавления К2Сr2O7 ледяную баню удаляли, и реакционную смесь оставляли нагреваться до комнатной температуры. Когда реакция начинала протекать слишком быстро, ледяную баню возвращали и добавляли остаток К2Сr2O7. Реакционную смесь перемешивали при 60°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, выливали на лед и экстрагировали ЕtOАс (3×). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 649 мг (74%) целевого продукта, который использовали непосредственно без дополнительной очистки.
Стадия Ж: Получение метил-4-хлор-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилата
Раствор 4-хлор-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты (390 мг; 2,07 ммоль) и концентрированной HCl (0,10 мл) в МеОН (6 мл) кипятили с обратным холодильником в течение 8 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением неочищенного вещества, которое перерастворяли в ЕtOАс. Органический слой промывали водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% гексанов до 10, до 20, до 30, до 50% ЕtOАс в гексанах), что позволило получить 72 мг (17%) целевого продукта.
Стадия 3: Получение метил-4-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилата
Перемешанный раствор метил-4-хлор-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилата (72 мг; 0,35 ммоль), 2-фтор-4-метилтиоанилина (69 мг; 0,44 ммоль), Pd(OAc)2 (10 мг; 0,044 ммоль), BINAP (2,2'-бис-(дифенилфосфино)-1,1'-бинафтил) (40 мг; 0,064 ммоль) и Сs2СО3 (197 мг; 0,60 ммоль) в толуоле (1,5 мл) закрывали в сосуде в атмосфере N2. Перемешивали в течение 10 минут при комнатной температуре и затем нагревали при 80°С в течение 16 часов при перемешивании. Реакционную смесь охлаждали до комнатной температуры и разбавляли ЕtOАс. Осадок отфильтровывали и промывали ЕtOАс. Фильтрат промывали водой. Органический слой отделяли, и водный слой экстрагировали ЕtOАс. Объединенные органические слои сушили над МgSO4, фильтровали, концентрировали с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% CH2Cl2 до 1% МеОН в CH2Cl2) с последующей дополнительной колоночной флэш-хроматографией на силикагеле (от 10 до 15, до 20% ЕtOАс в CH2Cl2), что позволило получить 48 мг (42%) целевого продукта.
Стадия И: Получение 4-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридазин-3-карбоксамида
К раствору метил-4-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидро-пиридазин-3-карбоксилата (25 мг; 0,077 ммоль) и O-(2-винилокси-этил)-гидроксиламина (24 мг; 0,23 ммоль) в THF (2 мл) при 0°С добавляли LiHMDS (0,54 мл; 0,54 ммоль, 1 М в THF). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь гасили насыщенным водным NаНСО3 и разбавляли ЕtOАс. Органический слой промывали рассолом, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% CH2Cl2 до 1,5% МеОН в СН2Сl2), что позволило получить 30 мг (99%) целевого продукта.
Стадия К: Получение 4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамида
К раствору 4-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридазин-3-карбоксамида (30 мг; 0,077 ммоль) в ЕtOН/THF (2 мл/2 мл) добавляли 1 н. водную HCl (0,15 мл; 0,15 ммоль, 1 н. водный раствор) при комнатной температуре. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь нейтрализовали до рН 7, разбавляли ЕtOАс (3×), промывали водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% ЕtOАс до 100% СН2Сl2, до 2,5, до 3, до 5% МеОН в СН2Сl2), что позволило получить 6 мг (22%) целевого продукта. MS АРСI (-) (масс-спектрометрия, химическая ионизация при атмосферном давлении, регистрация отрицательных ионов) m/z 367 (М-1); 1H ЯМР (400 МГц, CD3OD) δ 7.35 (t, 1H), 7.18 (dd, 1H), 7.14 (dd, 1H), 5.92 (s, 1H), 4.06 (t, 2H), 3.79 (t, 2H), 3.74 (s, 3Н), 2.51 (s, 3Н).
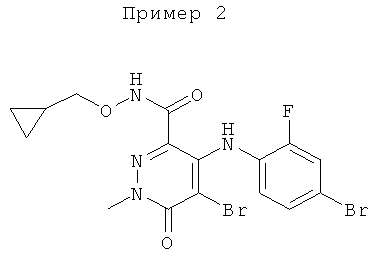
5-Бром-4-(4-бром-2-фторфениламино)-N-(циклопропилметокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
Стадия А: Получение метил-4-(2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилата
Указанное в заголовке соединение получили с выходом 61% по методике, описанной выше в Примере 1 (Стадия 3), используя метил-4-хлор-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилат (109 мг; 0,54 ммоль; получен как описано выше в Примере 1 (Стадии А-Ж)) и 2-фторанилин (0,053 мл; 0,54 ммоль).
Стадия Б: Получение метил-5-бром-4-(4-бром-2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилата
Смесь метил-4-(2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилата (88 мг; 0,32 ммоль) и NBS (59 мг; 0,33 ммоль) в DMF (1,5 мл) перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь разбавляли ЕtOАс и промывали водой (2×). Органический слой сушили над МgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% CH2Cl2 до 0,5% МеОН в CH2Cl2) с последующей дополнительной колоночной флэш-хроматографией на силикагеле (30% ЕtOАс в CH2Cl2) с получением 80 мг смеси метил-5-бром-4-(2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилата и метил-5-бром-4-(4-бром-2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилата. Эту смесь подвергали повторному бромированию. К этой смеси добавляли DMF (1,5 мл), затем NBS (29 мг; 0,22 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 2,5 часа при комнатной температуре. Добавляли дополнительные 15 мг NBS, и реакционную смесь перемешивали в течение еще 20 часов при комнатной температуре. Реакционную смесь разбавляли ЕtOАс и промывали водой (2×). Органический слой сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (30% ЕtOАс в CH2Cl2), что позволило получить 62 мг (64%) целевого продукта.
Стадия В: Получение 5-бром-4-(4-бром-2-фторфениламино)-N-(циклопропилметокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамида
Указанное в заголовке соединение получили с выходом 40% по методике, описанной в Примере 1 (Стадия И), используя метил-5-бром-4-(4-бром-2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксилат (31 мг; 0,071 ммоль) и O-циклопропилметил-гидроксиламин (20 мг; 0,23 ммоль). MS APCI (-) m/z 487, 489, 491 (М-1, образец с Вr); 1H ЯМР (400 МГц, CD3OD) δ 7.38 (dd, 1H), 7.31 (dd, 1H), 7.05 (t, 1H), 3.82 (s, 3H), 3.65 (d, 2H), 1.13 (m, 1H), 0.58 (q, 2H), 0.31 (q, 2H).
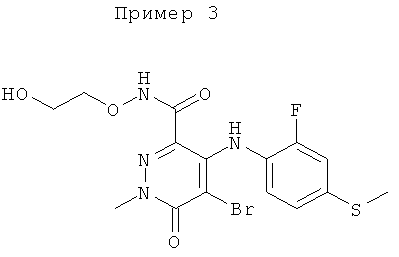
4-(2-Фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
Стадия А: Получение N-метилпропионогидразида
К раствору метилгидразина (27,6 мл; 508 ммоль) и каталитического количества DMAP (4-(N,N-диметиламино)пиридин) в CH2Cl2 (130 мл) при 0°С добавляли раствор ацетилхлорида (15,0 мл; 169 ммоль) в CH2Cl2 (30 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Белое твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали вакуумной дистилляцией, что позволило получить 8,25 г (48%) целевого продукта (63-66°С при 0,14 мМ Нg (18,7 Па)).
Стадия Б: Получение диэтил-2-(2-метил-2-пропионилгидразоно)малоната
Раствор N-метилпропионогидразида (18,78 г; 183,9 ммоль) и диэтилкетомалоната (56,1 мл; 368 ммоль) в толуоле (136 мл) кипятили с обратным холодильником с использованием ловушки Дина-Старка в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% гексанов до 5, до 10% ЕtOАс в гексанах), что позволило получить 23 г (49%) целевого продукта.
Стадия В: Получение этил-4-гидрокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксилата
К раствору LiHMDS (0,78 мл; 0,78 ммоль, 1М раствор в THF) в THF (1 мл) при -78°С добавляли раствор диэтил-2-(2-метил-2-пропионилгидразоно)-малоната (50 мг; 0,19 ммоль) в THF (1 мл). Полученную смесь медленно нагревали до -40°С и перемешивали в течение 1,5 часа при -40°С. Реакционную смесь гасили 10% водной HCl и разбавляли водой. Полученную смесь экстрагировали ЕtOАс (2×). Объединенные органические слои промывали водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% гексанов до 20% ЕtOАс в гексанах), что позволило получить 25 мг (61%) целевого продукта.
Стадия Г: Получение этил-4-хлор-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксилата
Смесь этил-4-гидрокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксилата (1,85 г; 8,72 ммоль) и РОСl3 (9 мл) нагревали в течение 16 часов при 85°С. РОСl3 удаляли при пониженном давлении. Затем неочищенное вещество гасили водой со льдом. Смесь нейтрализовали насыщенным водным NаНСО3 (рН приблизительно 6-7) и экстрагировали ЕtOАс (3×). Объединенные органические слои сушили над МgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% гексанов до 5, до 10, до 20% ЕtOАс в гексанах), что позволило получить 1,72 г (86%) целевого продукта.
Стадия Д: Получение этил-4-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксилата
Указанное в заголовке соединение получили с выходом 81% по методике, описанной в Примере 1 (Стадия 3), используя этил-4-хлор-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксилат (500 мг; 2,17 ммоль) и 2-фтор-4-метилтиоанилин (375 мг; 2,38 ммоль).
Стадия Е: Получение 4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамида
Указанное в заголовке соединение получали с выходом 78% (2 стадии) по методикам, описанным в Примере 1 (Стадии И и К), используя этил-4-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксилат (50 мг; 0,14 ммоль) и O-(2-винилокси-этил)-гидроксиламин (44 мг; 0,43 ммоль). MS APCI (-) m/z 381 (М-1); 1H ЯМР (400 МГц, CD3OD) δ 7.10 (dd, 1H), 7.03 (dd, 1H), 6.87 (t, 1H), 3.99 (t, 2H), 3.79 (s, 3H), 3.74 (t, 2H), 2.47 (s, 3H), 1.74 (s, 3H).
Следующие далее соединения получили по методике, которая описана в Примере 1 (Стадия И), с использованием этил-4-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксилата и соответствующего гидроксиламина.
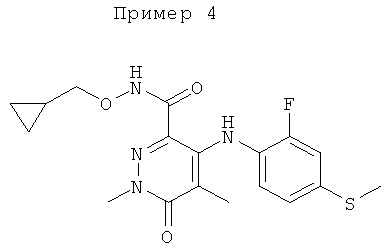
N-(Циклопропилметокси)-4-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 391 (М-1); 1H ЯМР (400 МГц, CD3OD) δ 7.09 (dd, 1H), 7.03 (dd, 1H), 6.86 (t, 1H), 3.78 (s, 3H), 3.71 (d, 2H), 2.47 (s, 3H), 1.75 (s, 3H) 1.16 (m, 1H), 0.58 (m, 2H), 0.31 (m, 2H).
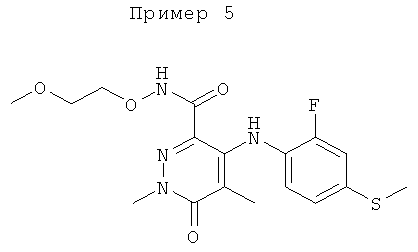
4-(2-Фтор-4-(метилтио)фениламино)-N-(2-метоксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 395 (M-1); 1Н ЯМР (400 МГц, CD3OD) δ 7.10 (dd, 1H), 7.03 (d, 1H), 6.87 (t, 1H), 4.05 (t, 2H), 3.78 (s, 3Н), 3.64 (t, 2H), 3.37 (s, 3H), 2.47 (s, 3H), 1.74 (s, 3H).
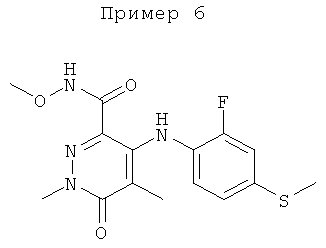
4-(2-Фтор-4-(метилтио)фениламино)-N-метокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 351 (M-1); 1H ЯМР (400 МГц, СD3OD) δ 7.10 (d, 1H), 7.04 (d, 1H), 6.87 (t, 1H), 3.78 (s, 3H), 3.76 (s, 3H), 2.47 (s, 3H), 1.74 (s, 3H).
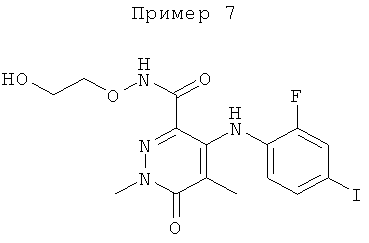
4-(2-Фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
Стадия А: Получение 4-гидрокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты
К раствору LiHMDS (331 мл; 331 ммоль, 1 М раствор в THF) в THF (430 мл) при -78°С добавляли раствор диэтил-2-(2-метил-2-пропионилгидразоно)-малоната (21,40 г; 82,86 ммоль), полученный по методике, описанной в Примере 3 (Стадия Б), в THF (10 мл). Полученную смесь медленно нагревали до -40°С в течение 1 часа и перемешивали в течение 1,5 часа при -40°С. К реакционной смеси при -40°С добавляли воду (500 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления THF. Полученную водную смесь гасили 6 н. водной НСl при 0°С и подкисляли до рН 1-2. Полученную смесь перемешивали в течение 16 часов при комнатной температуре. Осадок отфильтровывали и растирали с CH2Cl2, что позволило получить 7,21 г (47%) целевого продукта. Фильтрат экстрагировали ЕtOАс (3×). Объединенные органические слои промывали водой, сушили над МgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое растирали с СН2Сl2, что позволило дополнительно получить 3,56 г (23%) целевого продукта. Водный слой вновь экстрагировали ЕtOАс (3×). Объединенные органические слои промывали водой, сушили над МgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое растирали с CH2Cl2, что позволило дополнительно получить 1,32 г (9%) целевого продукта. Суммарно получили 12,09 г (79%) целевого продукта.
Стадия Б: Получение 4-хлор-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты
Смесь 4-гидрокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты (876 мг; 4,76 ммоль) и РОСl3 (4,5 мл) нагревали в течение 24 часов при 85°С. РОСl3 удаляли при пониженном давлении. Неочищенное вещество гасили льдом. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. После удаления твердых веществ фильтрованием водный фильтрат экстрагировали ЕtOАс (3×). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Извлеченное вещество объединяли с твердыми веществами, выделенными ранее, и растирали с эфиром, что позволило получить 577 мг (60%) целевого продукта.
Стадия В: Получение 4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты
К суспензии 4-хлор-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты (200 мг; 0,99 ммоль) и 2-фтор-4-йоданилина (478 мг; 1,97 ммоль) в THF (6,5 мл) при -78°С медленно добавляли раствор LiHMDS (3,00 мл; 3,00 ммоль, 1 М раствор в THF). После завершения добавления полученную смесь медленно нагревали до комнатной температуры и перемешивали в течение 4 часов. Реакционную смесь гасили 6 н. водной HCl (8 мл) при 0°С, нагревали до комнатной температуры и перемешивали в течение 1,5 часа. Осадок отфильтровывали, промывали водой и эфиром и растирали с эфиром, что позволило получить 158 мг (38%) целевого продукта.
Стадия Г: Получение 4-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамида
К суспензии 4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты (41 мг; 0,10 ммоль) и HOBt (28 мг; 0,21 ммоль) в DMF (1,5 мл) добавляли EDCI (40 мг; 0,21 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 1,5 часа. К активированному сложному эфиру при комнатной температуре добавляли O-(2-винилокси-этил)-гидроксиламин (21 мг; 0,20 ммоль) и TEA (триэтиламин) (0,030 мл; 0,22 ммоль). После перемешивания в течение 1,5 часа реакционную смесь разбавляли ЕtOАс и промывали насыщенным водным NH4Cl, рассолом, насыщенным водным NaHCO3 (2×) и рассолом. Органический слой отделяли, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридазин-3-карбоксамида, который использовали непосредственно без дополнительной очистки. Указанное в заголовке соединение получили по методике, описанной выше в Примере 1 (Стадия К), используя неочищенный 4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридазин-3-карбоксамид (выход 40% за две стадии). MS APCI (-) m/z 461 (М-1); 1H ЯМР (400 МГц, СD3OD) δ 7.52 (dd, 1Н), 7.44 (d, 1H), 6.63 (t, 1H), 3.98 (t, 2H), 3.80 (s, 3H), 3.74 (t, 2H), 1.78 (s, 3Н).
Следующие далее соединения получили по методикам, которые описаны ранее в Примере 7 (Стадии В и Г) с использованием соответствующих анилинов и гидроксиламина. В некоторых случаях в конце может потребоваться стадия удаления защиты. Такие удаления защиты могут быть выполнены стандартными, известными в литературе методами.
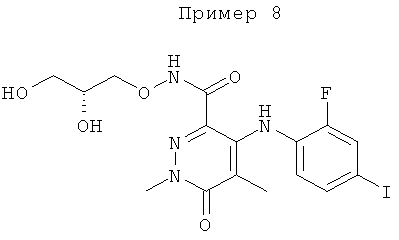
(R)-N-(2,3-Дигидроксипропокси)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 491 (М-1); 1H ЯМР (400 МГц, СD3OD) δ 7.52 (dd, 1H), 7.44 (d, 1H), 6.63 (t, 1H), 4.02 (m, 1H), 3.88 (m, 2H), 3.80 (s, 3H), 3.59 (m, 2H), 1.77 (s, 3Н).
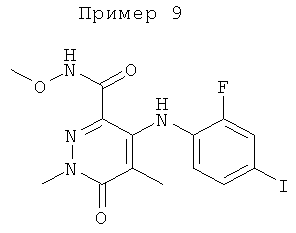
4-(2-Фтор-4-йодфениламино)-N-метокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 431 (М-1); 1H ЯМР (400 МГц, CD3OD) δ 7.52 (dd, 1H), 7.44 (d, 1H), 6.63 (t, 1H), 3.79 (s, 3Н), 3.75 (s, 3Н), 1.77 (s, 3H).
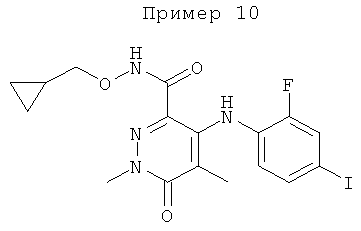
N-(Циклопропилметокси)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 471 (М-1); 1H ЯМР (400 МГц, CD3OD) δ 7.51 (dd, 1H), 7.44 (d, 1H), 6.62 (t, 1H), 3.79 (s, 3Н), 3.70 (d, 2H), 1.78 (s, 3Н), 1.15 (m, 1H), 0.57 (q, 2H), 0.30 (q, 2H).
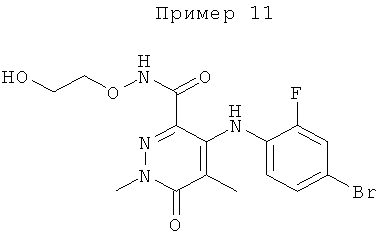
4-(4-Бром-2-фторфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
Стадия А: Получение диэтил-2-(2-метилгидразоно)малоната
К раствору диэтилкетомалоната (95 г; 546 ммоль) в ЕtOН (600 мл) (3-горлая колба емкостью 2 л, оснащенная термопарой, трубопроводом для подачи N2, холодильником и механической мешалкой) добавляли MeNHNH2 (32 мл; 600 ммоль) одной порцией при комнатной температуре. Реакционную смесь нагревали до 60°С (внутренняя температура, нагревали с использованием нагревающей сетки) и перемешивали в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного вещества вместе с твердыми осажденными веществами, которое очищали через набивку силикагеля (3:2, гексаны:ЕtOАс), что позволило получить 81 г (74%) целевого продукта.
Стадия Б: Получение диэтил-2-(2-метил-2-пропионилгидразоно)малоната
К раствору 2-(2-метилгидразоно)малоната (100 г; 494 ммоль) в THF (1 л) при 0°С через капельную воронку добавляли LiHMDS (643 мл; 643 ммоль) в течение 45 минут. Реакционную смесь перемешивали в течение 45 минут при 0°С. Добавляли пропионилхлорид (51,6 мл; 593 ммоль) одной порцией. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 20 часов. Реакционную смесь гасили насыщенным водным NH4Cl (85 мл) и водой (85 мл). Реакционную смесь концентрировали при пониженном давлении и дополнительно добавляли воду (300 мл). Полученную смесь экстрагировали ЕtOАс (3×250 мл). Объединенные органические слои промывали насыщенным водным NаНСО3 (2×250 мл), затем рассолом (250 мл), сушили над МgSO4, фильтровали и концентрировали при пониженном давлении с получением 112 г (88%) неочищенного продукта, который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия В: Получение 4-гидрокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты
К раствору LiHMDS (331 мл; 331 ммоль, 1 М раствор в THF) в THF (430 мл) при -78°С добавляли раствор 2-(2-метил-2-пропионилгидразоно)малоната (21,40 г; 82,86 ммоль) в THF (10 мл). Полученную смесь медленно нагревали до -40°С в течение 1 часа и перемешивали в течение 1,5 часа при -40°С. К реакционной смеси добавляли воду (500 мл) при -40°С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении, гасили 6 н. водной HCl при 0°С и подкисляли до рН 1-2. Полученную смесь перемешивали в течение 16 часов при комнатной температуре. Осадок отфильтровывали и растирали с CH2Cl2, что позволило получить 7,21 г (47%) целевого продукта. Фильтрат экстрагировали ЕtOАс (3×). Объединенные органические слои промывали водой, сушили над МgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое растирали с CH2Cl2, что позволило дополнительно получить 3,56 г (23%) целевого продукта. Водный слой вновь экстрагировали ЕtOАс (3×). Объединенные органические слои промывали водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое растирали с CH2Cl2, что позволило дополнительно получить 1,32 г (9%) целевого продукта. Суммарно получили 12,09 г (79%) целевого продукта.
Стадия Г: Получение 4-хлор-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты
Смесь 4-гидрокси-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты (35,4 г; 192 ммоль), каталитического количества DMF (3 капли) и РОСl3 (178 мл; 1,92 моль) нагревали в течение 2 суток при 90°С, и затем РОСl3 удаляли при пониженном давлении. Неочищенное вещество гасили льдом, и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Осадок, выпавший из раствора, отфильтровывали и промывали эфиром. Собранный осадок растирали с эфиром, что позволило получить 11,7 г (30%) целевого продукта. Фильтрат экстрагировали ЕtOАс (2×). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который растирали с эфиром и сушили при пониженном давлении, что позволило дополнительно получить 9,56 г (24%) целевого продукта. Суммарно получили 21,29 г (55%) целевого продукта.
Стадия Д: Получение 4-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты
К раствору 4-бром-2-фторанилина (22,6 г; 116 ммоль) в THF (165 мл) при -78°С медленно добавляли раствор LiHMDS (174 мл; 174 ммоль, 1М раствор в THF). Полученную смесь перемешивали в течение 1 часа при -78°С. К этой смеси добавляли 4-хлор-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновую кислоту (11,0 г; 54,4 ммоль) в виде твердого вещества при -78°С. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 21 часа. Реакционную смесь гасили и подкисляли 10% водной HCl (250 мл) при 0°С. К этой смеси добавляли воду (100 мл), ЕtOАс (350 мл) и рассол (50 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. Органический слой отделяли, и кислотный водный слой экстрагировали ЕtOАс (2×300 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое растирали с эфиром (5×), фильтровали, промывали эфиром и сушили при пониженном давлении, что позволило получить 14,51 г (75%) целевого продукта.
Стадия Е: Получение 4-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридазин-3-карбоксамида
К суспензии 4-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоновой кислоты (14,51 г; 40,74 ммоль) и HOBt (11,01 г; 81,48 ммоль) в DMF (165 мл) добавляли EDCI (15,62 г; 81,48 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 1,5 часа. К активированному сложному эфиру при комнатной температуре добавляли O-(2-(винилокси)этил)гидроксиламин (8,36 мл; 81,48 ммоль) и TEA (11,36 мл; 81,48 ммоль). После перемешивания в течение 1,5 часа реакционную смесь разбавляли ЕtOАс и промывали насыщенным водным NH4Cl, рассолом, насыщенным водным NаНСО3 (2×) и рассолом. Органический слой отделяли, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который использовали непосредственно без дополнительной очистки.
Стадия Ж: Получение 4-(4-бром-2-фторфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамида
Смесь 4-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридазин-3-карбоксамида (17,98 г; 40,75 моль) и 6 н. водной HCl (13,58 мл; 81,50 ммоль) в ЕtOН/THF (50 мл/50 мл) перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и разбавляли водой (50 мл). Полученную смесь экстрагировали ЕtOАс (2×). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной флэш-хроматографией на силикагеле (от 100% CH2Cl2 до 2,5% МеОН в CH2Cl2), что позволило получить 9,41 г (56% за две стадии) целевого продукта. MS APCI (-) m/z 413, 415 (М-1, образец с Вr); 1H ЯМР (400 МГц, CD3OD) δ 7.38 (dd, 1H), 7.27 (d, 1H), 6.79 (t, 1H), 3.99 (t, 2H), 3.80 (s, 3H), 3.74 (t, 2H), 1.77 (s, 3H).
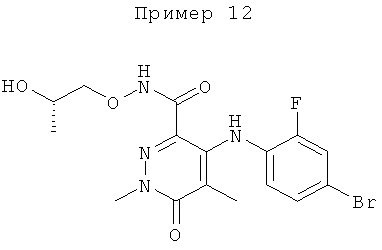
(S)-4-(4-Бром-2-фторфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 427, 429 (М-1, образец с Вr); 1H ЯМР (400 МГц, CD3OD) δ 7.39 (dd, 1H), 7.27 (dd, 1H), 6.79 (t, 1H), 3.98 (m, 1H), 3.84 (dd, 1H), 3.80 (s, 3H), 3.72 (dd, 1H), 1.78 (s, 3H), 1.15 (d, 3H).
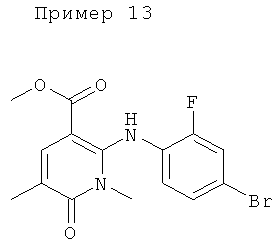
Метил-2-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Стадия А: Получение 2-хлор-6-оксо-1,6-дигидро-пиридин-3-карбоновой кислоты
2-Хлор-6-оксо-1,6-дигидро-пиридин-3-карбоновую кислоту получили из дихлорникотиновой кислоты (3,00 г; 15,6 ммоль, Aldrich) по методике, описанной в патенте США №3682932, с получением 1,31 г (48%) целевого продукта.
Стадия Б: Получение метилового эфира 2-хлор-1-метил-6-оксо-1,6-дигидро-пиридин-3-карбоновой кислоты
К раствору 2-хлор-6-оксо-1,6-дигидро-пиридин-3-карбоновой кислоты (0,644 г; 3,71 ммоль) в DMF (20 мл) добавляли гидрид лития (95%; 0,078 г; 9,28 ммоль), и реакционную смесь перемешивали в течение 40 минут в атмосфере N2. Затем добавляли метилйодид (0,508 мл; 1,16 г; 8,16 ммоль), и реакционную смесь перемешивали в течение дополнительных 45 минут. Реакционную смесь гасили 2М HCl до тех пор, пока рН не достигал 6-7. Реакционную смесь разбавляли ЕtOАс и насыщенным NaCl, и слои разделяли. Водный слой вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4) и концентрировали при пониженном давлении с получением неочищенного желтого твердого вещества. HPLC-анализ показал два продукта в соотношении 4:1, которые разделяли колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, от 15:1 до 10:1) с получением 0,466 г (62%) чистого целевого продукта в виде белого кристаллического твердого вещества. Также выделили минорный продукт в виде бледно-желтого кристаллического твердого вещества и идентифицировали его как региоизомер метилового эфира 2-хлор-6-метокси-никотиновой кислоты.
Стадия В: Получение метил-5-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К раствору метил-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,100 г; 0,496 ммоль) в DMF (5 мл) добавляли N-бромсукцинимид (0,177 г; 0,992 ммоль), и реакционную смесь перемешивали в течение 4 часов при комнатной температуре в атмосфере N2. Реакционную смесь гасили насыщенным бисульфитом натрия, затем разбавляли ЕtOАс и H2O, и слои разделяли. Водный слой вновь экстрагировали ЕtOАс (2×). Объединенные органические слои сушили (Nа2SO4) и концентрировали при пониженном давлении с получением желтого твердого вещества с количественным выходом.
Стадия Г: Получение метил-2-хлор-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К суспензии метил-5-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,400 г; 1,43 ммоль) и 1,1'-бис(дифенилфосфино)-ферроцендихлорпалладия(II) (0,0587 г; 0,0713 ммоль) в диоксане (8 мл) при 0°С в атмосфере N2 добавляли диметилцинк (0,713 мл; 1,43 ммоль, 2М раствор в толуоле). Реакционную смесь сразу нагревали до 100°С в течение 30 минут. Реакционную смесь охлаждали до 0°С и гасили МеОН (0,800 мл). Реакционную смесь разбавляли ЕtOАс и промывали 1М HCl. Водный слой вновь экстрагировали ЕtOАс (1×). Объединенные органические слои промывали насыщенным NaCl, сушили (Na2SO4) и концентрировали при пониженном давлении до темно-желтой смолы. Очистка колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 15:1) дала 0,164 г (53%) чистого целевого продукта в виде желтого кристаллического твердого вещества.
Стадия Д: Получение метил-2-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К раствору 4-бром-2-фторбензоламина (0,058 г; 0,31 ммоль) в THF (2 мл) при -78°С в атмосфере N2 добавляли по каплям бис(триметилсилил)амид лития (0,56 мл; 0,56 ммоль, 1М раствор в гексанах). Реакционную смесь перемешивали в течение одного часа при -78°С. Затем по каплям добавляли метил-2-хлор-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилат (0,060 г; 0,28 ммоль) в виде раствора в THF (1 мл), и реакционную смесь перемешивали в течение 25 минут при -78°С. Реакционную смесь гасили добавлением H2O, pH регулировали, используя 0,1М HCl, затем разбавляли ЕtOАс и насыщенным NaCl, и слои разделяли. Водный слой вновь экстрагировали ЕtOАс (1×). Объединенные ЕtOАс-слои сушили (Nа2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 20:1) дала 0,086 г (84%) чистого целевого продукта в виде белого кристаллического твердого вещества. MS ESI (+) m/z 371, 373 (M+, образец с Вr); 1H ЯМР (400 МГц, CDCl3) δ 9.57 (s, 1Н), 7.79 (s, 1H), 7.32 (d, 1H), 7.18 (d, 1H), 6.58 (t, 1H), 3.85 (s, 3Н), 3.29 (s, 3H), 2.14 (s, 3H).
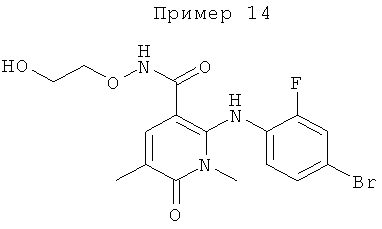
2-(4-Бром-2-фторфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Стадия А: Получение 2-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридин-3-карбоксамида
К раствору метил-2-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,060 г; 0,16 ммоль) в THF (2 мл) добавляли O-(2-винилокси-этил)-гидроксиламин (0,042 мл; 0,41 ммоль). Раствор охлаждали до 0°С и по каплям добавляли бис(триметилсилил)амид лития (0,81 мл; 0,81 ммоль, 1М раствор в гексанах). Реакционную смесь нагревали до комнатной температуры. После перемешивания в течение 35 минут реакционную смесь гасили добавлением насыщенного NаНСО3 и распределяли между ЕtOАс и насыщенным NaCl. Слои разделяли, органический слой сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией (метиленхлорид/МеОН, 20:1) дала 0,067 г (94%) чистого целевого продукта в виде не совсем белого кристаллического твердого вещества.
Стадия Б: Получение 2-(4-бром-2-фторфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида
К раствору 2-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)-этокси)-1,6-дигидропиридин-3-карбоксамида (0,067 г; 0,150 ммоль) в этаноле (2 мл) добавляли водную 2М HCl (0,380 мл; 0,760 ммоль). Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. рН реакционной смеси регулировали, используя 1М NaOH. Реакционную смесь разбавляли ЕtOАс и H2O. Органический слой отделяли и промывали насыщенным NaCl. Объединенные водные слои вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4) и концентрировали при пониженном давлении с получением 0,060 г (94%) чистого целевого продукта в виде не совсем белого кристаллического твердого вещества. MS ESI (+) m/z 414, 416 (M+, образец с Br); 1H ЯМР (400 МГц, CDCl3) δ 9.80 (s, 1H), 8.44 (s, 1H), 7.31 (d, 1H), 7.19 (d, 1H), 6.59 (t, 1H), 4.05 (m, 2H), 3.85 (m, 1H), 3.75 (m, 2H), 3.29 (s, 3H), 2.15 (s, 3H).
Следующие соединения получили, используя способы, описанные в Примерах 13 и 14. В некоторых случаях, например в Примере 14, в конце может потребоваться стадия удаления защиты. Такие удаления защиты могут быть выполнены стандартными, известными в литературе методами.
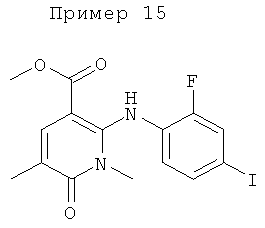
Метил-2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Метил-2-хлор-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилат превращали в метил-2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилат по методике, описанной на Стадии Д Примера 13, используя 2-фтор-4-йодбензоламин, с получением целевого продукта в виде белого кристаллического твердого вещества. MS ESI (+) m/z 417 (М+1); 1H ЯМР (400 МГц, CDCl3) δ 9.56 (s, 1H), 7.79 (s, 1H), 7.49 (d, 1H), 7.36 (d, 1H), 6.43 (t, 1H), 3.85 (s, 3H), 3.30 (s, 3H), 2.15 (s, 3H).
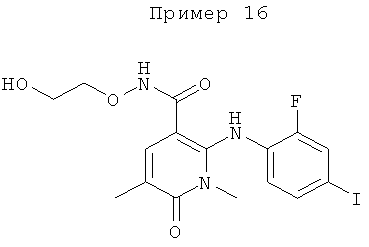
2-(2-Фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Стадия А: Получение 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридин-3-карбоксамида
К раствору метил-2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,500 г; 1,20 ммоль) в THF (60 мл) добавляли O-(2-винилокси-этил)-гидроксиламин (0,149 г; 1,44 ммоль). Раствор охлаждали до 0°С и по каплям добавляли бис(триметилсилил)амид лития (4,81 мл; 4,81 ммоль) (1М раствор в гексанах). Реакционную смесь нагревали до комнатной температуры. После перемешивания в течение 10 минут реакционную смесь гасили добавлением 1М HCl и распределяли между ЕtOАс и насыщенным NaCl. Слои разделяли, и органический слой сушили (Na2SO4) и концентрировали при пониженном давлении с получением неочищенного желтого твердого вещества, которое использовали без очистки на следующей стадии.
Стадия Б: Получение 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида
К раствору неочищенного 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридин-3-карбоксамида (0,585 г; 1,20 ммоль) в этаноле (10 мл) добавляли водную 2М HCl (3 мл). Реакционную смесь перемешивали в течение 45 минут при комнатной температуре. рН реакционной смеси доводили до рН 7, используя 1М NaOH. Реакционную смесь разбавляли ЕtOАс и Н2O. Органический слой отделяли и промывали насыщенным NaCl. Объединенные водные слои вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией на силикагеле (метиленхлорид/МеОН, 15:1) дала 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридин-3-карбоксамид (0,421 г; 76% за две стадии) в виде бледно-желтого твердого вещества. MS ESI (+) m/z 462, образец (М+1); 1H ЯМР (400 МГц, CDCl3) δ 9.77 (s, 1H), 8.50 (s, 1H), 7.47 (d, 1H), 7.36 (d, 1H), 6.43 (t, 1H), 4.04 (br s, 2H), 3.85 (br s, 1H), 3.74 (br s, 2H), 3.29 (s, 3H), 2.14 (s, 3H).
Пример 16А
Получение Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида
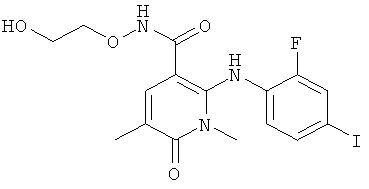
Стадия 1: Получение метилового эфира 2-(2-фтор-4-йод-фениламино)-1,5-диметил-6-оксо-1,6-дигидро-пиридин-3-карбоновой кислоты
К перемешиваемому раствору 2-фтор-4-йоданилина (182 г; 0,77 моль) в THF (5,25 л) при -45°С в атмосфере азота добавляли 1М раствор бис(триметилсилил)амида лития в гексанах (1260 г) в течение 28 минут при температуре от -43 до -41,6°С. Через 1 час добавляли метиловый эфир 2-хлор-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (155 г; 0,72 моль) в THF (1,05 л) в течение 43 минут. Смесь выдерживали в течение 1 часа 55 минут при -46°С, затем оставляли нагреваться до -13°С и гасили водой (186 мл; 10,3 моль) в течение 5 минут, поддерживая температуру между -13°С и -11°С. Смесь затем оставляли нагреваться до 0°С в течение 30 минут. Затем добавляли 2М HCl в течение 1 часа до тех пор, пока не достигали рН 7-8 (добавляли 1855 мл). После стояния в течение ночи смесь оставляли нагреваться до температуры окружающей среды и добавляли раствор хлорида натрия (1 л, 15% масс./об.). Нижний (водный слой) отбрасывали, а THF-слой концентрировали дистилляцией до приблизительно 1,4 л. К смеси при приблизительно 52°С в течение 1 часа 15 минут добавляли изогексан (4,65 л), и затем смесь охлаждали до 20°С в течение 3 часов. После выдержки в течение 1 часа при 20°С смесь охлаждали до 0°С и выдерживали при этой температуре в течение ночи. Реакционную смесь затем фильтровали, и твердое вещество промывали холодным изогексаном (5°С) (2×1,25 л). Твердое вещество сушили в вакуумном сушильном шкафу при 45°С с получением метилового эфира 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (248 г; 0,60 моль; выход 83%). 1H ЯМР (D6-DMSO): δ 7.75 (d, 1H, J 1 Гц, ArH), 7.68 (dd, 1H, J 11, 2 Гц, ArH), 7.42 (d, 1H, J 8.5 Гц, ArH), 6.62 (~t, 1H, J 8.5 Гц, ArH), 3.69 (s, 3Н, ОСН3), 3.22 (s, 3H, NСН3), 2.03 (s, 3H, АrСН3).
Стадия 2: Получение (2-винилокси-этокси)-амида 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты
К перемешиваемому раствору метилового эфира 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (221 г; 0,53 моль) и O-(2-винилоксиэтил)-гидроксиламина (63 г; 0,61 моль) в THF (2,85 л), в атмосфере азота, добавляли 1М раствор бис(триметилсилил)амида лития в гексанах (1431 г) в течение 55 минут, поддерживая температуру от -14,7 до -12,4°С. После выдержки в течение 2 часов при -15°С к смеси добавляли воду (165 мл; 9,2 моль), затем 2М раствор HCl (1,98 л), который добавляли в течение 20 минут. Смесь затем оставляли нагреваться до 22°С, и нижнюю водную фазу (2,25 л) отделяли и отбрасывали. Органическую фазу промывали раствором хлорида натрия (15% масс./масс., 1100 мл), и объем уменьшали до приблизительно 1,75 л отгонкой 2,25 л растворителя при давлении окружающей среды. К смеси в течение 2,5 часа добавляли изогексан (3,35 л), поддерживая температуру приблизительно при 58°С. Спустя еще 1 час при этой температуре смесь охлаждали до 20°С, выдерживали в течение 1 часа, затем охлаждали до 0°С и выдерживали при этой температуре в течение ночи. Добавляли дополнительное количество изогексана (500 мл), и смесь выдерживали в течение 1 часа, затем еще добавляли изогексан (500 мл). После выдержки в течение 45 минут при 0°С суспензию фильтровали, и твердое вещество промывали охлажденным изогексаном (1,1 л), затем сушили в вакуумном сушильном шкафу при 30°С с получением (2-винилоксиэтокси)-амида 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (190 г; 0,39 моль; выход 74%). 1H ЯМР (D6-DMSO): δ 7.63 (dd, 1H, J 11, 2 Гц, ArH), 7.52 (s, 1H, ArH), 7.38 (d, 1H, J 8.5 Гц, ArH), 6.55-6.46 (m, 2H, ArH/OCH=CH2), 4.18 (dd, 1H, J 14, 2 Гц, ОСН=СН2), 3.99 (dd, 1H, J 7, 2 Гц, ОСН=СН2), 3.90-3.88 (m, 2H, ОСН2), 3.81-3.79 (m, 2H, ОCH2), 3.25 (s, 3Н, NСН3), 2.02 (s, 3Н, АrСН3).
Стадия 3: Получение Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида
К перемешиваемому раствору (2-винилоксиэтокси)-амида 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (170 г; 0,35 моль) в атмосфере азота в THF (850 мл) добавляли 2М HCl (318 мл) в течение 15 минут при 17-22°С. Через 1 час реакция завершилась (что показала HPLC), и добавляли 2М раствор гидроксида натрия (318 мл) в течение 10 минут, поддерживая температуру приблизительно 22°С. рН смеси составил приблизительно 8. Затем смесь распределяли с MIBK (метилизобутилкетон) (1,02 л), и нижний водный слой отделяли и отбрасывали. Затем объем органического раствора уменьшали дистилляцией при давлении окружающей среды и при температуре рубашки 85-95°С. После удаления 750 мл растворителя скорость дистилляции снижалась значительно, и смесь охлаждали до приблизительно 22°С. К смеси добавляли кристаллическую Форму 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида (1 г; затравка, полученная как описано в Примере 16 В), затем этилацетат (170 мл). Через 5 минут смесь начала кристаллизоваться, и добавляли изогексан (1,7 л) при 23-25°С в течение 50 минут. Суспензию выдерживали при 25°С в течение 80 минут и затем фильтровали. Твердое вещество промывали изогексаном (680 мл), затем сушили в вакуумном сушильном шкафу при 30°С с получением указанного в заголовке вещества (147 г; 0,31 моль; выход 89%). 1H ЯМР (D6-DMSO): δ 7.63 (dd, 1H, J 11, 2 Гц, АrН), 7.55 (s, 1H, ArH), 7.38 (d, 1H, J 8.5 Гц, ArH), 6.52 (~t, 1H, J 8.5 Гц, ArH), 4.91-4.35 (bs, 1H, ОН), 3.74 (~t, 2H, J 5 Гц, ОСН2), 3.51 (~t, 2H, J 5 Гц, ОСН2), 3.25 (s, 3Н, NCH3), 2.02 (s, 3Н, АrСН3). MS (ESI) (+) m/z484 (27%, [M+Na]+), 462 (100%, [M+H]+), 385 (8%), 100 (26%).
Стадия 4: Получение Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида
К перемешиваемой суспензии продукта со Стадии 3 (123 г) в этилацетате (2,0 л) при 50°С добавляли Форму 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида (4,9 г) (полученную, как описано на Стадии 5), и остаточное вещество смывали в сосуд этилацетатом (0,45 л). Смесь выдерживали при этой температуре в течение 64 часов. Анализ образца показал, что данное вещество главным образом представляло собой Форму 2. Спустя еще 1 час температуру смеси поднимали до 60°С и после 6 часов выдерживания при этой температуре дополнительно добавляли 3,25 г Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида (полученной, как описано в Примере 16Г) и промывали этилацетатом (100 мл). Перемешивание при 60°С продолжали в течение следующих 16 часов, после чего анализ показал, что некоторое количество Формы 2 оставалось. Затем объем смеси уменьшали отгонкой растворителя (удаляли 780 мл) при температуре в смеси 52°С и 400 мбар (40 кПа). Перемешивание при 60°С затем продолжали в течение ночи, и смесь анализировали повторно, однако анализ показал, что некоторое количество Формы 2 все еще оставалось. Спустя дополнительные 7 часов в реактор помещали дополнительный турбулизатор, и перемешивание продолжали до следующего дня. Затем добавляли дополнительное количество этилацетата (0,5 л) для того, чтобы способствовать эффективности перемешивания, и смесь выдерживали в течение следующих 2 часов при 60°С. Было обнаружено, что отобранный в этот момент образец представлял собой Форму 1. В целом, время, затраченное на превращение из Формы 2 в Форму 1, составило 143 часа. Вещество выдерживали в течение ночи при 50°С, затем охлаждали до 12°С и отфильтровывали. Осадок на фильтре промывали этилацетатом (400 мл) при 12°С, затем сушили в вакуумном сушильном шкафу в течение выходных дней (68 часов) при 35°С с получением Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида (109 г).
Пример 16Б
Получение Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида
К быстро перемешиваемой смеси (2-винилоксиэтокси)-амида 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (4,2 г; 8,52 ммоль) (полученного согласно Примеру 16А, Стадия 2) в этилацетате (126,00 мл) добавляли хлористый водород (17,05 мл; 17,0493 г; 17,05 ммоль). Через 2 часа осталось менее 1% исходного вещества (по данным HPLC-анализа), и фазы оставляли стоять до расслоения. Нижнюю водную фазу отделяли и отбрасывали, а органическую фазу промывали хлоридом натрия (42 мл, 15% масс./об.; затем 2×25 мл, 9% масс./об.). Затем объем уменьшали отгонкой растворителя (44 мл) при атмосферном давлении (температура головного погона 65°С). Раствор затем охлаждали до 70°С и добавляли Форму 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида (40,3265 мг), полученную согласно Примеру 16А, Стадия 4. Смесь перемешивали в течение 20 часов при 70°С. Температуру снижали до 24°С в течение 4 часов 15 минут и затем снижали до 1°С в течение 1 часа. Затем суспензию фильтровали, осадок промывали холодным этилацетатом (17 мл), и твердое вещество сушили в вакуумном сушильном шкафу при 45°С с получением Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида (3,15 г; 76%).
Пример 16В
Получение Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида
Смесь (2-винилокси-этокси)-амида 2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидро-пиридин-3-карбоновой кислоты (500 мг; 915 мкмоль) и хлористого водорода (1 мл) в тетрагидрофуране (5 мл) перемешивали в течение ночи. Затем добавляли гидроксид натрия (1М; 2,00 мл) и еще через 10 минут к смеси добавляли метилизобутилкетон (3 мл) и этилацетат (3 мл). Слои разделяли, и органический раствор промывали 50% рассолом (4 мл), затем упаривали (приблизительно половина вещества была потеряна в результате утечки). Остаток переносили в метилизобутилкетон (3 мл) и этилацетат (1 мл), и смесь нагревали до температуры дефлегмации. После охлаждения до 50°С смесь становилась мутной и добавляли изогексан (5 мл). Это привело к кристаллизации твердого вещества, и смесь охлаждали до 20°С с последующим добавлением дополнительного количества изогексана (5 мл). Твердое вещество затем отфильтровывали, промывали изогексаном (1 мл) и сушили в вакуумном сушильном шкафу при 40°С с получением указанного в заголовке соединения 140 мг.
Пример 16Г
Получение Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида
Конечный продукт из Примера 16 (25 мг) помещали в реакционную пробирку Syn 10 (Radleys) вместе с магнитным стержнем, и вещество растворяли в метаноле путем добавления одной аликвоты (1 мл) метанола, предварительно нагретого до 50°С, при перемешивании. В реакционную пробирку добавляли еще 5 мг метанола для того, чтобы при охлаждении обеспечить получение пересыщенного раствора. Когда большая часть твердого вещества растворилась, полученный раствор фильтровали через фильтр Pall 0,45 мкм PTFE Acrodisc CR13 во вторую пробирку Syn 10 при 50°С. Пробирку затем охлаждали до 0°С при скорости 3°С/мин и выдерживали при 0°С до тех пор, пока вещество не кристаллизовалось. Образцы отфильтровывали, сушили отсасыванием, затем выдерживанием в условиях окружающей среды. Твердые вещества осторожно удаляли с фильтровальной бумаги и исследовали методом XRPD.
Пример 16Д
Дифракция рентгеновских лучей на порошке (XRPD)
Картины дифракции рентгеновских лучей на порошке Формы 1 и Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида определяли путем помещения образца кристаллического вещества на держатель, представляющий собой пластинку монокристалла кремния (SSC) Siemens, и распределения образца в тонкий слой с помощью предметного стекла микроскопа. Образец вращали при 30 оборотах в минуту (для улучшения статистики счета) и облучали рентгеновскими лучами, генерируемыми длиннофокусной медной трубкой, работающей при 40 кВ и 40 мА, с длиной волны 1,5406 ангстрем, используя порошковый рентгеновский дифрактометр Bruker D5000 (Bruker AXS, Banner Lane Coventry CV4 9GH). Коллимированный пучок рентгеновских лучей пропускали через щель с автоматической расходимостью, установленной на 20 В, а отраженное излучение направляли через антирассеивающую щель 2 мм и приемную щель в 0,2 мм. Образец подвергали воздействию в течение 1 секунды с шагом 2-тета 0,02 градуса (непрерывный режим сканирования) в диапазоне от 2 градусов до 40 градусов 2-тета в режиме тета-тета. Прибор был оснащен сцинтилляционным счетчиком в качестве детектора. Получение контрольных данных и экспериментальных данных осуществляли с помощью дисплейного терминала Dell Optiplex 686 NT 4.0, работающего с программным обеспечением Diffract+. Данные собирали в диапазоне 2-тета от 2 до 40 градусов с шагом 2-тета 0,02 градуса, 4 с на шаг.
Специалист осведомлен, что может быть получена картина дифракции рентгеновских лучей на порошке, которая имеет одну или более ошибок измерения в зависимости от условий измерения (таких как оборудование, подготовка образца или используемое устройство). В частности, общеизвестно, что интенсивности на картине дифракции рентгеновских лучей на порошке могут колебаться в зависимости от условий измерения и подготовки образца. Например, специалисту понятно, что на относительную интенсивность пиков могут оказывать влияние, например, частицы с размером более 30 микрон и неунитарными соотношениями размеров, что может влиять на анализ образцов. Специалисту также понятно, что на положение отражений может влиять точная высота, на которой образец находится в дифрактометре, и нуль-калибровка дифрактометра. Плоскостность поверхности образца также может производить небольшой эффект. Следовательно, специалист в данной области поймет, что данные дифракционных картин, представленные в этом описании, не должны истолковываться как абсолютные (дополнительную информацию смотри в Jenkins, R & Snyder, R.L. "Introduction to X-Ray Powder Diffractometry" John Wiley & Sons, 1996). Поэтому следует иметь в виду, что кристаллическая форма 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида не ограничивается кристаллами, которые дают картины дифракции рентгеновских лучей на порошке, идентичные картинам дифракции рентгеновских лучей на порошке, представленным на Фиг.10-13, и любые кристаллы, дающие картины дифракции рентгеновских лучей на порошке по существу такие же, как показано на Фиг.10-13, находятся в пределах объема настоящего изобретения. Специалист в области дифракции рентгеновских лучей на порошке способен оценить существенную идентичность картин дифракции рентгеновских лучей на порошке.
Пример 16Е
Дифференциальная сканирующая калориметрия
Анализ методом дифференциальной сканирующей калориметрии (DSC) проводили на Формах 1 и 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, используя Mettler DSC820e. Образцы обычно менее 5 мг вещества, помещенные в алюминиевую кювету на 40 мкл с проколотой крышкой, нагревали в температурном диапазоне от 25°С до 325°С при постоянной скорости нагревания 10°С в минуту. В качестве продувочного газа использовали азот при скорости потока 100 мл в минуту.
Результаты показывают, что Форма 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида демонстрирует большую, острую эндотерму с температурой начала 169,7°С вследствие плавления (Фиг.15), а Форма 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида имеет большую, острую эндотерму с температурой начала 154,3°С вследствие плавления (Фиг.14). После плавления наблюдается большое экзотермическое событие вследствие разложения. Следует иметь в виду, что значения температуры начала и/или температуры пика при DSC могут незначительно варьировать от прибора к прибору, от метода к методу или от образца к образцу, поэтому приведенные значения не должны истолковываться как абсолютные.
(S)-2-(4-Бром-2-фторфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Стадия А: Метил-2-(4-бром-2-фторфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксилат превращали в (S)-2-(4-бром-2-фторфениламино)-N-(2-(трет-бутилдиметилсилилокси)пропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид по методике, описанной на Стадии А Примера 14.
Стадия Б: К раствору (S)-2-(4-бром-2-фторфениламино)-N-(2-(трет-бутилдиметилсилилокси)пропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида (0,037 г; 0,0682 ммоль) в THF (1,00 мл) добавляли 1М HCl (0,682 мл; 0,682 ммоль). Реакционную смесь перемешивали в течение одного часа при комнатной температуре. Реакционную смесь разбавляли ЕtOАс и промывали насыщенным NаНСО3 (3×), насыщенным NaCl (1×), сушили (Nа2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией (метиленхлорид/метанол, 30:1) дала 0,020 г (69%) чистого целевого продукта в виде желтого твердого вещества. MS ESI (+) m/z 428, 430 (M+, образец с Вr); 1H ЯМР (400 МГц, CD3OD) δ 7.55 (s, 1H), 7.40 (d, 1H), 7.24 (d, 1H), 6.68 (t, 1H), 3.86 (m, 1H), 3.71 (m, 1H), 3.58 (m, 1H), 3.40 (s, 3Н), 2.12 (s, 3Н), 1.10 (d, 3H).
Метил-2-(4-бром-2-фторфениламино)-5-этил-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Стадия А. Получение метил-2-хлор-5-этил-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
Метил-5-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат превращали в метил-2-хлор-5-этил-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат, как описано на Стадии Г Примера 13, используя диэтилцинк (1М в гексанах), с получением целевого продукта в виде желтого кристаллического твердого вещества.
Стадия Б. Метил-2-(4-бром-2-фторфениламино)-5-этил-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Метил-2-хлор-5-этил-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат превращали в метил-2-(4-бром-2-фторфениламино)-5-этил-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат, как описано на Стадии Д Примера 13. MS ESI (+) m/z 383, 385 (M+, образец с Вr); 1H ЯМР (400 МГц, CDCl3) δ 9.59 (s, 1H), 7.76 (s, 1H), 7.32 (d, 1H), 7.18 (d, 1H), 6.59 (t, 1H), 3.86 (s, 3Н), 3.28 (s, 3Н), 2.56 (q, 2H), 1.22 (t, 3H).
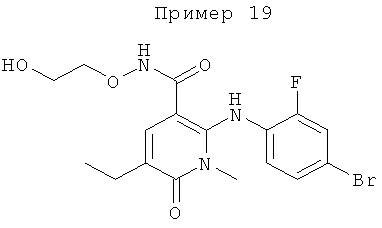
2-(4-Бром-2-фторфениламино)-5-этил-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Метил-2-(4-бром-2-фторфениламино)-5-этил-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата связывали и лишали защиты, как описано в Примере 14, с получением целевого продукта в виде желтого твердого вещества. MS APCI (+) m/z 428, 430 (M+, образец с Вr); 1H ЯМР (400 МГц, DMSO-d6) δ 11.51 (br s, 1H), 9.54 (br s, 1H), 7.57 (d, 1H), 7.47 (s, 1H), 7.25 (d, 1H), 6.69 (t, 1H), 4.67 (br s, 1H), 3.74 (m, 2H), 3.50 (m, 2H), 3.24 (s, 3H), 2.43 (q, 2H), 1.14 (t, 3H).
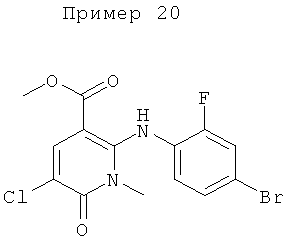
Метил-2-(4-бром-2-фторфениламино)-5-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
К раствору метил-2-(4-бром-2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата в DMF добавляли N-хлорсукцинимид. Реакционную смесь перемешивали при комнатной температуре в течение 25 минут и затем гасили насыщенным бисульфитом натрия. Реакционную смесь разбавляли H2O и распределяли между смесью ЕtOАс/диэтиловый эфир и насыщенным NaCl. Слои разделяли, и водный слой вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 15:1) дала целевой продукт в виде белого твердого вещества. MS ESI (+) m/z 389, 391, 393 (М+, образец с Cl, Br). 1H ЯМР (400 МГц, CDCl3) δ 9.88 (s, 1H), 8.13 (s, 1H), 7.34 (d, 1H), 7.24 (d, 1H), 6.69 (t, 1H), 3.87 (s, 3H), 3.29 (s, 3H).
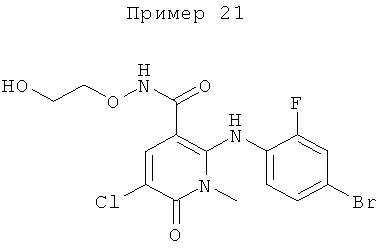
2-(4-Бром-2-фторфениламино)-5-хлор-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Метил-2-(4-бром-2-фторфениламино)-5-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата связывали и лишали защиты, как описано в Примере 14, с получением целевого продукта в виде бледно-желтого твердого вещества. MS APCI (+) m/z 434, 436, 438 (М+, образец с Cl, Br); 1H ЯМР (400 МГц, DMSO-d6) δ 11.56 (br s, 1H), 9.75 (br s, 1H), 7.91 (s, 1H), 7.57 (d, 1H), 7.26 (d, 1Н), 6.89 (t, 1H), 4.68 (br s, 1H), 3.70 (m, 2H), 3.50 (m, 2H), 3.28 (s, 3H).
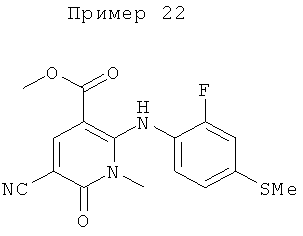
Метил-5-циано-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Стадия А: Получение метил-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К раствору, приготовленному из метил-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата и 2-фтор-4-(метилтио)бензоламина, в THF (5 мл) при -78°С в атмосфере N2 по каплям добавляли бис(триметилсилил)амид лития (1М раствор в гексанах). Реакционную смесь перемешивали в течение одного часа при -78°С. Затем по каплям добавляли метил-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат в виде раствора в THF, и реакционную смесь перемешивали в течение одного часа при -78°С. Реакционную смесь гасили добавлением Н2O, и рН доводили до рН 7 насыщенным NH4Cl и затем разбавляли ЕtOАс. Органический слой отделяли и промывали насыщенным NaCl, сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 15:1) дала целевой продукт.
Стадия Б: Получение метил-5-бром-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К раствору метил-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата добавляли N-бромсукцинимид. Реакционную смесь перемешивали при комнатной температуре в течение 25 минут и затем гасили насыщенным бисульфитом натрия. Реакционную смесь разбавляли Н2О и распределяли между смесью ЕtOАс/диэтиловый эфир и насыщенным NaCl. Слои разделяли, и водный слой вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 15:1) дала метил-5-бром-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат.
Стадия В: Метил-5-циано-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Смесь метил-5-бром-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,020 г; 0,050 ммоль), трис(дибензилиденацетон)-дипалладия(0) (0,046 г; 0,050 ммоль), 1,1'-бис(дифенилфосфин)-ферроцена (0,055 г; 0,100 ммоль) и Zn(CN)2 (0,006 г; 0,055 ммоль) нагревали при 120°С в течение 2 часов. Реакционную смесь разбавляли ЕtOАс и H2O, и слои разделяли. ЕtOАс-слой промывали насыщенным NН4Сl и насыщенным NaCl, сушили (Na2SO4) и концентрировали при пониженном давлении до темно-желтой смолы. Очистка колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 10:1) дала 0,005 г (29%) чистого целевого продукта в виде желтого твердого вещества. MS APCI (-) m/z 346 (М-1); 1H ЯМР (400 МГц, CDCl3) δ 10.84 (s, 1H), 8.39 (s, 1H), 6.95-7.06 (m, 3H), 3.90 (s, 3H), 3.17 (s, 3H), 2.50 (s, 3H).
Следующие соединения получили по методикам, описанным в приведенных выше Примерах, если не указано иное.
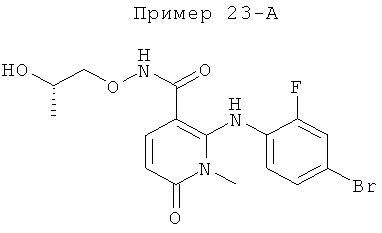
(S)-2-(4-Бром-2-фторфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Стадия А: Получение 2-хлор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты
2-Хлор-6-оксо-1,6-дигидропиридин-3-карбоновую кислоту получили из дихлор-никотиновой кислоты (3,00 г; 15,6 ммоль, Aldrich) по методике, описанной в патенте США №3682932 (1972), с получением 1,31 г (48%) целевого продукта.
Стадия Б: Получение метил-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К раствору 2-хлор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (0,644 г; 3,71 ммоль) в DMF (20 мл) добавляли гидрид лития (95%, 0,078 г; 9,28 ммоль), и реакционную смесь перемешивали в течение 40 минут в атмосфере N2. Затем добавляли метилйодид (0,508 мл; 1,16 г; 8,16 ммоль), и реакционную смесь перемешивали в течение дополнительных 45 минут. Реакционную смесь гасили 2М HCl до тех пор, пока рН не становился 6-7. Реакционную смесь разбавляли ЕtOАс и насыщенным NaCl, и слои разделяли. Водный слой вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4) и концентрировали при пониженном давлении с получением неочищенного желтого твердого вещества. HPLC-анализ показал наличие двух продуктов с соотношением 4:1, которые разделяли колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, от 15:1 до 10:1) с получением 0,466 г (62%) чистого целевого продукта в виде белого кристаллического твердого вещества. Также выделяли минорный продукт в виде бледно-желтого кристаллического твердого вещества и идентифицировали его как региоизомер метил-2-хлор-6-метоксиникотината.
Стадия В: Получение метил-2-(4-бром-2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К раствору 4-бром-2-фторанилина (0,192 г; 1,01 ммоль) в THF (5 мл) при -78°С в атмосфере N2 по каплям добавляли бис(триметилсилил)амид лития (1,50 мл; 1,50 ммоль; 1М раствор в гексанах). Реакционную смесь перемешивали в течение одного часа при -78°С. Затем по каплям добавляли метил-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (0,202 г; 1,00 ммоль) в виде раствора в THF (5 мл), и реакционную смесь перемешивали в течение одного часа при -78°С. Реакционную смесь гасили добавлением H2O, и рН доводили до рН 7 насыщенным NН4Сl и затем разбавляли ЕtOАс. Органический слой отделяли и промывали насыщенным NaCl, сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 15:1) дала 0,232 г (65%) чистого целевого продукта в виде белого кристаллического твердого вещества.
Стадия Г: Получение (S)-2-(4-бром-2-фторфениламино)-N-(2-(трет-бутилдиметилсилилокси)пропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида
К раствору метил-2-(4-бром-2-фторфениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,050 г; 0,14 ммоль) и (S)-O-(2-(трет-бутилдиметилсилилокси)пропил)гидроксиламина (0,072 г; 0,35 ммоль) в THF (1,50 мл) при 0°С медленно добавляли бис(триметилсилил)амид лития (0,70 мл; 0,70 ммоль). После добавления реакционную смесь перемешивали в течение 1 часа при комнатной температуре и затем гасили насыщенным NаНСО3. Реакционную смесь распределяли между ЕtOАс и насыщенным NaCl. Слои разделяли, и водный слой вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении с получением неочищенного коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия Д: Получение (S)-2-(4-бром-2-фторфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида
К раствору (S)-2-(4-бром-2-фторфениламино)-N-(2-(трет-бутилдиметил-силилокси)пропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида (0,074 г; 0,14 ммоль) в THF (1,50 мл) добавляли 1М водную HCl (1,4 мл; 1,4 ммоль). Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь разбавляли ЕtOАс и промывали насыщенным водным NаНСО3 (3×) и насыщенным водным NaCl. Органический слой сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении с получением неочищенного белого твердого вещества. Очистка неочищенного продукта растиранием с Et2O и выделение полученного твердого вещества дали (S)-2-(4-бром-2-фторфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид (0,030 г; 52% за две стадии) в виде белого твердого вещества. MS ESI (+) m/z 414, 416 (M+) образец с Br; 1H ЯМР (400 МГц, CD3OD) δ 7.65 (d, 1Н), 7.42 (dd, 1Н), 7.28 (m, 1H), 6.81 (t, 1H), 6.28 (d, 1H), 3.88 (m, 1H), 3.70 (dd, 1H), 3.58 (dd, 1H), 3.38 (s, 3Н), 1.11 (d, 3Н).
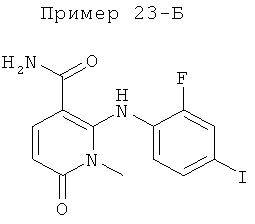
2-(2-Фтор-4-йодфениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 388, образец (М+1); 1H ЯМР (400 МГц, CDCl3) δ 10.8 (s, 1H), 7.47 (d, 2H), 7.39 (d, 1H), 6.54 (t, 1H), 6.26 (d, 1H), 5.59 (br s, 2H), 3.24 (s, 3Н).
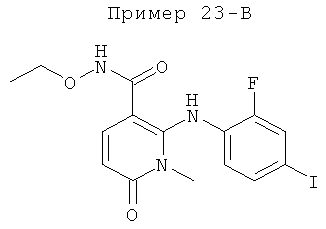
N-Этокси-2-(2-фтор-4-йодфениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 432, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.4 (br s, 1H), 9.83 (br s, 1H), 7.66 (dd, 1H), 7.58 (d, 1H), 7.43 (d, 1H), 6.65 (t, 1H), 6.18 (d, 1H), 3.70 (q, 2H), 3.21 (s, 3Н), 1.10 (t, 3Н).
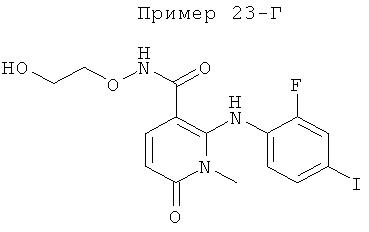
2-(2-Фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 448, образец (М+1); 1H ЯМР (400 МГц, CD3OD) δ 7.66 (d, 1H), 7.56 (m, 1H), 7.46 (m, 1H), 6.65 (t, 1H), 6.28 (d, 1H), 3.85 (t, 2H), 3.67 (t, 2H), 3.36 (s, 3Н).
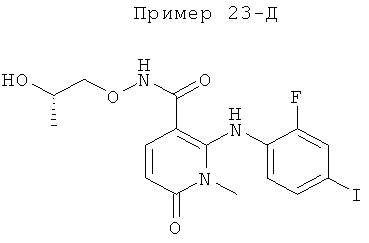
(S)-2-(2-Фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 462, образец (М+1); 1H ЯМР (400 МГц, CD3OD) δ 7.66 (d, 1H), 7.56 (d, 1H), 7.46 (d, 1H), 6.65 (t, 1H), 6.28 (d, 1H), 3.85 (m, 1H), 3.67 (m, 1H), 3.57 (m, 1H), 3.38 (s, 3Н), 1.11 (d, 3Н).
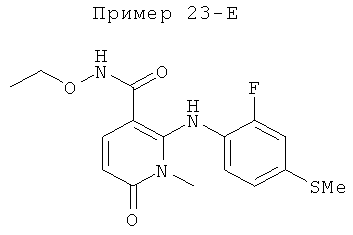
N-Этокси-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид.
MS APCI (+) m/z 352, образец (М+1); 1H ЯМР (400 МГц, CD3OD) δ 7.64 (d, 1H), 7.12 (dd, 1H), 7.05 (m, 1H), 6.86 (t, 1H), 6.21 (d, 1H), 3.85 (q, 2H), 3.32 (s, 3H), 2.47 (s, 3H), 1.22 (t, 3H).
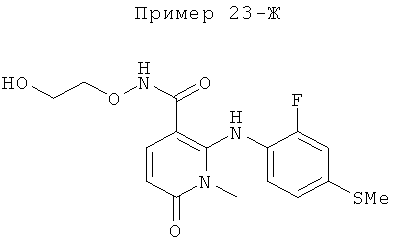
2-(2-Фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 368, образец (М+1); 1H ЯМР (400 МГц, CDCl3) δ 10.28 (s, 1H), 8.48 (s, 1H), 7.38 (d, 1H), 7.00 (m, 1H), 6.96 (m, 1H), 6.79 (t, 1H), 6.19 (d, 1H), 4.04 (m, 2H), 3.88 (m, 1H), 3.75 (m, 2H), 3.22 (s, 3H), 2.48 (s, 3H).
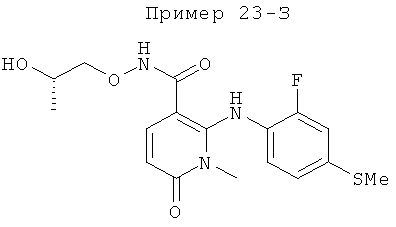
(S)-2-(2-Фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 382, образец (М+1); 1H ЯМР (400 МГц, CD3OD) δ 7.64 (d, 1H), 7.12 (d 1H), 7.04 (d, 1H), 6.85 (t, 1H), 6.21 (d, 1H), 4.01 (m, 1H), 3.90 (m, 1H), 3.71 (m, 1H), 3.60 (m, 1H), 3.32 (s, 3H), 2.47 (s, 3H), 1.10 (d, 3H).
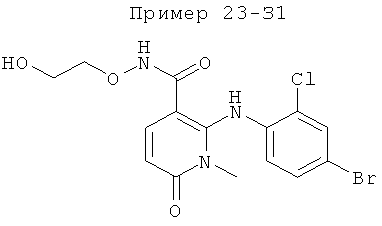
2-(4-Бром-2-хлорфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
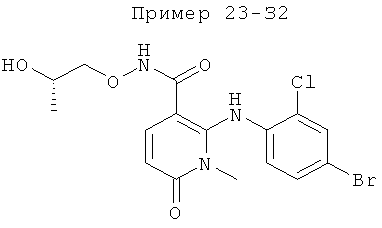
(S)-2-(4-Бром-2-хлорфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
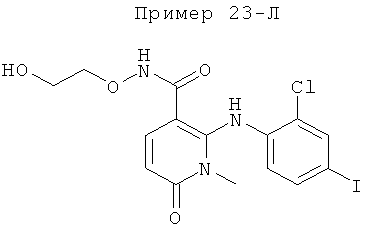
2-(2-Хлор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 464, 466 (M+, образец с Cl); 1H ЯМР (400 МГц, DMSO-d6) δ 11.59 (brs, 1H), 10.06 (brs, 1H), 7.86 (d, 1H), 7.64 (d, 1H), 7.54 (dd, 1H), 6.53 (d, 1H), 6.21 (d, 1H), 4.67 (t, 1H), 3.78 (t, 2H), 3.52 (m, 2H), 3.13 (s, 3H).
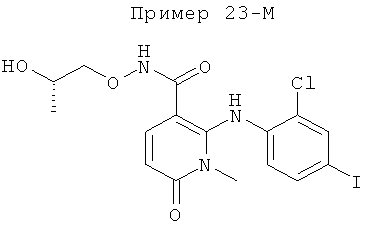
(S)-2-(2-Хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 478, 480 (M+, образец с Cl); 1H ЯМР (400 МГц, DMSO-d6) δ 11.59 (s, 1H), 9.99 (s, 1H), 7.86 (d, 1H), 7.64 (d, 1H), 7.54 (dd, 1H), 6.53 (d, 1H),6.21 (d, 1H), 4.73 (m, 1H), 3.75 (m, 1H), 3.58 (m, 2H), 3.14 (s, 3H), 1.02 (d, 3H).
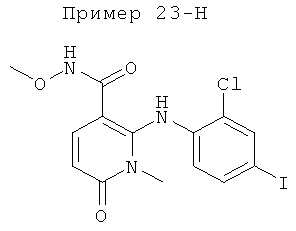
2-(2-Хлор-4-йодфениламино)-N-метокси-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
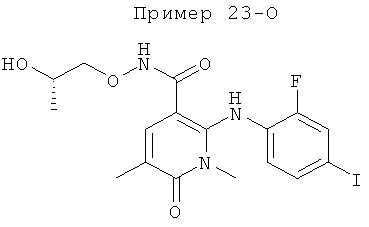
(S)-2-(2-Фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 476, образец (М+1); 1H ЯМР (400 МГц, CDCl3) δ 9.79 (s, 1H), 8.53 (s, 1H), 7.46 (d, 1H), 7.35 (m, 1H), 6.44 (t, 1H), 4.15 (m, 1H), 3.92 (dd, 1H), 3.69 (dd, 1H), 3.28 (s, 3H), 2.14 (s, 3H), 1.14 (d, 3H).
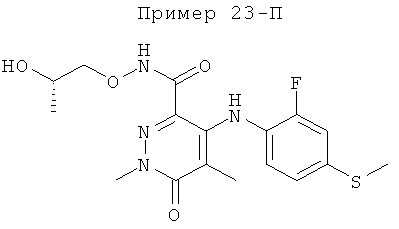
(S)-4-(2-Фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 395 (М-1); 1Н ЯМР (400 МГц, СD3ОD) δ 7.10 (dd, 1H), 7.03 (d, 1H), 6.87 (t, 1H), 4.00 (m, 1H), 3.85 (dd, 1H), 3.79 (s, 3H), 3.72 (dd, 1H), 2.47 (s, 3H), 1.75 (s, 3H), 1.16 (d, 3H).
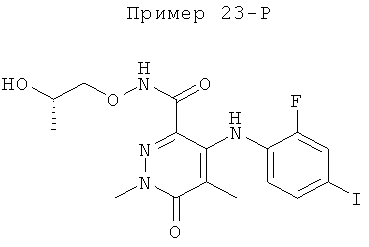
(S)-4-(2-Фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 475 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.52 (dd, 1H), 7.44 (dd, 1H), 6.63 (t, 1H), 3.98 (m, 1H), 3.84 (dd, 1H), 3.79 (s, 3Н), 3.72 (dd, 1H), 1.78 (s, 3H), 1.16 (d, 3H).
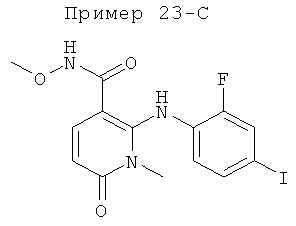
2-(2-Фтop-4-йoдфeнилaминo)-N-мeтoкcи-1-мeтил-6-oкco-1,6-дигидpoпиpидин-3-карбоксамид
Следующие соединения получили по методикам, которые описаны в приведенных выше Примерах.
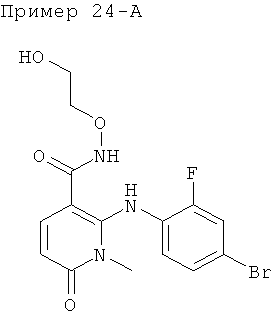
2-(4-Бром-2-фторфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
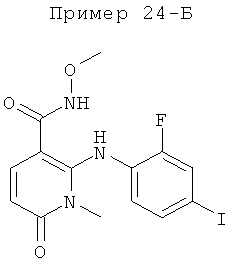
2-(2-Фтор-4-йодфениламино)-N-метокси-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
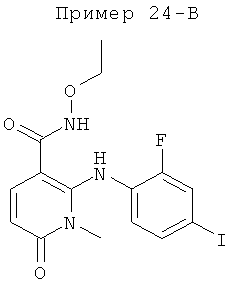
N-Этокси-2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
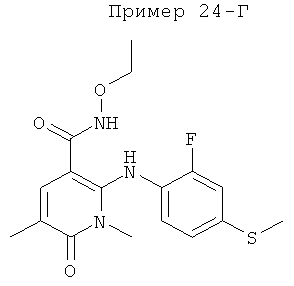
N-Этокси-2-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 366, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.38 (br s, 1H), 9.79 (br s, 1H), 7.54 (s, 1H), 7.23 (dd, 1H), 6.99 (dd, 1H), 6.73 (t, 1H), 3.76 (q, 2H), 3.19 (s, 3Н), 2.46 (s, 3Н), 2.01 (s, 3H), 1.12 (t, 3Н).
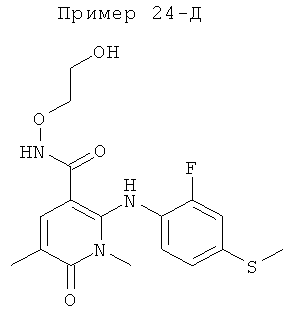
2-(2-Фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 382, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.48 (brs, 1H), 9.78 (brs, 1H), 7.56 (s, 1H), 7.23 (dd, 1H), 6.99 (m, 1H), 6.73 (t, 1H), 4.68 (brs, 1H), 3.76 (t, 2H), 3.51 (t, 2H), 3.19 (s, 3H), 2.46 (s, 3H), 2.01 (s, 3H).
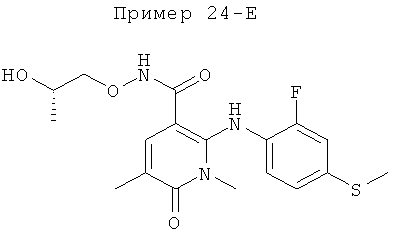
(S)-2-(2-Фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 396, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.48 (br s, 1H), 9.68 (br s, 1H), 7.55 (s, 1H), 7.23 (dd, 1H), 6.99 (dd, 1H), 6.73 (t, 1H), 4.73 (d, 1H), 3.74 (m, 1H), 3.56 (d, 2H), 3.20 (s, 3H), 2.46 (s, 3H), 2.01 (s, 3H), 1.02 (d, 3H).
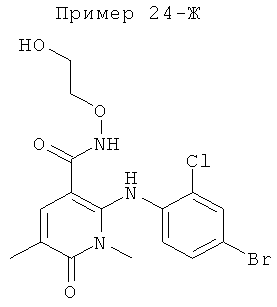
2-(4-Бром-2-хлорфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
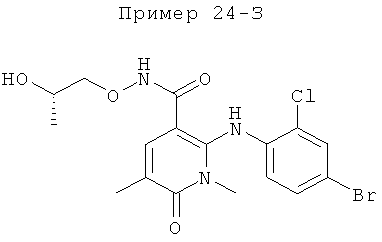
(S)-2-(4-Бром-2-хлорфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
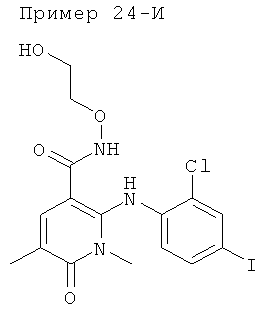
2-(2-Хлор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Детектировано: MS APCI (+) m/z 478, 480 (М+, образец с Cl); 1H ЯМР (400 МГц, CD3OD) δ 7.79 (d, 1H), 7.59 (s, 1H), 7.52 (dd, 1H), 6.39 (d, 1H), 3.89 (t, 2H), 3.67 (t, 2H), 3.34 (s, 3Н), 2.13 (s, 3Н).
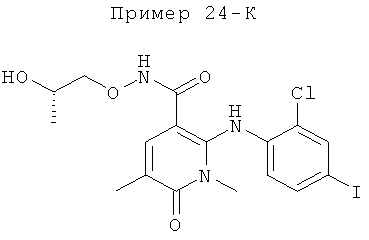
(S)-2-{2-Хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
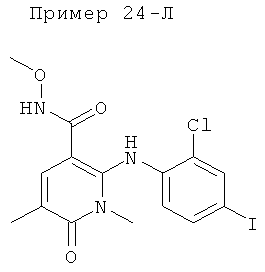
2-(2-Хлор-4-йодфениламино)-N-метокси-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
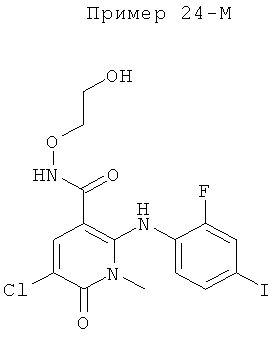
5-Хлор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 482, 484 (M+, образец с Cl); 1H ЯМР (400 МГц, DMSO-d6) δ 11.56 (br s, 1Н), 9.69 (br s, 1Н), 7.89 (s, 1H), 7.64 (dd, 1H), 7.40 (dd, 1H), 6.72 (t, 1H), 4.66 (t, 1H), 3.67 (t, 2H), 3.49 (m, 2H), 3.28 (s, 3Н).
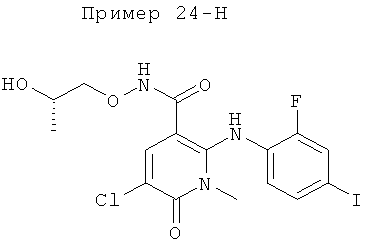
(S)-5-Хлор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
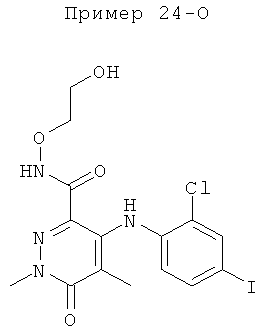
4-(2-Хлор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 477, 479 (М-1, образец с Cl); 1H ЯМР (400 МГц, CD3OD) δ 7.77 (d, 1H), 7.54 (dd, 1H), 6.51 (d, 1H), 4.01 (t, 2H), 3.81 (s, 3Н), 3.75 (t, 2H), 1.74 (s, 3Н).
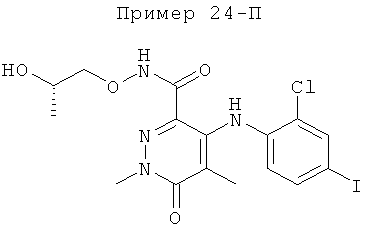
(S)-4-(2-Хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 491, 493 (М-1, образец с Cl); 1H ЯМР (400 МГц, СD3OD) δ 7.77 (d, 1Н), 7.54 (dd, 1H), 6.51 (d, 1H), 4.00 (m, 1H), 3.87 (dd, 1H), 3.80 (s, 3H), 3.75 (dd, 1H), 1.74 (s, 3H), 1.16 (d, 3H).
4-(4-Бром-2-хлорфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 429, 431, 433 (М-1, образец с Br, Cl); 1H ЯМР (400 МГц, CD3OD) δ 7.62 (d, 1H), 7.38 (dd, 1H), 6.67 (d, 1H), 4.02 (t, 2H), 3.81 (s, 3H), 3.75 (t, 2H), 1.73 (s, 3H).
(S)-4-(4-Бром-2-хлорфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
Дополнительные соединения по настоящему изобретению включают соединения общих формул Ia, IVa, IVб, IVв, IVг, IVд, IVe и IVж, которые представлены в следующих далее Таблицах 1-8.

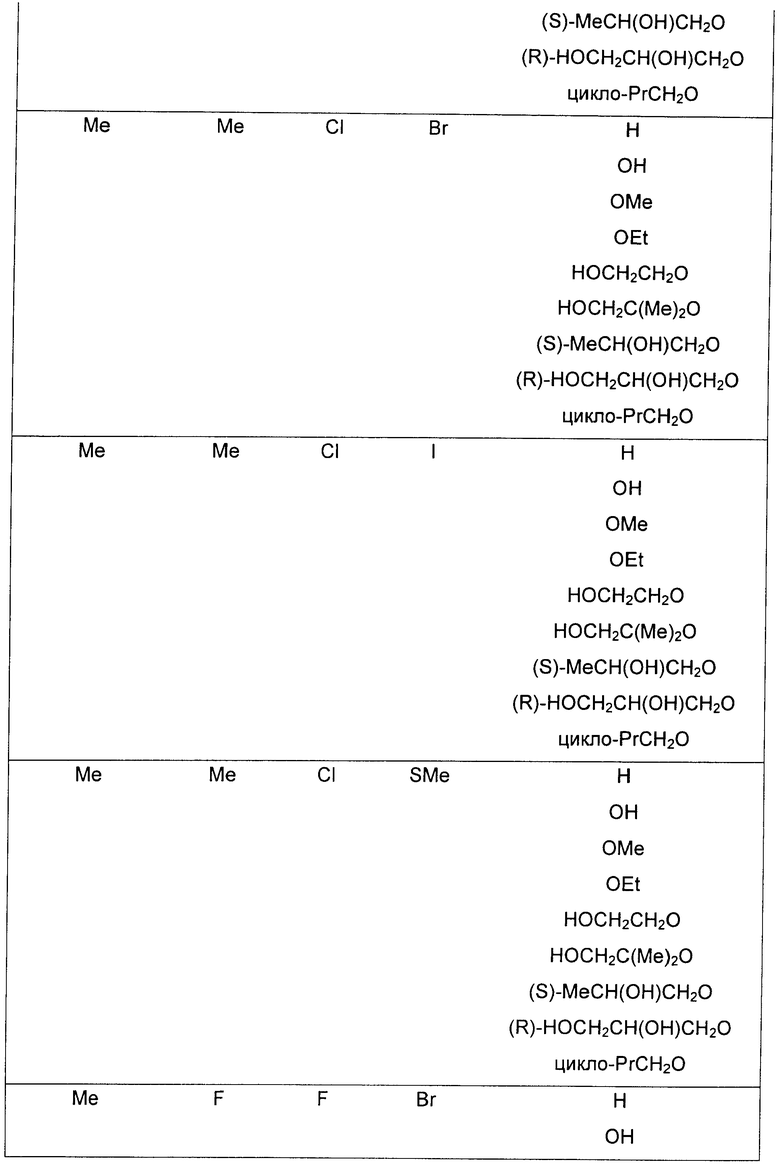

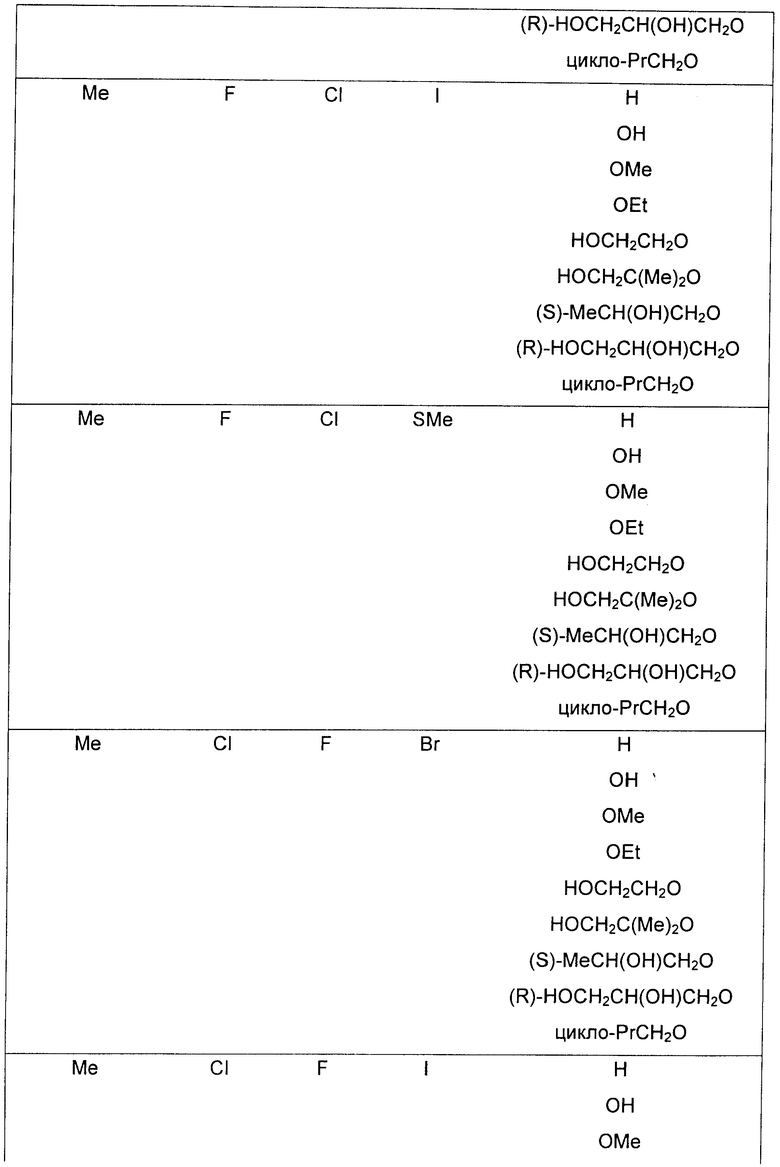
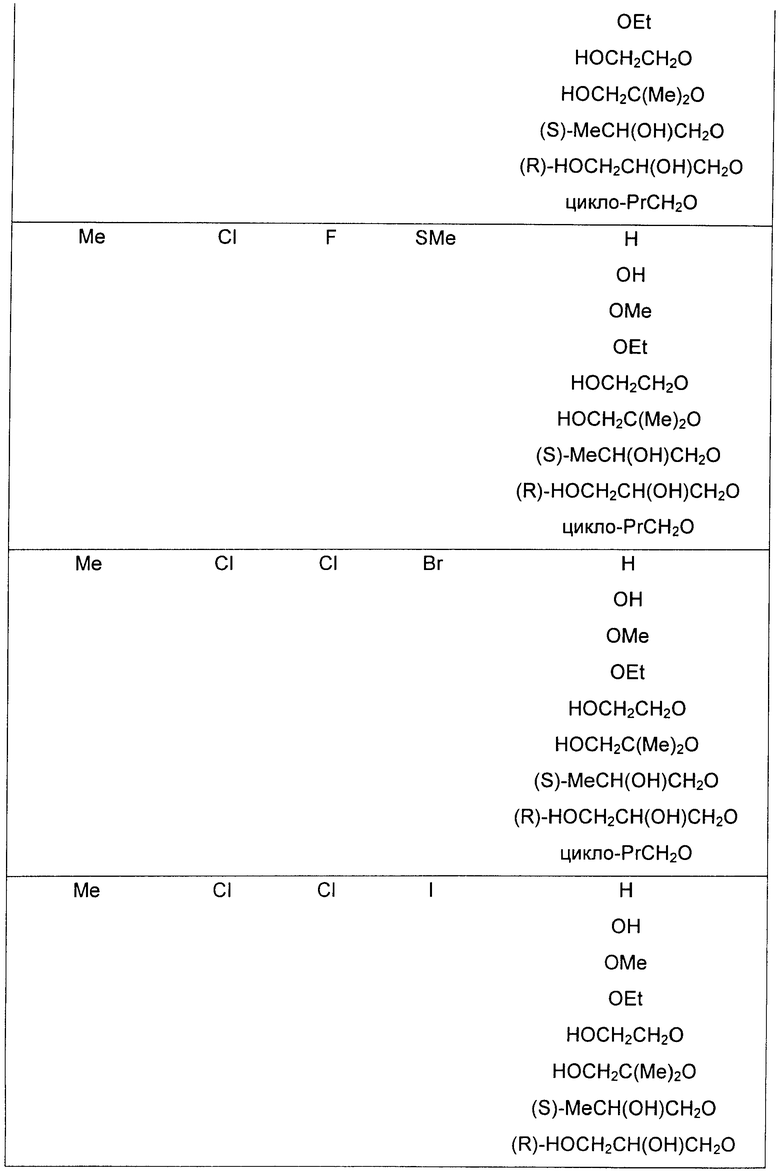


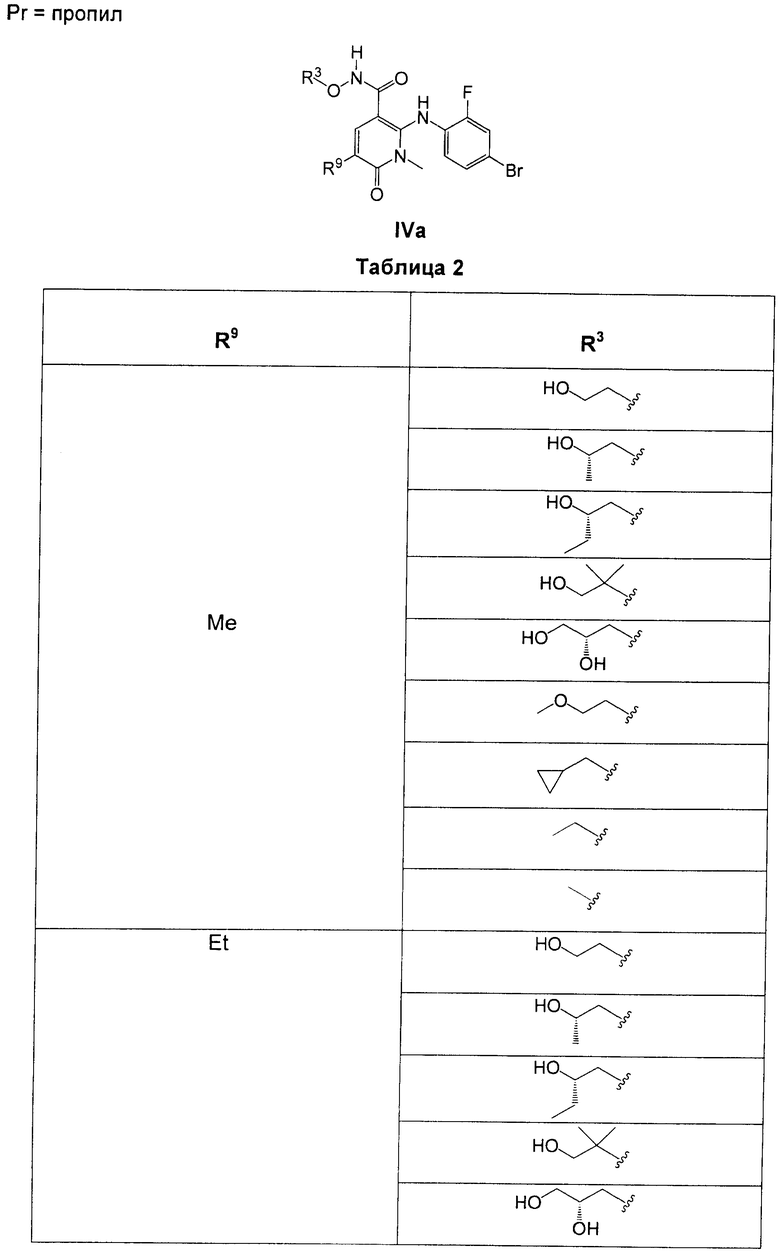
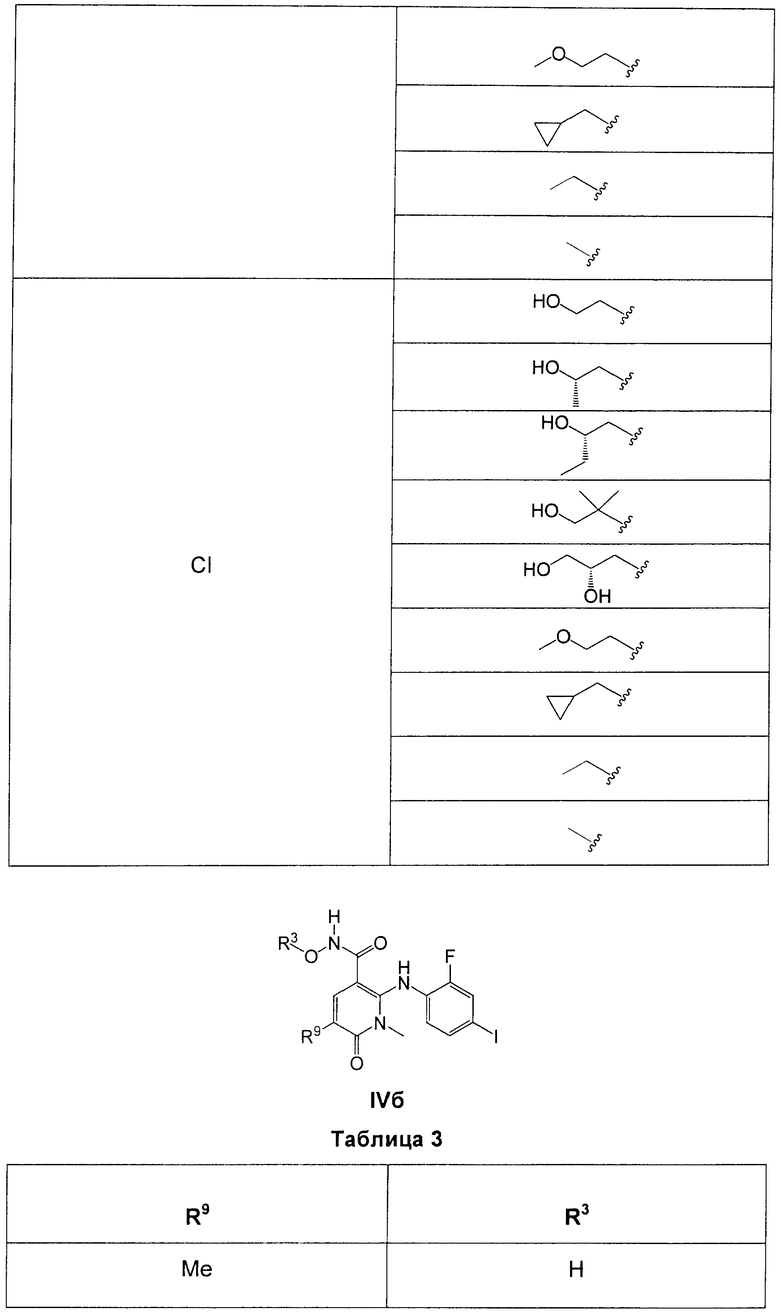
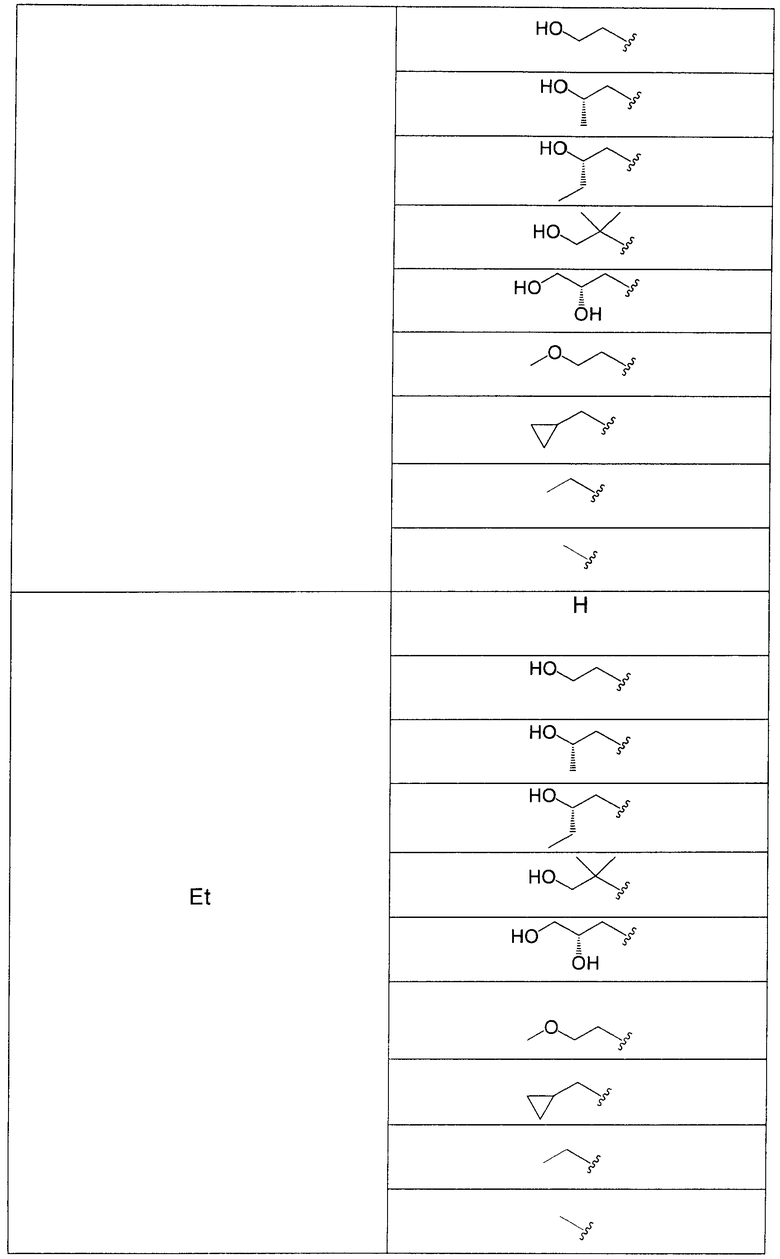
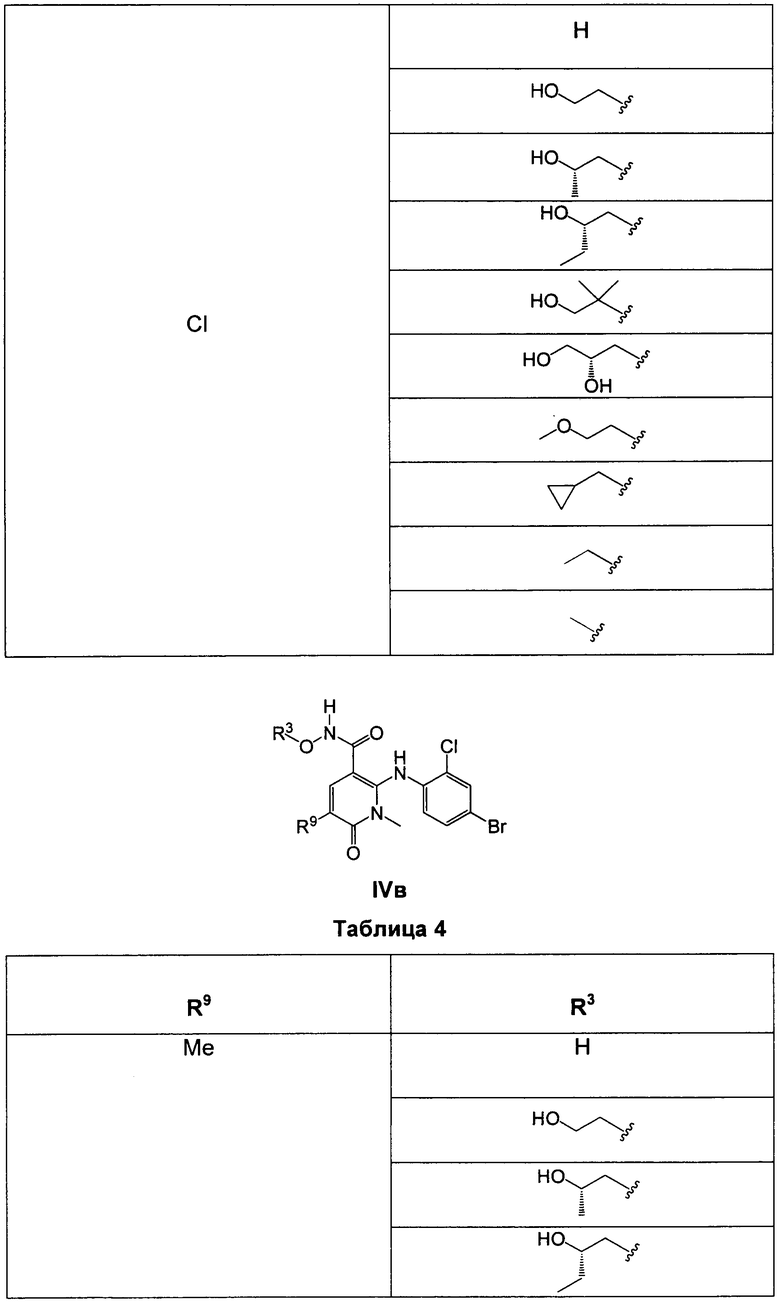
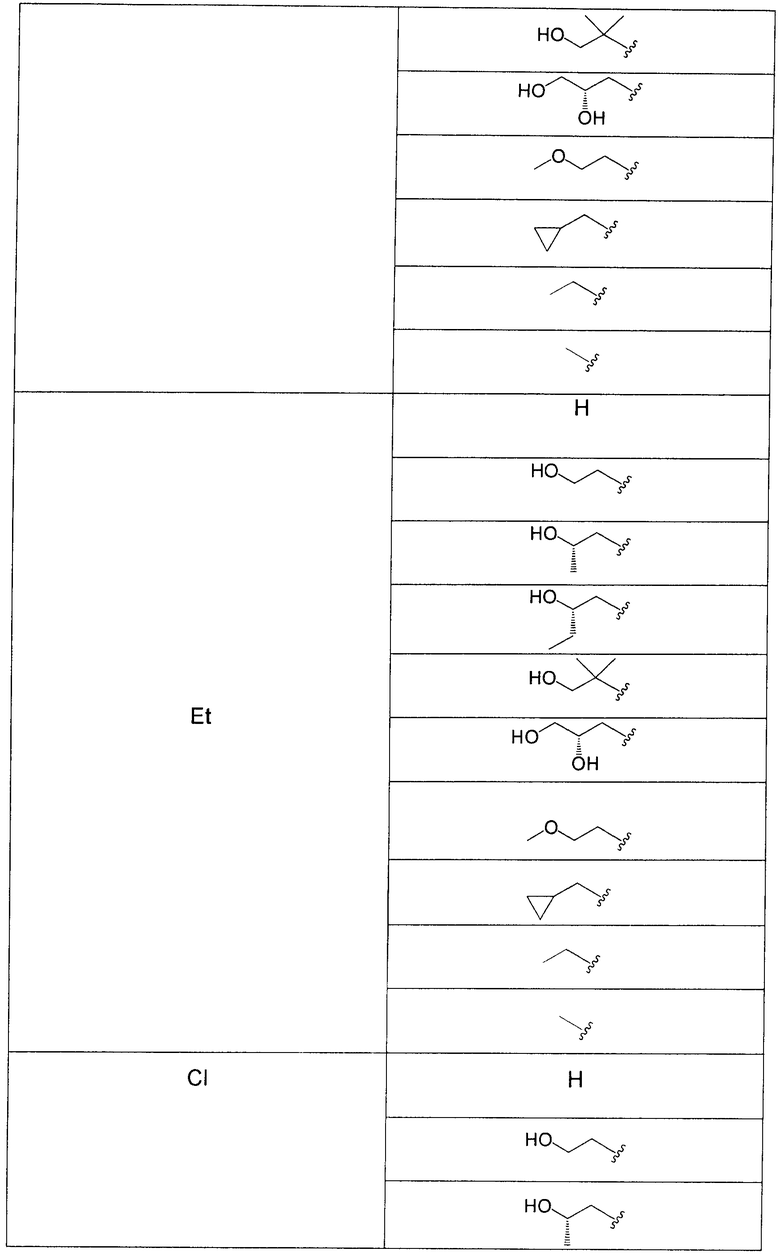
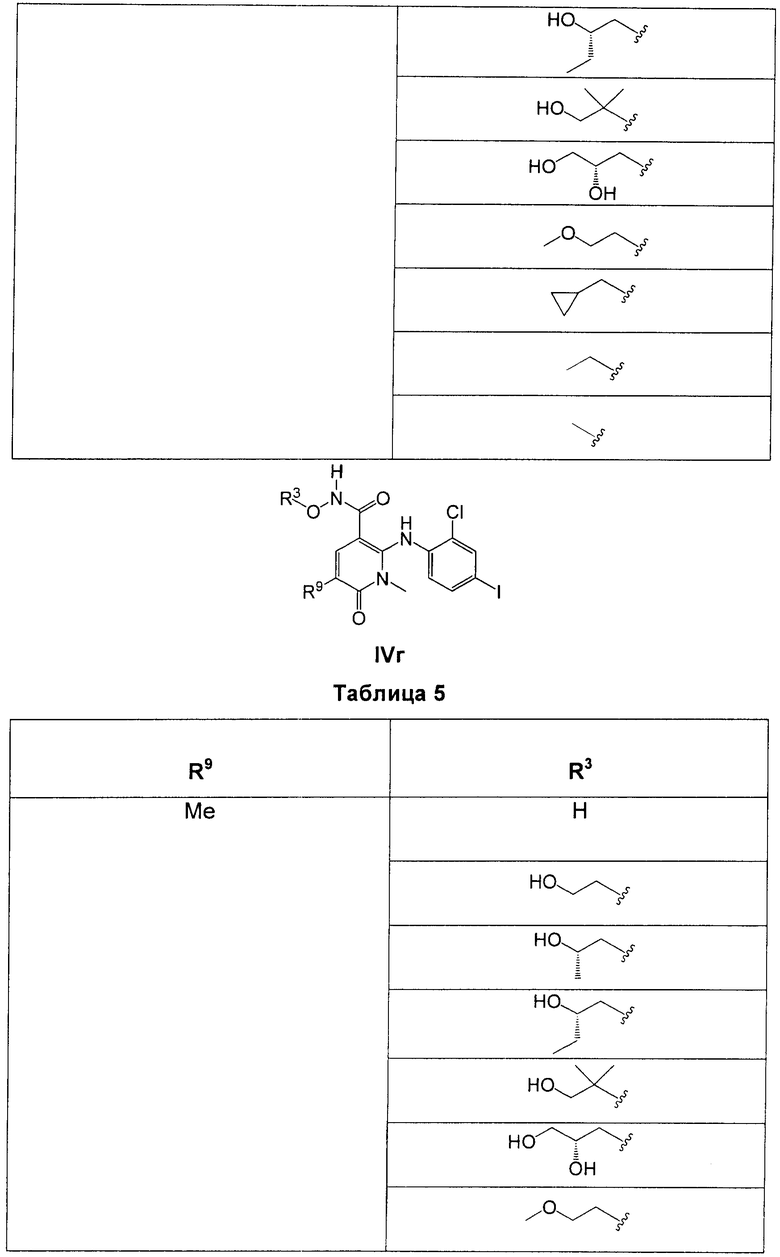
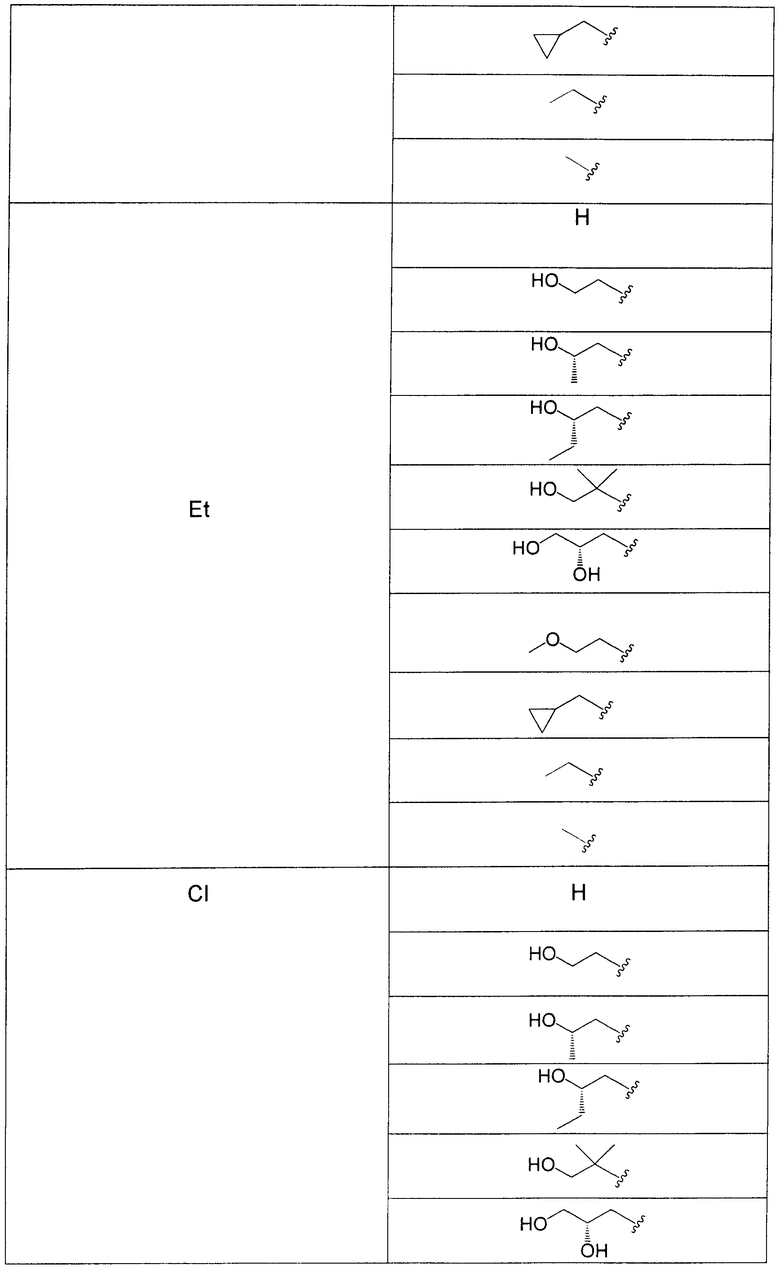
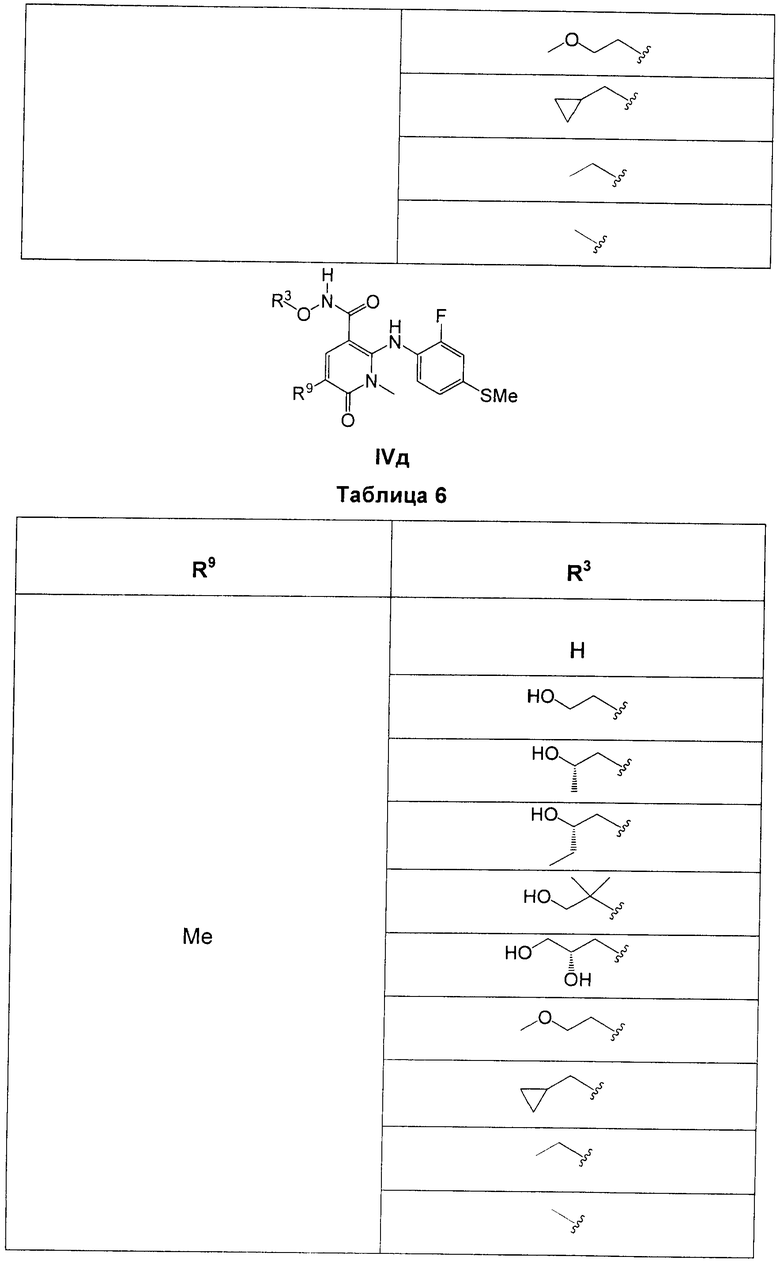
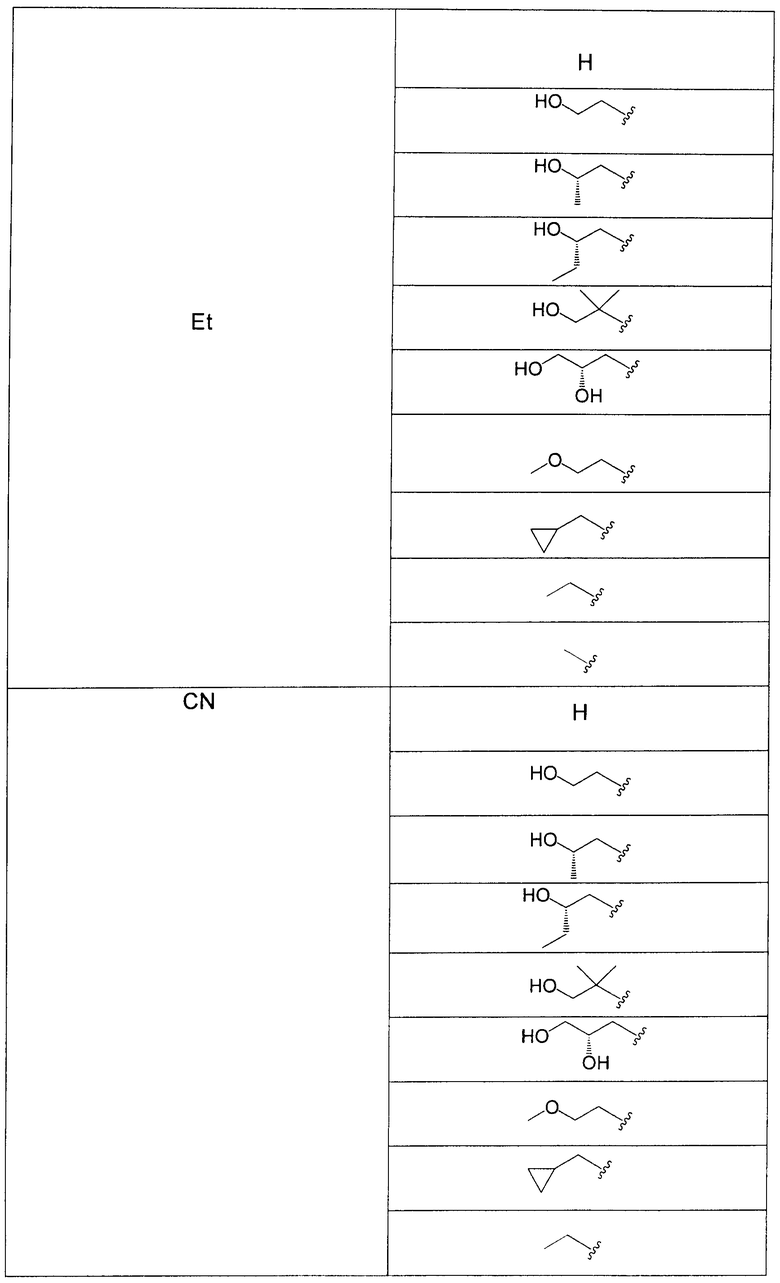
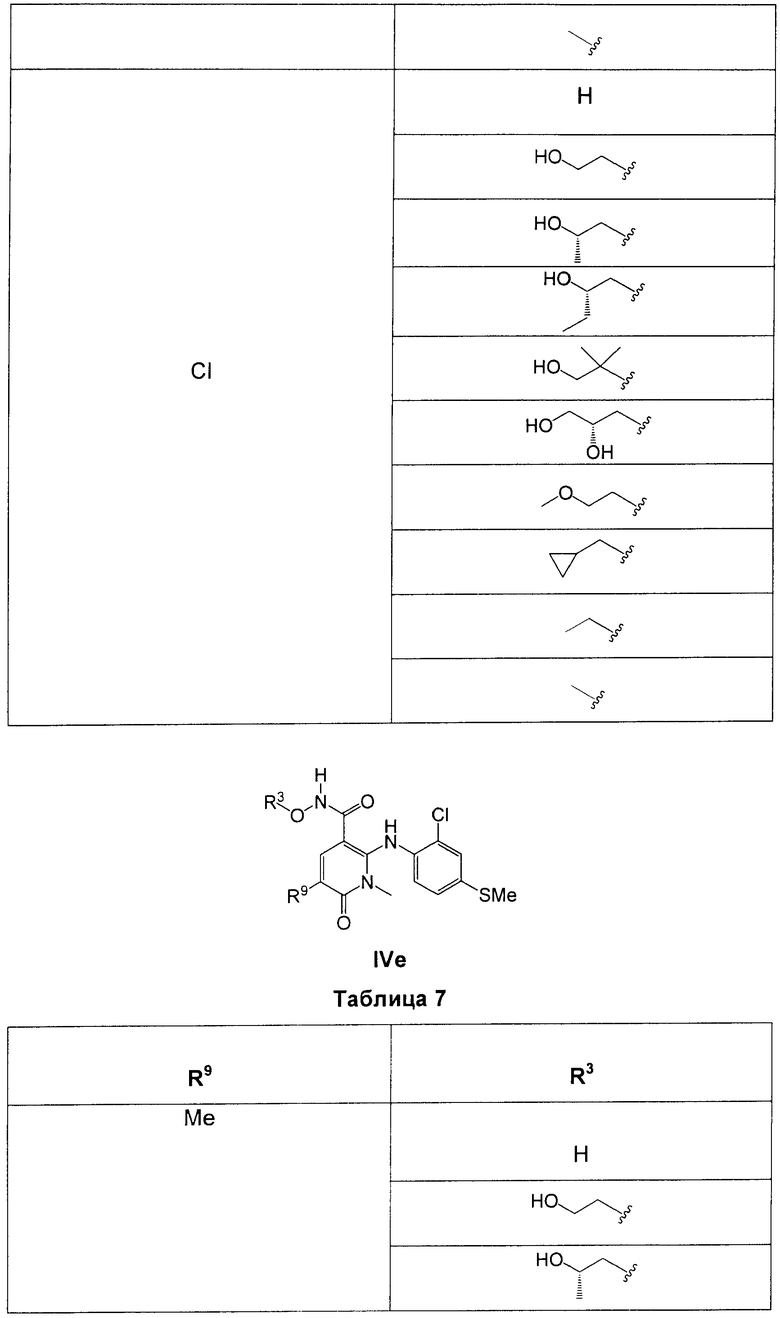
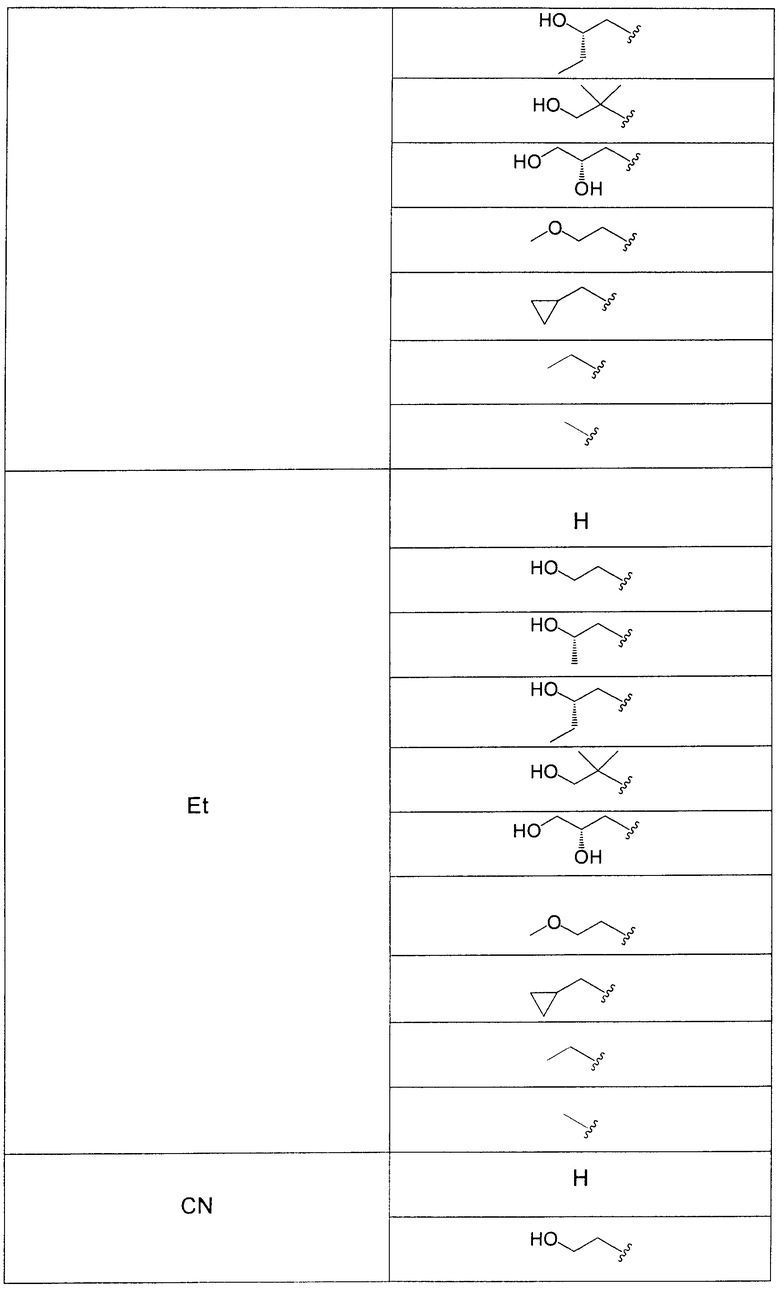
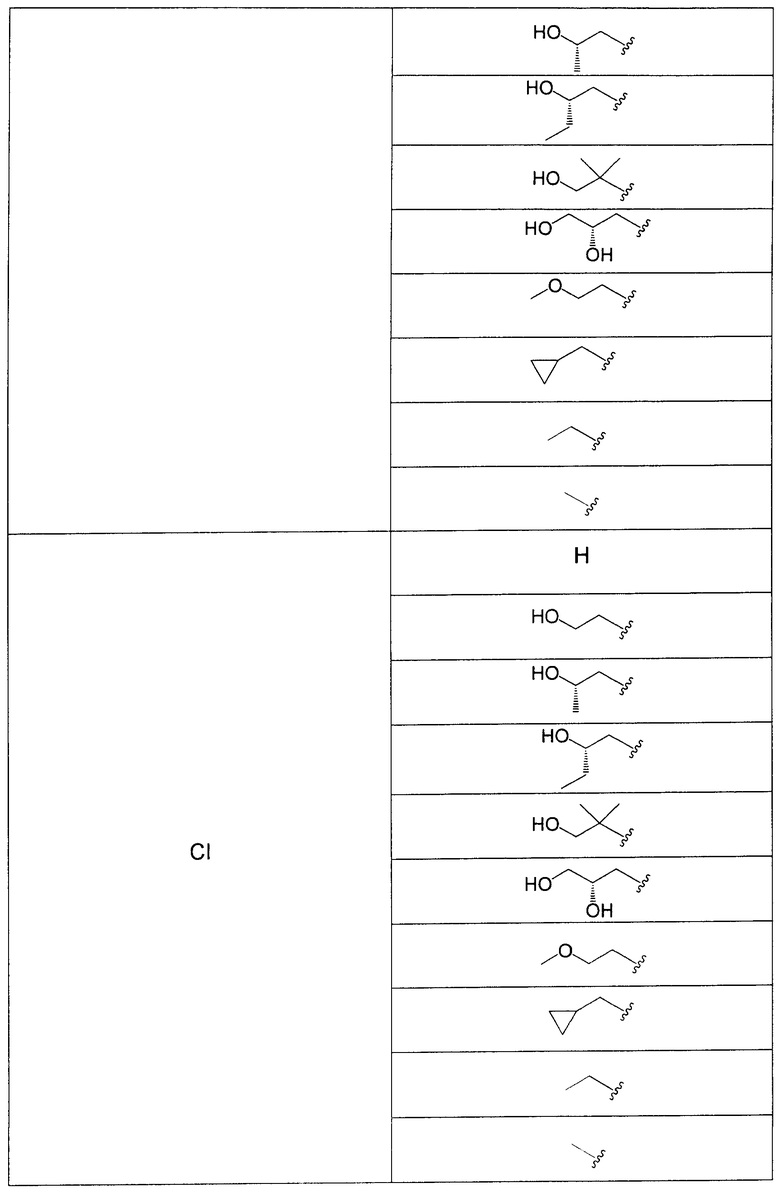
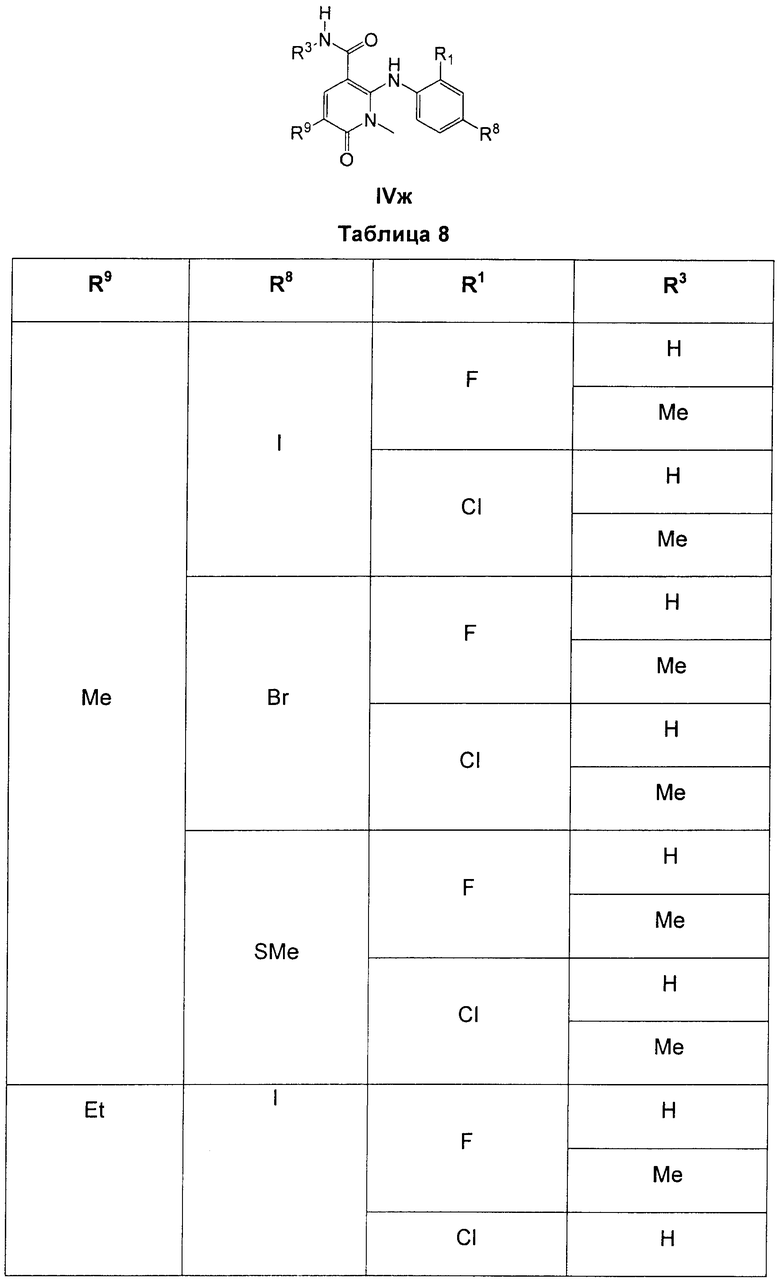
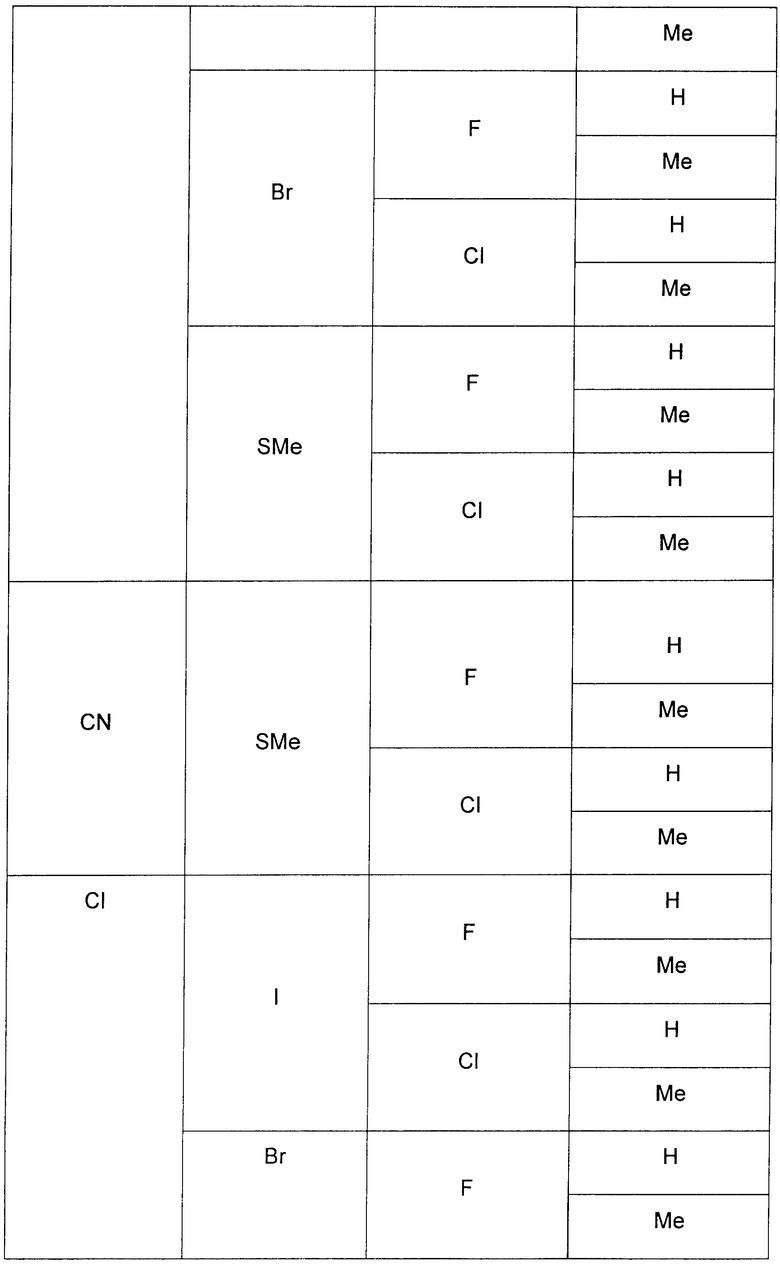
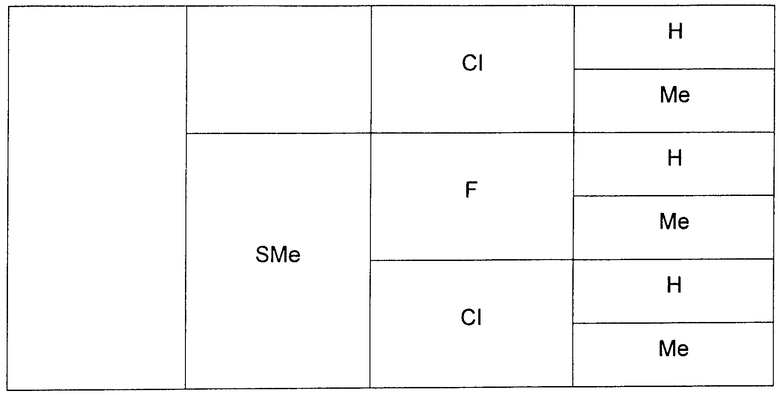
Дополнительные примеры по изобретению включают следующие далее примеры, которые могут быть получены описанными выше способами, если не указано иное.
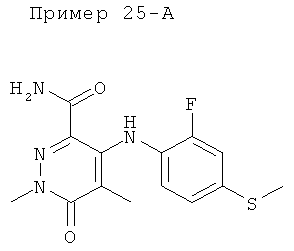
4-(2-Фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 321 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.09 (dd, 1H), 7.04 (d, 1H), 6.87 (t, 1H), 3.81 (s, 3Н), 2.48 (s, 3H), 1.70 (s, 3H).
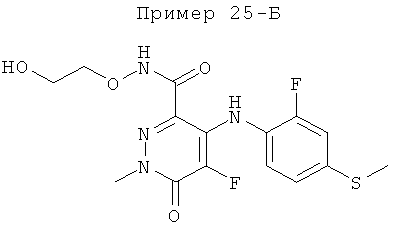
5-Фтор-4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 385 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.14 (td, 1H), 7.07 (m, 2H), 4.05 (t, 2H), 3.79 (s, 3Н), 3.78 (t, 2H), 2.49 (s, 3H).
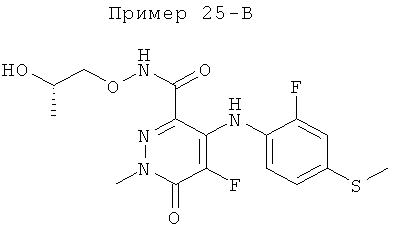
(S)-5-Фтор-4-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 399 (M-1); 1H ЯМР (400 МГц, СD3ОD) δ 7.14 (td, 1H), 7.07 (m, 2H), 4.04 (m, 1H), 3.93 (dd, 1H), 3.81 (m, 1H), 3.80 (s, 3H), 2.49 (s, 3H), 1.18 (d, 3Н).
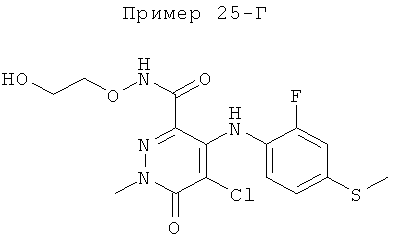
5-Хлор-4-(2-Фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 401, 403 (M-1, образец с Cl); 1H ЯМР (400 МГц, CD3OD) δ 7.06 (m, 3Н), 3.94 (t, 2H), 3.81 (s, 3H), 3.73 (t, 2H), 2.49 (s, 3H).
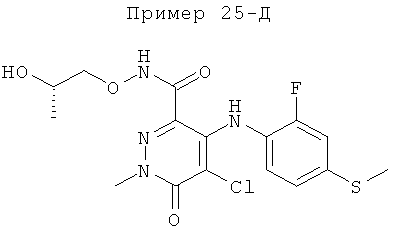
(S)-5-Xлop-4-(2-фтop-4-(мeтилтиo)фeнилaминo)-N-(2-гидроксипpoпoкcи)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 415, 417 (M-1, образец с Cl); 1H ЯМР (400 МГц, СD3OD) δ 7.06 (m, 3Н), 3.98 (m, 1H), 3.81 (m, 1H), 3.80 (s, 3Н), 3.69 (dd, 1H), 2.49 (s, 3Н), 1.16 (d, 3H).
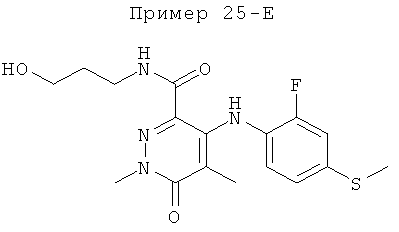
4-(2-Фтор-4-(метилтио)фениламино)-N-(3-гидроксипропил)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 379 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.09 (dd, 1H), 7.03 (d, 1H), 6.86 (t, 1H), 3.81 (s, 3Н), 3.64 (t, 2H), 3.43 (t, 2H), 2.47 (s, 3H), 1.80 (m, 2H), 1.71 (s, 3H).
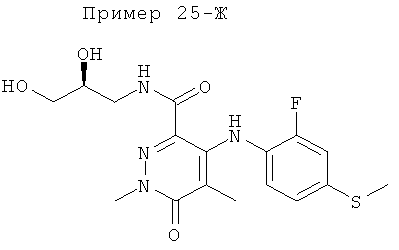
(S)-N-(2,3-Дигидроксипропил)-4-(2-фтор-4-(метилтио)фениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 395 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.10 (dd, 1H), 7.03 (dd, 1H), 6.86 (t, 1H), 3.81 (s, 3Н), 3.80 (m, 1H), 3.51 (m, 3Н), 3.37 (dd, 1H), 2.47 (s, 3Н), 1.71 (s, 3H).
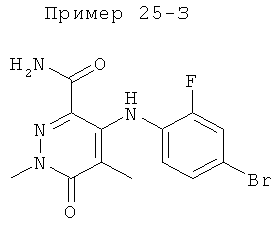
4-(4-Бром-2-фторфeнилaминo)-1,5-диметил-6-oкco-1,6-дигидpoпиpидaзин-3-карбоксамид
MS APCI (-) m/z 353, 355 (M-1, образец с Вr); 1H ЯМР (400 МГц, СD3OD) δ 7.38 (dd, 1H), 7.27 (m, 1H), 6.80 (t, 1H), 3.82 (s, 3Н), 1.72 (s, 3Н).
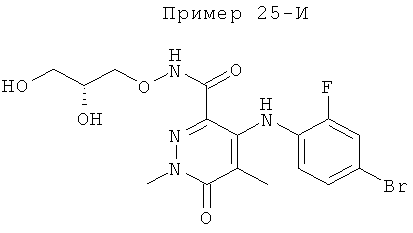
(R)-4-(4-Бром-2-фторфениламино)-N-(2,3-дигидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 443, 445 (M-1, образец с Вr); 1H ЯМР (400 МГц, CD3OD) δ 7.39 (dd, 1Н), 7.27 (m, 1Н), 6.79 (t, 1H), 4.03 (m, 1H), 3.89 (m, 2H), 3.80 (s, 3H), 3.59 (m,2H), 1.77 (s, 3H).
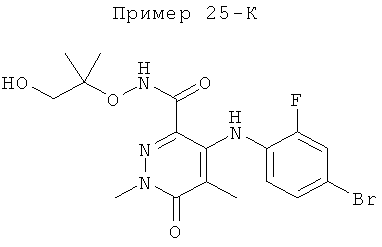
4-(4-Бром-2-фторфениламино)-N-(1-гидрокси-2-метилпропан-2-илокси)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 441, 443 (M-1, образец с Вr); 1H ЯМР (400 МГц, CD3OD) δ 7.38 (dd, 1H), 7.27 (d, 1H), 6.79 (t, 1H), 3.81 (s, 3H), 3.38 (s, 2H), 1.78 (s, 3H), 1.25 (s, 6H).
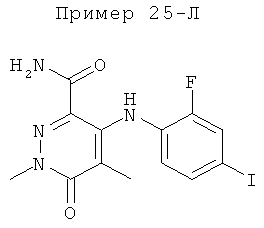
4-(2-Фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 401 (M-1); 1H ЯМР (400 МГц, DMSO-d6) δ 9.75 (s, 1H), 8.25 (s, 1H), 7.90 (s, 1H), 7.65 (s, 1H), 7.43 (s, 1H), 6.63 (t, 1H), 3.71 (s, 3H), 1.63 (s, 3H).
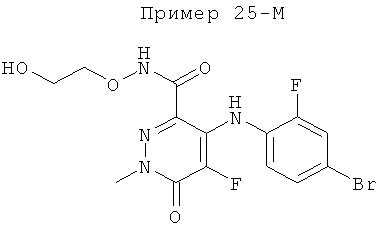
4-(4-Бром-2-фторфениламино)-5-Фтор-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 417, 419 (M-1, образец с Br); 1H ЯМР (400 МГц, CDCl3) δ 9.66 (br. s, 1H), 9.30 (br. s, 1H), 7.28 (m, 2H), 6.97 (td, 1H), 4.11 (t, 2H), 3.84 (t, 2H), 3.82 (s, 3H), 3.51 (t, 1H).
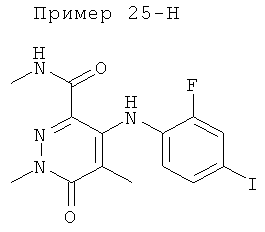
4-(2-Фтор-4-йодфениламино)-N,1,5-триметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 415 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.52 (dd, 1H), 7.44 (m, 1H), 6.61 (t, 1H), 3.81 (s, 3H), 2.87 (s, 3H), 1.74 (s, 3H).
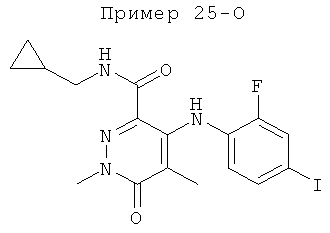
N-(Циклопропилметил)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 455 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.52 (dd, 1H), 7.44 (dd, 1H), 6.62 (t, 1H), 3.83 (s, 3H), 3.18 (d, 2H), 1.75 (s, 3H), 1.06 (m, 1H), 0.51 (dd, 2H), 0.27 (dd, 2H).
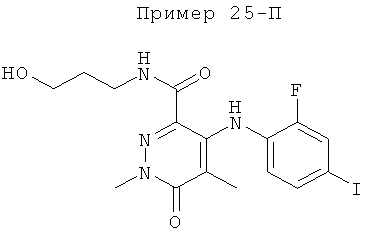
4-(2-Фтор-4-йодфениламино)-N-(3-гидроксипропил)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 459 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.52 (dd, 1H), 7.44 (dd, 1H), 6.62 (t, 1H), 3.81 (s, 3Н), 3.63 (t, 2H), 3.43 (t, 2H), 1.79 (m, 2H), 1.74 (s, 3Н).
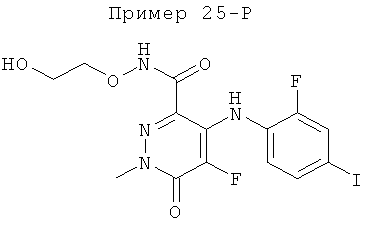
5-Фтор-4-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 465 (M-1); 1H ЯМР (400 МГц, СD3OD) δ 7.55 (dd, 1H), 7.50 (d, 1H), 6.95 (td, 1H), 4.05 (t, 2H), 3.80 (s, 3Н), 3.78 (t, 2H).
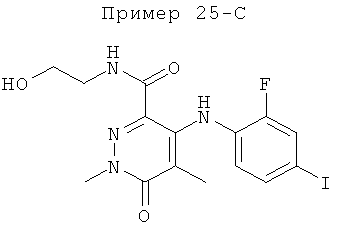
4-(2-Фтор-4-йодфениламино)-N-(2-гидроксиэтил)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 445 (M-1); 1H ЯМР (400 МГц, СD3OD) δ 7.52 (dd, 1H), 7.44 (dd, 1H), 6.62 (t, 1H), 3.82 (s, 3Н), 3.68 (t, 2H), 3.46 (t, 2H), 1.74 (s, 3Н).
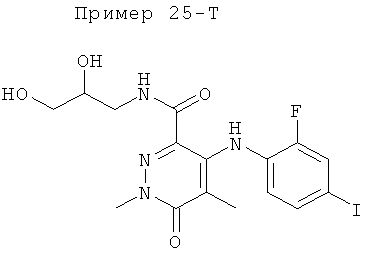
N-(2,3-Дигидроксипропил)-4-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 475 (М-1); 1H ЯМР (400 МГц, СD3OD) δ 7.52 (dd, 1H), 7.44 (dd, 1H), 6.62 (t, 1H), 3.82 (s, 3H), 3.80 (m, 1H), 3.52 (m, 3Н), 3.36 (dd, 1H), 1.74 (s, 3Н).
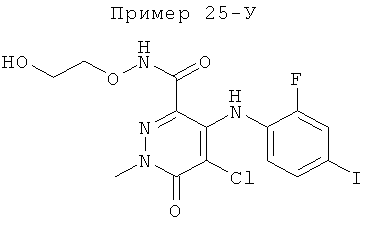
5-Хлор-4-(2-Фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 481, 483 (М-1, образец с Cl); 1H ЯМР (400 МГц, CD3OD) δ 7.53 (dd, 1H), 7.49 (d, 1H), 6.88 (t, 1H), 3.97 (t, 2H), 3.81 (s, 3Н), 3.74 (t, 2H).
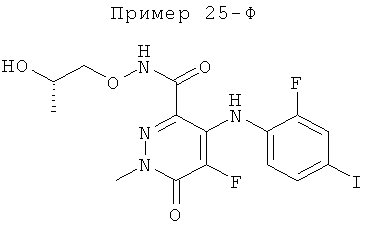
(S)-5-Хлор-4-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 495, 496 (М-1, образец с Cl); 1H ЯМР (400 МГц, CD3OD) δ 7.53 (dd, 1H), 7.49 (d, 1H), 6.88 (t, 1H), 3.99 (m, 1H), 3.83 (m, 1H), 3.81 (s, 3Н), 3.71 (dd, 1H), 1.17 (d, 3H).
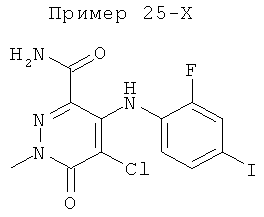
5-Хлор-4-(2-фтор-4-йодфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 421, 423 (M-1, образец с Cl); 1H ЯМР (400 МГц, CDCl3/CD3OD) δ 7.56 (td, 1Н), 7.46 (m, 1Н), 6.82 (t, 1H), 3.87 (s, 3H).
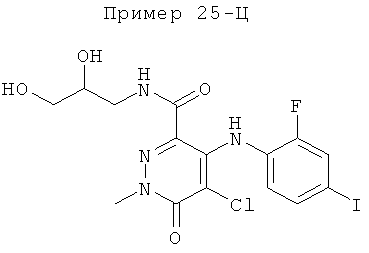
5-Хлор-N-(2,3-дигидроксипропил)-4-(2-фтор-4-йодфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (+) m/z 497, 499 (М+1, образец с Cl); 1H ЯМР (400 МГц, CD3OD) δ 7.53 (dd, 1Н), 7.49 (d, 1Н), 6.86 (t, 1Н), 3.84 (s, 3H), 3.80 (m, 1Н), 3.55 (d, 2H), 3.50 (m, 1H), 3.37 (dd, 1Н).
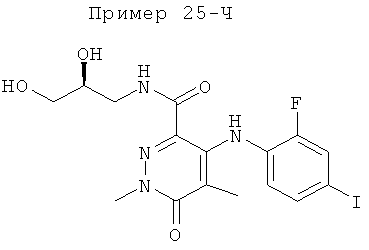
(S)-N-(2,3-Дигидрокcипpoпил)-4-(2-фтop-4-йoдфeнилaминo)-1,5-димeтил-6-oкco-1,6-дигидропиридазин-3-карбоксамид
MS APCI (-) m/z 475 (M-1); 1H ЯМР (400 МГц, CD3OD) δ 7.52 (dd, 1Н), 7.44 (d, 1Н), 6.62 (t, 1Н), 3.82 (s, 3H), 3.80 (m, 1Н), 3.52 (m, 3H), 3.36 (dd, 1Н), 1.74 (s, 3H).
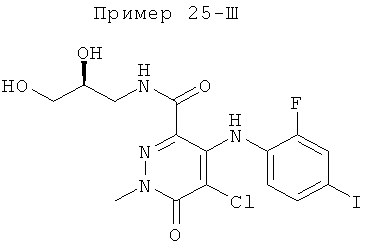
(S)-5-Хлор-N-(2,3-дигидроксипропил)-4-(2-фтор-4-йодфениламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-карбоксамид
MS APCI (+) m/z 497, 499 (М+1, образец с Cl); 1H ЯМР (400 МГц, CD3OD) δ 7.52 (dd, 1H), 7.48 (d, 1H), 6.86 (t, 1H), 3.84 (s, 3H), 3.80 (m, 1H), 3.55 (d, 2H), 3.51 (d, 1H), 3.37 (dd, 1H).
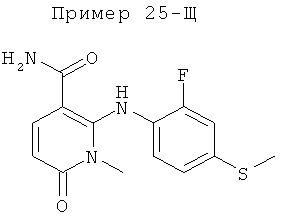
2-(2-Фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 308, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.38 (s, 1H), 7.92 (br s, 1H), 7.89 (d, 1H), 7.45 (br s, 1H), 7.25 (dd, 1H), 7.04 (dd, 1H), 6.88 (t, 1H), 6.09 (d, 1H), 3.07 (s, 3H), 2.48 (s, 3H).
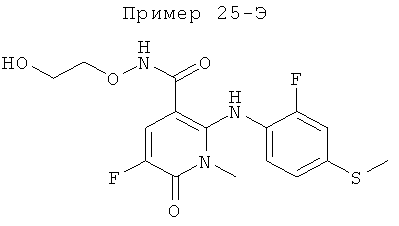
5-Фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
Стадия А: Получение 2-хлор-5-фтор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты
Смесь 2,6-дихлор-5-фторникотиновой кислоты (15,00 г; 71,43 ммоль, синтез по Ланкастеру) и 2 н. NaOH (178,6 мл; 357,2 ммоль) перемешивали при температуре дефлегмации в течение 2 часов и затем при комнатной температуре в течение 16 часов. Реакционную смесь охлаждали до 0°С и подкисляли 12 н. HCl (32,74 мл; 392,9 ммоль). Смесь охлаждали в течение 30 минут на ледяной бане, твердое вещество отфильтровывали и промывали H2O. Готовили суспензию выделенного твердого вещества в теплом ЕtOН, фильтровали и затем промывали теплым ЕtOН. Твердые вещества собирали и сушили под вакуумом в течение ночи с получением целевого продукта (6,4 г; 47%) в виде бежевого твердого вещества.
Стадия Б: Получение метил-2-хлор-5-фтор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К раствору 2-хлор-5-фтор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (6,37 г; 33,26 ммоль) в DMF (250 мл) при 0°С добавляли LiH (95%; 0,661 г; 83,14 ммоль). Реакционную смесь перемешивали в течение 45 минут и затем добавляли йодметан (4,56 мл; 73,16 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем гасили 2М HCl до тех пор, пока рН реакционной смеси не достигал 6-7. Реакционную смесь разбавляли ЕtOАс и насыщенным NaCl, и слои разделяли. Водный слой вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4) и концентрировали при пониженном давлении с получением неочищенного желтого масла. HPLC-анализ показал присутствие двух продуктов в сотношении 5:1, которые разделяли колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 15:1) с получением целевого продукта (5,40 г; 74%) в виде бледно-желтого твердого вещества. Также выделили минорный продукт в виде бледно-желтого кристаллического твердого вещества и идентифицировали его как региоизомер метил-2-хлор-5-фтор-6-метоксиникотината.
Стадия В: Получение метил-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
К раствору 2-фтор-4-(метилтио)анилина (0,236 г; 1,50 ммоль) в THF (10 мл) при -78°С в атмосфере N2 по каплям добавляли бис(триметилсилил)амид лития (3,42 мл; 3,42 ммоль, 1 М раствор в гексанах). После завершения добавления реакционную смесь перемешивали в течение одного часа при -78°С. Затем по каплям добавляли метил-2-хлор-5-фтор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (0,300 г; 1,37 ммоль) в виде раствора в THF (5 мл), и реакционную смесь перемешивали в течение 30 минут при -78°С.Реакционную смесь гасили добавлением 1М HCl до тех пор, пока рН реакционной смеси не достигал 5, а затем разбавляли ЕtOАс и насыщенным NaCl. Органический слой отделяли, сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией (метиленхлорид/ЕtOАс, 15:1) дала чистый целевой продукт (0,359 г; 75%) в виде белого твердого вещества.
Стадия Г: Получение 5-фтор-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридин-3-карбоксамида
К смеси метил-5-фтор-2-(2-фтор-4-(метилтио)фениламино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,100 г; 0,294 ммоль) и O-(2-(винилокси)этил)-гидроксиламина (0,045 мл; 0,441 ммоль) в THF (2 мл) при 0°С по каплям добавляли бис(триметилсилил)амид лития (1,18 мл; 1,18 ммоль, 1 М раствор в гексанах). Реакционную смесь перемешивали в течение 20 минут, гасили 1М HCl, а затем распределяли между ЕtOАс и насыщенным NaCl. Слои разделяли, и водный слой вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении с получением неочищенного желтого твердого вещества, которое использовали на следующей стадии без очистки.
Стадия Д: Получение 5-фтор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1.6-дигидропиридин-3-карбоксамида
К раствору 5-фтор-2-(2-фтор-4-(метилтио)фениламино)-1 -метил-6-оксо-N-(2-(винилокси)этокси)-1,6-дигидропиридин-3-карбоксамида (0,121 г; 0,294 ммоль) в ЕtOН (3 мл) добавляли 2М HCl (0,75 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. рН реакционной смеси доводили до рН 7, используя 1 М NaOH. Реакционную смесь разбавляли ЕtOАс и Н2O. Органический слой отделяли и промывали насыщенным NaCl. Объединенные водные слои вновь экстрагировали ЕtOАс (1×). Объединенные органические слои сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка колоночной флэш-хроматографией на силикагеле (метиленхлорид/МеОН, 15:1) дала 5-фтор-2-(2-фтор-4-(метилтио)-фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид (0,079 г; 70% за две стадии) в виде белого твердого вещества. MS ESI (+) m/z 386, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.54 (br s, 1H), 9.65 (br s, 1H), 7.65 (d, 1H), 7.23 (dd, 1H), 6.99 (dd, 1H), 6.81 (t, 1H), 4.67 (t, 1H), 3.74 (t, 2H), 3.51 (q, 2H), 3.25 (s, 3Н), 2.46 (s, 3Н).
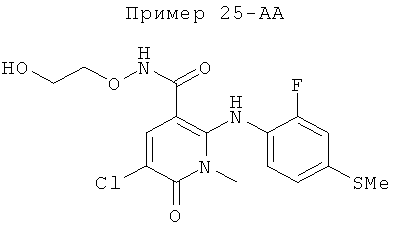
5-Хлор-2-(2-фтор-4-(метилтио)фениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 402, 404 (M+, образец с Cl); 1H ЯМР (400 МГц, DMSO-d6) δ 11.59 (brs, 1H), 10.00 (brs, 1H), 7.93 (s, 1H), 7.23 (dd, 1H), 7.01 (dd, 1H), 6.93 (t, 1H), 4.66 (t, 1H), 3.73 (t, 2H), 3.51 (m, 2H), 3.24 (s, 3Н), 2.47 (s, 3Н).
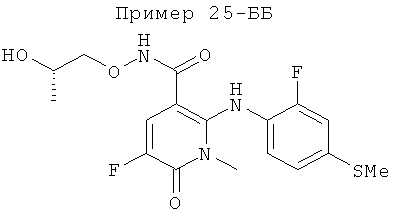
(S)-5-фтор-2-(2-фтop-4-(метилтио)фениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 400, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.54 (brs, 1H), 9.61 (brs, 1H), 7.64 (d, 1H), 7.22 (dd, 1H), 6.99 (dd, 1H), 6.81 (t, 1H), 4.73 (s, 1H), 3.73 (m, 1H), 3.54 (d, 2H), 3.25 (s, 3Н), 2.46 (s, 3Н), 1.01 (d, 3Н).
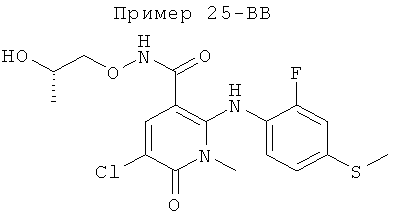
(S)-5-Xлop-2-(2-фтop-4-(мeтилтиo)фeнилaминo)-N-(2-гидрoкcипpoпoкcи)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 416, 418 (М+, образец с Cl); 1H ЯМР (400 МГц, DMSO-d6) δ 11.59 (br s, 1H), 9.94 (br s, 1H), 7.92 (s, 1H), 7.23 (dd, 1H), 7.01 (dd, 1H), 6.94 (t, 1H), 4.71 (d, 1H), 3.75 (m, 1H), 3.54 (d, 2H), 3.24 (s, 3Н), 2.47 (s, 3H), 1.02 (d, 3H).
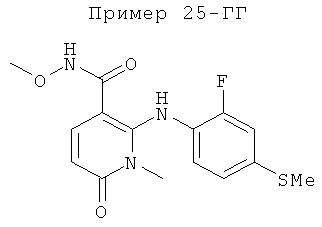
2-(2-Фтор-4-(метилтио)фениламино)-N-метокси-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 338, образец (М+1); 1H ЯМР (400 МГц, CDCl3) δ 10.33 (s, 1H), 8.39 (s, 1H), 7.40 (d, 1H), 7.02 (dd, 1H), 6.96 (dd, 1H), 6.75 (t, 1H), 6.20 (d, 1H), 3.83 (s, 3Н), 3.23 (s, 3H), 2.47 (s, 3H).
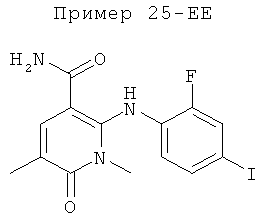
2-(2-Фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 402, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 10.75 (s, 1H), 7.85 (br s, 1H), 7.78 (s, 1H), 7.66 (d, 1H), 7.40 (m, 2H), 6.54 (t, 1H), 3.13 (s, 3H), 2.00 (s, 3H).
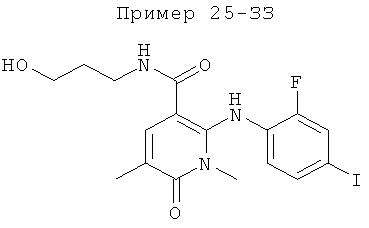
2-(2-Фтор-4-йодфениламино)-N-(3-гидроксипропил)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 460, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 10.34 (s, 1H), 8.27 (t, 1H), 7.72 (s, 1H), 7.64 (dd, 1H), 7.38 (dd, 1H), 6.50 (t, 1H), 4.41 (t, 1H), 3.17 (s, 5H), 2.01 (s, 3H), 1.55 (s, 2H).
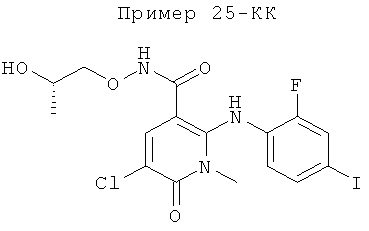
(S)-5-Хлор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 460, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.54 (br s, 1H), 9.62 (br s, 1H), 7.86 (s, 1H), 7.62 (dd, 1H), 7.38 (dd, 1H), 6.69 (t, 1H), 4.69 (m, 1H), 3.46 (m, 2H), 3.27 (s, 3H), 0.99 (d, 3H).
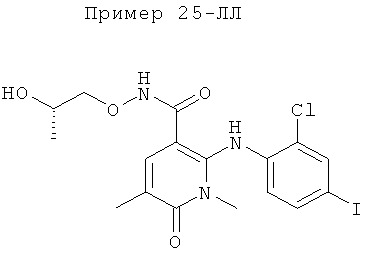
(3)-2-(2-Хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 492, образец (М+1); 1H ЯМР (400 МГц, CD3OD) δ 7.79 (d, 1H), 7.58 (m, 1H), 7.52 (dd, 1H), 6.39 (d, 1H), 3.87 (m, 1H), 3.73 (dd, 1H), 3.62 (dd, 1H), 3.35 (s, 3H), 2.13 (s, 3H), 1.10 (d, 3H).
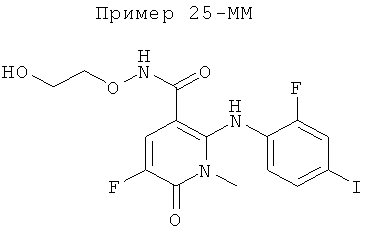
5-Фтор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS ESI (+) m/z 466, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.53 (br s, 1H), 9.37 (br s, 1H), 7.64 (dd, 1H), 7.62 (d, 1H), 7.37 (dd, 1H), 6.61 (t, 1H), 4.68 (t, 1H), 3.69 (t, 2H), 3.49 (q, 2H), 3.30 (s, 3H).
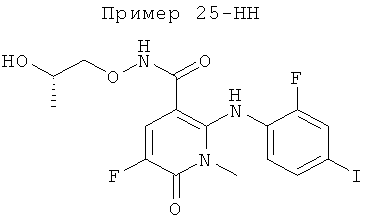
(S)-5-Фтор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид
MS APCI (+) m/z 480, образец (М+1); 1H ЯМР (400 МГц, DMSO-d6) δ 11.49 (br s, 1H), 9.48 (br s, 1H), 7.61 (m, 2H), 7.36 (m, 1H), 6.59 (t, 1H), 4.77 (br s, 1H), 3.69 (m, 1H), 3.49 (s, 1H), 3.48 (d, 1H), 3.29 (s, 3H), 0.99 (d, 3H).
Приведенное описание рассматривается только как иллюстрация принципов изобретения. Кроме того, поскольку многочисленные модификации и изменения будут очевидны специалистам в данной области, нежелательно ограничивать данное изобретение точными продемонстрированными конструкцией и способом, которые описаны выше. Соответственно все подходящие модификации и эквиваленты могут считаться подпадающими под объем изобретения, которое определено следующей ниже формулой изобретения.
Слова «содержат», «содержащий», «включают», «включающий» и «включает», когда они используются в этом описании и в следующей ниже формуле изобретения, предназначены для точного определения наличия изложенных признаков, целых чисел, компонентов или стадий, однако они не отрицают наличия или добавления одного или более других признаков, целых чисел, компонентов, стадий или их групп.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ МЕК И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2500673C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА | 2012 |
|
RU2632915C2 |
| ИНГИБИТОРЫ ФЕРМЕНТОВ | 2014 |
|
RU2674028C2 |
| ПРОЛЕКАРСТВО ТРИАЗОЛОНОВОГО СОЕДИНЕНИЯ | 2011 |
|
RU2581369C2 |
| ПИРИДОНОВЫЕ И АЗАПИРИДОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2011 |
|
RU2617405C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ЗОНДЫ ВИЗУАЛИЗАЦИИ БЕЛКА ГЕНТИНГТИНА | 2016 |
|
RU2721419C2 |
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2815314C2 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
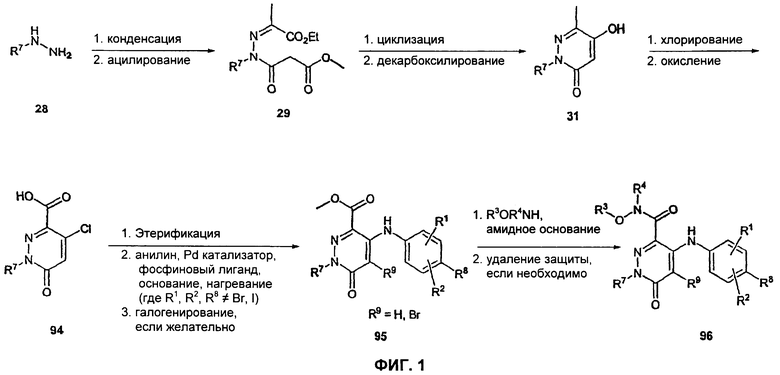
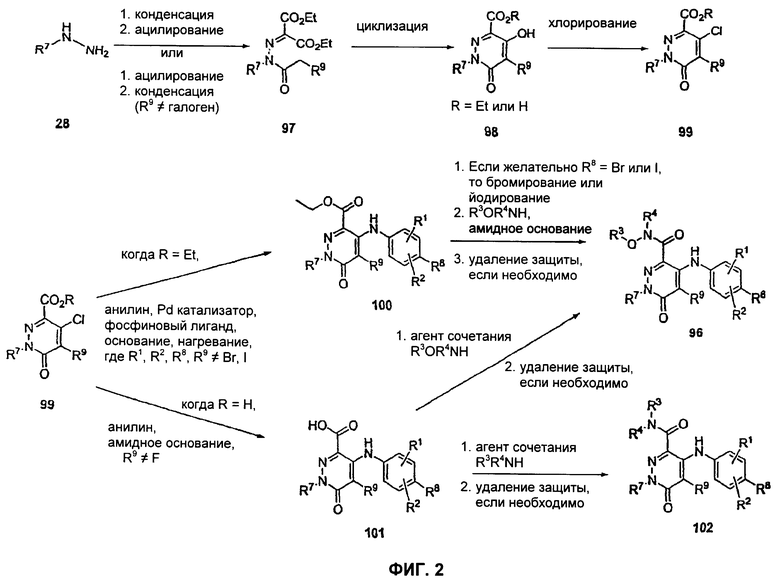
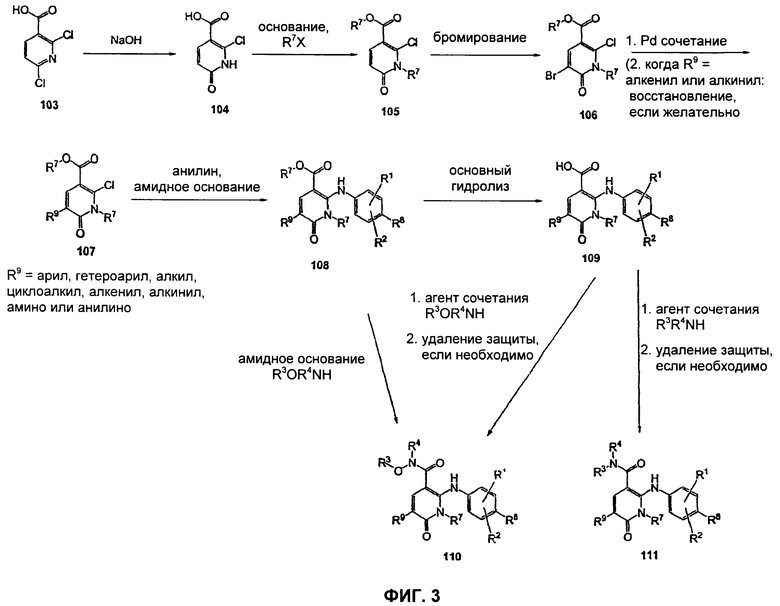
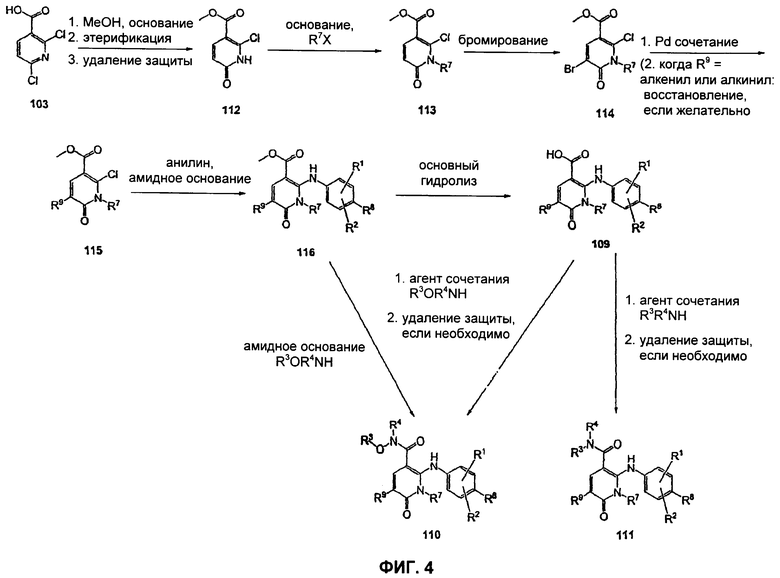
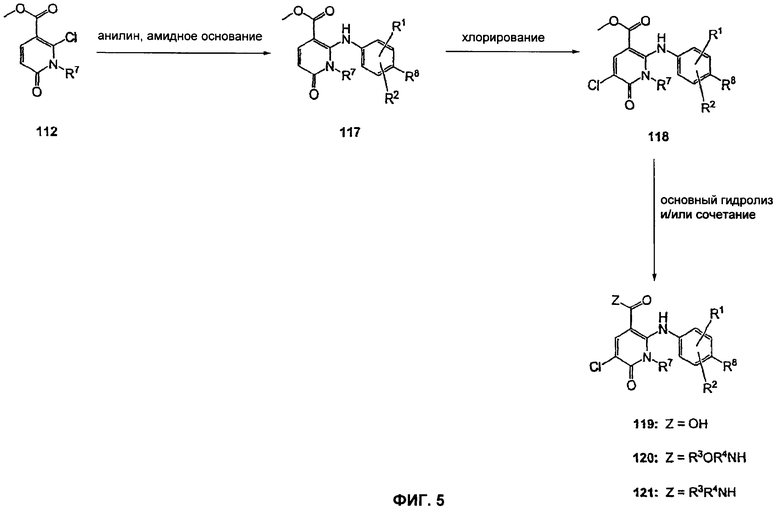
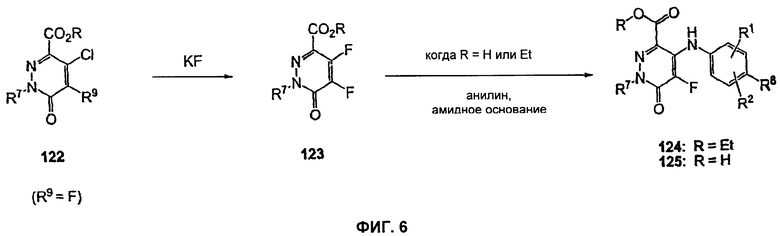
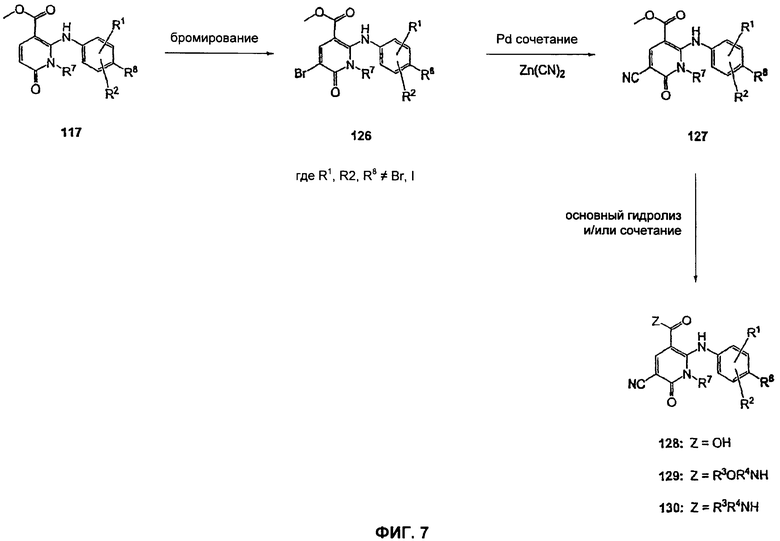
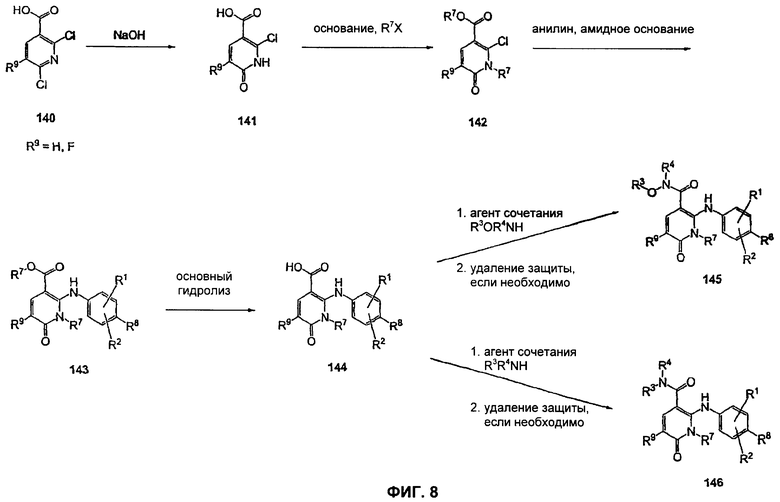
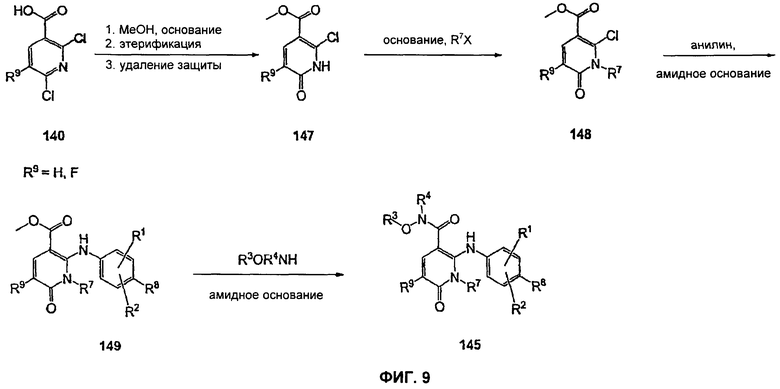
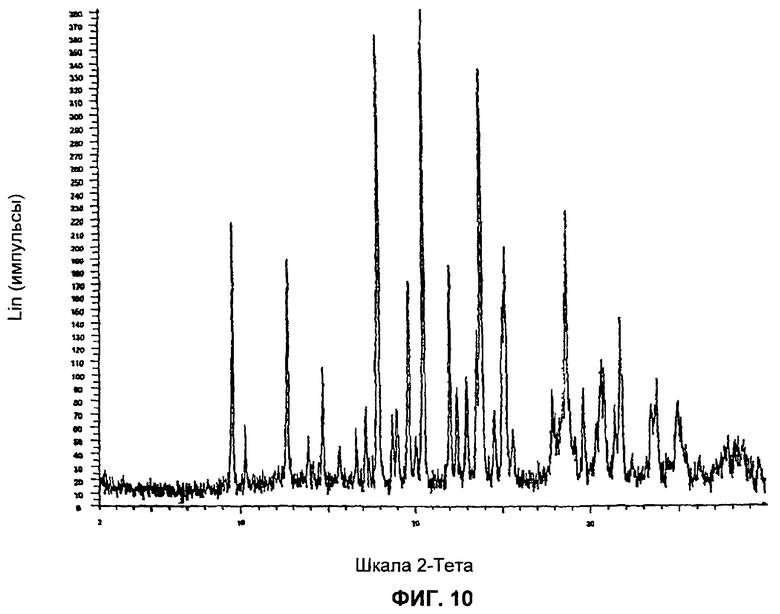
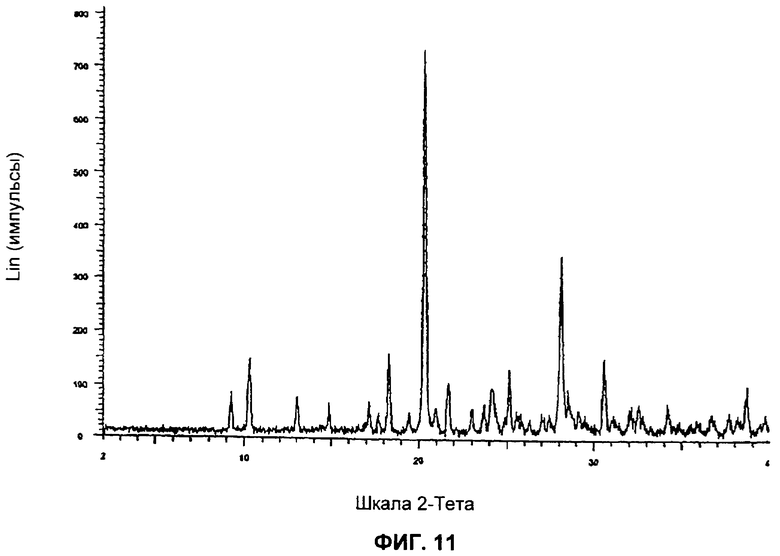
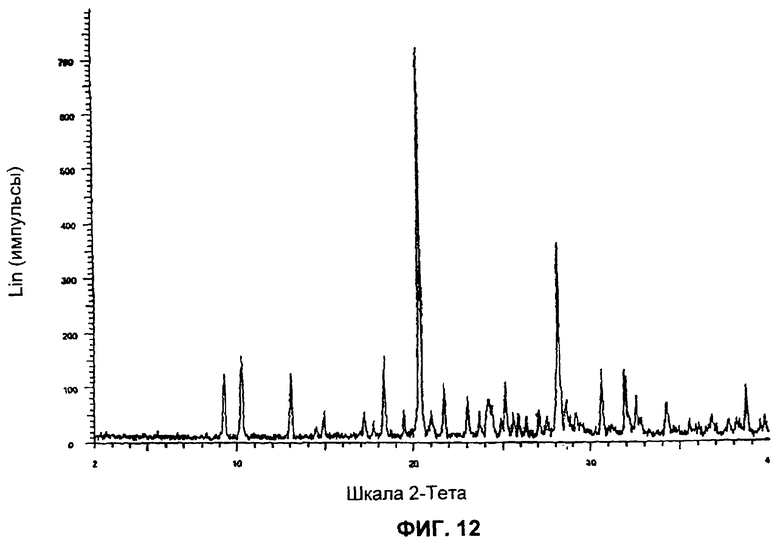
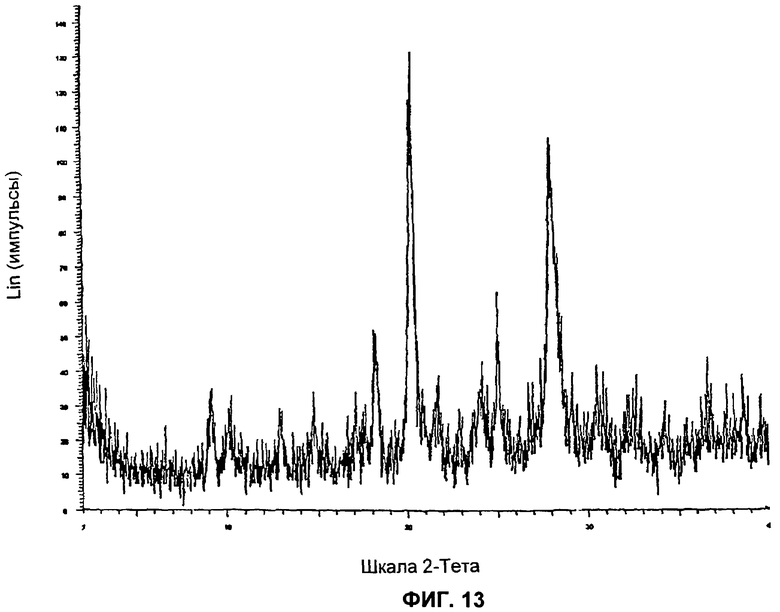
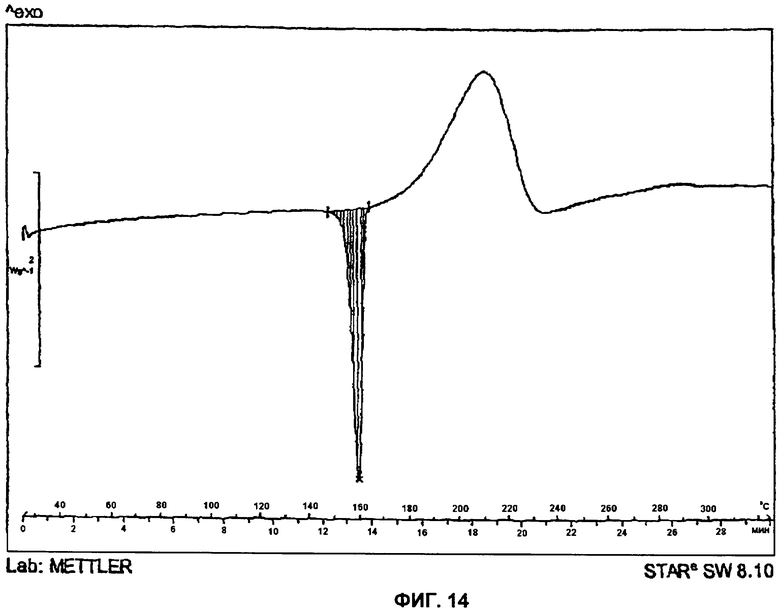
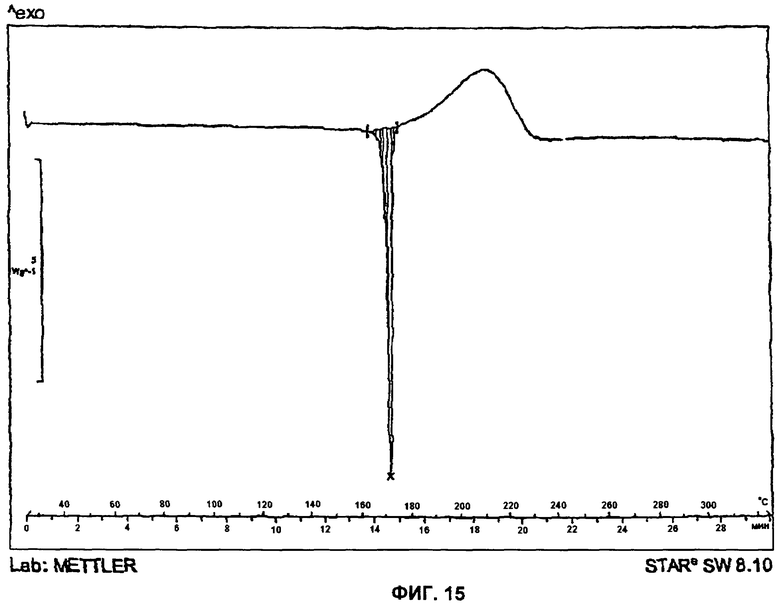
Изобретение относится к соединению, имеющему формулу IV:
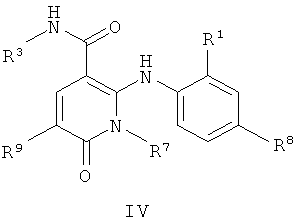
где R1 представляет собой С1 или F; R3 представляет собой H, Me, Et, ОН, МеО-, ЕtO-, НОСН2CH2О-, HOCH2C(Me)2O-, (S)-MeCH(OH)CH2O-, (R)-НОСН2СН(ОН)СН2O-, циклопропил-СН2O-, НОСН2СН2-,
 ,
,  ,
,  ,
,  ,
,  или
или 
R7 представляет собой метил или этил, которые возможно замещены одним или более чем одним F; R8 представляет собой Вr, 1 или SMe; и R9 представляет собой Н, С1-С4алкил, С1 или CN, где указанный алкил возможно замещен одной или более группами, независимо выбранными из F или CN, при условии, что когда а) R1 представляет собой F, R8 представляет собой Вr, R9 представляет собой Н, и R7 представляет собой либо Me, либо Et, тогда R3 не может представлять собой HOCH2CH2O; б) R1 представляет собой F, R8 представляет собой I, R9 представляет собой Н, и R3 представляет собой МеО, тогда R7 не может представлять собой Me; в) R1 представляет собой F, R8 представляет собой Me, R9 представляет собой Н, и R3 представляет собой НОСH2СН2О, тогда R7 не может представлять собой Me; и г) R1 представляет собой F, R8 представляет собой Вr, R9 представляет собой Н, и R3 представляет собой циклопропил-СН2O, тогда R7 не может представлять собой Me, а также к применению этого соединения в изготовлении лекарственного средства для лечения гиперпролиферативного расстройства или воспалительного состояния и к фармацевтической композиции, которая ингибирует МЕК. Технический результат: получены и описаны новые соединения, полезные в лечении гиперпролиферативных заболеваний, таких как рак и воспаление. 6 н. и 11 з.п. ф-лы, 8 табл., 15 ил.
1. Соединение, включая таутомеры, разделенные энантиомеры и их фармацевтически приемлемые соли, имеющее формулу IV:
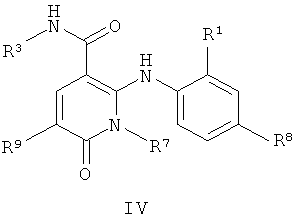
где R1 представляет собой Сl или F;
R3 представляет собой Н, Me, Et, ОН, МеО-, ЕtO-, НОСН2СН2О-, НОСН2С(Ме)2O-, (S)-MeCH(OH)CH2O-, (R)-HOCH2CH(OH)CH2O-, циклопропил-СН2O-, НОСН2СН2-,
 ,
,  ,
, 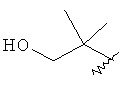 ,
, 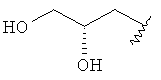 ,
,  или
или 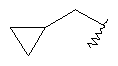
R7 представляет собой метил или этил, которые возможно замещены одним или более чем одним F;
R8 представляет собой Вr, I или SMe; и
R9 представляет собой Н, С1-С4алкил, Сl или CN, где указанный алкил возможно замещен одной или более группами, независимо выбранными из F или CN, при условии, что когда
а) R представляет собой F, R8 представляет собой Br, R9 представляет собой Н, и R7 представляет собой либо Me, либо Et, тогда R3 не может представлять собой НОСН2СН2О;
б) R1 представляет собой F, R8 представляет собой I, R9 представляет собой Н, и R3 представляет собой МеО, тогда R7 не может представлять собой Me;
в) R1 представляет собой F, R8 представляет собой SMe, R9 представляет собой Н, и R3 представляет собой НОСН2СН2О, тогда R7 не может представлять собой Me; и
г) R1 представляет собой F, R8 представляет собой Br, R9 представляет собой Н, и R3 представляет собой циклопропил-СН2О, тогда R7 не может представлять собой Me.
2. Соединение по п.1, где R9 представляет собой Н, Me, Et, Cl или CN.
3. Соединение, имеющее формулу VI:
или его фармацевтически приемлемая соль,
где R1 представляет собой Cl или F;
R3 представляет собой Н, HOCH2CH2O или (S)-MeCH(OH)CH2O; и
R9 представляет собой Н, Me, F или Cl.
4. Соединение по п.3, где R1 представляет собой F, R3 представляет собой НОСН2СН2О-, и R9 представляет собой метил, или его фармацевтически приемлемая соль.
5. Соединение по п.3, которое представляет собой:
2-(2-хлор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
2-(2-фтор-4-йодфениламино)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
2-(2-хлор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
5-хлор-2-(2-фтор-4-йодфениламино)-N-2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид;
(S)-2-(2-хлор-4-йодфениламино)-N-(2-гидроксипропокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид; или
(S)-5-хлор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамид.
6. Соединение по п.3, выбранное из:
5-фтор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида; и
(S)-5-фтор-2-(2-фтор-4-йодфениламино)-N-(2-гидроксипропокси)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксамида.
7. Соединение по п.3, которое представляет собой 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамид.
8. Кристаллическая форма соединения формулы XI
по существу в виде Формы 2 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, характеризующаяся картиной дифракции рентгеновских лучей с характеристическими пиками при примерно 9,5 и 12,6 по шкале 2θ.
9. Кристаллическая форма соединения формулы XI по п.8, характеризующаяся картиной дифракции рентгеновских лучей с характеристическими пиками при примерно 9,5; 12,6; 14,7 и 19,6 по шкале 2θ.
10. Кристаллическая форма соединения формулы XI по п.8, характеризующаяся картиной дифракции рентгеновских лучей, по существу такой, как показано на Фиг.10.
11. Кристаллическая форма соединения формулы XI
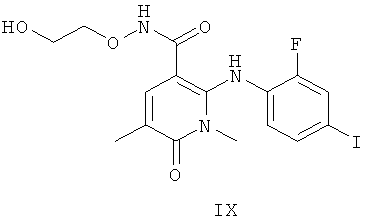
по существу в виде Формы 1 2-(2-фтор-4-йодфениламино)-N-(2-гидроксиэтокси)-1,5-диметил-6-оксо-1,6-дигидропиридин-3-карбоксамида, характеризующаяся картиной дифракции рентгеновских лучей с характеристическими пиками при примерно 9,2 и 13,0 по шкале 2θ.
12. Кристаллическая форма соединения формулы XI по п.11, характеризующаяся картиной дифракции рентгеновских лучей с характеристическими пиками при примерно 9,2; 13,0; 18,3; 21,0 и 21,7 по шкале 2θ.
13. Кристаллическая форма соединения формулы XI по п.11, характеризующаяся картиной дифракции рентгеновских лучей, по существу такой, как показано на Фиг.11.
14. Соединение по любому из пп.1, 3, 8 и 11 для применения в качестве лекарственного средства, которое ингибирует МЕК (киназа митоген-активируемых ERK (киназ, регулируемых внеклеточными сигналами)).
15. Соединение по любому из пп.1, 3, 8 и 11 для применения в качестве лекарственного средства для лечения гиперпролиферативного расстройства или воспалительного состояния.
16. Применение соединения по любому из пп.1, 3, 8 и 11 в изготовлении лекарственного средства для лечения гиперпролиферативного расстройства или воспалительного состояния.
17. Фармацевтическая композиция, которая ингибирует МЕК, содержащая соединение по любому из пп.1, 3, 8 и 11 вместе с фармацевтически приемлемым носителем.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ПИРИМИДИНАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ (ЛЕЧЕНИЯ) ОПУХОЛИ | 1994 |
|
RU2135491C1 |

