Настоящее изобретение относится к ингибиторам калликреина плазмы и к содержащим их фармацевтическим композициям и применению таких производных.
Предшествующий уровень техники изобретения
Ингибиторы по настоящему изобретению представляют собой ингибиторы калликреина плазмы и имеют ряд терапевтических применений, особенно в лечении проницаемости сосудов сетчатки, ассоциированной с диабетической ретинопатией и диабетическим макулярным отеком.
Калликреин плазмы представляет собой трипсиноподобную сериновую протеазу, которая может высвобождать кинины из кининогенов (см. K. D. Bhoola et al., "Kallikrein-Kinin Cascade", Encyclopedia of Respiratory Medicine, p483-493; J. W. Bryant et al., "Human plasma kallikrein-kinin system: physiological and biochemical parameters" Cardiovascular and haematological agents in medicinal chemistry, 7, p234-250, 2009; K. D. Bhoola et al., Pharmacological Rev., 1992, 44, 1; and D. J. Campbell, "Towards understanding the kallikrein-kinin system: insights from the measurement of kinin peptides", Brazilian Journal of Medical and Biological Research 2000, 33, 665-677). Он является необходимым членом внутреннего каскада коагуляции крови, хотя его роль в указанном каскаде не включает высвобождение брадикинина или ферментативное расщепление. Калликреин плазмы кодируется единственным геном и синтезируется в печени. Он секретируется гепатоцитами в виде неактивного прекалликреина плазмы, который циркулирует в плазме в виде гетеродимерного комплекса, связанного с высокомолекулярным кининогеном, который активируется с образованием активного калликреина плазмы. Кинины являются сильными медиаторами воспаления, которые действуют посредством G-белок-связанных рецепторов и антагонистов кининов (таких как антагонисты брадикинина), ранее изучаемых как потенциальные терапевтические средства для лечения ряда заболеваний (F. Marceau and D. Regoli, Nature Rev., Drug Discovery, 2004, 3, 845-852).
Считают, что калликреин плазмы играет роль при ряде воспалительных расстройств. Основным ингибитором калликреина плазмы является ингибитор серпин С1-эстеразы. Пациенты, у которых имеется генетический дефицит ингибитора C1 эстеразы, страдают от врожденного ангионевротического отека (HAE), который приводит к периодическим отекам лица, рук, глотки, желудочно-кишечного тракта и гениталий. Пузыри, образующиеся во время острых эпизодов, содержат высокий уровень калликреина плазмы, который отщепляет высокомолекулярный кининоген, высвобождая брадикинин, приводящий к повышенной сосудистой проницаемости. Показано, что лечение крупномолекулярным ингибитором калликреина плазмы эффективно воздействует на HAE путем предотвращения высвобождения брадикинина, который вызывает повышенную проницаемость сосудов (A. Lehmann "Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery" Expert Opin. Biol. Ther. 8, p1187-99).
Калликреин-кининовая система плазмы патологически избыточна у пациентов с прогрессирующим диабетическим макулярным отеком. Недавно было опубликовано, что калликреин плазмы участвует в дисфункции сосудов сетчатки у крыс с диабетом (A. Clermont et al. "Plasma kallikrein mediates retinal vascular dysfunction and induces retinal thickening in diabetic rats" Diabetes, 2011, 60, p1590-98). Более того, введение ингибитора калликреина плазмы ASP-440 улучшало и проницаемость сосудов сетчатки, и патологический кровоток сетчатки у крыс с диабетом. Следовательно, ингибитор калликреина плазмы должен обладать способностью в качестве лечения уменьшать проницаемость сосудов сетчатки, ассоциированную с диабетической ретинопатией и диабетическим макулярным отеком.
Другие осложнения диабета, такие как кровоизлияние в головной мозг, нефропатия, кардиомиопатия и нейропатия, все из которых ассоциированы с калликреином плазмы, могут также расцениваться как цели для ингибитора калликреина плазмы.
Синтетические и низкомолекулярные ингибиторы калликреина плазмы были описаны ранее, например, Garrett et al. ("Peptide aldehyde." J. Peptide Res. 52, p62-71 (1998)), T. Griesbacher et al. ("Involvement of tissue kallikrein but not plasma kallikrein in the development of symptoms mediated by endogenous kinins in acute pancreatitis in rats" British Journal of Pharmacology 137, p692-700 (2002)), Evans ("Selective dipeptide inhibitors of kallikrein" WO03/076458), Szelke et al. ("Kininogenase inhibitors" WO92/04371), D. M. Evans et al. (Immunolpharmacology, 32, p115-116 (1996)), Szelke et al. ("Kininogen inhibitors" WO95/07921), Antonsson et al. ("New peptides derivatives" WO94/29335), J. Corte et al. (”Six membered heterocycles useful as serine protease inhibitors” WO2005/123680), J. Stürzbecher et al. (Brazilian J. Med. Biol. Res 27, p1929-34 (1994)), Kettner et al. (US 5,187,157), N. Teno et al. (Chem. Pharm. Bull. 41, p1079-1090 (1993)), W. B. Young et al. ("Small molecule inhibitors of plasma kallikrein" Bioorg. Med. Chem. Letts. 16, p2034-2036 (2006)), Okada et al. ("Development of potent and selective plasmin and plasma kallikrein inhibitors and studies on the structure-activity relationship" Chem. Pharm. Bull. 48, p1964-72 (2000)), Steinmetzer et al. ("Trypsin-like serine protease inhibitors and their preparation and use" WO08/049595), Zhang et al. ("Discovery of highly potent small molecule kallikrein inhibitors" Medicinal Chemistry 2, p545-553 (2006)), Sinha et al. ("Inhibitors of plasma kallikrein" WO08/016883), Shigenaga et al. (“Plasma Kallikrein Inhibitors” WO2011/118672), and Kolte et al. (“Biochemical characterization of a novel high-affinity and specific kallikrein inhibitor”, British Journal of Pharmacology (2011), 162(7), 1639-1649). Также, Steinmetzer et al. (“Serine protease inhibitors” WO2012/004678) описывают циклические пептидные аналоги, которые являются ингибиторами человеческого плазмина и калликреина плазмы.
К настоящему времени не одобрены низкомолекулярные синтетические ингибиторы калликреина плазмы для медицинского применения. Молекулы, описанные в известной области техники, страдают от ограничений, таких как плохая селективность относительно связанных ферментов, таких как KLK1, тромбин и другие сериновые протеазы, и плохая пероральная биодоступность. Крупные белковые ингибиторы калликреина плазмы представляют риски анафилактических реакций, как сообщалось для Экаллантида (Ecallantide). Следовательно, сохраняется необходимость в соединениях, которые селективно ингибируют калликреин плазмы, которые не вызывают анафилаксии, и которые являются перорально доступными. Кроме того, огромное множество молекул в известной области техники содержат высокополярную и ионизируемую гуанидиновую или амидиновую функциональную группу. Хорошо известно, что такие функциональные группы могут ограничивать проницаемостью кишечника и, следовательно, пероральную биодоступность. Например, сообщают Tamie J. Chilcote and Sukanto Sinha (“ASP-634: An Oral Drug Candidate for Diabetic MacularEdema”, ARVO 2012 May 6th-May 9th, 2012, Fort Lauderdale, Florida, Presentation 2240), что ASP-440, бензамидин, страдает от плохой пероральной биодоступности. Дополнительно сообщают, что абсорбция может быть улучшена путем создания пролекарства, такого как ASP-634. Однако, хорошо известно, что пролекарства могут иметь несколько недостатков, например, плохую химическую стабильность и потенциальную токсичность инертных носителей или неожиданных метаболитов. В другом сообщении индоламиды заявлены как соединения, которые могут преодолеть проблемы, ассоциированные с лекарственными средствами, обладающими плохой или неадекватной ADME-токсичностью и физико-химическими свойствами, не проявляя и не заявляя ингибирования калликреина плазмы (Griffioen et al, “Indole amide derivatives and related compounds for use in the treatment of neurodegenerative diseases”, WO2010, 142801).
BioCryst Pharmaceuticals Inc. сообщили об обнаружении перорально доступного ингибитора калликреина плазмы BCX4161 (“BCX4161, An Oral Kallikrein Inhibitor: Safety and Pharmacokinetic Results Of a Phase 1 Study In Healthy Volunteers”, Journal of Allergy and Clinical Immunology, Volume 133, Issue 2, Supplement, February 2014, page AB39 and “A Simple, Sensitive and Selective Fluorogenic Assay to Monitor Plasma Kallikrein Inhibitory Activity of BCX4161 in Activated Plasma”, Journal of Allergy and Clinical Immunology, Volume 133, Issue 2, Supplement February 2014, page AB40). Однако дозы для людей являются относительно большими, в настоящее время исследуются в исследованиях для подтверждения механизма действия в дозировках 400 мг три раза в сутки.
Существует только несколько сообщений об ингибиторах калликреина плазмы, которые не содержат гуанидиновой или амидиновой функциональных групп. Одним примером является Brandl et al. (“N-((6-amino-piridin-3-yl)methyl)-heteroaryl-carboxamides as inhibitors of plasma kallikrein” WO2012/017020), который описывает соединения, которые несут аминопиридиновую функциональную группу. Пероральная эффективность в моделях на крысах продемонстрирована в относительно высоких дозах 30 мг/кг и 100 мг/кг, но фармакокинетический профиль не представлен. Следовательно, до настоящего времени неизвестно, обеспечивают ли такие соединения достаточную пероральную биодоступность или эффективность для продвижения в клинику. Другими примерами являются Brandl et al. (“Aminopiridine derivatives as plasma kallikrein inhibitors” WO2013/111107) и Flohr et al. (“5-membered heteroarylcarboxamide derivatives as plasma kallikrein inhibitors” WO2013/111108). Однако ни в одном из указанных документов не сообщают каких-либо in vivo данных и, следовательно, еще не известно, обеспечивают ли указанные соединения достаточную пероральную биодоступность или эффективность для продвижения в клинику.
Следовательно, остается необходимость в разработке новых ингибиторов калликреина плазмы, которые обладают способностью лечить широкое множество расстройств, в частности для уменьшения проницаемости сосудов сетчатки, ассоциированной с диабетической ретинопатией и диабетическим макулярным отеком. Предпочтительные соединения обладают хорошим фармакокинетическим профилем и в частности подходят в качестве лекарственных средств для перорального введения.
Сущность изобретения
Настоящее изобретение относятся к ряду амидов, которые являются ингибиторами калликреина плазмы. Указанные соединения демонстрируют хорошую селективность в отношении калликреина плазмы и являются потенциально применимыми в лечении нарушений остроты зрения, диабетической ретинопатии, макулярного отека, врожденного ангиоотека, диабета, панкреатита, кровоизлияния в головной мозг, нефропатии, кардиомиопатии, нейропатии, воспалительных заболеваний кишечника, артрита, воспаления, септического шока, гипотонии, рака, респираторного дистресс синдрома взрослых, диссеминированного сосудистого свертывания, операциях сердечно-легочного шунтирования и кровотечений из послеоперационных ран. Изобретение дополнительно относится к фармацевтическим композициям ингибиторов, к применению композиций в качестве терапевтических средств и к способам лечения с использованием указанных композиций.
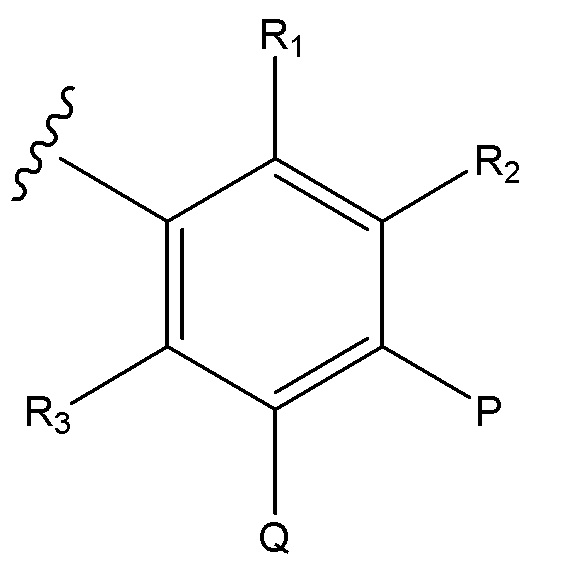
В одном аспекте настоящее изобретение обеспечивает соединения по формуле I
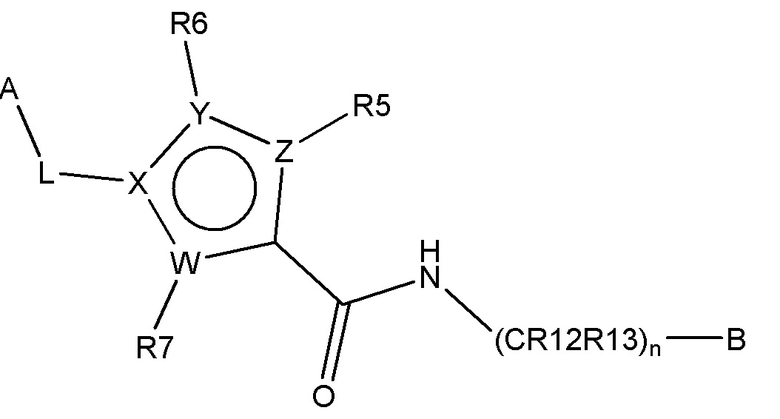
Формула (I)
где
B представляет собой
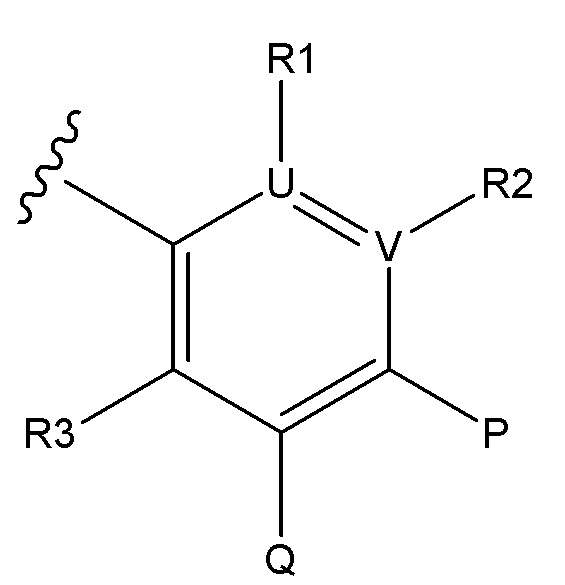
или B представляет собой сшитый 6,5- или 6,6-гетероароматический бициклический цикл, содержащий N, и, необязательно, один или два дополнительных гетероатома, независимо выбираемых из N, O и S, которые являются необязательно моно-, ди или тризамещенными заместителем, выбираемым из алкил, алкокси, OH, галогена, CN, COOR8, CONR8R9, CF3 и NR8R9;
n представляет собой 0, 1 или 2;
W, X, Y и Z независимо выбирают из C, C(R16)-C, C(R16)=C, C=N, N, O и S, так что цикл, содержащий W, X, Y и Z, представляет собой шестичленный ароматический гетероцикл;
где,
R5, R6 и R7 независимо отсутствуют или независимо выбирают из H, алкил, алкокси, галоген, OH, арил, гетероарил, -NR8R9, CN, COOR8, CONR8R9, -NR8COR9 и CF3; и
R16 независимо выбирают из H, алкил, алкокси, галоген, OH, NR8R9, арил, гетероарил и CF3;
A выбирают из арил, гетероарил, и группы заместителя, выбираемой из формулы (A), (B), (C), и (D):
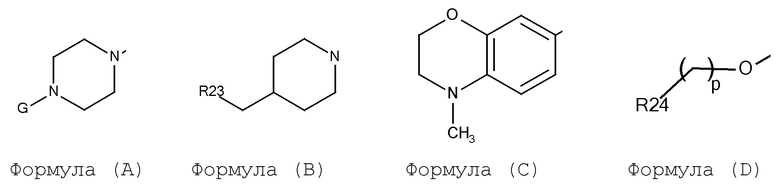
где:
G выбирают из H, алкил, циклоалкил, CO-арил, SO2-арил, (CH2)m-арил, и (CH2)m-гетероарил;
m выбирают из 0 и 1;
p выбирают из 0, 1, 2 и 3;
R23 выбирают из арил и гетероарил;
R24 выбирают из арил и гетероарил;
L представляет собой линкер, выбираемый из ковалентной связи, -(CHR17)-, -(CH2)1-10-, -O-(CH2)2-10-, -(CH2)1-10-O-(CH2)1-10-, -(CH2)1-10-NH-(CH2)1-10-, -CONH-(CH2)1-10-, -CO-, и -SO2-;
U и V независимо выбирают из C и N, так что ароматическим циклом, содержащим U и V, является фенил, пиридин или пиразин;
R1 отсутствует, когда U является N;
R2 отсутствует, когда V является N;
или, при наличии, R1 и R2 независимо выбирают из H, алкил, алкокси, CN, галогена и CF3;
R3 выбирают из H, алкил, алкокси, CN, галоген и CF3;
P представляет собой H и Q представляет собой -C(R18)(R19)NH2, или P представляет собой -C(R18)(R19)NH2 и Q представляет собой H;
R8 и R9 независимо выбирают из H и алкил;
R12 и R13 независимо выбирают из H и алкил, или могут вместе образовывать циклоалкил;
R17 выбирают из алкил и OH;
R18 и R19 независимо выбирают из H и алкил, или могут вместе образовывать циклоалкил или циклический эфир;
алкил представляет собой линейный насыщенный углеводород, имеющий до 10 атомов углерода (C1-C10) или разветвленный насыщенный углеводород от 3 до 10 атомов углерода (C3-C10); алкил может необязательно быть замещенным 1 или 2 заместителями, независимо выбираемыми из (C1-C6)алкокси, OH, CN, CF3, COOR10, CONR10R11, фтор, фенил и NR10R11;
циклоалкил представляет собой моноциклический насыщенный углеводород из 3-7 атомов углерода;
циклический эфир представляет собой моноциклический насыщенный углеводород от 4 до 7 атомов углерода, где один из атомов цикла заменен атомом кислорода;
алкокси представляет собой линейный O-связанный углеводород от 1 до 6 атомов углерода (C1-C6) или разветвленный O-связанный углеводород от 3 до 6 атомов углерода (C3-C6); алкокси может необязательно быть замещенной 1 или 2 заместителями, независимо выбираемыми из OH, OCH3, CN, CF3, COOR10, CONR10R11, фтора и NR10R11;
арил представляет собой фенил, бифенил или нафтил; арил может быть необязательно замещенным 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, метилендиокси, этилендиокси, OH, галоген, CN, морфолинил, пиперидинил, гетероарил, -(CH2)0-3-O-гетероарил, арилb, -O-арилb, -(CH2)1-3-арилb, -(CH2)1-3-гетероарил, -COOR10, -CONR10R11, -(CH2)1-3-NR14R15, CF3 и -NR10R11;
арилb представляет собой фенил, бифенил или нафтил, который может быть необязательно замещенным 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, OH, галогена, CN, морфолинил, пиперидинил, -COOR10, -CONR10R11, CF3 и NR10R11;
гетероарил представляет собой 5, 6, 9 или 10 членный моно- или бициклический ароматический цикл, содержащий, когда возможно, 1, 2 или 3 членов цикла, независимо выбираемые из N, NR8, S и O; гетероарил может быть необязательно замещенным 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, OH, галогена, CN, арил, морфолинил, пиперидинил, -(CH2)1-3-арил, гетероарилb, -COOR10, -CONR10R11, CF3 и -NR10R11;
гетероарилb представляет собой 5, 6, 9 или 10 членный моно- или бициклический ароматический цикл, содержащий, когда возможно, 1, 2 или 3 члена цикла, независимо выбираемые из N, NR8, S и O; где гетероарилb может быть необязательно замещенным 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, OH, галоген, CN, морфолинил, пиперидинил, арил, -(CH2)1-3-арил, -COOR10, -CONR10R11, CF3 и NR10R11;
R10 и R11 независимо выбирают из H и алкил; или R10 и R11 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероцикл, который может быть насыщенным или ненасыщенным с 1 или 2 двойными связями и который может быть необязательно моно- или ди-замещенным заместителями, выбираемыми из оксо, алкил, алкокси, COOR8, OH, F и CF3;
R14 и R15 независимо выбирают из алкил, арилb и гетероарилb; или R14 и R15 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероцикл, который может быть насыщенным или ненасыщенным с 1 или 2 двойными связями и необязательно может быть оксо-замещенным;
и их таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереоизомеры, рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В другом аспекте настоящее изобретение обеспечивает пролекарство соединения по формуле (I), как определено в настоящем описании, или его фармацевтически приемлемую соль.
В еще одном аспекте настоящее изобретение обеспечивает N-оксид соединения по формуле (I), как определено в настоящем описании, или его пролекарство или его фармацевтически приемлемую соль.
Понимают, что определенные соединения по настоящему изобретению могут существовать в сольватированной, например, гидратированной, а также, несольватированных формах. Понимают, что настоящее изобретение охватывает все такие сольватированные формы.
В одном аспекте настоящее изобретение обеспечивает соединения по формуле Ia
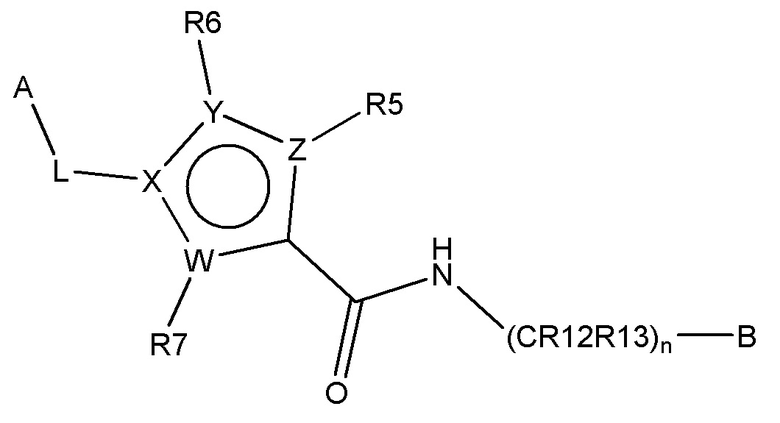
Формула (Ia)
где
B представляет собой
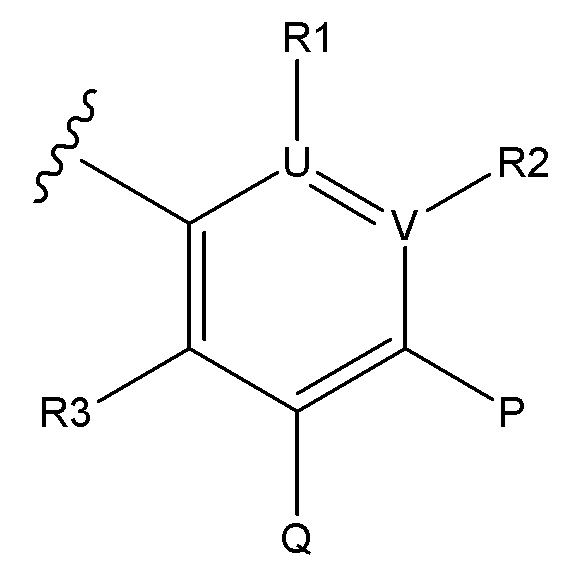
или B является сшитым 6,5- или 6,6-гетеродимерным бициклическим циклом, содержащим N и, необязательно, один или два дополнительных гетероатома, независимо выбираемых из N, O и S, который является необязательно моно-, ди или тризамещенным заместителем, выбираемым из алкил, алкокси, OH, галоген, CN, COOR8, CONR8R9, CF3 и NR8R9;
n составляет 0, 1 или 2;
W, X, Y и Z независимо выбирают из C(R16)-C, C(R16)=C, C, N, O и S, так что цикл, содержащий W, X, Y и Z, представляет собой шестичленный ароматический гетероцикл;
где,
R5, R6 и R7 независимо отсутствуют или независимо выбирают из H, алкил, галоген, OH, арил, гетероарил и CF3; и
R16 независимо выбирают из H, алкил, алкокси, галоген, OH, арил, гетероарил и CF3;
A выбирают из арил, гетероарил, и неароматического пяти- или шестичленного цикла, содержащего N или NR10, и необязательно содержащего один или два дополнительных гетероатома, выбираемых из N, O и S, где указанный цикл является необязательно сшитым с фенилом;
L представляет собой линкер, выбираемый из ковалентной связи, -(CH2)1-10-, -O-(CH2)2-10-, -(CH2)1-10-O-(CH2)1-10-, -(CH2)1-10-NH-(CH2)1-10-, -CONH-(CH2)1-10-, -CO-, и -SO2-;
U и V независимо выбирают из C и N, так что ароматический цикл, содержащий U и V, представляет собой фенил, пиридин или пиразин;
R1 отсутствует, когда U является N;
R2 отсутствует, когда V является N;
или, при наличии, R1 и R2 независимо выбирают из H, алкил, алкокси, CN, галоген и CF3;
R3 выбирают из H, алкил, алкокси, CN, галоген и CF3;
P представляет собой H и Q представляет собой -C(R18)(R19)NH2, или P представляет собой -C(R18)(R19)NH2 и Q представляет собой H;
R8 и R9 независимо выбирают из H и алкил;
R12 и R13 независимо выбирают из H и алкил, или могут вместе образовывать циклоалкил;
R18 и R19 независимо выбирают из H и алкил, или могут вместе образовывать циклоалкил или циклический эфир;
алкил представляет собой линейный насыщенный углеводород, имеющий до 10 атомов углерода (C1-C10) или разветвленный насыщенный углеводород от 3 до 10 атомов углерода (C3-C10); алкил может необязательно быть замещенным 1 или 2 заместителями, независимо выбираемыми из (C1-C6)алкокси, OH, CN, CF3, COOR10, CONR10R11, фтор и NR10R11;
циклоалкил представляет собой моноциклический насыщенный углеводород от 3 до 7 атомов углерода;
циклический эфир представляет собой моноциклический насыщенный углеводород от 4 до 7 атомов углерода, где один из атомов цикла замещен атомом кислорода;
алкокси представляет собой линейный O-сшитый углеводород от 1 до 6 атомов углерода (C1-C6) или разветвленный O-сшитый углеводород от 3 до 6 атомов углерода (C3-C6); алкокси может необязательно быть замещенным 1 или 2 заместителями, необязательно выбираемыми из OH, CN, CF3, COOR10, CONR10R11, фтор и NR10R11;
арил представляет собой фенил, бифенил или нафтил; арил может быть необязательно замещенным 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, метилендиокси, этилендиокси, OH, галоген, CN, морфолинил, пиперидинил, гетероарил, -(CH2)0-3-O-гетероарил, арилb, -O-арилb, -(CH2)1-3-арилb, -(CH2)1-3-гетероарил, -COOR10, -CONR10R11, -(CH2)1-3-NR14R15, CF3 и -NR10R11;
арилb представляет собой фенил, бифенил или нафтил, который может быть необязательно замещенным 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, OH, галоген, CN, морфолинил, пиперидинил, -COOR10, -CONR10R11, CF3 и NR10R11;
гетероарил представляет собой 5, 6, 9 или 10 членный моно- или дициклический ароматический цикл, содержащий, когда возможно, 1, 2 или 3 членов цикла, независимо выбираемые из N, NR8, S и O; гетероарил может быть необязательно замещенным 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, OH, галоген, CN, арил, морфолинил, пиперидинил, -(CH2)1-3-арил, гетероарилb, -COOR10, -CONR10R11, CF3 и -NR10R11;
гетероарилb представляет собой 5, 6, 9 или 10 членный моно- или бициклический ароматический цикл, содержащий, когда возможно, 1, 2 или 3 членов цикла, независимо выбираемые из N, NR8, S и O; где гетероарилb может быть необязательно замещен 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, OH, галоген, CN, морфолинил, пиперидинил, арил, -(CH2)1-3-арил, -COOR10, -CONR10R11, CF3 и NR10R11;
R10 и R11 независимо выбирают из H и алкил; или R10 и R11 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероциклический цикл, который может быть насыщенным или ненасыщенным 1 или 2 двойными связями;
R14 и R15 независимо выбирают из алкил, арилb и гетероарилb; или R14 и R15 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероциклический цикл, который может быть насыщенным или ненасыщенным с 1 или 2 двойными связями, и необязательно может быть оксо замещенным;
и его таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереоизомеры и их рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В одном аспекте изобретение включает подгруппу соединений по формуле I
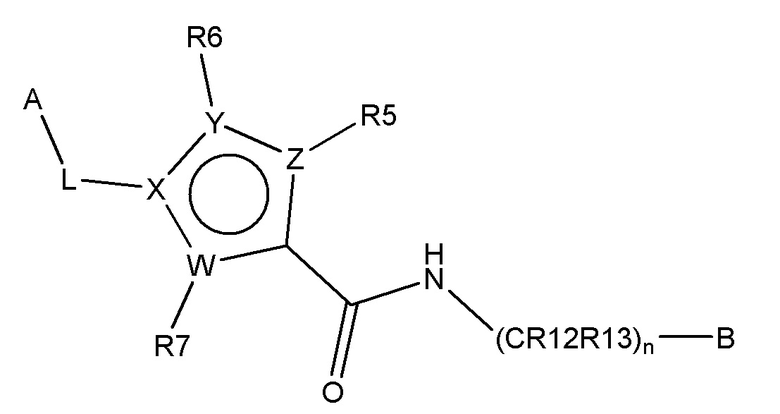
Формула (I)
где
L представляет собой линкер, выбираемый из ковалентной связи, -(CHOH)-, и -(CH2)1-6-;
B представляет собой
или B является сшитым 6,5- или 6,6-гетероароматическим бициклическим циклом, содержащим N и, необязательно, один или два дополнительных гетероатома, независимо выбираемые из N, O и S, который является необязательно моно-, ди или тризамещенным заместителем, выбираемым из алкил, алкокси, OH, галоген, CN, COOR8, CONR8R9, CF3 и NR8R9;
и где A, W, X, Y, Z, R1, R2, R3, R5, R6, R7, R8, R9, R12, R13, R17, алкил, алкокси и n являются, как определено в соответствии с формулой (I) или формулой (Ia) выше,
и их таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереоизомеры и рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В одном аспекте изобретение включает подгруппу соединений по формуле (I), как определено формулой (II),
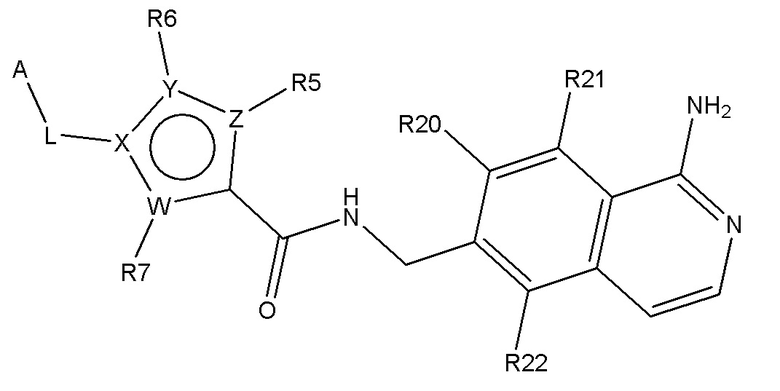
Формула (II)
где R20, R21 и R22 независимо выбирают из H, алкил, COOR8, CONR8R9, OH, алкокси, NR8R9, F и Cl; и где A, L, W, X, Y, Z, R5, R6, R7, алкил, алкокси, R8 и R9 являются, как определено в соответствии с формулой (I) или формулой (Ia) выше;
и их таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереоизомеры и рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В одном аспекте изобретение включает подгруппу соединений по формуле (I), как определено по формуле (II),
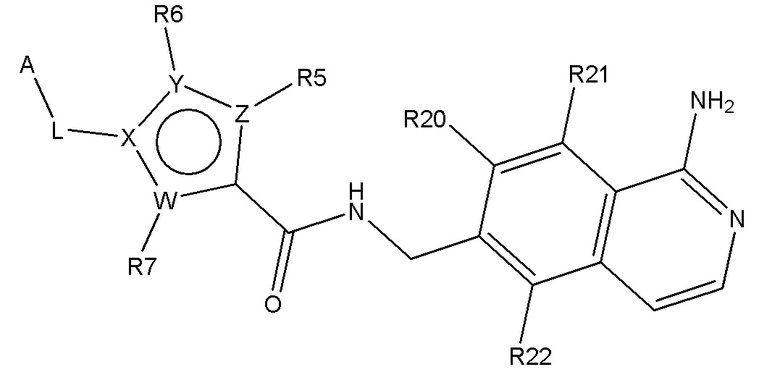
Формула (II)
где R20, R21 и R22 независимо выбирают из H, алкил, COOR8, CONR8R9, OH, алкокси, NR8R9, F и Cl;
L представляет собой линкер, выбираемый из -(CHOH)-, и -(CH2)1-6-;
и где A, W, X, Y, Z, R5, R6, R7, алкил, алкокси, R8 и R9 являются, как определено в соответствии с формулой (I) или формулой (Ia) выше;
и их таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереоизомеры и рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В одном аспекте изобретение включает подгруппу соединений по формуле (I), как определено формулой (III),
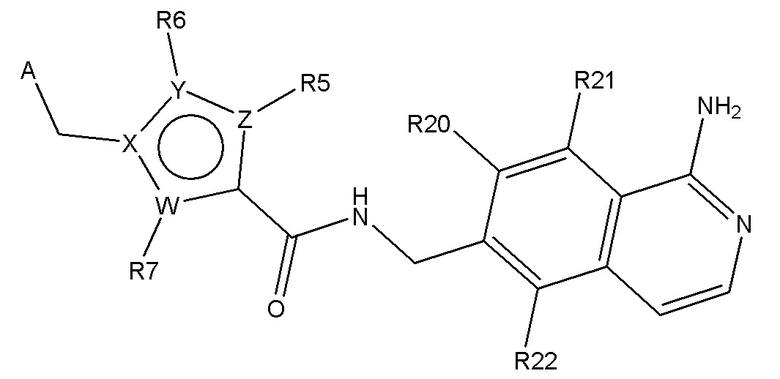
Формула (III)
где R20, R21 и R22 независимо выбирают из H, алкил, COOR8, CONR8R9, OH, алкокси, NR8R9, F и Cl; и где A, W, X, Y, Z, R5, R6, R7, алкил, алкокси, R8 и R9 являются, как определено в соответствии с формулой (I) или формулой (Ia) выше;
и их таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереоизомеры и рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В одном аспекте изобретение включает подгруппу соединений по формуле (I), как определено формулой (IV),
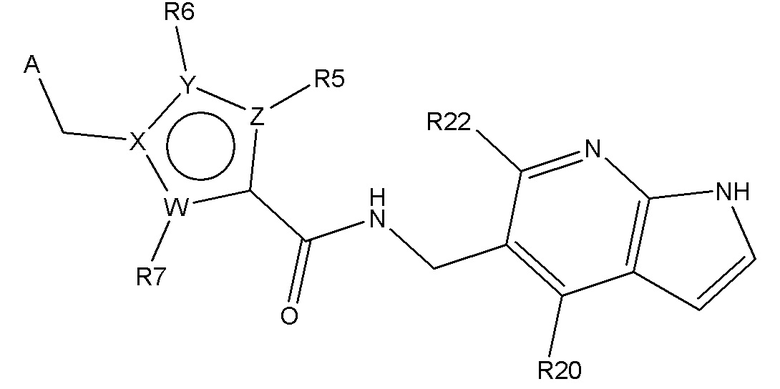
Формула (IV)
где R20 и R22 независимо выбирают из H, алкил, COOR8, CONR8R9, OH, алкокси, NR8R9, F и Cl; и где A, W, X, Y, Z, R5, R6, R7, алкил, алкокси, R8 и R9 являются, как определено в соответствии с формулой (I) или формулой (Ia) выше;
и их таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереоизомеры и рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В одном аспекте изобретение включает подгруппу соединений по формуле (I), как определено формулой (V),
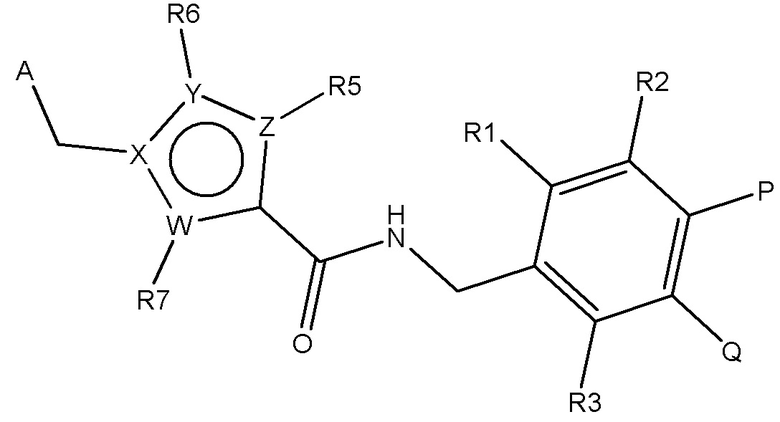
Формула (V)
где A, W, X, Y, Z, R1, R2, R3, R5, R6, R7, P и Q являются, как определено в соответствии с формулой (I) или формулой (Ia) выше;
и их таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереоизомеры и рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
Настоящее изобретение также включает следующие аспекты и их комбинации:
Соединения по формуле (I), где B является сшитым 6,5- или 6,6-гетероароматическим бициклическим циклом, содержащим N и, необязательно, один или два дополнительных гетероатома, независимо выбираемых из N, O и S, которые представляют собой необязательно моно-, ди или тризамещенные заместителем, выбираемым из алкил, алкокси, OH, галоген, CN, COOR8, CONR8R9, CF3 и NR8R9; где R8 и R9 независимо выбирают из H и алкил; где, когда B является сшитым 6,5-гетероароматическим аза-бициклом, он связан с -(CR12R13)n- посредством 6-членного компонента цикла.
Соединения по формуле (I), где B является сшитым 6,5 или 6,6-гетероароматическим бициклическим циклом, содержащим один, два или три атома N, который является необязательно моно- замещенным заместителем, выбираемым из алкил, алкокси, OH, галоген, CN, COOR8, CONR8R9, CF3 и NR8R9; где R8 и R9 независимо выбирают из H и алкил.
Соединения по формуле (I) где, B является сшитым 6,6-гетероароматическим аза-бициклом, который является необязательно монозамещенным заместителем, выбираемым из алкил, алкокси, OH, галогена, CN, COOR8, CONR8R9, CF3 и NR8R9; где R8 и R9 независимо выбирают из H и алкил.
Соединения по формуле (I), где B является сшитым 6,5- или 6,6-гетероароматическим бициклическим циклом, содержащим N и, необязательно, один или два дополнительных гетероатома, независимо выбираемых из N и O, который является необязательно монозамещенным заместителем, выбираемым из алкил, алкокси, OH, галоген, CN, COOR8, CONR8R9, CF3 и NR8R9; где R8 и R9 независимо выбирают из H и алкил.
Соединения по формуле (I), где B является сшитым 6,6-гетероароматическим аза-бициклом, который является необязательно монозамещенным заместителем, выбираемым из алкил, алкокси, OH, и NR8R9; где R8 и R9 независимо выбирают из H и алкил.
Соединения по формуле (I), где B является сшитым 6,6- гетероароматическим аза-бициклом, который является необязательно монозамещенным NR8R9; где R8 и R9 независимо выбирают из H и алкил.
Соединения по формуле (I), где B является необязательно моно-, ди или тризамещенным изохинолинилом, где указанный заместитель(и) выбирают из алкил, алкокси, OH, F, Cl, CN, COOR8, CONR8R9, CF3 и NR8R9; где R8 и R9 независимо выбирают из H и алкил.
Соединения по формуле (I) где, B является необязательно монозамещенным изохинолинилом; где указанный необязательный заместитель выбирают из алкил, алкокси, OH, и NR8R9; где R8 и R9 независимо выбирают из H и алкил.
Соединения по формуле (I), где B является необязательно замещенным 1H-пирроло[2,3-b]пиридином, где указанный необязательный заместитель(и) выбирают из алкил, алкокси, OH, F, Cl, CN, COOR8, CONR8R9, CF3 и NR8R9 и где R8 и R9 независимо выбирают из H и алкил.
Соединения по формуле (I), где B является:
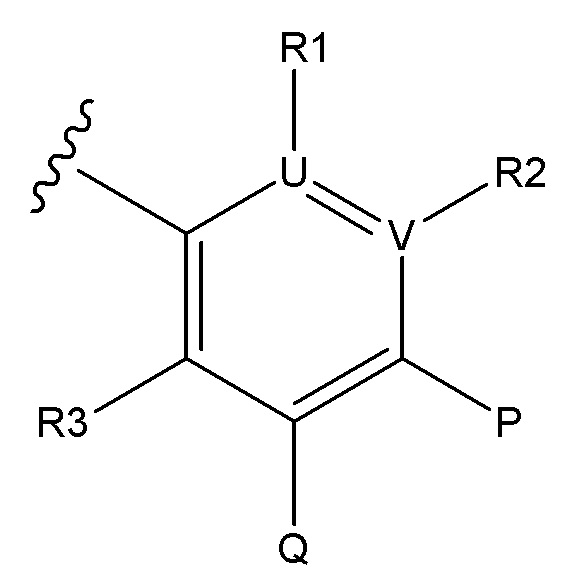
и где R1, R2, R3, P, Q, U и V являются, как определено в соответствии с формулой (I) или формулой (Ia) выше.
Соединения по формуле (I) где, B является:
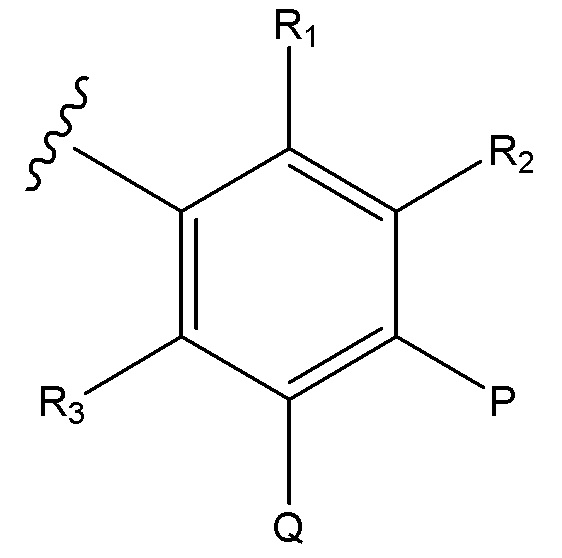
и где R1, R2, R3, P и Q являются, как определено в соответствии с формулой (I) или формулой (Ia) выше.
Соединения по формуле (I), где B является:
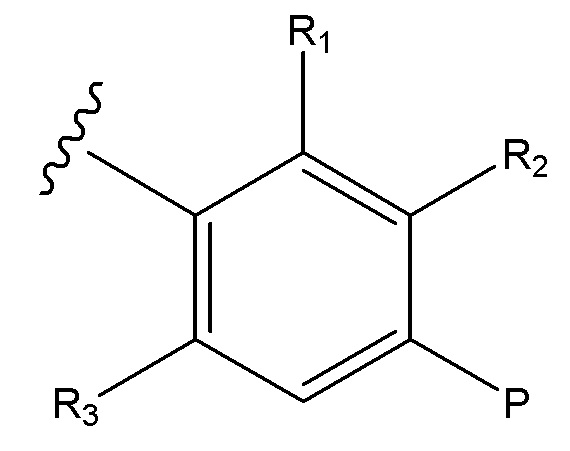
и где R1, R2, R3 и P являются, как определено в соответствии с формулой (I) или формулой (Ia) выше.
Предпочтительными являются соединения по формуле (I), где B является необязательно монозамещенным изохинолинилом где указанным необязательным заместителем является NR8R9; где R8 и R9 независимо выбирают из H и алкил.
Более предпочтительными являются соединения по формуле (I), где B является необязательно монозамещенным изохинолинилом, где указанным необязательным заместителем является NR8R9; и где R8 и R9 являются H.
Соединения по формуле (I), где U и V независимо выбирают из C и N, так что ароматический цикл, содержащий U и V, представляет собой фенил, пиридин или пиразин.
Соединения по формуле (I), где U и V независимо выбирают из C и N, так что ароматический цикл, содержащий U и V, представляет собой фенил или пиридин.
Предпочтительными являются соединения по формуле (I), где U и V являются C, так что ароматический цикл, содержащий U и V, представляет собой фенил.
Соединения по формуле (I), где R1 отсутствует, когда U является N, R2 отсутствует, когда V является N; и где, при наличии, R1 и R2 независимо выбирают из H, алкил, алкокси, CN, галоген и CF3.
Соединения по формуле (I) или формуле (V) где, при наличии, R1 и R2 независимо выбирают из H, метил, метокси, Cl, F и CF3.
Соединения по формуле (I) или формуле (V) где, при наличии, R1 и R2 независимо выбирают из H, метил и F.
Соединения по формуле (I) или формуле (V) где, при наличии, R1 выбирают из H и метил.
Соединения по формуле (I) или формуле (V) где, при наличии, R2 выбирают из H и F.
Соединения по формуле (I) или формуле (V) где, R3 выбирают из H, алкил, алкокси, CN, галоген и CF3;
Соединения по формуле (I) или формуле (V) где, R3 выбирают из H и алкил.
Предпочтительными являются соединения по формуле (I) или формуле (V), где R3 выбирают из H и метил.
Предпочтительными являются соединения по формуле (I) или формуле (V) где, P представляет собой -C(R18)(R19)NH2 и Q представляет собой H.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III) или формуле (IV), где R20, R21 и R22 независимо выбирают из H и алкил.
Предпочтительными являются соединения по формуле (II) или формуле (III), где R20, R21 и R22 являются H.
Предпочтительными являются соединения по формуле (IV), где R20 и R22 являются H.
Соединения по формуле (I), где R12 и R13 независимо выбирают из H и алкил, или могут вместе образовывать циклоалкил.
Предпочтительными являются соединения по формуле (I), где R12 и R13 независимо выбирают из H и метил.
Наиболее предпочтительными являются соединения по формуле (I), где R12 и R13 являются H.
Соединения по формуле (I) где, n составляет 0, 1 или 2.
Соединения по формуле (I) где, n составляет 1 или 2.
Предпочтительными являются соединения по формуле (I), где n составляет 1.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где W, X, Y и Z независимо выбирают из C=N, C(R16)-C, C(R16)=C, C, N, O и S, так что цикл, содержащий W, X, Y и Z, является шестичленным ароматическим гетероциклом.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где W, X, Y и Z независимо выбирают из C=N, C, C(R16)-C, C(R16)=C и N, так что цикл, содержащий W, X, Y и Z является шестичленным ароматическим гетероциклом; где R16 выбирают из H, алкил и OH.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V) где, W, X, Y и Z независимо выбирают из C, C(R16)-C, C(R16)=C и N, так что цикл, содержащий W, X, Y и Z представляет собой шестичленный ароматический гетероцикл; где R16 выбирают из H, алкил и OH.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где W является C или N.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где X выбирают из C, C(R16)-C, C(R16)=C или N.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где X выбирают из C(R16)-C или C(R16)=C и R16 является H; Y является N; и W и Z являются C.
Наиболее предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V) где, W, X, Y и Z образуют шестичленный ароматический гетероцикл, выбираемый из:
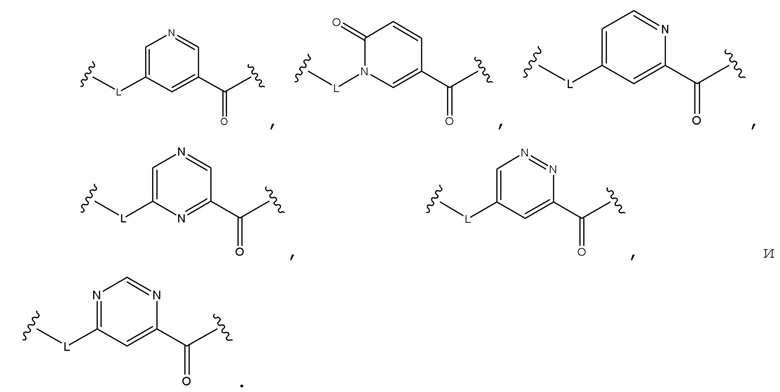
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R5, R6 и R7 независимо отсутствуют, или независимо выбирают из H, алкил, алкокси, галоген, OH, арил, гетероарил, -NR8R9, CN, COOR8, CONR8R9, -NR8COR9 и CF3.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R5, R6 и R7 независимо отсутствуют и их независимо выбирают из H, алкил, алкокси, галоген, OH, арил, гетероарил и CF3.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R5 выбирают из H, алкил и OH; и где R6 и R7 независимо отсутствуют или H.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III) или формуле (IV), где R5 выбирают из H, метил и OH; R6 отсутствует; и R7 является H.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R14 и R15 независимо выбирают из алкил, арилb и гетероарилb; или R14 и R15 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероцикл, который может быть насыщенным или ненасыщенным с 1 или 2 двойными связями, и необязательно может быть оксо замещенным.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R14 и R15 независимо выбирают из алкил и гетероарилb; или R14 и R15 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероцикл, который может быть насыщенным или ненасыщенным с 1 или 2 двойными связями, и необязательно может быть оксо замещенным.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R14 и R15 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероцикл, который может быть насыщенным или ненасыщенным с 1 или 2 двойными связями, и необязательно может быть оксо замещенным.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R16 независимо выбирают из H, алкил, алкокси, галоген, OH, NR8R9, арил, гетероарил и CF3.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R16 независимо выбирают из H и алкокси и OH.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R16 является H.
Соединения по формуле (I) или формуле (II), где L является линкером, выбираемым из ковалентной связи, -(CHR17)-, -(CH2)1-10-, -O-(CH2)2-10-, -(CH2)1-10-O-(CH2)1-10-, -(CH2)1-10-NH-(CH2)1-10-, -CONH-(CH2)1-10-, -CO-, и -SO2-.
Соединения по формуле (I) или формуле (II), где L представляет собой линкер, выбираемый из ковалентной связи, -(CH2)1-10-, -O-(CH2)2-10-, -(CH2)1-10-O-(CH2)1-10-, -(CH2)1-10-NH-(CH2)1-10-, -CONH-(CH2)1-10-, -CO-, и -SO2-.
Соединения по формуле (I) или формуле (II), где L представляет собой -(CH2)1-6- или -(CHR17)-.
Предпочтительными являются соединения по формуле (I) или формуле (II), где L представляет собой -(CH2)1-6- или -(CHOH)-.
Наиболее предпочтительными являются соединения по формуле (I) или формуле (II), где L представляет собой -CH2-.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A выбирают из арил, гетероарил, и группу заместитель выбирают из формулы (A), (B), (C), и (D):
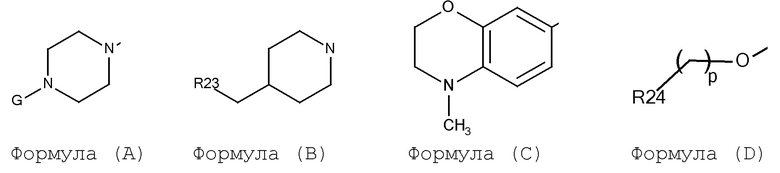
где:
G выбирают из H, алкил, циклоалкил, CO-арил, SO2-арил, (CH2)m-арил, и (CH2)m-гетероарил;
m выбирают из 0 и 1;
p выбирают из 0, 1, 2 и 3;
R23 выбирают из арил и гетероарил;
R24 выбирают из арил и гетероарил;
где алкил, циклоалкил, арил и гетероарил являются, как определено в соответствии с формулой (I) или формулой (Ia) выше.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A выбирают из арил, гетероарил, и заместитель выбирают из формулы (C) и (D).
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A выбирают из арил и гетероарил, каждый необязательно замещенный, как установлено выше в соответствии с формулой (I) или формулой (Ia) выше.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A представляет собой гетероарил, необязательно замещенный 1, 2 или 3 заместителями, независимо выбираемыми из алкил, алкокси, OH, галоген, CN, арил, морфолинил, пиперидинил, -COOR10, -CONR10R11, CF3 и -NR10R11; где R10 и R11 выбирают из H и алкил, или R10 и R11 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероцикл, который может быть насыщенным или ненасыщенным с 1 или 2 двойными связями; и где алкил, алкокси и арил являются, как определено в соответствии с формулой (I) или формулой (Ia) выше.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A представляет собой гетероарил, необязательно замещенный заместителем, выбираемым из алкил, алкокси, OH, галоген, CN, арил, морфолинил и пиперидинил; и где алкил, алкокси и арил являются, как определено в соответствии с формулой (I) или формулой (Ia) выше.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A представляет собой гетероарил, замещенный фенилом.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A является тиазолилом, замещенным фенилом.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A представляет собой фенил, замещенный гетероарилом, -(CH2)1-3-гетероарилом и -(CH2)1-3-NR14R15; и где гетероарил, R14 и R15 определяют в соответствии с формулой (I) или формулой (Ia) выше.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где A выбирают из:
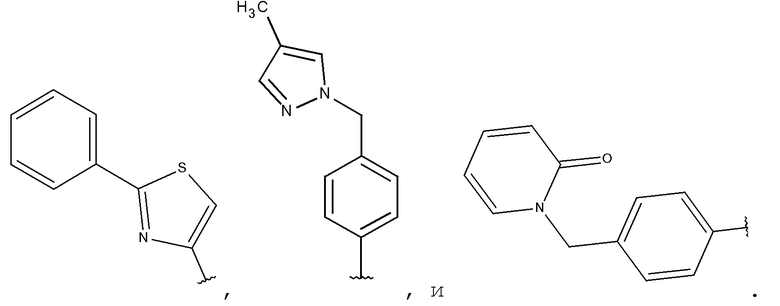
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где p выбирают из 0, 1, 2 и 3.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где p составляет 2.
Соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R23 и R24 независимо выбирают из арил и гетероарил; где арил и гетероарил являются как определено в соответствии с формулой (I) или формулой (Ia) выше.
Предпочтительными являются соединения по формуле (I), формуле (II), формуле (III), формуле (IV) или формуле (V), где R23 и R24 представляют собой гетероарил; где гетероарил является как определено в соответствии с формулой (I) или формулой (Ia) выше.
В одном аспекте изобретение включает соединение, выбираемое из:
и их фармацевтически приемлемые соли и сольваты.
Терапевтическое применение
Как упомянуто ранее, соединения по настоящему изобретению являются сильными и селективными ингибиторами калликреина плазмы. Следовательно, они являются применимыми в лечении патологических состояний, для которых причиной является повышенная активность калликреина плазмы.
Соответственно, настоящее изобретение обеспечивает соединение по формуле (I) для применения в медицине.
Настоящее изобретение также обеспечивает применение соединения по формуле (I) в получении лекарственного препарата для лечения или профилактики заболевания или состояния, в которое вовлечена активность калликреина плазмы.
Настоящее изобретение также обеспечивает соединение по формуле (I) для применения в лечении или профилактике заболевания или состояния, в которое вовлечена активность калликреина плазмы.
Настоящее изобретение также обеспечивает способ лечения заболевания или состояния, в которое вовлечена активность калликреина плазмы, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по формуле (I).
В одном аспекте заболевания или состояния в которые вовлечена активность калликреина плазмы, включают нарушение остроты зрения, диабетическую ретинопатию, диабетический макулярный отек, врожденный ангиоотек, диабет, панкреатит, кровоизлияние в головной мозг, нефропатию, кардиомиопатию, нейропатию, воспалительные заболевания кишечника, артрит, воспаление, септический шок, гипотензию, рак, респираторный дистресс синдром взрослых, диссеминированное сосудистое свертывание, операцию сердечно-легочного шунтирования и кровотечение после оперативных вмешательств.
В другом аспекте заболевание или состояние, в которое вовлечена активность калликреина плазмы, представляет собой проницаемость сосудов сетчатки, ассоциированную с диабетической ретинопатией и диабетическим макулярным отеком.
Комбинированная терапия
Соединения по настоящему изобретению можно вводить в комбинации с другими терапевтическими средствами. Подходящая комбинированная терапия включает соединение по формуле (I) в комбинации с одним или более средствами, выбираемыми из средств, которые ингибируют тромбоцитарный фактор роста (PDGF), эндотелиальный фактор роста (VEGF), интегрин альфа5бета1, стероиды, другие средства, которые ингибируют калликреин плазмы, и другие ингибиторы воспаления. Специфически примеры терапевтических средств, которые могут быть скомбинированы с соединениями по настоящему изобретению, включают описанные в EP2281885A и S. Patel in Retina, 2009 Jun;29(6 Suppl):S45-8.
Когда используется комбинированная терапия соединения по настоящему изобретению, и указанные комбинированные агенты могут существовать в одной или разных фармацевтических композициях, их можно вводить отдельно, последовательно или одновременно.
В другом аспекте соединения по настоящему изобретению можно вводить в комбинации с лазерным лечением сетчатки. Известна комбинация лазерной терапии с интравитреальной инъекцией ингибитора VEGF для лечения диабетического макулярного отека (Elman M, Aiello L, Beck R, et al. “Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema”. Ophthalmology. 27 April 2010).
Определения
Термин "алкил" включает насыщенный углеводородный остаток, включающий:
- линейные группы до 10 атомов углерода (C1-C10), или до 6 атомов углерода (C1-C6), или до 4 атомов углерода (C1-C4). Примеры таких алкильных групп включают, без ограничения C1-метил, C2-этил, C3-пропил и C4-н-бутил.
- разветвленные группы от 3 до 10 атомов углерода (C3-C10), или до 7 атомов углерода (C3-C7), или до 4 атомов углерода (C3-C4). Примеры таких алкильных групп включают, без ограничения, C3-изо-пропил, C4-втор-бутил, C4-изо-бутил, C4-трет-бутил и C5-нео-пентил,
каждый необязательно замещенный, как указано выше.
Циклоалкил представляет собой моноциклический насыщенный углеводород от 3 до 7 атомов углерода; где циклоалкил может быть необязательно замещенным заместителем, выбираемым из алкил, алкокси и NR10R11; где R10 и R11 независимо выбирают из H и алкил, или R10 и R11 вместе с азотом, к которому они прикреплены, образуют 4-, 5-, 6- или 7-членный гетероцикл, который может быть насыщенным или ненасыщенным с 1 или 2 двойными связями. Циклоалкильные группы могут содержать от 3 до 7 атомов углерода, или от 3 до 6 атомов углерода, или от 3 до 5 атомов углерода, или от 3 до 4 атомов углерода. Примеры подходящих моноциклических циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин "алкокси" включает O-сшитые углеводородные остатки, включающие:
- линейные группы от 1 до 6 атомов углерода (C1-C6), или от 1 до 4 атомов углерода (C1-C4). Примеры таких алкоксигрупп включают, без ограничения, C1-метокси, C2-этокси, C3-н-пропокси и C4-н-бутокси.
- разветвленные группы от 3 до 6 атомов углерода (C3-C6) или от 3 до 4 атомов углерода (C3-C4). Примеры таких алкокси групп включают, без ограничения, C3-изо-пропокси, и C4-втор-бутокси и трет-бутокси,
каждый необязательно замещенный, как указано выше.
Если не указано иначе, галоген выбирают из Cl, F, Br и I.
Арил определен выше. Обычно арил необязательно замещен 1, 2 или 3 заместителями. Необязательные заместители выбирают из таковых, указанных выше. Примеры подходящих арильных групп включают фенил и нафтил (каждый необязательно замещенный как указано выше). Предпочтительно арил выбирают из фенила, замещенного фенила (замещенного как указано выше), и нафтила.
Гетероарил является, как указано выше. Примеры подходящих гетероарильных групп включают тиенил, фуранил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиридазинил, пиримидинил, пиразинил, индолил, бензимидазолил, бензотриазолил, хинолинил и изохинолинил (необязательно замещенный, как указано выше). Предпочтительно гетероарил выбирают из пиридила, бензотиазола, индола, N-метилиндола, тиазола, замещенного тиазола, тиофенил, фурила, пиразина, пиразола и замещенного пиразола; где заместители представлены выше.
Термин "N-связанный", например, как в "N-связанный гетероциклоалкил", обозначает, что гетероциклоалкильная группа связана с остатком молекулы посредством атома азота цикла.
Термин "O-связанный", например, как в "O-связанный углеводородный остаток", обозначает, что углеводородный остаток прикреплен к остатку молекулы посредством атома кислорода.
В группах, таких как-COOR*, "-" обозначает точку прикрепления группы заместителя к остатку молекулы.
"Фармацевтически приемлемая соль" обозначает физиологически или токсикологически переносимую соль и включает, когда необходимо, фармацевтически приемлемые аддитивные соли оснований и фармацевтически приемлемые аддитивные соли кислот. Например, (i) когда соединение по изобретению содержит одну или более кислотных групп, например, карбоксигруппы, фармацевтически приемлемые аддитивные соли оснований, которые могут быть получены, включают натрий, калий, кальций, магния и аммония соли, или соли с органическими аминами, такие как диэтиламин, N-метилглюкамин, диэтаноламин или аминокислоты (например, лизин) и подобные; (ii) когда соединение по изобретению содержит основную группу, такую как аминогруппа, фармацевтически приемлемые аддитивные соли кислот, которые могут быть получены, включают гидрохлориды, гидробромиды, сульфаты, фосфаты, ацетаты, цитраты, лактаты, тартраты, мезилаты, сукцинаты, оксалаты, фосфаты, эзилаты, тозилаты, бензолсульфонаты, нафталандисульфонаты, малеаты, адипаты, фумараты, гиппураты, камфораты, ксинафоаты, п-ацетамидобензоаты, дигидроксибензоаты, гидроксинафтоаты, сукцинаты, аскорбаты, олеаты, бисульфаты и подобные.
Также могут быть получены гемисоли кислот и оснований, например, гемисульфат и гемикальциевые соли.
Для обзора подходящих солей см. "Handbook of Pharmaceutical Salts: Properties, Selection and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
“Пролекарство” относится к соединению, которое преобразуется in vivo посредством метаболизма (например, посредством гидролиза, восстановления или окисления) в соединение по изобретению. Подходящие группы для получения пролекарств описаны в ‘The Practice of Medicinal Chemistry, 2nd Ed. pp561-585 (2003) и в F. J. Leinweber, Drug Metab. Res., 1987, 18, 379.
Соединения по изобретению могут существовать и в сольватированных и в несольватированных формах. Термин 'сольват' используют в настоящем описании для описания молекулярного комплекса, включающего соединение по изобретению и стехиометрическое количество молекул одного или более фармацевтически приемлемых растворителей, например, этанола. Термин 'гидрат' используют, когда растворителем является вода.
Когда соединения по изобретению существуют в одной или более геометрических, оптических, энантиомерных, диастереомерных и таутомерных формах, включая, без ограничения цис- и транс-формы, E- и Z-формы, R-, S- и мезо-формы, кето-, и енольные формы. Если не указано иначе, ссылка на определенное соединение включает все такие изомерные формы, включая рацемические и другие их смеси. Когда необходимо, такие изомеры могут быть отделены от их смесей путем применения или адаптации известных методов (например, хроматографических методик и методик рекристаллизации). Когда необходимо, такие изомеры могут быть получены путем применения или адаптации известных методов (например, асимметричного синтеза).
В контексте настоящего изобретения ссылки в настоящем описании на "лечение" включают ссылки на терапевтическое, паллиативное и профилактическое лечение.
Общие методы
Соединения по формуле (I) необходимо оценивать в отношении их биофармацевтических свойств, таких как растворимость и стабильность раствора (по pH), проницаемость и др., с целью выбора наиболее соответствующей лекарственной формы и пути введения для лечения определенного показания. Их можно вводить отдельно или в комбинации с одним или более другими соединениями по изобретению или в комбинации с одним или более другими лекарственными средствами (или в виде любой их комбинации). Обычно их вводят в виде композиции в сочетании с одним или более фармацевтически приемлемыми вспомогательными веществами. Термин ’вспомогательное вещество’ используют в настоящем описании с целью обозначения любого ингредиента, иного, чем соединение (я) по изобретению, которое может влиять или на функциональные (т.е., регуляция скорости высвобождения лекарственного средства) и/или нефункциональные (т.е. вспомогательные добавки или разбавитель) характеристики композиций. Выбор вспомогательного вещества в большой степени зависит от факторов, таких как определенный путь введения, действие вспомогательного вещества на растворимость и стабильность, и природа лекарственной формы.
Соединения по изобретению, предназначенные для фармацевтического применения, можно вводить в виде твердого вещества или жидкости, например, таблетки, капсулы или раствора. Фармацевтические композиции, подходящие для доставки соединений по настоящему изобретению и методов для их получения, очевидны специалисту в области техники. Такие композиции и методы для их получения могут быть обнаружены, например, в in Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
Соответственно, настоящее изобретение обеспечивает фармацевтическую композицию, включающую соединение по формуле (I) и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
Для лечения состояний, таких как повышенная проницаемость сосудов сетчатки, ассоциированная с диабетической ретинопатией и диабетическим макулярным отеком, соединения по изобретению можно вводить в форме, подходящей для инъекции в область глаз пациента, в частности, в форме, подходящей для интра-витреальной инъекции. Считают, что композиции, подходящие для такого применения, имеют форму стерильных растворов соединения по изобретению в подходящем водном носителе. Композиции можно вводить пациенту под наблюдением лечащего врача.
Соединения по изобретения можно вводить непосредственно в кровоток, в подкожную ткань, в мышцы или во внутренние органы. Подходящие средства для парентерального введения включают внутривенное, интраартериальное, интраперитонеальное, интратекальное, интравентрикулярное, интрауретральное, интрастернальное, интракраниальное, внутримышечное, интрасиновиальное и подкожное. Подходящие устройства для парентерального введения включают иглу (включая микроиглу) инжекторы, безигольные инжекторы и инфузионные методики.
Парентеральные композиции обычно являются водными или масляными растворами. Когда раствор является водным, вспомогательные вещества, такие как сахара (включая, но, не ограничиваясь, глюкозу, маннит, сорбит и др.), соли, углеводы и буферы (предпочтительно до pH от 3 до 9), но, для некоторых применений, они могут быть более соответственно подходящими для рецептирования в виде стерильного неводного раствора или в виде сухой формы для применения в сочетании с подходящим носителем, такими как стерильная, апирогенная вода.
Парентеральные композиции могут включать импланты, полученные из биоразлагаемых полимеров, таких как полиэфиры (т.е. полимолочная кислота, полилактид, полилактид-ко-гликолид, поликапро-лактон, полигидроксибутират), полиортоэфиры и полиангидридаы. Такие композиции можно вводить посредством хирургических разрезов в подкожную ткань, мышечную ткань или непосредственно в специфические органы.
Получение парентеральных композиций в стерильных условиях, например, путем лиофилизации, может быть легко осуществлено с использованием стандартных фармацевтических методик, хорошо известных специалисту в области техники.
Растворимость соединений по формуле (I), используемых в получении парентеральных растворов, может быть увеличена с использованием соответствующих методик рецептирования, таких как включение со-растворителей и/или средств, увеличивающих растворимость, таких как поверхностно-активные вещества, мицеллярные структуры и циклодекстрины.
В одном варианте осуществления изобретения соединения по изобретению можно вводить перорально. Пероральное введение может включать глотание, так что соединение попадает в желудочно-кишечный тракт и/или буккальное, лингвальное или сублингвальное введение, посредством которого соединение попадает в кровоток непосредственно из полости рта.
Композиции, подходящие для перорального введения, включают твердые пробки, твердые микрочастицы, полутвердые и жидкие (включая многофазовые или диспергированные системы), такие как таблетки; мягкие или твердые капсулы, содержащие мульти- или наночастицы, жидкости, эмульсии или порошки; пастилки (включая заполненные жидкостью); жевательные резинки; гели; быстроразлагаемые лекарственные формы; пленки; суппозитории; спреи; и буккальные/мукоадгезивные пластыри.
Композиции, подходящие для перорального введения, также могут быть созданы для доставки соединений по изобретению путем немедленной доставки или с замедленной скоростью, где профиль высвобождения может быть отложенным, пульсовым, контролируемым, длительным или модифицированным таким образом, чтобы оптимизировать терапевтическую эффективность указанных соединений. Средства для доставки соединений с замедленной скоростью известны в области техники и включают полимеры с замедленным высвобождением, которые могут быть рецептированы с указанными соединениями для регуляции их высвобождения.
Примеры полимеров, замедляющих скорость, включают разлагаемые и неразлагаемые полимеры, которые могут быть использованы для высвобождения указанных соединений путем диффузии или комбинации диффузии и эрозии полимера. Примеры полимеров, замедляющих скорость, включают гидроксипропил метилцеллюлозу, гидроксипропилцеллюлозу, метилцеллюлозу, этилцеллюлозу, карбоксиметилцеллюлозу натрия, поливиниловый спирт, поливинилпирролидон, ксантановую камедь, полиметакрилаты, полиэтиленоксид и полиэтиленгликоль.
Жидкостные (включая многофазовые и диспергированные системы) композиции включают эмульсии, растворы, сиропы и эликсиры. Такие композиции могут быть представлены в виде наполнителей в мягких или твердых капсулах (состоящих, например, из желатина или гидроксипропилметилцеллюлозы), и обычно включают носитель, например, воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу и подходящее масло и один или более эмульгирующих агентов и/или суспендирующих агентов. Жидкие композиции также могут быть получены путем восстановления твердого вещества, например, из саше.
Соединения по изобретению также могут быть использованы в быстрорастворяющихся, быстроразрушающихся лекарственных формах, таких как описанные в Liang and Chen, Expert Opinion in Therapeutic Patents, 2001, 11 (6), 981-986.
Композиции таблеток обсуждаются в Pharmaceutical Dosage Forms: Tablets, Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
Для введения пациентам людям общая суточная доза соединений по изобретению обычно находится в диапазоне от 0,01 мг до 1000 мг, или от 0,1 мг до 250 мг, или от 1 мг и 50 мг в зависимости, конечно, от пути введения.
Общую дозировку можно вводить в одной или нескольких дозах и может, по решению врача, находиться в обычном диапазоне, представленном в настоящем описании. Такие дозировки предназначены для среднего пациента человека, имеющего массу около 60 кг - 70 кг. Врач легко сможет определить дозы для пациентов, чья масса не попадает в указанный диапазон, таких как младенцы и пожилые.
Методы синтеза
Соединения по настоящему изобретению могут быть получены в соответствии с методиками, проиллюстрированными специфическими примерами, представленными ниже. Более того путем иллюстрации методик, описанных в настоящем описании, обычный специалист в области техники может легко получить дополнительные соединения, которые попадают в рамки настоящего изобретения, заявленного в настоящем описании. Соединения, проиллюстрированные в примерах, однако не должны рассматриваться как единственные, которые расцениваются как изобретение. Примеры дополнительно иллюстрируют подробности получения соединений по настоящему изобретению. Специалист в области техники понимает, что известные варианты состояний и процессов следующих методик получения могут быть использованы для получения указанных соединений.
Соединения по изобретению могут быть выделены в форме их фармацевтически приемлемых солей, таких как таковые, описанные ранее выше.
Может быть необходимо защищать реакционноспособные функциональные группы (например, гидрокси, амино, тио или карбокси) в промежуточных соединениях, используемых в получении соединений по изобретению во избежание их нежелательного участия в реакции, приводящего к образованию соединений. Могут быть использованы обычные защитные группы, например, таковые, описанные в T. W. Greene and P. G. M. Wuts in “Protective groups in organic chemistry” John Wiley and Sons, 4th Edition, 2006. Например, обычной амино-защитной группой, подходящей для применения в настоящем описании, является трет-бутокси карбонил (Boc), которая легко удаляется путем обработки кислотой, такой как трифторуксусная кислота или соляная кислота, в органическом растворителе, таком как дихлорметан. Альтернативно амино-защитной группой может быть группа бензилоксикарбонил (Z), которая может быть удалена путем гидрирования с катализатором палладием в атмосфере водорода, или группа 9-фторенилметилоксикарбонил (Fmoc) которая может быть удалена растворами вторичных органических аминов, таких как диэтиламин или пиперидин в органических растворителях. Карбоксильные группы являются обычно защищенными в виде сложных эфиров, таких как метил, этил, бензил или трет-бутил, которые все могут быть удалены путем гидролиза в присутствии оснований, таких как гидроксид лития или натрия. Бензил-защитные группы также могут быть удалены путем гидрирования с катализатором палладием в атмосфере водорода, тогда как трет-бутильные группы могут также быть удалены посредством трифторуксусной кислоты. Альтернативно, трихлорэтилэфирную защитную группу удаляют с цинком в уксусной кислоте. Обычная гидрокси-защитная группа, подходящая для применения в настоящем описании, представляет собой метиловый эфир, условия депротектирования включают нагревание в колбе с обратным холодильником в 48% водном HBr в течение 1-24 часов, или путем перемешивания с трибромидом борана в дихлорметане в течение 1-24 часов. Альтернативно, когда гидроксигруппу защищают как бензиловый эфир, условия депротектирования включают гидрирование с катализатором палладием в атмосфере водорода.
Соединения по общей формуле (I) могут быть получены с использованием обычных методик синтеза. Например, амин может быть связан с использованием стандартных условий пептидного связывания с активированной альфа карбоновой кислотой. При наличии, дополнительная амин-функциональная группа может быть соответственно амино-защищена стандартной защитной группой, такой как трет-бутилоксикарбонил (Boc), бензилоксикарбонил (Z) или 9-фторенилметилоксикарбонил (Fmoc). Активирующей группой может быть N-гидроксисукцинимид. Применение такой группы хорошо известно в области техники. Другие стандартные методы пептидного связывания включают реакцию кислот с аминами в присутствии гидроксибензотриазола и карбодиимида, такого как водорастворимый карбодиимид, или гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламина или гексафторфосфат бензотриазол-1-ил-окси-трис-пирролидинoфосфония или гексафторфосфат бром-трипиролидинофосфония или гексафторфосфат 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония (V) (HATU) в присутствии органических оснований, таких как триэтиламин, диизопропилэтиламин или N-метилморфолин. На типичной второй стадии защитную группу, если есть, удаляют с использованием стандартных методов, как описано ранее.
Амин может типично быть алкилированным или ацилированным. Ацилирование может проводиться посредством обработки ацилирующим агентом, таким как ацилхлорид, например, ацетилхлорид или бензоилхлорид, в присутствии основания, обычно основания третичного амина, такого как триэтиламин или диизопропилэтиламин. Алкилирование может обычно проводиться посредством обработки алкилгалоидом или восстановительным алкилированием. Обычно в методике восстановительного алкилирования амину позволяют реагировать с альдегидом или кетоном в присутствии подходящего восстанавливающего агента, такого как цианоборогидрид натрия или ацетоксиборогидрид натрия в подходящем растворителе, таком как метанол, при комнатной температуре.
Нитрильное соединение обычно может быть восстановлено посредством гидрирования. Преобразование может быть достигнуто в одной стадии или путем прямого восстановления нитрила посредством гидрирования в подходящем растворителе, таком как метанол, в присутствии подходящего катализатора, такого как палладий на активированном угле, в присутствии кислоты, такой как соляная кислота, или восстановления с помощью подходящего борогидрида в присутствии подходящего переходного металла, такого как хлорид кобальта или никеля, в подходящем растворителе, таком как метанол, при комнатной температуре. Альтернативно, амин, защищенный трет-бутоксикарбонилом (Boc), может быть выделен (с использованием, например, метода, как описано в S. Caddick et al., Tetrahedron Lett., 2000, 41, 3513) и впоследствии депротектирован стандартными средствами, описанными ранее, для получения амина.
Примеры
Изобретение проиллюстрировано следующими неограничивающими примерами, в которых используют следующие сокращения и определения:
Все реакции проводили в атмосфере азота, если не указано иначе.
1H ЯМР спектры записывали на спектрометре Bruker (400 МГц) с контролем растворителем дейтерием и при кт.
Молекулярные ионы получали с использованием ЖХМС, которую проводили с использованием колонки Chromolith Speedrod RP-18e, 50×4,6 мм, с линейным градиентом 10%-90% 0,1% HCO2H/MeCN в 0,1% HCO2H/H2O в течение 13 мин, скоростью тока 1,5 мл/мин, или с использованием Agilent, X-Select, кислого, 5-95% MeCN/вода в течение 4 мин. Данные собирали с использованием масс спектрометра Thermofinnigan Surveyor MSQ с ионизацией электронным лучом в сочетании с ЖХ системой Thermofinnigan Surveyor.
Химические наименования создавали с использованием программного обеспечения Autonom, представленного как часть ISIS исходного пакета из систем MDL Information Systems, или в форме IUPAC с использованием программного обеспечения Chemaxon.
Когда продукты очищают хроматографией с испарительной колонкой, ‘диоксид кремния’ относится к силикагелю для хроматографии, 0,035-0,070 мм (сито 220-440) (например, Merck silica gel 60), и применяемому давлению азота до 10 фунтов на кв. дюйм ускоренной колонки элюции. Очистку препаративной ВЭЖХ с обращенной фазой проводили с использованием насосной системы с бинарным градиентом Waters 2525 со скоростью тока обычно 20 мл/мин с использованием фотодиодного лучевого детектора Waters 2996.
Все растворители и коммерческие реагенты использовали, как получали.
Способы I-V ниже описывают синтез промежуточных продуктов, применяемых в получении примеров.
I. Гидрохлорид 6-Аминометилизохинолин-1-иламина
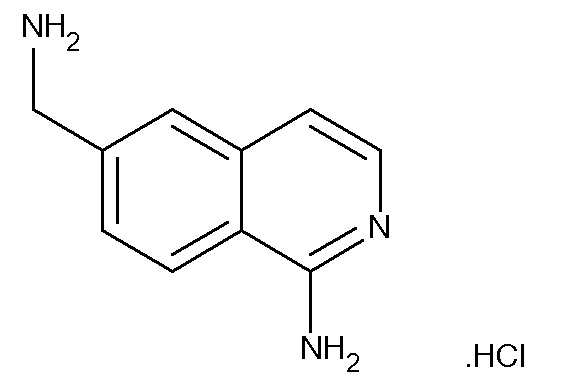
A. 2-((E)-2-Диметиламиновинил)терефтогалогеннитриловый эфир
Метилтерефтгалогеннитрил (1,42 г, 9,99 ммоль) и реагент Бредерика (3,48 г, 19,98 ммоль) растворяли в DMF (15 мл). Реакционную смесь нагревали при 75°C в атмосфере азота в течение 72 ч, после указанного времени растворитель удаляли в вакууме. Растирание с петролейным эфиром давало ярко-желтое твердое вещество, идентифицированное как 2-((E)-2-диметиламиновинил)терефтгалогеннитриловый эфир (1,88 г, 0,95 ммоль, 95%).
1H ЯМР (CD3OD) δ: 3,20 (6H, с), 5,34 (1H, d, J=13,4 Гц), 7,21 (1H, dd, J=8,0 Гц, 1,4 Гц), 7,9 (1H, d, 13,4 Гц), 7,61 (1H, d, J=8,0 Гц), 7,94 (1H, d, J =1,2 Гц).
B. 1-Амино-2-(2,4-диметоксибензил)-1,2-дигидроизохинолин-6-карбонитрил
2-((E)-2-Диметиламиновинил)терефтгалогеннитриловый эфир (1,85 г, 9,38 ммоль) растворяли в 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидиноне (5 мл) и добавляли 2,4-диметоксибензиламин (2,35 г, 14,07 ммоль). Реакционную смесь нагревали до 75°C в атмосфере азота. Через 3 ч реакционную смесь охлаждали и добавляли диэтиловый эфир/петр. эфир (15:85). Желтое твердое вещество фильтровали, сушили в вакууме и идентифицировали как 1-амино-2-(2,4-диметоксибензил)-1,2-дигидроизохинолин-6-карбонитрил (2,65 г, 8,38 ммоль, 89%).
[M+H]+=320,0.
1H ЯМР (CD3OD) δ: 3,85 (3H, s), 3,92 (3H, s), 5,02 (2H, s), 6,39 (1H, d, J=7,4 Гц), 6,57 (1H, dd, J=8,4 Гц, 2,4 Гц), 6,66 (1H, d, 2,4 Гц), 7,18 (1H, d, 8,4 Гц), 7,24(1H, d, 7,4 Гц), 7,72 (1H, dd, J=8,5 Гц, 1,4 Гц), 7,93 (1H, s), 8,45 (1H, d, J=8,5 Гц).
C. 1-Аминоизохинолин-6-карбонитрил
1-Амино-2-(2,4-диметоксибензил)-1,2-дигидроизохинолин-6-карбонитрил (1,6 г, 5,0 ммоль) растворяли в анизоле (17 мл) и трифтоуксусной кислоте (20 мл). Реакционную смесь нагревали при 105°C в атмосфере азота в течение 12 ч, после чего реакционную смесь охлаждали, добавляли диэтиловый эфир/петр. эфир (3:7), полученное твердое вещество фильтровали в вакууме и идентифицировали как 1-аминоизохинолин-6-карбонитрил (770 мг, 4,54 ммоль, 91%).
[M+H]+=170,0
1H ЯМР (CD3OD) δ: 7,23-7,25 (1H, d, J=6,9 Гц), 7,65 (1H, d, J=6,8 Гц), 8,11 (1H, dd, J=8,7 Гц, 1,6 Гц), 8,33 (1H, s), 8,45 (1H, d, J=8,7 Гц).
D. Трет-бутиловый эфир (1-Аминоизохинолин-6-илметил)карбамовой кислоты
1-Аминоизохинолин-6-карбонитрил (200 мг, 1,18 ммоль) растворяли в метаноле (20 мл). Полученный раствор охлаждали до 0°C. Добавляли гексагидрат хлорида никеля (II) (28 мг, 0,12 ммоль) и ди-третбутил дикарбонат (516 г, 2,36 ммоль) с последующим борогидридом натрия (313 г, 8,22 ммоль) по частям. Реакционную смесь перемешивали при 0°C до комнатной температуры в течение 3 дней. MeOH удаляли выпариванием. Остаток растворяли в CHCl3 (70 мл), промывали насыщ. NaHCO3 (1×30 мл), водой (1×30 мл), солевым раствором (1×30 мл), сушили (Na2SO4) и выпаривали в вакууме для получения желтого масла, идентифицированного как (1-амино-изохинолин-6-илметил)карбамовой кислоты трет-бутиловый эфир (110 мг, 0,4 ммоль, 34%).
[M+H]+=274,1.
E. Гидрохлорид 6-Аминометилизохинолин-1-иламина
Трет-бутиловый эфир (1-Аминоизохинолин-6-илметил)карбамовой кислоты (110 мг, 0,40 ммоль) растворяли в 4M HCl в диоксане (40 мл). Через 18 ч при комнатной температуре растворитель удаляли в вакууме для получения бледно коричневого твердого вещества, идентифицированного как гидрохлорид 6-аминометилизохинолин-1-иламина (67 мг, 0,39 ммоль, 96%).
[M+H]+=174,3.
II. C-(4,6-Диметил-1H-пирроло[2,3-b]пиридин-5-ил)метиламина гидрохлорид
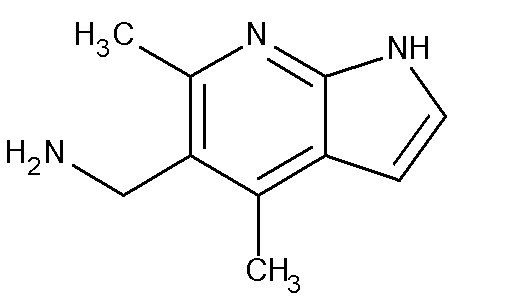
A. 1-трет-Бутил-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрил
Смесь 5-амино-1-трет-бутил-1H-пиррол-3-карбонитрила (2,6 г, 15,93 ммоль) и пентан-2,4-диона (1,595 г, 15,93 ммоль,) растворяли в этаноле (80 мл) и добавляли концентрированную HCl (0,2 мл). Реакционную смесь нагревали в колбе с обратным холодильником в течение 18 ч. Смесь концентрировали в вакууме и сырой продукт очищали колоночной хроматографией, элюируя с шагами градиента 95:5 до 9:1 петр. эфир/этилацетат для получения желтого масла, идентифицированного как 1-трет-бутил-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрил (3,05 г, 13 ммоль, 84% выход).
[M+H]+=228,4.
1H ЯМР: (CDCl3), δ: 1,81 (9H, s), 2,58 (3H, s), 2,70 (3H, s), 6,84 (1H, s), 7,75 (1H, s).
B. 5-бром-1-трет-бутил-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрил
Раствор 1-трет-бутил-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрила (2,820 г, 12,4 ммоль) в дихлорметане (50 мл) в атмосфере N2 охлаждали до, по меньшей мере, -5°C (лед/NaCl, 3:1). Затем добавляли 1,3-дибром-5,5-диметилгидантоин (1,774 г, 6,203 ммоль) и реакционную смесь перемешивали при -5°C или ниже. После перемешивания при -5°C добавляли дополнительный 1,3-дибром-5,5-диметилгидантоин (88 мг, 0,31 ммоль) и перемешивание продолжали при -5°C в течение дополнительных 3 ч. Реакционную смесь гасили Na2SO3 (водн) до нагревания реакционной смеси до кт. Добавляли 1M NaOH и слои разделяли. Водную фазу экстрагировали дихлорметаном (2×10 мл), комбинированные органические экстракты промывали солевым раствором (2×10 мл) и концентрировали в вакууме. Сырой продукт очищали хроматографией с испарительной колонкой на силикагеле, элюируя петр. эфир/этилацетатом 95:5. Фракции, содержащие продукт, концентрировали и остаток рекристаллизовывали из этилацетата/петр. эфира для получения белого твердого вещества, идентифицированного как 5-бром-1-трет-бутил-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрил (3,19 г, 10,42 ммоль, 84% выход).
[M+H]+=305,7.
1H ЯМР: (CDCl3), δ: 1,81 (9H, s), 2,78 (3H, s), 2,82 (3H, s), 7,78 (1H, s).
C. 5-Бром-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрил
5-Бром-1-(трет-бутил)-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрил (2,1 г, 6,87 ммоль) добавляли по частям к перемешиваемой суспензии трихлорида алюминия (2,75 г, 20,6 ммоль) в хлорбензоле (160 мл). После добавления смесь нагревали до 100°C в течение ночи, получая черный липкий раствор. Через 24 ч. реакционной смеси позволяли остыть, затем вливали в воду (300 мл) и дихлорметан (300 мл). Смесь аккуратно обрабатывали конц. HCl (135 мл) и смесь перемешивали в течение 10 мин, затем фильтровали, промывая водой и дихлорметаном. Полученное твердое вещество сушили в вакууме в присутствии CaCl2 в течение выходных для получения бледно серого твердого вещества, идентифицированного как 5-бром-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрил (1,56 мг, 6,16 ммоль, 90% выход).
D. 5-Бром-4,6-диметил-1H-пирроло[2,3-b]пиридин
Суспензию 5-бром-4,6-диметил-1H-пирроло[2,3-b]пиридин-3-карбонитрила (1,56 г, 6,16 ммоль) в конц. соляной кислоте, 37% (235 мл) нагревали в колбе с обратным холодильником в течение ночи. Дополнительно добавляли конц. HCl (100 мл) и реакционную смесь нагревали в колбе с обратным холодильником в течение дополнительных 20 ч. Смесь охлаждали и вливали в ледяную воду (1 л) и нейтрализовали 2Н NaOH до pH 9, образуя осадок. Его фильтровали, промывали водой, затем сушили в вакууме в присутствии CaCl2 для получения серого твердого вещества, идентифицированного как 5-бром-4,6-диметил-1H-пирроло[2,3-b]пиридин (1,3 г, 5,72 ммоль, 92% выход).
[M+H]+=225,1.
1H ЯМР: (CDCl3), δ: 2,66 (3H, s), 2,82 (3H, s), 6,49 (1H, dd, J=3,5, 2,1 Гц), 7,29 (1H, dd, J=3,4, 2,7 Гц), 11,14 (1H, br.s).
E. 4,6-Диметил-1H-пирроло[2,3-b]пиридин-5-карбонитрил
5-Бром-4,6-диметил-1H-пирроло[2,3-b]пиридин (1,3 г, 5,72 ммоль) растворяли в N,N-диметилацетамиде (20 мл). Раствор дегазировали N2 до добавления цинкового порошка (45 мг, 0,693 ммоль), ацетата цинка (127 мг, 0,693 ммоль), 1,1'-бис(дифенилфосфино)ферроцена (128 мг, 0,23 ммоль), Zn(CN)2 (339 мг, 2,888 ммоль) и трис(дибензилиденацетон)дипалладия(0) (106 мг, 0,116 ммоль). Реакционную смесь нагревали при 120°C в течение 48 ч. После охлаждения до кт реакционную смесь разводили этилацетатом и промывали 2M NH4OH и солевым раствором. Органический слой сушили над MgSO4 и фильтровали. После концентрирования в вакууме сырой продукт очищали хроматографией с испарительной колонкой на силикагеле, элюируя 9:1, 8:2, 7:3, 1:1. (петр. эфир/этилацетат). Фракции собирали и концентрировали в вакууме. Желтое твердое вещество растирали в диэтиловом эфире для получения не совсем белого твердого вещества, идентифицированного как 4,6-диметил-1H-пирроло[2,3-b]пиридин-5-карбонитрил (660 мг, 3,83 ммоль, 67% выход).
[M+H]+=172,1.
1H ЯМР: (CDCl3), δ: 2,76 (3H, s), 2,86 (3H, s), 6,59 (1H, dd, J=3,5, 2,0 Гц), 7,36 (1H, dd, J=3,5, 2,4 Гц), 10,86 (1H, br.s).
F. Трет-бутиловый эфир (4,6-Диметил-1H-пирроло[2,3-b]пиридин-5-илметил)карбамовой кислоты
4,6-Диметил-1H-пирроло[2,3-b]пиридин-5-карбонитрил (610 мг, 3,56 ммоль) растворяли в метаноле (75 мл). Полученный раствор охлаждали до 0°C. Добавляли гексагидрат хлорида никеля (II) (85 мг, 0,36 ммоль) и ди-третбутил дикарбонат (1,56 г, 7,13 ммоль) с последующим борогидридом натрия (943 мг, 24,94 ммоль) по частям. Реакционную смесь перемешивали при 0°C до комнатной температуры в течение 18 ч. MeOH удаляли выпариванием. Остаток растворяли в CHCl3 (70 мл), промывали насыщ. NaHCO3 (1×30 мл), водой (1×30 мл) и солевым раствором (1×30 мл), сушили (Na2SO4) и выпаривали в вакууме для получения желтого масла. Очищали хроматографией с испарительной колонкой, (диоксид кремния), элюент 40% петр. эфир, 60% EtOAc для получения белого твердого вещества, идентифицированного как трет-бутиловый эфир (4,6-диметил-1H-пирроло[2,3-b]пиридин-5-илметил)карбамовой кислоты (710 мг, 2,56 ммоль, 72% выход).
[M+H]+=276,1.
1H ЯМР: (CDCl3), 1,49 (9H, s), 2,61 (3H, s), 2,71 (3H, s), 4,46 (1H, br.s), 4,51 (2H, d, J=4,4 Гц), 6,50 (1H, dd, J=3,5, 2,0 Гц), 7,25 (1H, dd, J=3,4, 2,5 Гц), 9,64 (1H, br.s).
G. Гидрохлорид C-(4,6-Диметил-1H-пирроло[2,3-b]пиридин-5-ил)метиламина
Трет-бутиловый эфир 4,6-Диметил-1H-пирроло[2,3-b]пиридин-5-илметил)карбамовой кислоты (710 мг, 2,56 ммоль) растворяли в 4M HCl в диоксане (10 мл). Через 2 ч при кт растворитель удаляли в вакууме для получения желтого твердого вещества, идентифицированного как гидрохлорид C-(4,6-диметил-1H-пирроло[2,3-b]пиридин-5-ил)метиламина (360 мг, 2,00 ммоль, 80% выход).
[M+H]+=176,4.
1H ЯМР: (DMSO-d6), 2,53 (3H, s), 2,60 (3H, s), 3,94 (2H, s), 4,76 (2H, br.s), 6,43 (1H, d, J=2,3 Гц), 7,28 (1H, dd, J=3,2, 1,9 Гц), 11,32 (1H, br.s).
III. Трет-бутиловый эфир (4-Аминометил-3,5-диметилбензил)карбамовой кислоты
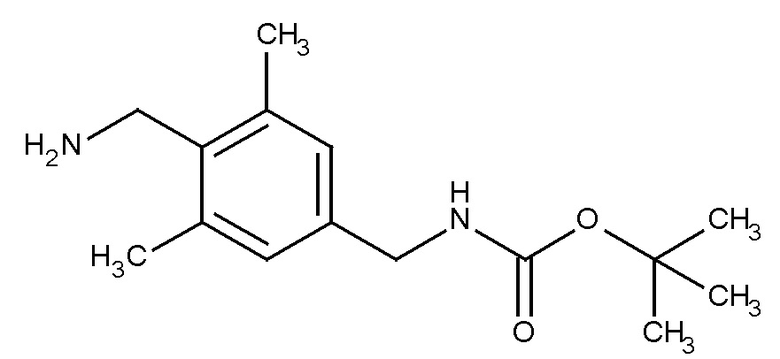
A. Трет-бутиловый эфир (4-Бром-2,6-диметилбензил)карбамовой кислоты
4-Бром-2,6-диметилбензонитрил (2,5 г, 11,9 ммоль) растворяли в метаноле (150 мл). Полученный раствор охлаждали до 0°C. Добавляли гексагидрат хлорида никеля (II) (238 мг, 1,19 ммоль) и ди-третбутил дикарбонат (5,19 г, 23,80 ммоль) с последующим борогидридом натрия (3,15 г, 83,30 ммоль) по частям. Реакционную смесь перемешивали при 0°C до комнатной температуры в течение 3 дней. MeOH удаляли путем выпаривания. Остаток растворяли в CHCl3 (70 мл), промывали насыщ. NaHCO3 (1×30 мл), водой (1×30 мл), солевым раствором (1×30 мл), сушили (Na2SO4) и выпаривали в вакууме для получения бесцветного масла, идентифицированного как трет-бутиловый эфир (4-бром-2,6-диметилбензил)карбамовой кислоты (3,0 г, 9,55 ммоль, 80%).
B. Трет-бутиловый эфир (4-Циано-2,6-диметилбензил)карбамовой кислоты
К дегазированному раствору трет-бутилового эфира (4-бром-2,6-диметилбензил)карбамовой кислоты (3,0 г, 9,55 ммоль) в N,N-диметилацетамиде (30 мл) добавляли порошок цинка (75 мг, 1,15 ммоль), ацетат цинка (210 мг, 1,15 ммоль), 1,1'-бис(дифенилфосфино) ферроцен (635 мг, 1,15 ммоль), цианид цинка (560 мг, 4,77 ммоль), и трис(дибензилиденацетон) дипалладия(0) (524 мг, 0,57 ммоль). Реакционную смесь нагревали до 120°C в течение 4 ч. После чего реакционную смесь охлаждали до комнатной температуры и добавляли еще 1,1'-бис(дифенилфосфино) ферроцен (423 мг, 0,77 ммоль) и трис(дибензилиденацетон) дипалладия(0) (350 мг, 0,38 ммоль), и реакционную смесь нагревали при 120°C в течение дополнительных 28 ч. Реакционную смесь охлаждали до кт, фильтровали через целит и промывали этилацетатом (250 мл). Фильтрат промывали насыщ. NaHCO3 (1×30 мл), водой (1×30 мл), солевым раствором (1×30 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали хроматографией с испарительной колонкой, (диоксид кремния), элюент 80% петр. эфир (60-80°C), 20% EtOAc для получения не совсем белого твердого вещества, идентифицированного как трет-бутиловый эфир (4-циано-2,6-диметилбензил)карбамовой кислоты (630 мг, 2,42 ммоль, 25%).
[M+H]+=261,06.
C. Гидрохлорид 4-Аминометил-3,5-диметилбензонитрила
Трет-бутиловый эфир (4-Циано-2,6-диметилбензил)карбамовой кислоты (630 мг, 2,42 ммоль) растворяли в 4M HCl в диоксане (10 мл). Через один час при комнатной температуре растворитель удаляли в вакууме для получения бледно коричневого твердого вещества, идентифицированного как гидрохлорид 4-аминометил-3,5-диметилбензонитрила (470 мг, 2,39 ммоль, 99%).
D. Бензиловый эфир (4-Циано-2,6-диметилбензил)карбамовой кислоты
Гидрохлорид 4-Аминометил-3,5-диметилбензонитрила (470 мг, 2,39 ммоль) растворяли в дихлорметане (50 мл) и раствор охлаждали до 0°C. Добавляли N,N-диизопропилэтиламин (679 мг, 5,26 ммоль) с последующим добавлением бензилхлорформата (489 мг, 2,87 ммоль). Через один час при 0°C до комнатной температуры реакционную смесь разводили хлороформом, полученный раствор промывали насыщ. NaHCO3 (1×30 мл), водой (1×30 мл), солевым раствором (1×30 мл), сушили (Na2SO4) и выпаривали в вакууме для получения коричневого масла, идентифицированного как бензиловый эфир (4-циано-2,6-диметилбензил)карбамовой кислоты (700 мг, 2,38 ммоль, 99%).
[M+H]+=295,04.
E. Бензиловый эфир [4-(трет-бутоксикарбониламинометил)-2,6-диметилбензил]карбамовой кислоты
Бензиловый эфир (4-Циано-2,6-диметилбензил)карбамовой кислоты (700 мг, 2,38 ммоль) растворяли в метаноле (75 мл). Полученный раствор охлаждали до 0°C. Добавляли гексагидрат хлорида никеля (II) (57 мг, 0,24 ммоль) и ди-третбутил дикарбонат (1,04 г, 4,76 ммоль) с последующим борогидридом натрия (630 мг, 16,65 ммоль) по частям. Реакционную смесь перемешивали при 0°C до комнатной температуры в течение 3 дней. MeOH удаляли путем выпаривания. Остаток растворяли в CHCl3 (70 мл), промывали насыщ. NaHCO3 (1×30 мл), водой (1×30 мл), солевым раствором (1×30 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали хроматографией с испарительной колонкой, (диоксид кремния), элюент 65% петр. эфир (60-80°C), 35% EtOAc для получения не совсем белого твердого вещества, идентифицированного как бензиловый эфир [4-(трет-бутоксикарбониламинометил)-2,6-диметилбензил]карбамовой кислоты (600 мг, 1,51 ммоль, 63%).
m/z=421,05 (M+Na).
F. Трет-бутиловый эфир (4-Аминометил-3,5-диметилбензил)карбамовой кислоты
Бензиловый эфир [4-(трет-бутоксикарбониламинометил)-2,6-диметилбензил]карбамовой кислоты (600 мг, 1,51 ммоль) растворяли в метаноле (60 мл). Полученный раствор гидрировали над 10% Pd/C (100 мг) при атмосферном давлении и комнатной температуре в течение одного часа, после которого катализатор фильтровали и промывали метанолом (30 мл), комбинированные фильтраты выпаривали в вакууме для получения белого твердого вещества, идентифицированного как трет-бутиловый эфир (4-аминометил-3,5-диметилбензил)карбамовой кислоты (350 мг, 1,32 ммоль, 88%).
m/z=287,07 (M+Na).
IV. 5-((2-Фенилтиазол-4-ил)метил)никотиновая кислота
A. Метил 5-((2-фенилтиазол-4-ил)метил)никотинат
Во флакон для микроволновой печи добавляли: (5-(метоксикарбонил)пиридин-3-ил)бороновую кислоту (540 мг, 2,089 ммоль), карбонат калия (412 мг, 2,98 ммоль), 4-(бромметил)-2-фенилтиазол (379 мг, 1,492 ммоль), THF (3 мл) и воду (0,3 мл). Смесь дегазировали в течение 10 мин до добавления катализатора Pd(Ph3)4 (172 мг, 0,149 ммоль). Нагревали в микроволновой печи CEM Discover при 80°C/300 В в течение 20 мин. Реакционную смесь разводили EtOAc (80 мл) и водой (50 мл). Органические экстракты смешивали и промывали насыщенным солевым раствором (50 мл) и затем сушили над MgSO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали хроматографией (20-60% EtOAc в изогексанах) для получения метил 5-((2-фенилтиазол-4-ил)метил)никотината (184 мг, 0,545 ммоль, 36,6% выход) в виде желтого вязкого масла.
[M+H]+=311,1.
B. 5-((2-Фенилтиазол-4-ил)метил)никотиновая кислота
К перемешиваемому раствору метил 5-((2-фенилтиазол-4-ил)метил)никотината (189 мг, 0,609 ммоль) в THF (4 мл) и MeOH (2 мл) добавляли NaOH 2M (913 мкл, 1,827 ммоль) и оставляли при кт в течение 1,5 ч. Реакционную смесь подкисляли путем добавления уксусной кислоты (3 мл) и растворитель удаляли в вакууме. Азеотропировали толуолом (2×30 мл) для удаления уксусной кислоты для получения 5-((2-фенилтиазол-4-ил)метил)никотиновой кислоты (98 мг, 0,298 ммоль, 48,9% выход) в виде не совсем белого твердого вещества.
[M+H]+=297,1.
V. 1-(4-(Бромметил)бензил)-4-метил-1H-пиразол
A. (4-((4-Метил-1H-пиразол-1-ил)метил)фенил)метанол
В круглодонную колбу под N2 добавляли: (4-(хлорметил)фенил)метанол (10,04 г, 60,9 ммоль), 4-метил-1H-пиразол (5,05 мл, 60,9 ммоль) и сухой MeCN (100 мл). Затем добавляли карбонат калия (9,26 г, 67,0 ммоль) и белую суспензию нагревали до 60°C в течение 18 ч. Летучие вещества удаляли в вакууме. Остаток распределяли между EtOAc (100 мл) и водой (150 мл). Водный слой нейтрализовали до pH 7 с помощью 1 Н HCl и экстрагировали EtOAc (2×100 мл). Комбинированные органические слои промывали водой (100 мл), солевым раствором (50 мл) затем сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой продукт очищали хроматографией (10-80% EtOAc в изогексанах) для получения (4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанола (2,9 г, 14,05 ммоль, 23,07% выход) в виде свободно текучего маяла, которое отвердевало при стоянии.
[M+H]+=203,2.
B. 1-(4-(Бромметил)бензил)-4-метил-1H-пиразол
В колбу в N2 добавляли: (4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанол (250 мг, 1,236 ммоль), трифенилфосфин (373 мг, 1,421 ммоль) и сухой DCM (5,0 мл). Охлаждали на ледяной бане до добавления пербромметана (451 мг, 1,360 ммоль). Перемешивали при кт в течение 1 ч. Концентрировали в вакууме и очищали хроматографией (0-20% EtOAc в изогексанах) для получения 1-(4-(бромметил)бензил)-4-метил-1H-пиразола (0,33 г, 1,182 ммоль, 96% выход) в виде масла, которое отвердевало при стоянии до белого твердого вещества.
[M+H]+=265,1/267,1.
Пример 1
N-[(1-Аминоизохинолин-6-ил)метил]-5-({4-[(4-метилпиразол-1-ил)метил]фенил}метил)пиридин-3-карбоксамид
A. Метиловый эфир 5-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)никотиновой кислоты
В сухую колбу в атмосфере N2 добавляли: 4,4,4’,4’,5,5,5’,5’-октаметил-2,2’-би(1,3,2-диоксаборолан) (0,441 г, 1,736 ммоль), метил 5-бромникотинат (0,25 г, 1,157 ммоль), ацетат калия (0,341 г, 3,47 ммоль) и сухой диоксан (10 мл). Реакционную смесь гасили в атмосфере азота в течение 5 минут до добавления Pd(dppf)Cl2 (0,085 г, 0,116 ммоль) для получения ярко красного раствора. Реакционную смесь нагревали до 80°C в течение 16 ч. Реакционную смесь распределяли между EtOAc (50 мл) и насыщ. водн. NH4Cl (30 мл). Водный слой экстрагировали EtOAc (2×30 мл). Комбинированные органические слои промывали водой (30 мл), затем солевым раствором (20 мл) и сушили (MgSO4), фильтровали и концентрировали в вакууме до коричневого масла, которое использовали на следующей стадии без дополнительной очистки.
B. Метиловый эфир 5-[4-(4-Метилпиразол-1-илметил)бензил]никотиновой кислоты
Во флакон для микроволновой печи добавляли: метил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)никотинат (0,5 г, 0,760 ммоль), карбонат калия (0,150 г, 1,086 ммоль), 1-(4-(бромметил)бензил)-4-метил-1H-пиразол (0,144 г, 0,543 ммоль), THF (10 мл) и воду (1,0 мл). Растворитель дегазировали за 10 мин до добавления Pd(PPh3)4 (0,063 г, 0,054 ммоль). Реакционную смесь нагревали в микроволновой печи при 80°C в течение 20 мин, после указанного времени ЖХ-МС показала полную конверсию в продукт. Реакционную смесь распределяли между EtOAc (50 мл) и водой (20 мл). Органический слой отделяли и промывали водой (20 мл), солевым раствором (20 мл) затем сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (12 г RediSep, 0-80% EtOAc в изогексанах) и продукт сушили в вакууме (40°C) в течение 1 ч. Метиловый эфир 5-[4-(4-Метил-пиразол-1-илметил)-бензил]никотиновой кислоты идентифицировали как коричневое вещество (151 мг, 0,460 ммоль, 85% выход).
[M+H]+=322,2.
C. 5-[4-(4-Метилпиразол-1-илметил)-бензил]никотиновая кислота
В круглодонную колбу добавляли: метил 5-(4-((4-метил-1H-пиразол-1-ил)метил)бензил)никотинат (0,19 г, 0,591 ммоль), THF (7,0 мл), MeOH (3,0 мл), затем гидроксид лития в воде (0,042 г, 1,774 ммоль) в воде (3,0 мл). Светло-коричневый раствор нагревали до 65°C в течение 1 часа. Реакционную смесь распределяли между EtOAc (20 мл) и водой (20 мл). Водный слой концентрировали в вакууме до черного масла, подкисляли до pH 3 с помощью 1 Н HCl и экстрагировали EtOAc (4×30 мл). Комбинированные органические слои промывали водой (25 мл), солевым раствором (20 мл), сушили (Na2SO4), фильтровали, затем концентрировали в вакууме до бледно желтого твердого вещества. Продукт сушили в вакууме (40°C) в течение ночи для получения 5-[4-(4-метил-пиразол-1-илметил)-бензил]никотиновой кислоты в виде бледно-желтого порошка (0,145 г, 0,462 ммоль, 78% выход).
[M+H]+=308,2.
D. N-(1-Аминоизохинолин-6-илметил)-5-[4-(4-метилпиразол-1-илметил)бензил]никотинамид
Во флакон добавляли: 5-(4-((4-метил-1H-пиразол-1-ил)метил)бензил)никотиновую кислоту (45 мг, 0,146 ммоль), 6-(аминометил)изохинолин-1-амин (26,6 мг, 0,154 ммоль), HATU (61,2 мг, 0,161 ммоль) и DCM (3,0 мл) для получения оранжевой суспензии. Затем добавляли DIPEA (77 мкл, 0,439 ммоль) для получения бледно оранжевого раствора. Перемешивали при комнатной температуре в течение 2 ч, в течение указанного времени образовывался оранжевый осадок. ЖХ-МС показала полную конверсию с желаемый продукт. Реакционную смесь распределяли между DCM (10 мл) и насыщ. NH4Cl (20 мл). MeOH (1 мл) добавляли для улучшения растворимости. Водный слой экстрагировали DCM (10 мл) и комбинированные органические слои промывали водой (10 мл) и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Сырой продукт очищали хроматографией на колонке RediSep (12 г колонка, 0-10% MeOH (1% NH3) в DCM). Продукт сушили в осушителе в течение ночи для получения не совсем белого твердого вещества (57 мг, 0,121 ммоль, 82% выход), идентифицированного как N-(1-амино-изохинолин-6-илметил)-5-[4-(4-метил-пиразол-1-илметил)бензил]никотинамид
[M+H]=463,3.
1H ЯМР (DMSO-d6) δ: 1,97 (3H, s), 4,01 (2H, s), 4,60 (2H, d, J=5,9 Гц), 5,18 (2H, s), 6,71 (2H, br. s), 6,86 (1H, d, J=5,6 Гц), 7,13 (2H, d, J=8,2 Гц), 7,21-7,24 (3H, m), 7,40 (1H, dd, J=1,7, 8,6 Гц), 7,50 (1H, t, J=0,8 Гц), 7,56 (1H, br. s), 7,76 (1H, d, J=5,8 Гц), 8,06 (1H, t, J=2,1 Гц), 8,13 (1H, d, J=8,6 Гц), 8,63 (1H, d, J=2,1 Гц), 8,91 (1H, d, J=2,1 Гц), 9,27 (1H, t, J=5,9 Гц).
Пример 2
5-({4-[(4-Метилпиразол-1-ил)метил]фенил}метил)-N-{7H-пирроло[2,3-b]пиридин-3-илметил}пиридин-3-карбоксамид
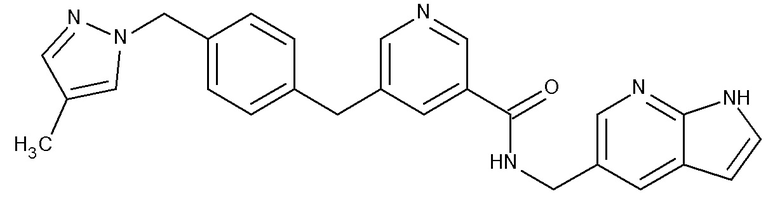
Во флакон добавляли: 5-(4-((4-метил-1H-пиразол-1-ил)метил)бензил)никотиновую кислоту (48 мг, 0,156 ммоль), гидрохлорид (1H-пирроло[2,3-b]пиридин-5-ил)метанамина (28,7 мг, 0,156 ммоль), HATU (65,3 мг, 0,172 ммоль) и DCM (3,0 мл) для получения белой суспензии. Затем добавляли DIPEA (82 мкл, 0,469 ммоль) для получения бесцветного раствора. Реакционную смесь перемешивали при кт в течение 2 ч, в течение указанного времени цвет менялся на оранжевый. ЖХ-МС показала полную конверсию в желаемый продукт. Реакционную смесь распределяли между DCM (10 мл) и насыщ. NH4Cl (20 мл). MeOH (1 мл) добавляли для улучшения растворимости. Водный слой экстрагировали DCM (10 мл), после этого комбинированные органические слои промывали водой (10 мл) и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Сырой продукт очищали хроматографией RediSep (12 г колонка, 0-10% MeOH (NH3) в DCM) и сушили в осушителе в течение ночи. Продукт выделяли в виде белого твердого вещества и идентифицировали как 5-[4-(4-метил-пиразол-1-илметил)бензил]-N-(1H-пирроло[2,3-b]пиридин-5-илметил)никотинамид (57 мг, 0,128 ммоль, 82% выход).
m/z 437,3 (M+H)+ (ES+) при 1,49
1H ЯМР (DMSO-d6) δ: 1,97 (3H, s), 3,99 (2H, s), 4,55 (2H, d, J=5,8 Гц), 5,17 (2H, s), 6,41 (1H, dd, J=1,9, 3,4 Гц), 7,12 (2H, d, J=8,2 Гц), 7,21-7,23 (3H, m), 7,44 (1H, dd, J=2,7, 3,2 Гц), 7,50 (1H, t, J=0,7 Гц), 7,88 (1H, d, J=1,6 Гц), 8,03 (1H, t, J=2,1 Гц), 8,20 (1H, d, J=2,0 Гц), 8,60 (1H, d, J=2,0 Гц), 8,87 (1H, d, J=2,0 Гц), 9,17 (1H, t, J=5,6 Гц), 11,57 (1H, s).
Пример 3
N-[(1-Аминоизохинолин-6-ил)метил]-4-({4-[(4-метилпиразол-1-ил)метил]фенил}метил)пиридин-2-карбоксамид
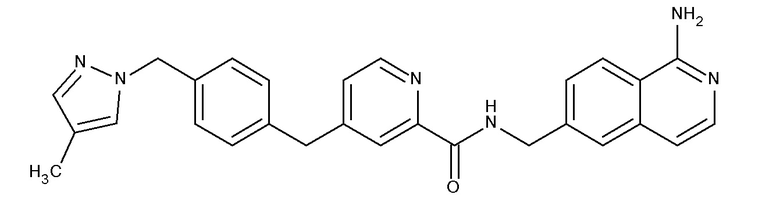
A. Метиловый эфир 4-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)пиридин-2-карбоновой кислоты
В колбу для печи добавляли: метил 4-бромпиколинат (0,5 г, 2,314 ммоль), 4,4,4’,4’,5,5,5’,5’-октаметил-2,2’-би(1,3,2-диоксаборолан) (0,705 г, 2,78 ммоль), ацетат калия (0,681 г, 6,94 ммоль) и сухой диоксан (20 мл). Растворитель дегазировали (N2) в течение 10 минут до добавления PdCl2(dppf) (0,085 г, 0,116 ммоль). Темно красный раствор нагревали до 80°C (темп. основы) в течение 20 ч. Реакционную смесь распределяли между EtOAc (100 мл) и насыщ. водн. NH4Cl (50 мл). Водный слой экстрагировали EtOAc (2×30 мл). Комбинированные органические слои промывали водой (3×30 мл), затем солевым раствором (20 мл), сушили (MgSO4) и фильтровали. Концентрирование в вакууме давало коричневое масло, идентифицированное как метиловый эфир 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиридин-2-карбоновой кислоты (1,0 г, 2,281 ммоль, 99% выход). Продукт использовали в последующей реакции без дополнительной очистки.
[M+H]+=182,1.
B. Метиловый эфир 4-[4-(4-Метилпиразол-1-илметил)бензил]пиридин-2-карбоновой кислоты
Во флакон для микроволновой печи добавляли: метил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиколинат (0,77 г, 1,756 ммоль), карбонат калия (0,347 г, 2,509 ммоль), 1-(4-(бромметил)бензил)-4-метил-1H-пиразол (0,333 г, 1,254 ммоль), THF (10,0 мл) и воду (0,5 мл). Растворитель дегазировали с помощью N2 в течение 10 мин до добавления Pd(PPh3)4 (0,072 г, 0,063 ммоль). Реакционную смесь нагревали до 80°C в микроволновой печи в течение 35 минут. Реакционную смесь распределяли между EtOAc (50 мл) и водой (30 мл). Водный слой экстрагировали EtOAc (2×30 мл). Комбинированные органические слои промывали водой (3×20 мл), затем солевым раствором (20 мл) и сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой материал очищали колоночной хроматографией (40 г RediSep, сухая нагрузка. 10-100% EtOAc в изогексанах). Продукт элюировали при 100% EtOAc для получения коричневого твердого вещества, идентифицированного как метиловый эфир 4-[4-(4-метилпиразол-1-илметил)бензил]пиридин-2-карбоновой кислоты (0,111 г, 0,321 ммоль, 25,6% выход).
[M+H]+=322,2.
C. 4-[4-(4-Метилпиразол-1-илметил)бензил]пиридин-2-карбоновая кислота
В круглодонную колбу добавляли: метил 4-(4-((4-метил-1H-пиразол-1-ил)метил)бензил)пиколинат (0,19 г, 0,503 ммоль), THF (6,0 мл), MeOH (2,0 мл) и гидроксид лития (0,036 г, 1,508 ммоль) в виде раствора в воде (2,0 мл). Полученный коричневый раствор нагревали до 65°C в течение 1 часа. ЖХ-МС показала полную конверсию в желаемую кислоту. Реакционную смесь концентрировали в вакууме, затем распределяли между EtOAc (20 мл) и водой (20 мл). Воду подкисляли до pH 4-5 с помощью 1 Н HCl, и слой экстрагировали с помощью EtOAc (3×30 мл). Продукт тщательно растворяли. Комбинированные органические слои промывали водой (25 мл), затем концентрировали в вакууме для получения белого твердого вещества. Твердое вещество азеотропировали толуолом (2×20 мл) для удаления воды. Продукт сушили в вакуумной печи (40°C) в течение выходных для получения не совсем белого твердого вещества, идентифицированного как 4-[4-(4-метилпиразол-1-илметил)бензил]пиридин-2-карбоновая кислота (0,3 г, 0,436 ммоль, 87% выход).
[M+H]+=308,2.
D. (1-аминоизохинолин-6-илметил)амид 4-[4-(4-Метилпиразол-1-илметил)бензил]пиридин-2-карбоновой кислоты
Во флакон добавляли: 4-(4-((4-метил-1H-пиразол-1-ил)метил)бензил)пиколиновую кислоту (50 мг, 0,065 ммоль), 6-(аминометил)изохинолин-1-амин (11,27 мг, 0,065 ммоль), HATU (27,2 мг, 0,72 ммоль) и сухой DCM (2,5 мл). Затем добавляли DIPEA (114 мкл, 0,651 ммоль) и реакционную смесь перемешивали при комнатной температуре. ЖХ-МС через 1 час показала полную конверсию в желаемый продукт. Реакционную смесь распределяли между DCM (20 мл) и насыщ. водн. NH4Cl (20 мл). Водный слой экстрагировали DCM (2×20 мл). Комбинированные органические слои промывали водой (20 мл), затем солевым раствором (20 мл) и сушили (Na2SO4), фильтровали, затем концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (4 г RediSep, сухая нагрузка, 0-10% MeOH (1% NH3) в DCM). Чистый продукт сушили в вакууме в течение ночи для получения бесцветного стекла, идентифицированного как (1-аминоизохинолин-6-илметил)амид 4-[4-(4-метилпиразол-1-илметил)бензил]пиридин-2-карбоновой кислоты (7,7 мг, 0,013 ммоль, 20,47% выход).
[M+H]+=463,2.
1H ЯМР (DMSO-d6) δ: 1,98 (3H, s), 4,05 (2H, s), 4,60 (2H, d, J=6,4 Гц), 5,19 (2H, s), 5,75 (2H, s), 6,90 (1H, d, J=6,0 Гц), 7,13-7,15 (2H, m), 7,22-7,25 (2H, m), 7,47 (1H, dd, J=1,7, 5,0 Гц), 7,50-7,51 (1H, m), 7,57 (1H, br. s), 7,71 (1H, d, J=6,0 Гц), 7,87-7,88(1H, m), 8,17 (1H, d, J=8,6 Гц), 8,54 (1H, dd, J=0,6, 4,9 Гц), 9,43 (1H, t, J=6,6 Гц).
Пример 4
4-({4-[(4-Метилпиразол-1-ил)метил]фенил}метил)-N-{7H-пирроло[2,3-b]пиридин-3-илметил}пиридин-2-карбоксамид
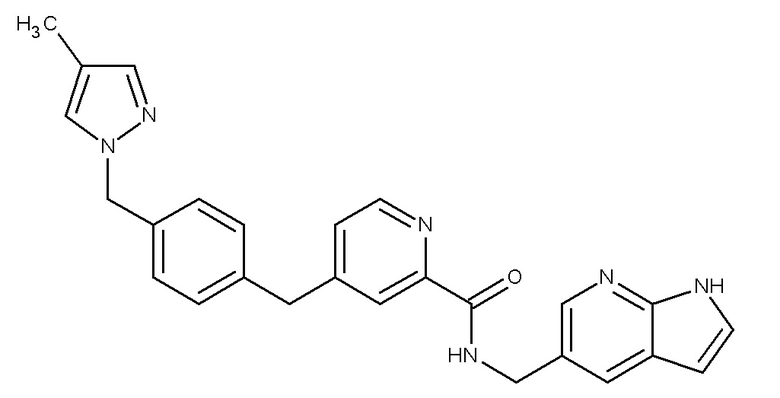
Во флакон добавляли: 4-(4-((4-метил-1H-пиразол-1-ил)метил)бензил)пиколиновую кислоту, HCl (100 мг, 0,145 ммоль), гидрохлорид (1H-пирроло[2,3-b]пиридин-5-ил)метанамина (28,0 мг, 0,153 ммоль), HATU (60,8 мг, 0,160 ммоль) и сухой DCM (2,5 мл). Затем добавляли DIPEA (254 мкл, 1,454 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. ЖХ-МС показала конверсию в желаемое соединение, так реакционная смесь распределялась между DCM (10 мл) и насыщ. NH4Cl (10 мл). Водный слой экстрагировали DCM (3×10 мл). Комбинированные органические слое промывали водой (20 мл), солевым раствором (10 мл) затем сушили (Na2SO4), фильтровали и концентрировали в вакууме до масла. Сырой продукт очищали колоночной хроматографией (4 г RediSep, сухая нагрузка, 0-10% MeOH (1% NH3) в DCM). Продукт сушили в течение ночи в вакууме (40°C) для получения стекловидного твердого вещества, идентифицированного как (1H-пирроло[2,3-b]пиридин-5-илметил)амид 4-[4-(4-метил-пиразол-1-илметил)-бензил]пиридин-2-карбоновой кислоты (11 мг, 0,020 ммоль, 13,86% выход).
[M+H]+=437,20.
1H ЯМР (DMSO-d6) δ: 1,98 (3H, s), 4,03 (2H, s), 4,54 (2H, d, J=6,4 Гц), 5,19 (2H, s), 6,39 (1H, dd, J=1,9, 3,4 Гц), 7,12-7,14 (2H, m), 7,21-7,23 (3H, m), 7,41-7,44 (2H, m), 7,51-7,52 (1H, m), 7,86-7,88 (2H, m), 8,20 (1H, d, J=2,0 Гц), 8,50 (1H, dd, J=0,6, 4,9 Гц), 9,30 (1H, t, J=6,4 Гц), 11,54 (1H, br. s).
Пример 5
N-[(1-Аминоизохинолин-6-ил)метил]-6-оксо-1-({4-[(2-оксопиридин-1-ил)метил]фенил}метил)пиридин-3-карбоксамид
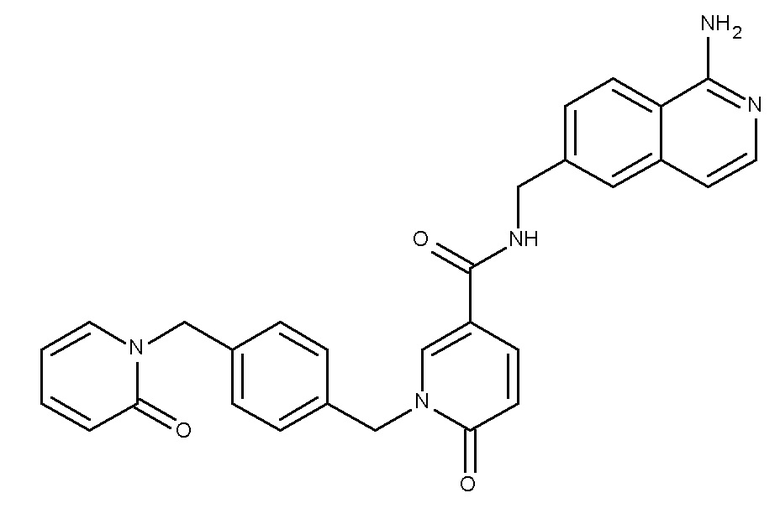
A. Метиловый эфир 6-Оксо-1-[4-(2-оксо-2H-пиридин-1-илметил)бензил]-1,6-дигидропиридин-3-карбоновой кислоты
В круглодонную колбу в атмосфере азота добавляли: метил 1-(4-(хлорметил)бензил)-6-оксо-1,6-дигидропиридин-3-карбоксилат (0,45 г, 1,234 ммоль), пиридин-2-ол (0,129 г, 1,357 ммоль), карбонат калия (0,341 г, 2,468 ммоль) и сухой MeCN (8 мл) для получения золотисто оранжево окрашенной реакционной смеси. Реакционную смесь нагревали до 88°C (темп. пластины основания) в течение ночи. После завершения реакционную смесь распределяли между EtOAc (50 мл) и водой (30 мл). Водный слой экстрагировали EtOAc (2×20 мл). Комбинированные органические слои промывали водой (20 мл), солевым раствором (20 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Сырой продут очищали хроматографией на RediSep (12 г колонка, 0-10% EtOH в EtOAc), для получения порошкообразного белого твердого вещества, идентифицированного как метиловый эфир 6-оксо-1-[4-(2-оксо-2H-пиридин-1-илметил)бензил]-1,6-дигидропиридин-3-карбоновой кислоты (0,37 г, 1,035 ммоль, 84% выход).
[M+H]+=351,2 (M+H)+.
B. 6-Оксо-1-[4-(2-оксо-2H-пиридин-1-илметил)бензил]-1,6-дигидропиридин-3-карбоновая кислота
В круглодонную колбу добавляли: метил 6-оксо-1-(4-((2-оксопиридин-1(2H)-ил)метил)бензил)-1,6-дигидропиридин-3-карбоксилат (0,37 г, 1,035 ммоль), THF (1,0 мл), MeOH (1,0 мл) и гидроксид лития (0,124 г, 5,17 ммоль) в виде раствора в воде (2,0 мл). Реакционную смесь нагревали до 65°C в течение 16 ч, после указанного времени ЖХ-МС показала полную конверсию в желаемое соединение. Летучие вещества удаляли в вакууме и остаток распределяли между водой (20 мл) и EtOAc (20 мл). Водный слой концентрировали в вакууме досуха. Остаток заново растворяли в воде (3,0 мл) и подкисляли до pH 3-4 с помощью 1 Н HCl для осаждения продукта. Суспензию перемешивали при комнатной температуре в течение 30 минут до сбора твердого вещества фильтрацией, промывали водой (2×3,0 мл) и сушили отсасыванием в течение 15 минут, затем в вакуумной печи (40°C) в течение выходных дней. Продукт выделяли в виде белого твердого вещества, идентифицированного как 6-оксо-1-[4-(2-оксо-2H-пиридин-1-илметил)-бензил]-1,6-дигидро-пиридин-3-карбоновая кислота (0,28 г, 0,816 ммоль, 79% выход).
[M+H]+=33,1 (M+H)+.
C. (1-аминоизохинолин-6-илметил)амид 6-Оксо-1-[4-(2-оксо-2H-пиридин-1-илметил)бензил]-1,6-дигидропиридин-3-карбоновой кислоты
Во флакон добавляли: 6-оксо-1-(4-((2-оксопиридин-1(2H)-ил)метил)бензил)-1,6-дигидропиридин-3-карбоновую кислоту (50 мг, 0,149 ммоль), дигидрохлорид 6-(аминометил)изохинолин-1-амина (40,2 мг, 0,164 ммоль), гексафторфосфат(V) 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония (65,0 мг, 0,171 ммоль) и сухой DCM (2,5 мл) для получения белой суспензии. Затем добавляли N-этил-N-изопропилпропан-2-амин (104 мкл, 0,595 ммоль) и полученную суспензию перемешивали при комнатной температуре в течение 2 ч. ЖХ-МС показала конверсию в продукт. Реакционную смесь распределяли между DCM (30 мл) и насыщ. водн. NH4Cl (20 мл). Экстрагировали DCM/IPA (20:1, 2×20 мл). Комбинированные органические слои промывали водой (20 мл) и солевым раствором (20 мл) затем сушили (Na2SO4), фильтровали и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией (12 г RediSep, сухая нагрузка, 0-10% MeOH(1% NH3) в DCM). Продукт сушили в вакууме (40°C) в течение 6 ч для получения бледно желтого твердого вещества, идентифицированного как (1-аминоизохинолин-6-илметил)амид 6-оксо-1-[4-(2-оксо-2H-пиридин-1-илметил)бензил]-1,6-дигидропиридин-3-карбоновой кислоты (32 мг, 0,062 ммоль, 42,0% выход).
[M+H]=492,3.
1H ЯМР (DMSO-d6) δ: 4,56 (2H, d, J=5,8 Гц), 5,06 (2H, s), 5,13 (2H, s), 6,21 (1H, dt, 1,3, 6,7 Гц), 6,38 (1H, br d, J=9,1 Гц), 6,46 (1H, d, J=9,5 Гц), 6,87-6,89 (3H, m), 7,24-7,29 (4H, m), 7,38-7,42 (2H, m), 7,55 (1h, br s), 7,74-7,76 (2H, m), 7,93 (1H, dd, J=2,6, 9,5 Гц), 8,15 (1H, d, J=8,6 Гц), 8,48 (1H, d, J=2,5 Гц), 8,89 (1H, t, J=5,8 Гц).
Пример 6
6-Оксо-1-({4-[(2-оксопиридин-1-ил)метил]фенил}метил)-N-{7H-пирроло[2,3-b]пиридин-3-илметил}пиридин-3-карбоксамид
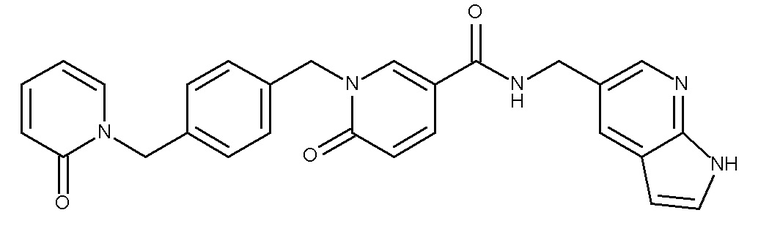
Во флакон добавляли: 6-оксо-1-(4-((2-оксопиридин-1(2H)-ил)метил)бензил)-1,6-дигидропиридин-3-карбоновую кислоту (75 мг, 0,223 ммоль), (1H-пирроло[2,3-b]пиридин-5-ил)метанамин (36,1 мг, 0,245 ммоль), гексафторфосфат(V) 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония (98 мг, 0,256 ммоль) и сухой DCM (3,0 мл) для получения белой суспензии. Затем добавляли N-этил-N-изопропилпропан-2-амин (97 мкл, 0,557 ммоль) для получения бледно желтого мутного раствора. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Далее добавляли (1H-пирроло[2,3-b]пиридин-5-ил)метанамин (9,85 мг, 0,067 ммоль) и перемешивание продолжали в течение 1 ч и образовывалась суспензия коричневого цвета. Реакционную смесь разводили MeOH (5,0 мл) и выделяли посредством SCX захвата и высвобождения. Затем сырой материал очищали колоночной хроматографией RediSep (12 г диоксида кремния, сухая нагрузка, 0-10% MeOH(1% NH3) в DCM). Продукт сушили в вакуумной печи (40°C) в течение выходных дней для получения бледно желтого твердого вещества, идентифицированного как (1H-пирроло[2,3-b]пиридин-5-илметил)амид 6-оксо-1-[4-(2-оксо-2H-пиридин-1-илметил)-бензил]-1,6-дигидропиридин-3-карбоновой кислоты (80 мг, 0,167 ммоль, 74,8% выход).
[M+H]+=466,2.
1H ЯМР (DMSO-d6) δ: 4,50 (2H, d, J=5,6 Гц), 5,05 (2H, s), 5,11 (2H, s), 6,21 (1H, dt, J=1,4, 6,7 Гц), 6,37-6,41 (2H, m), 6,43 (1H, d, 9,5 Гц), 7,23-7,27 (4H, m), 7,40 (1H, dq, J=2,1, 9,2 Гц), 7,44 (1H, t, J=2,8 Гц), 7,74 (1H, dd, J=1,6, 6,8 Гц), 7,87 (1H, d, J=1,6 Гц), 7,90 (1H, dd, J=2,6, 9,5 Гц), 8,19 (1H, d, J=2,0 Гц), 8,44 (1H, d, J=2,5 Гц), 8,76 (1H, t, J=5,6 Гц), 11,58 (1H, s).
Пример 7
N -((1-Аминоизохинолин-6-ил)метил)-5-(гидрокси(4-((4-метил-1H-пиразол-1-ил)метил)фенил)метил)никотинамид
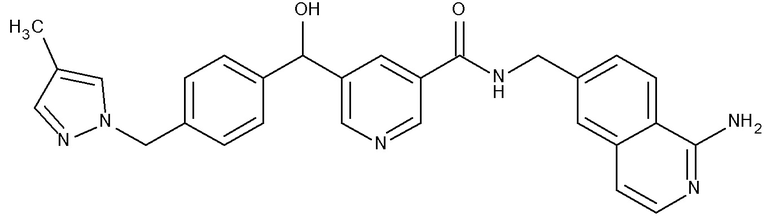
A. 4-((4-Метил-1H-пиразол-1-ил)метил)бензальдегид
Раствор (4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанола (2,54 г, 12,56 ммоль) в DCM (85 мл) обрабатывали оксидом марганца (IV), активировали (21,84 г, 251 ммоль) и смеси позволяли перемешиваться при температуре окружающей среды в течение ночи. Смесь фильтровали через подушечку целита, промывая DCM (200 мл), затем концентрировали в вакууме для получения 4-((4-метил-1H-пиразол-1-ил)метил)бензальдегида (2,04 г, 9,68 ммоль, 77% выход) в виде прозрачного масла.
[M+H]+=201,2 (M+H)+.
B. (5-Бромпиридин-3-ил)(4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанол
Бутиллитий, 2,5M в гексанах (4,08 мл, 10,19 ммоль) добавляли по каплям к охлажденному раствору (-78°C) 3,5-дибромпиридина (2,390 г, 10,09 ммоль) в сухом эфире (50 мл), поддерживая температуру -70°C. Смесь перемешивали в течение 15 минут и по каплям добавляли раствор 4-((4-метил-1H-пиразол-1-ил)метил)бензальдегида (2,04 г, 10,19 ммоль) в сухом эфире (5 мл), поддерживая температуру ниже -70°C. Смесь перемешивали в течение 15 минут и затем позволяли нагреться до температуры окружающей среды в течение 1 часа. Смеси позволяли перемешиваться при температуре окружающей среды в течение ночи. Смесь охлаждали на ледяной бане и гасили добавлением насыщенного водного раствора NH4Cl (50 мл). Слои разделяли и водную фазу экстрагировали EtOAc (3×50 мл). Комбинированные органические слои сушили (MgSO4), фильтровали и концентрировали. Сырой материал очищали хроматографией с испарительной колонкой, нагружая в DCM, элюируя с градиентом от 0 до 100% EtOAc/изогексанов для получения (5-бромпиридин-3-ил)(4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанола (1,30 г, 3,27 ммоль, 32,4% выход) в виде липкой желтой смолы при сушке.
[M+H]+=358,1/360,1 (M+H)+.
C. (5-Бромпиридин-3-ил)(4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанон
Раствор (5-бромпиридин-3-ил)(4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанола (1,08 г, 2,261 ммоль) в хлороформе (35 мл) обрабатывали оксидом марганца (IV), активировали (3,93 г, 45,2 ммоль), и смеси позволяли перемешиваться при температуре окружающей среды в течение ночи. Смесь фильтровали через целит и концентрировали в вакууме. Сырой остаток очищали хроматографией с испарительной колонкой, нагружая в DCM, элюируя с градиентом от 0 до 70% EtOAc/изогексанов для получения указанного в заголовке соединения.
[M+H]+=356,1/358,1 (M+H)+.
D. 5-(4-((4-Метил-1H-пиразол-1-ил)метил)бензоил)никотинонитрил
Перемешиваемый раствор (5-бромпиридин-3-ил)(4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанона (0,500 г, 1,404 ммоль) в безводном DMA (9 мл) обрабатывали дицианоцинком (0,379 г, 3,23 ммоль) и дегазировали путем барботирования N2. Загружали Pd(PPh3)4 (0,081 г, 0,070 ммоль) и смесь дополнительно дегазировали N2, затем нагревали до 110°C (температура бани Drysyn) в течение 5 ч, затем при температуре окружающей среды в течение ночи. Смесь дегазировали N2 в течение 10 минут, затем дополнительно добавляли Pd(PPh3)4 (0,081 г, 0,070 ммоль) и реакционную смесь нагревали при 110°C в течение 2 ч. Смесь охлаждали и обрабатывали дополнительно дицианоцинком (0,379 г, 3,23 ммоль) и Pd(PPh3)4 (0,081 г, 0,070 ммоль), затем перемешивали и нагревали при 110°C в течение 3 ч и до 120°C в течение 2 ч, затем при температуре окружающей среды в течение выходных дней. Смесь разводили DCM (50 мл) и фильтровали через Celite, промывая DCM (100 мл). Растворители удаляли в вакууме и остаток очищали хроматографией с испарительной колонкой, нагружая в DCM, элюируя с градиентом от 0 до 50% EtOAc/DCM, для получения указанного в заголовке соединения (353 мг).
[M+H]+=303,2 (M+H)+.
E. 5-(4-((4-Метил-1H-пиразол-1-ил)метил)бензоил)никотиновая кислота
Раствор 5-(4-((4-метил-1H-пиразол-1-ил)метил)бензоил)никотинонитрила (248 мг, 0,574 ммоль) в THF (3 мл) и воду (1 мл) обрабатывали гидроксидом лития (68,8 мг, 2,87 ммоль) и смесь нагревали до 80°C в течение 21 ч. Органические растворители удаляли в вакууме и остаток распределяли между EtOAc (15 мл) и водой (10 мл, при pH 10). Водный слой экстрагировали дополнительно EtOAc (10 мл) и водный слой доводили до pH 3 с помощью 1M HCl. Водный слой ре-экстрагировали EtOAc (3×15 мл) и комбинированные органические слой сушили (MgSO4), фильтровали и концентрировали для получения 5-(4-((4-метил-1H-пиразол-1-ил)метил)бензоил)никотиновой кислоты (138 мг, 0,408 ммоль, 71,1% выход) в виде желтой пены.
[M+H]+=322,1 (M+H)+.
F. N-((1-Аминоизохинолин-6-ил)метил)-5-(4-((4-метил-1H-пиразол-1-ил)метил)бензоил)никотинамид
Сцинтилляционный флакон заполняли 5-(4-((4-метил-1H-пиразол-1-ил)метил)бензоил)никотиновой кислотой (134 мг, 0,417 ммоль), 6-(аминометил)изохинолин-1-амин дигидрохлоридом (113 мг, 0,459 ммоль), гексафторфосфатом (V) 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония (174 мг, 0,459 ммоль), сухим DCM (3 мл) и DMF (0,3 мл). Затем добавляли N,N-диизопропилэтиламин (291 мкл, 1,668 ммоль) и смеси позволяли перемешиваться при температуре окружающей среды в течение ночи. Реакционную смесь концентрировали в вакууме и очищали SCX (~3,5 г), промывая MeOH, элюируя 1% NH3/MeOH. Сырой остаток очищали хроматографией с испарительной колонкой, нагружая в DCM (метка MeOH), элюируя с градиентом 0-7% MeOH/DCM (содержащим 0,3% NH3). Фракции, содержащие продукт, комбинировали и повторно очищали хроматографией с испарительной колонкой, нагружая в DCM, элюируя с градиентом 0-30% EtOH/EtOAc. Самые чистые фракции комбинировали для получения N-((1-аминоизохинолин-6-ил)метил)-5-(4-((4-метил-1H-пиразол-1-ил)метил)бензоил)никотинамида (34 мг, 0,070 ммоль, 16,77% выход) в виде бледно желтого порошка.
[M+H]+=477,3 (M+H)+.
G. N -((1-Аминоизохинолин-6-ил)метил)-5-(гидрокси(4-((4-метил-1H-пиразол-1-ил)метил)фенил)метил)никотинамид
Раствор N-((1-аминоизохинолин-6-ил)метил)-5-(4-((4-метил-1H-пиразол-1-ил)метил)бензоил)никотинамида (50 мг, 0,105 ммоль) в безводном MeOH (1,5 мл) обрабатывали борогидридом натрия (11,91 мг, 0,315 ммоль) и смеси позволяли перемешиваться при температуре окружающей среды в течение 2,5 ч. ЖХМС показала чистую конверсию в желаемое соединение. Растворители удаляли в вакууме и остаток распределяли между EtOAc (30 мл) и водой (20 мл). Водный слой экстрагировали EtOAc (30 мл) и комбинированные органические слои промывали солевым раствором (20 мл), сушили (MgSO4), фильтровали и концентрировали. Сырой продукт очищали хроматографией с испарительной колонкой, нагружая в DCM (следы MeOH), элюируя с градиентом 0-10% MeOH/DCM (содержащего 0,3% NH3), для получения N-((1-аминоизохинолин-6-ил)метил)-5-(гидрокси(4-((4-метил-1H-пиразол-1-ил)метил)фенил)метил)никотинамида (34 мг, 0,070 ммоль, 67,0% выход) в виде прозрачного стекла.
[M+H]+=479,3.
1H ЯМР (DMSO-d6) δ: 1,97 (3H, s), 4,61 (2H, d, J=5,8 Гц), 5,19 (2H, s), 5,83 (1H, d, J=3,9 Гц), 6,19 (1H, d, J=3,9 Гц), 6,72 (2H, s), 6,86 (1H, d, J=5,8 Гц), 7,13-7,19 (2H, m), 7,21 (1H, s), 7,33-7,44 (3H, m), 7,51 (1H, s), 7,56 (1H, s), 7,76 (1H, d, J=5,8 Гц), 8,14 (1H, d, J=8 Гц), 8,20 (1H, t, J=2,1 Гц), 8,70 (1H, d, J=2,1 Гц), 8,93 (1H, d, J=2,1 Гц), 9,32 (1H, t, J=5,9 Гц).
Пример 44
N-[(1-Аминоизохинолин-6-ил)метил]-5-({4-[(2-фторфенил)метил]пиперазинил-1-ил}метил)пиридин-3-карбоксамид
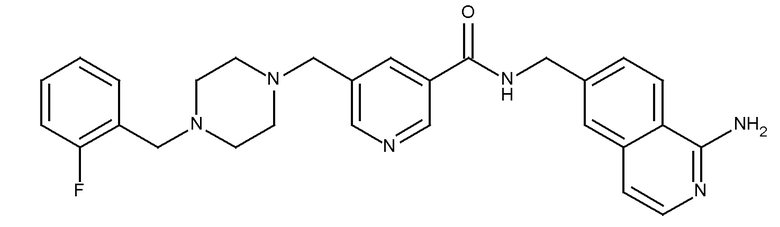
A. Трет-Бутил 4-((5-(метоксикарбонил)пиридин-3-ил)метил)пиперазин-1-карбоксилат
Метил 5-бромникотинат (2,95 г, 13,67 ммоль), диацетоксипалладий (0,153 г, 0,683 ммоль), калий (4-boc-пиперазин-1-ил)метилтрифторборат (5,022 г, 16,40 ммоль), карбонат цезия (11,13 г, 34,2 ммоль), и добавляли X-Phos (0,652 г, 1,367 ммоль), растворенный в THF (40 мл) и воде (10 мл). Полученную смесь продували N2 в течение 10 минут, перемешивали и нагревали при 70°C в/н. Смесь разводили водой (10 мл) и экстрагировали EtOAc (3×30 мл). Комбинированные органические слои сушили (MgSO4), фильтровали и концентрировали. Сырой продукт очищали хроматографией с испарительной колонкой (EtOAc в i-Hex 0-100%, содержащий 1% Et3N) для получения трет-бутил 4-((5-(метоксикарбонил)пиридин-3-ил)метил)пиперазин-1-карбоксилата (4,81 г, 13,62 ммоль, 100% выход) в виде бледно коричневого твердого вещества.
[M+H]+=336,1.
B. Метил 5-(пиперазин-1-илметил)никотинат
К перемешиваемому раствору трет-бутил 4-((5-(метоксикарбонил)пиридин-3-ил)метил)пиперазин-1-карбоксилата (4,81 г, 14,34 ммоль) в DCM (10 мл) при кт добавляли трифторуксусную кислоту (10 мл, 130 ммоль). Полученный раствор перемешивали при кт в течение 5 ч. Реакционную смесь разводили толуолом (20 мл) и нагружали на SCX (28 г), промывая MeOH и элюируя 1% NH3 в MeOH. Растворитель выпаривали при пониженном давлении для получения метил 5-(пиперазин-1-илметил)никотината (3,55 г, 14,34 ммоль, 100% выход) в виде бледно желтого твердого вещества.
[M+H]+=236,0 (M+H)+.
C. Метил 5-((4-(2-фторбензил)пиперазин-1-ил)метил)никотинат
К перемешиваемому раствору 2-фторбензальдегида (79 мг, 0,638 ммоль) и метил 5-(пиперазин-1-илметил)никотината (150 мг, 0,638 ммоль) в DCM (4 мл) добавляли каплю уксусной кислоты и оставляли при кт в течение 1 часа. К нему добавляли метил 5-(пиперазин-1-илметил)никотинат (150 мг, 0,638 ммоль) и оставляли при кт в течение ночи. Реакционную смесь разводили DCM (5 мл) и добавляли NaHCO3(водн) (5 мл). Слои разделяли и водный слой экстрагировали DCM (2×5 мл); комбинированные органические слои сушили (Na2SO4), фильтровали и выпаривали при пониженном давлении для получения метил 5-((4-(2-фторбензил)пиперазин-1-ил)метил)никотината (160 мг, 0,410 ммоль, 64,3% выход) в виде бледно желтого густого масла.
[M+H]+=344,1.
D. 5-((4-(2-Фторбензил)пиперазин-1-ил)метил)никотиновая кислота
К перемешиваемому раствору метил 5-((4-(2-фторбензил)пиперазин-1-ил)метил)никотината (147 мг, 0,428 ммоль) в THF (2 мл) и воде (1 мл) при кт добавляли гидроксид лития (51,3 мг, 2,140 ммоль). Полученный раствор перемешивали при кт в течение ночи. Реакционную смесь нагружали на SCX (2 г), промывая MeOH и элюируя 1% NH3 в MeOH. Растворитель выпаривали при пониженном давлении для получения 5-((4-(2-фторбензил)пиперазин-1-ил)метил)никотиновой кислоты (130 мг, 0,395 ммоль, 92% выход) в виде бесцветного липкого масла.
[M+H]+=330,0.
E. N-[(1-Аминоизохинолин-6-ил)метил]-5-({4-[(2-фторфенил)метил]пиперазин-1-ил}метил)пиридин-3-карбоксамид
К перемешиваемому раствору 5-((4-(2-фторбензил)пиперазин-1-ил)метил)никотиновой кислоты (118 мг, 0,358 ммоль), 6-(аминометил)изохинолин-1-амин дигидрохлорида (97 мг, 0,394 ммоль), HATU (163 мг, 0,430 ммоль) в DCM (2 мл) добавляли диизопропилэтиламин (250 мкл, 1,433 ммоль). Полученную смесь перемешивали при кт в течение ночи. Реакционную смесь разводили EtOAc (20 мл) и промывали 1M NaOH(водн) (2×10 мл). Органические слои сушили (Na2SO4), фильтровали и выпаривали при пониженном давлении для получения сырого соединения, которое очищали хроматографией с испарительной колонкой (EtOH в EtOAc 0-50%), дважды для получения N-((1-аминоизохинолин-6-ил)метил)-5-((4-(2-фторбензил)пиперазин-1-ил)метил)никотинамида (39,3 мг, 0,079 ммоль, 22,07% выход) в виде бледно желтого твердого вещества.
m/z 485,1 (M+H)+.
1H ЯМР (DMSO-d6) δ: 2,20-2,47 (8H, m), 3,51 (2H, s), 3,55 (2H, s), 4,63 (2H, d, J=5,8 Гц), 6,72 (2H, s), 6,87 (1H, d, J=5,6 Гц), 7,10-7,19 (2H, m), 7,26-7,34 (1H, m), 7,35-7,45 (2H, m), 7,58 (1H, s), 7,76 (1H, d, J=5,8 Гц), 8,12-8,17 (2H, m), 8,61 (1H, d, J=2,0 Гц), 8,97 (1H, d, J=2,1 Гц), 9,32 (1H, t, J=5,9 Гц.
Пример 77
N-[(1-Аминоизохинолин-6-ил)метил]-5-{[2-(пирролидин-1-ил)пиридин-4-ил]метил}пиридин-3-карбоксамид
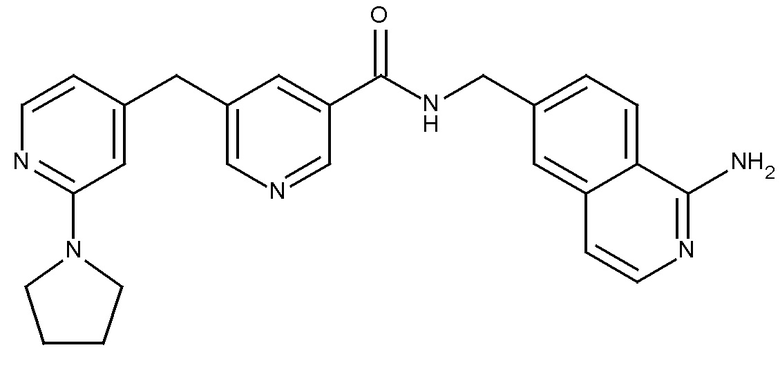
A. 4-(хлорметил)-2-фторпиридин
В 500 мл колбу загружали 2-фтор-4-метилпиридин (25 г, 225 ммоль), N-хлорсукцинимид (45,1 г, 337 ммоль), бензоилпероксид, Luperox (1,453 г, 4,50 ммоль), уксусную кислоту (1 мл, 17,47 ммоль) и ацетонитрил (132 мл, 2527 ммоль). Реакционную смесь аккуратно нагревали в колбе с обратным холодильником, получая бледно желтый раствор, который оставляли перегоняться в течение 5 часов, затем при температуре окружающей среды в течение ночи. Смесь распределяли между водой (20 мл) и EtOAc (30 мл). Солевой раствор (30 мл) добавляли для получения двух слоев. Их затем разделяли и дополнительно ре-экстрагировали EtOAc (2×30 мл). Комбинированные органические слои промывали солевым раствором (30 мл), сушили (MgSO4), фильтровали и концентрировали. При охлаждении осадок фильтровали, промывая DCM (40 мл), затем снова концентрировали в вакууме. Остаток повторно очищали хроматографией с испарительной колонкой, нагружая в минимальном количестве DCM, элюируя с градиентом 0-15% EtOAc/изогексанов (удерживая при 6% для элюирования продукта) для получения 4-(хлорметил)-2-фторпиридина (11,8 г, 78 ммоль, 34,6% выход) в виде прозрачного масла.
B. Метил 5-((2-фторпиридин-4-ил)метил)никотинат
Раствор 4-(хлорметил)-2-фторпиридина (2,012 г, 13,82 ммоль) и метил 5-(4,4,5,5-тетраметил-1,3,2-диокссаборолан-2-ил)никотината (4,00 г, 15,20 ммоль) в THF (40 мл) и воды (1 мл) обрабатывали карбонатом калия (3,82 г, 27,6 ммоль) и смесь дегазировали N2 в течение 5 минут. Добавляли Pd(Ph3)4 катализатор (1,597 г, 1,382 ммоль) и смесь коротко дегазировали при нагревании до 90°C (температура бани Drysyn) в течение 2 часов. Реакционную смесь распределяли между EtOAc (150 мл) и водой (75 мл). Водный слой экстрагировали дополнительно EtOAc (2×70 мл) и комбинированные органические слои промывали солевым раствором (75 мл), сушили (MgSO4), фильтровали и концентрировали. Сырой материал очищали хроматографией, нагружая в DCM, элюируя с градиентом 10-70% EtOAc/изогексанов (удерживая при 55% для элюции продукта) для получения метил 5-((2-фторпиридин-4-ил)метил)никотината (1,99 г, 8,00 ммоль, 57,9% выход) в виде желтой смолы.
m/z=247,1 (M+H)+.
C. 5-((2-фторпиридин-4-ил)метил)никотинат аммония
Раствор метил 5-((2-фторпиридин-4-ил)метил)никотината (2,45 г, 9,95 ммоль) в MeOH (25 мл) и THF (60 мл) обрабатывали водой (20 мл) и гидроксидом лития (0,286 г, 11,94 ммоль), затем перемешивали при температуре окружающей среды в течение ночи. Дополнительно добавляли гидроксид лития (0,286 г, 11,94 ммоль) и смесь перемешивали при температуре окружающей среды в течение ночи. Большую часть органических растворителей удаляли в вакууме и добавляли воду (25 мл). pH доводили до ~5 и смесь очищали непосредственно SCX (45 г), промывая MeOH, элюируя 1% NH3/MeOH. Выделенный продукт растирали с DCM (30 мл) и фильтровали для получения 5-((2-фторпиридин-4-ил)метил)никотината аммония (1,85 г, 7,35 ммоль, 73,9% выход) в виде белого порошка.
m/z=233,1 (M+H)+.
D. N-((1-Аминоизохинолин-6-ил)метил)-5-((2-фторпиридин-4-ил)метил)никотинамид
Смесь 5-((2-фторпиридин-4-ил)метил)никотината аммония (1,40 г, 5,62 ммоль), дигидрохлорида 6-(аминометил)изохинолин-1-амина (1,521 г, 6,18 ммоль) и гексафторфосфата (V) 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония (2,349 г, 6,18 ммоль) в смеси безводного DCM (20 мл) и безводный DMF (2 мл) обрабатывали N,N-диизопропилэтиламином (5,28 мл, 30,3 ммоль) и полученную суспензию обрабатывали ультразвуком коротко до перемешивания при температуре окружающей среды в течение ночи. Растворители удаляли в вакууме. Остаток распределяли между EtOAc (50 мл, следы MeOH для растворимости) и насыщенным водным NH4Cl (50 мл). Водный слой экстрагировали дополнительно EtOAc (6×50 мл) и комбинированные органические слои сушили (MgSO4), фильтровали и концентрировали. Сырой материал очищали хроматографией с испарительной колонкой, загружая в DCM (следы MeOH), элюируя с градиентом 0-30% EtOH/EtOAc для получения N-((1-аминоизохинолин-6-ил)метил)-5-((2-фторпиридин-4-ил)метил)никотинамидa в виде бледно желтого порошка.
m/z=388,2 (M+H)+.
E. N-[(1-Аминоизохинолин-6-ил)метил]-5-{[2-(пирролидин-1-ил)пиридин-4-ил]метил}пиридин-3-карбоксамид
Смесь N-((1-аминоизохинолин-6-ил)метил)-5-((2-фторпиридин-4-ил)метил)никотинамида (100 мг, 0,258 ммоль) и пирролидина (424 мкл, 5,16 ммоль) в безводных диоксанах (200 мкл) нагревали вместе при 90°C в течение 5 часов. Растворители удаляли в вакууме и остаток очищали хроматографией с испарительной колонкой нагружая в DCM, элюируя с градиентом 0-7,5% MeOH/DCM (содержащем 0,3% NH3). Соединение растворяли в минимальном количестве DCM и Et2O добавляли к осадку. Полученную смесь обрабатывали ультразвуком и перемешивали в течение ~30 минут, затем фильтровали для получения N-((1-аминоизохинолин-6-ил)метил)-5-((2-(пирролидин-1-ил)пиридин-4-ил)метил)никотинамида (73 мг, 0,165 ммоль, 63,8% выход) в виде белого порошка.
(M+H)+=439,1.
1H ЯМР (DMSO-d6) δ: 1,90 (4H, m), 3,33 (4H, m), 3,93 (2H, s), 4,61 (2H, d, J=5,9 Гц), 6,37 (1H, s), 6,41 (1H, dd, J=5,1, 1,4 Гц), 6,74 (2H, s), 6,86 (1H, d, J=5,9 Гц), 7,40 (1H, dd, J=8,6, 1,7 Гц), 7,56 (1H, s), 7,76 (1H, d, J=5,8 Гц), 7,94 (1H, d, J=5,1 Гц), 8,10 (1H, t, J=2,2 Гц), 8,13 (1H, d, J=8,6 Гц), 8,67 (1H, d, J=2,1 Гц), 8,94 (1H, d, J=2,1 Гц), 9,32 (1H, t, J=5,9 Гц).
Соединения в следующих таблицах синтезировали, как описано для примеров 1-7 и 44 и 77.
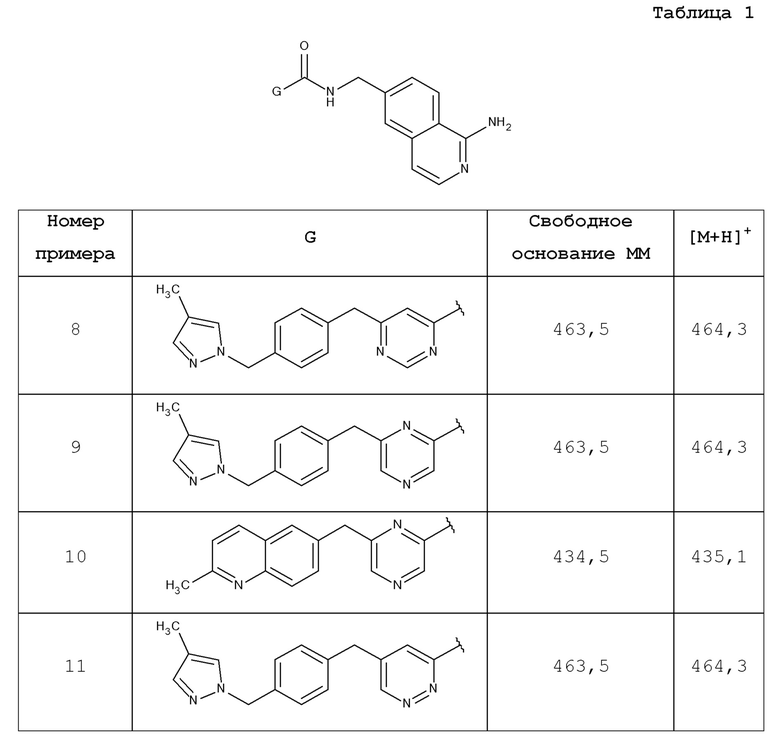
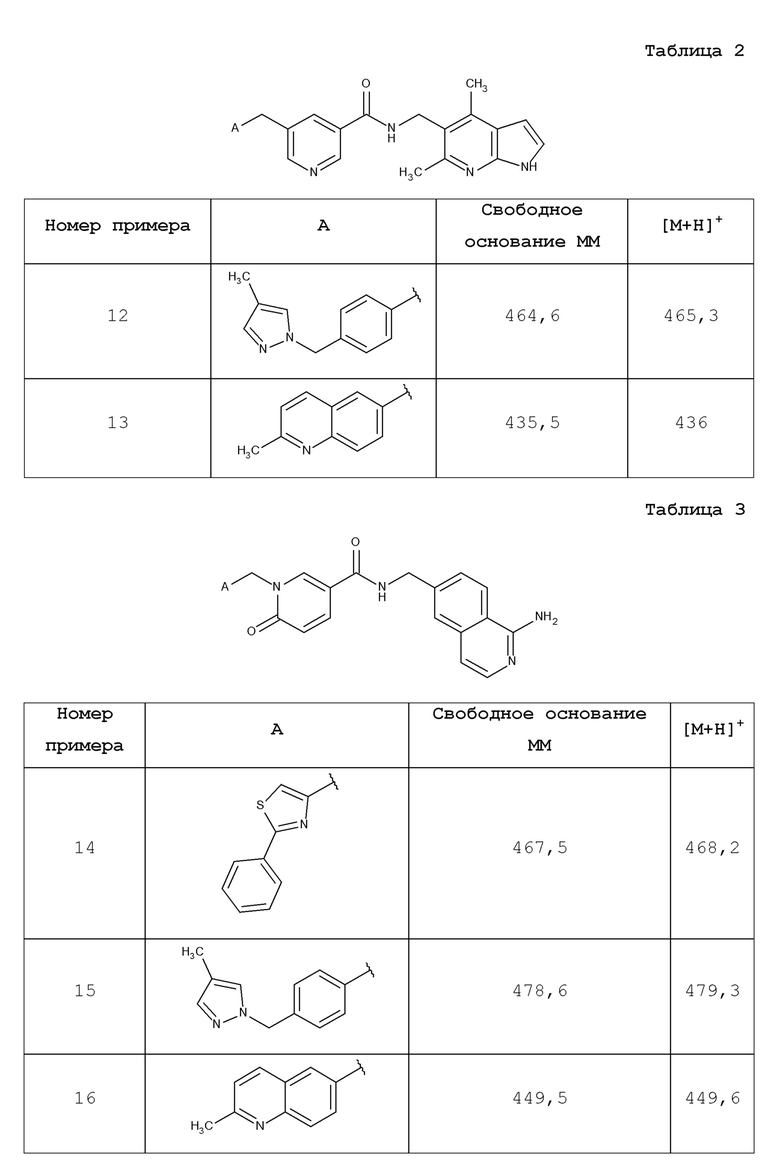
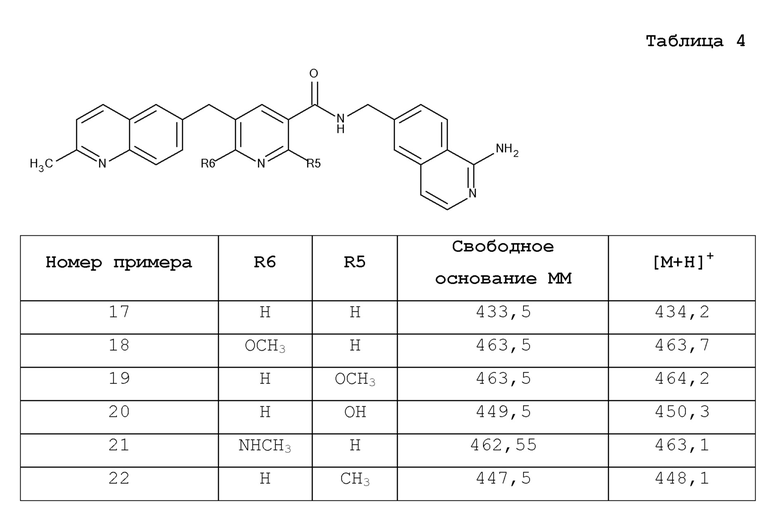
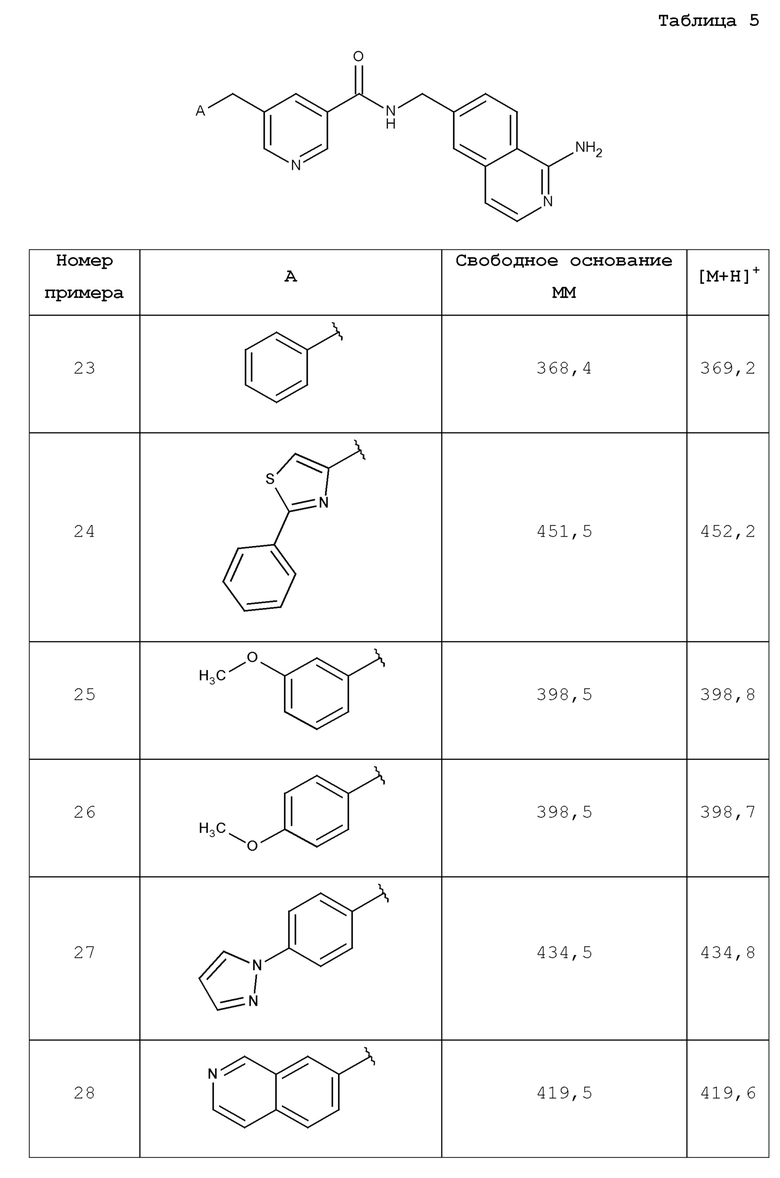
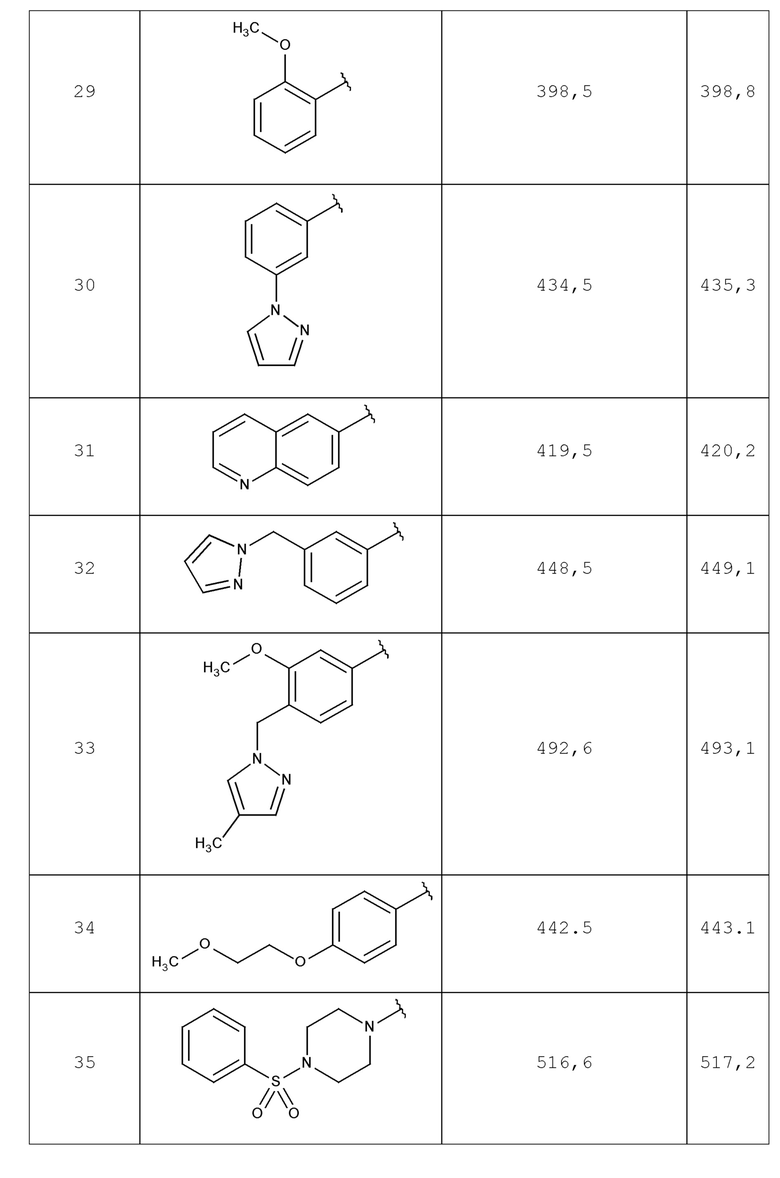
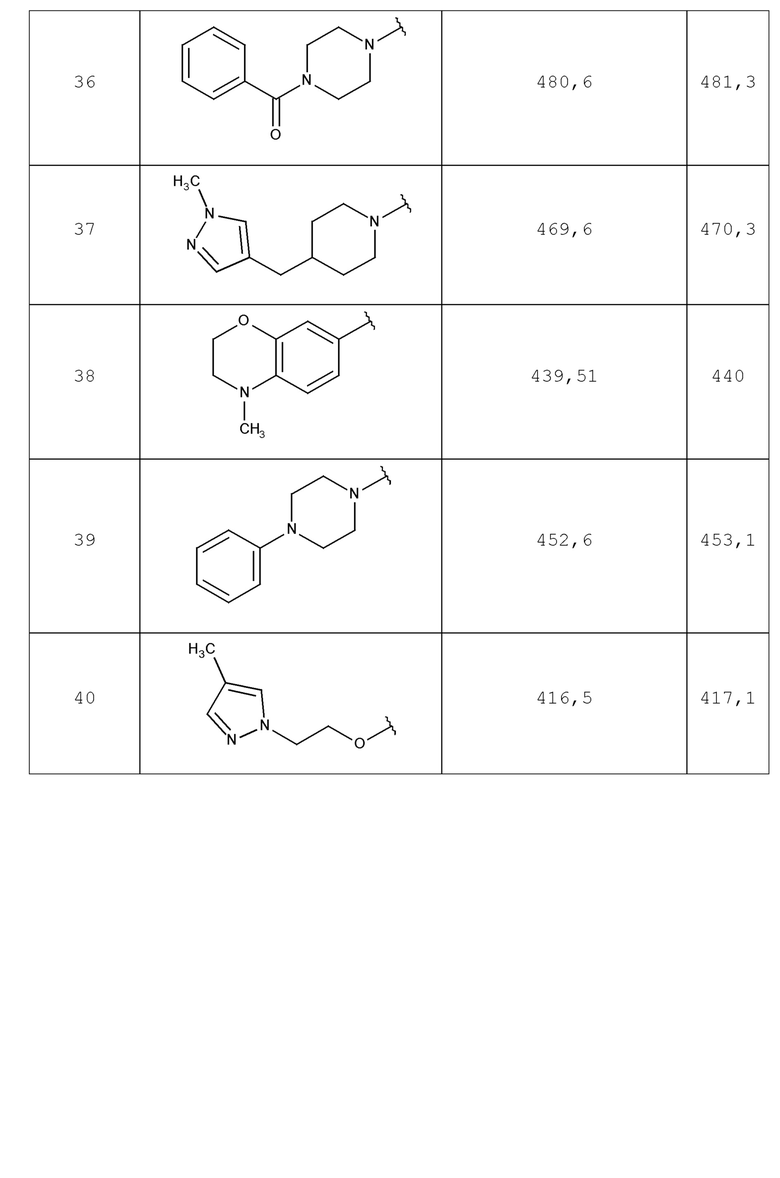
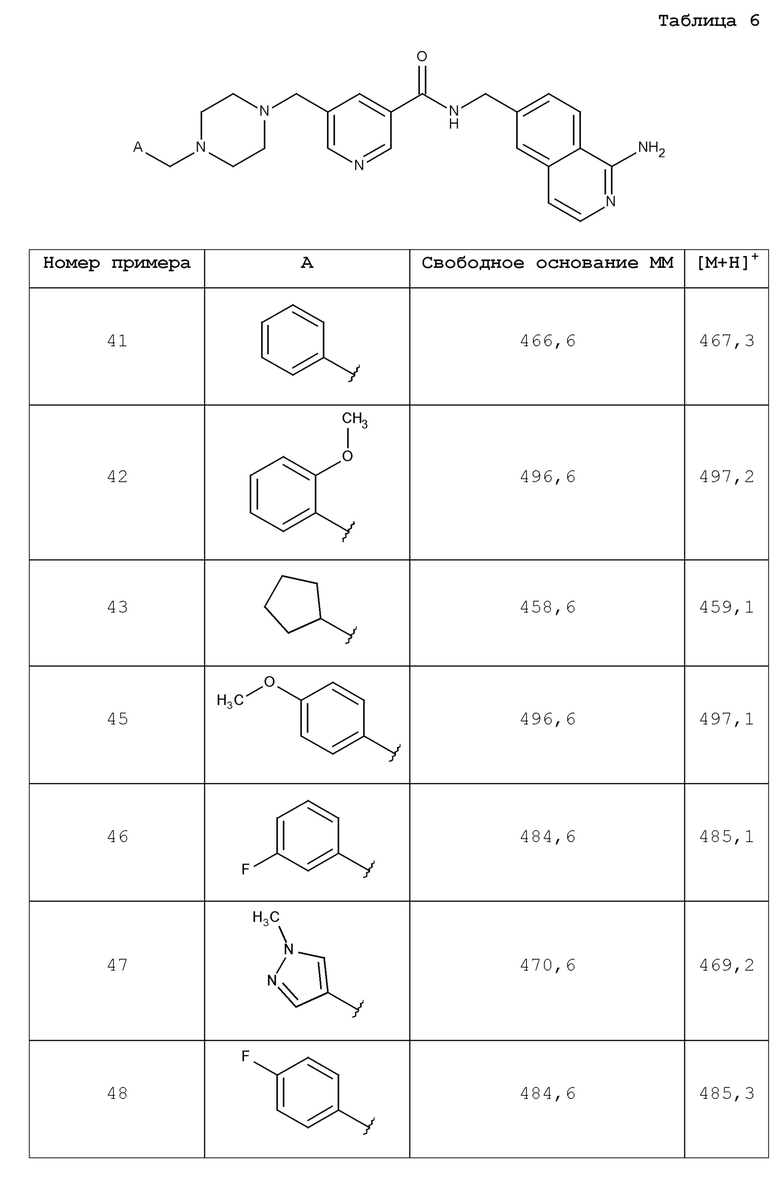
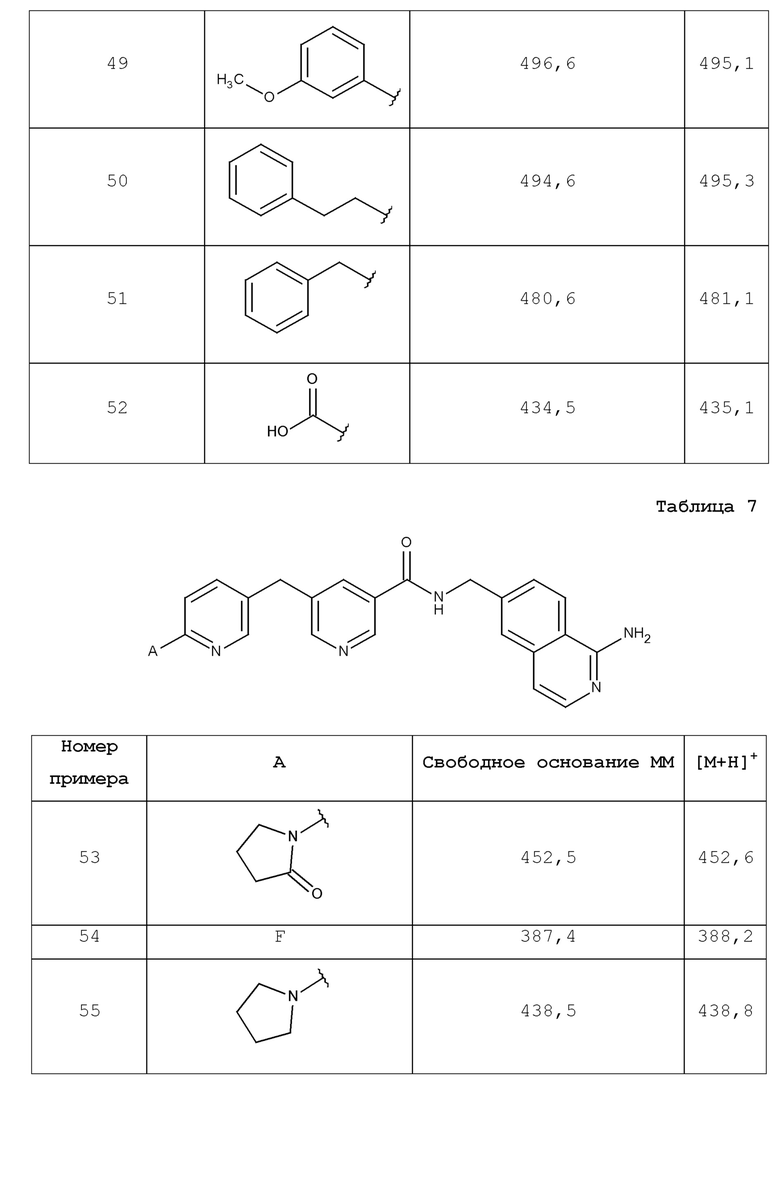
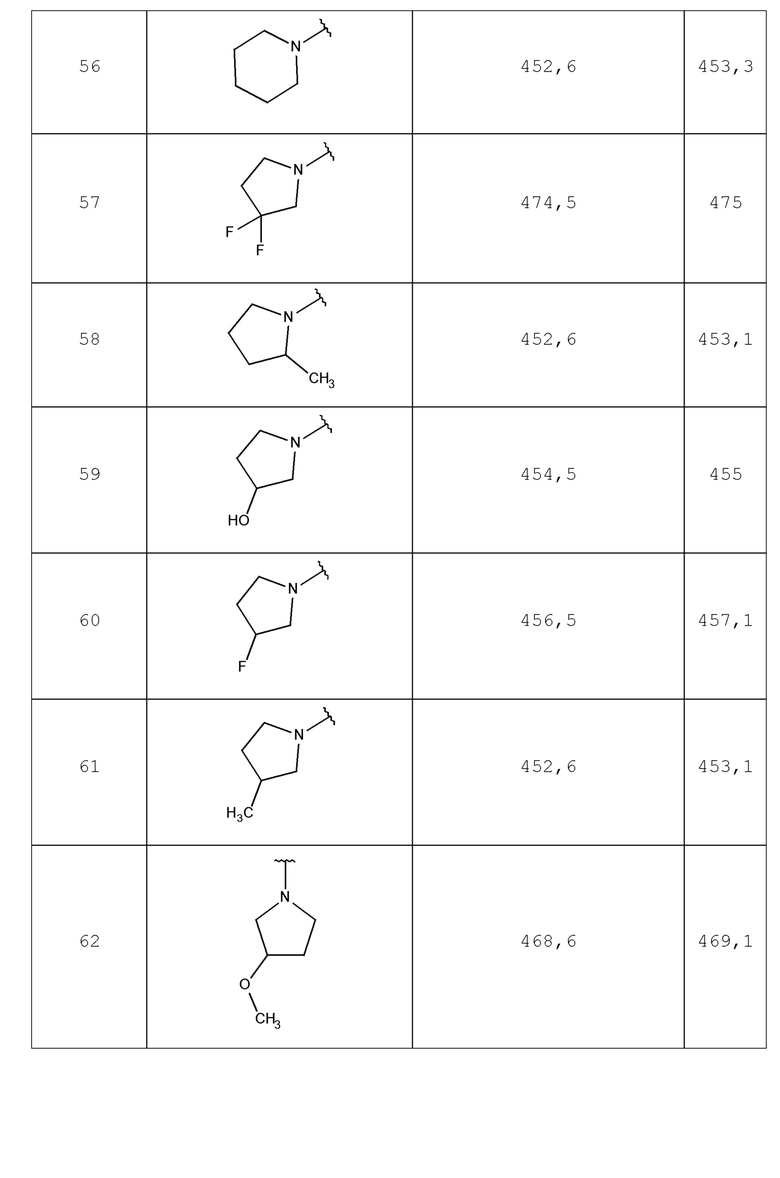
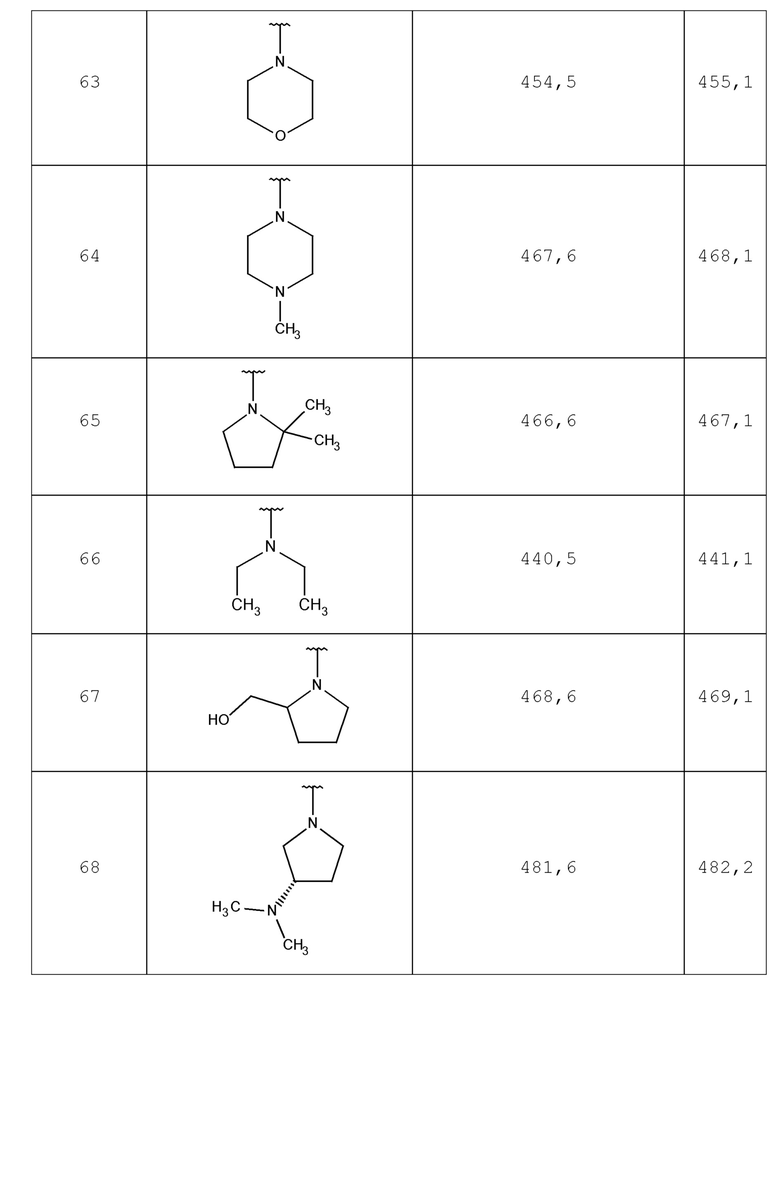
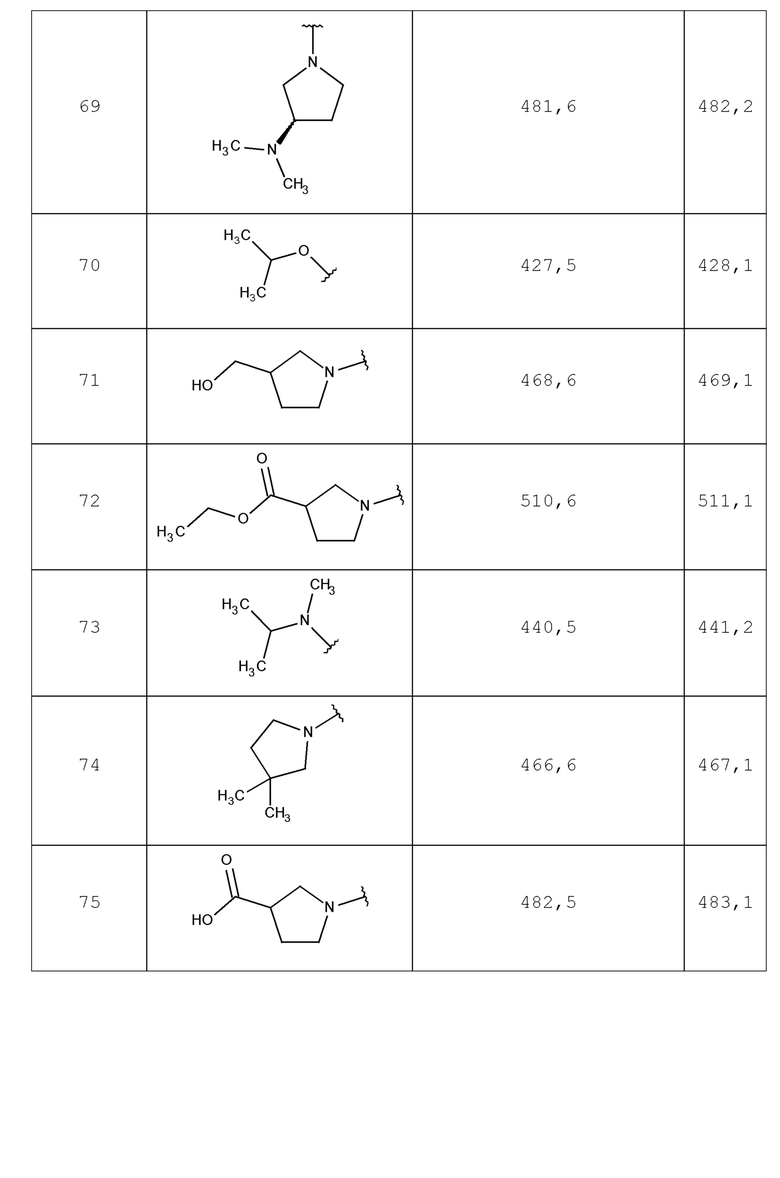
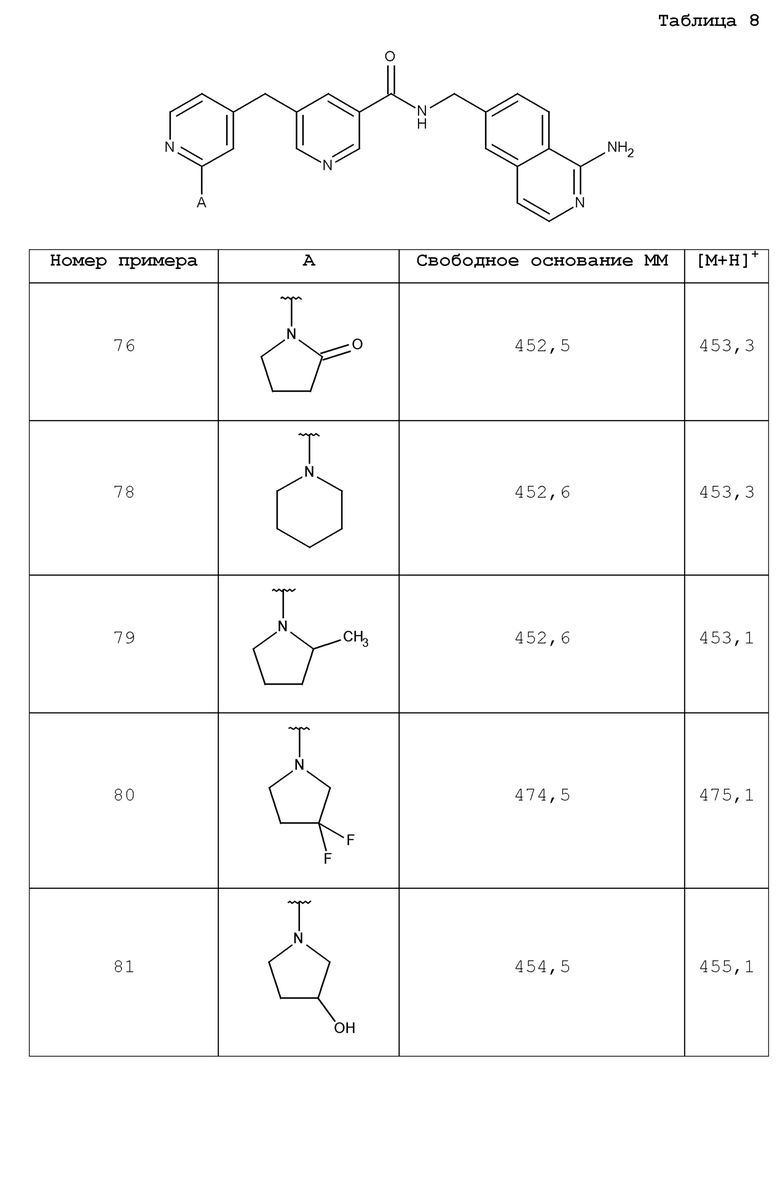
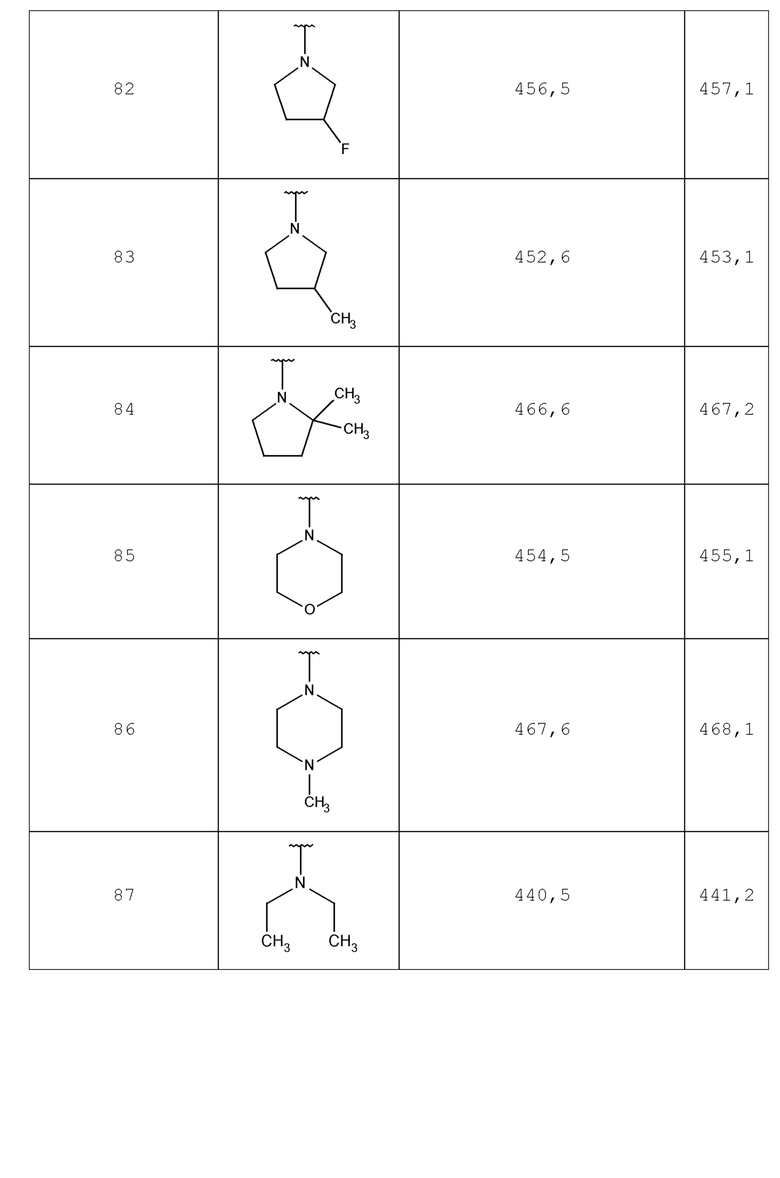
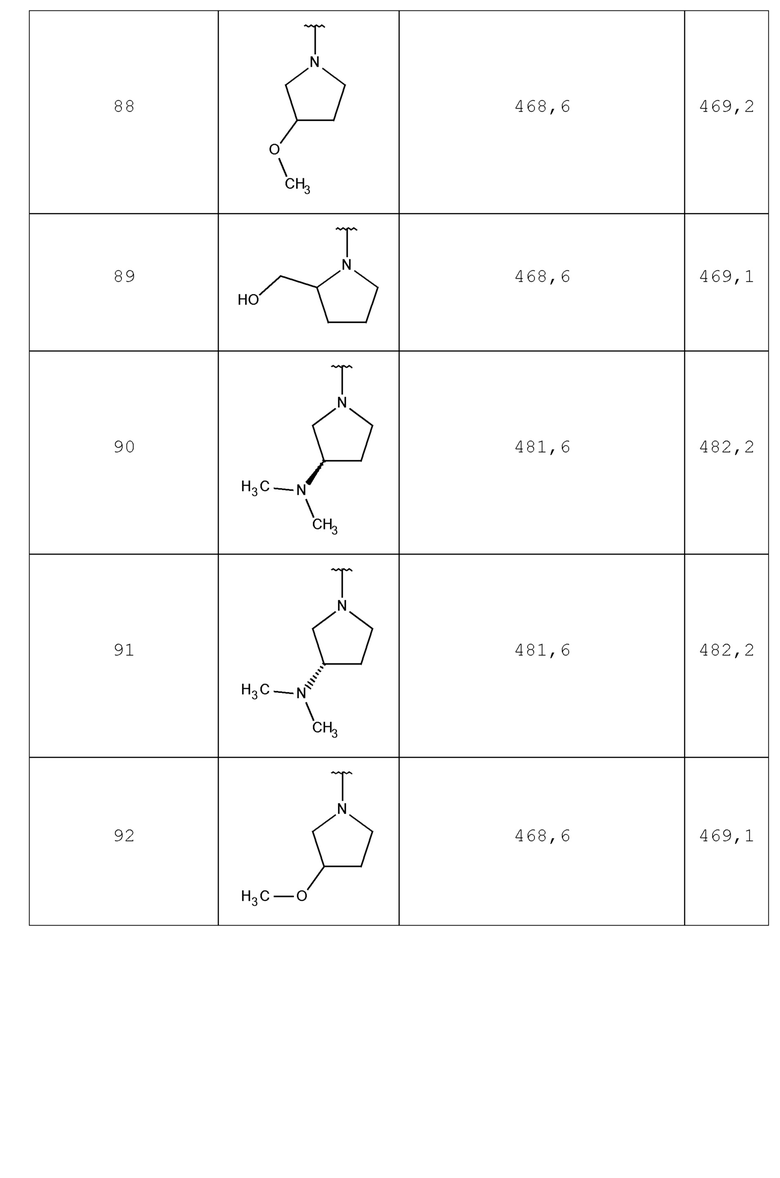
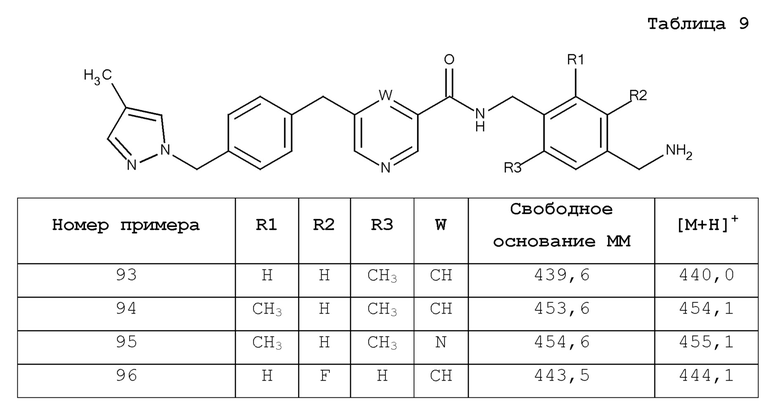
Данные 1H ЯМР примеров (растворитель DMSO-d6)
Биологические методы
Способность соединений по формуле (I) ингибировать калликреин плазмы может быть определена с использованием следующих биологических исследований:
Определение ИК50 для калликреина плазмы
Ингибирующую активность калликреина плазмы in vitro определяли с использованием стандартных опубликованных методов (см., например, Johansen et al., Int. J. Tiss. Reac. 1986, 8, 185; Shori et al., Biochem. Pharmacol., 1992, 43, 1209; Stürzebecher et al., Biol. Chem. Hoppe-Seyler, 1992, 373, 1025). Человеческий калликреин плазмы (Protogen) инкубировали при 37° с фторогенным субстратом H-DPro-Phe-Arg-AFC и различными концентрациями тестируемого соединения. Остаточную ферментативную активность (начальная скорость реакции) определяли путем измерения изменения оптического поглощения при 410 нм, и определяли значение ИК50 для тестируемого соединения.
Данные, полученные из указанных исследований, показаны в таблице 12 ниже. Обычно, но не исключительно, предпочтительные соединения демонстрируют ИК50 менее чем 200 нM.
Выбранные соединения дополнительно скринировали в отношении ингибирующей активности относительно связанного фермента KLK1. Способность соединений по формуле (I) ингибировать KLK1 может быть определена с использованием следующего биологического исследования:
Определение ИК50 для KLK1
Ингибирующую активность KLK1 in vitro определяли с использованием стандартных опубликованных методов (см., например, Johansen et al., Int. J. Tiss. Reac. 1986, 8, 185; Shori et al., Biochem. Pharmacol., 1992, 43, 1209; Stürzebecher et al., Biol. Chem. Hoppe-Seyler, 1992, 373, 1025). Человеческий KLK1 (Callbiochem) инкубировали при 37oC с фторогенным субстратом H-DVal-Leu-Arg-AFC и различными концентрациями тестируемого соединения. Остаточную ферментативную активность (исходная скорость реакции) определяли путем измерения изменений оптического поглощения при 410 нм и определяли значения ИК50 для тестируемого соединения.
Данные, полученные из указанного анализа, показаны в таблице 13 ниже:
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КАЛЛИКРЕИНА ПЛАЗМЫ | 2014 |
|
RU2712621C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2016 |
|
RU2744766C2 |
| НОВЫЕ СОЕДИНЕНИЯ КОНДЕНСИРОВАННЫХ ГЕТЕРОЦИКЛИЛ-КАРБОНОГИДРАЗОНОИЛДИЦИАНИДОВ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2826002C1 |
| ИНГИБИТОРЫ БРОМОДОМЕНА | 2017 |
|
RU2741808C2 |
| МОНОЦИКЛИЧЕСКОЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ | 2014 |
|
RU2645352C2 |
| Соединения, полезные в качестве пестицидов, и промежуточные соединения, композиции и способы, связанные с ними | 2016 |
|
RU2821715C2 |
| АРИЛ-N-АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2815493C2 |
| ФЕНИЛ/ПИРИДИЛ-N-ФЕНИЛ/ПИРИДИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2803216C2 |
| (6S,9AS)-N-БЕНЗИЛ-6-[(4-ГИДРОКСИФЕНИЛ)МЕТИЛ]-4,7-ДИОКСО-8-({ 6-[3-(ПИПЕРАЗИН-1-ИЛ)АЗЕТИДИН-1-ИЛ]ПИРИДИН-2-ИЛ} МЕТИЛ)-2-(ПРОП-2-ЕН-1-ИЛ)-ОКТАГИДРО-1H-ПИРАЗИНО[2,1-C][1,2,4]ТРИАЗИН-1-КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2669805C2 |
| ПРОИЗВОДНОЕ N-(АЗААРИЛ)ЦИКЛОЛАКТАМ-1-КАРБОКСАМИДА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2765785C2 |
Изобретение относится к соединению по формуле I, в которой B представляет собой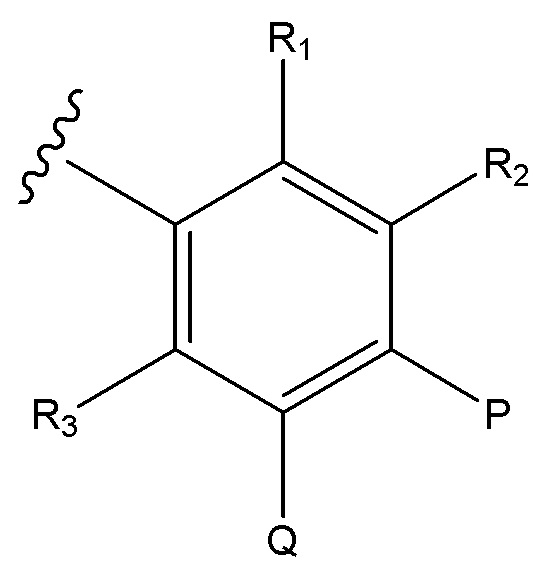 или B является конденсированным 6,5- или 6,6-гетероароматическим бициклом, содержащим N и, необязательно, один дополнительный гетероатом, выбираемый из N, который является необязательно моно- или дизамещенным заместителем, выбираемым из алкила и NR8R9; W, X, Y и Z независимо выбирают из C, C(R16)-C, C(R16)=C, C=N и N, так что цикл, содержащий W, X, Y и Z, представляет собой шестичленный ароматический гетероцикл; или цикл, содержащий W, X, Y и Z, представляет собой
или B является конденсированным 6,5- или 6,6-гетероароматическим бициклом, содержащим N и, необязательно, один дополнительный гетероатом, выбираемый из N, который является необязательно моно- или дизамещенным заместителем, выбираемым из алкила и NR8R9; W, X, Y и Z независимо выбирают из C, C(R16)-C, C(R16)=C, C=N и N, так что цикл, содержащий W, X, Y и Z, представляет собой шестичленный ароматический гетероцикл; или цикл, содержащий W, X, Y и Z, представляет собой 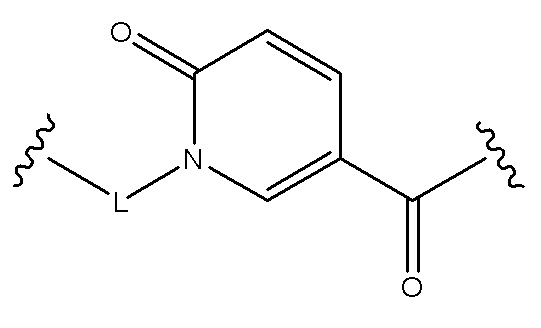 , R5, R6 и R7 независимо отсутствуют или независимо выбирают из H, алкила, алкокси, OH и -NR8R9; A выбирают из арила, гетероарила и заместителя формулы (A), (B), (C) и (D). Значения остальных заместителей указаны в формуле изобретения. Изобретение относится также к фармацевтической композиции, к применениям соединения, к способу лечения заболевания или состояния. Технический результат: получены новые соединения, обладающие активностью ингибировать плазменный калликреин. 6 н. и 21 з.п. ф-лы, 13 табл., 96 пр.
, R5, R6 и R7 независимо отсутствуют или независимо выбирают из H, алкила, алкокси, OH и -NR8R9; A выбирают из арила, гетероарила и заместителя формулы (A), (B), (C) и (D). Значения остальных заместителей указаны в формуле изобретения. Изобретение относится также к фармацевтической композиции, к применениям соединения, к способу лечения заболевания или состояния. Технический результат: получены новые соединения, обладающие активностью ингибировать плазменный калликреин. 6 н. и 21 з.п. ф-лы, 13 табл., 96 пр.
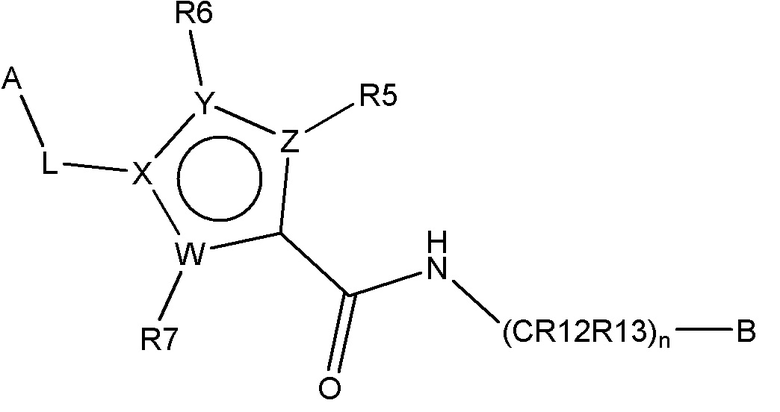 Формула (I)
Формула (I)
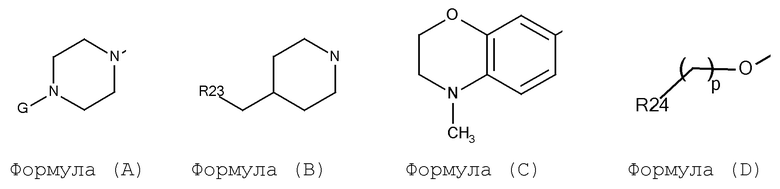
1. Соединение по формуле I
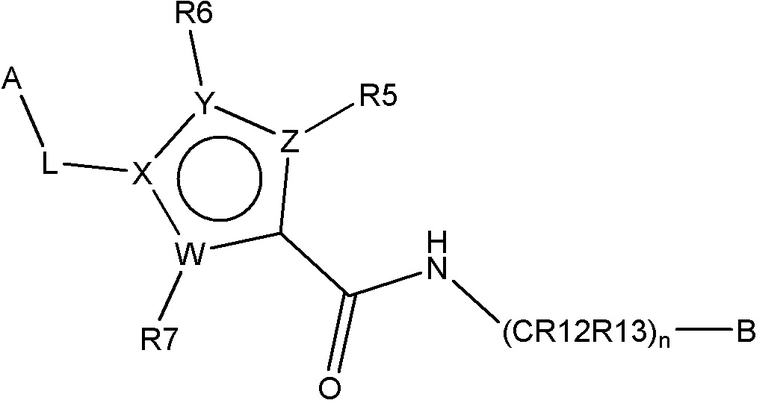
Формула (I)
где
B представляет собой
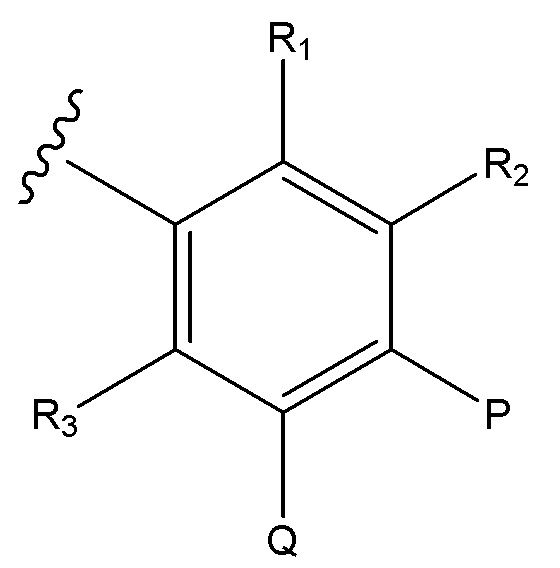
или B является конденсированным 6,5- или 6,6-гетероароматическим бициклом, содержащим N и, необязательно, один дополнительный гетероатом, выбираемый из N, который является необязательно моно- или дизамещенным заместителем, выбираемым из алкила и NR8R9;
n равно 1;
W, X, Y и Z независимо выбирают из C, C(R16)-C, C(R16)=C, C=N и N, так что цикл, содержащий W, X, Y и Z, представляет собой шестичленный ароматический гетероцикл;
или цикл, содержащий W, X, Y и Z, представляет собой
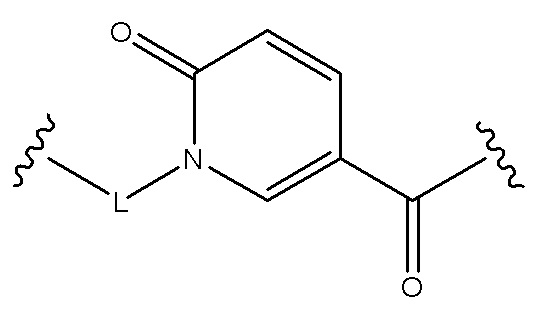
R5, R6 и R7 независимо отсутствуют или независимо выбирают из H, алкила, алкокси, OH и -NR8R9;
R16 независимо выбирают из H, алкокси и NR8R9;
A выбирают из арила, гетероарила и заместителя формулы (A), (B), (C) и (D):
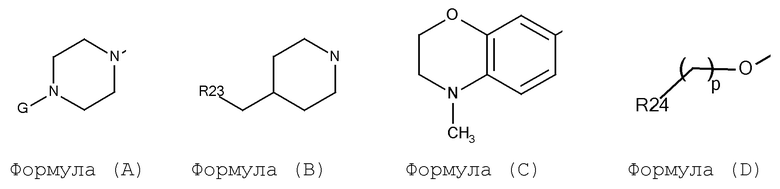
где:
G выбирают из алкила, CO-арила, SO2-арила, (CH2)m-арила и (CH2)m-гетероарила;
m выбирают из 0 и 1;
p выбирают из 0, 1, 2 и 3;
R23 представляет собой гетероарил;
R24 представляет собой гетероарил;
L представляет собой линкер, выбираемый из -(CHR17)- и -(CH2)1-6;
R1 и R2 независимо выбирают из H, алкила и галогена;
R3 выбирают из H и алкила;
P представляет собой -C(R18)(R19)NH2 и Q представляет собой H;
R8 и R9 независимо выбирают из H и алкила;
R12 и R13 независимо выбирают из Н;
R17 представляет собой OH;
R18 и R19 представляют собой H;
алкил, представляющий линейный насыщенный углеводород, имеющий до 6 атомов углерода (C1-C6), или разветвленный насыщенный углеводород из 3-6 атомов углерода (C3-C6); алкил может необязательно быть замещенным 1 заместителем, выбираемым из OH, COOR10, фенила и NR10R11;
алкокси представляет собой линейный O-связанный углеводород от 1 до 6 атомов углерода (C1-C6) или разветвленный O-связанный углеводород от 3 до 6 атомов углерода (C3-C6); алкокси может необязательно быть замещенным 1 заместителем, выбираемым из OCH3;
арил представляет собой фенил, арил может быть необязательно замещенным 1 или 2 заместителями, независимо выбираемыми из алкокси, галогена, гетероарила, -(CH2)1-3-гетероарила, -(CH2)1-3-NR14R15 и -NR10R11;
гетероарил представляет собой 5-, 6-, 9- или 10-членный моно- или бициклический ароматический цикл, содержащий, когда возможно, 1 или 2 члена цикла, независимо выбираемых из N, NR8 и S; гетероарил может быть необязательно замещенным 1 заместителем, независимо выбираемым из алкила, алкокси, OH, галогена, арила, морфолинила, пиперидинила, - и -NR10R11;
R10 и R11 независимо выбирают из H и алкила; или R10 и R11 вместе с азотом, к которому они прикреплены, образуют 5- или 6-членный гетероцикл, который может быть насыщенным или ненасыщенным с 2 двойными связями и который может быть необязательно моно- или дизамещенным заместителями, выбираемыми из оксо, алкила, алкокси, COOR8, OH и F;
R14 и R15 вместе с азотом, к которому они присоединены, образуют 5- или 6-членный гетероцикл, который является ненасыщенным с 2 двойными связями и необязательно может быть оксозамещенным;
и его фармацевтически приемлемые соли.
2. Соединение по п. 1, где B выбирают из необязательно моно- или дизамещенного изохинолинила и необязательно замещенного 1H-пирроло[2,3-b]пиридина, где указанные необязательные заместитель(и) выбирают из алкила и NR8R9, где алкил, R8 и R9 являются, как определено в п. 1.
3. Соединение по п. 1, где B представляет собой:
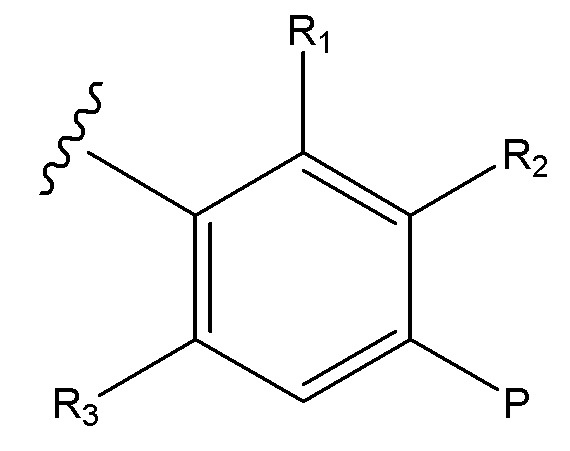
где
R1 и R2 независимо выбирают из H, алкил и галогена;
R3 выбирают из H и алкила;
P представляет собой -CH2NH2; и
алкил определен в п. 1.
4. Соединение по п. 1 или 2, где B представляет собой необязательно монозамещенный изохинолинил, где указанный необязательный заместитель представляет собой NR8R9; и где R8 и R9 являются, как определено в п. 1.
5. Соединение по п. 1 или 2, где B является необязательно монозамещенным изохинолинилом, где указанным необязательным заместителем является NR8R9; и где R8 и R9 представляют собой H.
6. Соединение по п. 1 или 2, где B является необязательно замещенным 1H-пирроло[2,3-b]пиридином, где указанным необязательным заместителем является NR8R9; и где R8 и R9 являются, как определено в п. 1.
7. Соединение по любому из пп. 1-3, где L представляет собой -(CH2)1-6- или -(CHOH)-.
8. Соединение по любому из пп. 1-3, где L представляет собой -CH2-.
9. Соединение по п. 1, как определено формулой (III),
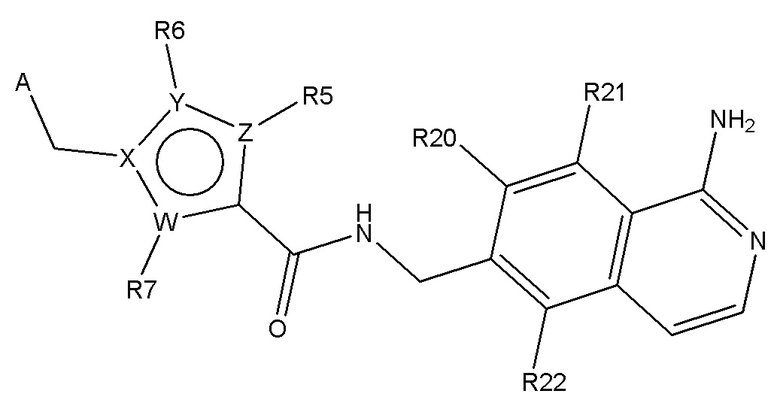
формула (III)
и где R20, R21 и R22 независимо выбирают из H, алкила и NR8R9; и где A, W, X, Y, Z, R5, R6, R7, алкил, R8 и R9 являются, как определено в п. 1.
10. Соединение по любому из пп. 1-3, где W, X, Y и Z независимо выбирают из C=N, C, C(R16)-C, C(R16)=C и N, так что цикл, содержащий W, X, Y и Z, представляет собой шестичленный ароматический гетероцикл; где R16 представляет собой H.
11. Соединение по любому из пп. 1-3, где W, X, Y и Z образуют шестичленный ароматический гетероцикл, выбираемый из:
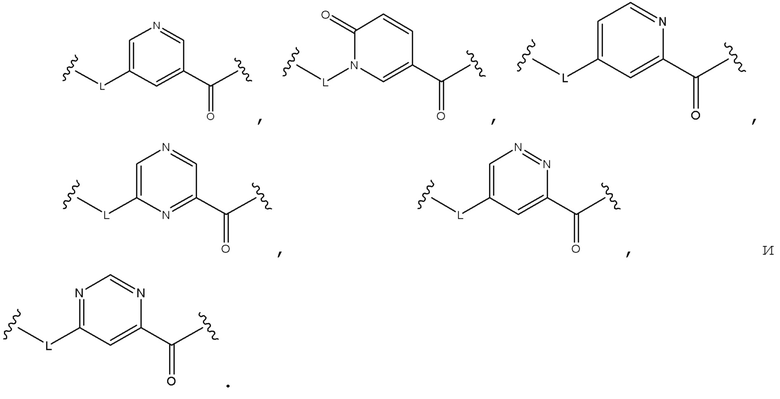
12. Соединение по любому из пп. 1-3, где A представляет собой гетероарил, замещенный фенилом, где фенил является необязательно замещенным, как определено в п. 1; или A представляет собой фенил, замещенный гетероарилом, -(CH2)1-3-гетероарилом или -(CH2)1-3-NR14R15, где гетероарил, R14 и R15 являются, как определено в п. 1.
13. Соединение по любому из пп. 1-3, где A выбирают из:
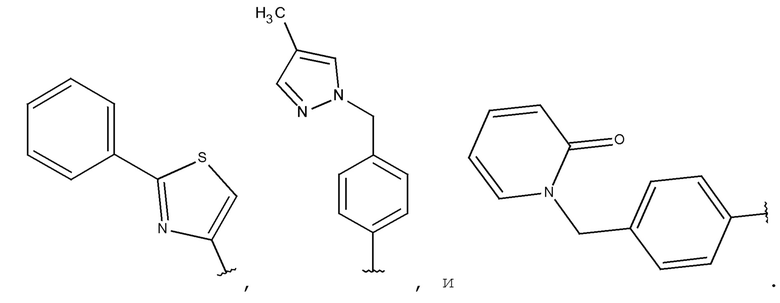
14. Соединение по п. 1, выбираемое из:
и его фармацевтически приемлемые соли.
15. Фармацевтическая композиция, обладающая активностью ингибировать плазменный калликреин, включающая терапевтически эффективное количество соединения, как заявлено в любом из пп. 1-14, и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
16. Применение соединения по любому из пп. 1-14 в медицине для лечения или профилактики заболевания или состояния, в которое вовлечена активность калликреина.
17. Применение соединения, как заявлено в любом из пп. 1-14, для изготовления лекарственного препарата для лечения или профилактики заболевания или состояния, в которое вовлечена активность плазменного калликреина.
18. Способ лечения заболевания или состояния, в которое вовлечена активность калликреина плазмы, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения, как заявлено в любом из пп. 1-14.
19. Применение соединения по любому из пп. 1-14 в способе лечения заболевания или состояния, в которое вовлечена активность плазменного калликреина.
20. Применение по п. 17 или 19, где заболевание или состояние, в которое вовлечена активность плазменного калликреина, выбирают из нарушения остроты зрения, диабетической ретинопатии, диабетического макулярного отека, врожденного ангиоотека, диабета, кровоизлияния в головной мозг, диссеминированного внутрисосудистого свертывания, коагуляции крови во время операции сердечно-легочного шунтирования и кровотечения из послеоперационных ран.
21. Применение по п. 17 или 19, где заболевание или состояние, в которое вовлечена активность плазменного калликреина, представляет собой проницаемость сосудов сетчатки, ассоциированную с диабетической ретинопатией и диабетическим макулярным отеком.
22. Применение по п. 17 или 19, где заболевание или состояние, в которое вовлечена активность плазменного калликреина, представляет собой диабетический макулярный отек.
23. Применение по п. 17 или 19, где заболевание или состояние, в которое вовлечена активность плазменного калликреина, представляет собой врожденный ангиоотек.
24. Способ по п. 18, где заболевание или состояние, в которое вовлечена активность плазменного калликреина, выбирают из нарушения остроты зрения, диабетической ретинопатии, диабетического макулярного отека, врожденного ангиоотека, диабета, кровоизлияния в головной мозг, диссеминированного внутрисосудистого свертывания, свертывания крови во время операции сердечно-легочного шунтирования и кровотечения из послеоперационных ран.
25. Способ по п. 18, где заболевание или состояние, в которое вовлечена активность плазменного калликреина, представляет собой проницаемость сосудов сетчатки, ассоциированную с диабетической ретинопатией и диабетическим макулярным отеком.
26. Способ по п. 18, где заболевание или состояние, в которое вовлечена активность плазменного калликреина, представляет собой диабетический макулярный отек.
27. Способ по п. 18, где заболевание или состояние, в которое вовлечена активность плазменного калликреина, представляет собой врожденный ангиоотек.
| WO 2008016883 A2, 07.02.2008 | |||
| WO 2012017020 A1, 09.02.2012 | |||
| WO 2012142308 A1, 18.10.2012 | |||
| WO 2013111108 A1, 01.08.2013 | |||
| EA 201200917 A1, 28.12.2012 | |||
| КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛЬПАИНОВ | 2007 |
|
RU2485114C2 |

