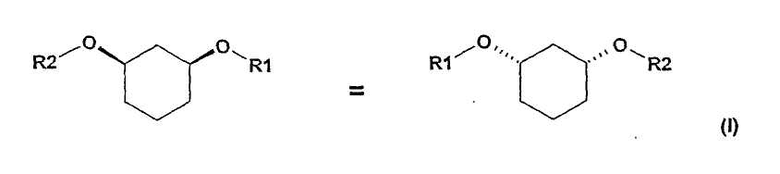
Изобретение относится к способу получения хиральных, нерацемических 1,3-дизамещенных циклогексанoлов формулы (I) в цис-конфигурации
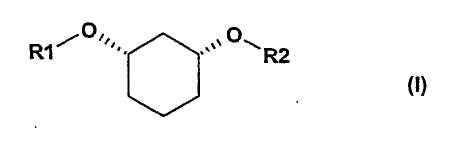
Различно замещенные, имеющие цис-конфигурацию 1,3-дизамещенные производных циклогексана (соединения формулы (I) при R1≠R2,) являются центральными структурными элементами или предшественниками описанных в WO 03/020269 лекарственных активных веществ, которые в целом подходят для лечения нарушений липидного обмена, например, диабета II типа, синдрома X и др.
Описанные в патентной заявке WO 03/020269 способы синтеза нерацемических, имеющих цис-конфигурацию производных 1,3-циклогексана, не годятся в качестве технологического процесса: так, например, алкилирование посредством NaH/ДМФ в многокилограммовом масштабе с надежностью не осуществимо (C&EN, 13 сентября, 1982,5). Кроме того, алкилирование по способу Bu2SnO в масштабе опытной установки требует недопустимо высоких затрат; отделение соединений олова от желаемого продукта с применением хроматографических методов разделения также очень затруднительно и чаще всего неполное. Утилизация соединений цинка является следующей проблемой или экономическим фактором. Разделение энантиомеров (расщепление рацематов) путем хроматографии на хиральной фазе также является трудоемким и слишком дорогим. Помимо этого, для хроматографического разделения энантиомеров требуется, чтобы рацемическое соединение имело хорошую химическую чистоту, что часто можно достичь только путем дополнительной, заранее проводимой хроматографии.
Другие описанные в литературе методы синтеза цис-1,3-циклогександиольных структурных элементов или производных, как, например, раскрытие эпоксициклогексана (P.Crotti, V. Di Bussolo, L.Favero, M.Pineschi, F.Marianucci, G.Renzi, G.Amici, G.Roselli, Tetrahedron 2000, 56, 7513-7524 и цитированная литература) или катализированное металлами гидроборирование производных циклогексена (J.A.Brinkmann, T.T.Nguyen, J.R.Sowa, J., Org. Lett. 2000, 2, 981-983; C.E.Garrett, G.C.Fu, J. Org. Chem. 1998, 63, 1370-1371), являются в отношении регио- и стереоселективности в подавляющем большинстве неудовлетворительными. Помимо этого, общее количество стадий заметно более высокое. Поэтому в качестве промышленных способов они не годятся.
Синтез производных цис-1,3-циклогександиола из цис,цис-1,3,5-циклогексантриола или производных цис,цис-1,3,5-циклогексантриола (L.Dumortier, M.Carda, J. Van der Eycken, G.Snatzke, M. Vandewalle, Tetrahedron: Asymmetry 1991, 2, 789-792; H.Suemune, K.Matsuno, M.Uchida, K.Sakai, Tetrahedron: Asymmetry 1992, 3, 297-306) из-за большого числа этапов также очень затратен и неэкономичен и поэтому не подходит для промышленного применения.
Ферментативная реакция цис/транс смесей 1,3-циклогександиола с S-этилтиооктаноатом также не годится в качестве промышленного способа. Помимо практически неизбежной загазованности при обращении с соединениями серы и того факта, что для достижения требуемой степени превращения высвобождающийся этантиол должен непрерывно удаляться, описанная реакция приводит к смеси 9 изомерных форм или производных циклогександиола, а именно: непрореагировавшим изомерам (S,S)-диол, (R,R)-диол и (R,S)-диол, кроме того, моноацилированным продуктам (S,S)-монооктаноат, (R,R)-монооктаноат и (R,S)-монооктаноат, и в-третьих, группе диацилированных продуктов (S,S)-диоктаноат, (R,R)-диоктаноат и (R,S)-диоктаноат. Оптически активный, моноацилированный (R,S)-монооктаноат, имеющий цис-конфигурации, составляет во фракции моноацилированных циклогександиолов долю всего примерно 12%. Проводимое в препаративном масштабе получение и выделение этого продукта не описано, однако ввиду количественного соотношения и описанных проблем разделения не может быть экономичным. Кроме того, известно, что частично ацилированные ди- или полигидроксисоединения склонны к миграции ацильных групп. Если этот случай имеет место, например, в ходе очистки (R,S)-монооктаноата (например, при хроматографии на силикагеле или при водной экстракции) или в ходе последующей реакции (например, при алкилировании свободных гидроксильных групп), то это ведет к заметному уменьшению оптической чистоты, или к рацемизации.
Имеющие цис-конфигурирацию (R,S)-диолы или диацилированные (R,S)-соединения оптически не активны и поэтому не представляют интереса.
Поэтому задачей настоящего изобретения является разработка способа, который не имеет указанных недостатков.
Задачей настоящего изобретения является способ получения хиральных, нерацемических соединений формулы I
с R1
в которой означают:
и
отличающийся тем, что проводят:
A)
a) алкилирование (Alk-R 2 /Alk-SG)
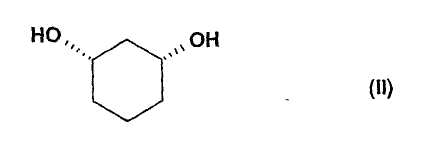
Цис-1,3-циклогександиол формулы (II)
подвергают взаимодействию с соединением формулы (III)

где
R2 определен выше, и
X1 означает Cl, Br, I, OMs (O-мезил), OTs (O-тозил), OTf (O-трифлат);
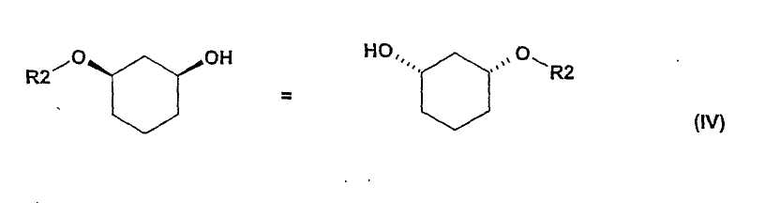
в присутствии основания в подходящем растворителе, с получением рацемического соединения формулы (IV)
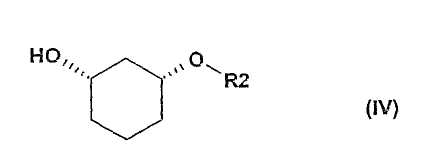
в которой R2 определен выше,
b1) ферментативное образование сложного эфира (EB)+разделение (T)
полученное соединение формулы (IV) подвергают стереоселективному ферментативному образованию сложного эфира (EB), причем спирты в органическом растворителе, как например, дихлорметан, смешивают с донором ацильных групп, как например, виниловым эфиром R6-O-CH=CH2 или ангидридом кислоты R6-O-R6, где R6 определен выше, и ферментом, и полученную смесь перемешивают при температуре от -20 до 80°C, и по окончании реакции один стереомер присутствует в виде сложного эфира формулы (V)
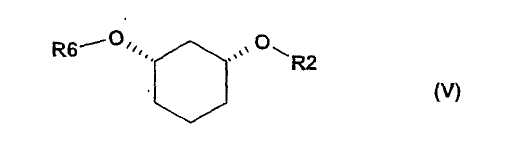
где
R6 означает C(=O)-(C1-C16)-алкил, C(=O)-(C2-C16)-алкенил, C(=O)-(C3-C16)-алкинил, C(=O)-(C3-C16)-циклоалкил, причем один или несколько атомов углерода могут быть заменены атомами кислородa и могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, CN, NO2, гидрокси, метокси, этокси, фенил и CO-O(C1-C4)-алкил, CO-O(C2-C4)-алкенил, которые в свою очередь могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3 , и
R2 определен выше,
и другой стереомер остается в неизмененном виде, как спирт формулы (IV), и поэтому за счет использования их различающихся химических или физико-химических свойств (например, значения Rf или разницы растворимости в воде или других растворителях) они могут быть отделены друг от друга (разделение T), например, путем простой хроматографии на силикагеле, экстракцией (например, смесью гептан/метанол или органический растворитель/вода) или также путем проводимых далее химических последовательных реакций, например, спирта, в которых сложный эфир не участвует,
или
b2) ферментативное расщепление сложного эфира [=химическая этерификация (CV)+ферментативное расщепление (ES)]+разделение (T)
полученные соединения формулы (IV) подвергают стереоселективному ферментативному расщеплению сложного эфира, причем рацемические спирты сначала путем химической этерификации (CV), например, посредством хлорангидрида кислоты R6-Cl или ангидрида кислоты R6-O-R6, в присутствии основания, как например, триэтиламин, преобразуют в рацемический сложный эфир формулы (V)
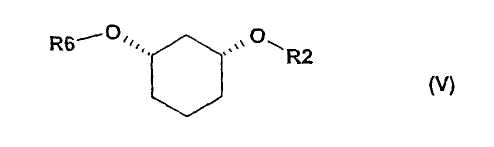
где R6 и R2 определены выше,
который затем путем проведения стереоселективного ферментативного расщепления сложного эфира (ES) вводят в гомогенную или гетерогенную, водную, водно-органическую или органическую среду и при температуре 10-80°C в присутствии фермента вводят в реакцию, в случае гидролиза - с водой, а в случае алкоголиза - со спиртом, как например, н-бутанолом, по завершении которой один стереомер присутствует как спирт формулы (IV), а другой остается неизмененным в виде сложного эфира формулы (V), и тем самым они могут быть отделены друг от друга как описано в пункте b1), причем
энантиомеры формулы (IV), получающиеся как спирт, перерабатываются далее как описано в пункте d),
или
c) химический гидролиз (CH)
энантиомеры формулы (V), получающиеся как сложный эфир, могут быть омылены с получением химически энантиомерных спиртов по известному способу, и
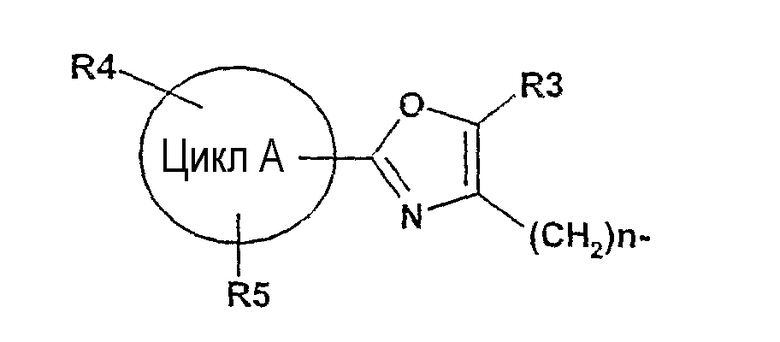
d) алкилирование (Alk-R 1 )
далее подвергают взаимодействию с соединениями формулы (VI)
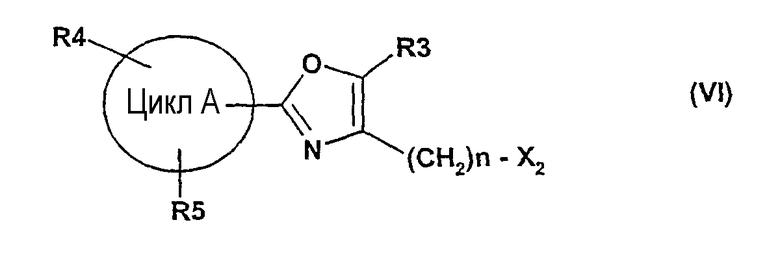
где
цикл А, R3, R4, R5 и n определены выше и
X2 означает Cl, Br, I, OTs, OMs, OTf;
в присутствии основания в подходящем растворителе с получением соединения формулы (I), и
e) отщепление защитной группы SG (AbSG)
если R2 означает OH-защитную группу (SG), и R2 определен, как описано выше, соединения формулы (Ia)
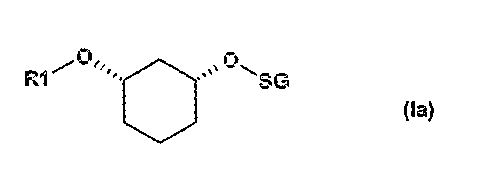
где R1 и SG определены выше,
путем отщепления защитной группы по известному способу, как например, в случае отщепления SG=бензилоксиметил или SG=бензил-гидрированием на Pd/C, или в случае отщепления SG=пара-метоксибензил - например, посредством DDQ (2,3-дихлор-5,6-дицианобензохинон), или в случае отщепления SG=трет-бутил-диметилсилил - например, посредством Bu4NF, переводят в соединения формулы (VII)
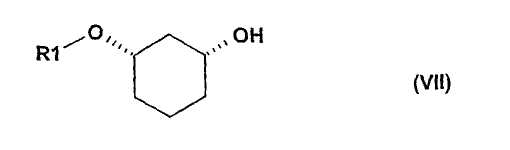
где R1 определен выше,
f) алкилирование (Alk-R 2 )
которое затем подвергают взаимодействию с соединениями формулы (III),
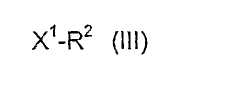
где X1 и R2 определены выше,
в присутствии основания в подходящем растворителе с получением соединения формулы (I), продукта или энантиомерной формы,
причем возможно также последовательность отдельных стадий реакции, которые описаны выше в пункте A):
A) Alk-R2→EB+T/CV+ES+T [→CH]→Alk-R1 [→AbSG→Alk-R2]→
продукт/энантиомерная форма
изменить на
B) Alk-R1→EB+T/CV+ES+T [→CH]→Alk-R2 [→AbSG→Alk-R2]→
продукт/энантиомерная форма
или на
C) Alk-SG→EB+T/CV+ES+T→CH→Alk-R2→AbSG→Alk-R1→
продукт/энантиомерная форма
или
D) Alk-SG→EB+T/CV+ES+T→Alk-R1→AbSG→Alk-R2→
продукт/энантиомерная форма.
Далее на схемах I-IV представлены возможные варианты способа:
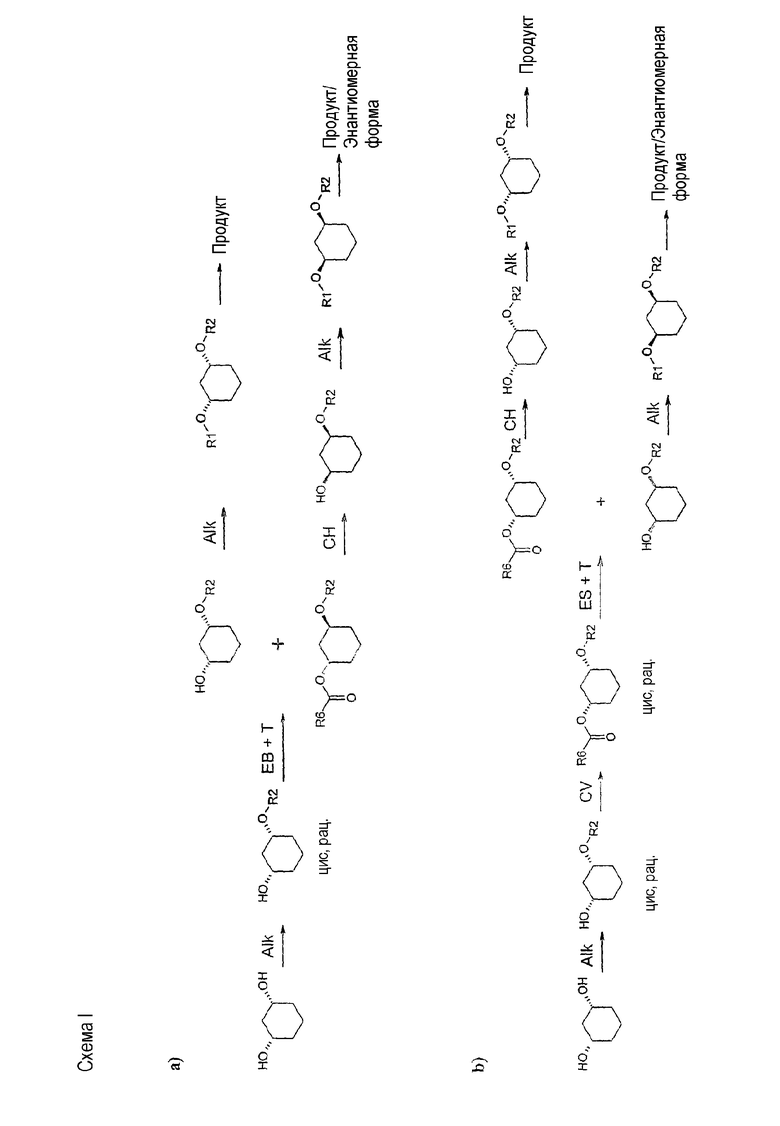

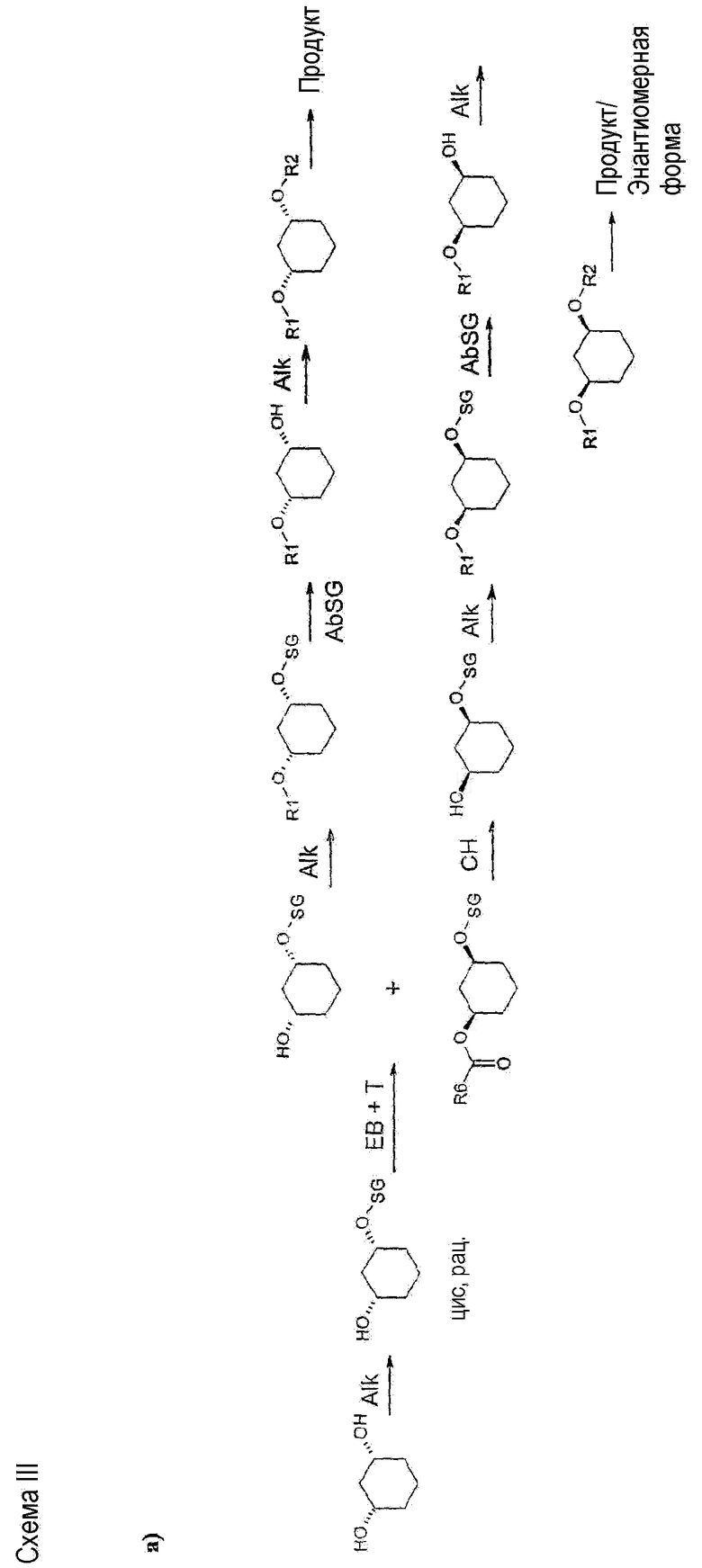
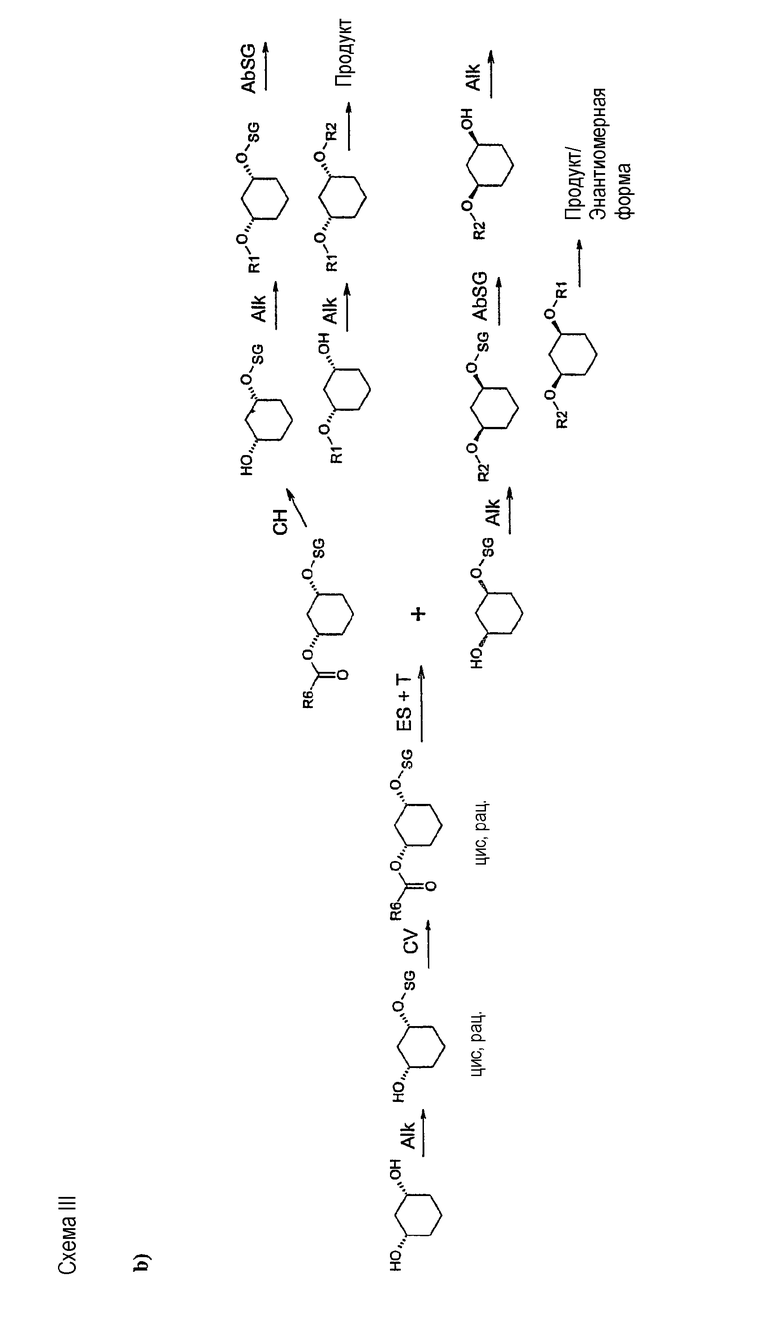
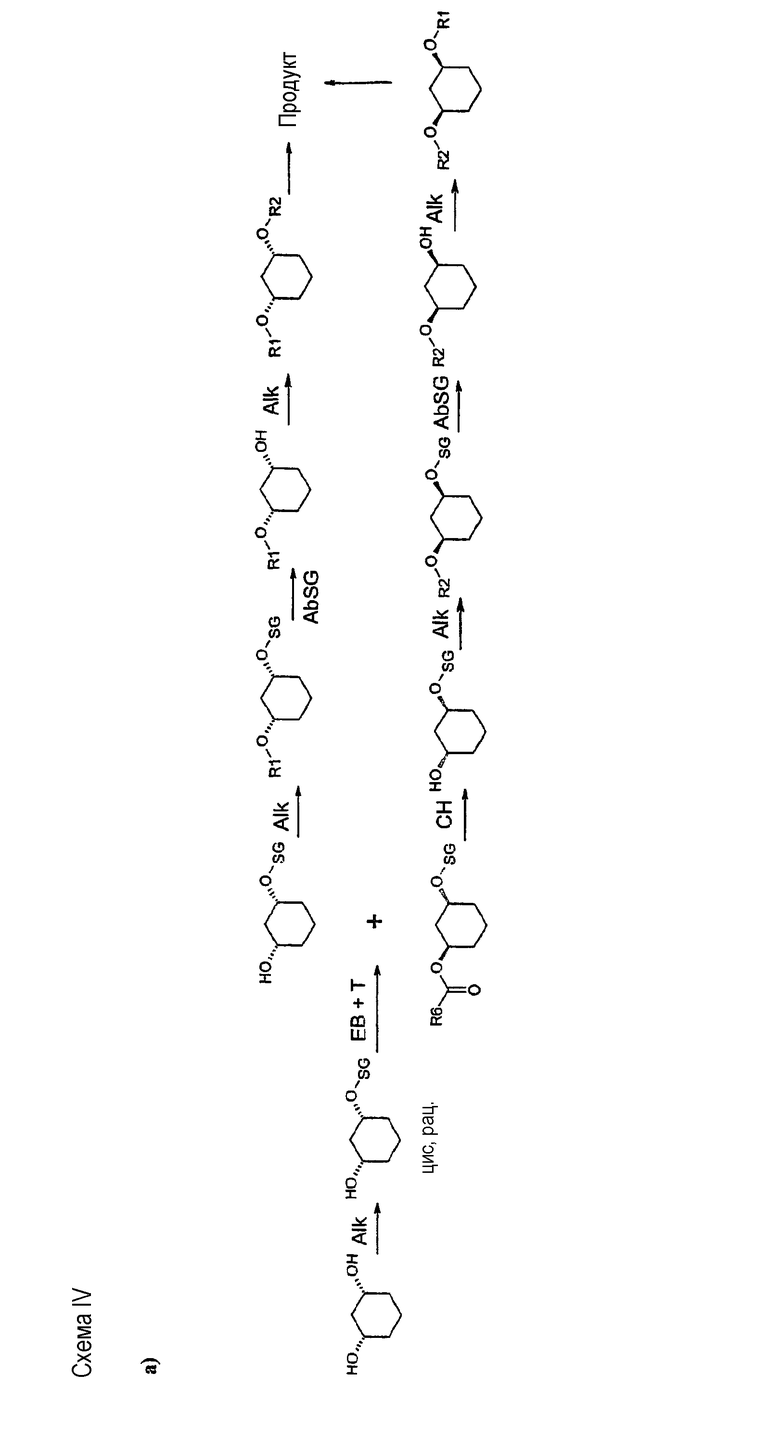
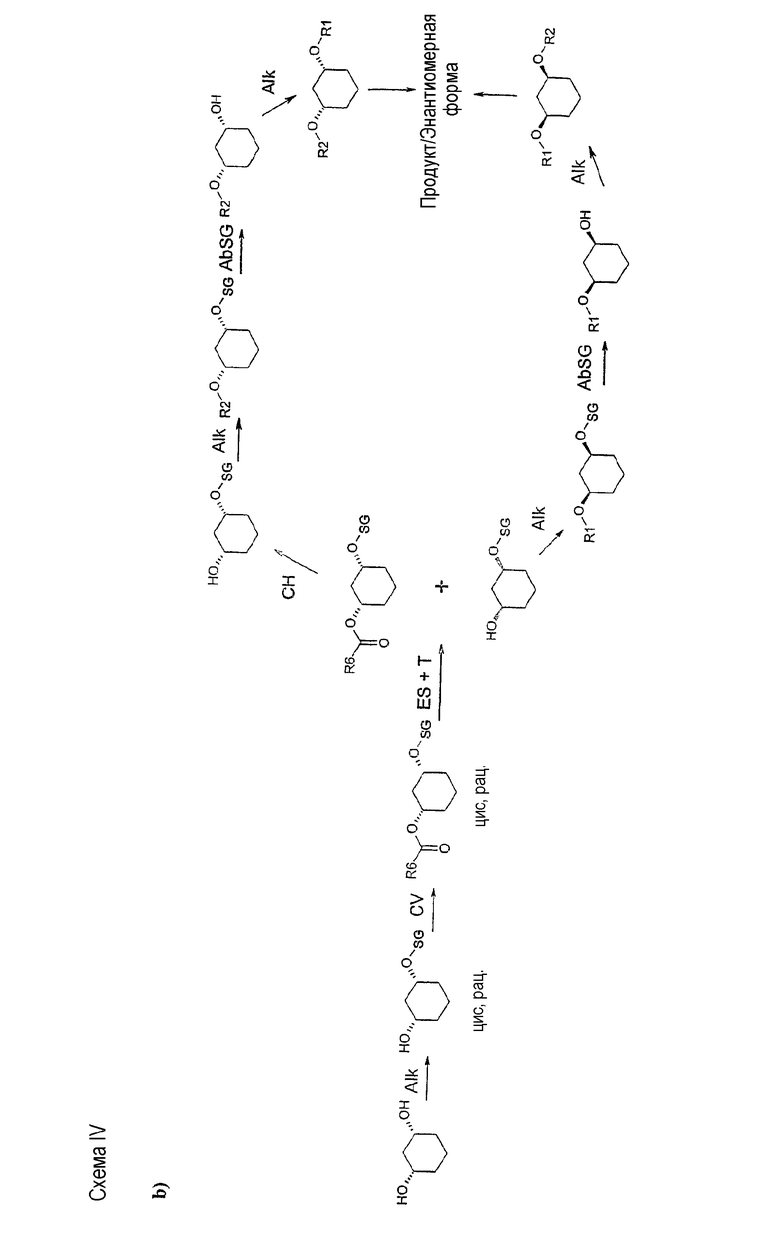
Способ согласно изобретению является экономичным, простым и быстрым. Способ полностью предотвращает риск миграции ацильных групп, он не требует эквимолярных количеств оптически чистых исходных или вспомогательных веществ, дорогих реагентов, разделения рацематов путем хроматографии на хиральных фазах, непропорционально больших количеств растворителя и дорогостоящих стадий обработки.
Типичные для отщепления рацематов потери 50% могут быть уменьшены применением обоих энантиомеров и изменением последовательности алкилирования. Предпочтителен так называемый энантиоконвергентный способ (смотри схему IV или способы C) и D)), при котором действуют, например, следующим образом: алкилирование цис-1,3-циклогександиола формулы (II) соединением формулы (III), причем R2 в качестве SG выбирают так, чтобы SG в ходе дальнейшего синтеза можно было опять легко и селективно удалить, таким образом, SG является, например, бензилом, или пара-метоксибензилом, или трет-бутил-диметилсилилом; полученное соединение формулы (IV) подвергают стереоселективному ферментативному образованию или расщеплению сложного эфира (см. выше) и после прошедшего разделения оба: непрореагировавший спирт и сложный эфир, - по отдельности и различными путями переводят в одинаковый оптически чистый продукт, тем, что спирт (как описано в первой части) преобразуют, например, соединением формулы (VI), в соединение формулы (Ia), которое затем путем отщепления SG-группы преобразуют в соединение формулы (VII), и его затем с помощью соединения формулы (III) с R2, какой желателен в продукте, преобразуют в соединение формулы (I), а изомерный сложный эфир, напротив, путем простого расщепления сложного эфира переводят в соединение формулы (IV), которое затем сначала с помощью соединения формулы (III) с R2, какой желателен в продукте, преобразуют в соединение формулы (VIII),
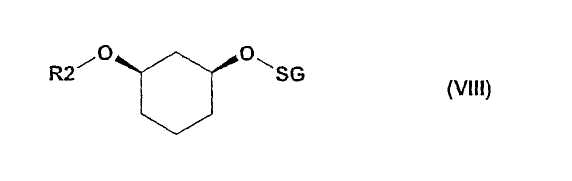
которое, в свою очередь, отщеплением SG-группы преобразуют в соединение формулы (IV)
а его затем с помощью соединения формулы (VI) преобразуют в соединение формулы (I).
Предпочтительно используют соединения формулы (III)
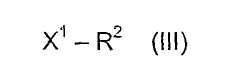
в которых
X1 означает Cl, Br, I, OMs или OTs,
Особенно предпочтительны такие соединения, в которых
X1 означает Cl, Br, или I.
Предпочтителен способ получения соединений формулы (I),
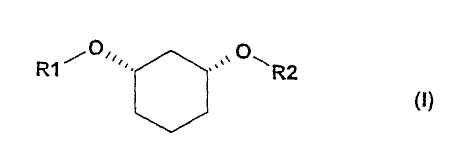
в которой:
R1 является
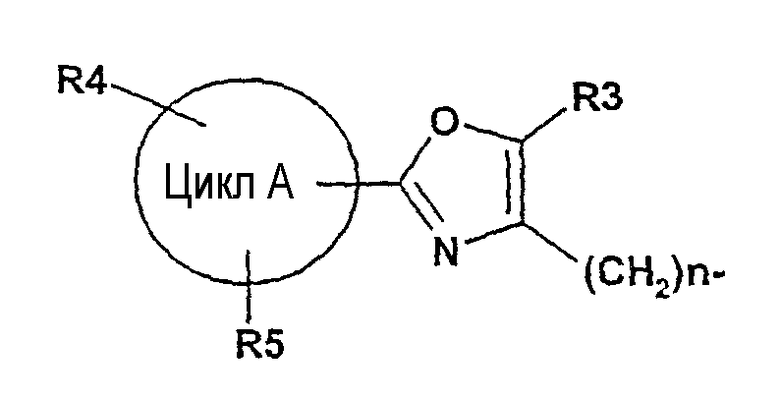
где
Особенно предпочтителен способ получения соединения формулы (I),
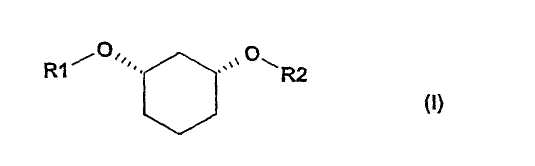
в которой:
R1 является

где
Алкильные остатки в заместителях R2, R3, R4 и R5 могут быть как линейными, так и разветвленными.
Под гетероароматическим циклом понимаются как моно-, так и бициклические кольца максимум с 4 гетероатомами, в частности, такие, которые содержат до 4 атомов азотa и/или 1 кислород или 1 атом серы, как, например: фуран, тиофен, тиазол, оксазол, тиадиазол, триазол, пиридин, триазин, хинолин, изохинолин, индол, бензотиофен, бензофуран, бензотриазол. Ароматические циклы могут быть моно- или бициклическими, а также конденсированными, как, например, нафтил, бензо[1,3]диоксол, дигидро-бензо[1,4]-диоксин.
Рацемические, имеющие цис-конфигурацию 1,3-производные циклогексана формулы (IV) и формулы (VII) получают моноалкилированием цис-циклогександиола (соединения формулы II), но оно может быть получено также восстановительным раскрытием соответствующего ацеталя (R.Hunter et al., J. Org. Chem. 1993, 85, 6756), кроме того, так называемым восстановительным образованием простого эфира исходя из простых силиловых эфиров и альдегидов или кетонов (J.S.Bajwa, X.Jiang, J.Slade, K.Prasad, O.Repic, T.J.Blacklock, Tetrahedron Lett. 2002, 43, 6709-6713).
Реагенты алкилирования формулы III имеются в продаже или могут быть получены по известным из литературы методам, например, путем радикального галогенирования боковых цепей (см. литературный обзор R.C.Larock, Comprehensive Organic Transformations, S. 313, 1989 VCH Publishers, Inc.) или из спиртов или получаемых из них производных (см. литературный обзор R.C.Larock, Comprehensive Organic Transformations, S. 353-363, 1989 VCH Publishers, Inc.).
Далее, известно (см. J. Chem.Soc. 1925, 127, 2275-2297; J. Chem. Soc. 1922, 121, 2202-2215) получение различных бромидов 2-бромометилбензойной кислоты путем радикального бромирования, которые затем могут быть переведены путем последующих реакций со спиртами в сложный эфир бромометилбензойной кислоты, относящийся к группе реагентов алкилирования формулы (III).
Реагенты алкилирования формулы (VI) или спирты X2=OH, которые могут служить в качестве предшественников, имеются в продаже или могут быть получены по известным из литературы методам [a) The Chemistry of Heterocyclic Compounds (Ed.: A.Weissberger, E.C.Taylor): Oxazoles (Ed.: I.J.Turchi); b) Methoden der Organischen Chemie, Houben-Weyl 4. Auflage, Hetarene III, Teilband 1; c) I.Simit, E. Chindris, Arch. Pharm. 1971, 304, 425; d) Y.Goto, M.Yamazaki, M.Hamana, Chem. Pharm. Bull. 1971, 19(10), 2050-2057].
Реагенты алкилирования формул III и VI в присутствии основания приводят в реакцию с 1,3-циклогександиолом или производными 1,3-циклогександиола. Подходящими основаниями являются, например, гидроксиды, как KOH, карбонаты, как Cs2CO3, алкоголяты, как KOtBu, а также такие соединения, как LDA, BuLi, LiHMDS, KH, NaH и NaHMDS. Подходящими растворителями являются, например, ТГФ, MTBE, DME, NMP, ДМФ и хлорбензол.
Для разделения рацематов спиртов их вводят в органические растворители, как например, диметоксиэтан (DME), метил-трет-бутиловый эфир (MTBE), диизопропиловый эфир (DIPE), ТГФ, н-гексан, циклогексан, толуол, хлорбензол, ацетон, диметилформамид (ДМФ), дихлорметан, 1,2-дихлорэтан и трет-бутанол, добавляют ацильные доноры, как винилацетат, винилпропионат, винилбутират, 2,2,2-трифторэтил-2H,2H-перфтордеканоат, этоксивинилацетат, п-нитро- или п-хлорфенилацетат, сложный эфир оксима, ацетангидрид, ангидрид пропионовой кислоты, ангидрид янтарной кислоты, ангидрид глутаровой кислоты, ангидрид изо-валериановой кислоты, 2,2,2-трихлорэтилбутират, 2,2,2-трифторэтил-2H,2H-перфтордеканоат, и затем реакционную смесь смешивают с подходящим ферментом и перемешивают при температуре от -20 до 80°C. Доля сорастворителей в растворе составляет предпочтительно 10-90%, но при необходимости ферментативная реакция проводится также предпочтительно в чистом доноре ацильных групп, например винилацетате, без сорастворителей.
Для разделения рацематов производных сложного эфира, например, ацетила, пропионила, бутирила или глутарила, эти производные в однородной или гетерогенной, водной, водно-органической или органической среде в присутствии подходящего фермента подвергают стереоселективному гидролизу или алкоголизу (например, н-бутанолом) при температуре 10-80°C, при необходимости в присутствии сорастворителей (см. выше) и буфера, причем реакционная смесь предпочтительно содержит 2-50 вес.% сложного эфира.
Получение вышеназванных производных сложного эфира может проводиться по известным из литературы методам, например путем взаимодействия спирта с хлорангидридами кислоты, как ацетилхлорид, или ангидридами, как ацетангидрид, в присутствии амина, как например, триэтиламина или пиридина (см. литературный обзор R.C. Larock, Comprehensive Organic Transformations, S. 978, 1989 VCH Publishers, Inc.).
По окончании реакции продукты или энантиомеры можно разделить простыми способами, например путем экстракции по известным из литературы методам [a) T.Yamano, F.Kikumoto, S.Yamamoto, K.Miwa, M.Kawada, T.Ito, T.Ikemoto, K.Tomimatsu, Y.Mizuno, Chem. Lett. 2000, 448; b) B.Hungerhoff, H.Sonnenschein, F.Theil, J. Org. Chem. 2002, 67, 1781] или путем применения хроматографических методов.
Следующие методы состоят в том, чтобы по окончании ферментативной реакции заметно повысить водорастворимость оставшихся спиртов путем дериватизации, например, ацилированием циклическими ангидридами, как, например, ангидридом глутаровой кислоты, или переводом в холиновый эфир [a) H.Kunz, M.Buchholz, Chem. Ber. 1979, 112, 2145; b) M.Schelhaas, S.Glomsda, M.Hänsler, H.-D.Jakubke, H.Waldmann, Angew. Chem. 1996, 108, 82], и таким образом достичь разделения водонерастворимых или плохо растворимых сложных эфиров путем экстракции. После разделения дериватизацию спиртов можно опять прекратить путем химического или ферментативного омыления.
Особенно интересная возможность разделения энантиомеров состоит в том, чтобы в случае ферментативного ацилирования так выбрать ацильные доноры, чтобы ацилированный энантиомер был заметно лучше растворим в воде, чем непрореагироваший спирт. Подходящими донорами ацильных групп являются, например, циклические ангидриды, как ангидрид янтарной кислоты. По окончании ферментативного ацилирования продукт ацилирования имеет свободную карбоксильную группу, которая делает возможным быстрое разделение продукта путем водной экстракции в основных растворах, например, в насыщенном водном растворе NaHCO3.
При ферментативном разделении рацематов путем отщепления сложного эфира действуют предпочтительно так, что сложный эфир формулы (I), например, при R1=COCH3, COCH2CH3 или COCH2CH2CH2COOH, смешивают в водо- или спиртосодержащем растворе с эстеразой или липазой и перемешивают.
Может быть благоприятным добавлять в указанный раствор буфер, например фосфатный буфер или TRIS[=трис-(гидроксиметил)-метиламин]-буфер. Добавка может быть, например, 0,01-1,0 молярной. Благоприятный диапазон буфера составляет pH 5-10.
В качестве фермента предпочтительно применяются гидролазы из печени млекопитающих, как например, липаза из поджелудочной железы свиней (Fluka) или из микроорганизмов, как например, липаза B из Candida antarctica (Roche Diagnostics), липаза OF из Candida rugosa (Meito Sangyo), липаза SL из Pseudomonas cepacia (Meito Sangyo), липаза L-10 из Alcaligenes spec. (Roche Diagnostics) и липаза QL из Alcaligenes spec. (Meito Sangyo). Если под применяемыми сложными эфирами имеются в виду производные глутаровой кислоты, как например, моно-(3-бензилоксициклогексиловый)-эфир глутаровой кислоты, может быть выгодным вместо вышеуказанных липаз применять глутарил-7-ACA-ацилазу (Roche Diagnostics).
Особенно предпочтительна липаза B из Candida antarctica (Roche Diagnostics), причем может предпочтительно применяться свободный фермент или иммобилизованная форма фермента, например, один из трех имеющихся в настоящее время в продаже продуктов.
Каждый из указанных ферментов может применяться в свободной или иммобилизованной форме (Immobilized Biocatalysts, W.Hartmeier, Springer Verlag Berlin, 1988). Количество фермента выбирается произвольно в зависимости от скорости реакции или от желаемого времени реакции и типа фермента (например, свободный или иммобилизованный) и легко устанавливается простым предварительным испытанием.
Повторное извлечение фермента можно проводить путем сушки вымораживанием. Разделение (и при необходимости последующее извлечение) фермента можно облегчить иммобилизацией.
Путем подходящего проведения реакции всегда удается получить оптически чистым по меньше мере один энантиомер. Если желательно получить оптически чистый сложный эфир, степень превращения в случае ферментативного образования сложного эфира быть ниже (или равной) 50%, а в случае ферментативного гидролиза или алкоголиза - выше (или равной) 50%. Если желателен оптически чистый спирт, то степень превращения в случае катализованного ферментами образования сложного эфира должна быть выше (или равной) 50%, а в случае гидролиза или алкоголиза должна быть ниже (или равной) 50%.
Определение степени превращения ферментативной реакции проводится или по ВЭЖХ непосредственно из реакционной смеси или путем расчета из оптической чистоты продуктов реакции (сложного эфира и кислоты), которую определяли непосредственно из реакционной смеси по ВЭЖХ на хиральной фазе.
Нижеследующими примерами настоящее изобретение поясняется более подробно.
Примеры:
Все выделенные продукты или смеси сырых продуктов идентифицировали посредством 1H-ЯМР и масс-спектроскопией или по ВЭЖХ.
Оптическую чистоту сложных эфиров и спиртов определяли по ВЭЖХ, например, на Chiralpak AD 250×4,6 (Daicel) или Chiracel OD 250×4,6.
К схеме Ia:
Пример 1
Синтез рацемического метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
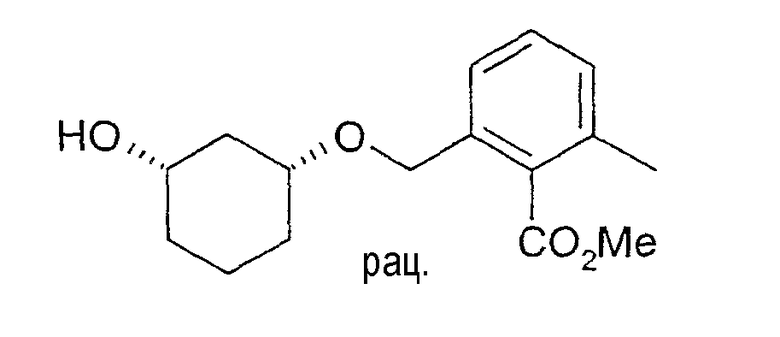
500 г (4,3 моль) цис-1,3-циклогександиола растворяют в 5 л NMP и смешивают с 336 г (3,0 моль) трет-бутилата калия (KOtBu). Внутреннюю температуру повышают до 28°C. Перемешивают 30 мин, затем охлаждают до -5°C и по каплям смешивают с 370 г (приблизительно 94%-ным, примерно 1,4 моль) метилового эфира 2-бромометил-6-метилбензойной кислоты, который можно получить, например, метанолизом бромида 2-бромометил-6-метилбензойной кислоты или бромированием метилового эфира 2,6-диметилбензойной кислоты, перемешивают 30 мин и затем разбавляют 5 л воды. После трехкратной промывки, используя по 3 л н-гептана, и отвода раствора н-гептана, оставшуюся водную фазу экстрагируют четыре раза, используя по 2,5 л MTBE. Очищенные MTBE-фазы один раз промывают 5 л воды, сушат Na2SO4 и затем выпаривают при пониженном давлении. Получают 234 г желаемого соединения в виде желтоватого масла, которое без дальнейшей очистки применяют на следующей реакции (например, разделения рацематов); 1H-ЯМР (CDCl3), δ=1,27 (м, 1H), 1,45 (м, 1H), 1,55 (м, 1H), 1,74 (м, 1H), 1,83 (м, 1H), 2,05 (м, 1H), 2,34 (с, 3H), 3,47 (м, 1H), 3,72 (м, 1H), 3,91 (с, 3H), 4,58 (дд, 2H), 7,15 (д, 1H), 7,20 (д, 2H), 7,27 (м, 1H).
Пример 2
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
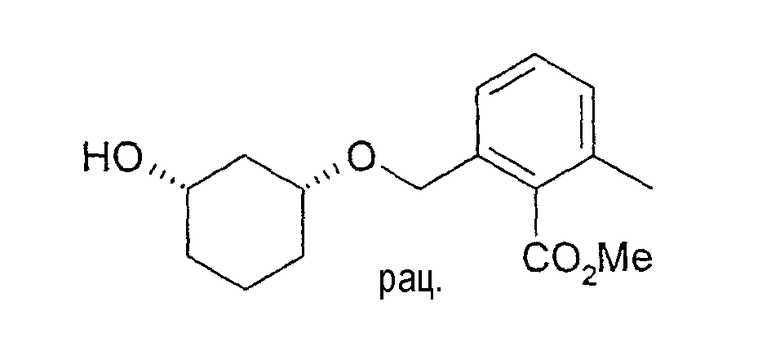
490 г сырого рацемического метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты (см. пример 1) растворяют в 3,1 л метиленхлорида и 850 мл винилацетата, смешивают с 18 г Novozym 435 и перемешивают при 21-24°C. Через 28 ч добавляют еще 2 г Novozym 435. Через полных 44 ч реакцию заканчивают отфильтровыванием фермента, и фильтрат выпаривают при пониженном давлении, получают 540 г. Хроматография остатка на примерно 6 кг силикагеля (этиловый эфир уксусной кислоты/н-гептан 1:1) дает 184 г (1R,3S)-метилового эфира 2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты; чистота>98% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA); 1H-ЯМР (CDCl3), δ=1,27 (м, 1H), 1,45 (м, 1H), 1,55 (м, 1H), 1,74 (м, 1H), 1,83 (м, 1H), 2,05 (м, 1H), 2,34 (с, 3H), 3,47 (м, 1H), 3,72 (м, 1H), 3,91 (с, 3H), 4,58 (дд, 2H), 7,15 (д, 1H), 7,20 (д, 2H), 7,27 (м, 1H)), и 239 г (1S,3R)-ацетата (чистота 93%, ВЭЖХ на Chiralcel OD/20 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 3
Синтез 4-йодметил-2-(3-метоксифенил)-5-метилоксазола
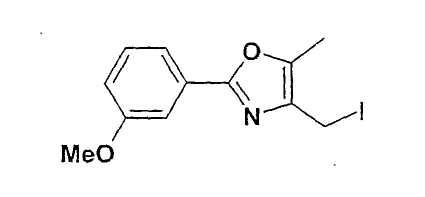
150,0 г (0,63 моль) 4-хлорметил-2-(3-метоксифенил)-5-метилоксазола растворяют в 2,7 л ТГФ и смешивают с 106 г (0,71 моль) NaI. Перемешивают 4 ч и оставляют на ночь, отсасывают соли, фильтрат концентрируют в вакууме. Через примерно 1-2 часа желаемый йодид становится твердым, выход: 216 г, т.пл. 58-59°C. 1H-ЯМР (CDCl3): δ=2,30 (с, 3H), 3,88 (с; 3H), 4,34 (с, 2H), 6,97 (дд, 1H), 7,34 (т, 1H), 7,52 (д, 1H), 7,58 (д, 1H).
Пример 4
Синтез (1R,3S)-метилового эфира 2-{3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты

184 г (0,66 моль) (1R,3S)-метилового эфира 2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты (см. пример 2) растворяют в 2,2 л t-BuOMe. К этому добавляют 88,0 г (примерно 55%, 1,8 ммоль) NaH и перемешивают 45 минут при 20-22°C. Добавляют 282 г (83,8 ммоль) 4-йодметил-2-(3-метоксифенил)-5-метилоксазола (см. пример 3), перемешивают 8 часов при 22°C и оставляют на ночь. Перемешивают еще 4 ч и затем при охлаждении осторожно добавляют сначала 200 мл, позднее еще 1,5 л воды. Органическую фазу отделяют, сушат (Na2SO4) и концентрируют при пониженном давлении. Получают 383 г сырого продукта, который выделяют путем хроматографии на примерно 6 кг силикагеля (дихлорметан/ацетон 19:1), выход: 199 г желтоватого масла; 1H-ЯМР
(CDCl3), δ=1,15-1,32 (м, 4H), 1,81 (м, 1H), 2,00 (м, 1H), 2,07 (м, 1H), 2,34 (с, 3H), 2,40 (с, 3H), 2,51 (м, 1H), 3,27 (м, 1H), 3,37 (м, 1H), 3,87 (с, 3H), 3,90 (м, 3H), 4,48 (с, 2H), 4,60 (с, 2H), 6,96 (м, 1H), 7,12-7,35 (м, 4H), 7,53 (с, 1H), 7,58 (д, 1H).
Пример 5
Синтез (1R,3S)-2-{3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты

199 г (0,41 моль) (1R,3S)-метилового эфира 2-{3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты (см. пример 4) растворяют в 2 л этанола. Добавляют 250 мл 33%-ного NaOH и кипятят 15 часов с обратным холодильником. Этанол отгоняют в вакууме, остаток растворяют примерно в 2 л воды и дважды промывают метил-трет-бутиловым эфиром, используя по 500 мл. Водную фазу при охлаждении подкисляют концентрированной соляной кислотой до pH 1 и получаемый маслянистый продукт экстрагируют 1,5 л этилового эфира уксусной кислоты. Раствор этилового эфира уксусной кислоты сушат и концентрируют в вакууме. Остаток растворяют в 1,2 л DIPE при примерно 40°C. После кристаллизации и сушки в вакууме при 60°C получают 132,5 г желаемой карбоновой кислоты; т.пл. 103-105°C; чистота>98% (ВЭЖХ на Chiralpak AD-H 250x4,6; 1 мл/мин, гептан/EtOH/CH3CN 90:7:1+0,1% TFA); 1H-ЯМР (CDCl3), δ=1,14-1,38 (м, 4H), 1,80 (м, 1H), 1,93 (м, 2H), 2,41(с, 3H), 2,44 (с, 3H), 2,61 (м, 1H), 3,40 (м, 2H), 3,86 (с, 3H), 4,53 (с, 2H), 4,68 (дд, 2H), 6,98 (дд, 1H), 7,17-7,36 (м, 4H), 7,55 (с, 1H), 7,61 (д, 1H).
Пример 6
Синтез 4-йодметил-2-(4-метилфенил)-5-метилоксазола
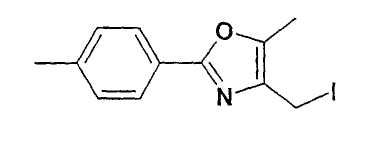
6,0 г 4-хлорметил-2-(4-метилфенил)-5-метилоксазола растворяют в 120 мл ТГФ и смешивают с 4,18 г (27,9 ммоль) NaI. Перемешивают 3,5 ч, добавляют еще 1,5 г NaI и нагревают до 35°C. Через 30 минут отсасывают соли и фильтрат концентрируют в вакууме; выход: 10,1 г, т.пл. 104-105°C; 1H-ЯМР (CDCl3): δ=2,29 (с, 3H), 2,39 (с, 3H), 4,34 (с, 2H), 7,24 (д, 2H), 7,88 (д, 2H).
Пример 7
Синтез (1R,3S)-метилового эфира 2-{3-[2-(4-метилфенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты
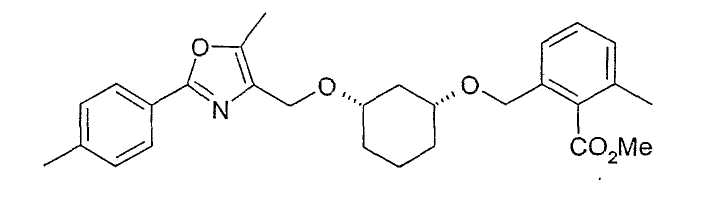
36,0 г (0,129 моль) (1R,3S)-метилового эфира 2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты (см. пример 2) растворяют в 430 мл tBuOMe. Добавляют 17,2 г (примерно 55%, 0,35 моль) NaH и перемешивают 30 минут при 23°C. Добавляют 55,1 г (0,166 моль) 4-йодметил-2-(4-метилфенил)-5-метилоксазола (пример 6). После 6 часов перемешивания и выстаивания в течение 2 дней добавляют при охлаждении 400 мл воды и отделяют органическую фазу. После сушки (Na2SO4) и концентрирования сырой продукт (75 г) выделяют путем хроматографии на силикагеле (примерно 1 кг) (дихлорметан/ацетон 19:1), выход: 42 г диалкилированного производного 1,3-циклогександиола в виде желтоватого масла; 1H-ЯМР (CDCl3), δ=1,16-1,31 (м, 4H), 1,80 (м, 1H), 1,97-2,1 (м, 2H), 2,34 (с, 3H), 2,39 (с, 3H), 2,40 (с, 3H), 2,52 (м, 1H), 3,27 (м, 1H), 3,37 (м, 1H), 3,89 (с, 3H), 4,47 (с, 2H), 4,59 (с, 2H), 7,13 (д, 1H), 7,20-7,28 (м, 4H), 7,88 (д, 1H).
Пример 8
Синтез (1R,3S)-2-{3-[2-(4-метилфенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты

42,0 г (0,09 моль) (1R,3S)-метилового эфира 2-{3-[2-(4-метилфенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты (см. пример 7) растворяют в 420 мл этанола. Добавляют 45 мл 33%-ного NaOH и нагревают примерно 20 часов с обратным холодильником. Этанол отгоняют в вакууме, остаток растворяют в 500 мл воды и раствор четыре раза промывают простым эфиром MTB, используя по 100 мл. Водную фазу при охлаждении подкисляют концентрированной соляной кислотой (pH 1) и получаемый маслянистый продукт экстрагируют этиловым эфиром уксусной кислоты. Раствор этилового эфира уксусной кислоты сушат и концентрируют в вакууме. Остаток растворяют в 250 мл DIPE в тепле. При охлаждении начинается кристаллизация. По окончании кристаллизации и сушки в вакууме при 60°C получают 28,4 г желаемой карбоновой кислоты; т.пл. 117-119°C; чистота>98% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 90:7:1+0,1% TFA); 1H-ЯМР (CDCl3), δ=1,14-1,36 (м, 4H), 1,80 (м, 1H), 1,91 (м, 2H), 2,39 (с, 3H), 2,40 (с, 3H), 2,46 (с, 3H), 2,64 (м, 1H), 3,40 (м, 2H), 4,54 (с, 2H), 4,68 (дд, 2H), 7,17-7,30 (м, 5H), 7,91 (д, 2H).
Пример 9
Синтез 4-йодметил-2-(3-метилфенил)-5-метилоксазола

6,0 г 4-хлорметил-2-(4-метилфенил)-5-метилоксазола растворяют в 120 мл ТГФ и смешивают с 4,5 г (30 ммоль) NaI. Перемешивают 5 ч и оставляют на ночь. Отделение твердой фазы и концентрирование фильтрата в вакууме дает 10,2 г желаемого йодида; т.пл. 32°C; 1H-ЯМР (CDCl3): δ=2,30 (с, 3H), 2,40 (с, 3H), 4,34 (с, 2H), 7,24 (д, 1H), 7,32 (т, 1H), 7,77 (д, 1H), 7,83 (д, 1H).
Пример 10
Синтез (1R,3S)-метилового эфира 2-{3-[2-(3-метилфенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты
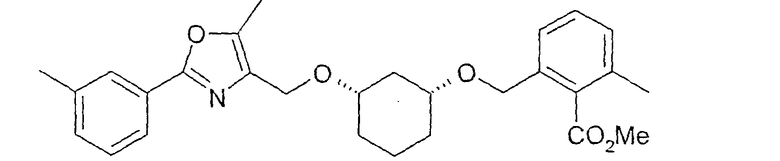
36,0 г (0,129 моль) (1R,3S)-метилового эфира 2-(3-гидроксициклогексилоксиметил)-6-метилбензойной (см. пример 2), растворяют в 430 мл tBuOMe. Добавляют 17,19 г (примерно 55%, 0,35 моль) NaH и перемешивают 30 минут при 20-22°C. Добавляют 55,1 г (0,166 моль) 4-йодметил-2-(3-метилфенил)-5-метилоксазола (см. пример 9). После 6 часов перемешивания и выдерживания в течение 2 суток добавляют при охлаждении 400 мл воды и отделяют органическую фазу. После сушки (Na2SO4) и концентрирования сырой продукт (75 г) выделяют путем хроматографии на силикагеле (1,2 кг) (дихлорметан/ацетон 19:1), выход: 49 г (1R,3S)-метилового эфира 2-{3-[2-(3-метилфенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты; 1H-ЯМР (CDCl3), δ=1,13-1,31 (м, 4H), 1,80 (м, 1H), 1,97-2,1 (м, 2H), 2,34 (с, 3H), 2,40 (с, 3H), 2,41 (с, 3H), 2,52 (м, 1H), 3,27 (м, 1H), 3,37 (м, 1H), 3,90 (с, 3H), 4,48 (с, 2H), 4,59 (с, 2H), 7,12-7,33 (м, 4H), 7,78 (д, 1H), 7,84 (с, 1H).
Пример 11
Синтез (1R,3S)-2-{3-[2-(3-метилфенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты
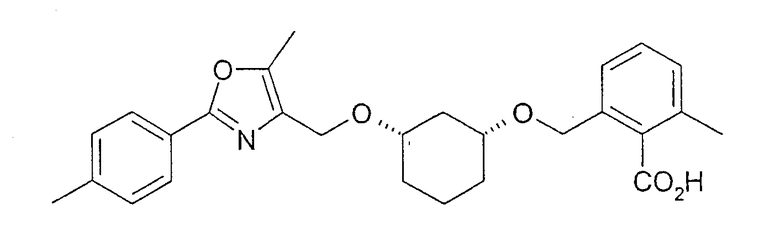
49,0 г (0,09 моль) (1R,3S)-метилового эфира 2-{3-[2-(3-метилфенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты (см. пример 10) растворяют в 500 мл этанола. Добавляют 50 мл 33%-ного NaOH и нагревают примерно 14 часов с обратным холодильником. Этанол отгоняют в вакууме, остаток растворяют в 500 мл воды и раствор трижды промывают метил-трет-бутиловым эфиром, используя по 150 мл. Водную фазу при охлаждении подкисляют концентрированной соляной кислотой (pH 1) и маслянистый продукт экстрагируют этиловым эфиром уксусной кислоты. Раствор этилового эфира уксусной кислоты сушат и концентрируют в вакууме. Остаток растворяют в 250 мл DIPE в тепле. При охлаждении начинается кристаллизация. По окончании кристаллизации и сушки в вакууме при 60°C получают 29,9 г желаемой карбоновой кислоты; т.пл. 109-111°C; чистота>98% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 90:7:1+0,1% TFA); 1H-ЯМР (CDCl3), δ=1,14-1,36 (м, 4H), 1,80 (м, 1H), 1,93 (м, 2H), 2,40 (с, 2х3H), 2,45 (с, 3H), 2,64 (м, 1H), 3,40 (м, 2H), 4,53 (с, 2H), 4,68 (дд, 2H), 7,17-7,34 (м, 5H), 7,81 (д, 1H), 7,85 (с, 1H).
К схеме IIa:
Пример 12
Разделение рацематов цис-3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола
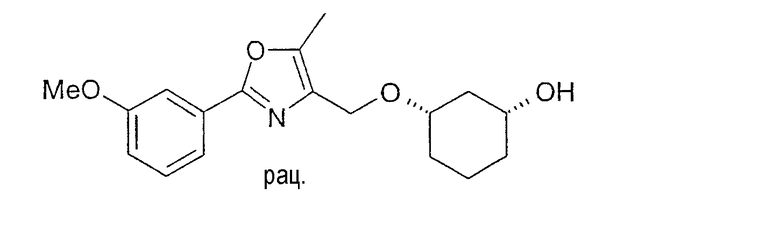
24,9 г рацемического цис-3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола (полученного алкилированием цис-1,3-циклогександиола 4-йодметил-2-(3-метоксифенил)-5-метилоксазолом) растворяют в 100 мл винилацетата, смешивают с 1,0 г Chirazyme L-2, лиофилизованного, и перемешивают при 20-23°C. Через примерно 30 минут фермент отфильтровывают и раствор концентрируют в вакууме, сырой продукт: 25,8 г. После хроматографии на силикагеле (н-гептан/этиловый эфир уксусной кислоты 10:1 - 0:1) получают 13,7 г (1S,3R)-3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола и 11,3 г (1R,3S)-ацетильного соединения.
Пример 13
Получение (1R,3S)-3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола
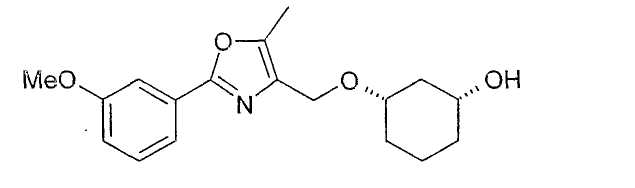
11,2 г (1R,3S)-ацетата из примера 12 растворяют в примерно 100 мл MeOH, смешивают с 0,5 мл NaOMe (30%-ного) и перемешивают при 20-23°C. Через 3,5 ч нейтрализуют концентрированной уксусной кислотой, вводят в этиловый эфир уксусной кислоты, промывают NaHCO3, сушат над Na2SO4 и концентрируют в вакууме. После фильтрации через силикагель (н-гептан/этиловый эфир уксусной кислоты 10:1-0:1) получают 8,8 г (1R,3S)-3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола чистотой 92% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 90:7:1+0,1% TFA).
Пример 14
Синтез (1R,3S)-метилового эфира 2-{3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты

1,4 г (4,4 ммоль) (1R,3S)-3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексанола (см. пример 13) вводят в 15 мл tBuOMe, при 24-27°C смешивают с 1,20 г (10,7 ммоль) KOtBu и перемешивают примерно 30 минут. Охлаждают до 0-5°C, по каплям добавляют 1,89 г (примерно 94%-ного, примерно 7,4 ммоль) метилового эфира 2-бромометил-6-метилбензойной кислоты и сначала перемешивают 30 минут при 0-5°C. В отсутствии дополнительного охлаждения реакционная смесь через 1,5 часа имеет температуру примерно 20°C. После перемешивания в течение ночи и добавления примерно 200 мг KOtBu реакцию заканчивали поcле еще одного часа перемешивания при 22°C. Отгонка растворителя в вакууме, распределение остатка между водой и tBuOMe и сушка содержащей продукт органической фазы дает после концентрирования в вакууме 1,6 г (1R,3S)-метилового эфира 2-{3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты в виде желтоватого масла; 1H-ЯМР (CDCl3), δ=1,15-1,32 (м, 4H), 1,81 (м, 1H), 2,00 (м, 1H), 2,07 (м, 1H), 2,34 (с, 3H), 2,40 (с, 3H), 2,51 (м, 1H), 3,27 (м, 1H), 3,37 (м, 1H), 3,87 (с, 3H), 3,90 (с, 3H), 4,48 (с, 2H), 4,60 (с, 2H), 6,96 (м, 1H), 7,12-7,35 (м, 4H), 7,53-7,60 (м, 2H).
Пример 15
Синтез (1S,3R)-метилового эфира 2-{3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты
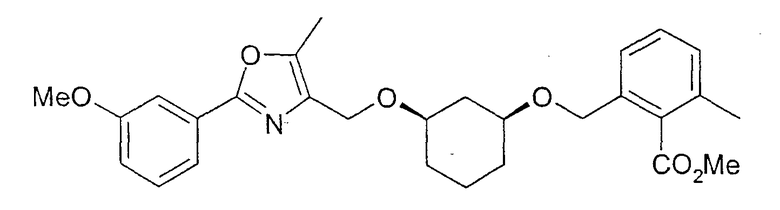
Исходя из (1S,3R)-3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола из примера 12, удается аналогично примеру 14 путем алкилирования получить (1S,3R)-метиловый эфир 2-{3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты; данные 1H-ЯМР совпадают с данными примера 14.
Пример 16
Разделение рацематов цис-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексан-1-ола, получение (1S,3R)-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексан-1-ола
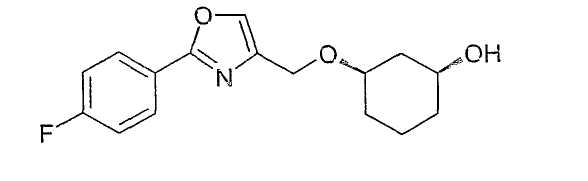
30 мг рацемического цис-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексан-1-ола вводят в примерно 3 мл дихлорметана, смешивают с 60 мг п-нитрофенилацетата и 10 мг Novozyme 435, перемешивают при 20-23°C. Через 70 ч отфильтровывают иммобилизованный фермент. Анализ оптической чистоты непосредственно из концентрированной реакционной смеси дает для (1S,3R)-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексан-1-ола чистоту>95% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, ацетонитрил), а для (1R,3S)-ацетата чистоту 95% (ВЭЖХ на Chiralpak AD 250×4,6, 1 мл/мин, ацетонитрил). Для выделения (1S,3R)-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексан-1-ола сырую смесь выделяют путем хроматографии на силикагеле (ЭА/н-гептан); выход 12 мг, чистота 96%.
Пример 17
Синтез (1S,3R)-метилового эфира 2-{3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты
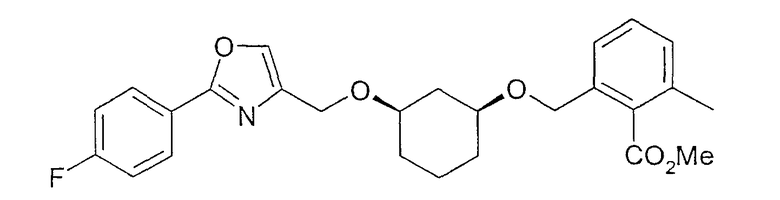
Исходя из (1S,3R)-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексан-1-ола из примера 16 путем алкилирования 2-бромометил-6-метилбензойной кислоты с метиловым эфиром получают (1S,3R)-метиловый эфир 2-{3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты (см. пример 35).
Пример 18
Разделение рацематов 3-[2-(4-метилфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола
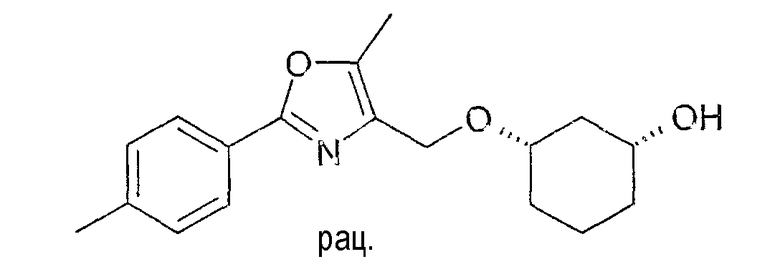
(причем R4=p-Me-, R5=H и R3=Me)
16,3 г рацемического 3-(2-(4-метилфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола растворяют в 100 мл винилацетата, смешивают с 1,9 г Chirazyme L-2, лиофилизованного, и перемешивают при 20-23°C. Через примерно 30 минут фермент отфильтровывают и раствор концентрируют в вакууме, сырой продукт: 16,6 г. После хроматографии на силикагеле (н-гептан/этиловый эфир уксусной кислоты 10:1-0:1) получают 8,6 г (1S,3R)-3-[2-(4-метилфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола и 6,8 г (1R,3S)-ацетата.
Пример 19
Получение (1R,3S)-3-[2-(4-метилфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола
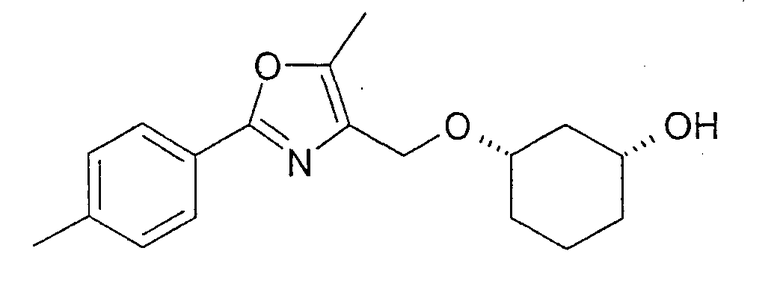
6,8 г (1R,3S)-ацетильного соединения из примера 18 растворяют примерно в 65 мл MeOH, смешивают с 0,32 мл NaOMe (30%-ного) и перемешивают при 20-23°C. Через 4 ч нейтрализуют уксусной кислотой, концентрируют в вакууме, вводят в этиловый эфир уксусной кислоты, промывают NaHCO3, сушат (Na2SO4) и концентрируют в вакууме. После фильтрации через силикагель (н-гептан/этиловый эфир уксусной кислоты 10:1-0:1) получают 8,8 г желаемого (1R,3S)-3-[2-(4-метилфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола с чистотой 95% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 90:7:1+0,1% TFA).
Пример 20
Разделение рацематов цис-3-[2-фенил-5-метилоксазол-4-илметокси]-циклогексан-1-ола

2,0 г рацемического цис-3-[2-фенил-5-метилоксазол-4-илметокси]-циклогексан-1-ола растворяют в 50 мл винилацетата, смешивают с 0,1 г Chirazyme L-2, лиофилизованного, и перемешивают при 20-23°C. Через примерно 5 ч фермент отфильтровывают и раствор концентрируют в вакууме. После хроматографии на силикагеле (н-гептан/этиловый эфир уксусной кислоты 2:1-1:2) получают 1,0 г (1S,3R)-3-[2-фенил-5-метилоксазол-4-илметокси]-циклогексан-1-ола в виде светло-желтой твердой фазы и 0,96 г ацетилированного (1R,3S)-соединения в виде бесцветного масла.
Пример 21
Получение (1R,3S)-3-[2-фенил-5-метилоксазол-4-илметокси]-циклогексан-1-ола
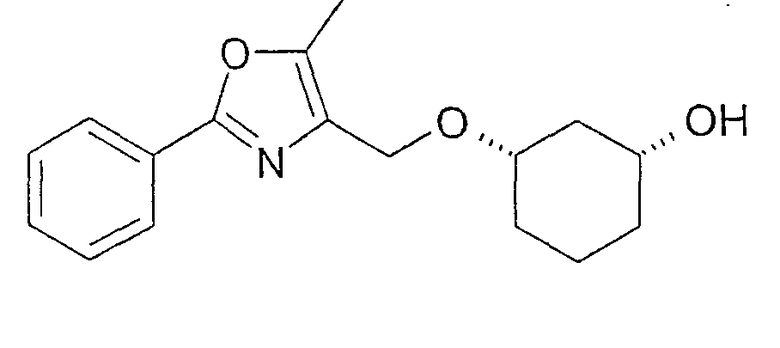
0,96 г (1R,3S)-ацетильного соединения из примера 20 растворяют примерно в 5-10 мл MeOH, смешивают с 0,1 мл NaOMe (30%-ного) и перемешивают при 20-23°C. Через 3 ч нейтрализуют уксусной кислотой и концентрируют в вакууме, вводят в этиловый эфир уксусной кислоты, промывают насыщенным NaHCO3, сушат (MgSO4) и концентрируют в вакууме. После фильтрации через силикагель (н-гептан/этиловый эфир уксусной кислоты 10:1 - 0:1) получают 0,84 г желаемого (1R,3S)-3-[2-фенил-5-метилоксазол-4-илметокси]-циклогексан-1-ола с чистотой 95% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 22
Разделение рацематов цис-3-[2-(4-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола
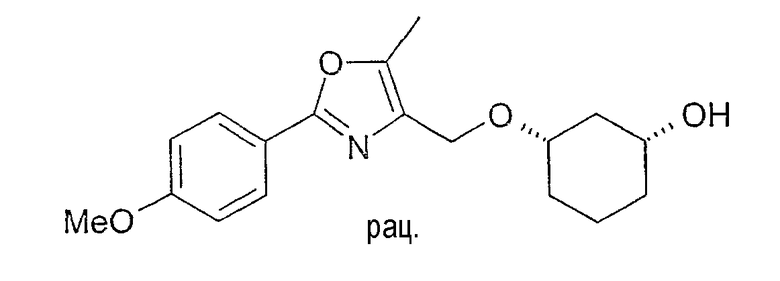
2,0 г рацемического цис-3-[2-(4-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола растворяют в 50 мл винилацетата, смешивают с 0,1 г Chirazyme L-2, лиофилизованного, и перемешивают при 20-23°C. Через примерно 5 ч фермент отфильтровывают и раствор концентрируют в вакууме. После хроматографии на силикагеле (н-гептан/этиловый эфир уксусной кислоты 2:1-1:2) получают 1,16 г (1S,3R)-3-[2-(4-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола и 0,79 г (1R,3S)-ацетата.
Пример 23
Получение (1R,3S)-3-[2-(4-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола
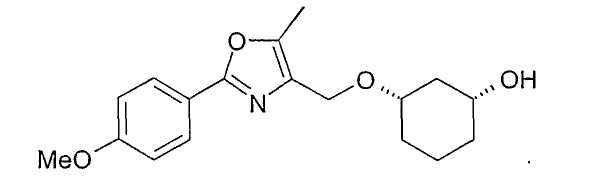
0,79 г ацетата из примера 22 растворяют в примерно 5-10 мл MeOH, смешивают с 0,1 мл NaOMe (30%-ного) и перемешивают при 20-23°C. Через 3 ч нейтрализуют разбавленной уксусной кислотой и концентрируют в вакууме, вводят в этиловый эфир уксусной кислоты, промывают насыщенным NaHCO3, сушат (MgSO4) и концентрируют в вакууме. После фильтрации через силикагель (н-гептан/этиловый эфир уксусной кислоты 10:1-0:1) получают 0,84 г (1R,3S)-3-[2-(4-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола в виде желтого масла с чистотой 92% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 90:7:1+0,1% TFA).
Пример 24
Разделение рацематов цис-3-[2-(4-фторфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола
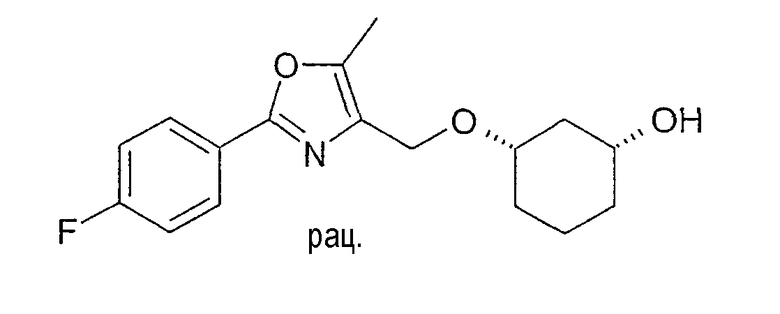
1,70 г рацемического цис-3-[2-(4-фторфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола растворяют в 50 мл винилацетата, смешивают с 0,1 г Chirazyme L-2, лиофилизованного, и перемешивают при 20-23°C. Через примерно 1,5 ч фермент отфильтровывают, и раствор концентрируют в вакууме. После хроматографии на силикагеле (н-гептан/этиловый эфир уксусной кислоты 5:1-1:1) получают 1,0 г (1S,3R)-3-[2-(4-фторфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола и 0,75 г (1R,3S)-ацетата.
Пример 25
Получение (1R,3S)-3-[2-(4-фторфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола
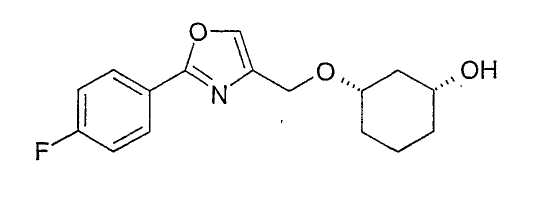
0,75 г ацетата из примера 24 растворяют в примерно 30 мл MeOH, смешивают с 0,2 мл NaOMe (30%-ного) и перемешивают при 20-23°C. Через 1 ч нейтрализуют разбавленной уксусной кислотой и концентрируют в вакууме, вводят в этиловый эфир уксусной кислоты, промывают насыщенным NaHCO3, сушат (MgSO4) и концентрируют в вакууме, выход: 0,59 г (1R,3S)-3-[2-(4-фторфенил)-5-метилоксазол-4-илметокси]-циклогексан-1-ола в виде белой твердой фазы с чистотой 94% (ВЭЖХ на Chiralpak OD/19 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 110:2:1+0,05% TFA).
К схеме IIb:
Пример 26
Стереоселективный гидролиз 3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексилового эфира уксусной кислоты, (получение (1R,3S)-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексан-1-ола
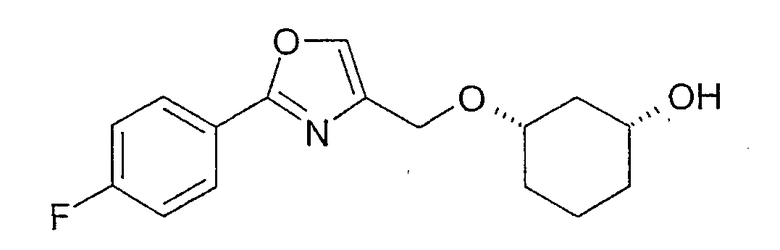
Примерно 10 мг рацемического 3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексилового эфира уксусной кислоты (полученного реакцией 3-бензилоксициклогексан-1-ола с ангидридом уксусной кислоты, аналогично синтезу моно-(3-бензилоксициклогексилового)-эфира глутаровой кислоты, см. пример 39) вводят в 2 мл фосфатного буфера (0,1M, pH 7,0) и 2 мл DME и при 20-23°C перемешивают примерно 20-24 ч с примерно 5 мг Chirazyme L-2, лиофилизованного. Реакционную смесь экстрагируют дихлорметаном. Органическую фазу смешивают с толуолом и выпаривают в вакууме. Определение оптической чистоты дает для (1R,3S)-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексан-1-ола чистоту 99,4% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, ацетонитрил) и для (1S,3R)-ацетата чистоту 98,9% (ВЭЖХ на Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 110:5:1+0,1% TFA).
К схеме IIIа
Пример 27
Синтез рацемического цис-3-бензилоксициклогексан-1-ола

150,0 г (1,29 моль) цис-1,3-циклогександиола растворяют в 1,5 л NMP, смешивают с 111,6 г (0,99 моль) трет-бутилата калия (KOtBu) и перемешивают при 25-27°C. Через примерно 30 минут охлаждают до 0°C и по каплям добавляют 78,1 г (0,46 моль) бензилбромида. Перемешивают 15 мин при примерно 0°C и затем добавляют 1,5 л воды. После трехкратного промывания 700 мл н-гептана и отведения раствора н-гептана, водный раствор четырежды экстрагируют MTBE, используя по 500 мл. Концентрированные MTBE-фазы дважды промывают водой, используя по 1 л, сушат (Na2SO4) и затем выпаривают при пониженном давлении. Получают 48,0 г желаемого соединения в виде прозрачного желтого масла; 1H-ЯМР (CDCl3), δ=1,29 (м, 1H), 1,43-1,93 (м, 6H), 2,06 (м, 1H), 2,55 (с (шир.), 1H), 3,56 (м, 1H), 3,74 (шир., 1H), 4,55 (дд, 2H), 7,25-7,36 (м, 5H).
Пример 28
Разделение рацематов 3-бензилоксициклогексан-1-ола
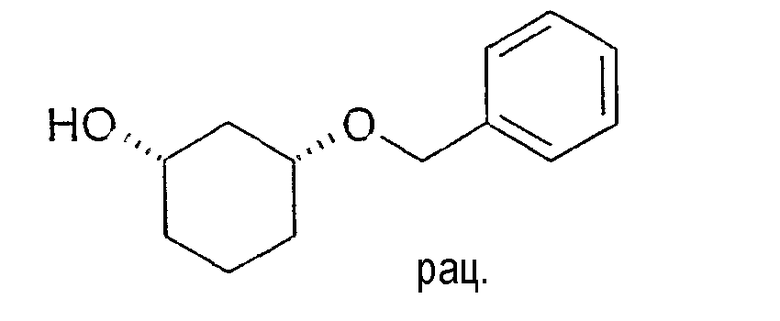
20,3 г цис-3-бензилоксициклогексан-1-ола растворяют в 35 мл винилацетата и 125 мл метиленхлорида, смешивают с 2,0 г Novozym 435 и перемешивают 6 ч при 20-23°C. После выстаивания в течение ночи фермент отфильтровывают. Отбирают пробу и выпаривают в вакууме. Энантиомерный остаток (1S,3R)-3-бензилоксициклогексан-1-ола составляет>99% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), энантиомерный остаток (1R,3S)-ацетата составляет 78% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 29
Разделение рацематов 3-бензилоксициклогексан-1-ола
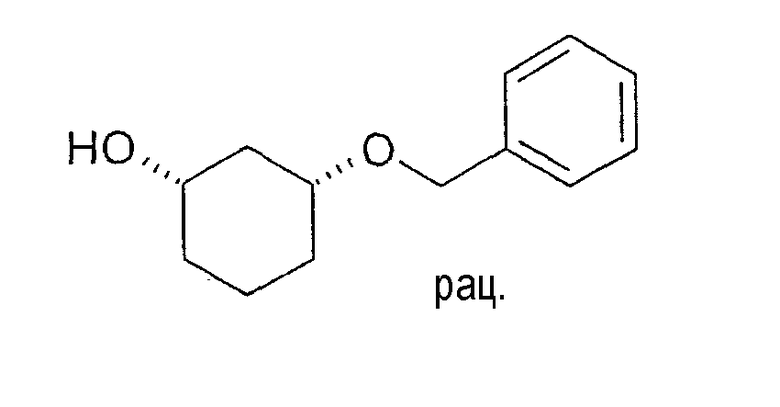
100,0 г цис-3-бензилоксициклогексан-1-ола растворяют в 170 мл винилацетата и 630 мл метиленхлорида, смешивают с 5,0 г Novozym 435 и перемешивают 26 ч при 20-23°C. Фермент отфильтровывают, отбирают пробу и выпаривают в вакууме. Энантиомерный остаток (1S,3R)-3-бензилоксициклогексан-1-ола составляет >99% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), энантиомерный остаток (1R,3S)-ацетата составляет 90% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 30
Выделение (1S,3R)-3-бензилоксициклогексан-1-ола, разделение смеси ацетата и спирта с помощью пиридин-SO3
1,9 г сырой смеси ацетат/спирт из стереоселективного ферментативного ацетилирования 3-бензилоксициклогексан-1-ола (из примера 29) в 10 мл пиридина и 2 мл ДМФ перемешивают при 20-22°C с 2 г пиридин-SO3. Через 4 ч превращение бензилциклогексанола в пиридиновую соль эфира серной кислоты является почти количественным. Реакционную смесь разбавляют 40 мл воды и дважды экстрагируют с помощью примерно 20 мл MTBE. MTBE-фазы количественно содержат неизмененный (1R,3S)-ацетат. Оставшуюся, не содержащую ацетата водную фазу выпаривают в вакууме. Остаток смешивают с MTBE, продукт сульфатирования становится твердым; выход: 2,7 г.
2,7 г пиридиновой соли эфира серной кислоты и (1S,3R)-бензилциклогексан-1-ола перемешивают 2 ч при 55°C в 45 мл ТГФ, 4 мл воды и 1 мл концентрированной серной кислоты. Разбавляют 40 мл воды, добавляют примерно 10 мл MTBE, фазы разделяют и водную фазу один раз экстрагируют MTBE. Очищенные органические фазы сушат (Na2SO4) и выпаривают; выход: 640 мг светло-желтого масла. Данные ЯМР совпадают с данными, указанными в примере 16; проверка оптической чистоты дает чистоту >99%.
Пример 31
Выделение (1S,3R)-3-бензилоксициклогексан-1-ола, разделение смеси ацетата и спирта путем экстракции
10 г сырой смеси ацетат/спирт из примера 29 вводят в примерно 90 мл метанола и примерно 70 мл воды и трижды промывают н-гептаном, используя примерно по 50 мл. Очищенные гептановые фазы (содержат преимущественно ацетат) экстрагируют 50 мл смеси метанол/вода 1:1. Очищенные водные фазы еще раз промывают н-гептаном. После концентрирования водной фазы получают 3,6 г желаемого (1S,3R)-3-бензилоксициклогексан-1-ола, концентрирование очищенной гептановой фазы дает 5,5 г (1R,3S)-ацетата.
Пример 32
Синтез 4-йодметил-2-(4-фторфенил)-оксазола
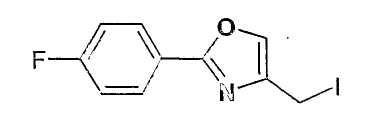
4,0 г (18,9 ммоль) 4-хлорметил-2-(4-фторфенил)-оксазола растворяют в 80 мл ТГФ и смешивают с 3,18 г (21,2 ммоль) NaI . Перемешивают 3 ч при 20-23°C и примерно 12 ч при 50°C, отсасывают соли и фильтрат концентрируют в вакууме, выход: 6,1 г. Продукт кристаллизован; т.пл. 100-102°C; 1H-ЯМР (CDCl3): δ=4,34 (с, 2H), 6,97 (дд, 1H), 7,14 (м, 2H), 7,68 (с, 1H), 8,03 (м, 2H).
Пример 33
Синтез (1S,3R)-4-(3-бензилоксициклогексил-1-оксиметил)-2-(4-фторметил)-оксазола
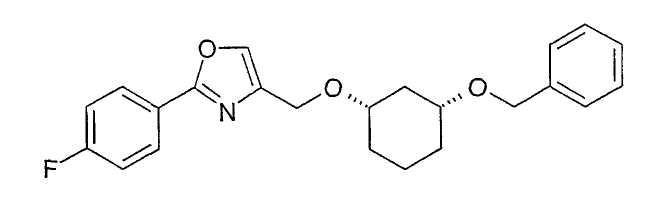
2,0 г (9,7 ммоль) (1S,3R)-3-бензилоксициклогексан-1-ола растворяют в 35 мл tBuOMe. Добавляют 1,3 г (примерно 55%, 43,7 ммоль) NaH и перемешивают 60 минут при 22°C. Добавляют 3,9 г (12,9 ммоль) 4-йодметил-2-(4-фторметил)-оксазола (пример 32) и перемешивают примерно 3 часа при 22-23°C. После выдерживания в течение ночи перемешивают еще 11 ч при 22-23°C. При охлаждении добавляют воду (примерно 30 мл) и отделяют органическую фазу. Сушка (Na2SO4), концентрирование (выход сырого продукта: 4,5 г) и хроматография на силикагеле (дихлорметан/ацетон 19:1) дают 2,4 г желаемого, имеющего цис-конфигурацию, оптически чистого, диалкилированного производного 1,3-циклогександиола в виде белой твердой фазы; т.пл. 61-62°C; 1H-ЯМР (CDCl3), δ=1,11-1,39 (м, 4H), 1,82 (м, 1H), 2,07 (м, 2H), 2,55 (м, 1H), 3,38 (м, 2H), 4,55 (с, 2H), 4,57 (с, 2H), 7,13 (м, 2H), 7,25-7,35 (м, 5H), 7,63 (с, 1H), 8,02 (м, 2H).
Пример 34
Синтез (1R,3S)-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексанол путем гидрирования

2,4 г (1S,3R)-4-(3-бензилоксициклогексил-1-оксиметил)-2-(4-фторметил)-оксазола растворяют в примерно 40 мл метанола, смешивают с Pd/C, взятого на кончике шпателя (10%, с 50% воды), при 20-23°C и нормальном давлении и гидрируют примерно 8 часов. Отфильтровывание катализатора и концентрирование оставшегося раствора дает 1,8 г желаемого, имеющего цис-конфигурацию, моноалкилированного производного 1,3-циклогександиола в виде масла, которое кристаллизуется при добавлении DIPE; выход 1,6 г; т.пл. 81-82°C; 1H-ЯМР (CDCl3), δ=1,25-2,14 (м, 9H), 3,63 (м, 1H), 3,75 (м, 1H), 4,55 (дд, 2H), 7,13 (м, 2H), 7,64 (с, 1H), 8,02 (м, 2H); МС (DCl): 292,3 (100%).
Пример 35
Синтез (1R,3S)-метилового эфира 2-{3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты

0,8 г (2,75 ммоль) (1R,3S)-3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексанола (из примера 34) вводят в 10 мл tBuOMe, смешивают с 0,78 г (6,95 ммоль) KOtBu и перемешивают примерно 30 минут при 22-27°C. Охлаждают до 0-5°C, по каплям добавляют 1,24 г (примерно 94%-ного, примерно 4,8 ммоль) метилового эфира 2-бромометил-6-метилбензойной кислоты, перемешивают сначала 2 часа при 3°C и еще один час при 20°C. Оставляют перемешиваться на ночь при 18-21°C, затем растворитель отгоняют. Остаток распределяют между водой и tBuOMe. Органическую фазу сушат (Na2SO4) и концентрируют в вакууме; выход: 1,04 г (1R,3S)-метилового эфира 2-{3-[2-(4-фторфенил)-оксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты в виде желтоватого масла; 1H-ЯМР (CDCl3), δ=1,15-1,32 (м, 4H), 1,82 (м, 1H), 1,98-2,1 (м, 2H), 2,34 (с, 3H), 2,50 (м, 1H), 3,27 (м, 1H), 3,39 (м, 1H), 3,90 (с, 3H), 4,54 (с, 2H), 4,60 (с, 2H), 7,11-7,30 (м, 5H), 7,63 (с, 1H), 8,02 (м, 2H).
Пример 36
Синтез (1R,3S)-4-(3-бензилоксициклогексил-1-оксиметил)-2-(3-метоксифенил)-5-метилоксазола
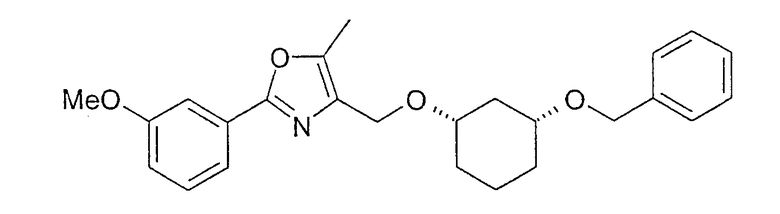
4,6 г (22,3 ммоль) (1R,3S)-3-бензилоксициклогексан-1-ола растворяют в 70 мл хлорбензола. Добавляют 6,6 г (58,8 ммоль) KOtBu, перемешивают 30 минут при 22°C и затем добавляют 10,3 г (31,3 ммоль) 4-йодметил-2-(3-метоксифенил)-5-метилоксазола. Температуру повышают до 35°C. Реакционную смесь немного охлаждают и перемешивают еще 2 часа при 22-23°C. После отгонки хлорбензола в вакууме остаток распределяют между tBuOMe и водой. Органическую фазу сушат (Na2SO4) и концентрируют в вакууме; выход сухого продукта: 10,6 г. Субстанцию без дополнительной очистки применяют в следующей реакции (гидрирование, см. пример 37).
Пример 37
Синтез (1R,3S)-(3-[2-(3-метоксифенил)-оксазол-4-илметокси]-циклогексанола путем гидрирования
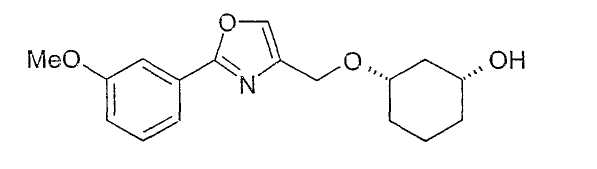
10,5 г (1R,3S)-4-(3-бензилоксициклогексил-1-оксиметил)-2-(3-метоксифенил)-оксазола растворяют примерно в 120 мл метанола, смешивают с 2 г Pd/C (10%-ный с 50% воды) и гидрируют при 20-23°C и нормальном давлении в течение ночи. Отфильтровывание катализатора и концентрирование оставшегося раствора, распределение между MTB-эфиром и водой и сушка органической фазы дает 6,4 г желаемого, имеющего цис-конфигурацию моноалкилированного производного 1,3-циклогександиола в виде желтого масла. 1 г субстанции разделяют путем хроматографии на силикагеле (этиловый эфир уксусной кислоты); получают 0,8 г бесцветного масла; 1H-ЯМР (CDCl3), δ=1,25-1,90 (м, 7H), 2,12 (м, 2H), 2,41 (с, 3H), 3,61 (м, 1H), 3,75 (м, 1H), 3,87 (с, 3H), 4,48 (дд, 2H), 6,96 (д, 1H), 7,33 (т, 1H), 7,53 (с, 1H), 7,58 (д, 1H); МС(ES+): 318,27 (83%), 243,18 (100%).
Пример 38
Синтез (1R,3S)-метилового эфира 2-{3-[2-(3-метоксифенил)-5 метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты
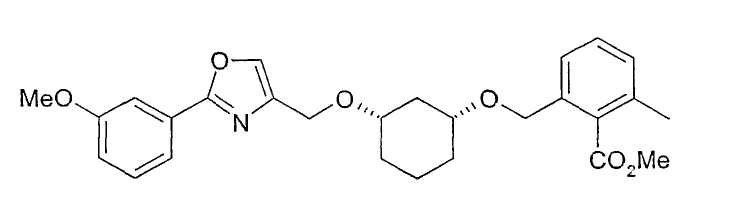
136 мг (0,4 ммоль) (1R,3S)-(3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексанола (из примера 37, гидрирование) растворяют в 1 мл хлорбензола, при 24-27°C смешивают с 120 мг (1,07 ммоль) KOtBu и перемешивают примерно 30 минут. Охлаждают до 0-5°C, по каплям добавляют 189 мг (примерно 94%-ного, примерно 0,78 ммоль) метилового эфира 2-бромометил-6-метилбензойной кислоты и перемешивают сначала 30 минут при 0-5°C. В отсутствие дополнительного охлаждения через 1,5 часа температура реакционной смеси составляет примерно 20°C. После выдерживания в течение ночи и добавления примерно 20 мг KOtBu реакцию заканчивают после дополнительного перемешивания при 22°C в течение 1 часа. Отгонка хлорбензола в вакууме, распределение остатка между водой и tBuOMe и сушка органической фазы дают после концентрирования в вакууме 160 мг (1R,3S)-метилового эфира 2-{3-[2-(3-метоксифенил)-5-метилоксазол-4-илметокси]-циклогексил-1-оксиметил}-6-метилбензойной кислоты в виде желтоватого масла; 1H-ЯМР (CDCl3), δ=1,15-1,32 (м, 4H), 1,81 (м, 1H), 2,00 (м, 1H), 2,06 (м, 1H), 2,34 (с, 3H), 2,40 (с, 3H), 2,51 (м, 1H), 3,27 (м, 1H), 3,36 (м, 1H), 3,87 (с, 3Н), 3,90 (м, 3H), 4,48 (с, 2H), 4,60 (с, 2H), 6,96 (м, 1H), 7,12-7,35 (м, 4H), 7,53-7,60 (м, 2H).
К схеме IIIb
Пример 39
Синтез моно-(3-бензилоксициклогексилового)-эфира глутаровой кислоты

3,0 г 3-бензилоксициклогексан-1-ола, 2,15 г ангидрида глутаровой кислоты и 3,03 г триэтиламина перемешивают в 25 мл метиленхлорида при 21-23°C. После полного превращения добавляют воду, экстрагируют эфиром и сушат над MgSO4. После концентрирования в вакууме получают 4,3 г желаемого соединения; 1H-ЯМР (CDCl3), δ=1,20-1,28 (м, 4H), 1,82 (м, 1H), 1,90-1,97 (м, 3H), 2,05 (м, 1H), 2,32-2,42 (м, 5H), 3,39 (м, 1H), 4,55 (дд, 2H), 4,69 (м, 1H), 7,25-7,33 (м, 5H), 8,7 (шир., 1H).
Пример 40
Стереоселективный гидролиз моно-(3-бензилокси-циклогексилового)-эфира глутаровой кислоты, получение (1R,3S)-3-бензилоксициклогексан-1-ола
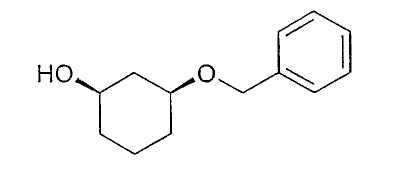
20 мг рацемического моно-(3-бензилоксициклогексилового)-эфира глутаровой кислоты (из примера 39) распределяют в 2 мл фосфатного буфера, pH 8, и 3-5 каплях DME, смешивают с 3-5 мг Novozym 435 и перемешивают при 21-23°C. По достижении примерно 50%-ного превращения реакционный раствор распределяют между насыщенным водным раствором NaHCO3 и этиловым эфиром уксусной кислоты. Фазу этилового эфира уксусной кислоты сушат и концентрируют, выход: 5 мг (1R,3S)-3-бензилоксициклогексан-1-ола, энантиомерный остаток составляет >95% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 41
Синтез (1R,3S)-4-(3-бензилоксициклогексил-1-оксиметил)-2-(4-фторфенил)-оксазола
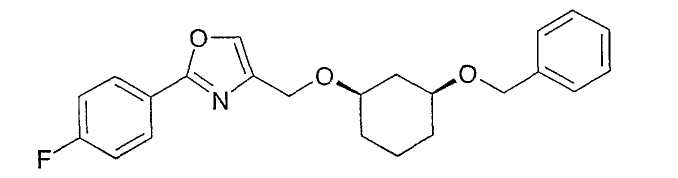
Исходя из (1R,3S)-3-бензилоксициклогексан-1-ола (см. пример 40) алкилированием с 4-йодметил-2-(4-фторфенил)-оксазолом (см. пример 32) получают (1R,3S)-4-(3-бензилоксициклогексил-1-оксиметил)-2-(4-фторфенил)-оксазол (сравни пример 33).
Другие примеры алкилирования цис-1,3-циклогександиола
Пример 42
Синтез рацемического метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
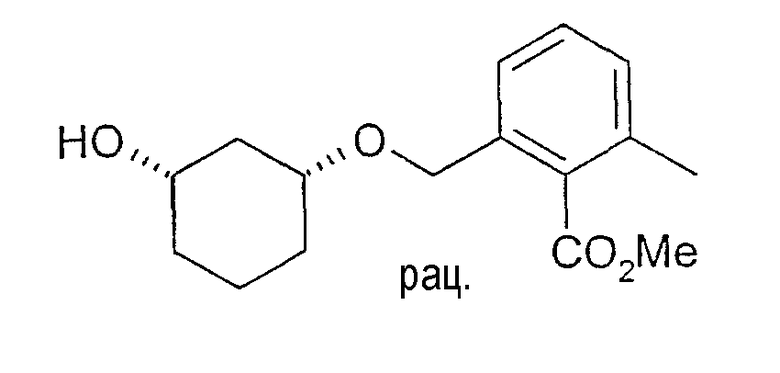
5 г (42,8 ммоль) цис-1,3-циклогександиола растворяют в 50 мл диметоксиэтана (DME), смешивают с 3,36 г (30 ммоль) трет-бутилата калия (KOtBu) при 20-23°C и перемешивают. Через примерно 30 минут охлаждают до 5°C и по каплям добавляют 3,7 г (примерно 50%-ного) метилового эфира 2-бромометил-6-метилбензойной кислоты, который можно получить, например, метанолизом бромида кислоты (бромид 2-бромометил-6-метилбензойной кислоты) или бромированием метилового эфира 2,6-диметилбензойной кислоты. Перемешивают 1 ч при 5-10°C и затем в течение ночи при 20-23°C. Добавляют воду и метил-трет-бутиловый эфир (MTBE), интенсивно перемешивают, фазы разделяют, водную фазу еще раз промывают MTBE, а очищенные органические фазы концентрируют в вакууме. Остаток выделяют путем хроматографии на силикагеле (этиловый эфир уксусной кислоты/н-гептан 1:1). Получают 600 мг желаемого соединения в виде желтоватого масла, 1H-ЯМР (CDCl3), δ=1,27 (м, 1H), 1,45 (м, 1H), 1,55 (м, 1H), 1,74 (м, 1H), 1,83 (м, 1H), 2,05 (м, 1H), 2,34 (с, 3H), 3,47 (м, 1H), 3,72 (м, 1H), 3,91 (с, 3H), 4,58 (дд, 2H), 7,15 (д, 1H), 7,20 (д, 2H), 7,27 (м, 1H).
Пример 43
Синтез рацемического метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты

10,0 г (86 ммоль) цис-1,3-циклогександиола вводят в 150 мл метил-трет-бутилового эфира (MTBE), смешивают с 6,72 г (59,9 ммоль) трет-бутилат калия (KOtBu) при примерно 20°C и перемешивают. Через примерно 30 минут суспензию охлаждают до 5°C и по каплям добавляют 7,4 г (примерно 50%-ного) метилового эфира 2-бромометил-6-метилбензойной кислоты, который можно получить, например, метанолизом бромида кислоты (бромид 2-бромометил-6-метилбензойной кислоты) или бромированием метилового эфира 2,6-диметилбензойной кислоты. Перемешивают 1 ч при 0-5°C, нагревают до 20-23°C и оставляют перемешиваться на ночь. Добавляют воду, интенсивно перемешивают, фазы разделяют, органическую фазу еще раз промывают водой и затем органическую фазу концентрируют в вакууме. Остаток (4,6 г) выделяют путем хроматографии на силикагеле (этиловый эфир уксусной кислоты/н-гептан 1:1). Получают 1,2 г желаемого соединения в виде желтоватого масла, 1H-ЯМР (CDCl3), δ=1,27 (м, 1H), 1,45 (м, 1H), 1,55 (м, 1H), 1,74 (м, 1H), 1,82 (м, 1H), 2,05 (м, 1H), 2,34 (с, 3H), 3,46 (м, 1H), 3,72 (м, 1H), 3,91 (с, 3H), 4,58 (дд, 2H), 7,15 (д, 1H), 7,20 (д, 2H), 7,27 (м, 1H).
Пример 44
Синтез рацемического метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
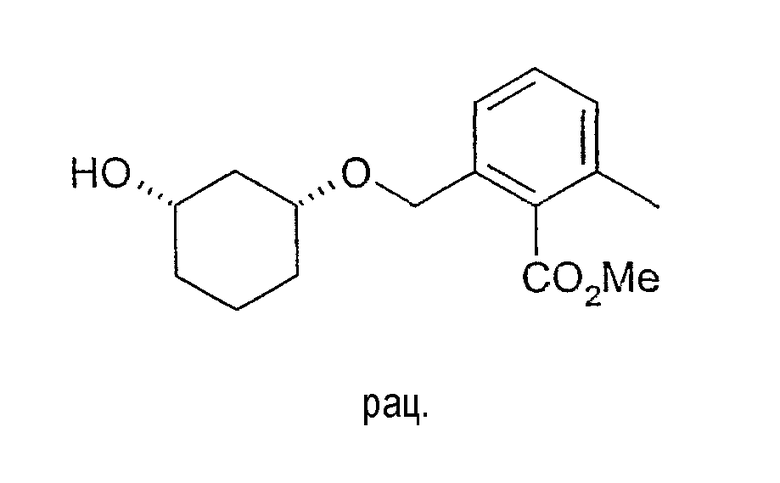
5 г (42,8 ммоль) цис-1,3-циклогександиола растворяют в 40 мл хлорбензола и 10 мл 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинона (DMPU, диметилпропиленмочевина), смешивают с 3,36 г (30 ммоль) трет-бутилат калия (KOtBu) при 20-23°C и перемешивают. Через 10-15 минут охлаждают до 15-20°C и по каплям добавляют 3,7 г (примерно 50%-ного) метилового эфира 2-бромометил-6-метилбензойной кислоты. Перемешивают 1,5 ч при 20°C и добавляют воду. Органическую фазу выделяют и концентрируют при пониженном давлении. Остаток вводят в смесь NMP/вода и для удаления примесей дважды промывают н-гептаном. Затем для выделения продукта дважды экстрагируют MTBE. Очищенные MTBE-фазы промывают водой, сушат (Na2SO4) и концентрируют в вакууме. Остаток (1,2 г) выделяют путем хроматографии на силикагеле (этиловый эфир уксусной кислоты/н-гептан 1:1). Получают 580 г желаемого соединения в виде желтоватого масла, 1H-ЯМР (CDCl3), δ=1,27 (м, 1H), 1,45 (м, 1H), 1,55 (м, 1H), 1,74 (м, 1H), 1,83 (м, 1H), 2,05 (м, 1H), 2,34 (с, 3H), 3,47 (м, 1H), 3,72 (м, 1H), 3,91 (с, 3H), 4,58 (дд, 2H), 7,15 (д, 1H), 7,20 (д, 2H), 7,27 (м, 1H).
Дальнейшие примеры отщепления рацематов путем стереоселективного ферментативного образования сложного эфира (EB)
Пример 45
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
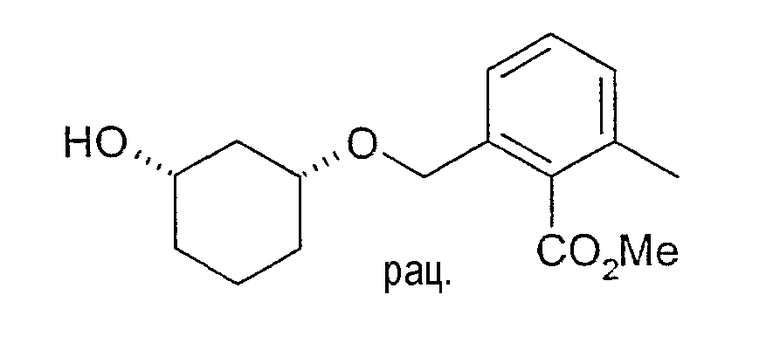
730 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 5 мл метиленхлорида и 2 мл винилацетата, нагревают до 38°C и смешивают с 100 мг Novozym 435.
Через примерно 5 ч реакцию заканчивают путем отфильтровывания фермента и по ВЭЖХ определяют оптическую чистоту образованного ацетата и непрореагировавшего спирта (ВЭЖХацетат: Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5; ВЭЖХспирт: Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA). Определение оптической чистоты дает для (3S,1R)-метилового эфира 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты чистоту 98% и для (3R,1S)-ацетата чистоту 86%.
Пример 46
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты
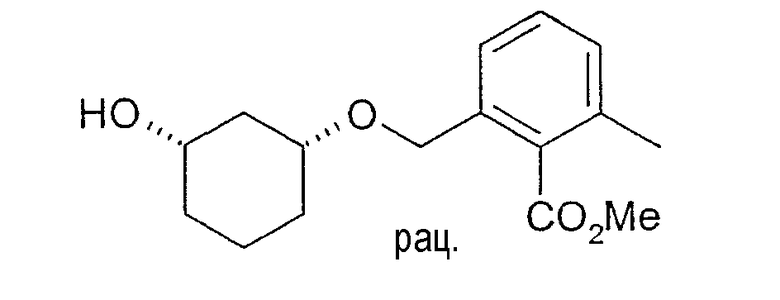
20 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 2 мл хлорбензола и 1 мл винилацетата, смешивают при 22-25°C с 8 мг Chirazyme L-2, лиофилизованного (Roche), и перемешивают. Через примерно 6 ч реакцию оканчивают отфильтровыванием фермента и по ВЭЖХ определяют оптическую чистоту образованного ацетата и непрореагировавшего спирта (ВЭЖХацетат: Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5; ВЭЖХспирт Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA): (3S,1R)-метиловый эфир 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты: чистота 84%, (3R,1S)-ацетат: чистота 95%.
Пример 47
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты
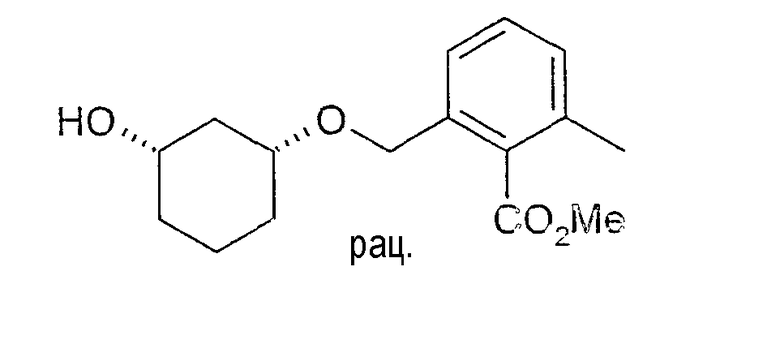
1,0 г метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 10 мл 1,2-дихлорэтана и 2 мл винилпропионата, смешивают с 25 мг Chirazyme L-2, лиофилизованного (Roche) и перемешивают 40 ч при 21-24°C. Отфильтровывание фермента, концентрирование фильтрата в вакууме и хроматография остатка на силикагеле (этиловый эфир уксусной кислоты/н-гептан 1:1) дает 0,49 г (3R,1S)-пропионата с чистотой 92% (ВЭЖХ на Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5), 1H-ЯМР (CDCl3), δ=1,13 (т, 3H), 1,15-1,36 (м, 4H), 1,79 (м, 1H), 1,91(м, 1H), 2,01(м, 1H), 2, 30 (квинтет, 2H), 2,34 (с, 3H), 2,35 (м, 1H), 3,34 (м, 1H), 3,90 (с, 3H), 4,58 (дд, 2H), 4,67 (м, 1H), 7,14 (д, 1H), 7,19 (д, 1H) 7,26 (м, 1H), а также 0,3 г непрореагировавшего (3S,1R)-метилового эфира 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты с чистотой 98% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), 1H-ЯМР (CDCl3), δ=1,27 (м, 1H), 1,45 (м, 1H), 1,55 (м, 1H), 1,74 (м, 1H), 1,83 (м, 1H), 2,05 (м, 1H), 2,34 (с, 3H), 3,47 (м, 1H), 3,72 (м, 1H), 3,91 (с, 3H), 4,58 (дд, 2H), 7,15 (д, 1H), 7,20 (д, 2H), 7,27 (м, 1H).
Пример 48
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты
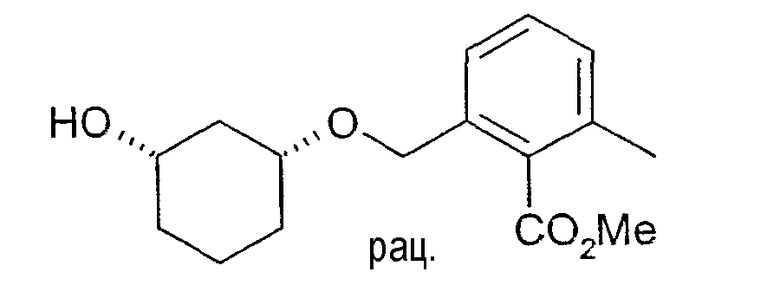
10 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 1 мл винилацетата, смешивают с примерно 4-6 мг липазы TL (Pseud. stutzeri, Meito Sangyo) и перемешивают при 22-25°C. По достижении степени превращения>50% реакцию заканчивают путем отфильтровывания фермента, и определяют оптическую чистоту непрореагировавшего (3S,1R)-метилового эфира 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты: чистота >98% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 49
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
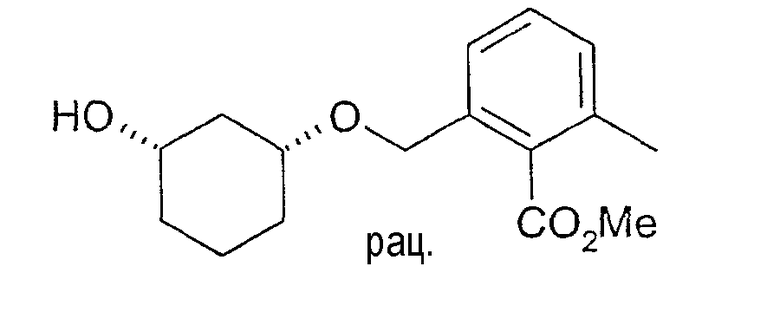
3,9 г рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 25 мл метиленхлорида и 10 мл винилацетата, нагревают до 45°C и смешивают с 250 мг Novozym 435.
По достижении степени превращения примерно 45% реакцию заканчивают путем отфильтровывания фермента и реакционную смесь концентрируют. Хроматография остатка на силикагеле (этиловый эфир уксусной кислоты/н-гептан 1:1) дает 1,9 г (3R,1S)-ацетата (чистота>95%, ВЭЖХ на Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5) и 1,9 г непрореагировавшего (3S,1R)-метилового эфира 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты (чистота 82%, ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 50
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
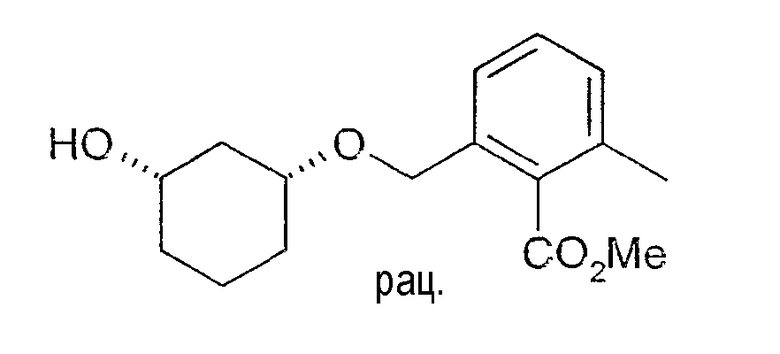
20 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 2 мл толуола и 1 мл винилацетата, при 20-23°C смешивают с 6-8 мг Chirazyme L-2, лиофилизованного (Roche), и перемешивают. По достижении степени превращения примерно 45% реакцию заканчивают путем отфильтровывания фермента и определяют оптическую чистоту образованного (3R,1S)-ацетата: чистота 94% (ВЭЖХ на Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 51
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
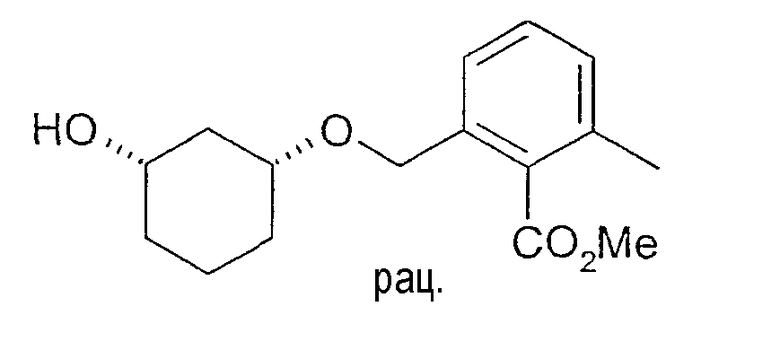
10 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 1 мл винилацетата, смешивают с примерно 4-6 мг липазы QL (Alcaligenes spec., Meito Sangyo) и перемешивают при 20-23°C. По достижении степени превращения примерно 52% реакцию заканчивают путем отфильтровывания фермента и определяют оптическую чистоту образованного ацетата и непрореагировавшего спирта, чистота ацетата: 91% (ВЭЖХ на Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5), чистота (3S,1R)-метилового эфира 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты: >98% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 52
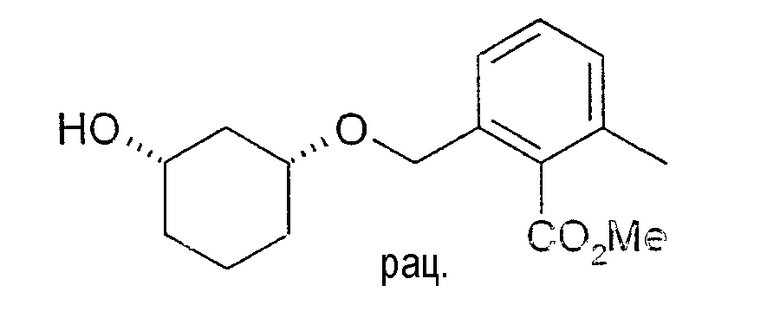
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
10 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 1 мл винилацетата, смешивают примерно с 4-6 мг липазы SL (Pseud. cepacia, Meito Sangyo) и перемешивают при 20-23°C. По достижении степени превращения примерно 52% реакцию заканчивают путем отфильтровывания фермента и определяют оптическую чистоту образованного ацетата и непрореагировавшего спирта; чистота ацетата: 90% (ВЭЖХ на Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5), чистота (3S,1R)-метилового эфира 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты: >95% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 53
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
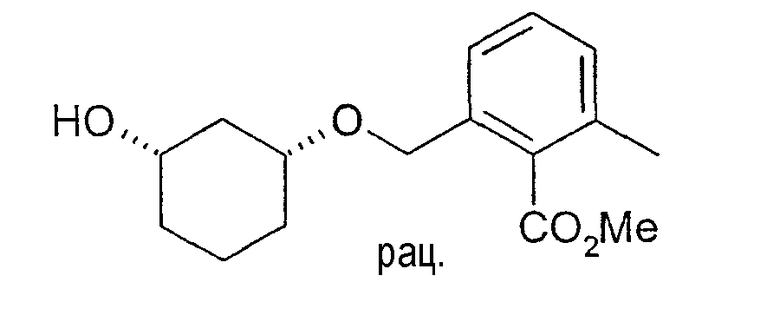
39 г метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 250 мл метиленхлорида и 50 мл винилацетата, нагревают до 45°C и смешивают с 1,0 г Novozym 435. Через 25 ч добавляют еще 0,5 г Novozym 435. Еще через 6,5 ч фермент отфильтровывают и реакционную смесь концентрируют. Хроматография остатка на 630 г силикагеля (этиловый эфир уксусной кислоты/н-гептан 1:1) дает 18,2 г (3S,1R)-метилового эфира 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты (чистота>98%, ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), 1H-ЯМР (CDCl3), δ=1,27 (м, 1H), 1,45 (м, 1H), 1,55 (м, 1H), 1,74 (м, 1H), 1,83 (м, 1H), 2,05 (м, 1H), 2,34 (с, 3H), 3,47 (м, 1H), 3,72 (м, 1H), 3,91 (с, 3H), 4,58 (дд, 2H), 7,15 (д, 1H), 7,20 (д, 2H), 7,27 (м, 1H).
Пример 54
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
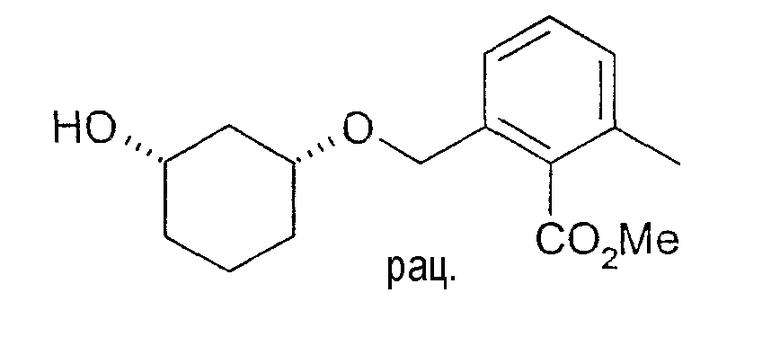
20 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 2 мл ТГФ и 1 мл винилацетата, при 20-23°C смешивают с 6-8 мг Chirazyme L-2, лиофилизованного (Roche), и перемешивают. Через примерно 6 ч реакцию заканчивают путем отфильтровывания фермента и по ВЭЖХ определяют оптическую чистоту образованного ацетата и непрореагировавшего спирта (ВЭЖХацетат: Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5; ВЭЖХспирт: Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA): чистота (3S,1R)-метилового эфира 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты: 89%, чистота (3R,1S)-ацетата: 95%.
Пример 55
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты

Примерно 15 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 2 мл трет-бутанола и 1 мл винилацетата, при 20-23°C смешивают с примерно 6 мг Novozym 435 и перемешивают. Через примерно 24 ч реакцию заканчивают путем отфильтровывания фермента и по ВЭЖХ определяют оптическую чистоту образованного ацетата и непрореагировавшего спирта; (3R,1S)-ацетат: чистота 91% (ВЭЖХ: Chiralcel OD 250×4,6, 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5), (3S,1R)-метиловый эфир 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты - чистота 96% (ВЭЖХ: Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 56
Разделение рацематов метилового эфира цис-2-(3-гидроксициклогексилоксиметил)-6-метилбензойной кислоты
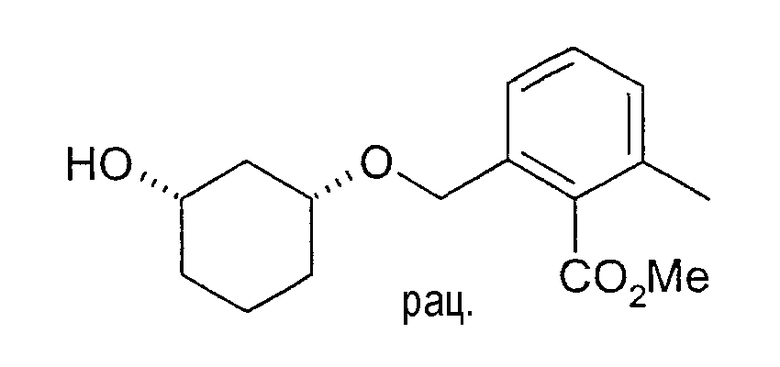
10 мг рацемического метилового эфира цис-2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты растворяют в 1 мл винилацетата, смешивают с примерно 4-6 мг липазы TL (Pseud. stutzeri, Meito Sangyo) и перемешивают при 20-23°C. По достижении степени превращения>50% реакцию заканчивают путем отфильтровывания фермента и определяют оптическую чистоту непрореагировавшего спирта: (3S,1R)-метиловый эфир 2-(3-гидроксициклогексил-1-оксиметил)-6-метилбензойной кислоты - чистота>98% (ВЭЖХ на Chiralpak AD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 57
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
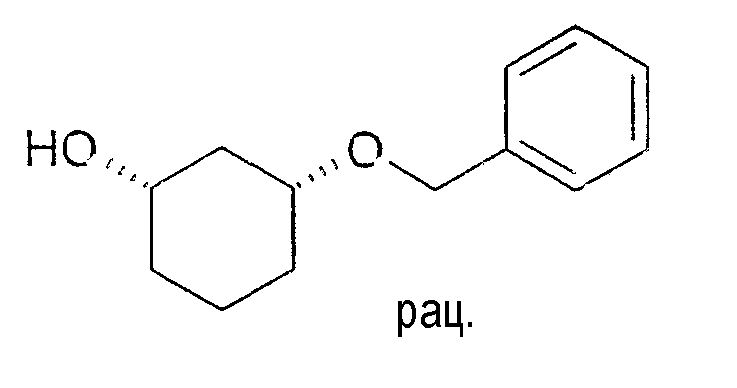
35-40 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 0,5-1 мл винилацетата и 2-3 мл метиленхлорида, смешивают с примерно 8-10 мг Novozym 435 и перемешивают при 22-25°C. Через 4 дня реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота спирта (1S,3R)-3-бензилоксициклогексан-1-ола составляет>98% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), чистота (1R,3S)-ацетата составляет 82% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 58
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
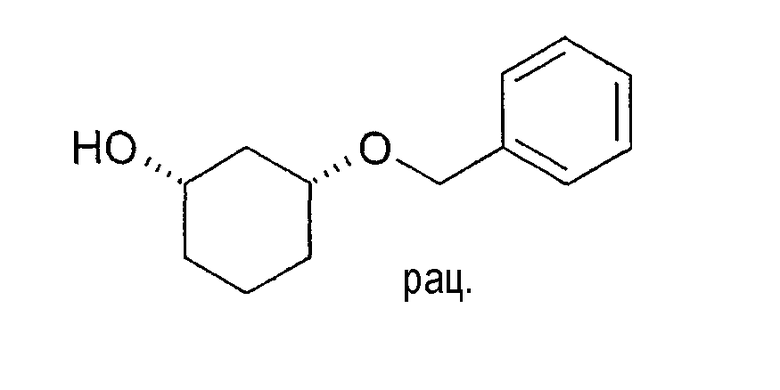
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл ТГФ, смешивают с примерно 5 мг липазы L-10 и перемешивают при 22-25°C. По достижении степени превращения ≥50% реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет ≥90% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 59
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
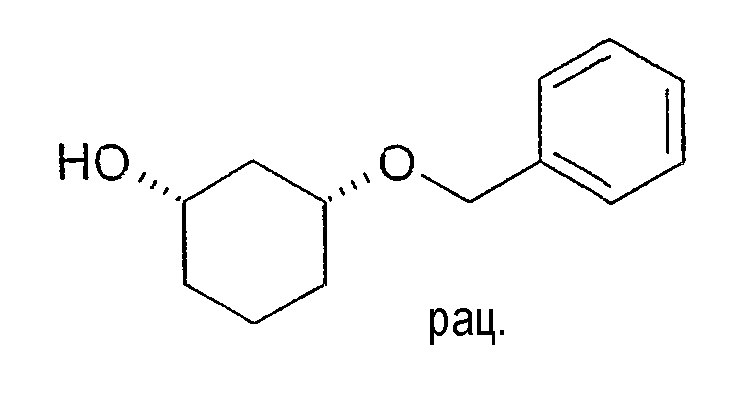
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл хлорбензола, смешивают с 10 мг Novozym 435 и перемешивают при 22-25°C. Через 4 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота спирта (1S,3R)-3-бензилоксициклогексанола составляет 68% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), чистота энантиомерного ацетата составляет 95% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 60
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл циклогексана, смешивают с примерно 5 мг липазы QL и перемешивают при 22-25°C. Через 24 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 94% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 61
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл толуола, смешивают с 10 мг Novozym 435 и перемешивают при 22-25°C. Через 4 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 70% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), чистота (1R,3S)-ацетата равна 95% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 62
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл циклогексана, смешивают с примерно 10 мг Novozym 435 и перемешивают при 22-25°C. Через примерно 4 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 95% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), чистота (1R,3S)-ацетата равна 90% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 63
Разделение рацематов цис-3-бензилоксициклогексан-1-ола

10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл циклогексана, смешивают с примерно 5 мг липазы L-10 и перемешивают при 22-25°C. Через 24 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет>95% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 64
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
Примерно 10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл ТГФ, смешивают с 10 мг Novozym 435 и перемешивают при 22-25°C. Через 4 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексанола составляет 73% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), чистота (1R,3S)-ацетата равна 94% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 65
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
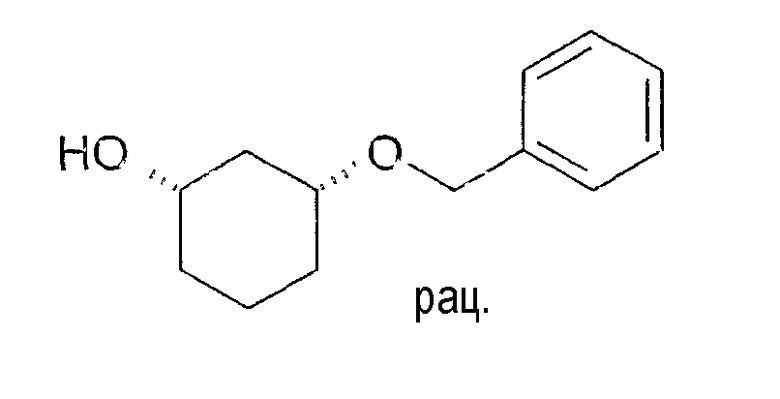
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл хлорбензола, смешивают с примерно 5 мг липазы L-10 и перемешивают при 22-25°C. По достижении степени превращения ≥50% реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет ≥92% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 66
Разделение рацематов цис-3-бензилоксициклогексан-1-ола

Примерно 10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл этилового эфира уксусной кислоты, смешивают с примерно 10 мг Novozym 435 и перемешивают при 22-25°C. Через 4 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 77% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), чистота (1R,3S)-ацетата равна 93% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 67
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
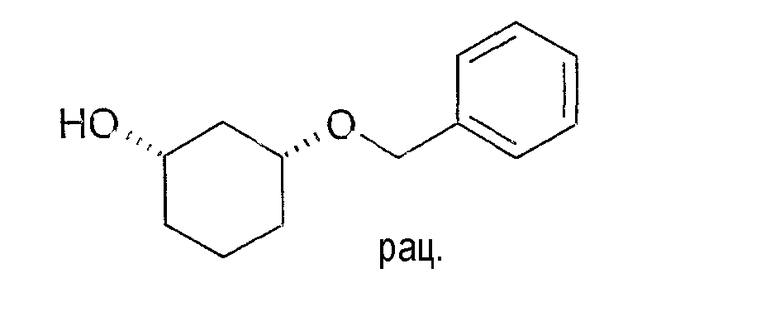
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл хлорбензола, смешивают с примерно 5 мг липазы SL и перемешивают при 22-25°C. По достижении степени превращения ≥50% реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет ≥87% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 68
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
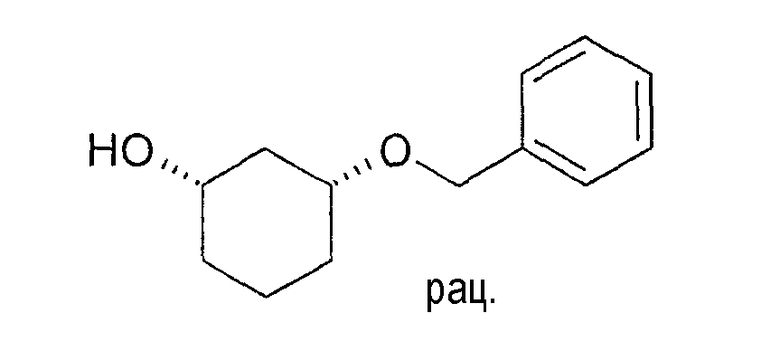
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл диизопропилового эфира, смешивают с 10 мг Novozym 435 и перемешивают при 22-25°C. Через 4 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 90% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), чистота (1R,3S)-ацетата равна 90% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 69
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
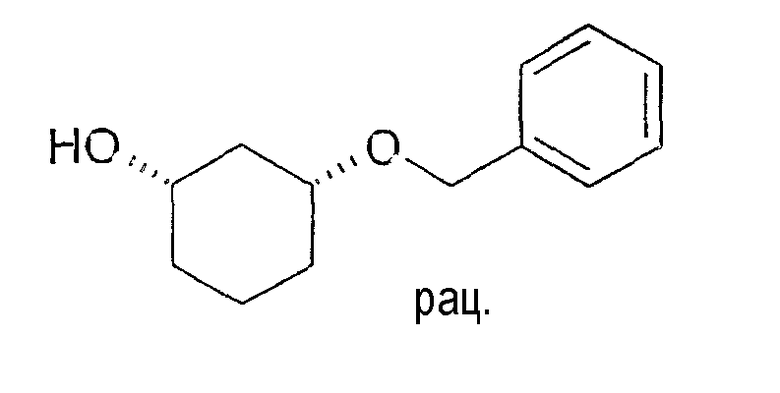
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл MTBE, смешивают с 10 мг Novozym 435 и перемешивают при 22-25°C. Через 4 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 93% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1 TFA), чистота (1R,3S)-ацетата равна 89% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 70
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
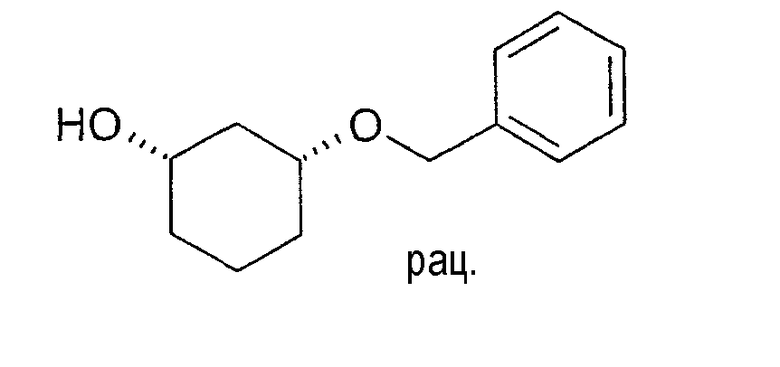
10 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 1 мл винилацетата и 3 мл циклогексана, смешивают с примерно 5 мг липазы SL и перемешивают при 22-25°C. Через 24 часа реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет >90% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 71
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
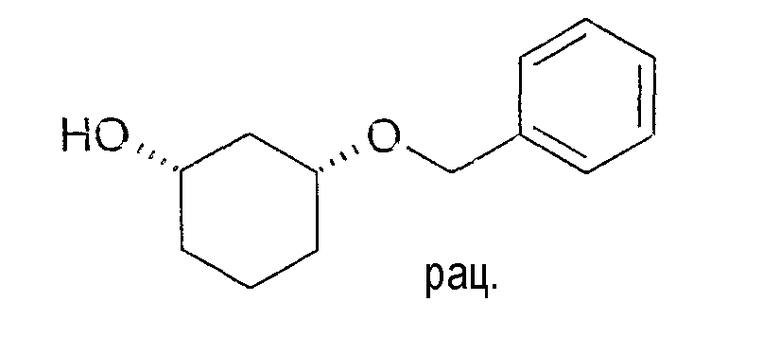
27 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 3 мл метиленхлорида, смешивают с 65 мг ангидрида изо-валериановой кислоты и 11 мг Novozym 435 и перемешивают при 22-25°C. По достижении степени превращения 45-50% реакцию заканчивают путем отфильтровывания фермента. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 87% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), энантиомерный остаток (1R,3S)-производного изо-валериановой кислоты составляет>95% (ВЭЖХ на Chiralcel OD 250×4,6;1 мл/мин, гептан/EtOH/CH3CN 100:1:0,5).
Пример 72
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
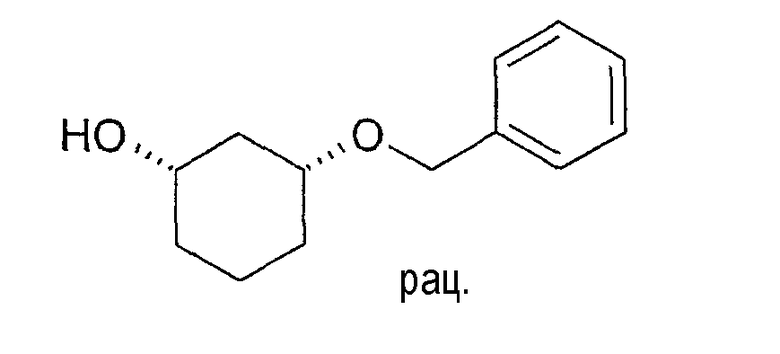
200 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 3 мл хлорбензола, смешивают с 100 мг ангидрида янтарной кислоты и 10 мг Chirazyme L-2, лиофилизованного, и перемешивают при 25-27°C. Через 29 часов реакцию заканчивают путем отфильтровывания фермента. Из концентрированной пробы определяют оптическую чистоту как непрореагировавшей основы, так и образованного продукта ацилирования. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет >98% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), оптическая чистота производного янтарной кислоты составляет 94% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 73
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
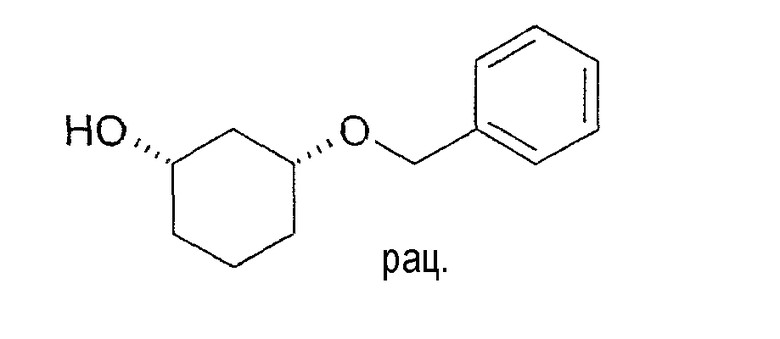
200 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 3 мл DME, смешивают с 100 мг ангидрида янтарной кислоты и 10 мг Chirazyme L-2, лиофилизованного, и перемешивают при 25-27°C. Через 29 часов реакцию заканчивают путем отфильтровывания фермента. Из концентрированной пробы определяют оптическую чистоту как непрореагировавшей основы, так и образованного продукта ацилирования. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет >95% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), оптическая чистота производного янтарной кислоты составляет >97% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 74
Разделение рацематов цис-3-бензилоксициклогексан-1-ола

200 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 3 мл ТГФ, смешивают с 100 мг ангидрида янтарной кислоты и 10 мг Chirazyme L-2, лиофилизованного, и перемешивают при 25-27°C. Через 29 ч реакцию заканчивают путем отфильтровывания фермента. Из концентрированной пробы определяют оптическую чистоту как непрореагировавшей основы, так и образованного продукта ацилирования. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 84% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), оптическая чистота производного янтарной кислоты составляет>95% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 75
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
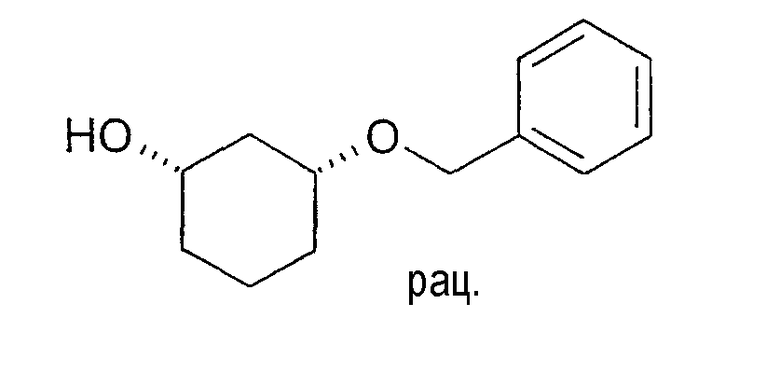
200 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 3 мл метиленхлорида, смешивают с 100 мг ангидрида янтарной кислоты и 10 мг Chirazyme L-2, лиофилизованного, и перемешивают при 25-27°C. Через 29 ч реакцию заканчивают путем отфильтровывания фермента. Из концентрированной пробы определяли оптическую чистоту как непрореагировавшей основы, так и образованного продукта ацилирования. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет >98% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), оптическая чистота производного янтарной кислоты составляет 88% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 76
Разделение рацематов цис-3-бензилоксициклогексан-1-ола
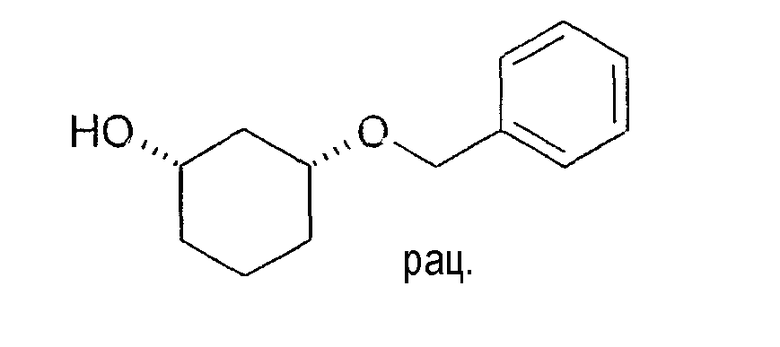
200 мг рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 3 мл ацетона, смешивают с 100 мг ангидрида янтарной кислоты и 10 мг Chirazyme L-2, лиофилизованного, и перемешивают при 25-27°C. Через 29 ч реакцию заканчивают путем отфильтровывания фермента. Из концентрированной пробы определяют оптическую чистоту как непрореагировавшей основы, так и образованного продукта ацилирования. Оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет >99% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA), оптическая чистота производного янтарной кислоты составляет 78% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Пример 77
Разделение рацематов цис-3-бензилоксициклогексан-1-ола, разделение спирта и производного янтарной кислоты
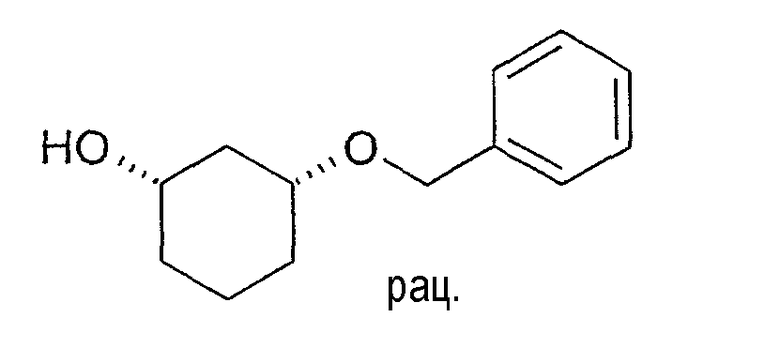
8,15 г (39,5 ммоль) рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 120 мл ТГФ, смешивают с 3,9 г (39,0 ммоль) ангидрида янтарной кислоты и 390 мг Chirazyme L-2, лиофилизованного, и перемешивают при 22-25°C. По достижении степени превращения примерно 40% реакцию заканчивают путем отфильтровывания фермента. Фильтрат концентрируют в вакууме. Остаток вводят в tBuOMe и трижды интенсивно экстрагируют насыщенным водным раствором NaHCO3, используя по 100 мл. Органическую фазу сушат (MgSO4) и концентрируют в вакууме; выход: 4,4 г; оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет 70% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA). Оптическая чистота растворенного в водной фазе производного янтарной кислоты составляет >99% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA).
Водный раствор производного янтарной кислоты гидролизуют химически путем обработки концентрированным раствором едкого натра. Образованный (1R,3S)-3-бензилоксициклогексан-1-ол экстрагируют tBuOMe; выход: 2,9 г.
Пример 78
Разделение рацематов цис-3-бензилоксициклогексан-1-ола, разделение спирта и производного янтарной кислоты
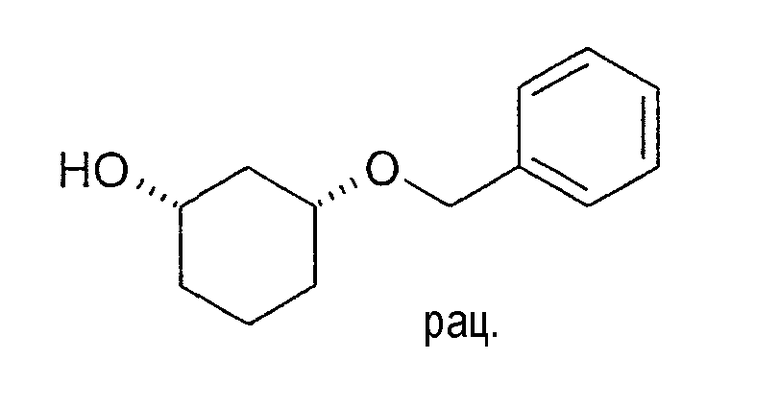
5,06 г (24,5 ммоль) рацемического цис-3-бензилоксициклогексан-1-ола растворяют в 75 мл ТГФ, смешивают с 2,52 г (25,2 ммоль) ангидрида янтарной кислоты и 3,1 мг Novozym 435 и перемешивают при 22-25°C. Через 28,5 ч реакцию заканчивают путем отфильтровывания фермента. Фильтрат концентрируют в вакууме до примерно 15 мл. Остаток смешивают с 30 мл воды и оставшийся ТГФ отгоняют в вакууме. Добавляют 15 мл насыщенного водного раствора NaHCO3, 15 мл воды и 30 мл метиленхлорида и смесь 15-30 мин интенсивно перемешивают. После разделения фаз экстрагируют сначала 90 мл насыщенного водного раствора NaHCO3 и 150 мл воды, затем еще 15 мл насыщенного водного раствора NaHCO3 и 30 мл воды, дважды промывают водой, используя по 30 мл. Органическую фазу сушат (MgSO4) и концентрируют в вакууме; выход: 2,52 г (50%), 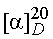 +12,1° (c=1,0, MeOH); оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет >99% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA). Очищенные водные фазы смешивают с 20 мл уксусной кислоты (ледяной уксус) и дважды экстрагируют метиленхлоридом, используя по 20 мл. Органическую фазу сушат (MgSO4) и концентрируют. Выход: 3,55 г (47%) производного янтарной кислоты, оптическая чистота составляет>99% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA); 1H-ЯМР (CDCl3), δ=1,2-1,43 (м, 4H), 1,82 (м, 1H), 1,93 (м, 1H), 2,04 (м, 1H), 2,40 (м, 1H), 2,58-2,70 (м, 4H), 3,39 (м, 1H), 4,54 (м, 2H), 4,71 (м, 1H), 7,23-7,36 (м, 5H).
+12,1° (c=1,0, MeOH); оптическая чистота (1S,3R)-3-бензилоксициклогексан-1-ола составляет >99% (ВЭЖХ на Chiralpak AD-H 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA). Очищенные водные фазы смешивают с 20 мл уксусной кислоты (ледяной уксус) и дважды экстрагируют метиленхлоридом, используя по 20 мл. Органическую фазу сушат (MgSO4) и концентрируют. Выход: 3,55 г (47%) производного янтарной кислоты, оптическая чистота составляет>99% (ВЭЖХ на Chiralcel OD 250×4,6; 1 мл/мин, гептан/EtOH/CH3CN 25:1:0,5+0,1% TFA); 1H-ЯМР (CDCl3), δ=1,2-1,43 (м, 4H), 1,82 (м, 1H), 1,93 (м, 1H), 2,04 (м, 1H), 2,40 (м, 1H), 2,58-2,70 (м, 4H), 3,39 (м, 1H), 4,54 (м, 2H), 4,71 (м, 1H), 7,23-7,36 (м, 5H).
Пример 79
Получение рацемического цис-3-(трет-бутилдиметилсиланилокси)-циклогексанола
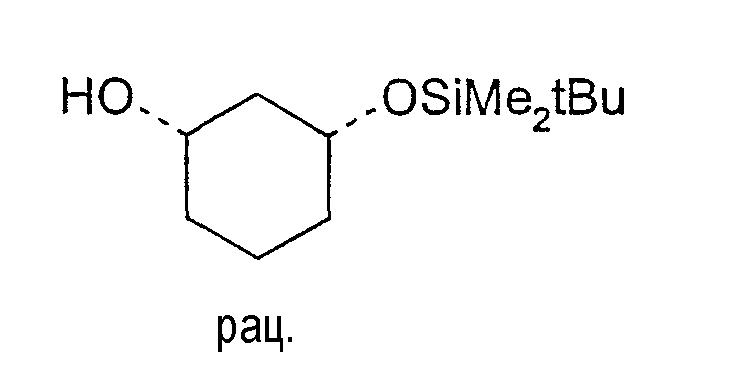
К охлажденному до 10°C раствору 1,3-циклогександиола (20,05 г, 0,173 моль), Et3N (28,79 мл, 1,2 экв.) и DMAP (0,844 г, 0,04 экв.) в CH2Cl2 (600 мл) медленно добавляют TBDMSCI (28,62 г, 1,1 экв.). После 18-часового перемешивания при 20-23°C реакционную смесь промывают H2O (2×100 мл). Органическую фазу промывают насыщенным NH4Cl (2×100 мл), сушат над MgSO4 и концентрируют в вакууме. Хроматография на силикагеле (н-гептан/ЭА 20:1) дает 18,77 г (47%) желаемого моносилилового эфира; 1H-ЯМР (CDCl3), δ=0,0-0,1 (м, 6H), 0,8-0,9 (м, 9H), 1,2-2,0 (м, 8H), 3,2 (с, шир., 1H), 3,8 (м, 1H), 3,95 (м, 1H).
Пример 80
Получение (1S,3R)-цис-3-(трет-бутилдиметилсиланилокси)-циклогексанола
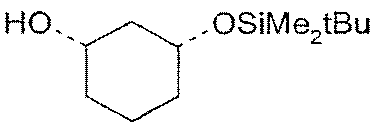
770 мг рацемического цис-3-(трет-бутилдиметилсиланилокси)-циклогексанола растворяют в 10 мл ацетона, смешивают с 0,36 мл винилацетата и 400 мг Novozym 435 и перемешивают при 21-24°C. Через 47 ч (степень превращения примерно 50%) реакцию заканчивают путем отфильтровывания фермента, и раствор выпаривают в вакууме. Хроматография части на силикагеле (н-гептан/ЭА 3:1) дает (1S,3R)-цис-3-(трет-бутилдиметилсиланилокси)-циклогексанола с +12,8° (c=1,0, MeOH).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДИАРИЛЦИКЛОАЛКИЛПРОИЗВОДНЫХ | 2005 |
|
RU2414459C2 |
| ДИАРИЛЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ PPAR-АКТИВАТОРОВ | 2002 |
|
RU2330846C2 |
| СОЕДИНЕНИЯ | 2016 |
|
RU2725616C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ПОВЫШЕНИЯ АКТИВНОСТИ CFTR | 2015 |
|
RU2767460C2 |
| ВТОРИЧНЫЕ АМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕНИНА | 2007 |
|
RU2425032C2 |
| ЗАМЕЩЕННЫЕ КАРБОНОВЫМИ КИСЛОТАМИ ОКСАЗОЛОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ PPAR-АЛЬФА И ГАММА-АКТИВАТОРОВ ПРИ ЛЕЧЕНИИ ДИАБЕТА | 2002 |
|
RU2278859C2 |
| 3-КАРБОНИЛАМИНО-5-ЦИКЛОПЕНТИЛ-1H-ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИТОРОВ CDK2 | 2020 |
|
RU2797889C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЯНУС-КИНАЗЫ 3 | 2006 |
|
RU2434013C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
Изобретение относится к вариантам способа получения хирального нерацемического соединения формулы I
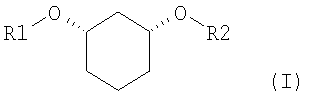
где R1 обозначает 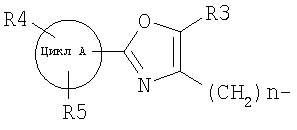 .
.
Значения остальных радикалов указаны в формуле изобретения. Соединение формулы I получают в несколько стадий, в качестве исходного вещества используют цис-1,3-циклогександиол. Одной из ключевых стадий является ферментативное образование сложного эфира или ферментативное расщепление сложного эфира. 4 н. и 4 з.п. ф-лы.
1. Способ получения хирального нерацемического соединения формулы I
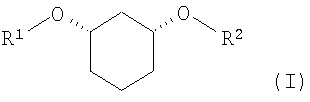
где R1
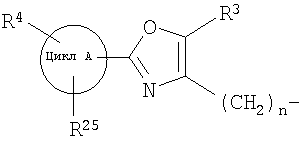
в которой означают
цикл А - фенил;
R3 - H, F, Cl, Br, ОН, NO2, CF3, OCF3, (С1-С6)-алкил, (С3-С8)-циклоалкил, фенил;
R4, R5 - H, F, Cl, Br, ОН, NO2, CF3, OCF3, OCF2H, OCF2-CF3, OCF2-CHF2, SCF3, O-фенил, (С1-С6)-алкил, O-(С1-С6)-алкил, O-(С1-С6)-алкил-O-(С1-С3)-алкил;
n - от 1 до 3; и
R2 - (C1-C8)-алкил, причем в алкильных группах одна или несколько СН2-групп может быть заменена О, СО, S, SO или SO2, и алкил может быть от одного до трех раз замещен F, Cl, Br, CF3, CN, NO2, NHAc, NHBoc, NH-СО-С(СН3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, CO-бензокси, СО-О(С1-С6)-алкилом, тетразолом, тиазолидин-2,4-дионом, индолом и фенилом, причем тиазолидин-2,4-дион и фенил, в свою очередь, могут быть замещены F, Cl, Br, CF3, CN, NO2, NHAc, NHTs, NHBoc, NHCbz, NH-CO-C(CH3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, (C1-С6)-алкилом, O-(С1-С6)-алкилом или тетразолом, отличающийся тем, что проводят следующие стадии:
а) алкилирование (Alk-R2/Alk-SG)цис-1,3-циклогександиол формулы (II)

соединением формулы (III)

в которой R2 определен выше, или означает ОН-защитную группу (SG), как например, бензилоксиметил, бензил, пара-метоксибензил или трет-бутил-диметилсилил;
X1 означает Cl, Br, I, OMs, OTs, OTf;
в подходящем растворителе в присутствии основания с получением рацемического соединения формулы (IV)
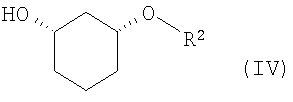
в которой R2 определен выше,
b1) ферментативное образование сложного эфира (ЕВ), на которой полученное соединение формулы (IV) подвергают стереоселективному ферментативному ацилированию с образованием сложного эфира (ЕВ), причем спирты смешивают в органическом растворителе с соответствующим ацильным донором и ферментом, выбранным из группы, включающей Novozym, Chirazyme, липазы и полученную смесь перемешивают при температуре от -20 до 80°С с получением по окончании реакции одного стереомера в виде сложного эфира формулы (V)
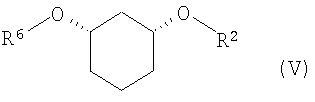
где R6 означает С(=O)-(С1-С6)-алкил, С(=O)-(С2-С6)-алкенил, С(=O)-(С3-С7)-алкинил, С(=O)-(С3-С7)-циклоалкил, причем один или несколько атомов углерода могут быть заменены на атомы кислорода и могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, CN, NO2, гидрокси, метокси, этокси, фенил и СО-O(С1-С4)-алкил, СО-O(С2-С4)-алкенил, которые в свою очередь могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, и
R2 определен выше,
и другого стереоизомера, который остается неизмененным, в виде спирта формулы (IV), или
b2) ферментативное расщепление сложного эфира [включающий химическую этерификацию (CV)+ферментативное отщепление (ES)], на которой рацемическое соединение формулы (IV) подвергают сначала химической этерификации (CV), хлорангидридом кислоты R6-Cl или ангидридом кислоты R6-O-R6, в присутствии основания с получением рацемического сложного эфира формулы (V)
где R6 и R2 определены выше,
который затем подвергают стереоселективному ферментативному расщеплению сложного эфира (ES) в гомогенной или гетерогенной, водной, водно-органической или органической среде в присутствии фермента, выбранного из группы, включающей Novozym, Chirazyme, липазу, при температуре 10-80°С и вводят в реакцию, в случае гидролиза - с водой, а в случае алкоголиза - со спиртом, по завершении которой один стереомер находится в виде спирта формулы (IV), а другой в виде оставшегося неизмененным сложного эфира формулы (V),
разделение (Т), согласно которому смесь сложного эфира формулы (V) и спирта формулы (IV), полученного на стадии b1) или b2) разделяют друг от друга (разделение Т) благодаря различию в химических и физико-химических свойствах;
c) химический гидролиз (СН)
получаемый в виде сложного эфира, на стадиях b1) или b2), энантиомер формулы (V) омыляют с получением химически энантиомерного спирта формулы IV,
d) алкилирование (Alk-R1)
получаемый в виде спирта на стадиях с), b1) или b2) энантиомер формулы (IV) подвергают взаимодействию с соединением формулы (VI)
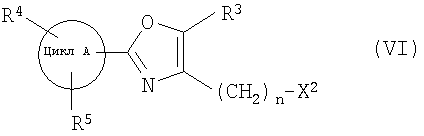
где цикл A, R3, R4, R5 и n определены выше и
X2 означает Cl, Br, I, OTs, OMs, OTf;
в подходящем растворителе в присутствии основания с получением соединения формулы (I), в случае, когда R2 означает защитную группу, проводят следующие дополнительные стадии:
е) отщепление защитной группы SG (AbSG)
соединение формулы (Ia)
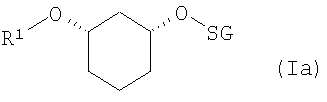
где R1 и SG определены выше,
путем отщепления защитной группы переводят в соединение формулы (VII)
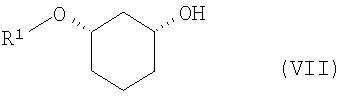
где R1 определен выше,
f) алкилирование (Alk-R2)
полученное на предыдущей стадии соединение формулы (VII) подвергают взаимодействию с соединением формулы (III),

где X1 и R2 определены выше (для соединения формулы (I)),
в подходящем растворителе в присутствии основания с получением соединения формулы (I).
2. Способ получения хирального нерацемического соединения формулы I
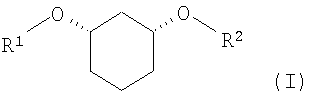
где R1
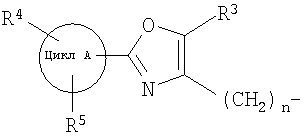
в которой означают
цикл А - фенил;
R3 - Н, F, Cl, Br, ОН, NO2, CF3, OCF3, (С1-С6)-алкил, (С3-С8)-циклоалкил, фенил;
R4, R5 - Н, F, Cl, Br, ОН, NO2, CF3, OCF3, OCF2H, OCF2-CF3, OCF2-CHF2, SCF3, O-фенил, (С1-С6)-алкил, O-(С1-С6)-алкил, O-(С1-С6)-алкил-O-(С1-С3)-алкил;
N - от 1 до 3; и
R2 - (C1-C8)-алкил, причем в алкильных группах одна или несколько СН2-групп может быть заменена О, СО, S, SO или SO2, и алкил может быть от одного до трех раз замещен F, Cl, Br, CF3, CN, NO2, NHAc, NHBoc, NH-CO-C(CH3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-О(С1-С6)-алкилом, тетразолом, тиазолидин-2,4-дионом, индолом и фенилом, причем тиазолидин-2,4-дион и фенил, в свою очередь, могут быть замещены F, Cl, Br, CF3, CN, NO2, NHAc, NHTs, NHBoc, NHCbz, NH-СО-С(СН3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, (C1-С6)-алкилом, O-(С1-С6)-алкилом или тетразолом,
отличающийся тем, что проводят следующие стадии:
а) алкилирование (Alk-R1) цис-1,3-циклогександиола формулы (II)
соединением формулы (VI)
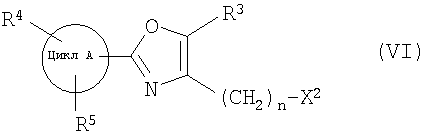
где цикл А, R3, R4, R5 и n определены выше и
X2 означает Cl, Br, I, OTs, OMs, OTf;
в подходящем растворителе в присутствии основания с получением рацемического соединения формулы (VII)
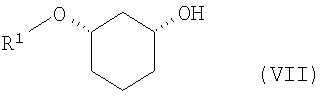
в которой R1 определен выше,
b1) ферментативное образование сложного эфира (ЕВ), на которой полученное соединение формулы (VII) подвергают стереоселективному ферментативному ацилированию с образованием сложного эфира (ЕВ), причем спирты смешивают в органическом растворителе с соответствующим ацильным донором и ферментом, выбранным из группы, включающей Novozym, Chirazyme, липазы и полученную смесь перемешивают при температуре от -20 до 80°С с получением по окончании реакции одного стереомера в виде сложного эфира формулы (V)'
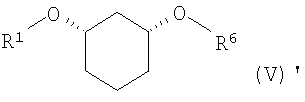
где R6 означает С(=O)-(С1-С6)-алкил, С(=O)-(С2-С6)-алкенил, С(=O)-(С3-С7)-алкинил, С(=O)-(С3-С7)-циклоалкил, причем один или несколько атомов углерода могут быть заменены на атомы кислорода и могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, CN, NO2, гидрокси, метокси, этокси, фенил и СО-O(С1-С4)-алкил, СО-O(С2-С4)-алкенил, которые в свою очередь могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, и
R1 определен выше,
и другого стереоизомера, который остается неизмененным, в виде спирта формулы (VII), или
b2) ферментативное расщепление сложного эфира [включающий химическую этерификацию (CV) + ферментативное отщепление (ES)], на которой рацемическое соединение формулы (VII) подвергают сначала химической этерификации (CV), хлорангидридом кислоты R6-Cl или ангидридом кислоты R6-O-R6, в присутствии основания с получением рацемического сложного эфира формулы (V)'
где R6 и R1 определены выше,
который затем подвергают стереоселективному ферментативному расщеплению сложного эфира (ES) в гомогенной или гетерогенной, водной, водно-органической или органической среде в присутствии фермента, выбранного из группы, включающей Novozym, Chirazyme, липазу, при температуре 10-80°С и вводят в реакцию, в случае гидролиза - с водой, а в случае алкоголиза - со спиртом, по завершении которой один стереомер находится в виде спирта формулы (VII), а другой - в виде оставшегося неизмененным сложного эфира формулы (V)',
разделение (Т), согласно которому смесь сложного эфира формулы (V)' и спирта формулы (VII), полученного на стадии b1) или b2), разделяют друг от друга (разделение Т) благодаря различию в химических и физико-химических свойствах;
с) химический гидролиз (СН),
получаемый в виде сложного эфира, на стадиях b1) или b2), энантиомер формулы (V)' омыляют с получением химически энантиомерного спирта формулы (VII),
d) алкилирование (Alk-R2),
получаемый в виде спирта на стадиях с), b1) или b2) энантиомер формулы (VII) подвергают взаимодействию с соединением формулы (III)

в которой R2 определен выше, или означает ОН-защитную группу (SG), как, например, бензилоксиметил, бензил, пара-метоксибензил или трет-бутил-диметилсилил;
X1 означает Cl, Br, I, OMs, OTs, OTf;
в подходящем растворителе в присутствии основания с получением соединения формулы (I);
в случае, когда R2 означает защитную группу проводят следующие дополнительные стадии:
e) отщепление защитной группы SG (AbSG)
соединение формулы (Ia)
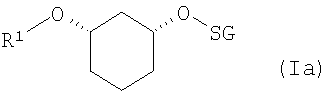
где R1 и SG определены выше,
путем отщепления защитной группы переводят в соединение формулы (VII)
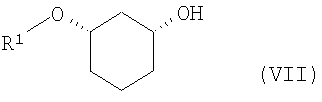
где R1 определен выше,
f) алкилирование (Alk-R2)
полученное на предыдущей стадии соединение формулы (VII) подвергают взаимодействию с соединением формулы (III),
где X1 и R2 определены выше (для соединения формулы (I)),
в подходящем растворителе в присутствии основания с получением соединения формулы (I).
3. Способ получения хирального нерацемического соединения формулы I

где R1

в которой означают
цикл А - фенил;
R3 - H, F, Cl, Br, ОН, NO2, CF3, OCF3, (С1-С6)-алкил, (С3-С8)-циклоалкил, фенил;
R4, R5 - H, F, Cl, Br, ОН, NO2, CF3, OCF3, OCF2H, OCF2-CF3, OCF2-CHF2, SCF3, O-фенил, (С1-С6)-алкил, O-(С1-С6)-алкил, O-(С1-С6)-алкил-O-(С1-С3)-алкил;
N -от 1 до 3; и
R2 - (C1-C8)-алкил, причем в алкильных группах одна или несколько СН2-групп может быть заменена О, СО, S, SO или SO2, и алкил может быть от одного до трех раз замещен F, Cl, Br, CF3, CN, NO2, NHAc, NHBoc, NH-СО-С(СН3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, тетразолом, тиазолидин-2,4-дионом, индолом и фенилом, причем тиазолидин-2,4-дион и фенил, в свою очередь, могут быть замещены F, Cl, Br, CF3, CN, NO2, NHAc, NHTs, NHBoc, NHCbz, NH-CO-C(CH3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, (С1-С6)-алкилом, O-(С1-С6)-алкилом или тетразолом,
отличающийся тем, что проводят следующие стадии:
а) алкилирование (Alk-SG) цис-1,3-циклогександиола формулы (II)
соединением формулы (III)

в которой SG означает ОН-защитную группу, как, например, бензилоксиметил, бензил, пара-метоксибензил или трет-бутил-диметилсилил;
X1 означает Cl, Br, I, OMs, OTs, OTf;
в подходящем растворителе в присутствии основания с получением рацемического соединения формулы (IV)'
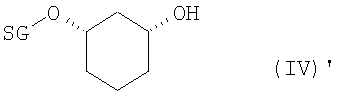
в которой SG определен выше,
b1) ферментативное образование сложного эфира (ЕВ), на которой полученное соединение формулы (IV)' подвергают стереоселективному ферментативному ацилированию с образованием сложного эфира (ЕВ), причем спирты смешивают в органическом растворителе с соответствующим ацильным донором и ферментом, выбранным из группы, включающей Novozym, Chirazyme, липазы и полученную смесь перемешивают при температуре от -20 до 80°С с получением по окончании реакции одного стереомера в виде сложного эфира формулы (V)''
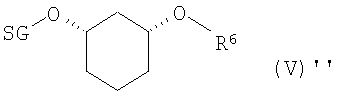
где
R6 означает С(=O)-(С1-С6)-алкил, С(=O)-(С2-С6)-алкенил, С(=O)-(С3-С7)-алкинил, С(=O)-(С3-С7)-циклоалкил, причем один или несколько атомов углерода могут быть заменены на атомы кислорода и могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, CN, NO2, гидрокси, метокси, этокси, фенил и СО-O(С1-С4)-алкил, СО-О(С2-С4)-алкенил, которые в свою очередь могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, и
SG определен выше,
и другого стереоизомера, который остается неизмененным, в виде спирта формулы (IV)', или
b2) ферментативное расщепление сложного эфира [включающий химическую этерификацию (CV) + ферментативное отщепление (ES)], на которой рацемическое соединение формулы (IV)' подвергают сначала химической этерификации (CV), хлорангидридом кислоты R6-Cl или ангидридом кислоты R6-O-R6, в присутствии основания с получением рацемического сложного эфира формулы (V)''
где R6 и R2 определены выше,
который затем подвергают стереоселективному ферментативному расщеплению сложного эфира (ES) в гомогенной или гетерогенной, водной, водно-органической или органической среде в присутствии фермента, выбранного из группы, включающей Novozym, Chirazyme, липазу, при температуре 10-80°С и вводят в реакцию, в случае гидролиза - с водой, а в случае алкоголиза - со спиртом, по завершении которой один стереомер находится в виде спирта формулы (IV)', а другой в виде оставшегося неизмененным сложного эфира формулы (V)'',
разделение (Т), согласно которому смесь сложного эфира формулы (V)'' и спирта формулы (IV)', полученного на стадии b1) или b2), разделяют друг от друга (разделение T) благодаря различию в химических и физико-химических свойствах;
c) химический гидролиз (СН)
получаемый в виде сложного эфира, на стадиях b1) или b2), энантиомер формулы (V)'' омыляют с получением химически энантиомерного спирта формулы (IV)',
d) алкилирование (Alk-R2)
получаемый в виде спирта на стадиях с), b1) или b2) энантиомер формулы (IV)' подвергают взаимодействию с соединением формулы соединением формулы (III)

в которой R2 определен выше,
X1 означает Cl, Br, I, OMs, OTs, OTf;
в подходящем растворителе в присутствии основания с получением рацемического соединения формулы (Ia)'
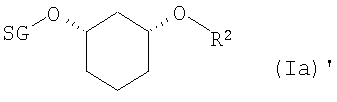
в которой R2 определен выше,
е) отщепление защитной группы SG (AbSG)
соединение формулы (Ia)'
где R2 и SG определены выше,
путем отщепления защитной группы переводят в соединение формулы (IV)
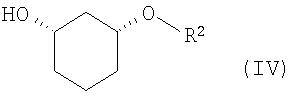
где R2 определен выше,
f) алкилирование (Alk-R1)
полученное на предыдущей стадии соединение формулы (IV) подвергают взаимодействию с соединением формулы (VI)
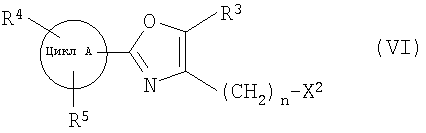
где цикл A, R3, R4, R5 и n определены выше и
X2 означает Cl, Br, I, OTs, OMs, OTf;
в подходящем растворителе в присутствии основания с получением соединения формулы (I).
4. Способ получения хирального нерацемического соединения формулы I
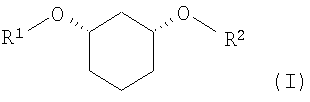
где R1
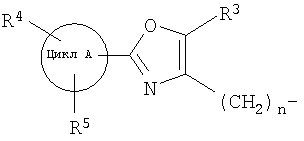
в которой означают
цикл А - фенил;
R3 - H, F, Cl, Br, ОН, NO2, CF3, OCF3, (С1-С6)-алкил, (С3-С8)-циклоалкил, фенил;
R4, R5 - H, F, Cl, Br, ОН, NO2, CF3, OCF3, OCF2H, OCF2-CF3, OCF2-CHF2, SCF3, O-фенил, (С1-С6)-алкил, O-(С1-С6)-алкил, O-(С1-С6)-алкил-O-(С1-С3)-алкил;
n - от 1 до 3; и
R2 - (C1-C8)-алкил, причем в алкильных группах одна или несколько СН2-групп может быть заменена О, СО, S, SO или SO2, и алкил может быть от одного до трех раз замещен F, Cl, Br, CF3, CN, NO2, NHAc, NHBoc, NH-СО-С(СН3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, тетразолом, тиазолидин-2,4-дионом, индолом и фенилом, причем тиазолидин-2,4-дион и фенил, в свою очередь, могут быть замещены F, Cl, Br, CF3, CN, NO2, NHAc, NHTs, NHBoc, NHCbz, NH-СО-С(СН3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, (С1-С6)-алкилом, O-(С1-С6)-алкилом или тетразолом,
отличающийся тем, что проводят следующие стадии:
а) алкилирование (Alk-SG) цис-1,3-циклогександиола формулы (II)
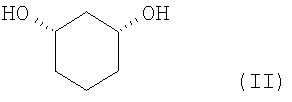
соединением формулы (III)
в которой SG означает ОН-защитную группу, как, например, бензилоксиметил, бензил, пара-метоксибензил или трет-бутил-диметилсилил;
X1 означает Cl, Br, I, OMs, OTs, OTf;
в подходящем растворителе в присутствии основания с получением рацемического соединения формулы (IV)'
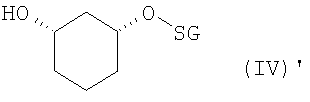
в которой SG определен выше,
b1) ферментативное образование сложного эфира (ЕВ), на которой полученное соединение формулы (IV)' подвергают стереоселективному ферментативному ацилированию с образованием сложного эфира (ЕВ), причем спирты смешивают в органическом растворителе с соответствующим ацильным донором и ферментом, выбранным из группы, включающей Novozym, Chirazyme, липазы и полученную смесь перемешивают при температуре от -20 до 80°С с получением по окончании реакции одного стереомера в виде сложного эфира формулы (V)''
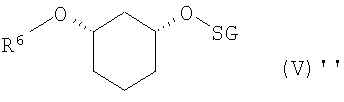
где R6 означает С(=O)-(С1-С6)-алкил, С(=O)-(С2-С6)-алкенил, С(=O)-(С3-С7)-алкинил, С(=O)-(С3-С7)-циклоалкил, причем один или несколько атомов углерода могут быть заменены на атомы кислорода и могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, CN, NO2, гидрокси, метокси, этокси, фенил и СО-O(С1-С4)-алкил, СО-O(С2-С4)-алкенил, которые в свою очередь могут быть замещены 1-3 заместителями из группы F, Cl, Br, CF3, и
SG определен выше,
и другого стереоизомера, который остается неизмененным, в виде спирта формулы (IV)', или
b2) ферментативное расщепление сложного эфира [включающий химическую этерификацию (CV) + ферментативное отщепление (ES)], на которой рацемическое соединение формулы (IV)' подвергают сначала химической этерификации (CV), хлорангидридом кислоты R6-Cl или ангидридом кислоты R6-O-R6, в присутствии основания с получением рацемического сложного эфира формулы (V)''
где R6 и R2 определены выше,
который затем подвергают стереоселективному ферментативному расщеплению сложного эфира (ES) в гомогенной или гетерогенной, водной, водно-органической или органической среде в присутствии фермента, выбранного из группы, включающей Novozym, Chirazyme, липазу, при температуре 10-80°С и вводят в реакцию, в случае гидролиза - с водой, а в случае алкоголиза - со спиртом, по завершении которой один стереомер находится в виде спирта формулы (IV)', а другой в виде оставшегося неизмененным сложного эфира формулы (V)'',
разделение (Т), согласно которому смесь сложного эфира формулы (V)'' и спирта формулы (IV)', полученного на стадии b1) или b2) разделяют друг от друга (разделение Т) благодаря различию в химических и физико-химических свойствах;
c) химический гидролиз (СН)
получаемый в виде сложного эфира, на стадиях b1) или b2), энантиомер формулы (V)'' омыляют с получением химически энантиомерного спирта формулы IV',
d) алкилирование (Alk-R1) получаемый в виде спирта на стадиях с), b1) или b2) энантиомер формулы (IV)' подвергают взаимодействию с соединением формулы соединением формулы (VI)
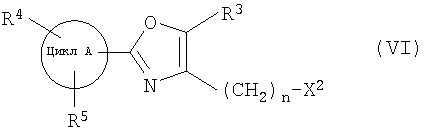
где цикл A, R3, R4, R5 и n определены выше и
X2 означает Cl, Br, I, OTs, OMs, OTf;
в подходящем растворителе в присутствии основания с получением соединения (Ia)
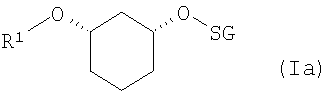
е) отщепление защитной группы SG (AbSG)
соединение формулы (Ia)
где R1 и SG определены выше,
путем отщепления защитной группы переводят в соединение формулы (VII)
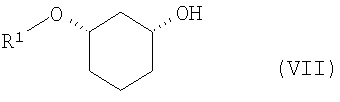
где R1 определен выше,
f) алкилирование (Alk-R2)
полученное на предыдущй стадии соединение формулы (VII) подвергают взаимодействию с соединением формулы (III)
в которой R2 определен выше,
X' означает Cl, Br, I, OMs, OTs, OTf;
в подходящем растворителе в присутствии основания с получением соединения формулы (I).
5. Способ по пп.1-4, отличающийся тем, что используют соединение формулы (III) или (III)',
или
в котором X1 означает Cl, Вr, I, OMs или OTs.
6. Способ по п.5, отличающийся тем, что используют соединение формулы (III) или (III)',
или
в котором X1 означает Cl, Br, или I.
7. Способ по пп.1-4, отличающийся тем, что получают соединение формулы (I)
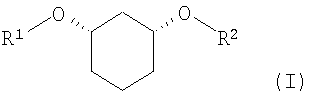
в котором
R1 означает

где цикл А - фенил,
R3 - Н, CF3, (С1-С6)-алкил, (С3-С8)-циклоалкил, фенил;
R4, R5 - Н, F, Br, CF3, OCF3, (С1-С6)-алкил, O-(С1-С6)-алкил;
n - от 1 до 2; и
R2 - (C1-C9)-алкил, причем в алкильных группах одна или несколько СН2-групп может быть заменена на О, СО, S, SO или SO2, и алкил может быть от одного до трех раз замещен F, Cl, Br, CF3, CN, NO2, NHAc, NHBoc, NH-CO-C(CH3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, тетразолом, тиазолидин-2,4-дионом, индолом и фенилом, причем тиазолидин-2,4-дион и фенил в свою очередь могут быть замещены F, Cl, Br, CF3, CN, NO2, NHAc, NHTs, NHBoc, NHCbz, NH-CO-C(CH3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, (С1-С6)-алкилом, O-(С1-С6)-алкилом или тетразолом.
8. Способ по п.7, отличающийся тем, что получают соединение формулы (I), в котором:
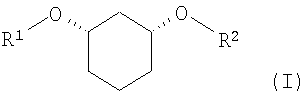
в котором R1 означает
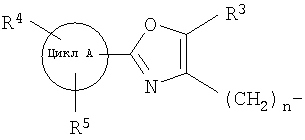
где цикл А - фенил,
R3 - (С1-С4)-алкил;
R4, R5 - H, (С1-С4)-алкил, O-(С1-С4)-алкил;
n - 1; и
R2 - (С1-С8)-алкил, причем в алкильных группах одна или несколько СН2-групп может быть заменена на О, СО, S, SO или SO2, и алкил может быть от одного до трех раз замещен F, Cl, Br, CF3, CN, NO2, NHAc, NHBoc, NH-CO-C(CH3)3, гидроксилом, OCF3, O-(C1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, тетразолом, тиазолидин-2,4-дионом, индолом и фенилом, причем тиазолидин-2,4-дион и фенил в свою очередь могут быть замещены F, Cl, Br, CF3, CN, NO2, NHAc, NHTs, NHBoc, NHCbz, NH-CO-C(CH3)3, гидроксилом, OCF3, O-(С1-С6)-алкилом, СООН, СО-бензокси, СО-O(С1-С6)-алкилом, (С1-С6)-алкилом, O-(С1-С6)-алкилом или тетразолом.
| Способ получения транс-4-(транс-4-н-алкилциклогексил)-1-алкоксициклогексанов | 1990 |
|
SU1816753A1 |
| Suemune et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

