ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому производному ариламидина и его соли, обладающих противогрибковой активностью, и противогрибковому агенту, включающему то же самое в качестве активного ингредиента.
УРОВЕНЬ ТЕХНИКИ
Серьезный глубокий микоз, такой как инвазивный кандидоз, часто может представлять собой болезнь со смертельным исходом. Полагают, что основной защитный механизм организма хозяина в отношении грибка первоначально приписывают неспецифическому иммунитету нейтрофилами. Поскольку этот механизм защиты функционирует нормально, риск инфекции грибками ограничен. Однако в последние годы риск развития глубокого микоза возрос из-за увеличения числа пациентов, предрасположенных к заболеваниям, компрометирующим иммунную систему организма, таким как злокачественные опухоли или СПИД, передозировки противоопухолевыми средствами или иммунодепрессивными средствами, интенсивное применение антибактериальных антибиотиков или стероидных гормонов и длительное применение внутривенной гипералиментации и венной катетеризации (Непатентный документ 1).
Только 7 агентов для такого глубокого микоза известны, а именно амфотерицин В, флуцитозин, миконазол, флуконазол, интраконазол, микафунгин и вориканазол. Амфотерицин В обладает сильным противогрибковым действием, но его клиническое применение ограничено из-за проблемы побочных эффектов, например почечная токсичность. Флуцитозин имеет проблему развития толерантности и редко сегодня применяется отдельно. Микафунгин имеет слабую активность в отношении Cryptococcus spp. Другие агенты обычно называют азольными противогрибковыми средствами и наиболее часто применяют сейчас вследствие благоприятного баланса между эффективностью и безопасностью, хотя их фунгицидные действия обычно бывают в общем ниже, чем у амфотерицина В (Непатентный документ 2).
Недавно флуконазол-резистентный Сandida albicans был обнаружен с высокой частотой, полученный из ротоглоточного кандидоза пацентов со СПИДом, которые получали повторное введение доз флуконазола. Кроме того, многие из резистентных штаммов показали перекрестную резистентность к интраконазолу и другим азольным агентам. Кроме того, выделение резистентных штаммов у пациентов без СПИДа, которые обнаруживали хронический кожно-слизистый кандидоз, было доложено (Непатентный документ 3). Результат резистентности имеет серьезный импульс к лечению быстро возрастающего числа пациентов с глубоким микозом (Непатентный документ 3).
С другой стороны, производное ариламидина, обладающее противогрибковой активностью, известно. (Патентные документы 1 и 2).
Патентный документ 1: WO-A-03-074476.
Патентный документ 2: WO-A-2006-003881.
Непатентный документ 1: Rinsho to Biseibutsu (Clinics and Microorganisms), vol.17, p.265-266, 1990.
Непатентный документ 2: Rinsho to Biseibutsu (Clinics and Microorganisms), vol.21, p.277-283, 1994.
Непатентный документ 3: Rinsho to Biseibutsu (Clinics and Microorganisms), vol.28, p.51-58, 2001.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Противогрибковый агент сильно желательный, который действует на основании различного механизма действия из механизмов существующих агентов, является эффективным в отношении азол-агент-резистентных грибков, имеет мало побочных эффектов и хорошо абсорбируется перорально.
При таких обстоятельствах настоящие изобретатели интенсивно изучали и обнаружили, что производное ариламидина, представленное общей формулой [1] или его соль:
[Формула 1]

(где R1 и R2 одинаково или различно представляют необязательно замещенную С3-С4 алкильную группу), является превосходящим при пероральной абсорбции, эффективным в отношении азол-агент-резистентных грибков и уменьшало побочные эффекты, таким образом выполняя настоящее изобретение.
ЭФФЕКТ ИЗОБРЕТЕНИЯ
Соединения настоящего изобретения обладают сильной активностью в отношении грибков, включая азол-агент-резистентные грибки, являются превосходящими при пероральной абсорбции, уменьшали взаимодействие с другими агентами, являются высоко безопасными и пригодными в качестве противогрибкового агента.
ЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение описано ниже более детально.
В настоящем описании, если не определено иначе, атом галогена означает атом фтора, атом хлора, атом брома и атом иода; низшая алкильная группа означает С1-6 алкильную группу с прямой или разветвленной цепью, такую как метил, этил, пропил, изопропил, бутил, сек-бутил, изобутил, трет-бутил, пентил и изопентил; С3-4 алкильная группа означает пропильную, изопропильную, бутильную, сек-бутильную, изобутильную и трет-бутильную группу; аралкильная группа означает ар-С1-6алкильную группу, такую как бензил, дифенилметил, тритил, фенэтил и нафтилметил; алкоксиалкильная группа означает С1-6алкилокси-С1-6алкильную группу, такую как метоксиметил и 1-этоксиэтил; аралкилоксиалкильная группа означает ар-С1-6алкилокси-С1-6алкильную группу, такую как бензилоксиметил и фенэтилоксиметил;
алкансульфонильная группа означает С1-6алкансульфонильную группу, такую как метансульфонильная, этансульфонильная и пропансульфонильная; арилсульфонильная группа означает, например, бензолсульфонильную, толуолсульфонильную и нафталинсульфонильную группу; алкансульфонилокси группа означает С1-6алкансульфонилокси группу, такую как метансульфонилокси и этансульфонилокси; арилсульфонилокси группа означает, например, бензолсульфонилокси и толуолсульфонилокси группу;
ацильная группа означает, например, формильную группу, С2-12алканоильную группу с прямой или разветвленной цепью, такую как ацетильная, пропионильная и изовалерьяновая, ар-С1-6алкилкарбонильную группу, такую как бензилкарбонильная, ароильную группу, такую как бензоильная и нафтоильная, гетероциклическую карбонильную группу, такую как никотиноильная, теноильная, пирролидинокарбонильная и фуроильная, карбокси-С1-6алкилкарбонильную группу, такую как 3-карбоксипропаноильная и 4-карбоксибутаноильная, С1-6алкилоксикарбонильную С1-6алкилкарбонильную, такую как 3-(метоксикарбонил)пропаноильная и 4-(метоксикарбонил)бутаноильная, сукцинильную группу, глутарильную группу, малеоильную группу, фталоильную группу и α-аминоалканоильную группу с прямой или разветвленной цепью, чей N-конец необязательно защищен и который получен из аминокислоты (примеры аминокислот включают: глицин, аланин, валин, лейцин, изолейцин, серин, треонин, цистеин, метионин, аспарагиновая кислота, глютаминовая кислота, аспарагин, глютамин, аргинин, лизин, гистидин, гидроксилизин, фенилаланин, тирозин, триптофан, пролин и гидроксипролин);
алкилоксикарбонильная группа означает С1-12алкилоксикарбонильную группу, такую как метоксикарбонильная, этоксикарбонильная, 1,1-диметилпропоксикарбонильная, изопропоксикарбонильная, 2-этилгексилоксикарбонильная, трет-бутоксикарбонильная и трет-пентилоксикарбонильная; аралкилоксикарбонильная группа означает ар-С1-6алкилоксикарбонильную группу, такую как бензилоксикарбонильная и фенэтилоксикарбонильная; арилоксикарбонильная группа означает фенилоксикарбонильную группу; кислородсодержащая гетероциклическая группа означает группу, такую как тетрагидрофурильная и тетрагидропиранильная; гетероциклическая оксикарбонильная группа означает группу, такую как 2-фурфурилоксикарбонил и 8-хинолилоксикарбонил; замещенная силильная группа означает, например, группу, такую как триметилсилильная, триэтилсилильная и трибутилсилильная.
Каждая из вышеупомянутых групп далее необязательно замещена одной или более группами, выбранными из атома галогена, гидроксильной группы, карбоксильной группы и низшей алкильной группы.
Аминозащитная группа включает все стандартные группы, которые пригодны в качестве защитных групп для аминогруппы, например, ацильную группу, алкилоксикарбонильную группу, аралкилоксикарбонильную группу, арилоксикарбонильную группу, аралкильную группу, алкоксиалкильную группу, аралкилоксиалкильную группу, алкансульфонильную группу, арилсульфонильную группу и замещенную силильную группу.
Гидроксилзащитная группа включает все стандартные группы, которые пригодны в качестве защитных групп для аминогруппы, например, ацильную группу, алкилоксикарбонильную группу, аралкилоксикарбонильную группу, гетероциклическую оксикарбонильную группу, аралкильную группу, кислород-содержащую гетероциклическую группу, алкоксиалкильную группу, аралкилоксиалкильную группу, алкансульфонильную группу, арилсульфонильную группу и замещенную силильную группу.
Уходящая группа включает, например, атом галогена, алкансульфонилокси группу и арилсульфонилокси группу.
Соль соединения общей формулы [1] включает, например, соль с неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, фосфорная кислота и серная кислота; соль с органической карбоксильной кислотой, такой как муравьиная кислота, трихлоруксусная кислота, L-винная кислота, малеиновая кислота, фумаровая и трифторуксусная кислота; и соль с сульфоновой кислотой, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, мезитиленсульфоновая кислота и нафталинсульфоновая кислота.
Предпочтительная соль соединения общей формулы [1] включает фармацевтически приемлемую соль.
Возможный заместитель для необязательно замещенной С3-4 алкильной группы R1 и R2 включает атом галогена, гидроксильную группу и карбоксильную группу.
Предпочтительное соединение настоящего изобретения включает следующие соединения:
соединение, в котором R1 представляет собой предпочтительно С3-4 алкильную группу, более предпочтительно пропильную, изопропильную или бутильную группу и далее предпочтительно бутильную группу;
соединение, в котором R2 представляет собой предпочтительно С3-4 алкильную группу, более предпочтительно пропильную, изопропильную или бутильную группу и далее предпочтительно бутильную группу;
соединение, в котором R1 и R2 являются одинаковыми, представляет собой предпочтительное.
Способ получения соединений настоящего изобретения описан.
Соединения настоящего изобретения можно получить комбинацией открыто известных способов, например, следующим способом получения.
[Способ получения 1]
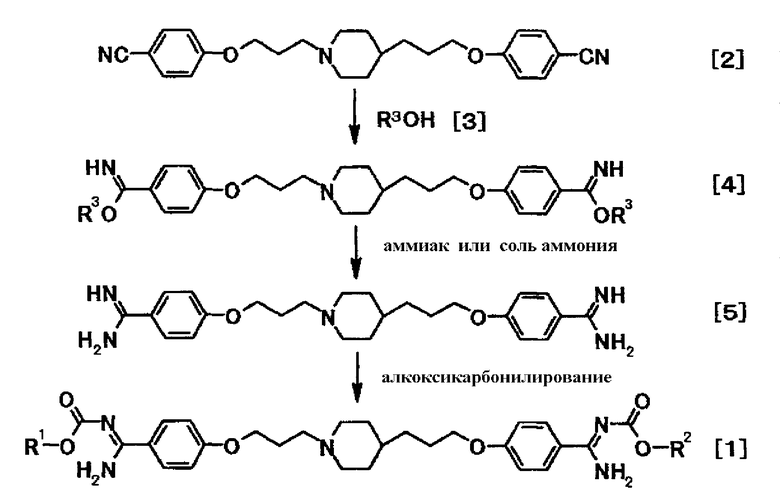
где R3 представляет низшую алкильную группу; и R1 и R2 представляют собой, как описано выше.
(1-1)
Соединение общей формулы [4] можно получить реакцией соединения формулы [2] с соединением общей формулы [3] в присутствии кислоты.
Растворитель, используемый в реакции, в частности не ограничен до такой степени, как он неблагоприятно влияет на реакцию. Примеры растворителя включают: спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метилен хлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон и 2-бутанон; эфиры, такие как этилацетат; и карбоксильные кислоты, такие как уксусная кислота. Эти растворители можно применять в комбинации. Соединение общей формулы [3] можно использовать в качестве растворителя.
Примеры кислот, используемых в реакции, включают хлороводород, бромоводород, хлорную кислоту, п-толуолсульфоновую кислоту и метансульфоновую кислоту. Такая кислота может быть применена от 1 до 200-кратных молей, предпочтительно от 5 до 100-кратных молей для количества соединения формулы [2].
В реакции используемое количество соединения общей формулы [3] может быть от 2 до 1000-кратных молей для количества соединения формулы [2], и соединение общей формулы [3] предпочтительно используют в качестве растворителя.
Реакцию могут проводить при от -30 до 150°С, предпочтительно при от 10 до 50°С в течение от 30 минут до 24 часов.
(1-2)
Соединение формулы [5] может быть получено реакцией соединения общей формулы [4] c аммиаком или солью аммония.
Растворитель, используемый в реакции, в частности не ограничен до такой степени, как он неблагоприятно влияет на реакцию. Примеры растворителя включают: спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метилен хлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид; гетероароматические соединения, такие как пиридин; и воду. Эти растворители можно использовать в комбинации.
Примеры соли аммония включают, например, хлорид аммония, бромид аммония и ацетат аммония. Используемое количество аммиака или соли аммония может быть от 3 до 100-кратных молей, предпочтительно от 3 до 10-кратных молей для количества соединения общей формулы [4].
Реакцию могут проводить при от 0 до 150°С, предпочтительно при от 20 до 120°С в течение от 1 минуты до 24 часов.
(1-3)
Соединение общей формулы [1] можно получить подверганием соединения формулы [5] реакции алкоксикарбонилирования с реагирующим производным в присутствии или отсутствии основания.
Растворитель, используемый в реакции, в частности не ограничен до такой степени, как он неблагоприятно влияет на реакцию. Примеры растворителя включают: амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метилен хлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон, метилизобутилкетон и 2-бутанон; эфиры, такие как этилацетат; карбоксильные кислоты, такие как уксусная кислота; гетероароматические соединения, такие как пиридин; и воду. Эти растворители можно применять в комбинации.
Примеры реагирующих производных включают: эфиры хлормуравьиной кислоты, такие как пропиловый эфир хлормуравьиной кислоты, изопропиловый эфир хлормуравьиной кислоты, бутиловый эфир хлормуравьиной кислоты и изобутиловый эфир хлормуравьиной кислоты; активные эфиры, такие как 4-нитрофенил пропил карбонат, 4-нитрофенил изопропил карбонат, бутил 4-нитрофенил карбонат, изобутил 4-нитрофенил карбонат, пропил 1Н-имидазол-1-карбоксилат, бутил 1Н-имидазол-1-карбоксилат, изопропил 1Н-имидазол-1-карбоксилат и изобутил 1Н-имидазол-1-карбоксилат. Эти реагирующие производные можно использовать без выделения после получения реакционной системы.
Примеры основания, которые могут необязательно использовать в реакции, включают: алкоголяты металлов, такие как метилат натрия, этилат натрия, трет-бутилат калия и трет-бутилат натрия; неорганические основания, такие как гидроксид натрия, гидроксид калия, бикарбонат натрия, карбонат натрия, карбонат калия, гидрид натрия и гидрид калия; и органические основания, такие как триэтиламин, N,N-диизопропилэтиламин, 1,8-диазабицикло-[5,4,0]-ундек-7-ен (DBU) и пиридин.
Используемое количество реагирующего производного и основания может быть от 2 до 100-кратных молей, предпочтительно от 2 до 10-кратных молей для количества соединения формулы [5].
Реакцию можно проводить при от -20 до 100°С, предпочтительно при от 20 до 80°С в течение от 1 минуты до 24 часов.
[Способ получения 2]

где R4 представляет ацильную, низшую алкильную или аралкильную группу, которая необязательно замещенная; и R1 и R2 представляют собой, как определено выше.
Соединение формулы [6] можно получать из соединения формулы [2]. Затем соединение формулы [6] можно алкилировать или ацилировать для получения соединения общей формулы [7]. Далее восстановлением соединения формулы [6] соединение формулы [5] можно получить. Соединение формулы [5] можно также получить восстановлением соединения общей формулы [7]. Эти реакции можно проводить согласно или основываясь на способах, описанных в Tetrahedron, vol.51, p.12047-12068 (1995); Synthetic Communication, vol.26, p.4351-4367 (1996); Synthesis, vol.16, p.2467-2469 (2003); Heterocycles, vol.60, p.1133-1145 (2003); и Bioorganic and Medicinal Chemistry Letter, vol.12, p.1203-1208 (2002), и т.д. Затем соединение формулы [5] можно алкоксикарбонилировать для получения соединения общей формулы [1].
Затем серии реакций описаны ниже более детально.
(2-1)
Соединение формулы [6] можно получить реакцией соединения формулы [2] с гидроксиламином или его солью в присутствии или отсутствии основания.
Растворитель, используемый в реакции, в частности не ограничен до такой степени, как он неблагоприятно влияет на реакцию. Примеры растворителя включают: спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метилен хлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон и 2-бутанон; гетероароматические соединения, такие как пиридин; и воду. Эти растворители можно использовать в комбинации.
Примеры основания, которые могут необязательно использовать в реакции, включают: алкоголяты металлов, такие как метилат натрия, этилат натрия, трет-бутилат калия и трет-бутилат натрия; неорганические основания, такие как гидроксид натрия, гидроксид калия, бикарбонат натрия, карбонат натрия, карбонат калия, гидрид натрия и гидрид калия; и органические основания, такие как триэтиламин и пиридин.
Используемое количество основания может быть от 2 до 100-кратных молей, предпочтительно от 2 до 20-кратных молей для соединения формулы [2].
Примеры соли гидроксиламина включают гидрохлорид и сульфат.
Используемое количество гидроксиламина и его соли может быть от 2 до 100-кратных молей, предпочтительно от 2 до 20-кратных молей для количества соединения формулы [2].
Реакцию могут проводить при от 0 до 150°С, предпочтительно при от 50 до 150°С в течение от 1 минуты до 24 часов.
(2-2)
Соединение общей формулы [7] можно получить реакцией соединения формулы [6] с реагирующим производным или алкилирующим агентом в присутствии или отсутствии основания.
Растворитель, используемый в реакции, в частности не ограничен до такой степени, как он неблагоприятно влияет на реакцию. Примеры растворителя включают: амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метилен хлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон и 2-бутанон; эфиры, такие как этилацетат; карбоксильные кислоты, такие как уксусная кислота; гетероароматические соединения, такие как пиридин; и воду. Эти растворители можно использовать в комбинации.
Примеры реагирующих агентов включают: ангидриды кислот, такие как ацетилформилоксид, уксусный ангидрид, трихлоруксусный ангидрид и трифторуксусный ангидрид; смешанные ангидриды кислот органической карбоксильной кислоты, такой как уксусная кислота и моноалкильных эфиров муравьиной кислоты, таких как этиловый эфир хлормуравьиной кислоты и изобутиловый эфир хлормуравьиной кислоты; смешанные ангидриды кислот органической карбоксильной кислоты, такой как уксусная кислота и органических кислот, таких как триметилуксусная кислота; хлориды кислот, такие как ацетил хлорид, трихлорацетил хлорид и трифторацетил хлорид; бромиды кислот, такие как ацетил бромид; активные эфиры, такие как п-нитрофениловый эфир, N-гидроксисукцинимидный эфир и N-гидроксифталимидный эфир. Эти реагирующие производные можно использовать без выделения после получения в реакционной системе.
Реагирующие производные могут быть получены в реакционной системе, используя связующее вещество. Примеры связующего вещества включают: карбодиимиды, такие как N,N'-дициклогексилкарбодиимид и N-этил-N'-(3-диметиламинопропил)карбодиимид; карбонилы, такие как карбонилдиимидазол; азиды кислоты, такие как дифенилфосфорил азид; цианиды кислоты, такие как диэтилфосфорил цианид; 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин; О-бензотриазол-1-ил-1,1,3,3-тетраметилмочевины гексафторфосфат; и О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилмочевины гексафторфосфат.
Примеры алкилирующего агента включают: галиды алкилов, такие как метил иодид или этил иодид; галиды аралкилов, такие как бензил хлорид и бензил бромид; и сульфаты эфиров, таких как диметил сульфат.
Примеры основания, которые можно необязательно использовать в реакции, включают: алкоголяты металлов, такие как метилат натрия, этилат натрия, трет-бутилат калия и трет-бутилат натрия; неорганические основания, такие как гидроксид натрия, гидроксид калия, бикарбонат натрия, карбонат натрия, карбонат калия, гидрид натрия и гидрид калия; и органические основания, такие как триэтиламин и пиридин.
Используемое количество реагирующего агента, алкилирующего агента и основания может быть от 2 до 100-кратных молей, предпочтительно от 2 до 10-кратных молей для количества соединения формулы [6].
Реакцию можно проводить при от -20 до 100°С, предпочтительно при от 0 до 50°С в течение от 1 минуты до 24 часов.
(2-3)
Соединение формулы [5] можно получать подверганием соединения формулы [6] реакции восстановления. Кроме того, соединение формулы [5] можно также получать подверганием соединения общей формулы [7] реакции восстановления.
Примеры используемых реакций восстановления включают реакцию каталитического гидрирования, используя металлический катализатор, и восстановление, используя металл и кислоту, например, цинк-уксусную кислоту.
Когда соединение формулы [6] или соединение общей формулы [7] подвергают реакции каталитического гидрирования, используемый растворитель, в частности, не ограничен до такой степени, как он неблагоприятно влияет на реакцию. Примеры растворителя включают: спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метилен хлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; нитрилы, такие как ацетонитрил, сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон и 2-бутанон; эфиры, такие как этилацетат; карбоксильные кислоты, такие как уксусная кислота; гетероароматические соединения, такие как пиридин; и воду. Эти растворители можно использовать в комбинации.
Примеры металлических катализаторов включают: палладиевые катализаторы, такие как палладий на углероде, оксид палладия, гидроксид палладия и черный палладий; никелевые катализаторы, такие как никель Ренея; и оксид платины. Используемое количество катализатора может быть от 0,001 до 1-кратным (в/в), предпочтительно от 0,01 до 0,5-кратным (в/в) для количества соединения формулы [6] или соединения общей формулы [7].
Примеры восстанавливающего агента, отличные от водорода, включают муравьиную кислоту; формиаты, такие как формиат натрия, формиат аммония и формиат триэтиламмония; циклогексен; и циклогексадиен. Используемое количество может быть от 2 до 100-кратных молей, предпочтительно от 2 до 10-кратных молей для количества соединения формулы [6] или соединения общей формулы [7].
Давление водорода для реакции каталитического гидрирования соединения формулы [6] может быть атмосферным давлением до 30 атм, предпочтительно от 2 до 10 атм.
Давление водорода для реакции каталитического гидрирования соединения формулы [7] может быть атмосферным давлением.
Реакцию можно проводить при от 0 до 200°С, предпочтительно при от 0 до 100°С в течение от 1 минуты до 24 часов.
(2-4)
Соединение общей формулы [1] могут получать подверганием соединения формулы [5] реакции алкоксикарбонилирования с реагирующим производным в присутствии или отсутствии основания. Реакцию можно проводить, основываясь на способе получения 1-3.
[Способ получения 3]

где R5 представляет низшую алкильную или аралкильную группу, которая необязательно замещенная; и R1, R2 и R3 представляют собой, как определено выше.
Соединение общей формулы [9] можно получить из соединения общей формулы [4]. Восстановлением соединения общей формулы [9] соединение формулы [5] можно получить. Затем соединение формулы [5] можно алкоксикарбонилировать для получения соединения общей формулы [1].
Затем серии этих реакций описаны ниже детально.
(3-1)
Соединение общей формулы [9] можно получить реакцией соединения общей формулы [4] c соединением общей формулы [8] или его соль.
Примеры соединения общей формулы [8] включают O-метилгидроксиламин и О-бензилгидроксиламин.
Примеры соли соединения общей формулы [8] включают гидрохлорид и сульфат.
Реакцию можно проводить, основываясь на способе получения 1-2.
(3-2)
Соединение формулы [5] можно получить восстановлением соединения общей формулы [9]. Реакцию можно проводить, основываясь на способе получения 2-3.
(3-3)
Соединение общей формулы [1] можно получить подверганием соединения формулы [5] реакции алкоксикарбонилирования с реагирующим производным в присутствии или отсутствии основания. Реакцию можно проводить, основываясь на способе получения 1-3.
В вышеупомянутых способах получения соединения в виде сольватов, гидратов и различных форм кристаллов можно использовать.
Способ получения соединения формулы [2], которое представляет собой исходное вещество для получения соединений настоящего изобретения, описан ниже. Соединение формулы [2] можно получить комбинацией открыто известных способов, например, следующим способом получения.
[Способ получения А]
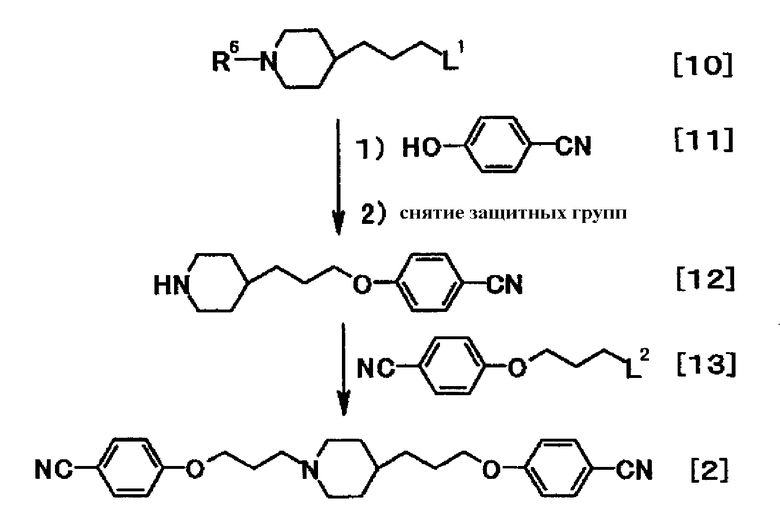
где R6 представляет аминозащитную группу; и L1 и L2 стоят для уходящих групп.
Примеры соединения общей формулы [10] включают бензил 4-(3-бромпропил)пиперидин-1-карбоксилат (J.Med.Chem., vol.46, p.2606-2620 (2003)), трет-бутил 4-(3-бромпропил)-1-пиперидинкарбоксилат (Tetrahedron, vol.55, p.11619-11639 (1999)) и 3-[N-[(трет-бутокси)карбонил]пиперидин-4-ил]пропил иодид (J. Med.Chem., vol.37, p.2537-2551 (1994)). Далее подобное можно синтезировать, используя исходное вещество трет-бутил 4-(3-гидроксипропил)-1-пиперидинкарбоксилат, и т.д. комбинацией открыто известных способов.
(A-1)
Соединение формулы [12] можно получить реакцией соединения общей формулы [10] с соединением формулы [11] в присутствии или отсутствии основания, с последующим снятием защитных групп.
Растворитель, используемый в реакции, в частности, не ограничен до такой степени, как он неблагоприятно влияет на реакцию. Примеры растворителя включают: спирты, такие как метанол, этанол, 2-пропанол и 2-метил-2-пропанол; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метилен хлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон и 2-бутанон; эфиры, такие как этилацетат; гетероароматические соединения, такие как пиридин; и воду. Эти растворители можно использовать в комбинации.
Примеры основания, которые можно необязательно использовать в реакции, включают: алкоголяты металлов, такие как метилат натрия, этилат натрия, трет-бутилат калия и трет-бутилат натрия; неорганические основания, такие как гидроксид натрия, гидроксид калия, бикарбонат натрия, карбонат натрия, карбонат калия, гидрид натрия и гидрид калия; и органические основания, такие как триэтиламин, N,N-диизопропилэтиламин и пиридин.
Используемое количество основания может быть от 1 до 10-кратных молей, предпочтительно от 1 до 3-кратных молей для количества соединения общей формулы [10].
Количество соединения формулы [11] может быть от 1 до 20-кратных молей, предпочтительно от 1 до 5-кратных молей для количества соединения общей формулы [10].
Реакцию можно проводить при от 0 до 200°С, предпочтительно при от 0 до 150°С в течение от 1 минуты до 24 часов.
Удаление аминозащитной группы, обозначенной в качестве R6, можно проводить согласно или основываясь на способе, описанном в «Protective groups in organic synthesis» (third edition, p.494-653 (1999)) или т.п.
(A-2)
Соединение формулы [2] можно получить реакцией соединения формулы [12] c соединением общей формулы [13]. Реакцию можно проводить согласно способу получения А-1.
[Способ получения В]

где R7 представляет атом водорода или гидроксил защитную группу; и L1 и L2 являются описанными выше.
В качестве соединения общей формулы [14] 3-(4-пиперидинил)-1-пропанол известно. Далее соединение общей формулы [14] можно получить, используя в качестве исходного соединения трет-бутил 4-(3-гидроксипропил)-1-пиперидинкарбоксилат и т.п. и комбинацией открыто известных способов.
(В-1)
Соединение формулы [15] можно получить реакцией соединения общей формулы [13] c соединением общей формулы [14] с последующим снятием защитных групп при необходимости. Реакцию можно проводить, основываясь на способе получения А-1.
Удаление гидроксилзащитной группы, обозначенной в качестве R7, можно проводить согласно или основываясь на способе, описанном в «Protective groups in organic synthesis» (third edition, p.494-653 (1999)) или т.п.
(В-2)
Соединение общей формулы [16] можно получить превращением гидроксильной группы соединения формулы [15] в уходящую группу.
Когда уходящая группа представляет собой алкансульфонилокси группу или арилсульфонилокси группу, соединение формулы [15] может реагировать в присутствии или отсутствии основания с алкансульфонил хлоридом, таким как метансульфонил хлорид или арилсульфонил хлоридом, таким как п-толуолсульфонил хлорид.
Примеры основания, которые можно необязательно использовать в реакции, включают: алкоголяты металлов, такие как метилат натрия, этилат натрия, трет-бутилат калия и трет-бутилат натрия; неорганические основания, такие как гидроксид натрия, гидроксид калия, бикарбонат натрия, карбонат натрия, карбонат калия, гидрид натрия и гидрид калия; и органические основания, такие как триэтиламин, N,N-диизопропилэтиламин и пиридин.
Используемое количество алкансульфонил хлорида или арилсульфонил хлорида, так же как основания, может быть от 1 до 10-кратных молей, предпочтительно от 1 до 3-кратных молей для количества соединения формулы [15].
Когда уходящая группа представляет собой атом галогена, соединение формулы [15] может реагировать, например, с тионил хлоридом, тионил бромидом, трибромидом бора и тетрабромидом-трифенилфософином углерода.
Используемое количество таких реагентов может быть от 1 до 10-кратных молей, предпочтительно от 1 до 3-кратных молей для количества соединения формулы [15].
Растворитель, используемый в реакции, в частности, не ограничен до такой степени, как он неблагоприятно влияет на реакцию. Примеры растворителя включают: амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и 1-метил-2-пирролидон; галогенированные углеводороды, такие как метилен хлорид, хлороформ и дихлорэтан; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диоксан, тетрагидрофуран, анизол, диметиловый эфир диэтиленгликоля, диэтиловый эфир диэтиленгликоля и монометиловый эфир этиленгликоля; нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид; и гетероароматические соединения, такие как пиридин. Эти растворители можно использовать в комбинации.
(В-3)
Соединение формулы [2] можно получить реакцией соединения общей формулы [16] c соединением формулы [11]. Реакцию можно проводить, основываясь на способе получения А-1.
[Способ получения С]

где L3 представляет уходящую группу; и L2 представляет собой, как определено выше.
Примеры соединений общей формулы [17] включают 3-хлор-1-пропанол и 3-бром-1-пропанол.
(C-1)
Соединение формулы [18] можно получить реакцией соединения формулы [11] c соединением общей формулы [17]. Реакцию можно проводить, основываясь на способе получения А-1.
(С-2)
Соединение общей формулы [13] можно получить превращением гидроксильной группы соединения формулы [18] в уходящую группу. Реакцию можно проводить, основываясь на способе получения В-2.
Когда соединение настоящего изобретения применяют в качестве лекарственного средства, средства состава, обычно используемые для состава, например эксципиент, носитель и разбавитель, можно смешать соответствующим образом. Лекарственное средство можно перорально или парентерально вводить обычным способом в форме таблетки, капсулы, порошка, сиропа, гранулы, пилюли, суспензии, эмульсии, жидкости, порошкового состава, суппозитории, глазных капель, капель для носа, ушных капель, пластыря, мази или инъекции. Способ введения, дозировку и частоту введения можно выбрать соответствующим образом в зависимости от возраста, веса тела и симптомов пациента. Обычно для взрослого дозу от 0,01 до 1,000 мг/кг в сутки можно вводить, разделенную от 1 до нескольких фракций, перорально или парентерально (например, инъекцией, капельницей или ректальным введением).
Для выяснения применимости соединения настоящего изобретения следующие тесты проводили.
В качестве Сравнительных Соединений соединение, описанное в Примере 91 WO-A-03-074476, и соединения, описанные в Примерах 32 и 33 WO-A-2006-003881, применяли.
Сравнительное соединение 1 (WO-A-03-074476, Пример 91)
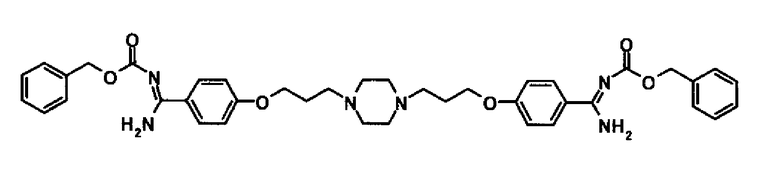
Сравнительное соединение 2 (WO-A-2006-003881, Пример 32)

Сравнительное соединение 3 (WO-A-2006-003881, Пример 33)

Тестовый пример 1: Тест, использующий модель инфекции мышиного кандидоза (пероральное введение)
В качестве Тестируемых Соединений соединения Примера 1, Примера 2, Примера 3 и Примера 4 использовали.
Сandida albicans TIMM 1623 культивировали при 35°С за ночь на слое среды декстрозного агара Сабуро и полученную культуру суспендировали в стерильный физиологический солевой раствор, который затем разбавляли для получения раствора инокулята.
Мышам-самцам (4-недельного возраста, 5 мышей/группа) вводили внутрибрюшинно 200 мг/кг циклофосфамида за 4 дня до инфицирования и 100 мг/кг на следующий день после инфицирования для получения кратковременного нарушенного состояния. Приготовленный раствор инокулята Сandida albicans TIMM 1623 в количестве 0,2 мл вводили в вену хвоста каждой мыши для индуцирования инфицирования (около 3×104 КОЕ/мышь). Тестируемые соединения растворяли в 0,1 мл/л соляной кислоты и раствор разбавляли стерилизованной водой и вводили перорально в дозе 3 мг/кг веса тела мыши. Это лечение начинали 2 часа после инфицирования и проводили один раз ежедневно в течение 7 дней. Группе, не получающей Тестируемые Соединения, равное количество стерильного физиологического солевого раствора вводили. Жизнеспособность мышей наблюдали и регистрировали в течение 14 дней после инфицирования.
В результате мыши в группе, не получающей Тестируемые Соединения, все умерли, тогда как 80% или более мышей в группах, получающих соединения Примера 1, Примера 2, Примера 3 и Примера 4, выжили.
Соединения Примера 1, Примера 2, Примера 3 и Примера 4 проявили превосходную терапевтическую эффективность.
Тестовый пример 2: Тест, использующий модель инфекции мышиного кандидоза (подкожное введение).
В качестве Тестируемого Соединения соединение Примера 3 использовали.
Мышам-самцам (4-недельного возраста, 5 мышей/группа) вводили внутрибрюшинно 200 мг/кг циклофосфамида за 4 дня до инфицирования и 100 мг/кг на следующий день после инфицирования для получения кратковременного нарушенного состояния. Сandida albicans TIMM 1623, культивируемый на SDA при 35°С, суспендировали в стерильный физиологический солевой раствор для приготовления суспензии при 1,5×105 клеток/мл. Каждые 0,2 мл раствора вводили в вену хвоста каждой мыши для индуцирования инфицирования (около 3×104 КОЕ/мышь). Тестируемые соединения растворяли в небольшом количестве 0,1 моль/л соляной кислоты и раствор разбавляли стерильным физиологическим солевым раствором для получения 0,01 мг/мл раствора. Раствор вводили подкожно в дозе 10 мл/кг веса тела мыши (0,1 мг/кг веса тела). Введения проводили один раз 2 часа после инфицирования и один раз ежедневно в течение следующих идущих подряд 3 дней, в сумме 4 раза. Группе, не получающей Тестируемые Соединения, равное количество стерильного физиологического солевого раствора вводили. Жизнеспособность мышей наблюдали и регистрировали в течение 21 дня после инфицирования.
В результате мыши в группе, не получающей Тестируемые Соединения, все умерли, тогда как 80% в группе, получающей соединение Примера 3, выжили.
Соединение Примера 3 проявило превосходную терапевтическую эффективность.
Тестовый пример 3: Тест, использующий модель мышиного аспергиллеза (пероральное введение)
В качестве тестируемых соединений соединение Примера 3 и Сравнительное соединение 1 использовали.
Споры Aspergillus fumigatus IFM46895 культивировали на среде картофельного агара с декстрозой при 30°С в течение недели. Извлеченные споры суспендировали в стерильный физиологический солевой раствор, содержащий 0,05% Твина 80, который затем был разбавлен для получения раствора инокулята.
Мышам-самцам (4-недельного возраста, 5 мышей/группа) вводили внутрибрюшинно 200 мг/кг циклофосфамида за 4 дня до инфекции и 100 мг/кг на следующий день после инфекции для получения кратковременного нарушенного состояния. Каждые 0,2 мл раствора инокулята вводили в вену хвоста каждой мыши для индуцирования инфицирования (около 1×105 КОЕ/мышь). Тестируемые соединения растворяли в небольшом количестве 0,1 моль/л соляной кислоты и раствор разбавляли стерилизованной дистиллированной водой для получения 1 мг/мл раствора. Раствор вводили перорально в дозе 10 мл/кг веса тела мыши (10 мг/кг веса тела). Введения проводили 1 раз 2 часа после инфекции и один раз ежедневно в течение следующих 6 дней, в сумме 7 раз. Группе, не получающей Тестируемые Соединения, равное количество стерильного физиологического солевого раствора вводили. Жизнеспособность мышей наблюдали и регистрировали в течение 21 дня после инфицирования.
В результате мыши в группе, не получающей Тестируемого Соединения, все умерли, 20% мышей в группе, получающей Сравнительное Соединение 1, выжили, тогда как 80% мышей в группе, получающей соединение Примера 3, выжили.
Соединение Примера 3 проявило превосходную терапевтическую эффективность.
Тестовый пример 4: Тест, использующий модель мышиного аспергиллеза (подкожное введение)
В качестве Тестируемого Соединения соединение Примера 3 использовали.
Споры Aspergillus fumigatus IFM46895 культивировали на среде картофельного агара с декстрозой при 30°С в течение недели. Извлеченные споры суспендировали в стерильный физиологический солевой раствор, содержащий 0,05% Твина 80, которые был разбавлен для получения раствора инокулята.
Мышам-самцам (4-недельного возраста, 5 мышей/группа) вводили внутрибрюшинно 200 мг/кг циклофосфамида за 4 дня до инфицирования и 100 мг/кг на следующий день после инфицирования для получения кратковременного нарушенного состояния. Каждые 0,2 мл раствора инокулята вводили в вену хвоста каждой мыши для индуцирования инфицирования (около 1×105 КОЕ/мышь). Тестируемое Соединение растворяли в небольшом количестве 0,1 моль/л соляной кислоты и раствор разбавляли стерильным физиологическим солевым раствором для получения 0,3 мг/мл раствора. Раствор вводили подкожно в дозе 10 мл/кг веса тела мыши (0,3 мг/кг веса тела). Введения проводили один раз 2 часа после инфекции и один раз ежедневно в течение следующих 6 дней, в сумме 7 раз. Группе, не получающей Тестируемого соединения, равное количество стерильного физиологического солевого раствора вводили. Жизнеспособность мышей наблюдали и регистрировали в течение 21 дня после инфицирования.
В результате мыши в группе, не получающей Тестируемого Соединения, все умерли, тогда как 60% в группе, получающей соединение Примера 3, выжили.
Соединение Примера 3 проявило превосходную терапевтическую эффективность.
Тестовый пример 5: Тест торможения роста на клетках Vero
В качестве тестируемых соединений соединения Примера 1, и Примера 2, и Сравнительного Примера 1 использовали.
Цитотоксичность соединений оценивали, используя клетки Vero. Соответствующие Тестовые Соединения растворяли в диметилсульфоксиде (ДМСО) для получения растворов 10 мг/мл. Растворы разбавляли с Е'MEM с 10% FBS до конечной концентрации 50 мкг/мл и помещали в 96-луночный планшет. Клетки суспендировали в Е'MEM с 10% FBS и сеяли на 96-луночный планшет при 3000 клеток/лунка и затем культивировали в CO2 инкубаторе при 37°С в течение 3 дней. Рост клеток Vero оценивали анализом, используя 2,3-бис-(2-метокси-4-нитро-5-сульфофенил)-5-[(фениламино)карбонил]-2Н-тетразолиум (внутренняя соль) мононатриевую соль («ХТТ»). А именно ХТТ раствор, содержащий 1 мг/мл ХТТ и 25 мкмоль/л феназин метосульфата (PMS), добавляли в каждую лунку. После инкубирования в СО2 инкубаторе в течение 2 часов абсорбцию при 450 нм (стандарт при 655 нм) соответствующих лунок измеряли на аппарате для прочтения микропланшетов. T/C вычисляли из отношения абсорбции контроля (без Соединения) и соответствующих лунок. Результаты показаны в Таблице 1.
Соединение настоящего изобретения было значительно превосходящим по безопасности Сравнительное Соединение 1.
Тестовый пример 6: Изучение повторной внутривенной дозы токсичности на мышах.
В качестве Тестируемых Cоединений соединение Примера 3, Сравнительное Соединение 2 и Сравнительное соединение 3 использовали.
Изучение повторной внутривенной дозы токсичности проводили, используя ICR штамм мышей-самцов (6-недельного возраста, 5 мышей/группа). Введение растворов готовили добавлением 3-кратного молярного количества соляной кислоты к соответствующему Тестируемому Соединению и далее добавление стерильного физиологического солевого раствора. Соединения Примера 3 и Сравнительное Соединение 2, соответственно 25 мг/кг и Сравнительное Соединение 1 6,25 мг/кг вводили в вену хвоста один раз ежедневно в течение 3 дней. Контрольной группе вводили стерильный физиологический солевой раствор.
В день 1 после завершения введения каждой мыши давали эфирный наркоз. Образцы крови брали из брюшной вены, используя медицинский шприц, содержащий гепарин в качестве антикоагулянта (Ново-Гепарин 1.000 единиц для инъекции, Aventis Pharma Ltd.), и образцы центрифугировали (3,300 об/мин, 4°С, 10 мин; Kubota Model 5900) для получения плазмы. Биохимические анализы крови по отношению к аспартат-аминотрансферазе (АСТ) и аланин-аминотрансферазе (АЛТ) образцов проводили согласно JSCC согласованному способу измерения. Показатели для Тестируемых Соединений и Сравнительных Соединений вычисляли, основываясь на значениях контроля (введение стерильного физиологического солевого раствора), как 100.
Никаких отклонений от нормы не наблюдали в АСТ и АЛТ для соединения Примера 3. С другой стороны, для Сравнительных Соединений 2 и 3 увеличения в АСТ и АЛТ наблюдали, показывая наличие нарушений печени.
Соединение настоящего изобретения превосходило по безопасности Сравнительное Соединение 2 и Сравнительное Соединение 3.
Тестовый пример 7: Изучение острой токсичности на мышах (пероральное введение)
100 мг/мл суспензии соединения Примера 3 получали с 0,1 моль/л соляной кислоты. Раствор Тестируемого Соединения перорально вводили мышам-самцам (6-недельного возраста, 2 мыши/группа) 10 мл/кг (1000 мг/кг веса тела) и мышей наблюдали в течение 2-х дней после введения.
В результате все мыши выжили в течение 2-х дней после введения.
Тестовый пример 8: Изучение острой токсичности на мышах (внутривенное введение)
Соединение Примера 3 растворяли в небольшом количестве 0,1 мол/л соляной кислоты и раствор разбавляли стерильным физиологическим солевым раствором для получения раствора 5 мг/мл. Раствор Тестируемого Соединения вводили внутривенно мышам-самцам (4-недельного возраста, 2 мыши/группа) 10 мл/кг (50 мг/кг веса тела) и мышей наблюдали в течение 2-х дней после введения.
В результате все мыши выжили в течение 2-х дней после введения.
Тестовые примеры 7 и 8 показали, что соединение настоящего изобретения было превосходящим по безопасности.
Тестовый пример 9: Ингибиторные эффекты на печеночный фермент, метаболизирующий лекарственные средства, на людях
(1) Ингибиторный эффект на CYP2D6
Ингибиторные эффекты соединения Примера 3, Сравнительного Примера 1, Сравнительного Примера 2 и Сравнительного Примера 3 печеночный фермент CYP2D6, метаболизирующий лекарственные средства, сравнивали. Микросому, полученную из клеток насекомых, экспрессирующих человеческий CYP2D6, использовали и субстрат был 3-[2-(N,N-диэтил-N-метиламмоний)этил]-7-метокси-4-метилкумарин иодид. Реакцию проводили в фосфатно-буферном растворе (100 ммоль/л, pH 7,4), включающем конечные концентрации 20 нмоль/л для фермента, 1,5 мкмоль/л для субстрата, 1,55 ммоль/л для никотинамид аденин динуклеотид фосфата окисленная форма (НАДФ+), 3,3 ммоль/л для глюкозо-6-фосфата, 3,3 ммоль/л для хлорида магния и 0,4 един/мл для глюкозо-6-фосфат дегидрогеназы (G6PDH). Концентрации соответствующих соединений в реакционном растворе получали серией 3-кратного разбавления с конечной концентрацией в диапазоне от 72 до 0,0329 мкмоль/л. Реакционные растворы инкубировали при 37°С в течение 30 мин. Затем реакцию завершали 80% раствором ацетонитрила (содержащий трис при конечной концентрации 0,1 моль/л) и активность ферментов определяли измерением флуоресценции с длиной волны 465 нм, используя длину волны возбуждения 400 нм. Ингибиторный эффект выражали как IC50. Хинидин использовали в качестве положительного контроля.
Соединение Примера 3 не проявляло ингибиторный эффект на CYP2D6 до 72 мкмоль/л. Сравнительное Соединение 1 с IC50 0,68 мкмоль/л ингибировало человеческий CYP2D6 сильно. Сравнительное Соединение 2 и Сравнительное Соединение 3 ингибировали человеческий CYP2D6.
(2) Ингибиторный эффект на CYP2С19
Ингибиторные эффекты соединения Примера 3 и Сравнительного Примера 1 на печеночный фермент CYP2С19, метаболизирующий лекарственные средства, сравнивали. Микросому, полученную из клеток насекомых, экспрессирующих человеческий CYP2С19, использовали. Дибензилфлуоресцеин использовали в качестве субстрата. Реакцию проводили в фосфатно-буферном растворе (100 ммоль/л, pH 7,4), включающем конечные концентрации 15 нмоль/л для фермента, 1,0 мкмоль/л для субстрата, 1,55 ммоль/л для никотинамид аденин динуклеотид фосфата окисленная форма (НАДФ+), 3,3 ммоль/л для глюкозо-6-фосфата, 3,3 ммоль/л для хлорида магния и 0,4 един/мл для глюкозо-6-фосфат дегидрогеназы (G6PDH). Концентрации соответствующих соединений в реакционном растворе получали серией 3-кратного разбавления с конечной концентрацией в диапазоне от 72 до 0,0329 мкмоль/л. Реакционные растворы инкубировали при 37°С в течение 30 мин. Затем реакцию завершали 2 моль/л водным раствором гидроксида натрия и реагент далее инкубировали при 37°С в течение 2 часов. Активность ферментов определяли измерением флуоресценции с длиной волны 535 нм, используя длину волны возбуждения 485 нм. Ингибиторный эффект представляли как IC50. Транилципромин использовали в качестве положительного контроля.
Соединение Примера 3 не проявляло ингибиторный эффект на активность CYP2С19 до 72 мкмоль/л. Тогда как Сравнительное Соединение 1 ингибировало человеческий CYP2С19 сильно с IC50 4,36 мкмоль/л.
(3) Ингибиторный эффект на CYP3А4
Ингибиторные эффекты соединения Примера 3 и Сравнительного Примера 1 на печеночный фермент CYP2С19, метаболизирующий лекарственные средства, сравнивали. Микросому, полученную из клеток насекомых, экспрессирующих человеческий CYP3А4, использовали. Дибензилфлуоресцеин использовали в качестве субстрата. Реакцию проводили в фосфатно-буферном растворе (100 ммоль/л, pH 7,4), включающем конечные концентрации 2,5 нмоль/л для фермента, 1,0 мкмоль/л для субстрата, 1,55 ммоль/л для никотинамид аденин динуклеотид фосфата окисленная форма (НАДФ+), 3,3 ммоль/л для глюкозо-6-фосфата, 3,3 ммоль/л для хлорида магния и 0,4 един/мл для глюкозо-6-фосфат дегидрогеназы (G6PDH). Концентрации соответствующих соединений в реакционном растворе получали серией 3-кратного разбавления с конечной концентрацией в диапазоне от 72 до 0,0329 мкмоль/л. Реакционные растворы инкубировали при 37°С в течение 15 мин. Затем реакцию завершали 2 моль/л водным раствором гидроксида натрия и реагент далее инкубировали при 37°С в течение 2 часов. Активность ферментов определяли измерением флуоресценции с длиной волны 535 нм, используя длину волны возбуждения 485 нм. Ингибиторный эффект представляли как IC50. Клотримазол использовали в качестве положительного контроля.
Соединение Примера 3 с IC50 45,4 мкмоль/л ингибировало человеческий CYP3А4 слабо. Тогда как Сравнительное Соединение 1 ингибировало человеческий CYP3А4 сильно с IC50 4,73 мкмоль/л.
Соединение настоящего изобретения показало слабый ингибиторный эффект на различные печеночные ферменты, метаболизирующие лекарственные средства, имеющие ограниченный риск лекарственного взаимодействия с другими агентами, и было превосходящим по безопасности по сравнению со сравнительными соединениями.
ПРИМЕРЫ
Настоящее изобретение сейчас будет описано посредством Справочных Примеров и Примеров, но настоящее изобретение не будет этим ограничиваться.
В дальнейшем соотношение концентраций элюента всегда выражают в объемном соотношении и носитель колоночной хроматографии представляет собой BW Silica Gel BW-127ZH (Fuji Silysia Chemical Ltd.), если не определено иначе.
Аббревиатуры в Примерах имеют следующие значения соответственно:
Ac: ацетил, Ме: метил, Ms: метансульфонил, DMSO-d6: дейтерированный диметилсульфоксид.
Ссылочный Пример 1

В суспензию 9,42 г трет-бутилата калия в 100 мл N,N-диметилформамида 10,0 г 4-цианофенола и 7,02 мл 3-хлор-1-пропанола добавляли при охлаждении воду и суспензию перемешивали при 100°С в течение 1 часа. В реакционную смесь после охлаждения до комнатной температуры 200 мл воды и 200 мл этилацетата добавляли. Органический слой отделяли, промывали 5% водным раствором карбонатом калия и насыщенным водным раствором хлорида натрия последовательно и высушивали над безводным сульфатом магния с последующим удалением растворителя под вакуумом. Полученное маслянистое вещество 11,9 г растворяли в 100 мл диоксана. К смеси 9,28 мл триэтиламина добавляли и 5,15 мл метансульфонил хлорида добавляли по каплям при охлаждении на льду в течение 8 минут, которую затем перемешивали при комнатной температуре в течение 10 минут. Реакционную смесь после добавления по каплям 100 мл воды перемешивали при комнатной температуре в течение 45 минут. Твердое вещество собирали фильтрацией и промывали 100 мл воды и 50 мл 2-пропанола для получения 12,3 г 3-(4-цианофенокси)пропил метансульфоната в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,27 (2H, тт, J=6,0, 6,0 Гц) 3,02 (3Н, с), 4,15 ((2Н, т, J=6,0 Гц), 4,45 (2H, т, J=6,0 Гц), 6,93-6,99 (2Н, м), 7,57-7,61 (2Н, м).
Ссылочный Пример 2
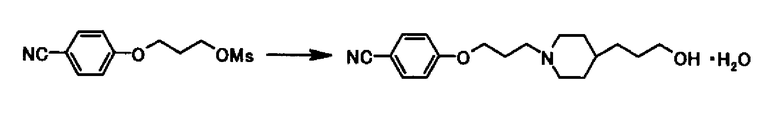
В раствор 50,0 г 3-(4-цианофенокси)пропил метансульфоната в 250 мл N,N-диметилформамида, 32,5 г иодида калия, 32,9 г бикарбоната натрия и 37,0 г 3-(4-пиперидинил)-1-пропанол гидрохлорида добавляли при комнатной температуре, который затем перемешивали при 70°С в течение 7 часов. К реакционной смеси после охлаждения до комнатной температуры 250 мл воды и 150 мл толуола добавляли и затем соляную кислоту добавляли до установления рН 1,0. Водный слой отделяли, устанавливали рН до 10 20% водным раствором гидроксида натрия и перемешивали при комнатной температуре в течение 15 минут и при охлаждении на льду в течение 30 минут. Твердое вещество собирали фильтрацией и промывали дважды 50 мл воды и дважды 50 мл толуола для получения 52,3г 4-{3-[4-(3-гидроксипропил)-1-пиперидинил]пропокси}бензонитрил моногидрата в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,20-1,75 (10H, м), 1,85-2,05 (4Н, м) 2,46-2,50 (2Н, м), 2,90-2,94 ((2Н, м), 3,64 (2H, т, J=6,6 Гц), 4,06 (2H, т, J=6,3 Гц), 6,92-6,96 (2Н, м), 7,55-7,59(2Н, м).
Ссылочный Пример 3
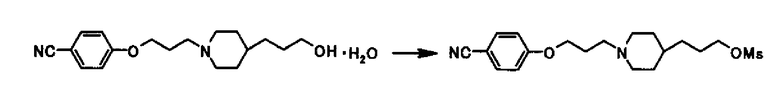
Раствор 96,2 г 4-{3-[4-(3-гидроксипропил)-1-пиперидинил]пропокси}бензонитрил моногидрата в 870 мл тетрагидрофурана нагревали для выпаривания 480 мл тетрагидрофурана при атмосферном давлении. К раствору 36,4 г триэтиламина добавляли при охлаждении водой, затем 36,1 г метансульфонил хлорида добавляли по каплям на протяжении 10 минут и раствор перемешивали при комнатной температуре в течение 20 минут. После добавления 6,07 г триэтиламина и 6,87 г метансульфонил хлорида раствор перемешивали при комнатной температуре в течение 20 минут, к которому 3,03 г триэтиламина и 3,44 г метансульфонил хлорида дополнительно добавляли и раствор перемешивали при комнатной температуре в течение 20 минут. К раствору 192 мл 2-пропанола затем добавляли и 670 мл воды добавляли по каплям на протяжении 25 минут при охлаждении на льду. После перемешивания при той же температуре в течение 30 минут твердое вещество собирали фильтрацией и промывали дважды 100 мл 50% (об/об) водного раствора 2-пропанола для получения 93,4 г 3-{1-[3-(4-цианофенокси)пропил]-4-пиперидинил}пропил метансульфоната в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,18-1,38 (5H, м), 1,55-1,82 (4Н, м) 1,88-2,05 (4Н, м), 2,44-2,52 (2Н, м), 2,88-2,96 (2Н, м), 3,01 (3Н, с), 4,06 (2H, т, J=6,3 Гц), 4,22 (2H, т, J=6,6 Гц), 6,92-6,96 (2Н, м), 7,56-7,59(2Н, м).
Ссылочный Пример 4
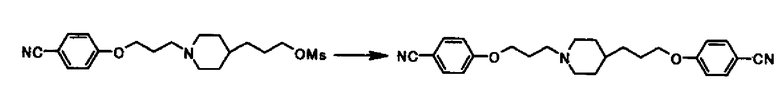
В раствор 91,9 г 3-{1-[3-(4-цианофенокси)пропил]-4-пиперидинил}пропил метансульфоната в 460 мл диметилсульфоксида, 66,9 г карбоната калия и 28,8 г 4-цианофенола добавляли при комнатной температуре и раствор перемешивали при 60°С в течение 2 часов. В реакционную смесь после охлаждения 640 мл воды добавляли по каплям в течение 20 минут и затем смесь перемешивали при комнатной температуре в течение 35 минут и после охлаждения водой в течение 30 минут. Твердое вещество собирали фильтрацией и промывали дважды 180 мл воды и затем 360 мл 2-пропанола для получения 90,0 г 4-(3-{4-[3-(4-цианофенокси)пропил]-1-пиперидинил}пропокси)бензонитрила в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,20-1,45 (5H, м), 1,65-2,05 (8Н, м) 2,40-2,55 (2Н, м), 2,85-3,00 (2Н, м), 3,99 (2H, т, J=6,5 Гц), 4,06 (2H, т, J=6,3 Гц), 6,93 (2H, д, J=8,8 Гц), 6,94 (2H, д, J=8,8 Гц), 7,57 (2H, д, J=8,8 Гц), 7,57 (2H, д, J=8,8 Гц).
Ссылочный Пример 5
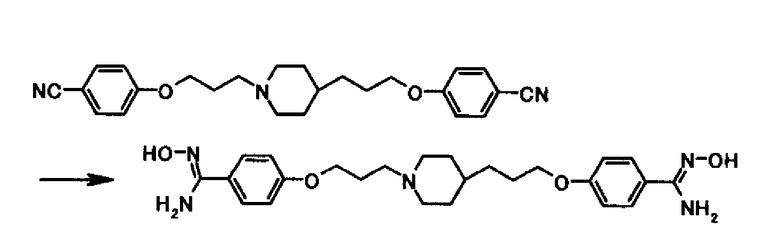
В суспензию 12,6 г 4-(3-{4-[3-(4-цианофенокси)пропил]-1-пиперидинил}пропокси)бензонитрила в 126 мл диметилсульфоксида 19,1 мл 50% водного раствора гидроксиламина добавляли и суспензию перемешивали при 50°С в течение 19 часов. К смеси после охлаждения до комнатной температуры 260 мл воды добавляли по каплям на протяжении 50 минут, которую затем перемешивали при комнатной температуре и после охлаждения водой в течение 2 часов. Твердое вещество собирали фильтрацией для получения 15,0 г 4-{3-[4-(3-{4-[амино(гидроксиламино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-гидроксибензамидина в виде белого твердого вещества.
1Н-ЯМР (DMSO-d6) δ: 1,05-1,40 (5H, м), 1,60-1,80 (4Н, м), 1,80-1,90 (4Н, м), 2,35-2,45 (2Н, м), 2,80-2,90 (2Н, м), 3,96 (2H, т, J=6,5 Гц), 4,01(2H, т, J=6,5 Гц), 5,65-5,75 (4Н, м) 6,85-6,95 (4Н, м), 7,55-7,65 (4Н, м), 9,43 (1H, c), 9,43 (1H, c).
Ссылочный Пример 6

В суспензию 1,07 г 4-{3-[4-(3-{4-[амино(гидроксиламино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-гидроксибензамидина в 10 мл уксусной кислоты 0,64 мл уксусного ангидрида добавляли при комнатной температуре и суспензию перемешивали при комнатной температуре в течение 40 минут. Смесь после добавления 0,10 г 5% палладия на углероде перемешивали в атмосфере водорода в течение 2 часов 15 минут. Смесь фильтровали для удаления нерастворимых частиц и после добавления 4 мл 6,0 моль/л соляной кислоты смесь отфильтровывали снова для удаления нерастворимых частиц и растворитель удаляли выпариванием под вакуумом. К полученному осадку 5,0 моль/л водного раствора гидроксида натрия добавляли для установления рН 12,5, затем твердое вещество собирали фильтрацией для получения 0,61 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина в виде белого твердого вещества.
1Н-ЯМР (DMSO-d6) δ: 1,00-1,40 (5H, м), 1,60-1,80 (4Н, м), 1,80-1,95 (4Н, м), 2,35-2,45 (2Н, м), 2,80-2,90 (2Н, м), 3,98 (2H, т, J=6,5 Гц), 4,03 (2H, т, J=6,3 Гц), 6,30-7,20(4Н, ушир.), 6,85-7,00 (4Н, м), 7,65-7,80 (4H, м).
Ссылочный Пример 7
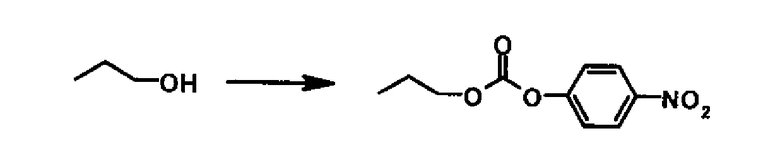
В раствор 0,75 г пропанола и 1,90 мл триэтиламина в 10 мл тетрагидрофурана раствор 2,50 г 4-нитрофенил хлорформиата в 15 мл тетрагидрофурана добавляли по каплям при охлаждении на льду. После перемешивания при комнатной температуре в течение 20 мин этилацетат и воду добавляли в реакционную смесь. Органический слой отделяли, промывали водой и насыщенным водным раствором хлорида натрия последовательно, высушивали над безводным сульфатом магния с последующим удалением растворителя выпариванием под вакуумом. К осадку добавляли гексан и нерастворимые частицы удаляли фильтрацией. После удаления растворителя выпариванием под вакуумом 2,59 г 4-нитрофенил пропил карбоната получали в виде светло-желтого маслянистого вещества.
1Н-ЯМР (CDCl3) δ: 1,03 (3H, т, J=7,4 Гц) 1,71-1,85 (2Н, м), 4,26 ((2Н, т, J=6,7 Гц), 7,39 (2H, д, J=9,0 Гц), 8,28 (2H, д, J=9,0 Гц).
Ссылочный Пример 8
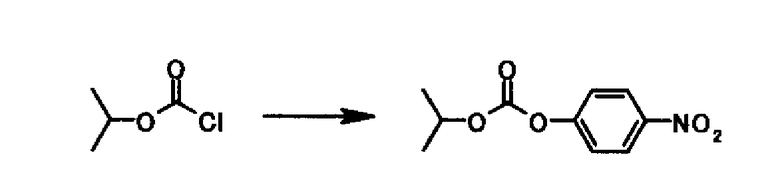
В раствор 3,00 г 4-нитрофенола и 3,31 мл триэтиламина в 30 мл тетрагидрофурана 2,46 мл изопропил хлорформиата добавляли по каплям при охлаждении на льду. В реакционную смесь после перемешивания при той же температуре в течение 10 минут этилацетат и воду добавляли. Органический слой отделяли, промывали насыщенным водным раствором хлорида натрия, высушивали над безводным сульфатом магния с последующим удалением растворителя выпариванием под вакуумом. Осадок растворяли в 50 мл этилацетата, промывали 5% водным раствором карбоната калия и насыщенным водным раствором хлорида натрия последовательно и высушивали над безводным сульфатом магния. После удаления растворителя выпариванием под вакуумом 3,00 г 4-нитрофенил изопропил карбоната получали в виде светло-желтого твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,41 (6H, д, J=6,3 Гц) 4,96-5,07 (1Н, м), 7,36-7,41 (2H, м), 8,25-8,30 (2H, м).
Ссылочный Пример 9

В раствор 3,00 г 4-нитрофенола и 3,31 мл триэтиламина в 30 мл тетрагидрофурана 2,75 мл бутил хлорформиата добавляли по каплям при охлаждении на льду. В реакционную смесь после перемешивания при той же температуре в течение 10 минут этилацетат и воду добавляли. Органический слой отделяли, промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом магния. После удаления растворителя выпариванием под вакуумом 4,60 г бутил 4-нитрофенил карбоната получали в виде светло-желтого маслянистого вещества.
1Н-ЯМР (CDCl3) δ: 0,99 (3H, т, J=7,4 Гц), 1,41-1,52 (2Н, м), 1,70-1,80 (2Н, м), 4,30 (3H, т, J=6,6 Гц), 7,36-7,41 (2H, м), 8,26-8,31 (2H, м).
Ссылочный Пример 10
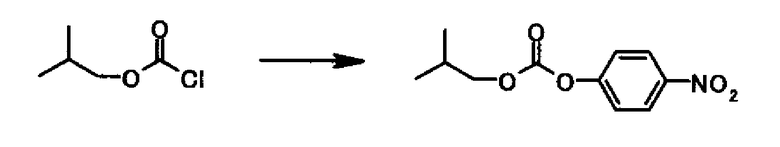
Подобно Ссылочному Примеру 9 из 3,00 г 4-нитрофенола и 2,80 мл изобутил хлорформиата 5,63 г изобутил 4-нитрофенил карбоната получали в виде светло-желтого маслянистого вещества.
1Н-ЯМР (CDCl3) δ: 1,02 (6H, д, J=6,6 Гц), 2,02-2,13 (1Н, м), 4,08 (2H, д, J=6,6 Гц), 7,39 (3H, д, J=9,1 Гц), 8,28 (2H, д, J=9,1 Гц).
Пример 1

В раствор 1,71 г 4-нитрофенил пропил карбоната в 15 мл N,N-диметилформамида 1,50 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина добавляли при комнатной температуре и раствор перемешивали при той же температуре в течение 4 часов. Хлороформ и воду добавляли в реакционную смесь. Органический слой отделяли, промывали водой, дважды 5% водным раствором карбоната калия и насыщенным водным раствором хлорида натрия последовательно и высушивали над безводным сульфатом магния с последующим удалением растворителя выпариванием под вакуумом. Полученный осадок очищали через силикагель колоночной хроматографии (элюент: хлороформ:метанол=4:1). Полученное твердое вещество растворяли в хлороформе, промывали 5% водным раствором карбоната калия и насыщенным водным раствором хлорида натрия последовательно и высушивали над безводным сульфатом магния. После удаления растворителя выпариванием под вакуумом 1,25 г 4-{3-[4-(3-{4-[амино(пропоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(пропоксикарбонил)бензамидина получали в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 0,99 (6H, т, J=7,4 Гц), 1,22-1,45 (5Н, м), 1,66-1,86 (8Н, м), 1,90-2,04 (4Н, м), 2,46-2,54 (2Н, м), 2,90-2,98 (2Н, м), 3,99 (2H, т, J=6,5 Гц), 4,06 (2H, т, J=6,3 Гц), 4,11 (4H, т, J=7,0 Гц), 6,88-6,96 (4Н, м), 7,82-7,88 (4H, м).
Пример 2
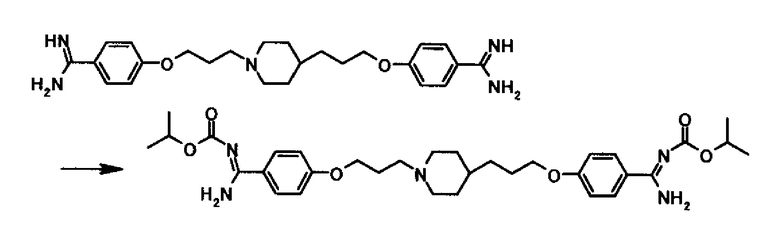
Подобно Примеру 1 из 1,71 г 4-нитрофенил изопропил карбоната и 1,50 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина 1,35 г 4-{3-[4-(3-{4-[амино(изопропоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(изопропоксикарбонил)бензамидина получали в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,20-1,46 (5H, м), 1,34 (12Н, д, J=6,3 Гц), 1,56-1,86 (4Н, м), 1,88-2,04 (4Н, м), 2,46-2,54 (2Н, м), 2,90-2,98 (2Н, м), 3,99 (2H, т, J=6,5 Гц), 4,06 (2H, т, J=6,3 Гц), 4,94-5,04 (2H, м), 6,88-6,96 (4Н, м), 7,80-7,88 (4H, м).
Пример 3-1

Подобно Примеру 1 из 1,82 г бутил 4-нитрофенил карбоната и 1,50 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина получали 1,39 г 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(бутоксикарбонил)бензамидина в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 0,95 (6H, т, J=7,3 Гц), 1,20-1,50 (9Н, м), 1,60-2,05 (12Н, м), 2,45-2,54 (2Н, м), 2,90-3,00 (2Н, м), 3,99 (2H, т, J=6,6 Гц), 4,06 (2H, т, J=6,3 Гц), 4,16 (4H, т, J=6,8 Гц), 6,88-6,96 (4Н, м), 7,82-7,88 (4Н, м).
Пример 3-2
В раствор 1,82 г бутил 4-нитрофенил карбоната в 15 мл N,N-диметилформамида 1,50 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина добавляли при комнатной температуре и раствор перемешивали при той же температуре в течение 2 часов. Хлороформ и воду добавляли в реакционную смесь. Органический слой отделяли, промывали дважды 5% водным раствором карбоната калия и насыщенным водным раствором хлорида натрия последовательно и высушивали над безводным сульфатом магния с последующим удалением растворителя выпариванием под вакуумом. Полученный осадок очищали через силикагель колоночной хроматографии (элюент: хлороформ:метанол=4:1). Полученное твердое вещество растворяли в хлороформе, промывали дважды 5% водным раствором карбоната калия и насыщенным водным раствором хлорида натрия последовательно и высушивали над безводным сульфатом магния. После удаления растворителя выпариванием под вакуумом получали 1,39 г 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(бутоксикарбонил)бензамидина в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 0,95 (6H, т, J=7,3 Гц), 1,20-1,50 (9Н, м), 1,60-2,05 (12Н, м), 2,45-2,54 (2Н, м), 2,90-3,00 (2Н, м), 3,99 (2H, т, J=6,6 Гц), 4,06 (2H, т, J=6,3 Гц), 4,16 (4H, т, J=6,8 Гц), 6,88-6,96 (4Н, м), 7,82-7,88 (4Н, м).
Пример 4

В раствор 1,82 г изобутил 4-нитрофенил карбоната в 15 мл N,N-диметилформамида 1,50 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина добавляли при комнатной температуре и раствор оставляли реагировать при той же температуре в течение 17 часов. Хлороформ и воду добавляли в реакционную смесь. Органический слой отделяли, промывали водой, 5% водным раствором карбоната калия и насыщенным водным раствором хлорида натрия последовательно и высушивали над безводным сульфатом магния с последующим удалением растворителя выпариванием под вакуумом. Полученный осадок очищали через силикагель колоночной хроматографии (элюент: хлороформ:метанол= 4:1). Полученный осадок растворяли в хлороформе, промывали 5% водным раствором карбоната калия и насыщенным водным раствором хлорида натрия последовательно и высушивали над безводным сульфатом магния. После удаления растворителя выпариванием под вакуумом получали 1,43 г 4-{3-[4-(3-{4-[амино(изобутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(изобутоксикарбонил)бензамидина в виде белого твердого вещества.
1Н-ЯМР (CDCl3) δ: 0,99 (12H, д, J=6,8 Гц), 1,20-1,45 (5Н, м), 1,55-2,12 (10Н, м), 2,46-2,53 (2Н, м), 2,90-3,00 (2Н, м), 3,94 (4H, д, J=6,8 Гц), 3,99 (2H, т, J=6,5 Гц), 4,06 (2H, т, J=6,3 Гц), 6,88-6,96 (4Н, м), 7,80-7,90 (4Н, м).
Пример на Состав 1
100 мг соединения, полученного в Примере 1 и 18 г хлорида натрия, добавляли к 1,8 л для инъекции. рН устанавливали до 4 соляной кислотой. После растворения соединения воду для инъекции добавляли для получения 2 л. Раствор отфильтровывали через мембранный фильтр 0,22 мкм и 100 мл полученного раствора заполняли и герметизировали в ампулы для получения инъекции.
Пример на Состав 2
Смесь 500 мг соединения, полученного в Примере 1, 350 мг лактозы, 250 мг кукурузного крахмала и 400 мг кристаллической целлюлозы (торговая марка: Сeolus PH101, Asahi Kasei Chemicals Corp.), 0,6 мл 5% водного раствора гидроксипропилцеллюлозы и воды добавляли и смесь перемешивали. Полученную смесь высушивали при 60°С и примешивали 100 мг кросповидона (торговая марка: Kollidon CL, BASF), 100 мг безводной осветленной кремниевой кислоты и 20 мг стеарата магния. Таблетку круглой формы и диаметром 8 мм получали прессованием 175 мг смеси.
Пример на Состав 3
Смесь 500 мг соединения, полученного в Примере 1, 200 мг лактозы, 530 мг кукурузного крахмала, 0,6 мл 5% водного раствора гидроксипропилцеллюлозы и воды добавляли и смесь перемешивали. Полученную смесь высушивали при 60°С и примешивали 70 мг кросповидона (торговая марка: Kollidon CL, BASF), 180 мг кристаллической целлюлозы (торговая марка: Сeolus PH302, Asahi Kasei Chemicals Corp.) и 20 мг стеарата магния. В желатиновую капсулу Тип 3 150 мг смеси заполняли для получения капсулированной формы.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Соединения по настоящему изобретению обладают высокой активностью в отношении грибков, включая грибки, устойчивые к азольному агенту, и, следовательно, являются пригодными в качестве противогрибковых агентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ АРИЛАМИДИНОВОЕ ПРОИЗВОДНОЕ, ЕГО СОЛЬ И ПРОТИВОГРИБКОВОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ТАКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2359959C2 |
| НОВОЕ ПРОИЗВОДНОЕ АРИЛАМИДИНА ИЛИ ЕГО СОЛЬ | 2003 |
|
RU2299195C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ПРОИЗВОДНОЕ ФЕНИЛАМИДИНА, И СПОСОБ ПРИМЕНЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ В КОМБИНАЦИИ С ПРОТИВОГРИБКОВЫМ СРЕДСТВОМ | 2007 |
|
RU2429843C2 |
| НОВЫЕ КРИСТАЛЛЫ 4-{3-[4-(3-{4-[АМИНО(БУТОКСИКАРБОНИЛ-ИМИНО)МЕТИЛ]ФЕНОКСИ}ПРОПИЛ)-1-ПИПЕРИДИНИЛ]ПРОПОКСИ}-N'-(БУТОКСИКАРБОНИЛ)БЕНЗАМИДИНА | 2008 |
|
RU2456272C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ И СПОСОБ ПРИМЕНЕНИЯ ПРОТИВОГРИБКОВОГО СРЕДСТВА В КОМБИНАЦИИ | 2006 |
|
RU2396955C2 |
| Соединение, обладающее агонистической активностью в отношении GPR119, способ его получения и фармацевтическая композиция, содержащая его в качестве эффективного компонента | 2015 |
|
RU2670197C1 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| ЗАМЕЩЕННОЕ ПИРИДИНОВОЕ СОЕДИНЕНИЕ | 2011 |
|
RU2572606C2 |
| НОВОЕ ЦИАНОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2010 |
|
RU2533115C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2366659C2 |
Изобретение относится к новым производным ариламидина, представленным общей формулой, или к их солям:

где R1 и R2 одинаково или различно представляют необязательно замещенную С3-4алкильную группу. Изобретение также относится к 4-{3-[4-(3-{4-[амино(пропоксикарбонилимино)метил] фенокси} пропил)-1-пиперидинил]пропокси}-N'-(пропоксикарбонил)бензамидину, к 4-{3-[4-(3-{4-[амино(изопропоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(изопропоксикарбонил)бензамидину, к 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(бутоксикарбонил)бензамидину, а также к противогрибковому агенту. Технический результат - получение новых биологически активных соединений, которые обладают противогрибковой активностью. 5 н.п. и 1 з.п. ф-лы, 1 табл.
1. Производное ариламидина, представленное общей формулой, или его соль:
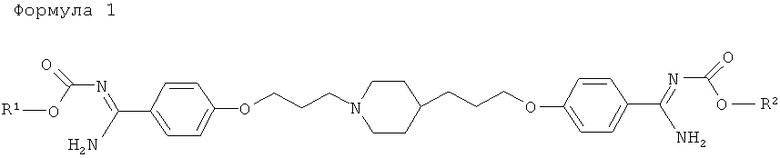
где R1 и R2 одинаково или различно представляют необязательно замещенную С3-4 алкильную группу.
2. Производное ариламидина или его соль по п.1, в котором R1 и R2 представляют одинаково С3-4 алкильную группу.
3. 4-{3-[4-(3-{4-[амино(пропоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(пропоксикарбонил)бензамидин или его соль.
4. 4-{3-[4-(3-{4-[амино(изопропоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(изопропоксикарбонил)бензамидин или его соль.
5. 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(бутоксикарбонил)бензамидин или его соль.
6. Противогрибковый агент, содержащий производное ариламидина или его соль по любому одному из пп.1-5.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| RU 98106118 A, 27.01.2000. | |||

