Область техники
Настоящее изобретение относится к фармацевтической комбинации, которая применима при лечении грибковых инфекций, вызванных грибковыми патогенами, и которая содержит ариламидиновое производное или его соль и одно или несколько средств, выбранных из числа азольных противогрибковых средств, полиеновых противогрибковых средств, кандиновых противогрибковых средств, фторпиримидиновых противогрибковых средств и иммунодепрессантов. Настоящее изобретение также относится к способу применения комбинации средств для лечения грибковых инфекций.
Уровень техники
Тяжелая глубокая грибковая инфекция, такая как инвазивный кандидоз, часто может являться смертельным заболеванием. В прошлом считалось, что основным защитным механизмом со стороны организма-хозяина против грибов, таких как Candida, является неспецифическая иммунизация нейтрофилами. Когда такой защитный механизм функционирует нормально, имеется небольшой риск заражения грибами. Однако в последние годы опасность заболевания глубокой грибковой инфекцией повышается из-за возросшего числа пациентов с основными заболеваниями, снижающими иммунологическую функцию организма, таких как злокачественные опухоли (в частности, злокачественные опухоли органов кроветворения, такие как острый лейкоз или злокачественная лимфома) и СПИД, частого применения противораковых средств или иммунодепрессантов, интенсивного применения антибактериальных антибиотиков или стероидных гормонов, длительного применения избыточного питания через центральную вену или венозной катетеризации и т.п. (непатентный документ 1).
Средств, применяемых для лечения такой глубокой грибковой инфекции, очень немного по сравнению с используемыми антибактериальными средствами, и они включают только амфотерицин В, флуцитозин, миконазол, флуконазол, итраконазол, вориконазол, микафунгин и т.п.
Соответственно, существует возрастающая потребность в безопасных и эффективных средствах против условно-патогенных грибковых инфекций, вызванных грибковыми патогенами, такими как Candida, Cryptococcus и Aspergillus.
Хотя средства, которые применяют в настоящее время, например амфотерицин В, обладают исключительно сильным фунгицидным действием, они имеют проблему побочного действия, такого как нефротоксичность, так что их клиническое применение ограничено. В настоящее время редко используют один флуцитозин, поскольку это средство имеет проблемы, например развитие резистентности. Микафунгин имеет низкую активность против Cryptococcus. Азолы, такие как флуконазол и вориконазол, в настоящее время используют наиболее часто из-за их баланса между эффективностью и безопасностью, хотя их фунгицидная активность ниже активности амфотерицина В (непатентные документы 2 и 3).
Malassezia, который является патогенным грибом поверхностных грибковых инфекций, представляет собой грибковый патоген, который, как полагают, является причиной или обостряющим фактором кожных болезней, таких как tinea versicolor, себорейная экзема и атопический дерматит. Поэтому использование противогрибковых средств для лечения таких заболеваний могло бы быть эффективным. Однако противогрибковые средства с превосходной противогрибковой активностью против Malassezia ограничиваются всего несколькими противогрибковыми средствами, такими как кетоконазол и итраконазол. Кроме того, имеются сообщения о возобновлении вспышек заболеваний сразу же после окончания лечения, что означает, что нельзя гарантировать удовлетворительное лечебное действие (непатентный документ 4).
Способы применения комбинаций противогрибковых средств используются в таких целях, как усиление лечебного действия (непатентный документ 5). Также развиваются исследования в направлении комбинаций противогрибковых средств (патентные документы 1, 2 и 3). Однако число сочетаемых средств ограничено, что означает, что нельзя гарантировать удовлетворительное лечебное действие.
С другой стороны, известны ариламидиновые производные, обладающие противогрибковой активностью (патентный документ 4).
Патентный документ 1: патент Японии № 3288051.
Патентный документ 2: JP-A-11-504931.
Патентный документ 3: JP-A-2003-527314.
Патентный документ 4: публикация Международного патента № WO03/074476.
Непатентный документ 1: Rinsho to Biseibutsu (Clinics and Microorganisms), Vol.17, pp.265-266, 1990.
Непатентный документ 2: Rinsho to Biseibutsu (Clinics and Microorganisms), Vol.21, pp.277-283, 1994.
Непатентный документ 3: Rinsho to Biseibutsu (Clinics and Microorganisms), Vol.30, pp.595-614, 2003.
Непатентный документ 4: Nippon Ishinkin Gakkai Zasshi (Japanese Journal of Medical Mycology), Vol.46, pp.163-167, 2005.
Непатентный документ 5: Shinzaisei Shinkinsho no Shindan & Chiryo Gaidorain (Guidelines for the Diagnosis and Treatment of Deep Mycosis), p.20, p.29, 2003 (Ishiyaku Pub. Inc.).
Раскрытие изобретения
Требуется фармацевтическая комбинация, которая применима при лечении грибковых инфекций и которая обладает сильной противогрибковой активностью с незначительным побочным действием, и способ применения комбинации противогрибковых средств.
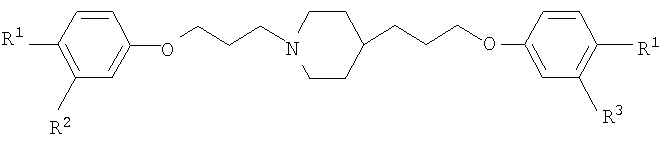
При таком условии в результате интенсивного исследования авторы настоящего изобретения обнаружили, что фармацевтическая комбинация, содержащая ариламидиновое производное, представленное приведенной далее общей формулой [1]
[формула 1]

где R1 представляет амидиногруппу, которая может быть замещена гидроксильной группой, которая может быть защищена ацильной группой, амидиногруппу, которая может быть замещена алкоксигруппой, которая может быть замещенной, или амидиногруппу, которая может быть замещена аралкилоксигруппой, которая может быть замещенной; и R2 и R3 могут быть одинаковыми или различными и представляют атом водорода или атом галогена; или его соль и одно или несколько противогрибковых средств, выбранных из числа азольных противогрибковых средств, полиеновых противогрибковых средств, кандиновых противогрибковых средств и фторпиримидиновых противогрибковых средств, обладает сильной противогрибковой активностью и применима при лечении грибковых инфекций, и что способ применения комбинации таких противогрибковых средств применим при лечении грибковых инфекций, посредством чего пришли к настоящему изобретению.
Кроме того, авторы настоящего изобретения обнаружили, что фармацевтическая комбинация, содержащая ариламидиновое производное, представленное общей формулой [1], или его соль, и иммунодепрессанты обладают сильной противогрибковой активностью и применима при лечении грибковых инфекций и кожных болезней, таких как атопический дерматит, и что способ применения комбинации таких средств применим при лечении грибковых инфекций и кожных болезней, таких как атопический дерматит, посредством чего пришли к настоящему изобретению.
Фармацевтическая комбинация, содержащая ариламидиновое производное или его соль, обладающие противогрибковой активностью, и одно или несколько противогрибковых средств, выбранных из числа азольных противогрибковых средств, полиеновых противогрибковых средств, кандиновых противогрибковых средств и фторпиримидиновых противогрибковых средств, обладает сильной противогрибковой активностью и применима при лечении грибковых инфекций. Способ применения комбинации таких средств применим как превосходный способ лечения грибковых инфекций.
Кроме того, фармацевтическая комбинация, содержащая ариламидиновое производное или его соль и иммунодепрессанты, обладает сильной противогрибковой активностью и применима при лечении грибковых инфекций и кожных болезней, таких как атопический дерматит. Способ применения комбинации таких средств применим при лечении грибковых инфекций и кожных болезней, таких как атопический дерматит.
Наилучший способ осуществления изобретения
Теперь настоящее изобретение будет описано подробнее.
В настоящем описании, если не указано иное, термин "атом галогена" относится к атому фтора, атому хлора, атому брома или атому иода; термин "алкильная группа", например, относится к линейной или разветвленной C1-12-алкильной группе, такой как метил, этил, пропил, изопропил, бутил, втор-бутил, изобутил, трет-бутил, пентил, изопентил, гексил, гептил и октил; термин "низшая алкильная группа", например, относится к линейной или разветвленной C1-6-алкильной группе, такой как метил, этил, пропил, изопропил, бутил, втор-бутил, изобутил, трет-бутил, пентил и изопентил; термин "алкенильная группа", например, относится к линейной или разветвленной С2-12-алкенильной группе, такой как винил, аллил, пропенил, изопропенил, бутенил, изобутенил, пентенил, гексенил, гептенил и октенил; термин "арильная группа", например, относится к такой группе, как фенил и нафтил; термин "аралкильная группа", например, относится к ар-С1-6-алкильной группе, такой как бензил, дифенилметил, тритил, фенетил и нафтилметил;
термин "алкоксигруппа", например, относится к линейной или разветвленной С1-6-алкилоксигруппе, такой как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентилокси и изопентилокси; термин "аралкилоксигруппа", например, относится к ар-С1-6-алкилоксигруппе, такой как бензилокси, дифенилметилокси, тритилокси, фенетилокси и нафтилметилокси; термин "алкоксиалкильная группа", например, относится к С1-6-алкилокси-С1-6-алкильной группе, такой как метоксиметил и 1-этоксиэтил; термин "аралкилоксиалкильная группа", например, относится к ар-С1-6-алкилокси-С1-6-алкильной группе, такой как бензилоксиметил и фенетилоксиметил;
термин "ацильная группа", например, относится к формильной группе, линейной или разветвленной С2-12-алканоильной группе, такой как ацетил, пропионил и изовалерил, и ар-С1-6-алкилкарбонильной группе, такой как бензилкарбонил, ароильной группе, такой как бензоил и нафтоил, гетероциклической карбонильной группе, такой как никотиноил, теноил, пирролидинокарбонил и фуроил, сукцинильной группе, глутарильной группе, малеоильной группе, фталоильной группе или линейной или разветвленной α-аминоалканоильной группе, у которой N-конец может быть защищен, образованной от аминокислоты (включая, например, глицин, аланин, валин, лейцин, изолейцин, серин, треонин, цистеин, метионин, аспарагиновую кислоту, глутаминовую кислоту, аспарагин, глутамин, аргинин, лизин, гистидин, гидроксилизин, фенилаланин, тирозин, триптофан, пролин или гидроксипролин);
термин "алкилоксикарбонильная группа", например, относится к линейной или разветвленной С1-12-алкилоксикарбонильной группе, такой как метоксикарбонил, этоксикарбонил, 1,1-диметилпропоксикарбонил, изопропоксикарбонил, 2-этилгексилоксикарбонил, трет-бутоксикарбонил и трет-пентилоксикарбонил; термин "аралкилоксикарбонильная группа", например, относится к ар-С1-6-алкилоксикарбонильной группе, такой как бензилоксикарбонильная и фенетилоксикарбонильная группа; термин "арилоксикарбонильная группа", например, относится к такой группе, как фенилоксикарбонил; термин "гетероциклическая оксикарбонильная группа", например, относится к такой группе, как 2-фурфурилоксикарбонил и 8-хинолилоксикарбонил;
термин "арилтиогруппа", например, относится к такой группе, как фенилтио; термин "алкансульфонильная группа", например, относится к С1-6-алкансульфонильной группе, такой как метансульфонил, этансульфонил и пропансульфонил; термин "арилсульфонильная группа", например, относится к такой группе, как бензолсульфонил, толуолсульфонил и нафталинсульфонил; термин "аралкилиденовая группа", например, относится к такой группе, как бензилиден и нафтилметилен; термин "циклоалкилиденовая группа", например, относится к такой группе, как циклопентилиден и циклогексилиден; термин "диалкиламиноалкилиденовая группа", например, относится к такой группе, как N,N-диметиламинометилен и N,N-диэтиламинометилен; термин "азотсодержащая гетероциклилалкилиденовая группа", например, относится к такой группе, как 3-гидрокси-4-пиридилметилен;
термин "кислородсодержащая гетероциклилалкильная группа", например, относится к такой группе, как 5-метил-2-оксо-2Н-1,3-диоксол-4-илметил; термин "кислородсодержащая гетероциклическая группа", например, относится к такой группе, как тетрагидрофурил и тетрагидропиранил; термин "серосодержащая гетероциклическая группа", например, относится к такой группе, как тетрагидротиопиранил; термин "диарилфосфорильная группа", например, относится к такой группе, как дифенилфосфорил; термин "диаралкилфосфорильная группа", например, относится к такой группе, как дибензилфосфорил; и термин "замещенная силильная группа", например, относится к такой группе, как триметилсилил, триэтилсилил и трибутилсилил.
Каждая из вышеописанных групп также может быть замещена одной или несколькими группами, выбранными из числа атома галогена, аминогруппы, которая может быть защищенной, гидроксильной группы, которая может быть защищенной, нитрогруппы, низшей алкильной группы, алкенильной группы, алкоксигруппы, аралкилоксигруппы, арильной группы, ацильной группы и оксогруппы.
Группы, защищающие аминогруппу, охватывают все обычные группы, которые можно использовать как защитные группы для аминогруппы, и включают, например, ацильную группу, алкилоксикарбонильную группу, аралкилоксикарбонильную группу, арилоксикарбонильную группу, аралкильную группу, алкоксиалкильную группу, аралкилоксиалкильную группу, арилтиогруппу, алкансульфонильную группу, арилсульфонильную группу, аралкилиденовую группу, циклоалкилиденовую группу, диалкиламиноалкилиденовую группу, азотсодержащую гетероциклилалкилиденовую группу, кислородсодержащую гетероциклилалкильную группу, диарилфосфорильную группу, диаралкилфосфорильную группу и замещенную силильную группу.
Группы, защищающие гидроксильную группу, охватывают все обычные группы, которые можно использовать как защитные группы для гидроксильной группы, и включают, например, ацильную группу, алкилоксикарбонильную группу, аралкилоксикарбонильную группу, гетероциклилоксикарбонильную группу, алкильную группу, алкенильную группу, аралкильную группу, кислородсодержащую гетероциклическую группу, серосодержащую гетероциклическую группу, алкоксиалкильную группу, аралкилоксиалкильную группу, алкансульфонильную группу, арилсульфонильную группу и замещенную силильную группу.
Примеры соединений, представленных общей формулой [1], используемых в настоящем изобретении, включают соединения, указанные далее.
[Формула 2]
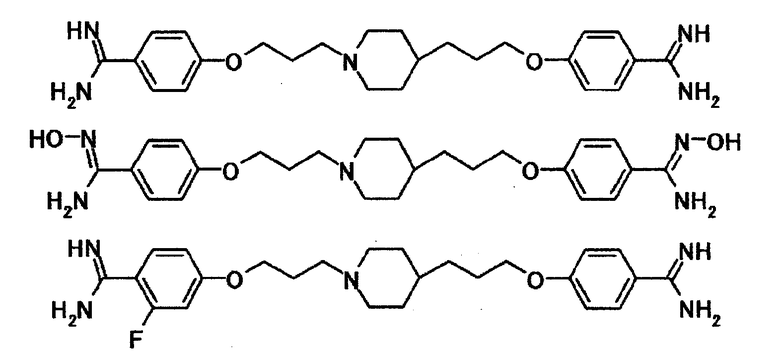
Для соединения настоящего изобретения предпочтительные примеры соединений, представленных общей формулой [1], включают соединения, указанные далее.
В качестве соединения, представленного общей формулой [1], предпочтительными являются соединения, в которых R1 представляет собой амидиногруппу, которая может быть замещена гидроксильной группой, и более предпочтительными являются соединения, в которых R1 представляет собой амидиногруппу.
Предпочтительными являются соединения, в которых R2 и R3 представляют собой атомы водорода.
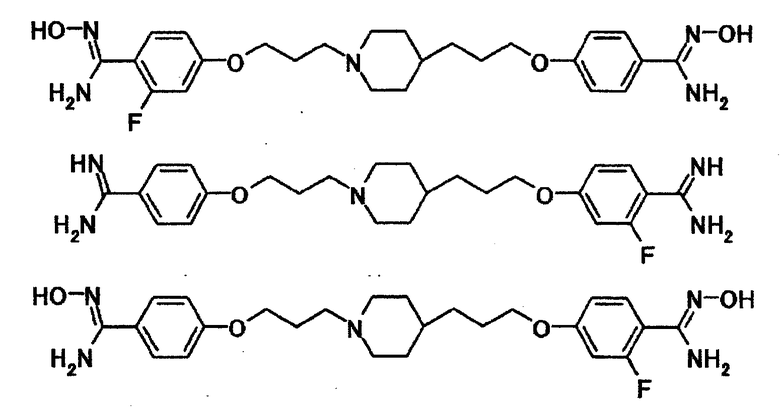
Конкретно в качестве соединения, представленного общей формулой [1], даже более предпочтительными являются соединения, указанные далее.
[Формула 3]

В качестве соединения, представленного общей формулой [1], даже более предпочтительными являются соединения, указанные далее.
[Формула 4]
В случае, когда существуют сольваты или гидраты соединения, представленного общей формулой [1], или его соли, можно использовать такие сольваты или гидраты. Кроме того, также можно использовать кристаллы, имеющие различные формы.
Примеры солей соединения, представленного общей формулой [1], включают соли неорганических кислот, таких как хлороводородная кислота, бромоводородная кислота и серная кислота; соли органических карбоновых кислот, таких как винная кислота, муравьиная кислота, уксусная кислота, лимонная кислота, трихлоруксусная кислота и трифторуксусная кислота; соли сульфоновых кислот, таких как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, мезитиленсульфоновая кислота и нафталинсульфоновая кислота; и соли фосфорной кислоты.
Предпочтительные соли соединения, представленного общей формулой [1], включают фармакологически приемлемые соли и более предпочтительными являются гидрохлориды.
Хотя соединение общей формулы [1], используемое в настоящем изобретении, можно получить обычными способами, его также можно получить способами, описанными, например, в публикации Международного патента № WO2006/003881.
Примеры азольных противогрибковых средств, используемых в настоящем изобретении, включают противогрибковые средства на основе триазола, такие как флуконазол, фосфлуконазол, итраконазол, вориконазол, позаконазол, равуконазол, BMS-379224, BAL-8557 и CS-758, а также противогрибковые средства на основе имидазолов, такие как кетоконазол, миконазол, бифоназол, ланоконазол и луликоназол.
Предпочтительные примеры азольных противогрибковых средств включают противогрибковые средства на основе триазола, такие как флуконазол, фосфлуконазол, итраконазол, вориконазол, позаконазол, равуконазол, BMS-379224, BAL-8557 и CS-758. Более предпочтительными являются флуконазол, фосфлуконазол, вориконазол и итраконазол и даже более предпочтительными являются флуконазол, вориконазол и итраконазол.
Примеры полиеновых противогрибковых средств, используемых в настоящем изобретении, включают амфотерицин В и его липосомные композиции (например, Abelcet (торговое наименование) или AmBisome (торговое наименование)), нистатин, трихомицин, SPK-843 и пимарицин.
Предпочтительные примеры полиеновых противогрибковых средств включают амфотерицин В и его липосомные композиции (например, Abelcet (торговое наименование) или AmBisome (торговое наименование)).
Примеры кандиновых противогрибковых средств, используемых в настоящем изобретении, включают микафунгин, каспофунгин, анидулафунгин и аминокандин.
Предпочтительным примером кандиновых противогрибковых средств, используемых в настоящем изобретении, является микафунгин.
Примером фторпиримидиновых противогрибковых средств, используемых в настоящем изобретении, является флуцитозин.
Примеры иммунодепрессантов, используемых в настоящем изобретении, включают макролидные соединения, такие как рапамицин, циклоспорин и такролим (tacrolimus).
Предпочтительным примером иммунодепрессантов является такролим.
Способ введения ариламидинового производного, представленного общей формулой [1], или его соли особо не ограничивается, и ариламидиновое производное или его соль можно вводить внутривенно, перорально, внутримышечно, подкожно или каким-либо другим способом введения. Кроме того, ариламидиновое производное, представленное общей формулой [1], или его соль можно вводить одновременно, раздельно или в особом порядке с азольными противогрибковыми средствами, полиеновыми противогрибковыми средствами, кандиновыми противогрибковыми средствами, фторпиримидиновыми противогрибковыми средствами и иммунодепрессантами.
Фармацевтические композиции по настоящему изобретению обнаруживают превосходное действие против грибов, таких как Candida, Cryptococcus, Aspergillus и Malassezia. Фармацевтическая композиция по настоящему изобретению обнаруживает особенно превосходное действие против Candida, таких как Candida albicans, Candida glabrata, Candida guilliermondii, Candida kefyr, Candida krusei, Candida parapsilosis, Candida stellatoidea, Candida tropicalis и Candida lusitaniae; Cryptococcus, таких как Cryptococcus neoformans; Aspergillus, таких как Aspergillus clavatus, Aspergillus flavus, Aspergillus fumigatus, Aspergillus nidulans, Aspergillus niger, Aspergillus terreus, Aspergillus versicolor и Aspergillus restrictus; и Malassezia, таких как Malassezia furfur, Malassezia pachydermatis, Malassezia sympodialis и Malassezia slooffiae.
Фармацевтическая композиция по настоящему изобретению эффективна при предупреждении и лечении различных грибковых инфекций, таких как кандидоз, криптококкоз, аспергиллез и малассезиаз.
С помощью фармацевтической композиции по настоящему изобретению можно лечить тяжелые грибковые инфекции. Кроме того, так как сильное противогрибковое действие проявляется даже если снижено количество каждого из средств, которые вводят, можно уменьшить побочное действие соответствующих средств.
Кроме того, фармацевтическая композиция, содержащая иммунодепрессанты, обнаруживает превосходное действие против поверхностных грибов, таких как Malassezia, которые, как полагают, являются причиной или обостряющим фактором при кожных нарушениях, таких как атопический дерматит. Кроме того, иммунодепрессанты, которые являются компонентом фармацевтической композиции, проявляют превосходное действие против кожных нарушений, таких как атопический дерматит. Поэтому фармацевтическая композиция, содержащая иммунодепрессанты, применима в качестве фармацевтической композиции для лечения грибковых инфекций и в случае лечения кожных нарушений против атопического дерматита или подобных заболеваний.
Примеры
Теперь настоящее изобретение будет описываться подробнее со ссылками на приведенные далее примеры испытаний. Однако настоящее изобретение не ограничивается этими примерами.
Соответствующие аббревиатуры имеют следующие значения:
FLCZ - флуконазол; MCFG - микафунгин; АМРН-В - амфотерицин В; ITCZ - итраконазол; 5-FC - флуцитозин; VLCZ - вориконазол; KCZ - кетоконазол; и ТАС - такролим.
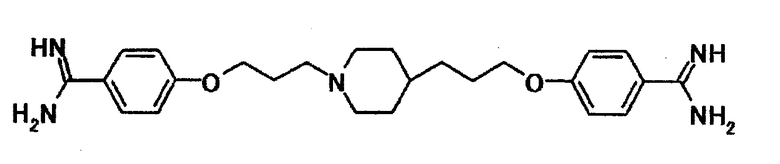
В качестве испытываемого соединения выбирают соединение, указанное далее. Химическая структурная формула указанного соединения приводится ниже.
[Формула 5]
В качестве средств выбирают флуконазол (коммерчески доступный продукт), микафунгин (коммерчески доступный продукт), амфотерицин В (коммерчески доступный продукт), итраконазол (коммерчески доступный продукт), вориконазол (коммерчески доступный продукт), кетоконазол (коммерчески доступный продукт), такролим (коммерчески доступный продукт) и флуцитозин (Sigma).
Пример испытаний 1. Испытания in vitro (Aspergillus и Cryptococcus)
Испытание грибов на чувствительность осуществляют с использованием метода разведения в питательной среде.
Среда, используемая в испытании на чувствительность, состоит из RPMI1640 (Sigma) (RPMI/MOPS), доведенной до рН 7,0 0,165 моль/л раствором морфолинпропансульфоновой кислоты (MOPS) и 1,0 моль/л раствором гидроксида натрия.
Испытываемое соединение растворяют в небольшом количестве 0,1 моль/л соляной кислоты. Раствор разбавляют стерильной водой и затем получают раствор предварительно установленной концентрации с использованием RPMI/MOPS.
Средства растворяют в небольшом количестве стерильной воды или диметилсульфоксида, и затем полученные растворы доводят до предварительно установленной концентрации с использованием RPMI/MOPS.
Каждый инокулят получают разведением суспензии Cryptococcus neoformans, АТСС 90112, который выращивают в течение двух суток при 35°С на агаровой пластине Sabouraud, или разведением суспензии конидий Aspergillus fumigatus, TIMM0063, суспендированных в стерильном физиологическом растворе, до предварительно установленной концентрации с использованием RPMI/MOPS (конечная концентрация каждого раствора приблизительно 1 х 103 клеток/мл и приблизительно 1 х 104 КОЕ/мл). Наконец получают микропланшет, содержащий предварительно установленные концентрации испытываемого соединения и соответствующих средств, а также среду и грибы. Планшет культивируют при 35°С в течение 48-72 часов. Измеряют поглощение при 630 нм до и после культивирования с использованием автоматического спектрофотометра. Наименьшую концентрацию, при которой наблюдают 50% ингибирование роста по сравнению с ростом контроля, к которому не добавляют испытываемое вещество, определяют как IC50.
Индекс FIC вычисляют, сравнивая IC50 для испытываемого соединения с показателями для флуконазола, микафунгина, амфотерицина В, итраконазола и флуцитозина, когда их используют по отдельности и когда используют в сочетании, и оценивая противогрибковую активность соответствующей комбинации средств с испытываемым соединением методом разграфленной доски (checkerboard method). За индекс FIC принимают минимальное значение величин, определенных согласно соотношению (величина IC50 при использовании в комбинации с испытываемым соединением / величина IC50 при использовании отдельно с испытываемым соединением) + (величина IC50 при использовании в комбинации со средством / величина IC50 при использовании отдельно со средством).
В случаях, когда индекс FIC равен 0,5 или меньшей величине, определяют, что имеется сильный синергический эффект как результат сочетания двух средств (Antimicrobial Agents and Chemotherapy, Vol.39, p.1691, 1995).
Результаты комбинирования испытываемого соединения с соответствующими средствами против Aspergillus fumigatus, TIMM0063 приводятся в таблице 1.
Сильный синергический эффект подтверждается для испытываемого соединения с микафунгином, амфотерицином В и флуцитозином.
Результаты комбинирования испытываемого соединения с соответствующими средствами против Cryptococcus neoformans, АТСС 90112, приводятся в таблице 2.
Сильный синергический эффект подтверждается для испытываемого соединения с флуконазолом, итраконазолом, амфотерицином В и флуцитозином.
Пример испытаний 2. Испытание in vitro (Malassezia)
Испытание грибов на чувствительность осуществляют с использованием метода разведения в питательной среде.
Среда, используемая в испытании на чувствительность, состоит из смеси (m RPMI/MOPS) RPMI1640 (Sigma), доведенной до рН 7,0 0,165 моль/л раствором 3-(N-морфолино)пропансульфоновой кислоты (MOPS) и 1,0 моль/л раствором гидроксида натрия, глюкозы, окисл. желчи, глицерина и твина 20 (Wako Pure Chemical Industries, Ltd.).
Испытываемое соединение растворяют в небольшом количестве 0,1 моль/л соляной кислоты. Раствор разбавляют стерильной водой и затем получают в предварительно установленной концентрации с использованием m RPMI/MOPS. Итраконазол, кетоконазол и такролим растворяют в диметилсульфоксиде, и затем полученные растворы доводят до предварительно установленной концентрации с использованием m RPMI/MOPS.
Инокулят получают суспендированием Malassezia furfur, NBRC0656, который выращивают в течение двух суток при 30°С на агаровой среде 103 (среда, образованная путем получения смеси 1% глюкозы, 0,5% пептона, 0,3% дрожжевого экстракта и 0,3% солодового экстракта, доведением до рН 5,6 с использованием 1,0 моль/л соляной кислоты, загрузкой в смесь 1% оливкового масла и 1,5% агарового порошка, последующей стерилизацией полученной смеси паром высокого давления), до предварительно установленной концентрации с использованием m RPMI/MOPS (конечная концентрация раствора приблизительно 1 х 103 клеток/мл). Наконец получают микропланшет, содержащий предварительно установленные концентрации испытываемого соединения, соответствующих средств, среду и грибы. Планшет культивируют при 30°С в течение 70-72 часов.
Наименьшую концентрацию, при которой рост грибов после окончания культивирования не наблюдают, определяют как МIC.
Индекс FIC вычисляют, сравнивая МIC для испытываемого соединения с показателями для итраконазола, кетоконазола и такролима, когда их используют по отдельности и когда используют в сочетании, и оценивая противогрибковую активность соответствующей комбинации средств с испытываемым соединением методом разграфленной доски. За индекс FIC принимают минимальное значение величин, определенных согласно соотношению (величина МIC при использовании в комбинации с испытываемым соединением / величина МIC при использовании отдельно с испытываемым соединением) + (величина МIC при использовании в комбинации со средством / величина МIC при использовании отдельно со средством).
Результаты комбинирования испытываемого соединения с соответствующими средствами против Malassezia furfur, NBRC0656, приводятся в таблице 3.
Сильный синергический эффект подтверждается для испытываемого соединения с итраконазолом, кетоконазолом и такролимом.
Пример испытаний 3. Испытание in vivo (Candida)
Активность in vivo оценивают с использованием мышиной модели системного заражения с использованием Candida albicans.
Мышам (в возрасте четырех недель (при заражении), самцы мышей ICR, 10 мышей на группу) вводят интраперитонеально циклофосфамид за 4 дня до заражения (200 мг/кг) и на следующий день после заражения (100 мг/кг). Инокулят получают суспендированием Candida albicans, TIMM1623, на агаровой пластине Sabourand, который культивировали в течение одной ночи при 35°С, в стерильном физиологическом растворе, подсчитывая число клеток с использованием биологического микроскопа и последующим разбавлением стерильным физиологическим раствором. Мышей инокулируют внутривенно в их хвосты 0,2 мл инокулята для того, чтобы вызвать заражение (приблизительно 3 х 104 КОЕ/мышь).
Испытываемое соединение растворяют в небольшом количестве 0,1 моль/л соляной кислоты, и полученный раствор разбавляют стерильным физиологическим раствором.
Флуконазол и микафунгин подготавливают с использованием стерильного физиологического раствора.
Амфотерицин В растворяют в 5% глюкозе.
Испытываемое соединение (0,0313 мг/кг), флуконазол (0,25 мг/кг), амфотерицин В (0,1 мг/кг) и микафунгин (0,25 мг/кг) вводят подкожно через 2 часа после заражения и затем один раз в день в течение 6 дней (всего 7 раз). Осуществляют испытание в случае, когда каждое из средств вводят одно и в комбинации, вводя каждое из средств сразу же после того, как введено испытываемое соединение.
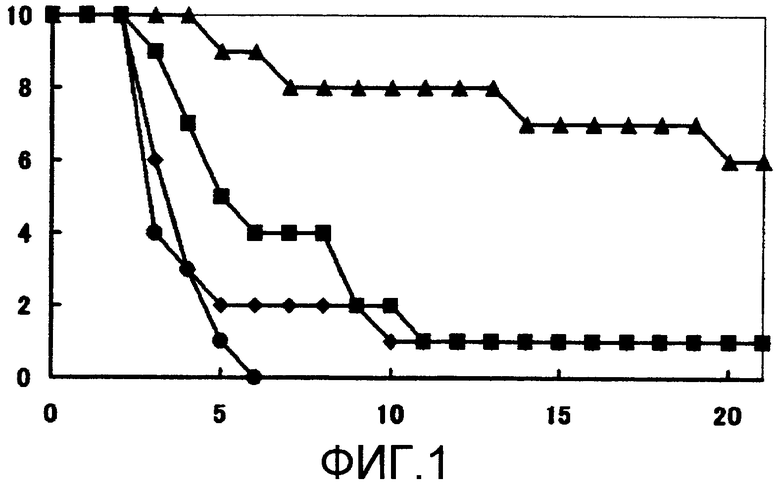
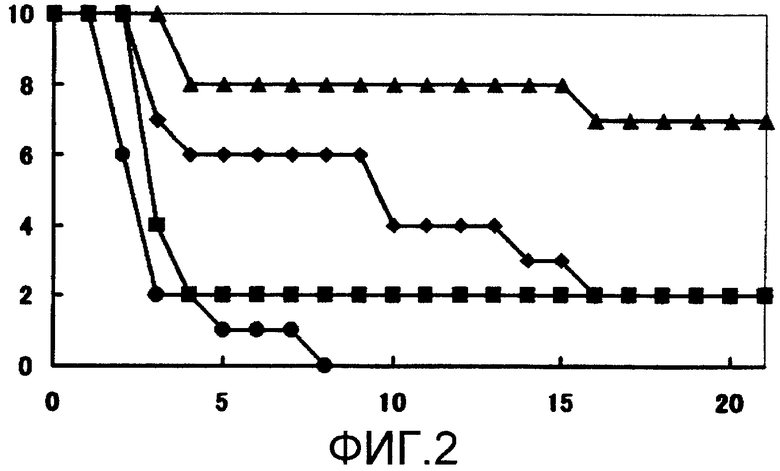
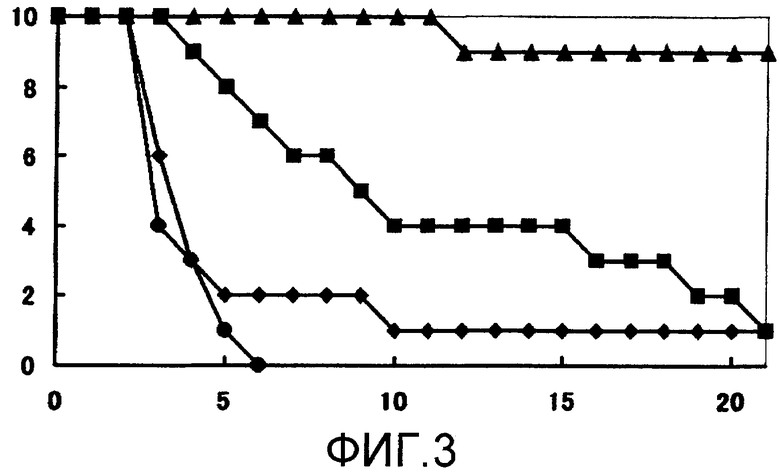
Устанавливают время выживания для групп, которым испытываемое соединение или каждое из средств вводили по одному, и время выживания для групп, которым испытываемое соединение вводили в комбинации с каждым из средств, для того, чтобы определить, имеется ли какое-либо значимое различие при 5% двустороннем уровне значимости относительно времени выживания группы, которой не вводили какие-либо средства, с использованием log рангового критерия Каплана-Мейера (множественное групповое сравнение). Результаты испытания показаны на фиг. 1-3.
Обнаружено, что ни одна из групп, которым вводили единственное средство, не имеет значимого (p > 0,05) эффекта продления жизни по сравнению с группой, которой не вводили какие-либо средства. С другой стороны, обнаружено, что в группах с комбинированным введением испытываемого соединения и флуконазола, испытываемого соединения и амфотерицина В и испытываемого соединения и микафунгина имеется значимый (p ≤ 0,0001) эффект продления жизни по сравнению с группой, которой не вводили какие-либо средства.
На мышиной модели системного заражения Candida albicans обнаружено, что комбинированное введение испытываемого соединения и флуконазола, испытываемого соединения и амфотерицина В и испытываемого соединения и микафунгина проявляет превосходное лечебное действие.
Пример испытаний 4. Испытание in vivo (Aspergillus)
Активность in vivo оценивают с использованием мышиной модели системного заражения с использованием Aspergillus fumigatus.
Как для инокулята, мышам (в возрасте четырех недель (при заражении), самцы мышей ICR, 10 мышей на группу) вводят интраперитонеально циклофосфамид за 4 дня до заражения (200 мг/кг) и на следующий день после заражения (100 мг/кг). Суспензию конидий Aspergillus fumigatus, TIMM0063, разводят в стерильном физиологическом растворе, содержащем 0,05% твина 80 (произведен Difco Laboratories) для того, чтобы получить инокулят. Мышей инокулируют внутривенно в их хвосты 0,2 мл инокулята для того, чтобы вызвать заражение (приблизительно 4 х 104 КОЕ/мышь).
Испытываемое соединение растворяют в небольшом количестве 0,1 моль/л соляной кислоты, и полученный раствор разбавляют стерильным физиологическим раствором.
Вориконазол растворяют в 50 мг/мл водном растворе сульфобутилового эфира β-циклодекстриннатрия (SIDIX).
Флуцитозин суспендируют в 0,5% растворе метилцеллюлозы.
Испытываемое соединение (0,1 мг/кг) и вориконазол (5 мг/кг) вводят подкожно, а флуцитозин (50 мг/кг) вводят перорально через 2 часа после заражения и затем один раз в день в течение 6 дней (всего 7 раз). Осуществляют испытание в случае, когда каждое из средств вводят одно и в комбинации, вводя каждое из средств сразу же после того, как введено испытываемое соединение. Эффективность в испытании на чувствительность оценивают по выживаемости через 15 дней после заражения.
Результаты испытания комбинации испытываемого соединения и вориконазола против Aspergillus fumigatus показаны в таблице 4. Результаты испытания комбинации испытываемого соединения и флуцитозина против Aspergillus fumigatus показаны в таблице 5.
На мышиной модели системного заражения Aspergillus fumigatus комбинированное введение испытываемого соединения и вориконазола и испытываемого соединения и флуцитозина показывает превосходное лечебное действие.
Пример испытаний 5. Испытание in vivo (Cryptococcus)
Активность in vivo оценивают с использованием мышиной модели системного заражения с использованием Cryptococcus neoformans.
Мышам (в возрасте четырех недель (при заражении), самцы мышей ICR, 10 мышей на группу) вводят интраперитонеально циклофосфамид за 4 дня до заражения (200 мг/кг) и на следующий день после заражения (100 мг/кг). Инокулят получают суспендированием в стерильном физиологическом растворе Cryptococcus neoformans, АТСС90112, на матрасе SDA, который культивировали в течение одной ночи при 35°С, подсчитывая число клеток с использованием биологического микроскопа, и последующим разведением стерильным физиологическим раствором. Мышей инокулируют внутривенно в их хвосты 0,2 мл инокулята для того, чтобы вызвать заражение (приблизительно 8 х 104 КОЕ/мышь).
Испытываемое соединение растворяют в небольшом количестве 0,1 моль/л соляной кислоты, и полученный раствор разбавляют стерильным физиологическим раствором.
Флуконазол подготавливают с использованием стерильного физиологического раствора.
Амфотерицин В растворяют в 5% водном растворе глюкозы.
Испытываемое соединение (0,125 мг/кг), флуконазол (5 мг/кг) и амфотерицин В (0,25 мг/кг) вводят подкожно через 2 часа после заражения и затем один раз в день в течение 6 дней (всего 7 раз). Осуществляют испытание в случае, когда каждое из средств вводят одно и в комбинации, вводя каждое из средств сразу же после того, как введено испытываемое соединение. Эффективность в испытании на чувствительность оценивают по выживаемости через 22 дня после заражения.
Результаты испытания комбинации испытываемого соединения с флуконазолом против Cryptococcus neoformans показаны в таблице 6. Результаты испытания комбинации испытываемого соединения с амфотерицином В против Cryptococcus neoformans показаны в таблице 7.
На мышиной модели системного заражения Cryptococcus neoformans комбинированное введение испытываемого соединения и флуконазола и комбинированное введение испытываемого соединения и амфотерицина В показывает превосходное лечебное действие.
Из вышеприведенных результатов ясно, что сочетание ариламидинового производного, представленного общей формулой [1], или его соли с различными противогрибковыми средствами или подобными средствами показывает синергические противогрибковую активность и лечебное действие и эффективно при лечении грибковых инфекций, вызванных грибковыми патогенами.
Далее ариламидиновое производное, представленное общей формулой [1], или его соль, используемые в настоящем изобретении, будут описываться с обращением к примерам получения и примерам композиций лекарственных средств.
Отношение концентраций компонентов смеси в элюенте определяется отношением способности растворяться. Если не указано иное, носителем для колоночной хроматографии на силикагеле является силикагель B.W. BW-127ZH, Fuji Silysia Chemical Ltd.
Соответствующие аббревиатуры имеют следующие значения:
Ас - ацетил; Вос - трет-бутоксикарбонил; Et - этил; ДМСО-d6 - дейтерированный диметилсульфоксид.
Пример получения 1
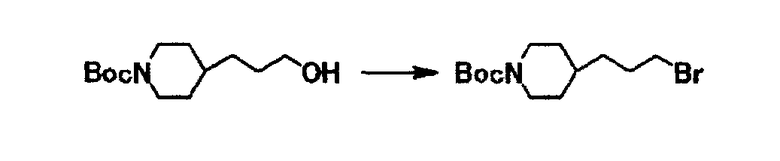
К раствору 10,7 г трет-бутил-4-(3-гидроксипропил)-1-пиперидинкарбоксилата в тетрагидрофуране (110 мл) при охлаждении водой добавляют 19,0 г тетрабромида углерода и затем к полученной смеси добавляют 15,0 г трифенилфосфина в течение 13 минут. Полученную смесь перемешивают при комнатной температуре в течение 2 часов 30 минут и оставляют на 13 часов. К реакционной смеси добавляют воду, этилацетат и насыщенный водный раствор хлорида натрия. Органический слой отделяют, промывают насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния, и затем отгоняют растворитель при пониженном давлении. Полученный остаток очищают с использованием колоночной хроматографии на силикагеле (элюент гексан:этилацетат = 3:1) и получают 13,2 г трет-бутил-4-(3-бромпропил)-1-пиперидинкарбоксилата в виде бесцветного масла.
1Н-ЯМР (CDCl3) δ величина: 1,00-1,20 (2Н, м), 1,20-1,50 (3Н, м), 1,45 (9Н, с), 1,60-1,70 (2Н, м), 1,80-1,95 (2Н, м), 2,60-2,75 (2Н, м), 3,40 (2Н, т, J=6,8 Гц), 3,90-4,25 (2Н, м).
Пример получения 2

К раствору 13,2 г трет-бутил-4-(3-бромпропил)-1-пиперидинкарбоксилата в диметилсульфоксиде (130 мл) при комнатной температуре добавляют 5,13 г 4-цианофенола и 11,9 г карбоната калия и затем полученную смесь перемешивают при той же температуре в течение 26 часов. Реакционную смесь добавляют к смеси толуола и воды. Органический слой отделяют, промывают насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния, затем отгоняют растворитель при пониженном давлении и получают 14,5 г трет-бутил-4-[3-(4-цианофенокси)пропил]-1-пиперидинкарбоксилата в виде белого вещества в твердой форме.
1Н-ЯМР (CDCl3) δ величина: 1,05-1,20 (2Н, м), 1,40-1,50 (3Н, м), 1,46 (9Н, с), 1,65-1,75 (2Н, м), 1,75-1,90 (2Н, м), 2,60-2,80 (2Н, м), 3,99 (2Н, т, J=6,3 Гц), 4,00-4,20 (2Н, м), 6,93 (2Н, д, J=8,7 Гц), 7,58 (2Н, д, J=8,7 Гц).
Пример получения 3

К раствору 14,0 г трет-бутил-4-[3-(4-цианофенокси)пропил]-1-пиперидинкарбоксилата в хлороформе (100 мл) при охлаждении водой добавляют по каплям 40 мл трифторуксусной кислоты в течение 10 минут. Смесь перемешивают при той же температуре в течение 20 минут и затем перемешивают при комнатной температуре в течение 35 минут. После отгонки растворителя при пониженном давлении добавляют хлороформ и воду. К полученной смеси добавляют водный раствор гидроксида натрия для доведения рН до 13,0. Органический слой отделяют, и водный слой экстрагируют хлороформом. Органический слой и экстракт объединяют, затем объединенный раствор промывают водным раствором гидроксида натрия и сушат над карбонатом калия, затем отгоняют растворитель при пониженном давлении и получают 10,3 г 4-[3-(4-пиперидинил)пропокси]бензонитрила в виде бледно-желтого вещества в твердой форме.
1Н-ЯМР (CDCl3) δ величина: 1,05-1,20 (2Н, м), 1,35-1,45 (3Н, м), 1,65-1,90 (4Н, м), 2,50-2,65 (2Н, м), 3,00-3,15 (2Н, м), 3,99 (2Н, т, J=6,6 Гц), 4,78 (1Н, с), 6,93 (2Н, д, J=9,0 Гц), 7,58 (2Н, д, J=9,0 Гц).
Пример получения 4
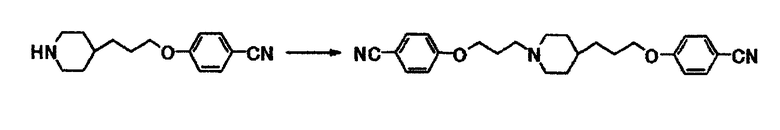
К раствору 10,2 г 4-[3-(4-пиперидинил)пропокси]бензонитрила в N,N-диметилформамиде (150 мл) при комнатной температуре последовательно добавляют 11,2 г карбоната калия и 9,72 г 4-(3-бромпропокси)бензонитрила и затем полученную смесь перемешивают при той же температуре в течение 18 часов. К реакционной смеси добавляют толуол и воду. Выпавшее в осадок вещество собирают фильтрацией и получают 13,7 г 4-(3-{4-[3-(4-цианопропокси)пропил]-1-пиперидинил}пропокси)бензонитрила в виде белого вещества в твердой форме.
1Н-ЯМР (CDCl3) δ величина: 1,20-1,45 (5Н, м), 1,65-2,05 (8Н, м), 2,40-2,55 (2Н, м), 2,85-3,00 (2Н, м), 3,99 (2Н, т, J=6,5 Гц), 4,06 (2Н, т, J=6,3 Гц), 6,93 (2Н, д, J=8,8 Гц), 6,94 (2Н, д, J=8,8 Гц), 7,57 (2Н, д, J=8,8 Гц), 7,57 (2Н, д, J=8,8 Гц).
Пример получения 5

Раствор 1,12 г трет-бутил-4-(3-бромпропил)-1-пиперидинкарбоксилата в 2-бутаноне (7,6 мл) добавляют к смеси 0,50 г 2-фтор-4-гидроксибензонитрила и 0,56 г карбоната калия в 2-бутаноне (7,0 мл) и затем полученную смесь кипятят с обратным холодильником в течение 6 часов 30 минут. После охлаждения до комнатной температуры реакционную смесь добавляют к смеси этилацетата и воды. Органический слой отделяют, промывают водой и сушат над безводным сульфатом магния, и затем отгоняют растворитель при пониженном давлении. Полученный остаток очищают с использованием колоночной хроматографии на силикагеле (элюент гексан:этилацетат = 4:1) и получают 0,72 г трет-бутил-4-[3-(4-циано-3-фторфенокси)пропил]-1-пиперидинкарбоксилата в виде бесцветного масла.
1Н-ЯМР (CDCl3) δ величина: 1,05-1,20 (2Н, м), 1,35-1,45 (3Н, м), 1,46 (9Н, с), 1,65-1,75 (2Н, м), 1,75-1,90 (2Н, м), 2,60-2,75 (2Н, м), 3,99 (2Н, т, J=6,3 Гц), 4,00-4,20 (2Н, м), 6,65-6,80 (2Н, м), 7,45-7,54 (1Н, м).
Пример получения 6
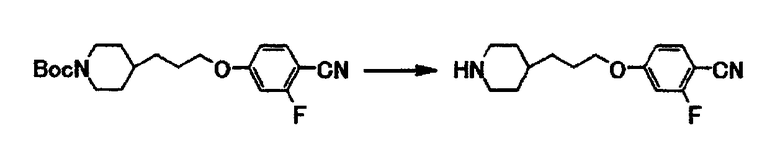
К раствору 0,66 г трет-бутил-4-[3-(4-циано-3-фторфенокси)пропил]-1-пиперидинкарбоксилата в метиленхлориде (5,5 мл) при охлаждении льдом в течение 2 минут добавляют по каплям 1,8 мл трифторуксусной кислоты и полученную смесь перемешивают при комнатной температуре в течение 6 часов. Растворитель отгоняют при пониженном давлении и к полученному остатку добавляют хлороформ и 1,0 моль/л водный раствор гидроксида натрия. Органический слой отделяют и сушат над безводным сульфатом магния, и затем отгоняют растворитель при пониженном давлении. Полученный остаток очищают с использованием колоночной хроматографии на силикагеле (элюент хлороформ:метанол = 4:1) и получают 0,28 г 2-фтор-4-[3-(4-пиперидинил)пропокси]бензонитрила в виде бледно-желтого масла.
1Н-ЯМР (CDCl3) δ величина: 1,05-1,20 (2Н, м), 1,30-1,45 (3Н, м), 1,50-1,75 (2Н, м), 1,75-1,90 (2Н, м), 2,50-2,65 (2Н, м), 3,00-3,15 (2Н, м), 3,98 (2Н, т, J=6,5 Гц), 6,69 (1Н, дд, J=11,0, 2,3 Гц), 6,75 (1Н, дд, J=8,5, 2,3 Гц), 7,50 (1Н, дд, J=8,5, 8,5 Гц).
Пример получения 7
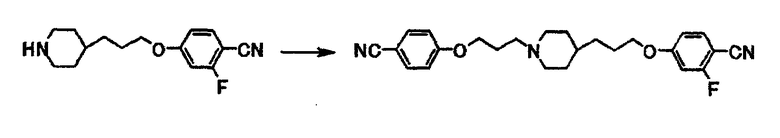
К раствору 0,10 г 2-фтор-4-[3-(4-пиперидинил)пропокси]бензонитрила в N,N-диметилформамиде (2,0 мл) при комнатной температуре последовательно добавляют 0,10 г карбоната калия и 0,13 г 4-(3-бромпропокси)бензонитрила и полученную смесь перемешивают при той же температуре в течение 13 часов. К реакционной смеси добавляют этилацетат, воду и толуол. Органический слой отделяют и сушат над безводным сульфатом натрия, и затем отгоняют растворитель при пониженном давлении. Полученный остаток очищают с использованием колоночной хроматографии на силикагеле (элюент хлороформ:метанол = 4:1) и получают 68 мг 4-(3-{1-[3-(4-цианофенокси)пропил]-4-пиперидинил}пропокси)-2-фторбензонитрила в виде белого вещества в твердой форме.
1Н-ЯМР (CDCl3) δ величина: 1,20-1,45 (5Н, м), 1,65-2,05 (8Н, м), 2,40-2,55 (2Н, м), 2,85-3,00 (2Н, м), 3,98 (2Н, т, J=6,5 Гц), 4,06 (2Н, т, J=6,3 Гц), 6,69 (1Н, дд, J=11,0, 2,4 Гц), 6,74 (1Н, дд, J=8,8, 2,4 Гц), 6,94 (2Н, д, J=8,7 Гц), 7,45-7,55 (1Н, м), 7,57 (2Н, д, J=8,7 Гц).
Пример получения 8
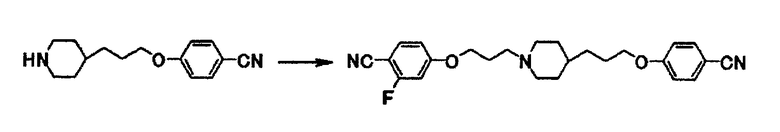
Так же, как описано в примере получения 7, используют 0,12 г 4-[3-(4-пиперидинил)пропокси]бензонитрила и 0,15 г 4-(3-бромпропокси)-2-фторбензонитрила и получают 0,10 г 4-(3-{4-[3-(4-цианофенокси)пропил]-1-пиперидинил}пропокси)-2-фторбензонитрила в виде белого вещества в твердой форме.
1Н-ЯМР (CDCl3) δ величина: 1,20-1,35 (3Н, м), 1,35-1,45 (2Н, м), 1,60-2,05 (8Н, м), 2,40-2,50 (2Н, м), 2,85-3,00 (2Н, м), 3,99 (2Н, т, J=6,5 Гц), 4,06 (2Н, т, J=6,3 Гц), 6,70-6,80 (2Н, м), 6,93 (2Н, д, J=9,0 Гц), 7,45-7,55 (1Н, м), 7,57 (2Н, д, J=9,0 Гц).
Пример получения 9
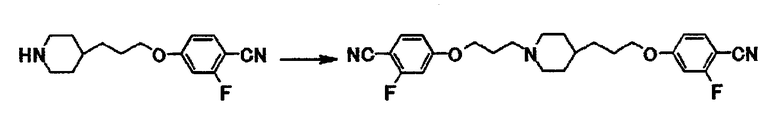
К раствору 0,26 г 2-фтор-4-[3-(4-пиперидинил)пропокси]бензонитрила и 0,21 г 4-(3-хлорпропокси)-2-фторбензонитрила в диметилсульфоксиде (4,0 мл) добавляют 0,88 мл N-этилдиизопропиламина и затем полученную смесь перемешивают при 80-90°С в течение 8 часов 15 минут. Реакционную смесь охлаждают до комнатной температуры, затем к смеси добавляют воду и затем экстрагируют этилацетатом. Экстракт дважды промывают водой и сушат над безводным сульфатом магния, и затем отгоняют растворитель при пониженном давлении. Полученный остаток очищают с использованием колоночной хроматографии на силикагеле (элюент хлороформ:метанол = 10:1) и получают 0,25 г 4-(3-{1-[3-(4-циано-3-фторфенокси)пропил]-4-пиперидинил}пропокси)-2-фторбензонитрила в виде коричневого вещества в твердой форме.
1Н-ЯМР (CDCl3) δ величина: 1,20-1,45 (5Н, м), 1,65-2,05 (8Н, м), 2,40-2,50 (2Н, м), 2,85-3,00 (2Н, м), 3,98 (2Н, т, J=6,5 Гц), 4,06 (2Н, т, J=6,3 Гц), 6,65-6,80 (4Н, м), 7,45-7,55 (2Н, м).
Пример получения 10
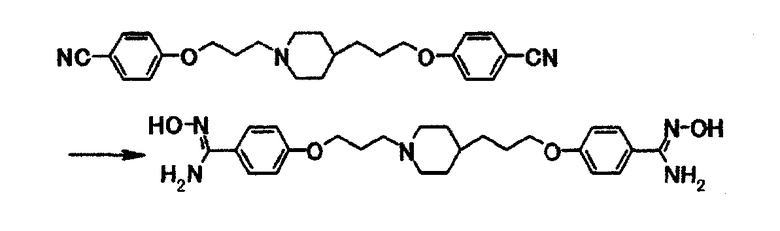
К суспензии 12,6 г 4-(3-{4-[3-(4-цианофенокси)пропил]-1-пиперидинил}пропокси)бензонитрила в диметилсульфоксиде (126 мл) добавляют 19,1 мл 50% водного раствора гидроксиламина и затем полученную смесь перемешивают при 50°С в течение 19 часов. Смесь охлаждают до комнатной температуры, затем к смеси добавляют по каплям в течение 50 минут 260 мл воды, затем смесь перемешивают при комнатной температуре в течение 30 минут и затем при охлаждении водой в течение 2 часов. Выпавшее в осадок вещество собирают фильтрацией и получают 15,0 г 4-{3-[4-(3-{4-[амино(гидроксиимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-гидроксибензамидина в виде белого вещества в твердой форме.
1Н-ЯМР (ДМСО-d6) δ величина: 1,05-1,40 (5Н, м), 1,60-1,80 (4Н, м), 1,80-1,90 (4Н, м), 2,35-2,45 (2Н, м), 2,80-2,90 (2Н, м), 3,96 (2Н, т, J=6,5 Гц), 4,01 (2Н, т, J=6,5 Гц), 5,65-5,75 (4Н, м), 6,85-6,95 (4Н, м), 7,55-7,65 (4Н, м), 9,43 (1Н, с), 9,43 (1Н, с).
Пример получения 11
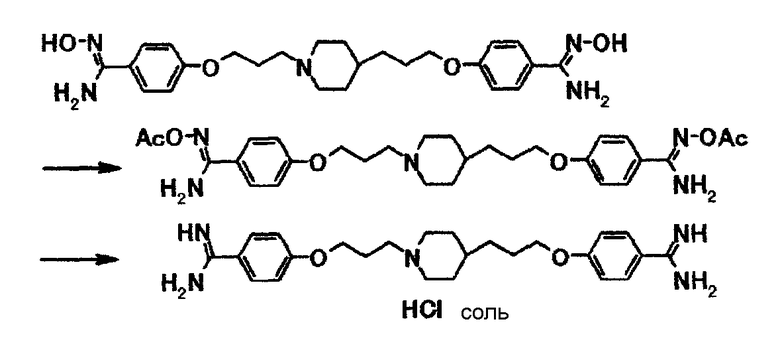
К суспензии 14,9 г 4-{3-[4-(3-{4-[амино(гидроксиимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-гидроксибензамидина в уксусной кислоте (150 мл) при комнатной температуре добавляют 5,97 мл уксусного ангидрида и затем полученную смесь перемешивают при комнатной температуре в течение 1 часа 20 минут. К полученной смеси добавляют 1,50 г 5% палладия-на-угле и затем смесь перемешивают в атмосфере водорода в течение 4 часов 40 минут. Нерастворимое вещество отфильтровывают и затем добавляют 55 мл 6,0 моль/л соляной кислоты. Растворитель отгоняют при пониженном давлении и к полученному остатку добавляют этанол. Твердое вещество собирают фильтрацией и получают 14,0 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина в виде белого вещества в твердой форме.
1Н-ЯМР (ДМСО-d6) δ величина: 1,30-1,45 (2Н, м), 1,45-1,70 (3Н, м), 1,70-1,90 (4Н, м), 2,15-2,30 (2Н, м), 2,80-3,00 (2Н, м), 3,10-3,20 (2Н, м), 3,45-3,55 (2Н, м), 4,10 (2Н, т, J=6,2 Гц), 4,19 (2Н, т, J=6,1 Гц), 7,15 (2Н, д, J=8,4 Гц), 7,16 (2Н, д, J=8,4 Гц), 7,84 (2Н, д, J=8,4 Гц), 7,86 (2Н, д, J=8,4 Гц). 8,90-9,00 (4Н, м), 9,15-9,30 (4Н, м), 10,60-10,80 (1Н, уш).
Пример получения 12
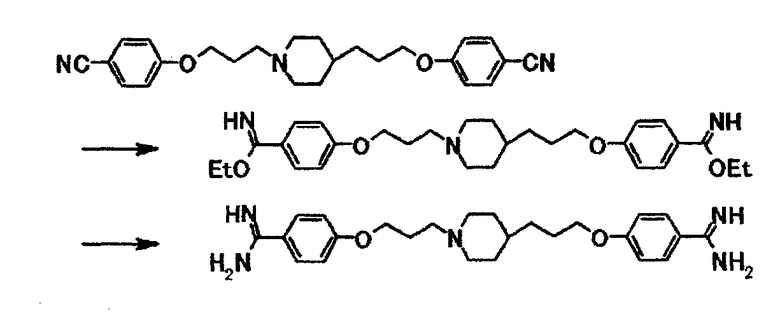
В суспензию 1,15 г 4-(3-{4-[3-(4-цианофенокси)пропил]-1-пиперидинил}пропокси)бензонитрила в этаноле (20 мл) при охлаждении льдом подают хлороводород и затем смесь перемешивают при комнатной температуре в течение 24 часов. Растворитель отгоняют при пониженном давлении и полученный остаток растворяют в 20 мл этанола. К смеси добавляют 1,54 г ацетата аммония и затем смесь кипятят с обратным холодильником в течение 3 часов 45 минут. Реакционную смесь охлаждают до комнатной температуры, добавляют к смеси воду и затем отгоняют этанол при пониженном давлении. К полученному остатку добавляют хлороформ и затем к смеси добавляют 5,0 моль/л водный раствор гидроксида натрия для доведения рН до 12,5. Выпавшее в осадок вещество собирают фильтрацией и получают 1,13 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина в виде белого вещества в твердой форме.
1Н-ЯМР (ДМСО-d6) δ величина: 1,00-1,40 (5Н, м), 1,60-1,80 (4Н, м), 1,80-1,95 (4Н, м), 2,35-2,45 (2Н, м), 2,80-2,90 (2Н, м), 3,98 (2Н, т, J=6,5 Гц), 4,03 (2Н, т, J=6,3 Гц), 6,30-7,20 (4Н, уш), 6,85-7,00 (4Н, м), 7,65-7,80 (4Н, м).
Пример получения 13

К суспензии 0,50 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина в этаноле (10 мл) при комнатной температуре добавляют 1,77 мл 2,6 моль/мл раствора хлороводорода в этаноле и затем смесь перемешивают при комнатной температуре в течение 4 часов 15 минут. Выпавшее в осадок вещество собирают фильтрацией и получают 0,49 г гидрохлорида 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина в виде бесцветного вещества в твердой форме.
Спектральные данные 1Н ЯМР, полученные в ДМСО-d6, согласуются с величинами, полученными в примере получения 11.
Пример получения 14
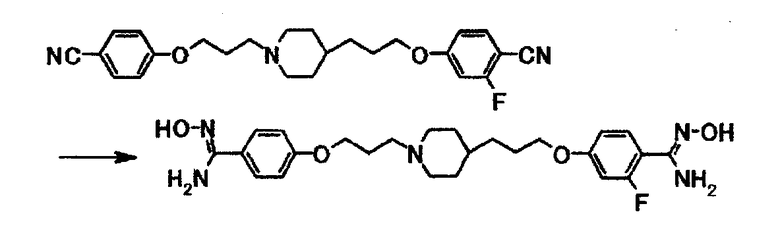
К суспензии 67 мг 4-(3-{1-[3-(4-цианофенокси)пропил]-4-пиперидинил}пропокси)-2-фторбензонитрила в диоксане (3,0 мл) добавляют 1,0 мл 50% водного раствора гидроксиламина и затем смесь кипятят с обратным холодильником в течение 2 часов. Смесь охлаждают до комнатной температуры, затем к смеси добавляют по каплям 10 мл воды и затем перемешивают при охлаждении льдом в течение 30 минут. Выпавшее в осадок вещество собирают фильтрацией и получают 63 мг 4-{3-[1-(3-{4-[амино(гидроксиимино)метил]фенокси}пропил)-4-пиперидинил]пропокси}-2-фтор-N'-гидроксибензамидина в виде бледно-желтого вещества в твердой форме.
1Н-ЯМР (ДМСО-d6) δ величина: 1,00-1,40 (5Н, м), 1,60-1,80 (4Н, м), 1,80-1,95 (4Н, м), 2,35-2,45 (2Н, м), 2,80-2,90 (2Н, м), 3,98 (2Н, т, J=6,4 Гц), 4,00 (2Н, т, J=6,0 Гц), 5,60-5,80 (4Н, м), 6,70-6,85 (2Н, м), 6,90 (2Н, д, J=8,8 Гц), 7,35-7,45 (1Н, м), 7,58 (2Н, д, J=8,8 Гц), 9,43 (1Н, с), 9,50 (1Н, с).
Пример получения 15

К суспензии 56 мг 4-{3-[1-(3-{4-[амино(гидроксиимино)метил]фенокси}пропил)-4-пиперидинил]пропокси}-2-фтор-N'-гидроксибензамидина в уксусной кислоте (2,0 мл) при комнатной температуре добавляют 0,043 мл уксусного ангидрида и затем полученную смесь перемешивают при той же температуре в течение одного часа. К полученной смеси добавляют 5,0 мг 5% палладия-на-угле и затем смесь перемешивают в атмосфере водорода в течение 2 часов. Нерастворимое вещество отфильтровывают и затем отгоняют растворитель при пониженном давлении. К остатку добавляют 6,0 моль/л соляную кислоту и воду и затем отгоняют растворитель при пониженном давлении. Полученный остаток очищают с использованием колоночной хроматографии на силикагеле (силикагель ODS-AM120-S50 от YMC, элюент вода). Полученный остаток растворяют в 5,0 мл воды и затем к раствору добавляют 5,0 моль/л водный раствор гидроксида натрия для доведения рН до 12,2. Раствор перемешивают при охлаждении льдом в течение 20 минут, выпавшее в осадок вещество собирают фильтрацией и получают 43 мг 4-{3-[1-(3-{4-[амино(имино)метил]фенокси}пропил)-4-пиперидинил]пропокси}-2-фторбензамидина в виде белого вещества в твердой форме.
1Н-ЯМР (ДМСО-d6) δ величина: 1,05-1,40 (5Н, м), 1,60-2,05 (8Н, м), 2,30-2,45 (2Н, м), 2,80-2,90 (2Н, м), 3,98 (2Н, т, J=6,5 Гц), 4,02 (2Н, т, J=6,3 Гц), 6,20-6,70 (4Н, уш), 6,75-6,85 (2Н, м), 6,92 (2Н, д, J=8,4 Гц), 7,45-7,55 (1Н, м), 7,21 (2Н, д, J=8,4 Гц).
Пример получения 16
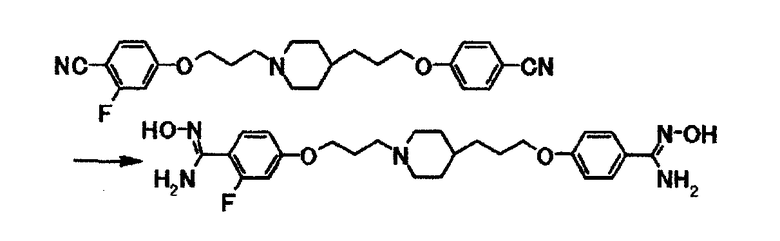
Как описано в примере получения 14, используют 0,10 г 4-(3-{4-[3-(4-цианофенокси)пропил]-1-пиперидинил}пропокси)-2-фторбензонитрила и получают 0,11 г 4-{3-[4-(3-{4-[амино(гидроксиимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-2-фтор-N'-гидроксибензамидина в виде белого вещества в твердой форме.
1Н-ЯМР (ДМСО-d6) δ величина: 1,00-1,40 (5Н, м), 1,60-1,75 (4Н, м), 1,75-1,90 (4Н, м), 2,30-2,40 (2Н, м), 2,80-2,90 (2Н, м), 3,96 (2Н, т, J=6,5 Гц), 4,03 (2Н, т, J=6,3 Гц), 5,65-5,80 (4Н, м), 6,75-6,90 (2Н, м), 6,90 (2Н, д, J=8,9 Гц), 7,35-7,45 (1Н, м), 7,58 (2Н, д, J=8,9 Гц), 9,43 (1Н, с), 9,50 (1Н, с).
Пример получения 17
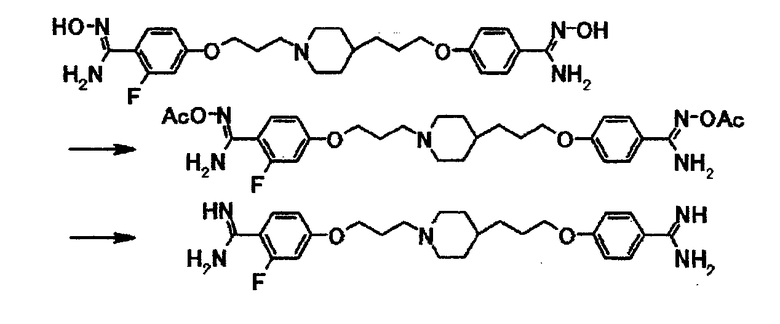
Как описано в примере получения 15, используют 90 мг 4-{3-[4-(3-{4-[амино(гидроксиимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-2-фтор-N'-гидроксибензамидина и получают 34 мг 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-2-фторбензамидина в виде белого вещества в твердой форме.
1Н-ЯМР (ДМСО-d6) δ величина: 1,05-1,40 (5Н, м), 1,60-1,90 (8Н, м), 2,30-2,45 (2Н, м), 2,80-2,90 (2Н, м), 3,98 (2Н, т, J=6,5 Гц), 4,03 (2Н, т, J=6,0 Гц), 6,30-6,75 (4Н, уш.), 6,75-6,85 (2Н, м), 6,93 (2Н, д, J=8,7 Гц), 7,45-7,55 (1Н, м), 7,71 (2Н, д, J=8,7 Гц).
Пример получения 18
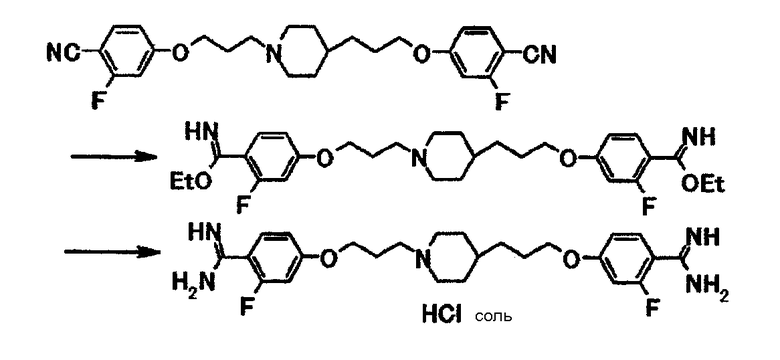
В суспензию 0,10 г 4-(3-{1-[3-(4-циано-3-фторфенокси)пропил]-4-пиперидинил}пропокси)-2-фторбензонитрила в этаноле (10 мл) при охлаждении льдом подают хлороводород и затем смесь перемешивают при той же температуре в течение 1 часа 10 минут и при комнатной температуре в течение 17 часов. Растворитель отгоняют при пониженном давлении и полученный остаток суспендируют в 5,0 мл этанола, к полученной смеси добавляют 44 мг ацетата аммония и затем смесь кипятят с обратным холодильником в течение 5 часов 30 минут. Растворитель отгоняют при пониженном давлении и полученный остаток растворяют в 8,0 мл 1,0 моль/л соляной кислоты, и затем отгоняют растворитель при пониженном давлении. Полученный остаток очищают с использованием колоночной хроматографии на силикагеле (силикагель ODS-AM120-S50 от YMC, элюент вода) и получают 46 мг гидрохлорида 4-{3-[1-(3-{4-[амино(имино)метил]-3-фторфенокси}пропил)-4-пиперидинил]пропокси}-2-фторбензамидина в виде белого вещества в твердой форме.
1Н-ЯМР (ДМСО-d6) δ величина: 1,30-1,45 (2Н, м), 1,50-1,70 (3Н, м), 1,70-1,90 (4Н, м), 2,20-2,30 (2Н, м), 2,80-2,95 (2Н, м), 3,10-3,20 (2Н, м), 3,40-3,55 (2Н, м), 4,10 (2Н, т, J=6,0 Гц), 4,20 (2Н, т, J=5,7 Гц), 6,95-7,05 (2Н, м), 7,05-7,15 (2Н, м), 7,60-7,75 (2Н, м), 9,20-9,50 (8Н, м), 10,95-11,10 (1Н, уш).
Пример 1 композиции лекарственного средства
В воде для инъекций растворяют 1,25 г соединения, полученного в примере получения 11, и 5,0 г D-маннита и получают общее количество 100 мл. Раствор фильтруют через 0,22-мкм мембранный фильтр, 10 мл полученного раствора лекарственного средства упаковывают в ампулу и запаивают, затем стерилизуют паром и получают средство для инъекций.
Промышленная применимость
Фармацевтическая композиция по настоящему изобретению, обладающая противогрибковой активностью, содержащая ариламидиновое производное или его соль и одно или несколько средств, выбранных из числа азольных противогрибковых средств, полиеновых противогрибковых средств, кандиновых противогрибковых средств, фторпиримидиновых противогрибковых средств и иммунодепрессантов, обладает сильной противогрибковой активностью и применима при лечении грибковых инфекций, вызванных грибковыми патогенами. Кроме того, способ лечения по настоящему изобретению применим как превосходный способ лечения грибковых инфекций.
Краткое описание чертежей
Фиг.1 представляет собой кривые выживания по результатам с использованием FLCZ вместе с испытываемым соединением (пример испытаний 2). Заштрихованные кружочки обозначают результаты для группы, которой не вводили какие-либо средства; заштрихованные квадраты обозначают результаты для группы, которой вводили 0,25 мг/кг FLCZ; заштрихованные ромбы обозначают результаты для группы, которой вводили 0,0313 мг/кг испытываемого соединения; и заштрихованные треугольники обозначают результаты для группы, которой вводили 0,0313 мг/кг испытываемого соединения и 0,25 мг/кг FLCZ.
Фиг.2 представляет собой кривые выживания по результатам с использованием АМРН-В вместе с испытываемым соединением (пример испытаний 2). Заштрихованные кружочки обозначают результаты для группы, которой не вводили какие-либо средства; заштрихованные квадраты обозначают результаты для группы, которой вводили 0,1 мг/кг АМРН-В; заштрихованные ромбы обозначают результаты для группы, которой вводили 0,0313 мг/кг испытываемого соединения; и заштрихованные треугольники обозначают результаты для группы, которой вводили 0,0313 мг/кг испытываемого соединения и 0,1 мг/кг АМРН-В.
Фиг.3 представляет собой кривые выживания по результатам с использованием MCFG вместе с испытываемым соединением (пример испытаний 2). Заштрихованные кружочки обозначают результаты для группы, которой не вводили какие-либо средства; заштрихованные квадраты обозначают результаты для группы, которой вводили 0,25 мг/кг MCFG; заштрихованные ромбы обозначают результаты для группы, которой вводили 0,0313 мг/кг испытываемого соединения; и заштрихованные треугольники обозначают результаты для группы, которой вводили 0,0313 мг/кг испытываемого соединения и 0,25 мг/кг MCFG.
Изобретение относится к области фармацевтики и касается фармацевтической комбинации для лечения грибковых инфекций, содержащей ариламидиновое производное формулы (1) и одно или несколько средств, выбранных из азольного противогрибкового средства, полиенового противогрибкового средства, кандинового противогрибкового средства и фторпиримидинового противогрибкового средства, и способу применения ариламидинового производного формулы (1). Комбинация обладает сильной противогрибковой активностью. 2 н. и 28 з.п. ф-лы, 3 ил., 7 табл.
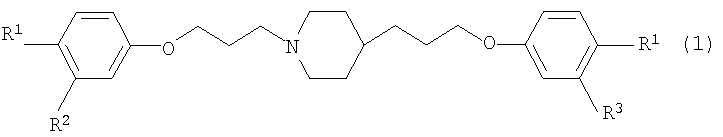
1. Фармацевтическая комбинация для лечения грибковых инфекций, содержащая ариламидиновое производное, представленное приведенной далее общей формулой
где R1 представляет амидиногруппу, которая может быть замещена гидроксильной группой, которая может быть защищена ацильной группой, амидиногруппу, которая может быть замещена алкоксигруппой, которая может быть замещенной, или амидиногруппу, которая может быть замещена аралкилоксигруппой, которая может быть замещенной; R2 и R3 могут быть одинаковыми или различными и представляют атом водорода или атом галогена, или его соль; и одно или несколько средств, выбранных из числа азольных противогрибковых средств, полиеновых противогрибковых средств, кандиновых противогрибковых средств, фторпиримидиновых противогрибковых средств и иммунодепрессантов.
2. Фармацевтическая комбинация по п.1, где R1 представляет амидиногруппу, которая может быть замещена гидроксильной группой, и R2 и R3 представляют собой атомы водорода.
3. Фармацевтическая комбинация по п.1 или 2, где средства представляют собой одно или несколько средств, выбранных из числа азольных противогрибковых средств, полиеновых противогрибковых средств, кандиновых противогрибковых средств и фторпиримидиновых противогрибковых средств.
4. Фармацевтическая комбинация по п.3, где средства представляют собой одно или несколько средств, выбранных из числа азольных противогрибковых средств.
5. Фармацевтическая комбинация по п.3, где средства представляют собой одно или несколько средств, выбранных из числа полиеновых противогрибковых средств.
6. Фармацевтическая комбинация по п.3, где средства представляют собой одно или несколько средств, выбранных из числа кандиновых противогрибковых средств.
7. Фармацевтическая комбинация по п.3, где средства представляют собой одно или несколько средств, выбранных из числа фторпиримидиновых противогрибковых средств.
8. Фармацевтическая комбинация по п.4, где азольные противогрибковые средства представляют собой противогрибковые средства на основе триазола.
9. Фармацевтическая комбинация по п.8, где противогрибковыми средствами на основе триазола являются флуконазол, вориконазол или итраконазол.
10. Фармацевтическая комбинация по п.5, где полиеновыми противогрибковыми средствами являются амфотерицин В или его липосомная композиция.
11. Фармацевтическая комбинация по п.6, где кандиновым противогрибковым средством является микафунгин.
12. Фармацевтическая комбинация по п.1 или 2, где средства представляют собой одно или несколько средств, выбранных из числа иммунодепрессантов.
13. Фармацевтическая комбинация по п.12, где иммунодепрессантом является такролим.
14. Фармацевтическая комбинация по п.1, где грибковая инфекция вызвана грибковым патогеном, выбранным из числа Candida, Cryptococcus, Aspergillus и Malassezia.
15. Фармацевтическая комбинация по п.14, где грибковая инфекция вызвана грибковым патогеном, выбранным из числа Candida, Cryptococcus и Aspergillus.
16. Способ применения ариламидинового производного, представленного общей формулой
где R1 представляет амидиногруппу, которая может быть замещена гидроксильной группой, которая может быть защищена ацильной группой, амидиногруппу, которая может быть замещена алкоксигруппой, которая может быть замещенной, или амидиногруппу, которая может быть замещена аралкилоксигруппой, которая может быть замещенной; и R2 и R3 могут быть одинаковыми или различными и представляют атом водорода или атом галогена, или его соли, в комбинации с одним или несколькими средствами, выбранными из числа азольных противогрибковых средств, полиеновых противогрибковых средств, кандиновых противогрибковых средств, фторпиримидиновых противогрибковых средств и иммунодепрессантов, для лечения грибковых инфекций, вызванных грибковыми патогенами.
17. Способ по п.16, где R1 представляет собой амидиногруппу, которая может быть замещена гидроксильной группой, и R2 и R3 представляют собой атомы водорода.
18. Способ по п.16 или 17, где средства представляют собой одно или несколько средств, выбранных из числа азольных противогрибковых средств, полиеновых противогрибковых средств, кандиновых противогрибковых средств и фторпиримидиновых противогрибковых средств.
19. Способ по п.18, где средства представляют собой одно или несколько средств, выбранных из числа азольных противогрибковых средств.
20. Способ по п.18, где средства представляют собой одно или несколько средств, выбранных из числа полиеновых противогрибковых средств.
21. Способ по п.18, где средства представляют собой одно или несколько средств, выбранных из числа кандиновых противогрибковых средств.
22. Способ по п.18, где средства представляют собой одно или несколько средств, выбранных из числа фторпиримидиновых противогрибковых средств.
23. Способ по п.19, где азольные противогрибковые средства представляют собой противогрибковые средства на основе триазола.
24. Способ по п.23, где противогрибковыми средствами на основе триазола являются флуконазол, вориконазол или итраконазол.
25. Способ по п.20, где полиеновыми противогрибковыми средствами являются амфотерицин В или его липосомная композиция.
26. Способ по п.21, где кандиновым противогрибковым средством является микафунгин.
27. Способ по п.16 или 17, где средства представляют собой одно или несколько средств, выбранных из числа иммунодепрессантов.
28. Способ по п.27, где иммунодепрессантом является такролим.
29. Способ по п.16, где грибковые инфекции вызваны грибковыми патогенами, выбранными из числа Candida, Cryptococcus, Aspergillus и Malassezia.
30. Способ по п.29, где грибковые инфекции вызваны грибковыми патогенами, выбранными из числа Candida, Cryptococcus и Aspergillus.
| WO 03074476, A1, 12.09.2003 | |||
| RU 2004129725, A, 10.05.2006. |

