Область изобретения
Настоящее изобретение относится к соединению, обладающему агонистической активностью в отношении GPR119, способу его получения и фармацевтической композиции, содержащей его в качестве эффективного компонента.
Предшествующий уровень техники
Метаболическое заболевание относится к синдрому, сопровождающемуся факторами риска, такими как ожирение, диабет, гипертриглицеридемия, гипертензия, другие сердечно-сосудистые заболевания и нарушение гемостаза. Согласно III редакции доклада по лечению взрослых (ATP, Adult Treatment Programm) Национальной образовательной программы по холестерину США (NCEP, US National Cholesterol Education Program), опубликованной в 2001 г., диагноз метаболического синдрома может быть поставлен при проявлении у пациента по меньшей мере трех из следующих пяти факторов риска: 1) абдоминального ожирения, определяемого как окружность талии 40 дюймов (102 см) или более у мужчин и 35 дюймов (88 см) или более у женщин, 2) гипертриглицеридемии, определяемой как концентрация триглицеридов 150 мг/дл или более, 3) ЛПВП-холестерина (холестерина липопротеинов высокой плотности) 40 мг/дл или менее у мужчин и 50 мг/дл или менее у женщин, 4) гипертензии, определяемой как артериальное давление 130/85 мм Hg или более и 5) концентрация глюкозы натощак 110 мг/дл или более.
За счет увеличения числа тучных людей и людей, ведущих малоподвижный образ жизни, распространенность сахарного диабета быстро растет во всем мире, и согласно данным Международной федерации диабета (МФД) ожидается, что число пациентов с сахарным диабетом резко возрастет с 246 миллионов в 2007 г. до 435 миллионов в 2030 г.
Инкретины представляют собой гормоны кишечника, секретируемые из энтероэндокринных клеток в кровь в течение нескольких минут после еды, которые включают в себя глюкагоноподобный пептид 1 (GLP-1) и глюкозозависимый инсулинотропный пептид (GIP). GLP-1 представляет собой пептидный гормон, обладающий коротким периодом полувыведения менее 2 минут, который секретируется посредством стимуляции L-клеток тонкого кишечника после приема пищи, в результате чего происходит индукция секреции инсулина в бета-клетках поджелудочной железы. Поэтому предположили, что существенное лечение возможно посредством улучшения функции бета-клеток, которое невозможно при существующих терапевтических средствах для лечения сахарного диабета (Baggio LL., Drucker DJ., Gastroenterology, 2007(132):2131-2157). Соответственно, недавно проведено множество исследований на лекарственных средствах, действующих непосредственно на рецептор GLP-1 или увеличивающих секрецию эндогенного GLP-1 (Gallwitz В., Handb Exp Pharmacol, 2011(203):53-74; Gallwitz В., Expert Opin Investig Drugs, 2011(20):723-32; Jones RM et al., Expert Opin Ther Pat, 2009(19):1339-1359).
Поскольку рецептор GLP-1 является одним из сопряженных с G-белком рецепторов класса В (GPCR), его третичная белковая структура не идентифицирована. В связи с тем, что GPCR класса В имеют уникальный центр взаимодействия, в котором N-конец рецептора связывается с лигандом для определения его сродства, их признают мишенью лекарственного средства, для которого трудно разработать низкомолекулярный синтетический лиганд (Dong М et al., Mol Endocrinol, 2008(22): 1489-1499; Hoare SR., Drug Discov Today, 2005(10):417-427).
Активация сопряженного с G-белком рецептора 119 (GPR119) приводит к секреции GLP-1 ( Diabet Obes Metab, 2011(13):158-166). GPR119 является членом GPCR класса А, и, следовательно, в отличие от класса В, является лекарственно-ориентированной мишенью для разработки низкомолекулярных лигандов. Описано, что агонисты GPR119 стимулируют секрецию GLP-1 в тонком кишечнике и прямо или косвенно повышают секрецию инсулина в бета-клетках поджелудочной железы (Lauffer LM. et al., Diabetes, 2009(58): 1058-1066; Chu ZL. et al., Endocrinology, 2008(149):2038-2047; Yoshida S. et al., Biochem Biophys Res Commun, 2010(400):745-751). Повышение секреции инсулина после активации GPR119 отчасти связывают с усиленным биосинтезом инсулина с последующей активацией промотора гена инсулина (Yoshida S. et al., Diabetes Obes Metab, 2011(13):34-41). Кроме того, Guo Z. et al. недавно сообщили, что при активации GPR119 низкомолекулярным соединением возрастает пролиферация бета-клеток поджелудочной железы с повышением эффективности после трансплантации островков Лангерганса (Guo Z. et al., Transplant Proc, 2011(43):3217-20). Предположили, что, помимо функции гликемического контроля, GPR119 обладает важной функцией в распознавании концентрации жиров, вводимых извне в эпителиальные клетки тонкого кишечника, поддерживая гомеостаз жира in vivo (Schwartz TW. et al, Trends in Pharmacological Sciences, 2012 in press, doi.10.1016/j.tips.2012.03.014). При активации низкомолекулярным соединением активация GPR119 приводит к подавлению всасывания жира в тонком кишечнике и к улучшению метаболизма липидов, что указывает на то, что агонист GPR119 обладает терапевтическим потенциалом в отношении дислипидемии (Brown KK. et al., 631-Р and Nunez DJ. et al., 1084-P in 72nd Scientific Session of American Diabetes Association, Philadelphia, PA). Недавно, согласно работе Hu YW et al., показано, что GPR119 играет важную роль в гомеостазе холестерина и в иммунной реакции иммунных клеток (Hu YW et al., J Lipid Res, 2014(55):681-97). После того как было показано, что активация GPR119 эффективно ингибирует повышенную постпрандиальную концентрацию триглицеридов, обладает эффективностью в повышении концентрации ЛПВП-холестерина и снижении концентрации ЛПНП-холестерина (холестерина липопротеинов низкой плотности), поддерживает гомеостаз холестерина и контролирует иммунную реакцию, повысился его потенциал как превосходной мишени для лекарственного средства, способного улучшать сердечно-сосудистую безопасность, в качестве терапевтического средства для лечения сахарного диабета. Кроме того, поскольку наблюдали, что селективные низкомолекулярные агонисты GPR119, такие как PSN632408, ингибируют потребление пищи и уменьшают прибавление массы тела и массы жира у крыс, питающихся кормом с высоким содержанием жира, GPR119 стал известен в качестве мишени, связанной с ожирением и родственными ему метаболическими заболеваниями (Overton НА. et al., Cell Metabolism, 2006(3):167-175).
Diabet Obes Metab, 2011(13):158-166). GPR119 является членом GPCR класса А, и, следовательно, в отличие от класса В, является лекарственно-ориентированной мишенью для разработки низкомолекулярных лигандов. Описано, что агонисты GPR119 стимулируют секрецию GLP-1 в тонком кишечнике и прямо или косвенно повышают секрецию инсулина в бета-клетках поджелудочной железы (Lauffer LM. et al., Diabetes, 2009(58): 1058-1066; Chu ZL. et al., Endocrinology, 2008(149):2038-2047; Yoshida S. et al., Biochem Biophys Res Commun, 2010(400):745-751). Повышение секреции инсулина после активации GPR119 отчасти связывают с усиленным биосинтезом инсулина с последующей активацией промотора гена инсулина (Yoshida S. et al., Diabetes Obes Metab, 2011(13):34-41). Кроме того, Guo Z. et al. недавно сообщили, что при активации GPR119 низкомолекулярным соединением возрастает пролиферация бета-клеток поджелудочной железы с повышением эффективности после трансплантации островков Лангерганса (Guo Z. et al., Transplant Proc, 2011(43):3217-20). Предположили, что, помимо функции гликемического контроля, GPR119 обладает важной функцией в распознавании концентрации жиров, вводимых извне в эпителиальные клетки тонкого кишечника, поддерживая гомеостаз жира in vivo (Schwartz TW. et al, Trends in Pharmacological Sciences, 2012 in press, doi.10.1016/j.tips.2012.03.014). При активации низкомолекулярным соединением активация GPR119 приводит к подавлению всасывания жира в тонком кишечнике и к улучшению метаболизма липидов, что указывает на то, что агонист GPR119 обладает терапевтическим потенциалом в отношении дислипидемии (Brown KK. et al., 631-Р and Nunez DJ. et al., 1084-P in 72nd Scientific Session of American Diabetes Association, Philadelphia, PA). Недавно, согласно работе Hu YW et al., показано, что GPR119 играет важную роль в гомеостазе холестерина и в иммунной реакции иммунных клеток (Hu YW et al., J Lipid Res, 2014(55):681-97). После того как было показано, что активация GPR119 эффективно ингибирует повышенную постпрандиальную концентрацию триглицеридов, обладает эффективностью в повышении концентрации ЛПВП-холестерина и снижении концентрации ЛПНП-холестерина (холестерина липопротеинов низкой плотности), поддерживает гомеостаз холестерина и контролирует иммунную реакцию, повысился его потенциал как превосходной мишени для лекарственного средства, способного улучшать сердечно-сосудистую безопасность, в качестве терапевтического средства для лечения сахарного диабета. Кроме того, поскольку наблюдали, что селективные низкомолекулярные агонисты GPR119, такие как PSN632408, ингибируют потребление пищи и уменьшают прибавление массы тела и массы жира у крыс, питающихся кормом с высоким содержанием жира, GPR119 стал известен в качестве мишени, связанной с ожирением и родственными ему метаболическими заболеваниями (Overton НА. et al., Cell Metabolism, 2006(3):167-175).
Подводя итог, поскольку низкомолекулярное лекарственное средство, активирующее GPR119, обладает эффективным гипогликемическим действием и положительно воздействует на бета-клетки поджелудочной железы, это обусловливает его ценность в качестве терапевтического средства для лечения сахарного диабета 2-го типа, улучшающего метаболизм липидов, который является постоянным фактором сердечно-сосудистого риска. Среди современных ведущих материалов клиническая разработка JNJ-38431055 и GSK1292263 приостановлена из-за утраты эффективности или недостаточной эффективности при многократном введении, однако, МВХ-2982 все еще находится в клинической разработке фазы II.
На основе вышеуказанного, авторы настоящего изобретения провели исследование терапевтического средства для лечения метаболического заболевания, такого как диабет, распространенность которого быстро растет во всем мире, и синтезировали новые низкомолекулярные соединения, активирующие GPR119, для которых было идентифицировано эффективное гипогликемические действие и положительное воздействие на бета-клетки поджелудочной железы, и за счет этого было завершено настоящее изобретение.
Раскрытие изобретения
Техническая задача
Настоящее изобретение было выполнено с целью разработки нового соединения, обладающего агонистической актиьвностью в отношении GPR119.
Кроме того, настоящее изобретение было выполнено с целью разработки способа получения этого нового соединения, обладающего агонистической активностью в отношении GPR119.
Кроме того, настоящее изобретение было выполнено с целью разработки фармацевтической композиции, содержащей это новое соединение в качестве эффективного ингредиента и полезной для лечения или предотвращения метаболического заболевания.
Техническое решение
Далее настоящее изобретение будет описано подробно.
В иллюстративном воплощении настоящего изобретения предложено соединение, представленное приведенной ниже химической формулой 1:

где
A представляет собой оксадиазол, дигидрооксазол, тиазол или тиадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила и С1-С6 спирта, где алкильная или спиртовая группа возможно замещена водородом, галогеном или С1-С6 алкоксигруппой;
В представляет собой пиридин, пиримидин, пиразин или оксадиазол, возможно замещенный одним более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечной или разветвленной С1-С6 алкильной, С1-С6 спиртовой, С1-С6 алкокси и оксадиазольной групп, где алкильная, спиртовая, алкокси или оксадиазольная группа возможно замещена водородом, галогеном или С1-С6 алкильной или С1-С6 алкоксигруппой; и
X независимо представляет собой F, Cl, Br или I, предпочтительно F; или его изомер, или его фармацевтически приемлемая соль.
Согласно одному воплощению настоящего изобретения в химической формуле 1
A может представлять собой 


 или
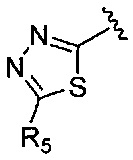
или  где R1-R6 независимо представляют собой один или более чем один заместитель, выбранный из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила и С1-С6 спирта, где алкильная или спиртовая группа возможно замещена водородом, галогеном или С1-С6 алкоксигруппой.
где R1-R6 независимо представляют собой один или более чем один заместитель, выбранный из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила и С1-С6 спирта, где алкильная или спиртовая группа возможно замещена водородом, галогеном или С1-С6 алкоксигруппой.
Согласно одному воплощению настоящего изобретения в химической формуле 1
В может представлять собой 
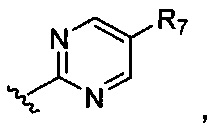

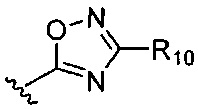 или
или 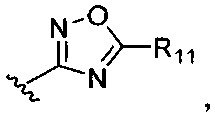 где R7-R11 возможно замещены одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечной или разветвленной С1-С6 алкильной, С1-С6 спиртовой, С1-С6 алкокси и оксадиазольной групп, где С1-С6 алкильная, С1-С6 спиртовая, С1-С6 алкокси или оксадиазольная группа возможно замещена водородом, галогеном, С1-С6 алкильной или С1-С6 алкоксигруппой.
где R7-R11 возможно замещены одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечной или разветвленной С1-С6 алкильной, С1-С6 спиртовой, С1-С6 алкокси и оксадиазольной групп, где С1-С6 алкильная, С1-С6 спиртовая, С1-С6 алкокси или оксадиазольная группа возможно замещена водородом, галогеном, С1-С6 алкильной или С1-С6 алкоксигруппой.
Более предпочтительно согласно одному воплощению настоящего изобретения может быть предложено соединение, где в химической формуле 1 A представляет собой С1-С6 алкил, например, оксадиазол замещен изопропильной группой; В представляет собой пиримидин, замещенный С1-С6 алкильной, например этильной группой; и X представляет собой галоген, например F; или его изомер, или его фармацевтически приемлемая соль.
Термин «галоген» при использовании в настоящем описании относится к фтору, хлору, брому или йоду.
Термин «алкил» при использовании в настоящем описании относится к прямоцепочечному или разветвленному углеводородному остатку, если не указано иное. Примеры С1-С6 алкила включают метил, этил, пропил, изопропил, бутил, изобутил, пентил, гексил и тому подобное.
Термин «алкокси» при использовании в настоящем описании включает алкил-кислородный радикал, в котором алкил является таким, как определено выше, если не указано иное. Примеры С1-С6 алкокси включают метокси, этокси, пропокси, бутокси, пентокси и тому подобное.
Термин «гетероцикл» или «гетероциклический» при использовании в настоящем описании относится к 5-13-членному гетероароматическому или неароматическому соединению, содержащему от 1 до 3 гетероатомов, выбранных из группы, состоящей из N, O и S, если не указано иное.
Более предпочтительно, согласно одному воплощению настоящего изобретения соединение, представленное приведенной выше химической формулой 1, может быть выбрано из группы, состоящей из следующих соединений:
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4,5-дигидрооксазола,
(R)-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4-метил-4,5-дигидрооксазола,
(S)-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4-метил-4,5-дигидрооксазола,
(S)-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-4,5-дигидрооксазола,
(R)-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-4,5-дигидрооксазола,
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5,5-диметил-4,5-дигидрооксазола,
(R)-(2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4,5-дигидрооксазол-5-ил)метанола,
(S)-(2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4,5-дигидрооксазол-5-ил)метанола,
(R)-3-(2-(4-(3-(3,5-дифтор-4-(5-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)пиримидин-5-ил)-5-изобутил-1,2,4-оксадиазола,
(R)-5-(4-(3-(3,5-дифтор-4-(4-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазола,
(S)-5-(4-(3-(3,5-дифтор-4-(5-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазола,
5-(4-(3-(4-(5,5-диметил-4,5-дигидрооксазол-2-ил)-3,5-дифторфенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазола,
3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,2,4-оксадиазола,
3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-пропил-1,2,4-оксадиазола,
3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазола,
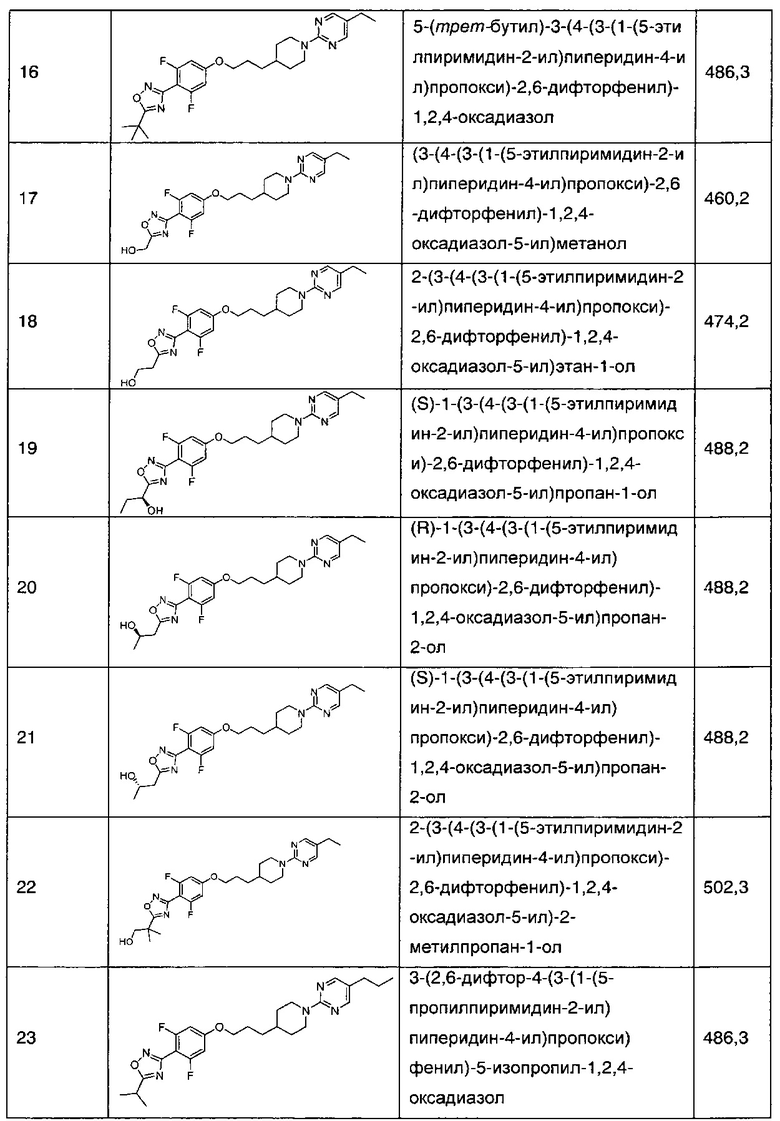
5-(трет-бутил)-3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазола,
(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)метанола,
2-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)этан-1-ола,
(S)-1-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)пропан-1-ола,
(R)-1-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)пропан-2-ола,
(S)-1-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)пропан-2-ола,
2-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)-2-метилпропан-1-ола,
3-(2,6-дифтор-4-(3-(1-(5-пропилпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-пентилпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-метоксипиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-изопропоксипиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазола,
3-(4-(3-(1-(5-хлорпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазола,
3-(4-(3-(1-(5-бромпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-этил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазола,
5-(втор-бутил)-3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-(метоксиметил)-1,2,4-оксадиазола,
(S)-1-(3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-1,2,4-оксадиазол-5-ил)пропан-1-ола,
2-(3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-1,2,4-оксадиазол-5-ил)-2-метилпропан-1-ола,
3-(4-(3-(1-(5-хлорпиразин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиридин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,2,4-оксадиазола,
3-(2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазола,
(3-(2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)фенил)-1,2,4-оксадиазол-5-ил)метанола,
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,3,4-оксадиазола,
2-этил-5-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,3,4-оксадиазола,
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-оксадиазола,
5-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-N-изопропил-1,3,4-оксадиазол-2-амина,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,3,4-оксадиазола,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-этил-1,3,4-оксадиазола,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-оксадиазола,
2-(4-(3-(1-(5-хлорпиразин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,3,4-оксадиазола,
2-(4-(3-(1-(5-хлорпиразин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-этил-1,3,4-оксадиазола,
2-(4-(3-(1-(5-хлорпиразин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-оксадиазола,
5-(4-(3-(3,5-дифтор-4-(5-метил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-пропил-1,2,4-оксадиазола,
5-(4-(3-(3,5-дифтор-4-(5-этил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-пропил-1,2,4-оксадиазола,
5-(4-(3-(3,5-дифтор-4-(5-изопропил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-пропил-1,2,4-оксадиазола,
5-(4-(3-(3,5-дифтор-4-(5-метил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазола,
5-(4-(3-(4-(5-этил-1,3,4-оксадиазол-2-ил)-3,5-дифторфенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазола,
5-(4-(3-(3,5-дифтор-4-(5-изопропил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазола,
5-(4-(3-(3,5-дифтор-4-(5-метил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-(2,2,2-трифторэтил)-1,2,4-оксадиазола,
3-(4-(3-(3,5-дифтор-4-(5-изопропил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-5-изопропил-1,2,4-оксадиазола,
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-тиадиазола,
2-(2,6-дифтор-4-(3-(1-(5-пропилпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазола,
2-(2,6-дифтор-4-(3-(1-(5-пентилпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазола,
2-(2,6-дифтор-4-(3-(1-(5-фторпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазола,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазола,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиридин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазола и
4-этил-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)тиазола.
В то же время, соединение, представленное химической формулой 1, может иметь асимметрический атом углерода, и при наличии асимметрического углеродного центра может существовать в виде оптического изомера, диастереомера или рацемата, и все формы изомеров, включая эти, также могут находиться в пределах объема соединения в соответствии с одним из воплощений настоящего изобретения.
Кроме того, фармацевтически приемлемая соль соединения, представленного химической формулой 1, или фармацевтически приемлемая соль изомеров соединения, представленного химической формулой 1, может также находится в пределах объема описанного выше одного из воплощений изобретения. Например, неограничивающие примеры фармацевтически приемлемой соли соединения, представленного химической формулой 1, или его изомера может включать в себя соль с неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, фосфорная кислота или серная кислота; соль с органической карбоновой кислотой, такой как уксусная кислота, трифторуксусная кислота, лимонная кислота, малеиновая кислота, щавелевая кислота, янтарная кислота, бензойная кислота, винная кислота, фумаровая кислота, миндальная кислота, аскорбиновая кислота или яблочная кислота, или соль с сульфоновой кислотой, такой как метансульфоновая кислота или пара-толуолсульфоновая кислота; соль с щелочным металлом, таким как натрий, калий или литий; соль с различными кислотами, известными как способные к образованию ионов других фармацевтически приемлемых кислот, или тому подобными.
Соединение в пределах объема соединения описанной выше химической формулы 1 может обладать превосходной агонистической активностью в отношении GPR119 и, соответственно, обладать гипогликемическим действием и оказывать положительное воздействие на бета-клетки поджелудочной железы, в результате чего его можно более эффективно использовать для лечения различных метаболических заболеваний.
Как описано выше, авторы настоящего изобретения недавно синтезировали соединение химической формулы 1, обладающее агонистической активностью в отношении GPR119, и фармацевтическая композиция, содержащая соединение, обладающее агонистической активностью в отношении сопряженного с G-белком рецептора (GPR119), может обладать эффективным гипогликемическим действием и оказывать положительное воздействие на бета-клетки поджелудочной железы, а также оказывать эффект улучшения метаболизма липидов, который представляет собой хронический фактор сердечно-сосудистого риска, в результате чего она эффективна в лечении и/или профилактике метаболического заболевания.
Агонистическая активность в отношении GPR119 может повышать секрецию глюкагоноподобного пептида 1 (GLP-1) или стабильность секретируемого GLP-1, обладающего эффективностью против ожирения и против сахарного диабета, опосредованной действием эндогенного инкретина.
Соответственно, в другом воплощении настоящего изобретения предложена фармацевтическая композиция, содержащая указанное выше соединение, его изомер или его фармацевтически приемлемую соль в качестве эффективного компонента. Более предпочтительно, фармацевтическая композиция может быть предназначена для лечения или предотвращения метаболического заболевания. Более предпочтительно, метаболическое заболевание может быть выбрано из группы, состоящей из диабета, ожирения, гипертензии, сердечно-сосудистого заболевания, расстройства гемостаза и дислипидемии.
Фармацевтическую композицию, содержащую соединение, представленное химической формулой 1, его изомер или его фармацевтически приемлемую соль в качестве эффективного компонента, можно использовать в форме обычного лекарственного препарата. Лекарственный препарат можно вводить в различных композициях, например в лекарственной форме для перорального и парентерального введения, и в зависимости от использования лекарственная форма может быть определена различным образом.
При включении композиции в различные лекарственные формы для перорального и парентерального введения ее можно изготавливать при использовании общепринятых эксципиентов, таких как наполнитель, разбавитель, объемообразующий агент, связующее вещество, увлажняющий агент, разрыхлитель, поверхностно-активное вещество.
Твердый препарат для перорального введения может включать таблетки, пилюли, порошки, гранулы, капсулы и тому подобное, и твердый препарат можно изготавливать путем смешивания соединения, представленного химической формулой 1, его изомера или его фармацевтически приемлемой соли с по меньшей мере одним эксципиентом, например крахмалом, карбонатом кальция, сахарозой или лактозой, желатином и тому подобным. Кроме того, кроме простого эксципиента можно использовать смазывающее вещество, такое как стеарат магния и тальк.
Кроме того, жидкий препарат для перорального введения может представлять собой суспензии, пероральные жидкости, эмульсии, сиропы и тому подобное и включать различные эксципиенты, например увлажняющий агент, подсластитель, ароматическое вещество, консервант и тому подобное в дополнение к воде и жидкому парафину, которые являются простыми общепринятыми разбавителями.
Препарат для парентерального введения включает стерильный водный раствор, неводный растворитель, суспензию, эмульсию, лиофилизированный препарат, суппозиторий и тому подобное. В качестве неводного растворителя и растворителя суспензии можно использовать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, инъекционный сложный эфир, такой как этилолеаьшт, и тому подобное. В качестве основы суппозитория можно использовать витепсол, макрогол, твин 61, масло какао, лауриновое масло, глицерожелатин и тому подобное.
Кроме того, в фармацевтической композиции по настоящему изобретению, содержащей соединение, представленное химической формулой 1, его изомер или его фармацевтически приемлемую соль в качестве эффективного компонента, эффективное количество может находиться в диапазоне дозы от приблизительно 0,1 до приблизительно 1000 мг. Дозировку или дозу можно вводить в различных дозированных формах и различными способами, например, в отдельных дозировках от одного до нескольких раз в день в зависимости от массы тела, возраста, пола, состояния здоровья, режима питания пациента, времени введения, способа введения, скорости выведения и тяжести заболевания.
В то же время, еще в одном другом воплощении настоящего изобретения предложен способ получения соединения химической формулы 1 описанного выше одного из воплощений, включающий введение группы В в азотную группу пиперидина соединения приведенной ниже химической формулы 2 с получением соединения приведенной ниже химической формулы 4; и введение соединения приведенной ниже химической формулы 12 в гидроксильную группу соединения химической формулы 4:

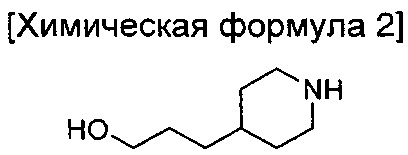
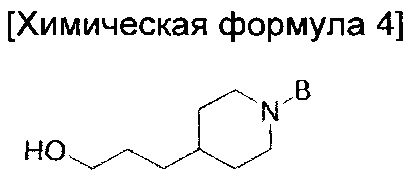
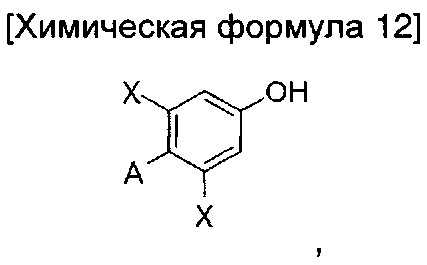
где
A представляет собой оксадиазол, дигидрооксазол, тиазол или тиадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила и С1-С6 спирта, где алкильная или спиртовая группа возможно замещена водородом, галогеном или С1-С6 алкоксигруппой;
В представляет собой пиридин, пиримидин, пиразин или оксадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечной или разветвленной С1-С6 алкильной, С1-С6 спиртовой, С1-С6 алкокси и оксадиазольной группы, где алкильная, спиртовая, алкокси или оксадиазольная группа возможно замещена водородом, галогеном или С1-С6 алкильной группой или С1-С6 алкоксигруппой; и
X независимо представляет собой F, Cl, Br или I.
В способе получения соединения химической формулы 1 порядок реакций стадии введения группы В в азотную группу пиперидина соединения химической формулы 2 и стадии введения соединения химической формулы 12 в гидроксильную группу соединения химической формулы 4 не ограничен, и, следовательно, сначала можно вводить соединение химической формулы 12 в гидроксильную группу соединения химической формулы 2, а можно сначала вводить группу В в азотную группу пиперидина.
Предпочтительно стадия введения соединения химической формулы 12 в гидроксильную группу соединения химической формулы 4 может включать взаимодействие соединения химической формулы 4 и соединение приведенной ниже химической формулы 12а; и преобразование A' в A:

где
A' представляет собой цианогруппу, карбоксильную группу, сложноэфирную группу, кетоновую группу или галоген.
Более предпочтительно, стадия взаимодействия соединения химической формулы 4 и соединения химической формулы 12а может включать введение метансульфонильной группы в гидроксильную группу соединения химической формулы 4; и взаимодействие с соединения химической формулы 4, в которое введена метансульфонильная группа, с соединением химической формулы 12а.
Стадия введения метансульфонильной группы в гидроксильную группу соединения химической формулы 4 может включать взаимодействие соединения химической формулы 4 с соединением, выбранным из группы, состоящей из метансульфонилхлорида, пара-толуолсульфонилхлорида и трихлорметансульфонилхлорида.
При взаимодействии соединения химической формулы 4 с метансульфонилхлоридом, пара-толуолсульфонилхлоридом или трихлорметансульфонилхлоридом метансульфонильная группа может быть введена в гидроксильную группу химической формулы 4, и более предпочтительно можно использовать метансульфонилхлорид. Условия в описанной выше реакции, такие как температура реакции и время реакции, можно соответствующим образом контролировать в зависимости от количества реагентов, условий окружающей среды и тому подобного, однако, метансульфонильная группа может быть более эффективно введена, например, путем взаимодействия при температуре от -10 до 10°C или при температуре приблизительно 0°C в течение от 10 минут до 3 часов в растворителе, представляющем собой дихлорметан (метиленхлорид, MX).
Далее, соединение химической формулы 4, в которое введена метансульфонильная группа, можно подвергать взаимодействию с соединением химической формулы 12а.
В частности, соединение химической формулы 4, в котором метансульфонильная группа введена в гидроксильную группу на предшествующей стадии, можно подвергать реакции сочетания с гидроксильной группой соединения химической формулы 12а посредством проведения реакции с соединением химической формулы 12а.
Реакцию сочетания можно проводить в присутствии одного или более чем одного основания, выбранного из группы, состоящей из карбоната натрия, карбоната кальция, карбоната калия и карбоната цезия; и одного или более растворителей, выбранных из группы, состоящей из метилсульфоксида, диметилформамида, N-метилпирролидин-2-она, тетрагидрофурана и 1,4-диоксана. В качестве основания предпочтительно можно использовать карбонат калия, а в качестве растворителя можно предпочтительно использовать диметилформамид. Условия, такие как температура реакции и время реакции сочетания, можно соответствующим образом контролировать в зависимости от количества реагентов, условий окружающей среды и тому подобного, однако, например, ее можно проводить в температурном диапазоне от 50°C до 100°C в течение от 5 до 24 часов.
Далее, группу A' химической формулы 12а можно преобразовать в группу A. Стадию преобразования можно проводить, используя подходяший способ в зависимости от вида группы А, и более конкретно, используя описанный ниже способ.
Соединение, где A представляет собой 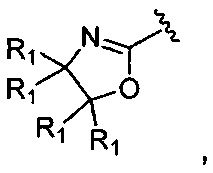 может быть получено способом, включающим:
может быть получено способом, включающим:
окисление соединения химической формулы 12а, где A' представляет собой карбоксильную группу или сложноэфирную группу, с получением карбоновой кислоты; взаимодействие карбоновой кислоты с аминоэтанолом приведенной ниже химической формулы 13 с введением соединения приведенной ниже химической формулы 14 в группу A' химической формулы 12а; и замыкание кольца соединения, полученного на предшествующей стадии.

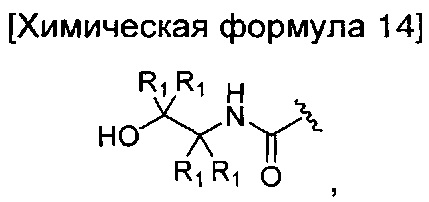
где R1 является таким, как определено в химической формуле 1.
Стадию окисления можно проводить в метиловом спирте, этиловом спирте, тетрагидрофуране, 1,4-диоксане или тому подобном в качестве растворителя путем использования водного раствора гидроксида натрия, водного раствора гидроксида калия или тому подобного, и реакцию можно проводить при температуре от 0°C до 80°C в течение от 1 до 5 часов. Затем проводят подкисление водным раствором HCl.
Кроме того, взаимодействие с аминоэтанолом можно проводить в метиловом спирте, этиловом спирте, тетрагидрофуране, 1,4-диоксане или тому подобном в качестве растворителя путем добавления гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC.HCl) и гидроксибензотриазола, и реакцию проводят при температуре от 10 до 40°C в течение от 5 минут до 3 часов, а затем добавляют триэтиламин и аминоэтанол, и реакцию проводят при температуре от 10 до 40°C в течение от 1 до 10 часов.
Затем стадию замыкания кольца можно проводить путем взаимодействия трифенилфосфина и 2,3-дихлор-5,6-дициано-1,4-бензохинона при температуре от 10 до 40°C в течение от 0 минут до 3 часов в растворителе, представляющем собой дихлорметан (MX).
Кроме того, соединение, где A представляет собой 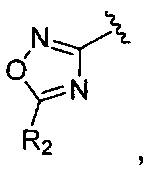 может быть получено способом, включающим:
может быть получено способом, включающим:
взаимодействие соединения химической формулы 12а, где A' представляет собой цианогруппу, с гидроксиламином с введением соединения приведенной ниже химической формулы 15 в группу A' химической формулы 12а; и взаимодействие соединения, полученного на предшествующей стадии, и соединения приведенной ниже химической формулы 16.
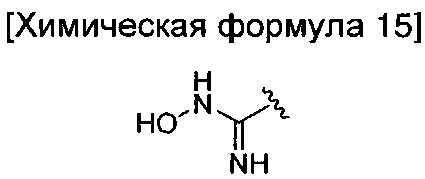
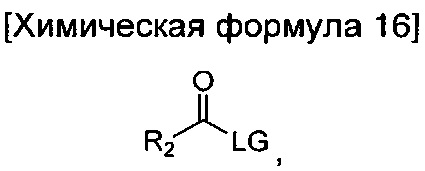
где R2 идентичен R2 химической формулы 1; и LG представляет собой уходящую группу.
Взаимодействие с гидроксиламином можно проводить в метиловом спирте, этиловом спирте, тетрагидрофуране или 1,4-диоксане в качестве растворителя при температуре от 80 до 150°C в течение от 1 до 10 часов.
Кроме того, взаимодействие соединения химической формулы 16 можно проводить путем осуществления реакции при температуре от 10 до 40°C в течение от 10 минут до 3 часов в растворителе, представляющем собой дихлорметан (MX), вместе с триэтиламином, а затем второй реакции при температуре от 100 до 200°C в течение от 1 до 10 часов. LG в химической формуле 16 представляет собой функциональную группу для отщепления в ходе реакции, и более конкретно может представлять собой галоген, и еще более конкретно Cl, но не ограничена им.
Кроме того, соединение, где A представляет собой 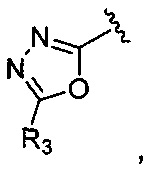 может быть получено способом, включающим:
может быть получено способом, включающим:
окисление соединения химической формулы 12а, где A' представляет собой карбоксильную группу или сложноэфирную группу, с получением карбоновой кислоты; взаимодействие карбоновой кислоты с гидразином с введением соединения приведенной ниже химической формулы 17 в группу A' химической формулы 12а; и взаимодействие соединения, полученного на предшествующей стадии, с соединением приведенной ниже химической формулы 18 или 19.

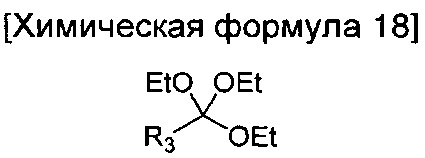
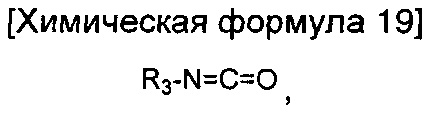
где R3 является таким, как определено в химической формуле 1.
Стадию окисления можно проводить в метиловом спирте, этиловом спирте, тетрагидрофуране, 1,4-диоксане или тому подобном в качестве растворителя путем использования водного раствора гидроксида натрия, водного раствора гидроксида калия или тому подобного, и реакцию можно проводить при температуре от 0°C до 80°C в течение от 1 до 5 часов. Затем проводят подкисление водным раствором HCl.
Взаимодействие с гидразином можно проводить в дихлорметане (MX) в качестве растворителя путем добавления гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC.HCl) и гидроксибензотриазола, и реакцию осуществляют при температуре от 10 до 40°C в течение от 5 минут до 3 часов, а затем добавляют гидразин, и реакцию осуществляют при температуре от 10 до 40°C в течение от 1 до 10 часов.
Кроме того, стадию взаимодействия с соединением химической формулы 18 можно проводить путем добавления реагента, полученного на предшествующей стадии, к раствору соединения химической формулы 18, и реакцию осуществляют при температуре от 100 до 200°C в течение от 1 до 10 часов, и стадию взаимодействия с соединением химической формулы 19 можно проводить путем растворения соединения химической формулы 17, полученного на предшествующей стадии, в водном растворе, а затем добавления триэтиламина и соединения химической формулы 19, и реакцию выполняют при температуре от 100 до 200°C в течение от 1 до 12 часов.
Корме того, соединение, где A представляет собой  может быть получено способом, включающим:
может быть получено способом, включающим:
окисление соединения химической формулы 12a, где A' представляет собой карбоксильную группу или сложноэфирную группу, с получением карбоновой кислоты; взаимодействие карбоновой кислоты с гидразидом с введением соединения приведенной ниже химической формулы 20 в группу A' химической формулы 12а; и взаимодействие соединения, полученного на предшествующей стадии, с соединением приведенной ниже химической формулы 21 (реагентом Лоуссона).
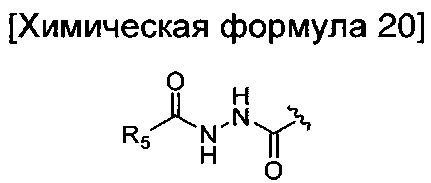
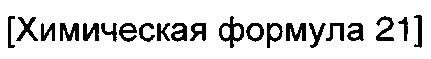
где R5 является таким, как определено в химической формуле 1.
Стадию окисления можно проводить в метиловом спирте, этиловом спирте, тетрагидрофуране, 1,4-диоксане или тому подобном в качестве растворителя путем использования водного раствора гидроксида натрия, водного раствора гидроксида калия или тому подобного, и реакцию можно проводить при температуре от 0°C до 80°C в течение от 1 до 5 часов. Затем проводят подкисление водным раствором HCl.
Стадию взаимодействия с гидразидом можно проводить в дихлорметане (MX) в качестве растворителя путем добавления гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC.HCl) и гидроксибензотриазола, и реакцию осуществляют при температуре от 10 до 40°C в течение от 5 минут до 3 часов, а затем добавляют гидразид, и реакцию осуществляют при температуре от 10 до 40°C в течение от 1 до 18 часов.
Кроме того, стадию взаимодействия с соединением химической формулы 21 можно проводить путем растворения соединения химической формулы 20, полученного на предшествующей стадии, в ксилоле, а затем добавления соединения химической формулы 21, и реакцию выполняют при температуре от 100 до 200°C в течение от 10 минут до 2 часов.
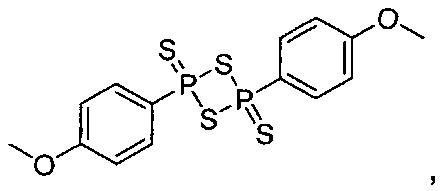
Кроме того, соединение, где A представляет собой  может быть получено способом, включающим:
может быть получено способом, включающим:
окисление соединения химической формулы 12а, где A' представляет собой карбоксильную группу или сложноэфирную группу, с получением карбоновой кислоты; взаимодействие карбоновой кислоты и тионилхлорида с введением структуры приведенной ниже химической формулы 22 в группу A' химической формулы 12а; преобразование структуры химической формулы 22 в амидную структуру приведенной ниже химической формулы 23; взаимодействие полученного соединения с соединением химической формулы 21 (реагентом Лоуссона) с преобразованием в соединение, имеющее структуру тиоамида, приведенной ниже химической формулы 24; и взаимодействие полученного соединения с соединением приведенной ниже химической формулы 25.
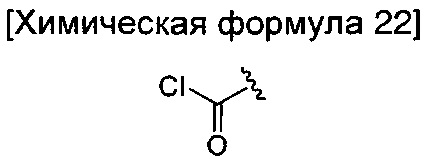
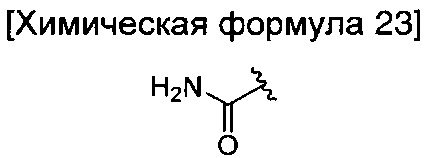
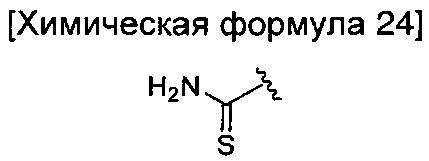
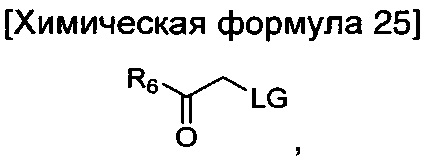
где R6 является таким, как определено в химической формуле 1; и LG представляет собой уходящую группу.
Стадию окисления можно проводить в метиловом спирте, этиловом спирте, тетрагидрофуране, 1,4-диоксане или тому подобном в качестве растворителя путем использования водного раствора гидроксида натрия, водного раствора гидроксида калия или тому подобного, и реакцию можно проводить при температуре от 0°C до 80°C в течение от 1 до 5 часов. Затем проводят подкисление водным раствором HCl.
Стадию введения соединения химической формулы 22 можно осуществлять путем добавления тионилхлорида в растворителе дихлорметане при температуре от 0°C до 80°C в течение от 1 до 5 часов.
Стадию введения соединения химической формулы 23
Полезный эффект изобретения
Новое соединение, его изомер или его фармацевтически приемлемая соль по настоящему изобретению обладает агонистической активностью в отношении GPR119 и, следовательно, его можно с пользой применять при лечении и/или профилактике метаболического заболевания, такого как сахарный диабет. Более конкретно посредством агонистической активности GPR119 может быть получено эффективное гликемическое действие и положительное воздействие на бета-клетки поджелудочной железы, а также может быть улучшен метаболизм липидов, представляющий хронический фактор сердечно-сосудистого риска.
Способ осуществления изобретения
Далее настоящее изобретение будет подробно описано приведенными ниже примерами с целью обеспечения понимания изобретения. Однако эти примеры предназначены только для иллюстрации настоящего изобретения, и объем настоящего изобретения не ограничен ими. Примеры настоящего изобретения приведены с целью более полного объяснения настоящего изобретения специалисту в данной области техники.
В соответствии с одним иллюстративным воплощением изобретения пример способа получения соединения химической формулы 1, включающего введение группы В в азотную группу пиперидина соединения химической формулы 2 с получением соединения химической формулы 4 и введения соединения химической формулы 12 в гидроксильную группу соединения химической формулы 4 кратко изложен в приведенных ниже схемах реакций 1-3.
Однако на этих схемах реакций 1-3 представлен только кратко изложенный пример способа получения соединения по настоящему изобретению и способ получения других воплощений изобретения не ограничен этим примером.

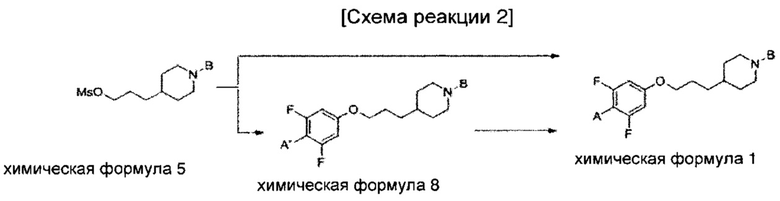
Соединения, синтезированные в приведенных ниже препаративных примерах, идентифицировали на основании спектра ядерного магнитного резонанса и масс-спектрометрии.
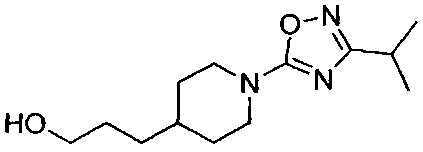
<Препаративный пример 1> Получение (R)-5-(4-(3(3,5-дифтор-4-(4-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазола
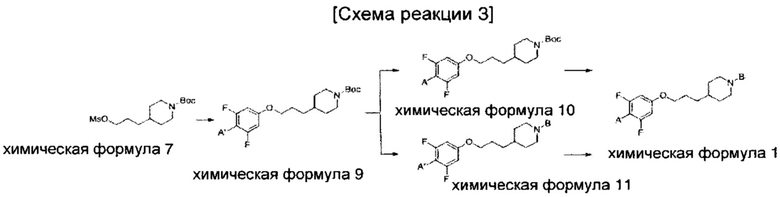
(Стадия 1-1) Получение 4-(3-гидроксипропил)пиперидин-1-карбонитрил (Химическая формула 3)
Гидрохлорид 3-(пиперидин-4-ил)пропан-1-ола химической формулы 2 (10 г, 69,8 ммоль) растворяли в смешанном растворе дихлорметана (MX, 75,0 мл) и воды (55,0 мл); к нему добавляли бикарбонат натрия (NaHCO3, 16,36 г, 195,0 ммоль); затем к нему добавляли цианистый бромид (6,48 г, 61,2 ммоль); и проводили перемешивание при комнатной температуре в течение 15 часов. Добавляли избыточное количество водного раствора хлорида аммония; проводили экстракцию дихлорметаном; а затем проводили промывание соляным раствором. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 4-(3-гидроксипропил)пиперидин-1-карбонитрил, при количественном выходе, который использовали в следующей реакции без очистки.
[М+1]+=169,1 m/z (ионизация электрораспылением (ИЭР)).
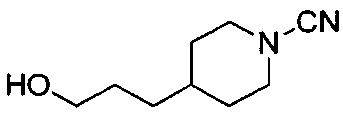
(Стадия 1-2) Получение N-гидроксиизобутилимидамида
Изобутиронитрил (химическая формула 15, 6,22 г, 90 ммоль) растворяли в этаноле (125 мл) и добавляли к нему 50% водный раствор гидроксиламина (18 мл) и гидроксид натрия (5,4 г, 135 ммоль). Реакционный раствор нагревали в условиях перемешивания до образования флегмы в течение 2 часов, а затем концентрировали при пониженном давлении, разбавляли водой и экстрагировали этилацетатом (ЭА). Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, N-гидроксиизобутилимидамид, при количественном выходе, который использовали в следующей реакции без очистки.
[М+1]+=103,1 m/z (ИЭР).
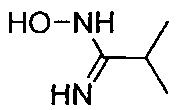
(Стадия 1-3) Получение 3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропан-1-ол
4-(3-Гидроксипропил)пиперидин-1-карбонитрил химической формулы 3, синтезированный на описанной выше стадии 1-1 (11,67 г, 69,4 ммоль), и N-гидроксиизобутилимидамид, синтезированный на описанной выше стадии 1-2 (8,5 г, 83,0 ммоль), растворяли в диэтиловом эфире (150 мл), а затем добавляли 1 М раствор хлорида цинка в диэтиловом эфире (90 мл, 90 ммоль) и перемешивали при комнатной температуре в течение 40 минут. Перемешанный реакционный раствор нагревали до 100°C до выпаривания 100 мл или более диэтилового эфира, а затем к нему добавляли этанол (200 мл). Затем к нему добавляли по каплям концентрированную соляную кислоту (4,21 мл, 139 ммоль), перемешивание проводили при 100°C в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении, разбавляли водой и экстрагировали ЭА. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, 3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропан-1-ола (14,3 г, 56,4 ммоль), при выходе 81%.
1H ЯМР (400 МГц, CDCl3) δ 4.09 (d, 2Н, J=12.8 Гц), 3.62 (t, 2Н, J=6.8 Гц), 2.99 (t, 2Н, J=13.2 Гц), 2.85 (m, 1Н, J=6.8 Гц), 1.75 (d, 2Н, J=12.4 Гц), 1.56 (m, 2Н), 1.46 (m, 1Н), 1.33 (m, 2Н), 1.25 (d, 6Н, J=6.8 Гц), 1.20 (m, 2Н); [М+1]+=254,2 m/z (ИЭР).
(Стадия 1-4) Получение 3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропилметансульфоната
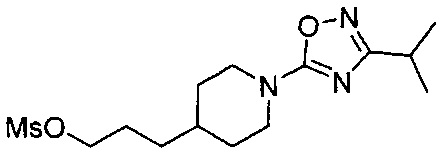
3-(1-(3-Изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропан-1-ол, синтезированный на описанной выше стадии 1-3 (108,9 г, 0,43 моль), растворяли в MX и охлаждали до 0°C. К реакционному раствору медленно добавляли по каплям триэтиламин (89,1 мл, 0,64 моль) и метансульфонилхлорид (39,7 мл, 0,51 моль). Реакционный раствор перемешивали при комнатной температуре в течение 1 часа, разбавляли MX и промывали водой. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропилметансульфонат, при количественном выходе.
[М+1]+=332,2 m/z (ИЭР).
(Стадия 1-5) Получение метил-2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пропокси)бензоата
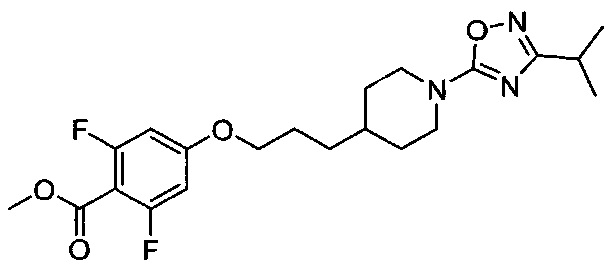
Метил-2,6-дифтор-4-гидроксибензоат (111,9 г, 0,59 моль) растворяли в N,N-диметилформамиде (ДМФ, 2 л), и к реакционному раствору добавляли 3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропилметансульфонат, синтезированный на описанной выше стадии 1-4 (165,7 г, 0,50 моль), и карбонат калия (K2CO3, 205,6 г, 1,49 моль). Реакционный раствор перемешивали при 60°C в течение 18 часов, а затем разбавляли водой и экстрагировали ЭА. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, метил-2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пропокси)бензоат, при выходе 85%.
[M+1]+=424,2 m/z (ИЭР).
(Стадия 1-6) Получение 2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)бензойной кислоты

Соединение, полученное на описанной выше стадии 1-5, метил-2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пропокси)бензоат (88,9 г, 0,21 моль), растворяли в растворителе, представляющем собой 1,4-диоксан (1,5 л), а затем добавляли по каплям 2 н. водный раствор NaOH (312 мл, 0,62 моль). Реакционный раствор перемешивали при 80°C в течение 3 часов, а затем разбавляли водой, и к нему добавляли 2 н. водный раствор HCl (800 мл) для подкисления раствора. Смешанный раствор экстрагировали ЭА (1,7 л), а затем из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)бензойную кислоту при выходе 97%.
[M+1]+=410,2 m/z (ИЭР).
(Стадия 1-7) Получение (R)-2,6-дифтор-N-(2-гидроксипропил)-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)бензамида
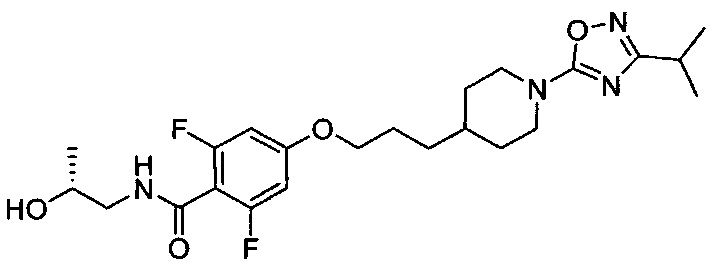
2,6-Дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)бензойную кислоту, полученную на описанной выше стадии 1-6 (0,41 г, 0,001 моль), растворяли в ТГФ, и к нему добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC.HCl, 0,38 г, 0,002 моль) и моногидрат гидроксибензотриазола (HOBt..H2O, 0,27 г, 0,002 моль). После перемешивания при комнатной температуре в течение 1 часа добавляли триэтиламин (0,42 мл, 0,003 моль) и (R)-1-аминопропан-2-ол (0,38 г, 0,005 моль). После перемешивания при комнатной температуре в течение 4 часов проводили разбавление водой и экстракцию ЭА. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, (R)-2,6-дифтор-N-(2-гидроксипропил)-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)бензамид, при выходе 85%.
[M+1]+=467,2 m/z (ИЭР).
(Стадия 1-8) Получение (R)-5-(4-(3(3,5-дифтор-4-(4-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазол (Препаративный пример 1)

Описанный выше 2,6-дифтор-N-(2-гидроксиэтил)-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)бензамид (42,9 мг, 0,092 ммоль) растворяли в MX, а затем к реакционному раствору добавляли трифенилфосфин (PPh3, 36,2 мг, 0,138 ммоль) и 2,3-дихлор-5,6-дициано-1,4-бензохинон (DDQ, 31,3 мг, 0,138 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 1 часа, разбавляли ЭА и промывали водой. Смешанный раствор экстрагировали ЭА, а затем из органического растворителя удаляли влагу с помощью MgSO4, органический слой фильтровали и концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, (R)-5-(4-(3(3,5-дифтор-4-(4-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазола, при выходе 85%.
1H ЯМР (600 МГц, CDCl3) δ 6.45 (d, 2Н, J=9.6 Гц), 4.48 (dd, 1Н, J=9.0 Гц, 8.4 Гц), 4.37 (m, 1Н), 4.12 (d, 2Н, J=12.6 Гц), 3.94 (m, 2Н), 3.01 (td, 2Н, J=13.2, 2.4 Гц), 2.86 (q, 1Н, J=7.2 Гц), 1.79 (m, 4Н), 1.50 (m, 1Н), 1.41 (m, 2Н), 1.36 (d, 3H, J=6.6 Гц), 1.26 (d, 6H, J=7.2 Гц), 1.26 (td, 2H, J=18.6 Гц, 4.2 Гц); [М+1]+=449,2 m/z (ИЭР).
<Препаративный пример 2> Получение 3-(2,6-Дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,2,4-оксадиазола
(Стадия 2-1) Получение 3-(1-(5-бромпиримидин-2-ил)пиперидин-4-ил)пропан-1-ола
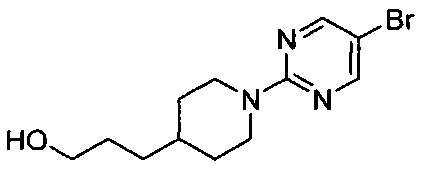
3-(Пиперидин-4-ил)пропан-1-ол химической формулы 2 (10 г, 69,8 ммоль) и 5-бром-2-хлорпиримидин (13,5 г, 69,8 ммоль) растворяли в N,N-диметилформамиде (ДМФ, 10 мл), а затем к нему добавляли карбонат калия (K2CO3, 10,6 г, 76,8 ммоль), и реакцию проводили при 80°C в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, разбавляли водой, экстрагировали этилацетатом (ЭА, 150 мл), а затем промывали соляным раствором. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, 3-(1-(5-бромпиримидин-2-ил)пиперидин-4-ил)пропан-1-ола, при выходе 82%.
1H ЯМР (400 МГц, CDCl3) δ 8.24 (s, 2Н), 4.64 (d, 2Н, J=15.2 Гц), 3.66-3.61 (m, 2Н), 2.86-2.79 (m, 2Н), 1.75 (d, 2Н, J=12.4 Гц), 1.63-1.56 (m, 2Н), 1.53-1.50 (m, 1Н), 1.34-1.27 (m, 2Н), 1.18-1.11 (m, 2Н); [М+1]+=300,1 m/z (ИЭР).
(Стадия 2-2) Получение 2-(4-(3-гидроксипропил)пиперидин-1-ил)пиримидин-5-карбонитрила
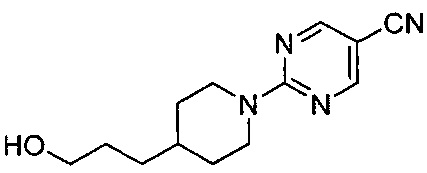
Цианид меди (KCN, 222 г, 3,0 моль) и йодид меди (CuI, 22 г) добавляли к N-метил-2-пирролидону (NMP, 750 мл), а затем нагревали до 160°C. К реакционному раствору медленно добавляли 3-(1-(5-бромпиримидин-2-ил)пиперидин-4-ил)пропан-1-ол химической формулы 9, синтезированный на описанной выше стадии 2-1 (222,0 г, 0,90 моль), растворенный в NMP (750 мл). После перемешивания в течение 3 часов реакционный раствор разбавляли ЭА и промывали водой (7500 мл). Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 2-(4-(3-гидроксипропил)пиперидин-1-ил)пиримидин-5-карбонитрил, при выходе 83%.
1H ЯМР (400 МГц, CDCl3) δ 8.46 (s, 2Н), 4.84-4.81 (m, 2Н), 3.67-3.64 (m, 2H), 2.95-2.90 (m, 2H), 1.84-1.82 (m, 2H), 1.64-1.59 (m, 2H), 1.36-1.33 (m, 2H), 1.29-1.27 (m, 1H), 1.20-1.13 (m, 2H); [M+1]+=247.2 m/z (ИЭР).
(Стадия 2-3) Получение N-гидрокси-2-(4-(3-гидроксипропил)пиперидин-1-ил)пиримидин-5-карбоксиимидамида

2-(4-(3-Гидроксипропил)пиперидин-1-ил)пиримидин-5-карбонитрил синтезированный на описанной выше стадии 2-2 (150,0 г, 0,61 моль), растворяли в этаноле (1800 мл), а затем к нему медленно добавляли по каплям гидрат гидроксиламина (430 г, 6,09 моль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, а затем концентрировали при пониженном давлении и добавляли к ней воду (1000 мл), и реакционную смесь перемешивали при 0-10°C в течение 1 часа. Полученное твердое вещество фильтровали с получением желаемой формы соединения, N-гидрокси-2-(4-(3-гидроксипропил)пиперидин-1-ил)пиримидин-5-карбоксиимидамида при выходе 85%.
[М+1]+=280,2 m/z (ИЭР).
(Стадия 2-4) Получение 2-(4-(3-гидроксипропил)пиперидин-1-ил)-N-((3-метилбутаноил)окси)пиримидин-5-карбоксиимидамида
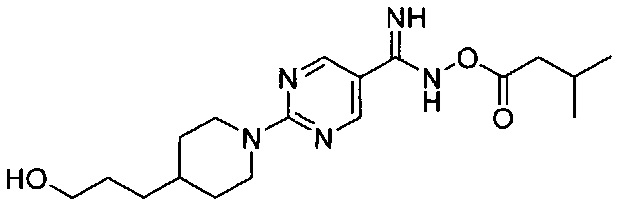
N-гидрокси-2-(4-(3-гидроксипропил)пиперидин-1-ил)пиримидин-5-карбоксиимидамид, синтезированный на описанной выше стадии 2-3 (144,1 г, 0,516 моль), растворяли в пиримидине (3000 мл), а затем к нему медленно добавляли по каплям изовалериановую кислоту (96,1 г, 0,516 моль) при 0-5°C. Реакционный раствор перемешивали в течение 30 минут с получением желаемой формы соединения, 2-(4-(3-гидроксипропил)пиперидин-1-ил)-N-((3-метилбктаноил)окси)пиримидин-5-карбоксиимидамид, который использовали в следующей реакции без очистки.
(Стадия 2-5) Получение 3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропан-1-ола

Реакционный раствор 2-(4-(3-гидроксипропил)пиперидин-1-ил)-N-((3-метилбутаноил)окси)пиримидин-5-карбоксиимидамида, синтезированного на описанной выше стадии 2-4, нагревали при перемешивании с обратным холодильником в течение 18 часов. Реакционный раствор концентрировали при пониженном давлении, к нему добавляли по каплям воду (2500 мл) при комнатной температуре в течение 30 минут, а затем реакционный раствор перемешивали при 0-5°C в течение 1 часа. Полученное твердое вещество фильтровали с получением желаемого соединения, 3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропан-1-ола, при выходе 94%.
1H ЯМР (600 МГц, CDCl3) δ 8.89 (s, 2Н), 4.86 (d, 2Н, J=13.2 Гц), 3.66 (t, 2Н, J=13.2 Гц), 2.95-2.90 (m, 2Н), 2.80 (d, 2Н, J=7.2 Гц), 2.28-2.24 (m, 1Н), 1.81 (d, 2Н, J=11.4 Гц), 1.65-1.61 (m, 2Н), 1.60-1.36 (m, 1Н), 1.35-1.22 (m, 2Н), 1.22-1.15 (m, 2Н), 1.04 (d, 6H, J=6.0 Гц); [M+1]+=345,2 m/z (ИЭР).
(Стадия 2-6) Получение 3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропилметансульфоната
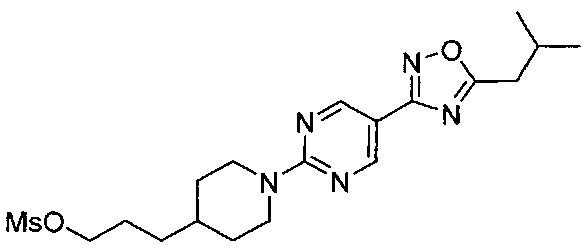
3-(1-(5-(5-Изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропан-1-ол, синтезированный на описанной выше стадии 2-5 (146,9 г, 0,43 моль), растворяли в MX и охлаждали до 0°C. К реакционному раствору медленно добавляли по каплям триэтиламин (89,1 мл, 0,64 моль) and метансульфонилхлорид (39.7 мл, 0.51 моль). Реакционный раствор перемешивали при комнатной температуре в течение 1 часа, разбавляли ЭА и промывали водой. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропилметансульфоната, при количественном выходе.
1H ЯМР (600 МГц, CDCl3) δ 8.90 (s, 2Н), 4.87 (d, 2Н, J=13.8 Гц), 4.24 (t, 2Н, J=13.2 Гц), 3.01 (s, 3H), 2.94-2.90 (m, 2Н), 2.80 (d, 2Н, J=7.2 Гц), 2.27-2.25 (m, 1Н), 1.83-1.79 (m, 4Н), 1.59 (m, 1Н), 1.41-1.37 (m, 2Н), 1.21-1.18 (m, 2Н), 1.04 (d, 6Н, J=6.0 Гц); [М+1]+=242,2 m/z (ИЭР).
(Стадия 2-7) Получение 2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)бензонитрила
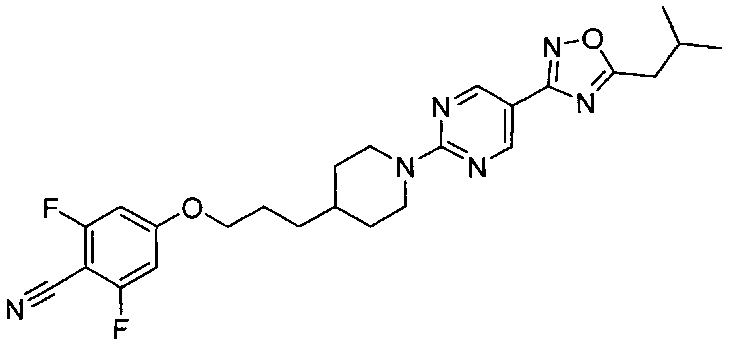
2,6-Дифтор-4-гидроксибензонитрил (6,6 г, 0,042 моль) растворяли в N,N-диметилформамиде (ДМФ, 0,3 л), и к реакционному раствору добавляли 3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропилметансульфонат, синтезированный на описанной выше стадии 2-6 (15 г, 0,035 моль) и карбонат калия (K2CO3, 14,7 г, 0,11 моль). Реакционный раствор перемешивали при 60°C в течение 18 часов, а затем разбавляли водой и экстрагировали ЭА. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, 2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)бензонитрила, при выходе 85%.
1H ЯМР (600 МГц, CDCl3) δ 8.88 (s, 2Н), 6.44 (d, 2Н, JHF=10.2 Гц), 4.86 (d, 2Н, J=13.2 Гц), 3.95-3.93 (m, 2Н), 3.89 (s, 3H), 2.94-2.89 (m, 2Н), 2.79 (d, 2Н, J=7.8 Гц), 2.27-2.22 (m, 1Н), 1.85-1.80 (m, 4Н), 1.61-1.59 (m, 1Н), 1.43-1.39 (m, 2Н), 1.23-1.19 (m, 2Н), 1.02 (d, 6Н, J=7.2 Гц); [М+1]+=516,3 m/z (ИЭР).
(Стадия 2-8) Получение 2,6-дифтор-N-гидрокси-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)бензимидамид
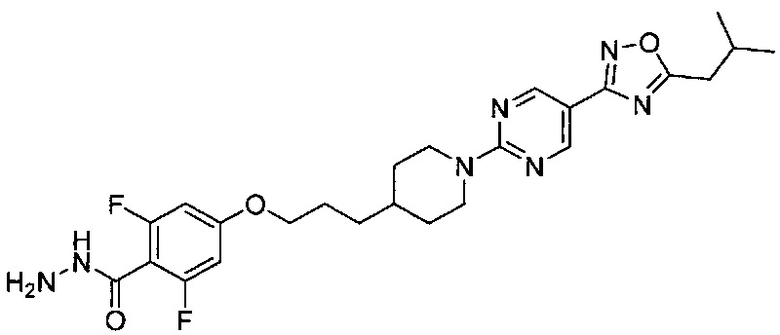
2,6-Дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)бензонитрил, синтезированный на описанной выше стадии 2-7 (5,7 г, 11,81 ммоль), растворяли в этаноле (68 мл), а затем к нему добавляли 50% водный раствор гидроксиламина (7,24 мл, 118,1 ммоль). Реакционный раствор перемешивали при 100°C в течение 5 часов, затем охлаждали до комнатной температуры и концентрировали до 1/10 объема. К концентрату добавляли по каплям воду (38 мл), затем перемешивание проводили в течение 1 часа, и полученное твердое вещество отфильтровывали, в результате чего получили желаемую форму соединения, 2,6-дифтор-N-гидрокси-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)бензимидамида, при количественном выходе.
1H ЯМР (600 МГц, CDCl3) δ 8.88 (s, 2Н), 6.44 (d, 2Н, JHF=10.2 Гц), 4.86 (d, 2Н, J=13.2 Гц), 3.95-3.93 (m, 2Н), 3.89 (s, 3H), 2.94-2.89 (m, 2Н), 2.79 (d, 2Н, J=7.8 Гц), 2.27-2.22 (m, 1Н), 1.85-1.80 (m, 4Н), 1.61-1.59 (m, 1Н), 1.43-1.39 (m, 2Н), 1.23-1.19 (m, 2Н), 1.02 (d, 6Н, J=7.2 Гц); [М+1]+=516,3 m/z (ИЭР).
(Стадия 2-9) Получение 3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,2,4-оксадиазол (пример 2)
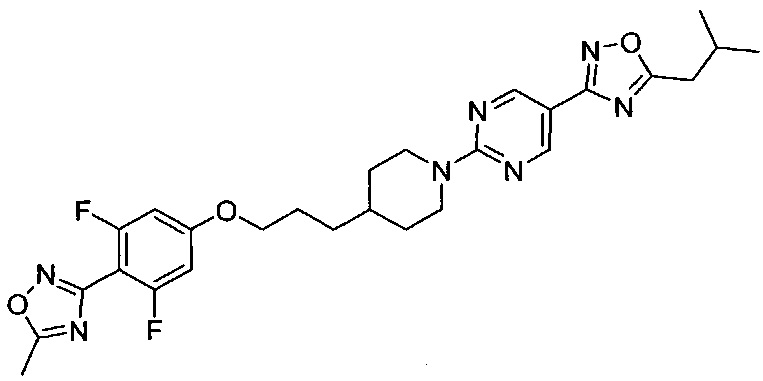
2,6-Дифтор-N-гидрокси-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)бензимидамид, полученный на описанной выше стадии 2-8 (70 мг, 0,14 ммоль) растворяли в N,N-диметилформамиде (ДМФ, 4 мл), а затем к нему добавляли по каплям триэтиламин (0,023 мл, 0,16 ммоль) и ацетилхлорид (0,013 мл, 0,16 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 1 часа, а затем дополнительно перемешивали при 140°C в течение 3 часов. После охлаждения до комнатной температуры раствор разбавляли водой и экстрагировали ЭА. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, 3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,2,4-оксадиазола, при выходе 85%.
1H ЯМР (600 МГц, CDCl3) δ 8.88 (s, 2Н), 6.55 (d, 2Н, JHF=10.2 Гц), 4.86 (d, 2Н, J=13.8 Гц), 3.95-3.93 (m, 2Н), 3.97 (t, 2Н, J=13.2 Гц), 2.94-2.89 (m, 2Н), 2.79 (d, 2Н, J=7.8 Гц), 2.64 (s, 3H), 2.25-2.23 (m, 1Н), 1.85-1.81 (m, 4Н), 1.56 (m, 1Н), 1.44-1.40 (m, 2H), 1.21-1.19 (m, 2H), 1.02 (d, 6H, J=6.7 Гц); [M+1]+=540,2 m/z (ИЭР).
<Препаративный пример 3> Получение 2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,3,4-оксадиазола
(Стадия 3-1) Получение метил-4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензоата
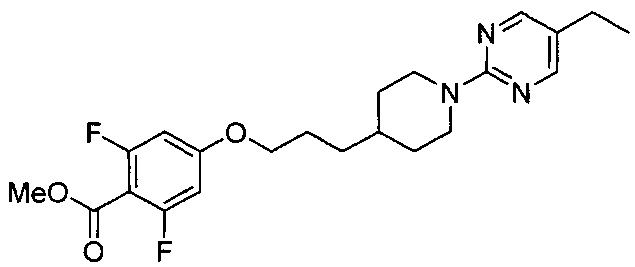
Метил 2,6-дифтор-4-гидроксибензоат (1,72 г, 9,16 ммоль) растворяли в N,N-диметилформамиде (ДМФ, 30 мл), а затем к реакционному раствору добавляли 3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропил метансульфонат (3,3 г, 10,08 ммоль) и карбонат калия (K2CO3, 3,8 г, 27,5 ммоль). Реакционный раствор перемешивали при 65°C в течение 12 часов, а затем разбавляли водой и экстрагировали ЭА. Из органического растворителя удаляли влагу с помощью MgSO4, органический слой фильтровали и концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, метил-4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензоата при выходе 93%.
1H ЯМР (400 МГц, CDCl3) δ 8.13 (s, 2Н), 6.43 (d, 2Н, J=10.8 Гц), 4.68 (d, 2Н, J=12.8 Гц), 3.93 (t, 2Н, J=6.4 Гц), 3.88 (s, 3H), 2.82 (t, 2Н, J=12.8 Гц), 2.42 (q, 2Н, J=7.6 Гц), 1.79 (m, 4Н), 1.53 (m, 1Н), 1.38 (m, 2Н), 1.19 (m, 2Н), 1.15 (t, 3H, J=7.6 Гц); [М+1]+=420,2 m/z (ИЭР).
(Стадия 3-2) Получение 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил-пропокси)-2,6-дифторбензойной кислоты
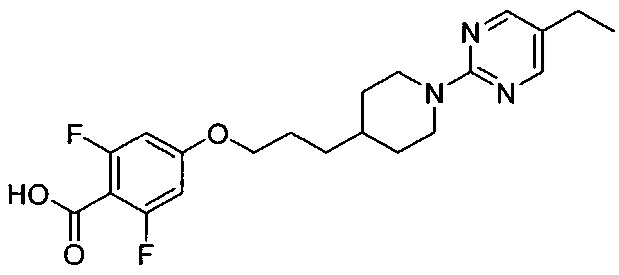
Метил-4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензоат, синтезированный на описанной выше стадии 3-1 (14,26 г, 34 ммоль) растворяли в этаноле (250 мл), а затем к нему добавляли 2 н. водный раствор гидроксида натрия (85 мл, 170 ммоль). Реакционный раствор перемешивали при 70°C в течение 15 часов, а затем разбавляли водой, и добавляли к нему 2 н. водный раствор HCl для подкисления раствора. Смешанный раствор экстрагировали ЭА, из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензойной кислоты при количественном выходе.
1H ЯМР (600 МГц, CDCl3) δ 8.15 (s, 2Н), 6.48 (d, 2Н, J=8.0 Гц), 4.69 (d, 2Н, J=8.8 Гц), 3.79 (t, 2Н, J=4.4 Гц), 2.87 (t, 2Н, J=8.4 Гц), 2.46 (q, 2Н, J=4.4 Гц), 1.85 (m, 2Н), 1.84 (d, 2Н, J=8.4 Гц), 1.57 (m, 1Н), 1.42 (m, 2Н), 1.24 (m, 2Н), 1.20 (m, 3H); [М+1]+=406,2 m/z (ИЭР).
(Стадия 3-3) Получение 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензогидразида
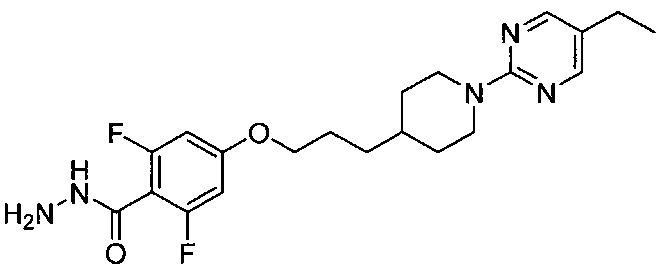
4-(3-(1-(5-Этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензойную кислоту, синтезированную на описанной выше стадии 3-2 (12,16 г, 30 ммоль), растворяли в дихлорметане (300 мл), а затем к нему добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (11,5 г, 60 ммоль) и гидроксибензотриазол (9,19 г, 60 ммоль) и перемешивали при комнатной температуре в течение 30 минут. Затем к нему добавляли по каплям гидрат гидразина (65%, 2,73 мл, 36 ммоль), а затем дополнительно перемешивали в течение 15 минут. Смешанный раствор экстрагировали дихлорметаном, из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензогидразида, при количественном выходе, который использовали в следующей реакции без очистки.
[М+1]+=420,2 m/z (ИЭР).
(Стадия 3-4) Получение 2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,3,4-оксадиазола (Препаративный пример 3)
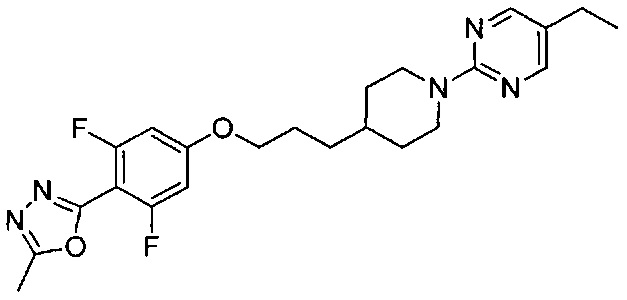
4-(3-(1-(5-Этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензогидразид, полученный на описанной выше стадии 3-3 (12,6 г), растворяли в триэтилортоацетате (50 мл), а затем перемешивали при 120°C в течение 6 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле, в результате чего получили желаемую форму соединения, 2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,3,4-оксадиазола (9,09 г, 20,49 ммоль), при выходе 68%.
1H ЯМР (600 МГц, CDCl3) δ 8.15 (appr-s, 2Н), 6.57 (appr-d, 2Н, J=10.2 Гц), 4.69 (d, 2Н, J=11.4 Гц), 3.98 (t, 2Н, J=6.0 Гц), 2.85 (appr-t, 2Н, J=5.4 Гц), 2.61 (s, 3H), 2.44 (q, 2Н, J=7.8 Гц), 1.84 (m, 3H), 1.78 (d, 2Н, J=12.0 Гц), 1.41 (m, 2Н), 1.22 (m, 2Н), 1.18 (t, 3H, J=7.8 Гц); [М+1]+=444,2 m/z (ИЭР).
<Препаративный пример 4> Получение 2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-тиадиазола
(Стадия 4-1) Получение 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифтор-N'-изобутирилбензогидразида

4-(3-(1-(5-Этилпиримидин-2-ил)пиперидин-4-ил-пропокси)-2,6-дифторбензойную кислоту, синтезированную на описанной выше стадии 3-2 <Препаративного примера 3>, растворяли в дихлорметане (4 мл), а затем к ней добавляли EDC (70,9 мг, 0,37 ммоль) и HOBt⋅H2O (56,7 мг, 0,37 ммоль). После активации при комнатной температуре в течение 1 часа добавляли по каплям изобутирогидразид (37,8 мг, 0,37 ммоль), и перемешивание проводили в течение 18 часов. После завершения реакции реакционную смесь фильтровали через целлит, а затем концентрировали при пониженном давлении с получением желаемой формы соединения, 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифтор-N'-изобутирилбензогидразида, при выходе 88%.
[М+1]+=490,3 m/z (ИЭР).
(Стадия 4-2) Получение 2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-тиадиазола (Препаративный пример 4)
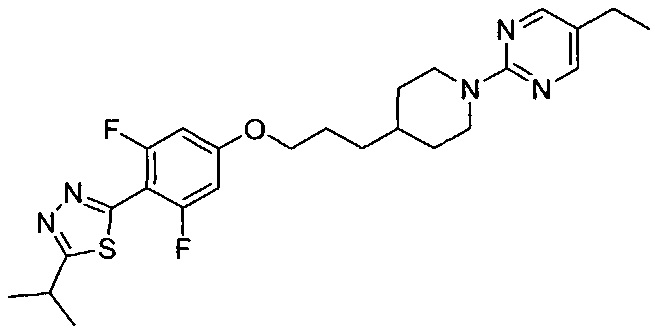
4-(3-(1-(5-Этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифтор-N'-изобутирилбензогидразид, полученный на описанной выше стадии 4-1 (22 мг, 0,05 ммоль), растворяли в ксилоле (4 мл), добавляли к нему реагент Лоуссона (27,3 мг, 0,07 ммоль), и перемешивание проводили при 140°C в течение 30 минут. После завершения реакции проводили разбавление водой и экстракцию этилацетатом. Из органического растворителя удаляли влагу с помощью MgSO4, органический слой фильтровали и концентрировали при пониженном давлении, а затем остаток очищали колоночной хроматографией на силикагеле, в результате чего получили желаемую форму соединения, 2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-тиадиазола, при выходе 44%.
1H ЯМР (600 МГц, CDCl3) δ 8.14 (s, 2Н), 6.56 (d, 2Н, J=10.4 Гц), 4.68 (d, 2Н, J=13.2 Гц), 3.96 (t, 2Н, J=12.8 Гц), 3.50 (m, 1Н), 2.83 (td, 2Н, J=12.4 Гц, 1.6 Гц), 2.42 (m, 2Н), 1.81 (m, 4Н), 1.52 (m, 1Н), 1.44 (d, 6Н, J=10.0 Гц), 1.40 (m, 2Н), 1.20 (m, 2Н), 1.16 (m, 3H); [М+1]+=488,3 m/z (ИЭР).
<Препаративный пример 5> Получение 4-этил-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)тиазола
(Стадия 5-1) Получение 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензоилхлорида
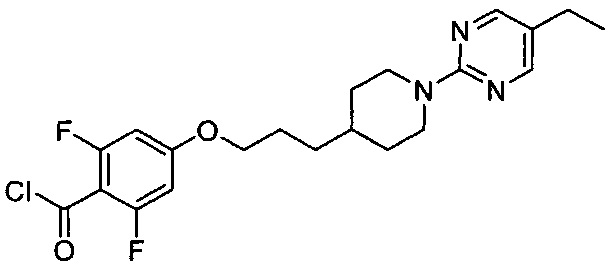
4-(3-(1-(5-Этилпиримидин-2-ил)пиперидин-4-ил-пропокси)-2,6-дифторбензойную кислоту, синтезированную на описанной выше стадии 3-2 <Препаративного примера 3> (1,39 г, 3,42 ммоль), растворяли в дихлорметане (15 мл), затем к нему добавляли по каплям тионилхлорид (0,75 мл, 10,27 ммоль), и перемешивание проводили при 65°C в течение 4 часов. После завершения реакции проводили разбавление водой, экстракцию дихлорметаном, а затем промывание соляным раствором. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензоилхлорида, при количественном выходе.
[М+1]+=424,2 m/z (ИЭР).
(Стадия 5-2) Получение 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензамида
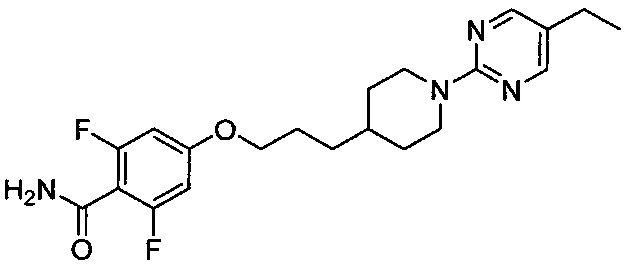
4-(3-(1-(5-Этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензоилхлорид, синтезированный на описанной выше стадии 5-1 (1,46 г, 3,44 ммоль), растворяли в бензоле (10 мл), затем к нему добавляли гидроксид натрия (0,83 г, 20,65 ммоль) и хлорид аммония (0,55 г, 10,32 ммоль) и перемешивали в течение 2 часов. После завершения реакции проводили разбавление водой, экстракцию этилацетатом, а затем промывание соляным раствором. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении. Концентрированный остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензамида, при выходе 84%.
1H ЯМР (400 МГц, CDCl3) δ 8.14 (s, 2Н), 6.46 (dd, 2Н, J=5.2 Гц, 15.6 Гц), 6.03 (s, 1Н), 5.83 (s, 1Н), 4.68 (d, 2Н, J=13.2 Гц), 3.93 (t, 2Н, J=12.8 Гц), 2.83 (m, 2Н), 2.43 (m, 2Н), 1.82 (m, 4Н), 1.63 (m, 1Н), 1.54 (m, 1Н), 1.39 (m, 2Н), 1.16 (m, 4Н); [М+1]+=405,2 m/z (ИЭР).
(Стадия 5-3) Получение 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензотиоамида
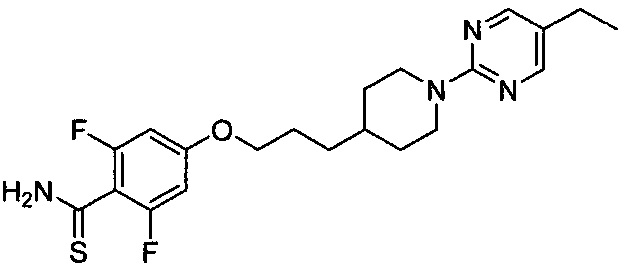
4-(3-(1-(5-Этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензамид, синтезированный на описанной выше стадии 5-2 (1,17 г, 2,88 ммоль), растворяли в тетрагидрофуране (ТГФ, 10 мл), затем к нему добавляли реагент Лоуссона (1,75 г, 4,32 ммоль) и перемешивали при 50°C в течение 3 часов. После завершения реакции проводили разбавление водой и экстракцию этилацетатом. Из органического растворителя удаляли влагу с помощью MgSO4, органический слой фильтровали и концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, 4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензотиоамида, при выходе 32%.
[M+1]+=421,2 m/z (ИЭР).
(Стадия 5-4) Получение 4-этил-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)тиазола (Препаративный пример 5)
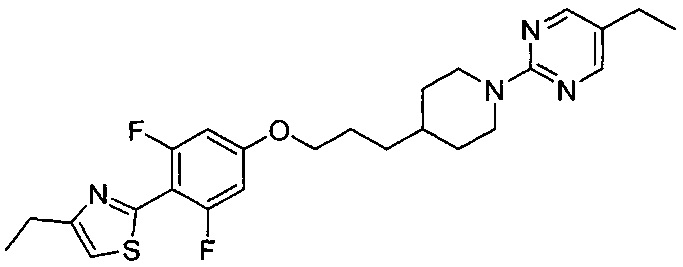
4-(3-(1-(5-Этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторбензотиоамид (0,38 г, 0,91 ммоль), соединение, синтезированное на описанной выше стадии 5-3, растворяли в этаноле (6 мл), а затем к нему добавляли по каплям 1-бромбутан-2-он (3,31 мкл, 0,91 ммоль) при комнатной температуре. Реакционную смесь перемешивали с обратным холодильником при 100°C. После завершения реакции растворитель концентрировали при пониженном давлении, и органический слой экстрагировали, используя воду и этилацетат. Из органического растворителя удаляли влагу с помощью MgSO4, органический слой фильтровали и концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, 4-этил-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)тиазола, при выходе 62%.
1H ЯМР (400 МГц, CDCl3) δ 8.11 (s, 2Н), 6.98 (s, 1Н), 6.49 (d, 2Н, J=15.6 Гц), 4.66 (d, 2Н, J=13.2 Гц), 3.91 (m, 2Н), 2.82 (m, 4Н), 2.40 (m, 2Н), 1.78 (m, 4Н), 1.51 (m, 1Н), 1.37 (m, 2Н), 1.29 (m, 3H), 1.18 (m, 2Н), 1.13 (m, 3H); [М+1]+=473,2 m/z (ИЭР).
<Препаративный пример 6> Получение 3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазола
(Стадия 6-1) Получение 2,6-дифтор-N',4-дигидробензимидамида

2,6-Дифтор-4-гидроксибензонитрил (3,0 г, 19,3 ммоль) растворяли в этаноле (12 мл), а затем к реакционному раствору добавляли 50% водный раствор гидроксиламина (NH2OH, 12,6 г, 193,0 ммоль). Реакционный раствор перемешивали с обратным холодильником в течение 3 часов, затем концентрировали при пониженном давлении для удаления растворителя, к нему добавляли воду, и проводили фильтрование, в результате чего получили желаемую форму соединения, 2,6-дифтор-N',4-дигидробензимидамида, при выходе 75%.
[М+1]+=189,0 m/z (ИЭР).
(Стадия 6-2) Получение 3,5-дифтор-4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенола
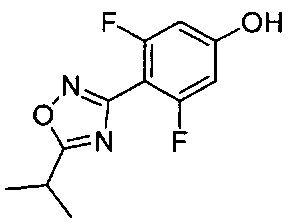
2,6-Дифтор-N',4-дигидробензимидамид (2,6 г, 10,6 ммоль), соединение, синтезированное на описанной выше стадии 6-1, растворяли в 1,4-диоксане (80 мл), а затем к реакционному раствору добавляли ангидрид изомасляной кислоты (1,7 г, 10,6 ммоль). Реакционный раствор перемешивали в течение 1 часа, к нему добавляли сульфат магния (MgSO4, 2,6 г) и перемешивание с обратным холодильником в течение 18 часов. Реакционный раствор концентрировали при пониженном давлении, затем остаток очищали колоночной хроматографией на силикагеле и добавляли к нему дополнительное количество эфира, затем проводили фильтрование, в результате чего получили желаемую форму соединения, 3,5-дифтор-4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенола, при выходе 48%.
1H ЯМР (400 МГц, ДМСО-d6) δ 11.07 (brs, 1Н), 6.68 (d, 2Н, J=14.8 Гц), 3.37 (m, 1Н), 1.38 (d, 6Н, J=6.8 Гц)
(Стадия 6-3) Получение 3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропан-1-ола
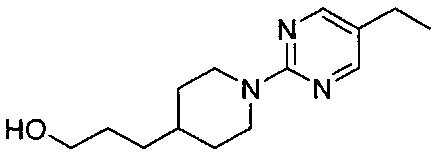
2-хлор-5-этилпиримидин (1,0 г, 7,0 ммоль) растворяли в N,N-диметилформамиде (ДМФ, 15 мл), а затем к реакционному раствору добавляли 3-(пиперидин-4-ил)пропан-1-ол (1,1 г, 7,7 ммоль) и карбонат калия (K2CO3, 2,9 г, 21,0 ммоль). Реакционный раствор перемешивали при 65°C в течение 12 часов, а затем разбавляли водой и экстрагировали ЭА. Из органического растворителя удаляли влагу с помощью MgSO4, органический слой фильтровали и концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением желаемой формы соединения, 3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропан-1-ола, при выходе 75%.
1H ЯМР (400 МГц, CDCl3) δ 8.15 (s, 2Н), 4.67 (d, 2Н, J=13.6 Гц), 2.87 (m, 2Н), 2.83 (t, 2Н, J=12.6 Гц), 2.44 (q, 2Н, J=7.6 Гц), 1.46-1.38 (m, 9Н), 1.21 (t, 3H, J=7.6 Гц)
(Стадия 6-4) Получение 3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропилметансульфоната
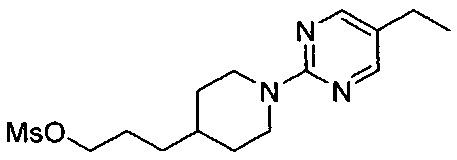
3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропан-1-ол, синтезированный на описанной выше стадии 6-3 (1,0 г, 4,0 ммоль), растворяли в MX и охлаждали до 0°C. К реакционному раствору медленно добавляли по каплям триэтиламин (0,6 г, 6,0 ммоль) и метансульфонилхлорид (0,6 г, 4.8 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 1 часа, разбавляли MX и промывали водой. Из органического растворителя удаляли влагу с помощью MgSO4, и органический слой фильтровали и концентрировали при пониженном давлении, в результате чего получили желаемую форму соединения, 3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропилметансульфоната, при количественном выходе.
1H ЯМР (400 МГц, CDCl3) δ 8.13 (s, 2Н), 4.69 (d, 2Н, J=13.2 Гц,), 4.22 (t, 2Н, J=6.8 Гц), 2.98 (s, 3H), 2.84 (t, 2Н, J=13.2 Гц), 2.45 (q, 2Н, J=7.6 Гц), 1.82 (m, 4Н), 1.55 (m, 2Н), 1.37 (m, 2Н), 1.20 (t, 3H, J=7.6 Гц)
(Стадия 6-5) Получение 3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазола (Препаративный пример 6)
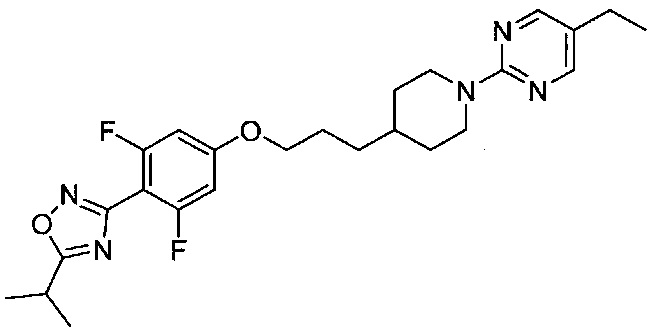
3,5-Дифтор-4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенол, синтезированный на описанной выше стадии 6-2 (10 г, 4,1 ммоль) растворяли в N,N-диметилформамиде (ДМФ, 15 мл), а затем к реакционному раствору добавляли 3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропилметансульфонат, синтезированный на описанной выше стадии 6-4 (1,2 г, 3,7 ммоль), и карбонат калия (K2CO3, 1,7 г, 12,4 ммоль). Реакционный раствор перемешивали при 65°C в течение 17 часов, а затем разбавляли водой и экстрагировали ЭА. Из органического растворителя удаляли влагу с помощью MgSO4, органический слой фильтровали и концентрировали при пониженном давлении, а затем остаток очищали колоночной хроматографией на силикагеле, в результате чего получили желаемую форму соединения, 3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазола, при выходе 73%.
1H ЯМР (400 МГц, CDCl3) δ 8.14 (s, 2Н), 6.54 (d, 2Н, J=9.6 Гц), 4.67 (d, 2Н, J=13.2 Гц), 3.96 (t, 2Н, J=6.6 Гц), 3.30 (m, 1Н), 2.83 (m, 2Н), 2.43 (q, 2Н, J=7.4 Гц), 1.83 (m, 2Н), 1.77 (m, 2Н), 1.52 (m, 1Н), 1.44 (d, 6Н, J=7.2 Гц), 1.39 (m, 2Н), 1.21 (m, 2Н), 1.16 (t, 3H, J=7.4 Гц)
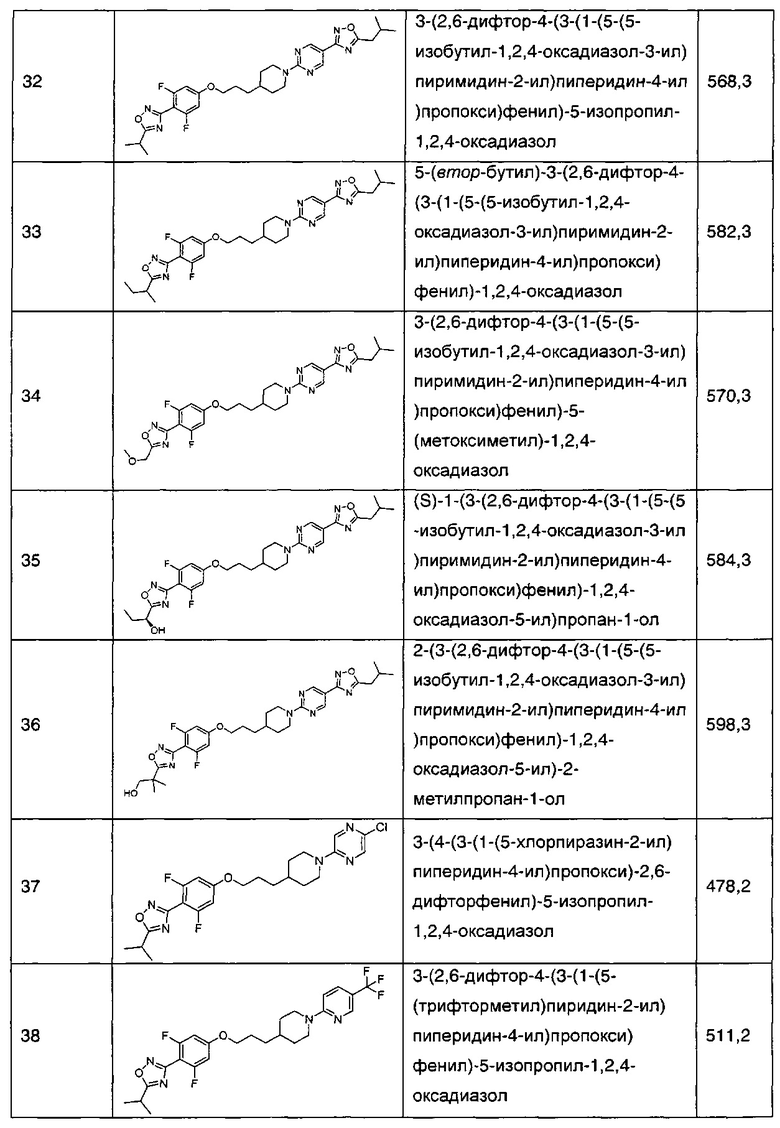
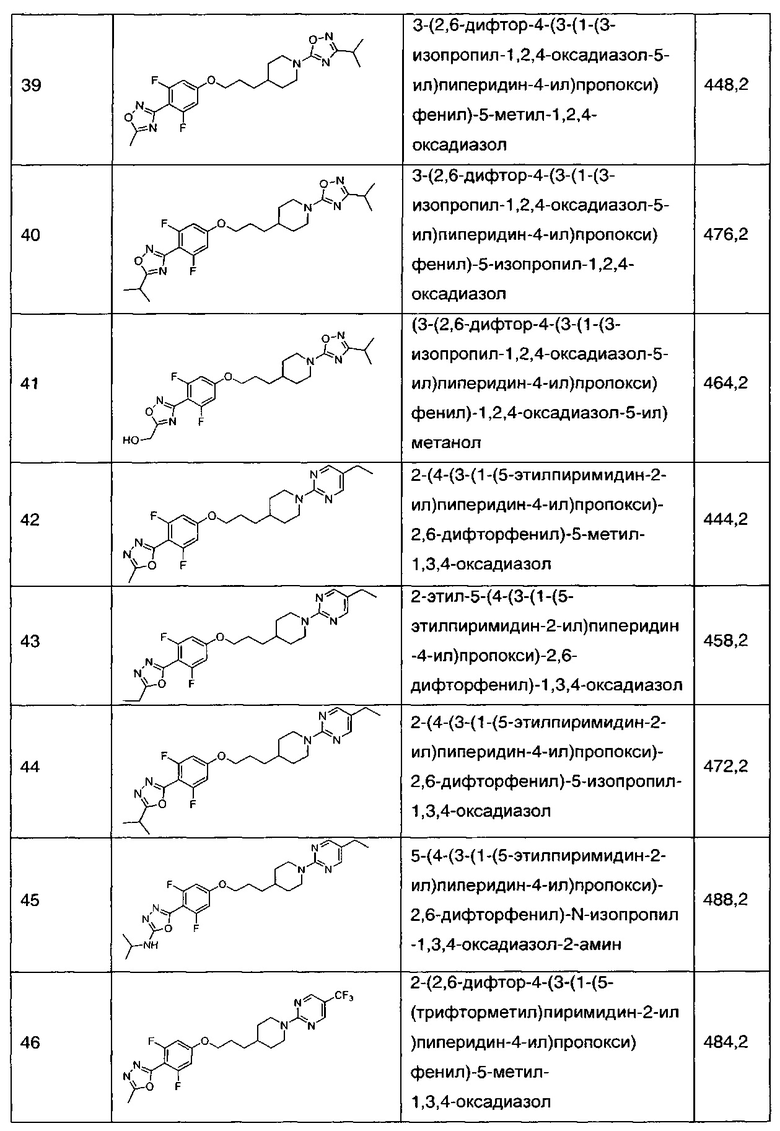
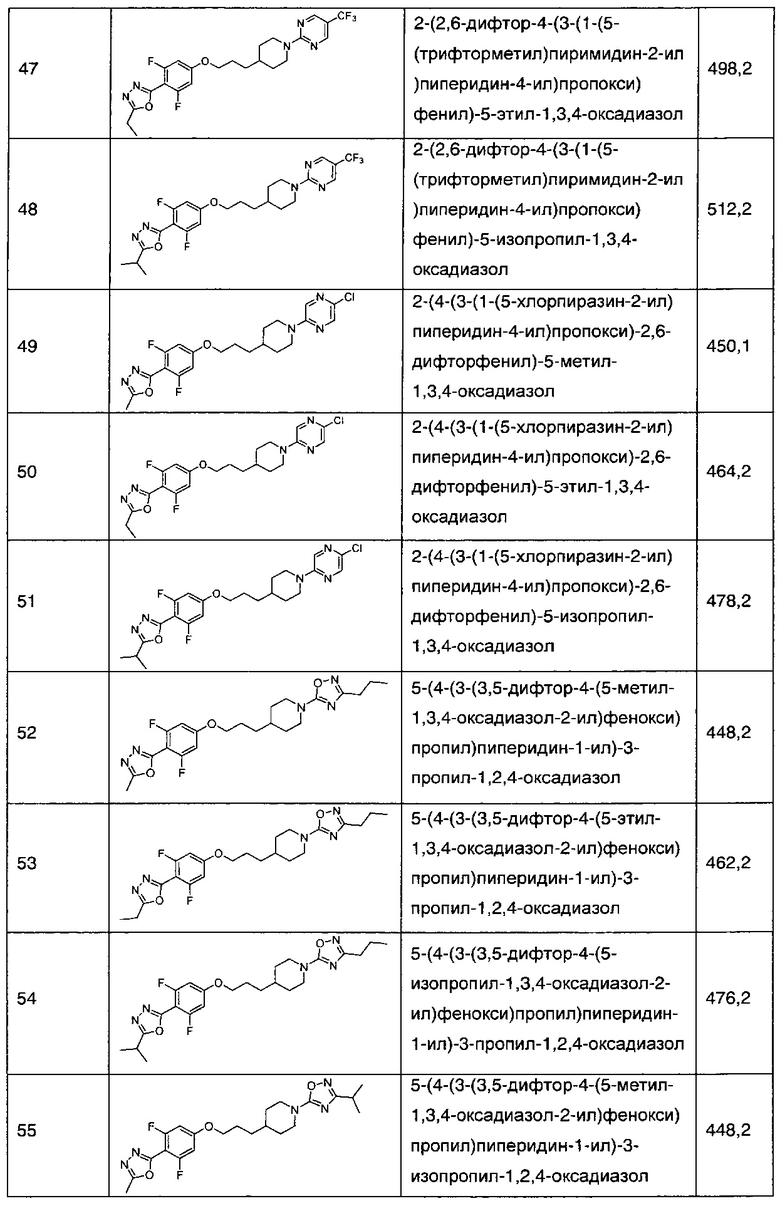
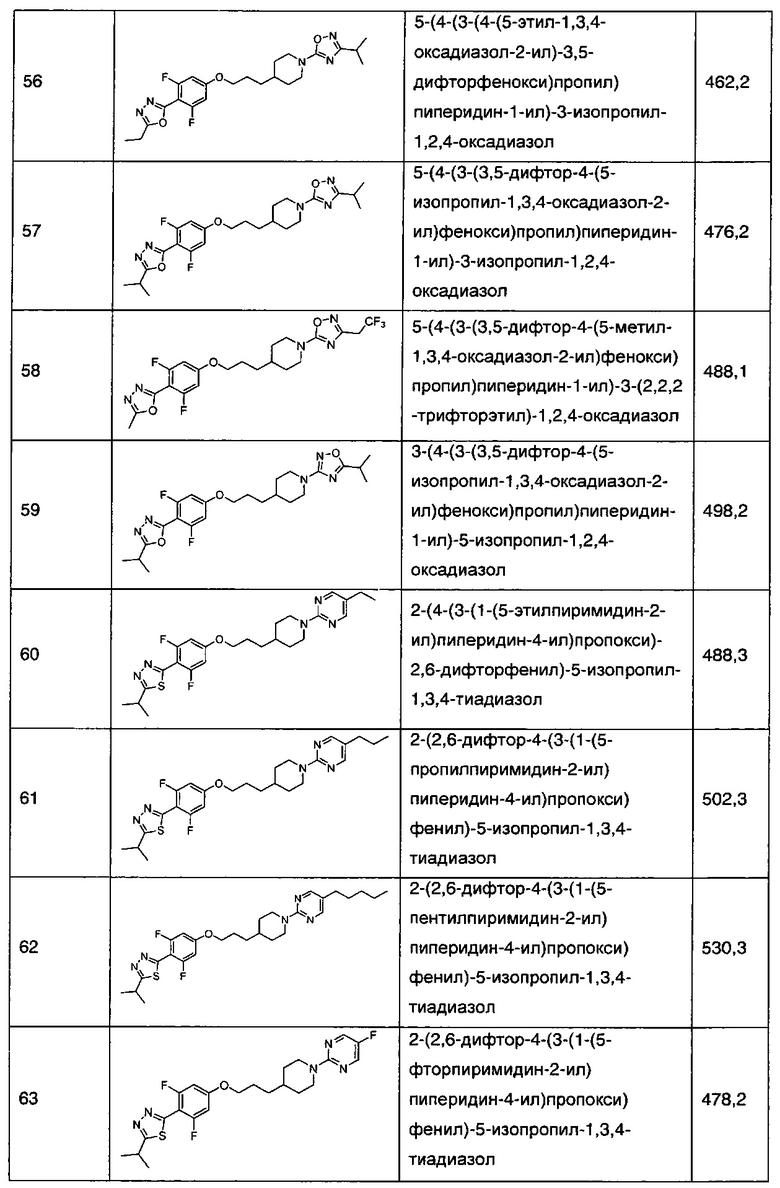
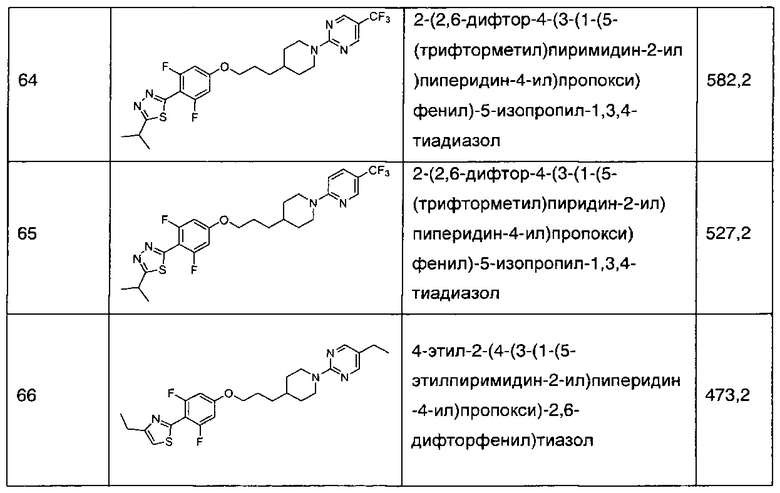
В соответствии со способами получения приведенных выше примеров реагент, соответствующий каждому заместителю каждого примера, использовали для получения соединений примеров 1-67 в приведенной ниже таблице 1.



<Экспериментальный пример 1> Анализ активации GPR119 человека
GPR119 человека в течение опредленного времени экспрессировали в клетках, в связи с этим определяли количество циклического аденозин-3',5'-монофосфата (сАМР), увеличивающееся при активации GPR119 посредством соединения по настоящему изобретению, с использованием препарата от Cysbio методом гомогенной флуоресценции с временным разрешением (HTRF, homogeneous time resolved fluorescence), и такое количественное определение использовали для обозначения эффективности активации GPR119.
Осуществляли гиперэкспрессию экспрессионного вектора GPR119 человека (Origene) в эпителиальных клетках почки хомяка (HEK293) (АТСС), и клетки стабилизировали в течение 48 часов. Клетки предварительно обрабатывали в течение 10 минут раствором 11,1 мМ глюкозы, 0,1% бычьего сывороточного альбумина и 0,5 мМ IBMX (3-изобутил-1-метилксантина), представляющего собой ингибитор фосфодиэстеразы, добавленным к буферному раствору KRBH (бикарбонатному раствору Кребса-Рингера с HEPES; Hou ZQ. et al., Mol Cell Endocrinol, 2008 (291): 71-78). Затем клетки обрабатывали тем же раствором, содержащим лекарственное средство, в течение 60 минут, затем супернатант удаляли и проводили количественное определение увеличения концентрации сАМР в клетках с использованием набора Cysbio сАМР HiRange.
Что касается максимальной эффективности тестируемых соединений, проводили многократную оценку концентраций соединений по настоящему изобретению, оценивая, таким образом, относительный уровень активации (%) от максимального эффекта олеоилэтаноламида (OEA), представляющего собой эндогенный лиганд GPR119.
Результаты представлены в таблице 2, из которой видно, что 67 соединений по примерам обладают превосходными активностями, при этом максимальные активности по меньшей мере эквивалентны ОЕА и составляют от 1 нМ до 10 нМ.

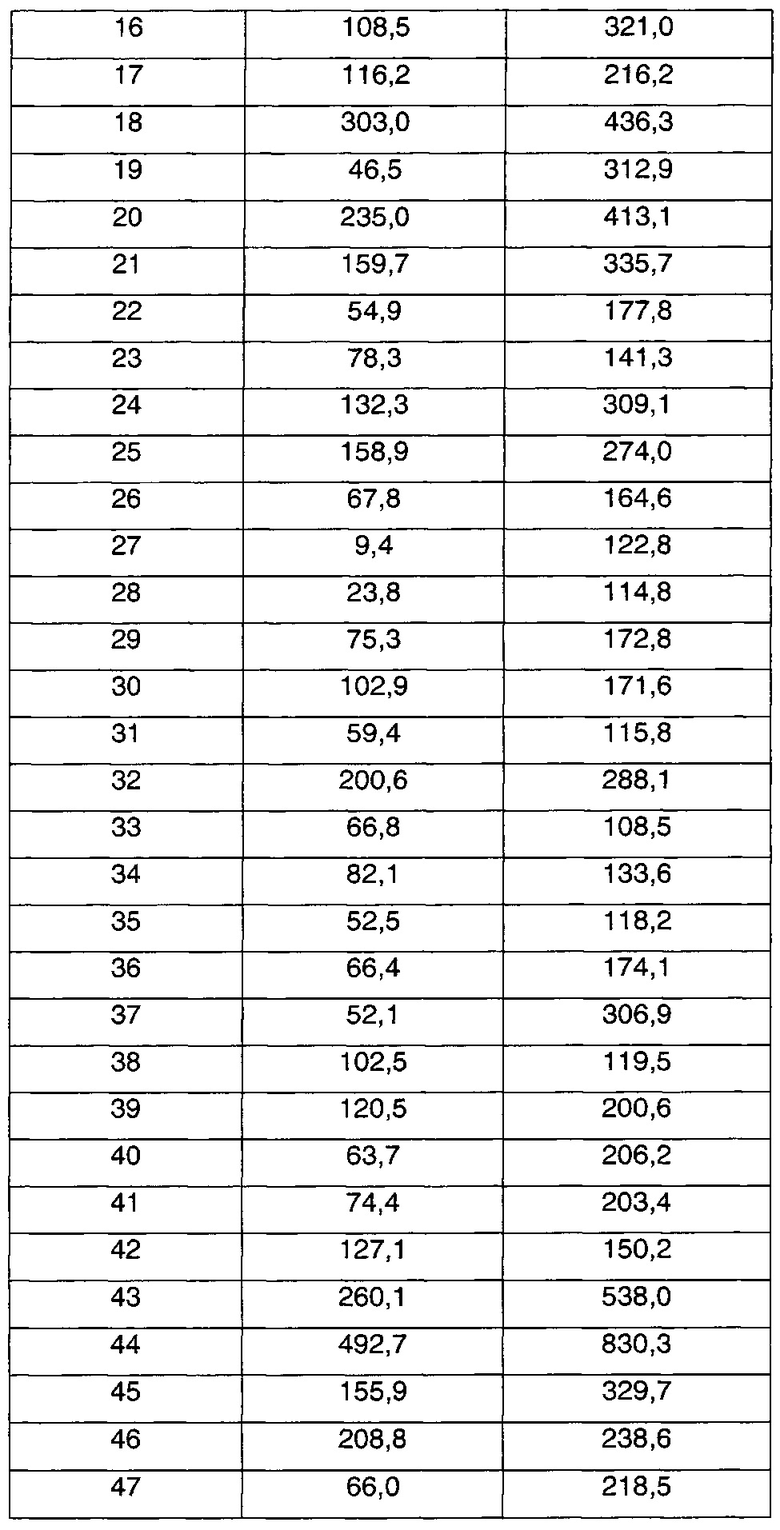
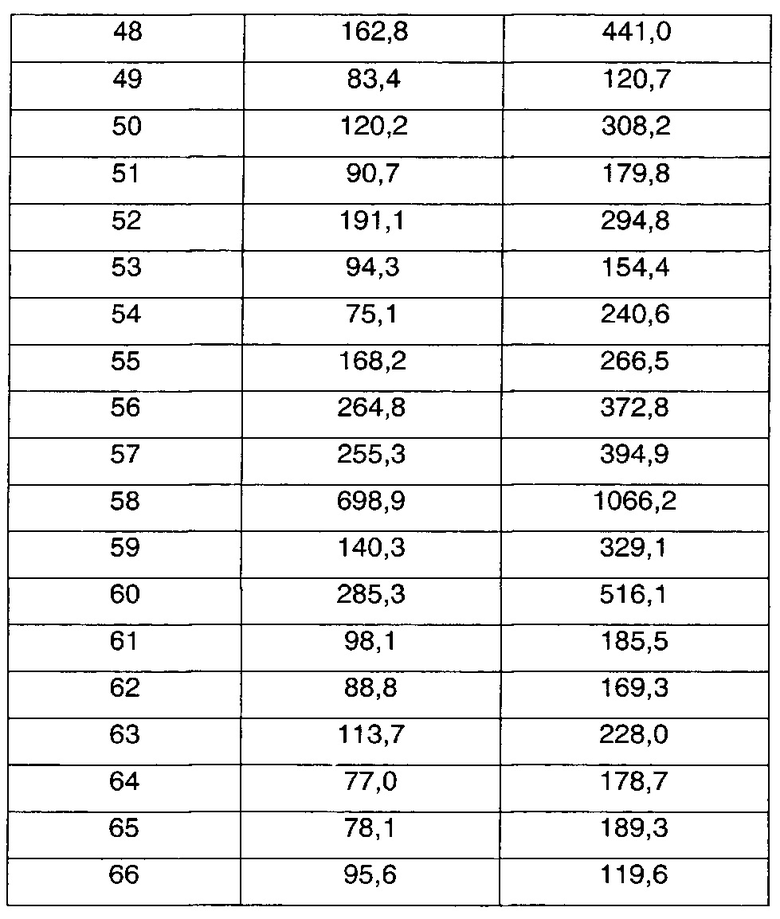
<Экспериментальный пример 2> Анализ эффекта улучшения толерантности к глюкозе у мышей
В качестве одного из индикаторов антидиабетического эффекта, проводили оценку эффекта улучшения толерантности к глюкозе описанных выше соединений у самцов лабораторных мышей 7-недельного возраста (мыши C57BL/6) в качестве эффекта улучшения способности к постпрандиальному гликемическому контролю.
Лабораторные мыши не получали пищу, начиная за день до эксперимента, в течение 16-17 часов. Соединение по настоящему изобретению вводили перорально за 30 минут до введения глюкозы, и через 30 минут перорально вводили раствор глюкозы (2 г/кг/10 мл). Лекарственное средство готовили путем суспендирования в 10% растворе гелуцира (Gelucire). В моменты времени, непосредственно предшествующие введению лекарственного средства, непосредственно предшествующие введению раствора глюкозы, а также через 15 минут, 30 минут, 60 минут, 90 минут и 120 минут после введения глюкозы измеряли концентрации глюкозы в цельной крови из хвостовой вены, используя глюкометр для крови (AccuChek Active, Roche Diagnostics), и вычисляли площадь под кривой концентрации глюкозы в крови в зависимости от времени. Из вычисленной площади под кривой концентрации глюкозы в крови вычитали площадь под кривой концентрации глюкозы в крови отрицательной контрольной группы, которой не вводили раствор глюкозы, и активность, ингибирующую повышение концентрации глюкозы в крови, относительно контрольной группы, которой вводили только 10% раствор гелуцира и раствор глюкозы, вычисляли в процентном отношении, посредством чего оценивали эффективность лекарственного средства по улучшению толерантности к глюкозе.
Результаты представлены в таблице 3, в которой эффект улучшения толерантности к глюкозе, представленный при дозе 10 мг/кг, приведен путем классификации на три группы, менее 30%, от 30% до 40% и более 40%. Статистически значимое действие по улучшению толерантности к глюкозе было идентифицировано для 22 соединений, на которых был проведен эксперимент, и среди этих соединений у 16 соединений была представлена превосходная активность in vivo 30% ингибирующей дозы 10 мг/кг или менее. В приведенной ниже таблице 3 A представляет ингибирующую активность более 40%, B представляет ингибирующую активность от 30% до 40%, и C представляет ингибирующую активность менее 30%.
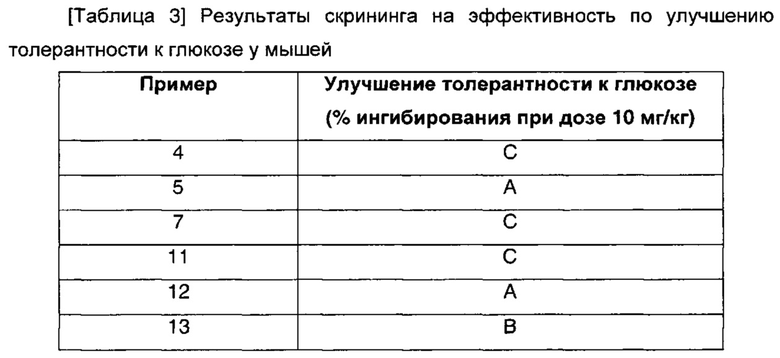
Как показано в приведенных выше таблицах 2 и 3, было подтверждено, что новые соединения, синтезированные в примерах 1-67, их изомеры или их фармацевтически приемлемые соли обладают агонистической активностью в отношении GPR119. Кроме того, было подтвержден превосходный эффект улучшения толерантности к глюкозе для многих соединений примеров, на которых проводили эксперимент. Соответственно, ожидают, что указанные выше соединения по примерам обладают высоким терапевтическим эффектом или профилактическим эффектом в отношении метаболических заболеваний, таких как ожирение, диабет, гипертензия, сердечно-сосудистые заболевания, гемостатическое расстройство, дислипидемия и тому подобное.
Настоящее изобретение подробно описано в конкретных разделах, и очевидно, что такой конкретный метод является только предпочтительным иллюстративным воплощением изобретения для специалиста в данной области техники без ограничения объема настоящего изобретения. Таким образом, объем настоящего изобретения по существу определен прилагаемой формулой изобретения и ее эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ НЕАЛКОГОЛЬНОЙ ЖИРОВОЙ ИНФИЛЬТРАЦИИ ПЕЧЕНИ, СОДЕРЖАЩАЯ ЛИГАНД GPR119 В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2019 |
|
RU2768943C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ НЕАЛКОГОЛЬНОГО СТЕАТОГЕПАТИТА | 2021 |
|
RU2803733C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ ДИАБЕТА И СВЯЗАННЫХ С НИМ МЕТАБОЛИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2021 |
|
RU2809286C1 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| КОНДЕНСИРОВАННЫЕ КОЛЬЦЕВЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TRK | 2015 |
|
RU2708674C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ GPR119 | 2008 |
|
RU2443699C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2814288C1 |
| Гетероциклическое соединение в качестве ингибитора протеинкиназы | 2018 |
|
RU2783723C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ВЕЩЕСТВА - АГОНИСТЫ GPR119 | 2017 |
|
RU2734500C2 |
| СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ ОКСАДИАЗОЛ, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2789456C2 |
Изобретение относится к новому соединению формулы 1 или его фармацевтически приемлемой соли
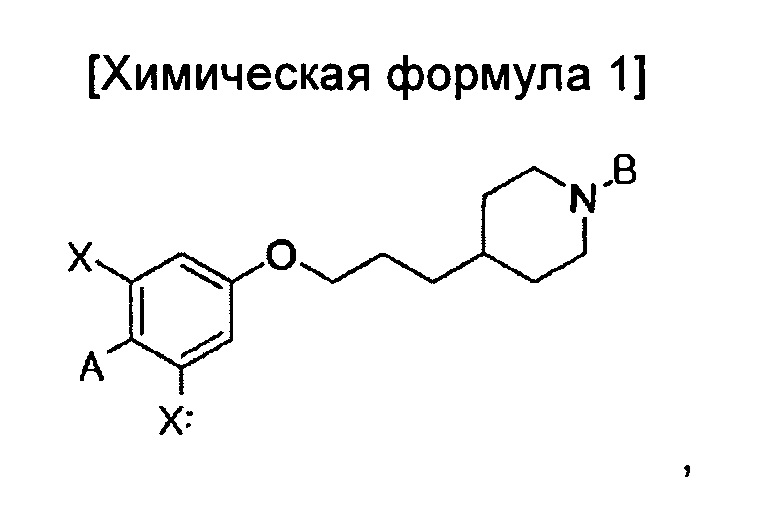
где А представляет собой оксадиазол, дигидрооксазол, тиазол или тиадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила и С1-С6 спирта, где прямоцепочечная или разветвленная С1-С6 алкильная или С1-С6 спиртовая группа возможно замещена водородом, галогеном или С1-С6 алкоксигруппой; В представляет собой пиридин, пиримидин, пиразин или оксадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечной или разветвленной С1-С6 алкильной, С1-С6 спиртовой, С1-С6 алкокси и оксадиазольной групп, где прямоцепочечная или разветвленная С1-С6 алкильная, С1-С6 спиртовая, С1-С6 алкокси или оксадиазольная группа возможно замещена водородом, галогеном, С1-С6 алкильной группой или С1-С6 алкоксигруппой; и X независимо представляет собой F, Cl, Br или I, обладающему агонистической активностью в отношении GPR119. 3 н. и 16 з.п. ф-лы., 3 табл., 6 пр.
1. Соединение, представленное приведенной ниже химической формулой 1, его изомер или его фармацевтически приемлемая соль
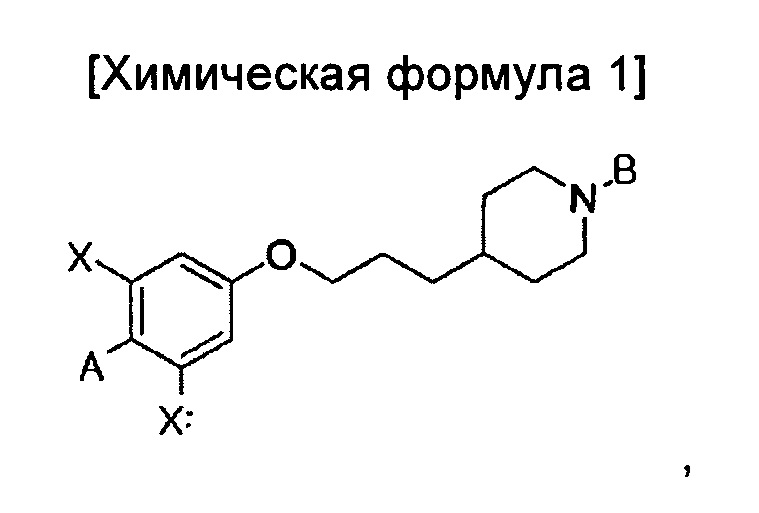
где
А представляет собой оксадиазол, дигидрооксазол, тиазол или тиадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила и С1-С6 спирта, где прямоцепочечная или разветвленная С1-С6 алкильная или С1-С6 спиртовая группа возможно замещена водородом, галогеном или С1-С6 алкоксигруппой;
В представляет собой пиридин, пиримидин, пиразин или оксадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечной или разветвленной С1-С6 алкильной, С1-С6 спиртовой, С1-С6 алкокси и оксадиазольной групп, где прямоцепочечная или разветвленная С1-С6 алкильная, С1-С6 спиртовая, С1-С6 алкокси или оксадиазольная группа возможно замещена водородом, галогеном, С1-С6 алкильной группой или С1-С6 алкоксигруппой; и
X независимо представляет собой F, Cl, Br или I.
2. Соединение по п. 1, его изомер или его фармацевтически приемлемая соль, где в химической формуле 1 А представляет собой дигидрооксазол, тиазол или тиадиазол; и В представляет собой пиримидин, пиридин или оксадиазол.
3. Соединение по п. 1, его изомер или его фармацевтически приемлемая соль, где А представляет собой оксадиазол; и В представляет собой пиридин, пиримидин, пиразин или оксадиазол.
4. Соединение по п. 1, его изомер или его фармацевтически приемлемая соль,
где А представляет собой  или
или  , и
, и
R1-R3, R5 и R6 независимо представляют собой один или более чем один заместитель, выбранный из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила и С1-С6 спирта, где алкильная или спиртовая группа возможно замещена водородом, галогеном или С1-С6 алкоксигруппой.
5. Соединение по п. 1, его изомер или его фармацевтически приемлемая соль, где В представляет собой 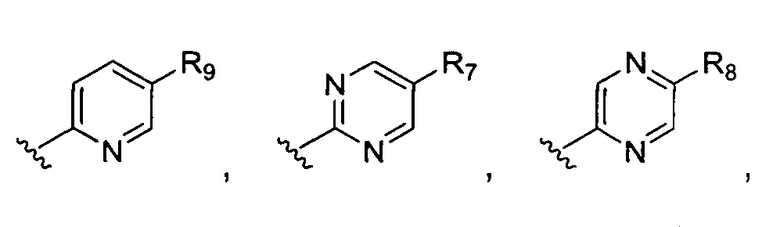
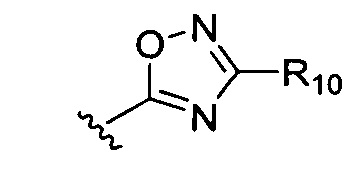 или
или 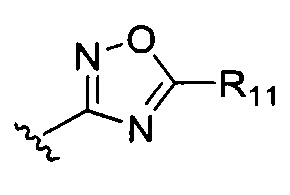 , и
, и
R7-R11 возможно замещены одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечной или разветвленной С1-С6 алкильной, С1-С6 спиртовой, С1-С6 алкокси и оксадиазольной групп, где прямоцепочечная или разветвленная С1-С6 алкильная, С1-С6 спиртовая, С1-С6 алкокси или оксадиазольная группа возможно замещена водородом, галогеном, С1-С6 алкильной группой или С1-С6 алкоксигруппой.
6. Соединение по п. 1, его изомер или его фармацевтически приемлемая соль, где X представляет собой фтор; или его изомер.
7. Соединение по п. 1, его изомер или его фармацевтически приемлемая соль, где А представляет собой оксадиазол, замещенный прямоцепочечным или разветвленным С1-С6 алкилом; В представляет собой пиримидин, замещенный прямоцепочечным или разветвленным С1-С6 алкилом; и X представляет собой F.
8. Соединение по п. 1, его изомер или его фармацевтически приемлемая соль, выбранное из группы, состоящей из следующих соединений:
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4,5-дигидрооксазол,
(R)-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4-метил-4,5-дигидрооксазол,
(S)-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4-метил-4,5-дигидрооксазол,
(S)-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-4,5-дигидрооксазол,
(R)-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-4,5-дигидрооксазол,
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5,5-диметил-4,5-дигидрооксазол,
(R)-(2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4,5-дигидрооксазол-5-ил)метанол,
(S)-(2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-4,5-дигидрооксазол-5-ил)метанол,
(R)-3-(2-(4-(3-(3,5-дифтор-4-(5-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)пиримидин-5-ил)-5-изобутил-1,2,4-оксадиазол,
(R)-5-(4-(3-(3,5-дифтор-4-(4-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазол,
(S)-5-(4-(3-(3,5-дифтор-4-(5-метил-4,5-дигидрооксазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазол,
5-(4-(3-(4-(5,5-диметил-4,5-дигидрооксазол-2-ил)-3,5-дифторфенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазол,
3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,2,4-оксадиазол,
3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-пропил-1,2,4-оксадиазол,
3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазол,
5-(трет-бутил)-3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол,
(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)метанол,
2-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)этан-1-ол,
(S)-1-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)пропан-1-ол,
(R)-1-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)пропан-2-ол,
(S)-1-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)пропан-2-ол,
2-(3-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,2,4-оксадиазол-5-ил)-2-метилпропан-1-ол,
3-(2,6-дифтор-4-(3-(1-(5-пропилпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-пентилпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-метоксипиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-изопропоксипиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазол,
3-(4-(3-(1-(5-хлорпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазол,
3-(4-(3-(1-(5-бромпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-этил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазол,
5-(втор-бутил)-3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-(метоксиметил)-1,2,4-оксадиазол,
(S)-1-(3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-1,2,4-оксадиазол-5-ил)пропан-1-ол,
2-(3-(2,6-дифтор-4-(3-(1-(5-(5-изобутил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-1,2,4-оксадиазол-5-ил)-2-метилпропан-1-ол,
3-(4-(3-(1-(5-хлорпиразин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиридин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,2,4-оксадиазол,
3-(2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,2,4-оксадиазол,
(3-(2,6-дифтор-4-(3-(1-(3-изопропил-1,2,4-оксадиазол-5-ил)пиперидин-4-ил)пропокси)фенил)-1,2,4-оксадиазол-5-ил)метанол,
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,3,4-оксадиазол,
2-этил-5-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-1,3,4-оксадиазол,
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-оксадиазол,
5-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-N-изопропил-1,3,4-оксадиазол-2-амин,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-метил-1,3,4-оксадиазол,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-этил-1,3,4-оксадиазол,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-оксадиазол,
2-(4-(3-(1-(5-хлорпиразин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-метил-1,3,4-оксадиазол,
2-(4-(3-(1-(5-хлорпиразин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-этил-1,3,4-оксадиазол,
2-(4-(3-(1-(5-хлорпиразин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-оксадиазол,
5-(4-(3-(3,5-дифтор-4-(5-метил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-пропил-1,2,4-оксадиазол,
5-(4-(3-(3,5-дифтор-4-(5-этил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-пропил-1,2,4-оксадиазол,
5-(4-(3-(3,5-дифтор-4-(5-изопропил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-пропил-1,2,4-оксадиазол,
5-(4-(3-(3,5-дифтор-4-(5-метил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазол,
5-(4-(3-(4-(5-этил-1,3,4-оксадиазол-2-ил)-3,5-дифторфенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазол,
5-(4-(3-(3,5-дифтор-4-(5-изопропил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазол,
5-(4-(3-(3,5-дифтор-4-(5-метил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-3-(2,2,2-трифторэтил)-1,2,4-оксадиазол,
3-(4-(3-(3,5-дифтор-4-(5-изопропил-1,3,4-оксадиазол-2-ил)фенокси)пропил)пиперидин-1-ил)-5-изопропил-1,2,4-оксадиазол,
2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)-5-изопропил-1,3,4-тиадиазол,
2-(2,6-дифтор-4-(3-(1-(5-пропилпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазол,
2-(2,6-дифтор-4-(3-(1-(5-пентилпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазол,
2-(2,6-дифтор-4-(3-(1-(5-фторпиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазол,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиримидин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазол,
2-(2,6-дифтор-4-(3-(1-(5-(трифторметил)пиридин-2-ил)пиперидин-4-ил)пропокси)фенил)-5-изопропил-1,3,4-тиадиазол и
4-этил-2-(4-(3-(1-(5-этилпиримидин-2-ил)пиперидин-4-ил)пропокси)-2,6-дифторфенил)тиазол.
9. Фармацевтическая композиция, обладающая агонистической активностью в отношении GPR119, содержащая соединение по любому из пп. 1-8, его изомер или его фармацевтически приемлемую соль в качестве эффективного компонента в эффективном количестве.
10. Фармацевтическая композиция по п. 9, предназначенная для лечения или предотвращения метаболического заболевания.
11. Фармацевтическая композиция по п. 10, где метаболическое заболевание выбрано из группы, состоящей из диабета, ожирения, гипертензии, сердечно-сосудистого заболевания, расстройства гемостаза и дислипидемии.
12. Способ получения соединения, представленного следующей химической формулой 1, включающий:
введение группы В в азотную группу пиперидина соединения приведенной ниже химической формулы 2 с получением соединения приведенной ниже химической формулы 4; и
введение соединения приведенной ниже химической формулы 12 в гидроксильную группу соединения химической формулы 4
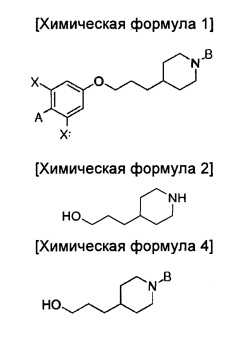
где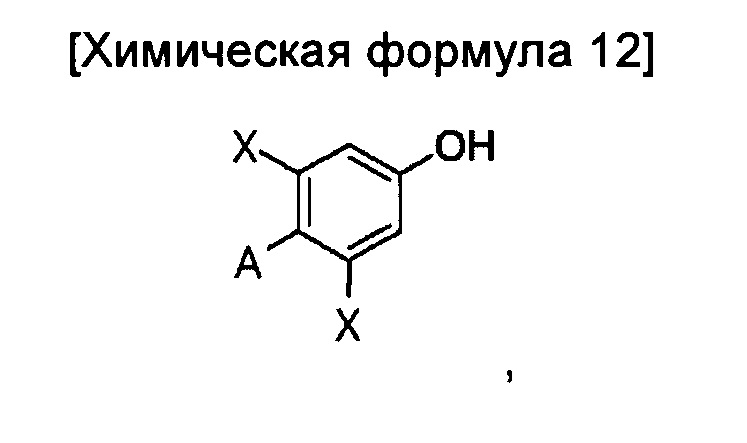
А представляет собой оксадиазол, дигидрооксазол, тиазол или тиадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила и С1-С6 спирта, где прямоцепочечная или разветвленная С1-С6 алкильная или С1-С6 спиртовая группа возможно замещена водородом, галогеном или С1-С6 алкоксигруппой;
В представляет собой пиридин, пиримидин, пиразин или оксадиазол, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из водорода, галогена, прямоцепочечного или разветвленного С1-С6 алкила, С1-С6 спирта, С1-С6 алкокси и оксадиазола, где прямоцепочечная или разветвленная С1-С6 алкильная, С1-С6 спиртовая, С1-С6 алкокси или оксадиазольная группа возможно замещена водородом, галогеном, С1-С6 алкильной группой или С1-С6 алкоксигруппой; и
X независимо представляет собой F, Cl, Br или I.
13. Способ по п. 12, где введение соединения химической формулы 12 в гидроксильную группу соединения химической формулы 4 включает:
взаимодействие соединения химической формулы 4 и соединения приведенной ниже химической формулы 12а; и преобразование А' в А

где
А' представляет собой цианогруппу, карбоксильную группу, сложноэфирную группу, кетоновую группу или галоген.
14. Способ по п. 13, где взаимодействие соединения химической формулы 4 и соединения химической формулы 12а включает:
введение метансульфонильной группы в гидроксильную группу соединения химической формулы 4; и
взаимодействие соединения химической формулы 4, имеющего введенную в него метансульфонильную группу, с соединением химической формулы 12а.
15. Способ по п. 13, где преобразование А' в А включает:
когда А' представляет собой карбоксильную группу или сложноэфирную группу, окисление А' до карбоновой кислоты;
взаимодействие карбоновой кислоты и аминоэтанола приведенной ниже химической формулы 13 с введением соединения приведенной ниже химической формулы 14 в А' химической формулы 12а
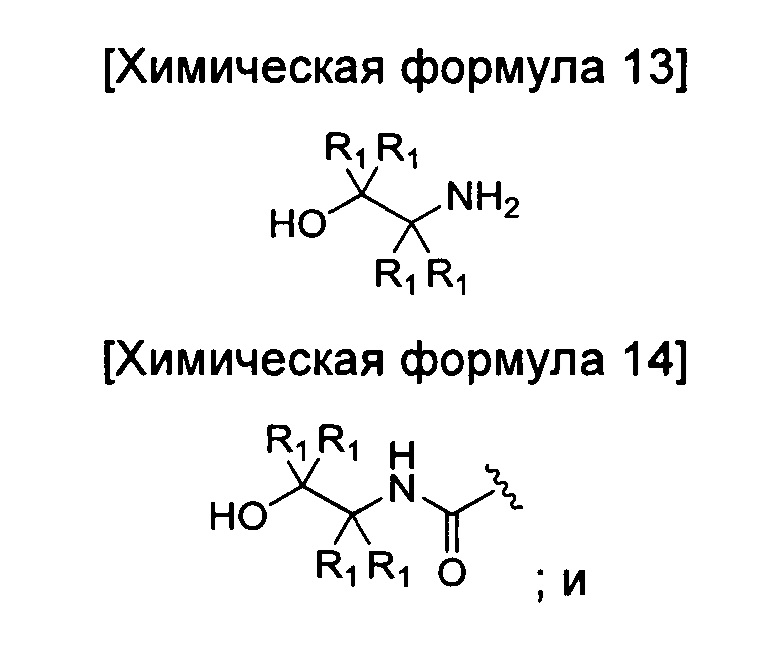
замыкание кольца соединения, полученного на предшествующей стадии,
с образованием соединения, где А представляет собой  , где R1
, где R1
независимо представляет собой водород или гидрокси-замещенный или незамещенный С1-С6 алкил.
16. Способ по п. 13, где преобразование А' в А включает:
когда А' представляет собой цианогруппу, взаимодействие А' с гидроксиламином с введением соединения приведенной ниже химической формулы 15 в А' химической формулы 12а
[Химическая формула 15]

взаимодействие соединения, полученного на предшествующей стадии, и соединения приведенной ниже химической формулы 16 с образованием
соединения, где А представляет собой 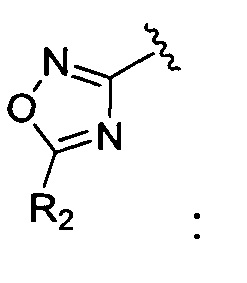 где
где 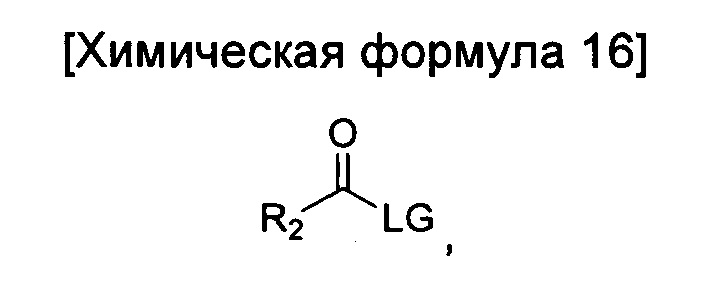
R2 представляет собой С1-С6 алкил, возможно замещенный водородом или С1-С4 алкокси, или С1-С6 спирт; и
LG представляет собой уходящую группу.
17. Способ по п. 13, где преобразование А' в А включает:
когда А' представляет собой карбоксильную группу или сложноэфирную группу, окисление А' до карбоновой кислоты;
взаимодействие карбоновой кислоты и гидразина с введением соединения приведенной ниже химической формулы 17 в А' химической формулы 12а

взаимодействие соединения, полученного на предшествующей стадии, с соединением приведенной ниже химической формулы 18 или 19 с
образованием соединения, где А представляет собой 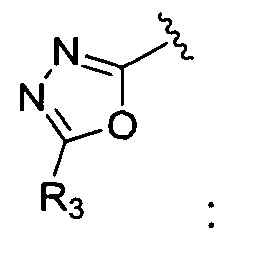

где
R3 представляет собой С1-С6 алкил или С1-С4 алкиламин.
18. Способ по п. 13, где преобразование А' в А включает:
окисление А' с получением карбоновой кислоты, если А' представляет собой карбоксильную группу или сложноэфирную группу;
взаимодействие карбоновой кислоты с гидразином или гидразидом с введением соединения приведенной ниже химической формулы 20 в А' химической формулы 12а
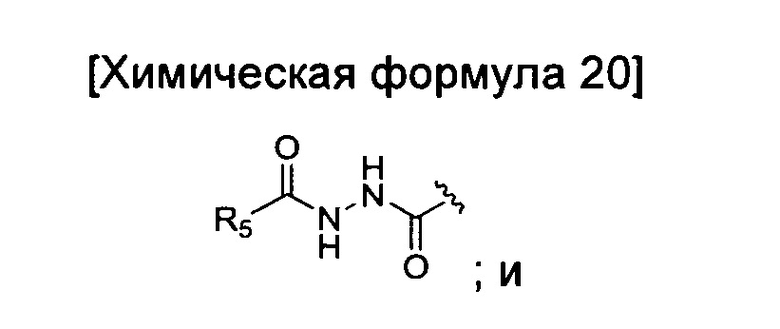
взаимодействие соединения, полученного на предшествующей стадии, с соединением приведенной ниже химической формулы 21 с образованием
соединения, где А представляет собой 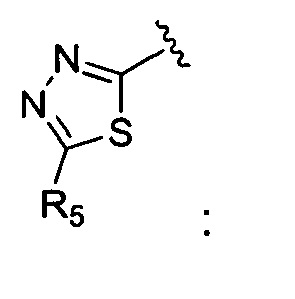
где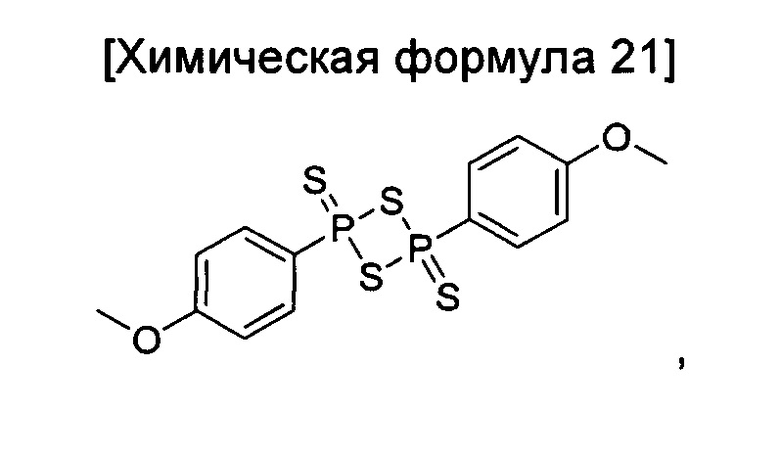
R5 представляет собой С1-С6 алкил.
19. Способ по п. 13, где преобразование А' в А включает:
когда А' представляет собой карбоксильную группу или сложноэфирную группу,
(а) окисление А' с получением карбоновой кислоты;
(б) взаимодействие карбоновой кислоты, полученной на стадии (а), с тионилхлоридом с введением структуры химической формулы 22 в А' химической формулы 12а;
(в) преобразование структуры химической формулы 22 соединения, полученного на стадии (б), в амидную структуру химической формулы 23;
(г) взаимодействие соединения, полученного на стадии (в), с соединением химической формулы 21 с получением соединения, имеющего тиоамидную структуру химической формулы 24; и
(д) взаимодействие соединения, полученного на стадии (г), с соединением химической формулы 25 с образованием соединения, где А
представляет собой 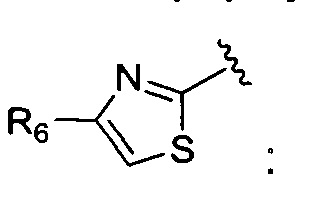

где
R6 представляет собой С1-С6 алкил.
| WO 2010004347 A1, 14.01.2010 | |||
| WO 2013062835 A1, 02.05.2013 | |||
| WO 2008081204 A1, 10.07.2008 | |||
| WO 2008081205 A1, 10.07.2008 | |||
| WO 2007003962 A2, 11.01.2007 | |||
| WO 2004041813 A1, 21.05.2004 | |||
| ПРОИЗВОДНЫЕ ПИРИДАЗИНАМИНОВ, А ТАКЖЕ ИХ СОЛИ ИЛИ СТЕРЕОХИМИЧЕСКИЕ ИЗОМЕРНЫЕ ФОРМЫ, АНТИВИРУСНАЯ КОМПОЗИЦИЯ И ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 1990 |
|
RU2051149C1 |

