Область изобретения
Настоящее изобретение относится к новым кристаллам 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(бутоксикарбонил)бензамидина, разработанным в качестве противогрибкового средства.
Предшествующий уровень техники
4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]-фенокси}пропил)-1-пиперидинил]пропокси}-N'-(бутоксикарбонил)бензамидин (называемый в описании далее "соединение А") обладает сильной противогрибковой активностью, включая устойчивые к азолу грибы, и является превосходным для пероральной абсорбции, слабо взаимодействует с другими лекарствами и имеет высокую безопасность, и является полезным для противогрибкового средства (патентный документ 1).
Кристаллы соединения А, получаемые способами, упомянутыми в патентном документе 1, называют "кристаллами типа II".
[Патентный документ 1] Международная патентная публикация WO2007/074868, брошюра.
Сущность изобретения
Проблемы, которые должны быть решены изобретением
Кристалл соединения А, имеющий превосходные свойства в качестве лекарственного вещества, в особенности удобен в обращении, является весьма ожидаемым.
Способ решения проблемы
При этих обстоятельствах авторы настоящего изобретения провели интенсивное исследование и, как результат, нашли, что кристаллы соединения А, имеющие пики в области 5,8, 18,2, 20,9 и 24,7° угла дифракции 2θ на порошковой рентгенограмме (называемые в описании далее "кристаллы типа II"); кристаллы соединения А, имеющие пики в области 8,7, 12,0, 22,2 и 24,3° угла дифракции 2θ на порошковой рентгенограмме (называемые в описании далее "кристаллы типа III"); кристаллы соединения А, имеющие пики в области 9,8 и 23,5 угла дифракции 2θ на порошковой рентгенограмме (называемые в описании далее "кристаллы типа IV"), являются превосходными в качестве лекарственного вещества, так как: (1) после утряски их плотность является высокой, (2) их трудно зарядить электричеством; (3) они удобны в обращении; (4) они имеют хорошую формуемость при прессовании; (5) они трудно слипаются и (6) возможно их промышленное производство, таким образом, заявители реализовали изобретение.
Эффект изобретения
Кристаллы по настоящему изобретению: (1) имеют высокую плотность после утряски, (2) они трудно электролизуются, (3) они удобны в обращении, (4) имеют хорошую формуемость при прессовании, (5) они трудно слипаются, и (6) промышленное производство которых возможно, поэтому они являются пригодными в качестве лекарственного вещества.
Лучший способ осуществления изобретения
Настоящее изобретение описано подробно далее.
Настоящее изобретение относится к кристаллам типа II, имеющим пики в областях 5,8, 18,2, 20,9 и 24,7° угла дифракции 2θ на порошковой рентгенограмме, кристаллам типа III, имеющим пики в областях 8,7, 12,0, 22,2 и 24,3° угла дифракции 2θ на порошковой рентгенограмме и кристаллам типа IV, имеющим пики в областях 9,8 и 23,5° угла дифракции 2θ на порошковой рентгенограмме.
Эти кристаллы по настоящему изобретению являются совершенно неизвестными до настоящего времени, совсем не описаны в патентном документе 1 и являются новыми кристаллами. Кроме того, характеристические пики порошковой рентгенограммы могут изменяться в зависимости от условий измерения. Поэтому пики порошковой рентгенограммы соединений по настоящему изобретению не интерпретируются строго.
Объясняется способ получения соединений по настоящему изобретению.
Кристаллы типа II, например, могут быть получены способом изготовления, показанным, как изложено ниже.
Способ изготовления 1
Кристаллы типа II могут быть получены путем суспендирования и перемешивания кристаллов типа I в растворителях.
В качестве растворителей, используемых при этом способе получения, используют кетоны, такие как метилэтилкетон и метилизобутилкетон; спирты, такие как 2-пропанол и бутанол; сложные эфиры, такие как этилацетат и бутилацетат; простые эфиры, такие как 1,4-диоксан; алифатические углеводороды, такие как гептан и циклогексан; S-оксиды, такие как диметилсульфоксид; ароматические углеводороды, такие как толуол; нитрилы, такие как ацетонитрил; амиды, такие как N,N-диметилформамид и N-метилпирролидон; и вода. Эти растворители могут быть использованы в комбинации.
Количество используемого растворителя желательно составляет от 1 до 100 объемов (об./мас.) на единицу массы кристаллов типа I и, предпочтительно, от 5 до 10 объемов (об./мас.).
Температура перемешивания составляет желательно от 50 до 150°C и, предпочтительно, от 70 до 120°C.
Время перемешивания желательно составляет от 0,1 ч до 5 ч и, предпочтительно, от 0,5 ч до 3 ч.
Согласно способу, описанному выше, кристаллы типа II могут быть получены путем использования кристаллов типа III или кристаллов типа IV, оба раскрыты в описании далее, вместо кристаллов типа I.
Кристаллы типа III, например, могут быть получены способом, показанным, как изложено ниже.
Способ изготовления 2
Кристаллы типа III могут быть получены путем суспендирования и перемешивания кристаллов типа I в водных растворителях.
В качестве растворителей, используемых при этом получении, используют кетоны, такие как метилэтилкетон; спирты, такие как бутанол; сложные эфиры, такие как этилацетат; простые эфиры, такие как тетрагидрофуран; ароматические углеводороды, такие как толуол; и галогенированные углеводороды, такие как хлороформ. Эти растворители могут быть использованы в комбинации. Количество используемого водного растворителя желательно составляет от 1 до 100 объемов (об./мас.) на единицу массы кристаллов типа I и, предпочтительно, от 2 до 10 объемов (об./мас.).
Соотношение растворителя и воды желательно находится в интервале, в котором отношение (растворитель)/(вода) составляет от 99/1 до 30/70, и предпочтительно в интервале, в котором отношение (растворитель)/(вода) составляет от 90/10 до 50/50.
Температура перемешивания составляет желательно от 10 до 40°C и предпочтительно от 20 до 30°C.
Время перемешивания желательно составляет от 0,1 часа до 30 суток и предпочтительно от одного часа до 14 суток.
Согласно способу, описанному выше, кристаллы типа III могут быть получены путем использования кристаллов типа II вместо кристаллов типа I.
Способ изготовления 3
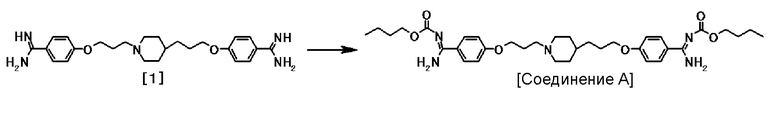
Кристаллы типа III могут быть получены кристаллизацией после того, как соединение формулы [1] прореагировало с реакционноспособным производным в присутствии или отсутствии основания.
(1) Соединение А может быть получено реакцией соединения формулы [1] с реакционноспособным производным в присутствии или отсутствии основания.
В качестве растворителей, используемых в этой реакции, используют, например, кетоны, такие как метилэтилкетон; сложные эфиры, такие как этилацетат; простые эфиры, такие как тетрагидрофуран; ароматические углеводороды, такие как толуол; галогенированные углеводороды, такие как хлороформ; амиды, такие как N,N-диметилформамид; и вода. Эти растворители могут быть использованы в комбинации.
В качестве реакционноспособных производных используют, например, бутилхлороформиат, бутил-4-нитрофенилкарбонат и бутил-1Н-имидазол-1-карбоксилат. Эти реакционноспособные производные могут быть использованы после получения in situ без выделения.
Для оснований, используемых в этой реакции, если желательно, используют, например, алкоксиды металлов, такие как метоксид натрия, этоксид натрия, трет-бутоксид калия и трет-бутоксид натрия; неорганические основания, такие как гидроксид натрия, гидроксид калия, кислый карбонат натрия, карбонат натрия, карбонат калия, гидрид натрия и гидрид калия; и органические основания, такие как триэтиламин, N,N-диизопропилэтиламин, 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU) и пиридин.
Количество реакционноспособного производного и основания может быть 2-100-кратным (в молях), желательно 2-10-кратным (в молях) в расчете на соединение формулы (1).
Эта реакция может быть проведена при температуре от -20 до 100°C, желательно от 20 до 80°C в течение времени от одной минуты до 24 часов.
Эта реакция может быть проведена предпочтительно при температуре от 20 до 80°C в течение времени от одной минуты до 7 часов.
(2) После реакции полученное соединение А экстрагируют из реакционной смеси обычными методами.
В качестве используемых экстрагирующих растворителей используют, например, кетоны, такие как метилэтилкетон; сложные эфиры, такие как этилацетат; простые эфиры, такие как тетрагидрофуран; ароматические углеводороды, такие как толуол; и галогенированные углеводороды, такие как хлороформ. Эти растворители могут быть использованы в комбинации.
Температура экстракции не является особо лимитированной, но желательно составляет от 50 до 80°C.
(3) Кристаллы типа III могут быть получены кристаллизацией после добавления затравки кристаллов типа III к экстрактному раствору.
Условием кристаллизации является желательно охлаждение от 50-80°C до 0-10°C в течение от 12 до 24 часов.
Кристаллы типа IV, например, могут быть получены способом, показанным, как изложено далее.
Способ изготовления 4
Кристаллы типа IV могут быть получены суспендированием и перемешиванием кристаллов типа II в растворителе.
В качестве растворителей, используемых при этом получении, используют кетоны, такие как ацетон.
Количество используемого растворителя желательно составляет от 1 до 100 объемов (об./мас.) на единицу массы кристаллов типа II, и предпочтительно от 2 до 30 объемов (об./мас.).
Температура перемешивания желательно составляет от 0 до 10°C.
Время перемешивания желательно составляет от 1 до 30 суток, и предпочтительно от 7 до 30 суток.
В случае, когда соединения по настоящему изобретению (кристаллы типа II, кристаллы типа III и кристаллы типа IV) используют в качестве лекарственного средства, они могут быть использованы отдельно или в смеси.
При использовании в качестве лекарственного средства, соединение по изобретению может быть, как правило, тщательно смешано с фармацевтическими вспомогательными веществами, такими как эксципиент, носитель и разбавитель, которые используют при приготовлении лекарственных средств, и может быть введено перорально или парентерально в форме таблеток, капсул, порошков, сиропов, гранул, пилюль, суспензий, эмульсий, растворов, порошковых препаратов, свечей, глазных капель, капель для носа, ушных капель, пластырей, мазей или инъекций согласно обычным способам. Кроме того, способ введения, дозировка и частота приема лекарственного средства могут быть должным образом выбраны в зависимости от возраста, массы тела и симптомов пациента. Обычно от 0,01 до 1,00 мг/кг соединения может быть введено перорально или парентерально (например, инъекцией, капельной инфузией или ректальным введением) взрослому за один прием или несколькими раздельными порциями в день.
Далее, полезность соединений по настоящему изобретению объяснена следующими испытаниями.
В качестве материала для испытаний были использованы соединения по настоящему изобретению (кристаллы типа II, кристаллы типа III и кристаллы типа IV) и кристаллы типа I.
Тестовый пример 1
Плотность после утряски
Испытуемые материалы, пропущенные через сито (18 меш), заполняли мерный цилиндр и измеряли массу W (г) засыпанных испытуемых материалов. Далее, мерный цилиндр, содержащий испытуемый материал, механически постукивали 180 раз с помощью прибора для измерения характеристик порошка (тестер порошка PT-E, Hosokawa Micron Corporation). После постукивания измеряли объем V1 (мл) уплотненного испытуемого материала. Плотность после утряски (г/мл) рассчитывали по следующей формуле. Результаты показаны в таблице 1.
Плотность после утряски (г/мл)=W/V1
г/мл
Плотность после утряски соединений по настоящему изобретению была выше, чем плотность кристаллов типа I.
Тестовый пример 2
Поверхностный потенциал
Следующее испытание проводили в термогидростате (25°C, относительная влажность 50%).
Испытуемые материалы устанавливали на чашку для образцов (SUS304, поверхность загрунтована buff400, объем: около 6 мл, глубина: 3 мм, цилиндрическая, верхняя сторона открыта) и заряжали коронным разрядом (напряжение короны: -4 кВ) от прибора приложенного напряжения в течение 2 минут (расстояние между прибором приложенного напряжения и чашкой для образцов: 30 мм). После зарядки измеряли поверхностный потенциал испытуемого материала (расстояние между датчиком и образцом: 5 мм). Результаты показаны в таблице 2.
Термогидростат: KCL-2000, EYELA CORPORATION.
Прибор для измерения поверхностного потенциала: SK-200, KEYENCE CORPORATION
Прибор приложенного напряжения: SJ-G036, KEYENCE CORPORATION
Регистратор данных: AD-DIO Card Bus, Interface Corporation
кВ
Поверхностный потенциал соединений по настоящему изобретению был ниже, чем потенциал кристаллов типа I, и следовательно, соединениям по настоящему изобретению трудно зарядиться статическим электричеством.
Тестовый пример 3
Тест на прилипание
Таблетки (плоской формы, диаметр 8,5 мм), содержащие 200 мг испытуемого вещества, готовили, используя анализатор процесса таблетирования (Tab Flex TAB-10, OKADA SEIKO CO., LTD.) (усилие прессования: 10 кН). Далее, измеряли давление скребка, во время удаления таблеток скребком. Результаты показаны в таблице 3.
Давление скребка на соединениях по настоящему изобретению было меньше, чем давление для кристаллов типа I. Было предположено, что соединения по настоящему изобретению не прилипают (осложнения при таблетировании из-за адгезии порошков к металлу) в сравнении с кристаллами типа I.
Тестовый пример 4
Тест на прилипание к металлической чашке
5 г испытуемых материалов, пропущенных через сито (18 меш), помещали в металлическую чашку (SUS304, внутренний диаметр: 65 мм, объем: около 200 мл) и затем испытуемые материалы удаляли переворачиванием чашки. Определяли массу испытуемых материалов, прилипших к поверхности чашки. Результаты показаны в таблице 4.
мг
Количество прилипшего вещества для соединений по настоящему изобретению было меньше, чем для кристаллов типа I. Было предположено, что соединения по настоящему изобретению не прилипают к металлической чашке.
Тестовый пример 5
Тест на формуемость прессованием
Таблетки (плоской формы, диаметр 8,5 мм), содержащие 200 мг испытуемого вещества, готовили, используя анализатор процесса таблетирования (Tab Flex TAB-10, OKADA SEIKO CO., LTD.) (усилие прессования: 5 и 10 кН). Далее, измеряли прочность таблеток на излом, используя тестер прочности таблеток (PC-30, OKADA SEIKO CO., LTD.). Результаты показаны в таблице 5.
5 кН
10 кН
Прочность таблеток с кристаллами типа I не изменялась даже, если повышалось усилие прессования. С другой стороны, прочность таблеток, содержащих соединения по настоящему изобретению была выше, чем содержащих кристаллы типа I. Кроме того, прочность таблеток, содержащих соединения по настоящему изобретению возрастала, когда повышалось усилие прессования. Формуемость прессованием таблеток, содержащих соединения по настоящему изобретению, превосходила таковую для кристаллов типа I.
Тестовый пример 6
Тест на модели мыши, инфицированной Candida (пероральное введение)
Candida albicans TIMM1623, который был получен из инкубированных на протяжении ночи культур, выращенных в чашках с декстрозным агаром Sabouraud (SDA) при 35°C, суспендировали в стерильном физиологическом растворе и затем разбавляли, чтобы приготовить суспензию высеваемого организма.
Преходящую иммуносупрессию у мышей (возраст 4 недели, 5 мышей на группу) вызывали внутрибрюшинным введением 200 мг/кг циклофосфамида за 4 суток до инфицирования и 100 мг/кг циклофосфамида через 1 сутки после инфицирования. Инфекцию вводили внутривенной инокуляцией 0,2 мл клеточной суспензии Candida albicans TIMM1623 (около 3×104 КОЕ/мышь) через латеральную хвостовую вену. Испытуемое соединение растворяли в 0,1 моль/л хлористоводородной кислоте, разбавляли стерилизованной водой и вводили перорально 1 мг/кг массы тела мыши. Лечение начинали через 2 часа после инфицирования и проводили один раз в сутки в течение 4 суток. Эквивалентное количество стерилизованного физиологического раствора вводили группе, которой не вводили испытуемые соединения. Наблюдали число выживших мышей и регистрировали его на протяжении 14 суток.
В результате в группе, которой не вводили испытуемое соединение, умерли все мыши, но 80% мышей выжило в группе, которой вводили соединения ссылочного примера 1, примера 1, примера 2 и примера 4.
Соединения ссылочного примера 1, примера 1, примера 2 и примера 4 имели превосходный терапевтический эффект.
Далее, настоящее изобретение объясняется со ссылками на ссылочные примеры и примеры, но не ограничивается ими.
Условия измерений для порошковой дифракции рентгеновских лучей:
Антикатод: Cu, напряжение трубки: 40 кВ, ток трубки: 40 мА.
Значения для пиков спектров ИК-поглощения, которые были использованы для того, чтобы различать кристаллы, перечислены.
Ссылочный пример 1
(Получение кристаллов типа I, патентный документ 1, пример 3-2)
К раствору 1,82 г бутил-4-нитрофенилкарбоната в N,N-диметилформамиде (15 мл) добавляли при комнатной температуре 1,50 г 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил}-1-пиперидинил]пропокси}бензамидина, и смесь перемешивали при той же температуре в течение 2 часов. К реакционной смеси добавляли хлороформ и воду. Органический слой отделяли, промывали последовательно 5% водным раствором карбоната калия (2 раза) и насыщенным водным раствором хлорида натрия, и затем сушили над безводным сульфатом магния, после чего отгоняли растворитель при пониженном давлениим.
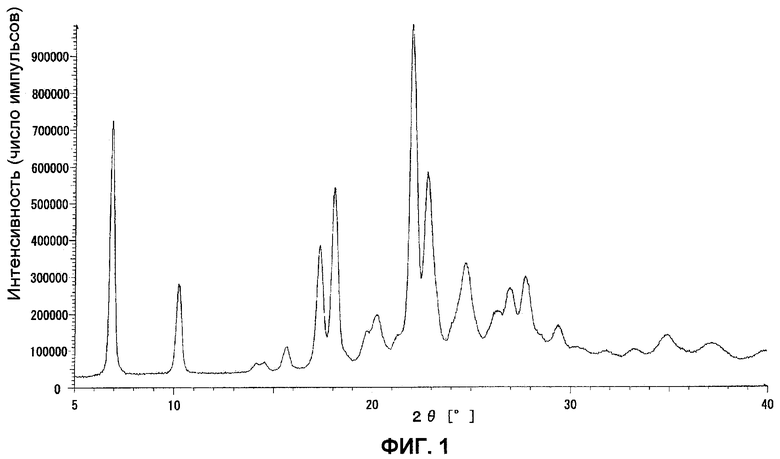
Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ:метанол=4:1). Полученное в результате твердое вещество растворяли в хлороформе, промывали последовательно 5% водным раствором карбоната калия (2 раза) и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния, после чего отгоняли растворитель при пониженном давлении, получая 1,39 г кристаллов типа I 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N'-(бутоксикарбонил)бензамидина в виде белого твердого вещества.
1H-ЯМР (CDCl3) значение δ: 0,95 (6H, т, J=7,3 Гц), 1,20-1,50 (9H, м), 1,60-2,05 (12H, м), 2,45-2,54 (2H, м), 2,90-3,00 (2H, м), 3,99 (2H, т, J=6,6 Гц), 4,06 (2H, т, J=6,3 Гц), 4,16 (4H, т, J=6,8 Гц), 6,88-6,96 (4H, м), 7,82-7,88 (4H, м).
Данные порошковой рентгенограммы показаны в таблице 6, и дифрактограмма показана на фиг.1.
ИК (ATR):1075, 1026 см-1
Пример 1
(Получение кристаллов типа II)
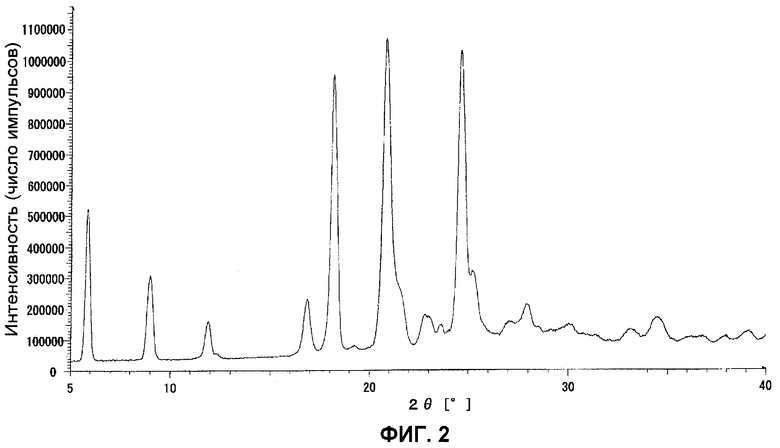
К этилацетату (1630 мл) добавляли 163 г кристаллов типа I и кипятили с обратным холодильником в течение 30 минут. После того, как реакционная смесь охлаждалась до 60-65°C, ее перемешивали при этой температуре в течение 30 минут. Твердое вещество отфильтровывали, собирали и сушили на воздухе, получив 139 г кристаллов типа II.
Данные порошковой рентгенограммы показаны в таблице 7, и дифрактограмма показана на фиг.2.
ИК (ATR): 1071, 1048 см-1
Пример 2
(Получение кристаллов типа III)
Метилэтилкетон (435 мл) и воду (435 мл) добавляли к 87,2 г кристаллов типа I, и смесь перемешивали при комнатной температуре в течение 24 часов.
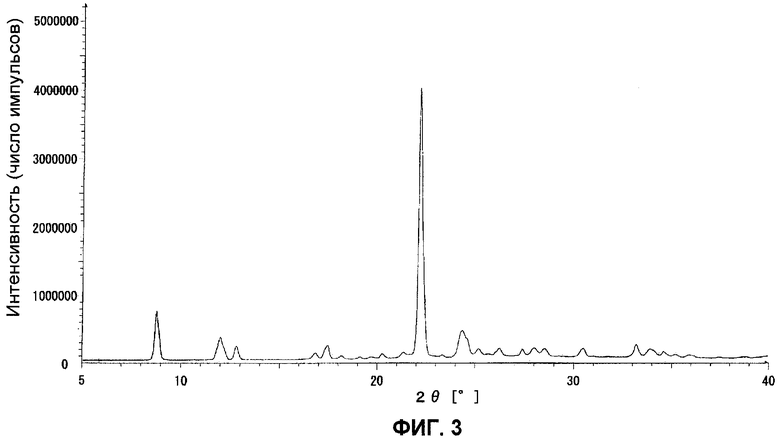
Твердое вещество отфильтровывали, собирали и сушили на воздухе, получив 69,8 г кристаллов типа III.
Данные порошковой рентгенограммы показаны в таблице 8, и дифрактограмма показана на фиг.3.
ИК (ATR): 1072, 1054, 1018 см-1
Пример 3
(Получение кристаллов типа III)
К метилэтилкетоновому (1800 мл) раствору 231 г имидазола добавляли 232 г бутилхлороформата при комнатной температуре и смесь затем оставляли при этой температуре на ночь. К реакционной смеси добавляли воду (1440 мл), 360 г пентагидрата гидрохлорида 4-{3-[4-(3-{4-[амино(имино)метил]фенокси}пропил)-1-пиперидинил]пропокси}бензамидина и этилацетат (360 мл) и смесь кипятили с обратным холодильником в течение 3 часов. Органический слой отделяли при 60-70°C, промывали водой и к органическому слою добавляли метилэтилкетон (720 мл). Нерастворимое вещество отфильтровывали при 60-70°C и лепешку промывали метилэтилкетоном (720 мл). Фильтрат и промывные жидкости объединяли, нагревали и растворяли. Добавляли затравку кристаллов типа III при 40-45°C и смесь перемешивали при той же температуре в течение 2 часов. После того, как смесь перемешивали до охлаждения до 5°C в течение 14 часов, твердое вещество отфильтровывали и собирали, получив 333 г кристаллов типа III.
ИК и картина порошковой рентгенограммы соответствовали значениям примера 2.
Пример 4
(Получение кристаллов типа IV)
Ацетон (4 мл) добавляли к 0,20 г кристаллов типа II и перемешивали при 5-10°C в течение недели. Твердое вещество отфильтровывали, собирали и сушили на воздухе, получив 0,17 г кристаллов типа IV.
Данные порошковой рентгенограммы были показаны в таблице 9, и дифрактограмма показана на фиг. 4.
ИК (ATR): 1094, 1070, 1056, 1019 см-1
Краткое описание чертежей
ФИГ.1 представляет порошковую рентгенограмму кристаллов типа I.
ФИГ.2 представляет порошковую рентгенограмму кристаллов типа II.
ФИГ.3 представляет порошковую рентгенограмму кристаллов типа III.
ФИГ.4 представляет порошковую рентгенограмму кристаллов типа IV.
Применимость в промышленности
Кристаллы по настоящему изобретению: (1) плотность после утряски которых высока, (2) которые трудно зарядить электричеством, (3) которые удобнее в обращении, (4) которые имеют хорошую формуемость при прессовании, (5) которые трудно слипаются и (6) промышленное производство которых возможно, являются полезными для лекарственного вещества.

Изобретение описывает новые кристаллы 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N′-(бутоксикарбонил)бензамидина. Технический результат: кристаллы типа II, кристаллы типа III и кристаллы типа IV 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)-метил]фенокси}-пропил)-1-пиперидинил]пропокси}-N′-(бутоксикарбонил)бензамидина являются превосходными в качестве лекарственных соединений для противогрибковых средств вследствие того, что они более удобны в обращении, т.к., например, имеют более хорошую формуемость при прессовании. 3 н.п. ф-лы, 4 пр., 9 табл., 4 ил.
1. Кристаллы 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N′-(бутоксикарбонил)бензамидина, имеющие пики в области 5,8, 18,2, 20,9 и 24,7° угла дифракции 2θ на порошковой рентгенограмме.
2. Кристаллы 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N′-(бутоксикарбонил)бензамидина, имеющие пики в области 8,7, 12,0, 22,2 и 24,3° угла дифракции 2θ на порошковой рентгенограмме.
3. Кристаллы 4-{3-[4-(3-{4-[амино(бутоксикарбонилимино)метил]фенокси}пропил)-1-пиперидинил]пропокси}-N′-(бутоксикарбонил)бензамидина, имеющие пики в области 9,8 и 23,5° угла дифракции 2θ на порошковой рентгенограмме.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| НОВОЕ ПРОИЗВОДНОЕ АРИЛАМИДИНА ИЛИ ЕГО СОЛЬ | 2003 |
|
RU2299195C2 |

