Область техники
Данное изобретение относится в общем к области фармакологии, неврологии и психиатрии и к способам лечения состояний расстройства сна - бодрствования. Более конкретно, данное изобретение предусматривает способы применения некоторых карбаматных соединений для лечения расстройства сна - бодрствования, включая чрезмерную дневную сонливость и патологическую сонливость.
Уровень техники
Чрезмерная дневная сонливость (EDS) или патологическая сонливость относится к чрезмерной сонливости в течение дня, связанной с широким кругом расстройств сна и бодрствования. Эти расстройства могут быть первичными расстройствами сна, такими как нарколепсия, или они могут быть результатом некоторых других медицинских состояний, которые вредно влияют на особенности сна.
Чрезмерная дневная сонливость (EDS) является основной жалобой пациентов в клиниках сна, она касается до 12% всего населения. Действие EDS может быть изнуряющим и даже угрожающим жизни. Пациенты с EDS могут проявлять психосоциальный стресс, пониженную активность на работе или в школе и подвержены повышенному риску несчастных случаев. Дифференциальный диагноз EDS требует объективных обследований, таких как полисомнография и множественный тест латентности сна.
Существуют четыре основные причины EDS: (1) патологические аномалии центральной нервной системы (ЦНС), такие как нарколепсия и идиопатическая гиперсомния ЦНС; (2) качественные или количественные дефекты сна, такие как синдром апноэ во сне, обструктивный синдром апноэ во сне, недостаточный сон ночью из-за, например, хронической и острой боли, возникающей из-за различных состояний, включая болезнь Паркинсона, недержание мочи, усталость от рассеянного склероза, ADHD, болезнь Альцгеймера, большая депрессия, биполярное расстройство и ишемическая болезнь сердца; (3) неотрегулированное взаимодействие сердечного ритмоводителя циркадного ритма организма с окружающей средой (например, при перелетах на реактивных самолетах или сменной работе); и (4) лекарства, которые могут увеличивать сонливость или терапевтически, или как побочный эффект.
В зависимости от этиологии стратегия преодоления EDS включает увеличение времени пребывания в постели, дремоту, хирургическое вмешательство, различные медицинские приспособления (например, оральные приспособления, непрерывное положительное давление в дыхательных путях) и фармакотерапию.
Усталость и чрезмерная сонливость также являются общими симптомами большой депрессии и других перепадов настроения, таких как биполярное расстройство, и могут быть вредными побочными эффектами, связанными с лечением антидепрессантами, или могут быть остаточными симптомами неадектватного лечения антидепрессантами. Кроме того, пациенты иногда страдают от побочных эффектов, касающихся сна, связанных с прекращением лечения антидепрессантами.
Нарколепсия является распространенной причиной EDS и представляет собой неврологическое расстройство, которое впервые было установлено более ста лет назад Gélineau, J.В. (De la narcolepsy, Gazette des Hopitaux Paris (1880) 53: 626-628). Нарколепсия является хроническим расстройством, характеризующимся прерывистыми периодами сна, устойчивой чрезмерной дневной сонливостью и аномальным быстрым движением глаз во сне («REM»), например началом периодов «быстрого» сна, катаплексией, параличом во сне, гипнагогическими галлюцинациями, или и тем, и другим. Большинство пациентов, страдающих от нарколепсии, имеют также нарушенный ночной сон.
Обзор проявлений нарколепсии см. в Chokroverty, S. (ed), Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, 2 2nd edition, Butterworth Heinemann, Boston, Mass. USA 1999; Aldrich, M., Sleep Medicine, Oxford University Press, New York, N.Y. USA 1999; Vgnotzas, A.N. et al., Annu. Rev. Med. (1999) 50: 387-400; и Guillenminault, С., Narcolepsy Syndrome in Principles and Practice of Sleep Medicine, 2nd edition (Kryger M. H., et al. (eds), (W.B.Saunders Philadelphia, Pa. USA 1989), pages 338-246).
Симптомы нарколепсии включают чрезмерную дневную сонливость (EDS), гипнагогические и гипнопомпические галлюцинации (галлюцинации во время перехода ко сну от бодрствования и обратно, соответственно), катаплексию (внезапная и обратимая потеря мышечного тонуса), паралич во сне (неспособность двигаться при засыпании или пробуждении) и «быстрый» сон в начале сна (Guilleminault, С.1989). У нарколептиков сон возникает в неподходящее время и в опасных и затрудняющих ситуациях. Хотя общее время сна является почти нормальным, ночной сон нарушается частыми пробуждениями (Mitler, M. et al., Psych. Clin. N. Amer. (1987) 10: 593-606).
Катаплексия, временный, частичный или полный паралич из-за внезапной потери мышечного тонуса с неповрежденным сознанием обычно является результатом внезапных сильных эмоций, таких как эмоции, сопровождающие смех, гнев и смущение. У некоторых пациентов состояние катаплексии или периоды повторяющейся потери мышечного тонуса возникают и могут длиться в течение нескольких часов и дней.
Нарколепсия описана и у других животных и была наиболее хорошо изучена у собак (Foutz, A.S., et al., (1979) Sleep 1: 413-421; Nishino, S. and Mignot, E. (1997) Prog. Neurobiol. 52: 27-78; Cederberg, R., et al., (1998) Vet. Rec. 142, 31-36). Нарколепсия у собак породы доберман-пинчер и лабрадор передается как аутосомальный рецессивный признак очевидно одного гена с полным проникновением, canarc-1 (Foutz, A.S., et al., (1979) Sleep 1:413-421; Baker, T.L. and Dement W.C. (1985), Canine narcolepsy-cataplexy syndrome: evidence for an inherited monoaminergic-cholinergic imbalance in Brain Mechanisms of Sleep, D.J. McGinty, R.Drucker-Colin, A.Morrison, и P.L.Parmeggiani, eds. (New York: Raven Press), pages 199-233).
Большое количество физиологических и фармакологических исследований показало тесное сходство между нарколепсией у людей и собак (Baker, Т.L. and Dement W.С. (1985) и Nishino S. and Mignot E. (1997). Эти животные имели все основные симптомы, характеризующие нарколепсию у людей, включая случаи катаплексии.
Собаки-нарколепсики также характеризуются чрезмерной дневной сонливостью и прерывающимися периодами сна (Kaitin, K.I., et al., Electroenceph. Clin. Neurophysiol. (1986) 64: 447-454). Cholinergic antagonists block cataplexy in both canine and human narcoleptics (Delashaw et al., (1979) Exp.Neurology 66: 745-757). al-блокаторы (такие как празоцин) обостряют катаплексию у собак и людей и могут вызывать status cataplecticus у тех и других (Mignot, E. et al. (1988) Brain Res. 444: 184-188; Guilleminault, С. et al. (1988) The Lancet 2: 511).
Лекарства, применяемые для лечения катаплексии и чрезмерной сонливости у людей, эффективны также и в случае собак-нарколептиков (Baker and Dement, 1985). Нарколепсия у людей обычно не развивается до юности, но может возникать в возрасте трех лет и в 45 лет и позже (Yoss and Daly (1960) Pediatrics 25: 1025-1033; Billard, (1985) Ann. Clin. Res. 17: 220-226). Возникновение катаплексии как предвестника начала нарколепсии/катаплексии развивается у собак в возрасте от 4 до 24 недель.
Примерно 250000 американцев больны нарколепсией (Aldrich M.S., New Eng. J. Med. (1990) 323: 389-394). Хотя описаны случаи семейной нарколепсии, большинство этих случаев являются спорадическими и полагают, что эта болезнь является мультигенной и зависящей от окружающей среды (Honda, Y., and Matsuki, K., Genetic Aspects of Narcolepsy in Handbook of Sleep Disorders, M.Thorpy (ed.) (Marcel Dekker, Inc., New York, N.Y. 1990) pages 217-234). Одним предрасполагающим генетическим фактором является специфический HLA-DQ аллель, HLA-DQB 1*0602 (Matsuki, K., et al., (1992) Lancet 339: 1052; Mignot, E. et al. (1994) Sleep 17: S60-S67; Mignot, E. (1998) Neurology 50 S16-S22). Примерно 95% нарколептиков имеют этот HLA гаплотип по сравнению с 30% всего населения (Aldrich M. S., New Eng. J. Med. (1990) 323: 389-394). В случае некоторых HLA ассоциированных болезней, таких как юношеский диабет, глютеновая болезнь, системная красная волчанка и ревматоидный артрит, сообщалось об аутоиммунном механизме (Sinha A., et al., Science (1990) 248: 1380-1388); однако до сих пор все попытки определить достоверность аутоиммунной гипотезы потерпели неудачу (Mignot, E. et al., Adv. Neuroimmunol (1995) 5:23-37).
Недавно появились сообщения, что нарколепсия связана с дисфункцией недавно обнаруженной гипокретиновой (Hcrt) (орексин) белковой системы. Это сообщение основывалось на делиции в транскриптах гена гипокретинового рецептора 2 (Hcrt2) у доберманов и лабрадоров, страдающих от нарколепсии (Lin L., et al., Cell (1999) 97: 365-376). Chemelli и др. создали мышей, нокаутированных по Hcrt, которые имели аномалии сна, похожие на аспекты нарколепсии (Chemelli R.M. et al., Cell (1999) 98: 437-451).
Нарколепсия требует длительного лечения симптомов (Fry J., Neurology (1998) 50 (2 Suppl. 1): S8-15). Вмешательство может быть не фармакологическим, таким как изменение образа жизни, и фармакологическим, для облегчения симптомов дневной сонливости, катаплексии, паралича во сне, гипнагогических галлюцинаций и/или гипнопомпических галлюцинаций.
Фармакологическое лечение нарколепсии зависит от использования стимулянтов центральной нервной системы (ЦНС) для увеличения бодрствования или для снижения количества и серьезности приступов катаплексии или гипнагогических галлюцинаций. Стимулянты ЦНС могут быть эффективны для уменьшения сонливости при нарколепсии; однако необходимы чрезвычайно высокие дозы для восстановления активности до нормального уровня (Mitler M. et al., Sleep (1993) 16: 306-317). Такие дозы могут вызвать очень опасные побочные эффекты. Из-за этих побочных эффектов большинство больных нарколепсией применяют стимулянты только в случае абсолютной необходимости или постоянно применяют низкие дозы, не способные восстановить нормальные уровни активности. Можно иногда применять периодически «каникулы без лекарств» для поддержания эффективности стимулянтов (Mitler M.S. et al., Sleep (1994) 17: S103-S106). Могут быть эффективными периоды дремоты, перемежающиеся с периодами бодрствования (Aldrich M.S., Neurology (1992) 42 (S6): 34-43). Иногда катаплексию можно эффективно лечить трициклическими антидепрессантами или селективными ингибиторами повторного поглощения серотонина (SSRI's). И трициклические антидепрессанты, и SSRI's действуют с образованием метаболитов, которые активируют норадренергические рецепторы (Nishino, S., et al., Sleep (1993) 16: 706-712; Mignot, E., et al., Psychopharmacology (1993) 113: 76-82). Даже при таком лечении несчастные случаи у нарколептиков, вызванные сонливостью и катаплексией, являются распространенными и обучение нарколептиков затруднено (Broughton W.A. and Broughton R.J.Sleep (1994) 17: S45-S49).
Чрезмерная дневная сонливость (EDS) или патологическая сонливость, вызванная нарколепсией или другими причинами, калечит и является опасной, так как вызывает периоды внезапного сна, пониженное внимание и ошибки в поведении. EDS, независимо от причины, связана с различными несчастными случаями на транспорте и на производстве и вызывает снижение работоспособности и тяжелое недомогание. Терапевтический агент, который уменьшает или устраняет EDS, имел бы важное значение не только для отдельных пациентов, но и для общественного здоровья и безопасности.
Сущность изобретения
Данное изобретение относится к способу лечения расстройств сна у субъекта, включая чрезмерную дневную сонливость (EDS) или патологическую сонливость, путем введения субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I):
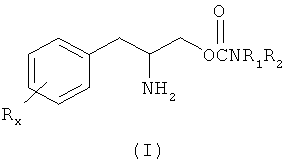
или его фармацевтически приемлемых соли или сложного эфира,
где Rx выбран из группы, содержащей водород, низший алкил с количеством углеродных атомов от 1 до 8, галоген, выбранный из фтора, хлора, брома и йода, алкокси с количеством атомов углерода от 1 до 3, нитро, гидрокси, трифторметил и тиоалкокси с количеством атомов углерода от 1 до 3;
х является целым числом от 1 до 3, при условии, что R может быть таким же или другим, когда х равен 2 или 3;
R1 и R2 могут быть одинаковыми или отличаться друг от друга и независимо выбираться из группы, содержащей водород, низший алкил с числом атомов углерода от 1 до 8, арил, арилалкил, циклоалкил с числом атомов углерода от 3 до 7; R1 и R2 могут быть соединены с целью образования 5-7 членного гетероцикла, замещенного членом, выбранным из группы, содержащей водород, алкильную и арильную группы, при этом циклическое соединение может содержать от 1 до 2 атомов азота и от 0 до 1 атомов кислорода, в котором атомы азота непосредственно не связаны друг с другом или с атомом кислорода.
Варианты данного изобретения включают способ лечения чрезмерной дневной сонливости (EDS) у субъекта, включающий стадию введения субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества энантиомера формулы (I), практически не содержащего других энантиомеров или энантиомерной смеси, в которой преобладает один энантиомер формулы (I):
или его фармацевтически приемлемых соли или сложного эфира,
где Rx выбран из группы, содержащей водород, низший алкил с количеством углеродных атомов от 1 до 8, галоген, выбранный из фтора, хлора, брома и йода, алкокси с количеством атомов углерода от 1 до 3, нитро, гидрокси, трифторметил и тиоалкокси с количеством атомов углерода от 1 до 3;
х является целым числом от 1 до 3, при условии, что R может быть таким же или другим, когда х равен 2 или 3;
R1 и R2 могут быть одинаковыми или отличаться друг от друга и независимо выбираться из группы, содержащей водород, низший алкил с числом атомов углерода от 1 до 8, арил, арилалкил, циклоалкил с числом атомов углерода от 3 до 7; R1 и R2 могут быть соединены с целью образования 5-7 членного гетероцикла, замещенного членом, выбранным из группы, содержащей водород, алкильную и арильную группы, при этом циклическое соединение может содержать от 1 до 2 атомов азота и от 0 до 1 атомов кислорода, в котором атомы азота непосредственно не связаны друг с другом или с атомом кислорода. Предпочтительно, чтобы Rx, R1 и R2 все были выбраны из водорода.
Предпочтительно, соединение, в котором один энантиомер, выбранный из группы, состоящей из соединений формулы I, преобладает до содержания, равного примерно 90% или более.
Более предпочтительно, когда энантиомер, выбранный из группы, состоящей из соединений формулы I, содержится в преобладающем количестве до примерно 98% или более.
Варианты данного изобретения включают применение для приготовления лекарственного препарата для лечения EDS энантиомера, выбранного из группы соединений формулы (I):
или его фармацевтически приемлемой соли или сложного эфира,
где Rx выбран из группы, содержащей водород, низший алкил с количеством углеродных атомов от 1 до 8, галоген, выбранный из фтора, хлора, брома и йода, алкокси с количеством атомов углерода от 1 до 3, нитро, гидрокси, трифторметил и тиоалкокси с количеством атомов углерода от 1 до 3;
х является целым числом от 1 до 3, при условии, что R может быть таким же или другим, когда х равен 2 или 3;
R1 и R2 могут быть одинаковыми или отличаться друг от друга и независимо выбираться из группы, содержащей водород, низший алкил с числом атомов углерода от 1 до 8, арил, арилалкил, циклоалкил с числом атомов углерода от 3 до 7; R1 и R2 могут быть соединены с целью образования 5-7 членного гетероцикла, замещенного членом, выбранным из группы, содержащей водород, алкильную и арильную группы, при этом циклическое соединение может содержать от 1 до 2 атомов азота и от 0 до 1 атомов кислорода, в котором атомы азота непосредственно не связаны друг с другом или с атомом кислорода.
Варианты данного изобретения предусматривают способ, включающий применение энантиомера формулы I, практически не содержащего других энантиомеров, который является энантиомером формулы Ib, (R)-(бета-аминобензопропил)карбаматом или (О-карбамоил-(D)-фенилаланинолом), или энантиомерной смеси, в которой энантиомер формулы Ib, (R)-(бета-аминобензопропил)карбамат или (О-карбамоил-(D)-фенилаланинол) преобладает:
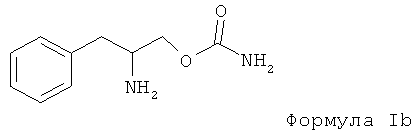
В соединении формулы Ib, являющемся (R)-(бета-аминобензопропил)-карбаматом или (О-карбамоил-(D)-фенилаланинолом), энантиомер формулы Ib, (R)-(бета-аминобензопропил)карбамат или (О-карбамоил-(D)-фенилаланинол) содержится в преобладающем количестве, равном примерно 90% или более. Более предпочтительным является соединение, в котором энантиомер формулы Ib, (R)-(бета-аминобензопропил)карбамат или (O-карбамоил-(D)-фенилаланинол) содержится в преобладающем количестве до примерно 98% или более.
Варианты данного изобретения включают способы, когда причина EDS выбирается из группы, включающей патологические аномалии центральной нервной системы (ЦНС), удар, нарколепсию, идеопатическую гиперсомнию, недостаток сна, синдром апноэ во сне, обструктивный синдром апноэ во сне, недостаточный ночной сон, хроническую боль, острую боль, болезнь Паркинсона, недержание мочи, усталость от рассеянного склероза, расстройство гиперфункции дефицита внимания (ADHD), болезнь Альцгеймера, большую депрессию, биполярное расстройство, ишемическую болезнь сердца, неотрегулированность взаимодействия сердечного циркадного ритмоводителя с окружающей средой, нарушение биологических ритмов организма при перелетах на реактивных самолетах, сменную работу и седативные лекарства.
Подробное описание изобретения
Настоящее изобретение частично основано на обнаружении того факта, что карбаматы фенилалкиламина с формулой I обладают новыми и уникальными фармакологическими свойствами. Такие соединения были испытаны на животных и на человеке в целях достижения активизирующего и возбуждающего эффекта. Хотя точный механизм воздействия еще не полностью понятен, полагают, что такие соединения действуют по другим механизмам, чем большинство других известных стимулирующих лекарств в целях получения их активизирующих и возбуждающих подобных эффектов. При испытаниях на животных лечение карбаматом фенилалкиламина с формулой I при дозировке 30 мг/кг сильно увеличивало активное бодрствование за счет времени, потраченного на дремоту, глубокий сон и «быстрый» сон, в течение первых 3-4 часов после приема. Обратный эффект наблюдался через 4-10 часов после приема соединения, так как увеличение времени пребывания в глубоком сне постепенно уменьшалось через часы после этого. Кроме того, соединение с формулой I воздействовало на другие параметры сна - бодрствования; конкретнее, оно значительно увеличило количество переходов от дремоты и быстрого сна к бодрствованию, а также пролонгировало скрытое состояние начала «быстрого» сна.
По этим двум причинам соединения формулы I особенно приемлемы для использования при лечении EDS и других заболеваний, при которых желательно увеличивать продолжительность времени бодрствования. Таким образом, такие соединения могут безопасно использоваться в целях достижения эффективного лечения EDS несмотря на точную причину указанного нарушения сна.
Характерно, что дозы соединения формулы I могут начинаться с 10-25 мг/день и увеличиваться с приростом около 10-25 мг/день еженедельно до появления побочных эффектов или адекватного ответа, полученного с максимальной дозой в пределах от 500 мг/день до 2000 мг/день.
Одно соединение с формулой I содержит (D) энантиомер с указанной ниже формулой, где Rx=R1=R2 = водород, в указанной ниже формуле аминогруппа расположена внизу
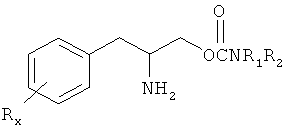
Это соединение является (R) энантиомером согласно формуле и, следовательно, является карбаматом (R)-(бета-аминобензолпропила). Это соединение является правовращающимся энантиомером и таким образом может быть определено как O-карбамоил-(D)-фенилаланинол и в данной заявке отнесено к «испытуемому соединению». Два химических названия могут использоваться равнозначно в этом определении.
Это соединение было испытано на многих животных и на человеке и показало результаты, включающие значительное увеличение активного бодрствования за счет времени дремоты, глубокого сна и «быстрого» сна в течение первых 3-4 часов после приема. Дополнительно, это соединение значительно увеличило количество переходов из дремоты и быстрого сна в состояние бодрствования, а также пролонгировало скрытое состояние начала быстрого сна. Это соединение также показывает стимулирующее и возбуждающее воздействие в модели Spontaneous Locomotor Activity у мышей и крыс.
Таким образом, в некоторых вариантах настоящее изобретение направлено на способ предотвращения или уменьшения серьезности EDS. Способ включает введение субъекту, в случае необходимости, терапевтически эффективного количества соединения, выбранного из группы, которая содержит карбаматы фенилалкиламина со следующей формулой I:
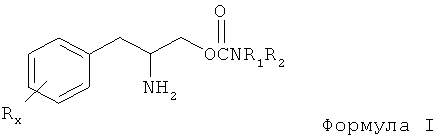
или его энантиомер, диастереомер, рацемат или их смеси, или его фармацевтически приемлемую соль или эфир,
где Rx выбран из группы, содержащей водород, низший алкил с количеством углеродных атомов от 1 до 8, галоген, выбранный из фтора, хлора, брома и йода, алкокси с количеством атомов углерода от 1 до 3, нитро, гидрокси, трифторметил и тиоалкокси с количеством атомов углерода от 1 до 3;
х является целым числом от 1 до 3, при условии, что R может быть таким же или другим, когда х равен 2 или 3;
R1 и R2 могут быть одинаковыми или отличаться друг от друга и независимо выбираться из группы, содержащей водород, низший алкил с числом атомов углерода от 1 до 8, арил, арилалкил, циклоалкил с числом атомов углерода от 3 до 7; R1 и R2 могут быть соединены с целью образования 5-7 членного гетероцикла, замещенного членом, выбранным из группы, содержащей водород, алкильную и арильную группы, при этом циклическое соединение может содержать от 0 до 2 атомов азота и от 0 до 1 атомов кислорода, в котором атомы азота непосредственно не связаны друг с другом или с атомом кислорода, и его фармацевтически приемлемые соли или эфиры.
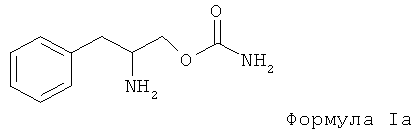
Настоящий способ также включает применение соединения, выбранного из группы соединений формулы I, где Rx, R1 и R2 предпочтительно выбраны из водорода, эта формула Iа указана ниже:
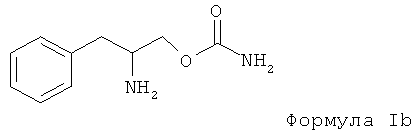
Настоящий способ также включает применение (D)-энантиомера, выбранного из группы, содержащей соединения с формулой I или энантиомерную смесь, в которой (D)-энантиомер выбран из группы, в которой преобладает соединение с формулой 1а, в которой Rx, R1 и R2 предпочтительно выбраны из водорода, то есть является O-карбамоил-(D)-фенилаланинолом. Формула Ib указана ниже (отметим - в формуле Ib, т.е. в (D)-энантиомере, как показано, аминная группа на хиральном углероде сориентирована внутрь плоскости):
В энантиомерных смесях доминирует один энантиомер, выбранный из группы, содержащей соединения с формулой I, предпочтительно, энантиомер, выбранный из группы, содержащей соединения с формулой I, преобладает до величины приблизительно 90% или более. Более предпочтительно, энантиомер, выбранный из группы, содержащей соединения с формулой I, преобладает до величины приблизительно 98% или более.
Соединения с формулой I могут быть синтезированы с применением методов, известных специалистам. Соли и эфиры соединений с формулой I могут быть получены путем обработки соединения приемлемой минеральной или органической кислотой (НХ) в приемлемом растворителе или путем других методов, известных специалистам в этой области.
Детали вышеуказанных реакционных схем для синтезирования соединений с формулой I, а также представленные примеры получения специфических соединений описаны в патенте США №5705640, патенте США №5756817, патенте США №5955499, патенте США №6140532.
Из формулы I очевидно, что некоторые соединения по настоящему изобретению имеют, по меньшей мере, один или возможно больше асимметричных углеродных атомов. Подразумевается, что настоящее изобретение включает в свой объем стереохимически чистые изомерные формы соединений, а также их рацематы. Стереохимически чистые изомерные формы могут быть получены путем применения известных в этой области методов. Диастереоизомеры могут быть выделены путем применения методов физического разделения, таких как фракционная кристаллизация и хроматографические способы, а энантиомеры могут быть отделены один от другого путем селективной кристаллизации диастереомерных солей с оптически активными кислотами или основаниями или путем хиральной хроматографии. Чистые стереоизомеры также могут быть получены синтетически из соответствующих стереохимически чистых исходных материалов или с применением стереоселективных реакций.
Во время любых процессов получения соединений по настоящему изобретению может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы в любых молекулах. Это может быть достигнуто путем применения стандартных защитных групп, подобных тем, которые описаны в Protective Groups in Organic Chemistry, ed. J.F.W.McOmie, Plenum Press, 1973; и Т.W.Greene & P.G.M.Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, 1999. Защитные группы могут быть удалены на стандартной соответствующей стадии с применением методов, известных в этой области техники.
Другие варианты по настоящему изобретению включают использование для получения медикаментов для лечения EDS одного из соединений или энантиомерных смесей, описанных выше, или их фармацевтически приемлемых солей или эфиров.
Определения
Для удобства некоторые термины, употребленные в данном описании, примерах и формуле изобретения, собраны в данном разделе. Должно быть понятно, что данное изобретение не ограничено конкретной технологией, протоколами, видами животных и описанными реагентами, так как все они могут разниться. Также должно быть понятно, что использованная здесь терминология применена в целях описания только конкретных вариантов и не ограничивает объем настоящего изобретения, который будет лимитирован только приложенной формулой изобретения.
Примененный здесь термин «чрезмерная дневная сонливость» (EDS) будет использоваться равнозначно с термином «патологическая сонливость» и будет означать состояние, когда субъект в течение дня чувствует себя сонливым и имеет трудности с сопротивлением засыпанию независимо от того, имел ли он или не имел время для сна ночью. Чрезмерная сонливость определена как сонливость, встречающаяся в ситуации, когда субъект должен бодрствовать и быть активным. Клинически симптомы EDS могут быть количественно определены и измерены различными путями, которые включают, но не ограничивают: the Multiple Sleep Latency Test (MSLT) (см., Carskadon MA and Dement WC, Sleep 1982; 5 Suppi 2: S67-72), the Maintenance of Wakefulness Test (MWT) (см., Mitler MM, et al. Electroencephalogr Clin. Neurophysiol, 1982; 53(6): 658-61) или the Stanford Sleepiness Scale (SSS) (см., Hoddes E et al., Psychophysiology, 1973; 10(4): 431-6) (см. также, Arand D et al., Sleep, 2005; 28(1); 123-144). Причины EDS многочисленны и использование здесь термина EDS не намерено подразумевать какую-либо конкретную причину или этиологию. Люди с EDS часто дремлют, бывают сонными или засыпают в ситуациях, когда они нуждаются в бодрствовании или хотят быть полностью бодрыми. Диагноз может быть поставлен, когда симптомы EDS сильно мешают способности субъекта концентрироваться и исполнять ежедневные задачи и рутинные дела, такие как работа, семейные обязанности, вождение машины или выполнение других опасных механических или обычных жизненных операций.
Использованный здесь термин «психическое расстройство» или «психическая болезнь» относится к тем, которые определены в Diagnostic and Statistical Manual (DSM IV), American Psychological Association (АРА). Такие психические расстройства включают, но не ограничиваясь, аффективные расстройства, большую депрессию и родственные депрессивные расстройства. Примеры аффективных расстройств включают перепады настроения, маниакальное расстройство, большую депрессию и биполярное аффективное расстройство. Перепады настроения включают, но не ограничиваясь, депрессивные расстройства, включая большую депрессию с психическими проявлениями и без психических проявлений, дистимическое расстройство, биполярные расстройства (I и II) и циклотимические расстройства.
Использованный здесь термин «субъект» относится к животным, предпочтительно, млекопитающим, и, наиболее предпочтительно, к человеку, который явился объектом лечения, наблюдения или изучения.
Использованный здесь термин «терапевтически эффективное количество» означает то количество активного соединения или фармацевтического агента, которое вызывает биологический или медицинский ответ в тканях у животных или у человека, который замечается исследователем, ветеринаром, врачом или другим клиницистом и который облегчает одно или более проявлений или симптомов болезни или расстройства, которые подвергаются лечению.
Термин «профилактически эффективное количество» предназначен для обозначения того количества фармацевтического препарата, которое будет предотвращать или уменьшать риск проявления биологического или медицинского ответа (который хотят предотвратить) в системе, в тканях у животных и у человека исследователи, ветеринары, врачи или другие клиницисты.
Термин «фармацевтически приемлемые соли или эфиры» будет означать нетоксичные соли или эфиры соединений, использованных в настоящем изобретении, которые обычно получаются путем взаимодействия свободной кислоты с приемлемым органическим или неорганическим основанием или свободного основания с приемлемой органической или неорганической кислотой. Примеры таких солей включают, но не ограничиваясь, ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, кальций, кальциевую соль этилендиаминтетрауксусной кислоты, камсилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, соль этилендиаминтетрауксусной кислоты, эдисилат, эстолат, эсилат, фумарат, глуцептат, глюконат, глютамат, гликоллиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, олеат, оксалат, памоат, пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, калий, соль салициловой кислоты, натрий, стеарат, основную уксуснокислую соль, соль янтарной кислоты, таннат, тартрат, теоклат, тозилат, триэтиодид, валерат.
Термины «субъект» и «пациент» использованы здесь равнозначно и они обозначают любое млекопитающее, включающее, но не ограничивающее, человека, включая человека или субъекта, которому могут быть введены композиции по настоящему изобретению. Термин «млекопитающее» включает людей и приматов, а также подопытных животных, таких как кролики, крысы, мыши, и других животных.
Использованный здесь термин «пациент, нуждающийся в лечении» будет относится к любому субъекту или пациенту, который постоянно имеет или у которого прогрессирует любой из вышеуказанных синдромов или расстройств, включая любое состояние или расстройство, при котором субъект затрачивает избыточное количество времени на спящее состояние или не способен поддерживать удовлетворительную степень бодрствования в течение дня, когда бодрствование необходимо или желательно, или другое расстройство, при котором текущее клиническое состояние пациента или его прогнозы могут выиграть от введения одного или более соединений формулы I одного или в сочетании с другим терапевтическим вмешательством, включая, но не ограничивая, другой препарат.
Использованный здесь термин «лечащий» или «лечение» относится к любым показаниям успеха в предотвращении или улучшении травмы, патологии или состояния, включающим любые объективные или субъективные параметры, такие как ослабление, ремиссия, уменьшение симптомов или большая переносимость травмы, патологии или состояния пациентом, замедление скорости дегенерации или прогрессирования, или ухудшение болезни; облегчение последней стадии ухудшения; или улучшение физического или психического состояния субъекта. Лечение или облегчение симптомов может быть основано на объективных или субъективных параметрах, включающих результаты физического обследования, изучения сна, неврологического обследования и/или психиатрической оценки. Соответственно термин «лечащий» или «лечение» включает введение соединений или агентов по настоящему изобретению с целью достижения повышенной активности или сниженного стремления в желании уснуть. В некоторых примерах лечение соединениями по настоящему изобретению будет проводиться в сочетании с другими соединениями с целью достижения повышенной активности или уменьшенной необходимости во сне или предотвращения заторможенности или остановки прогрессирования EDS.
Использованный здесь термин «терапевтический эффект» относится к эффективному обеспечению указанного выше действия.
Использованный здесь термин «терапевтический эффективное количество» означает достаточное количество одного или более соединений по настоящему изобретению с целью достижения терапевтического эффекта, как было определено выше, у субъекта или пациента в случае необходимости в таком нейрозащитном лечении.
Использованный здесь термин «сопутствующее введение» или «комбинированное введение» соединения, терапевтического агента или известного лекарства вместе с соединением по настоящему изобретению означает введение известного препарата или лекарства и в дополнение одного или более соединения по настоящему изобретению в такой момент, когда и известное лекарство, и это соединение обеспечат терапевтический эффект. В некоторых случаях терапевтический эффект будет синергическим. Такое сопутствующее введение может включать одновременное (т.е. в то же самое время), предварительное или последующее введение известного лекарства по отношению к введению соединения по настоящему изобретению. Сотрудник с обычной квалификацией в этой области не будет иметь трудностей в определении соответствующего времени, последовательности и дозы для введения конкретных лекарств и соединений по настоящему изобретению.
Кроме того, в некоторых вариантах соединения по настоящему изобретению будут применяться либо одни, либо в сочетании с одним или более терапевтическими лекарственными препаратами, как было указано выше, или их солями или эфирами для получения лекарства в целях лечения EDS или родственных состояний у пациента или субъекта в случае необходимости.
Использованный здесь термин «C1-C4 алкил» относится к замещенным или незамещенным алифатическим углеводородам с числом атомов углерода от 1 до 4. Конкретно, включенными в определение «алкил» являются те алифатические углеводороды, которые выборочно замещены. В предпочтительном варианте по настоящему изобретению C1-C4 алкил является незамещенным или замещенным фенилом.
Использованный здесь термин «испытуемое соединение» (tc) или.«ИСПЫТУЕМОЕ СОЕДИНЕНИЕ» (ТС) означает гидрохлоридную соль (R)-(бета-аминобензолпропил)карбамата, который может быть также назван О-карбамоил-(D)-фенилаланинолом. Это соединение является (R) энантиомером с формулой Ib и также является правовращающим энантиомером. Испытуемое соединение обозначается также как R228060 в Таблицах 1-4.
Использованный здесь термин «фенил», используется ли он самостоятельно или как часть другой группы, определен как замещенное или незамещенное ароматическое углеводородное кольцо, имеющее 6 атомов углерода. Конкретно, включенными в определение «фенил» являются те фенильные группы, которые выборочно замещены. Например, в предпочтительном варианте по настоящему изобретению «фенильная группа» является либо незамещенной, либо замещенной галогеном, C1-C4 алкилом, C1-C4 алкокси, амино, нитро или цианом.
В этой области известны методы определения терапевтически и профилактически эффективных доз для данной фармацевтической композиции. Например, для лечения EDS соединения по настоящему изобретению могут применяться для ежедневной дозы в размере около от 1 до 1000 мг обычаю от 1 до 3 раз в день для среднего взрослого человека. Однако эффективное количество может варьироваться в зависимости от конкретного применяемого соединения, метода введения, эффективности препарата и прогрессирования заболевания. Кроме того, факторы, связанные с подвергающимся лечению конкретным пациентом, такие как возраст пациента, вес, диета, продолжительность введения, приведут к необходимости регулирования доз.
Соединения могут вводиться субъекту любым известным способом введения, который включает, но не ограничиваясь, внутривенный, оральный, подкожный, внутримышечный, интрадермальный и парентеральный. В зависимости от способа введения соединения формулы I могут быть приготовлены в любой форме. Например, формы, приемлемые для орального введения, включают твердые формы, такие как пилюли, гелевые капсулы, таблетки, каплеты, капсулы (включающие составы для немедленного высвобождения, высвобождения во времени и для пролонгированного высвобождения), гранулы и порошки. Формы, приемлемые для орального введения, также включают жидкие формы, такие как растворы, сиропы, эликсиры, эмульсии и суспензии. Кроме того, формы, приемлемые для парентерального введения, включают стерильные растворы, эмульсии и суспензии.
Для того чтобы приготовить фармацевтические композиции по настоящему изобретению, одно или более соединений формулы I или их соли, как активный ингредиент, тщательно смешиваются с фармацевтическим носителем в соответствии с известными методами приготовления фармацевтических составов. Необходимы носители и фармацевтические инертные наполнители, включающие, но не ограничивающие, связующие, суспендирующие агенты, смазки, отдушки, подсластители, консерванты, красители и покрытия. Для приготовления композиций для оральных дозированных форм может быть использован любой из обычных фармацевтических носителей. Например, для приготовления жидких оральных форм приемлемые носители и добавки включают воду, гликоли, масла, спирты, отдушки, консерванты, красители и т.п.; для приготовления твердых оральных форм приемлемые носители и добавки включают крахмалы, сахара, разбавители, гранулирующие агенты, смазки, связующие, дезинтегрирующие агенты и т.п.
Для парентерального использования носители обычно включают стерильную воду или физиологический раствор, хотя могут быть включены другие ингредиенты, например для целей повышения растворимости или консервации. Впрыскиваемые суспензии также могут быть приготовлены, когда могут применяться соответствующие жидкие носители, суспендирующие агенты и т.п.
Из-за легкости их введения таблетки и капсулы представляют собой наиболее благоприятные оральные дозированные стандартные формы, в которых применяются твердые фармацевтические носители. По желанию таблетки могут иметь сахарное или энтеропокрытие, нанесенное стандартными способами. Могут быть изготовлены свечи, в которых в качестве носителя применяют масло какао. Таблетки или пилюли могут иметь покрытие или скомпонованы другим способом с целью придания лекарственной форме свойств пролонгированного действия. Например, таблетки или пилюли могут содержать внутри и снаружи дозированный компонент, причем последний будет в виде конверта, покрывающего первый. Два компонента могут быть разделены слоем энтеропокрытия, который служит препятствием для разрушения в желудке и способствует продвижению внутреннего компонента в двенадцатиперстную кишку или задерживает высвобождение. Множество материалов может использоваться для таких энтерослоев и покрытий, такие материалы включают ряд полимерных кислот с такими материалами, как шеллак, гексадециловый спирт и ацетат целлюлозы.
Активное лекарство может доставляться путем использования моноклональных антител в качестве индивидуальных носителей, к которым присоединяются молекулы компонентов. Активное лекарство также может сочетаться с растворимыми полимерами в качестве целевых носителей лекарства. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, поли- гидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Более того, активное лекарство может сочетаться с классом биоразлагаемых полимеров, используемых для получения лекарства с контролируемым высвобождением, например такие как полимолочная кислота, полигликолевая кислота, сополимеры полимолочной и полигликолевой кислот, полиэпсилон-капролактон, полигидроксимасляная кислота, полиортоэфиры, полиацетали, полидигидропираны, полицианакрилаты и сшитые или амфипатические блоксополимеры в виде гидрогелей.
Предпочтительно, такие композиции приготовлены в виде стандартных лекарственных форм, таких как таблетки, пилюли, капсулы, порошки, гранулы, стерильные парентеральные растворы или суспензии, дозированные аэрозоли или жидкие распылители, капли, ампулы, устройства с автоматическим впрыском или свечи для орального, парентерального введения, введения через нос, введения под язык или через прямую кишку и введение путем ингаляции или вдыхания.
Альтернативно, композиция может быть в форме, приемлемой для введения один раз в неделю или один раз в месяц, например, нерастворимая соль активного компонента, такая как деканоат, может быть применена с целью получения депо для внутримышечной инъекции.
Фармацевтические композиции по настоящему изобретению будут содержать в каждой стандартной дозе, например таблетке, капсуле, порошке, инъекции, чайной ложке, свече и т.п., такое количество активного ингредиента, которое необходимо для доставки эффективной дозы, как было описано выше. Например, фармацевтические композиции по настоящему изобретению могут содержать на единицу лекарственной формы от приблизительно 10 до приблизительно 1000 мг активного вещества. Предпочтительно, интервал составит от приблизительно 25 до приблизительно 200 мг активного вещества.
В некоторых вариантах по настоящему изобретению карбаматы, которые приемлемы для использования на практике по настоящему изобретению, будут вводиться либо самостоятельно, либо в сочетании с, по меньшей мере, одним или более другими соединениями или терапевтическими агентами, например с другими агентами, которые имеют тенденцию увеличивать возбудимость или активность. В этих вариантах настоящее изобретение предлагает методы лечения или предупреждения EDS у пациента. Этот метод включает стадию введения пациенту, в случае необходимости лечения, эффективного количества одного из карбаматов, описанных в настоящей заявке, в сочетании с эффективным количеством одного или более других соединений или терапевтических агентов, которые имеют способность производить полезные комбинированные эффекты, такие как способность увеличивать активирующее воздействие соединений по настоящему изобретению. Должно быть понятно, что заместители и положения замещения в соединениях по настоящему изобретению могут быть выбраны специалистом в области получения соединений, которые химически устойчивы и которые могут быть легко синтезированы способами, известными в этой области техники, а также способами по настоящему изобретению.
Настоящее изобретение включает применение выделенных энантиомеров формулы I. В одном предпочтительном варианте фармацевтическая композиция, содержащая выделенный S-энантиомер формулы I, использовалась в целях лечения пациента. В другом предпочтительном варианте фармацевтическая композиция, содержащая выделенный R-энантиомер формулы I, использовалась в для лечения пациента.
Настоящее изобретение также включает использование смесей энантиомеров с формулой I. В одном аспекте настоящего изобретения один энантиомер будет превалировать. Энантиомер, который доминирует в смеси, является энантиомером, который содержится в смеси в количестве большем, чем любые другие энантиомеры в смеси, например в количестве более 50%. В одном аспекте один энантиомер будет доминировать в количестве 90% или в количестве 91%, 92%, 93%, 94%, 95%, 96%, 97% или 98% или более. В одном предпочтительном варианте энантиомер, который доминирует в композиции, содержащей соединение формулы I, является S-энантиомером с формулой I.
Настоящее изобретение обеспечивает способы использования энантиомеров и энантиомерных смесей соединений формулы I. Карбаматный энантиомер с формулой I содержит асимметричный хиральный углерод в положении бензила, который является вторым алифатическим углеродом, примыкающим к фенильному кольцу.
Выделенный энантиомер является, по существу, свободным от соответствующего энантиомера. Таким образом, выделенный энантиомер относится к соединению, которое выделяется путем применения методов разделения или получения не содержащим соответствующий энантиомер.
Использованный здесь термин «по существу не содержащий» означает, что соединение получено со значительно большим количеством одного энантиомера. В предпочтительных вариантах соединение включает, по меньшей мере, около 90 вес.% предпочтительного энантиомера. В других вариантах настоящего изобретения соединение включает, по меньшей мере, около 99 вес.% предпочтительного энантиомера. Предпочтительные энантиомеры могут быть выделены из рацемических смесей путем применения методов, которые известны специалистам, включающих жидкостную хроматографию высокого разрешения (ЖХВР) и образование и кристаллизацию хиральных солей, или предпочтительные энантиомеры могут быть получены с применением методов, описанных в настоящей заявке.
Карбаматные соединения в качестве фармацевтических веществ
Настоящее изобретение предусматривает рацемические смеси, энантиомерные смеси и изолированные энантиомеры с формулой I как фармацевтические вещества. Карбаматы используются как фармацевтические вещества для лечения EDS и сопутствующих ему состояний у субъекта.
В общем, карбаматы по настоящему изобретению могут вводиться как фармацевтические композиции с применением методов, которые известны в области введения терапевтических лекарств, включающих оральные, буккальные, местные, системные (например, через кожу, через нос или с помощью свечей) или парентеральные (например, внутримышечные, подкожные или внутривенные инъекции). Введение соединений непосредственно в нервную систему может включать, например, способы введения внутримозговые, внутрижелудочковые, в желудочки и в мозг, интратекальные, внутриполостные, внутрипозвоночные или околопозвоночные, путем доставки через внутричерепные или внутримозговые иглы или катетеры с насосами и без насосов.
Композиции могут иметь форму таблеток, пилюль, капсул, полутвердых форм, порошков, составов с пролонгированным высвобождением, растворов, суспензий, эмульсий, сиропов, эликсиров, аэрозолей или любых других соответствующих композиций и содержать, по меньшей мере, одно соединение по настоящему изобретению в сочетании, по меньшей мере, с одним фармацевтически приемлемым носителем. Приемлемые носители хорошо известны специалистам в этой области техники, и методы образования композиций могут быть найдены в таких стандартных изданиях, как Alfonso A R; Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton P A, 1985. Приемлемые жидкие носители, особенно для растворов для инъекций, включают воду, водный физиологический раствор, водный раствор декстрозы и гликоли.
Карбаматы могут быть приготовлены в виде водных суспензий. Водные суспензии по настоящему изобретению могут содержать карбамат в смеси с подходящими носителями для получения водных суспензий. Такие носители могут включать, например, суспендирующий агент, такой как карбокси-метилцеллюлоза натрия, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, смола трагаканта, смола акации, и диспергирующие или увлажняющие агенты, такие как встречающийся в природе фосфатид (например, лецитин), продукт конденсации окиси алкилена с жирной кислотой (например, полиоксиэтиленстеарат), продукты конденсации окиси этилена с алифатическим спиртом с длинной цепочкой (например, оксицетанол гептадекаэтилена), продукт конденсации окиси этилена с частичным эфиром, полученным из жирной кислоты и гекситола (например, моноолеат полиоксиэтиленсорбита) или продукт конденсации окиси этилена с частичным эфиром, полученным из жирной кислоты и ангидрида гекситола (например, моноолеат полиоксиэтиленсорбитана).
Водные суспензии могут также содержать один или более консервантов, таких как этил- или н-пропил-п-гидроксибензоат, один или более окрашивающих агентов, одну или более отдушек, один или более подсластителей, таких как сахароза, аспартам или сахарин. Составы могут быть подобраны по осмолярности.
Масляные суспензии могут быть приготовлены путем суспендирования карбамата в растительном масле, таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло, или в минеральном масле, таком как жидкий парафин, или в их смеси. Масляные суспензии могут содержать загущающий агент, такой как пчелиный воск, твердый парафин или гексадециловый спирт. Для получения съедобного орального продукта могут быть добавлены подсластители, такие как глицерин, сорбит или сахароза. Эти продукты могут быть защищены путем добавления антиоксиданта, такого как аскорбиновая кислота. Пример масляного носителя для инъекций смотри в публикации Minto, J. Pharmacol. Exp. Ther. 281: 93-102, 1997. Фармацевтические составы по настоящему изобретению могут быть также в виде масляно-водных эмульсий. Масляная фаза может быть растительным маслом или минеральным маслом, как было описано выше, или их смесью.
Приемлемые эмульгирующие агенты включают встречающиеся в природе смолы, такие как смола акации и смола трагаканта, встречающиеся в природе фосфатиды, такие как соевый лецитин, эфиры или частичные эфиры, полученные из жирных кислот и ангидридов гекситола такие как, моноолеат сорбитана и продукты конденсации таких частичных эфиров с окисью этилена, такие как моноолеат полиоксиэтиленсорбитана. Эмульсия может также содержать подсластители и отдушки как при приготовлении сиропов, так и эликсиров. Такие композиции также могут содержать антиэмульгатор, консервант или окрашивающий агент.
Выбор соединения одного или в сочетании с другими приемлемыми компонентами может быть сделан при составлении аэрозолей (то есть они могут быть «распылены») при введении путем ингаляции. Аэрозольные композиции могут помещены в приемлемые сжатые пропелленты, такие как дихлордифтор-метан, пропан, азот и т.п.
Композиции по настоящему изобретению, которые приемлемы для парентерального введения, такого как, например, введение в сочленения (в суставы), внутривенное, внутримышечное, подкожное введение, введение через брюшину и через кожу, могут включать водные и неводные, изотонические стерильные инъекционные растворы, которые могут содержать антиокислители, буферные агенты, антибактериальные агенты и растворенные вещества, что делает композицию изотонической с кровью предполагаемого субъекта, и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты, растворяющие и загущающие агенты, стабилизаторы и консерванты. Среди приемлемых носителей и растворителей, которые могут быть применены, могут быть вода и раствор Рингера, изотонический хлорид натрия. Кроме того, стерильные нелетучие масла могут стандартно применяться как растворитель или суспендирующая среда. Для этой цели может быть использовано любое жидкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, могут применяться жирные кислоты, такие как олеиновая кислота, для приготовления препарата для инъекции. Эти растворы являются стерильными и обычно свободными от нежелаемых веществ.
Когда соединения являются достаточно растворимыми, они могут непосредственно растворяться в нормальном физиологическом растворе с растворителями и без применения приемлемых органических растворителей, таких как пропиленгликоль или полиэтиленгликоль. Дисперсии хорошо диспергированных соединений могут быть приготовлены в водном крахмале или растворе карбоксиметилцеллюлозы натрия, или в приемлемом масле, таком как арахисовое масло. Эти составы могут быть стерильными за счет применения стандартных хорошо известных методов стерилизации. Композиции могут содержать фармацевтически приемлемые вспомогательные вещества, которые требуются для приближения к физиологическим условиям, такие как агенты для регулирования рН, буферные агенты, агенты для регулирования токсичности, например ацетат натрия, хлорид натрия, хлорид калия, хлорид кальция, лактат натрия и т.п.
Концентрация карбамата в этих композициях может лежать в широких пределах и будет определяться в основном на основании объемов жидкости, вязкостей, веса тела и т.п. в соответствии с конкретным выбранным способом введения и состоянием пациента. Для IV введения композиция может быть приготовлена стерильной для инъекций, например стерильная впрыскиваемая водная или масляная суспензия. Эта суспензия может быть приготовлена в соответствии с известной практикой использования таких приемлемых диспергирующих или увлажняющих и суспендирующих агентов. Стерильной впрыскиваемой композицией также может быть стерильный впрыскиваемый раствор или суспензия в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как 1,3-бутандиол. Композиции могут быть представлены в единичной дозе или в виде герметичных контейнеров с многократной дозой, таких как ампулы и пузырьки. Растворы для инъекции и суспензии могут быть приготовлены из стерильных порошков, гранул и таблеток описанным выше способом.
Карбамат, приемлемый для применения настоящего изобретения на практике, может и, предпочтительно, должен вводиться орально. Количество соединения в композиции по настоящему изобретению может лежать в широких пределах в зависимости от типа композиции, размера единичной дозы, вида носителя и других факторов, хорошо известных специалистам в данной области. В целом, конечная композиция может содержать, например, от 0,000001 вес.% (вес.%) до 50 вес.% карбамата, предпочтительно, от 0,00001 вес.% до 25 вес.%, остальное является носителем или носителями.
Фармацевтические композиции для орального введения могут быть образованы с использованием фармацевтически приемлемых носителей, которые хорошо известны в области форм, приемлемых для орального введения. Такие носители приспособлены к фармацевтическим композициям, которые вводятся в единичные лекарственные формы, такие как таблетки, пилюли, порошки, драже, капсулы, жидкости, лепешки, гели, сиропы, кашицы, суспензии и т.д., приемлемые для проглатывания пациентом.
Композиции, приемлемые для орального введения, могут состоять из: (а) жидких растворов, таких как эффективное количество фармацевтической композиции, суспендированное в разбавителях, таких как вода, физиологический раствор или PEG 400; (б) капсул, пакетиков или таблеток, каждый из которых содержит предварительно определенное количество активного ингредиента в виде жидкостей, твердых веществ, гранул или в желатине; (в) суспензии в соответствующей жидкости и (г) приемлемых эмульсий.
Фармацевтические композиции для орального применения могут быть получены через комбинацию соединений с твердым носителем по настоящему изобретению выборочного измельчения образующейся смеси и переработки смеси гранул после добавления приемлемых дополнительных соединений, по желанию, с целью получения ядер таблеток и драже. Приемлемыми твердыми носителями являются углеводы или белковые наполнители, которые включают, но не ограничиваясь, сахара, включающие лактозу, сахарозу, маннит или сорбит; крахмал из кукурузы, пшеницы, риса, картофеля или других растений; целлюлозу, такую как метилцеллюлозу или гидроксиметилцеллюлозу, гидроксипропил-метилцеллюлозу или карбоксиметилцеллюлозу натрия; и смолы, включающие гуммиарабик и смолу трагаканта; а также белки, такие как желатин и коллаген.
По желанию, могут быть добавлены размягчающие или растворяющие агенты, такие как поперечно сшитый поливинилпирролидон, агар, альгиновая кислота или ее соль, такая как альгинат натрия. Таблеточные формы могут включать одно или более соединений, таких как лактоза, сахароза, маннит, сорбит, фосфаты кальция, кукурузный крахмал, картофельный крахмал, микрокристаллическую целлюлозу, желатин, коллоидную двуокись кремния, тальк, стеарат магния, стеариновую кислоту и другие носители, красящие вещества, наполнители, связующие, разбавители, буферные агенты, увлажнители, консерванты, отдушки, красители, дезинтегрирующие агенты и фармацевтически совместимые носители. Формы лепешек могут содержать активный ингредиент со вкусовой добавкой, например сахарозой, а также таблетки, содержащие активный ингредиент на инертной основе, такой как желатин и глицерин или сахарозу и эмульсии акации, гели и т.п., содержащие в дополнение к активному ингредиенту носители, известные в этой области техники.
Соединения по настоящему изобретению могут также вводиться в виде свечей через прямую кишку. Эти комбинации могут быть приготовлены путем смешения лекарства с приемлемым нераздражающим носителем, который при обычной температуре является твердым веществом и жидким при температуре прямой кишки и будет плавиться в кишечнике для высвобождения лекарства. Такими материалами являются масло какао и полиэтиленгликоли.
Соединения по настоящему изобретению могут также вводиться через нос, глаза, вагину и прямую кишку, включая свечи, составы для вдыхания, порошки и аэрозольные составы (например, стероидные вещества для ингаляции, см. публикацию Rohatagi, J. Clin. Pharmacol. 35: 1187-1193, 1995; Tjwa, Am. Allergy Asthma Immunol. 75: 107-111, 1995).
Соединения по настоящему изобретению могут доставляться через кожу топическим способом, как приготовленные в виде наклеек, растворов, суспензий, эмульсий, гелей, кремов, мазей, паст, желе, красок, порошков и аэрозолей.
Инкапсулирующие материалы могут также применяться с соединениями по настоящему изобретению и термин «композиция» может включать активный ингредиент в сочетании с инкапсулирующим материалом, таким как композиция с другим или без другого носителя. Например, соединения по настоящему изобретению могут также доставляться в виде микросфер для медленного высвобождения в организме. В одном варианте микросферы могут вводиться путем подкожных инъекций лекарства (например, мифепристон), содержащие микросферы, которые медленно подкожно высвобождают лекарство (см., например, публикацию Rao, J. Biomater. Sci. Polym. Ed. 7: 623-645, 1995); в виде биоразлагаемых и инъецируемых гелевых композиций (см., например, публикацию Gao, Pharm. Res. 12: 857-863, 1995); или в виде микросфер для орального введения (см., например, публикацию Eyies, J. Pharm. Pharmacol. 49: 669-674, 1997). Как подкожный способ введения, так и способ введения в кожу позволяют обеспечить постоянную доставку в течение недель или месяцев. Облатка также может быть использована для доставки соединений по настоящему изобретению.
В другом варианте соединения по настоящему изобретению могут доставляться путем использования липосом, которые сливаются с клеточной мембраной или эндоцитозируются, то есть при применении лигандов, присоединенных к липосомам, которые присоединяются к поверхности белкового рецептора мембраны клетки, что приводит к эндоцитозу. Активное лекарство может быть также введено в форме систем доставки липосом, таких как маленькие униламеллярные пузырьки, большие униламеллярные пузырьки и мульти-ламеллярные пузырьки. Липосомы могут быть получены из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолин.
При использовании липосом, особенно, когда поверхность липосомы несет лиганды специально для целевых клеток или другим способом, предпочтительно, направлена на специальный орган, можно нацелить доставку карбамата к целевым клеткам in vivo (см., например, Al-Muhammed, J. Microencapsul. 13: 293-306, 1996; Chonn, Curr. Opin. Biotechnol. 6: 698-708, 1995; Ostro, Аm. J. Hosp. Phann. 46: 1576-1587, 1989).
Фармацевтические композиции по настоящему изобретению могут содержать соли, которые могут быть образованы со многими кислотами, включая, но не ограничивая, соляную, серную, уксусную, молочную, винную, яблочную, янтарную кислоту и т.д. Соли имеют тенденцию быть более растворимыми в водных или других протонных растворителях, которые соответствуют свободным основным формам. В других случаях предпочтительной формой может быть лиофилизированный порошок, который может содержать, например, любой или все из следующих веществ: от 1 ммол до 50 ммол гистидина, от 0,1% до 2% сахарозы, от 2% до 7% маннита при величине рН от 4,5 до 5,5, которые объединены с буфером до применения.
Фармацевтически приемлемые соли и эфиры относятся к солям и эфирам, которые фармацевтически приемлемы и обладают желаемыми фармакологическими свойствами. Такие соли включают соли, которые могут быть образованы, когда протоны кислот присутствуют в соединениях, которые способны к взаимодействию с неорганическими или органическими основаниями. Приемлемые неорганические соли включают соли, образованные со щелочными металлами, например с натрием, калием, магнием, кальцием и алюминием. Приемлемые органические соли включают соли, образованные с органическими основаниями, такими как аминовые основания, например этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п. Фармацевтически приемлемые соли могут также включать соли присоединения кислот, образованные по реакции аминогрупп в родительском соединении с неорганическими кислотами (например, хлористоводородной и бромисто-водородной кислотами) и органическими кислотами (например, уксусной, лимонной, малеиновой кислотами, алкан- и арен-сульфокислотами, такими как метансульфоновая кислота и бензолсульфоновая кислота). Фармацевтически приемлемые эфиры включают эфиры, образованные из карбокси-, сульфонилокси- и фосфоноксигрупп, присутствующих в соединениях. Когда присутствуют две кислотные группы, фармацевтически приемлемая соль или эфир могут быть монокислотой-моно солью или эфиром, или дисолью, или диэфиром; и также, когда присутствуют более чем две кислотные группы, несколько или все такие группы могут образовывать соль или этерифицироваться.
Соединения, указанные в данном изобретении, могут присутствовать в несолевой или неэтерифицированной форме, или в солевой или этерифицированной форме, и наименование таких соединений, как подразумевается, включает как оригинальное (несолевое или неэтерифицированное) соединение, так и его фармацевтически приемлемые соли и эфиры. Настоящее изобретение включает фармацевтически приемлемую солевую и эфирную формы с формулой I. Более чем одна кристаллическая форма энантиомера с формулой I может иметь место как таковая и быть включена в настоящее изобретение.
Фармацевтическая композиция по настоящему изобретению может выборочно содержать в добавление к карбамату, по меньшей мере, один другой терапевтический агент, полезный при лечении EDS. Например, карбаматы формулы I могут быть физически объединены с другими активирующими или стимулирующими соединениями в фиксированных комбинациях доз с целью упрощения их введения.
Методы образования фармацевтических композиций описаны в многочисленных публикациях, таких как Pharmaceutical Dosage Forms: Tablets. Second Edition. Revised and Expanded. Volumes 1-3, edited by Lieberman et al; Pharmaceutical Dosage Forms: Parenteral Medications. Volumes 1-2, edited by Avis et al.; and Pharmaceutical Dosage Forms: Disperse Systems. Volumes 1-2, edited by Lieberman et al; опубликованные Marcel Dekker, Inc.
Фармацевтические композиции обычно производятся стерильными, фактически изотоническими и в полном соответствии со всеми правилами Good Manufacturing Practice (GMP) of The US Food and Drug Administration.
Схемы приема
Настоящее изобретение представляет способы лечения EDS и связанных с ним состояний у млекопитающих с применением карбаматов. Количество карбаматов, необходимое для обеспечения лечения EDS и связанных с ним состояний, определяется как терапевтически или фармацевтически эффективная доза. Схема приема и количества, эффективные для этого, например дозировка и схема приема, будут зависеть от множества факторов, включающих стадию болезни, физическое состояние пациента, возраст и т.п. При составлении схемы приема для пациента способ введения будет также приниматься во внимание.
Специалист в этой области будет иметь возможность без ненужного исследования, имея опыт и данное описание, определить терапевтически эффективное количество конкретного замещенного карбамата для практического применения настоящего изобретения, см., например, Lieberman, Pharmaceutical Dosage Forms: (Vols 1-3, 1992); Lloyd, 1999, The art, Science and Technology of Pharmaceutical Compounding; and Pickar, 1999, Dosage Calculations). Терапевтически активная доза является той дозой, при которой любые токсические или приносящие вред побочные эффекты активного агента, перекрываются в клинических условиях терапевтически полезными эффектами. Кроме того, должно быть отмечено, что для каждого конкретного субъекта должны быть оценены и подобраны во времени специальные схемы приема в соответствии с индивидуальной потребностью и профессиональной оценкой лица, осуществляющего введение или контроль введения соединения.
В целях лечения композиции или соединения, описанные в настоящей заявке, могут вводиться субъекту путем доставки в виде единичного болюса, через непрерывную доставку в течение продолжительного периода времени, или в режиме повторяющегося введения (например, ежечасно, ежедневно или еженедельно повторяющиеся схемы введения). Фармацевтические композиции по настоящему изобретению могут вводиться, например, один или более раз в день, 3 раза в неделю или еженедельно. В одном варианте данного изобретения фармацевтические композиции по настоящему изобретению вводятся орально один или два раза в день.
В этом контексте терапевтически эффективная доза биологически активного (активных) агента (-ов) может включать повторные дозы при пролонгированной схеме лечения, что принесет клинически значимые результаты лечения EDS и связанных состояний. Определение эффективных доз в этом контексте обычно основывается на опытах на моделях животных и последующих клинических испытаниях на людях и руководствуется расчетом эффективных доз и схемам введения, которые значительно уменьшают возникновение или развитие выявленных симптомов или состояний у субъекта. Приемлемые модели в этом контексте включают, например, модели мышей, крыс, свиней, кошек, приматов и других животных, известные в этой области.
Альтернативно, эффективные дозы могут определяться с использованием моделей in vitro (например, иммунологические и гистопатологические анализы).
Используя такие модели, проводят только обычные расчеты и подбор для того, чтобы определить подходящую концентрацию и дозу для введения терапевтически эффективного количества биологически активного агента (агентов), (например, количества, которые эффективны для достижения желаемой реакции при введении через нос, через кожу, внутривенно или внутримышечно).
В приведенном варианте по настоящему изобретению единичные лекарственные формы приготавливаются для стандартных схем введения. В этом случае композиция может быть легко физически разделена на более мелкие дозы. Например, единичные дозы могут быть приготовлены в виде пакетиков порошков, пузырьков или ампул, предпочтительно, в форме капсул или таблеток.
Активное соединение в таких единичных лекарственных формах композиции может находится в количестве, например, от около 10 мг до около 1 г или более, для одного или многих ежедневных введений в соответствии с конкретной необходимостью пациента. Начиная схему лечения с минимальной дозы около одного грамма, содержание карбамата в крови можно использовать, чтобы определить большую или меньшую дозу.
Эффективное введение карбаматов по настоящему изобретению может быть, например, проведено с оральной или парентеральной дозой от около 0,01 мг/кг/дозу до около 150 мг/кг/дозу. Предпочтительно, введение будет проведено с дозой от около 0,1 мг/кг/дозу до около 25 мг/кг/дозу, более предпочтительно, от около 0,2 мг/кг/дозу до около 18 мг/кг/дозу. Поэтому терапевтически эффективное количество активного ингредиента, содержащегося в единичной дозе, как это описано в данной заявке, может быть, например, от около 1 мг/день до около 7000 мг/день для субъекта, имеющего, например, средний вес 70 кг.
Методы по настоящему изобретению также предусматривают наборы для проведения лечения EDS и сопутствующих состояний. После того, как фармацевтическая композиция, содержащая один или более карбаматов по настоящему изобретению с возможным добавлением одного или более других соединений для получения терапевтического эффекта, была приготовлена в приемлемом носителе, она может быть помещена в соответствующий контейнер и замаркирована для обеспечения лечения EDS и сопутствующих состояний. Кроме того, другая фармацевтическая композиция, содержащая, по меньшей мере, еще один терапевтический агент, может быть помещена в контейнер, а также замаркирована для использования при лечении указанной болезни. Такая маркировка может включать, например, инструкции относительно количества, частоты и способа введения каждой фармацевтической композиции.
Хотя описанное выше изобретение было подробно описано путем приведения примеров в целях четкого понимания, специалистам будет очевидно, что могут быть сделаны некоторые изменения и модификации и могут быть применены без дополнительных исследований в объеме прилагаемой формулы изобретения. Следующие примеры обеспечивают иллюстрацию специфических аспектов изобретения и не предполагают наличие ограничений.
Пример.
Цель изучения.
Настоящее исследование предпринято с целью определения влияния (D)- или (R)-энантиомера фенилалкиламинокарбамата с формулой I, особенно, О-карбамоил-(D)-фенилаланинола, который также может быть назван как (R)-(бета-аминобензолпропил)карбамат, показанный выше в виде формулы Ib, и определенный в данной заявке как «ИСПЫТУЕМОЕ СОЕДИНЕНИЕ», на состояние сон - бодрствование у крыс после его быстрого введения и сравнения с влиянием амфетамина и кокаина.
Для того чтобы охарактеризовать профиль активности испытуемого соединения в процессе сна - бодрствования у крыс, животным постоянно имплантировали электроды для записи кортикальной электроэнцефалограммы, активности шейных мышц под действием электрического тока и движения глаз, при этом одновременно регистрировалось движение всего тела. Во-вторых, достигаемые эффекты сравнивали с эффектами, полученными при применении двух эталонных психостимулирующих лекарств, кокаина и амфетамина. На основе таких полисомнографических записей можно надежно обнаруживать изменения в процессе сна - бодрствования. Последующий анализ характера изменений позволил предсказать класс психотропных агентов, который больше всего напоминает исследуемое соединение (см. Ruigt G.S. et al. (1993) Neuropsycho-biology, 28(3): 138-153).
Материалы и способы
Животные
Опыты проводили на взрослых самцах крыс Spraque Dawley, поставляемых Harian (Borchen, Germany), весом 240-260 г во время операции. Животных помещали в плексигласовые клетки (25×33×18 см), которые подходят к IVС-кормушкам (индивидуально вентилируемые клетки = IVC), расположенным в камере с затухающим звуком. Крыс снабжали микрочипом с целью идентификации и выдерживали в контролируемых условиях во время исследования: 22±2°С, относительная влажность 60%, 12: 12 цикл свет - темнота (освещение от 12 ч до 00 ч; интенсивность света - 100 люкс) при снабжении обычной лабораторной пищей и водопроводной водой в свободном доступе. Все процедуры были одобрены комитетом охраны животных.
Хирургическое вмешательство.
В условиях анестезии путем ингаляции изофурана крыс помещали в стереотаксическое устройство. Овальную часть скальпа удаляли и череп очищали от периоста. В черепной кости просверливали три небольшие полости без перфорации твердой мозговой оболочки для помещения трех фиксирующих винтов из нержавеющей стали (диаметр 1 мм) для полиграфической записи фронтальной и париетальной электроэнцефалограмм (EEG). Два электрода помещали стереотаксически на каждую сторону сагиттального шва (АР + 2 мм, L - 2 мм; и АР - 6 мм, L - 3 мм от брегмы), в то время как третий (эталонный) электрод ввинчивали над мозжечком. Резцовая пластинка была расположена на расстоянии около 5 мм под центром ушной складки согласно стереотаксическому атласу Paxinos G. & Watson С. The Rat Brain in Stereotaxic Coordinates, Academic Press, San Diego, CA, USA (1998).
Для записи электроокулограммы (EOG) и электромиограммы (EMG) проволочки из нержавеющей стали помещали в периорбитальное пространство и вставляли в задней части шеи соответственно. Электроды (проволочки из нержавеющей стали, 7N51465T5TL7, 51/46 Teflon Bilaney, Germany) были связаны с штифтом (Future Electronics: 0672-2-15-15-30-27-10-0) при помощи небольшой вставки (track pins; Dataflex: TRP-1558-0000) и были вставлены в коннектор с 8 отверстиями. Затем электроды были зафиксированы дентальным клеем в черепе.
Животным давали привыкнуть к обстановке, по меньшей мере, в течение одной недели.
Методика записи сна и фармакологический тест.
Через 10 дней после хирургического вмешательства животные адаптировались в течение двух недель к процедуре записи в своих клетках. Крыс с регулярными промежутками соединяли кабелем с вращающимся шарнирным соединением, позволяющим осуществлять свободные движения во время записи EEG, EOG и EMG.
Во время испытаний использовали только тех крыс, которые отвечали требуемым критериям, то есть имели вес 300-700 г, было хорошее качество полиграфического сигнала, период промывки длился, по меньшей мере, 14 дней в случае повторного использования животного и два следующих друг за другом опыта были удачными. Для каждого соединения осуществляли две записи EEG для 32 оперированных животных, которых случайно распределяли по условиям на 4 группы (8 крыс в каждой группе).
Первую операцию записи начинали в 14.00 ч и продолжали 16 ч после введения физиологического раствора (n=32 крысы). Вторую операцию записи проводили в течение того же промежутка времени после введения физиологического раствора и различных доз ИСПЫТУЕМОГО СОЕДИНЕНИЯ (1, 3 и 10 мг/кг), кокаина (3, 10 и 30 мг/кг интраперитонеально) или амфетамина (3, 10 и 30 мг/кг интраперитонеально). Все соединения растворяли в физиологическом растворе и вводили в объеме 10 мл/кг веса. В качестве контроля вводили эквивалентный объем физиологического раствора. Мониторинг EEG, BOG, EMG и движения крыс проводили в течение 16 ч. Получение данных осуществляли при частоте 200 Гц. Все сигналы проходили через систему биполярного записывающего прибора (Embia), разработанную MedCare (Iceland), в компьютер, применяли программу Somnologica, MedCare, Iceland, которая превращала компьютер в полиграфическое устройство для записи сигналов.
Анализ состояния сон - бодрствование.
Автоматизированная система анализа сна у крыс непрерывно работала в течение 16 ч после инъекции соединения. Изучение состояния сон - бодрствование проводили автономно в автоматическом режиме в течение 30 мин на основе значений доменов 5 EEG частоты (δ: 0,4-4 Гц; Θ: 4,2-8 Гц; α: 8,2-12 Гц; σ: 12,2-14 Гц; β: 14,2-30 Гц), интегрированных EMG, EOG и активности движений крыс.
Различительный анализ использует правила классификации для последней стадии сна каждого конкретного периода EEG. Шесть стадий сна классифицировал как активное бодрствование (AW), пассивное бодрствование (PW), легкий сон с медленными волнами (ISWS), глубокий сон с медленными волнами (dSWS), промежуточную стадию (IS) или сон с быстрыми движениями глазных яблок (REMS). Вкратце различные стадии активности характеризовали как: AW, низковольтную быструю EEG активность, высоковольтную EMG активность, активность с многочисленными движениями глаз и высокую активность организма; PW, низковольтную быструю EEG активность, от высокой до умеренной EMG активность, многочисленные движения глаз и неподвижность тела; ISWS, высоковольтные медленные кортикальные волны, прерываемые низковольтными быстрыми волнами, и пониженную EMG активность; непрерывную активность с медленными волнами и высокой амплитудой на EEG в отсутствие EMG, EOG и активности организма; IS: временная веретенообразная активность с тета-ритмом, отсутствие EOG и движений тела; REMS; низковольтные быстрые кортикальные волны с регулярным тета-ритмом, наличие быстрых движений глаз и отсутствие движения мышц и тела.
Показатели были синхронизированы во времени с EEG сигналом, и система рассчитывала автоматически различные параметры сна - бодрствования, такие как количество времени пребывания в каждом состоянии, количество и продолжительность эпизодов в каждом состоянии активности, латентность для ISWS, dSWS и REMS и число переходов от одного состояния к другому. Для каждой фазы сна латентность определялась как время между началом записи и возникновением первого периода сна, продолжающегося, по меньшей мере, 30 с.
Статистический анализ
Время, проведенное в каждом состоянии активности (AW, PW, ISWS, dSWS, IS и REMS) выражали в %% от времени записи. Статистический анализ полученных данных проводили непараметрическим анализом вариабельности в 30-минутные периоды с последующим определением суммы рангов Уилкоксона - Манна - Уитни по сравнению с контрольной группой.
Действие ИСПЫТУЕМОГО СОЕДИНЕНИЯ
Прием ИСПЫТУЕМОГО СОЕДИНЕНИЯ приводил к значительным изменениям в распределении периодов сна - бодрствования.
Легкое изменение структуры состояния сон - бодрствование наблюдалось во все 16 ч записи после введения самой низкой дозы соединения (3 мг/кг интраперитонеально). Наблюдали увеличение всего периода легкого сна (+26%, р<0,05) и повышение стремления к бодрствованию от легкого сна, а также от глубокого сна (+46%, р<0,001; +15%, р<0,05, соответственно), что свидетельствовало о фрагментации сна после приема этой дозы соединения (р<0,05) (см. Таблицу 4).
При дозе 10 мг/кг и.п. ИСПЫТУЕМОЕ СОЕДИНЕНИЕ приводило к изменениям в структуре сна - бодрствования, связанным со значительным увеличением общей продолжительности легкого сна (+24%, р<0,05) и значительным увеличением числа переходов от «быстрого» (REM) сна к активному бодрствованию (+16%, р<0,05) (см. Таблицы 2 и 4).
Во время первых 90 мин записи наблюдалось значительное уменьшение периода глубокого сна в увеличением времени активного бодрствования (р<0,05).
В случае самой большой дозы (30 мг/кг и.п.) испытуемое соединение вызывало ярко выраженные изменения в распределении цикла сон - бодрствование. Происходили заметное увеличение общего времени активного бодрствования (+19%, р<0,05), уменьшение общего времени пассивного бодрствования (-29%, р<0,05), легкого сна (-20%, р<0,05), а также «быстрого» сна (-25%, р<0,05) в течение 16 ч после инъекции во время записи (см. Таблицу 2).
Кроме того, по сравнению с общим временем сна ИСПЫТУЕМОЕ СОЕДИНЕНИЕ вызывало увеличение времени, проводимого в глубоком сне, и уменьшало продолжительность «быстрого» сна (р<0,05) (см. Таблицу 4).
Значительное увеличение периода активного бодрствования наблюдали в течение первых 3 ч после введения ИСПЫТУЕМОГО СОЕДИНЕНИЯ (р<0,01). Вместе с этим происходило значительное уменьшение времени, проведенного во сне, например легком сне (р<0,01), глубоком сне (р<0,01) и «быстром» (REM) сне (р<0,01), сопровождаемое обратным эффектом, в частности увеличением периода глубокого сна через 3 ч после введения ИСПЫТУЕМОГО СОЕДИНЕНИЯ. Последнее продолжалось примерно 7 ч во время записи в светлое время. Следует отметить, что начало активности ИСПЫТУЕМОГО СОЕДИНЕНИЯ было почти немедленным, а именно примерно через первые 30 мин после его введения.
Значительное увеличение общего времени активного бодрствования и уменьшение периода пассивного бодрствования, легкого сна и REM сна были вызваны увеличением (+19%, р<0,05) и уменьшением (-30%, р<0,05; -23%, р<0,05, -24%, р<0,01) числа этих стадий сон - бодрствование соответственно. Однако средняя продолжительность этих стадий сон - бодрствование не изменилась. Как показывают результаты (см. Таблицу 4), ИСПЫТУЕМОЕ СОЕДИНЕНИЕ с дозой 30 мг/кг приводило к увеличению числа переходов от легкого сна и «быстрого» сна к бодрствованию (р<0,05) и это предполагает наличие фрагментации сна. Изучение латентности сна выявило значительные изменения после введения ИСПЫТУЕМОГО СОЕДИНЕНИЯ. Это СОЕДИНЕНИЕ с дозой 10 и 30 мг/кг привело к значительному увеличению латентности «быстрого» (REM) сна.
Действие кокаина.
Основные модификации структуры сна после введения кокаина наблюдались при самой большой испытанной дозе, а именно уменьшение общего времени REM сна (-18%) и увеличение общей продолжительности активного бодрствования (+14%) в течение 16 ч записи после лечения. Кроме того, при различных испытанных дозах кокаин не влиял на общую продолжительность сна, а также на количество переходов от сна к бодрствованию.
Кокаин в дозе 10 мг/кг приводил к значительному увеличению продолжительности активного бодрствования в течение трех часов после лечения (0,5 ч: +111%, р<0,001; 1 ч: +500%, р<0,01; 1,5 ч: +312%, р<0,01; 2 ч: +120%, р<0,001; 2,5 ч: +166%, р<0,001; 3 ч: +77%, р<0,001). Одновременно, время, проведенное в легком сне, а также в глубоком сне, уменьшилось в течение первых 2 ч записи (0,5 ч: -100% и 90%, р<0,001; 1 ч: -99% и -100%, р<0,001; 1,5 ч: -87% и -99%, р<0,001; 2 ч: -25% и -70, р<0,05 и р<0,001 соответственно). Продолжительность REM («быстрого») сна значительно снизилась в течение первых 3 ч после введения (0,5 ч, 1 ч, 1,5 ч: каждый, -100%, р<0,001; 2 ч: -87%, р<0,001; 2,5 ч: -47%, р<0,00; 3 ч: -78%, р<0,001).
Повышенная продолжительности активного бодрствования после введения кокаина с дозой 10 мг/кг возникла из-за увеличения числа периодов (0,5 ч: +111%, р<0,001; 1 ч: +500%, р<0,001; 1,5 ч: +312%, р<0,001; 2 ч: +119%, р<0,001; 2,5 ч: +166%, р<0,05; 3 ч: +77%, р<0,001), в то время как средняя продолжительность этого состояния не изменилась.
Уменьшение времени пребывания в легком сне и глубоком сне в течение первых 2 ч записи было обусловлено уменьшением числа периодов этих состояний (0,5 ч: -100% и -100%, р<0,001; 1 ч: -99% и -100%, р<0,01; 1,5 ч: -87% и -100%, р<0,001; 2 ч: -22% и -38%, р<0,05 соответственно). Точно также уменьшение продолжительности REM («быстрого») сна в течение первых 3 ч возникло вследствие уменьшения числа периодов этого состояния (0,5, 1 ч, 15 ч: каждый, -100%, р<0,001; 2 ч: -87%, р<0,001; 2,5 ч: -47%, р<0,00; 3 ч: -78%, р<0,001) соответственно.
Как показано в Таблице 1, латентность начала REM сна изменялась в зависимости от дозы (р<0,05).
Действие амфетамина
В течение всего периода записи (16 ч) амфетамин в дозе 1, 3 и 10 мг/кг приводил к значительным изменениям структуры сон - бодрствование. В зависимости от дозы амфетамин увеличивал общее время, проведенное в состоянии активного бодрствования (+27%, р<0,05; +47%, р<0,001; +66%, р<0,001), глубокого сна (+73%, р<0,05; +91%, р<0,05; +66%, р<0,001) и уменьшал общее время легкого сна (-35%, р<0,05; -49%, р<0,05; -51%, р<0,001) и REM сна (-4%, -22%, р<0,05; -41%, р<0,001) соответственно (см. Таблицу 2). Более того, по сравнению с носителем амфетамин в дозе 3 и 10 мг/кг пропорционально снижал общее время сна (р<0,01) и время легкого сна, в то время как соединение приводило к увеличению доли глубокого сна по сравнению с общим временем сна (р<0,05) (см. Таблицу 3). Значительное увеличение периода активного бодрствования и глубокого сна после введения 1, 3 и 10 мг/кг амфетамина возникло вследствие увеличения числа периодов активного бодрствования (+27%, р<0,05; +47%, р<0,001; +66%, р<0,001 соответственно) и периодов глубокого сна (+73%, р<0,001; +91%, р<0,001; +66%, р<0,001). Хотя средняя продолжительность активного бодрствования не менялась при различных дозах соединения, средняя продолжительность стадии глубокого сна уменьшалась после введения 3 и 10 мг/кг соединения (~ 19% р<0,05; -30%, р<0,05).
Уменьшение общего времени легкого SWS и RЕМ сна после введения 1, 3 и 10 мг/кг амфетамина было вызвано уменьшением числа легкого сна (-35%, р<0,05; -49%, р<0,001; -51%, р<0,001 соответственно) и периодов RЕМ сна (-4%, -22%, р<0,05; -41%, р<0,001 соответственно). Средняя величина уменьшения стадии RЕМ сна уменьшилась (-16%, р<0,05; -23%, р<0,05; -36%, р<0,05, соответственно), в то время как этот параметр незначительно менялся в случае легкого сна. Амфетамин способствовал увеличению периода активного бодрствования четко в зависимости от дозы в течение 3, 4 и 6 ч (1, 3 и 10 мг/кг соответственно, р<0,05). При этом зависимое от дозы снижение продолжительности легкого, глубокого и RЕМ сна наблюдалось в течение 3, 4 и 6 ч (р<0,05 соответственно) после введения.
Амфетамин оказывает двухфазное действие на продолжительность стадии глубокого сна, а именно она значительно уменьшалась в течение 3-6 ч после инъекции и затем возрастала как, вероятно, обратный эффект во время периода записи при освещении.
Как указано в Таблице 1, амфетамин значительно влиял на параметры сна, удлиняя латентность начала стадии сна (р<0,001).
Результаты
После введения ИСПЫТУЕМОГО СОЕДИНЕНИЯ в дозе 3 и 10 мг/кг отмечались незначительные изменения состояния активности. Однако введение ИСПЫТУЕМОГО СОЕДИНЕНИЯ в дозе 30 мг/кг сильно увеличивало период активного бодрствования за счет времени легкого сна, глубокого сна и RЕМ сна в течение первых 3-4 ч после введения. Обратный эффект был отмечен через 4-10 ч после введения соединения как увеличение времени глубокого сна, которое затем постепенно уменьшалось с течением времени. Более того, ИСПЫТУЕМОЕ СОЕДИНЕНИЕ влияло на другие параметры сна - бодрствования; более конкретно, оно значительно повышало число переходов от легкого сна и REM сна к бодрствованию, а также удлиняло период латентности начала REM сна. Кокаин в дозе 1 и 3 мг/кг лишь слегка влиял на структуру сна - бодрствования. В противоположность этому, введение кокаина в дозе 10 мг/кг значительно повышало период активного бодрствования и уменьшало периоды коротковолнового сна и REM сна в течение первых 3-4 ч после инъекции соединения. Все периоды латентности сна увеличивались. Увеличенный в зависимости от дозы амфетамина период бодрствования и уменьшенные периоды всех стадий сна наблюдались в течение 3-8 ч после введения. Для стадии глубокого сна отмечали четко выраженный зависимый от дозы обратный эффект. Кроме того, значительно увеличивались периоды латентности всех стадий сна.
Данные исследования показывают, что почти сразу же после интраперитонеальной инъекции ИСПЫТУЕМОГО СОЕДИНЕНИЯ оно было активным в течение, по меньшей мере, 4 ч с пиком активности примерно через 2 ч после введения. При применении самой низкой дозы, равной 3 мг/кг, были отмечены только незначительные изменения структуры состояний сна - бодрствования. Изменения параметров сна наблюдалось в случае средней (10 мг/кг) дозы и более выражено при более высокой испытанной дозе, равной 30 мг/кг. Изменения в распределении периодов сна - бодрствования, которые были наиболее очевидными в течение первых 3 ч периода записи, характеризовались значительным увеличением времени активного бодрствования, в то время как время, проведенное в состоянии пассивного бодрствования, легкого сна, глубокого сна и REM сна, уменьшилось. Интересно, что ИСПЫТУЕМОЕ СОЕДИНЕНИЕ приводило к обратному эффекту восстановления глубокого сна, связанного со значительным увеличением времени, проведенного в этом состоянии до 7 ч.
Результаты, полученные во время этого сравнительного исследования, четко показывают, что ИСПЫТУЕМОЕ СОЕДИНЕНИЕ в дозе 30 мг/кг имеет свойства психостимулянта в начале введения и затем приводит к увеличению предрасположенности ко сну, что показывает увеличение периода глубокого сна и активное косвенное влияние на гомеостаз сна.
Полный профиль действия ИСПЫТУЕМОГО СОЕДИНЕНИЯ в дозе 30 мг/кг был очень похож на профиль, наблюдаемый после введения амфетамина с самой низкой дозой, равной 1 мг/кг, по эффекту, размеру и продолжительности.
Следовательно, ИСПЫТУЕМОЕ СОЕДИНЕНИЕ на крысах обладает центральным действием сразу же после инъекции, что выражается в изменениях структуры состояний сна - бодрствования с функциональным пиком эффекта примерно через 2 ч после и.п. введения. Результаты показывают, что действие ИСПЫТУЕМОГО СОЕДИНЕНИЯ является двухфазным, то есть первоначально увеличение периода бодрствования и уменьшение периода сна сменялось увеличением продолжительности (глубокого) сна, наиболее вероятно являющимся обратным эффектом, которое длилось 4-10 ч. Эти данные очень похожи на действие на структуру состояний сна - бодрствования, наблюдаемое после введения психостимулирующих лекарств, более конкретно амфетамина, в самой низкой испытанной дозе (а именно, 1 мг/кг, и.п.). Следовательно, можно предположить, что ИСПЫТУЕМОЕ СОЕДИНЕНИЕ обладает стимулирующими свойствами сразу же после введения.
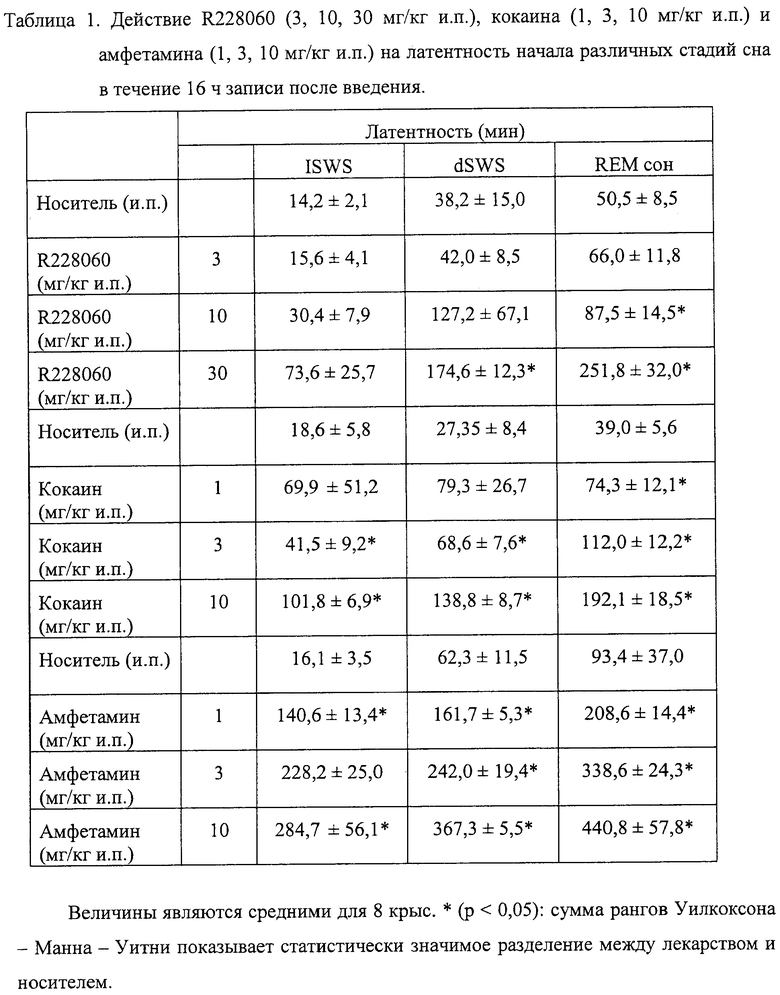
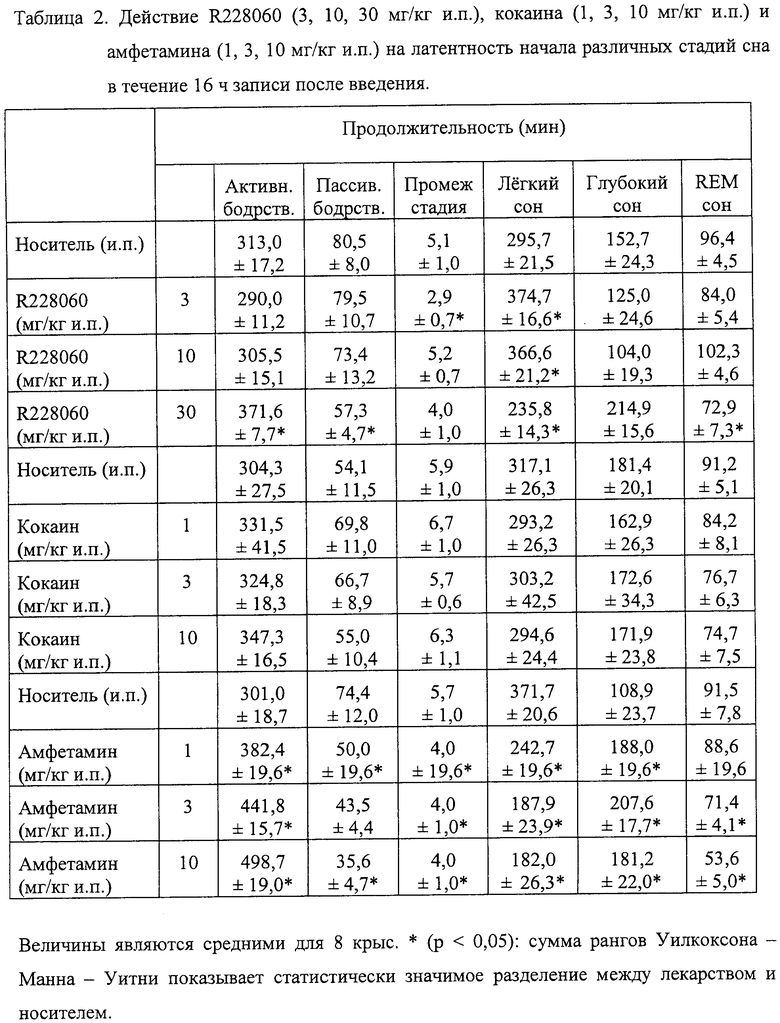
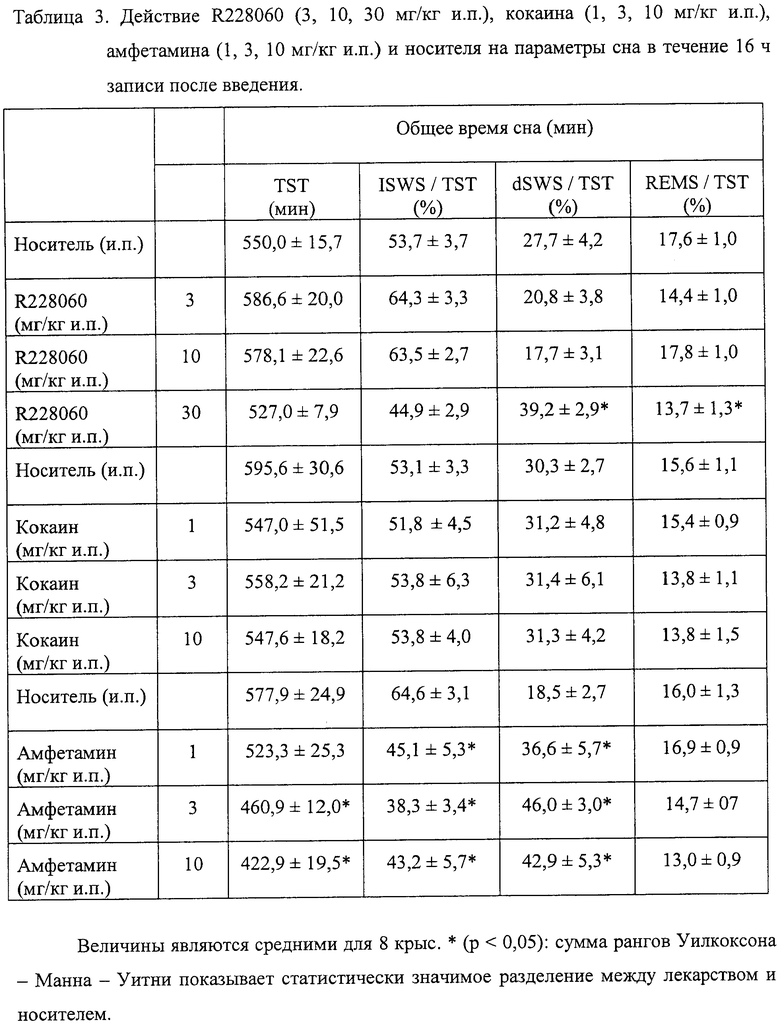
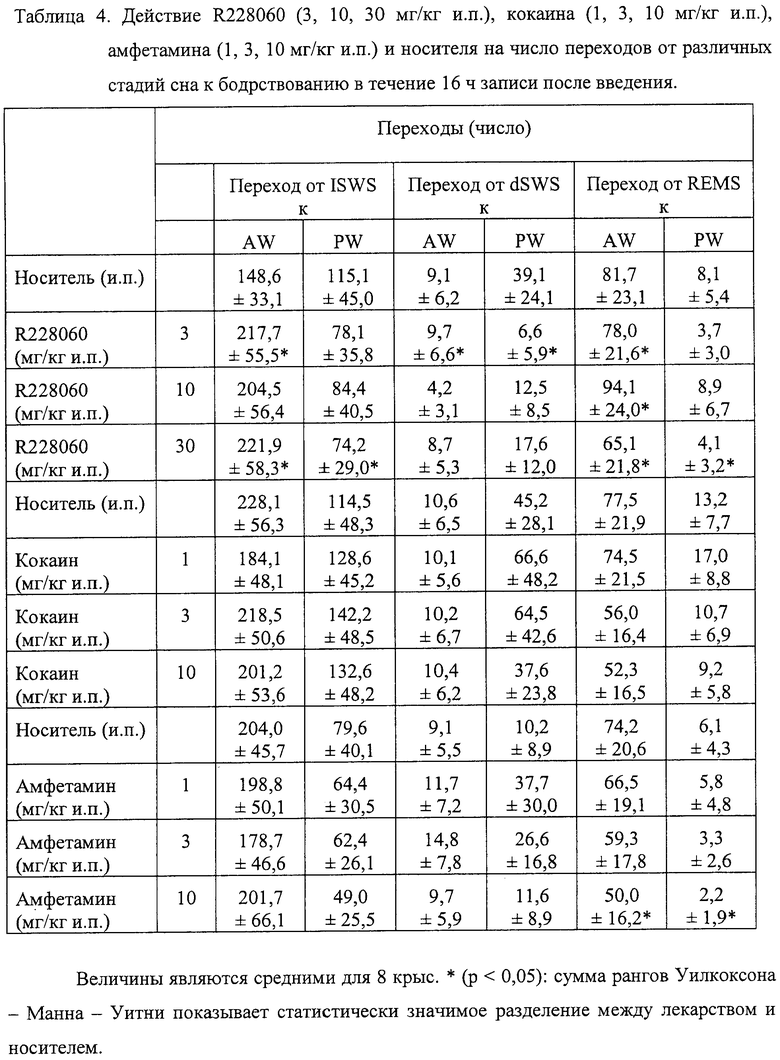
Данное изобретение не ограничено конкретными вариантами, описанными в данной заявке, которые приведены лишь для иллюстрации отдельных аспектов данного изобретения. Не выходя за рамки изобретения, специалисты в данной области могут осуществить многие модификации и изменения данного изобретения. Из приведенного выше описания специалистам очевидны функционально эквивалентные способы и средства в дополнение к перечисленным в данной заявке. Такие модификации и изменения должны входить в объем изобретения, определяемый формулой изобретения с учетом эквивалентов.
Изобретение относится к медицине и предназначено для лечения чрезмерной дневной сонливости (EDS). Вводят терапевтически эффективное количество (R) энантиомера бета-аминобензопропил карбамата или энантиомерной смеси, в которой (R) энантиомер преобладает. Способ позволяет увеличить период активного бодрствования и увеличить время глубокого сна. 5 з.п. ф-лы, 4 табл.
1. Способ лечения чрезмерной дневной сонливости у субъекта, включающий введение субъекту терапевтически эффективного количества (R) энантиомера формулы Ib (бета-аминобензопропил) карбамата или энантиомерной смеси, в которой (R) энантиомер преобладает
2. Способ по п.1, отличающийся тем, что (R) энантиомер преобладает в количестве, равном до примерно 90% или более.
3. Способ по п.1, отличающийся тем, что (R) энантиомер преобладает в количестве, равном до примерно 98% или более.
4. Способ по п.1, отличающийся тем, что чрезмерная дневная сонливость выбрана из группы, включающей патологические аномалии центральной нервной системы (ЦНС), удар, нарколепсию, идиопатическую гиперсомнию ЦНС, дефицит сна, синдром апноэ во сне, обструктивный синдром апноэ во сне, недостаточный ночной сон, хроническую боль, острую боль, болезнь Паркинсона, недержание мочи, усталость от рассеянного склероза, синдром гиперактивности и дефицита внимания (ADHD), болезнь Альцгеймера, большую депрессию, биполярное расстройство, ишемическую болезнь сердца, несогласованность работы циркадного ритмоводителя с окружающей средой, нарушение биологических ритмов организма при перелетах на реактивных самолетах, расстройства, вызванные сменной работой и седативными лекарствами.
5. Способ по п.4, отличающийся тем, что причиной является нарколепсия.
6. Способ по п.1, отличающийся тем, что терапевтически эффективное количество энантиомера (R)-(бета-аминобензопропил)карбамата составляет от примерно 3 мг/кг/дозу до примерно 30 мг/кг/дозу.
| WO 9624577, 08.02.1996 | |||
| WO 9815526, 16.04.1998 | |||
| МОДАФИНИЛ С ОПРЕДЕЛЕННЫМ РАЗМЕРОМ ЧАСТИЦ | 1995 |
|
RU2171674C2 |
| МОСОЛОВ С.Н | |||
| Современные антидепрессанты: механизмы действия и клиническое применение | |||
| Фарматека, 2003, №4 | |||
| Неврология и психиатрия, с.31-40 | |||
| BLACK J | |||
| Medications for the treatment of narcolepsy | |||
| Expert Opin | |||
| Emerg | |||
| Drags., 2001, Oct., №6, c.239-47, он-лайн, | |||

