Область техники
Данное изобретение относится к способу получения каталитической композиции по меньшей мере из одного неблагородного металла VIII группы и по меньшей мере одного металла группы VIB Периодической таблицы. Каталитическая композиция, которая также является предметом данного изобретения, обладает высокой удельной активностью в реакциях гидроочистки легких и средних фракций, предпочтительно таких реакциях гидроочистки углеводородных потоков, как гидродесульфирование (HDS), гидроденитрификация (HDN) и гидродеароматизация (HDA).
Уровень техники
Правительственные агентства по защите окружающей среды требуют, чтобы все больше топлив содержали среди прочего пониженные количества компонентов предшественников, загрязняющих атмосферу, таких как сера и азот, и низкие количества металлов - никеля и ванадия. Кроме того, чтобы максимально сохранить запасы нефти, необходимо обрабатывать все более и более тяжелые фракции, так что содержание указанных веществ, загрязняющих атмосферу, в полученных топливах возрастает; поэтому необходимо разрабатывать новые каталитические процессы и материалы, с помощью которых можно более эффективно удалять эти загрязняющие вещества из углеводородов или ископаемого топлива, с тем чтобы минимизировать выбросы газов в атмосферу и тем самым удовлетворить все более жестким экологическим требованиям.
Наиболее эффективными промышленными способами удаления загрязняющих веществ из ископаемого топлива являются способы гидроконверсии, которые применимы практически ко всем нефтяным фракциям, таким как газолин, дизельное топливо, сырье для каталитического крекинга (FCC) и промежуточные дистиллаты. В конкретном случае данного изобретения считается, что из легких и промежуточных нефтяных фракций можно получать углеводороды с температурами кипения, равными или ниже 180°С, а из промежуточных нефтяных фракций можно получать углеводороды с температурами кипения, равными или выше 180.1°С и ниже или равными 400°С.
В способах гидроконверсии легкие и промежуточные нефтяные фракции подвергают гидроочистке и/или гидрокрекингу в присутствии водорода. Способы гидроконверсии включают все способы, в которых часть углеводородов взаимодействует с водородом при высокой температуре и давлении, такие как: гидрирование, гидродесульфирование, гидроденитрификация, гидродеметаллирование, гидродеароматизация, гидроизомеризация и гидрокрекинг.
Используемые для этого катализаторы в основном состоят по меньшей мере из одного неблагородного металла VIII группы и по меньшей мере одного металла группы VIB Периодической таблицы, нанесенных на носитель с развитой удельной поверхностью, получаемый из оксидов металлов, таких как оксид алюминия, оксид кремния, оксид титана и/или их смеси, необязательно содержащие вторичные промоторы или добавки, такие как галогены, фосфор, бор и т.д. Катализаторы обычно готовят пропиткой носителя водными растворами, содержащими нужные соединения металлов, с последующей сушкой и прокаливанием. Способы получения катализаторов гидроочистки описаны в патентах США 5089462 и 2853257 и европейских патентах ЕР 0448117 и ЕР 0469675.
Обычно используемые носители содержат огнеупорное вещество, получаемое из оксида алюминия. Для гидродесульфирования предпочтительно используют алюмомолибденовые оксидные катализаторы, промотированные кобальтом, в то время как для гидроденитрификации и гидрирования ароматики (гидродеароматизации) наряду с гидродесульфированием фракций, подвергаемых гидроочистке, широко используют алюмомолибденовые оксидные катализаторы, промотированные никелем, обладающим высокой гидрирующей активностью.
Наиболее заметные успехи, достигнутые в последнее время в области катализаторов гидродесульфирования и гидроденитрификации и катализаторов гидроочистки в целом, касаются не содержащих носителя кобальтовых или никелевых молибдено-вольфрамовых фаз (США 6534437, США 6582590). Эти объемные Ni-Mo-W-О или Co-Mo-W-О катализаторы обладают высокой удельной поверхностью примерно 100 и 200 м2/г. Активность этих катализаторов в гидродесульфировании изучали на примере модельной молекулы дибензотиофена (DBT). Эти катализаторы проявляют удельную активность, измеренную как [молекул/г·с], которая выше активности традиционных катализаторов, нанесенных на оксид алюминия. Эти катализаторы обладают высокой плотностью, так что на единицу объема реактора приходится большее количество вещества. Таким образом, активность, отесенная к объему катализатора, оказывается примерно в четыре раза более высокой по сравнению с традиционными промышленными никель-молибденовыми катализаторами, нанесенными на оксид алюминия.
Синтез ненанесенных катализаторов на основе металлов VIII и VIB групп Периодической таблицы был описан ранее (Catal. Lett. 10 (1991)181; J. Thermal Anal. 40 (1993) 1253). Эти катализаторы применяли в основном для окисления углеводородов, например, для окислительного дегидрирования пропана и/или парциального окисления пропилена до акролеина и акриловой кислоты. Известно осаждение аммонийной фазы NiMoO4·mNH3, nH2O. Недавно эту методику осаждения применили для приготовления катализаторов дегидросульфирования и гидроочистки разных нефтяных фракций.
Предложенная на сегодня структура осадка, образующегося из аммонийного комплекса, представляет собой фазу молибдата никеля аммония со структурой гидроталькита стехиометрического состава (NH4)HNi2(ОН)2(MoO4)2. Это вещество обладает слоистой структурой, содержащей молибдатные анионы в межслойном пространстве, связанные со слоями гидроксида никеля. Методика синтеза веществ этого типа описана в работе Appl. Catal. 72, 321-329 (1991) и Solid State Ionics 63-65 (1993) 731-35.
В патентах США 6156696 В и 6162350 В описаны методики получения каталитической композиции по меньшей мере из одного неблагородного металла VIII группы, который может быть никелем или кобальтом, и по меньшей мере двух металлов VIB группы. Общая приведенная формула имеет вид (X)b(Mo)c(W)dOz, где X представляет собой неблагородный металл VIII группы (Ni или Со) и мольное соотношение b/(c+d) имеет значения 0.5-3, a z=[2b+6(c+d)]2. Дифрактограммы этих веществ имеют вид, типичный для аморфного вещества, с очень широкими пиками, отвечающими межплоскостным расстояниям 2.53 и 1.7 Å. Замещение атомов молибдена на атомы вольфрама в этом веществе позволяет получить аморфную или микрокристаллическую структуру, которая после прокаливания кристаллизуется с образованием неизвестной структуры, имеющей диффракционный пик 2-тета при 53.82° с полушириной от 1.3 до 1.7°. Для получения оптимального выхода при осаждении солей необходимо, чтобы по меньшей мере одна из солей частично растворялась во время осаждения. Полученные катализаторы смешивают с оксидом алюминия и экструдируют, получая высокоактивные катализаторы гидродесульфирования, гидроденитрификации и гидроочистки различных нефтяных фракций.
Другая стратегия синтеза объемных катализаторов гидродесульфирования заключается в термическом разложении тиометаллатов аммония. В патенте США 4243554 предложены промотированные кобальтом и никелем катализаторы на основе дисульфида молибдена, обладающие высокой удельной поверхностью, которые можно получить термическим разложением нескольких тиомолибдатов аммония формулы (NH4)2[MoOxS4-x], где х равен 2. Разложение тиосолей протекает в присутствии раствора углеводорода, содержащего соединения серы, при высоком давлении водорода и при температуре 300-800°С.
С другой стороны, разложение этих солей в присутствии углеводорода позволяет получить углеродсодержащий катализатор на основе сульфида молибдена, который ответственен за образование активных центров и высокую активность в гидродесульфировании (Berhault et. al. J.Catal. 198, 9-19 (2001)). В патенте США 4508847 раскрыта каталитическая композиция MoS2-xCz, где z представляет собой содержание углерода и находится в пределах 0.01<z<3, а х представляет собой содержание серы и находится в пределах 0.01<х<0.5. Этот катализатор получают при взаимодействии предшественника молибдена, такого как тиомолибдат аммония или тиовольфрамат аммония; молибдата аммония или вольфрамата аммония тиомолибдатов, молибдатов, тиовольфраматов, заместителей вольфрамата аммония - с потоком серы, водорода и углеводородов при температурах 150-600°С. Эти катализаторы обладают высокой удельной поверхностью и могут быть промотированы другими металлами типа кобальта и/или никеля с образованием высокоактивных катализаторов гидроочистки; они более активны, чем катализаторы с теми же металлами, нанесенными на оксид алюминия. Однако в методике, описанной в данном патенте, углерод образуется в основном за счет карбонизации углеводорода, присутствующего при разложении предшественника.
Введение органического соединения в качестве источника углерода в неорганические соли молибдена или прямое сульфидирование органических солей молибдена не только способствует образованию карбид-сульфидов металла, таких как MoSxCz, но также обеспечивает полное сульфидирование молибдена до MoS2, что приводит к более высокой плотности активных центров в катализаторе (Farag Н. Energy & Fuel, 16 (2002) 944-950). Это описано в патентах США 4528089 и 4650563, где раскрыта методика получения катализатора - дисульфида молибдена, содержащего углерод, которая заключается в термообработке соли-предшественника в присутствии серы и в отсутствие кислорода. Соль-предшественник имеет общую формулу ML (MoxW1-xS4), где М представляет собой один или несколько двухвалентных металлов-промоторов, таких как Ni, Со, Zn, Сu или их смесь; х находится в пределах от 0 до 1; и L представляет собой один или несколько нейтральных органических комплексов, которые могут действовать как хелатные полидентатные азотсодержащие лиганды. Полученные таким образом катализаторы обладают более высокой активностью в реакциях гидроочистки, чем катализаторы, получаемые из традиционных предшественников, таких как кобальт-молибден на оксиде алюминия, даже когда величина их удельной поверхности не очень велика.
Патенты США 4581125 и 4514517 относятся к катализатору на основе дисульфида молибдена, который получают терморазложением соли-предшественника, содержащей углерод, которая может иметь состав (NR4)2[M(WS4)2] или (NR4)x[M(MoS4)2]. Термическое разложение проводят в отсутствие кислорода и в присутствии серы и водорода при температуре выше 150°С. Группа (NR4) содержит углерод и является замещенным катионом аммония, где R может быть алкильной или арильной группой. М является металлическим промотором и тесно взаимодействует через ковалентные связи с анионом (MoS4)= или y(WS4)= и может быть никелем, кобальтом или железом; х равен 2, если М является никелем, и у равен 3, если М является кобальтом или железом. В идеальном случае для достижения максимальной каталитической активности катализатор надо получать в присутствии углеводородов.
Для повышения удельной поверхности катализаторов, полученных разложением тиосолей, в патенте США 6156693 описан способ гидротермальной обработки предшественника - соли тетратиомолибдата аммония, который растворен в высококипящем растворителе и воде под давлением водорода при температурах 350-400°С. Присутствие воды эффективно влияет на формирование активных центров; однако для получения более активного катализатора MoS2 после разложения тетратиомолибдата аммония ее следует удалить.
В патенте США 2005/0059545 А1 описан способ получения путем гидротермальной обработки углеродсодержащих катализаторов на основе сульфида молибдена и/или катализаторов на основе вольфрама. Способ заключается в гидротермальной обработке предшественника - соли тетратиомолибдата аммония AxMoS4, где А является ионом аммония, ионом тетраалкиламмония (х=2) или ионом диаммония (х=1), в присутствии соли промотора, который может быть никелем, кобальтом, железом или рутением. Полученный катализатор Ni/CoMoS2-xCx, где х принимает значения от 0 до 1, активируют в атмосфере H2S/H2 при высокой температуре перед реакцией гидродесульфирования.
Введение органической добавки, такой как хелатный или металлоорганический комплекс, в раствор для пропитки катализаторов гидродесульфирования, наносимых на оксид алюминия, промотирует оптимальное сульфидирование активных металлических компонентов и максимальное промотирование дисульфида молибдена, способствуя его диспергированию и формированию активных центров с высокой плотностью, которое обусловливает увеличение каталитической активности в реакциях гидроочистки.
Патент США 6566296 В2 относится к каталитической композиции из МoО3 в концентрациях 10-30 мас.%, WO3 в концентрациях 30 и 50 мас.%, NiO в концентрациях 30 и 50 мас.% и Al2O3 в концентрациях 0 и 20 мас.%. Каталитическую композицию получают соосаждением солей, из которых по меньшей мере одна соль остается в твердом состоянии или частично растворенной; затем их смешивают с оксидом алюминия для формирования экструдатов. Полученные экструдаты пропитывают органическим соединением, таким как диэтиленгликоль, или соединением с замещенной аминогруппой NR4, где R может содержать до 10 атомов углерода. Другие добавки, которые можно использовать в качестве источника углерода при пропитке экструдатов, включают гликоли, сахарин, полисахариды и этилендиаминтетрауксусную кислоту (EDTA). При такой стратегии введения органического соединения в качестве источника углерода достигается значительное увеличение каталитической активности этих катализаторов в реакциях гидроочистки.
Введение вольфрама в объемные катализаторы гидродесульфирования повышает их каталитическую активность по сравнению с катализаторами, содержащими только молибден (патент США 6534437). Средняя объемная плотность этих катализаторов значительно выше, чем у традиционных катализаторов, и это приводит к тому, что для заполнения объема данного реактора требуется большее количество катализатора. В случае объемных катализаторов заметно возрастает стоимость катализаторов, что стимулировало поиск новых альтернатив для замещения атомов молибдена другими менее плотными металлами, такими как хром и марганец (патенты США 6635599 В1, 6783663 В1).
Известен ряд нанесенных и ненанесенных катализаторов реакций гидроочистки, основные фазы которых содержат металлы - кобальт, никель, железо, молибден и вольфрам - в виде сульфидов. Однако существует необходимость в разработке более эффективных каталитических систем для очистки топлив от загрязняющих веществ или уменьшения их концентрации. В данном изобретении предложен способ получения каталитической композиции по меньшей мере из одного неблагородного металла VIII группы и по меньшей мере одного металла группы VIB и, кроме того, углерода, образующегося из органического соединения во время синтеза катализатора, который способствует сульфидированию и повышает каталитическую активность в реакциях гидроочистки.
Способ и катализатор, которые являются предметом данного изобретения, применяют в реакциях гидроочистки, которые включают гидродесульфирование, гидроденитрификацию и гидрирование ароматики, поскольку эти катализаторы применяют в способах переработки нефти для получения чистых топлив, удаления серы и азота в разных углеводородных фракциях и погонах и уменьшения содержания ароматики в топливах. Их можно также применять для гидроочистки тяжелых фракций типа вакуумного мазута и тяжелой нефти.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к новой высокоактивной каталитической композиции для реакций гидроконверсии различных углеводородных фракций, содержащихся в нефти, предпочтительно для реакций гидродесульфирования, гидроденитрификации и гидрирования ароматики. Катализатор, который является предметом данного изобретения, состоит по меньшей мере из одного неблагородного металла VIII группы, по меньшей мере одного элемента группы VIB и одного органического компонента в качестве предшественника углерода, тесно связанного с активными металлическими фазами.
Поэтому одной из целей настоящего изобретения является получение каталитической композиции, содержащей по меньшей мере один металл группы VIB, из растворимой в полярном растворителе соли предшественника, который может быть молибденом или вольфрамом, предпочтительно молибденом; по меньшей мере один неблагородный металл VIII группы, вводимый в виде соли предшественника - кобальта, никеля, железа или рутения, предпочтительно никеля и/или кобальта, и органическое соединение в качестве предшественника углерода, которое может быть поверхностно-активным реагентом, таким как ионное, катионное, анионное или нейтральное поверхностно-активное вещество, или четвертичной солью замещенного аммония (NR4 +), где R может быть алкильной или арильной группой, содержащей 1-8 атомов углерода в алкильной цепи.
Другой целью данного изобретения является способ получения катализаторов, который заключается в полном растворении солей предшественников в полярном растворителе, таком как вода, поддержании рН раствора в интервале от 5 до 14 путем добавления основания, которое может быть гидроксидом аммония, карбонатом аммония или гидроксидом четвертичной аммонийной соли, такой как тетрабутиламмоний гидроксид. Как только образовался полностью растворенный комплекс, добавляют органическое вещество; в случае, если использованное основание не является гидроксидом четвертичной аммонийной соли, оно состоит из поверхностно-активного реагента, такого как ионное, катионное, анионное или нейтральное поверхностно-активное вещество. После этого при упаривании избыточного растворителя комплекс кристаллизуется, и получают смешанный оксогидроксид металла VIB группы и металл VIII группы, содержащий углерод или органическое соединение.
Другой целью настоящего изобретения является термообработка полученного вещества, которую никогда нельзя проводить в окислительной газовой атмосфере при температурах выше 200°С. Указанный способ состоит в термообработке при температурах 200-1000°С в токе инертного газа, такого как азот, гелий или аргон, при которой получают смешанный оксид никеля-молибдена, содержащий углерод, если выбранные металлы представляли собой молибден и никель; или молибдат кобальта, смешанный оксид молибдена-кобальта, содержащий углерод, если были выбраны молибден и кобальт; или смешанный оксид никеля, кобальта и молибдена или смешанный молибдат никеля и кобальта, если выбраны два металла VIII группы, и молибден.
Другая цель настоящего изобретения представляет собой способ сульфидирования для получения сульфидной формы катализаторов, который заключается в контактировании сухого вещества и/или вещества, термообработанного в инертной атмосфере при температурах 200-600°С, с потоком H2S, разбавленного 0.5 и 30 об.% водорода. Другой способ состоит в прямом контакте сухого вещества - оксогидроксида кобальта-никеля-молибдена - или термообработанного вещества - молибдата никеля или молибдата кобальта или молибдата никеля и кобальта - с потоком жидкого углеводорода, содержащего органические соединения серы, обогащенным диметилдисульфидом (DMDS) в концентрации 0.1 и 5 мас.% серы, при температуре 200 и 600°С и давлении водорода 1-100 кг/см2. При этом способе окисленные фазы активных компонентов превращаются в активные сульфидные фазы. Присутствие углерода способствует сульфидированию, промотированию дисульфида молибдена и образованию на поверхности высокой концентрации активных форм.
Активные формы, включенные в данное изобретение, состоят из сульфидных фаз по меньшей мере одного неблагородного металла VIII группы и по меньшей мере одного металла группы VIB, содержащих углерод.
Другая цель настоящего изобретения состоит в интегрировании каталитической композиции, которая также является целью настоящего изобретения, с неорганическим веществом, таким как связующее, для формирования экструдатов, причем связующее может представлять собой оксид алюминия, оксид титана, оксид циркония, оксид кремния или их смесь в соотношении 0-50 мас.% оксида соответствующего металла в расчете на общее содержание присутствующих оксидов.
Еще одной целью данного изобретения является получение катализатора в виде экструдата с такой же объемной плотностью, как у традиционных нанесеннных катализаторов, благодаря объединению со связующим, имеющим низкую плотность, при формировании экструдатов.
Другая цель настоящего изобретения включает способ гидроочистки углеводородной фракции, в котором используют катализаторы в сульфидной фазе, при контакте с жидкой порцией углеводородной фракции в типичных условиях гидроочистки.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
В целях лучшего понимания каталитической композиции для гидроконверсии нефтяной фракции можно обратиться к следующим фигурам:
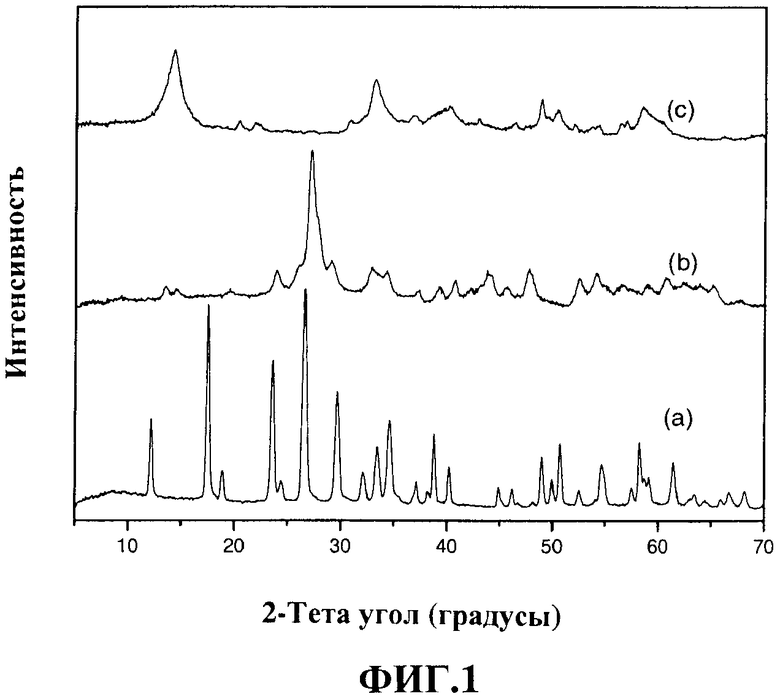
На фигуре 1 представлена дифрактограмма, относящаяся к примеру 1, типичная для сухой каталитической композиции (а), дифрактограмма термически обработанной смеси бета- и альфа-фаз молибдата никеля (b) и сульфидированной фазы (с), где показаны пики, характерные для смеси сульфидов металлов VIII и VIB групп, которые являются предметом данного изобретения.
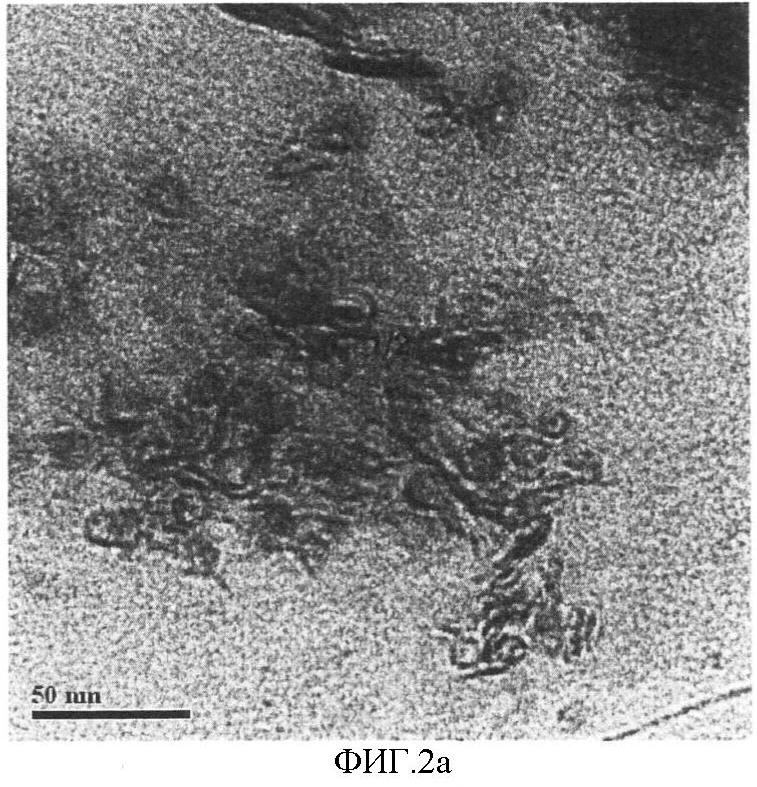
На фигуре 2 представлены полученные методом просвечивающей электронной микроскопии микрофотографии сульфидного материала, приготовленного в примере 1; такие микрофотографии характерны для каталитической композиции, которая является предметом данного изобретения: а) микрофотография показывает морфологию металлосульфидных высокоплотных наносфер металла VIB группы и наносфер металла VIII группы, b) электронная микрофотография высокого разрешения показывает полые наносферы, содержащие 2-10 структурных слоев.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к способу получения каталитической композиции, состоящей по меньшей мере из одного неблагородного металла VIII группы и по меньшей мере одного металла VIB группы, а также органического компонента в качестве предшественника углерода, находящегося в тесном взаимодействии с металлами. Данный способ включает приготовление, смешение и реакцию растворов, содержащих соли предшественника, с раствором, содержащим органический компонент, и затем его кристаллизацию при удалении избытка растворителя. Для достижения тесного взаимодействия металлов с органическим компонентом все компоненты должны полностью растворяться до начала процесса кристаллизации.
Для получения каталитической композиции, которая является предметом данного изобретения, существенно, чтобы все компоненты полностью растворились в добавленном растворе, который содержит органическое соединение или поверхностно-активный реагент. Способ включает смешение и реакцию растворов, которые содержат металлические компоненты катализаторов, с раствором, содержащим органическое соединение или поверхностно-активный реагент, с образованием комплекса металлов, который полностью растворяется в полярном растворителе и затем кристаллизуется из растворителя или путем упаривания. Термин «полностью растворяется» в данном контексте означает, что при смешении растворов солей предшественников, содержащих поверхностно-активный реагент, не образуется ни суспензии, ни осадка, т.е. образуется прозрачный раствор.
Каталитическая композиция, которая является предметом данного изобретения, содержит по меньшей мере один неблагородный металл VIII группы и по меньшей мере один металл VIB группы. Металлами VIB группы могут быть молибден, вольфрам, хром или их смесь, предпочтительно молибден и вольфрам и более конкретно молибден. Неблагородными металлами VIII группы могут быть железо, кобальт, рутений, никель, предпочтительно никель и кобальт. Это также может быть комбинация металлов, такая как никель-кобальт-молибден, никель-кобальт-вольфрам, никель-кобальт-молибден-вольфрам или никель-кобальт-молибден-вольфрам-хром, предпочтительно никель-кобальт-молибден.
Мольное соотношение металлов VIB/VIII групп, используемых в способе данного изобретения, находится в пределах от 0.1 до 10, предпочтительно от 0.4 до 2 и более конкретно от 0.5 до 1.5. В случае, когда в качестве металлов VIB группы каталитическая композиция содержит молибден и вольфрам, мольное соотношение Mo/W находится в пределах от 10 до 1, предпочтительно от 10 до 5. Тот факт, что элементы VIB группы представлены только молибденом, заслуживает особого внимания по экономическим причинам, т.к. введение вольфрама повышает плотность катализатора и стоимость этого металла значительно в большей степени, чем в случае молибдена. Если в качестве металлов VIII группы каталитическая композиция содержит никель и кобальт, то мольное соотношение Ni/Co будет находиться в пределах от 0.05 до 20, предпочтительно от 0.1 до 10.
При использовании воды в качестве полярного растворителя солями-предшественниками компонентов - металлов VIB группы - могут быть: ацетилацетонат молибдена, молибдат аммония, молибденовая кислота, вольфрамат аммония, вольфрамовая кислота, предпочтительно метавольфрамат аммония и/или гептамолибдат аммония.
При использовании воды в качестве полярного растворителя солями-предшественниками компонентов - металлов VIII группы - могут быть: нитрат, хлорид, ацетилацетонат, ацетат, сульфат, гидроксид, предпочтительно нитрат и/или хлорид. В случае, когда металлом VIII группы является никель, солями-предшественниками могут быть: нитрат никеля, хлорид никеля, ацетат никеля, цитрат никеля, предпочтительно нитрат и/или хлорид никеля. Когда металлом VIII группы является кобальт, солями-предшественниками могут быть: нитрат кобальта, хлорид кобальта, ацетат кобальта, цитрат кобальта, предпочтительно нитрат и/или хлорид кобальта. Растворы можно готовить отдельно, т.е. каждую соль-предшественник металлического компонента можно растворять независимо в полярном растворителе, который может быть водой или спиртом или их смесью, и затем смешать их с образованием раствора, содержащего все полностью растворенные металлические компоненты в заданных концентрациях для каждого образца катализатора. Необязательно растворы можно готовить путем растворения солей-предшественников в той же емкости для получения прозрачного раствора, который содержит металлические компоненты в полностью растворенном состоянии. При использовании нерастворимого предшественника металлического компонента следует найти подходящие условия для полного включения нерастворимого твердого вещества путем повышения температуры или введения добавок основания или кислоты, с тем чтобы все металлы, участвующие в синтезе катализатора, находились в полностью растворенном состоянии. С другой стороны, раствор, содержащий органическое соединение или поверхностно-активный реагент, готовят отдельно путем растворения четвертичной аммониевой соли или ионного или нейтрального поверхностно-активного реагента в полярном растворителе, таком как вода, спирт или их смесь.
При приготовлении раствора, содержащего по меньшей мере один металл VIB группы, который может быть молибденом, вольфрамом и/или хромом, предпочтительно молибденом или вольфрамом и более предпочтительно только молибденом, соль-предшественик этого металла, такую как гептамолибдат аммония, молибденовую кислоту, ацетилацетонат молибдена, триоксид молибдена и/или метавольфрамат аммония, вольфрамовую кислоту, триоксид вольфрама, предпочтительно гептамолибдат аммония или метавольфрамат аммония, растворяют в полярном растворителе.
Раствор, содержащий по меньшей мере один неблагородный металл VIII группы, такой как кобальт, никель, железо, рутений и т.д., предпочтительно никель и/или кобальт, готовят путем растворения соли-предшественника, такой как нитрат, хлорид, ацетилацетонат, ацетат, сульфат, гидроксид или карбонат никеля и/или кобальта, предпочтительно нитрат, хлорид или ацетат, в полярном растворителе, который может быть водой, спиртом или их смесями.
Раствор, содержащий органическое соединение в качестве предшественника углерода, готовят растворением четвертичной аммониевой соли или твердого поверхностно-активного реагента в полярном растворителе. Поверхностно-активный реагент, такой как катионный, анионный или нейтральный поверхностно-активный реагент, предпочтительно катионный или нейтральный, нужен для достижения оптимального взаимодействия между анионами неорганических компонентов. Поверхностно-активный реагент может быть солью тетраалкиламмония (NR4 +), где R является алкильной группой и соответствует насыщенной углеводородной цепи или алкильным группам, содержащим 1-8 атомов углерода каждая. Все алкильные группы могут содержать одинаковое число атомов углерода или они могут иметь разные величины. Ионы тетраалкиламмония могут быть галогенированными, замещенными хлором или бромом; или могут быть гидроксилированными, т.е. замещенными гидроксильными группами (ОН). Поверхностно-активный реагент может быть катионным с более длинной алкильной группой формулы R'NR3, где R' соответствует длинной цепи насыщенных углеводородов или алкильной группе, содержащей 12-20 атомов углерода, a R является другой алкильной группой, которая соответствует короткой углеводородной цепи, содержащей 1-8 атмомов углерода.
Растворы, содержащие металлические компоненты и органическое соединение, смешивают при непрерывном перемешивании; затем добавляют концентрированный раствор основания, такого как гидроксид аммония, карбонат аммония и/или гидроксид натрия или калия, предпочтительно гидроксид аммония, до тех пор пока рН раствора не достигнет величины 5-14, предпочтительно 8-12. Затем температуру раствора повышают до 50-200°С, предпочтительно 60-100°С, а щелочной раствор добавляют для поддержания всех компонентов в полностью растворенном состоянии путем перемешивания в течение времени от 5 мин до 24 час, предпочтительно от 5 мин до 10 час, для того чтобы достичь тесного взаимодействия всех металлических компонентов и органического соединения - предшественника углерода. Пребывание всех соединений металлов в полностью растворенном состоянии в полярном растворителе в присутствии органического соединения позволяет достичь максимального взаимодействия между ними, и они реагируют друг с другом с образованием смешанного комплекса неорганического и органического компонентов с последующей кристаллизацией.
В случае, когда полярный растворитель является водой, предпочтительно, чтобы температура реакции была ниже температуры кипения, т.е. 60-100°С, для того чтобы реакция протекала в открытом сосуде при атмосферном давлении или в системе с обратным холодильником. В случае, когда температура выше температуры кипения воды, реакцию следует проводить в закрытом сосуде при автогенном давлении, таком как в автоклаве, где кристаллизацию компонентов приготовленных растворов следует проводить в гидротермальных условиях и автогенном давлении.
Для способа получения, который является предметом настоящего изобретения, важно адекватно выбирать температуру реакции и рН для поддержания всех металлических компонентов, участвующих в получении катализатора, в полностью растворенном состоянии; рН в интервале 5-14, предпочтительно 8-12 и более предпочтительно 8-10. Время реакции составляет от 5 мин до 24 час, предпочтительно от 5 мин до 10 час и более предпочтительно от 5 мин до 5 час.
По истечении времени, необходимого для оптимального взаимодействия и реакции между всеми компонентами, полностью прозрачный или полностью идеальный раствор поступает на кристаллизацию, которая заключается в удалении избыточных ионов аммония и испарении полярного растворителя, предпочтительно воды. После испарения избыточного растворителя образуется осадок, который может быть зеленого или пурпурного цвета в зависимости от того, преобладает ли в нем никель или кобальт. Выход вещества зависит от степени упаривания, и при адекватном регулировании процесса кристаллизации можно получить выход более 99%.
Кристаллизация из раствора, содержащего все металлы, участвующие в получении катализатора, может быть мгновенной или постепенной, и ее можно проводить непрерывно или периодически. По этой причине способ пригоден для широкомасштабного применения, такого как в периодическом реакторе и/или в испарителе.
При проведении кристаллизации путем постепенного испарения необходима последующая стадия разделения твердого вещества и жидкости для отделения осадка фильтрованием и/или центрифугированием. Небольшие количества металла остаются растворенными в маточном растворе, и, чтобы не потерять это количество металла вместе с маточным раствором, образовавшимся после выделения, следует повторить эту операцию, что является предметом настоящего изобретения. Полученное твердое вещество в сухом или влажном состоянии сушат в статических условиях в сушильной камере или в токе горячего воздуха или путем непрерывной сушки при температуре 50-300°С, предпочтительно 80-150°С.
Для получения катализаторов нужной геометрической формы данное изобретение также предлагает добавлять неорганический оксид или смешанный оксид в качестве связующего, выбранный из группы имеющихся связующих, таких как оксид алюминия, оксид титана, оксид алюминия-титана, цеолиты, оксид кремния и алюмосиликат, которые являются устойчивыми носителями для катализаторов гидроочистки. Термин «связующее или связующий реагент» относится к неорганическому оксиду, способному объединять или связывать частицы вещества в виде экструдатов, таблеток или сфер для придания им нужной формы при использовании в каталитическом процессе с неподвижным слоем.
Связующий реагент можно добавлять на разных стадиях способа получения катализатора, который является предметом настоящего изобретения. При желании его можно добавить в виде соли-предшественника неорганического оксида во время растворения солей-предшественников металлов VIII и VIB групп. Соли-предшественники неорганических оксидов, выбранные в качестве связующих, включают нитраты, хлориды, ацетаты, сульфаты, гидроксиды и т.д. Указанные соли-предшественники будут осаждаться сразу после добавления щелочного раствора, содержащего гидрокисд и/или карбонат аммония и/или натрия. С другой стороны, связующее можно ввести сразу по завершении стадии реакции путем добавления связующего в виде порошка в раствор, содержащий комплекс металлических и органических компонентов; затем проводят кристаллизацию путем испарения растворителя в присутствии связующего. Предпочтительно добавлять связующее к сухим кристаллическим частицам катализатора, содержащим металлические компоненты, выбранные из металлов VIII и VIB групп. Конкретно связующее может быть в сухом или влажном состоянии, пептизированным с помощью неорганической или органической кислоты, такой как азотная кислота, соляная кислота, фосфорная кислота, борная кислота, серная кислота, лимонная кислота, уксусная кислота и т.п., предпочтительно фосфорная кислота, лимонная кислота, азотная кислота и/или уксусная кислота. При смешении обеих частей, т.е. связующего и частиц катализатора в сухом состоянии, пептизацию связующего проводят, добавляя разбавленную органическую и неорганическую кислоту. Затем частицы влажного связующего и катализатора тщательно перемешивают в месильной машине Мюллера до совершенно гомогенной и однородной пасты с соответствующими реологическими свойствами, которую можно экструдировать.
Связующее или связующий реагент можно выбрать из веществ, традиционно используемых в качестве связующих в катализаторах гидроочистки. Кроме того, их можно выбирать из группы оксидов, гидроксидов, оксогидроксидов и т.д., таких как оксид кремния, алюмосиликат, оксиды алюминия (бемит, псевдобемит, байерит, гиббсит) и/или оксиды алюминия типа гамма-, эта-, тета- и хи-оксидов алюминия, оксиды циркония, оксиды циркония-алюминия, оксиды циркония-кремния, цеолиты с различными структурами: ZSM-5, бета, Y, X и т.д. Можно также их выбрать из кремнеалюминатов семейства МСМ. Также можно выбирать из оксидов титана с разными структурами, таких как анатаз, рутил и брокит, смешанных кислых титанатов и/или титанатов натрия с нанотрубчатой и/или нановолокнистой морфологией и развитой поверхностью, оксидов титана-алюминия, оксидов титана-циркония, оксидов титана-кремния и т.д. Предпочтительно, чтобы связующими были оксид алюминия, оксид циркония, оксид кремния, алюмосиликат и оксид титана и/или смеси этих компонентов. Более предпочтительно, чтобы они представляли собой оксид алюминия, оксид циркония, оксид титана, оксид алюминия-титана и оксид циркония-титана. Использование оксидов титана, таких как оксогидроксиды титана, кислые титанаты и/или смешанные кислые и/или средние титанаты натрия с нановолокнистой и/или нанопластинчатой морфологией, развитой поверхностью и низкой плотностью, особенно предпочтительно в данном изобретении; и/или использование связующих на основе оксида алюминия как с нановолокнистой, так и/или нанопластинчатой морфологией и низкой плотностью. Величина поверхности выбранного связующего обычно находится в интервале 20-700 м2/г, предпочтительно 150-500 м2/г, по данным физической адсорбции азота по методу БЭТ, и объем пор находится в интервале 0.05-2.5 см2/г, предпочтительно 0.1-2 см2/г.
Композиция связующего в конечной композиции катализатора составляет 0-50 мас.% от всех компонентов катализатора в окисленной форме, предпочтительно 0-40 мас.% и более предпочтительно 0-20 мас.%. Однако состав связующего следует подбирать в зависимости от способа применения, активности и механической прочности конечной композиции катализатора. Способ получения катализатора, который является предметом данного изобретения, включает формование экструдатов, таблеток, шариков и/или микросфер, предпочтительно формование экструдатов и/или таблеток, которые можно использовать в способе с неподвижным слоем. Сформованные частицы катализатора могут быть цилидрическими и иметь радиус в интервале 1/20-1/8 дюйма или быть трех- или четырехлопастными. Экструзию проводят в механическом экструдере с головками, имеющими отверстия разных диаметров для получения экструдатов разного размера, как уже было указано. Полученные экструдаты оставляют на период от 5 мин до 12 час, предпочтительно от 5 мин до 5 час. Затем их сушат в сушильной камере на воздухе или в токе теплого воздуха при температуре 50-300°С, предпочтительно 80-150°С, или в осушителе непрерывного действия с регулируемой атмосферой при температуре 50-300°С. На этой стадии катализаторы приобретают форму экструдатов, таблеток или сфер, которые можно не прокаливать или прокалить для перевода в окисленную форму. Катализатор содержит органическое соединение в виде поверхностно-активного реагента, от которого после термообработки остается углеродный остаток, стабилизирующий экструдаты и придающий им повышенную механическую прочность. Эту операцию никогда нельзя проводить в кислороде или на воздухе. Термообработку следует проводить в инертной атмосфере, например, в токе азота, аргона, гелия или их смеси. Эту термообработку следует проводить при температуре 200-1000°С, предпочтительно 300-600°С и более предпочтительно 300-500°С, в течение времени от 0.1 до 24 час, предпочтительно 0.5-10 час. Ток инертного газа пропускают через катализатор во время термообработки со скоростью 0.01-5 л/г.мин, предпочтительно 0.01-1 л/г мин. Сухие экструдаты можно не подвергать термообработке и направлять прямо на стадию сульфидирования, которая также является предметом данного изобретения.
Способ получения каталитической композиции, который является предметом данного изобретения, включает стадию сульфидирования. Эта стадия включает контакт частиц катализатора, не важно до или после термообработки в инертной атмосфере, с жидким или газовым потоком, содержащим соединения серы, и потоком газообразного водорода. Используемые для сульфидирования соединения серы могут включать сероводород, диметилсульфид, дисульфид углерода, тиофены, полисульфиды, бензотиофены или поток углеводородов, содержащих органические соединения серы. Можно необязательно сульфидировать каталитическую композицию, содержащую сухой уголь или уголь, термически обработанный в инертной атмосфере. Для получения сульфидированного вещества сульфидирование в газовой фазе включает создание потока H2S с содержанием 0.5 и 30 об.%, остальное водород, над неподвижным слоем каталитической композиции при температуре 200-600°С, предпочтительно 250-500°С. Сульфидирование в жидкой фазе включает контакт каталитической композиции с жидким потоком углеродородов, содержащим 0.1-5 мас.% серы в виде сероорганических соединений или соединений, обогащенных диметилсульфидом, тиофеном или CS2, и потоком водорода при температуре 200-600°С, предпочтительно 250-500°С, и при давлении 1-100 кг/см2.
Для применения углеводородов в реакциях гидроочистки в реакторе с неподвижным слоем сульфидирование следует проводить «in situ» до или после термообработки. Предпочтительно сульфилировать «in situ» непосредственно сухую каталитическую композицию, которая включает органический компонент, содержащий углерод, и/или после стабилизации путем термообработки в инертной атмосфере. После термообработки в инертной атмосфере каталитическая композиция, которая является предметом настоящего изобретения, содержит по меньшей мере один неблагородный металл VIII группы и по меньшей мере один неблагородный металл VIB группы в окисленном состоянии и, кроме того, углерод. Если металлом VIB группы является молибден, что является предпочтительным для данного изобретения, на дифрактограммах наблюдаются характеристические пики молибдатов никеля и/или кобальта с бета- и/или альфа-структурой или смеси двух фаз, как показано на фигуре 1. Если каталитическая композиция состоит из вольфрама и/или молибдена или вольфрама с высокой концентрацией, на дифрактограммах наблюдаются широкие пики, типичные для аморфного вещества, которые нельзя отнести к фазам, приведенным на картах Объединенного комитета по стандартам порошковой дифракции (Joint Committee on Powder Diffraction Standards, JCPDS).
Если неблагородным металлом VIII группы является никель, а элементом VIB группы молибден, на дифрактограмме видны характеристические пики альфа- и/или бета-фаз молибдата никеля или пики, соответствующие обеим фазам. Альфа-фаза имеет моноклинную структуру тетраэдрической симметрии, в которой молибден имеет координационное число четыре, а никель имеет координационное число шесть и находится в октаэдрической симметрии, в то время как бета-фаза имеет орторомбическую структуру, в которой молибден и никель имеют координационное число шесть и октаэдрическую симметрию. Если неблагородный металл VIII группы является кобальтом, а элементом VIB группы является молибден, пики на дифрактограмме соответствуют альфа- и/или бета-фазе молибдата кобальта или смеси двух фаз. В случае, когда неблагородные компоненты VIII группы представляют собой смесь никеля и кобальта, пики на дифрактограмме соответствуют альфа- и/или бета-фазам смешанного молибдата никеля-кобальта или их смеси.
После термообработки в инертной атмосфере катализаторы, содержащие углерод, по меньшей мере один неблагородный металл VIII группы и по меньшей мере один металл VIB группы, имеют удельную поверхность 50-300 м2/г, предпочтительно 70-150 м2/г, объем пор 0.05-1.5 см3/г, предпочтительно 0.1-0.7 см3/г. Диаметр пор в катализаторах после термообработки в инертной атмосфере, определенный по физической адсорбции азота, составляет 3-20 нм, предпочтительно 3-10 нм.
Текстурные свойства катализаторов в значительной степени зависят от количества связующего или реагента, использованного для формования экструдатов. В свою очередь, количество добавленного связующего зависит от требуемой активности каталитической композиции и находится в интервале 0-50% от общей массы компонентов катализатора в окисленной форме, предпочтительно 0-40 мас.% и более предпочтительно 0-20 мас.%. Однако состав связующего следует подбирать в зависимости от способа применения катализатора, а также от активности и нужной механической прочности конечной каталитической композиции.
Важно, чтобы частицы катализатора были равномерно и прочно распределены в связующем, который является связующей средой для частиц катализатора и дает возможность сформовать их в виде экструдатов, таблеток и/или сфер таким образом, чтобы их можно было испытать в неподвижном слое непрерывного способа гидроочистки. Таким образом, в присутствии описанной выше композиции связующего катализаторы проявляют механическую прочность по отношению к боковому раздавливающему усилию по меньшей мере 2 фунт/мм, предпочтительно 4 фунт/мм или больше.
Каталитическая композиция в сульфидной форме, которая является предметом настоящего изобретения, состоит из сульфидов по меньшей мере одного неблагородного металла VIII группы и одного металла VIB группы и, кроме того, остаточного углерода, образовавшегося из органического соединения, введенного на стадии кристаллизации. При добавлении связующего она также включает, помимо сульфидов и углерода, неорганический оксид, выбранный из группы веществ, или традиционные носители катализаторов гидроочистки.
После термообработки и последующего сульфидирования или после прямого сульфидирования каталитическая композиция содержит углерод в концентрации 0-10 мас.%, предпочтительно 0.05-5 мас.% в расчете на общую композицию оксидов и/или серы. Углерод может образовать соединения типа сульфид-карбида в очень низкой концентрации или может присутствовать в виде аморфного углерода.
В результате сульфидирования по меньшей мере 60% окисленных фаз по меньшей мере одного неблагородного металла VIII группы и по меньшей мере одного металла VIB группы образуют сульфиды, предпочтительно по меньшей мере 80% металлов и более предпочтительно по меньшей мере 95% металлов, входящих в каталитическую композицию. Присутствие органического или углеродного компонента способствует сульфидированию металлов, входящих в каталитическую композицию.
На дифрактограмме каталитической композиции в сульфидированной фазе наблюдаются пики кристаллических фаз, которые можно отнести к смешанным сульфидам неблагородного металла VIII группы и металла VIB группы, как показано на фигуре 1(с).
Методом электронной просвечивающей микроскопии было установлено, что каталитическя композиция, которая является предметом настоящего изобретения, состоит из высокоплотных частиц с искривленной слоистой структурой, образующих фуллереновые 2-20-слойные наночастицы, как видно на фигуре 2(а, b).
Активные сульфилированные материалы, полученные как прямым сульфидированием каталитической композиции, так и путем сульфидирования окисленных форм после предварительной термообработки каталитической композиции, содержащей по меньшей мере один неблагородный металл VIII группы и по меньшей мере один металл VIB группы и остаточный углерод, были испытаны в качестве катализаторов реакции гидроочистки углеводородов, предпочтительно гидродесульфирования и гидроденитрификации легких и промежуточных нефтяных фракций.
В конкретном случае данного изобретения авторы считают, что легкие и промежуточные нефтяные фракции включают углеводороды с температурой кипения 180°С или ниже, а промежуточные нефтяные фракции содержат углеводороды с температурами кипения 180.1°С и выше, а также 400°С или ниже.
В случае реакции гидродесульфирования, которая является одним из направлений применения каталитической композиции по данному изобретению, использовали модельную молекулу дибензотиофена (DBT), что не ограничивает возможностей ее применения, поскольку указанное соединение представляет собой молекулярную модель реакции гидродесульфирования легких и промежуточных нефтяных фракций (М.Daage and R.R.Chianelli, J. Catal. 149, 414-427 (1994)).
Для минимизации контакта сульфилированных катализаторов с окружающей средой, что могло бы привести к их дезактивации из-за образования сульфатов, сульфидированные вещества приводили в контакт с углеводородом, который содержал модельную молекулу DBT, в герметичной камере и затем вносили в автоклав. Предпочтительно добавлять катализатор в инертной атмосфере азота, гелия или аргона, с тем чтобы избежать насколько возможно его сульфатирования.
Количество DBT в реакционной смеси, которую надо гидродесульфировать, регулируют так же, как в предыдущем способе. Т.е. количество углеводорода взвешивают на аналитических весах высокой точности, затем добавляют требуемое количество DBT, от которого надо очистить углеводород, и затем добавляют катализатор в инертной атмосфере в герметичной камере, таким образом избегая его контакта с окружающей средой. При гидродесульфировании легкой или промежуточной нефтяной фракции катализатор смешивают непосредственно с 10 мл нужной фракции, к которой перед началом реакции гидродесульфирования добавляют органический растворитель, доводя объем смеси до 100 мл.
Реакцию гидродесульфирования проводят в периодическом режиме в трехфазном каталитическом реакторе-автоклаве; реакционную смесь вводят вместе с испытуемым катализатором либо в виде раствора DBT в углеводороде, либо в виде нефтяной фракции, которая подвергается гидродесульфированию.
После загрузки в реакторе для удаления воздуха создают давление инертного газа, такого как азот, гелий и/или аргон, предпочтительно азот. Затем начинают перемешивать с помощью механической мешалки со скоростью 100-2000 об/мин, предпочтительно 800-1500 об/мин, во избежание влияния внутренней диффузии. После этого температуру в реакторе повышают до конечной рабочей температуры в интервале 300-400°C, предпочтительно 320-360°С. Наконец, устанавливают рабочее давление 40-100 кг/см2, предпочтительно 50-70 кг/см2, путем добавления водорода.
После установления условий реакции, описанных в предыдущем параграфе, определяют каталитические свойства, такие как конверсия, скорость реакции, селективность и выход продукта, путем периодического отбора проб. При этом принимали модель псевдопервого порядка реакции по реагирующему углеводороду и нулевого порядка по водороду, т.к. его подавали в большом избытке. Для идентификации и количественной оценки полученных в реакции продуктов отобранные жидкие пробы анализировали на газовом хроматографе Varian 3400 СХ с пламенно-ионизационным детектором и капиллярной колонкой Ultra 2 Capillary Column (неподвижная фаза - 5% поперечно-сшитого фенилметилсиликона). При реальной загрузке общую концентрацию серы определяли на газовом хроматографе HP sievers с хемилюминесцентным детектором (SCD) и капиллярной колонкой. Каталитическую активность образцов катализаторов, которые являются предметами данного изобретения, оценивали по удельной константе скорости реакции (К), выраженной в [л/г с].
Для сравнения определяли каталитическую активность промышленного катализатора, содержащего те же активные компоненты, нанесенные на носитель с развитой поверхностью на основе оксида алюминия, для которого также определили удельную константу скорости реакции. Так была получена константа скорости реакции для катализатора сравнения, названная константой сравнения, которая служит для определения относительной активности катализаторов, составляющих предмет данного изобретения, в виде соотношения активности испытуемых катализаторов и активности катализатора сравнения. Указанное соотношение представляет собой константу скорости реакции на катализаторе, деленную на константу скорости на промышленном катализаторе сравнения.
При использовании DBT в качестве модельной молекулы реакции гидродесульфирования образцы катализаторов - объектов данного изобретения - проявили селективность 30-40% по бифенилу (BP) за три-четыре часа реакции и селективность 60-70% по циклогексилфенилу (СНР) и бициклогексилу (ВСН) - продуктам гидрирования ароматических колец, которые входят в состав BP, что указывает на высокую гидрирующую активность этих катализаторов. Указанную активность в гидрировании определяли как отношение селективностей по продуктам реакции путем деления селективности по СНР плюс селективность по ВСН на селективность по BP: [(SCHP+SBCH)/SBP]; гидрирующая активность катализаторов, которые являются предметом данного изобретения, оказалась значительно выше активности традиционных катализаторов на основе тех же металлов, нанесенных на оксид алюминия. Было показано, что присутствие углерода в каталитической композиции способствует не только увеличению скорости гидродесульфирования, но также и повышению активности в гидрировании. Когда в углеродсодержащую каталитическую композицию, которая является предметом данного изобретения, добавляют связующее, активность в гидродесульфировании несколько снижается, а гидрирующая активность резко уменьшается. Соотношение селективностей [(SCHP+SBCH)/SBP], составлявшее 1-3, предпочтительно 1.5-2.5, уменьшается до значения 0.05-0.5. Обычно в промышленных катализаторах на основе тех же металлов это соотношение равно 0.1-0.3.
Кроме того, присутствие углерода в каталитической композиции, содержащей по меньшей мере один неблагородный металл VIII группы и по меньшей мере один металл VIB группы, а также связующее или связующий реагент, способствует десульфирующей активности и промотирует гидрирущую активность. Поэтому присутствие углерода в каталитической композиции, которая является предметом данного изобретения, оказалось жизненно важным для приготовления высокоактивных катализаторов, на которых достигается высокая степень гидродесульфирования, и получения топлив с очень низким содержанием серы.
Наконец, в примере 12 активность каталитической композиции настоящего изобретения в гидроочистке показана для первичного светлого газойля (PLGO) с высоким содержанием азота (250 и 500 м.д.), свойства которого приведены в таблице 3. Каталитическая композиция, которая составляет предмет данного изобретения, проявляет каталитическую активность примерно в 5 раз большую, чем каталитическая активность традиционного промышленного катализатора с теми же металлами, нанесенными на оксид алюминия, как показано в таблице 4. Содержание серы в полученном продукте уменьшилось до концентрации ниже 30 м.д. по массе серы при проведении испытаний на объемной скорости 2.5 час-1, а когда объемную скорость уменьшили до 1.5 час-1, содержание серы в части продукта понизилось до концентрации ниже 10 м.д. по массе. Поэтому применение каталитической композиции, которая является предметом данного изобретения, для гидродесульфирования легких и промежуточных фракций, представляет собой альтернативу для получения топлив с ультранизким содержанием серы.
ПРИМЕРЫ
Пример 1
11.93 г гептамолибдата аммония [(NH4)6Мо7О24], 21.18 г нитрата никеля [Ni(NO3)2·6Н2О] и 0.33 г цетилтриметиламмоний бромида (СТАВ) растворили в 150 мл деионизированной воды. Получили прозрачный раствор зеленого цвета с рН 4-6; затем добавили примерно 125 мл 28 об.% раствора гидроксида аммония до достижения рН 8-10; раствор остается прозрачным и его цвет меняется с зеленого на голубой. Затем раствор нагрели до температуры 79-90°С, при этом испарился избыток воды (125 мл) и образовался зеленый осадок. Полученная суспензия имеет рН 7.3. Полученное вещество отфильтровали, промыли деионизанной водой и высушили при 120°С в течение 18 час.
Полученный порошок имеет кристаллическую структуру, характерную для смешанного оксогидроксида аммония, молибдена и никеля следующей формулы: (NH4)HNi2(ОН)2(МоО4)2, см. фигуру 1(a).
Затем 5 г вещества, приготовленного из оксогидроксида аммония, молибдена и никеля и содержащего органическое соединение, сульфидировали при 400°С в течение 2 час в кварцевой трубке со скоростью 100 мл/мин в потоке газовой смеси, состоящей из 10 об.% H2S и 90 об.% Н2. Затем твердому веществу дали остыть до комнатной температуры. В сульфидированной фазе вещество представляло собой смесь кристаллических фаз Ni2.5Mo6S6.7 и NiS1.19 (см. фигуру 1(c)).
Полученный смешанный сульфид молибдена и никеля имеет морфологию фуллерена, т.е. он состоит из наносфер с размерами 5-20 нм, состоящих из 2-10 структурных слоев.
Одну часть порошка сульфидированного вещества (сита Тайлера 80:100) испытали в реакции гидродесульфирования дибензотиофена, максимально возможно избегая контакта с окружающей средой. 100 мл раствора, приготовленного из 0.3 г DBT в 100 мл н-гексадекана, поместили в 500 мл автоклав, в который добавили 0.2 г сульфидированного вещества в качестве катализатора. Реактор закрыли и продули инертным газом. Затем температуру подняли до 320°С со скоростью нагрева 2-20°С/мин и установили давление водорода 56 кг/см2. Реакцию вели в течение 4-6 час, отбирая каждые 30 мин аликвоту реакционной смеси для анализа методом газовой хроматографии. Из таблицы 1 видно, что в конце 4 часа реакции конверсия DBT оказалась равной 96% и константа скорости реакции была равна 1.48×10-4 л/г.с.
Селективность по бифенилу (BP) оказалась равной 32.75 мол.%, а селективность по продуктам типа циклогексилфенила (СНР) и бициклогексила (ВСН) оказалась выше 67.25%. Такая высокая селективность по СНР и ВСН показывает, что катализатор обладает высокой гидрирующей активностью. Гидрирующую активность этого катализатора определяли как соотношение селективностей по продуктам реакции путем деления селективности по СНР плюс селективность по ВСН на селективность по BP [(SCHP+SBCH)/SBP]; указанное соотношение оказалось больше 2.05, что указывает на высокую гидрирующую активность.
Другую часть сульфидированного материала испытали в качестве катализатора гидродесульфирования первичного легкого газойля (PLGO) в автоклаве. Для этого эксперимента 20 мл PLGO с параметрами, указанными в таблице 1, разбавили 80 мл гексадекана; эту смесь поместили в 500 мл автоклав, добавив 2 г сульфидированного вещества в качестве катализатора, измельченного до размера частиц, проходящих через сита Тайлера 80-100 меш. Реактор закрыли, продули азотом и затем подняли температуру до 350°С со скоростью нагрева 2-20°С/мин. По достижении этой температуры установили давление водорода 70 кг/см2 и реакцию продолжали 6 час. Начальная концентрация серы в реакционной смеси составляла примерно 2688 м.д., а после 6 часов реакции жидкие продукты реакции содержали 35 м.д. серы. Определенная в этом тесте константа скорости реакции составила 3.42×10-6 л/г.с.
Пример 2
Для оценки влияния поверхностно-активного реагента, добавляемого в каталитическую композицию в качестве предшественника остаточного углерода, вещество, приготовленное в примере 1, прокалили при температуре 400°С на воздухе. Окисленная фаза полученного вещества имела кристаллическую структуру фазы β-NiMoO4 с примесью небольшого количества фазы α-NiMoO4 (см. фигуру 1(b)), и ее морфология представляла собой наночастицы смешанного оксида никеля и молибдена с поверхностью по БЭТ 105 м2/г, объемом пор 0.18 см3/г и средним диаметром пор 6.9 нм.
Вещество в окисленной форме подвергают сульфидированию. Для этого его нагревают со скоростью 4°С/мин до 400°С в кварцевой трубке в токе азота 100 мл/мин. По достижении 400°С ток азота заменяют на газовую смесь, состоящую из 10 об.% H2S и 90 об.% Н2, и продолжают продувку в течение 2 час.
Часть полученного сульфидированного порошка (сита Тайлера 80:100 меш) испытали в реакции гидродесульфирования дибензотиофена (DBT), максимально возможно избегая контакта с окружающей средой. 100 мл раствора, приготовленного из 0.3 г DBT в 100 мл н-гексадекана, поместили в 500 мл автоклав, в который добавили 0.2 г сульфидированного вещества в качестве катализатора. Реактор закрыли и продули инертным газом. Затем температуру подняли до 350°С со скоростью нагрева 2-20°С/мин и установили давление водорода 56 кг/см2. Реакцию вели в течение 4-6 час, отбирая каждые 30 мин аликвоту реакционной смеси для анализа методом газовой хроматографии. Из таблицы 1 видно, что в конце 4 часа реакции конверсия DBT оказалась равной 87% и константа скорости реакции была равна 9.34×10-5 л/г.с. Эти результаты значительно ниже, чем описанные в примере 1, где не применяли прокаливания или окисления, и органическое соединение, использованное в примере 1, образовало остаточный углерод при прогревании в инертной атмосфере.
Селективность по бифенилу (BP) оказалась равной 35.52 мол.%, а селективность по продуктам типа циклогексилфенила (СНР) и бициклогексила (ВСН) оказалась более 64.48 мол.%. Такая высокая селективность по СНР и ВСН показывает, что катализатор обладает высокой гидрирующей способностью, равной 1.82, определенной так же, как в примере 1; однако эта величина была ниже, чем полученная в примере 1, в котором каталитическая композиция содержит остаточный углерод.
Из примеров 1 и 2 можно сделать вывод, что добавка в катализатор поверхностно-активного реагента в качестве предшественника остаточного углерода способствует десульфированию и повышает скорость реакции и гидрирующую активность катализаторов.
Пример 3
Этот пример осуществлен по той же методологии, которая была описана в примере 1. В этом примере получали вещество без добавления органического компонента - предшественника углерода. Количества использованных солей были такими же, как в примере 1, без добавки СТАВ в качестве поверхностно-активного реагента. Полученный осадок отфильтровали и промыли нужным количеством деионизированной воды.
Полученное сухое вещество имело такую же кристаллическую структуру, как в примере 1; она соответствует аммиачному комплексу оксогидроксида никеля-молибдена, представленному на фигуре 1.
Вещество сульфидировали по методике, описанной в примере 1. Полученный порошок испытали в качестве катализатора по методике, описанной в примере 1, в реакции гидродесульфирования DBT и получили конверсию 96 мол.% за 6 час реакции с константой скорости реакции, равной 1.02×10-4 л/г.с. Поэтому этот катализатор оказался на 50% менее активным, чем полученный в примере 1 из композиции, в которую был добавлен поверхностно-активный реагент - предшественник остаточного углерода, и таким же активным, как в примере 2, в котором углерод был удален прокаливанием при 400°С на воздухе. Из этого сравнения сделан вывод о важности добавления поверхностно-активного реагента-предшественника остаточного углерода в ходе приготовления катализатора.
Тест с PLGO проводили в тех же условиях, что и в примере 1. После 6 час реакции был получен конечный продукт с содержанием серы менее 48 м.д. по массе; константа скорости реакции равна 2.85×10-6 л/г.с, что ниже константы скорости в таком же тесте примера 1.
Пример 4
Сухое вещество, полученное в примере 3, прокалили на воздухе по методике, описанной в примере 2. В окисленной фазе вещество имело такую же кристаллическую структуру, как приведенная в примере 2; однако поверхность по БЭТ была равна 92 м2/г, объем пор 0.132 см3/г и средний диаметр пор 5.1 нм. Вещество в окисленной форме сульфидировали. Для этого его нагревали со скоростью 4°С/мин до 400°С в кварцевой трубке в потоке азота 100 мл/мин. По достижении 400°С ток азота заменили на газовую смесь из 10 об.% H2S и 90 об.% Н2 и поддерживали ток в течение 2 час. Одну часть порошка сульфидированного вещества (сита Тайлера 80:100 меш) испытали в реакции гидродесульфирования дибензотиофена (DBT), максимально возможно избегая контакта с окружающей средой, по методике и в условиях, описанных в примере 1.
Как можно видеть в таблице 1, в конце 4 часа реакции конверсия DBT достигла 89%, а константа скорости реакции достигла значения 7.5×10-5 л/г.с. Эти результаты значительно ниже, чем полученные в примерах 1 и 3, где не было прокаливания, и значительно ниже результатов примера 2, где применили прокаливание, но исключили органическое соединение, вводимое во время смешения и на стадии реакции при приготовлении катализатора. Однако распределение селективностей образования продуктов было практически идентичным набюдаемому в примере 3, как следует из таблицы 1. То есть можно сказать, что оба катализатора обладают одинаковой гидрирующей способностью благодаря тому, что в этом случае во время прокаливания не удалили органическое соединение-предшественник.
Пример 5
В этом примере приготовили катализатор путем осаждения смешанного оксогидроксида никеля-молибдена в присутствии связующего или связующего реагента. Использовали оксид алюминия в виде бемита с величиной поверхности 243 м2/г, средним объемом пор 0.336 см3/г и средним диаметром пор 5.54 нм. Для этого, как и в примере 1, растворили 11.93 г нитрата никеля, 21.18 г гептамолибдата аммония и 0.33 г поверхностно-активного реагента (СТАВ) в 150 мл воды (рН 5). Раствор приобрел зеленый цвет, затем раствор нагрели до 90°С и добавили 125 мл гидроксида аммония (рН 10); прозрачный раствор стал голубым. Нагревание этого раствора при 90°С продолжили в течение 30 мин; затем добавили 68.25 г бемита (Catapal). Для выпаривания воды раствор с добавленным бемитом продолжали нагревать примерно три часа при указанной выше температуре и наблюдали изменение цвета с голубого на зеленый и затем на светло-зеленый. Полученный осадок оставили охлаждаться до комнатной температуры и промыли 4 раза по 600 мл дистиллированной воды. Наконец, полученный продукт сушили при температуре 120°С в течение 18 час.
После этого вещество прокалили по методике, описанной в примере 1. В окисленной фазе вещество дает дифракционную картину, типичную для гамма-оксида алюминия. Катализатор в окисленной форме содержит 16.4 мас.% МоО3, 7.17 мас.% NiO и 76.4 мас.% Al2O3. Это вещество имеет удельную поверхность 271 м2/г, определенную по физической адсорбции азота, объем пор 0.31 см3/г и диаметр пор 4.5 нм.
Часть сухого вещества сульфидировали по методике, описанной в примере 1, не исключая органического соединения - предшественника углерода. Его каталитическую активность определяли в реакции гидродесульфирования DBT по методике, описанной в примере 1. В конце 4 часа реакции конверсия составляла 98% и каталитическая активность достигла 1.27×10-4 л/г.с.
Селективность образования BP оказалась равной 79.07 мол.%, а селективность по гидрированным продуктам СНР и ВСН составила 20.93 мол.%. Гидрирующую активность определяли, как в примере 1, по соотношению селективностей [(SCHP+SBCH)/SBP], и она оказалась равной 0.26, что значительно меньше, чем в примере 1. Хотя скорость гидродесульфирования была высокой, гидрирующая способность оказалась низкой по сравнению с примерами 1-4, где в состав композиции не входил связующий компонент.
Пример 6
Пример 6 осуществили по методике примера 5. При этом во время приготовления раствора гептамолибдата аммония и нитрата никеля не добавляли поверхностно-активный реагент. Кристаллизацию провели в присутствии такого же связующего, как в примере 5.
Часть вещества прокалили на воздухе для получения его в окисленной форме. Дифракционная картина этого вещества в основном содержит пики, характерные для гамма-оксида алюминия. В окисленной фазе вещество содержит примерно 16.4 мас.% MoO3, 7.2 мас.% NiO и 76.4 мас.% Al2O3. Величина удельной поверхности, определенная по физической адсорбции азота, оказалась равной 260 м2/г, средний объем пор 0.29 см3/г и диаметр пор 4.5 нм. В сравнении с примером 5 это вещество обладало большей удельной поверхностью, чем в случае, когда его готовили в присутствии поверхностно-активного реагента.
Часть вещества сульфидировали по методике, описанной в примере 1. Его каталитическую активность определяли в реакции гидродесульфирования по методике, описанной в примере 1. После 4 час реакции конверсия на этом катализаторе составила 97% и константа скорости гидродесульфирования DBT была равна 1.06×10-4 л/г.с. Селективность образования BP была равна 89.04% и селективность по продуктам гидрирования, СНР и ВСН, оказалась равной 10.96%. Каталитическая активность этого материала была ниже активности такого же вещества, содержащего остаточный углерод, из примера 5. Кроме того, его гидрирующая активность снизилась до 0.12, т.к. селективность по продуктам гидрирования СНР и ВСН - была ниже. Сравнение примеров 5 и 6 свидетельствует о том, что введение органического соединения на стадии синтеза, такого как поверхностно-активное вещество, способствует гидродесульфированию и промотирует гидрирующую активность катализатора, даже если в него вводят связующее.
Пример 7
В этом примере определяли каталитическую активность промышленного катализатора, содержащего те же металлы, что и каталитическая композиция, которая является предметом данного изобретения; они были нанесены на традиционный оксид алюминия, используемый в катализаторах гидродесульфирования. Каталитическую активность в гидродесульфировании DBT определяли по методике, описанной в примере 1.
После 4 час реакции конверсия DBT составила 96%, а константа скорости была равна 1.12×10-4 л/г.с. Селективность образования продуктов гидрирования, в основном СНР, составила 9.53%, а селективность по BP составила 90.47%. Гидрирующая активность, определенная как соотношение селективностей [(SCHP+SBCH)/SBP], была равна 0.1.
Пример 8
Следуя методике, описанной в примере 1, катализатор готовили, заменив соль никеля на соль кобальта. Для этого взяли 11.93 г гептамолибдата аммония, 21.18 г нитрата кобальта [Со(NO3)2·6Н2О] и 0.33 г цетилтриметиламмоний бромида (СТАВ).
Полученный осадок имел ту же структуру, что и материал на основе никеля; только в этом случае аммиачный комплекс образовался из смешанного оксогидроксида кобальта-молибдена (NH4)HCO2(ОН)2(MoO4)2. Полученное вещество сульфидировали по методике, описанной в примере 1. Полученное сульфидированное вещество состояло из смеси сульфидированных фаз металлов: MoS2, CoxMoySz и Co4S3. В этом примере полученный порошок (сита Тайлера 80:100 меш) сульфидировали и в реакторе-автоклаве определяли его каталитическую активность в гидродесульфировании первичного легкого газойля (PLGO), разбавленного на 20 об.% гексадеканом, как описано в примере 1, но только в этом случае рабочее давление в реакции было меньше (56 кг/см2). В этой реакции после 6 час реакции получили продукт с 79 м.д. по массе серы, а константа скорости реакции была равна 2.19×10-6 л/г.с.
Пример 9
Вещество готовили по методике, описанной в примере 1. Для этого взяли то же количество гептамолибдата аммония (11.93 г), разделив количество соли второго металла на две части - 10.6 г нитрата никеля и 10.6 г нитрата кобальта. По той же методике получили осадок смешанного нитрата кобальта, никеля и молибдена. Осадок промыли и высушили, как в примере 1.
Полученное твердое вещество имело структуру смешанного оксогидроксида никеля, кобальта и молибдена с аммиаком типа (NH4)HCoNi(ОН)2(MoO4)2. Вещество с указанной структурой сульфидировали по методике, описанной в примере 1, и каталитическую активность порошка (80:100 меш) определяли в гидродесульфировании углеводородной фракции (PLGO) в виде разбавленного PLGO, как описано в примере 1. В этой реакции получили продукт, содержащий 106 м.д. серы, а константа скорости реакции составила 1.84×10-6 л/г.с.
Пример 10
Для выяснения влияния концентрации поверхностно-активного реагента, добавленного в ходе синтеза, в двух следующих примерах варьировали количество добавленного поверхностно-активного реагента. Катализатор готовили по методике, описанной в примере 1, используя те же количества солей-предшественников и растворителя; изменяли только количество добавленного поверхностно-активного реагента, которое составляло 1.65 г СТАВ. Осадок промыли и высушили, как в примере 1.
Полученное твердое вещество имело структуру смешанного оксогидроксида никеля, кобальта и молибдена с аммиаком, такого как (NH4)HNi2(ОН)2(MoO4)2. Вещество с такой структурой сульфилировали по методике, описанной в примере 1, и каталитическую активность полученного порошка (сита Тайлера 80:100 меш) определяли в гидродесульфировании PLGO, разбавленного, как описано в примере 1. Полученные результаты представлены в таблице 2. В этой реакции получили продукт, содержащий 43 м.д. серы, а константа скорости реакции, равная 3.02×10-6 л/г.с, была несколько меньше константы скорости в примере 1. Однако катализатор проявил очень высокую активность.
Пример 11
Следуя способу, описанному в примере 1, с теми же веществами, как в примере 10, в этом примере увеличили количество поверхностно-активного вещества СТАВ до 3.3 г, используя те же количества солей и растворителей, как в примере 1.
Полученное твердое вещество имело такую же структуру, как в предыдущих примерах с соответствующими металлами.
Полученное вещество сульфилировали по методике, описанной в примере 1, и каталитическую активность порошка (сита Тайлера 80:100 меш) определяли в гидродесульфировании углеводородной фракции (PLGO), используя разбавленный PLGO, как описано в примере 1. Полученные результаты представлены в таблице 2. В этой реакции получили продукт, содержащий 40 м.д. серы, а константа скорости реакции 3.08×10-6 л/г.с была несколько меньше, чем в примере 1, и была близка к активности катализатора, синтезированного в примере 10. Сравнивая результаты оценки катализаторов, полученных в примерах 1, 10 и 11, можно сделать вывод, что количество поверхностно-активного реагента не оказывает заметного влияния на скорость гидродесульфирования, вероятно, потому, что способ синтеза включает стадию промывки и в твердом веществе остается только органическое соединение, прочно связанное с анионами металлов, а концентрация анионов не изменяется с увеличением концентрации поверхностно-активного вещества.
Пример 12
Каталитическую композицию готовили по методике, описанной в примере 1. Полученный осадок - оксогидроксид никеля-молибдена - смешали с 20 мас.% оксида алюминия (бемита) в расчете на общую массу каталитической композиции. Затем добавили водный 3 мас.% раствор азотной кислоты, растворенной в дистиллированной воде, до получения пасты. Влажной пасте дали постоять в закрытом сосуде и затем экструдировали. Полученный экструдат сушили при 120°С по методике, описанной в примере 1.
Экструдаты подвергли термообработке в атмосфере азота при 400°С для удаления и возможной карбонизации органического соединения СТАВ, добавленного во время смешения и реакций растворов в ходе синтеза. Полученная каталитическая композиция содержит 16 мас.% Al2O3, 55 мас.% MoO3 и 37 мас.% NiO и менее 1.5% углерода. После термообработки в инертной атмосфере каталитическая композиция имела удельную поверхность 135 м2/г, средний объем пор 03 см3/г и средний диаметр пор 6 нм.
Одну часть этого материала тестировали в установке с неподвижным слоем и непрерывным потоком под давлением, в которой 5 мл вещества (4.5 г) поместили в реактор диаметром 1.4 см и длиной 48 см. После этого катализатор сульфидировали, пропуская через него углеводородную фракцию (первичный легкий газойль, PLGO), параметры которой приведены в таблице 3 и к которой добавили диметилсульфид (DMDS) для повышения содержания S на 10000 м.д. Таким образом, конечное общее содержание серы составило примерно 2.5 мас.%. Сульфидирование проводили при 27 кг/см2. Использованная для сульфидирования объемная скорость (LHSV) была равна 1.5 час-1 при соотношении Н2/углеводород, равном 333 м3/м3. Температуру слоя катализатора повышали от комнатной температуры до 135°С со скоростью 40°С/час. Затем скорость изменили на 28°С/час и подняли температуру до 343°С и поддерживали ее в течение 3 час. Затем систему выдержали в условиях давления, объемной скорости и соотношения Н2/углеводород, близких к условиям тестирования (70 кг/см2, LHSV=2.5 час-1 и 445 м3/м3), подавая углеводородное сырье (PLGO), которое подвергали гидрочистке, и оставили для стабилизации на 96 час при 343°С. Затем температуру подняли до первой температуры тестирования (350°С) и работали до того момента, когда содержание S в суспензии (анализатор Antek и газовый хроматограф с хемилюминесцентным детектором) стало практически постоянным. По окончании операции реактор готовили для следующей тестируемой температуры.

Изобретение относится к способу получения каталитической композиции, состоящей по меньшей мере из одного неблагородного металла VIII группы и по меньшей мере одного металла VIB группы. Описан способ получения каталитической композиции для гидроочищения потоков углеводородов, состоящей по меньшей мере из одного неблагородного металла VIII группы и по меньшей мере одного металла VIB группы и из четвертичной аммонийной соли в качестве предшественника углерода, которые находятся в полностью растворенном состоянии в растворителе, который включает следующие стадии: а) растворение и смешение солей-предшественников в растворителе, который является водой, спиртом, таким как метанол, этанол, пропанол, бутанол, и/или водно-спиртовой смесью; b) растворение органического компонента и/или поверхностно-активного реагента в растворителе; с) смешивание растворов, полученных на стадиях (а) и (b); d) добавление основного раствора гидроксида и/или карбоната аммония к раствору стадии (с) до получения рН 5-14, предпочтительно 8-12; е) повышение температуры раствора, полученного на стадии (d), до 50-200°С, предпочтительно 60-100°С; f) гомогенизацию раствора со стадии (е); g) кристаллизацию раствора, полученного на стадии (f), путем выпаривания растворителя; h) фильтрование или центрифугирование закристаллизованной суспензии, полученной на стадии (g), для разделения кристаллов и маточного раствора в том случае, когда выпаривание не было закончено; i) промывку твердого вещества, полученного на стадии (h), достаточным количеством деионизированной воды и/или водно/спиртовой смеси; j) сушку твердого вещества, полученного на стадии (i), при температуре 50-300°С, предпочтительно 80-150°С, к) термообработку твердого вещества, полученного на стадии (j), в инертной атмосфере, такой как азот, гелий, аргон и т.п., при температуре 200-1000°С, предпочтительно 300-600°С; 1) сульфидирование вещества, полученного на стадии (i) или (j), в токе газа, содержащего 10 об.% сероводорода, или углеводородной фракции, содержащей по меньшей мере 0.2 мас.% серы, в токе водорода при температуре 200-600°С, предпочтительно 250-500°С, и давлении 1-100 кг/см2. Также описана каталитическая композиция, полученная вышеописанным способом, которая включает по меньшей мере один неблагородный металл VIII группы, по меньшей мере один неблагородный металл VIB группы и углерод; и, возможно, содержащая неорганический оксид в качестве связующего, который может находиться в окисленном состоянии и/или быть частично восстановленным и/или сульфидированным. Технический результат - получение каталитической композиции, которая обладает высокой удельной активностью в реакциях гидроочистки легких и средних фракций. 2 н.п. и 35 з.п. ф-лы, 4 табл., 2 ил.
1. Способ получения каталитической композиции для гидроочищения потоков углеводородов, состоящей по меньшей мере из одного неблагородного металла VIII группы и по меньшей мере одного металла VIB группы и из четвертичной аммонийной соли в качестве предшественника углерода, которые находятся в полностью растворенном состоянии в растворителе, который включает следующие стадии:
a) растворение и смешение солей-предшественников в растворителе, который является водой, спиртом, таким как метанол, этанол, пропанол, бутанол, и/или водно-спиртовой смесью;
b) растворение органического компонента и/или поверхностно-активного реагента в растворителе;
c) смешивание растворов, полученных на стадиях (а) и (b);
d) добавление основного раствора гидроксида и/или карбоната аммония к раствору стадии (с) до получения рН 5-14, предпочтительно 8-12;
e) повышение температуры раствора, полученного на стадии (d) до 50-200°С, предпочтительно 60-100°С;
f) гомогенизацию раствора со стадии (е);
g) кристаллизацию раствора, полученного на стадии (f), путем выпаривания растворителя;
h) фильтрование или центрифугирование закристаллизованной суспензии, полученной на стадии (g), для разделения кристаллов и маточного раствора в том случае, когда выпаривание не было закончено;
i) промывку твердого вещества, полученного на стадии (h), достаточным количеством деионизированной воды и/или водно-спиртовой смеси;
j) сушку твердого вещества, полученного на стадии (i), при температуре 50-300°С, предпочтительно 80-150°С;
k) термообработку твердого вещества, полученного на стадии (j), в инертной атмосфере, такой как азот, гелий, аргон и т.п., при температуре 200-1000°С, предпочтительно 300-600°С;
l) сульфидирование вещества, полученного на стадии (i) или (j), в токе газа, содержащего 10 об.% сероводорода, или углеводородной фракции, содержащей по меньшей мере 0,2 мас.% серы, в токе водорода при температуре 200-600°С, предпочтительно 250-500°С, и давлении 1-100 кг/см2.
2. Способ по п.1, в котором соли-предшественники всех металлов VIII группы и VIB группы полностью растворены в жидкости, используемой в качестве растворителя.
3. Способ по п.1, в котором полностью растворенные соли-предшественники смешивают с раствором, содержащим органическое соединение.
4. Способ по п.1, в котором используемый растворитель представляет собой воду, и/или спирт, и/или спиртово-водную смесь.
5. Способ по п.1, в котором предшественник углерода представляет собой четвертичный аммонийный поверхностно-активный реагент, такой как катионное поверхностно-активное вещество, определяемое формулой [R4N]+X-, где R - алкильная группа, а Х - галоген и/или гидроксил.
6. Способ по п.1, в котором неблагородный металл VIII группы включает никель, кобальт и/или железо или смесь этих металлов.
7. Способ по п.6, в котором никель и/или кобальт представляют 100 мас.% неблагородных металлов VIII группы в расчете на оксиды.
8. Способ по п.1, в котором металл VIB группы представляет собой молибден, и/или вольфрам, и/или хром.
9. Способ по п.8, в котором молибден составляет по меньшей мере 50 мас.% выбранных металлов VIB группы, предпочтительно по меньшей мере 80 мас.% и более предпочтительно 100 мас.% металлов VIB группы.
10. Способ по п.1, который включает стадию введения основного раствора, который можно выбрать из раствора гидроксида аммония, и/или карбоната аммония, и/или мочевины, или четвертичной аммонийной соли, для поддержания рН 5-14, предпочтительно 8-12.
11. Способ по п.1, в котором стадия гомогенизации протекает при таких значениях температуры и рН, при которых все выбранные соли-предшественники находятся в полностью растворенном состоянии.
12. Способ по п.1, в котором стадия гомогенизации растворов протекает за время от 5 мин до 24 ч, предпочтительно от 5 мин до 10 ч.
13. Способ по п.1, в котором стадию кристаллизации из раствора проводят путем частичного упаривания растворителя в кристаллизаторе и/или полного упаривания растворителя в испарителе.
14. Способ по п.1, в котором стадию кристаллизации из раствора проводят путем испарения растворителя при температуре 80-200°С.
15. Способ по п.1, в который в случае неполного упаривания и/или кристаллизации дополнительно включает стадии фильтрования и/или центрифугирования и рециркуляции маточных растворов.
16. Способ по п.1, в котором термообработку в инертной атмосфере проводят при температуре 200-1000°С, предпочтительно 300-600°С, в токе азота, гелия, аргона или их смеси со скоростью по меньшей мере 0,01 л/г·мин.
17. Способ по п.1, в котором термообработку не следует проводить в окислительной атмосфере.
18. Способ по п.1, в котором сульфидируют непосредственно сухое вещество без предварительной термообработки.
19. Способ по п.1, в котором сульфидируют вещество после термообработки в инертной атмосфере согласно п.16.
20. Способ по п.2, в котором сульфидируют либо сухое, либо термически обработанное вещество в токе газа, состоящего из смеси сероводорода (H2S), дисульфида углерода (C2S), диметилсульфида (DMDS), тиофена и т.п. и водорода, при температуре 200-600°С, предпочтительно 250-500°С, при атмосферном давлении.
21. Способ по п.1, в котором сульфидируют либо сухое, либо термообработанное вещество в присутствии жидкой углеводородной фракции, содержащей по меньшей мере 0,1 мас.% серы, и водорода при давлениях 1-100 кг/см2 и температурах 200-600°С, предпочтительно 250-500°С.
22. Способ по п.1, который включает введение неорганического оксида типа связующего, такого как оксид алюминия, оксид кремния, оксид титана, алюмосиликат, оксид алюминия-оксид титана, оксид циркония, оксид циркония-оксид титана, оксид циркония-оксид алюминия, аморфные алюминаты кремния и/или кристаллические вещества, глины и т.п., на стадии гомогенизации смеси солей-предшественников.
23. Способ по п.1, в котором неорганический оксид типа связующего добавляют после стадии кристаллизации к влажному и/или сухому веществу.
24. Способ по п.1, в котором неорганический оксид типа связующего добавляют после термической обработки и/или сульфидирования.
25. Способ по п.1, в котором формуют экструдаты вещества, содержащего неорганический оксид типа связующего, добавленный на любой из стадий по пп.22-24.
26. Каталитическая композиция, полученная способом по п.1, которая включает по меньшей мере один неблагородный металл VIII группы, по меньшей мере один неблагородный металл VIB группы и углерод; и, возможно, содержащая неорганический оксид в качестве связующего, который может находиться в окисленном состоянии и/или быть частично восстановленным и/или сульфидированным.
27. Каталитическая композиция по п.26, в которой неблагородный металл VIII группы представляет собой никель и/или кобальт или их смесь, концентрация которых находится в интервале 20-50 мас.% от общей композиции составляющих оксидов.
28. Каталитическая композиция по п.26, в которой металл VIB группы предпочтительно представляет собой молибден и/или вольфрам, композиция которых составляет 30-50% от общей композиции, определенной как оксиды составляющих металлов.
29. Каталитическая композиция по п.28, в которой металл VIB группы предпочтительно является молибденом.
30. Каталитическая композиция по п.26, в которой углерод находится в аморфном состоянии или образует карбиды с металлами VIB и VIII групп, концентрация которых составляет 0,05-5 мас.% от общей композиции составляющих оксидов.
31. Каталитическая композиция по п.26, в которой металлы VIII и VIB групп составляют 50-100 мас.% от всей каталитической композиции в виде оксидов.
32. Каталитическая композиция по п.26, дифракционная картина которой соответствует фазе альфа- и бета-молибдатов металла VIII группы в случае, когда металл, выбранный из VIB группы, является молибденом, и/или вольфрамату металла VIII группы в случае, когда выбранный металл является вольфрамом.
33. Каталитическая композиция по п.26, дифракционная картина которой соответствует фазе альфа- и бета-молибдатов никеля или молибдату кобальта или молибдату никеля и кобальта в случае, когда металл, выбранный из VIB группы, является молибденом, и/или вольфрамату или вольфрамату кобальта или вольфрамату никеля в случае, когда металл, выбранный из VIB группы, является вольфрамом.
34. Каталитическая композиция по п.26, которая содержит сульфиды по меньшей мере одного неблагородного металла VIII группы и по меньшей мере одного металла VIB группы и/или смешанные сульфиды металлов VIII и VIB групп.
35. Каталитическая композиция по п.34, в которой по меньшей мере часть сульфидов металлов VIB группы и сульфидов металлов VIII группы имеет морфологию фуллереновых наносфер, состоящих из 2-20 структурных слоев.
36. Каталитическая композиция по п.26, обладающая высокой активностью в гидродесульфировании, гидрировании и гидроденитрификации в ходе гидроочистки углеводородных фракций.
37. Каталитическая композиция по п.26, обладающая высокой активностью в гидродесульфировании и гидроденитрификации при получении топлив с ультранизким содержанием серы и азота.
| WO 2002004117 A1, 17.01.2002 | |||
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ГИДРООЧИСТКИ НЕФТЯНЫХ ФРАКЦИЙ | 1994 |
|
RU2074025C1 |
| SU 1557742 A1, 10.03.1997 | |||
| US 6635599 B1, 21.10.2003 | |||
| WO 2004000456 A2, 31.12.2004 | |||
| US 20030111391 A1, 19.06.2003. | |||

