Ссылка на предшествующую заявку
Настоящая заявка заявляет преимущество предварительной заявки №60/728174, поданной 19 октября 2005, включенной в настоящее описание путем ссылки.
Уровень техники
Настоящее изобретение относится к функционализированным или «модифицированным по концам цепей» эластомерным полимерам, их применению при получении эластомерных композиций и изделиям из них. В частности, изобретение относится к применению так называемых «сульфанилсиланов» в качестве модификаторов концов цепей для «живущих» анионных эластомерных полимеров. Концы полимерных цепей являются реакционноспособными вследствие присутствия в них ненасыщенных участков основной полимерной цепи эластомерного полимера и/или наполнителей или других компонентов, содержащихся в эластомерной композиции. Данные модифицированные эластомерные полимеры находят применение в производстве вулканизованных эластомерных композиций, обладающих относительно низкими гистерезисными потерями. Такие композиции могут быть использованы в производстве многих изделий, включая протекторы шин, обладающие низким сопротивлением качению, наряду с хорошим сочетанием других желательных физико-механических и химических свойств, например, хорошим сопротивлением скольжению, износостойкостью, пределом прочности при разрыве и способностью к переработке.
Считается, что основной причиной гистерезисных потерь в вулканизованных эластомерных полимерах являются свободные концы полимерных цепей, то есть участок цепи эластомерного полимера между последней поперечной связью и концом полимерной цепи. Этот свободный конец полимера не участвует эффективно ни в каком процессе эластичного восстановления, и в результате теряется энергия, передаваемая на данную часть полимерной цепи. Данная диссипированная энергия приводит к заметному гистерезису в условиях динамической деформации. Гистерезисные потери эластомерной полимерной композиции связывают с величиной tg δ при 60°C. Как правило, вулканизованные композиции на основе эластомерных полимеров, обладающие относительно малой величиной tg δ при 60°C, являются предпочтительными как имеющие более низкие гистерезисные потери. В шинах это проявляется в более низком сопротивлении качению и повышенной экономии топлива.
Одним общепринятым подходом в снижении гистерезисных потерь является снижение числа свободных концов цепей эластомерных полимеров. Описаны различные способы в открытой литературе, включающие использование «сшивающих агентов», таких как тетрахлорид олова, которые могут функционализировать конец полимерной цепи и взаимодействовать с ненасыщенными частями основной полимерной цепи и/или другими компонентами эластомерной композиции, такими как наполнитель. Примеры таких методов, наряду с другими документами по рассматриваемому вопросу, включают: патент США 3281383; 3244664 и 3692874 (например, тетрахлорсилан); патент США 3978103; патент США 4048206: 4474908; патент США 6777569 (блокированные меркаптосиланы) и патент США 3078254 (многозамещенный галогеном углеводород, такой как 1,3,5-три(бромметил)бензол); патент США 4616069 (соединение олова с органическим аминосоединением или аминным соединением); и патент США 2005/0124740.
«Синтез полимера с концевыми функциональными группами методом живущей анионной полимеризации», Journal of Macromolecular Chemistry and Physics, 197, (1996), 3135-3148, описывает синтез содержащих полистирол и полиизопрен живущих полимеров с гидрокси- (-ОН) и меркапто- (-SH) функциональными концевыми группами, полученных взаимодействием живущего полимера с галогеналканами, содержащими простые силилэфирные и простые силилтиоэфирные группы. Трет-бутилдиметилсилильная (TBDMS) группа является предпочтительной защитной группой для групп -ОН и -SH в реакциях обрыва цепи, потому что, как найдено, и соответствующие простые силилэфиры, и простые тиоэфиры являются стабильными и совместимыми с анионными живущими полимерами.
В патенте США №6579949 описано использование серосодержащих соединений аналогичного класса, включающих трет-бутилдиметилсилил-3-хлор-1-пропилсульфид (Cl-(CH2)3-S-Si-(CH2)2C(CH3)3), для получения резиновых изделий, обладающих низкими гистерезисными потерями. Более конкретно, представленные серосодержащие соединения взаимодействуют с живущими полимерами анионного инициирования с получением полимеров с модифицированными концами цепей, которые затем смешивают с наполнителями, вулканизующими агентами, ускорителями, масляными наполнителями и другими различными добавками для производства шин с низкими гистерезисными потерями. Трет-бутилдиметилсилилпропилсульфидный конец нельзя легко удалить в условиях стандартной полимеризации, но защитная трет-диметилсилильная группа расщепляется при реакции с добавками, содержащими H+, F-, или соединением цинка перед процессом вулканизации или в процессе вулканизации, оставляя, таким образом, меркапто («тиольную») группу взаимодействовать (по меньшей мере, 20 процентов) с ненасыщенными сегментами основной цепи других эластомерных полимеров. К сожалению, в результате реакции модификации конца цепи образуется хлорид лития. Хлоридные ионы, присутствующие в реакционной системе, сильно ускоряют коррозию технологического оборудования.
В патенте США №6229036 раскрыт широкий класс сульфанилсиланов, полученных взаимодействием меркаптосиланов с хлорсиланами, и их использование в качестве сшивающих агентов в резиновых смесях для получения протекторов, имеющих низкое сопротивление качению и хорошее сцепление с влажным асфальтом. Описаны многие сульфанилсиланы, включая (EtO)3-Si-(CH2)3-S-Si-(CH3)3 и (MeO)3-Si-(CH2)3-S-Si-(C2H5)3. Согласно данной ссылке эластомерные полимеры получают с использованием традиционных методов обрыва цепи, а затем смешивают с оксидными наполнителями и от 0,1 до 15 массовых процентов (относительно наполнителя) сульфанилсиланового сшивающего агента, а затем вулканизуют с получением резинотехнического продукта. Таким образом, в отличие от подхода, описанного в патенте США №6579949, сульфанилсилановый сшивающий агент не использован в качестве модификатора конца цепи для живущего полимера, а только объединен с эластомерным полимером после реакции обрыва цепи в процессе компаундирования. Данный подход невыгоден из-за сложности распределения сшивающего агента в резиновой смеси в процессе ее компаундирования. То есть, в отличие от типичной низковязкой, основанной на использовании растворителя среды, связанной с большинством процессов анионной полимеризации, среда в процессе изготовления резиновой смеси обычно является высоковязкой и не содержит растворителя, что приводит к менее равномерному распределению сшивающего агента по объему смеси. Как следствие этого, взаимодействие содержащего функциональные группы полимера с материалом наполнителя и/или ненасыщенными сегментами основной полимерной цепи является менее полным. Если модифицирующее соединение вводят в полимер, содержащий исключительно оборванные полимерные цепи, то невозможно эффективно объединить (или осуществить взаимодействие) концы цепей этого полимера с другими полимерными цепями, или с наполнителями, при использовании модифицирующего соединения. Кроме того, невозможно эффективно объединить или соединить полимер с наполнителями или другими полимерными цепями.
Краткое изложение сущности изобретения
Изобретение относится к модифицированному по концу цепи эластомерному полимеру, включающему продукт взаимодействия
i) живущего анионного эластомерного полимера и
ii)силансульфидного модификатора, представленного формулой
(RO)x(R)ySi-R'-S-SiR3,
где:
Si означает кремний; S означает серу; О означает кислород;
x представляет собой целое число, выбранное из 1, 2 и 3;
y представляет собой целое число, выбранное из 0, 1 и 2; где х+y=3;
R являются одинаковыми или различными и представляют собой (С1-С16)алкил; и R' представляет собой арил, алкиларил или (С1-С16)алкил.
В другом аспекте настоящее изобретение также относится к вулканизованной композиции на основе эластомерного полимера, включающей продукт взаимодействия следующих компонентов:
1) наполнителя;
2) вулканизующего агента и
3) модифицированного по концу цепи эластомерного полимера, где модифицированный по концу цепи эластомерный полимер представляет собой продукт взаимодействия:
i) живущего анионного эластомерного полимера и
ii) силансульфидного модификатора, представленного формулой:
(RO)x(R)ySi-R'-S-SiR3,
где:
Si означает кремний; S означает серу; О означает кислород;
x представляет собой целое число, выбранное из 1, 2 и 3;
y представляет собой целое число, выбранное из 0, 1 и 2; при этом х+y=3;
R являются одинаковыми или различными и представляют собой (С1-С16)алкил; и
R' представляет собой арил, алкиларил или (С1-С16)алкил.
В другом аспекте настоящее изобретение относится к способу получения вулканизованной композиции на основе эластомерного полимера, включающему объединение следующих компонентов:
1) наполнителя;
2) вулканизующего агента и
3) модифицированного по концу цепи эластомерного полимера, который представляет собой продукт взаимодействия:
i) живущего анионного эластомерного полимера и
ii) силан-сульфидного модификатора, представленного формулой:
(RO)x(R)ySi-R'-S-SiR3,
где:
Si означает кремний; S означает серу; О означает кислород;
x представляет собой целое число, выбранное из 1, 2 и 3;
y представляет собой целое число, выбранное из 0, 1 и 2; при этом х+y=3;
R являются одинаковыми или различными и представляют собой (С1-С16)алкил; и
R' представляет собой арил, алкиларил или (С1-С16)алкил.
Изобретение также относится к модифицированному по концу цепи эластомерному полимеру, включающему продукт взаимодействия:
i) живущего анионного эластомерного полимера и
ii) силансульфидного модификатора, представленного формулой:
GJMSi-A-S-SiTXZ,
где:
Si означает кремний; S означает серу; G означает (C1-С16)алкокси;
J и М являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода (Н), (С1-С16)алкила, (С1-С16)алкокси, (С7-С16)арила, (С7-С16)алкиларила и -А-S-SiTXZ (где A, T, X и Z определены ниже);
А представляет собой арил, алкиларил, (С7-С16)алкиларил или (С1-С16)алкил, который может быть линейным или разветвленным, насыщенным или ненасыщенным и может быть замещен (С1-С4)алкилом, (С1-С4)алкокси, (С7-С16)арилом, (С7-С16)аралкилом, нитрилом, амином, NO2, тиоалкилом, -A-S-SiTXZ (где A, T, X и Z определены ниже); и
группы Т, Х и Z являются одинаковыми или различными, и каждая независимо выбрана из группы, состоящей из водорода (Н), (С1-С16)алкила, (С1-С16)алкокси, (С7-С16)арила, (С7-С16)алкиларила и -S-А-SiMJG (A, M, J и G определены в описании). Настоящее изобретение также относится к вулканизованным композициям на основе эластомерного полимера, содержащим данный модифицированный по концу цепи эластомерный полимер, и способам их получения.
Подробное описание изобретения
Настоящее изобретение включает модифицированный по концу цепи полимер, содержащий продукт взаимодействия живущего анионного полимера и силансульфидного модификатора, представленного формулой 1, и более предпочтительно формулой 2, каждая из которых представлена ниже. Изобретение дополнительно включает способы получения таких модифицированных по концам цепей полимеров, их применение для получения вулканизованных композиций на основе эластомерного полимера и изделия, полученные из данных композиций, такие как пневматические шины, шинные протекторы, ремни, обувь и тому подобное.
Представленные композиции демонстрируют более низкую величину tg δ при 60°C при сохранении хороших технологических свойств и хороший баланс физико-механических свойств, включая одно или несколько из следующих свойств: износостойкость, предел прочности при разрыве, условное напряжение при растяжении и относительное удлинение при разрыве. Представленные композиции могут быть использованы в производстве протекторов, обладающих пониженным сопротивлением качению при сохранении хорошего сцепления шин с влажной дорогой. Представленные композиции, в частности, особенно применимы в производстве шин, включая наполнители, такие как технический углерод, диоксид кремния, двухфазный наполнитель углерод-диоксид кремния и тому подобное.
Термин «эластомерные полимеры» означает эластомеры или каучуки, включая структурированные полимеры, которые, будучи поперечносшитыми, обладают свойствами, аналогичными свойствам вулканизованного натурального каучука (цис-1,4-полиизопрена), например, удлинением при растяжении и относительно быстрым сокращением приблизительно до исходной длины после снятия растяжения.
Использование литиевых инициаторов для полимеризации сопряженных диенов, триенов и моновинилалифатических и ароматических мономеров является хорошо известным фактом. Данные процессы полимеризации протекают по механизмам анионной полимеризации, где при взаимодействии мономеров, под действием нуклеофильного инициирования, происходит рост полимерной структуры. В процессе протекания полимеризации полимерная структура является ионной или «живущей». Таким образом, полимерная структура имеет, по меньшей мере, один реакционноспособный или «живущий» конец. Это представляет собой понятие термина «живущий», как использовано в настоящем документе, для описания представленных эластомерных полимеров.
Таким образом, как рассмотрено выше, термин «живущий анионный эластомерный полимер», как использовано в настоящем документе, относится к полимеру, содержащему полимерные цепи, в которых каждая цепь содержит реакционноспособную анионную концевую группу, расположенную, «по меньшей мере, на одном конце» полимерной цепи. Этот термин известен в данной области.
Как рассмотрено в настоящем документе, термины «модифицированный по концу цепи эластомерный полимер», «модифицированный по концу цепи полимер», «модифицированный по концу цепи эластомер» и аналогичные термины, используемые в данном описании, относятся к продукту взаимодействия «живущего анионного эластомерного полимера» с силансульфидным модификатором, как показано ниже формулой 1 или формулой 2. Одна, или более чем одна, полимерная цепь может взаимодействовать с одним силансульфидным модификатором (см. также формулу 5).
В одном варианте осуществления изобретения живущий анионный эластомерный полимер выбран из группы, состоящей из гомополимеров изопрена, гомополимеров бутадиена, сополимеров бутадиена со стиролом, сополимеров изопрена со стиролом, терполимеров бутадиена со стиролом и изопреном и их комбинаций. В другом варианте осуществления изобретения живущий анионный эластомерный полимер выбран из группы, состоящей из гомополимеров бутадиена и сополимеров бутадиена со стиролом.
Мономеры, используемые при получении представленных эластомерных полимеров, включают сопряженные олефины и олефины, выбранные из группы, включающей α-олефины, внутренние олефины, циклические олефины, полярные олефины и несопряженные диолефины. Подходящими сопряженными ненасыщенными мономерами являются предпочтительно сопряженные диены, такие как 1,3-бутадиен, 2-алкил-1,3-бутадиен, предпочтительно изопрен (2-метил-1,3-бутадиен), 2,3-диметил-1,3-бутадиен, 1,3-пентадиен, 2,4-гексадиен, 1,3-гексадиен, 1,3-гептадиен, 1,3-октадиен, 2-метил-2,4-пентадиен, циклопентадиен, 2,4-гексадиен, 1,3-циклооктадиен. Предпочтительными олефинами являются С2-20 α-олефины, включающие, без ограничения, длинноцепочечные макромолекулярные α-олефины, особенно ароматические винильные соединения. Предпочтительными ароматическими винильными соединениями являются стирол, включая С1-4алкилзамещенный стирол, такой как 2-метилстирол, 3-метилстирол, 4-метилстирол, 2,4-диметилстирол, 2,4,6-триметилстирол, α-метилстирол и стильбен, 2,4-диизопропилстирол, 4-трет-бутилстирол, винилбензилдиметиламин, (4-винилбензил)диметиламиноэтиловый простой эфир, N,N-диметиламиноэтилстирол, трет-бутоксистирол, винилпиридин и их смеси. Подходящие полярные олефины включают акринитрил, метакрилаты, метилметакрилат. Подходящие несопряженные олефины включают С4-20диолефины, особенно норборнадиен, этилиденнорборнен, 1,4-гексадиен, 1,5-гексадиен, 1,7-октадиен, 4-винилциклогексен, дивинилбензол, включая 1,2-дивинилбензол, 1,3-дивинилбензол и 1,4-дивинилбензол и их смеси. Предпочтительные сопряженные диены включают бутадиен, изопрен и циклопентадиен, и предпочтительные ароматические α-олефины включают стирол и 4-метилстирол.
Примеры применимых эластомерных полимеров включают гомополимеры сопряженных диенов, особенно бутадиена или изопрена, и статистические или блок-со- и терполимеры, по меньшей мере, одного сопряженного диена, особенно бутадиена или изопрена, по меньшей мере, с одним ароматическим α-олефином, особенно стиролом и 4-метилстиролом, ароматическим диолефином, особенно дивинилбензолом. Особенно предпочтительной является статистическая сополимеризация, необязательно терполимеризация, по меньшей мере, одного сопряженного диена, по меньшей мере, с одним ароматическим α-олефином и, необязательно, по меньшей мере, одного ароматического диолефина или алифатического α-олефина, особенно бутадиена или изопрена со стиролом, 4-метилстиролом и/или дивинилбензолом.
Предпочтительные модифицированные эластомерные полимеры (или модифицированные полимеры) включают модифицированный полибутадиен, модифицированный полиизопрен, модифицированный сополимер стирол-бутадиен, модифицированный сополимер стирол-изопрен, модифицированный сополимер бутадиен-изопрен и модифицированный сополимер изопрен-стирол. Более предпочтительные эластомеры (или полимеры) включают модифицированный полибутадиен и модифицированный сополимер стирол-бутадиен. Термины «модифицированные эластомерные полимеры» и «модифицированные полимеры» относятся к «модифицированным по концу цепи полимерам», как рассмотрено выше.
В одном варианте осуществления изобретения модифицированный эластомерный полимер выбран из группы, состоящей из модифицированных гомополимеров изопрена, модифицированных гомополимеров бутадиена, модифицированных сополимеров бутадиена со стиролом, модифицированных сополимеров изопрена со стиролом, модифицированных терполимеров бутадиена с изопреном и стиролом и их комбинаций. В другом варианте осуществления изобретения модифицированный эластомерный полимер выбран из группы, состоящей из модифицированных гомополимеров бутадиена и модифицированных сополимеров бутадиена со стиролом.
Как правило, полимеризация диенового мономера(ов) или сополимеризация диенового мономера(ов) с α-олефиновым мономером(ами) может быть осуществлена в условиях, хорошо известных в данной области для анионных реакций полимеризации живущего типа, таких как температуры от -50 до 250°C, предпочтительно от 0 до 120°C. Температура реакции может быть такой же, как и температура инициирования полимеризации. Полимеризация может быть эффективной при атмосферном давлении, при пониженном давлении или при повышенном давлении вплоть до, и даже выше, чем 500 МПа, непрерывной или периодической. Предпочтительно полимеризацию проводят при давлениях от 0,01 до 500 МПа, наиболее предпочтительно от 0,01 до 10 МПа и, в частности, от 0,1 до 2 МПа. Могут быть применены более высокие давления. В таком процессе высокого давления также может быть успешно использован инициатор согласно настоящему изобретению. Процессы полимеризации в растворе обычно протекают при более низких давлениях, предпочтительно ниже 10 МПа. Полимеризацию можно осуществлять в газовой фазе, а также в жидкой реакционной среде. Полимеризацию обычно проводят в условиях периодической, непрерывной или полунепрерывной полимеризации. Процесс полимеризации может быть проведен методом газофазной полимеризации (например, в псевдоожиженном слое или в реакторе с перемешиваемым слоем), методом полимеризации в растворе, где образованный полимер является по существу растворимым в реакционной смеси, методом суспензионной/дисперсионной полимеризации, где образованный полимер является по существу нерастворимым в реакционной смеси, или методом так называемого процесса полимеризации в массе, в котором избыток подлежащего полимеризации мономера используется в качестве реакционной среды.
Полимеризацию названных выше мономеров обычно инициируют анионным инициатором, таким как, без ограничений, металлорганическое соединение, содержащее, по меньшей мере, один атом лития, натрия, калия или магния, металлоорганические соединения, содержащие от 1 до примерно 20 атомов углерода. Предпочтительно металлорганическое соединение содержит, по меньшей мере, один атом лития, например, этиллитий, пропиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий, фениллитий, гексиллитий, 1,4-дилитио-н-бутан, 1,3-ди(2-литио-2-гексил)бензол, предпочтительно н-бутиллитий и втор-бутиллитий. Данные литийорганические инициаторы могут быть использованы отдельно или в комбинации в виде смеси двух или более различных типов. Количество используемого литийорганического инициатора меняется в зависимости от подлежащих полимеризации мономеров и от заданной молекулярной массы получаемого полимера; однако это количество обычно составляет от 0,1 до 5 ммоль, предпочтительно от 0,3 до 3 ммоль на 100 граммов мономера (всего полимеризуемого мономера).
Полярные координационные соединения необязательно могут быть добавлены к полимеризационной смеси для регулирования микроструктуры (содержания винильной связи) участка сопряженного диолефина «гомо-, сополимера или терполимера диолефинового типа» или для регулирования распределения по составу ароматического винильного соединения в «сополимере или терполимере на основе сопряженного диенового мономера» и, таким образом, например, может служить рандомизирующим компонентом. Полярными координационными соединениями являются, например, без ограничения, простые эфирные соединения, такие как простой диэтиловый эфир, ди-н-бутиловый простой эфир, простой диэтиловый эфир этиленгликоля, простой дибутиловый эфир этиленгликоля, простой диметиловый эфир диэтиленголиколя, простой диметиловый эфир пропиленгликоля, простой диэтиловый эфир пропиленгликоля, простой дибутиловый эфир пропиленгликоля, алкилтетрагидрофуриловые простые эфиры, такие как метилтетрагидрофуриловый простой эфир, этилтетрагидрофуриловый простой эфир, пропилтетрагидрофуриловый простой эфир, бутилтетрагидрофуриловый простой эфир, гексилтетрагидрофуриловый простой эфир, октилтетрагидрофуриловый простой эфир, тетрагидрофуран, 2,2-(бистетрагидрофурфурил)пропан, бистетрагидрофурфурилформаль, простой метиловый эфир тетрагидрофурфурилового спирта, простой этиловый эфир тетрагидрофурфурилового спирта, простой бутиловый эфир тетрагидрофурфурилового спирта, α-метокситетрагидрофуран, диметоксибензол и диметоксиэтан, и/или соединения третичного амина, такие как простой бутиловый эфир триэтиламина, пиридин, N,N,N',N'-тетраметилэтилендиамин, дипиперидиноэтан, простой метиловый эфир N,N-диэтилэтаноламина, простой этиловый эфир N,N-диэтилэтаноламина и N,N-диэтилэтаноламин. Полярное координационное соединение обычно будут добавлять в молярном отношении полярного координационного соединения к литиевому инициатору в интервале от примерно 0,012:1 до примерно 5:1, но обычно от примерно 0,1:1 до примерно 4:1, предпочтительно от 0,25:1 до примерно 3:1 и более предпочтительно от 0,5:1 до примерно 3:2.
Полимеризацию можно необязательно проводить с использованием олигомерного оксоланилалкана в качестве полярного координационного соединения. Примеры таких соединений представлены в патентах США №№6790921 и 6664328, каждый из которых включен в данное описание путем ссылки.
Полимеризация может необязательно включать активаторы для повышения реакционной способности инициатора, для статистического распределения ароматических винильных соединений, вводимых в полимер, или для обеспечения одноцепочечных ароматических винильных соединений, влияя, таким образом, на распределение по составу ароматических винильных соединений в «модифицированном сополимере или терполимере, содержащем сопряженный диен» по настоящему изобретению. Примеры подходящих активаторов включают алкоксиды натрия и калия или феноксиды калия, такие как изопропоксид калия, трет-бутоксид калия, трет-амилоксид калия, н-гептаоксид калия, бензилоксид калия, феноксид калия; калиевые соли карбоновых кислот, таких как изовалериановая кислота, каприловая кислота, лауриловая кислота, пальмитиновая кислота, стеариновая кислота, олеиновая кислота, линоленовая кислота, бензойная кислота, фталевая кислота или 2-этилгексановая кислота; калиевые соли органических сульфоновых кислот, таких как додецилбензолсульфоновая кислота, тетрадецилбензолсульфоновая кислота, гексадецилбензолсульфоновая кислота или октадецилбензолсульфоновая кислота; и калиевые соли органических фосфорных кислот, такие как диэтилфосфит, диизопропилфосфит, дифенилфосфит, дибутилфосфит и дилаурилфосфит. Данные соединения калия могут быть добавлены в количестве 0,005-0,5 моль на 1,0 грамм-атомный эквивалент литиевого инициатора. Если добавить менее 0,005 моль, то обычно не удается достичь достаточного эффекта. С другой стороны, если количество соединения калия составляет более чем примерно 0,5 моль, то существенно снижается производительность и эффективность реакции модификации конца цепи.
Вместе с инициатором полимеризации также может быть добавлен алкоксид щелочного металла для увеличения реакционной способности реакции полимеризации. Алкоксид щелочного металла может быть получен путем взаимодействия спирта и органического соединения щелочного металла. Данную реакцию можно провести в среде углеводородного растворителя в присутствии мономеров, предпочтительно сопряженных диолефиновых мономеров и ароматических винильных мономеров, перед сополимеризацией данных мономеров. Пояснительными примерами алкоксидов щелочных металлов являются алкоксиды металлов тетрагидрофурфурилового спирта, N,N-диметилэтаноламин, N,N-диэтилэтаноламин, 1-пиперазинэтаноламин или тому подобное. Органическим соединением щелочного металла предпочтительно может быть литийорганическое соединение, и оно может быть использовано в качестве реагента для соединения со спиртом с получением алкоксида щелочного металла. Например, можно использовать этиллитий, пропиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий и гексиллитий и их смеси. Из них предпочтительными являются н-бутиллитий и втор-бутиллитий. Молярное отношение спиртового соединения и литийорганического соединения должно составлять от 1:0,7 до 1:5,0, предпочтительно от 1:0,8 до 1:2,0 и более предпочтительно от 1:0,9 до 1:1,2. Если молярное отношение литийорганического соединения к спиртовому соединению составляет более чем 5,0, то эффект улучшения предела прочности при разрыве, износостойкости и гистерезиса является сомнительным. С другой стороны, молярное отношение литийорганического соединения менее чем 0,8 приводит к замедлению скорости полимеризации и существенно снижает производительность за счет низкой эффективности реакции модификации конца цепи.
Для дополнительного регулирования молекулярной массы полимера и свойств полимера может быть использован связывающий агент или сшивающий агент. Например, галогенид олова, галогенид кремния, алкоксид олова, алкоксид кремния или смесь вышеназванных соединений можно непрерывно вводить в реакционную смесь в процессе полимеризации в тех случаях, когда желательно асимметричное связывание. Данное непрерывное введение реагента обычно осуществляют в реакционной зоне, отдельной от зоны, где протекает полимеризация в массе. Сшивающий агент может быть добавлен в виде раствора в углеводороде, например, циклогексане, в полимеризационную смесь при соответствующем перемешивании для распределения и протекания реакции. Сшивающий агент обычно добавляют только после того, как достигнут высокой степени конверсии. Например, сшивающий агент обычно добавляют только после того, как конверсия мономера составит более чем примерно 85 процентов. Обычно предпочтительно, когда конверсия мономера достигает, по меньшей мере, примерно 90 процентов перед добавлением сшивающего агента. Обычные сшивающие агенты на основе галогенида включают тетрахлорид олова, тетрабромид олова, тетрафторид олова, тетраиодид олова, тетрахлорид кремния, тетрабромид кремния, тетрафторид кремния, тетраиодид кремния, также могут быть использованы тригалогениды олова и кремния или дигалогениды олова и кремния. Полимеры, сшитые тетрагалогенидами олова или кремния, имеют максимум четыре ответвления (или четыре сшитые полимерные цепи), тригалогенидами олова и кремния имеют максимум три ответвления и дигалогенидами олова и кремния имеют максимум два ответвления. В качестве сшивающих агентов также могут быть использованы гексагалогендисиланы или гексагалогендисилоксаны, приводящие к получению полимеров максимум с шестью ответвлениями. Пригодные для использования сшивающие агенты на основе галогенидов олова и кремния включают SnCl4, (R1)3SnCl, (R1)2SnCl2, R1SnCl3, SiCl4, (R1)3SiCl, (R1)2SiCl2, R1SiCl3, Cl3Si-SiCl3, Cl3Si-O-SiCl3, Cl3Sn-SnCl3, Cl3Sn-O-SnCl3. Примеры сшивающих агентов на основе алкоксидов олова и кремния включают Sn(OMe)4, Si(OMe)4, Sn(OEt)4 или Si(OEt)4. Наиболее предпочтительными сшивающими агентами являются SnCl4, SiCl4, Sn(OMe)4 и Si(OMe)4.
В одном варианте осуществления изобретения модифицированный по концу цепи эластомерный полимер дополнительно содержит, по меньшей мере, один сшивающий агент, выбранный из группы, включающей галогенид олова, алкоксид олова, галогенид кремния и алкоксид кремния.
Комбинация соединений олова и кремния, как описано выше, необязательно может быть использована для сшивания полимера. При использовании такой комбинации сшивающих агентов на основе соединений олова и кремния можно достичь улучшенных свойств шинных резин, таких как более низкий гистерезис. Особенно желательно использовать комбинацию сшивающих агентов на основе соединений олова и кремния в шинных протекторных смесях, которые содержат как диоксид кремния, так и технический углерод. В таких случаях молярное отношение соединения олова к соединению кремния, используемых для сшивания эластомерного полимера, обычно будет находиться в интервале от 20:80 до 95:5, более типично от 40:60 до 90:10 и предпочтительно от 60:40 до 85:15. Наиболее типично используется интервал от примерно 0,01 до 4,5 миллиэквивалентов сшивающего агента (соединения олова и кремния) на 100 граммов эластомерного полимера. Обычно предпочтительно использовать от примерно 0,01 до примерно 1,5 миллиэквивалентов сшивающего агента на 100 граммов полимера для получения желательной вязкости по Муни. При использовании большего количества наблюдается тенденция образования полимеров, содержащих концевые реакционноспособные группы или недостаточно сшитые. Используют от нуля до менее чем одного эквивалента олово- и/или кремнийсодержащей сшивающей группы на эквивалент литиевого инициатора, чтобы обеспечить последующую функционализацию остальной фракции живущего полимера. Например, если в качестве сшивающего агента используют тетрахлорид олова или кремния, или смесь этих соединений, то расходуется от 0 до менее чем 1,0 моль, предпочтительно от 0 до 0,8 моль и более предпочтительно от 0 до 0,6 моль сшивающего агента на каждые 4,0 моля концов цепей живущего литиевого полимера. Сшивающий агент может быть добавлен в раствор углеводорода, например, в циклогексан, в полимеризационную смесь в реакторе при подходящем перемешивании для обеспечения распределения и протекания реакции.
Для процессов полимеризации в растворе полимеризацию проводят в среде подходящего растворителя, в присутствии диспергаторов или разбавителя. Некоординационные инертные жидкости являются предпочтительными, включая, без ограничения, линейные и разветвленные углеводороды, такие как пропан, бутан, изобутан, пентан, гексан, гептан, октан, циклические и алициклические углеводороды, такие как циклогексан, циклогептан, метилциклогексан, метилциклогептан, ароматические и алкилзамещенные ароматические соединения, такие как бензол, толуол и ксилол, и изомеры вышеназванных соединений и их смеси, а также пентаметилгептан или фракции минерального масла, такие как легкий или стандартный бензин, лигроин, керосин или газойль. Также подходящими являются фторированные углеводородные жидкости, такие как перфторированные С4-10алканы. Кроме того, подходящие растворители, включающие жидкие олефины, которые могут действовать как мономеры или сомономеры в процессе полимеризации, включают пропилен, 1-бутен, 1-пентен, циклопентен, 1-гексен, 3-метил-1-пентен, 4-метил-1-пентен, бутадиен, изопрен, 1,4-гексадиен, 1,7-октадиент, 1-октен, 1-децен, стирол, дивинилбензол, этилиденнорборнен, аллилбензол, 2-метилстирол, 3-метилстирол, 4-метилстирол, 4-винилциклогексен и винилциклогексан. Подходящими являются также смеси растворителей. Также могут быть использованы ароматические углеводороды, например, бензол и толуол.
Термин «модификатор конца цепи», «модификатор концевой группы», «модифицирующий агент», «модифицирующее соединение» и просто «модификатор» все предназначены для обозначения силансульфидных соединений настоящего изобретения, описанных в данном документе со ссылкой на формулы 1 и 2, представленные ниже. Термины «модифицированный по концу цепи эластомерный полимер» и «модифицированный эластомерный полимер» оба предназначены для обозначения продукта взаимодействия живущего эластомерного полимера с модификатором конца цепи по настоящему изобретению.
Модификатор по настоящему изобретению включает соединения согласно формуле 1:
GLMSi-A-S-SiTXZ (Формула 1),
(Формула 1),
где:
Si представляет собой кремний; S представляет собой серу; G представляет собой (С1-С16)алкокси, предпочтительно (С1-С10)алкокси, более предпочтительно (С1-С6)алкокси и еще более предпочтительно (С1-С4)алкокси; и
J и M являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода (Н), (С1-С16)алкила, (С1-С16)алкокси, (С7-С16)арила, (С7-С16)алкиларила и -А-S-SiTXZ (где А, Т, Х и Z определены ниже), но предпочтительно независимо выбраны из (С1-С5)алкила и (С1-С5)алкокси.
А представляет собой арил, алкиларил, (С7-С16)алкиларил или (С1-С16)алкил, который может быть линейным или разветвленным, насыщенным или ненасыщенным и может быть замещен (С1-С4)алкилом, (С1-С4)алкокси, (С7-С16)арилом, (С7-С16)аралкилом, нитрилом, амином, NO2, тиоалкилом, -A-S-SiTXZ (где А, Т, Х и Z определены в настоящем описании), но предпочтительно представляет линейный или разветвленный (С1-С5)алкил.
В предпочтительном варианте осуществления изобретения А представляет собой (С1-С16)алкил, предпочтительно (С1-С12)алкил, более предпочтительно (С1-С8)алкил и наиболее предпочтительно (С1-С5)алкил. В другом варианте осуществления изобретения А представляет собой (С7-С16)алкиларил, предпочтительно (С7-С12)алкиларил, наиболее предпочтительно (С7-С10)алкиларил. Обозначения (С1-Cn) или (С7-Cn), где n означает верхний предел числа атомов углерода, как использовано в настоящем документе, относится к общему числу атомов углерода в группе «А».
В другом варианте осуществления изобретения А предпочтительно представляет собой (С1-С16)алкил, который не содержит гетероатома, такого как O, N, P или S, более предпочтительно (С1-С12)алкил, который не содержит гетероатома, такого как O, N, P или S, более предпочтительно (С1-С8)алкил, который не содержит гетероатома, такого как O, N, P или S, и наиболее предпочтительно (С1-С5)алкил, который не содержит гетероатома, такого как O, N, P или S. В другом варианте осуществления изобретения А представляет собой (С7-С16)алкиларил, который не содержит гетероатома, такого как O, N, P или S, более предпочтительно (С7-С12)алкиларил, который не содержит гетероатома, такого как O, N, P или S, и наиболее предпочтительно (С7-С10)алкиларил, который не содержит гетероатома, такого как O, N, P или S. Как рассмотрено выше, обозначение (С1-Cn) или (С7-Cn), где n означает верхний предел числа атомов углерода, как использовано в настоящем документе, относится к общему числу атомов углерода в группе «А».
В предпочтительном варианте осуществления изобретения группа А, когда она представляет собой алкил, содержит от трех до пяти атомов углерода.
Примеры групп А на основе (С7-С8)алкиларилов включают следующие структуры:

Группы Т, Х и Z являются одинаковыми или различными, и каждая независимо выбрана из группы, состоящей из водорода (Н), (С1-С16)алкила, (С1-С16)алкокси, (С7-С16)арила, (С7-С16)аралкила и -S-А-SiMJG (где A, M, J и G определены в данном описании), но предпочтительно T, X и Z независимо выбраны из (С1-С5)алкила и (С1-С5)алкокси, более предпочтительно каждая из групп T, X и Z представляет собой (С1-С5)алкильную группу. В другом варианте осуществления настоящего изобретения каждая из групп T, X и Z независимо представляет собой (С1-С16)алкил, более предпочтительно (С1-С12)алкил, еще более предпочтительно (С1-С8)алкил и наиболее предпочтительно (С1-С5)алкил.
Хотя в формуле 1 не показано, следует понимать, что соединения настоящего изобретения также могут включать их соответствующие аддукты с кислотами Льюиса (например, с молекулами растворителя тетрагидрофурана, простого диэтилового эфира, диметоксиэтана, соединенный координационными связями с атомами кремния).
В предпочтительном варианте осуществления изобретения модификатор, как изображено в формуле 1 (см. выше) и формуле 2 (см. ниже), не содержит галогенсодержащего фрагмента и более предпочтительно не содержит хлорида, который потенциально может образовывать корродирующие побочные продукты.
Термин «алкил», как подразумевают, включает как линейные углеводороды (например, метил (Ме), этил (Et), н-пропил (Pr), н-бутил (Bu), н-пентил, н-гексил и т.д.), так и разветвленные углеводородные группы (например, изопропил, трет-бутил и т.д.), и углеводорода на основе неароматических колец. Данные углеводородные группы могут быть необязательно замещены алкилом, алкокси, гидроксилом или другими гетероатомами, такими как азот, сера и фосфор, но предпочтительно не содержат заместителей, содержащих гетероатомы.
Термин «алкокси», как подразумевают, включает метокси (MeO), этокси (EtO), пропокси (PrO), бутокси (BuO), изопропокси, изобутокси, пентокси и тому подобное.
Термин «арил», как подразумевают, включает фенилы, бифенилы и другие бензоидные соединения, необязательно замещенные алкилом, алкокси, гидроксилом или другими гетероатомами, такими как азот, сера и фосфор. Арильные группы, определенные в формуле 1, предпочтительно не содержат гетероатомных заместителей, еще более предпочтительно содержат только одно ароматическое кольцо и наиболее предпочтительно содержат шесть атомов углерода в ароматическом кольце.
Термин «алкиларил», как подразумевают, включает арильную группу, связанную с алкильной группой. Обозначение (С7-С16) и аналогичные обозначения означают общее число атомов углерода в группе. Алкиларильные группы, определенные в формуле 1, предпочтительно не содержат гетероатомных заместителей, еще более предпочтительно содержат только одно ароматическое кольцо и наиболее предпочтительно содержат шесть атомов углерода в ароматическом кольце.
Более предпочтительно представленный модификатор выбран из класса соединений, определенных формулой 2:
(RO)x(R)ySi-R'-S-SiR3
 (Формула 2)
(Формула 2)
где:
О означает кислород, х представляет собой целое число, выбранное из 1, 2 и 3; y представляет собой целое число, выбранное из 0, 1 и 2; где x+y=3.
R являются одинаковыми или различными и представляют собой (С1-С16)алкил, предпочтительно (С1-С8)алкил и более предпочтительно (С1-С5)алкил, особенно включающий Me, Et, Pr и Bu; и R' представляет собой (С1-С16)алкил, предпочтительно (С1-С5)алкил.
R' представляет собой эквивалент группы «А» и, таким образом, определен согласно тому, как рассмотрено выше.
В предпочтительном варианте осуществления изобретения каждая из групп R является одинаковой или отличной и каждая независимо представляет собой (С1-С5)алкил, и R' представляет собой (С1-С5)алкил.
Хотя в формуле 2 не показано, но следует понимать, что настоящие соединения включают их соответствующие аддукты с кислотами Льюиса (например, с молекулами растворителя, такого как тетрагидрофуран, простой диэтиловый эфир, диметоксиэтан, соединенный координационными связями с атомами кремния). Конкретные предпочтительные соединения настоящего модификатора включают соединения (и их соответствующие аддукты с кислотами Льюиса, которые не показаны), представленные следующими формулами:
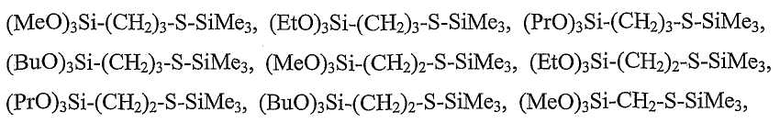







Модификаторы настоящего изобретения могут быть получены взаимодействием серосодержащего соединения согласно формуле 3:
(RO)x(R)ySi-R'-S-H (Формула 3),
(Формула 3),
где символы имеют те же значения, что даны при определении формулы 2, с соединением формулы 4:
QSiR3
 (Формула 4),
(Формула 4),
где Q представляет собой атом фтора, хлора или брома.
Модификатор настоящего изобретения включает сульфанилсилановые соединения, описанные в патенте США №6229036 (который в полной мере разрешен законом и включен в данное описание путем ссылки, включая способы получения сульфанилсилановых соединений). Из предложенных сульфанилсилановых соединений предпочтительными являются соединения без галогенов.
Модификатор можно вводить периодически (или через правильные или неравномерные промежутки времени) или непрерывно в процессе полимеризации, но предпочтительно его вводят со скоростью конверсии при полимеризации более чем 80%, более предпочтительно более чем 90%. Предпочтительно значительное количество концов полимерных цепей являются незакрытыми к началу реакции с модификатором; то есть, существуют концы живущих полимерных цепей, способные взаимодействовать с модификатором по реакции модификации концов полимерных цепей. Реакция модификации может протекать до, после или в процессе добавления сшивающего агента (если таковой используют). Предпочтительно реакция модификации завершается после добавления сшивающего агента (если таковой используют). В некоторых вариантах осуществления изобретения более одной трети концов полимерных цепей взаимодействует со сшивающим агентом(ами) перед добавлением модификатора. В некоторых вариантах осуществления изобретения никакой сшивающий агент не используют, и цепи живущего полимера взаимодействуют с модификатором. В ходе реакции модификации одна или более одной полимерной цепи могут взаимодействовать с модификатором. В результате одна или более одной полимерной цепи соединяются с функциональными группами, образованными модификатором. Модификатор может быть непосредственно введен в раствор полимера без разбавления; однако может выгодно проводить добавление модификатора в растворе, таком как инертный растворитель (например, циклогексан). Количество модификатора, добавляемого в полимеризационную смесь, меняется в зависимости от мономерных соединений, модифицирующих соединений, условий реакции и желательных конечных свойств, но обычно оно составляет от 0,05 до 5 моль-эквивалентов, предпочтительно от 0,1 до 2,0 моль-эквивалентов и наиболее предпочтительно от 0,2 до 1,5 моль-эквивалентов на моль-эквивалент щелочного металла в органическом соединении щелочного металла, требуемом в качестве инициатора полимеризации. Реакцию модификации можно проводить в температурном интервале от 0°C до 150°C, предпочтительно от 15°C до 100°C и даже еще более предпочтительно от 25°C до 80°C. Не существует ограничений на продолжительность реакции функционализации, однако с учетом экономических параметров процесса полимеризации реакция модификации обычно заканчивается примерно через 10-60 минут после добавления модификатора.
Полагают, что реакция модификации конца цепи приводит к получению модифицированного по концу цепи эластомерного полимера, представленного формулой 5:
(D)z(RO)x(R)ySi-R'-S-SiR3
 (формула 5),
(формула 5),
где D представляет собой эластомерный полимер, x представляет собой целое число, выбранное из 0, 1 и 2; y представляет собой целое число, выбранное из 0, 1 и 2; z представляет собой целое число, выбранное из 1, 2 и 3 и x+y+z=3, а все другие символы имеют значения, указанные ранее применительно к формуле 2. Хотя в формуле 5 не показано, но следует понимать, что представленное(ые) соединение(я) включает(ют) их соответствующие аддукты с кислотами Льюиса. В некоторых предпочтительных вариантах осуществления изобретения модифицированный по концу цепи полимер может быть частично сшит в результате реакции с вышеупомянутым(ыми) сшивающим(и) агентом(ами).
Не желая быть связанными теорией, авторы изобретения полагают, что триалкилсилильная группа (-SiR3) формулы 5 действует как защитная группа, которая предотвращает протекание нежелательной последующей реакции. Данная «защитная» триалкилсилильная группа (-SiR3) может быть удалена воздействием соединений, содержащих группы -ОН, таких как вода, спирты, анионные кислоты или органические кислоты (например, соляная кислота, серная кислота или карбоновые кислоты), с образованием, таким образом, «незащищенной» тиольной (-SH) группы. Такие условия обычно имеют место в процессе вулканизации. В зависимости от условия «переработки» полимера может присутствовать как незащищенный, так и/или защищенный модифицированный эластомерный полимер. Например, при удалении летучих соединений обработкой паром из раствора полимера, содержащего модифицированный полимер согласно формуле 5, будет удаляться некоторый процент защитных триалкилсилильных групп, приводя к накоплению незащищенной формы с тиольной (-SH) группой. Альтернативно, обработка методом, не предусматривающим использование воды, может обеспечить получение модифицированных полимеров согласно формуле 5.
Не желая быть связанными теорией, авторы изобретения полагают, что незащищенная тиольная (-SH) группа модифицированного эластомерного полимера является реакционноспособной как относительно ненасыщенных участков основной полимерной цепи, так и содержащихся наполнителей (таких как диоксид кремния и/или технический углерод). Полагают, что данное взаимодействие приводит к образованию связей или, в случае некоторых наполнителей, к электростатическим взаимодействиям, результатом которых является более однородное распределение наполнителя в композициях на основе эластомерного полимера.
Полученный модифицированный эластомерный полимер предпочтительно содержит сульфидные группы (например, тиольные) в количестве от 0,0010 до 0,20 или от 0,0020 до 0,20 ммоль/грамм эластомерного полимера, предпочтительно от 0,0010 до 0,10 ммоль/грамм, более предпочтительно от 0,0025 до 0,1 ммоль/грамм и даже еще более предпочтительно от 0,0025 до 0,05 или от 0,0030 до 0,05 ммоль/грамм полимера. В другом варианте осуществления изобретения сульфидные группы присутствуют в количестве менее чем или равном 0,20 ммоль/грамм эластомерного полимера, предпочтительно менее чем или равном 0,10 ммоль/грамм и более предпочтительно менее чем или равном 0,05 ммоль/грамм. В другом варианте осуществления изобретения сульфидные группы присутствуют в количестве более чем или равном 0,0010 ммоль/грамм эластомерного полимера, предпочтительно более чем или равном 0,0020 ммоль/грамм и более предпочтительно более чем или равном 0,0030 ммоль/грамм.
Для большинства областей применения модифицированный полимер предпочтительно представляет собой гомополимер, образованный из сопряженного диолефина, сополимер, образованный из сопряженного диолефинового мономера и ароматического винильного мономера, и/или терполимер на основе одного или двух типов сопряженных диолефинов и одного или двух типов ароматических винильных соединений. Более предпочтительно модифицированный полимер представляет собой сополимер сопряженного диолефинового мономера с ароматическим винильным мономером, например, сополимер бутадиена со стиролом с сульфидной группой (например, тиольной), связанной, по меньшей мере, с некоторыми концами полимерных цепей.
Предпочтительные модифицированные по концам цепей полимеры (или модифицированные эластомерные полимеры) включают, но не ограничиваются ими, модифицированный по концу цепи полибутадиен, модифицированный по концу цепи полиизопрен, модифицированные по концам цепей сополимеры бутадиен-стирол, модифицированные по концам цепей сополимеры бутадиен-изопрен, модифицированные по концам цепей сополимеры изопрен-стирол и модифицированные по концам цепей терполимеры бутадиен-изопрен-стирол. Из вышеназванных полимеров (или эластомерных полимеров) особенно предпочтительными являются модифицированный по концу цепи полибутадиен и модифицированные по концам цепей сополимеры бутадиен-стирол.
Хотя нет конкретных ограничений, касающихся содержания 1,2-связей и/или 3,4-связей (далее по тексту называемых «винильные связи») в области сопряженного диолефина эластомерного полимера, для большинства областей применения содержание винильных связей предпочтительно составляет от 10 до 90 процентов по массе и особенно предпочтительно от 15 до 80 процентов по массе. Если содержание винильных связей в эластомерном полимере составляет менее чем 10 процентов по массе, то полученный продукт может иметь низкое сопротивление проскальзыванию на мокрой дороге. Если содержание винильных связей в эластомерном полимере превышает 90 процентов по массе, то продукт может иметь ухудшенное сопротивление разрыву и износостойкость и относительно большие гистерезисные потери.
Хотя нет конкретных ограничений, касающихся количества ароматического винильного мономера, используемого в настоящем модифицированном эластомерном полимере, в большинстве областей применения ароматические винильные мономеры составляют от 5 до 60 процентов по массе от общего содержания мономеров и более предпочтительно от 10 до 50 процентов по массе. Значения менее чем 5 процентов по массе могут привести к понижению сопротивления проскальзыванию на мокрой дороге, износостойкости и предела прочности при разрыве, тогда как значения более чем 60 процентов по массе приводят к повышенным гистерезисным потерям. Модифицированным эластомерным полимером может быть блок- или статистический сополимер, но предпочтительно 40 процентов по массе или более звеньев ароматического винильного соединения соединены одинарной связью, а 10 процентов по массе или меньше представляют собой «блоки, в которых последовательно соединены восемь или более ароматических винильных соединений. Длину последовательно соединенных ароматических винильных звеньев можно измерить методом озонолиза-гельпроникающей хроматографии, разработанным Tanaka et al. (Polymer, Vol. 22, Pages 1721-1723 (1981)).
В зависимости от конкретного полимера и желательной конечной области применения, модифицированные полимеры согласно изобретению, как конечный продукт реакции полимера в массе, перед изготовлением резиновых смесей и вулканизацией предпочтительно имеют величину вязкости по Муни (ML 1+4, 100°C, измеренную в соответствии с методом ASTM D 1646 (2004)) в интервале от 20 до 150, предпочтительно от 30 до 100, при использовании вискозиметра Monsanto MV2000. Модифицированные полимеры могут необязательно включать наполнитель и/или масло и/или другие полимеры. Если вязкость по Муни (ML 1+4, 100°C) составляет менее чем 20, то износостойкость и гистерезисные потери ухудшаются. Кроме того, увеличиваются липкость и холодное течение эластомерного полимера, что приводит к трудностям при обращении с продуктом, низкой когезионной прочности и низкой стабильности при хранении. Если вязкость по Муни (ML 1+4, 100°C) полимера превышает 150, ухудшается перерабатываемость (введение наполнителя и выделение тепла в закрытом смесителе, образование шкурки на вальцах, скорость экструзии, стабильность размеров экструдата, гладкость и т.п.) и увеличиваются затраты на переработку.
В одном варианте осуществления изобретения модифицированный полимер, как конечный продукт реакции полимера в массе, перед изготовлением резиновых смесей и вулканизацией содержит масло и имеет вязкость по Муни (ML 1+4, 100°C, измеренную в соответствии с методом ASTM D 1644 (2004), как рассмотрено выше) в интервале от 20 до 150 и предпочтительно от 30 до 100. В другом варианте осуществления изобретения модифицированный полимер, как конечный продукт реакции полимера в массе, перед изготовлением резиновой смеси и вулканизацией не содержит наполнителя или масла и имеет вязкость по Муни (ML 1+4, 1000°C, измеренную в соответствии с методом ASTM D 1645 (2004), как рассмотрено выше) в интервале от 20 до 150 и предпочтительно от 30 до 100.
Предпочтительное молекулярно-массовое распределение модифицированного полимера по изобретению, как конечного продукта реакции полимера в массе, перед процессами изготовления резиновой смеси и вулканизации и перед введением масла, наполнителя или второго исходного эластомерного полимера, представленное отношением средневесовой молекулярной массы к среднечисловой молекулярной массе (Mw/Mn), предпочтительно находится в интервале значений от 1,3 до 3,0. Перерабатываемость полимера ухудшается, если Mw/Mn составляет менее 1,3. Плохая перерабатываемость не только увеличивает стоимость производства, но также ухудшает характеристики смешения компонентов, такие как недостаточное распределение наполнителей и других добавок, что может привести к низким физико-механическим свойствам. Если Mw/Mn составляет более чем 3,0, содержание низкомолекулярных компонентов увеличивается, и увеличиваются гистерезисные потери.
Модифицированные полимеры по изобретению (или модифицированные эластомерные полимеры) могут содержать комбинацию двух или нескольких отличительных признаков или вариантов осуществления, как рассмотрено в настоящем документе. К вышеупомянутому, особенно предпочтительными модифицированными полимерами являются следующие:
1) модифицированный полибутадиен с вязкостью по Муни в интервале от 30 до 80 и содержанием винильных связей в интервале от 5 до 30 процентов по массе, в расчете на долю сопряженного диолефина модифицированного эластомерного полимера, как рассмотрено выше,
2) модифицированный полибутадиен с вязкостью по Муни в интервале от 30 до 80 и содержанием винильных связей в интервале от 45 до 80 процентов по массе, в расчете на долю сопряженного диолефина модифицированного эластомерного полимера, как рассмотрено выше,
3) модифицированный сополимер бутадиен-стирол с вязкостью по Муни в интервале от 45 до 80, содержанием винильных связей в интервале от 50 до 80 процентов по массе, в расчете на долю сопряженного диолефина модифицированного эластомерного полимера, как рассмотрено выше, и содержанием стирола от 15 до 30 процентов по массе (в сополимере), имеющий 50 процентов по массе или больше стирольных звеньев, соединенных одинарной связью, и 10 процентов по массе или меньше, соединенных «в блоки» из восьми или более стирольных звеньев, и
4) модифицированный сополимер бутадиен-стирол с вязкостью по Муни в интервале от 45 до 80, содержанием винильных связей в интервале от 5 до 50 процентов по массе, в расчете на долю сопряженного диолефина модифицированного эластомерного полимера, как рассмотрено выше, и содержанием стирола от 30 до 55 процентов по массе (в сополимере), имеющий 40 процентов по массе или больше стирольных звеньев, соединенных одинарной связью, и 10 процентов по массе или меньше, соединенных «в блоки» из восьми или больше стирольных звеньев.
Вязкость по Муни измеряют так, как рассмотрено выше.
Масло для наполнения может быть использовано в комбинации с представленными эластомерными полимерами для снижения величины вязкости по Муни. Изобретение относится к композициям, содержащим модифицированный по концу цепи эластомерный полимер и масло для наполнения. Пригодные для использования масла для наполнения включают минеральные масла, которые представляют собой смеси масла ароматического типа, масла алициклического типа и масла алифатического типа, и подразделяются на масла для наполнения ароматического типа, масла для наполнения алициклического типа или масла для наполнения алифатического типа. Из них минеральные масла для наполнения ароматического типа, имеющие гравитационно-вязкостную постоянную (величину V.G.C.) 0,900-1,049 (ароматическое масло), и минеральное масло алициклического типа, имеющее величину V.G.C. 0,850-0,899 (нафтеновое масло), являются особенно предпочтительными для обеспечения оптимальных низкотемпературных гистерезисных потерь, обеспечивающих отличное сопротивление проскальзыванию на мокрой дороге. Такое наполнение модифицированного полимера настоящего изобретения маслом для наполнения обеспечивает равномерное распределение в полимере наполнителей, таких как технический углерод и диоксид кремния, и улучшает перерабатываемость и различные свойства вулканизованных изделий. Количество масла для наполнения, используемого в настоящем изобретении, составляет от 0 до 100 частей по массе, предпочтительно от 0 до 80 частей по массе и более предпочтительно от 0 до 70 частей по массе на 100 частей по массе модифицированного эластомерного полимера, как конечного блочного полимерного продукта реакции, перед процессами изготовления резиновой смеси и вулканизации. Если в раствор полимера добавляют масло для наполнения, то должен быть выбран момент добавления после модификации полимера или обрыва реакции полимеризации, например, после добавления модификатора или агента обрыва полимеризации. После добавления масла для наполнения получают маслонаполненный полимер путем отделения полимера от растворителя прямой сушкой или удалением летучих соединений обработкой паром, сушкой каучука с использованием вакуумной сушилки, сушки горячим воздухом или тому подобное. Например, в US 2005/0159513, опубликованной 31 июля 2005, предложена композиция на основе наполненного маслом каучука, включающая эластомерный полимер полимеризации в растворе в сочетании с кремний- или оловосодержащим сшивающим агентом и маслом с низким содержанием полициклических ароматических углеводородов.
В важном варианте осуществления настоящего изобретения модифицированный полимер согласно изобретению объединяют и осуществляют взаимодействие с наполнителем(ями) и вулканизующим агентом и необязательно дополнительными составляющими, включая, но не ограничиваясь ими, ускорители, сшивающие агенты и немодифицированные эластомерные полимеры (например, традиционные эластомерные полимеры, которые не взаимодействовали с модификатором по настоящему изобретению, но которые получены с обрывом реакции, как принято в данной области. Термин «композиция на основе эластомерного полимера» предназначен для описания продукта взаимодействия, полученного из данной комбинации. Полученную композицию на основе эластомерного полимера обычно формуют в желательную конфигурацию и вулканизуют с получением эластомерного изделия, такого как шина.
Модифицированные эластомерные полимеры или полимеры согласно изобретению (включая маслонаполненные варианты осуществления изобретения) предпочтительно составляют, по меньшей мере, 30 процентов по массе от общего количества содержащегося эластомерного полимера, более предпочтительно, по меньшей мере, 50 процентов по массе. Остальную часть эластомерного полимера составляет немодифицированный эластомерный полимер. Предпочтительный немодифицированный эластомерный полимер включает цис-1,4-изопреновый полимер, натуральный каучук, 3,4-изопреновый полимер, полимер на основе сополимера стирол/бутадиен, терполимер стирол/изопрен/бутадиен, цис-1,4-бутадиеновый полимер, транс-1,4-бутадиеновый полимер, бутадиеновые полимеры с содержанием винила от низкого до высокого (имеющие содержание винила 10-90 процентов), сополимеры акрилонитрил/бутадиен и хлоропреновые полимеры. Из них предпочтительными являются сополимер стирол-бутадиен, натуральные каучуки, полиизопрен и полибутадиен. Желательно, чтобы немодифицированные полимеры имели вязкость по Муни (ML 1+4, 100°C) в интервале от 20 до 200, предпочтительно от 25 до 150 (измеренную в соответствии с методом ASTM D 1646 (2004), как рассмотрено выше). Добавление немодифицированных полимеров в вышеуказанном интервале обеспечивает получение эластомерной композиции по настоящему изобретению с низкой себестоимостью без существенного ухудшения ее свойств.
Представленная эластомерная композиция предпочтительно включает наполнители, которые служат усиливающими агентами. Технический углерод, диоксид кремния, двухфазный наполнитель технический углерод-диоксид кремния, каолин, карбонат кальция, карбонат магния и тому подобное являются их примерами. Из них предпочтительным является совместное использование технического углерода и диоксида кремния, использование только двухфазных наполнителей технический углерод-диоксид кремния или совместное использование двухфазного наполнителя технический углерод-диоксид кремния и технического углерода и/или диоксида кремния. Технический углерод получен печным способом и имеет удельную поверхность по адсорбции азота 50-200 м2/г и по поглощению масла DBP 80-200 мл/100 грамм, например, предпочтительным является технический углерод марок FEF, HAF, ISAF или SAF. Особенно предпочтителен технический углерод с высокой агломерацией. Технический углерод обычно добавляют в количестве от 2 до 100 частей по массе, предпочтительно от 5 до 100 частей по массе, более предпочтительно от 10 до 100 частей по массе и даже еще более предпочтительно от 10 до 95 частей по массе на 100 частей по массе всего эластомерного полимера.
Примеры наполнителей на основе диоксида кремния включают диоксид кремния влажного способа получения, диоксид кремния сухого способа получения и диоксид кремния типа синтетического силиката. Диоксид кремния с частицами малого диаметра оказывает заметное усиливающее действие. Диоксид кремния с частицами малого диаметра, высокой агломерацией (т.е. который имеет большую удельную поверхность и высокую абсорбцию масла) отлично распределяется в композиции на основе эластомерного полимера, обеспечивает желательные свойства и превосходную перерабатываемость. Средний диаметр частиц диоксида кремния, в единицах диаметра первичной частицы, предпочтительно составляет от 5 до 60 мкм и более предпочтительно от 10 до 35 мкм. Кроме того, удельная поверхность частиц диоксида кремния (измеренная методом BET) предпочтительно составляет от 45 до 280 м2/г. Диоксид кремния добавляют в количестве от 10 до 100 частей по массе, предпочтительно от 30 до 100 частей по массе и даже еще более предпочтительно от 30 до 95 частей по массе на 100 частей по массе всего эластомерного полимера.
Технический углерод и диоксид кремния могут быть введены вместе; в таком случае общее количество вводимых технического углерода и диоксида кремния составляет от 30 до 100 частей по массе и предпочтительно от 30 до 95 частей по массе на 100 частей по массе всего эластомерного полимера. Поскольку данные наполнители гомогенно распределяются в эластомерной композиции, то повышенные количества (в пределах цитированных выше интервалов) приводят к получению композиций, обладающих отличной способностью к вальцеванию и экструдированию, а вулканизованные продукты обладают благоприятными гистерезисными потерями, сопротивлением качению, улучшенным сопротивлением проскальзыванию на мокрой дороге, износостойкостью и пределом прочности при разрыве.
Двухфазный наполнитель технический углерод-диоксид кремния может быть использован в настоящем изобретении либо независимо, либо в комбинации с техническим углеродом и/или диоксидом кремния. Двухфазный наполнитель технический углерод-диоксид кремния может оказывать такие действия, которые получены совместным использованием технического углерода и диоксида кремния, даже в том случае, когда его вводят отдельно. Двухфазный наполнитель технический углерод-диоксид кремния представляет собой так называемый покрытый диоксидом кремния технический углерод, полученный нанесением покрытия из диоксида кремния на поверхность технического углерода, и коммерчески доступен под торговой маркой CRX2000, CRX2002 или CRX2006 (продукты Cabot Co.). Двухфазный наполнитель технический углерод-диоксид кремния вводят в таких же количествах, которые указаны выше в отношении диоксида кремния. Двухфазный наполнитель технический углерод-диоксид кремния может быть использован в комбинации с другими наполнителями, например, техническим углеродом, диоксидом кремния, каолином, карбонатом кальция и карбонатом магния. Из данных наполнителей предпочтительно использование технического углерода и диоксида кремния, либо по отдельности, либо в комбинации.
Предпочтительно добавлять в полимерную композицию силановый сшивающий агент, если используют диоксид кремния или двухфазный наполнитель технический углерод-диоксид кремния. Обычное количество силанового сшивающего агента составляет от примерно 1 до примерно 20 частей по массе и предпочтительно от 5 до 15 частей по массе на 100 частей по массе общего количества диоксида кремния и/или двухфазного наполнителя технический углерод-диоксид кремния. Силановый сшивающий агент, который содержит в молекуле как функциональную группу, реакционноспособную по отношению к поверхности диоксида кремния, такую как, например, без ограничения, алкоксисилильная группа, так и функциональную группу, реакционноспособную по отношению к двойной связи углерод-углерод полимера, такую как полисульфидная группа, меркаптогруппа или эпоксигруппа, является предпочтительным и включает бис-(3-триэтоксисилилпропил)тетрасульфид, бис-(3-триэтоксисилилпропил)дисульфид, бис-(2-триэтоксисилилэтил)тетрасульфид, бис-(2-триэтоксисилилэтил)дисульфид, 3-меркаптопропилтриметоксисилан, 3-триэтоксисилилпропил-N, N-диметилтиокарбамоилтетрасульфид, 3-триэтоксисилилпропилбензотиазолтетрасульфид, 3-октаноилтио-1-пропилтриэтоксисилан (NXT силан © Crompton Corporation). Дополнительные примеры описаны в патентной заявке США 2006/0041063F, включенной в настоящее описание путем ссылки. Использование такого силанового сшивающего агента повышает эффект усиления, обусловленный совместным использованием технического углерода и/или диоксида кремния или использованием двухфазного наполнителя технический углерод-диоксид кремния.
В одном варианте осуществления изобретения модифицированный полимер (или модифицированный эластомерный полимер) содержит вулканизующий агент и/или ускоритель вулканизации. Серосодержащие соединения и пероксиды являются наиболее широко используемыми вулканизующими агентами. Ускоритель вулканизации типа сульфенамида, типа гуанидина или типа тиурама может быть использован вместе с вулканизующим агентом, в зависимости от необходимости. Необязательно могут быть введены другие добавки, такие как цинковые белила, активаторы вулканизации, противостарители, технологические добавки и тому подобное. Вулканизующий агент обычно вводят в полимерную композицию в количестве от 0,5 до 10 частей по массе, предпочтительно от 1 до 6 частей по массе на 100 частей по массе всего эластомерного полимера. Дополнительную информацию, касающуюся вулканизующих агентов, можно найти в Kirk-Othmer, Encyclopedia of Chemical technology 3rd Ed, Wiley Interscience, N.Y. 1982, volume 29, pp. 365-468, в частности, “Vulcanizing Agents and Auxiliary Materials” pp. 390-402.
В одном варианте осуществления изобретения вулканизованная композиция на основе эластомерного полимера согласно изобретению включает от 10 до 100 частей по массе наполнителя и от 0,5 до 10 частей по массе вулканизующего агента, оба в расчете на 100 частей по массе эластомерного полимера в композиции.
В другом варианте осуществления изобретение относится к протектору шины, включающему или образованному вулканизованной композицией на основе эластомерного полимера согласно изобретению. Еще в одном варианте осуществления изобретение относится к шине, включающей, по меньшей мере, один компонент, образованный вулканизованной композицией на основе эластомерного полимера согласно изобретению.
Композиция на основе эластомерного полимера по настоящему изобретению может быть приготовлена смешением описанных выше модифицированных эластомерных полимеров (включающих маслонаполненные типы), немодифицированных эластомерных полимеров (включающих маслонаполненные типы), наполнителей (технический углерод, диоксид кремния, двухфазный наполнитель технический углерод-диоксид кремния и т.д.), силановых сшивающих агентов и других добавок в резиносмесителе при 140-180°C. После охлаждения вводят вулканизующие агенты, такие как сера, ускорители вулканизации и тому подобное, и полученную смесь смешивают с использованием смесителя Бэнбери или открытых смесительных вальцов, формуют в желательную форму и вулканизуют при температуре 140-180°C с получением вулканизованного эластомерного продукта.
Вследствие того, что вулканизованные композиции на основе эластомерного полимера по настоящему изобретению характеризуются низким сопротивлением качению, низким тепловыделением в динамическом режиме и превосходным сопротивлением проскальзыванию на мокрой дороге, композиции на основе эластомерного полимера по настоящему изобретению хорошо подходят для использования при изготовлении шин, шинных протекторов, боковин шин и каркаса, а также в производстве других промышленных изделий, таких как ремни и ленты, рукава, виброизоляционные резины и обувь.
Настоящее изобретение будет пояснено более подробно с помощью примеров, которые не предназначены ограничивать сущность настоящего изобретения.
Примеры
Следующие примеры представлены для дополнительного пояснения изобретения, и их не следует рассматривать как ограничивающие его сущность. Примеры включают получение модификаторов согласно изобретению, наряду со сравнительными модификаторами, получение и испытания модифицированных эластомерных полимеров и получение и испытания композиций на основе эластомерных полимеров. Если не указано иное, все части и процентное содержание выражены по массе. Термин «в течение ночи» относится к промежутку времени приблизительно 16-18 часов, и «комнатная температура» относится к температуре примерно 20-25°C. Полимеризацию проводили в условиях, исключающих влагу и кислород, в атмосфере азота. Использовали различные методы испытания и измерения примеров. Представлено краткое описание данных методов.
Соотношение между содержанием 1,4-цис-, 1,4-транс- и 1,2-полидиеновых звеньев в бутадиеновом или изопреновом полимерах определяли методами ИК, 1Н-ЯМР- и 13С-ЯМР-спектроскопии (ЯМР (прибор Avance 400 (1H=400 МГц; 13С=100 МГц) Bruker Analytic GmbH). Содержание винильных звеньев в области сопряженного диолефина дополнительно определяли по спектру ИК поглощения (метод Morello, IFS 66 FT-IR спектрометр Bruker Analytic GmbH). Образцы для ИК спектроскопии готовили с использованием CS2 в качестве агента для набухания.
Содержание связанного стирола: калибровочную кривую получали снятием ИК спектра (IR(IFS 66 FT-IR спектрометр Bruker Analytik GmbH). Образцы для снятия ИК спектра готовили с использованием CS2 в качестве агента для набухания). Содержание стирола альтернативно измеряли методом ЯМР (ЯМР (прибор Avance 400 (1Н=400 МГц; 13С=100 МГц) Bruker Analytik GmbH).
Звено одноцепочечного ароматического винильного соединения (звено с ароматическим винильным соединением, соединенное одинарной связью) и звенья длинноцепочечного ароматического винильного соединения (звено, в котором соединены восемь или более ароматических винильных соединений) определяли методом ЯМР (ЯМР (прибор Avance 400 (1H=400 МГц, 13С=100 МГц) Bruker Analytik GmbH). В частности, общее содержание стирольных блоков определяли методом 1Н-ЯМР анализа сигналов в области от 6,2 до 6,9 м.д., отражающих все стирольные блоки, содержащие более трех стирольных звеньев (n≥3). Стирольные микроблоки с n=3-5 соответствовали сигналам в области от 6,7 до 6,9 м.д. и длинные блоки с n>6 и n=6 соответствовали сигналам от 6,2 до 6,7 м.д.
Молекулярно-массовое распределение (Mw/Mn) определяли по отношению к отнесенной к полистиролу средневесовой молекулярной массы (Mw) к среднечисловой молекулярной массе (Mn), которую измеряли гельпроникающей хроматографией (SEC с определением вязкости (универсальное калибрование) в ТГФ при комнатной температуре). Мр1 и Мр2 соответствовали молекулярной массе, измеренной на первом и втором максимальных пиках кривой ГПХ (GPC), соответственно, фракции с несвязанной молекулярной массой.
Температуру стеклования (Tg) определяли методом ДСК (DSC). ДСК (дифференциальная сканирующая калориметрия) измерения проводили с использованием калориметра DSC 2920 от TA Instruments.
Вязкость по Муни измеряли согласно методу ASTM D 1646 (2004) при продолжительности предварительного нагрева 1 минута и продолжительности работы ротора 4 минуты при температуре 100°C [ML1+4(100°C)].
Эффективность модификации сульфанилсиланами определяли по концентрации группы (-SiMe3) и группы (-Si-OMe), измеренной методом ЯМР (ЯМР (прибор Avance 400 (1H=400 МГц; 13С=100 МГц) Bruker Analytic GmbH). Сигнал (-Si-OMe) при 3,3-3,5 м.д. и сигнал (-SiMe3) при 0,1-0,2 м.д. Для определения эффективности модификации содержащим алкоксигруппу сульфанилсилановым соединением в процентах величину делили на среднечисловую молекулярную массу (Mn), измеренную методом ГПХ, так как измеренная величина представляет собой количество Si-C связей на единицу массы.
Эффективность модификации сульфанилсиланами также определяли по содержанию серы в сульфате. Методика предусматривает сжигание образца в автоматической печи (Combustor 02 компании GAMAB, Germany, Bad Dürrenberg) c последующей абсорбцией дымового газа в воде с 0,1% гидроксида гидразиния и последующим определением концентрации сульфата методом ионной хроматографии (Metrohm, колонка Dionex IonPac AS12A).
Предел прочности при разрыве, относительное удлинение при разрыве и условное напряжение при удлинении 300% (Modulus 300) измеряли согласно методу ASTM D 412 на приборе Zwick Z010.
Тепловыделение измеряли согласно методу ASTM D 623, метод A, на Doli 'Goodrich-Flexometer.
Tg δ (60°C) измеряли с использованием динамического спектрометра Eplexor 150N производства Gabo Qualimeter Testanlagen GmbH (Germany), прилагая деформацию в режиме динамического сжатия 0,2% с частотой 2 Гц при 60°C. Чем меньше индекс, тем ниже сопротивление качению (ниже=лучше). Tg δ (0°C) измеряли с использованием того же оборудования и условий нагрузки при 0°C. Чем больше индекс, тем лучше сопротивление проскальзыванию на мокрой дороге (выше=лучше).
Истирание по DIN измеряли согласно методу DIN 53516. Чем больше индекс, тем ниже износостойкость (меньше=лучше).
Измерение реологических свойств невулканизованных резиновых смесей согласно методу ASTM D 5289 c использованием ротационного реометра (MDR 2000 E) использовали для определения времени подвулканизации (TS) и времени вулканизации (TC). Значения «ТС 50» и «ТС 90» представляют собой соответствующие времена, требуемые для достижения 50 процентов и 90 процентов конверсии при реакции вулканизации. «TS 1» и «TS 2» представляют собой соответствующие времена, требуемые для увеличения крутящего момента на 1 дНм и 2 дНм больше соответствующего минимального крутящего момента (ML) в процессе вулканизации.
Как правило, чем выше величина относительного удлинения при разрыве, предела прочности при разрыве, условного напряжения при растяжении на 300% и tg δ при 0°C, тем лучше; тогда как чем ниже tg δ при 60°C, тепловыделение и истирание, тем лучше. Предпочтительно TS 1 составляет >1,5 минут, TS 2 составляет >2,5 минут, ТС 50 составляет от 3 до 8 минут и ТС 90 составляет от 8 до 19 минут.
Получение модификатора: В примерах использовали четыре модификатора. Структурная формула и способ получения (или источник получения) представлены ниже. Модификаторы 1 и 2 являются примерами модификаторов по настоящему изобретению, тогда как модификаторы 3 и 4 представляют собой сравнительные модификаторы.
Модификатор 1 представлен ниже формулой М1 и получен следующим образом.

В 250 мл колбу Шленка загружали 100 г циклогексана, 8,6 г (85 ммоль) триэтиламина и 13,12 г (80 ммоль) гамма-меркаптопропилтриметоксисилана [Silquest A-189] от Cromton GmbH. 17,9 г (165 ммоль) триметилхлорсилана разбавляли 50 г циклогексана и полученный раствор затем по каплям добавляли в колбу Шленка. Сразу же выпадал белый осадок триэтиламмонийхлорида. Суспензию перемешивали примерно в течение 24 часов при комнатной температуре, а затем еще в течение трех часов при 60°C. Белый осадок затем отделяли фильтрованием. Полученный бесцветный раствор перегоняли в вакууме с получением 16 г (67,7 ммоль) модификатора 1.
Модификатор 2 представлен ниже формулой М2 и получен следующим образом.

В 250 мл колбу Шленка загружали 100 г циклогексана, 8,6 г (85 ммоль) триэтиламина и 14,4 г (79,8 ммоль) гамма-меркаптопропил(метил)диметоксисилана от ABCR GmbH. 17,4 г (160 ммоль) триметилхлорсилана разбавляли 50 г циклогексана и полученный раствор затем по каплям добавляли в колбу Шленка. Сразу же выпадал белый осадок триэтиламмонийхлорида. Суспензию перемешивали примерно в течение 24 часов при комнатной температуре, а затем еще в течение трех часов при 60°C. Белый осадок затем отделяли фильтрованием. Полученный бесцветный раствор перегоняли в вакууме с получением 17,2 г (68,1 ммоль) модификатора 2.
Модификатор 3 представлен ниже формулой М3 и получен следующим образом.

В 250 мл колбу Шленка загружали 100 г циклогексана, 8,6 г (85 ммоль) триэтиламина и 8,85 г (80,0 ммоль) гамма-меркаптопропилхлорида от Aldrich GmbH. 17,4 г (160 ммоль) триметилхлорсилана разбавляли 50 г циклогексана и полученный раствор затем по каплям добавляли в колбу Шленка. Сразу же выпадал белый осадок триэтиламмонийхлорида. Суспензию перемешивали примерно в течение 24 часов при комнатной температуре, а затем еще в течение трех часов при 60°C. Белый осадок затем отделяли фильтрованием. Полученный бесцветный раствор перегоняли в вакууме с получением 11,9 г (65,3 ммоль) модификатора 3.
Модификатор 4 представлен ниже формулой М4 и получен следующим образом.

Гамма-меркаптопропилтриметоксисилан 4 [Silquest A-189] от Cromton GmbH.
Модификатор 1, представленный выше формулой М1, альтернативно получали следующим образом.
В 100 мл колбу Шленка загружали 25 мл тетрагидрофурана (ТГФ), 79,5 мг (10 ммоль) гидрида лития, а затем 1,80 г (10 ммоль) гамма-меркаптопропилтриметоксисилана [Silquest A-189] от Cromton GmbH. Реакционную смесь перемешивали в течение 24 часов при комнатной температуре, а затем еще в течение двух часов при 50°C. Затем 1,09 г (10 ммоль) триметилхлорсилана разбавляли 10 г ТГФ и полученный раствор затем по каплям добавляли в колбу Шленка. Выпадал в осадок хлорид лития. Суспензию перемешивали в течение примерно 24 часов при комнатной температуре, а затем в течение еще двух часов при 50°C. Растворитель ТГФ отгоняли в вакууме. Затем добавляли 30 мл циклогексана. Белый осадок затем отделяли фильтрованием. Циклогексановый растворитель удаляли в вакууме (при пониженном давлении). Полученный бесцветный жидкий раствор обладал 99-процентной чистотой по ГХ, и поэтому не требовалось дополнительной очистки.
Получали 2,2 г (9,2 ммоль) модификатора 1.
Модификатор 1, представленный выше формулой М1, в другом случае получали следующим образом.
В 100 мл колбу Шленка загружали 1,80 г (10 ммоль) гамма-меркаптопропилтриметоксисилана [Silquest A-189] от Cromton GmbH, 25 мл тетрагидрофурана (ТГФ), а затем 0,594 г (11 ммоль) метанолята натрия (NaOMe), растворенного в 10 мл ТГФ. Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Затем 1,09 г (10 ммоль) триметилхлорсилана разбавляли 10 г ТГФ и полученный раствор затем по каплям добавляли в колбу Шленка. В осадок выпадал хлорид натрия. Суспензию перемешивали в течение примерно 24 часов при комнатной температуре, а затем в течение еще двух часов при 50°C. Растворитель ТГФ удаляли в вакууме. Затем добавляли 30 мл циклогексана. Белый осадок затем отделяли фильтрованием. Циклогексановый растворитель удаляли в вакууме (при пониженном давлении). Полученный бесцветный жидкий раствор обладал 89% чистотой по ГХ. Дополнительная очистка состояла во фракционной перегонке.
Получали 1,7 г (7,2 ммоль) модификатора 1.
Гомополимеризация 1,3-бутадиена (примеры 1/1а и 2/2а)
Процессы полимеризации для примеров 1/1а и 2/2а проводили в двухлитровом стальном реакторе с двойными стенками, который продували азотом перед загрузкой органического растворителя, мономеров, полярного координационного соединения, инициатора и других компонентов. Полимеризационный реактор нагревали до 50°C, если не указано иное. Затем добавляли следующие компоненты в следующем порядке: циклогексановый растворитель (500 граммов); тетраметилендиамин (TMEDA) (45,0 ммоль) в качестве полярного координационного соединения, бутадиеновый мономер, и смесь перемешивали в течение одного часа. Добавляли N-бутиллитий (50,0 ммоль), чтобы инициировать реакцию полимеризации. Полимеризацию проводили при 50°C приблизительно в течение 2 часов, после чего часть полимерного раствора извлекали из реактора и отдельно перерабатывали, как описано ниже. После этого добавляли модификатор (1 или 2). Для примеров 1а и 2а модификатор не добавляли. Для обрыва реакции полимеризации полимерный раствор через один час помещали в отдельный стальной реактор с двойными стенками, содержащий 50 мл метанола и Irganox 1520 в качестве стабилизатора полимера (1 литр метанола содержал два грамма Irganox). Данную смесь перемешивали в течение 15 минут. Полимеризационный растворитель и другие летучие соединения затем удаляли в вакууме.
Примеры 1 и 1а. Реакцию полимеризации проводили с использованием 54,1 г (1,00 моль) бутадиена. После удаления 66,6% полимерного раствора в полимеризационный реактор добавляли 5,91 граммов (25,0 ммоль) модификатора 1. Такую же методику получения использовали для примера 1а, за исключением того, что не добавляли модификатор.
Примеры 2 и 2а. Реакцию полимеризации проводили с использованием 10,0 г (0,185 моль) бутадиена. После удаления 50% полимерного раствора в полимеризационный реактор добавляли 12,5 ммоль модификатора 2. Такую же методику получения использовали для примера 2а, за исключением того, что не добавляли модификатор.
-ОМе, мол.%
-SiMe3, ммоль/г полибу-тадиена
Исследование методом ГХ-МС примера 2 подтвердило наличие триметилсилильных групп (-SiMe3) (m/e=73) в трех различных полимерных фракциях, при времени удерживания 13,17 минут, 13,25 минут и 22,04. Фрагмент (-SiMe3) обнаружен в большинстве полимерных фракций, указывая на наличие, по меньшей мере, одной группы (-SiMe3) в большинстве полимерных цепей.
Отдельным исследованием было показано эффективное удаление защитной группы (-SiMe3) сначала получением гексадецилтриметилсилилсульфида, затем удалением группы (-SiMe3) с помощью HCl. Более конкретно, 5,1 г (20 ммоль) гексадецилтиола растворяли в 25 мл циклогексана. Затем добавляли триэтиламин, 2,15 г (21,25 ммоль), а затем 4,47 г (41,25 ммоль) хлортриметилсилана в 25 мл циклогексана. Полученную реакционную смесь перемешивали в течение 24 часов, а затем нагревали при 60°C в течение трех часов. Полученный раствор фильтровали, а циклогексановый растворитель удаляли в вакууме. Образовывался гексадецилтриметилсилилсульфид (выход: 6,6 г (20,0 ммоль). Группу (-SiMe3) подтверждали ЯМР спектроскопией (сигнал появлялся в спектре 1Н-ЯМР при 0,23 м.д.). Гексадецилтриметилсилилсульфид, 1 грамм (ммоль) растворяли в 15 мл циклогексана, добавляли 2 грамма соляной кислоты (36%) в 10 мл этанола и перемешивали в течение 15 часов при комнатной температуре. После удаления органического слоя разделением фаз и экстракцией органическую фазу сушили с использованием сульфата магния и фильтровали. Удаление органического растворителя и основного количества образовавшегося гексахлордисилоксанового побочного продукта в вакууме позволяло выделить гексадецилтиол. Как и ожидалось, сигнал от (-SiMe3) в спектре 1Н-ЯМР при 0,23 м.д. исчезал и появлялся новый сигнал от (-SiMe3) очень низкой интенсивности при 0,13 м.д., указывающий на присутствие гексахлордисилоксанового побочного продукта.
Сополимеризация 1,3-бутадиена со стиролом (примеры 3-18)
Сополимеризацию проводили в 20-литровом стальном реакторе с двойными стенками, который сначала продували азотом перед загрузкой органического растворителя, мономеров, полярного координационного соединения, инициатора полимеризации или других компонентов. Полимеризационный реактор нагревали до 40°C, если не указано иное. Затем загружали следующие компоненты в следующем порядке: циклогексановый растворитель (9000 граммов), бутадиеновый мономер, стирольный мономер, тетраметилэтилендиамин (TMEDA) и смесь перемешивали в течение одного часа, после чего титровали н-бутиллитием для удаления следов влаги или других примесей. Добавляли дополнительное количество н-бутиллития, чтобы начать реакцию полимеризации. Полимеризацию проводили в течение 80 минут, не позволяя температуре полимеризации превышать 60°C. После этого добавляли 0,5% от общего количества бутадиенового мономера, а затем добавляли тетрахлорид олова, если не указано иное. Смесь перемешивали в течение 20 минут. Затем добавляли 1,8% от общего количества бутадиенового мономера, с последующим добавлением модификатора (1, 2, 3 или 4), если не указано иное. Для завершения процесса полимеризации полимерный раствор переносили через 45 минут в отдельный стальной реактор с двойными стенками, содержащий 100 мл этанола и 1,4 г концентрированной HCl (c концентрацией 96%) и 5 г Irganox 1520 в качестве стабилизатора полимера. Данную смесь перемешивали в течение 15 минут. Полученный полимерный раствор затем обрабатывали паром в течение одного часа с удалением растворителя и других летучих соединений и сушили в термостате при 70°C в течение 30 минут и следующих одного-трех дней при комнатной температуре.
Состав полученного полимера и некоторые характеристики полимеров суммированы ниже в таблицах 2 и 3. Если не указано иное, количества выражены в ммолях. Примеры, полученные в идентичных условиях полимеризации (в том же полимеризационном реакторе, в тот же день, тем же оператором) обозначены идентичными буквами рядом с номером примера (например, 3А, 4А).
Использование прочерка «-» в представленных ниже таблицах указывает, что данный компонент не вводили. Сокращение «N.M.» означает, что измерений не проводили или что соответствующие результаты не получены.
Состав полимеров согласно примерам
Свойства полимера
Общее содержание стирольных блоков для примеров 12-18 составило ≤1% при общем содержании длинных блоков (более чем или равном 5 повторяющимся звеньям стирола) ≤5%, а остальную часть составляют микроблоки (от 2 до 4 повторяющихся звеньев стирола).
Полимерные композиции готовили объединением и смешиванием компонентов, перечисленных ниже в таблице 4, в смесителе Бэнбери объемом 350 куб.см, и вулканизовали их при 160°C в течение 20 минут. Данные процесса вулканизации и физико-механические свойства для каждого образца вулканизованной композиции представлены ниже в таблицах 5 и 6.
Состав полимерной композиции
Характеристики процесса вулканизации
Свойства полимерных композиций
Готовили дополнительные полимерные композиции объединением и смешиванием компонентов, перечисленных ниже в таблице 7, в резиносмесителе Бэнбери и вулканизовали их при 160°C в течение 20 минут. Условия процесса вулканизации и физические свойства каждого примера эластомерной композиции представлены в таблицах 8 и 9.
Состав полимерной композиции
Характеристики процесса вулканизации
Свойства полимерных композиций
Одной из важных областей применения настоящего изобретения является производство эластомерных полимерных композиций, имеющих более низкие величины tg δ при 60°C без ухудшения остальных физико-механических свойств и перерабатываемости, особенно tg δ при 0°C. Протекторы, изготовленные из эластомерных полимерных композиций, имеющих более низкие значения tg δ при 60°C, соответственно имеют более низкое сопротивление качению, тогда как композиции с более высоким значением tg δ при 0°C имеют соответственно лучшее сопротивление проскальзыванию на мокрой дороге.
В виде средства иллюстрации изобретения в качестве относительно простого модельного полимера использовали живущие низкомолекулярные полибутадиены. Как показано в таблице 1, полибутадиены примеров 1а и 2а имели молекулярные массы (Mw) 2350 и 520 г/моль соответственно. Данные полимеры не содержали модифицированных полимерных цепей, т.е. не содержали ни триметилсилильных групп (-SiMe), ни метоксигрупп (-OMe). Аналогичные полимеры (примеры 1 и 2) получали и модифицировали модификаторами 1 и 2 согласно настоящему изобретению. Данная модификация приводит к удвоению средневесовой молекулярной массы (Mw), подтверждая модификацию полимерных цепей по метоксисилильным группам модификатора. Как и ожидалось, в спектре 1Н-ЯМР обнаружено небольшое число метоксигрупп. Оба исследования примера 2 методом ГХ-МС анализа и исследование примера 1 методом пиролитического МС анализа показали присутствие триметилсилильных (-SiMe3) групп в виде фрагмента масс-спектра при m/e=73,2. Молярная концентрация серы и триметилсилильных групп в каждом из примеров находится в том же интервале, т.е. примерно 26 процентов модификатора 1 присоединено к концам полимерных цепей в примере 1, тогда как примерно 34 процента модификатора 2 присоединено к концам полимерных цепей примера 2.
Чтобы показать возможность эффективного удаления триметилсилильных групп из модифицированного триметилсилилсульфидными группами полимера, в качестве модельного соединения был выбран гексадецилтриметилсилилсульфид. Как показано выше, гексадецилтриметилсилилсульфид количественно превращался в гексадецилтиол после воздействия соляной кислоты при комнатной температуре. Полагают, что существование триметилсилильных групп временно предотвращает (т.е. защищает) инактивацию существенного количества живущих концов полимерных цепей в ходе реакции.
Как указано ранее, одной из существенных областей применения представленных модифицированных эластомерных полимеров является их применение для получения композиций на основе эластомерных полимеров и, в частности, для шинных протекторов, обладающих низким сопротивлением качению, как показано на примере композиций, обладающих относительно низкими величинами tg δ при 60°C, без существенного ухудшения сопротивления проскальзыванию на мокрой дороге, как показано величиной tg δ при 0°C. Как показано в таблице 6, полимерные композиции, полученные из эластомерных полимеров, модифицированных согласно настоящему изобретению (т.е. модификаторами 1 или 2), имели относительно низкое значение tg δ при 60°C и более высокое значение tg δ при 0°C, по сравнению с примерами их прототипов (обозначенных той же буквой, например, 5А и 6А), полученных без такой модификации. Кроме того, предел прочности при разрыве, условное напряжение при растяжении на 300% и относительное удлинение при разрыве модифицированных примеров значительно улучшены или, по меньшей мере, существенно не ухудшились.
Как показано примерами 18F и 19F, важно, что представленные живущие эластомерные полимеры модифицированы представленными модификаторами, а не просто добавлением модификатора в эластомерную композицию на стадии смешения после получения эластомерных полимеров и их модификации по концам цепей. В частности, как показано данными в таблице 9, добавление модификатора 1 в сравнимой концентрации, такой же, что использована для модификации концов цепей, в эластомерную композицию после обрыва полимера (как описано в патенте США 6229036) мало влияет на величины tg δ при 60°C или 0°C.
Как показано в таблице 6, тепловыделение в процессе вулканизации снижено за счет использования представленных модифицированных эластомерных полимеров. Считают, что данное снижение улучшает срок службы полученной композиции и увеличивает общую эластичность. Аналогично, предел прочности при разрыве и условное напряжение при растяжении на 300% улучшаются, что предполагает образование стабильной полимерной сетки с более высоким сопротивлением воздействию механического напряжения. Хотя величины относительного удлинения при разрыве несколько снижаются, но они все же остаются приемлемыми, учитывая улучшенные значения предела прочности при разрыве и величины tg δ.
Таблицы 5 и 8 показывают, что времена подвулканизации (TS) и времена вулканизации (TC) сравнимы с этими величинами для немодифицированных полимеров и обеспечивают хорошие технологические свойства.
Особенно важно, что вышеуказанные преимущества были установлены как для полимерных композиций, содержащих технический углерод, так и для полимерных композиций, содержащих диоксид кремния.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ | 2009 |
|
RU2504555C2 |
| СИЛАНСУЛЬФИДНЫЕ МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ | 2012 |
|
RU2617403C2 |
| ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ, МОДИФИЦИРОВАННЫЕ СУЛЬФИДОМ | 2007 |
|
RU2459844C2 |
| МОДИФИЦИРОВАННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2558597C2 |
| ПОЛИМЕРЫ, МОДИФИЦИРОВАННЫЕ АМИНОСИЛАНОМ | 2012 |
|
RU2609166C2 |
| МОДИФИЦИРОВАННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2012 |
|
RU2599723C2 |
| ВИНИЛСИЛАНЫ ДЛЯ ПРИМЕНЕНИЯ В ФУНКЦИОНАЛИЗИРОВАННЫХ ЭЛАСТОМЕРНЫХ ПОЛИМЕРАХ | 2013 |
|
RU2669799C2 |
| ПРОЦЕСС ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО ИНТЕРПОЛИМЕРА ИЛИ МОДИФИЦИРОВАННОГО ПОЛИМЕРА | 2009 |
|
RU2531821C2 |
| ПРИМЕНЕНИЕ ОПРЕДЕЛЕННЫХ АМИНОСИЛИЛЬНЫХ МОНОМЕРОВ В ПРОИЗВОДСТВЕ КАУЧУКА | 2018 |
|
RU2782265C2 |
| МУЛЬТИБЛОЧНЫЙ СОПОЛИМЕР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2387674C2 |
Изобретение относится к функционализированным эластомерным полимерам, их применению при получении эластомерных композиций и изделиям из них. Модифицированный по концам цепей полимер включает продукт взаимодействия живущего анионного эластомерного полимера и силансульфидного модификатора формулы: (RO)x(R)ySi-R'-S-SiR3. Изобретение также относится к вулканизованной композиции на основе эластомерного полимера и к способу получения такой композиции. Способ включает объединение наполнителя, вулканизующего агента, модифицированного по концам цепей эластомерного полимера и вулканизацию эластомерной полимерной композиции. Композиция применяется для изготовления изделий, таких как пневматические шины, шинные протекторы, ремни и тому подобное. Представленные композиции характеризуются меньшей величиной tg δ при 60°С при сохранении хороших технологических свойств и хорошего баланса физико-механических свойств, включающих износостойкость, предел прочности при разрыве, условное напряжение при растяжении и относительное удлинение при разрыве. 5 н. и 18 з.п. ф-лы, 9 табл.
1. Модифицированный по концам цепей эластомерный полимер, включающий продукт взаимодействия
i) живущего анионного эластомерного полимера и
ii) силансульфидного модификатора, представленного формулой
(RO)x(R)ySi-R'-S-SiR3,
где Si означает кремний; S означает серу; О означает кислород;
х означает целое число, выбранное из 1, 2 и 3;
у означает целое число, выбранное из 0, 1 и 2; при этом х+y=3;
R имеют одинаковые или различные значения и представляют собой (C1-C16) алкил; и R' представляет собой арил, алкиларил или (C1-C16) алкил.
2. Модифицированный по концам цепей эластомерный полимер по п.1, где R' представляет собой (C1-C16) алкил.
3. Модифицированный по концам цепей эластомерный полимер по п.1, где каждая из групп R является одинаковой или отличной, и каждая независимо представляет собой (С1-С5)алкил, и где R' представляет собой (С1-С5)алкил.
4. Модифицированный по концам цепей эластомерный полимер по п.1, где продукт взаимодействия дополнительно включает, по меньшей мере, один сшивающий агент, выбранный из группы, состоящей из галогенида олова, алкоксида олова, галогенида кремния и алкоксида кремния.
5. Модифицированный по концам цепей эластомерный полимер по п.1, где модифицированный эластомерный полимер выбран из группы, состоящей из модифицированных гомополимеров изопрена, модифицированных гомополимеров бутадиена, модифицированных сополимеров бутадиена со стиролом, модифицированных сополимеров изопрена со стиролом, модифицированных терполимеров бутадиена с изопреном и стиролом и их комбинаций.
6. Вулканизованная композиция на основе эластомерного полимера, включающая продукт взаимодействия следующих ингредиентов:
1) наполнителя;
2) вулканизующего агента и
3) модифицированного по концам цепей эластомерного полимера, где модифицированный по концам цепей эластомерный полимер представляет собой продукт реакции
i) живущего анионного эластомерного полимера и
(ii) силансульфидного модификатора, представленного формулой:
(RO)x(R)ySi-R'-S-SiR3,
где Si означает кремний; S означает серу; О означает кислород;
х означает целое число, выбранное из 1, 2 и 3;
y означает целое число, выбранное из 0, 1 и 2; при этом х+y=3;
R имеют одинаковые или различные значения и представляют собой (C1-C16) алкил; и R' представляет собой арил, алкиларил или (С1-С16)алкил.
7. Композиция по п.6, где R' представляет собой (С1-С16)алкил.
8. Композиция по п.6, дополнительно включающая масло.
9. Композиция по п.6, где каждая из групп R является одинаковой или отличной, и каждая представляет собой (С1-С5)алкил, и где R' представляет собой (C1-С5)алкил.
10. Композиция по п.6, где продукт взаимодействия дополнительно включает, по меньшей мере, один сшивающий агент, выбранный из группы, состоящей из галогенида олова, алкоксида олова, галогенида кремния и алкоксида кремния.
11. Композиция по п.6, где наполнитель включает диоксид кремния.
12. Композиция по п.6, где наполнитель включает технический углерод.
13. Композиция по п.6, где модифицированный по концам цепей эластомерный полимер выбран из группы, состоящей из модифицированных гомополимеров изопрена, модифицированных гомополимеров бутадиена, модифицированных сополимеров бутадиена со стиролом, модифицированных сополимеров изопрена со стиролом, модифицированных терполимеров бутадиена, с изопреном и стиролом и их комбинаций.
14. Композиция по п.6, включающая от 10 до 100 частей по массе наполнителя и от 0,5 до 10 частей по массе вулканизующего агента, оба в расчете на 100 частей по массе всего эластомерного полимера.
15. Протектор шины, включающий вулканизованную композицию на основе эластомерного полимера по п.6.
16. Способ получения вулканизованной композиции на основе эластомерного полимера, где указанный способ включает:
a) объединение следующих компонентов с получением эластомерной полимерной композиции:
1) наполнителя,
2) вулканизующего агента и
3) модифицированного по концам цепей эластомерного полимера, который является продуктом взаимодействия
i) живущего анионного эластомерного полимера и
ii) силансульфидного модификатора, представленного формулой
(RO)x(R)ySi-R'-S-SiR3,
где Si означает кремний; S означает серу; О означает кислород;
х означает целое число, выбранное из 1, 2 и 3;
y означает целое число, выбранное из 0, 1 и 2; при этом х+y=3;
R имеют одинаковые или различные значения и представляют собой (C1-С16)алкил; и R' представляет собой арил, алкиларил или (С1-С16)алкил; и
b) вулканизацию эластомерной полимерной композиции с получением вулканизованной эластомерной полимерной композиции.
17. Способ по п.16, где R' представляет собой (С1-С16)алкил.
18. Способ по п.16, где композиция дополнительно включает масло.
19. Композиция для получения резиновой смеси, включающая следующие компоненты: (а) модифицированный по концам цепей эластомерный полимер по п.1 и (b) масло.
20. Модифицированный по концам цепей эластомерный полимер по п.1, где модифицированный эластомерный полимер выбран из группы, состоящей из модифицированных гомополимеров бутадиена и модифицированных сополимеров бутадиена со стиролом.
21. Композиция по п.6, где модифицированный по концам цепей эластомерный полимер выбран из группы, состоящей из модифицированных гомополимеров бутадиена и модифицированных сополимеров бутадиена со стиролом.
22. Модифицированный по концам цепей эластомерный полимер по п.1, где живущий анионный эластомерный полимер выбран из группы, состоящей из гомополимеров изопрена, гомополимеров бутадиена, сополимеров бутадиена со стиролом, сополимеров изопрена со стиролом, терполимеров бутадиена с изопреном и стиролом и их комбинаций.
23. Композиция по п.6, где живущий анионный эластомерный полимер выбран из группы, состоящей из гомополимеров изопрена, гомополимеров бутадиена, сополимеров бутадиена со стиролом, сополимеров изопрена со стиролом, терполимеров бутадиена с изопреном и стиролом и их комбинаций.
| ЕР 0916699 А2, 19.05.1999 | |||
| ЕР 1486513 A1, 15.12.2004 | |||
| US 5151469 A1, 29.09.1992 | |||
| ЭЛАСТОМЕРНАЯ КОМПОЗИЦИЯ ДЛЯ ПРОТЕКТОРОВ ШИН | 1996 |
|
RU2190641C2 |
| US 6579949 B1, 17.06.2003 | |||
| US 6229036 B1, 08.05.2001. | |||

