Настоящее изобретение относится к способу непрерывного получения модифицированных эластомерных полимеров, включающему стадии (i) обеспечения наличия и полимеризации по меньшей мере одного сопряженного диенового мономера и, необязательно, одного или нескольких ароматических виниловых мономеров в присутствии инициатора анионной полимеризации и соединения в соответствии с формулой 1, например, 2,2-ди-(2-оксоланил)пропана или его производного для предоставления живого анионного полимера; и (ii) добавления "сульфанилсиланового" соединения к живому анионному полимеру.
Настоящее изобретение также относится к эластомерным полимерам, которые могут быть получены в соответствии со способом, указанным выше. В еще одном варианте реализации настоящее изобретение относится к композиции, содержащей эластомерный полимер и, необязательно, дополнительные компоненты, такие как масло, наполнитель и/или вулканизирующий агент. Настоящее изобретение дополнительно относится к способу получения сшитого эластомерного полимера, включающему стадию добавления вулканизирующего агента к эластомерному полимеру. Более того, описаны сшитые эластомерные полимеры, получаемые таким образом, также как изделия и композиции, содержащие эластомерный полимер, сшитый эластомерный полимер или и тот, и другой.
Живые анионные полимеры, в том числе живые сопряженные полидиены и сополимеры сопряженных диенов с ароматическими альфа-олефинами, в частности живой полибутадиен, живой полиизопрен или сополимеры бутадиена и/или изопрена с ароматическими альфа-олефинами, в том числе стирол, могут быть модифицированы "сульфанилсилановыми" соединениями для получения модифицированных эластомерных полимеров с измененными свойствами. Такие полимеры также иногда называют "модифицированными полимерами" или "функционализированными полимерами". Как описано в патенте США №2008/0287601 А1, данные модифицированные полимеры могут быть использованы в производстве резиновых смесей. Такие резиновые смеси служат в качестве сырья для различных применений, например, для производства шин. Было обнаружено, что сшитые или отвержденные резиновые смеси обладают сниженными значениями Tan δ (тангенс угла потерь) при 60°C без оказания негативного воздействия на значения Tan δ при 0°C. В частности, было обнаружено, что шины обладают низким сопротивлением качению, если они содержат резиновую смесь со сниженными значениями Tan δ при 60°C. Такие шины демонстрируют хорошее сокращение потребления топлива и, следовательно, удовлетворяют текущие потребности, такие, как снижение выбросов двуокиси углерода. Значения Tan δ при 0°C сшитой резиновой смеси соответствуют противоскользящим свойствам изделий на мокрой поверхности, таких, как шины, содержащие такую смесь. Несмотря на некоторые преимущества, тем не менее, существуют и недостатки, связанные с данными модифицированными полимерами предшествующих уровней техники.
При переработке и компаундировании эластомерных полимеров предшествующих уровней техники, например, с наполнителями, такими как диоксид кремния или технический углерод, было обнаружено, что данные полимеры обладают повышенной вязкостью по Муни (CMU; в дальнейшем также называемой как "вязкость смеси по Муни") конечной композиции, то есть композиции, содержащей модифицированный полимер и дополнительные соединения, такие как, например, диоксид кремния или технический углерод. Повышенные значения CMU, тем не менее, уменьшают скорости пропускания в процессе компаундирования полимеров и повышают потребление энергии при смешивании. В некоторых случаях смесительное оборудование даже не может полностью переработать материалы с такой высокой вязкостью.
Попытки предшествующих уровней техники решить данные проблемы с помощью исходных материалов сосредоточены на модифицированных полимерах со сниженными вязкостями по Муни (MU) до компаундирования. Полагали, что низкие значения MU модифицированных полимеров обеспечат приемлемо низкие значения CMU в конечной композиции. Тем не менее, компаундирование модифицированных полимеров с низкими вязкостями оказалась затруднительным. В частности, переработка таких полимеров на заводах по производству резиновых смесей может привести к образованию липких полимерных крошек, и конечный полимер, скорее всего, будет обладать повышенной хладотекучестью. В то время как липкие полимерные крошки имеют склонность к агломерации и их нельзя эффективно высушить; полимеры, которые обладают повышенной хладотекучестью, нельзя надлежащим образом упаковать и транспортировать. Более того, если полимеры нельзя эффективно высушить, будет трудно рассчитать точное количество полимера, необходимое в соответствии с рецептурами состава полимерных шин.
Таким образом, существует необходимость в предоставлении эластомерных полимеров с улучшенными технологическими характеристиками по сравнению с полимерами, полученными посредством обычных реакций модификации, и позволяющих предоставление сшитых (вулканизированных) полимерных композиций со сниженными значениями Tan δ при 60°C, в то время как значения Tan δ при 0°C все еще соответствуют требованиям. Более того, существует необходимость в полимерах, облегчающих получение изделий, таких как шины с низким сопротивлением качению и хорошими противоскользящими свойствами на мокрой поверхности. Существует дополнительная необходимость в полимерных композициях, содержащих "сульфанилсилановые" модифицированные полимеры, диоксид кремния и/или технический углерод, и обладающих значениями CMU, облегчающими стадии их переработки и компаундирования. В частности, существует необходимость в рецептурах резиновых смесей со сниженными значениями вязкости смеси по Муни (CMU) в их неотвержденном состоянии во время реактивного смешивания в сочетании с низкими потерями на гистерезис, как представлено значениями низкого образования тепла и низкого Tan δ при 60°C, а также хорошим сцеплением с дорогой, как представлено значениями высокого Tan δ при 0°C (сцепление шин с мокрым дорожным покрытием) и высокого Tan δ при -10°C (сцепление шин с обледенелым дорожным покрытием) отвержденных рецептур резиновых смесей. В целом, существует необходимость в предоставлении эффективного способа производства желаемых полимеров.
Таким образом, в первом аспекте настоящее изобретение относится к способу непрерывного получения эластомерного полимера. Таким образом, настоящее изобретение относится к непрерывному способу полимеризации, включающему следующие стадии:
(i) обеспечение наличия и полимеризация по меньшей мере одного сопряженного диенового мономера и, необязательно, одного или нескольких ароматических виниловых мономеров при температуре 110°C или ниже в органическом растворителе в присутствии активного инициатора и в присутствии соединения в соответствии с формулой 1:
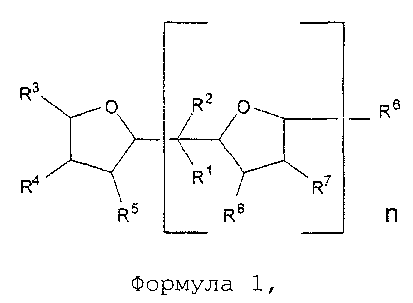
где О в формуле 1 представляет собой кислород, группы R1 и R2 в формуле 1 каждая независимо представляют собой водород или алкильную группу, предпочтительно - водород или С1-С4 алкильную группу; группы R3, R4, R5, R6, R7 и R8, каждую независимо, выбирают из водорода или алкильной группы, предпочтительно - водорода или C1-C6 алкильной группы; n является целым числом, выбранным из 1, 2, 3 или 4, предпочтительно 1;
и где активный инициатор представляет собой инициатор анионной полимеризации, а молярное отношение соединения в соответствии с формулой 1 к активному инициатору составляет от 0,15 до 10;
с получением живого анионного полимера; и
(ii) добавление первого количества соединения в соответствии с формулой 2 к живому анионному полимеру в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 2 к активному инициатору составляло от 0,21 или более:
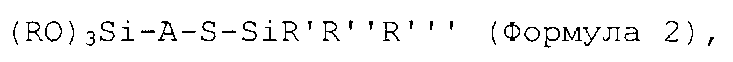
где S в формуле 2 представляет собой атом серы, Si представляет собой кремний, О представляет собой кислород; группы R каждую независимо выбирают из алкила, арила или аралкила, предпочтительно - из C1-C10алкила, C7-С17аралкила или C6-С16арила; группы R′, R″ и R″′ независимо друг от друга выбирают из алкила, аралкила или арила, предпочтительно - из C1-C10алкила, C6-C16арила или С7-С17аралкила; А представляет собой двухвалентную гидрокарбильную группу, предпочтительно - C1-C20алкилен или С7-С25аралкилен.
Термин "непрерывный способ", "непрерывная полимеризация" или аналогичные термины, как применяют в данном документе, относится к способу полимеризации, в котором растворитель, по меньшей мере один сопряженный диеновый мономер и, необязательно, один или более ароматический виниловый мономер, а также все ингредиенты, необходимые для выполнения реакции полимеризации, добавляют дозами в реактор в определенных долях в непрерывном режиме. Как правило, используют два или более последовательно соединенных полимеризатора. В предпочтительном варианте реализации изобретения стадии (i) и (ii), а также дополнительную стадию (iii), которая описана в данном документе ниже, осуществляют в реакционных котлах, соединенных последовательно. Стадия (i) может быть выполнена более чем в одном реакционном котле, но, предпочтительно, стадию (i) выполняют в одном реакционном котле. Предпочтительно поток растворителя, по меньшей мере одного сопряженного диенового мономера и, необязательно, одного или более ароматического винилового мономера, а также всех ингредиентов, необходимых для выполнения реакции полимеризации (в том числе инициатора анионной полимеризации и соединения в соответствии с формулой 1), регулируют таким образом, чтобы среднее время пребывания реакционной смеси в течение каждой из стадий (i), (ii) и, при необходимости, стадии (iii) находилось в диапазоне от 30 до 150 минут. Если отдельные стадии (i), (ii) и, при необходимости, (iii) проводят в отдельных, последовательных реакционных котлах, предпочтительным является, чтобы скорости потока регулировались таким образом, чтобы время пребывания в каждом реакционном котле составляло от 30 до 150 минут. В одном варианте реализации изобретения время пребывания может находиться в диапазоне от 30 до 90 минут. В другом варианте реализации изобретения время пребывания может находиться в диапазоне от 90 до 150 минут.
Мономеры, предоставленные на стадии (i) данного способа в соответствии с настоящим изобретением, включают по меньшей мере один сопряженный диеновый мономер. Репрезентативные сопряженные диеновые мономеры включают, без ограничения ими, 1,3-бутадиен, 2-алкил-1,3-бутадиен, изопрен(2-метил-1,3-бутадиен), 2,3-диметил-1,3-бутадиен, 1,3-пентадиен, 2,4-гексадиен, 1,3-гексадиен, 1,3-гептадиен, 1,3-октадиен, 2-метил-2,4-пентадиен, циклопентадиен, 2,4-гексадиен, 1,3-циклооктадиен, а также их комбинации. Предпочтительные сопряженные диены включают, без ограничения ими, 1,3-бутадиен, изопрен и их комбинации.
В дополнение по меньшей мере к одному сопряженному диеновому мономеру на стадии (i) дополнительно могут быть предоставлены другие способные к полимеризации мономеры. Подходящие примеры дополнительных мономеров включают, без ограничения ими, олефины, выбранные из α-олефинов, олефинов с внутренней двойной связью, циклических олефинов, полярных олефинов или несопряженных диолефинов. Предпочтительными дополнительными мономерами являются C2-20 α-олефины, в том числе, без ограничения ими, длинноцепочечные макромолекулярные α-олефины и ароматические виниловые соединения.
В предпочтительном варианте реализации настоящее изобретение относится к способу, в котором на стадии (i) один или более ароматических виниловых мономеров предлагаются в качестве дополнительного мономера. Репрезентативные примеры ароматических виниловых мономеров включают, без ограничения ими, стирол и его производные, в том числе, без ограничения ими, С1-4 алкилзамещенные стиролы, такие как 2-метилстирол, 3-метилстирол, 4-метилстирол, 2,4-диметилстирол, 2,4,6-триметилстирол, α-метилстирол и стильбен, 2,4-диизопропилстирол, 4-трет-бутилстирол, винилбензилдиметиламин, сложный (4-винилбензил)диметиламиноэтиловый эфир, N,N-диметиламиноэтилстирол, трет-бутоксистирол, винилпиридин и их смеси. Предпочтительные ароматические виниловые мономеры включают стирол, 4-метилстирол и их комбинации.
Подходящие полярные олефины включают акрилонитрил, метакрилаты и метилметакрилат. Подходящие несопряженные олефины включают С4-20диолефины, в особенности норборнадиен, этилиденнорборнен, 1,4-гексадиен, 1,5-гексадиен, 1,7-октадиен, 4-винилциклогексен и дивинилбензол, в том числе 1,2-дивинилбензол, 1,3-дивинилбензол и 1,4-дивинилбензол, а также их смеси.
В одном варианте реализации настоящего изобретения данный способ включает полимеризацию сопряженных диенов, предпочтительно 1,3-бутадиена или изопрена с целью получения гомополимера. В дополнительном варианте реализации изобретения данный способ включает полимеризацию по меньшей мере одного сопряженного диена, предпочтительно 1,3-бутадиена или изопрена по меньшей мере с одним сопряженным диеном; и/или по меньшей мере с одним ароматическим α-олефином, предпочтительно - по меньшей мере с одним ароматическим виниловым мономером, более предпочтительно - со стиролом или 4-метилстиролом; и/или по меньшей мере с одним ароматическим диолефином, предпочтительно - дивинилбензолом, для получения статистических или блочных со- или терполимеров.
Растворитель, используемый в соответствии с настоящим изобретением, представляет собой органический растворитель, подходящий для реакций анионной полимеризации. Реакции анионной полимеризации происходят в соответствии с механизмом анионной полимеризации. На протяжении всей полимеризации концевая группа цепи полимера является ионной или "живой". В одном варианте реализации изобретения полимеризационный растворитель выбирают из неполярных ароматических и неароматических растворителей, в том числе из бутана, пентана, циклогексана, гексана, гептана и октана. В предпочтительном варианте реализации изобретения растворитель выбирают из одного или более бутана, циклогексана, гексана и гептана. Предпочтительно, чтобы Содержание Сухого Вещества Мономеров составляло от 6 до 24 процентов по массе, более предпочтительно - от 10 до 18 процентов по массе и наиболее предпочтительно - от 12 до 15 процентов по массе в расчете на общую массу мономеров и растворителя. Термин "общее содержание сухого вещества мономеров", "содержание сухого вещества мономеров" или аналогичные термины, которые применяют в данном документе, относятся к общей массовой (или весовой) доле мономеров в расчете на общую массу растворителя и мономеров (например, бутадиена и стирола).
Инициатор, используемый для начала реакции полимеризации в соответствии с настоящим изобретением, представляет собой инициатор анионной полимеризации. Подходящие примеры инициаторов анионной полимеризации включают металлорганические соединения по меньшей мере с одним атомом лития, металлорганические соединения, содержащие неполярную C1-C20 углеводородную группу. Предпочтительно металлорганическое соединение представляет собой алкиллитиевое соединение (в дальнейшем также называемое как алкиллитий), такое как этиллитий, пропиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий, фениллитий, гексиллитий и 1,4-дилитио-н-бутан или смесь двух или более из данных соединений. Наиболее предпочтительно н-бутиллитий, втор-бутиллитий или их смесь используют в качестве инициатора анионной полимеризации в соответствии с настоящим изобретением.
Приведенные выше инициаторы могут быть использованы по отдельности или в комбинации в виде смеси из двух или более различных видов. Количество используемого инициатора варьируется в зависимости от полимеризируемых мономеров и также оно зависит от заданной молекулярной массы получаемого полимера; тем не менее, количество, как правило, составляет от 0,0124 моль инициатора на кг мономера (общее количество полимеризуемого мономера) и 0,00143 моль инициатора на кг мономера (общее количество полимеризуемого мономера), если заданная среднечисловая молекулярная масса Mn составляет от 80000 г/моль до 700000 г/моль.
Термин "активный инициатор" (в дальнейшем также называемый как "I*"), как применяют в данном документе, относится к количеству инициатора, которое принимает участие в реакции полимеризации и которое не деактивируется примесями, содержащимися в реакционной среде. Например, при полимеризации бутадиена и стирола в присутствии бутиллития в качестве инициатора анионной полимеризации содержание активного бутиллития (nI*[моль/мин]) рассчитывается в расчете на абсолютную среднечисловую молекулярную массу (Mn) живого анионного полимера, полученного на стадии (i), конверсию и общее количество заряженных мономеров
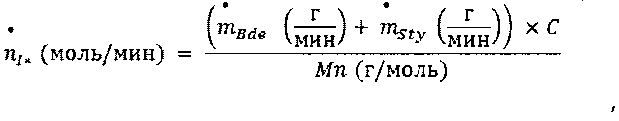
где С = конверсия мономера, и
Mn = среднечисловая молекулярная масса живого анионного полимера, полученного на стадии (i) перед добавлением соединения в соответствии с формулой 2.
Соединение в соответствии с формулой 1, приведенной выше, предпочтительно - 2,2-ди-(2-оксоланил)пропан или его производное, используют в соответствии с настоящим изобретением в качестве полярного соединения для регулирования распределения состава ароматического винилового мономера в качестве необязательного сомономера в полимере и, таким образом, служит в качестве случайного соединения. Соединение в соответствии с формулой 1, приведенной выше, предпочтительно - 2,2-ди-(2-оксоланил)пропан или его производное, также используют в соответствии с настоящим изобретением в качестве полярного соединения для регулирования распределения состава и количества 1,2-полибутадиеновых полимерных звеньев в полимере или необязательном сополимере и, таким образом, служит в качестве случайного соединения. Более того, соединение в соответствии с формулой 1, приведенной выше, предпочтительно - 2,2-ди-(2-оксоланил)пропан или его производное, также используют в соответствии с настоящим изобретением в качестве полярного соединения для увеличения скорости полимеризации. Не желая быть связанными теорией, также ожидается, что соединение в соответствии с формулой 1, приведенной выше, будет взаимодействовать с катионом, в частности, с катионом лития инициатора анионной полимеризации для регулирования добавления 1,4 сопряженного диенового мономера, в частности бутадиена. В предпочтительном варианте реализации изобретения группы R1 и R2 в формуле 1 каждая независимо представляют собой C1-C4 алкильную группу; а группы R3, R4, R5, R6, R7 и R8 в формуле 1 каждую независимо выбирают из водорода или C1-C6 алкильной группы. В некоторых вариантах реализации изобретения группы R3, R4, R5, R6, R7 и R8 каждая представляют собой водород. В предпочтительном варианте реализации изобретения n в формуле 1 равно 1. В альтернативном варианте реализации изобретения n выбирают из 2, 3 или 4. Если n в формуле 1 не равно 1, то, предпочтительно группа R3 представляет ту же группу, что и группа R6, группа R4 представляет ту же группу, что и группа R7, а группа R5 представляет собой такую же группу, что и группа R8.
В дополнительном предпочтительном варианте реализации изобретения соединение в соответствии с формулой 1, приведенной выше, представляет собой 2,2-ди-(2-оксоланил)пропан (в дальнейшем также применяют как аббревиатуру "DTHFP"), в дальнейшем также называемый как ди(тетрагидрофурил)пропан (в дальнейшем также применяют как аббревиатуру "DOP").
Соединение в соответствии с формулой 1 добавляют при молярном отношении соединения в соответствии с формулой 1 к активному инициатору в диапазоне от 0,15 до 10, предпочтительно при отношении - от 0,4 до 5, наиболее предпочтительно - от 0,8 до 3. Соединение в соответствии с формулой 1 может быть непосредственно добавлено к полимерному раствору без разбавления; тем не менее, может быть целесообразным добавление модификатора в раствор с использованием инертного растворителя, например, растворителя, который описан выше.
Было обнаружено, что применение соединения в соответствии с формулой 1 в качестве полярного модификатора в соответствии с настоящим изобретением позволяет случайное включение необязательного ароматического винилового мономера, например, стирола в полимерной цепи. Соответственно, содержание блоков ароматического винилового мономера, например, содержание блоков стирола, может быть низким, например, менее, чем 10 процентов по массе, предпочтительно - менее, чем 5 процентов по массе, более предпочтительно - менее, чем 3 процента по массе в расчете на массу ароматического винилового мономера в полимере, и в зависимости от молярного отношения соединения в соответствии с формулой 1 к активному инициатору. Низкое содержание блоков ароматического винилового мономера, в частности низкое содержание блоков стирола, как правило, требуется при производстве шин. Термин "концентрация блоков стирола", "содержание блоков стирола" или аналогичные термины, как применяют в данном документе, относится к массовой концентрации стирола в расчете на общее количество полимеризованного стирола в полимере, которое включено в качестве последовательностей стирола более, чем из шести звеньев стирола.
Стадию (i) предоставления и полимеризации мономеров, то есть по меньшей мере одного сопряженного диенового мономера и, необязательно, одного или более ароматического винилового мономера предпочтительно выполнять при температуре выше комнатной температуры (25°C). В более предпочтительном варианте реализации изобретения температура на стадии (i) находится в диапазоне от 40 до 110°C, наиболее предпочтительно - в диапазоне от 50 до 95°C. В другом предпочтительном варианте реализации изобретения температура на обеих стадиях (i) и (ii) находится в диапазоне от 55 до 90°C.
На стадии (ii) соединение в соответствии с формулой 2, приведенной выше, добавляют к живому анионному полимеру в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 2 к активному инициатору составляло от 0,21 или более, предпочтительно - от 0,22 или более, более предпочтительно - от 0,25 или более, еще более предпочтительно - от 0,28 или более и наиболее предпочтительно - от 0,3 или более.
В предпочтительном варианте реализации изобретения каждую группу R в формуле 2 независимо выбирают из C1-C10алкила, С7-С17аралкила или C6-C16арила; группы R′, R″ и R″′ в формуле 2 независимо друг от друга выбирают из C1-С10алкила, С6-С16арила или С7-С17аралкила; и А в формуле 2 предпочтительно представляет собой С1-С20алкилен или С7-С25аралкилен. В более предпочтительном варианте реализации изобретения группы R, R′, R″ и R″′ в формуле 2 независимо выбирают из метила, этила, изопропила, н-бутила, трет-бутила и втор-бутила; и двухвалентная группа А представляет собой алкиленовую группу, наиболее предпочтительно - линейную или разветвленную С1-С5 алкиленовую группу. Предпочтительные примеры соединений в соответствии с формулой 2 включают: (Meo)3Si-(CH2)3-S-SiMe3, (EtO)3Si-(CH2)3-S-SiMe3, (PrO)3Si-(CH2)3-S-SiMe3, (BuO)3Si-(CH2)3-S-SiMe3, (MeO)3Si-(CH2)2-S-SiMe3, (EtO)3Si-(CH2)2-S-SiMe3, (PrO)3Si-(CH2)2-S-SiMe3, (BuO)3Si-(CH2)2-S-SiMe3, (MeO)3Si-CH2-S-SiMe3, (EtO)3Si-CH2-S-SiMe3, (PrO)3Si-CH2-S-SiMe3, (BuO)3Si-CH2-S-SiMe3, (MeO)3Si-CH2-CMe2-CH2-S-SiMe3, (EtO)3Si-CH2-CMe2-CH2-S-SiMe3, (PrO)3Si-CH2-CMe2-CH2-S-SiMe3, (BuO)3Si-CH2-CMe2-CH2-S-SiMe3, ((MeO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (EtO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (PrO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (BuO)3Si-CH2-C(H)Me-CH2-S-SiMe3, (MeO)3Si-(CH2)3-S-SiEt3, (EtO)3Si-(CH2)3-S-SiEt3, (PrO)3Si-(CH2)3-S-SiEt3, (BuO)3Si-(CH2)3-S-SiEt3, (MeO)3Si-(CH2)2-S-SiEt3, (EtO)3Si-(CH2)2-S-SiEt3, (PrO)3Si-(CH2)2-S-SiEt3, (BuO)3Si-(CH2)2-S-SiEt3, (MeO)3Si-CH2-S-SiEt3, (EtO)3Si-CH2-S-SiEt3, (PrO)3Si-CH2-S-SiEt3, (BuO)3Si-CH2-S-SiEt3, (MeO)3Si-CH2-CMe2-CH2-S-SiEt3, (EtO)3Si-CH2-CMe2-CH2-S-SiEt3, (PrO)3Si-CH2-CMe2-CH2-S-SiEt3, (BuO)3Si-CH2-CMe2-CH2-S-SiEt3, ((MeO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (EtO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (PrO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (BuO)3Si-CH2-C(H)Me-CH2-S-SiEt3, (MeO)3Si-(CH2)3-S-SiMe2tBu, (EtO)3Si-(CH2)3-S-SiMe2tBu, (PrO)3Si-(CH2)3-S-SiMe2tBu, (BuO)3Si-(CH2)3-S-SiMe2tBu, (MeO)3Si-(CH2)2-S-SiMe2tBu, (EtO)3Si-(CH2)2-S-SiMe2tBu, (PrO)3Si-(CH2)2-S-SiMe2tBu, (BuO)3Si-(CH2)2-S-SiMe2tBu, (MeO)3Si-CH2-S-SiMe2tBu, (EtO)3Si-CH2-S-SiMe2tBu, (PrO)3Si-CH2-S-SiMe2tBu, (BuO)3Si-CH2-S-SiMe2tBu, (MeO)3Si-CH2-CMe2-CH2-S-SiMe2tBu, (EtO)3Si-CH2-CMe2-CH2-S-SiMe2tBu, (PrO)3Si-CH2-CMe2-CH2-S-SiMe2tBu, (BuO)3Si-CH2-CMe2-CH2-S-SiMe2tBu, ((MeO)3Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (EtO)3Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (PrO)3Si-CH2-C(H)Me-CH2-S-SiMe2tBu, (BuO)3Si-CH2-C(H)Me-CH2-S-SiMe2tBu.
Соединение в соответствии с формулой 2 (в дальнейшем также называемое как модификатор 1) предпочтительно добавляют при почти полной или при полной конверсии по меньшей мере одного сопряженного диенового мономера и, если это применимо, одного или более ароматического винилового мономера, предпочтительно - при коэффициенте конверсии полимеризации от 85 до 100% по массе в расчете на количество предусмотренных мономеров. Фраза "количество предусмотренных мономеров", "заложенное количество мономеров" или аналогичные термины, как применяют в данном документе, относится к количеству мономеров, предусмотренному в способе непрерывной полимеризации, например, в конкретном реакторе. В предпочтительном варианте реализации изобретения коэффициент конверсии составляет от 92,0 до 99,9% по массе, предпочтительно - от 94,0 до 99,9% по массе в расчете на количество предусмотренных мономеров. Термин "конверсия мономера", как применяют в данном документе, относится к конверсии мономера (например, к общей конверсии стирола и бутадиена), определяемой, например, на выходе из конкретного полимеризатора. Предпочтительно, чтобы значительное количество концевых групп цепи живого полимера не разрывалось до реакции с модификатором 1, то есть, чтобы концевые группы цепи присутствовали и могли вступать в реакцию с данным модификатором в реакции модификации концевой группы цепи полимера. В ходе реакции модификации одна или более одной полимерных цепей могут вступать в реакцию с модификатором 1, то есть с соединением в соответствии с формулой 2. В результате одна или более одной полимерных цепей связываются с функциональностью, полученной из соединения-модификатора.
Таким образом, применение соединения в соответствии с формулой 2 приводит к образованию "сульфанилсилановых" модифицированных полимеров. В одном варианте реализации изобретения данные полимеры обладают степенью модификации (моль % "сульфанилсилановой" модифицированной концевой группы цепи в расчете на общее количество полученных макромолекул), равной 20% или более, предпочтительно - 50% или более, наиболее предпочтительно - 80% или более.
Соединение в соответствии с формулой 2 может быть непосредственно добавлено к полимерному раствору без разбавления; тем не менее может быть целесообразным добавление модификатора в раствор с использованием инертного растворителя, например, растворителя, который описан выше.
В соответствии с условиями, описанными выше в данном документе, для стадии (i) можно получить высокую степень рандомизации и высокую степень жизненности. Считается, что высокая степень живых полимерных цепей обеспечивает высокую степень полимерных цепей для связи с соединением в соответствии с формулой 2 во время стадии (ii) и, необязательно, с соединением формулы 3 на стадии (iii), описанной ниже в данном документе. Не желая быть связанными теорией, считается, что данные условия (в том числе, например, относительно случайное распределение, например, звеньев стирола и винила, а также высокая степень полимерных цепей, модифицированных соединением формулы 2 и, необязательно, соединением формулы 3) обеспечивают эффективное снижение гистерезисной потери энергии, измеренное в качестве значений Tan δ при 60°C отвержденных полимерных композиций, содержащих "сульфанилсилановые" модифицированные полимеры, а также диоксид кремния и/или технический углерод. Более того, считается, что улучшенные технологические характеристики данных полимерных композиций, в том числе CMU, можно отнести к данным условиям. Дополнительно было обнаружено, что указанные выше условия позволяют относительно узкое распределение по молекулярной массе в конечном полимере. Например, было обнаружено, что конечный полимер может обладать распределением по молекулярной массе (MWD) от 1,7 до 2,5, предпочтительно - от 1,9 до 2,1, что можно отнести к высокой степени жизненности, о которой идет речь выше.
Было установлено, что выполнение способа, как описано выше, позволяет обеспечить модифицированный эластомерный полимер, демонстрирующий улучшенные технологические свойства по сравнению с другими эластомерными "сульфанилсилановыми" модифицированными полимерами. В частности, эластомерные полимеры в соответствии с настоящим изобретением демонстрируют улучшенные технологические характеристики во время реактивного смешивания, то есть в комбинации с наполнителями, такими как диоксид кремния или технический углерод, с получением кремний-наполненных резиновых или саженаполненных резиновых смесей. Данное улучшение может быть измерено как снижение вязкости смеси по Муни (CMU) неотвержденых композиций (в дальнейшем также называемых как "первая композиция"), содержащих эластомерные полимеры в соответствии с настоящим изобретением, а также наполнитель в сравнении с неотвержденными композициями, содержащими полимеры предшествующего уровня техники и наполнитель, при условии, что вязкость по Муни (ненаполненного) эластомерного полимера в соответствии с настоящим изобретением и вязкость по Муни (ненаполненного) эластомерного полимера предшествующего уровня техники является идентичной или по меньшей мере подобной. В альтернативном варианте улучшение может быть измерено как снижение разности вязкости смеси по Муни неотвержденной композиции, содержащей полимер и наполнитель (первая композиция), и вязкости по Муни ненаполненного эластомерного полимера (Улучшение Технологических Характеристик = ΔМуни = CMUпервая композиция - Muэластомерный полимер). Определенные вязкости смеси по Муни данных композиций позволяют повышенное пропускание во время реактивного смешивания. В то же время эластомерные полимеры, получаемые в соответствии со способом настоящего изобретения, оказываются полезными при получении сшитых эластомерных полимеров, как описано ниже.
В предпочтительном варианте реализации изобретения 1,2-бутадиен добавляют на указанной выше стадии (i) в качестве дополнительного компонента. Предпочтительно молярное отношение 1,2-бутадиена к активному инициатору составляет от 0,05 до 0,4, наиболее предпочтительно - от 0,1 до 0,2. Было обнаружено, что использование 1,2-бутадиена в комбинации с соединением в соответствии с формулой 1 предотвращает гелеобразование и засорение реактора и, таким образом, способствует эффективному процессу полимеризации. Засорение полимеризатора приводит к необходимости более частых чисток реактора.
В другом варианте реализации изобретения способ в соответствии с настоящим изобретением дополнительно включает дополнительную стадию (iii):
(iii) последовательного добавления второго количества соединения в соответствии с формулой 2 и/или добавления соединения в соответствии с формулой 3 в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 2 и/или формулой 3, добавляемых на стадии (iii), к активному инициатору составляло от 0,21 или более:

где группы R в формуле 3 независимо выбирают из алкильных, аралкильных или арильных групп, предпочтительно - из C1-C10алкила, С7-С17аралкила или C6-C16арила; группы R′, R″ и R″′ независимо выбирают из алкила, аралкила или арила, предпочтительно - из C1-C10алкила, C6-C16арила или С7-С17аралкила; группу R″″ выбирают из алкила, аралкила или арила, предпочтительно - из C1-С10алкила, C6-C16арила или C7-C17аралкила; А представляет собой двухвалентную гидрокарбильную группу, предпочтительно - C1-C20алкилен или С7-С25аралкилен.
Предпочтительно каждую группу R в формуле 3 независимо выбирают из C1-С10алкила, предпочтительно - из С1-С4алкила; С7-С17аралкила, предпочтительно - из С7-С10аралкила; или C6-С16арила, предпочтительно - из C6-C12арила; группы R′, R″, R″′ и R″″ независимо друг от друга выбирают из C1-С10алкила, предпочтительно - из C1-C6алкила; C6-C16арила, предпочтительно - из C6-C12арила; или С7-С17аралкила, предпочтительно - из С7-С10аралкила; и А представляет собой C1-C20алкилен или С7-С25аралкилен. Наиболее предпочтительно каждую из R, R′, R″, R″′ и R″″ в приведенной выше формуле 3 выбирают из метила, этила, изопропила, н-пропила, н-бутила, изобутила, трет-бутила, фенила и бензила; и А представляет собой -(CH2)m-, где m представляет собой целое число, выбранное из 1, 2, 3, 4, 5 или 6, предпочтительно - из 3, 4, 5 или 6.
Добавление "второго количества" соединения в соответствии с формулой 2 следует понимать в контексте приведенной выше стадии (ii), которая включает обязательное добавление соединения в соответствии с формулой 2 в количестве ("первое количество"), определенном выше. В предпочтительном варианте реализации изобретения стадия (iii) включает добавление соединения в соответствии с формулой 3. Если стадия (iii) не включает добавление второго количества соединения в соответствии с формулой 2, то соединение формулы 3 добавляют в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 3 к активному инициатору составляло от 0,21 или более, предпочтительно - от 0,22 или более, более предпочтительно - от 0,25 или более, еще более предпочтительно - от 0,28 или более и наиболее предпочтительно - от 0,3 или более. Тем не менее, если необязательная стадия (iii) включает добавление второго количества соединения в соответствии с формулой 2 без добавления соединения в соответствии с формулой 3, то это второе количество представляет собой такое количество, чтобы молярное отношение соединения в соответствии с формулой 2 к активному инициатору составляло от 0,21 или более. Тогда в данном последнем случае общее количество соединения в соответствии с формулой 2 является результатом стадий (ii) и (iii) таким образом, чтобы общее молярное отношение соединения в соответствии с формулой 2, как на стадиях (ii) и (iii), к активному инициатору составляло от 0,42 или более. В предпочтительном варианте реализации изобретения стадию (iii) добавления второго количества соединения в соответствии с формулой 2 и/или добавления соединения в соответствии с формулой 3 выполняют посредством добавления избыточного количества одного или обоих из данных соединений по отношению к молярному количеству полимерных цепей, которые все еще живы после стадии (ii).
На выходе из полимеризатора или ряда из двух или более полимеризаторов получают раствор эластомерного полимера, который предпочтительно смешан с антиоксидантом.
Модифицированный полимер, полученный согласно способу в соответствии с настоящим изобретением, может быть обработан, по сути, известным способом. В целом активный или неактивный остаток соединения инициатора, остатка соединения Формулы 2 и, необязательно, Формулы 3 деактивируют в некоторый момент во время переработки полимера, предпочтительно - после завершения стадии (ii) или (iii) способа, по сути, известным образом, например, с помощью воды, органических кислот, анионных кислот или спирта. Более того использование воды, органических кислот, анионных кислот или спирта делает металлические органические остатки, полученные из соединения инициатора полимеризации или металлированные концевые группы цепи полимера менее активными и, следовательно, менее опасными. Удаление остатков, полученных из соединения инициатора и из соединений формул 1, 2 и, необязательно, 3 иногда может быть опущено, в частности, если количество компонентов инициатора и соединений в соответствии с Формулами 1, 2 и, необязательно, 3 очень небольшое. Тем не менее, при желании уровень остатков, полученных из инициатора, из соединения формулы 1, из соединения Формулы 2 и/или из соединения в соответствии с формулой 3 можно уменьшить в полимере известным образом, например, посредством промывки. После стадии деактивации может следовать стадия десорбции (удаление органического растворителя (растворителей) из полимера).
Предпочтительно десорбировать полимерный раствор паром до сушки эластомерного полимера. В то время как десорбция паром может быть выполнена в смесительном баке, содержащем кипящую воду, сушка может быть выполнена, например, посредством сжатия и нагрева, например, в отделе, состоящем из двух экструдеров. В альтернативном варианте растворители могут быть удалены при пониженном давлении.
После сушки эластомерный полимер может быть сжат в любую желаемую форму, предпочтительно - в кипы. В альтернативном варианте или впоследствии эластомерный полимер может быть смешан или скомпаундирован, например, с наполнителями. Смешивание и компаундирование эластомерных полимеров, например, с наполнителями, в качестве дополнительного варианта, может быть выполнено до удаления растворителя.
Таким образом, во втором аспекте настоящее изобретение относится к эластомерному полимеру, получаемому в соответствии с приведенным выше способом.
В одном варианте реализации настоящего изобретения можно использовать пластификатор, такой как масло, в комбинации с эластомерным полимером для уменьшения значений вязкости или вязкости по Муни, или для улучшения технологических характеристик и различных эксплуатационных свойств полимерных композиций после вулканизации.
Масло (масла) можно добавить к эластомерному полимеру до окончания процесса получения полимера или в качестве отдельного компонента после полимеризации, например, после десорбции паром. Предпочтительно масло добавляют в количестве от 10 до 50 частей по массе масла на 100 частей по массе полимера. Репрезентативные примеры и классификации масел указаны ниже.
Несмотря на то, что нет никаких определенных ограничений, касающихся содержания 1,2-связей и/или 3,4-связей (в дальнейшем также называемое как "содержание винила") сопряжения диолефиновой части эластомерного полимера для большинства областей применения, содержание винила эластомерного полимера предпочтительно составляет от 10 до 90 процентов по массе. Термин "содержание винила", как применяют в данном документе, относится к массовой (или весовой) доле, например, бутадиена и изопрена, которая включена в полимерную цепь в 1,2 и в положении 1,2 или 3,4, соответственно, и основана на части, например, бутадиена и/или изопрена (общее количество полимеризованного бутадиена и/или изопрена) в полимере. В более предпочтительном варианте реализации изобретения содержание винила находится в диапазоне от 20 до 80 процентов по массе. Если содержание винильных связей в полимере составляет менее 10 процентов по массе, то полученные в результате продукты могут обладать худшим сопротивлением проскальзыванию на мокрой дороге. Если содержание винила в эластомерном полимере превышает 90 процентов винильных связей по массе, полученные в результате продукты могут обладать сниженной прочностью при растяжении и износоустойчивостью, а также относительно большими потерями на гистерезис.
В одном варианте реализации изобретения способ в соответствии с настоящим изобретением включает гомополимеризацию 1,3-бутадиена или изопрена при отсутствии стирола с получением гомополимера, то есть полибутадиена или полиизопрена. Тем не менее, в более предпочтительном варианте реализации изобретения способ включает сополимеризацию, например, 1,3-бутадиена и стирола с получением стирол-бутадиеновых сополимеров. Массовое отношение стирола к 1,3-бутадиену предпочтительно составляет от 0 до 50, более предпочтительно - от 5 до 45, наиболее предпочтительно - от 10 до 40 процентов по массе. В одном варианте реализации изобретения содержание стирола в сополимере составляет от 5 до 25 процентов по массе. В другом варианте реализации изобретения содержание стирола в сополимере составляет от 25 до 45 процентов по массе. Термин "содержание стирола", как применяют в данном документе, относится к массовой (или весовой) доле стирола в полимере относительно массы полимера.
В еще одном варианте реализации настоящего изобретения эластомерный полимер обладает распределением по молекулярной массе (MWD) от 1,7 до 2,5, предпочтительно - от 1,9 до 2,1. Предпочтительно эластомерный полимер обладает среднечисловой молекулярной массой (Mn) в диапазоне от 80000 до 700000 г/моль, более предпочтительно - от 110000 до 300000 г/моль.
Несмотря на зависимость от определенного полимера и желаемой конечной цели применения, модифицированные эластомерные полимеры по настоящему изобретению предпочтительно обладают вязкостью по Муни (ML 1+4, 100°C, как измерено в соответствии с ASTM D 1646 (2004)) в диапазоне от 0 до 150, предпочтительно - от 15 до 100 и более предпочтительно - в диапазоне от 30 до 90 с применением прибора Monsanto MV2000. Если вязкость по Муни (ML 1+4, 100°C) полимера составляет более 150 MU, это негативно сказывается на технологических характеристиках (включение наполнителя и образование тепла во внутреннем смесителе, полосы на вальцах, скорость экструзии, разбухание экструдируемого потока экструдируемой заготовки, гладкость и т.д.), так как компаундирующее оборудование, используемое при производстве шин, не предназначено для работы с марками резиновых смесей с такой высокой вязкостью по Муни, а также увеличивает стоимость переработки. В некоторых случаях вязкость по Муни (ML 1+4, 100°C) менее, чем 15 или даже иногда менее, чем 30 не может быть предпочтительной в связи с увеличением липкости и хладотекучести несшитого эластомерного полимера, что приводит к сложной транспортировке, плохой когезионной прочности и плохой размерной стабильности во время хранения.
В третьем аспекте настоящее изобретение относится к композиции, содержащей указанный выше эластомерный полимер. В предпочтительном варианте реализации изобретения композиция содержит масло и/или наполнитель. В еще одном варианте реализации изобретения композиция дополнительно содержит необязательные добавки. Другими словами, эластомерный полимер или композиция, содержащая эластомерный полимер, может быть смешана с одним или более наполнителем, при необходимости, с маслом и, при необходимости, с другими добавками.
В предпочтительном варианте реализации изобретения эластомерный полимер объединяют и он вступает в реакцию с наполнителем (наполнителями), выбранным из группы, включающей диоксид кремния, двухфазный углерод-кремнеземный наполнитель, технический углерод, наполнитель из углеродных нанотрубок, лигнин, стеклонаполнитель, слоистые силикаты, такие как, например, магадиит, в некоторых предпочтительных вариантах реализации - включающей диоксид кремния в качестве основного компонента наполнителя и, необязательно, дополнительные ингредиенты, в том числе, без ограничения ими, вспомогательные технологические добавки, масла, вулканизирующие агенты и силановые аппреты с образованием первой полимерной композиции.
В одном варианте реализации изобретения первая композиция дополнительно содержит по меньшей мере один дополнительный, модифицированный или немодифицированный, эластомерный полимер, необязательно, содержащий масло. Примеры предпочтительных немодифицированных эластомерных полимеров приведены в Международной патентной публикации № WO 2009/148932 (номер дела патентного поверенного 66798), а особенно предпочтительными являются стирол-бутадиеновый сополимер, натуральные каучуки, полиизопрен и полибутадиен. Желательно, чтобы немодифицированные полимеры обладали вязкостью по Муни (ML 1+4, 100°C, как измерено в соответствии с ASTM D 1646 (2004), как описано выше) в диапазоне от 20 до 200, предпочтительно - от 25 до 150. Заявленная композиция предпочтительно содержит наполнители, которые служат в качестве армирующих агентов.
Масло (масла) может быть добавлено к полимеру до окончания процесса получения полимера и в качестве отдельного компонента первой полимерной композиции в соответствии с настоящими идеями. Для репрезентативных примеров и классификации масел смотри Международную патентную заявку №PCT/US09/045553 и публикацию патентной заявки США №2005/0159513, каждая из которых включена в данный документ в виде ссылки в полном объеме. Репрезентативные масла включают, без ограничения ими, MES (Мягкий Экстракционный Сольват), TDAE (Обработанный Ароматический Экстракт Дистиллята), RAE (Остаточный Ароматический Экстракт), в том числе, без ограничения ими, T-RAE и S-RAE, DAE, в том числе T-DAE и NAP (легкие и тяжелые нафтеновые масла), в том числе, без ограничения ими, Nytex 4700, Nytex 8450, Nytex 5450, Nytex 832, Tufflo 2000 и Tufflo 1200. Помимо этого натуральные масла, в том числе, без ограничения ими, растительные масла можно использовать в качестве масел для наполнения. Репрезентативные масла также включают функционализированные вариации упомянутых выше масел, в частности эпоксидированных или гидроксилированных масел. Упомянутые выше масла содержат различные концентрации полициклических ароматических соединений, парафинов, нафтенов и ароматических углеводородов, а также обладают различными температурами стеклования. Упомянутые выше типы масел были охарактеризованы (Kautschuk Gummi Kunststoffe, том 52, страницы 799-805). В некоторых вариантах реализации изобретения MES, RAE и TDAE представляют собой масла для наполнения для резиновых смесей.
При необходимости вспомогательные технологические добавки могут быть добавлены к первой полимерной композиции в соответствии с настоящими идеями. Вспомогательные
технологические добавки, как правило, добавляют для уменьшения вязкости первой полимерной композиции. В результате период смешивания уменьшается и/или уменьшается количество смесительных стадий и, следовательно, потребляется меньше энергии, и/или достигается более высокая пропускаемость в ходе процесса экструзии резиновой смеси. Репрезентативные вспомогательные технологические добавки, которые, при необходимости, могут быть использованы в качестве компонента в первых полимерных композициях в соответствии с настоящими идеями, описаны в публикации Rubber Handbook, SGF, The Swedish Institution of Rubber Technology 2000 и у Werner Kleemann, Kurt Weber, Elastverarbeitung-Kennwerte und Berechnungsmethoden, Deutscher Verlag für Grundstoffindustrie (Leipzig, 1990), каждая из которых включена в данный документ в виде ссылки в полном объеме. Примеры репрезентативных вспомогательных технологических добавок, которые, при необходимости, могут быть использованы в качестве компонента в первых полимерных композициях в соответствии с настоящими идеями, могут быть классифицированы следующим образом:
(A) жирные кислоты, в том числе, без ограничения ими, олеиновая кислота, Priolene и стеариновая кислота, Pristerene;
(B) соли жирных кислот, в том числе, без ограничения ими, Aktiplast GT, PP, ST, Т, Т-60, 8, F; Deoflow S; Kettlitz Dispergator FL, FL Plus; Dispergum 18, С, E, K, L, N, T, R; Polyplastol 6, 15, 19, 21, 23; Struktol A50P, A60, EF44, EF66, EM16, EM50, WA48, WB16, WB42, WS180, WS280 и ZEHDL;
(C) диспергаторы и вспомогательные технологические добавки, в том числе, без ограничения ими, Aflux 12, 16, 42, 54, 25; Deoflow A, D; Deogum 80; Deosol Н; Kettlitz Dispergator DS, KB, OX; Kettlitz-Mediaplast 40, 50, Pertac/GR; Kettlitz-Dispergator SI; Struktol FL и WB 212; и
(D) диспергаторы для высокоактивных светлых наполнителей, в том числе, без ограничения ими, Struktol W33 и WB42.
Бифункционализированные силаны и монофункциональные силаны (также называемые в данном документе "силановые аппреты") также иногда называют вспомогательными технологическими добавками, но отдельно описаны ниже.
В некоторых вариантах реализации изобретения силановый аппрет (используемый для улучшения совместимости полимера и указанных наполнителей) добавляют к композиции, содержащей модифицированные полимеры, как описано в данном документе, а также диоксид кремния, слоистый силикат, например, без ограничения ими, магадиит или двухфазный углерод-кремнеземный наполнитель, который используют в качестве компонента наполнителя. Типичное количество добавляемых силановых аппретов составляет от около 1 до около 20 частей по массе, а в некоторых вариантах реализации изобретения - от около 5 до около 15 частей по массе на 100 частей по массе общего количества диоксида кремния и/или двухфазного углерод-кремнеземного наполнителя.
Силановые аппреты могут быть классифицированы в соответствии с Fritz Röthemeyer, Franz Sommer: Kautschuk Technologie, (Carl Hanser Verlag 2006):
(A) бифункционализированные силаны, в том числе, без ограничения ими, Si 230 (EtO)3Si(CH2)3Cl, Si 225 (EtO)3SiCH=CH2, A189 (EtO)3Si(CH2)3SH, Si 69 [(EtO)3Si(CH2)3S2]2, Si 264 (EtO)3Si-(CH2)3SCN и Si 363 (EtO)Si((CH2-CH2-O)5(CH2)12CH3)2(CH2)3SH) (Evonic Industries AG); и
(B) монофункциональные силаны, в том числе, без ограничения ими, Si 203 (EtO)3-Si-C3H7 и Si 208 (EtO)3-Si-C8H17.
Дополнительные примеры силановых аппретов приведены в Международной патентной заявке № PCT/US2009/045553 и включают, без ограничения ими, бис-(3-гидроксидиметилсилилпропил)тетрасульфид, бис-(3-гидроксидиметилсилилпропил)дисульфид, бис-(2-гидроксидиметилсилилэтил)тетрасульфид, бис-(2-гидроксидиметилсилилэтил)дисульфид, 3-гидроксидиметилсилилпропил-N,N-диметилтиокарбамоилтетрасульфид и 3-гидроксидиметилсилилпропилбензотиазола тетрасульфид.
Эластомерный полимер в соответствии с настоящими идеями может быть смешан с одним или более наполнителями с получением первой композиции. Наполнители служат в качестве армирующих агентов. Технический углерод, диоксид кремния, двухфазный углерод-кремнеземный наполнитель, глина (слоистые силикаты), карбонат кальция, карбонат магния, лигнин, углеродные нанотрубки, аморфные наполнители, такие как наполнители на основе частиц стекла, наполнители на основе крахмала и тому подобное, а также их комбинации являются примерами подходящих наполнителей. Примеры наполнителей описаны в Международной патентной заявке № PCT/US2009/045553, которая в полном объеме включена в настоящий документ в виде ссылки. В некоторых вариантах реализации изобретения применяют комбинированное использование технического углерода и диоксида кремния, использование только двухфазных углерод-кремнеземных наполнителей или комбинированное использование двухфазного углерод-кремнеземного наполнителя и технического углерода и/или диоксида кремния. Технический углерод получают печным способом, а в некоторых вариантах реализации изобретения используют удельную площадь поверхности адсорбции азота, равную 50-200 м2/г и адсорбцию DBP (дибутилфталат) масла, равную 80-200 мл/100 грамм; например, технический углерод класса FEF, HAF, ISAF или SAF. В некоторых вариантах реализации изобретения используют тип технического углерода с высокой агрегацией. Технический углерод, как правило, добавляют в количестве от 2 до 100 частей по массе, в некоторых вариантах реализации изобретения - от 5 до 100 частей по массе, в некоторых вариантах реализации изобретения - от 10 до 100 частей по массе, а в некоторых вариантах реализации изобретения - от 10 до 95 частей по массе на 100 частей по массе общего эластомерного полимера.
Примеры кремнекислотных наполнителей включают, без ограничения ими, диоксид кремния, полученный мокрым способом, диоксид кремния, полученный сухим способом, синтетический диоксид кремния силикатного типа и их комбинации. Диоксид кремния с малым диаметром частиц и высокой площадью поверхности обладает высоким армирующим эффектом. Диоксид кремния малого диаметра с высокой степенью агрегации (то есть с большой площадью поверхности и высокой способностью абсорбции масла) демонстрирует превосходную диспергируемость в эластомерной полимерной композиции, представляя желаемые свойства и высокие технологические характеристики. Средний диаметр частиц диоксида кремния, выраженный через диаметр первичных частиц, в некоторых вариантах реализации изобретения составляет от 5 до 60 нм и в некоторых вариантах реализации изобретения - от 10 до 35 нм. Более того, удельная площадь поверхности частиц диоксида кремния (измеренная по методу БЭТ) в некоторых вариантах реализации изобретения составляет от 35 до 300 м2/г. Для примеров диаметров кремнекислотных наполнителей, размеров частиц и поверхностей по методу БЭТ смотри Международную патентную заявку № PCT/US2009/045553. Диоксид кремния добавляют в количестве от 10 до 100 частей по массе, в некоторых вариантах реализации изобретения - от 30 до 100 частей по массе, а в некоторых вариантах реализации изобретения - от 30 до 95 частей по массе на 100 частей по массе общего эластомерного полимера. Кремнекислотные наполнители можно использовать в комбинациях с другими наполнителями, в том числе, без ограничения ими, с техническим углеродом, двухфазным углерод-кремнеземным наполнителем, глиной, карбонатом кальция, углеродными нанотрубками, карбонатом магния и их комбинациями.
Технический углерод и диоксид кремния можно добавлять вместе; в данном случае общее количество добавленного технического углерода и диоксида кремния составляет от 30 до 100 частей по массе, а в некоторых вариантах реализации изобретения - от 30 до 95 частей по массе на 100 частей по массе общего эластомерного полимера. Поскольку такие наполнители являются гомогенно-диспергированными в эластомерной композиции, увеличение количества (в пределах указанных выше диапазонов) приводит к тому, что композиции обладают превосходными технологическими характеристиками прокатки и экструзии, а также вулканизированные продукты обладают хорошими свойствами потери на гистерезис, сопротивлением качению, улучшенным сопротивлением проскальзыванию на мокрой дороге, износоустойчивостью и прочностью при растяжении.
Двухфазный углерод-кремнеземный наполнитель можно использовать либо независимо, либо в комбинации с техническим углеродом и/или диоксидом кремния в соответствии с настоящими идеями. Двухфазный углерод-кремнеземный наполнитель может обладать теми же эффектами, которые были получены при комбинированном использовании технического углерода и диоксида кремния, даже в том случае, если добавляют только его. Двухфазный углерод-кремнеземный наполнитель представляет собой так называемый технический углерод, покрытый диоксидом кремния, полученный посредством нанесения диоксида кремния на поверхность технического углерода, и является коммерчески доступным под торговой маркой CRX2000, CRX2002 или CRX2006 (продукты компании Cabot Co.). Двухфазный углерод-кремнеземный наполнитель добавляют в тех же количествах, как описано выше по отношению к диоксиду кремния. Двухфазный углерод-кремнеземный наполнитель может быть использован в комбинациях с другими наполнителями, в том числе, без ограничения ими, с техническим углеродом, диоксидом кремния, глиной, карбонатом кальция, углеродными нанотрубками, карбонатом магния и их комбинациями. В некоторых вариантах реализации изобретения используют технический углерод и диоксид кремния либо по отдельности, либо в комбинации.
Диоксид кремния, технический углерод или двухфазные углерод-кремнеземные наполнители, или их комбинации могут быть использованы в комбинации с природными наполнителями, в том числе, без ограничения ими, с крахмалом или лигнином.
Примеры наполнителей описаны в Международной патентной заявке № PCT/US2009/045553, которая включена в данный документ в виде ссылки в полном объеме.
В некоторых вариантах реализации изобретения диоксид кремния, включенный в резиновую смесь, имеет площадь поверхности, определяемую по адсорбции азота (в дальнейшем называемая как "N2A") от 150 до 300 м2/г. Диоксид кремния с N2A менее, чем 150 м2/г приводит к неблагоприятно низкому армирующему эффекту. Диоксид кремния с N2A более, чем 300 м2/г обеспечивает резиновую смесь с улучшенной вязкостью и ухудшенными технологическими характеристиками. В случае использования технического углерода, включенного в резиновую смесь, подходит N2A от 60 до 150 м2/г. Технический углерод с N2A менее, чем 60 м2/г приводит к низкому армирующему эффекту. Технический углерод с N2A более, чем 150 м2/г обеспечивает резиновую смесь с увеличенным потерями на гистерезис и ухудшенными
технологическими характеристиками.
В одном варианте реализации изобретения диоксид кремния добавляют в количестве от 10 до 100 частей по массе, в некоторых вариантах реализации изобретения - от 30 до 100 частей по массе, а в некоторых вариантах реализации изобретения - от 30 до 95 частей по массе на 100 частей по массе общего полимера.
В другом варианте реализации изобретения технический углерод добавляют в количестве от 2 до 100 частей по массе, в некоторых вариантах реализации изобретения - от 5 до 100 частей по массе, в некоторых вариантах реализации изобретения - от 10 до 100 частей по массе, а в некоторых вариантах реализации изобретения - от 10 до 95 частей по массе на 100 частей по массе общего полимера.
В еще одном варианте реализации изобретения технический углерод и диоксид кремния можно добавлять вместе; в данном случае общее количество добавленного технического углерода и диоксида кремния составляет от 30 до 100 частей по массе, а предпочтительно - от 30 до 95 частей по массе на 100 частей по массе общего полимера.
В дополнительном варианте реализации изобретения композиция, содержащая эластомерный полимер в соответствии с настоящим изобретением, дополнительно содержит вулканизирующий агент. Сера, серосодержащие соединения, действующие как доноры серы, системы серосодержащих ускорителей и пероксиды представляют собой наиболее распространенные вулканизирующие агенты. Примеры серосодержащих соединений, выступающих в качестве доноров серы, включают, без ограничения ими, дитиодиморфолин (DTDM), тетраметилтиурамдисульфид (TMTD), тетраэтилтиурамдисульфид (TETD) и дипентаметилентиурамтетрасульфид (DPTT). Примеры серосодержащих ускорителей включают, без ограничения ими, производные амина, производные гуанидина, продукты конденсации альдегидамина, тиазолы, тиурам сульфиды, дитиокарбаматы и тиофосфаты. Примеры пероксидов, используемых в качестве вулканизирующих агентов, включают, без ограничения ими, ди-трет-бутилпероксиды, ди-(трет-бутилперокситриметилциклогексан), ди-(трет-бутилпероксиизопропил)бензол, дихлорбензоилпероксид, дикумилпероксиды, трет-бутилкумилпероксид, диметил-ди(трет-бутилперокси)гексан и диметил-ди(трет-бутилперокси)гексин, и бутил-ди(трет-бутилперокси)валерат (Rubber Handbook, SGF, The Swedish Institution of Rubber Technology 2000). Дополнительные примеры и дополнительные сведения, касающиеся вулканизирующих агентов, можно найти у Kirk-Othmer, Encyclopedia of Chemical technology 3rd, Ed., (Wiley Interscience, N.Y. 1982), том 20, стр. 365-468, (в частности, "Vulcanizing Agents and Auxiliary Materials", стр. 390-402).
Ускоритель вулканизации сульфенамидного типа, гуанидинового типа или тиурамового типа может быть использован наряду с вулканизирующим агентом по мере необходимости. При необходимости могут быть добавлены другие добавки, такие как цинковые белила, вспомогательные вещества для вулканизации, противостарители, вспомогательные вещества для переработки и тому подобное. Вулканизирующий агент, как правило, добавляют к полимерной композиции в количестве от 0,5 до 10 частей по массе, а в некоторых предпочтительных вариантах реализации изобретения - от 1 до 6 частей по массе на 100 частей по массе общего эластомерного полимера. Примеры ускорителей вулканизации и количество ускорителя, добавленного по отношению к общему полимеру, приведены в Международной патентной заявке № WO 2009/148932. Системы серосодержащих ускорителей могут содержать оксид цинка или могут не содержать его. Предпочтительно в качестве компонента системы серосодержащих ускорителей применяют оксид цинка.
Таким образом, в еще одном аспекте настоящее изобретение относится к способу получения сшитого эластомерного полимера, включающему стадию (iv):
(iv) добавления вулканизирующего агента к эластомерному полимеру или к композиции, содержащей эластомерный полимер; и сшивания эластомерного полимера.
Стадия (iv) вулканизации эластомерного полимера или композиции, содержащей эластомерный полимер, может быть выполнена с использованием обычного оборудования для вулканизации и обычных вулканизирующих агентов, о которых идет речь выше.
Дополнительно настоящее изобретение относится к сшитым эластомерным полимерам, получаемым в соответствии с указанным выше способом, включающим стадию (iv), а также к композициям, содержащим указанный сшитый эластомерный полимер.
Более того, настоящее изобретение ориентировано на изделия, содержащие одну из композиций по настоящему изобретению. Другими словами, настоящее изобретение, таким образом, относится к изделиям, содержащим композиции, которые включают эластомерный полимер или сшитый эластомерный полимер, о которых идет речь выше. В предпочтительном варианте реализации изобретения изделие в соответствии с настоящим изобретением представляет собой протектор шины, боковину шины, конвейерную ленту, уплотнитель или рукав.
Было обнаружено, что вулканизированные эластомерные полимерные композиции по настоящему изобретению обладают низким сопротивлением качению, низким динамическим образованием тепла и превосходной характеристикой сопротивления проскальзыванию на мокрой дороге. Таким образом, полимерные композиции по настоящему изобретению, предпочтительно - композиции, содержащие модифицированные полимеры, как описано в данном документе, и наполнители, а также вулканизирующий агент и вулканизированные эластомерные полимерные композиции хорошо подходят для использования в производстве шин, протекторов шин, боковин и каркасов шин, а также других промышленных изделий, таких как ленты, рукава, гасители вибрации и компоненты обуви. Более того, полимеры в соответствии с настоящим изобретением демонстрируют улучшенные технологические характеристики (в том числе улучшенное включение и уменьшенное образование тепла в смесительном оборудовании, сокращение полос на вальцах, улучшенное поведение экструзии, сниженное разбухание экструдируемого потока экструдируемой заготовки, гладкость и т.д.) по сравнению с технологическими характеристиками композиций предшествующего уровня техники, содержащих традиционные модифицированные полимеры.
Помимо изложенного выше следует отметить, что модифицированные эластомерные полимеры в соответствии с настоящим изобретением могут быть использованы для модификации пластмасс, в частности, полистирола при производстве УПС (ударопрочный полистирол), сополимеров стиролакрилонитрила при производстве АБС (сополимеры акрилонитрила, бутадиена и стирола), полиуретана или поликарбоната.
Теперь настоящее изобретение будет описано более подробно с помощью следующих примеров.
СПОСОБЫ ИСПЫТАНИЯ
Эксклюзионная хроматография
И молекулярную массу, и распределение по молекулярной массе полимера измеряли с использованием эксклюзионной хроматографии (SEC) на основе полистирольных стандартов. Каждый образец полимера (9-11 мг) растворяли в тетрагидрофуране (10 мл) с образованием раствора. Раствор фильтровали с использованием 0,45-мкм фильтра. 100-мкл образец подавали в колонку хроматографа гель-проникающей хроматографии (система 1100 Hewlett Packard 1100 с 3-мя 10-мкм колонками "PLgel MIXED-B"). Определение коэффициента преломления использовали в качестве детектора для проведения анализа молекулярной массы. Молекулярную массу рассчитывали как для полистирола, основываясь на калибровке, выполненной с помощью Полистирольных стандартов EasiCal PS1 (Easy А и В) от Polymer Laboratories. Показатели среднечисловой молекулярной массы (Mn) и показатели средневесовой молекулярной массы (Mw) приведены на основе полистирольных стандартов. Распределение по молекулярной массе выражено как дисперсность D=Mw/Mn.
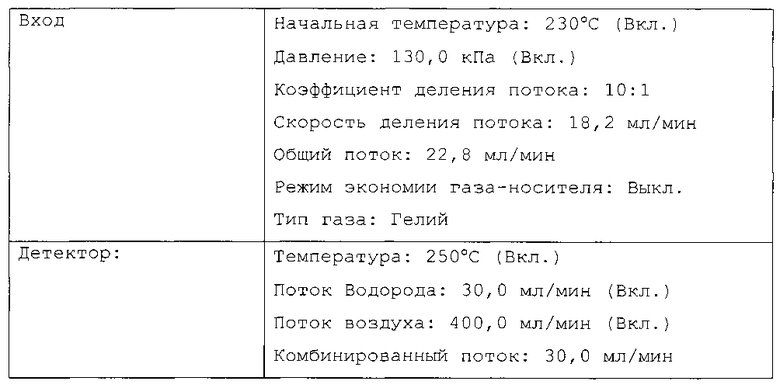
Газохроматографический анализ для измерения конверсии мономеров
Конверсию мономеров определяли посредством измерения концентрации остаточных мономеров в полимерном растворе в конце полимеризации посредством газовой хроматографии.
Около 0,5 г раствора полимера собирали в 20-мл пробирку и разводили 1:10 с использованием циклогексанового растворителя. Около 30 мг разбавленного образца взвешивали с помощью шприца в герметичной пробирке для парофазного анализа. Образец нагревали до 140°C в течение 10 минут. Паровую фазу анализировали посредством газовой хроматографии с использованием следующих условий, представленных в таблице А.

Количественный анализ выполняли посредством метода внешнего стандарта.
Максимальное содержание сухого вещества рассчитывают на основе общего количества подаваемых ингредиентов.
TSC макс=(г/минBde+г/минSty)/(TF)*100%.
TF (Общий Поток, г/мин)=(Bde (г/мин)+Sty (г/мин)+DOP (г/мин)+BuLi (г/мин)+1,2-Бутадиен (г/мин)+Циклогексан (г/мин)).
Bde=1,3-Бутадиен.
Sty=Стирол.
BuLi=Бутиллитий.
Результат газохроматографического анализа, как правило, дан в миллионных долях (ppm) остаточных мономеров, содержащихся в исходном растворе полимера.
Количество остаточных мономеров (RM) в "г/мин" может быть рассчитано по следующей формуле:
RM (г/мин)=((((г/минBde+г/минSty)/TSC макс)*100)/1000000)*(ppmBde+ppmSty))
или в альтернативном варианте
RM (г/мин)=(TF/1000000)*(ppmBde+ppmSty).
Конверсию мономера рассчитывали как:
С (%)=100-(RM/(г/минBd+г/минSt)*100.
1H-ЯМР
Содержание винила и общее содержание стирола измеряли с использованием 1H-ЯМР в соответствии с ISO 21561-2005 с использованием спектрометра ЯМР BRUKER Avance (400 МГц) и 5-мм двойного зонда. CDCl3/TMS использовали в качестве растворителя в массовом отношении 0,05% к 99,95%. Содержание блоков стирола, состоящего из более, чем 6 непрерывных стирольных звеньев, определяли в соответствии со способом, описанным Y. Tanaka с соавторами в Rubber Chemistry and Technology, 1981, 54, №4, 685-691 с использованием относительной интенсивности орто-протонов фенильного кольца, резонирующих выше, чем 6,7 миллионных долей.
Вязкость по Муни ML 1+4 (100°C)
Вязкость по Муни измеряли в соответствии с ASTM D1646 (2004) при времени предварительного нагрева, равному одной минуте, и времени работы ротора, равному четыре минуты, при температуре 100°C [ML 1+4 (100°C)] на MV2000 Е от Alpha-Technologies.
Газохроматографический анализ для измерения концентрации соединения в соответствии с формулой 2 и 3
Концентрацию соединения в соответствии с формулой 2 и 3 в растворе полимера измеряли посредством газовой хроматографии в условиях, изложенных в таблице В.
0,4-0,6 г раствора полимера собирали в 10 мл пробирку и разбавляли около 5 г циклогексана. Анализ проводят с использованием автоматического дозатора. В каждую пробирку добавляли 1 мл образца полимера. Выполняли калибровку с 3-мя различными растворами известной концентрации Модификатора 1.

Свойства вулканизированного соединения
Сопротивление качению
Значение величины Tan δ (60°C) измеряли с использованием динамического спектрометра Eplexor 150N производства компании Gabo Qualimeter Testanlage GmbH (Германия) с применением динамической деформации сжатия 0,2% с частотой 2 Гц при 60°C. Чем меньше данный показатель, тем ниже сопротивление качению.
Сцепление
Tan δ (0°C) измеряли при 0°C с использованием того же оборудования и режима нагрузки, как описано выше по отношению к сопротивлению качению. Чем больше данный индекс, тем лучше сопротивление проскальзыванию на мокрой дороге.
Износ
Износ Din измеряли в соответствии со стандартом DIN 53516. Чем больше данный индекс, тем ниже износоустойчивость.
ПРИМЕРЫ
Условия в реакторе
Следующие примеры были выполнены с использованием двух или трех химических проточных реакторов с мешалкой (CSTR), соединенных последовательно. Объем каждого реактора составлял 5,8 литров, и реактор был оборудован винтовой мешалкой, подходящей для смешивания растворов с высокой вязкостью, скорость мешалки во время всех испытаний составляла 200 оборотов в минуту. Желаемую температуру в стенках реактора регулировали с помощью внешней циркуляции воды. Реагенты, необходимые для полимеризации (стирола, 1,3-бутадиена, циклогексана, DOP (дитетрагидрофурилпропана), н-бутиллития и 1,2-бутадиена), подавали на входе в первый реактор массовыми расходомерами. Каждый расходомер регулировал желаемую подачу и гарантировал постоянный поток реагента. Циклогексан очищали посредством пропускания через колонку с оксидом алюминия.
В описании экспериментов термин "активный инициатор" (nBL, pm) относится к количеству инициатора (н-бутиллития), который принимает участие в реакции полимеризации, и который не деактивируется примесями, содержащимися в реакционной среде. Термин "избыточное количество инициатора" (nBL, ехс) относится к количеству инициатора, которое загружают для деактивации примесей, присутствующих в системе.
Все получившиеся в результате растворы полимеров десорбировали паром в течение одного часа, чтобы удалить растворитель и другие летучие вещества, и сушили в термостате при 70°C в течение 30 минут, а другой - в течение трех дней при комнатной температуре.
Реагенты
Циклогексан (дистиллированный) использовали в качестве растворителя. 1,3-бутадиен (Bde) (дистиллированный) и стирол (Sty) (высушенный с помощью CaH2) использовали в качестве мономеров. Дитетрагидрофурилпропан (DTHFP; DOP) и 1,2-бутадиен (1,2-Bde) разводили в циклогексане. Н-бутиллитий (nBL) использовали в качестве инициатора анионной полимеризации. Если явно не указано иное, (CH3O)3Si-(CH2)3-S-Si(CH3)2(С(CH3)3) использовали в качестве соединения в соответствии с формулой 2 (в дальнейшем также называемое как Модификатор 1) и (CH3O)2(CH3)Si-(CH2)3-S-Si(CH3)2(С(CH3)3) использовали в качестве соединения в соответствии с формулой 3 (в дальнейшем также называемое как Модификатор 2).
ПРИМЕР 1
Сополимеризацию стирола/бутадиена выполняли с использованием двух реакторов, соединенных последовательно. Первый реактор использовали для полимеризации, Модификатор 1 добавляли во второй реактор, молярное отношение Модификатора 1 к nBL, pm составляло 0,28.
В данном испытании использовали молярное отношение DOP к активному инициатору, равное 1,609. Температура первого реактора составляла 68°C, а температура второго реактора составляла 70°C. Поток общего количества ингредиентов и растворителя регулировали таким образом, чтобы время пребывания в каждом реакторе составило 100 минут. Скорости потока добавляемых ингредиентов в первом реакторе были следующими: Sty=1,39 г/мин, Bde=4,16 г/мин, Циклогексан=40,76 г/мин, nBL, pm=0,03848 ммоль/мин, nBL, ехс=0,003109, DOP=0,0619 ммоль/мин. Модификатор 1 добавляли во второй полимеризатор со скоростью потока, равной 0,01061 ммоль/мин (концентрация раствора Модификатора 1 0,00274 моль/л). На выходе из первого реактора получали конверсию мономера, равную 98,62%, а на выходе из второго реактора получали конверсию, равную 99,97%.
К раствору полимера на выходе из второго реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола). В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=217929 г/моль, Mw=461253 г/моль, D=2,116. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=305554 г/моль, Mw=623149 г/моль, D=2,039. В первом полимеризаторе для полимера получали следующие результаты: стирол=25,7%, винил=62,7% (1,2-полибутадиен, в расчете на фракцию бутадиена), блочный стирол (>6 стирольных звеньев): 1%.
Во втором полимеризаторе для полимера получали следующие результаты: стирол=25,8%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=62,9%, блочный стирол (>6 стирольных звеньев): 1%.
Вязкость по Муни ML 1+4 этого продукта составляла 94,2.
ПРИМЕР 2
Сополимеризацию стирола/бутадиена выполняли с использованием двух реакторов, соединенных последовательно. Первый реактор использовали для полимеризации. Модификатор 1 добавляли во второй реактор. Молярное отношение Модификатора 1 к nBL, pm снижали до 0,252.
В данном испытании использовали молярное отношение DOP к инициатору анионной полимеризации, равное 1,464. Температура первого реактора составляла 68°C, а температура второго реактора составляла 70°C. Следующие количества добавляли в первый полимеризатор: Sty=1,39 г/мин, Bde=4,16 г/мин, Циклогексан=40,76 г/мин, nBL, pm=0,01967 ммоль/мин; DOP=0,0288 ммоль/мин. Модификатор 1 добавляли во второй полимеризатор со скоростью потока, равной 0,00495 ммоль/мин (концентрация раствора Модификатора 1 0,00274 моль/л). Поток общего количества ингредиентов и растворителя регулировали таким образом, чтобы время пребывания в каждом реакторе составило 100 минут.
На выходе из первого реактора получали общую конверсию, равную 88,2%, а на выходе из второго реактора получали конверсию, равную 92,02%. К раствору полимера на выходе из второго реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола). В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=380968 г/моль, Mw=799801 г/моль, D=2,099. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=428465 г/моль, Mw=896831 г/моль, D=2,093. В первом полимеризаторе для полимера получали следующие результаты: стирол=25,4%, винил=60,8% (1,2-полибутадиен, в расчете на фракцию бутадиена), блочный стирол (>6 стирольных звеньев): 1%.
Во втором полимеризаторе для полимера получали следующие результаты: стирол=25,2%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=60,9%, блочный стирол (>6 стирольных звеньев): 1%. Вязкость по Муни ML 1+4 этого продукта составляла 115,4.
ПРИМЕР 3
Сополимеризацию стирола/бутадиена выполняли с использованием трех реакторов, соединенных последовательно. Первый реактор использовали для полимеризации. Модификатор 1 добавляли во второй реактор, а Модификатор 2 добавляли в третий реактор. Молярное отношение Модификатора 1 к nBL, pm составляло 0,388. Молярное отношение Модификатора 2 к nBL, pm составляло 1,163.
Более того, в данном примере использовали 1,2-бутадиен. Использовали молярное отношение DOP к nBL, pm, равное 1,697, и использовали молярное отношение 1,2-бутадиена к nBL, pm, равное 0,2114. Температура первого реактора составляла 68°C. Следующие количества добавляли в первый полимеризатор: Sty=1,39 г/мин, Bde=4,17 г/мин, Циклогексан=40,76 г/мин, nBL, pm=0,01892 ммоль/мин; DOP=0,0321 ммоль/мин, 1,2-бутадиен=0,004 ммоль/мин.
Модификатор 1 добавляли во второй полимеризатор со скоростью потока, равной 0,00734 ммоль/мин (концентрация раствора Модификатора 1 0,00273 моль/л). Модификатор 2 добавляли в третий реактор со скоростью потока, равной 0,02201 ммоль/мин (концентрация раствора Модификатора 2 0,0545 моль/л). Температура второго и третьего реактора составляла 70°C. Время пребывания в каждом реакторе составляло 100 минут.
На выходе из первого реактора получали конверсию мономера, равную 91,97%, на выходе из второго реактора получали конверсию, равную 96,7%, а на выходе из третьего реактора получали конверсию, равную 96,73%.
К раствору полимера на выходе из третьего реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола). В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=413244 г/моль, Mw=846182 г/моль, D=2,048. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=447261 г/моль, Mw=900845 г/моль, D=2,014. В третьем полимеризаторе для полученного полимера получали следующие результаты: Mn=470670 г/моль, Mw=934077 г/моль, D=1,985.
В первом полимеризаторе для полимера получали следующие результаты: стирол=24,9%, винил=60,8% (1,2-полибутадиен, в расчете на фракцию бутадиена), блочный стирол (>6 стирольных звеньев): 1%.
Во втором полимеризаторе для полимера получали следующие результаты: стирол=24,5%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=61,2%, блочный стирол (>6 стирольных звеньев): 1%.
В третьем полимеризаторе для полимера получали следующие результаты: стирол=24,2%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=61,5%, блочный стирол (>6 стирольных звеньев): 1%.
Вязкость по Муни ML 1+4 этого продукта составляла 112,6.
ПРИМЕР 4
Реакцию выполняли в 2 реакторах, соединенных последовательно. Первый реактор использовали для полимеризации, Модификатор 1 добавляли во второй реактор. Молярное отношение Модификатора 1 к nBL, pm составляло 0,644. В данном испытании использовали молярное отношение DOP к активному инициатору, равное 1,608. Температура первого реактора составляла 68°C, а температура второго реактора составляла 70°C.
Следующие количества добавляли в первый полимеризатор: Sty=1,041 г/мин, Bde=3,335 г/мин, Циклогексан=40,76 г/мин, nBL, pm=0,03849 ммоль/мин; DOP=0,0619 ммоль/мин. Модификатор 1 добавляли во второй полимеризатор со скоростью потока, равной 0,0248 ммоль/мин (концентрация раствора Модификатора 1 0,002739 моль/л). Время пребывания в каждом реакторе составляло 100 минут. На выходе из первого реактора получали конверсию мономера, равную 97,89%, а на выходе из второго реактора получали конверсию, равную 99,96%.
К раствору полимера на выходе из второго реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола).
В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=215666 г/моль, Mw=455748 г/моль, D=2,113. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=265780 г/моль, Mw=538284 г/моль, D=2,025.
В первом полимеризаторе для полимера получали следующие результаты: стирол=25,2%, винил=62,7% (1,2-полибутадиен, в расчете на фракцию бутадиена), блочный стирол (>6 стирольных звеньев): 1%. Во втором полимеризаторе для полимера получали следующие результаты: стирол=25,2%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=62,7%, блочный стирол (>6 стирольных звеньев): 1%.
Вязкость по Муни ML 1+4 этого продукта составляла 67,6.
ПРИМЕР 5
Сополимеризацию стирола/бутадиена выполняли с использованием трех реакторов, соединенных последовательно. Первые 2 реактора использовали для полимеризации. Модификатор 1 добавляли в третий реактор. Молярное отношение Модификатора 1 к nBL, pm составляло 0,296. Более того, в данном примере использовали 1,2-бутадиен. Использовали молярное отношение DOP к nBL, pm, равное 1,925, и использовали молярное отношение 1,2-бутадиена к nBL, pm, равное 0,148. Следующие количества добавляли в первый полимеризатор: Sty=4,724 г/мин, Bde=13,8 г/мин, Циклогексан=132,33 г/мин, nBL, pm=0,1256 ммоль/мин; DOP=0,2418 ммоль/мин, 1,2-бутадиен=0,01860 ммоль/мин.
Модификатор 1 добавляли в третий полимеризатор со скоростью потока, равной 0,03720 ммоль/мин (концентрация раствора Модификатора 1 0,005446 моль/л). Температура всех реакторов составляла 70°C. Время пребывания в каждом реакторе составляло 35 минут.
На выходе из первого реактора получали конверсию мономера, равную 91,04%, на выходе из второго реактора получали конверсию, равную 99,73%, а на выходе из третьего реактора получали конверсию, равную 99,94%.
К раствору полимера на выходе из третьего реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола). В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=205392 г/моль, Mw=397187 г/моль, D=1,934. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=215441 г/моль, Mw=403586 г/моль, D=1,873. В третьем полимеризаторе для полученного полимера получали следующие результаты: Mn=267466 г/моль, Mw=499838 г/моль, D=1,869.
В первом полимеризаторе для полимера получали следующие результаты: стирол=25,7%, винил=63,5% (1,2-полибутадиен, в расчете на фракцию бутадиена), блочный стирол (>6 стирольных звеньев): 1%.
Во втором полимеризаторе для полимера получали следующие результаты: стирол=25,7%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=63,8%, блочный стирол (>6 стирольных звеньев): 1%.
В третьем полимеризаторе для полимера получали следующие результаты: стирол=25,8%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=63,9%, блочный стирол (>6 стирольных звеньев): 1%.
Вязкость по Муни ML 1+4 этого продукта составляла 63,1.
ПРИМЕР 6
Сополимеризацию стирола/бутадиена выполняли с использованием трех реакторов, соединенных последовательно. Первые 2 реактора использовали для полимеризации. Модификатор 1 добавляли в третий реактор. Молярное отношение Модификатора 1 к nBL, pm составляло 0,259. Более того, в данном примере использовали 1,2-бутадиен. Использовали молярное отношение DOP к nBL, pm, равное 2,018, и использовали молярное отношение 1,2-бутадиена к nBL, pm, равное 0,155. Следующие количества добавляли в первый полимеризатор: Sty=4,724 г/мин, Bde=13,8 г/мин, Циклогексан=132,33 г/мин, nBL, pm=0,1198 ммоль/мин; DOP=0,2418 ммоль/мин, 1,2-бутадиен=0,01860 ммоль/мин.
Модификатор 1 добавляли в третий полимеризатор со скоростью потока, равной 0,031 ммоль/мин (концентрация раствора Модификатора 1 0,0310 моль/л). Температура всех реакторов составляла 70°C. Время пребывания в каждом реакторе составляло 35 минут.
На выходе из первого реактора получали конверсию мономера, равную 88,87%, на выходе из второго реактора получали конверсию, равную 99,43%, а на выходе из третьего реактора получали конверсию, равную 99,71%.
К раствору полимера на выходе из второго реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола). В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=121664 г/моль, Mw=409029 г/моль, D=1,923. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=222630 г/моль, Mw=408364 г/моль, D=1,834. В третьем полимеризаторе для полученного полимера получали следующие результаты: Mn=253857 г/моль, Mw=485285 г/моль, D=1,912.
В первом полимеризаторе для полимера получали следующие результаты: стирол=25,9%, винил=64,4% (1,2-полибутадиен, в расчете на фракцию бутадиена), блочный стирол (>6 стирольных звеньев): 1%.
Во втором полимеризаторе для полимера получали следующие результаты: стирол=26,2%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=64,4%, блочный стирол (>6 стирольных звеньев): 1%.
В третьем полимеризаторе для полимера получали следующие результаты: стирол=25,9%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=64,5%, блочный стирол (>6 стирольных звеньев): 1%.
Вязкость по Муни ML 1+4 этого продукта составляла 71,6.
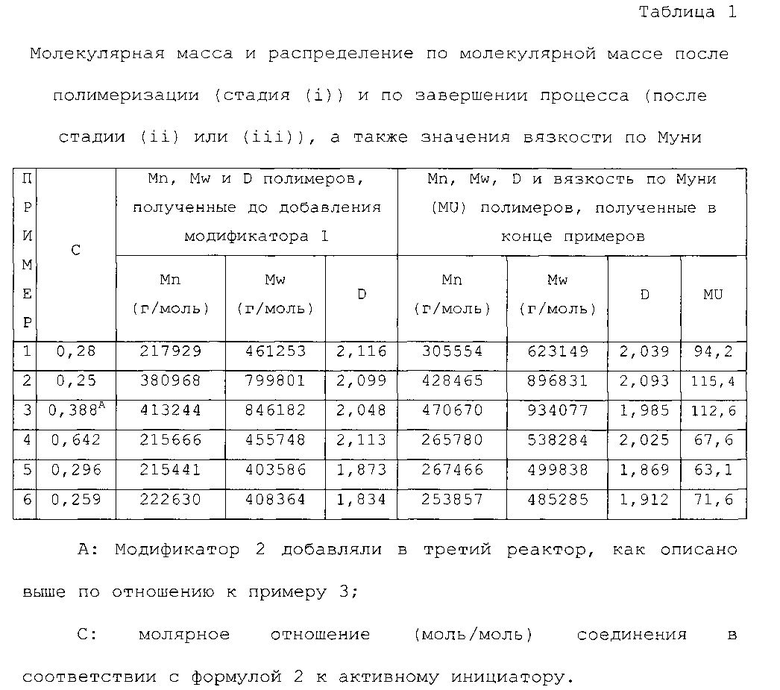
ПРИМЕР 7
Определение % молярного количества живых цепей
В следующем примере оценивают % молярного количества живых цепей. Эксперимент непрерывной полимеризации выполняли, как описано в предыдущих примерах. Молярный избыток Модификатора 1 по отношению к молярным долям активного инициатора добавляли во второй реактор. В данных условиях каждая живая цепь должна вступать в реакцию с одной молекулой Модификатора 1. Остаточное количество Модификатора 1 в полимерном растворе анализировали посредством газовой хроматографии. Молярное количество живых концевых групп цепи соответствует разности молярного количества загруженного Модификатора 1 и молярного количества Модификатора 1, обнаруженного в растворе полимера.
Реакцию выполняли в 2 реакторах, соединенных последовательно. Первый реактор использовали для полимеризации. Модификатор 1 добавляли во второй реактор. Молярный избыток Модификатора 1 добавляли во второй реактор: молярное отношение Модификатора 1 к nBL, pm составляло 1,19.
В данном испытании использовали молярное отношение DOP к инициатору анионной полимеризации, равное 1,605. Температура первого реактора составляла 68°C, а температура второго реактора составляла 70°C.
Следующие количества добавляли в первый полимеризатор: Sty=1,369 г/мин, Bde=4,19 г/мин, Циклогексан=40,77 г/мин, nBL, pm=0,018 ммоль/мин; DOP=0,0289 ммоль/мин. Модификатор 1 добавляли во второй полимеризатор со скоростью потока, равной 0,02146 ммоль/мин (концентрация раствора Модификатора 1 0,00275 моль/л). Время пребывания в каждом реакторе составляло 100 минут. Концентрация подачи Модификатора 1 в полимерный раствор составляла 127 миллионных долей.
На выходе из первого реактора получали конверсию мономера, равную 88,7%, а на выходе из второго реактора получали конверсию, равную 90,66%. К раствору полимера на выходе из второго реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола). В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=418924 г/моль, Mw=873412 г/моль, D=2,085. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=435858 г/моль, Mw=869482 г/моль, D=1,995.
Во втором полимеризаторе для полимера получали следующие результаты: стирол=25%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=61,0%, блочный стирол (>6 стирольных звеньев): 1%. В третьем полимеризаторе для полимера получали следующие результаты: стирол=24,9%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=61,3%, блочный стирол (>6 стирольных звеньев): 1%.
Количество Модификатора 1 в растворе полимера на выходе из второго реактора анализировали посредством газовой хроматографии, и оно составило 26 миллионных долей, что соответствует 0,003885 ммоль/мин Модификатора 1, обнаруженного в растворе полимера на выходе из второго реактора. Это означает, что 0,01757 ммоль/мин Модификатора 1 вступило в реакцию с живыми цепями и это количество также соответствует количеству цепей, которые все еще живы во втором реакторе. В данном эксперименте рассчитывают степень жизненности, равную 97,6%.
Полученные в данном испытании данные подтверждают, что очень высокая степень жизненности может быть получена в условиях реакции, описанных в настоящем изобретении.
ПРИМЕР 8 (сравнительный)
(Непрерывная полимеризация, линейная)
Реакцию выполняли в 2 реакторах, соединенных последовательно. Оба реактора использовали для полимеризации. В данном испытании использовали молярное отношение DOP к активному инициатору, равное 2,048, и молярное отношение 1,2-бутадиена к nBL, pm, равное 0,162. Температура первого реактора составляла 65°C, а температура второго реактора составляла 70°C. Следующие количества добавляли в первый полимеризатор: Sty=3,58 г/мин, Bde=11,04 г/мин, Циклогексан=107,25 г/мин, nBL, pm=0,07631 ммоль/мин; nBL, ехс=0,0151 ммоль/мин; DOP=0,1563 ммоль/мин, 1,2-бутадиен=0,01234 ммоль/мин. Время пребывания в каждом реакторе составляло 38 минут.
На выходе из первого реактора получали конверсию мономера, равную 84,12%, а на выходе из второго реактора получали конверсию, равную 99,01%.
К раствору полимера на выходе из второго реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола). В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=246650 г/моль, Mw=489634 г/моль, D=1,985. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=269100 г/моль, Mw=504031 г/моль, D=1,873.
В первом полимеризаторе для полимера получали следующие результаты: стирол=26,5%, винил=63,4% (1,2-полибутадиен, в расчете на фракцию бутадиена), блочный стирол (>6 стирольных звеньев): 1%. Во втором полимеризаторе для полимера получали следующие результаты: стирол=26,9%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=63,8%, блочный стирол (>6 стирольных звеньев): 1%.
Вязкость по Муни ML 1+4 этого продукта составляла 66,5.
ПРИМЕР 9 (сравнительный)
Сополимеризацию стирола/бутадиена выполняли с использованием двух реакторов, соединенных последовательно. Первый реактор использовали для полимеризации, Модификатор 1 добавляли во второй реактор, молярное отношение Модификатора 1 к nBL, pm составляло 0,202.
В данном испытании использовали молярное отношение DOP к активному инициатору, равное 1,764. Температура первого реактора составляла 68°C, а температура второго реактора составляла 70°C. Поток общего количества ингредиентов и растворителя регулировали таким образом, чтобы время пребывания в каждом реакторе составило 100 минут. Скорости потока добавляемых ингредиентов в первом реакторе были следующими: Sty=1,39 г/мин, Bde=4,16 г/мин, Циклогексан=40,76 г/мин, nBL, pm=0,03509 ммоль/мин, nBL, ехс=0,003109, DOP=0,0619 ммоль/мин. Модификатор 1 добавляли во второй полимеризатор со скоростью потока, равной 0,00707 ммоль/мин (концентрация раствора Модификатора 1 0,00274 моль/л). На выходе из первого реактора получали конверсию мономера, равную 97,40%, а на выходе из второго реактора получали конверсию, равную 99,97%.
К раствору полимера на выходе из второго реактора добавляли метанол в качестве агента обрыва цепи и IRGANOX 1520 (0,15 м.ч.) в качестве антиоксиданта. Полимеры, выходящие из первого и второго реакторов анализировали посредством гель-проникающей хроматографии (калибровкой полистирола). В первом полимеризаторе для полученного полимера получали следующие результаты: Mn=291033 г/моль, Mw=643178 г/моль, D=2,21. Во втором полимеризаторе для полученного полимера получали следующие результаты: Mn=299626 г/моль, Mw=671626 г/моль, D=2,242. В первом полимеризаторе для полимера получали следующие результаты: стирол=25,7%, винил=63% (1,2-полибутадиен, в расчете на фракцию бутадиена), блочный стирол (>6 стирольных звеньев): 1%.
Во втором полимеризаторе для полимера получали следующие результаты: стирол=25,7%, винил (1,2-полибутадиен, в расчете на фракцию бутадиена)=63%, блочный стирол (>6 стирольных звеньев): 1%.
Вязкость по Муни ML 1+4 этого продукта составляла 88,7.
ПРИМЕР 10 (сравнительный)
Сополимеризацию выполняли в 20-литровом стальном реакторе с двойной стенкой, который сначала продували азотом прежде, чем добавить органический растворитель, мономеры, полярное соединение-координатор, соединение-инициатор или другие компоненты. Температуру в полимеризаторе доводили до 40°C. Затем в следующем порядке добавляли следующие компоненты: растворитель циклогексан (9000 г); мономер 1,3-бутадиен (1514,35 г) и мономер стирол (414,52 г) и 20,5 ммоль тетраметилэтилендиамина (TMEDA) в качестве полярного координатора, TMEDA/nBL, pm=1,720. Полученную смесь перемешивали в течение одного часа. Чтобы начать полимеризацию, добавляли н-бутиллитий (11,92 ммоль). Полимеризацию выполняли в течение 80 минут, не позволяя, чтобы температура полимеризации превысила 60°C. После этого добавляли 7,75 г мономера 1,3-бутадиена с последующим добавлением тетрахлорида олова (0,903 ммоль). Полученную смесь перемешивали в течение 20 минут. После этого добавляли еще 27,9 г мономера 1,3-бутадиена с последующим добавлением (CH3O)3Si-(CH2)3-S-Si(CH3)3 в качестве соединения в соответствии с формулой 2 (10,16 ммоль, соединение в соответствии с формулой 2/nBL, pm=0,852). Для завершения процесса полимеризации полимерный раствор перемещали через 45 минут в отдельный стальной реактор с двойной стенкой, содержащий 100 мл этанола и 1,4 мл концентрированной HCl (концентрация 36%) и 5 г Irganox 1520 в качестве стабилизатора.
Для синтезированного полимера получали следующие результаты: Mn=342581 г/моль, Mw=496873 г/моль, D=1,263, стирол=21%, винил=62,4%, Вязкость по Муни=62,4.
ПРИМЕР 11 (сравнительный)
Сополимеризацию выполняли в 10-литровом стальном реакторе с двойной стенкой, который сначала продували азотом прежде, чем добавить органический растворитель, мономеры, полярное соединение-координатор, соединение-инициатор или другие компоненты. Температуру в полимеризаторе доводили до 40°C. Затем в следующем порядке добавляли следующие компоненты: циклогексан (5544 г); мономер 1,3-бутадиен (773,82 г); мономер стирол (210 г); тетраметилэтилендиамин в качестве полярного координатора (10,385 ммоль), TMEDA/nBL, pm=1,700. Полученную смесь перемешивали в течение одного часа. Чтобы начать полимеризацию, добавляли н-бутиллитий (6,109 ммоль). Полимеризацию выполняли в течение 80 минут, не позволяя, чтобы температура полимеризации превысила 60°C. После этого добавляли еще 14,18 г мономера 1,3-бутадиена с последующим добавлением (CH3O)3Si-(CH2)3-S-Si(CH3)3 в качестве соединения в соответствии с формулой 2 (7,262 ммоль, отношение соединения в соответствии с формулой 2 к nBL, pm=1,190). Для завершения процесса полимеризации полимерный раствор перемещали через 45 минут в отдельный стальной реактор с двойной стенкой, содержащий 100 мл этанола и 1,4 мл концентрированной HCl (концентрация 36%) и 5 г Irganox 1520 в качестве стабилизатора. Для синтезированного полимера получали следующие результаты: Mn=274259 г/моль, Mw=346417 г/моль, D=1,263, стирол=20,51%, винил=64,45%. Вязкость по Муни=65.
Приведенные ниже таблицы представляют обзор молекулярных отношений, задействованных в изложенных выше примерах (таблица 2), а также обобщают полимерные характеристики (таблица 3).
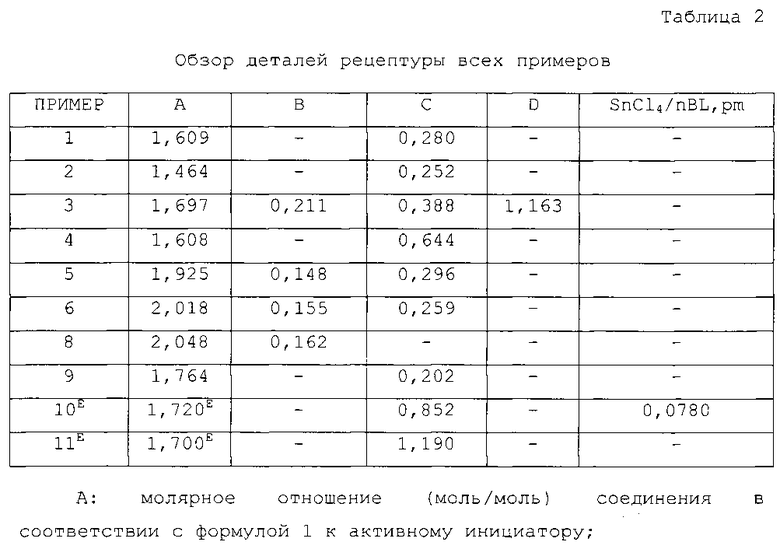

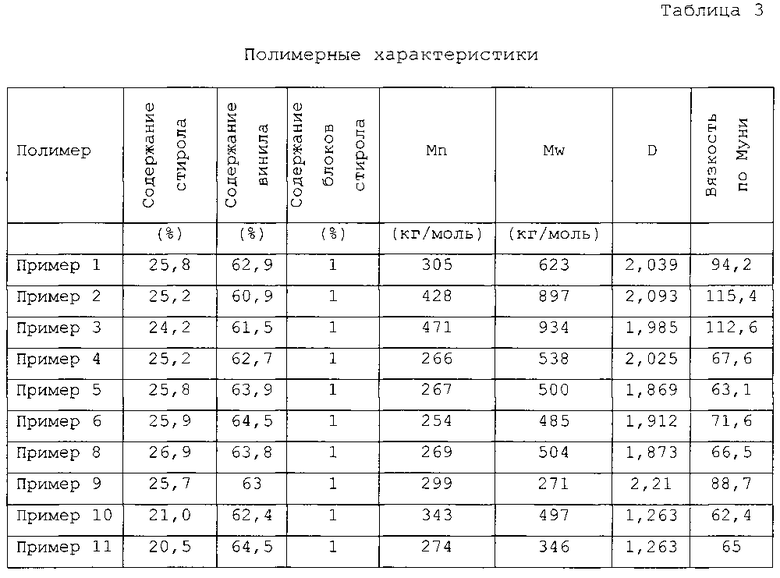
Получение композиций, содержащих наполнитель
Композиции были получены с использованием материалов растворного бутадиен-стирольного каучука (SSBR), описанных в изложенных выше примерах, за исключением примера 7.
Резиновые смеси на основе диоксида кремния
Полимерные композиции были получены посредством объединения и компаундирования 80 частей по массе каждого из эластомерных полимеров, полученных в примерах 1, 2, 3, 4, 5, 6, 8, 9, 10 и 11, с 20 частями по массе коммерчески доступного полибутадиена с высоким содержанием цис-звеньев и компонентов, перечисленных ниже в таблице 4, в 350 см3 смесителе Бенбери (Labstation 350S от Brabender GmbH & Co KG) с последующим процессом двухстадийного смешивания. Стадия 1: все компоненты, как указано в таблице 4, смешивали вместе в течение 5-7 минут при 60-80 оборотах в минуту за исключением компонентов вулканизирующего агента (то есть, серы, TBBS и DPG) с получением композиции стадии 1. Стадия 2: После этого добавляли вулканизирующий агент, и смесь перемешивали в течение дополнительных 2-3 минут при 30-50 оборотах в минуту с получением смесей стадии 2. Значения по Муни определяли для каждой из данных композиций ("смесь стадии 2") и указывали в приведенной ниже таблице 5 в качестве значений "Муни Смеси". После получения смесей стадии 2 начинали вулканизацию посредством нагревания смесей стадии 2 при 160°C в течение 20 минут. После вулканизации композиции оставляли, чтобы они достигли комнатной температуры с получением композиций "1А"-"11А". Физические свойства для каждой полученной таким образом композиции приведены в таблице 5. В данной таблице образцы "1А"-"11А" включительно обозначают композиции, полученные из полимеров, описанных выше в данном документе в примерах 1-11, за исключением примера 7.


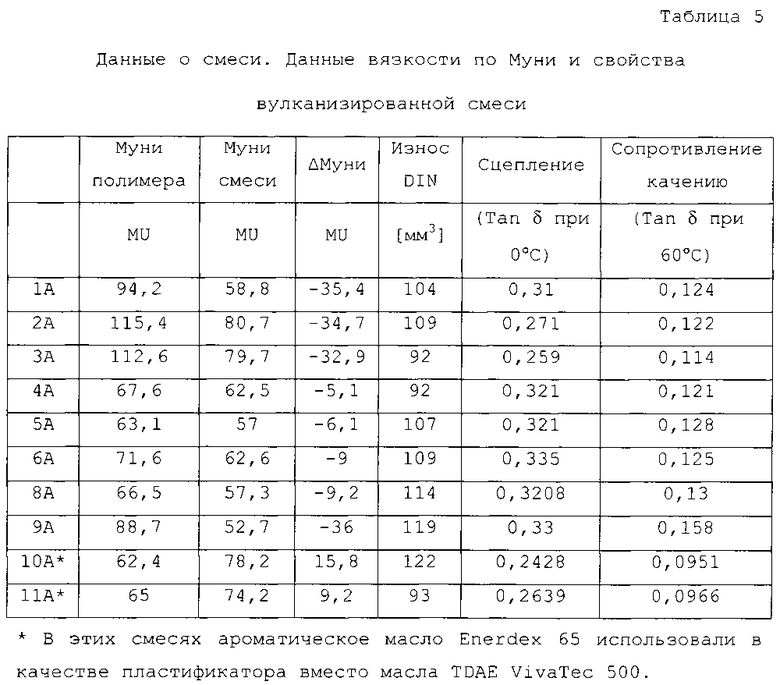
Значение "ΔМуни" представляет собой разность значений вязкости по Муни полимера, не содержащего растворителя ("Муни полимера"), и композиций, содержащих наполнитель ("Муни смеси"). Меньшие значения дельта по Муни связаны с лучшими технологическими свойствами.
Данные, представленные в таблице 5, демонстрируют, что композиции 1А, 2А, 3А, 4А, 5А и 6А, которые основаны на полимерах в соответствии с настоящим изобретением, обладают улучшенными технологическими характеристиками по сравнению с композициями 10А и 11А, которые основаны на полимерах, полученных в процессе полимеризации в реакторе периодического действия. В то же время свойства износа, сцепления и сопротивления качению улучшены по сравнению со сравнительными примерами 8А и 9А, которые основаны на полимерах 8 и 9, соответственно.
Таким образом, композиции в соответствии с настоящим изобретением объединяют положительные технологические свойства (как это наблюдалось в полимерах, которые были получены в процессах непрерывной полимеризации) с благоприятными свойствами продукта, такими как износ, сцепление и сопротивление качению. Композиции 1А, 2А, 3А, 4А, 5А и 6А демонстрируют в целом лучший баланс с точки зрения технологических свойств неотвержденных кремнийдиоксидных смесей и сопротивления качению отвержденных смесей.
Таким образом, способ по настоящему изобретению позволяет получать модифицированные эластомерные полимеры с улучшенными технологическими свойствами при реактивном смешивании модифицированных эластомерных полимеров с компонентами композиции, как представлено в Таблице 5. Не желая быть связанными теорией, авторы настоящего изобретения считают, что улучшенные технологические свойства являются результатом комбинации широкого распределения по молекулярной массе полимеров (по отношению к полимеру, полученному в процессах полимеризации в реакторе периодического действия) и эффективной модификации полимера, выполненной с использованием модификаторов 1 и/или 2.
В частности предпочтительные варианты реализации (EM) в соответствии с настоящим изобретением перечислены ниже в данном документе:
1. Способ непрерывного получения эластомерного полимера, включающий следующие стадии:
(i) обеспечение наличия и полимеризация по меньшей мере одного сопряженного диенового мономера и, необязательно, одного или нескольких ароматических виниловых мономеров при температуре 110°C или ниже в органическом растворителе в присутствии активного инициатора и соединения в соответствии с формулой 1:
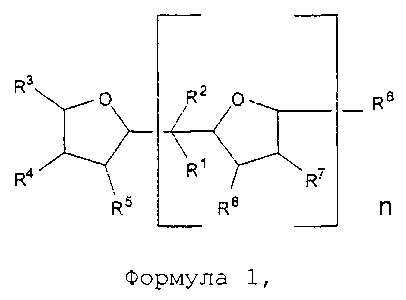
где О в формуле 1 представляет собой кислород, группы R1 и R2 в формуле 1 каждая независимо представляют собой водород или алкильную группу, предпочтительно - водород или C1-C4 алкильную группу; группы R3, R4, R5, R6, R7 и R8, каждую независимо, выбирают из водорода или алкильной группы, предпочтительно - водорода или C1-C6 алкильной группы; n является целым числом, выбранным из 1, 2, 3 или 4, предпочтительно 1;
и где активный инициатор представляет собой инициатор анионной полимеризации, а молярное отношение соединения в соответствии с формулой 1 к активному инициатору составляет от 0,15 до 10;
с получением живого анионного полимера; и
(ii) добавление первого количества соединения в соответствии с формулой 2 к живому анионному полимеру в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 2 к активному инициатору составляло от 0,21 или более:
где S в формуле 2 представляет собой атом серы, Si представляет собой кремний, О представляет собой кислород; группы R каждую независимо выбирают из алкила, арила или аралкила, предпочтительно - из C1-С10алкила, C7-C17аралкила или С6-С16арила; группы R′, R″ и R″′ независимо друг от друга выбирают из алкила, аралкила или арила, предпочтительно - из C1-С10алкила, C6-C16арила или C7-C17аралкила; А представляет собой двухвалентную гидрокарбильную группу, предпочтительно - С1-С20алкилен или С7-С25аралкилен.
2. Способ по EM 1, в котором стадия (ii) добавления соединения Формулы 2 к живому анионному полимеру начинается при конверсии мономера в диапазоне от 85 до 100 процентов по массе в расчете на количество предусмотренных мономеров.
3. Способ по любому из EM 1 или 2, где молярное отношение соединения в соответствии с формулой 1 к активному инициатору составляет от 0,4 до 5.
4. Способ по любому из предшествующих EM, включающий дополнительную стадию (iii):
(iii) последовательного добавления второго количества соединения в соответствии с формулой 2 и/или добавления соединения в соответствии с формулой 3 в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 2 и/или Формулой 3, добавленных на стадии (iii), к активному инициатору составляло от 0,21 или более:
где группы R в формуле 3 независимо выбирают из алкильных, аралкильных или арильных групп, предпочтительно - из C1-С10алкила, C7-C17аралкила или C6-C16арила; группы R′, R″ и R″′ независимо выбирают из алкила, аралкила или арила, предпочтительно - из C1-С10алкила, C6-C16арила или C7-C17аралкила; группу R″″ выбирают из алкила, аралкила или арила, предпочтительно - из C1-С10алкила, С6-С16арила или C7-C17аралкила; А представляет собой двухвалентную гидрокарбильную группу, предпочтительно - C1-C20алкилен или С7-С25аралкилен.
5. Способ по EM 1, где группы R1 и R2 в формуле 1 независимо выбирают из водорода или C1-С4алкила; каждую из групп R2, R3, R4, R5, R6, R7 и R8 независимо выбирают из водорода или C1-C6алкила; n равно 1; где в формуле 2 каждую группу R независимо выбирают из C1-С10алкила, C7-C17аралкила или C6-C16арила; группы R′, R″ и R″′ независимо друг от друга выбирают из C1-С10алкила, C6-С16арила или C7-C17аралкила; и А представляет собой C1-C20алкилен или С7-С25аралкилен; и где в формуле 3, каждую группу R независимо выбирают из C1-С10алкила, C7-C17аралкила или C6-C16арила; группы R′, R″, R″′ и R″″ независимо друг от друга выбирают из C1-C10алкила, C6-C16арила или C7-C17аралкила; и А представляет собой C1-C20алкилен или С7-С25аралкилен.
6. Способ по любому из предшествующих EM, где по меньшей мере один сопряженный диеновый мономер выбирают из 1,3-бутадиена, 2-алкил-1,3-бутадиена, 2-метил-1,3-бутадиена, 2,3-диметил-1,3-бутадиена, 1,3-пентадиена, 2,4-гексадиена, 1,3-гексадиена, 1,3-гептадиена, 1,3-октадиена, 2-метил-2,4-пентадиена, циклопентадиена, 2,4-гексадиена и/или 1,3-циклооктадиена.
7. Способ по любому из предшествующих EM, где органический растворитель выбирают из одного или более соединений из числа бутена, бутана, пентана, циклогексана, гексана, гептана, октана, бензола и толуола, предпочтительно - из одного или более из бутана, пентана, циклогексана, гексана и гептана.
8. Способ по любому из предшествующих EM, где инициатор анионной полимеризации содержит металлорганическое соединение, имеющее по меньшей мере один атом лития и содержащее неполярную С1-C20 углеводородную группу, или их смесь.
9. Способ по EM 8, где инициатор анионной полимеризации представляет собой алкиллитий, предпочтительно - н-бутиллитий, втор-бутиллитий или их смесь.
10. Способ по любому из предшествующих EM, где соединение в соответствии с формулой 1 представляет собой ди(тетрагидрофурил)пропан.
11. Способ по любому из предшествующих EM, где температура на стадии (i) находится в диапазоне от 40 до 110°C.
12. Способ по любому из предшествующих EM, где температура на стадии (i), стадии (ii) и необязательной стадии (iii) находится в диапазоне от 55 до 90°C.
13. Способ по любому из предшествующих EM, где стадию (i), стадию (ii) и необязательную стадию (iii) выполняют в реакционных котлах, соединенных последовательно.
14. Способ по EM 13, где по меньшей мере один сопряженный диеновый мономер, необязательно, ароматический виниловый мономер, инициатор анионной полимеризации и соединение в соответствии с формулой 1 добавляют в первый реакционный котел со скоростью потока таким образом, чтобы среднее время пребывания в каждой стадии (i) и (ii), а также в необязательной стадии (iii) составляло от 30 до 150 минут.
15. Способ по любому из предшествующих EM, где 1,2-бутадиен добавляют на стадии (i) в качестве дополнительного компонента.
16. Способ по EM 15, где молярное отношение 1,2-бутадиена к активному инициатору составляет от 0,05 до 0,4.
17. Способ по любому из предшествующих EM, где по меньшей мере один сопряженный диеновый мономер и, необязательно, один или более ароматический виниловый мономер представлены в таком количестве, чтобы общее содержание сухого вещества мономеров составляло от 6 до 24 процентов по массе в расчете на общую массу мономеров и растворителя.
18. Эластомерный полимер получают по любому из предшествующих EM.
19. Эластомерный полимер по EM 18 с содержанием винила от 20 до 80 процентов по массе и содержанием стирола от 0 до 50, предпочтительно - от 10 до 40 процентов по массе.
20. Эластомерный полимер по EM 18 и 19, обладающий распределением по молекулярной массе (MWD) от 1,7 до 2,5.
21. Эластомерный полимер по любому из EM 18-20, где полимер обладает среднечисловой молекулярной массой (Mn) от 80000 до 700000.
22. Эластомерный полимер по любому из EM 18-21, где полимер обладает Вязкостью по Муни ML 1+4 (100°C), равной от 15 до 100.
23. Композиция, содержащая эластомерный полимер по любому из EM 18-22.
24. Композиция по EM 23, дополнительно содержащая масло и/или наполнитель.
25. Композиция по EM 23 или 24, дополнительно содержащая вулканизирующий агент.
26. Композиция по любому из EM 23-25, где наполнитель выбирают из одного или более веществ из числа диоксида кремния и технического углерода.
27. Способ получения сшитого эластомерного полимера, включающий стадию (iv):
(iv) добавления вулканизирующего агента к эластомерному полимеру по любому из EM 18-22 или к композиции по любому из EM 23-26; и сшивания эластомерного полимера.
28. Сшитый эластомерный полимер получают по EM 27.
29. Композиция, содержащая сшитый эластомерный полимер по EM 28.
30. Изделие, содержащее композицию по любому из EM 23-26 или 29.
31. Изделие по EM 30, где изделие представляет собой протектор шины, боковину шины, конвейерную ленту, уплотнитель или рукав.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ СОПОЛИМЕРЫ С НИЗКИМ СОДЕРЖАНИЕМ ВИНИЛОВЫХ СВЯЗЕЙ | 2013 |
|
RU2661220C2 |
| ПОЛИМЕРЫ, МОДИФИЦИРОВАННЫЕ АМИНОСИЛАНОМ | 2012 |
|
RU2609166C2 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ СШИТЫЕ РЕЗИНОВЫЕ КОМПОЗИЦИИ | 2015 |
|
RU2701830C2 |
| ИНИЦИАТОРЫ ПОЛИМЕРИЗАЦИИ | 2013 |
|
RU2666359C2 |
| МОДИФИЦИРОВАННЫЕ ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ | 2009 |
|
RU2504555C2 |
| МОДИФИЦИРОВАННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2558597C2 |
| ШИНА ДЛЯ КОЛЕС ТРАНСПОРТНЫХ СРЕДСТВ | 2017 |
|
RU2731944C2 |
| СМЕСЬ ФУНКЦИОНАЛИЗИРОВАННЫХ ПОЛИМЕРОВ ДЛЯ ШИН | 2016 |
|
RU2716689C2 |
| СТИРОЛ-БУТАДИЕНОВЫЙ КАУЧУК С ВЫСОКИМ СОДЕРЖАНИЕМ ЗВЕНЬЕВ СТИРОЛА И ВИНИЛА И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2606129C2 |
| ЭЛАСТОМЕРНЫЕ ПОЛИМЕРЫ, МОДИФИЦИРОВАННЫЕ СУЛЬФИДОМ | 2007 |
|
RU2459844C2 |
Настоящее изобретение относится к способу непрерывного получения модифицированных эластомерных полимеров, включающему стадии: (i) обеспечение наличия и полимеризация по меньшей мере одного сопряженного диенового мономера, выбираемого из 1,3-бутадиена, 2-алкил-1,3-бутадиена, 2-метил-1,3-бутадиена, 2,3-диметил-1,3-бутадиена, 1,3-пентадиена, 2,4-гексадиена, 1,3-гексадиена, 1,3-гептадиена, 1,3-октадиена, 2-метил-2,4-пентадиена, циклопентадиена, 2,4-гексадиена и/или 1,3-циклооктадиена, и, необязательно, одного или нескольких ароматических виниловых мономеров при температуре 110°С или ниже в органическом растворителе в присутствии активного инициатора и соединения в соответствии с формулой 1, где О в формуле 1 представляет собой кислород, группы R1 и R2 в формуле 1 каждая независимо представляют собой водород или алкильную группу, предпочтительно - водород или C1-C4 алкильную группу; группы R3, R4, R5, R6, R7 и R8, каждую независимо, выбирают из водорода или алкильной группы, предпочтительно - водорода или С1-С6 алкильной группы; n является целым числом, выбранным из 1, 2, 3 или 4, предпочтительно 1; и где активный инициатор представляет собой инициатор анионной полимеризации, содержащий металлорганическое соединение, имеющее по меньшей мере один атом лития и содержащее неполярную C1-C20 углеводородную группу, или их смесь, а молярное отношение соединения в соответствии с формулой 1 к активному инициатору составляет от 0,15 до 10; с получением живого анионного полимера; и (ii) добавление первого количества соединения в соответствии с формулой 2 к живому анионному полимеру в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 2 к активному инициатору составляло от 0,21 или более, где S в формуле 2 представляет собой атом серы, Si представляет собой кремний, О представляет собой кислород; группы R каждую независимо выбирают из алкила, арила или аралкила, предпочтительно - из C1-C10 алкила, C7-C17 аралкила или C6-C16 арила; группы R′, R″ и R′″ независимо друг от друга выбирают из алкила, аралкила или арила, предпочтительно - из C1-С10 алкила, C6-C16 арила или С7-С17 аралкила; А представляет собой двухвалентную гидрокарбильную группу, предпочтительно - С1-С20 алкилен или С7-С25 аралкилен. Настоящее изобретение также относится к эластомерным полимерам, которые могут быть получены в соответствии со способом, указанным выше. Также настоящее изобретение относится к композиции, содержащей эластомерный полимер, к способу получения сшитого эластомерного полимера, к сшитым эластомерным полимерам, получаемым таким способом, а также к композии, содержащей сшитый эластомерный полимер и изделию, содержащей указанные выше композиции. Технический результат заключается в получении эластомерного полимера непрерывным способом с улучшенными технологическими характеристиками, такими как сопротивление к истиранию, скольжению и качению.7 н. и 8 з.п. ф-лы, 5 табл., 11 пр.
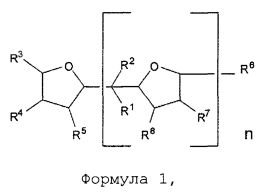
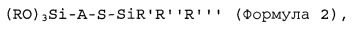
1. Способ непрерывного получения эластомерного полимера, включающий следующие стадии:
(i) обеспечение наличия и полимеризация по меньшей мере одного сопряженного диенового мономера, выбираемого из 1,3-бутадиена, 2-алкил-1,3-бутадиена, 2-метил-1,3-бутадиена, 2,3-диметил-1,3-бутадиена, 1,3-пентадиена, 2,4-гексадиена, 1,3-гексадиена, 1,3-гептадиена, 1,3-октадиена, 2-метил-2,4-пентадиена, циклопентадиена, 2,4-гексадиена и/или 1,3-циклооктадиена, и, необязательно, одного или нескольких ароматических виниловых мономеров при температуре 110°С или ниже в органическом растворителе в присутствии активного инициатора и соединения в соответствии с формулой 1:
где О в формуле 1 представляет собой кислород, группы R1 и R2 в формуле 1 каждая независимо представляют собой водород или алкильную группу, предпочтительно - водород или C1-C4 алкильную группу; группы R3, R4, R5, R6, R7 и R8, каждую независимо, выбирают из водорода или алкильной группы, предпочтительно - водорода или С1-С6 алкильной группы; n является целым числом, выбранным из 1, 2, 3 или 4, предпочтительно 1;
и где активный инициатор представляет собой инициатор анионной полимеризации, содержащий металлорганическое соединение, имеющее по меньшей мере один атом лития и содержащее неполярную C1-C20 углеводородную группу, или их смесь, а молярное отношение соединения в соответствии с формулой 1 к активному инициатору составляет от 0,15 до 10;
с получением живого анионного полимера; и
(ii) добавление первого количества соединения в соответствии с формулой 2 к живому анионному полимеру в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 2 к активному инициатору составляло от 0,21 или более
где S в формуле 2 представляет собой атом серы, Si представляет собой кремний, О представляет собой кислород; группы R каждую независимо выбирают из алкила, арила или аралкила, предпочтительно - из C1-C10 алкила, C7-C17 аралкила или C6-C16 арила; группы R′, R″ и R′″ независимо друг от друга выбирают из алкила, аралкила или арила, предпочтительно - из C1-С10 алкила, C6-C16 арила или С7-С17 аралкила; А представляет собой двухвалентную гидрокарбильную группу, предпочтительно - С1-С20 алкилен или С7-С25 аралкилен.
2. Способ по п. 1, отличающийся тем, что стадия (ii) добавления соединения Формулы 2 к живому анионному полимеру начинается при конверсии мономера в диапазоне от 85 до 100 процентов по массе в расчете на количество намеченного мономера.
3. Способ по п. 1 или 2, отличающийся тем, что способ включает дополнительную стадию (iii):
(iii) последовательного добавления второго количества соединения в соответствии с формулой 2 и/или добавления соединения в соответствии с формулой 3 в таком количестве, чтобы молярное отношение соединения в соответствии с формулой 2 и/или формулой 3, добавляемых на стадии (iii), к активному инициатору составляло от 0,21 или более
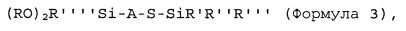
где группы R в формуле 3 независимо выбирают из алкильных, аралкильных или арильных групп, предпочтительно - из C1-С10 алкила, C7-C17 аралкила или C6-C16 арила; группы R′, R″ и R′″ независимо выбирают из алкила, аралкила или арила, предпочтительно - из C1-C10 алкила, C6-C16 арила или С7-С17 аралкила; группу R″″ выбирают из алкила, аралкила или арила, предпочтительно - из C1-C10 алкила, C6-C16 арила или C7-C17 аралкила; А представляет собой двухвалентную гидрокарбильную группу, предпочтительно - С1-С20 алкилен или С7-С25 аралкилен.
4. Способ по п. 1 или 2, отличающийся тем, что органический растворитель выбирают из одного или более соединений из числа бутена, бутана, пентана, циклогексана, гексана, гептана, октана, бензола и толуола, предпочтительно - из одного или более из бутана, пентана, циклогексана, гексана и гептана.
5. Способ по п. 1 или 2, отличающийся тем, что соединение в соответствии с формулой 1 представляет собой ди(тетрагидрофурил)пропан.
6. Эластомерный полимер, получаемый по любому из предшествующих пунктов.
7. Композиция, содержащая эластомерный полимер по п. 6.
8. Композиция по п. 7, дополнительно содержащая масло и/или наполнитель.
9. Композиция по п. 7 или 8, дополнительно содержащая вулканизирующий агент.
10. Композиция по п. 7 или п. 8, отличающаяся тем, что наполнитель выбирают из одного или более веществ из числа диоксида кремния и технического углерода.
11. Способ получения сшитого эластомерного полимера, включающий стадию (iv):
(iv) добавления вулканизирующего агента к эластомерному полимеру по п. 6 или к композиции по любому из пп. 7-10; и сшивания эластомерного полимера.
12. Сшитый эластомерный полимер, получаемый по п. 11.
13. Композиция, содержащая сшитый эластомерный полимер по п. 12.
14. Изделие, содержащее композицию по любому из пп. 7-10 или 13.
15. Изделие по п. 14, отличающееся тем, что изделие представляет собой протектор шины, боковину шины, конвейерную ленту, уплотнитель или рукав.
| WO 2007047943 A2 26.04.2007;WO 2011157742 A1 22.12.2011 | |||
| US 6229036 A1 08.05.2001 | |||
| EP 1505087 A1 09.02.2005 | |||
| СПОСОБ ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО ПОЛИМЕРА, МОДИФИЦИРОВАННЫЙ ПОЛИМЕР, ПОЛУЧЕННЫЙ ПО ДАННОМУ СПОСОБУ, И КАУЧУКОВАЯ КОМПОЗИЦИЯ, ЕГО СОДЕРЖАЩАЯ | 2007 |
|
RU2440384C2 |

