Настоящее изобретение относится к соединениям, композициям и способам модуляции in vitro и in vivo процессов, опосредуемых молекулами адгезии клеток. Описанные малые молекулы включают в себя 2,4,6-тригидроксифенильные субъединицы и эффективно модулируют функции, опосредуемые молекулами адгезии клеток.
Опосредуемые молекулами адгезии клеток функции являются частью комплексного каскада, приводящего к миграции циркулирующих белых кровяных клеток (лейкоцитов) из кровотока в окружающую ткань (трансмиграция). Физиологически трансмиграция лейкоцитов является критически важной для гомеостаза и иммунологического надзора или контроля в живых организмах, включая людей. Лимфоциты, например, конституитивно выходят из кровотока в лимфатические ткани для того, чтобы контролировать вредные антигены. Однако при патологических случаях, например локальном или системном воспалении и/или повреждении васкулярной системы, регуляция этого фундаментального процесса нарушается, по меньшей мере, частично вследствие повышенной поверхностной экспрессии Е- и Р-селектина. Следовательно, чрезмерная трансмиграция лейкоцитов приводит к патологическому клеточному инфильтрату с последующим повреждением ткани при некоторых клинически уместных условиях. Патологические состояния, такие как острое повреждение легких (ALI), острый респираторный дистресс-синдром (ARDS), бронхиальная астма (астма), хроническое обструктивное заболевание легких (COPD), псориаз, ревматоидный артрит и сепсис, все связаны с воспалением ткани, индуцированным и поддерживаемым патологически активированными лейкоцитами, инфильтрующими соответствующую ткань. Кроме того, инфильтрация эксагрегированных лейкоцитов способствует патогенезу ишемического-реперфузионного повреждения (IRI), связанного с трансплантацией органа, экстракорпоральным кровообращением или подкожной внутрипросветной ангиопластикой.
Для трансмиграции лейкоциты должны связываться со стенкой васкулярного эндотелия, чтобы диффундировать через клеточную стенку капилляра в окружающую ткань. Следовательно, лейкоциты должны скользить на клеточной оболочке и затем прилипать к клеточной оболочке эндотелия (начальный процесс скольжения или «связывания»). Это основное событие в трансмиграции опосредуется молекулами адгезии клеток семейства селектинов. Помимо непосредственного связывания с эндотелием лейкоциты могут прилипать к другим лейкоцитам, частицам-лейкоцитам, тромбоцитам или образованным из тромбоцитов частицам, которые уже прилипли к эндотелию.
Семейство селектинов молекул адгезии состоит из трех структурно-родственных, кальцийзависимых, связывающих углеводы белков клеточной поверхности, селектинов Е, P и L. Е-селектин экспрессируется только на воспаленном эндотелии, Р-селектин экспрессируется на воспаленном эндотелии, а также на тромбоцитах и L-селектин экспрессируется на лейкоцитах. Селектины состоят из аминоконцевого лектинового домена, домена, подобного эпидермальному фактору роста (EGF), изменяемого числа повторов, относящихся к рецептору комплемента, гидрофобного трансмембранного домена и С-концевого цитоплазматического домена. Предполагается, что связывающие взаимодействия, приводящие к адгезии лейкоцитов, опосредуются контактированием лектинового домена селектинов и лигандов различных углеводов на поверхности лейкоцитов. Все три селектина могут связываться с низкой аффинностью с углеводом сиалил-Lewisx(sLex), гликозильной частью, присутствующей на поверхности большинства лейкоцитов. Структурно-родственную гликозильную часть, сиалил-Lewisa(sLea), преимущественно обнаруживают на поверхности раковых клеток [K. Okazaki et al., J. Surg. Res., 1998, 78(1). 78-84; R. P. McEver et al., Glycoconjugate Journal, 1997, 14(5), 585-591]. В случае Р-селектина описан гликопротеиновый лиганд с явной высокой аффинностью [R.P. McEver, R.D. Cummings, J.Clin. Invest., 1997, 100, 485-492], так называемый гликопротеиновый лиганд-1 Р-селектина (PSGL-1), который способствует связыванию селектина с высокой аффинностью его sLex-частью, а также частями его пептидных компонентов, в особенности остатками сульфатированного тирозина [R.P. McEver, Ernst Schering Res. Found. Workshop, 2004, 44, 137-147]. PSGL-1 является одним из наиболее важных лигандов селектинов, связывающихся с самой высокой аффинностью с Р-селектином, но он связывается также с Е- и L-селектином [G. Constantin; Drug News Perspect; 2004; 17(9); 579-586]. Он является гомодимерным сиаломуцином, преимущественно экспрессируемым на лейкоцитах.
При воспалительных заболеваниях разрегулированная трансмиграция, по меньшей мере, частично опосредуется вследствие повышенной экспрессии Е- и Р-селектина на поверхности клеток. В противоположность их низкой базальной экспрессии экспрессия Е- и Р-селектина во время воспаления регулируется возрастающим образом, что приводит к значительному рекрутменту лейкоцитов в воспаленную ткань. Хотя для борьбы с инфекцией требуется опосредуемая селектином адгезия клеток, имеют место различные ситуации, в которых такая адгезия клеток является нежелательной или избыточной и приводит к серьезному повреждению ткани вместо заживления. В случае многих острых, а также хронических воспалительных нарушений [например, астмы, хронического обструктивного заболевания легких (COPD), псориаза и т.д.] показана связь между инфильтрацией активированных лейкоцитов в ткань одновременно с заметным повышением экспрессии в ткани соответствующих молекул адгезии, конкретно Е- и Р-селектина [Muller et al., J. Pathol, 2002, 198(2), 270-275; Di Stefano et al., Am. J. Respir. Crit. Care. Med., 1994, 149(3) 803-810; Terajima et al., Arch. Dermatol. Res., 1998, 290, 246-252].
Инфильтрация лейкоцитов может также играть роль в симптомах воспаления во время трансплантации и отторжения трансплантата. Кроме того, процесс свертывания крови дополнительно стимулируется связыванием лейкоцит-лейкоцит и лейкоцит-тромбоцит, которое имеет место, поскольку лейкоциты имеют как L-селектин, так и его соответствующий лиганд PSGL-1, и поэтому они могут взаимодействовать друг с другом через PSGL-1 и могут также связываться с тромбоцитами, которые содержат Р-селектин.
Следовательно, модуляция опосредуемой селектином адгезии клеток и других опосредуемых селектином функций, например активация лейкоцитов, предоставляет перспективную возможность препятствовать каскаду воспаления и останавливать его на очень ранней стадии. Являющиеся антагонистами селектина молекулы малого размера должны модулировать все три селектина одновременно как антагонисты пан-селектина, чтобы преодолеть различия между этими селектинами [M. Sperandio et al., Vascular Disease Prevention, 2004, 1, 185-195].
Помимо sLex/sLea природный, имеющий высокую аффинность лиганд PSGL-1 является другой темплатной структурой для разработки имеющих малые молекулы антагонистов селектинов. По сравнению с sLex/sLea PSGL-1 проявляет высокую аффинность для всех трех селектинов. Поэтому для обнаружения и детектирования новых лекарственных средств в виде малых молекул, которые конкурируют с PSGL-1 и подобными PSGL-1 лигандами, перспективной стратегией является разработка нового класса эффективных антагонистов пан-селектина для лечения воспалительных нарушений. Антагонисты селектина можно разработать с применением селектинов, а также с применением лиганда, подобного PSGL-1, в качестве темплатной структуры, поскольку предполагается, что они модулируют связывание между селектинами и PSGL-1 или другими лигандами с одинаковыми мотивами связывания.
Новые антагонисты селектинов, имеющие малые молекулы, могут встретиться с определенными требованиями для того, чтобы быть подобными лекарственным средствам и иметь потенциальную пероральную биологическую доступность. Термин подобие лекарственному средству описан в литературе [Lipinski; Adv. Drug Dev. Rev., 1997, 23, 3-25]. Предполагается, что, помимо других молекулярных свойств, пассивно транспортируемые молекулы имеют среднюю относительную молекулярную массу меньше чем 500, чтобы быть подобным лекарственному средству. Согласно этим правилам обычно соединения с относительной молекулярной массой меньше 500 или немного выше 500 считают соединениями с малыми молекулами. Маловероятно, что соединения с относительной молекулярной массой выше 500 являются перорально биологически доступными. Присутствие имеющих высокую полярность углеводных частей или пептидного компонента также не находится в соответствии с концепцией подобия лекарственным средствам [H. Ulbrich et al., Trends Pharmacol. Sci, 2003, 24(12), 640-647; D. Slee et al., J. Med. Chem., 2001, 44, 2094-2107]. То же самое является причиной разработки непригодных лекарственных средств на основе антител, поскольку они являются полипептидами, и поэтому пероральное введение является проблематичным. Кроме того, требуемые соединения должны быть стабильными во время прохождения через желудочно-кишечный тракт, так чтобы они могли усваиваться/абсорбироваться позднее клетками тонкой кишки. Это не имеет место в случае большинства гликозидных молекул и пептидных структур.
Были проведены различные исследования для разработки имеющих низкую молекулярную массу соединений с модулирующим действием на опосредуемые селектинами процессы. Эти соединения включают в себя дисалицилаты и С-гликозиды на основе дисалицилатов [WO 99/29706], бензиламиносульфоновые кислоты [WO 03/097658], дигликозилированные 1,2-диолы [WO 97/01569], замещенные 5-членные гетероциклы [WO 00/33836], маннопиранозилоксифенилбензойные кислоты [EP 0758243 В1], соединения на основе пиперазина (US 6432957В1], пептиды, являющиеся производными галловой кислоты [WO 2004/018502], производные галловой кислоты [C. C. M. Appeldoom et al., Circulation 2005, 111, 106-112; EP 1481669A1] и хинной кислоты [N. Kaila et al., J. Med. Chem. 2005, 48, 4346-4357]. Однако ни одно из этих соединений, создающих антагонизм селектинам, до сих пор не прошло успешно клинические испытания [S. J. Romano, Treat. Respir Med 2005, 4(2), 85-94; M. P. Schon, Therapeutics and Clinical Risk Management, 2005, 1(3), 201-208]. Это является результатом того факта, что многие из этих структур были разработаны на основе темплата sLex с низкой эффективностью. Возможно, что поэтому sLex-имитирующие структуры обнаруживают низкую эффективность. Другие соединения проявляют специфичность против различных членов семейства селектинов, но, проявляя антагонизм только для выбранных селектинов, могут не проявлять антагонизм для других селектинов [M. P. Schon, Therapeutics and Clinical Risk Management, 2005, 1(3), 201-208]. Кроме того, большинство соединений, разработанных до сих пор, имеют высокие молекулярные массы и часто содержат углеводы и/или пептиды, что придает им склонность к деградации и модификации под действием пептидаз и/или гликозидаз. Содержащие углеводы структуры имеют дополнительные недостатки, такие как высокая степень хиральности, аномерности и низкая вероятность переноса через липидные бислои. Подобные недостатки известны для содержащих пептиды соединений. Некоторые другие соединения, разработанные для создания антагонизма для опосредуемых селектинами процессов, содержат структуры пирогаллола и катехина. Эти структуры имеют склонность к процессам окисления [Kumamoto M. et al., Biosci. Biotechnol. Biochem., 2001, 65(1), 126-132], что делает фармацевтическую разработку этих соединений трудной. Кроме того, известно, что соединения со структурами пирогаллола, такие как галловая кислота, являются цитотоксическими [E. Sergediene et al., FEBS Letters, 1999, 462, 392-396] и индуцируют апоптоз [K. Satoh et al., Anticancer Research, 1997, 17, 2487-2490; N. Sakaguchi et al., Biochemical Pharmacology, 1998, 55, 1973-1981]. Основным соединением в области антагонистов селектинов является бимосиамоза [S. J. Romano, Treat. Respir Med 2005, 4(2), 85-94]. В настоящее время бимосиамоза [D. Bock et al., New Drugs, 2003, D04, 28, p.28; EP 0840606 B1] является наиболее продвинутым соединением в клинических исследованиях. Последние исследования подтверждают гипотезу, что бимосиамозу можно рассматривать как миметик PSGL-1 [E. Aydt, G. Wolff; Pathobiology; 2002-2003; 70; 297-301]. Это отличает бимосиамозу от других антагонистов селектинов. Однако она является имеющим высокую молекулярную массу соединением с углеводной структурой. По-видимому, у антагониста пан-селектина бимосиамозы отсутствует пероральная биологическая доступность. Некоторые наблюдения указывают на то, что бимосиамоза проявляет хорошую аффинность для Р-селектина и умеренную аффинность для Е- и L-селектина.
Имеется острая лекарственная потребность в новых, очень сильнодействующих антагонистах пан-селектина, которые модулируют опосредуемую селектином функцию, например селектинзависимую адгезию клеток, и в разработке способов, применяющих такие соединения для модуляции состояний, связанных с взаимодействием селектин-лиганд. Большинство из доступных противовоспалительных фармацевтических средств, которые являются доступными на рынке, включают в себя главным образом кортикостероиды или NSAID (нестероидные противовоспалительные лекарственные средства), имеющие несколько серьезных отрицательных/побочных действий и имеющие в качестве мишени различные стадии воспалительного каскада. В отличие от этого модуляция функции селектина является терапевтической концепцией, вмешивающейся в воспалительный каскад на очень ранней стадии. Почти все перспективные антагонисты селектинов до сих пор не стали продаваемыми лекарственными средствами главным образом вследствие низкой эффективности и/или высокой молекулярной массы, которая вызывает проблемы в их поведении абсорбция-распределение-метаболизм-экскреция (ADME), и тем самым в пероральной биологической доступности, требуемой для лечения большинства воспалительных нарушений, подобных ревматоидному артриту, септическому шоку, атеросклерозу, повреждении при реперфузии и многим другим.
Задачей изобретения является создание новых молекул небольшого размера, особенно негликозилированных/негликозидных и непептидных соединений, которые способны эффективно создавать антагонизм опосредуемым селектинами процессам и которые имеют меньше отрицательных побочных действий во время их применения, чем соединения известного уровня техники.
В отличие от большинства sLeX-имитирующих соединений, разработанных в данной области, соединения данного изобретения не восприимчивы к действию гликозидаз или пептидаз. Большинство из антагонистов селектина, разработанных до сих пор, структурно и биологически основаны на свойствах sLex или sLea. Поэтому эти полученные соединения проявляли низкую биологическую активность, подобную их темплатным структурам. Данное изобретение, однако, предоставляет новые, сильнодействующие, малые и подобные лекарственным средствам антагонисты пан-селектина, которые изобретены на основе биологических анализов in vitro, имитирующих PSGL-1 и подобные PSGL-1 лиганды или любые лиганды, имеющие sLex или sLea и тирозинсульфатные мотивы [N. V. Bovin; Biochem Soc Symp.; 2002;(69): 143-60. N. V. Bovin; Glycoconj. J; 1998; 15(5); 431-46. T.V. Pochechueva et al.; Bioorg Med Chem Lett; 2003; 13(10); 1709-12. G. Weitz-Schmidt et al.; Anal. Biochem.; 1996; 238; 184-190].
Настоящее изобретение относится к фармацевтическим композициям, включающим, по меньшей мере, одно соединение, имеющее общую структуру формулы (I), и фармацевтически приемлемый носитель, который является пригодным в медицине
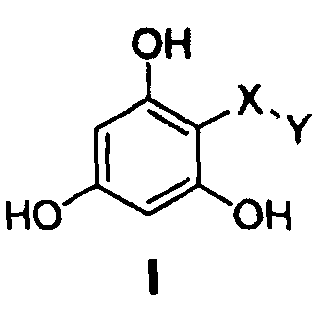
где символы и заместители имеют нижеследующие значения:
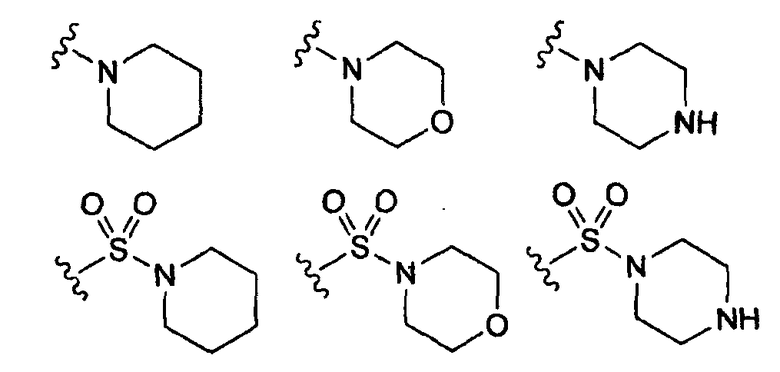
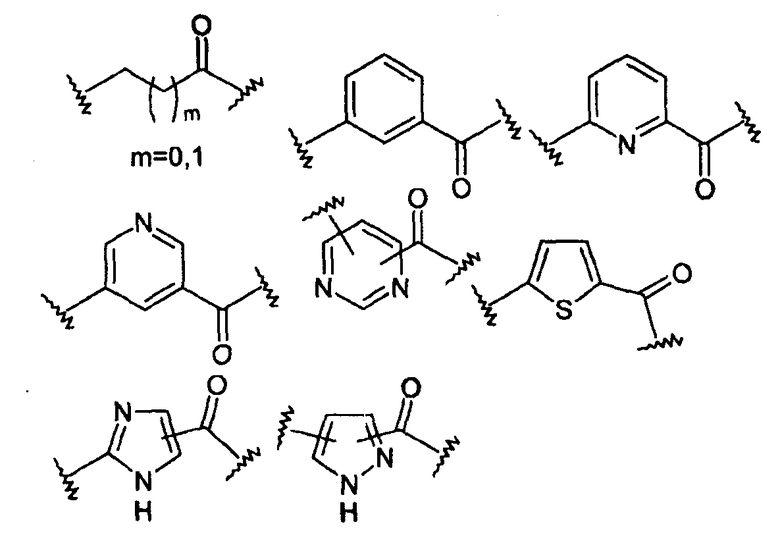
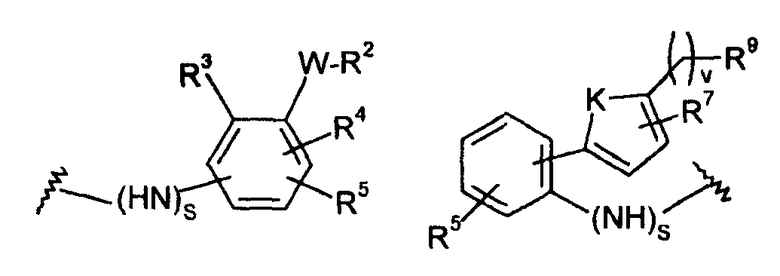
-Х- представляет собой
(а)
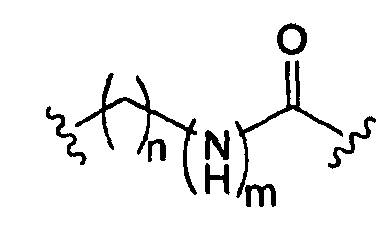
где m равно 0, 1; n равно целому числу от 1 до 3;
(b)
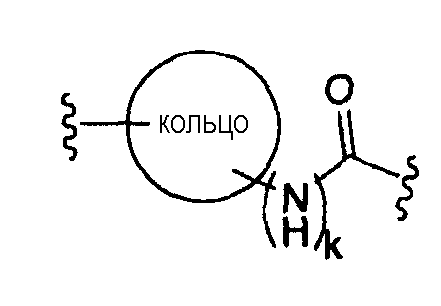
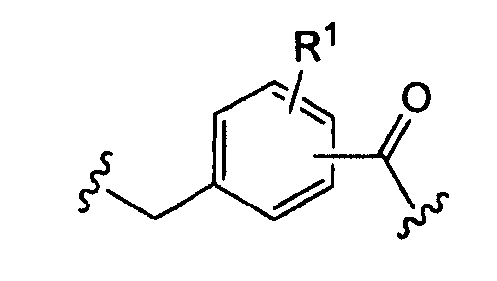
где «кольцо» представляет собой
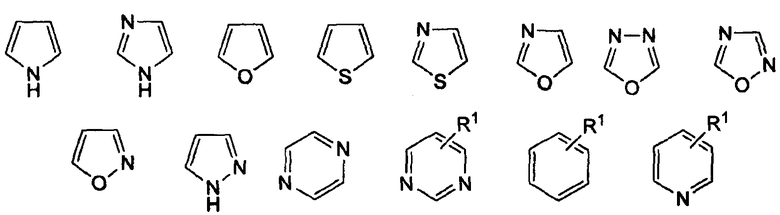
в котором R1 представляет собой Н, NO2, CF3, F, Cl, Br, I, CN, CH3, NH2, NHалкил, NHарил, NНацил и k равно 0, 1;
(с)
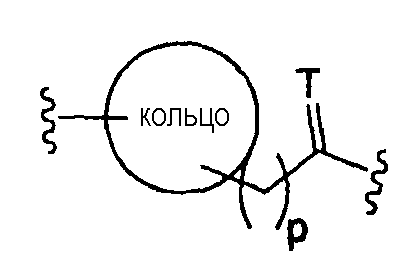
где Т представляет собой О, S или [H,H]; р равно 0, 1, 2;
(d)
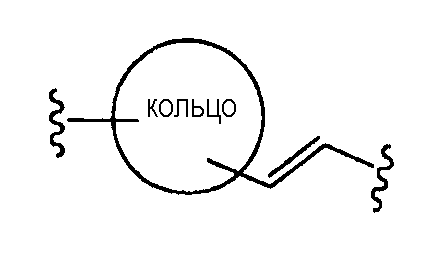
где двойная связь имеет Е- или Z-конфигурацию;
(е)
(f)
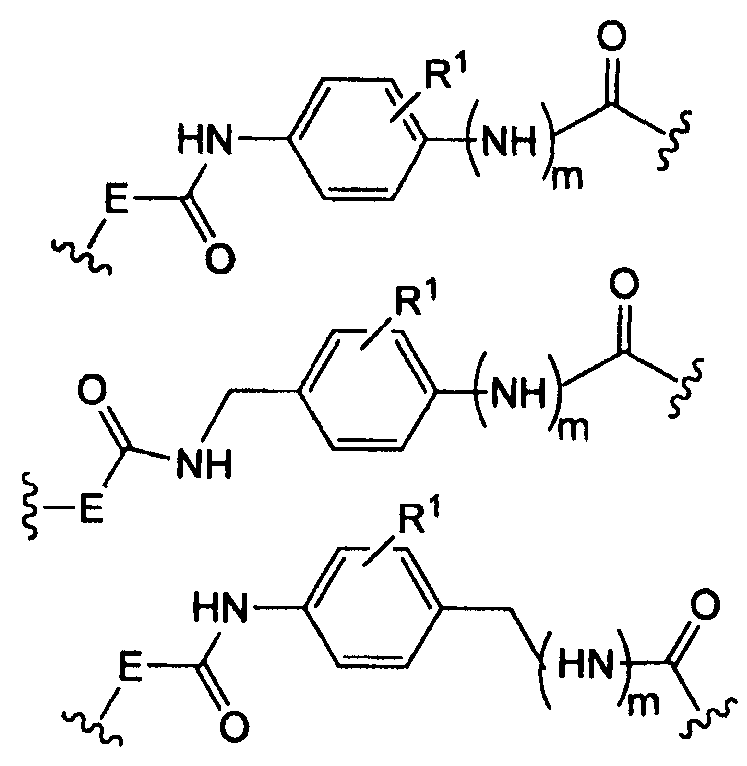
где -Е- представляет собой -(СН2-)qNH- и q равно 0, 1, 2, 3;
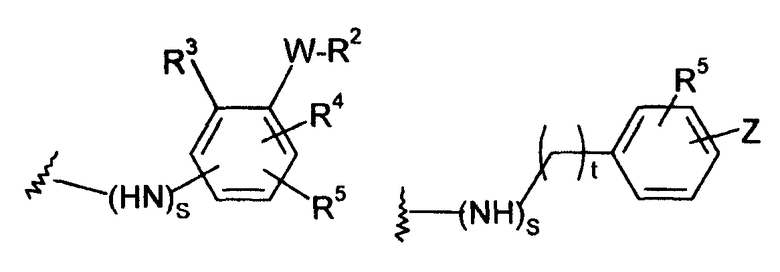
-Y представляет собой
(а)
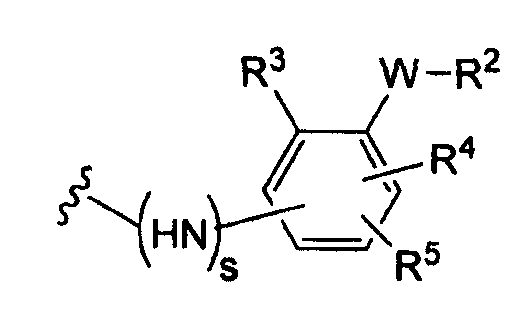
где s равно 0 или 1,
R2 представляет собой CO2H, CO2алкил, CO2арил, CO2NH2, CO2аралкил, SO3H, SO2NH2, PO(OH)2, 1-H-тетразолил-, CHO, COCH3, CH2OH, NH2, NHалкил, N(алкил)алкил', OCH3, CH2OCH3, SH, F, Cl, Br, I, CH3, CH2CH3, CN, CF3;
R3 независимо от R2 представляет собой H, CH3, CH2CH3, CF3, F, Cl, Br, I, CN, NO2 и
R4 независимо от R2 и R3 представляет собой H, CH3, CH2CH3, CF3, F, Cl, Br, I, CN, NO2, R2,
R5 представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2
и -W- представляет собой -(CH2-)V, цис-СН=СН- или транс-СН=СН-, где v равно 0, 1, 2,
в случае, когда -W- представляет собой цис-CH=CH- или транс-CH=CH-, R2 не должен быть NH2 или SH;
(b)
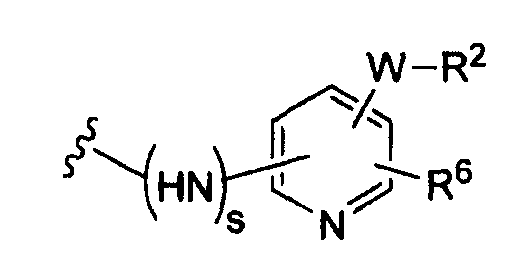
где R6 независимо от R2 представляет собой Н, F, Cl, Me, трет-Bu, CN, NH2;
(c)
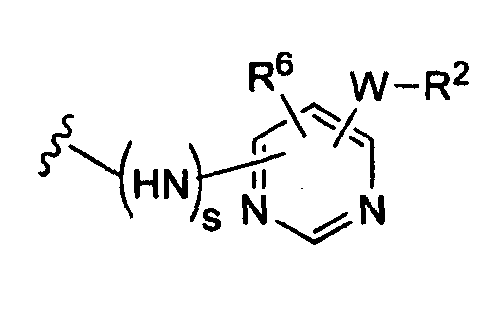
(d)
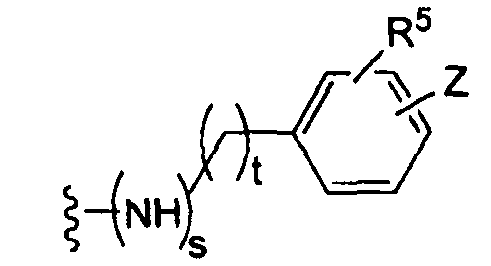
(е)
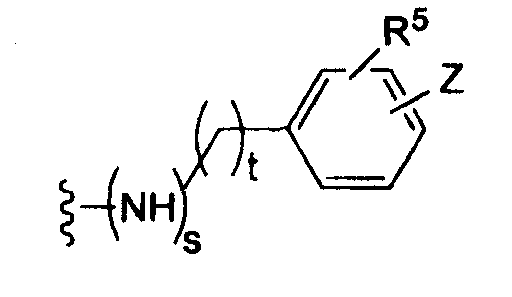
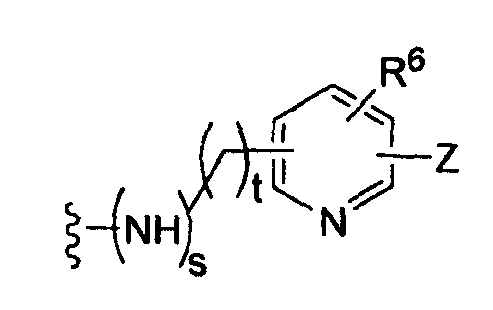
где t равно 0, 1, 2;
(f)
(g)
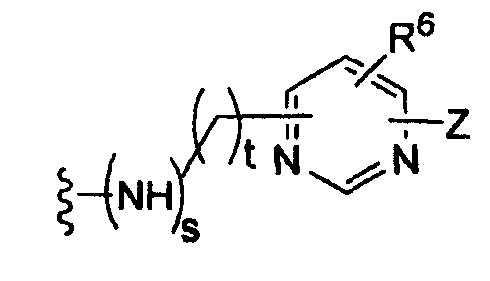
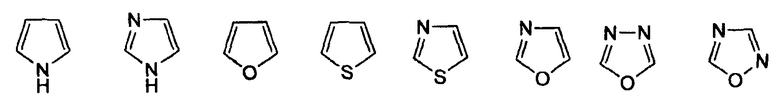
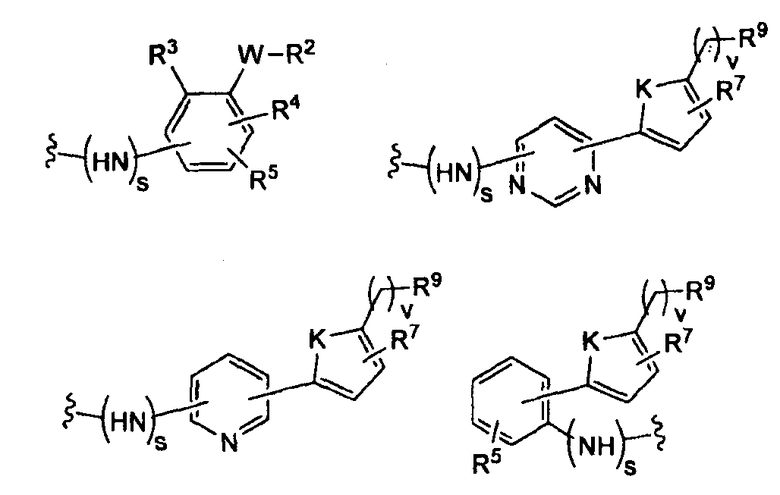
-Z представляет собой
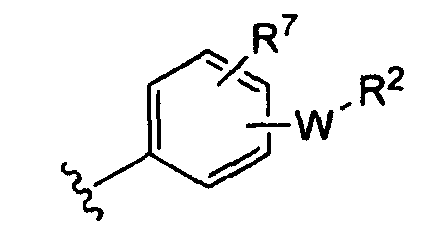
(i)
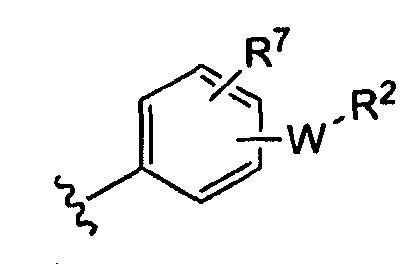
R7 независимо от R2 представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2,
(ii)
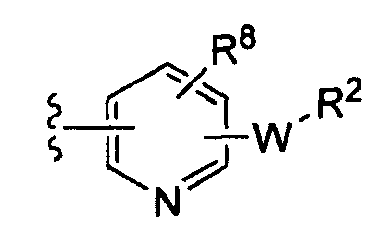
R8 независимо от R2 представляет собой H, F, Cl, Me, трет-Bu, CN, NH2,
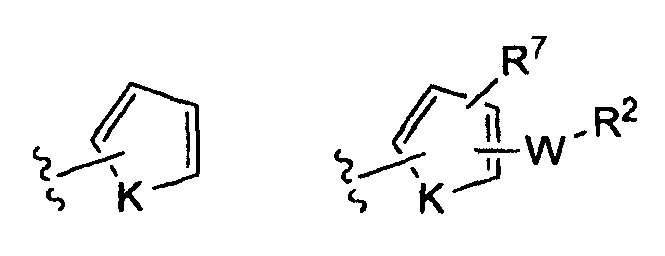
(iii)
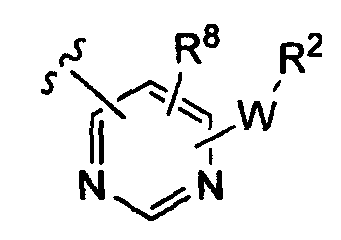
(iv)
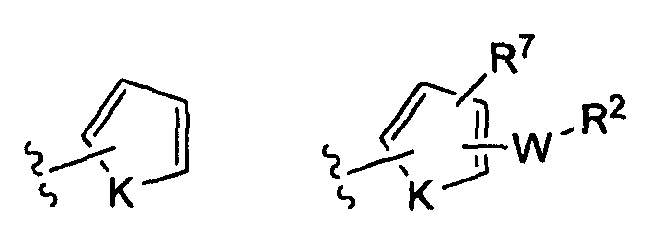
где K представляет собой NH, NMe, O, S,
(v)
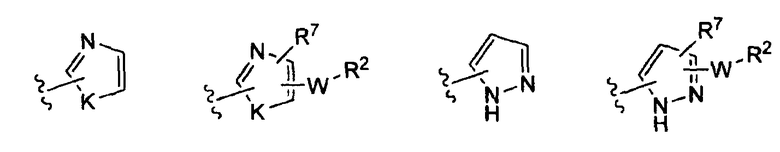
(vi)
(vii)
-W-R2,
или фармацевтически приемлемые соли, сложные эфиры или амиды и пролекарства вышеуказанных соединений формулы (I).
В следующем варианте осуществления изобретение относится к фармацевтическим композициям, включающим, по меньшей мере, одно соединение формулы (I) и фармацевтически приемлемый носитель, который является пригодным в медицине,
где символы и заместители имеют нижеследующие значения:
-Х- представляет собой
(а)
где m равно 0, 1; n равно целому числу от 1 до 3;
(b)
где «кольцо» представляет собой
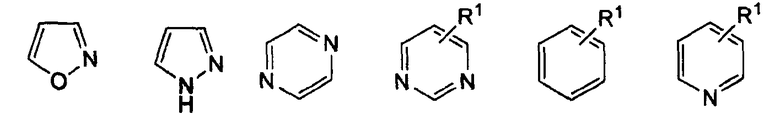
в котором R1 представляет собой Н, NO2, CF3, F, Cl, Br, I, CN, CH3, NH2, NHалкил, NHарил, NНацил и k равно 0, 1;
(с)
где Т представляет собой О, S или [H,H]; р равно 0, 1, 2;
-Y представляет собой
(а)
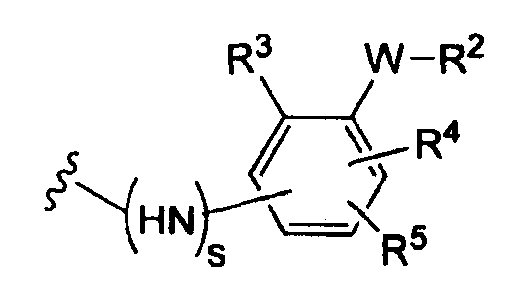
где s равно 0 или 1,
R2 представляет собой CO2H, CO2алкил, CO2арил, CO2NH2, CO2аралкил, SO3H, SO2NH2, PO(OH)2, 1-H-тетразолил-, CHO, COCH3, CH2OH, NH2, NHалкил, N(алкил)алкил', OCH3, CH2OCH3, SH, F, Cl, Br, I, CH3, CH2CH3, CN, CF3,
R3 независимо от R2 представляет собой H, CH3, CH2CH3, CF3, F, Cl, Br, I, CN, NO2 и
R4 независимо от R2 и R3 представляет собой H, CH3, CH2CH3, CF3, F, Cl, Br, I, CN, NO2, R2,
R5 представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2
и -W- представляет собой -(CH2-)V, цис-СН=СН- или транс-СН=СН- и v равно 0, 1, 2,
в случае, когда -W- представляет собой цис-CH=CH- или транс-CH=CH-, R2 не должен быть NH2 или SH;
(b)
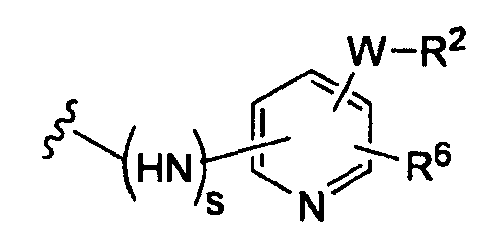
R6 независимо от R2 представляет собой Н, F, Cl, Me, трет-Bu, CN, NH2;
(c)
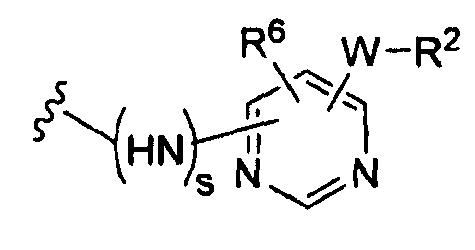
(е)
где t равно 0, 1, 2;
-Z представляет собой
(i)
R7 независимо от R2 представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2;
(iv)
где K представляет собой NH, NMe, O, S;
(v)
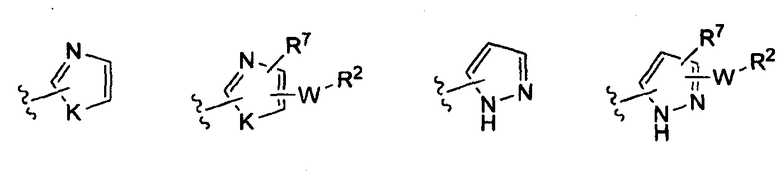
или фармацевтически приемлемые соли, сложные эфиры или амиды и пролекарства вышеуказанных соединений формулы (I).
Предпочтительные фармацевтические композиции включают в себя соединения формулы (II)

где -Y имеет значения, указанные выше, и где -Х'- представляет собой Х(а), Х(b), Х(c) и Х(d), которые имеют значения, указанные выше. Предпочтительными являются соединения формулы (II), где -Х'- представляет собой Х(а) или Х(b).
Следующие предпочтительные фармацевтические композиции включают в себя соединения формулы (А) или (В)
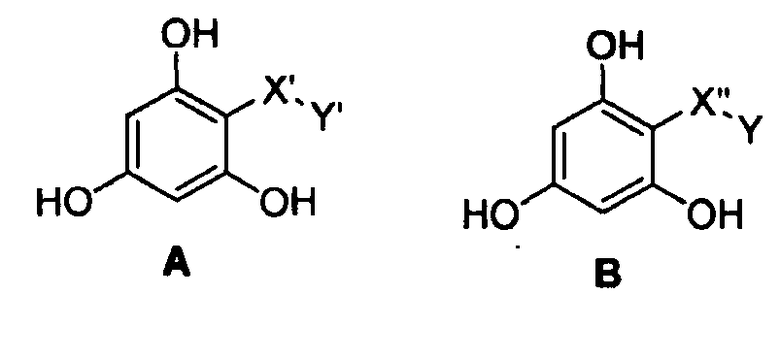
где -Х'- и -Y имеют значения, указанные выше, и где -X”- представляет собой
и где -Y' представляет собой
где все индексы, символы и заместители имеют значения, указанные выше.
В следующем варианте осуществления изобретение относится к фармацевтическим композициям, у которых соединения имеют формулу (А) или (В)
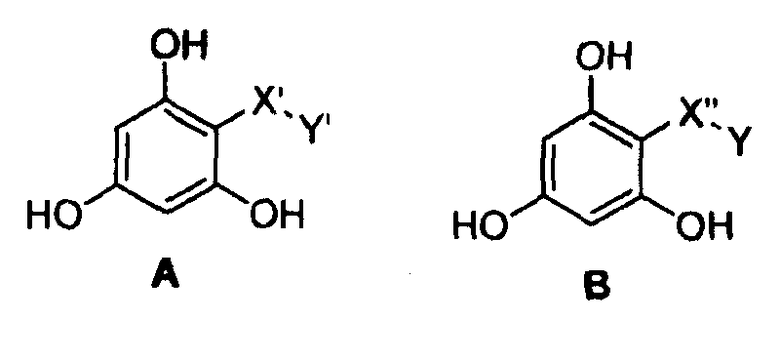
где -Х'- и -Y имеют значения, указанные выше, и где -Х”- представляет собой
и где -Y' представляет собой
где все другие индексы, символы и заместители имеют значения, указанные выше.
Особенно предпочтительные фармацевтические композиции включают в себя соединения формулы (С)
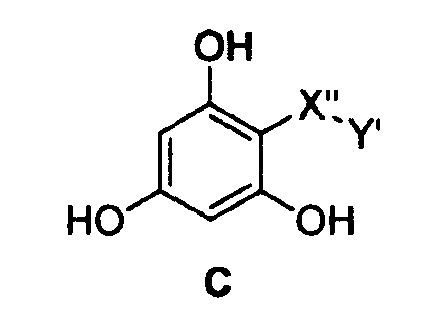
где -X”- и -Y' имеют значения, указанные выше.
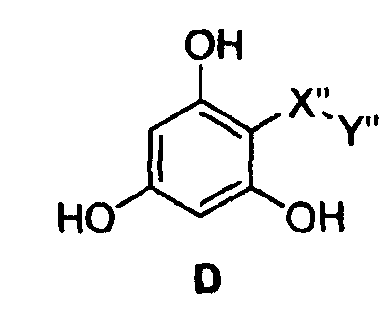
Самые предпочтительные фармацевтические композиции включают в себя соединения формулы (D)
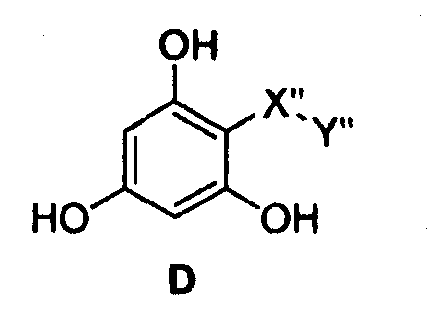
где -Х”- имеет значения, указанные выше, и -Y” представляет собой
где R9 представляет собой CO2H, CO2алкил, CO2арил, CO2NH2, CO2аралкил, CH2SO3H, CH2SO2NH2, CH2PO(OH)2, 1-H-тетразолил, CHO, COCH3, CH2OH, CH2NH2, СН2NHалкил, CH2N(алкил)алкил', CH2OCH3, CH2SH.
В следующем варианте осуществления изобретение относится к фармацевтической композиции, где соединения имеют формулу (D)
где -Х”- имеет значения, указанные выше, и -Y” представляет собой
где R9 представляет собой CO2H, CO2алкил, CO2арил, CO2NH2, CO2аралкил, CH2SO3H, CH2SO2NH2, CH2PO(OH)2, 1-H-тетразолил, CHO, COCH3, CH2OH, CH2NH2, СН2NHалкил, CH2N(алкил)алкил', CH2OCH3, CH2SH,
и все другие индексы, символы и заместители имеют значения, указанные выше.
Эти химические соединения (С) и (D) сами также являются новыми соединениями.
Все соединения, описываемые выше, обладают способностью модулировать адгезию клеток и модулируют связывание, опосредуемое селектином, а также PSGL-1-подобным лигандом. Соединения обладают способностью модулировать взаимодействие селектинов с sLex/sLea, также взаимодействие между селектинами и тирозинсульфатными остатками. Поэтому они являются пригодными для лечения острых и хронических воспалительных нарушений, а также других патологических состояний, в которых играют роль опосредуемые селектином процессы.
Термин «фармацевтический» включает в себя также диагностические применения.
Термин «фармацевтический» включает в себя также профилактические применения для предупреждения патологических состояний, в которых играют роль опосредуемые селектином процессы.
Термин «фармацевтический» включает в себя также применения, в которых соединения настоящего изобретения можно применять в качестве наполнителей для доставки лекарственного средства в орган-мишень для диагностических или терапевтических целей.
Изобретение относится к фармацевтическим композициям, включающим соединения формулы (I) и в предпочтительном варианте формулы (II).
В следующем предпочтительном варианте изобретение относится к фармацевтическим композициям, включающим, по меньшей мере, одно соединение формулы (А) или (В).
В особенно предпочтительном варианте изобретение относится к фармацевтическим композициям, включающим, по меньшей мере, одно соединение (С).
В самом предпочтительном варианте изобретение относится к фармацевтическим композициям, включающим, по меньшей мере, одно соединение (D).
Настоящее изобретение далее относится к способу модуляции связывания Р-селектина, L-селектина или Е-селектина с sLex или sLea и тирозинсульфатными остатками, включающим стадию введения пациенту эффективного количества, по меньшей мере, одного соединения, имеющего структуру формулы (I), для модуляции связывания Р-, Е- или L-селектина с sLex или sLea и тирозинсульфатом. Обнаружено, что соединения, имеющие формулу (I), показанную выше, действуют как модуляторы связывания Е-, Р- или L-селектина.
Применяемые в описании термины «алкил» будут означать одновалентную группу с неразветвленной цепью или разветвленной цепью из 1 или 2 или 3 или 4, или 5 или 6, или 7 или 8, или 9 или 10, или 11 или 12 атомов углерода, включающую в себя, но не ограничивающуюся перечисленным, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил и тому подобное. «Алкилы» не зависят друг от друга и могут быть разными или идентичными.
Термин «арил» будет означать карбоциклические и гетероциклические ароматические группы, включающие в себя, но не ограничивающиеся перечисленным, фенил, 1-нафтил, 2-нафтил, флуоренил, (1,2)-дигидронафтил, инденил, инданил, тиенил, бензотиенил, тиенопиридил и тому подобное.
Термин «аралкил» (называемый также арилалкил) будет означать арильную группу, присоединенную к алкильной группе, и включает в себя, но не ограничивается перечисленным, бензил, 1-нафтилметил, 2-нафтилметил, фторбензил, хлорбензил, бромбензил, иодбензил, алкоксибензил (где «алкокси» означает метокси, этокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси и тому подобное), гидроксибензил, аминобензил, нитробензил, гуанидинобензил, флуоренилметил, фенилметил (бензил), 1-фенилэтил, 2-фенилэтил, 1-нафтилэтил и тому подобное.
Термин «ацил» будет означать -(СНО), или -(С=О)-алкил, или -(С=О)-арил, или -(С=О)-аралкил и включает в себя, но не ограничивается перечисленным, формил, ацетил, н-пропионил, изопропионил, н-бутирил, изобутирил, пивалоил, бензоил, 4-нитробензоил и тому подобное.
Применяемый в описании термин «фармацевтически приемлемые соли, сложные эфиры, амиды и пролекарства» относится к таким карбоксилатным солям, аддитивным солям аминокислот, сложным эфирам, амидам и пролекарствам соединений настоящего изобретения, которые находятся в пределах объема исследовательской лекарственной оценки, являются подходящими для применения в контакте с тканями пациентов без чрезмерной токсичности, раздражения, аллергической реакции и тому подобное, имеют приемлемое отношение польза/риск и являются эффективными для предназначенного для них применения, а также, когда возможно, цвиттерионным формам соединений настоящего изобретения. Термин «соли» относится к относительно нетоксичным аддитивным солям соединений настоящего изобретения неорганических и органических кислот. Эти соли можно получить in situ во время последнего выделения и очистки соединений или отдельно реакцией очищенных соединений в их свободной форме с подходящей неорганической или органической кислотой или основанием и выделением таким образом образованных солей. Репрезентативные соли соединений настоящего изобретения включают в себя гидробромидные, гидрохлоридные, сульфатные, бисульфатные, нитратные, ацетатные, оксалатные, валератные, пальмитатные, стеаратные, лауратные, боратные, бензоатные, лактатные, фосфатные, тозилатные, цитратные, малеатные, фумаратные, сукцинатные, тартратные, нафтилатные, мезилатные, глюкогептаноатные, лактиобионатные, лаурилсульфонатные соли и тому подобное. Они могут включать в себя катионы на основе щелочных и щелочноземельных металлов, таких как натрий, литий, калий, кальций, магний и тому подобное, а также нетоксичные катионы аммония, четвертичного аммония и аминов, включающие в себя, но не ограничивающиеся перечисленным, катионы аммония, тетраметиламмония, тетраэтиламмония, метиламина, диметиламина, триметиламина, триэтиламина, этиламина и тому подобное.
Примеры фармацевтически приемлемых, нетоксичных сложных эфиров соединений данного изобретения включают в себя С1-, С2-, С3-, С4-, С5- и С6-алкиловые сложные эфиры, где алкильная группа имеет неразветвленную или разветвленную цепь. Приемлемые сложные эфиры включают в себя также С5-, С6- и С7-циклоалкиловые сложные эфиры, а также арилалкиловые сложные эфиры, такие как, но без ограничения указанным, бензиловый эфир. С1-, С2-, С3-, С4-, С5- и С6-алкиловые сложные эфиры являются предпочтительными. Сложные эфиры соединений настоящего изобретения можно получить согласно общепринятым методам.
Примеры фармацевтически приемлемых, нетоксичных амидов соединений данного изобретения включают в себя амиды, полученные из аммиака, первичных С1-, С2-, С3-, С4-, С5- и С6-алкиламинов и вторичных С1-, С2-, С3-, С4-, С5- и С6-диалкиламинов, где алкильные группы имеют неразветвленные или разветвленные цепи. В случае вторичных аминов амин может быть также в форме 5- или 6-членного гетероцикла, содержащего один атом азота. Предпочтительными являются амиды, полученные из аммиака, первичных С1-, С2- и С3-алкиламинов и вторичных С1-С2-диалкиламинов. Амиды соединений настоящего изобретения можно получить согласно общепринятым методам.
Термин «пролекарство» относится к одному или нескольким соединениям, которые быстро превращаются in vitro и превращаются из неактивного в активное состояние in vivo с образованием «родительского» соединения указанной выше формулы (I), например, гидролизом в крови или метаболизмом in vivo.
Предполагается также, что фармацевтически активные композиции могут содержать соединение настоящего изобретения или другие соединения, которые модулируют связывание Е-селектина, или Р-селектина, или L-селектина или конкурируют с таким связыванием.
Фармацевтически активные композиции настоящего изобретения включают в себя фармацевтически приемлемый носитель и соединение формулы (I), в результате чего фармацевтически приемлемым носителем может быть также лекарственно подходящая наночастица, дендример, липосома, микропузырек или полиэтиленгликоль (ПЭГ). Фармацевтические композиции настоящего изобретения могут включать в себя одно или несколько соединений, имеющих указанную выше структуру (I), в сочетании с одним или несколькими физиологически приемлемыми носителями, адъювантами или наполнителями, которые сообща называют в описании носителями, для парентеральной инъекции, для перорального введения в твердой или жидкой форме, для ректального или местного введения и тому подобное.
Композиции можно вводить людям и животным либо перорально, ректально, парентерально (внутривенно, внутримышечно, внутрикожно или подкожно), внутриполостным путем, интравагинально, внутрибрюшинно, местно (порошки, мази или капли) либо трансбуккально или ингаляцией (распылением или назальным спреем).
Композиции, подходящие для парентеральной инъекции, могут включать в себя физиологически приемлемые стерильные водные или неводные растворы, стабилизаторы, антиоксиданты, консерванты (например, аскорбиновую кислоту, сульфит натрия, гидросульфит натрия, бензиловый спирт, EDTA), дисперсии, суспензии или эмульсии и стерильные порошки для перевода их в стерильный инъецируемый раствор или дисперсию. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или наполнителей включают в себя воду, этанол, полиол (пропиленгликоль, полиэтиленгликоль, глицерин и тому подобное), их подходящие смеси, растительные масла (такие как оливковое масло или масло канолы) и инъецируемые органические сложные эфиры, такие как этилолеат. Подходящую текучесть можно поддерживать, например, применением покрытия, такого как лецитин, поддерживанием требуемого размера частиц в случае дисперсий и применением поверхностно-активных веществ.
Эти композиции могут содержать также адъюванты, такие как консервирующие, увлажняющие, эмульгирующие и диспергирующие агенты. Предотвращение действий микроорганизмов можно гарантировать различными антибактериальными и антигрибковыми агентами, например парабенами, хлорбутанолом, фенолом, сорбиновой кислотой и тому подобное. Может быть также желательно включение изотонических агентов, например сахаров, хлорида натрия и тому подобное. Пролонгированную абсорбцию инъецируемой фармацевтической формы можно достичь применением агентов, задерживающих абсорбцию, например моностеарата алюминия и желатина.
При желании и для более эффективного распределения соединения можно включать в системы для медленного или регулируемого по времени высвобождения или для доставки к органу-мишени, такие как полимерные матрицы, липосомы и микросферы. Их можно стерилизовать, например, фильтрованием через задерживающий бактерии фильтр или введением стерилизующих агентов в форме стерильной воды или некоторых других стерильных инъецируемых сред непосредственно перед применением.
Твердые лекарственные формы для перорального введения включают в себя капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение или пролекарство смешивают, по меньшей мере, с одним инертным обычным эксципиентом (или носителем), таким как цитрат натрия или дикальцийфосфат, или (i) наполнителями или разбавителями, такими как, например, крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, (ii) связывающими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь, (iii) увлажнителями, такими как, например, глицерин, (iv) дезинтегрирующими агентами, такими как, например, агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые комплексные силикаты и карбонат натрия, (v) замедлители растворения, такие как, например, парафин, (vi) ускорители абсорбции, такие как, например, соединения четвертичного аммония, (vii) смачивающие агенты, такие как, например, цетиловый спирт и моностеарат глицерина, (viii) адсорбенты, такие как, например, каолин и бентонит, и (ix) смазывающие вещества, такие как, например, тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль лекарственные формы могут включать в себя также буферные агенты.
Твердые композиции подобного типа можно также применять в качестве наполнителей в мягких и твердых наполняемых желатиновых капсулах с применением эксципиентов, таких как лактоза или молочные сахара, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное. Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, можно получить с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие, хорошо известные в данной области. Они могут содержать агенты, придающие композиции непрозрачность, и могут быть также такими композициями, которые высвобождают активное соединение или соединения в определенной части кишечного тракта замедленным образом. Примерами «заделывающих» композиций, которые можно применять, являются полимерные вещества и воски. Активные соединения могут быть также, если это приемлемо, в микрокапсулированной форме с одним или несколькими указанными выше эксципиентами.
Жидкие лекарственные формы для перорального введения включают в себя фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активных соединений жидкие лекарственные формы могут содержать инертные разбавители, обычно применяемые в данной области, такие как вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла, в частности хлопковое масло, арахисовое масло, кукурузное масло, оливковое масло, масло канолы, касторовое масло и кунжутное масло, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и эфиры жирных кислот сорбитана или смеси этих веществ и тому подобное. Помимо таких инертных разбавителей композиции могут включать в себя также адъюванты, такие как увлажняющие агенты, эмульгирующие и суспендирующие агенты, подслащивающие агенты, корригенты и отдушки.
Суспензии, помимо активных соединений, могут содержать суспендирующие агенты, например этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар, трагакант или смеси этих веществ и тому подобное.
Композициями для ректального введения предпочтительно являются суппозитории, которые можно получить смешиванием соединений настоящего изобретения с подходящими нераздражающими эксципиентами или носителями, такими как какао-масло, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при обычной температуре, но жидкими при температуре тела и поэтому плавятся в ректальной или вагинальной полости и высвобождают активный компонент. Лекарственные формы для местного введения соединения данного изобретения включают в себя мази, порошок, спреи и формы для ингаляции.
Активный компонент смешивают в стерильных условиях с физиологически приемлемым носителем и любыми необходимыми консервантами, буферами или пропеллентами, если они могут быть необходимы. Офтальмические препараты, глазные мази, суспензии, порошок и растворы также рассматриваются как включенные в объем данного изобретения.
Соединения настоящего изобретения можно также включить в липосомы или связать с ними или вводить в форме лимосом. Как известно в данной области, липосомы обычно получают из фосфолипидов или других липидных веществ. Липосомы образуют моно- или многослоистые гидратированные жидкие кристаллы, которые диспергируют в водной среде. Можно применять любой нетоксичный, физиологически приемлемый метаболизированный липид, способный образовывать липосомы. Настоящие композиции в форме липосом могут содержать, помимо связывающих селектин антагонистов настоящего изобретения, стабилизаторы, консерванты, эксципиенты и тому подобное. Предпочтительными липидами являются фосфолипиды и фосфатидилхолины (лецитины), как природные, так и синтетические. Методы образования липосом хорошо известны в данной области техники.
Непарентеральные лекарственные формы могут содержать также повышающий биологическую доступность агент (например, модуляторы ферментов, антиоксиданты), подходящий для защиты соединений от разрушения. Действительные уровни доз активного ингредиента в композиции настоящего изобретения можно варьировать так, чтобы получить количество активного ингредиента, которое является эффективным для достижения требуемой терапевтической реакции для конкретной композиции и метода введения. Следовательно, выбранный уровень дозы зависит от требуемого терапевтического действия, от пути введения, от требуемой продолжительности лечения и других факторов. Общая суточная доза соединений данного изобретения, введенная хозяину в виде одной или разделенных доз, может быть в диапазоне до 50 мг на кг массы тела. Стандартные лекарственные композиции могут содержать такие субмногократные количества их, которые можно применять для получения суточной дозы. Должно быть понятно, однако, что определенный уровень дозы для любого конкретного пациента, независимо от того, является ли он человеком или другим животным, будет зависеть от целого ряда факторов, включающих в себя массу тела, общее состояние здоровья, пол, режим питания, время и путь введения, скорости абсорбции и экскреции, комбинацию с другими лекарственными средствами и тяжесть конкретного подвергаемого лечению заболевания.
В частности, соединения настоящего изобретения можно применять для лечения целого ряда заболеваний, относящихся к воспалению и межклеточному распознаванию и адгезии. Например, соединения настоящего изобретения можно вводить пациенту для лечения хронического обструктивного заболевания легких (COPD), острого повреждения легких (ALI), при экстракорпоральном кровообращении, лечения острого респираторного дистресс-синдрома (ARDS), болезни Крона, септического шока, сепсиса, хронических воспалительных заболеваний, таких как псориаз, атопический дерматит и ревматоидный артрит, реперфузионного повреждения, которое имеет место после сердечных приступов, мозговых кровоизлияний, атеросклероза и трансплантации органов, травматического шока, недостаточности деятельности многих органов, аутоиммунных заболеваний, подобных рассеянному склерозу, при подкожной внутрипросветной ангиопластике, лечения астмы и воспалительного заболевания кишечника. В каждом случае эффективное количество соединений настоящего изобретения вводят либо как таковое, либо как часть фармацевтически активной композиции пациенту, нуждающемуся в таком лечении. Понятно также, что пациенту, нуждающемуся в таком введении, можно вводить комбинацию соединений. Соединения настоящего изобретения можно также вводить для лечения других заболеваний, которые связаны с межклеточной адгезией. Поскольку настоящие соединения модулируют связывание Е-селектина, или Р-селектина, или L-селектина, любое заболевание, которое связано с этим взаимодействием, можно потенциально лечить модуляцией такого связывающего взаимодействия.
Помимо обнаружения на некоторых лейкоцитах, sLea обнаружен на различных раковых клетках, в том числе раковых клетках легких и толстой кишки. Было высказано предположение, что адгезия клеток, включающая в себя участие sLea, может принимать участие в метастазировании некоторых раковых заболеваниях и антагонисты связывания sLea могут быть полезными при лечении некоторых форм рака.
Применение активных ингредиентов согласно изобретению в косметических или местных дерматологических композициях с эффективным содержанием активного ингредиента согласно изобретению неожиданно предоставляет возможность осуществления эффективного лечения, а также профилактики старения кожи, вызванного внешними и внутренними факторами.
Изобретение, в частности, относится к применению соединения формулы (I) или его стереоизомерной формы для получения косметической или дерматологической композиции.
Применяемое количество активного соединения или его стереоизомерной формы соответствует количеству, требуемому для получения желаемого результата при применении косметических или дерматологических композиций. Специалист в данной области способен определить это эффективное количество, которое зависит от применяемого производного, индивидуума, которому его наносят, и времени его применения. Для обеспечения порядка величины соединения формулы (I) или его стереоизомерной формы в косметических или дерматологических композициях согласно изобретению соединение формулы (I) или его стереоизомерную форму можно вводить в количестве от 0,001 мас.% до 40 мас.%, предпочтительно 0,005 мас.%-30 мас.% и, более предпочтительно от 0,01 мас.% до 20 мас.%.
Следующий аспект включает в себя косметические композиции, включающие в себя соединение формулы (I) или его стереоизомерную форму и, по меньшей мере, один косметически переносимый компонент, например косметически переносимый компонент для нанесений на кожу.
Количества различных компонентов физиологической среды косметической композиции согласно изобретению являются количествами, обычно применяемыми в рассматриваемых областях. Когда косметической композицией является эмульсия, содержание жирной фазы может составлять от 2 мас.% до 80 мас.% и предпочтительно от 5 мас.% до 50 мас.%, относительно общей массы косметической композиции.
Таким образом, косметическая композиция должна содержать нетоксичную физиологически приемлемую среду, которую можно наносить на кожу человека. Для местного нанесения на кожу косметическая композиция может быть в форме раствора, суспензии, эмульсии или дисперсии с более или менее текучей консистенцией и особенно жидкой или полужидкой консистенцией, полученной диспергированием жирной фазы в водной фазе (О/W) или наоборот (W/O), или же в форме геля. Можно также применять косметическую композицию в форме мусса или в форме спрея или аэрозоля, в таком случае включающую в себя находящийся под давлением пропеллент. Композиции могут быть также в форме лосьона для ухода за волосами, шампуня или кондиционера для волос, жидкого или твердого мыла, косметической маски или пенообразующего крема или геля для мытья волос. Они могут быть также в форме краски для волос или краски для бровей и ресниц.
Косметические композиции изобретения могут также включать в себя один или несколько других ингредиентов, обычно применяемых в рассматриваемых областях и выбранных из добавок к препаратам, например загустители водной фазы или масляной фазы или гелеобразующие агенты, красители, которые растворимы в среде косметической композиции, твердые частицы, такие как минеральные или органические наполнители или пигменты в форме микрочастиц или наночастиц, консерванты, душистые вещества, гидротопы или электролиты, нейтрализующие средства (подкисляющие или подщелачивающие агенты), пропелленты, анионогенные, катионогенные или амфотерные поверхностно-активные вещества, полимеры, в частности водорастворимые или вододиспергируемые анионогенные, неионогенные, катионогенные или амфотерные пленкообразующие полимеры, минеральные или органические соли, хелатирующие агенты; их смеси.
Косметические композиции можно применять для ингибирования микровоспалительного цикла. Поэтому настоящее изобретение относится также к косметическим композициям, включающим соединение формулы (I) или его стереоизомерную форму, которые применяют для косметического лечения или косметической профилактики микровоспалительных состояний.
Косметические композиции, включающие в себя соединение формулы (I) или его стереоизомерную форму, которые применяют для косметического лечения или косметической профилактики старения кожи, вызванной внутренними факторами, также являются предметом настоящего изобретения. Внутренними факторами, ответственными за старение кожи, являются генетически программированные детерминанты, включающие в себя возраст, гормональное состояние и физиологические факторы.
Помимо косметически неактивных ингредиентов косметические композиции настоящего изобретения могут включать в себя также один или несколько косметически активных ингредиентов с благотворным действием на кожу. Поэтому настоящее изобретение относится к косметическим композициям, включающим соединение формулы (I) или его стереоизомерную форму и, по меньшей мере, один дополнительный косметически активный ингредиент, например УФ защитное средство или белки.
Дерматологические композиции, включающие в себя соединение формулы (I) или его стереоизомерную форму и, по меньшей мере, один дерматологически переносимый компонент, например дерматологически переносимый компонент для применения для кожи, также являются предметом изобретения.
Дерматологически переносимые компоненты, которые можно применять для дерматологических композиций, описанных в данном описании, являются идентичными косметически переносимым компонентам, указываемым в данном изобретении.
Следующим вариантом осуществления данного изобретения являются дерматологические композиции, включающие в себя соединение формулы (I) или его стереоизомерную форму, которые применяют для дерматологического лечения, дерматологического диагноза или дерматологической профилактики микровоспалительных состояний.
В частности, изобретение включает в себя дерматологические композиции, включающие в себя соединение формулы (I) или его стереоизомерную форму, которые применяют для дерматологического лечения, дерматологического диагноза или дерматологической профилактики зуда и старения кожи, вызванного внешними факторами. Внешние факторы включают в себя в общем факторы окружающей среды; более конкретно, вызывающее старение кожи воздействие ультрафиолетовых лучей вследствие солнечного облучения, действия света или любого другого излучения, атмосферное загрязнение, раны, инфекции, травматизм, гипоксия, дым сигарет, гормональное состояние как реакция на внутренние факторы, нейропептиды, электромагнитные области, гравитация, образ жизни (например, избыточное потребление алкоголя), периодически повторяющиеся лицевые выражения, положения тела во время сна и физиологические стресс-факторы.
Помимо дерматологически неактивных ингредиентов дерматологические композиции могут включать в себя также дерматологически или фармацевтически активные ингредиенты. Поэтому настоящее изобретение относится также к дерматологическим композициям, включающим соединение формулы (I) или его стереоизомерную форму и, по меньшей мере, один дополнительный дерматологически или фармацевтически активный ингредиент. Дерматологически или фармацевтически активные ингредиенты, которые можно применять для дерматологических композиций, описанных в данном описании, определяют как косметически активные ингредиенты, указанные выше. Дерматологически или фармацевтически активные ингредиенты могут быть идентичными косметически активным ингредиентам, указываемым в данном изобретении.
Другим предметом настоящего изобретения являются дерматологические композиции, включающие в себя соединение формулы (I) или его стереоизомерную форму и, по меньшей мере, один дополнительный дерматологически или фармацевтически активный ингредиент, характеризующийся тем, что его применяют для дерматологического лечения, дерматологического диагноза или дерматологической профилактики микровоспалительных состояний.
В частности, настоящее изобретение относится к дерматологическим композициям, включающим соединение формулы (I) или его стереоизомерную форму и, по меньшей мере, один дополнительный дерматологически или фармацевтически активный ингредиент, характеризующийся тем, что его применяют для дерматологического лечения, дерматологического диагноза или дерматологической профилактики зуда и старения кожи, вызванных внешними факторами.
Старение кожи может быть вызвано комбинацией внутренних и внешних факторов. Поэтому настоящее изобретение относится также к дерматологическим композициям, включающим соединение формулы (I) или его стереоизомерную форму и, по меньшей мере, один дополнительный фармацевтически или косметически активный ингредиент, характеризующийся тем, что его применяют для косметического и дерматологического лечения и косметической и дерматологической профилактики старения кожи, вызванного комбинацией внутренних и внешних факторов.
Другим вариантом осуществления данного изобретения является способ получения косметической композиции смешиванием соединения формулы (I) или его стереоизомерной формы, по меньшей мере, одного косметически переносимого компонента и в конце концов дополнительных косметически активных ингредиентов.
В частности, предметом данного изобретения является способ получения косметической композиции смешиванием соединения формулы (I) или его стереоизомерной формы, по меньшей мере, одного косметически переносимого компонента и в конце концов дополнительных косметически активных ингредиентов, где композиция включает в себя от 0,01 мас.% до 20 мас.% соединения формулы (I) или его стереоизомерной формы в расчете на общую массу композиции.
Следующий аспект относится к способу получения дерматологической композиции смешиванием соединения формулы (I) или его стереоизомерной формы, по меньшей мере, одного дерматологически переносимого компонента и в конце концов дополнительных косметически активных ингредиентов.
Многие из соединений настоящего изобретения можно синтезировать согласно следующим общим синтетическим схемам.
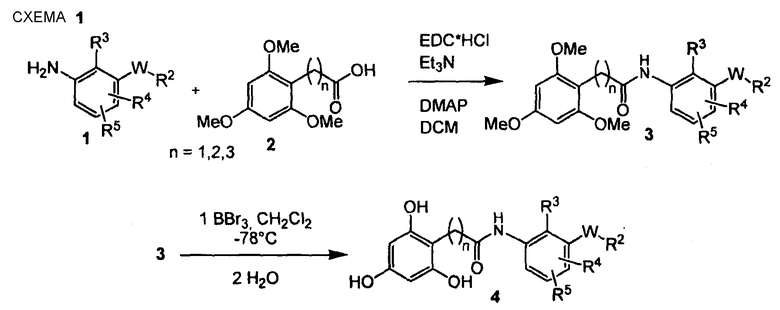
По схеме 1 анилин типа (1) подвергают взаимодействию в инертной атмосфере с N'-(3-диметиламинопропил)-N-этилкарбодиимидом (EDC), триэтиламином, 4-диметиламинопиридином (DMAP) и карбоновой кислотой типа (2) в дихлорметане, получая при этом амид типа (3). Амид типа (3) далее подвергают взаимодействию с трибромидом бора в дихлорметане при -78°С, получая при этом соответствующий 2,4,6-тригидроксифенильное соединение типа (4). Последовательность синтеза, показанная на схеме 1, приводящая к соединениям, подобным (4), пригодна не только для блоков построения Y-H, подобных (1), но ее обычно применяют для всех других блоков построения типа Y-H.
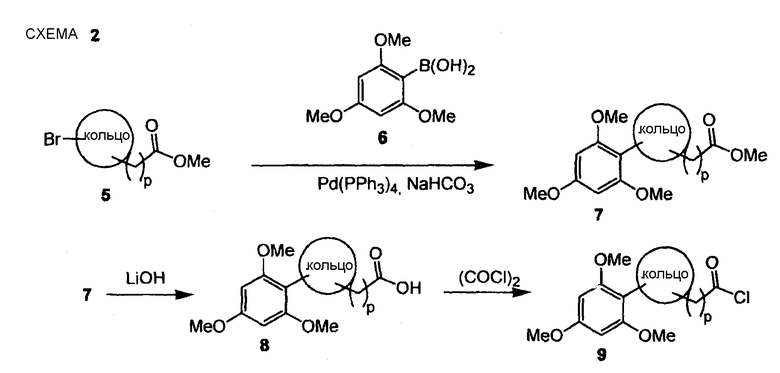
По схеме 2 бромированный ароматический или гетероароматический сложный эфир общего типа (5) подвергают взаимодействию в инертных условиях с 2,4,6-триметоксифенилбороновой кислотой (6) в основных условиях типа реакции Сузуки (Pd(PPh3)4 и водный бикарбонат натрия в диметоксиэтане, получая при этом биарил типа (7), который далее гидролизуют водным гидроксидом лития в ацетонитриле с получением соответствующей карбоновой кислоты (8), которую превращают в блок построения типа (9) взаимодействием с оксалилхлоридом в безводном дихлорметане.
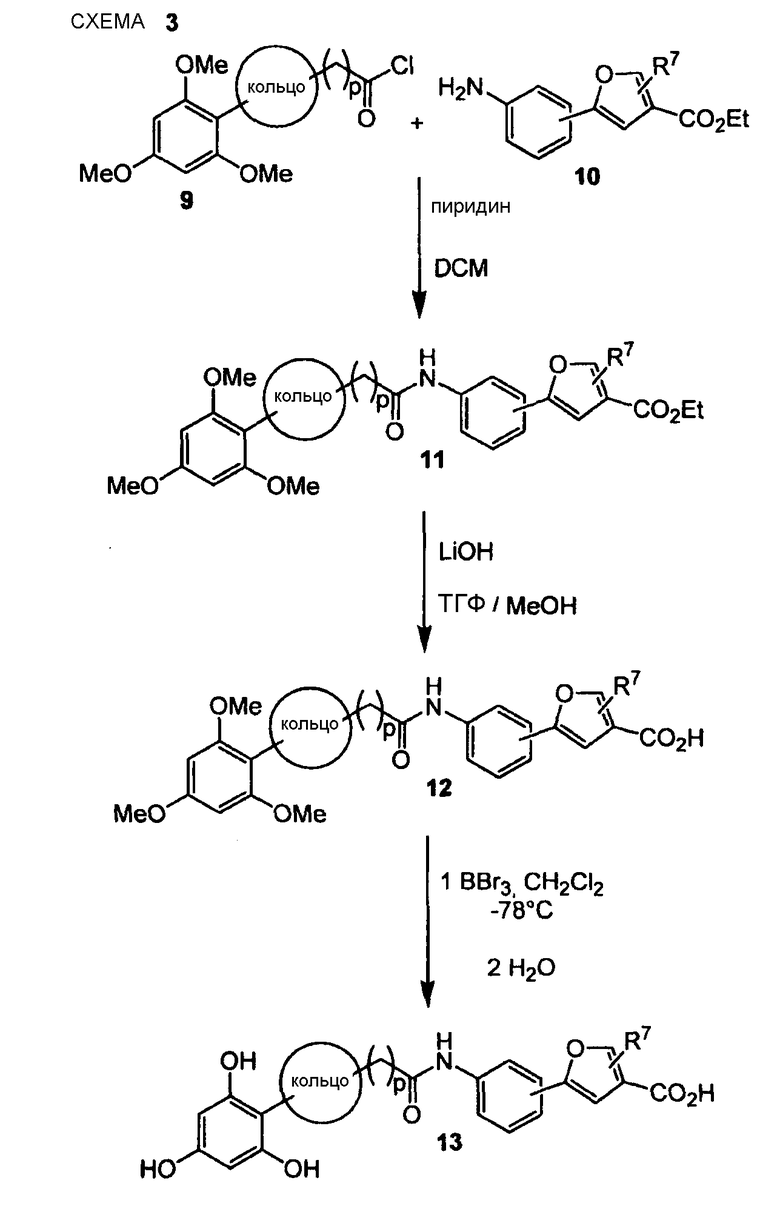
По схеме 3 хлорангидрид кислоты, подобный (9), подвергают взаимодействию с анилином общего типа (10) в основных условиях (пиридин в дихлорметане), получая при этом соответствующий анилид (11). В альтернативном случае для этой стадии реакции можно применять триэтиламин. Сложный эфир (11) гидролизуют LiOH в MeCN или ТГФ/МеОН с получением карбоновой кислоты, подобной (12), которую далее подвергают взаимодействию с трибромидом бора в дихлорметане при -78°С, получая при этом после последующей водной обработки соответствующие деметилированные кислоты типа (13). Последовательность синтеза, показанная на схеме 3, приводящая к соединениям, подобным (13), пригодна не только для блоков построения Y-H, подобных (10), но ее обычно применяют для всех других блоков построения типа Y-H.
Настоящее изобретение, кроме того, иллюстрируется нижеследующими репрезентативными примерами.
ПРИМЕР 1
3-[2-(2,4,6-Тригидроксифенил)ацетиламино]бензойная кислота (18)
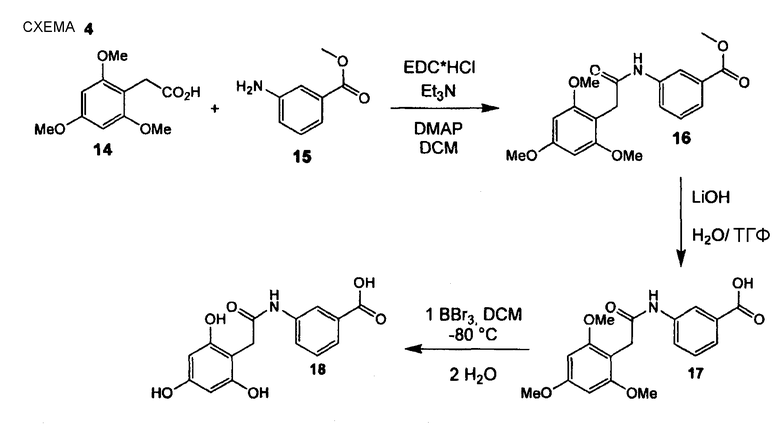
Стадия 1. (Нижеследующую реакцию проводят в безводной атмосфере N2). Гидрохлорид EDC (122 мг, 0,64 ммоль) и триэтиламин (89 мкл, 0,64 ммоль) растворяют в безводном дихлорметане (3,2 мл) и раствор перемешивают в течение 5 мин при комнатной температуре. Добавляют 2-(2,4,6-триметоксифенил)уксусную кислоту (14) (101 мг, 0,45 ммоль) и DMAP (8 мг, 0,6 ммоль) и смесь перемешивают в течение 10 мин. Добавляют сложный этиловый эфир (15) (70 мг, 0,42 ммоль) и реакционный раствор перемешивают на протяжении ночи при комнатной температуре. Реакционный раствор гидролизуют насыщенным водным NH4Cl с последующим гидролизом водой, слои разделяют, водный слой экстрагируют дихлорметаном (3 раза) и объединенные органические слои промывают водой и насыщенным раствором соли и сушат Na2SO4. Растворитель удаляют при пониженном давлении. Сырой продукт очищают препаративной радиальной хроматографией (силикагель 60PF, EtOAc/CyH, 1+1), получая при этом метиловый эфир 3-[2-(2,4,6-триметоксифенил)ацетиламино]бензойной кислоты (16) в виде белого твердого вещества (145 мг, 95%). [K. C. Nicolaou; P. S. Baran; Y.-L. Zhong; K. Sugita; J. Am. Chem. Soc.; 2002; 124; 10; 2212-2220]. 1H ЯМР (400 МГц, CDCl3): 3,68 (с, 2 H); 3,83 (с, 3 H); 3,84 (с, 6 H); 3,87 (с, 3 H); 6,18 (с, 2 H); 7,33 (т, 1 H, J = 8,0 Гц); 7,56 (ушир. с, 1 H); 7,69 (ушир. дд, 1 H, J = 7,8 Гц); 7,78 (т, 1 H, J = 1,8 Гц); 7,90 (дд, 1 H, J1 = 8,1 Гц, J2 = 1,3 Гц).
Стадия 2. Метиловый эфир 3-[2-(2,4,6-триметоксифенил)ацетиламино]бензойной кислоты (16) (140 мг, 0,39 ммоль) растворяют в ТГФ (25,0 мл) при комнатной температуре и добавляют 1 М водный LiOH (2,0 мл, 2,0 ммоль). Реакционную смесь перемешивают 40 час при комнатной температуре. Реакционную смесь гасят (охлаждающая баня) 2 М водной HCl. Смесь экстрагируют EtOAc (3×), объединенный органический слой промывают насыщенным раствором соли и сушат Na2SO4, получая при этом 3-[2-(2,4,6-триметоксифенил)ацетиламино]бензойную кислоту (17) (134 мг, 99%) в виде бежевого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 3,71 (с, 2 H); 3,84 (с, 3 H); 3,85 (с, 6 H); 6,29 (с, 2 H); 7,43 (т, 1 H, J = 7,8 Гц); 7,76 (д, 1 H, J = 7,8 Гц); 7,83 (д, 1 H, J= 8,1 Гц); 8,19 (ушир. с, 1 H).
Стадия 3. (Нижеследующую реакцию проводят в безводной атмосфере N2). 3-[2-(2,4,6-Триметоксифенил)ацетиламино]бензойную кислоту (17) (134 мг, 0,39 ммоль) растворяют в безводном DCM (5,3 мл), раствор охлаждают до -78°С и по каплям добавляют BBr3 (240 мкл, 2,55 ммоль). Реакционную смесь перемешивают в течение 30 мин при -78°С и после медленно нагревают в течение дополнительных 2 час при комнатной температуре. По каплям добавляют ледяную воду, слои разделяют и водный слой экстрагируют EtOAc (3 раза). Объединенный органический слой промывают насыщенным раствором соли и сушат Na2SO4. Сырой продукт очищают препаративной ОФ-ВЭЖХ (градиент, вода/CH3CN, 95:5 - 5:95), получая при этом 3-[2-(2,4,6-тригидроксифенил)ацетиламино]бензойную кислоту (18) (29 мг, 22%) в виде желтоватого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 3,69 (с, 2 H); 5,97 (с, 2 H); 7,43 (т, 1 H, J = 8,0 Гц); 7,76 (ушир. дд, 1 H, J1 = 7,6 Гц); 7,84 (ушир. дд, 1 H, J1 = 8,1 Гц); 8,17 (т, 1 H, J= 1,8 Гц).
ПРИМЕР 2
2-Метил-5-{4-[2-(2,4,6-тригидроксифенил)ацетиламино]фенил}фуран-3-карбоновая кислота (19)
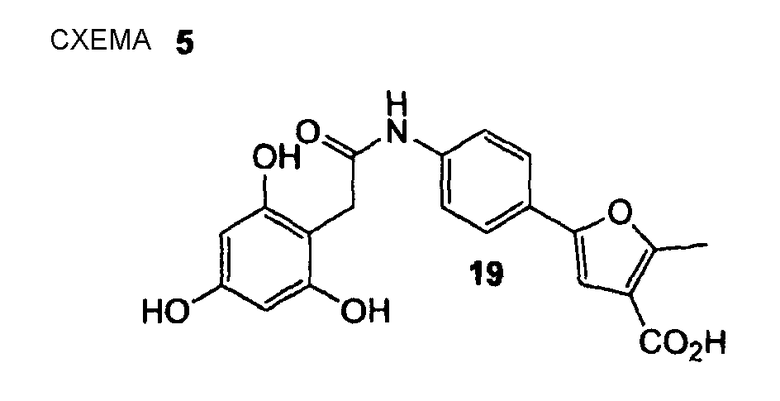
Согласно методике, описанной в примере 1, 2-метил-5-{4-[2-(2,4,6-тригидроксифенил)ацетиламино]фенил}фуран-3-карбоновую кислоту (19) получают в виде бежевого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 2,66 (с, 3 H); 3,68 (с, 2 H); 5,97 (с, 2 H); 6,89 (с, 1 H); 7,60 (дд, 2 H, J1 = 9,1 Гц, J2 = 2,3 Гц); 7,64 (дд, 2 H, J1 = 9,1 Гц, J2 = 2,3 Гц).
Ниже описано получение промежуточных соединений.
Метиловый эфир [5-(2-аминофенил)тиофен-2-ил]уксусной кислоты (44)
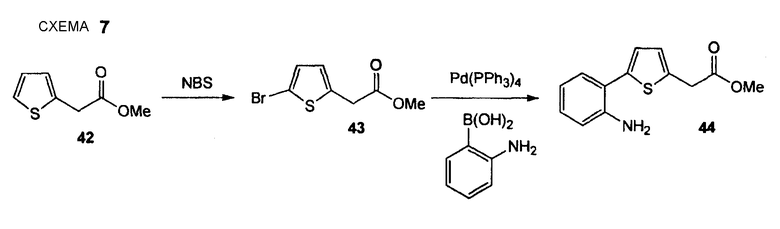
Стадия 1. (Нижеследующую реакцию проводят в безводной атмосфере N2). Метиловый эфир тиофен-2-илуксусной кислоты (42) (2,0 г, 12,8 ммоль) растворяют в безводном хлороформе (9,0 мл) и ледяной уксусной кислоте (9,0 мл), порциями добавляют N-бромсукцинимид (2,3 г, 13,0 ммоль) и смесь перемешивают в течение 3 дней при комнатной температуре. К реакционной смеси добавляют воду, слои разделяют и водный слой экстрагируют дихлорметаном. Объединенный органический слой промывают несколько раз 1 М водным NaOH и водой и один раз насыщенным раствором соли и сушат Na2SO4. Сырой продукт очищают препаративной радиальной хроматографией (CyH/EtOAc, 5+1], получая при этом метиловый эфир (5-бромтиофен-2-ил)уксусной кислоты (43) в виде желтого масла (2,46 г, 81%), который применяют без любой дополнительной очистки. 1H ЯМР (400 МГц, CDCl3): 3,71 (с, 3 H); 3,75 (с, 2 H); 6,67 (д, 1 H, J = 3,8 Гц); 6,88 (д, 1 H, J = 3,8 Гц).
Стадия 2. (Нижеследующую реакцию проводят в атмосфере N2). Этанол (3,7 мл), тетракис(трифенилфосфин)палладий(0) (289 мг, 0,25 ммоль) и декагидрат Na2CO3 (4,0 г, 14,0 ммоль), растворенный в воде (5,2 мл) последовательно добавляют к раствору гидрохлорида 2-аминобензолбороновой кислоты (910 мг, 5,25 ммоль) в толуоле (52 мл). Реакционную смесь осторожно (5 раз) дегазируют и снова промывают струей N2. Добавляют раствор метилового эфира (5-бромтиофен-2-ил)уксусной кислоты (43) (1,17 г, 5,0 ммоль) в толуоле (4,5 мл). Смесь снова дегазируют (5 раз) и перемешивают в течение 22 час при 100°С. Реакционный раствор распределяют между EtOAc и насыщенным раствором соли и отделенный водный слой экстрагируют EtOAc (3 раза). Объединенный органический слой промывают водой и насыщенным раствором соли и сушат его Na2SO4. Сырой продукт очищают препаративной радиальной хроматографией (CyH/EtOAc, 5+1), получая при этом метиловый эфир [5-(2-аминофенил)тиофен-2-ил]уксусной кислоты (44) в виде коричневого масла (634 мг, 51%). 1H ЯМР (400 МГц, CDCl3): 3,73 (с, 3 H); 3,83 (с, 2 H); 3,92-4,07 (ушир. с, 2 H); 6,74 (д, 1 H); 6,76 (тд, 1 H, J1 = 7,6 Гц, J2 = 1,3 Гц); 6,92 (д, 1 H, J = 3,5 Гц); 7,02 (д, 1 H, J = 3,5 Гц); 7,11 (тд, 1 H, J1 = 7,6 Гц, J2 = 1,5 Гц); 7,23 (дд, 14 H, J1 = 7,6 Гц, J2 = 1,5 Гц).
Метиловый эфир 5-(2-аминофенил)тиофен-2-карбоновой кислоты (47)
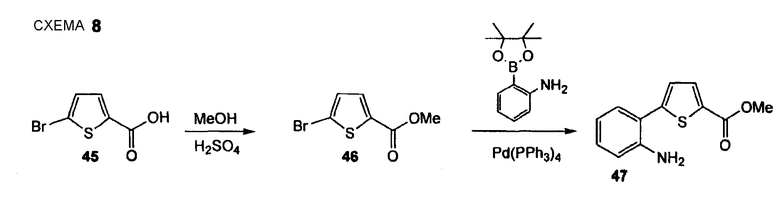
Стадия 1. LE23 5-Бромтиофен-2-карбоновую кислоту (45) (1,50 г, 7,24 ммоль) растворяют в метаноле (10 мл) и добавляют конц. серную кислоту (0,39 мл, 7,24 ммоль). Реакционную смесь перемешивают в течение 20 час при 75°С. Смесь охлаждают до комнатной температуре, растворитель удаляют при пониженном давлении и остаток растворяют в EtOAc. Этот органический слой промывают 3 раза 5% водным Na2CO3 и объединенный водный слой экстрагируют EtOAc. Объединенные органические слои промывают насыщенным раствором соли и сушат Na2SO4. Растворитель удаляют при пониженном давлении и остаток сушат без дополнительной очистки в вакууме, создаваемом масляным насосом, получая при этом сложный эфир (46) в виде белого твердого вещества (1,48 г, 92%). 1H ЯМР (400 МГц, CDCl3): 3,85 (с, 3 H); 7,05 (д, 1 H, J= 4,0 Гц); 7,53 (д, 1H, J= 4,0 Гц).
Стадия 2. LE29 (Нижеследующую реакцию проводят в атмосфере N2). Тетракис(трифенилфосфин)палладий(0) (510 мг, 0,45 ммоль) и сложный эфир (46) (1,97 г, 8,91 ммоль) растворяют в DME (16 мл), реакционную смесь осторожно дегазируют (5 раз) и промывают струей N2. Добавляют 2-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фениламин (2,15 г, 9,80 ммоль) и 1 М водный раствор NaHCO3 (27,0 мл, 27,0 ммоль), реакционную смесь снова осторожно дегазируют (5 раз) и промывают струей N2. Смесь перемешивают в течение 18 час при 95°С. Реакционный раствор распределяют между EtOAc и водой и отделенный водный слой экстрагируют EtOAc (3 раза). Объединенный органический слой промывают насыщенным раствором соли и его сушат Na2SO4. Сырой продукт очищают флэш-хроматографией (силикагель, CyH/EtOAc, 5+1], получая при этом метиловый эфир 5-(2-аминофенил)тиофен-2-карбоновой кислоты (47) в виде желтого твердого вещества (1,41 г, 67%). 1H ЯМР (400 МГц, CDCl3): 3,88 (с, 3 H); 4,00 (с, 2 H); 6,73-6,82 (м, 2 H); 7,13-7,21 (м, 2 H); 7,26 (дд, 1 H, J1 = 7,6 Гц, J2 = 1,0 Гц); 7,78 (д, 1 H, J = 3,8 Гц).
2',4',6'-Триметоксибифенил-3-карбонилхлорид (51)
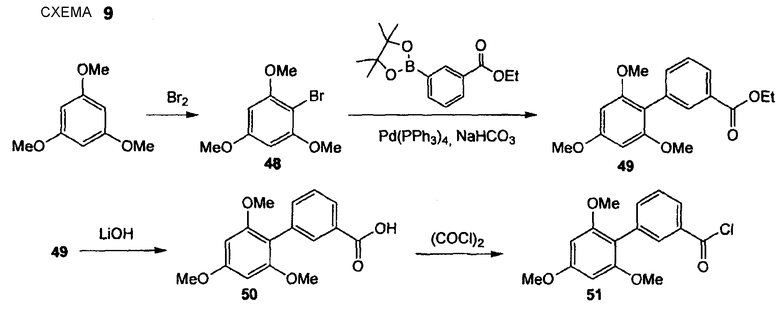
Стадия 1. KM03 1,3,5-Триметоксибензол (10,0 г, 59,46 ммоль) растворяют в безводном дихлорметане (100 мл), реакционную смесь охлаждают до -78°С, по каплям добавляют бром (3,0 мл, 59,44 ммоль), смесь перемешивают в течение 1 час при температуре между -70°С и -40°С. Раствор нагревают до 0°С и добавляют воду. Разделяют слои и водный слой экстрагируют EtOAc (3 раза). Объединенный органический слой промывают водой и насыщенным раствором соли и сушат Na2SO4. Растворитель удаляют и сырой продукт очищают перекристаллизацией из горячего EtOAc и циклогексана, получая при этом 2-бром-1,3,5-триметоксибензол (48) в виде белого твердого вещества (8,84 г, 60%). 1H ЯМР (400 МГц, CDCl3): 3,80 (с, 3 H); 3,86 (с, 6 H); 6,15 (с, 2 H).
Стадия 2: FR542 (Нижеследующую реакцию проводят в атмосфере N2). Pd(PPh3)4 (342 мг, 0,30 ммоль) и 2-бром-1,3,5-триметоксибензол (48) (2,44 г, 9,87 ммоль) растворяют в DME (20 мл) и раствор перемешивают в течение 15 мин при комнатной температуре. Добавляют этиловый эфир 3-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)бензойной кислоты (3,16 г, 11,45 ммоль) с последующим добавлением водного 1 М раствора бикарбоната натрия (29,6 мл, 29,6 ммоль). Реакционную смесь осторожно дегазируют, промывают струей N2 (5 раз) и перемешивают в течение 20 час при 100°С (кипячение с обратным холодильником). Реакционную смесь охлаждают до комнатной температуры, органический растворитель удаляют при пониженном давлении и остаток распределяют между водой и EtOAc. Водный слой экстрагируют EtOAc (3 раза), объединенный органический слой промывают водой и насыщенным раствором соли и сушат Na2SO4. Полученный сырой продукт очищают препаративной радиальной хроматографией (силикагель, EtOAc/CyH, 1+3), получая при этом бифенил (49) (2,41 г, 77%) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3): 1,35 (т, 3 H, J= 7,1 Гц); 3,70 (с, 6 H); 3,85 (с, 3 H); 4,35 (кв., 2 H, J = 7,8 Гц); 6,21 (с, 2 H); 7,42 (т, 1 H, J = 7,6 Гц); 7,95 (ушир. д, 1 H, J= 7,8 Гц); 7,99 (ушир. с, 1 H).
Стадия 3. FR543 Бифенил (49) (2,41 г, 7,62 ммоль) растворяют в MeCN (76 мл) и добавляют 1 М водный LiOH (38,0 мл, 38,0 ммоль). Реакционную смесь перемешивают в течение 4 час при кипячении с обратным холодильником. Охлажденную реакционную смесь (охлаждающая баня) гасят 1 М водным HCl (до установления рН приблизительно 3). Смесь экстрагируют EtOAc (3×), объединенный органический слой промывают водой и насыщенным раствором соли и сушат Na2SO4. Растворитель удаляют и остаток сушат без дополнительной очистки в вакууме, создаваемом масляным насосом, получая при этом карбоновую кислоту (50) в виде желтого твердого вещества (2,25 г, выход количеств.). 1Н ЯМР (400 МГц, CDCl3): 3,71 (с, 6 H); 3,85 (с, 3 H); 6,21 (с, 2 H); 7,45 (т, 1 H, J = 7,7 Гц); 7,56 (ушир. д, 1 H, J= 7,8 Гц); 8,00 (ушир. д, 1 H, J= 7,6 Гц); 8,06 (ушир. с, 1 H).
Стадия 4. DU011 (Нижеследующую реакцию проводят в безводной атмосфере N2). Карбоновую кислоту (50) (1,00 г, 3,50 ммоль) растворяют в безводном дихлорметане (23 мл) и добавляют безводный ДМФ (5 капель). Медленно добавляют оксалилхлорид (460 мкл, 5,25 ммоль) при поддержании температуры приблизительно при 20°С в водяной баней и смесь перемешивают в течение дополнительных 2 час при комнатной температуре. Растворитель удаляют и остаток сушат в вакууме, получая при этом неочищенный 2',4',6'-триметоксибифенил-3-карбонилхлорид (51) (1,10 г, выход количеств.) в виде желтого твердого вещества. Дополнительную очистку не проводят.
Метиловый эфир 5-(4-аминофенил)-2-метилфуран-3-карбоновой кислоты (54)
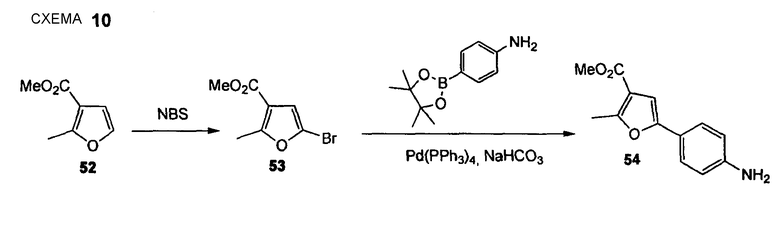
Стадия 1. DK001 (Нижеследующую реакцию проводят в отсутствие света). Метиловый эфир 2-метилфуран-3-карбоновой кислоты (52) (3,60 мл, 28,5 ммоль) растворяют в хлороформе (20 мл) и ледяной уксусной кислоте (20 мл) и порциями в раствор добавляют NBS (6,90 г, 38,8 ммоль) в течение периода 95 мин. Реакционную суспензию перемешивают в течение дополнительных 19 час при комнатной температуре. К реакционной смеси добавляют воду и водный слой экстрагируют дихлорметаном (2 раза), объединенный органический слой промывают 2 М водным NaOH, водой (3 раза) и насыщенным раствором соли и сушат Na2SO4, получая при этом метиловый эфир 5-бром-2-метилфуран-3-карбоновой кислоты (53) (4,90 г, 78%) в виде красно-коричневого масла. Дополнительную очистку не проводят. 1H ЯМР (400 МГц, CDCl3): 2,54 (с, 3 H); 3,80 (с, 3 H); 6,53 (с, 1 H).
Стадия 2: ДK002+003 (Нижеследующую реакцию проводят в атмосфере N2). Pd(PPh3)4 (1,26 г, 1,09 ммоль) и метиловый эфир 5-бром-2-метилфуран-3-карбоновой кислоты (53) (4,77 г, 21,77 ммоль) растворяют в DME (116 мл) и раствор перемешивают в течение 15 мин при комнатной температуре. Добавляют 4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фениламин (5,25 г, 23,96 ммоль) с последующим добавлением водного 1 М раствора бикарбоната натрия (65,4 мл, 65,3 ммоль). Реакционную смесь осторожно дегазируют, промывают струей N2 (5 раз) и перемешивают в течение 4 час при 95°С (кипячение с обратным холодильником). Реакционную смесь охлаждают до комнатной температуры, органический растворитель удаляют при пониженном давлении и остаток распределяют между водой и EtOAc. Водный слой экстрагируют EtOAc (3 раза), объединенный органический слой промывают водой и насыщенным раствором соли и сушат Na2SO4. Полученный сырой продукт очищают флэш-хроматографией (силикагель, EtOAc/CyH, 1+2), получая при этом метиловый эфир 5-(4-аминофенил)-2-метилфуран-3-карбоновой кислоты (54) (2,35 г, 46%) в виде желто-коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3): 2,60 (с, 3 H); 3,74 (ушир. с, 2 H); 3,82 (с, 3 H); 6,64 (с, 1 H); 6,67 (дт, 1 H, J1 = 8,6 Гц, J2 = 2,3 Гц); 7,42 (дт, 2 H, J1 = 8,8 Гц, J2 = 2,3 Гц).
2-Тиофен-2-илфениламин (55)
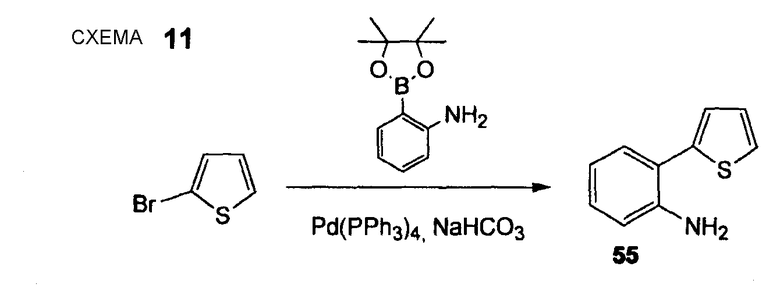
Стадия 1. АВ427 (Нижеследующую реакцию проводят в атмосфере N2). Тетракис(трифенилфосфин)палладий(0) (297 мг, 0,26 ммоль) и 2-бромтиофен (837 мг, 5,13 ммоль) растворяют в DME (42 мл), реакционную смесь осторожно дегазируют (5 раз) и промывают струей N2. После перемешивания 10 мин добавляют 2-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фениламин (1,24 г, 5,64 ммоль) и 1 М водный раствор NaHCO3 (15,4 мл, 15,4 ммоль), реакционную смесь снова осторожно дегазируют (5 раз) и промывают струей N2. Смесь перемешивают в течение 3 час при 95°С. Смесь охлаждают до комнатной температуры, растворитель удаляют при пониженном давлении и остаток распределяют между EtOAc и водой. Отделенный водный слой экстрагируют EtOAc (3 раза). Объединенный органический слой промывают насыщенным раствором соли и сушат Na2SO4. Сырой продукт очищают флэш-хроматографией (силикагель 60, CyH/EtOAc, 15+1), получая при этом 2-тиофен-2-илфениламин (55) в виде коричневого твердого вещества (825 мг, 92%). 1H ЯМР (400 МГц, CDCl3): 4,40-6,00 (м, 2 H); 6,88 (тд, 1 H, J1 = 7,6 Гц, J2 = 1,0 Гц); 6,93 (дд, 1 H, J1 = 8,0 Гц, J2 = 1,0 Гц); 7,07 (дд, 1 H, J1 = 5,3 Гц, J2 = 3,5 Гц); 7,17 (тд, 1 H, J1 = 8,0 Гц, J2 = 1,5 Гц) 7,22 (дд, 1 H, J1 = 3,5 Гц, J2 = 1,3 Гц); 7,30 (дд, 1 H, J1 = 7,6 Гц, J2 = 1,5 Гц); 7,33 (дд, 1 H, J1 = 5,3 Гц, J2 = 1,3 Гц).
Метиловый эфир [5-(3-аминофенил)тиофен-2-ил]уксусной кислоты (57)
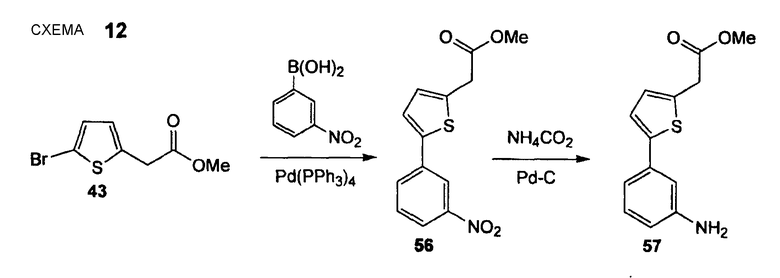
Стадия 1. FR544 (Нижеследующую реакцию проводят в атмосфере N2). Тетракис(трифенилфосфин)палладий(0) (1,12 г, 0,97 ммоль) и сложный эфир (43) (4,57 г, 19,44 ммоль) растворяют в толуоле (200 мл) и EtOH (20,0 мл), реакционную смесь осторожно дегазируют (5 раз) и промывают струей N2. Добавляют 3-нитрофенилбороновую кислоту (3,57 г, 21,38 ммоль) и 3 М водный раствор Na2CO3 (18,1 мл, 54,3 ммоль), реакционную смесь снова осторожно дегазируют (5 раз) и промывают струей N2. Смесь перемешивают в течение 18 час при 100°С. Реакционный раствор распределяют между EtOAc и водой и отделенный водный слой экстрагируют EtOAc (3 раза). Объединенный органический слой промывают насыщенным раствором соли и сушат Na2SO4. Полученный сырой продукт очищают препаративной радиальной хроматографией (силикагель, EtOAc/CyH, 1+5), получая при этом метиловый эфир [5-(3-нитрофенил)тиофен-2-ил]уксусной кислоты (56) в виде желтого твердого вещества (3,15 г, 58%). 1H ЯМР (400 МГц, CDCl3): 3,75 (с, 3 H); 3,85 (с, 2 H); 6,94 (ушир. д, 1 H, J = 3,8 Гц); 7,27 (д, 1 H, J = 3,8 Гц); 7,51 (т, 1 H, J = 8,0 Гц); 7,84 (ддд, 1 H, J1 = 7,8 Гц, J2 = 1,5 Гц, J3 = 0,8 Гц); 8,08 (ддд, 1 H, J, = 8,3 Гц, J2 = 2,1 Гц, J3 = 1,0 Гц); 8,39 (т, 1 H, J= 1,9 Гц).
Стадия 2. (Нижеследующую реакцию проводят в атмосфере N2). Метиловый эфир [5-(3-нитрофенил)тиофен-2-ил]уксусной кислоты (56) (3,15 мг, 11,35 ммоль) растворяют в МеОН (225 мл) и добавляют Pd на угле (содержание Pd 10% (мас./мас.), 1,20 г, 1,13 ммоль) с последующим добавлением NH4CO2H (7,15 г, 113,4 ммоль) при комнатной температуре. Реакционную смесь осторожно дегазируют (промывают струей N2) и перемешивают в течение 22 час при комнатной температуре. Реакционную смесь фильтруют через короткий слой целита и удаляют растворитель. Полученный сырой продукт очищают препаративной радиальной хроматографией (силикагель, EtOAc/CyH, 1+3), получая при этом метиловый эфир [5-(3-аминофенил)тиофен-2-ил]уксусной кислоты (57) (2,08 г, 74%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): 3,73 (с, 3 H); 3,81 (с, 2 H); 6,59 (дд, 1 H, J1 = 7,8 Гц, J2 = 2,0 Гц); 6,86 (ушир. д, 1 H, J = 3,5 Гц); 6,88 (т, 1 H, J = 1,9 Гц); 6,97 (ушир. д, 1 H, J = 7,6 Гц); 7,09 (д, 1 H, J = 3,5 Гц); 7,13 (т, 1 H, J = 7,7 Гц).
ПРИМЕР 3
2-Метил-5-{4-[(2',4',6'-тригидроксибифенил-3-карбонил)амино]фенил}фуран-3-карбоновая кислота (60) REV968
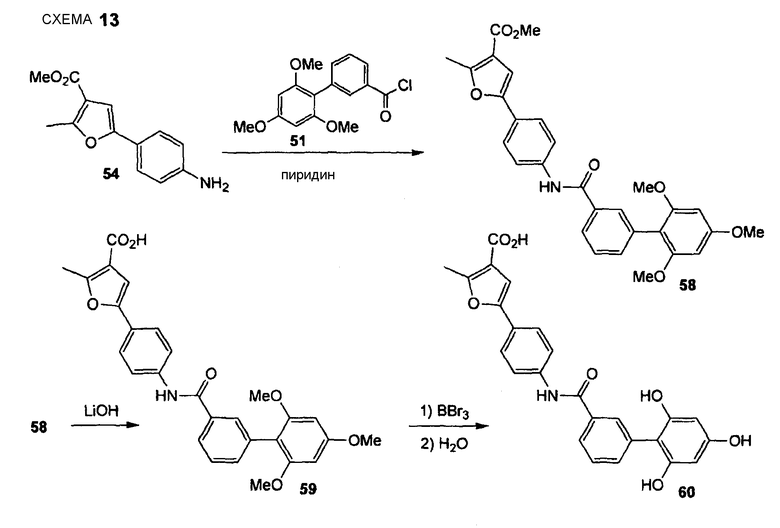
Стадия 1. АВ438 (Нижеследующую реакцию проводят в сухой атмосфере N2). Анилин (54) (150 мг, 0,65 ммоль) растворяют в безводном дихлорметане (10,0 мл), добавляют безводный пиридин (1,7 мл) и хлорангидрид карбоновой кислоты (51) (218 мг, 0,71 ммоль). Реакционную смесь перемешивают в течение 20 час при комнатной температуре. Реакционную смесь выливают в охлажденный льдом 1 М водный HCl, экстрагируют EtOAc (3×), объединенный органический слой промывают водой и насыщенным раствором соли и сушат Na2SO4. Полученный сырой продукт очищают препаративной радиальной хроматографией (силикагель, EtOAc/CyH, 1+5, затем 1+3, затем 1+1), получая при этом амид (58) в виде желтого твердого вещества (95 мг, 29%). 1H ЯМР (400 МГц, CDCl3): 2,63 (с, 3 H); 3,72 (с, 6 H); 3,83 (с, 3 H); 3,86 (с, 3 H); 6,23 (с, 2 H); 6,81 (с, 1 H); 7,46-7,53 (м, 2 H); 7,61 (д, 2 H, J = 8,5 Гц); 7,66 (д, 2 H, J = 8,6 Гц); 7,76-7,82 (м, 3 H).
Стадия 2. АВ439 Сложный эфир (58) (71 мг, 0,14 ммоль) растворяют в ТГФ (2,4 мл) и МеОН (0,6 мл) и добавляют 1 М водный LiOH (710 мкл, 0,71 ммоль). Реакционную смесь перемешивают в течение 24 час при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток распределяют между EtOAc и 1 М HCl. Водный слой отделяют и экстрагируют 3 раза EtOAc. Объединенный органический слой промывают водой и насыщенным раствором соли и сушат Na2SO4. Растворитель удаляют при пониженном давлении и остаток сушат без дополнительной очистки в вакууме, создаваемом масляным насосом, получая при этом неочищенный продукт (59) в виде бежевого твердого вещества (68 мг, 99%). 1H ЯМР (400 МГц, ДМСО-d6): 2,59 (с, 3 H); 3,67 (с, 6 H); 3,83 (с, 3 H); 6,34 (с, 2 H); 6,99 (с, 1 H); 7,38 (ушир. д, 1 H, J = 7,6 Гц); 7,48 (т, 1 H, 7,7 Гц); 7,67 (д, 2 H, J = 8,8 Гц); 7,77 (ушир. с, 1 H); 7,81-7,87 (м, 3 H); 10,29 (с, 1 H); 12,60 (ушир. с, 1 H).
Стадия 3. АВ440 (Нижеследующую реакцию проводят в безводной атмосфере N2). Карбоновую кислоту (59) (65 мг, 0,13 ммоль) суспендируют в безводном дихлорметане (2,7 мл) и безводном 1,2-дихлорэтане (2,0 мл), раствор охлаждают до -78°С и по каплям добавляют 1 М раствор BBr3 в дихлорметане (800 мкл, 0,80 ммоль). Реакционную смесь перемешивают в течение 10 мин при -78°С и после медленного нагревания перемешивают в течение дополнительных 4 час при комнатной температуре. Реакционную смесь охлаждают до 0°С, добавляют по каплям воду и дихлорметан с последующим добавлением EtOAc. Водный слой отделяют и экстрагируют 3 раза EtOAc. Объединенный органический слой промывают насыщенным раствором соли и сушат Na2SO4. Растворитель удаляют при пониженном давлении и сырой продукт очищают препаративной ОФ-ВЭЖХ (градиент, вода/CH3CN, 95:5 - 5:95), получая при этом 2-метил-5-{4-[(2',4',6'-тригидроксибифенил-3-карбонил)амино]фенил}фуран-3-карбоновую кислоту (60) (11 мг, 19%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 2,68 (с, 3 H); 6,03 (с, 2 H); 6,95 (с, 1 H); 7,52 (т, 1 H, J= 7,7 Гц); 7,61 (дт, 1 H, J1 = 7,6 Гц, J2 = 1,3 Гц), 7,71 (д, 2 H, J = 8,8 Гц); 7,81 (д, 2 H, J = 8,8 Гц); 7,83 (дт, 1 H, J1 = 8,1 Гц, J2 = 1,4 Гц), 7,95 (т, 1 H, J= 1,5 Гц).
ПРИМЕР 4
(5-{2-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]фенил}тиофен-2-ил)уксусная кислота (63) REV974
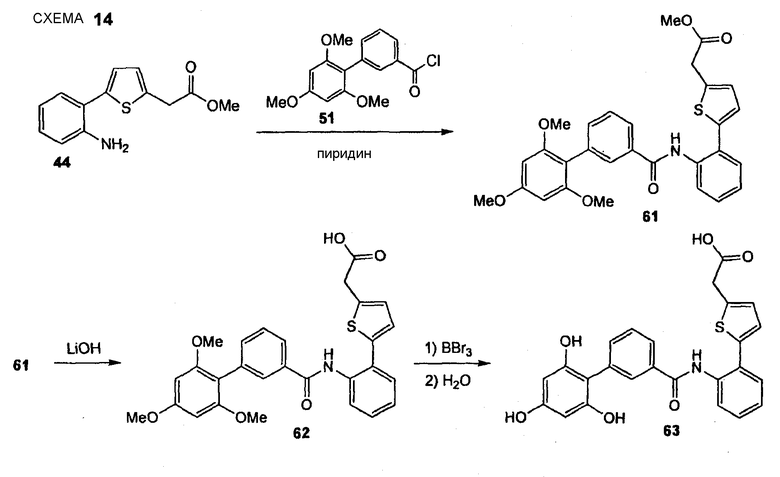
Стадия 1. FR600 (Нижеследующую реакцию проводят в безводной атмосфере N2). Анилин (44) (823 мг, 3,33 ммоль) растворяют в безводном дихлорметане (17,0 мл), добавляют безводный пиридин (680 мкл, 8,33 ммоль) и хлорангидрид карбоновой кислоты (51) (1,67 г, 4,32 ммоль). Реакционную смесь перемешивают в течение 20 час при комнатной температуре. Реакционную смесь выливают в охлажденный льдом 1 М водный HCl, экстрагируют EtOAc (3×), объединенный органический слой промывают насыщенным раствором соли и сушат Na2SO4. Сырой продукт очищают препаративной радиальной хроматографией (силикагель 60PF, CyH/EtOAc, 3+1), получая при этом амид (61) в виде желтого твердого вещества (722 мг, 41%). 1H ЯМР (400 МГц, CDCl3): 3,69 (с, 6 H); 3,72 (с, 3 H); 3,80 (с, 2 H); 3,86 (с, 3 H); 6,21 (с, 2 H); 6,92 (д, 1 H, J = 3,5 Гц); 7,01 (д, 1 H, J = 3,5 Гц); 7,14 (т, 1 H, J = 7,3 Гц); 7,36-7,42 (м, 2 H); 7,44 (д, 1 H, J = 7,8 Гц); 7,48 (ушир. д, 1 H, J = 7,6 Гц); 7,61 (ушир. д, 1 H, 7,3 Гц); 7,78 (ушир. с, 1 H); 8,35 (ушир. с, 1 H); 8,50 (д, 1 H, J = 8,3 Гц).
Стадия 2. FR601 Сложный эфир (61) (722 мг, 1,39 ммоль) растворяют в MeCN (14,0 мл) и добавляют 1 М водный LiOH (7,0 мл, 7,00 ммоль). Реакционную смесь перемешивают в течение 20 час при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток распределяют между EtOAc и 1 М HCl (1+1). Водный слой отделяют и экстрагируют 3 раза EtOAc. Объединенный органический слой промывают насыщенным раствором соли и сушат Na2SO4. Растворитель удаляют при пониженном давлении и остаток сушат без дополнительной очистки в вакууме, создаваемом масляным насосом, получая при этом неочищенный продукт (62) в виде желтого твердого вещества (714 мг, колич. выход). 1H ЯМР (400 МГц, CD3CN): 3,69 (с, 6 H); 3,80 (с, 2 H); 3,85 (с, 3 H); 6,30 (с, 2 H); 6,92 (д, 1 H, J = 3,5 Гц); 7,14 (д, 1 H, J = 3,8 Гц); 7,28 (тд, 1 H, J1 = 7,6 Гц, J2 = 1,3 Гц); 7,39 (тд, 1 H, J1 = 7,8 Гц, J2 = 1,3 Гц); 7,42-7,49 (м, 2 H); 7,54 (дд, 1 H, J1 = 7,8 Гц, J2 = 1,3 Гц); 7,69 (ушир. с, 1 H); 7,73 (дт, 1 H, J1 = 7,3 Гц, J2 = 1,8 Гц); 7,89 (д, 1 H, J = 7,6 Гц); 8,53 (ушир. с, 1 H).
Стадия 3. FR602 (Нижеследующую реакцию проводят в безводной атмосфере N2). Карбоновую кислоту (62) (700 мг, 1,39 ммоль) растворяют в безводном дихлорметане (28,0 мл), раствор охлаждают до -78°С и по каплям добавляют 1 М раствор BBr3 в дихлорметане (8,5 мл, 8,50 ммоль). Реакционную смесь перемешивают в течение 10 мин при -78°С и после медленного нагревания перемешивают в течение дополнительных 4 час при комнатной температуре. Реакционную смесь охлаждают до 0°С и по каплям добавляют воду и дихлорметан с последующим добавлением EtOAc. Водный слой отделяют и экстрагируют 3 раза EtOAc. Объединенный органический слой промывают насыщенным раствором соли и сушат Na2SO4. Растворитель удаляют при пониженном давлении и сырой продукт очищают препаративной ОФ-ВЭЖХ (градиент, вода/CH3CN, 95:5 - 5:95), получая при этом (5-{2-[(2',4',6'-тригидроксибифенил-3-карбонил)амино]фенил}тиофен-2-ил)уксусную кислоту (63) (146 мг, 22%) в виде бежевого твердого вещества. 1H ЯМР (400 МГц, CD3OD) 3,85 (с, 2 H); 6,02 (с, 2 H); 6,97 (д, 1 H, J = 3,5 Гц); 7,18 (д, 1 H, J = 3,5 Гц); 7,35 (тд, 1 H, J1 = 7,6 Гц, J2 = 1,3 Гц); 7,42 (тд, 1 H, J1 = 7,6 Гц, J2 = 1,5 Гц); 7,50 (т, 1 H, J = 7,7 Гц); 7,61 (ушир. д, 2 H, J = 7,6 Гц); 7,79 (ушир. д, 2 H, J = 7,6 Гц); 7,93 (ушир. с, 1 H).
ПРИМЕР 5
(5-{2-[2-(2,4,6-Тригидроксифенил)ацетиламино]фенил}тиофен-2-ил)уксусная кислота (25) REV989, DU27
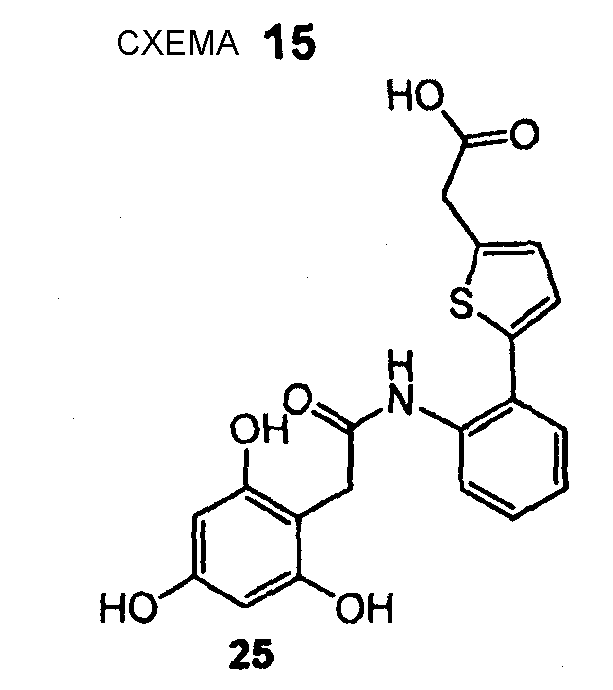
(5-{2-[2-(2,4,6-Тригидроксифенил)ацетиламино]фенил}тиофен-2-ил)уксусную кислоту (25) получают из амина (44) и хлорангидрида карбоновой кислоты (14) согласно методике, описанной в стадиях 1-3 примера 1. (5-{2-[2-(2,4,6-Тригидроксифенил)ацетиламино]фенил}тиофен-2-ил)уксусную кислоту (25) (25 мг, выход 16% после 3 стадий) получают в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 3,58 (с, 2 H); 3,88 (с, 2 H); 5,88 (с, 2 H); 6,67 (д, 1 H, J = 3,3 Гц); 6,84 (д, 1 H, J= 3,3 Гц); 7,14 (т, 1 H, J = 7,3 Гц); 7,31-7,37 (м, 2 H); 8,26 (д, 1 H, J = 8,6 Гц).
ПРИМЕР 6
5-{2-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]фенил}тиофен-2-карбоновая кислота (26) REV971, LE37
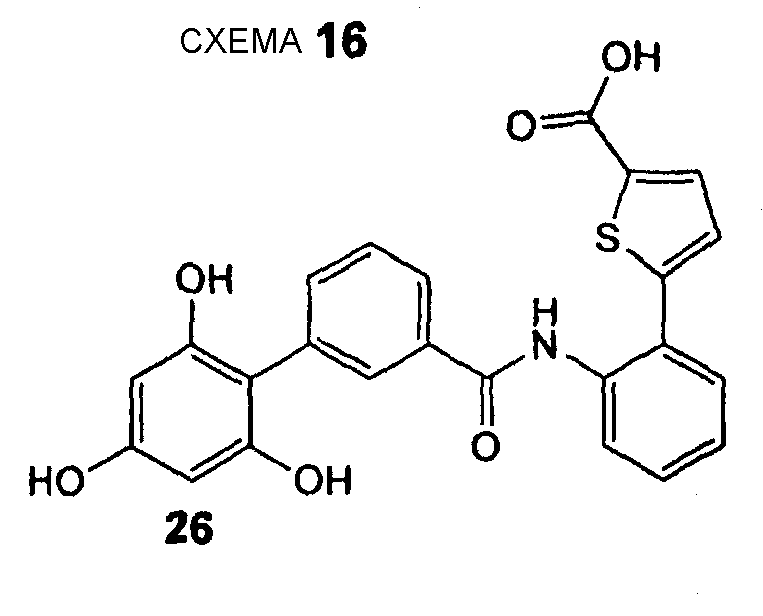
5-{2-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]фенил}тиофен-2-карбоновую кислоту (26) получают из амина (47) и хлорангидрида карбоновой кислоты (51) согласно методике, описанной выше в стадиях 1-3 примера 4. 5-{2-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]фенил}тиофен-2-карбоновую кислоту (26) (44 мг, выход 22% после 3 стадий) получают в виде белого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 6,02 (с, 2 H); 7,32-7,36 (м, 1 H); 7,41 (т, 1 H, J = 7,6 Гц); 7,46-7,53 (м, 2 H); 7,62 (дт, 1 H, J1 = 7,6 Гц, J2 = 1,3 Гц); 7,65-7,73 (м, 3 H); 7,80 (дт, 1 H, J1 = 8,1 Гц, J2 = 1,4 Гц); 7,96 (ушир. с, 1 H).
ПРИМЕР 7
(5-{3-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]фенил}тиофен-2-ил)уксусная кислота (64) REV996, КМ7
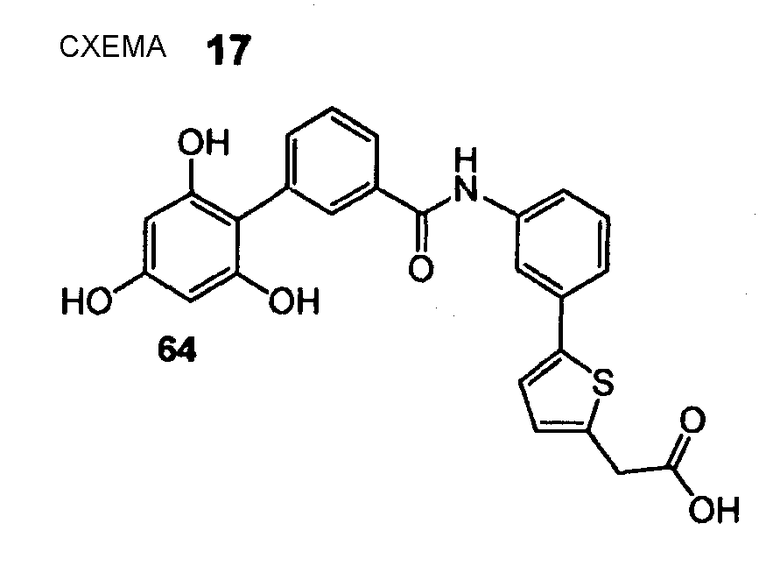
(5-{3-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]фенил}тиофен-2-ил)уксусную кислоту (64) получают из амина (57) и хлорангидрида карбоновой кислоты (51) согласно методике, описанной выше в стадиях 1-3 примера 4. (5-{3-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]фенил}тиофен-2-ил)уксусную кислоту (64) (3 мг, выход 4% после 3 стадий) получают в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 3,88 (с, 2 H); 6,03 (с, 2 H); 6,97 (д, 1 H, J = 3,5 Гц); 7,29 (д, 1 H, J = 3,8 Гц); 7,36-7,44 (м, 2 H) 7,53 (т, 1 H, J = 7,7 Гц); 7,61 (дт, 1 H, J1 = 7,6 Гц, J2 = 1,4 Гц); 7,66 (дт, 1 H, J1 = 7,3 Гц, J2 = 1,8 Гц), 7,85 (дт, 1 H, J1 = 7,8 Гц, J2 = 1,5 Гц); 7,96 (т, 1 H, J = 1,5 Гц); 8,05 (ушир. с, 1 H).
ПРИМЕР 8
(2-Тиофен-2-илфенил)амид 2',4',6'-тригидроксибифенил-3-карбоновой кислоты (65) REV965, AB437
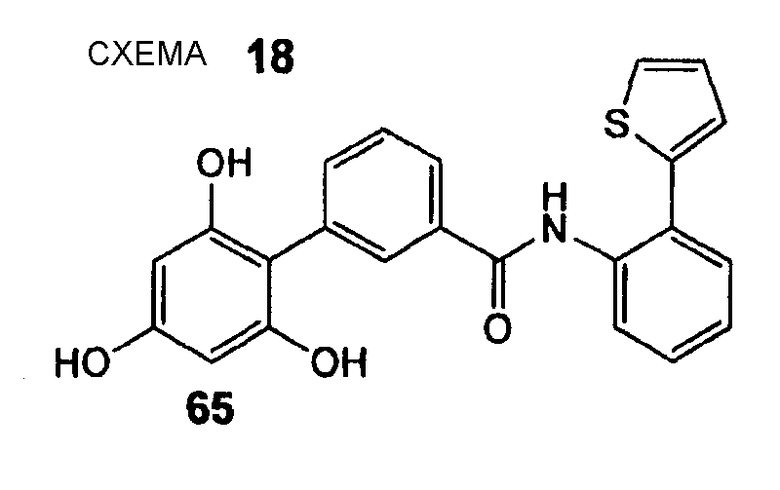
(2-Тиофен-2-илфенил)амид 2',4',6'-тригидроксибифенил-3-карбоновой кислоты (65) получают из амина (55) и хлорангидрида карбоновой кислоты (51) согласно методике, описанной выше в стадиях 1 и 3 примера 4. (2-Тиофен-2-илфенил)амид 2',4',6'-тригидроксибифенил-3-карбоновой кислоты (65) (12 мг, выход 9% после 2 стадий) получают в виде бежевого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 6,02 (с, 2 H); 7,13 (дд, 1 H, J1 = 5,1 Гц, J2 = 3,8 Гц); 7,35 (дд, 1 H, J1 = 3,5 Гц, J2 = 1,0 Гц); 7,38 (дд, 1 H, J1 = 7,6 Гц, J2 = 1,3 Гц); 7,41-7,48 (м, 2 H); 7,50 (д, 1 H, J = 7,6 Гц); 7,61 (дт, 1 H, J1 = 7,8 Гц, J2 = 1,4 Гц); 7,64 (дд, 1 H, J1 = 7,6 Гц, J2 = 1,5 Гц); 7,73 (дд, 1 H, J1 = 7,8 Гц, J2 = 1,3 Гц); 7,77 (дт, 1 H, J1 = 8,1 Гц, J2 = 1,4 Гц); 7,92 (т, 1 H, J = 1,5 Гц).
ПРИМЕР 9
(3-Трифторметилфенил)амид 2',4',6'-тригидроксибифенил-3-карбоновой кислоты (66) REV961, DU15
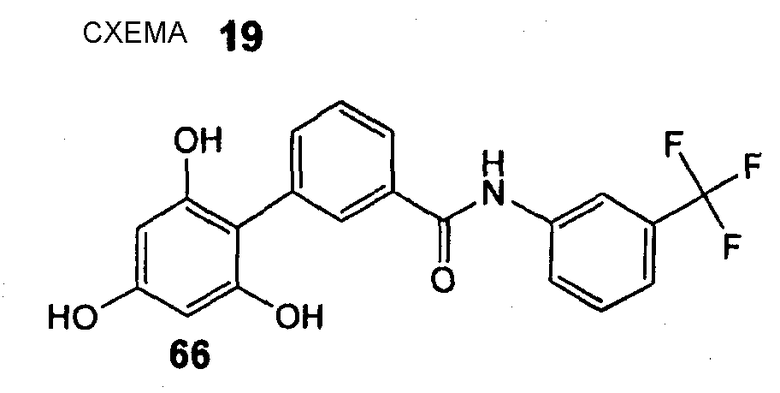
(3-Трифторметилфенил)амид 2',4',6'-тригидроксибифенил-3-карбоновой кислоты (66) получают из 3-трифторметилфениламина и хлорангидрида карбоновой кислоты (51) согласно методике, описанной выше в стадиях 1 и 3 примера 4. (3-Трифторметилфенил)амид 2',4',6'-тригидроксибифенил-3-карбоновой кислоты (66) (54 мг, выход 54% после 2 стадий) получают в виде бежевого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 6,02 (с, 2 H); 7,45 (ушир. д, 1 H, J = 7,6 Гц); 7,53 (т, 1 H, J = 7,7 Гц); 7,58 (т, 1 H, J = 8,0 Гц); 7,62 (дт, 1 H, J1 = 7,8 Гц, J2 = 1,4 Гц); 7,85 (ддд, 1 H, J1 = 7,8 Гц, J2 = 1,8 Гц, J3 = 1,3 Гц); 7,95-7,99 (м, 2 H); 8,21 (ушир. с, 1 H).
ПРИМЕР 10
3-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]бензойная кислота (67) REV1007, LE80
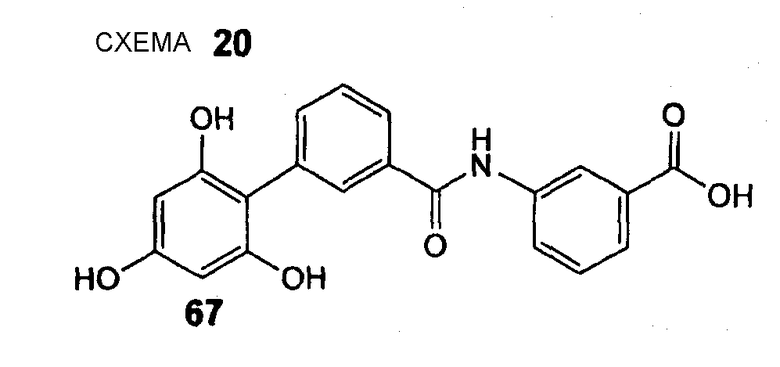
3-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]бензойную кислоту (67) получают из метилового эфира 3-аминобензойной кислоты и хлорангидрида карбоновой кислоты (51) согласно методике, описанной выше в стадиях 1-3 примера 3. 3-[(2',4',6'-Тригидроксибифенил-3-карбонил)амино]бензойную кислоту (67) (22 мг, выход 14% после 3 стадий) получают в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD): 6,02 (с, 2 H); 7,48 (т, 1 H, J = 8,0 Гц); 7,52 (т, 1 H, J = 7,8 Гц); 7,61 (ушир. д, 1 H, J = 7,8 Гц); 7,84 (т, 2 H, J = 7,2 Гц); 7,96 (ушир. с, 1 H); 8,01 (ушир. д, 1 H, J = 8,3 Гц); 8,34 (ушир. с, 1 H).
Соединения, указываемые в нижеследующей схеме 6, являются соединениями, конкретно называемыми в описании предпочтительными соединениями.
Анализ сиалил-Lewis X -тирозинсульфата (sLe X -TSA)
Соединения настоящего изобретения анализируют на молекулярном уровне на их способность ингибировать связывание химерных молекул Р-, L- или Е-селектина с sLeX и тирозинсульфатными остатками, связанными с полимерной матрицей как PSGL-1-замещение. Определяют выбранные величины IC50.
Титрационные микропланшеты выдерживают для покрытия на протяжении ночи в карбонатном буфере, рН 9,6, с козьим антителом против Fc-mAB человека (10 мкг/мл). После промывания в буфере для анализа (25 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота (HEPES), 150 мМ NaCl, 1 мМ CaCl2, рН 7,4) и блокирования (3% бычий сывороточный альбумин (BSA) в буфере для анализа) планшеты инкубируют в течение 2 час при 37°С с химерами Р-селектин-IgG человека (0,61 нМ, соответственно 150 нг/мл) или химерами L-селектин-IgG человека (0,61 нМ, соответственно 89 нг/мл), или химерами Е-селектин-IgG человека (0,61 нМ, соответственно 131 нг/мл). Образуют комплекс 5 мкл sLeX-тирозинсульфат-полиакриламида (1 мг/мл), содержащего 15% sLex, 10% тирозинсульфата и 5% биотина, при помощи 20 мкл раствора стрептавидин-пероксидаза (1 мг/мл) и 25 мкл буфера для анализа без CaCl2. Для применения в анализе комплекс лиганда разводят как 1:10000 в буфере для анализа и далее разводят как 1:1 варьируемыми количествами соединений в буфере для анализа, включающем в себя 2% ДМСО. Эту смесь добавляют к лункам, предварительно покрытым Е- или Р-селектином. После инкубации в течение 2 час при 37°С лунки промывают шесть раз в буфере для анализа, включающем в себя 0,005% монолаурата полиоксиэтиленсорбитана (твин 20), проявляют в течение 10-15 мин 20 мкл раствора субстрата 3,3',5,5'-тетраметилбензидин(ТМВ)/Н2О2 и реакцию останавливают 20 мкл 1 М H2SO4. Связанный комплекс лиганда sLex-тирозинсульфат определяют измерением оптической плотности при 450 нм по сравнению с 620 нм в планшет-ридере Fusion alpha-FP (приобретен у Packard Bioscience, Dreieich, Germany).
Анализ в проточной камере/адгезия и скольжение клеток в условиях потока
Для определения способности соединений ингибировать связывание клеток в динамических условиях, похожих на кровоток в кровеносном сосуде, в проточной камере проводят анализы, адресованные связыванию/тестирующие связывание клеток HL-60/различных клеточных линий с химерными молекулами Р-селектина, L-селектина и Е-селектина.
Прикрепление клеток в условиях потока определяют с применением системы параллельных проточных камер. Полистирольную чашку для культивирования 35 мм покрывают на 1 час при комнатной температуре буфером для сенсибилизации поверхности (50 мМ трис(гидроксиметил)аминометановый буфер (трис), 150 мМ NaCl, 2 мМ CaCl2; рН 7,4), содержащим химеры Е- или Р-селектин-IgG человека при концентрациях 2,5 мкг/мл или 10 мкг/мл соответственно. После удаления раствора для покрытия сайты неспецифического связывания блокируют в течение дополнительного часа 1% BSA в буфере для сенсибилизации поверхности при комнатной температуре. После промывания буфером для анализа (“Roswell Park Memorial Institute 1640” (RPMI 1640) + 10 мМ HEPES) чашку вставляют в камеру с параллельными ламинарными потоками (приобретена у Glycotech, Rockville, MD) и устанавливают на инвертированный фазово-контрастный микроскоп (приобретена у Olympus, Hamburg, Germany), оснащенный камерой CCD (JVC), которая соединена с РС. С применением перистальтического насоса (приобретен у Ismatec, Wertheim-Mondfeld, Germany) рециркулирующую систему уравновешивают буфером для анализа, содержащим 125 мкМ соединение или наполнитель-контроль (ДМСО). В камеру добавляют клетки (1 миллион/мл) и позволяют им распределиться в течение 2 минут при высокой скорости течения. Скорость течения затем снижают, что приводит к рассчитанному сдвигу потока 1 дина/см2. Видеопоследовательности 10 низких силовых полей регистрируют цифровым образом после 5 минут непрерывного течения. Процент ингибирования вычисляют из среднего числа клеток на поле, которые прилипли к поверхности покрытой чашки в присутствии соединения в сравнении с отсутствием соединения в независимых экспериментах.
Данные анализа в проточной камере для Е- и Р-селектина
Величины даны как нормализованные отношения в виде % ингибирования соединения, деленного на % ингибирование бимосиамозы.
[Отношение]
[Отношение]
[Отношение]
[Отношение]
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2006 |
|
RU2434006C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ НИТРОКАТЕХОЛА, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В КАЧЕСТВЕ ЛИГАНДОВ СЕЛЕКТИНА | 2006 |
|
RU2428979C2 |
| ДИ- И ТРИВАЛЕНТНЫЕ НЕБОЛЬШИЕ МОЛЕКУЛЫ ИНГИБИТОРОВ СЕЛЕКТИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СЕЛЕКТИНА | 1996 |
|
RU2165931C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2259202C2 |
| БЕНЗИЛАМИНОВЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2660421C2 |
| БЕНЗИЛАМИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ КАЛЛИКРЕИНА ПЛАЗМЫ | 2012 |
|
RU2607045C2 |
| ИНГИБИТОРЫ ПРОЛИЛГИДРОКСИЛАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2429226C9 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ПРОИЗВОДНЫЕ 8-МЕТОКСИ[1,2,4]ТРИАЗОЛО[1,5-a]ПИРИДИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 2002 |
|
RU2297416C2 |
Изобретение относится к химико-фармацевтической промышленности. Фармацевтические композиции, включающие в себя, по меньшей мере, одно соединение формулы (I), где -Х- представляет собой, например, группу формулы (II) и Y представляет собой, например, группу формулы III, или его фармацевтически приемлемые соли, сложные эфиры или амиды, или пролекарства и фармацевтически приемлемый носитель, который является пригодным в терапии, можно применять для модуляции in vitro и in vivo процессов связывания, опосредуемых связыванием Е-, Р- или L-селектина. 8 н. и 1 з.п. ф-лы, 3 табл.
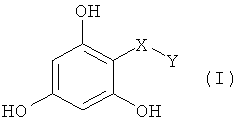
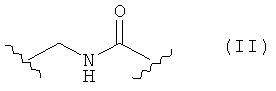
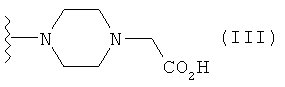
1. Фармацевтические композиции, включающие в себя, по меньшей мере, одно соединение формулы (I) и фармацевтически приемлемый носитель, который является пригодным в медицине,
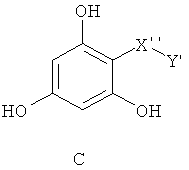
где символы и заместители имеют нижеследующие значения:
где -X″- представляет собой
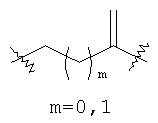
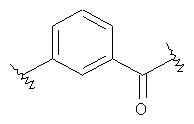
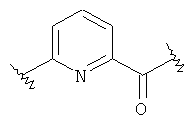
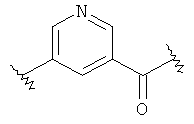
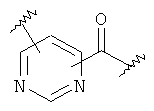
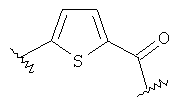
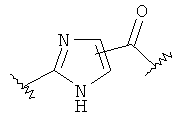
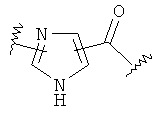
и где -Y′ представляет собой
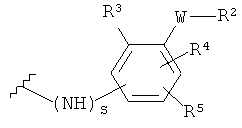
где s равно 0 или 1,
R2 представляет собой CO2H, СO2алкил, СO2арил, CO2NH2, СO2аралкил, SO3H, SO2NH2, РО(ОН)2, 1-Н-тетразолил-, СНО, СОСН3, СН2ОН, NH2, NНалкил, N(алкил)алкил′, ОСН3, СН2ОСН3, SH, F, Cl, Br, I, СН3, СН2СН3, CN, CF3;
R3 независимо от R2 представляет собой Н, СН3, СН2СН3, CF3, F, Cl, Br, I, CN, NO2 и
R4 независимо от R2 и R3 представляет собой Н, СН3, СН2СН3, CF3, F, Cl, Br, I, CN, NO2, R2,
R5 представляет собой Н, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2 и -W- представляет собой -(CH2-)v, цис-СН=СН- или транс-СН=СН-, где v равно 0, 1, 2,
в случае, когда -W- представляет собой цис-СН=СН- или транс-СН=СН-,
R2 не должен быть NH2 или SH; или
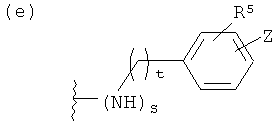
где t равно 0, 1, 2;
-Z представляет собой
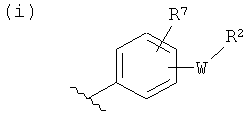
R7 независимо от R2 представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2,
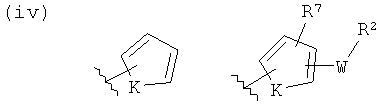
где K представляет собой NH, NMe, О, S,
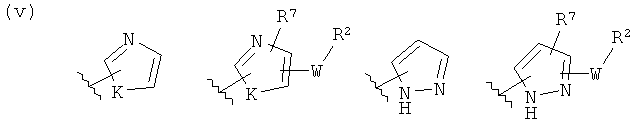
или фармацевтически приемлемые соли, сложные эфиры или амиды вышеуказанных соединений формулы (С).
2. Фармацевтические композиции по п.1, где соединения имеют формулу (D)
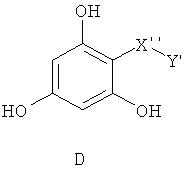
где -X″- имеет значения, указанные в п.1, и -Y″ представляет собой
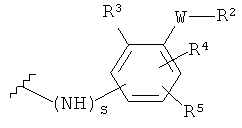
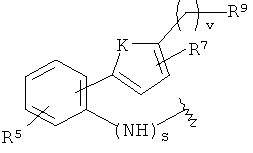
где R9 представляет собой СО2Н, СO2алкил, СO2арил, CO2NH2, СO2аралкил, CH2SO3H, CH2SO2NH2, CH2PO(OH)2, 1-Н-тетразолил, СНО, СОСН3, СН2ОН, CH2NH2, СН2NHалкил, CH2N(алкил)алкил′, СН2ОСН3, CH2SH,
где все индексы, символы и заместители имеют значения, указанные в п.1.
3. Химические соединения, имеющие общую структуру формулы (С) или (D) по п.1 или 2.
4. Применение соединений, имеющих структуру формулы (С) по п.1, для получения лекарственного средства для лечения хронического обструктивного заболевания легких (COPD), острого повреждения легких (ALI), при экстракорпоральном кровообращении, для лечения острого респираторного дистресс-синдрома (ARDS), болезни Крона, септического шока, сепсиса, хронических воспалительных заболеваний, таких как псориаз, атопический дерматит и ревматоидный артрит, и реперфузионного повреждения, которое имеет место после сердечных приступов, мозговых кровоизлияний, атеросклероза и трансплантации органов, травматического шока, недостаточности деятельности многих органов, аутоиммунных заболеваний, подобных рассеянному склерозу, при подкожной внутрипросветной ангиопластике, для лечения астмы и воспалительного заболевания кишечника.
5. Применение соединений, имеющих структуру формулы (С) по п.1, для получения лекарственного средства для лечения, диагностики или профилактики воспалительных нарушений.
6. Применение соединений, имеющих структуру формулы (С) по п.1, для получения наполнителя для доставки лекарственного средства к органу-мишени при диагностике или терапии.
7. Применение соединений, имеющих структуру формулы (С) по п.1, для получения косметической или дерматологической композиции.
8. Косметические композиции, включающие в себя, по меньшей мере, одно соединение формулы (С) по п.1 и, по меньшей мере, один косметически переносимый компонент.
9. Дерматологические композиции, включающие в себя, по меньшей мере, одно соединение формулы (С) по п.1 и, по меньшей мере, один дерматологически переносимый компонент.
| ЕР 1577289 А1, 21.09.2005 | |||
| М.Leech, et al., Endogenous glucocorticoids modulate neutrophil migration and synovial P-selectin but not neutrophil phagocytic or oxidative function in experimental arthritis., Clin Exp Immunol | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| Andrew C., Thomas В., The Role of E-Selectin, P-Selectin, and Very | |||

