Настоящее изобретение относится к соединениям, композициям и способам модулирования in vitro и in vivo процессов, опосредованных молекул клеточной адгезии. Описываемые малые молекулы являются ароматическими нитросоединениями, которые эффективно моделируют функции, опосредованные молекулами клеточной адгезии.
Функции, опосредованные молекулами клеточной адгезии являются частью сложного каскада, приводящего к миграции циркулирующих белых кровяных телец (лейкоцитов) из тока крови в окружающую ткань (трансмиграция). С точки зрения физиологии, трансмиграция лейкоцитов является исключительно важной для гомеостаза и иммунного надзора живых существ, включая людей. Лимфоциты, например, по своей природе покидают ток крови и переходят в лимфатические ткани для «патрулирования» в поисках вредных антигенов. Однако в обстоятельствах патологии, например, при местном или общем воспалении и/или повреждении сосудистой системы, этот основной процесс разупорядочивается, по меньшей мере, частично, в силу повышенной экспрессии E- и P-селектинов на поверхности. В результате избыточная трансмиграция лейкоцитов приводит к патологическим клеточным инфильтратам, с последующим повреждением ткани в некоторых клинически важных случаях. Такие болезненные состояния, как острое повреждение легких (ОПЛ), острый респираторный дистресс-синдром (ОРДС), бронхиальная астма (астма), хроническое обструктивное заболевание легких (ХОЗЛ), псориаз, ревматоидный артрит и сепсис, все связаны с воспалением тканей, вызванным и постоянно сохраняемым патологически активированной инфильтрацией лейкоцитов в соответствующие ткани. К тому же, чрезмерно увеличенная инфильтрация лейкоцитов способствует патогенезу ишемического/реперфузионного повреждений, связанных с трансплантацией органов, искусственным кровообращением или чрескожной транслюминальной ангиопластикой.
Для трансмиграции лейкоциты должны связаться со стенкой сосудистого эндотелия, чтобы продиффундировать через клеточную стенку в окружающую ткань. Таким образом, лейкоциты должны свернуться на стенке эндотелиальной клетки и затем связаться с ней (начальное сворачивание, или «привязывание»). Это первичное событие в трансмиграции протекает с участием молекул клеточной адгезии семейства селектинов. В дополнение к прямому связыванию с эндотелием, лейкоциты могут связываться с другими лейкоцитами, лейкоцитными частицами, тромбоцитами или образованными из тромбоцитов частицами, которые уже прикреплены к эндотелию.
Семейство селектинов молекул клеточной адгезии состоит из трех структурно родственных кальций-зависимых белков, связывающихся с поверхностью клетки посредством углеводных остатков, E-, P- и L-селектинов. E-селектин экспрессируется только на воспаленном эндотелии, P-селектин экспрессируется как на воспаленном эндотелии, так и на тромбоцитах, L-селектин экспрессируется на лейкоцитах. Селектины состоят из амино-терминированого лектинного домена, подобного эпидермальному фактору роста (ЭФР) домена, различного числа соотносящихся с комплементарным рецептором повторов, гидрофобного трансмембранного домена и углерод-терминированного цитоплазмического домена. Предполагается, что связывающие взаимодействия, приводящие к адгезии лейкоцитов, реализуются посредством контакта лектинного домена селектинов и различных углеводных лигандов на поверхности лейкоцитов.
Все три селектина могут связываться с углеводом сиалил Льюисx (sLex), присутствующим на поверхности большинства лейкоцитов глигозидным фрагментом, с низким сродством. Структурно родственный гликозидный фрагмент, сиалил Льюисa (sLea), преимущественно обнаружен на поверхности раковых клеток [K. Okazaki et al., J. Surg. Res., 1998, 78(1), 78-84; R. P. McEver et al., Glycoconjugate Journal, 1997, 14(5), 585-591]. В случае P-селектинов был описан гликопротеиновый лиганд с явно выраженным высоким сродством [R.P. McEver, R.D. Cummings, J.Clin.Invest., 1997, 100, 485-492], так называемый P-селектина гликопротеиновый лиганд-1 (PSGL-1), в котором высокое сродство связывания с селектином обеспечивается как его sLex-фрагментом, так и частью его пептидных компонентов, в частности сульфатированными тирозиновыми остатками [R.P.McEver, Ernst Schering Res. Found. Workshop, 2004, 44, 137-147]. PSGL-1 является одним из наиболее важных селектиновых лигандов, связываясь с высоким сродством с P-селектином, но он также связывается с E- и L-селектином [G. Constantin; Drug News Perspect; 2004; 17(9); 579-586]. Он является гомодимерным сиаломуцином, преимущественно экспрессированным на лейкоцитах.
При воспалительных заболеваниях разупорядоченная трансмиграция обеспечивается, по меньшей мере отчасти, посредством повышенной экспрессии E- и P- селектинов на поверхности клеток. В отличие от их низкой базальной экспрессии, экспрессия E- и P-селектинов повышается во время воспаления, приводя к существенному привлечению лейкоцитов в воспаленную ткань. Хотя клеточная адгезия, опосредованная селектинами, требуется для борьбы с инфекцией, существуют различные ситуации, в которых такая клеточная адгезия является нежелательной или избыточной, приводя к тяжелым повреждениям ткани вместо восстановления. В случае многих как острых, так и хронических нарушений (т.к. астма, хроническое обструктивное заболевание легких (ХОЗЛ), псориаз и т.д.) была показана связь инфильтрации активированных лейкоцитов в ткани с одновременным явным усилением экспрессии соответствующих молекул адгезии, в частности, E- и P-селектина, в тканях. [Muller et al., J. Pathol., 2002, 198(2), 270-275; Di Stefano et al., Am. J. Respir. Crit. Care. Med., 1994, 149(3), 803-810; Terajima et al., Arch. Dermatol. Res., 1998, 290, 246-252].
Инфильтрация лейкоцитов может также играть роль в воспалительных симптомах при отторжении трансплантата и пересаженных тканей. Также процесс свертывания крови дополнительно промотируется лейкоцит-лейкоцит и лейкоцит-тромбоцит связываниями, которые реализуются потому, что лейкоциты имеют как L-селектин, так и его соответствующий лиганд PSGL-1, и таким образом могут взаимодействовать с другими лейкоцитами через PSGL-1 и могут также связываться с тромбоцитами, которые несут P-селектин.
Следовательно, модулирование клеточной адгезии с участием селектина и других функций с их участием, например, активации лейкоцитов, предлагает многообещающую возможность вмешиваться в воспалительный каскад и останавливать его на очень ранней стадии. Для предотвращения возможного избыточного наличия селектинов низкомолекулярные антагонисты селектина должны модулировать все три селектина одновременно как пан-селектиновые антагонисты [M. Sperandio et al., Vascular Disease Prevention, 2004, 1, 185-195].
Помимо sLex/sLea природный лиганд высокого сродства PSGL-1 является еще одной структурой-шаблоном для разработки низкомолекулярных антагонистов селектина. По сравнению с sLex/sLea, PSGL-1 проявляет высокое сродство ко всем трем селектинам. Нахождение и обнаружение новых низкомолекулярных лекарственных средств, которые конкурируют с PSGL-1 и PSGL-1-подобными лигандами за связывание с селектином, является, таким образом, многообещающей стратегией разработки нового класса эффективных пан-селектиновых антагонистов для лечения воспалительных заболеваний.
Селектиновые антагонисты могут быть разработаны с использованием в качестве структуры-шаблона как селектинов, так и лигандов типа PSGL-1, т.к. они предназначены для модулирования связывания между селектинами и PSGL-1 или другими лигандами с похожими связывающими фрагментами.
Новые низкомолекулярные антагонисты селектина могут столкнуться с определенными требованиями для того, чтобы соответствовать понятию лекарственного средства и обладать потенциальной биодоступностью при пероральном введении. Термин соответствия лекарственному средству описан в литературе [Lipinski; Adv. Drug Dev. Rev., 1997, 23, 3-25]. Помимо иных молекулярных свойств для соответствия лекарству подразумевается, что пассивно транспортируемые молекулы в среднем имеют относительную молекулярную массу менее 500. Согласно этим правилам, общепринято определять соединения с относительной молекулярной массой менее 500 или несколько выше как малые молекулы (низкомолекулярные вещества). Соединения с относительной молекулярной массой выше 500 скорее всего не являются биодоступными при пероральном введении. Также с концепцией соответствия лекарственному средству не согласуется наличие высокополярных углеводных фрагментов или пептидных компонентов. [H. Ulbrich et al., Trends Pharmacol. Sci., 2003, 24(12), 640-647; D. Slee et al., J. Med. Chem., 2001, 44, 2094-2107]. То же самое имеет значение при разработке медикаментов на основе антител, поскольку они являются полипептидами и в силу этого пероральное введение является проблематичным. Более того, желаемые соединения должны оставаться стабильными при прохождении через желудочно-кишечный тракт, т.к. они должны быть поглощены/впитаны после клетками тонкого кишечника. Такая ситуация не имеет места в случае большинства гликозидных молекул и пептидных структур.
Некоторые нитрокатехолы, выступая как специфические ингибиторы катехол-О-метилтрансферазы (COMT), были предложены для лечения болезни Паркинсона [J. Axelrod et al., J. Biol. Chem., 1958, 233(3), 702-705; P. T. Mānnisto et al., Pharmacological Reviews, 1999, 51, 593-628], например толкапон, нитекапон и энтакапон [EP00426468]. Также нитрокатехолы описаны как обладающие кардиозащитным действием, вызванным хелатированием железа [N. Haramaki et al., Biochemical Pharmacology, 1995, 50, 839-843] или снижением уровня гомоцистеина в плазме [E. Nissinen et al., J. Neural Transm., 2005, 112, 1213-1221]. Нитекапон также заявлялся как перехватчик свободных радикалов [Y. J. Suzuki et al., Free Radical Biology & Medicine, 1992, 13, 517-525; L. Marcocci et al., Biochemistry and Molecular Biology International, 1994, 34, 531-541]. Некоторые наблюдения показывают, что энтакапон может защищать от вызываемого ангиотензином II повреждения почек [T. Helkamaa et al., J. Hypertens., 2003, 21, 2353-2363].
Однако вплоть до настоящего времени нитрокатехолы не были описаны в качестве модулирующих селектин соединений. Для разработки низкомолекулярных соединений с модулирующим процессы, опосредованные селектином действием были предприняты разнообразные исследования. Такие соединения включают дисалицилаты и С-глигозиды на основе дисалицилатов [WO 99/29706], бинзиламиносульфоновые кислоты [WO 03/097658], дигликозилированные 1,2-диолы [WO 97/01569], замещенные пятичленные гетероциклы [WO 00/33836], маннопиранозилоксифенилбензойные кислоты [EP0758243 B1], соединения на основе пиперазина [US6432957B1], производные галловой кислоты и пептидов [WO 2004/018502], галловую кислоту [C. C. M. Appeldoorn et al., Circulation, 2005, 111, 106-112; EP 1481669A1] и производные хинной кислоты [N. Kaila et al., J. Med. Chem., 2005, 48, 4346-4357].
Однако ни одно из этих селектин-антагонизирующих соединений не прошло успешно клинические испытания к настоящему времени. [S. J. Romano, Treat. Respir. Med., 2005, 4(2), 85-94; M. P. Schōn, Therapeutics and Clinical Risk Management, 2005, 1(3), 201-208]. Это является следствием того факта, что многие из этих структур были разработаны на основе слабосвязывающего шаблона sLex. Следовательно, sLex-миметические структуры, скорее всего, проявляют слабое действие. Другие соединения демонстрируют специфичность по отношению к различным членам семейства селектинов, однако антагонизация одного отдельного селектина может быть обойдено другими селектинами [M. P. Schōn, Therapeutics and Clinical Risk Management, 2005, 1(3), 201-208].
К тому же большинство соединений, разработанных к настоящему времени, обладают высокой молекулярной массой и зачастую содержат углеводы и/или пептиды, что делает их подверженными деградации и модификации пептидазами и/или гликозидазами. Углеводсодержащие структуры обладают и другими недостатками, такими как высокая степень хиральности, наличие аномерных центров и связанные с этим явления, низкая вероятность транспорта через липидный двойной слой. Подобные недостатки известны и для пептидсодержащих соединений. Некоторые другие соединения, разработанные для антагонизации процессов с участием селектина, содержат пирогаллольные субструктуры. Эти фрагменты склонны подвергаться окислительным процессам [Kumamoto M. et al., Biosci. Biotechnol. Biochem., 2001, 65(1), 126-132], что делает фармацевтическую разработку этих соединений затруднительной. К тому же известно, что соединения с пирогаллольными субструктурами, такие как галловая кислота, могут быть цитотоксичными [E. Sergediene et al., FEBS Letters, 1999, 462, 392-396] и вызывать апоптоз [K. Satoh et al., Anticancer Research, 1997, 17, 2487-2490; N. Sakaguchi et al., Biochemical Pharmacology, 1998, 55, 1973-1981].
Ведущим соединением среди антагонистов селектина является бимосиамоза (bimosiamose) [S. J. Romano, Treat. Respir. Med., 2005, 4(2), 85-94]. В настоящий момент бимосиамоза [D. Bock et al., New Drugs, 2003, D04, 28, p.28; EP 0 840 606 B1] является наиболее отлаженным соединением в клинических исследованиях. Недавние исследования подтвердили гипотезу, что бимосиамоза может рассматриваться как PSGL-1-миметик [E. Aydt, G. Wolff; Pathobiology; 2002-2003; 70; 297-301]. Это отличает бимосиамозу от других антагонистов селектина. Она, однако, имеет большую молекулярную массу и имеет углеводные структуры. Представляется, что пан-селектиновому антагонисту бимосиамозе недостает биодоступности при пероральном введении. Некоторые наблюдения свидетельствуют, что бимосиамоза демонстрирует хорошее сродство к P-селектину и умеренное сродство к E- и L-селектинам.
Имеется явно выраженная медицинская потребность в новых высокоэффективных пан-селектиновых антагонистах, которые модулируют функции, опосредованные селектинами, например селектин-зависимую клеточную адгезию, и в развитии способов вовлечения таких соединений для модулирования обстоятельств, связанных с взаимодействием лиганд-селектин. Большинство доступных противовоспалительных фармацевтических лечебных средств, доступных на рынке, включают, в основном, кортикостероиды или НСПВС (нестероидные противовоспалительные средства), имеющие несколько серьезных недостатков/побочных эффектов и направленные на различные стадии воспалительного каскада. В отличие от этого модулирование селектиновых функций является концепцией терапии, вмешивающейся в воспалительный каскад на очень ранней стадии. Почти все перспективные антагонисты селектина до настоящего времени не становились лекарственными средствами на рынке, в основном по причине низкой эффективности и/или большого молекулярного веса, что вызывает проблемы в их поведении в рамках абсорбции/распределения/метаболизма/выведения (АРМВ) и тем самым в биодоступности посредством перорального введения, требующейся для лечения большинства воспалительных заболеваний таких, как ревматоидный артрит, септический шок, атеросклероз, реперфузия и многие другие.
EP-A 1 577289 описывает ароматические соединения, гидроксилированные в фенильном кольце молекулы и которые могут быть использованы для лечения воспалительных заболеваний.
В WO 1997/01335 описаны ароматические соединения, связанные с углеводным остатком. Соединения, описанные в этом патенте, являются ингибиторами селектина и могут быть использованы как терапевтические средства, однако они структурно отличны от нитрозамещенных ароматических соединений по настоящему изобретению.
В EP-A 1 481669 описаны полигидроксифенолы, которые обладают P-селектин модулирующим действием.
Одной целью настоящего изобретения является предложение новых низкомолекулярных соединений, особенно негликозилированных/негликозидных и непептидных соединений, которые способны эффективно антагонизировать процессы, опосредованные селектинами и которые имеют меньшие отрицательные побочные эффекты в ходе их применения, чем соединения, известные из предшествующего уровня техники.
В отличие от большинства sLex-миметиков, разработанных в данной области, соединения по изобретению не подвержены действию гликозидаз или пептидаз. Большинство антагонистов селектина, разработанных к настоящему времени, основаны структурно и биологически на свойствах sLex или sLea. Получающиеся соединения показывают поэтому низкую биологическую активность, как и соединения-матрицы. Настоящее изобретение, однако, относится к новым мощным низкомолекулярным и соответствующим лекарственному средству пан-селектиновым антагонистам, которые были созданы на основе биологических in vitro исследований, имитируя PSGL-1 и PSGL-1-подобные лиганды или любые лиганды, несущие sLex или sLea и тирозинсульфатный фрагменты. [N. V. Bovin; Biochem. Soc. Symp.; 2002; (69): 143-60. N. V. Bovin; Glycoconj. J.; 1998; 15(5); 431-46. T. V. Pochechueva et al.; Bioorg. Med. Chem. Lett.; 2003 ; 13(10); 1709-12. G. Weitz-Schmidt et al.; Anal. Biochem.; 1996; 238; 184-190].
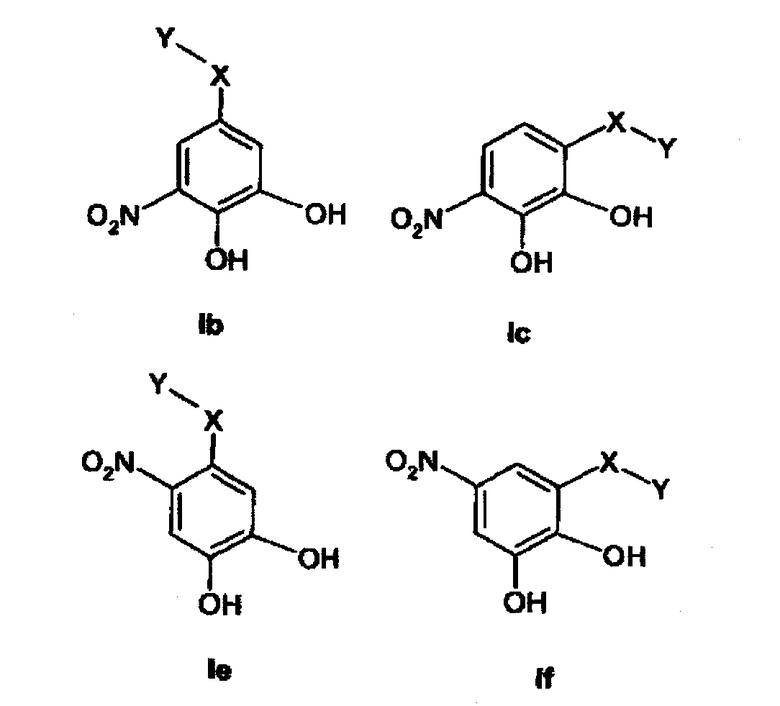
Настоящее изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение, описываемое формулой (Ia) или (Ib) или (Ic) или (Id) или (Ie) или (If), и фармацевтически приемлемый носитель, используемый в медицине,

где символы и заместители имеют следующее значение:
-X- представляет собой
(a)

m равно 0, 1; n равно целому числу от 1 до 3
(b)

где “ring” является
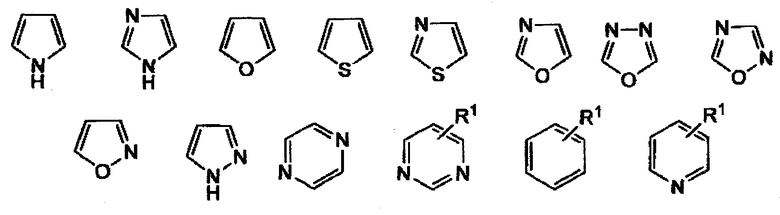
и R1 представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, NH2, NHалкил, NHарил, NHацил и k равно 0, 1,
(c)
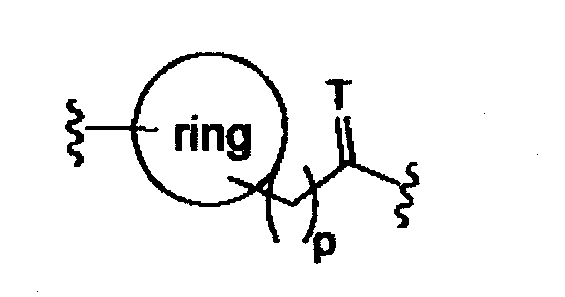
T представляет собой O, S или [H,H]; p равно 0, 1, 2,
(d)

двойная связь имеет E- или Z- конфигурацию
(e)

(f)
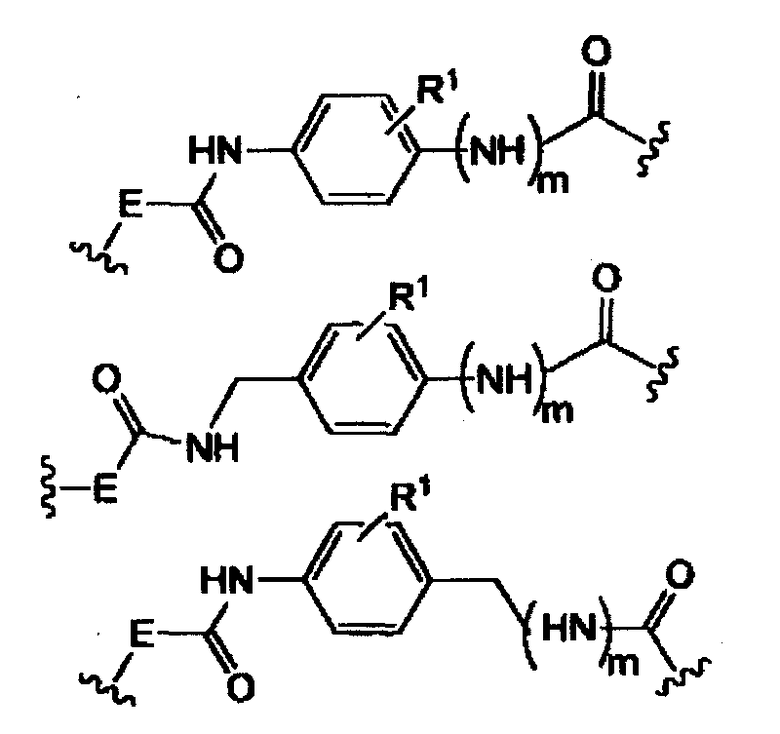
-E- представляет собой -(CH2-)qNH- и q равно 0, 1, 2, 3
-Y представляет собой
(a)

s равно 0 или 1,
R2 представляет собой CO2H, CO2алкил, CO2арил, CO2NH2, CO2аралкил, SO3H, SO2NH2, PO(OH)2, 1-H-тетразолил-, CHO, COCH3, CH2OH, NH2, NHалкил, N(алкил)алкил', OCH3, CH2OCH3, SH, F, Cl, Br, I, CH3, CH2CH3, CN, CF3,
R3, независимо от R2, представляет собой H, CH3, CH2CH3, CF3, F, Cl, Br, I, CN, NO2,
R4, независимо от R2 и R3, представляет собой H, CH3, CH2CH3, CF3, F, Cl, Br, I, CN, NO2, R2,
R5 представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2 и
-W- представляет собой -(CH2-)V, цис-CH=CH- или транс-CH=CH-, и v равно 0, 1, 2;
в случае, если -W- является цис-CH=CH- или транс-CH=CH-, R2 не должен быть NH2 или SH;
(b)

R6, независимо от R2, представляет собой H, F, Cl, Me, трет-Bu, CN, NH2,
(c)
(d)

(e)

t равно 0, 1, 2
(f)

(g)

-Z представляет собой
(i)

R7, независимо от R2, представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2,
(ii)

R8, независимо от R2, представляет собой H, F, Cl, Me, трет-Bu, CN, NH2,
(iii)
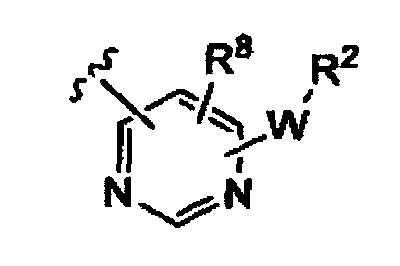
(iv)
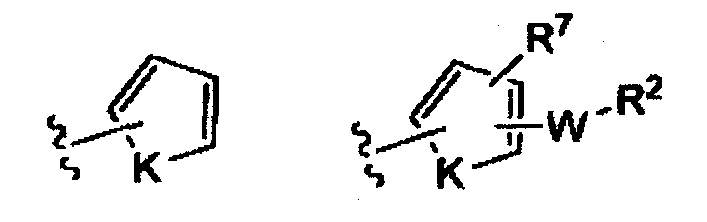
K представляет собой NH, NMe, O, S,
(v)

(vi)

(vii)
-W-R2
или фармацевтически приемлемые соли, сложные эфиры или амиды и пролекарства определенных выше соединений формулы (Ia) или (Ib) или (Ic) или (Id) или (Ie) или (If).
В предпочтительном варианте осуществления изобретения композиция включает по меньшей мере одно соединение формулы (Ib) или (Ic) или (Ie) или (If) и фармацевтически приемлемый носитель
где символы и заместители имеют следующее значение
-X- представляет собой
(a)

m равно 0, 1; n равно целому числу от 1 до 3
(b)

где «ring» является

и R1, представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, NH2, NHалкил, NHарил, NHацил и k равно 0, 1.
(e)

- Y представляет собой
(a)

s равно 0 или 1,
R2 представляет собой CO2H, CO2алкил, CO2арил, CO2NH2, CO2аралкил, SO3H, SO2NH2, PO(OH)2, 1-H-тетразолил-, CHO, COCH3, CH2OH, NH2, NHалкил, N(алкил)алкил', OCH3, CH2OCH3, SH, F, Cl, Br, I, CH3, CH2CH3, CN, CF3,
R3, независимо от R2, представляет собой H, CH3, CH2CH3, CF3, F, Cl, Br, I, CN, NO2 и
R4, независимо от R2 и R3, представляет собой H, CH3, CH2CH3, CF3, F, Cl, Br, I, CN, NO2, R2,
R5 представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2,
и -W- представляет собой -(CH2-)V, цис-CH=CH- или транс-CH=CH-, и v равно 0, 1, 2;
в случае, если -W- представляет собой цис-CH=CH- или транс-CH=CH-, R2 не должен быть NH2 или SH;
(e)
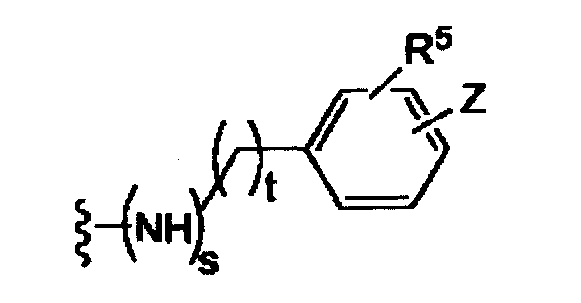
t равно 0, 1, 2
(f)
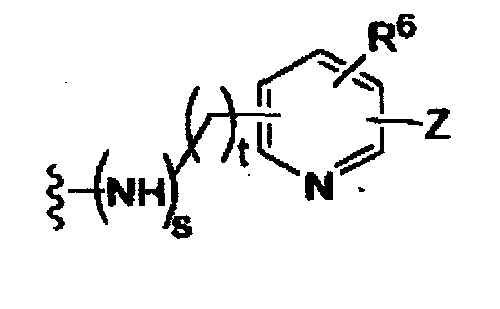
(g)
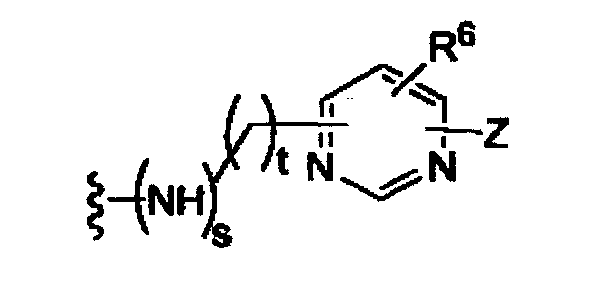
-Z представляет собой
(i)

R7, независимо от R2, представляет собой H, NO2, CF3, F, Cl, Br, I, CN, CH3, OCH3, SH, NH2,
(iv)
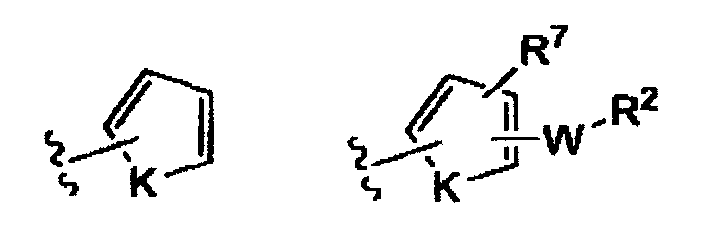
K представляет собой NH, NMe, O, S,
(v)
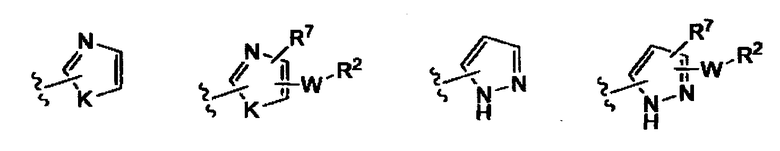
или фармацевтически приемлемые соли, сложные эфиры или амиды и пролекарства определенных выше соединений формулы (Ib) или (Ic) или (Ie) или (If).
Предпочтительные фармацевтические композиции включают соединения формул (IIa) или (IIb) или (IIc) или (IId) или (IIe) или (IIf)
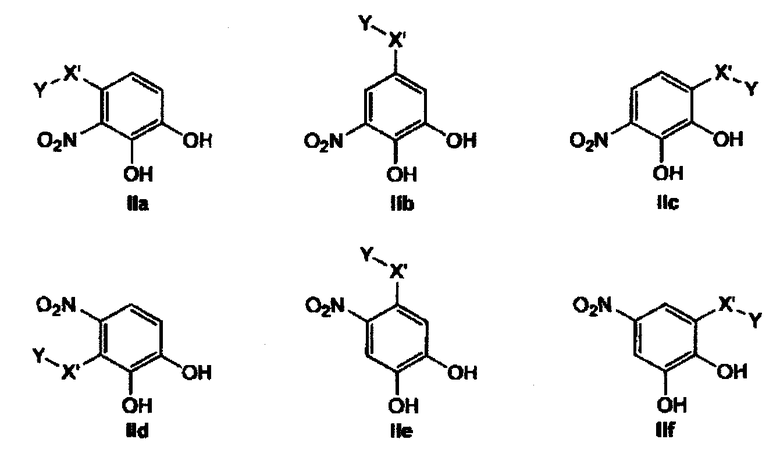
где -Y является таким, как определено выше и в которых -X'- является X(a), X(b), X(c) и X(d) как определено выше.
В еще одном варианте осуществления изобретение относится к композициям, содержащим соединения структур:

где -Y и -X'- как X(a), X(b) являются такими, как определено выше.
Другие предпочтительные фармацевтические композиции включают соединения формул (A1) или (A2) или (A3) или (A4) или (A5) или (A6) или (B1) или (B2) или (B3) или (B4) или (B5) или (B6)
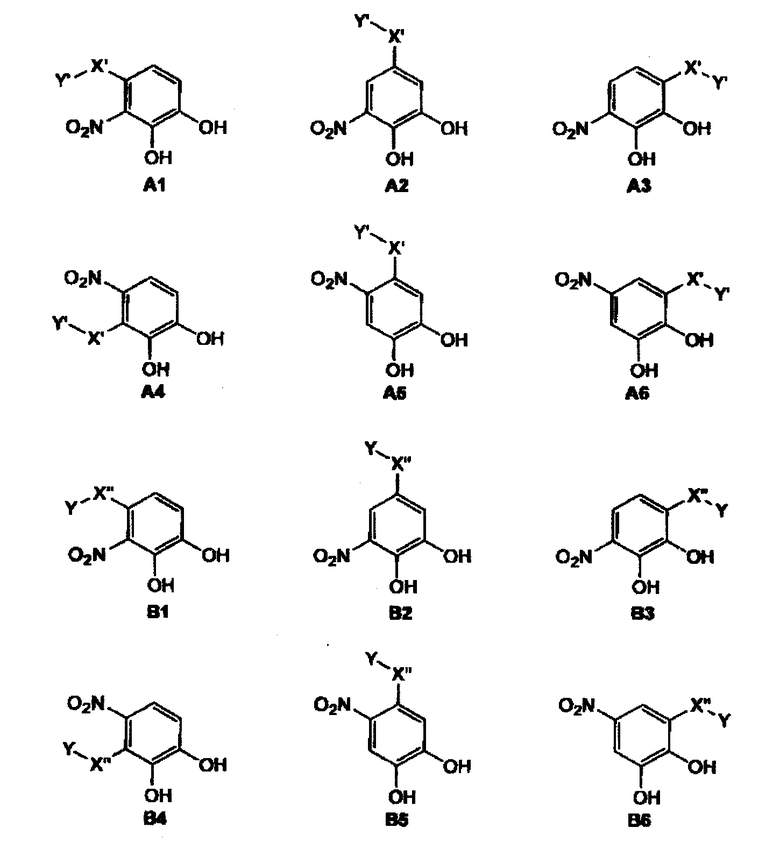
где -Y и -X'- являются такими, как определено выше и в которых -X”- является

и где Y' является

где все индексы, символы и заместители являются такими, как определено выше.
В еще одном варианте осуществления изобретение относится к фармацевтическим композициям, содержащим соединения формулы:

где -X'- и -Y являются такими, как определено выше и в которых -X”- является

и где Y' является
где все индексы, символы и заместители являются такими, как определено выше.
Особенно предпочтительные фармацевтические композиции содержат соединения формул (C1) или (C2) или (C3) или (C4) или (C5) или (C6)

где -X”- и -Y' являются такими, как определено выше.
Другая предпочтительная группа соединений соответствующей композиции имеет следующие структуры
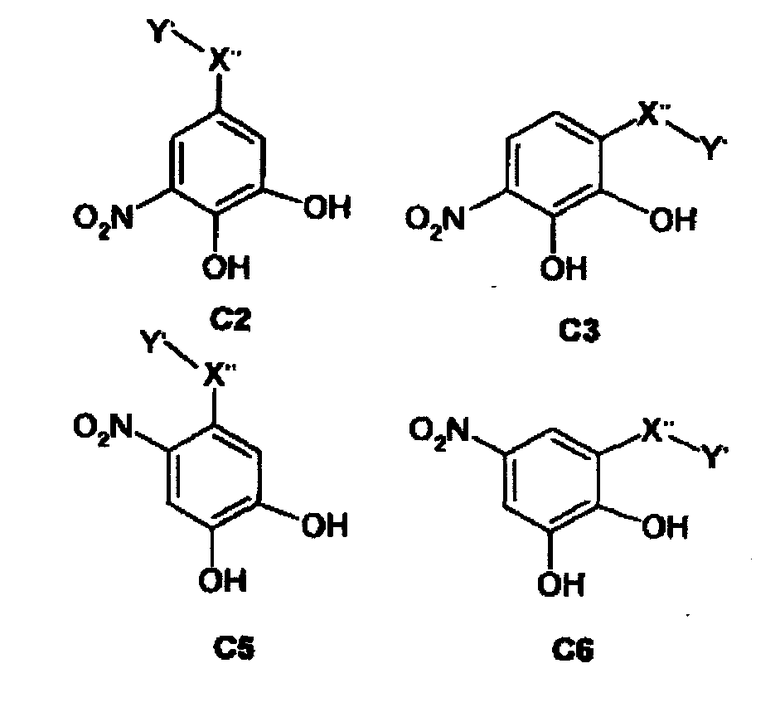
где -X”- и -Y' являются такими, как определено выше.
Наиболее предпочтительные фармацевтические композиции содержат соединения формул (D1) или (D2) или (D3) или (D4) или (D5) или (D6)

где -X”- является таким, как определено выше, и -Y” является
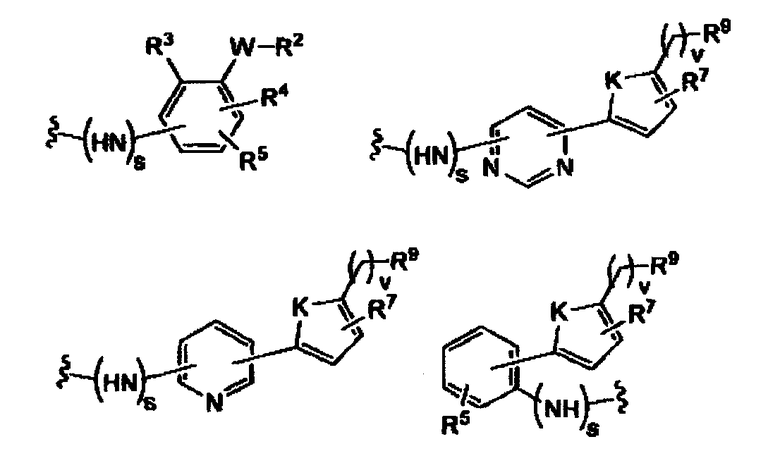
R9 представляет собой CO2H, CO2алкил, CO2арил, CO2NH2, CO2аралкил, CH2SO3H, CH2SO2NH2, CH2PO(OH)2, 1-H-тетразолил, CHO, COCH3, CH2OH, CH2NH2, CH2NHалкил, CH2N(алкил)алкил', CH2OCH3, CH2SH.
Также предпочтительными являются фармацевтические композиции, в которых соединения определены формулами (D2) или (D3) или (D5) или (D6).

где -X”- является таким, как определено выше, и -Y” является
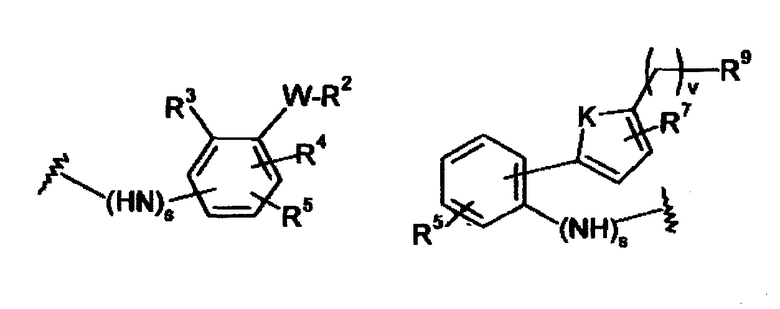
R9 представляет собой CO2H, CO2алкил, CO2арил, CO2NH2, CO2аралкил, CH2SO3H, CH2SO2NH2, CH2PO(OH)2, 1-H-тетразолил, CHO, COCH3, CH2OH, CH2NH2, CH2NHалкил, CH2N(алкил)алкил', CH2OCH3, CH2SH,
в которых все индексы, символы и заместители являются такими, как определено выше.
Эти химические соединения (C1), (C2), (C3), (C4), (C5), (C6), (D1), (D2), (D3), (D4), (D5) и (D6) являются также новыми соединениями сами по себе. Химические соединения, имеющие общую структуру формулы (C2) или (C3) или (C5) или (C6) или (D2) или (D3) или (D5) или (D6) являются предпочтительными.
Все описанные выше соединения проявляют способность модулированию клеточной адгезии и модулируют связывание, опосредованное селектином, а также аналога PSGL-1. Соединения имеют способность модулировать взаимодействие селектинов с sLex/sLea, а также взаимодействия между селектинами и тирозинсульфатными остатками. По сравнению с бимосиамозой, ведущим пан-селектиновым антагонистом, описанные здесь соединения показывают увеличенную биологическую активность. Поэтому они являются полезными для лечения острых и хронических воспалительных заболеваний, равно как и других медицинских состояний, в которых процессы, опосредованные селектином играют роль.
Термин «фармацевтический» включает также диагностические применения.
Термин «фармацевтический» включает также профилактические применения для предотвращения медицинских состояний, в которых играют роль процессы, опосредованные селектином.
Термин «фармацевтический» включает также применения, в которых соединения по настоящему изобретению могут быть использованы как носитель для введения лекарственного средства при диагностике или терапии.
Изобретение относится к фармацевтическим композициям, содержащим соединения в соответствии с формулами (Ia) или (Ib) или (Ic) или (Id) или (Ie) или (If) и в предпочтительном варианте в соответствии с формулами (IIa) или (IIb) или (IIc) или (IId) или (IIe) или (IIf).
В еще одном предпочтительном варианте изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение, соответствующее формуле (A1) или (A2) или (A3) или (A4) или (A5) или (A6) или (B1) или (B2) или (B3) или (B4) или (B5) или (B6).
В особенно предпочтительном варианте изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение, соответствующее формуле (C1) или (C2) или (C3) или (C4) или (C5) или (C6).
В предпочтительном варианте изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение, соответствующее формуле (D1) или (D2) или (D3) или (D4) или (D5) или (D6).
В еще одном варианте осуществления изобретения соединения, соответствующие формулам A2, A3, A5, A6, B2, B3, B5, B6, C2, C3, C5, C6, D2, D3, D5 или D6, используются в композиции.
Настоящее изобретение далее относится к способу модулирования связывания P-селектина, L-селектина или E-селектина c sLex или sLea и тирозинсульфатных остатков, включающий стадию введения пациенту эффективного количества по меньшей мере одного соединения, имеющего формулу (Ia) или (Ib) или (Ic) или (Id) или (Ie) или (If) для модулирования связывания P-, L- или E-селектина c sLex или sLea и тирозинсульфатным остатком. Было обнаружено, что соединения, имеющие формулу (Ia) или (Ib) или (Ic) или (Id) или (Ie) или (If), показанные выше, выступают в качестве модуляторов связывания P-, L- или E-селектина.
Используемый здесь термин «алкил» следует понимать как одновалентную, с неразветвленной или разветвленной цепочкой, группу из 1 или 2 или 3 или 4 или 5 или 6 или 7 или 8 или 9 или 10 или 11 или 12 углеродными атомами, включая, но не ограничиваясь перечисленным, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил и т.п. «Алкил» является независимым один от другого и может отличаться или совпадать.
Термин «арил» следует понимать как карбоциклические или гетероциклические ароматические группы, включая, но не ограничиваясь, фенил, 1-нафтил, 2-нафтил, флуоренил, 1,2-дигидронафтил, инденил, инданил, тиенил, бензотиенил, тиенопиридил и т.п.
Термин «аралкил», также называемый «арилалкил» следует понимать как арильную группу, прикрепленную к алкильной группе, включая, но не ограничиваясь, бензил, 1-нафтилметил, 2-нафтилметил, фторбензил, хлорбензил, бромбензил, иодбензил, алкоксибензил (где «алкокси» означает метокси, этокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси и т.п.), гидроксибензил, аминобензил, нитробензил, гуанидинобензил, флуоренилметил, фенилметил (бензил), 1-фенилэтил, 2-фенилэтил, 1-нафтилэтил и т.п.
Термин «ацил» следует понимать как -(CHO) или -(С=O)-арил или -(С=O)-алкил или -(С=О)-аралкил, включая, но не ограничиваясь, формил, ацетил, н-пропионил, изопропионил, н-бутирил, изобутирил, пивалоил, бензоил, 4-нитробензоил и т.п.
Используемый здесь термин «фармацевтически приемлемые соли, сложные эфиры, амиды и пролекарства» относится к тем солям карбоновых кислот, солям присоединения аминокислот, сложным эфирам, амидам и пролекарствам соединений по настоящему изобретению, которые, в рамках действующих медицинских представлений, подходят для использования при контакте с тканями пациента без чрезмерной токсичности, раздражающего действия, аллергического отклика и т.п., обладают соответствующим разумным отношением польза/риск, эффективны в назначаемом использовании, равно как и цвиттерионные формы, где они возможны, соединений по настоящему изобретению. Термин «соли» относится к солям присоединения относительно нетоксичных неорганических и органических кислот соединений по настоящему изобретению. Эти соли могут быть получены in situ во время конечного выделения и очистки соединений или же отдельным вовлечением очищенных соединений в их свободной форме в реакцию с соответствующей неорганической или органической кислотой или основанием и выделения таким образом полученной соли. Типичные соли соединений по изобретению включают гидробромид, гидрохлорид, сульфат, бисульфат, нитрат, ацетат, оксалат, валерат, пальмитат, стеарат, лаурат, борат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактиобионат, лаурилсульфонат и т.п. Они могут включать катионы щелочных и щелочноземельных металлов, таких как натрий, литий, калий, кальций, магний и т.п., также как нетоксичные катионы аммония, четвертичного аммония и аминов включая, но не ограничиваясь, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, этиламин и т.п.
Примеры фармацевтически приемлемых нетоксичных сложных эфиров соединений по настоящему изобретению включают C1, C2, C3, C4, C5 и C6 алкиловые сложные эфиры, в которых алкильная группа имеет прямую или разветвленную цепь. Приемлемые сложные эфиры также включают C5, C6 и C7 циклоалкиловые сложные эфиры, равно как и арилалкиловые эфиры, такие как, но не ограничиваясь, бензиловый. C1, C2, C3, C4, C5 и C6 алкиловые сложные эфиры являются предпочтительными. Сложные эфиры соединений по настоящему изобретению могут быть получены общепринятыми способами.
Примеры фармацевтически приемлемых нетоксичных амидов соединений по этому изобретению включают амиды-производные аммиака, первичных C1, C2, C3, C4, C5 и C6 алкиламинов и вторичных C1, C2, C3, C4, C5 и C6 диалкиламинов, в которых алкильная группа имеет прямую или разветвленную цепь. В случае вторичных аминов амин может быть в виде пяти- или шестичленного гетероцикла, содержащего один атом азота. Амиды - производные аммиака, C1, C2 и C3 первичные алкиламины и C1, C2 вторичные диалкиламины являются предпочтительными. Амиды соединений по настоящему изобретению могут быть получены с использованием общепринятых способов.
Термин «пролекарство» относится к одному или более соединений, которые легко трансформируются in vitro и из неактивного в активное состояние in vivo с образованием соединения, родственного вышеупомянутым формулам (Ia) или (Ib) или (Ic) или (Id) или (Ie) или (If), например, посредством гидролиза в крови или метаболизма in vivo.
Также предполагается, что фармацевтически активная композиция может содержать соединения по настоящему изобретению или другие соединения, которые модулируют или составляют конкуренцию в связывании E-селектина или P-селектина или L-селектина.
Фармацевтически активные композиции по настоящему изобретению включают фармацевтически приемлемый носитель и соединение по формуле (Ia) или (Ib) или (Ic) или (Id) или (Ie) или (If), в каком случае фармацевтически приемлемый носитель может также быть медицински подходящей наночастицей, дендримером, липосомой, микропузырьком или полиэтиленгликолем (ПЭГ). Фармацевтические композиции по настоящему изобретению могут включать одно или более соединений, имеющих вышеприведенные структуры (Ia) или (Ib) или (Ic) или (Id) или (Ie) или (If), составленные вместе с одним или более физиологически приемлемым носителем, адъювантом или наполнителем, которые все вместе описываются здесь как носители, для парентерального введения, для перорального введения в жидкой или твердой форме, для ректального или местного введения и т.п.
Композиции могут быть введены человеку или животным перорально, ректально, парентерально (внутривенно, внутримышечно, внутрикожно или подкожно), интрацистернально, вагинально, внутрибрюшно, местно (порошки, мази или капли) или буккально или ингаляционно (распыленным или в виде назальных спреев).
Композиции, подходящие для парентерального введения, могут включать физиологически приемлемые стерильные водные или неводные растворы, стабилизаторы, антиоксиданты, консерванты (например, аскорбиновую кислоту, сульфит натрия, гидросульфит натрия, бензиловый спирт, ЭДТА), дисперсии, суспензии или эмульсии и стерильные порошки для воссоздания в стерильный вводимый раствор или дисперсию. Примеры подходящих водных или неводных носителей, разбавителей, растворителей или носителей включают воду, этанол, полиол (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), подходящие смеси таковых, растительные масла (такие как оливковое или масло канола) и вводимые органические сложные эфиры, такие как этилолеат. Подобающая текучесть может поддерживаться, например, использованием покрытий, таких как лецитин, поддерживанием требуемого размера частиц в случае дисперсий и использованием сурфактантов.
Эти композиции могут также содержать такие вспомогательные вещества, как консервирующие, увлажняющие, эмульгирующие и диспергирующие вещества. Предохранение от действия микроорганизмов может быть обеспечено различными противомикробными и противогрибковыми веществами, например парабенами, хлорбутанолом, фенолом, (Е,Е)-Гекса-2,4-диеновая кислотой и т.п. Также может быть желательным включение изотонических веществ, например сахаров, хлорида натрия и т.п. Пролонгированное впитывание для закапываемых фармацевтических форм может быть осуществлено использованием веществ, замедляющих всасывание, например, моностеаратом алюминия и желатином.
Если желательно для более эффективного распределения, соединения могут быть включены в медленную или замедленного действия или направленную систему доставки, такую как полимерная матрица, липосомы и микросферы. Они могут быть стерилизованы, например, фильтрованием через бактерийудерживающий фильтр или включением стерилизующих веществ в стерильной воде или иной стерильной вводимой среде непосредственно перед использованием.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких лекарственных формах активный компонент или пролекарство смешивают с по меньшей мере одним общеупотребительным наполнителем (или носителем), таким как цитрат натрия или дикальция фосфат или (i) наполнителем или сухим разбавителем, таким как, например, крахмал, лактоза, сахароза, глюкоза, маннитол и кремниевая кислота, (ii) связывающим веществом, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик, (iii) влагоудерживающими средствами, такими как, например, глицерин, (iv) дезинтегрирующими веществами, такими как, например, агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, некоторые сложные силикаты и карбонат натрия, (v) замедлителями растворения, такими как, например, парафин, (vi) ускорителями абсорбции, такими как, например, четвертичные аммониевые соединения, (vii) увлажнителями, такими как цетиловый спирт и моностеарат глицерина, (viii) абсорбентами, такими как, например, каолин и бентонит, (ix) смазывающими веществами, такими как, например, тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и смесь таковых. В случае капсул, таблеток и пилюль, лекарственные формы могут содержать буферные вещества.
Твердые композиции такого же типа могут быть также использованы как наполнители в мягко- и твердонаполненных желатиновых капсулах с использованием таких наполнителей, как лактоза или сахара молока, также как и высокомолекулярные полиэтиленгликоли и т.п. Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть приготовлены с покрытием и оболочкой, такой как кишечнорастворимое покрытие и другие хорошо известные из уровня техники. Они могут содержать контрастные вещества, и могут быть также такой композиции, что они высвобождают активное соединение или соединения в определенной части желудочно-кишечного тракта отложенным способом. Примерами композиций для введения, которые могут быть использованы, являются полимерные вещества и воски. Активные соединения также могут существовать, если уместно, в микроинкапсулированном виде с одним или более из вышеупомянутых наполнителей.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие лекарственные формы могут содержать инертные разбавители, широко использующиеся в имеющемся уровне техники, такие как вода или другие растворители, солюбилизирующие вещества и эмульгаторы, такие как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла, в частности хлопковое масло, арахисовое масло, масло проращенного зерна, оливковое масло, масло канола, касторовое масло и масло из семян кунжута, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, или смеси этих соединений, и т.п. Помимо таких инертных разбавителей, композиции могут включать вспомогательные средства, такие как увлажняющие вещества, эмульгирующие и суспендирующие вещества, подсластители, ароматизаторы и отдушки.
Суспензии, наряду с активными соединениями, могут содержать суспендирующие вещества, например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтилен сорбита и сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар, трагакантовую камедь или смесь этих соединений, и т.п.
Композиции для ректального введения являются предпочтительно суппозиториями, которые могут быть получены смешением соединений по настоящему изобретению с подходящими нераздражающими наполнителями и носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при обычной температуре, но жидкими при температуре тела и поэтому плавятся в ректальной или вагинальной полости и высвобождают активный компонент. Лекарственные формы для местного применения соединения по этому изобретению включают мази, порошки, спреи и лекарственные формы для ингаляции.
Активный компонент смешивается в стерильных условиях с физиологически приемлемым носителем и любыми необходимыми консервантами, буферами или пропеллентами, при необходимости. Также рассматриваются офтальмологические рецептуры, глазные мази, суспензии, порошки и растворы, попадающие в рамки этого изобретения.
Соединения по настоящему изобретению могут также быть включены внутрь или связаны с липосомами или вводиться в виде липосом. Как известно из уровня техники, липосомы, как правило, образованы из фосфолипидов или других липидных соединений. Липосомы образуются одно- и многослойными гидратированными кристаллами, диспергированными в водной среде. Может быть использован любой нетоксичный, физиологически приемлемый метаболизируемый липид, способный образовывать липосомы. Настоящие композиции в виде липосом могут содержать, в дополнение к антагонисту связывания селектина по настоящему изобретению, стабилизаторы, консерванты, наполнители и т.п. Предпочтительными липидами являются фосфолипиды и фосфатидилхолины (лецитины) как природные, так и синтетические. Методы формирования липосом хорошо известны из уровня техники.
Лекарственные формы, не предназначенные для парентерального применения, могут также содержать вещество, усиливающее биодоступность (например, модуляторы ферментов, антиоксиданты), подходящие для предохранения соединений от разложения. Действительные уровни дозировки активного компонента в композиции по настоящему изобретению для частной композиции и метода назначения могут варьироваться таким образом, чтобы добиться количества активного компонента, которое эффективно для получения желаемого терапевтического отклика. Выбранные уровни дозировки, поэтому, зависят от желаемого терапевтического эффекта, способа введения, желаемой продолжительности лечения и других факторов. Полная дневная доза соединений по этому изобретению, введенная в организм однократно или разделенная на более мелкие дозы, может составлять до 50 мг на килограмм массы тела. Отдельные дозировки композиций могут включать такие делители, чтобы они могли быть использованы с доведением до дневной дозировки. Следует понимать, однако, что частный уровень дозировки для любого отдельно взятого пациента, будь то человек или другое животное, будет зависеть от многих условий, включая массу тела, общее состояние здоровья, половую диету, время и способ введения, скорость абсорбции и выведения, сочетания с другими лекарственными средствами и серьезность конкретного излечиваемого заболевания.
В частности, соединения по настоящему изобретению могут быть использованы для лечения широкого набора заболеваний, относящихся к воспалению и клеточно-клеточному распознаванию и адгезии. Например, соединения по настоящему изобретению могут быть назначены пациенту для лечения хронического обструктивного заболевания легких (ХОЗЛ), острого повреждения легких (ОПЛ), при искусственном кровообращении, острого респираторного дистресс-синдром (ОРДС), болезни Крона, септического шока, сепсиса, хронических воспалительных заболеваний, таких как псориаз, атопический дерматит и ревматоидный артрит, реперфузионных повреждений, которые происходят следом за инфарктами, инсультами, атеросклерозом и трансплантацией органов, травматического шока, функциональной недостаточности многих органов, автоиммунных заболеваний, таких как множественный склероз, при чрескожной транслюминальной ангиопластике, астме и воспалительной кишечной болезни. В каждом случае действенное количество соединений по изобретению вводится пациенту при необходимости такого лечения либо само по себе, либо как часть фармацевтически активной композиции. Также понимается, что при необходимости такого введение может быть введена комбинация соединений. Соединения по настоящему изобретению могут быть также введены для лечения других заболеваний, связанных с клеточно-клеточной адгезией. Поскольку настоящие соединения модулируют связывание E-селектина или P-селектина или L-селектина, любое заболевание, относящееся к этому взаимодействию потенциально, может лечиться модулированием этого связывающего взаимодействия.
В дополнение к обнаружению на некоторых белых кровяных тельцах, sLea был обнаружен на различных раковых клетках, включая клетки рака легких и толстой кишки. Было предположено, что клеточная адгезия, включающая sLea, может быть вовлечена в процесс метастаза при некоторых видах рака и антагонисты sLea-связывания могут быть полезны в лечении некоторых форм рака.
Использование активных компонентов по настоящему изобретению или косметических или местных дерматологических композиций с действенным содержанием активного компонента по изобретению, как ни удивительно, делает возможным эффективное лечение, а также профилактику старения кожи, вызванного внешними и внутренними факторами.
Изобретение в частности относится к использованию соединения формулы (Ia)-(If), в частности (Ib), (Ic), (Ie), или (If) или стереоизомерных форм таковых для получения косметической или дерматологической композиции.
Используемое количество активного компонента или его стереоизомерной формы связано с количеством, требуемым для получения желаемого результата с использованием косметических или дерматологических композиций. Специалист в данной области техники способен определить это эффективное количество, которое зависит от используемого производного, индивида, к которому оно применяется, и времени этого использования. Для грубой оценки, в косметических и дерматологических композициях по изобретению соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы могут быть введены в количествах, представляющих от 0,001 до 40 вес. %, предпочтительно от 0,005 до 30 вес. % и более предпочтительно от 0,01 до 20 вес. %.
Дополнительный аспект относится к косметическим композициям, содержащим соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы и по меньшей мере один косметически приемлемый компонент, например косметически приемлемый компонент для применения на кожу.
Количества различных компонентов физиологической среды косметической композиции по изобретению являются такими, какие, как правило, используются в рассматриваемых областях. Когда косметическая композиция является эмульсией, доля жировой фазы может варьироваться от 2 до 80 вес. %, предпочтительно от 5 до 50 вес. % по отношению к общему весу косметической композиции.
Таким образом, косметическая композиция должна содержать нетоксичную физиологически приемлемую среду, которая может быть нанесена на человеческую кожу. Для местного применения на кожу косметическая композиция может быть в виде раствора, суспензии, эмульсии или дисперсии более или менее жидкой консистенции и особенно жидкой или полужидкой консистенции, полученными диспергированием жировой (масляной) фазы и водной фазе (М/В) или наоборот (В/М), или, в качестве альтернативы, гелем. Также может быть предложена косметическая композиция в виде мусса или в форме спрея или аэрозоля, кроме того, содержащая находящийся под давлением пропеллент. Также композиции могут быть в форме лосьона для волос, шампуня или кондиционера для волос, жидкого или твердого мыла, лечебной маски, или пенного крема, или геля для чистки шевелюры. Также они могут быть в форме краски или туши для волос.
Косметические композиции по изобретению могут также включать один или более различных компонентов, обычно используемых в рассматриваемых областях, выбранных из добавок к рецептуре, таких как, например, загустители или желирующие компоненты на водной или масляной основе, красители, растворимые в среде косметической композиции, твердые частицы, такие как неорганические или органические наполнители, или красители в форме микрочастиц или наночастиц, консерванты отдушки, гидротопы или электролиты, нейтрализаторы (подкисляющие или защелачивающие вещества), пропелленты, анионные, катионные или амфотерные сурфактанты, полимеры, в частности водорастворимые или диспергируемые в воде анионные, неионные, катионные или амфотерные пленкообразующие полимеры, неорганические или органические соли, хелатирующие вещества; смесь таковых.
Косметические композиции могут быть использованы для ингибирования микровоспалительного цикла. Таким образом, настоящее изобретение также относится к косметическим композициям, содержащим соединения формулы (Ib), (Ic), (Ie), или (If), или их стереоизомерные формы, которые используются для косметического лечения или косметической профилактики микровоспалительных состояний.
Косметические композиции, содержащие соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы, которые используются для косметического лечения или косметической профилактики старения кожи, вызванного внешними или внутренними факторами, также являются предметом настоящего изобретения. Внутренние факторы, ответственные за старение кожи, являются генетически программируемыми определяющими факторами, включающими возраст, гормональный статус и физиологические факторы.
Помимо косметически неактивных компонентов косметические композиции по настоящему изобретению могут также включать один или более косметически активный компонент с благотворным действием на кожу. Таким образом, настоящее изобретение относится к косметическим композициям, содержащим соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы и по меньшей мере один дополнительный косметически активный компонент, например УФ-блокатор или белки.
Дерматологические композиции, содержащие соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы и по меньшей мере один дерматологически приемлемый компонент, например дерматологически приемлемый компонент для применений на кожу, также являются предметом изобретения.
Дерматологически приемлемые компоненты, которые могут быть использованы для дерматологических композиций, описанных здесь, являются идентичными косметически приемлемым компонентам как указано в этом изобретении.
Дополнительным вариантом осуществления этого изобретения являются дерматологические композиции, содержащие соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы, которые используются для дерматологического лечения, дерматологического диагностирования или дерматологической профилактики микровоспалительных состояний.
В частности, изобретение относится к дерматологическим композициям, содержащим соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы, которые используются для дерматологического лечения, дерматологического диагностирования или дерматологической профилактики зуда и старения кожи, вызванного внешними факторами. Внешние факторы включают факторы окружающей среды в целом; более частным образом, вызванное светом старение кожи в результате действия солнца, света или любого другого излучения, загрязнение атмосферы, раны, инфекции, травматизм, аноксия, сигаретный дым, гормональный статус как отклик на внешние факторы, нейропептиды, электромагнитные поля, гравитацию, образ жизни (например, избыточное потребление алкоголя), повторяющиеся выражения лица, положение сна и физиологические стресс-факторы.
В дополнение к дерматологически неактивным компонентам, дерматологические композиции также могут включать дерматологически или фармацевтически активные компоненты. Таким образом, настоящее изобретение относится к дерматологическим композициям, содержащим соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы и по меньшей мере один дополнительный дерматологически или фармацевтически активный компонент. Дерматологически или фармацевтически активные компоненты, которые могут быть использованы в описанных здесь дерматологических композициям, определяются как и косметически активные компоненты, определенные выше. Дерматологически или фармацевтически активные компоненты могут быть идентичны косметически активным компонентам как указано в этом изобретении.
Другим предметом настоящего изобретения являются дерматологические композиции, содержащие соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы и по меньшей мере один дополнительный дерматологически или фармацевтически активный компонент, отличающиеся тем, что он используется для дерматологического лечения, дерматологического диагностирования или дерматологической профилактики микровоспалительных состояний.
В частности, настоящее изобретение относится к дерматологическим композициям, содержащим соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы и по меньшей мере один дополнительный дерматологически или фармацевтически активный компонент, отличающиеся тем, что он используется для дерматологического лечения, дерматологического диагностирования или дерматологической профилактики зуда и старения кожи, вызванного внешними факторами.
Старение кожи может также быть вызвано комбинацией внутренних и внешних факторов. Таким образом, настоящее изобретение относится к дерматологическим композициям, содержащим соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерные формы и по меньшей мере один дополнительный фармацевтически или косметически активный компонент, отличающиеся тем, что он используется для косметического и дерматологического лечения и косметической и дерматологической профилактики старения кожи, вызванного сочетанием внутренних и внешних факторов.
Другим вариантом осуществления этого изобретения является процесс для получения косметической композиции смешением соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерной формы, по меньшей мере одного косметически приемлемого компонента и, наконец, дополнительных косметически активных компонентов.
В частности, процесс для получения косметической композиции путем смешения соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерной формы, по меньшей мере одного косметически приемлемого компонента и, наконец, дополнительных косметически активных компонентов, в каковом композиция включает от 0,01 до 20 вес. % соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерной формы, в расчете на общий вес композиции, являющегося предметом этого изобретения.
Дополнительный аспект относится к процессу для получения дерматологической композиции смешением соединения формулы (Ib), (Ic), (Ie), или (If) или их стереоизомерной формы, по меньшей мере одного дерматологически приемлемого компонента и, наконец, дополнительных фармацевтически активных компонентов.
Многие из соединений по настоящему изобретению могут быть синтезированы согласно следующим общим синтетическим схемам.

На схеме 1 анилин строения (1) в инертной атмосфере в дихлорметане вовлекают в реакцию с N'-(3-диметиламинопропил)-N-этилкарбодиимидом (EDC), триэтиламином, 4-диметиламинопиридином (DMAP) и карбоновой кислотой строения (2) с образованием амида строения (3). Амид строения (3) далее вовлекают в реакцию с трибромидом бора в дихлорметане при -78°С с образованием соответствующего 2,4,6-тригидроксифенила строения (4). Синтетическая последовательность, показанная на схеме 1, приводящая к такому соединению, как (4), не сводится только к таким Y-H строительным блокам, как (1), но может быть общим образом применена ко всем другим строительным блокам типа Y-H.

На схеме 2 хлорид кислоты строения (5) в основных условиях (пиридин в дихлорметане) вовлекают в реакцию с анилином общего строения (6) с образованием соответствующего анилида строения (7). Как вариант, на этой стадии может быть использован триэтиламин. Сложный эфир строения (7) гидролизуют действием LiOH в ацетонитриле или ТГФ/MeOH с образованием карбоновой кислоты строения (8), которую далее вовлекают в реакцию с трибромидом бора в дихлорметане при -78°С с образованием, после последующей водной обработки, соответствующих деметилированных кислот строения (9). Синтетическая последовательность, показанная на схеме 2 не сводится только к таким Y-H строительным блокам, как (6), но может быть общим образом применена ко всем другим строительным блокам типа Y-H.
Строительные блоки строения (2) или (5), как показано на предшествующих схемах, могут быть синтезированы как представлено на следующих схемах 3-9:
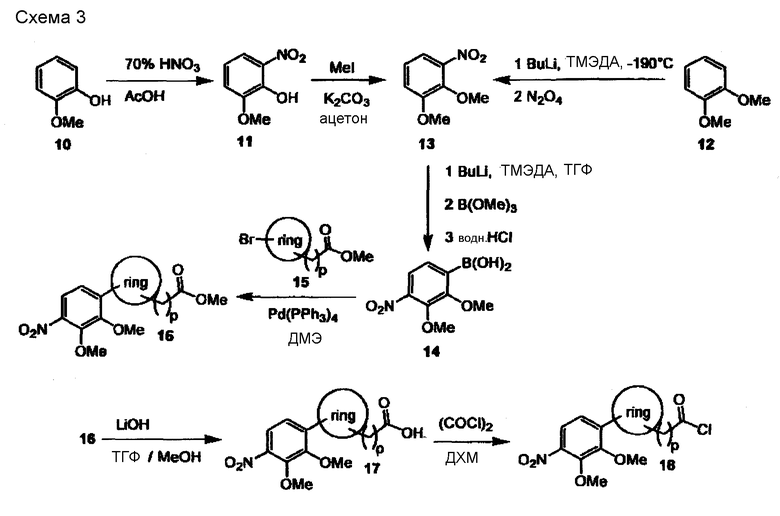
На схеме 3 гваякол (10) селективно нитруется 70% HNO3 в AcOH с образованием нитрогваякола (11), который далее метилируют метилиодидом и K2CO3 в ацетоне с образованием соответствующего вератрола (13). Как вариант, (13) доступен из вератрола (12) в результате реакции с BuLi и N,N,N',N'-тетраметилэтилендиамином (ТМЭДА) при низкой температуре с последующей обработкой тетраоксидом азота. Последующая реакция (13) с BuLi и ТМЭДА в ТГФ, а затем с триметилборатом и соляной кислотой дает бороновую кислоту (14). Бороновая кислота (14) вовлекается в реакцию с бромированным ароматическим или гетероароматическим сложным эфиром общего строения (15) в условиях сочетания Сузуки (Pd(PPh3)4 и водный бикарбонат натрия в диметоксиэтане) с образованием биарила строения (16). Биарил (16) далее гидролизуют водным гидроксидом лития в ТГФ и метаноле или ацетонитриле с образованием соответствующей карбоновой кислоты (17), которая превращается в строительный блок строения (18) по реакции с оксалилхлоридом в безводном дихлорметане.

На схеме 4 3-нитровератрол (13) реагирует с BuLi и (ТМЭДА) в ТГФ, а затем с бромидом меди (I) и этиловым эфиром бромуксусной кислоты с образованием замещенного этилового эфира фенилуксусной кислоты (19), который далее гидролизуют водным гидроксидом лития в ТГФ и метаноле или ацетонитриле с образованием соответствующей карбоновой кислоты (20).

На схеме 5 вератрол (12) селективно нитруется 70% HNO3 в AcOH с образованием 4-нитровератрола (21). Последующая реакция (21) с BuLi и ТМЭДА в ТГФ, а затем с триметилборатом и соляной кислотой дает бороновую кислоту (22). Бороновая кислота (22) вовлекается в реакцию с бромированным ароматическим или гетероароматическим сложным эфиром общего строения (15) в условиях сочетания Сузуки (Pd(PPh3)4 и водный бикарбонат натрия в диметоксиэтане) с образованием биарила строения (23). Биарил (23) далее гидролизуют водным гидроксидом лития в ТГФ и метаноле или ацетонитриле с образованием соответствующей карбоновой кислоты (24), которая превращается в строительный блок строения (25) по реакции с оксалилхлоридом в безводном дихлорметане.
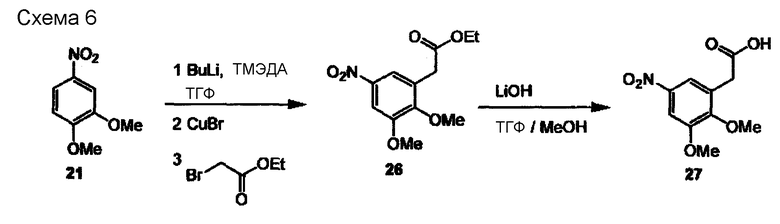
На схеме 6 представлено получение кислотного строительного блока (27). Синтез (27) аналогичен таковому, описанному на схеме 4

На схеме 7 4-бромгваякол (28) селективно нитруется 70% HNO3 в AcOH с образованием 4-бром-6-нитрогваякола (29), который далее метилируют метилиодидом и K2CO3 в ацетоне с образованием соответствующего 5-бром-3-нитровератрола (30). 5-бром-3-нитровератрол (30) вовлекается в реакцию с бороновой кислотой общего строения (31) в условиях сочетания Сузуки (Pd(PPh3)4 и водный бикарбонат натрия в диметоксиэтане) с образованием биарила строения (32). Биарил (32) далее гидролизуют водным гидроксидом лития в ТГФ и метаноле или ацетонитриле с образованием соответствующей карбоновой кислоты (33), которая превращается в строительный блок строения (34) по реакции с оксалилхлоридом в безводном дихлорметане.

Кислотный строительный блок (35), представленный на схеме 8, получен из 5-бром-3-нитровератрола (30) согласно схемам 4 и 6.

На схеме 9 3,4-диметоксифенол (36) селективно нитруется 70% HNO3 в AcOH с образованием 4,5-диметокси-2-нитрофенола (37), который далее переводят в соответствующий трифлат (38) действием ангидрида трифторметансульфоновой кислоты и пиридина в дихлорметане. Трифлат (38) вовлекается в реакцию с бороновой кислотой общего строения (31) в условиях сочетания Сузуки (Pd(PPh3)4 и водный фосфат калия в толуоле) с образованием биарила строения (39). Биарил (39) далее гидролизуют водным гидроксидом лития в ТГФ и метаноле или ацетонитриле с образованием соответствующей карбоновой кислоты (40), которая превращается в строительный блок строения (41) по реакции с оксалилхлоридом в безводном дихлорметане.
Пример А
Получение метилового эфира [5-(2-аминофенил)тиофен-2-ил]-уксусной кислоты (87)

Стадия 1: (следующая реакция проводится в атмосфере безводного азота). Растворяют метиловый эфир (тиофен-2-ил)уксусной кислоты (85) (2,0 г, 12,8 ммоля) в безводном хлороформе (9,0 мл) и ледяной уксусной кислоте (9,0 мл), порциями добавляют N-бромсукцинимид (2,3 г, 13,0 ммоля) и перемешивают смесь в течение 3 дней при комнатной температуре. Добавляют к реакционной массе воду, разделяют слои и водный слой экстрагируют дихлорметаном. Объединенные органические слои несколько раз промывают 1 М водным NaOH и водой и один раз насыщенным водным раствором NaCl и сушат Na2SO4. Полученный сырой продукт очищают препаративной радиальной хроматографией (CyH/EtOAc 5+1), получают метиловый эфир (5-бромтиофен-2-ил)уксусной кислоты (86) в виде желтого масла (2,46 г, 81 %), который используют без какой-либо дальнейшей очистки. 1Н-ЯМР (400 МГц, CDCl3): 3,71 (с, 3Н), 3,75 (с, 2Н), 6,67 (д, 1Н, J=3,8 Гц), 6,88 (д, 1Н, J=3,8 Гц).
Стадия 2: (следующую реакцию выполняют в атмосфере азота) Этанол (3,7 мл), тетракис(трифенилфосфин)палладий (0) (289 мг, 0,25 ммоля) и декагидрат карбоната натрия (4,0 г, 14,0 ммоля) растворяют в воде (5,2 мл) и затем добавляют к раствору гидрохлорида 2-аминофенилбороновой кислоты (910 мг, 5,25 ммоля) в толуоле (52 мл). Аккуратно дегазируют реакционную смесь (5 раз) и снова заполняют азотом. Добавляют раствор метилового эфира (5-бромтиофен-2-ил)уксусной кислоты (1,17 г, 5,0 ммоля) в толуоле (4,5 мл). Вновь реакционную смесь дегазируют (5 раз) и перемешивают при 100°С в течение 22 часов. Реакционную смесь распределяют между этилацетатом и насыщенным водным раствором NaCl и экстрагируют отделившийся водный слой этилацетатом (3 раза). Объединенные органические слои промывают водой, насыщенным водным раствором NaCl и сушат Na2SO4. Полученный сырой продукт очищают препаративной радиальной хроматографией (CyH/EtOAc 5+1), получают метиловый эфир [5-(2-аминофенил)-тиофен-2-ил] уксусной кислоты (87) в виде коричневого масла (634 мг, 51 %). 1Н-ЯМР (400 МГц, CDCl3): 3,73 (с, 3Н), 3,83 (с, 2Н), 3,92-4,07 (уш.с, 2Н), 6,74 (д, 1Н), 6,76 (тд, 1Н, J1=7,6 Гц, J2=1,3 Гц), 6,92 (д, 1Н, J=3,5 Гц), 7,02 (д, 1Н, J=3,5 Гц), 7,11 (тд, 1Н, J1 = 7,6 Гц, J2 = 1,5 Гц), 7,23 (дд, 14 Н, J1 = 7,6 Гц, J2 = 1,5 Гц).
Пример В
Получение метилового эфира 5-(2-аминофенил)-тиофен-2-карбоновой кислоты (90)
Стадия 1: LE23 Растворяют 5-бромтиофен-2-карбоновую кислоту (88) (1,50 г, 7,24 ммоля) в метаноле (10 мл) и добавляют концентрированную серную кислоту (0,39 мл, 7,24 ммоль). Реакционную смесь перемешивают при 75°С в течение 20 часов. Реакционную смесь охлаждают до комнатной температуры, удаляют растворитель при пониженном давлении вновь растворяют остаток в этилацетате. Промывают органический слой 5% водным NaOH 3 раза и экстрагируют объединенные водные слои этилацетатом. Промывают объединенные органические слои насыщенным водным раствором NaCl и сушат Na2SO4. Удаляют растворитель при пониженном давлении и без дальнейшей очистки сушат остаток в вакууме масляного насоса. Получают сложный эфир (89) в виде белого твердого вещества (1,48 г, 92%). 1Н-ЯМР (400 МГц, CDCl3): 3,88 (с, 3Н), 4,00 (уш.с, 2Н), 6,73-6,82 (м, 2Н), 7,13-7,21 (м, 2Н), 7,26 (дд, 1Н, J1 = 7,6 Гц, J2 = 1,0 Гц), 7,78 (д, 1Н, J=3,8).
Стадия 2: LE29 (следующую реакцию выполняют в атмосфере азота). Растворяют тетракис(трифенилфосфин)палладий (0) (510 мг, 0,45 ммоля) и сложный эфир (89) (1,97 г, 8,91 ммоля) в ДМЭ (16 мл), аккуратно дегазируют реакционную смесь (5 раз) и снова заполняют азотом. Добавляют 2-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фениламин (2,15 г, 9,80 ммоля) и 1М водный раствор NaHCO3 (27,0 мл, 27,0 ммоля), вновь реакционную смесь осторожно дегазируют (5 раз) и заполняют азотом. Реакционную смесь перемешивают при 95°С в течение 18 часов. Реакционную смесь распределяют между этилацетатом и водой и экстрагируют отделившийся водный слой этилацетатом (3 раза). Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Полученный сырой продукт очищают флэш-хроматографией (силикагель, CyH/EtOAc 5+1), получают метиловый эфир 5-(2-аминофенил)тиофен-2-карбоновой кислоты (90) в виде желтого твердого вещества (1,41 г, 67%). 1Н-ЯМР (400 МГц, CDCl3): 3,88 (с, 3Н), 4,00 (уш.с, 2Н), 6,73-6,82 (м, 2Н), 7,13-7,21 (м, 2Н), 7,26 (дд, 1Н, J1 = 7,6 Гц, J2 =1,0 Гц), 7,78 (д, 1Н, J=3,8 Гц).
Пример С
Получение 4',5'-диметокси-2'-нитробифенил-3-карбонилхлорида (95)

Стадия 1: DK006 (следующую реакцию выполняют в атмосфере азота). Растворяют тетракис(трифенилфосфин)палладий (0) (200 мг, 0,17 ммоля) и метил(3-бром)бензоат (91) (1,25 мг, 5,81 ммоля) в ДМЭ (12 мл), осторожно дегазируют реакционную смесь (5 раз) и снова заполняют азотом. Добавляют 3,4-диметоксифенилбороновую кислоту (1,25 г, 6,85 ммоля) и 1М водный раствор NaHCO3 (17,7 мл, 17,7 ммоля), вновь реакционную смесь осторожно дегазируют (5 раз) и заполняют азотом. Реакционную смесь перемешивают при 95°С в течение 2 часов. Реакционную смесь распределяют между этилацетатом и водой и экстрагируют отделившийся водный слой этилацетатом (4 раза). Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Полученный сырой продукт очищают флэш-хроматографией (силикагель, CyH/EtOAc 5+1), получают бифенил (92) в виде желтого твердого вещества (1,27 г, 80%). 1Н-ЯМР (400 МГц, CDCl3): 3,92 (с, 3Н), 3,93 (с, 3Н), 3,95 (с, 3Н), 6,94 (д, 1Н, J=8,3 Гц), 7,11 (д, 1Н, J=1,8 Гц), 7,16 (дд, 1Н, J1 = 8,3 Гц, J2 = 1,8 Гц), 7,47 (т, 1Н, J=7,7 Гц), 7,73 (дт, 1Н, J1 = 7,6 Гц, J2 =0,9 Гц), 7,96 (д, 1Н, J=7,8 Гц), 8,22 (с, 1Н).
Стадия 2: DK010 Растворяют бифенил (92) (653 мг, 2,39 ммоль) в ледяной уксусной кислоте (32 мл), добавляют 70 % азотную кислоту (1,18 мл, 26,30 ммоль) и перемешивают смесь при комнатной температуре в течение 30 минут. К охлажденной реакционной массе (0°С) медленно добавляют воду (12 мл), отфильтровывают осадок и остаток на фильтре осторожно промывают водой. Остаток на фильтре сушат без дополнительной очистки в вакууме масляного насоса и затем в эксикаторе, получают нитрованный бифенил (93) в виде желтого твердого вещества (644 мг, 98%). 1Н-ЯМР (400 МГц, CDCl3): 3,91 (с, 3Н), 3,94 (с, 3Н), 3,90 (с, 3Н), 6,75 (с, 1Н), 7,42-7,51 (м, 2Н), 7,60 (с, 1Н), 7,97 (т, 1Н, J=1,6 Гц), 8,05 (дт, 1Н, J1 = 7,1 Гц, J2 =1,6 Гц).
Стадия 3: DK011 Растворяют нитрованный бифенил (93) (1,28 г, 4,70 ммоль) в ацетонитриле (46 мл) и добавляют 1М водный LiOH (23,5 мл, 23,5 ммоля). Реакционную смесь перемешивают в течение 20 часов при комнатной температуре. Реакцию останавливают добавлением к реакционной смеси (охлаждающая баня) 1М водной HCl (до получения pH около 3). Смесь экстрагируют этилацетатом (трижды), объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Растворитель удаляют и сушат остаток без дополнительной очистки в вакууме масляного насоса. Получают карбоновую кислоту (94) в виде желтого твердого вещества (1,14 г, 80%). 1Н-ЯМР (400 МГц, ДМСО-d6): 3,89 (с, 3Н), 3,90 (с, 3Н), 7,01 (с, 1Н), 7,53-7,59 (м, 2Н), 7,64 (с, 1Н), 7,84 (уш.с, 1Н), 7,93-7,98 (м, 1Н).
Стадия 4: DK012 (следующую реакцию выполняют в атмосфере сухого азота). Растворяют карбоновую кислоту (94) (550 мг, 1,81 ммоля) в безводном дихлорметане (13 мл) и добавляют безводный ДМФА (1 мл). Медленно добавляют оксалилхлорид (237 мкл, 2,72 ммоль), поддерживая температуру около 20°С водяной баней, и перемешивают при комнатной температуре дополнительные 3 часа. Растворитель удаляют и остаток сушат в вакууме. Получают сырой 4',5'-диметокси-2'-нитробифенил-3-карбонилхлорид (95) в виде желтого твердого вещества. Дополнительно не очищают.
Пример D
Получение 2',3'-диметокси-5'-нитробифенил-3-карбонилхлорида (99)
Стадия 1: FR631 (следующую реакцию выполняют в атмосфере азота) Растворяют тетракис(трифенилфосфин)палладий (0) (217 мг, 0,19 ммоля) и метил(3-бром)бензоат (91) (1,35 мг, 6,28 ммоля) в ДМЭ (13 мл), осторожно дегазируют реакционную смесь (5 раз) и снова заполняют азотом. Добавляют 2,3-диметоксифенилбороновую кислоту (1,33 г, 7,28 ммоля) и 1М водный раствор NaHCO3 (19 мл, 19 ммоль), вновь реакционную смесь осторожно дегазируют (5 раз) и заполняют азотом. Реакционную смесь перемешивают при 100°С в течение 22 часов. Реакционную смесь распределяют между этилацетатом и водой и экстрагируют отделившийся водный слой этилацетатом (4 раза). Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Полученный сырой продукт очищают препаративной радиальной хроматографией (силикагель, CyH/EtOAc 10+1), получают бифенил (96) в виде бесцветного твердого вещества (1,50 г, 87%). 1Н-ЯМР (400 МГц, CDCl3): 3,57 (с, 3Н), 3,90 (с, 3Н), 3,91 (с, 3Н), 6,91-6,96 (м, 2Н), 7,11 (т, 1Н, J=8,0 Гц), 7,46 (т, 1Н, J=7,8 Гц), 7,75 (дт, 1Н, J1 = 7,8 Гц, J2 = 1,5 Гц), 8,00 (дт, 1Н, J1 = 7,8 Гц, J2 = 1,4 Гц), 8,19 (т, 1Н, J=1,5 Гц).
Стадия 2: FR632 Растворяют бифенил (96) (1,49 г, 5,47 ммоль) в ледяной уксусной кислоте (30 мл), добавляют 70% азотную кислоту (0,76 мл, 6,04 ммоль) и перемешивают смесь при комнатной температуре в течение 22 часов. Реакционную смесь выливают в воду со льдом, отфильтровывают осадок и остаток на фильтре осторожно промывают водой. Остаток на фильтре сушат без дополнительной очистки в вакууме масляного насоса и затем в эксикаторе, получают нитрованный бифенил (97) в виде желтого твердого вещества (1,29 мг, 74 %). 1Н-ЯМР (400 МГц, CDCl3): 3,71 (с, 3Н), 3,93 (с, 3Н), 3,99 (с, 3Н), 7,52 (т, 1Н, J=7,7 Гц), 7,71 (д, 1Н, J=7,8 Гц), 7,80 (д, 1Н, J=2,3 Гц), 7,91 (уш.д, 1Н, J=2,3 Гц), 8,07 (уш.д, 1Н, J=7,8 Гц), 8,18 (уш.с, 1Н).
Стадия 3: FR634 Растворяют нитрованный бифенил (97) (1,29 г, 4,07 ммоль) в ацетонитриле (100 мл) и добавляют 1М водный LiOH (41 мл, 41 ммоль). Реакционную смесь перемешивают в течение 22 часов при комнатной температуре. Реакцию останавливают добавлением к реакционной смеси (охлаждающая баня) 2М водной HCl (до получения pH около 3). Осадок отфильтровывают и остаток на фильтре промывают водой и однократно этилацетатом. Остаток на фильтре сушат без дополнительной очистки в вакууме масляного насоса и затем в эксикаторе, получают карбоновую кислоту (98) в виде бежевого твердого вещества (1,06 г, 86%). 1Н-ЯМР (400 МГц, ДМСО-d6): 3,71 (с, 3Н), 3,99 (с, 3Н), 7,61 (т, 1Н, J=7,7 Гц), 7,77 (уш.д, 1Н, J=7,6 Гц), 7,83 (д, 1Н, J=2,8 Гц), 7,89 (д, 1Н, J=2,8 Гц), 7,99 (уш.д, 1Н, J=7,6 Гц), 8,05 (т, 1Н, J=1,6 Гц), 13,14 (уш.с, 1Н).
Стадия 4: FR637 (следующую реакцию выполняют в атмосфере сухого азота). Растворяют карбоновую кислоту (98) (500 мг, 1,65 ммоля) в безводном дихлорметане (11 мл) и добавляют безводный ДМФА (5 мл). Медленно добавляют оксалилхлорид (220 мкл, 2,47 ммоль), поддерживая температуру около 20°С водяной баней, и перемешивают при комнатной температуре дополнительные 4 часа. Растворитель удаляют и остаток сушат в вакууме. Получают сырой 2',3'-диметокси-5'-нитробифенил-3-карбонилхлорид (99) в виде желтого твердого вещества. Дополнительно не очищают.
Пример E
Метиловый эфир 5-(4-аминофенил)-2-метилфуран-3-карбоновой кислоты (102)

Стадия 1: (следующая реакция проводится в темноте). Растворяют метиловый эфир 2-метилфуран-3-карбоновой кислоты (100) (3,60 мл, 28,5 ммоля) в хлороформе (20 мл) и ледяной уксусной кислоте (20 мл), порциями добавляют N-бромсукцинимид (6,90 г, 38,8 ммоля) в течение 95 минут. Перемешивают реакционную смесь в течение еще 19 часов при комнатной температуре. Добавляют к реакционной массе воду и водный слой экстрагируют дихлорметаном дважды, объединенные органические слои промывают 2 М водным NaOH, водой (3 раза) и насыщенным водным раствором NaCl и сушат Na2SO4. Получают метиловый эфир 5-бром-2-метилфуран-3-карбоновой кислоты (101) (4,90 г, 78%) в виде красно-коричневого масла. Дополнительно не очищают. 1Н-ЯМР (400 МГц, CDCl3): 2,54 (с, 3Н), 3,80 (с, 3Н), 6,53 (с, 1Н).
Стадия 2: (следующую реакцию выполняют в атмосфере азота). Растворяют Pd(PPh3)4 (1,26 г, 1,09 ммоля) и метиловый эфир 5-бром-2-метилфуран-3-карбоновой кислоты (101) (4,77 г, 21,77 ммоля) в ДМЭ (116 мл) и перемешивают при комнатной температуре в течение 15 минут. Добавляют 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фениламин (5,25 г, 23,96 ммоля) и затем 1М водный раствор бикарбоната натрия (65,4 мл, 65,3 ммоля). Реакционную смесь осторожно дегазируют и заполняют азотом (5 раз) и перемешивают при 95°С (с обратным холодильником) в течение 4 часов. Реакционную смесь охлаждают до комнатной температуры, удаляют органические растворители при пониженном давлении и остаток распределяют между этилацетатом и водой. Экстрагируют водный слой этилацетатом (3 раза), объединенные органические слои промывают водой, насыщенным водным раствором NaCl и сушат Na2SO4. Полученный сырой продукт очищают флэш-хроматографией (силикагель, EtOAc/CyH 1+2), получают метиловый эфир 5-(4-аминофенил)-2-метилфуран-3-карбоновой кислоты (102) (2,35 г, 46%) в виде желто-коричневого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): 2,60 (с, 3Н), 3,74 (уш.с, 2Н), 3,82 (с, 3Н), 6,64 (с, 1Н), 6,67(дт, 1Н, J1 =8,6 Гц, J2 = 2,3 Гц), 7,42 (дт, 2Н, J1 =8,8 Гц, J2 =2,3 Гц).
Пример F
(3,4-диметокси-5-нитрофенил)уксусная кислота (107)
Стадия 1: (следующая реакция проводится в темноте). Растворяют альдегид (103) (877 мг, 4,15 ммоля) в MeOH (30 мл), охлаждают раствор до 0°С, добавляют порциями натрийборгидрид (548 мг, 14,49 ммоля) в течение 40 минут. Реакционную смесь перемешивают еще 70 минут при комнатной температуре. Смесь охлаждают до 0°С, медленно добавляют 1 М HCl (20 мл) и удаляют растворитель. Остаток распределяют между этилацетатом и водой. Экстрагируют водный слой этилацетатом (3 раза), объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. После удаления растворителя получают бензиловый спирт (104) (876 мг, 99 %) в виде коричневого твердого вещества. Далее не очищают. 1Н-ЯМР (400 МГц, CDCl3): 3,92 (с, 3Н), 3,95 (с, 3Н), 4,68 (с, 2Н), 7,14 (д, 1Н, J=1,8 Гц), 7,29 (д, 1Н, J=1,8 Гц).
Стадия 2: (следующую реакцию проводят в трехгорлой колбе, снабженной обратным холодильником и капельной воронкой, в атмосфере сухого азота). Растворяют трибромид фосфора (800 мкл, 8,52 ммоля) в безводном толуоле (90 мл) и нагревают раствор до 80°С. Медленно добавляют суспензию бензилового спирта (104) (1,82 г, 8,52 ммоля) в безводном толуоле (80 мл) и перемешивают реакционную смесь еще 2 часа при 80°С. Охлаждают смесь на ледяной бане и к реакционной массе медленно добавляют ледяную воду. Реакционную смесь распределяют между этилацетатом и водой. Экстрагируют водный слой этилацетатом (3 раза), объединенные органические слои промывают водой и насыщенным водным раствором NaCl и сушат Na2SO4. После удаления растворителя получают бензилбромид (105) (2,23 г, 95 %) в виде коричневого твердого вещества. Далее не очищают. 1Н-ЯМР (400 МГц, CDCl3): 3,92 (с, 3Н), 3,96 (с, 3Н), 4,42 (с, 2Н), 7,10 (д, 1Н, J=2,0 Гц), 7,34 (д, 1Н, J=2,0 Гц).
Стадия 3: Растворяют бензилбромид (105) (2,22 г, 8,06 ммоля) в метаноле (44,0 мл) и воде (9,0 мл), добавляют цианид калия (787 мг, 12,09 ммоля) и перемешивают реакционную смесь еще 90 минут при 75°С (кипячение с обратным холодильником). Реакционную смесь распределяют между этилацетатом и водой. Экстрагируют водный слой этилацетатом (3 раза), объединенные органические слои промывают насыщенным водным раствором бикарбоната натрия (3 раза), насыщенным водным раствором NaCl и сушат Na2SO4. Полученный сырой продукт очищают препаративной радиальной хроматографией (силикагель, CyH/EtOAc 2+1), получают нитрил (106) в виде желтого твердого вещества (1,35 г, 75 %). 1Н-ЯМР (400 МГц, CDCl3): 3,74 (с, 2Н), 3,94 (с, 3Н), 3,96 (с, 3Н), 7,06 (д, 1Н, J=2,0 Гц), 7,26 (д, 1Н, J=2,0 Гц).
Стадия 4: Суспендируют нитрил (106) (1,35 г, 6,07 ммоля) в ледяной уксусной кислоте (53 мл) и воде (65 мл), медленно добавляют концентрированную серную кислоту (22 мл) и реакционную смесь кипятят с обратным холодильником при перемешивании еще 18 часов. Охлажденную реакционную смесь экстрагируют этилацетатом (5 раз), объединенные органические слои промывают водой (5 раз) и насыщенным водным раствором NaCl и сушат Na2SO4. После удаления растворителя получают (3,4-диметокси-5-нитрофенил)уксусную кислоту (107) (1,46 г, 99%) в виде желтого твердого вещества. Далее не очищают. 1Н-ЯМР (400 МГц, ДМСО-d6): 3,64 (с, 2Н), 3,83 (с, 3Н), 3,87 (с, 3Н), 7,30 (с, 2Н), 12,45 (уш.с, 1Н).
Пример 1
(5-{2-[2-(4,5-дигидрокси-2-нитрофенил)-ацетиламино]-фенил}тиофен-2-ил)уксусная кислота (110)

Стадия 1: (следующая реакция проводится в атмосфере сухого азота) Суспендируют EDC-гидрохлорид (58 мг, 0,30 ммоль) в безводном дихлорметане (1,0 мл), добавляют триэтиламин (0,042 мл, 0,30 ммоль) и перемешивают в течение 10 минут при комнатной температуре. Добавляют (4,5-диметокси-2-нитрофенил)уксусную кислоту (54 мг, 0,22 ммоля) и DMAP (8 мг, 0,06 ммоля) и перемешивают в течение 15 минут. Добавляют анилин (87) (50 мг, 0,20 ммоля) и перемешивают реакционную смесь 22 часа при 40°С. Реакционную смесь распределяют между дихлорметаном и водой (1+1), разделяют слои и экстрагируют водный слой дихлорметаном (3 раза). Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Сырой продукт очищают препаративной радиальной хроматографией (силикагель 60PF, CyH/EtOAc 3+1), получают продукт (108) в виде желто-белого твердого вещества (28 мг, 30%). 1Н-ЯМР (400 МГц, CDCl3): 3,75 (с, 3Н), 3,82 (с, 2Н), 3,92 (с, 2Н), 3,94 (с, 3Н), 3,96 (с, 3Н), 6,75 (д, 1Н, J=3,3 Гц), 6,80-6,84 (м, 2Н), 7,09 (т, 1Н, J=7,5 Гц), 7,27-7,35 (м, 2Н), 7,63 (с, 1Н), 7,89 (с, 1Н), 8,27 (д, 1Н, J=7,8 Гц).
Стадия 2: Растворяют сложный эфир (108) (49 мг, 0,10 ммоля) в метаноле (1,3 мл) и ТГФ (3,8 мл), добавляют 1М водный раствор LiOH (0,52 мл, 0,52 ммоля) и перемешивают 20 часов при 40°С. Удаляют растворитель при пониженном давлении и остаток распределяют между этилацетатом и 1М HCl (1+1). Отделяют водный слой и экстрагируют 3 раза этилацетатом. Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Удаляют растворитель при пониженном давлении и сушат остаток без дальнейшей очистки в вакууме масляного насоса. Получают сырой продукт (109) в виде светло-коричневого твердого вещества (48 мг, количественно). 1Н-ЯМР (400 МГц, CDCl3): 3,87 (с, 2Н), 3,92 (с, 2Н), 3,94 (с, 3Н), 3,96 (с, 3Н), 6,76 (д, 1Н, J=3,3 Гц), 6,82 (с, 1Н), 6,85 (д, 1Н, J=3,3 Гц), 7,09 (т, 1Н, J=7,7 Гц), 7,26-7,35 (м, 2Н), 7,63 (с, 1Н), 7,88 (уш.с, 1Н), 8,28 (д, 1Н, J=8,1 Гц).
Стадия 3: (следующая реакция проводится в атмосфере сухого азота). Растворяют карбоновую кислоту (109) (48 мг, 0,10 ммоля) в безводном дихлорметане (3,0 мл), охлаждают до -78°С (сухой лед/ацетон) и медленно добавляют 1М раствор BBr3 в дихлорметане (0,42 мл, 0,42 ммоля). Перемешивают реакционную смесь еще 30 минут при -78°С. Убирают охлаждающую баню и перемешивают реакционную смесь в течение 1,5 часов при 0°С и еще 2 часа при комнатной температуре. Охлаждают реакционную смесь до 0°С, медленно добавляют воду (1,00 мл) при интенсивном перемешивании. Реакционную смесь распределяют между этилацетатом и водой (1+1). Экстрагируют отделившийся водный слой этилацетатом (2 раза) и объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. После удаления растворителя при пониженном давлении и очистки сырого продукта препаративной ВЭЖХ на обращенной фазе (градиент, вода/ацетонитрил от 95:5 до 5:95) получают (5-{2-[2-(4,5-дигидрокси-2-нитрофенил)ацетиламино]-фенил}тиофен-2-ил)уксусную кислоту (110) в виде желто-коричневого твердого вещества (10 мг, 23 %). 1Н-ЯМР (400 МГц, CD3OD): 3,88 (с, 2Н), 3,96 (с, 2Н), 6,83 (с, 1Н), 6,91 (д, 1Н, J=3,3 Гц), 6,93 (д, 1Н, J=3,5 Гц), 7,23 (т, 1Н, J=7,5 Гц), 7,35 (т, 1Н, J=7,8 Гц), 7,45 (д, 1Н, J=7,8 Гц), 7,66 (с, 1Н), 7,85 (д, 1Н, J=8,1 Гц).
Пример 2
2-метил-5-{4-[(2'-нитро-4',5'-дигидроксибифенил-3-карбонил)амино]фенил}фуран-3-карбоновая кислота (113)
Стадия 1: (следующая реакция проводится в атмосфере сухого азота). Растворяют анилин (102) (25 мг, 0,11 ммоля) в безводном дихлорметане (700 мкл), добавляют безводный пиридин (22 мкл, 0,27 ммоля) и хлорид карбоновой кислоты (95) (45 мг, 0,14 ммоля). Перемешивают реакционную смесь 3,5 часа при комнатной температуре. Выливают реакционную смесь в охлажденную льдом 1М водную HCl, экстрагируют дихлорметаном (трижды), объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. После удаления растворителя получают амид (111) в виде желтого твердого вещества (51 мг, 91 %). Далее не очищают. 1Н-ЯМР (400 МГц, CDCl3): 2,64 (с, 3Н), 3,84 (с, 3Н), 3,96 (с, 3Н), 3,99 (с, 3Н), 6,78 (с, 1Н), 6,83 (с, 1Н), 7,44 (дт, 1Н, J1 = 8,1 Гц, J2 = 1,4 Гц), 7,53 (т, 1Н, J=7,7 Гц), 7,60-7,69 (м, 5Н), 7,79-7,83 (м, 2Н), 7,85 (дт, 1Н, J1 = 7,8 Гц, J2 = 1,4 Гц).
Стадия 2: растворяют сложный эфир (111) (51 мг, 0,10 ммоля) в ТГФ (2,0 мл) и метаноле (0,4 мл), добавляют 1М водный раствор LiOH (495 мкл, 0,49 ммоля). Перемешивают реакционную смесь 40 часов при 40°С. Удаляют растворитель при пониженном давлении и остаток распределяют между этилацетатом и 1М HCl (1+1). Отделяют водный слой и экстрагируют 3 раза этилацетатом. Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Удаляют растворитель при пониженном давлении и сушат остаток без дальнейшей очистки в вакууме масляного насоса. Получают сырой продукт (112) в виде желтого твердого вещества (35 мг, 70%). 1Н-ЯМР (400 МГц, ДМСО-d6): 2,58 (с, 3Н), 3,91 (с, 3Н), 3,92 (с, 3Н), 6,99 (уш.с, 1Н), 7,06 (с, 1Н), 7,53 (уш.д, 1Н, J=7,6 Гц), 7,59 (т, 1Н, J=7,7 Гц), 7,67 (с, 1Н), 7,68 (д, 2Н, J=8,6 Гц), 7,84 (д, 2Н, J=8,6 Гц), 7,93 (уш.с, 1Н), 7,98 (уш.д, 1Н, J=7,8 Гц), 10,38 (уш.c, 1Н).
Стадия 3: (следующая реакция проводится в атмосфере сухого азота). Растворяют карбоновую кислоту (112) (35 мг, 0,07 ммоля) в безводном дихлорметане (1,3 мл), охлаждают раствор до -78°С и по каплям добавляют 1М раствор BBr3 в дихлорметане (280 мкл, 0,28 ммоля). Перемешивают реакционную смесь 10 минут при -78°С и после медленного отогревания еще 2,5 часа при комнатной температуре. Охлаждают реакционную смесь до 0°С, по каплям добавляют воду и дихлорметан, затем EtOAc. Отделяют водный слой и экстрагируют 3 раза этилацетатом. Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. После удаления растворителя при пониженном давлении и очистки сырого продукта препаративной ВЭЖХ на обращенной фазе (градиент, вода/ацетонитрил от 95:5 до 5:95) получают 2-метил-5-{4-[(2'-нитро-4',5'-дигидроксибифенил-3-карбонил)амино]фенил}фуран-3-карбоновую кислоту (113) (8 мг, 24 %) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): 2,65 (с, 3Н), 6,82 (с, 1Н), 6,92 (с, 1Н), 7,48 (дт, 1Н, J1 = 7,8 Гц, J2 = 1,4 Гц), 7,57 (т, 1Н, J=7,7 Гц), 7,57 (с, 1Н), 7,69 (д, 2Н, J=8,8 Гц), 7,78 (д, 2Н, J=8,8 Гц), 7,89 (т, 1Н, J=1,6 Гц), 7,97 (дт, 1Н, J1 = 8,1 Гц, J2 = 1,4 Гц).
Пример 3
(5-{2-[(2',3'-дигидрокси-5'-нитробифенил-3-карбонил)-амино]фенил}тиофен-2-ил)уксусная кислота (116)
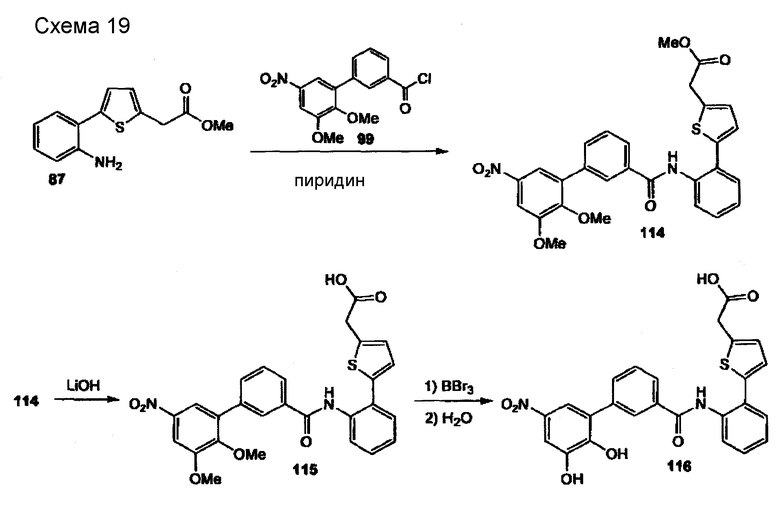
Стадия 1: (следующая реакция проводится в атмосфере сухого азота) Растворяют анилин (87) (100 мг, 0,40 ммоля) в безводном дихлорметане (3,0 мл), добавляют безводный пиридин (80 мкл, 1,01 ммоля) и хлорид карбоновой кислоты (99) (169 мг, 0,53 ммоля). Перемешивают реакционную смесь 20 часов при комнатной температуре. Выливают реакционную смесь в охлажденную льдом 1М водную HCl, экстрагируют дихлорметаном (трижды), объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Сырой продукт очищают препаративной радиальной хроматографией (силикагель 60PF, CyH/EtOAc 3+1), получают амид (114) в виде желтого твердого вещества (184 мг, 85%). 1Н-ЯМР (400 МГц, CDCl3): 3,69 (с, 3Н), 3,71 (с, 3Н), 3,86 (с, 2Н), 4,00 (с, 3Н), 6,89 (д, 1Н, J=3,3 Гц), 7,03 (д, 1Н, J=3,5 Гц), 7,17 (тд, 1Н, J1 = 7,6 Гц, J2 =1,0 Гц), 7,38-7,44 (м, 2Н), 7,53 (т, 1Н, J=7,7 Гц), 7,68 (дт, 1Н, J1 = 7,8 Гц, J2 = 1,3 Гц), 7,78 (дт, 1Н, J1 = 7,8 Гц, J2 =1,3 Гц), 7,80 (д, 1Н, J=2,5 Гц), 7,88 (д, 1Н, J=2,5 Гц), 7,91 (т, 1Н, J=3,0 Гц), 8,43 (уш.с, 1Н), 8,51 (д, 1Н, J=8,6 Гц).
Стадия 2: растворяют сложный эфир (114) (184 мг, 0,34 ммоля) в ацетонитриле (5,0 мл) и метаноле (2,0 мл), добавляют 1М водный раствор LiOH (1,7 мл, 1,7 ммоля). Перемешивают реакционную смесь 20 часов при 40°С. Удаляют растворитель при пониженном давлении и остаток распределяют между этилацетатом и 1М HCl (1+1). Отделяют водный слой и экстрагируют 3 раза этилацетатом. Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. Удаляют растворитель при пониженном давлении и сушат остаток без дальнейшей очистки в вакууме масляного насоса. Получают сырой продукт (115) в виде светло-желтого твердого вещества (183 мг, количественно). 1Н-ЯМР (400 МГц, CD3OD): 3,78 (с, 3Н), 3,85 (с, 2Н), 4,07 (с, 3Н), 6,98 (д, 1Н, J=3,5 Гц), 7,19 (д, 1Н, J=3,5 Гц), 7,38 (тд, 1Н, J1=7,5 Гц, J2 = 1,3 Гц), 7,44 (тд, 1Н, J1=7,7 Гц, J2= 1,5 Гц), 7,61-7,68 (м, 2Н), 7,73 (д, 1Н, J=8,1 Гц), 7,80 (дд, 1Н, J1 = 7,6 Гц, J2 = 1,3 Гц), 7,94-8,02 (м, 3Н), 8,10 (уш.с, 1Н).
Стадия 3: (следующая реакция проводится в атмосфере сухого азота). Растворяют карбоновую кислоту (115) (70 мг, 0,14 ммоля) в безводном дихлорметане (1,4 мл), охлаждают раствор до -78°С и по каплям добавляют 1М раствор BBr3 в дихлорметане (810 мкл, 0,81 ммоля). Перемешивают реакционную смесь 10 минут при -78°С и после медленного отогревания еще 2 часа при комнатной температуре. Охлаждают реакционную смесь до 0°С, по каплям добавляют воду и дихлорметан, затем EtOAc. Отделяют водный слой и экстрагируют 3 раза этилацетатом. Объединенные органические слои промывают насыщенным водным раствором NaCl и сушат Na2SO4. После удаления растворителя при пониженном давлении и очистки сырого продукта препаративной ВЭЖХ на обращенной фазе (градиент, вода/ацетонитрил от 95:5 до 5:95) получают (5-{2-[(2',3'-дигидрокси-5'-нитробифенил-3-карбонил)амино]фенил}тиофен-2-ил)уксусную кислоту (116) (12 мг, 18%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): 3,84 (с, 2Н), 6,99 (д, 1Н, J=2,8 Гц), 7,19 (д, 1Н, J=3,5 Гц), 7,37 (тд, 1Н, J1 =7,8 Гц, J2=1,5 Гц), 7,44 (тд, 1Н, J1=7,7 Гц, J2=1,5 Гц), 7,58-7,65 (м, 2Н), 7,72-7,76 (м, 2Н), 7,86-7,97 (м, 3Н), 8,16 (уш.с, 1Н).
Пример 4
4',5'-дигидрокси-2'-нитробифенил-3-карбоновой кислоты фениламид (117)
Схема 20
4',5'-дигидрокси-2'-нитробифенил-3-карбоновой кислоты фениламид (117) получен из анилина и хлорида карбоновой кислоты (95) согласно вышеописанной процедуре, стадии 1 и 3 в Примере 3. Получен 4',5'-дигидрокси-2'-нитробифенил-3-карбоновой кислоты фениламид (117) (2,4 мг, 10% выход на две стадии) в виде коричневого твердого вещества 1Н-ЯМР (400 МГц, CD3OD): 6,84 (с, 1Н), 7,19 (уш.т, 1Н, J=7,5 Гц), 7,39 (уш.д, 1Н, J=8,1 Гц), 7,41 (уш.д, 1Н, J=7,6 Гц), 7,48 (дт, 1Н, J1=7,6 Гц, J2=1,3 Гц), 7,57 (т, 1Н, J=7,7 Гц), 7,57 (с, 1Н), 7,70-7,75 (м, 2Н), 7,88 (т, 1Н, J=1,5 Гц). 7,97 (уш.д, 1Н, J=7,8 Гц).
Пример 5
(5-{2-[(4',5'-дигидрокси-2'-нитробифенил-3-карбонил)-амино]фенил}тиофен-2-ил)уксусная кислота (118)

(5-{2-[(4',5'-дигидрокси-2'-нитробифенил-3-карбонил)-амино]фенил}тиофен-2-ил)уксусная кислота (118) получена из анилина (87) и хлорида карбоновой кислоты (95) согласно описанной выше процедуре, стадии с 1 по 3 в Примере 3. Получена (5-{2-[(4',5'-дигидрокси-2'-нитробифенил-3-карбонил)амино]-фенил}тиофен-2-ил)уксусная кислота (118) (14 мг, 29% выход на три стадии) в виде желтого твердого вещества 1Н-ЯМР (400 МГц, CD3OD): 3,85 (с, 2Н), 6,83 (c, 1Н), 6,97 (д, 1Н, J=3,5 Гц), 7,17 (д, 1Н, J=3,5 Гц), 7,37 (тд, 1Н, J1 = 7,6 Гц, J2 = 1,5 Гц), 7,43 (тд, 1Н, J1 = 7,6 Гц, J2= 1,8 Гц), 7,47 (уш.д, 1Н, J=7,6 Гц), 7,55 (т, 1Н, J=7,6 Гц), 7,56 (с, 1Н), 7,64 (дд, 1Н, J1=7,3 Гц, J2=1,0 Гц), 7,69 (д, 1Н, J=7,6 Гц), 7,83 (уш.с, 1Н), 7,93 (уш.д, 1Н, J=7,6 Гц).
Пример 6
5-{2-[(4',5'-дигидрокси-2'-нитробифенил-3-карбонил)-амино]-фенил}тиофен-2-карбоновая кислота (119)

5-{2-[(4',5'-дигидрокси-2'-нитробифенил-3-карбонил)-амино]-фенил}тиофен-2-карбоновая кислота (119) получена из амина (90) и хлорида карбоновой кислоты (95) согласно описанной выше процедуре, стадии 1 и 3 в Примере 3. Получена 5-{2-[(4',5'-дигидрокси-2'-нитробифенил-3-карбонил)амино]фенил}тиофен-2-карбоновая кислота (119) (6 мг, 8% выход на две стадии) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): 6,84 (с, 1Н), 7,35 (д, 1Н, J=3,8 Гц), 7,41-7,63 (м, 6Н), 7,71 (дд, 1Н, J1=7,8 Гц, J2=1,3 Гц), 7,75 (д, 1Н, J=3,8 Гц), 7,86 (уш.с, 1Н), 7,93 (уш.д, 1Н, J=7,6 Гц).
Пример 7
5-{2-[(2',3'-дигидрокси-5'-нитробифенил-3-карбонил)-амино]-фенил}тиофен-2-карбоновая кислота (120)
5-{2-[(2',3'-дигидрокси-5'-нитробифенил-3-карбонил)амино]-фенил}тиофен-2-карбоновая кислота (120) получена из амина (90) и хлорида карбоновой кислоты (99) согласно описанной выше процедуре, стадии 1-3 в Примере 3. Получена (5-{2-[(2',3'-дигидрокси-5'-нитробифенил-3-карбонил)амино]фенил}тиофен-2-карбоновая кислота (120) (17 мг, 15% выход на три стадии) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): 7,36 (д, 1Н, J=4,0 Гц), 7,44 (тд, 1Н, J1=7,6 Гц, J2=1,5 Гц), 7,51 (тд, 1Н, J1=7,6 Гц, J2=1,5 Гц), 7,59-7,67 (м, 2Н), 7,69-7,75 (м, 3Н), 7,86-7,97 (м, 3Н), 8,21 (уш.с, 1Н).
Пример 8
(5-{2-[2-(3,4-дигидрокси-5-нитрофенил)-ацетиламино]-фенил}тиофен-2-ил)уксусная кислота (121)

(5-{2-[2-(3,4-дигидрокси-5-нитрофенил)ацетиламино]-фенил}тиофен-2-ил)уксусная кислота (121) получена из амина (87) и хлорида карбоновой кислоты (107) согласно описанной выше процедуре, стадии 1-3 в Примере 1. Получена ((5-{2-[2-(3,4-дигидрокси-5-нитрофенил)ацетиламино]фенил}тиофен-2-ил)уксусная кислота (121) (8 мг, 10% выход на три стадии) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): 3,65 (с, 2Н), 3,81 (c, 2Н), 6,83-6,89 (м, 2Н), 7,17 (уш.с, 1Н), 7,27 (тд, 1Н, J1=7,6 Гц, J2=1,5 Гц), 7,36 (тд, 1Н, J1=7,7 Гц, J2=1,5 Гц), 7,48 (дд, 1Н, J1=7,8 Гц, J2=1,5 Гц), 7,55 (с, 1Н), 7,69 (уш.д, 1Н, J=8,1 Гц).
Результаты из sLe x TSA: IC 50 данные для E-/P-/L-селектина
Данные из теста в проточной камере для E- и P-селектина
Значения даны в виде нормированных отношений процентного ингибирования соединения x, деленного на процентное ингибирование бимосиамозы
Соединения, отмеченные на следующей схеме 10 являются соединениями, отмеченными здесь как особенно предпочтительные соединения
sLe x -тирозинсульфат испытание:
Соединения по настоящему изобретению испытаны на молекулярном уровне на их способность ингибировать связывание P-, L- или E-селектинхимерных молекул с sLex и тирозинсульфатными остатками, закрепленными на полимерной основе в качестве заместителя PSGL-1. Определены избранные значения IC50.
Титрационный микропланшет на ночь покрывают карбонатным буфером (pH 9,6) с козьими притовочеловеческими Fc моноклональные антитела (10 мкг/мл). После промывания аналитическим буфером (25 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота (HEPES), 150 мМ NaCl, 1 мМ CaCl2 pH 7,4) и блокирования (3% альбумин бычьей сыворотки (BSA) в аналитическом буфере) планшеты выдерживают в течение 2 часов при 37°С с P-селектин-IgG-химерой человека (0,61 нМ, соответственно 150 нг/мл) или L-селектин-IgG-химерой человека (0,61 нМ, соответственно 89 нг/мл) или E-селектин-IgG-химерой человека (0,61 нМ, соответственно 131 нг/мл). 5 мкл sLex-тирозинсульфатполиакриламида (1 мг/мл), содержащего 15% sLex, 10% тирозинсульфата и 5% биотина, комплексуют 20 мкл раствора стрептавидинпероксидазы (1 мг/мл) и 25 мкл аналитического буфера без CaCl2. Для использования в испытании комплекс лиганда разбавляют в отношении 1 к 10000 в аналитическом буфере и далее разбавляют 1:1 различными количествами соединений в аналитическом буфере, содержащем 2% ДМСО. Эту смесь добавляют в лунки, предварительно покрытые E- или P-селектином. После выдерживания в течение 2 часов при 37°С лунки промывают шесть раз аналитическим буфером, содержащим 0,005% монолаурата полиоксиэтиленсорбитана (TWEEN 20), проявляют в течение 10-15 минут 20 мкл раствора 3,3',5,5'-тетраметилбензидина(ТМБ)/H2O2 и останавливают 20 мкл 1М H2SO4. Связанный комплекс лиганд/sLex-тирозинсульфат определяют измерением оптической плотности при 450 нм по сравнению с 620 нм в Fusion alpha-FP аппарате для чтения планшетов. (приобретен у Packard Bioscience, Драйях (Dreieich), Германия).
Испытание в проточной камере/клеточная адгезия и скатывание в проточных условиях
Для определения способности соединений ингибировать связывание клеток в динамических условиях, подобных току в кровеносных сосудах, были предприняты испытания в проточной камере, направленные на/испытывающие связывание HL-60 клеток/клеток различных линий с химерными молекулами P-, L- и E-селектина.
Прикрепление клеток в проточных условиях определяли с использованием параллельной системы проточных камер. 35-мм чашка для культивирования из полистирола на 1 час при комнатной температуре покрывалась покровным буфером (50 мМ трис-(гидроксиметил)аминометановый буфер (Трис), 150 мМ NaCl, 2 мМ CaCl2; pH 7,4), содержащим IgG-химеру человеческого E- или P-селектина в концентрации 2,5 мкг/мл или 10 мкг/мл, соответственно. После удаления покрывающего раствора неспецифические связывающие сайты блокируют при комнатной температуре в течение еще 1 часа 1% BSA в покровном буфере. После промывания аналитическим буфером (“Roswell Park Memorial Institute 1640” (RPMI 1640)+ 10 мМ HEPES) чашку устанавливают в параллельную камеру ламинарного потока (приобретенную от Glycotech, Rockville, MD) и монтируют к ивертированному фазовоконтрастному микроскопу (приобретенному от Olympus, Гамбург, Германия), снабженному подключенной к ПК камерой с управляемым компьютером дисплеем (JVC). С использованием перистальтического насоса (приобретенного от Ismatec, Вертхейм-Мондфелд (Wertheim-Mondfeld), Германия) рециркулирующая система уравнена аналитическим буфером, содержащим 125 мкМ соединения или контрольным наполнителем (ДМСО). В камеру добавляют клетки (1 миллион на мл) и дают распределиться в течение 2 минут при высокой скорости потока. Затем скорость потока уменьшают так, что в результате расчетный сдвиг течения составляет 1 дин/см2. Видеопоследовательности для 10 полей зрения под малым увеличением записываются в цифровой форме после пятиминутного постоянного потока. Процентное значение ингибирования рассчитывается из среднего числа клеток на область, связанную с покрытой поверхностью чашки, в присутствии и (по сравнению) в отсутствие соединения (в независимом эксперименте).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ФЛОРОГЛЮЦИНА, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ЛИГАНДА СЕЛЕКТИНА | 2006 |
|
RU2418584C2 |
| НОВЫЕ ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2006 |
|
RU2434006C2 |
| ИНГИБИТОРЫ АРГИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2011 |
|
RU2586219C2 |
| НОВОЕ ПРОИЗВОДНОЕ ОКСАЗОЛИДИНОНА И ВКЛЮЧАЮЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2012 |
|
RU2617408C2 |
| ТРИАРОМАТИЧЕСКИЕ АНАЛОГИ ВИТАМИНА D, ФАРМАЦЕВТИЧЕСКАЯ И КОСМЕТИЧЕСКАЯ КОМПОЗИЦИИ | 2000 |
|
RU2237651C2 |
| ИНДОЛЫ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АНДРОГЕНОВЫМИ РЕЦЕПТОРАМИ | 2003 |
|
RU2328484C2 |
| С-2'-МЕТИЛИРОВАННЫЕ ПРОИЗВОДНЫЕ ПАКЛИТАКСЕЛА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ | 2002 |
|
RU2287528C2 |
| СОЕДИНЕНИЯ, ОБРАЗУЮЩИЕ ПРОЛЕКАРСТВА | 2014 |
|
RU2667942C2 |
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
| БИЦИКЛИЧЕСКИЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1999 |
|
RU2188190C2 |
Изобретение относится к области фармацевтики и косметологии, более конкретно касается соединений, имеющих общую структуру формулы (С2) или (С5) или (С6) или (D2) или (D5) или (D6)


а также раскрывает косметические и фармацевтические композиции, их содержащие.
Композиции, содержащие описанные соединения, могут быть применены для модулирования in vitro и in vivo процессов связывания с посредничеством связывания Е-, Р- или L-селектина. 7 н. и 1 з.п. ф-лы, 2 табл.
1. Фармацевтическая композиция, обладающая способностью модулировать экспрессию селектина, содержащая по меньшей мере одно соединение, описываемое формулой (С2), или (С5), или (С6), и фармацевтически приемлемый носитель, используемый в медицине
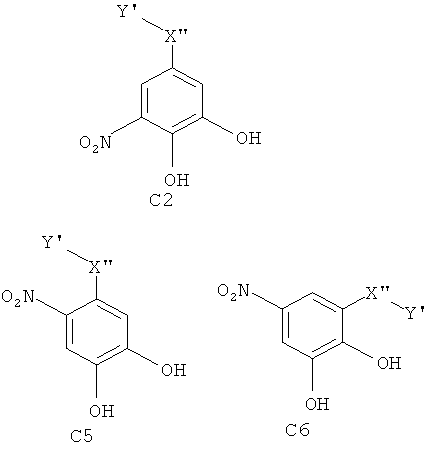
где символы и заместители имеют следующее значение:
X" представляет собой
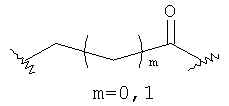

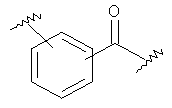
Y' представляет собой


m равно 0, 1;
s равно 1;
t равно 0, 1, 2;
R2 представляет собой СО2Н, СО2алкил, СF3;
R3 независимо от R2 представляет собой Н; и R4 независимо от R2 и R3 представляет собой Н;
R5 представляет собой Н; и
W представляет собой -(CH2-)v, и v означает 0, 1, 2;
Z представляет собой
(iv)

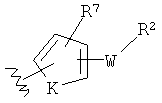
К представляет собой О, S,
R7 независимо от R2 представляет собой Н;
или фармацевтически приемлемые соли, сложные эфиры или амиды определенных выше соединений формулы (С2), или (С5), или (С6).
2. Фармацевтическая композиция по п.1, где соединение определяется формулой (D2), или (D5), или (D6)

где X" является таким, как определено в п.1, и Y" представляет собой


R9 представляет собой CO2H, СO2алкил,
где все индексы, символы и заместители являются такими, как определено в п.1.
3. Химическое соединение, имеющее общую структуру формулы (С2), или (С5), или (С6), или (D2), или (D5), или (D6) по п.1 или 2.
4. Применение соединения, имеющего структуру формулы (С2), или (С5), или (С6), как определено в п.1, для получения лекарственного средства для лечения хронического обструктивного заболевания легких (ХОЗЛ), острого повреждения легких (ОПЛ), при искусственном кровообращении, острого респираторного дистресс-синдрома (ОРДС), болезни Крона, септического шока, сепсиса, хронических воспалительных заболеваний, таких как псориаз, атопический дерматит и ревматоидный артрит, реперфузионных повреждений, которые происходят следом за инфарктами, инсультами, атеросклерозом и трансплантацией органов, травматического шока, функциональной недостаточности многих органов, аутоиммунных заболеваний, таких как множественный склероз, при чрезкожной транслюминальной ангиопластике, астме и воспалительной кишечной болезни.
5. Применение соединения, имеющего структуру формулы (С2), или (С5), или (С6), как определено в п.1, для получения лекарственного средства для лечения, диагностирования или профилактики воспалительных заболеваний.
6. Применение соединения, имеющего структуру формулы (С2), или (С5), или (С6), как определено в п.1, для получения косметической или дерматологической композиции.
7. Косметическая композиция, обладающая способностью модулировать экспрессию селектина, содержащая по меньшей мере одно соединение формулы (С2), или (С5), или (С6), как определено в п.1, и по меньшей мере один косметически приемлемый компонент.
8. Дерматологическая композиция, обладающая способностью модулировать экспрессию селектина, содержащая по меньшей мере одно соединение формулы (С2), или (С5), или (С6), как определено в п.1, и по меньшей мере один дерматологически приемлемый компонент.
| ДИ- И ТРИВАЛЕНТНЫЕ НЕБОЛЬШИЕ МОЛЕКУЛЫ ИНГИБИТОРОВ СЕЛЕКТИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СЕЛЕКТИНА | 1996 |
|
RU2165931C2 |
| MANNISTO Р Т ET AL.: "CATECHOL-O-METHYLTRANSFERASE (COMT): BIOCHEMISTRY, MOLECULAR BIOLOGY, PHARMACOLOGY, AND CLINICAL EFFICACY OF THE NEW SELECTIVE COMT INHIBITORS" PHARMACOLOGICAL REVIEWS, WILLIAMS AND WILKINS INC., BALTIMORE, MD, US, vol.51, no.4, 1999, pages 593-628 | |||
| FRIEDRICH, M ET AL.: "Pan-selectin antagonism | |||

