ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение в целом относится к сериям соединений, к фармацевтическим композициям, содержащим соединения, и к применению соединений и композиций в качестве терапевтических агентов. Более конкретно соединения по настоящему изобретению представляют собой тиофенильные и пирролильные азепиновые соединения. Эти соединения представляют собой лиганды серотонинового рецептора (5-HT2c) и могут быть применены для лечения болезней, расстройств и состояний, при которых модуляция активности серотониновых рецепторов (5-HT2c) является желательной (например, привыкание к наркотикам, беспокойство, депрессия, ожирение и другие).
УРОВЕНЬ ТЕХНИКИ
Серотонин участвует в ряде заболеваний, расстройств и состояний, которые возникают в центральной нервной системе, включая болезни, расстройства и состояния, относящиеся, например, ко сну, еде, восприятию боли, контролю температуры тела, контролю кровяного давления, депрессии, беспокойству, привыканию к наркотикам и шизофрении. Серотонин также играет важную роль в периферических системах, таких как желудочно-кишечная система, где он, как было найдено, опосредует разнообразные сократительные, секреторные и электрофизиологические эффекты.
Ввиду широкого распространения серотонина в пределах организма существует необходимость в лекарствах, которые воздействуют на серотонинэргические системы. В частности, агонисты, частичные агонисты и антагонисты серотонинэргических систем представляют интерес для лечения широкого диапазона расстройств, включая беспокойство, депрессию, гипертонию, мигрень, ожирение, маниакальные расстройства, шизофрению, аутизм, нейродегенеративные расстройства (например, болезнь Альцгеймера, Паркинсонизм и хорею Хантигтона) и вызываемую химиотерапией рвоту.
Основные классы серотониновых рецепторов (5-HT1-7) содержат от одного до семи отдельных рецепторов, которые были формально классифицированы. Смотри Glennon, et al., Neuroscience and Behavioral Reviews, 1990,14, 35; и D. Hoyer, et al. Phannacol, Rev, 1994, 46, 157-203.
Например, семейство 5-HT2 рецепторов содержит 5-HT2a, 5-HT2b и 5-HT2c подтипы, которые были сгруппированы совместно на основании первичной структуры, системы вторичного мессенджера и профиля функционирования. Все три 5-HT2 подтипа связаны с G-белком, активированной фосфолипазой C, в качестве основного механизма трансдукции, и имеют трансмембранную семидоменную структуру. Существуют различные отличия в распределении трех 5-HT2 подтипов в млекопитающих. 5-HT2b и 5-HT2a рецепторы широко распространены в периферической нервной системе, 5-HT2a также найден в мозге. 5-HT2c рецептор был найден только в центральной нервной системе и высоко экспрессирован во многих сегментах мозга человека. Смотри G. Baxter, et al. Trends in Pharmacol. Sci. 1995, 16, 105-110.
Подтип 5-HT2a был ассоциирован с эффектами, включая сужение кровеносных сосудов, агрегацию тромбоцитов и бронхоконстрикцию, также как определенными эффектами в ЦНС, в то время как подтип 5-HT2c был ассоциирован с заболеваниями, которые включают в себя депрессию, беспокойства, обсессивно-компульсивное расстройство, привыкание к наркотикам, панические расстройства, фобии, психиатрические синдромы и ожирение. Очень мало известно о фармакологической роли 5-HT2b рецептора. F. Jenck, et al., Exp. Opin. Invest. Drugs, 1998, 7, 1587-1599; M. Bos, et al., J. Med. Chem., 1997, 40, 2762-2769; J.R, Martin, et al., The Journal of Pharmacology and Experimental Therapeutics, 1998, 286, 913-924; S.M. Bromidge, et al., J. Med, Chem., 1998, 41, 1598-1612; G.A. Kennett, Drugs, 1998, 1, 4, 456-470; и A. Dekeyne, et al., Newophannacology, 1999, 38, 415-423.
WO 93/13105, патенты США №№5691330 и 5532240 впервые описывают производные тиофена; патент США №4414225 впервые описывает производные тиофена, фурана и пиррола; патент США №4575504 впервые описывает производные тиенотиазола; патент США №5258378 впервые описывает определенные пирролоазепиновые соединения; патенты США №№4414225 и 4904653 впервые описывают определенные азепиновые производные; WO 2005/019179 впервые описывает определенные бензазепины, WO 2005/003096, WO 2005/042490 и WO 2005/042491 впервые описывают производные бензазепина; WO 96/11201 впервые описывает производные фурана; WO2005/040169 впервые описывает определенные конденсированные пиррол- и пиразолсодержащие гетероциклические соединения, которые являются серотониновыми модуляторами; WO2004/024065 впервые описывает замещенные бициклические производные тиофена. Ни один из этих патентов или заявок на патент не описывает соединения по настоящему изобретению.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы:
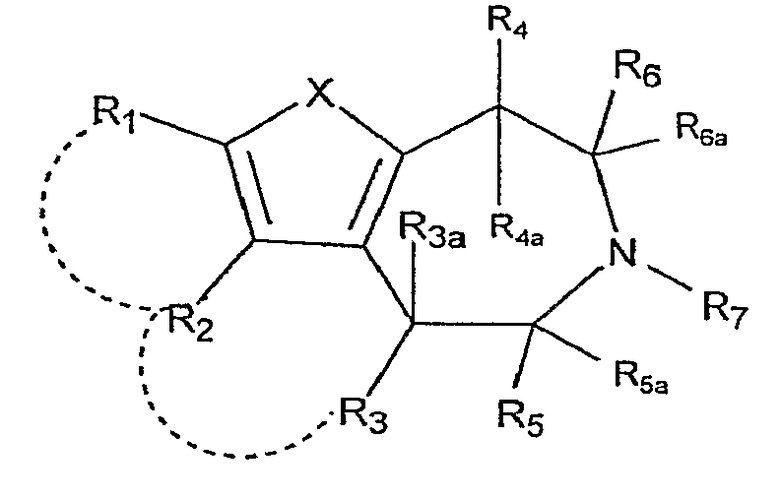 ,
,
где X представляет собой S или NR11;
R1 и R2 независимо выбраны из группы, состоящей из атома водорода, галогена, C1-8-алкила, C2-8-алкенила, C2-8-алкинила, пергалогеналкила, C1-8-алкилпергалогеналкила, -CN, OR8, SR8, -SO2R10, -C(=O)R10, -C(=O)NR8R9, -NR8CO2R10, -SO2NR8R9, -NR8SO2R10, арила или гетероарила, C1-8-алкиларила или гетероарила, C1-8-алкил-O-C1-8-алкила и C1-8-алкил-O-арила или гетероарила;
R1 и R2, взятые вместе с атомами, к которым они присоединены, могут образовывать 5-7-членный карбоцикл или гетероцикл, необязательно замещенный заместителями, вплоть до двух, выбранными из алкила, CF3 и -OR8;
R3 выбран из группы, состоящей из атома водорода, C1-8-алкила, OR8, арила и гетероарила;
R3a представляет собой атом водорода или C1-8-алкил; или R3 и R3a, взятые вместе, представляют собой -CH2CH2-;
R2 и R3, взятые вместе, образуют 5-7-членный карбоцикл или гетероцикл, необязательно замещенный заместителями, вплоть до двух, выбранными из алкила, CF3 и -OR8;
R4 представляет собой атом водорода, C1-8-алкил или OR8;
R4a представляет собой атом водорода, C1-8-алкил; или R4 и R4a, взятые вместе, представляют собой -CH2CH2-;
R5 выбран из группы, состоящей из атома водорода, C1-8-алкила, -C1-8-алкил-O-C1-8-алкила, C1-8-алкиларила или гетероарила и -C1-8-алкил-O-арила или гетероарила;
R5a представляет собой атом водорода или -C1-8-алкил;
R6 выбран из группы, состоящей из атома водорода; -C1-8-алкила, C1-8-алкил-O-C1-8-алкила, C1-9-алкиларила или гетероарила и -C1-8-алкил-O-арила или гетероарила;
R6a представляет собой атом водорода или -C1-8-алкил;
R7 выбран из группы, состоящей из атома водорода, -C1-8-алкила и -C1-8-алкиларила или гетероарила;
R8, R9 независимо выбраны из группы, состоящей из атома водорода, -C1-8-алкила, -C2-8-алкенила, -C2-8-алкинила, арила или гетероарила, -C1-8-алкиларила или гетероарила, -C1-8-алкил-O-C1-8-алкила и -C1-8-алкил-O-арила или гетероарила;
R8 и R9, взятые вместе с атомом, к которому они присоединены, образуют 5-7-членный гетероцикл;
R10 выбран из группы, состоящей из -C1-8-алкила, -C2-8-алкенила, -C2-8-алкинила, арила или гетероарила, -C1-8-алкиларила или гетероарила, -C1-8-алкил-O-C1-8-алкила и -C1-8-алкил-O-арила или гетероарила;
R11 выбран из группы, состоящей из атома водорода, -C1-8-алкила, -C1-8-алкил-O-C1-8-алкила, -SO2R10, -C(=O)R10, -C(=O)OR10, арила, гетероарила или C1-8-алкиларила, или гетероарила;
R11 и R1, вместе с атомом, к которым они присоединены, могут образовывать 5-7-членный гетероцикл, необязательно замещенный заместителями, вплоть до двух, выбранными из -C1-8-алкила, CF3 и -OR8; и
R11 и R4, вместе с атомами, к которым они присоединены, могут образовывать 5-7-членный гетероцикл, необязательно замещенный заместителями, вплоть до двух, выбранными из -C1-8-алкила, CF3 и -OR8;
где арил и гетероарил необязательно замещены заместителями, вплоть до двух, выбранными из -C1-8-алкила, галогена, CN, алкокси и их фармацевтически приемлемых солей.
Другой вариант осуществления по настоящему изобретению предоставляет фармацевтическую композицию, включающую в себя соединение формулы (I) или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
Следующий вариант осуществления по настоящему изобретению предоставляет способ лечения болезни, расстройства и/или состояния у млекопитающего (например, животного или человека), которое опосредовано 5-HT2c рецептором и желательна модуляция 5-HT2C функции. Способ включает в себя введение млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Следующий вариант осуществления по настоящему изобретению включает в себя способ модулирования функции 5-HT рецептора эффективным количеством соединения формулы (I) или его фармацевтически приемлемой соли.
Дополнительный вариант осуществления по настоящему изобретению предоставляет способ лечения или профилактики болезней, расстройств и/или состояний центральной нервной системы. Способ включает в себя введение млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Специфические заболевания, расстройства и/или состояния, по отношению к которым соединения формулы (I) могут обладать активностью, включают в себя ожирение, депрессию, шизофрению, беспокойство, обсессивно-компульсивное расстройство, привыкание к наркотикам, панические расстройства, расстройства сна, мигрень, диабет типа II, эпилепсия, фобии и психиатрические синдромы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Используют следующие определения, если явно не указано иного: применяемый здесь термин "алкил" включает в себя углеводородные группы с прямой и разветвленной цепью, содержащие указанное число атомов углерода, главным образом метил, этил, и прямые и разветвленные пропильные и бутильные группы. Термин "алкил" также включает в себя циклоалкил, то есть циклическую C3-C8-углеводородную группу, такую как циклопропил, циклобутил, циклопентил и циклогексил. Ссылка на индивидуальную группу или фрагмент, такую как "пропил", включает в себя только группу или фрагмент с прямой цепью. Изомер с разветвленной цепью, такой как "изопропил", обозначается специально.
Применяемый здесь термин "алкенил", отдельно или в комбинации, относится к замещенному или незамещенному алкенильному радикалу с прямой или разветвленной цепью, содержащему от 2 до 10 атомов углерода. Примеры таких радикалов включают в себя, без ограничения, этенил, E- и Z-пентенил, деценил и подобные.
Применяемый здесь термин "алкинил", отдельно или в комбинации, относится к замещенному или незамещенному алкинильному радикалу с прямой или разветвленной цепью, содержащему от 2 до 10 атомов углерода. Примеры таких радикалов включают в себя, без ограничения, этинил, пропинил, пропаргил, бутинил, гексинил, децинил и подобные.
Применяемый здесь термин "алкокси", отдельно или в комбинации, относится к остатку алкильного простого эфира, где термин "алкил" определен выше. Примеры пригодных алкоксирадикалов включают в себя, без ограничения, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, втор-бутокси, трет-бутокси и подобные.
Определенный здесь термин "гало" включает в себя фтор, хлор, бром или йод. Аналогично определенный здесь термин "галоген" включает в себя фтор, хлор, бром и йод.
Термин "амино", отдельно или в комбинации, включает в себя группу -NH2 или -NRaRb, где Ra и Rb независимо представляют собой атом водорода, алкил, алкиларил или арил.
Термин "арил", отдельно или в комбинации, определяется здесь как моноциклическая или бициклическая ароматическая группа (например, фенил или нафтил), которая может являться незамещенной или замещенной, например, одним или несколькими и, в частности, от одного до трех из следующих заместителей, выбранных из группы, состоящей из атома водорода, атом галогена, CN, NO2, CF3, N3, C1-6-алкила, OH, NRaRb, OC1-6-алкила, ORa, C(=O)NRaRb, C(=S)NRaRb, тетразолила, триазолила, амидинила, гуанидинила, тиогуанидинила, цианогуанадинила и арила, где Ra и Rb независимо представляют собой атом водорода, алкил, алкиларил или арил. В целом "арил" обозначает фенильную группу или орто-конденсированную бициклическую карбоциклическую группу, имеющую от девяти до десяти кольцевых атомов, из которых по меньшей мере одно кольцо является ароматическим (например, нафтил или тетрагидронафтил). Термин "арил" также сокращенно обозначается в различный химических структурах как "Ar."
Термин "гетероарил" определяется здесь как моноциклическая, бициклическая или трициклическая кольцевая система, содержащая один, два или три ароматических кольца и содержащая по меньшей мере один атом азота, кислорода или серы в ароматическом цикле, которая может являться незамещенной или замещенной, например, одним или несколькими, в частности от одного до трех, заместителями, выбранными из атома галогена, алкила, гидрокси, гидроксиалкила, алкокси, алкоксиалкила, галогеналкила, нитро, амино, алкиламино, ациламино, алкилтио, алкилсульфонила и алкилсульфонила. Примеры гетероарильных групп включают в себя, без ограничения, 2H-пирролил, 3H-индолил, 4H-хинолизинил, 4H-карбазолил, акридинил, бензо[b]тиенил, бензотиазолил, 13-карболинил, карбазолил, хроменил, циннолинил, дибензо[b,d]фуранил, фуразанил, фурил, имидазолил, имидизолил, индазолил, индолизинил, индолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазолил, нафтиридинил, нафто[2,3-b], оксазолил, перимидинил, фенантридинил, фенантролинил, фенарсазинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, птеридинил, пуринил, пиранил, пиразинил, пиразолил, пиридазинил, пиридил, пиримидинил, пирролил, хиназолинил, хинолил, хиноксалинил, тиадиазолил, тиантренил, тиазолил, тиенил, триазолил и ксантенил. В одном варианте осуществления термин "гетероарил" обозначает моноциклический ароматический цикл, содержащий пять или шесть кольцевых атомов, содержащих углерод и 1, 2, 3 или 4 гетероатома, независимо выбранных из группы, состоящей из непероксидного кислорода, серы и N(Z), где Z отсутствует или представляет собой атом водорода, O, C1-4-алкил, фенил или бензил. В другом варианте осуществления гетероарил обозначает орто-конденсированный бициклический гетероцикл, имеющий от около восьми до десяти кольцевых атомов и произведенные от него, особенно бензопроизводное или система, полученная путем приконденсирования к гетероциклу пропиленового или тетраметиленового дирадикала.
Термин "гетеро" или "гетероцикл" в целом представляет собой гетероциклическую группу, насыщенную или частично ненасыщенную, содержащую по меньшей мере один гетероатом, выбранный из группы, состоящей из кислорода, азота и серы, и необязательно замещенную C1-6-алкилом или C(=O)OR6. Типично "гетероцикл" представляет собой моноциклическую, бициклическую или трициклическую группу, содержащую один или несколько гетероатомов, выбранных из группы, состоящей из кислорода, азота и серы. "Гетероциклическая" группа также может содержать оксо-группу (=O), присоединенную к циклу. Неограничивающие примеры гетероциклических групп включают в себя 1,3-дигидробензофуран, 1,3-диоксолан, 1,4-диоксан, 1,4-дитиан, 2H-пиран, 2-пиразолин, 4H-пиран, хроманил, имидазолидинил, имидазолинил, индолинил, изохроманил, изоиндолинил, морфолин, пиперазинил, пиперидин, пиперидил, пиразолидин, пиразолидинил, пиразолинил, пирролидин, пирролин, хинуклидин и тиоморфолин.
В настоящее время предпочтительные соединения включают в себя:
2,2-Диметил-l-(3-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-пропан-1-он;
3-Бром-2-(2,2,2-трифторэтил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин;
2-Бензолсульфонил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин;
(R,S)-2-(2,2,2-Трифтор-1-метил-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин;
2-Этансульфонил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин;
(R,S)-1-Трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен;
(R,S)-3,3-Диметил-1-трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен;
2-(2,2,2-Трифтор-1,1-диметил-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин;
3-Бром-4-метил-2-(2,2,2-трифтор-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин;
2-Этансульфонил-3,4-диметил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин;
(R,S)-2,2-Диметил-l-(4-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-пропан-1-он;
1-(3-Бром-4-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-он;
(R,S)-2-(2,2,2-Трифтор-этил)-4,4a,5,6,7,8-гексагидро-3H-1-тиа-6-аза-циклопента[cd]азулен;
(R,S)-2-Бром-3,3-диметил-4,4a,5,6,7,8-гексагидро-3H-1-тиа-6-аза-циклопента[cd]азулен;
(R,S)-2-Бром-5-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин;
4-Метил-1-трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен;
3,3,4-Триметил-1-трифторметил-l,2,3,4,5,6,7,8-октагидро-9-тиа-6-азациклопента[a)азулен;
2,2-Диметил-1-(5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-пропан-1-он и
l-(3-Хлор-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-он.
Определенные соединения по изобретению могут существовать в различных изомерных формах (например, в виде энантиомеров и диастереомеров). Изобретение включает в себя все такие изомеры, как в чистой форме, так и в виде смесей, включая рацемические смеси. Также включены енольные формы.
Соединения по изобретению могут существовать как в несольватированной, так и в сольватированной формах, включая гидратированные формы, например полугидрат. В общем сольватированные формы с фармацевтически приемлемыми растворителями, такими как вода, этанол и подобные, эквивалентны несольватированным формам для целей изобретения.
Определенные соединения по изобретению также образуют фармацевтически приемлемые соли, например соли присоединения кислоты. Например, атомы азота могут образовывать соли с кислотами. Примерами пригодных кислот для образования соли являются соляная, серная, фосфорная, уксусная, лимонная, щавелевая, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, малеиновая, метансульфоновая и другие минеральные карбоновые кислоты, хорошо известные специалистам в данной области техники. Соли приготовляются путем приведения в контакт формы свободного основания с достаточным количеством желаемой кислоты для получения соли традиционным способом. Формы свободного основания могут быть регенерированы путем обработки соли пригодным разбавленным водным раствором основания, таким как разбавленный водный гидроксид калия, карбонат аммония и бикарбонат натрия. Формы свободного основания до некоторой степени отличаются от их соответствующих солевых форм в определенных физических свойствах, таких как растворимость в полярных растворителях, но соли с кислотой являются эквивалентными их соответствующим формам свободного основания для целей изобретения (смотри, например, S. M. Berge, et al., "Pharmaceutical Salts," J. Pharm. Sci., 66: 1-19 (1977), которая включена здесь посредством цитирования.
Применяемый здесь термин "композиция" предназначен для включения в себя продукта, содержащего определенные компоненты в определенных количествах, также как любой продукт, который возникает, прямо или непрямо, из комбинации определенных компонентов в определенных количествах.
Соединения по настоящему изобретению могут быть применены в форме фармацевтически приемлемых солей, произведенных из неорганических или органических кислот. Фраза "фармацевтически приемлемая соль" обозначает те соли, которые являются, в пределах объема тщательного медицинского обследования, пригодными для применения путем контакта с тканями людей и низших животных без чрезмерной токсичности, раздражения, аллергического отклика и подобного, и соответствуют приемлемому отношению преимущество/риск. Фармацевтически приемлемые соли являются хорошо известными в технике. Например, S. M. Berge et al. детально описывает фармацевтически приемлемые соли в J. Pharmaceutical Sciences 1977, 66: 1 et seq. Соли могут быть получены in situ во время конечного выделения и очистки соединений по изобретению или отдельно путем реакции свободной функции основания с пригодной органической кислотой. Репрезентативные соли присоединения кислоты включают в себя, без ограничения, ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, глицерофосфат, полусульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат (изотионат), лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмитоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, фосфат, глутамат, бикарбонат, пара-толуолсульфонат и ундеканоат. Кроме того, основные азотсодержащие группы могут быть кватернизованы такими реагентами, как низшие алкилгалогениды, такие как метил, этил, пропил и бутил хлориды, бромиды и йодиды; диалкил сульфаты, подобные диметил, диэтил, дибутил и диамил сульфатам; высшими алкилгалогенидами, такими как децил, лаурил, миристил и стеарил хлориды, бромиды и йодиды; арилалкил галогенидами, такими как бензил и фенэтилбромиды, и другими. Таким образом приготовляются водо- или маслорастворимые или диспергируемые продукты. Примеры кислот, которые могут быть использованы для получения фармацевтически приемлемых солей присоединения кислоты, включают в себя такие неорганические кислоты, как соляная кислота, бромоводородная кислота, серная кислота и фосфорная кислота, и такие органические кислоты, как щавелевая кислота, малеиновая кислота, янтарная кислота и лимонная кислота.
Соли присоединения основания могут быть получены in situ во время конечного выделения и очистки соединений по этому изобретению путем реакции фрагмента, содержащего карбоновую кислоту с пригодным основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла или аммиака или органического первичного, вторичного или третичного амина. Фармацевтически приемлемые соли включают в себя, без ограничения, катионы, основанные на щелочных металлах или щелочноземельных металлах, таких как соли лития, натрия, калия, кальция, магния и алюминия и подобные, и нетоксичные четвертичные аммониевые и аминные катионы, включая, помимо прочих, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламмоний, диметиламмоний, триметиламмоний, триэтиламмоний, диэтиламмоний и этиламмоний. Другие репрезентативные органические амины, которые могут быть применены для получения солей присоединения основания, включают в себя этилендиамин, этаноламин, диэтаноламин, пиперидин, пиперазин и подобные.
Формы дозировки для местного введения соединения по этому изобретению включают в себя порошки, спреи, мази и лекарственные формы для ингаляции. Активное соединение смешивается в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами, буферами или газами-вытеснителями, которые могут являться необходимыми. Глазные рецептуры, глазные мази, порошки и растворы также включаются в пределы объема этого изобретения.
Действительные уровни дозировки активных компонентов в фармацевтических композициях по этому изобретению могут варьироваться для получения количества активного соединения(ий), которое является эффективным для достижения желаемого терапевтического отклика у отдельного пациента, композиции и режима введения. Выбранный уровень дозировки будет зависеть от активности конкретного соединения, пути введения, тяжести состояния, которое излечивается, состояния и предшествующей медицинской истории пациента, который находится на лечении. Однако в пределах квалификации специалиста в данной области, начинать с доз соединения при уровнях меньших, чем требуется для достижения желаемого терапевтического эффекта, и постепенно увеличивать дозировку до тех пор, пока не достигнут желаемый эффект.
При применении при вышеупомянутых или других лечениях терапевтически эффективное количество одного из соединений по настоящему изобретению может быть применено в чистом виде или, если такие формы существуют, в виде фармацевтически приемлемой соли, эфира или форме пролекарства. Альтернативно соединение может быть введено в виде фармацевтической композиции, содержащей интересующее соединение в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями. Фраза "терапевтически эффективное количество" соединения по изобретению обозначает количество соединения, достаточное для лечения расстройств, при приемлемом отношении преимущество/риск, применимом к любому медикаментозному лечению. Будет понятно, однако, что общая дневная частота использования соединений и композиций по настоящему изобретению будет решаться лечащим врачом в пределах объема тщательного медицинского обследования. Специфический терапевтически эффективный уровень дозы для любого отдельного пациента будет зависеть от разнообразных факторов, включая излечиваемое расстройство и тяжесть расстройства; активности специфического применяемого соединения; специфической применяемой композиции; возраста, массы тела, общего здоровья, пола и диеты пациента; времени введения, пути введения и скорости выведения специфического применяемого соединения; продолжительности лечения; применяемых лекарств в комбинации или случайно одновременных со специфическим применяемым соединением и подобных факторов, хорошо известных в медицинской технике. Например, в пределах квалификации специалиста в данной области, начинать от доз соединения при уровнях меньших, чем требуется для достижения желаемого терапевтического эффекта, и постепенно увеличивать дозировку до тех пор, пока не достигнут желаемый эффект.
Общая дневная доза соединений по этому изобретению, вводимых человеку или низшему животному, может находиться в диапазоне от около 0,0001 до около 1000 мг/кг/день. Для целей орального введения более предпочтительные дозы могут находиться в диапазоне от около 0,001 до около 5 мг/кг/день. Если желательно, эффективная дневная доза может быть разделена на множество доз для целей введения; следовательно, композиции одиночной дозы могут содержать такие количества или их части для составления дневной дозы.
Настоящее изобретение также предоставляет фармацевтические композиции, которые включают в себя соединения по настоящему изобретению, рецептурированные совместно с одним или несколькими нетоксичными фармацевтически приемлемыми носителями. Фармацевтические композиции могут быть специально рецептурированы для орального введения в твердой или жидкой форме для парентеральной инъекции или для ректального введения.
Фармацевтические композиции по этому изобретению могут быть введены людям и другим млекопитающим перорально, ректально, парентерально, внутрь полости, интравагинально, интраперитонеально, местно (в виде порошков, мазей или капель), трансбуккально или в виде орального или назального спрея. Термин "парентерально", применяемый здесь, относится к способу введения, который включает в себя внутривенные, внутримышечные, внутрибрюшинные, в подложечную ямку, подкожные и внутрисуставные инъекции и инфузии.
В другом аспекте настоящее изобретение предоставляет фармацевтическую композицию, включающую в себя компонент по настоящему изобретению и физиологически приемлемый разбавитель. Настоящее изобретение включает в себя один или несколько соединений, как описано выше, рецептурированных в композициях совместно с одним или несколькими нетоксичными физиологически приемлемыми или пригодными разжижителями, носителями, адъювантами или разбавителями, которые обобщенно обозначаются здесь как разбавители, для, среди прочего, парентеральной инъекции, для внутриносового назначения, для орального введения в твердой или жидкой форме, для ректального или местного введения.
Композиции могут также высвобождаться через канал для локальной доставки в целевой ткани, путем внутрикоронарного стента (трубчатое устройство, составленное из тонкой проволочной сетки), или путем биоразлагаемого полимера. Соединения могут также образовывать комплекс с лигандами, такими как антитела, для направленной доставки.
Композиции, пригодные для парентеральной инъекции, могут включать в себя физиологически приемлемые, стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для восстановления влагосодержания в стерильных инъецируемых растворах или дисперсиях. Примеры пригодных водных и неводных носителей, разжижителей, растворителей или разбавителей включают в себя воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и подобные), растительные масла (такие как оливковое масло), инъецируемые органические эфиры, такие как этил олеат, и их пригодные смеси.
Эти композиции могут также содержать адъюванты, такие как консервирующие, смачивающие, эмульгирующие и диспергирующие агенты. Предотвращение действия микроорганизмов может быть обеспечено различными антибактериальными и противогрибковыми агентами, например парабенами, хлорбутаном, фенолом, сорбиновой кислотой и подобными. Также может быть желательно включение изотонических агентов, например сахаров, хлорида натрия и подобных. Продолжительная абсорбция инъецируемой фармацевтической формы может быть осуществлена путем применения агентов, замедляющих абсорбцию, например моностеарата алюминия и желатина.
Суспензии, в добавление к активным соединениям, могут содержать суспендирующие агенты, как например этоксилированные изостеариловые спирты, сорбит полиоксиэтилена и эфиры сорбита, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант, или смеси этих веществ и подобное.
В некоторых случаях, для того чтобы пролонгировать эффект лекарства, желательна медленная абсорбция лекарства из подкожной или внутримышечной инъекции. Это может быть достигнуто путем применения жидкой суспензии кристаллического или аморфного материала с плохой растворимостью в воде. Затем скорость абсорбции лекарство зависит от его скорости растворения, которая, в свою очередь, может зависеть от размера кристалла и кристаллической формы. Альтернативно продолжительная абсорбция парентерально введенной лекарственной формы достигается растворением или суспендированием лекарства в масляном носителе.
Инъецируемые формы типа депо приготовляются путем образования микрокапсульных матриц лекарства в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения лекарства к полимеру и природы конкретного применяемого полимера скорость высвобождения лекарства может контролироваться. Примеры других биоразлагаемых полимеров включают в себя поли(ортоэфиры) и поли(ангидриды). Инъецируемые рецептуры типа депо также приготовляются путем захвата лекарства в липосомы или микроэмульсии, которые являются совместимыми с тканями организма.
Инъецируемые рецептуры могут быть стерилизованы, например, путем фильтрования через задерживающий бактерии фильтр или путем включения стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другой стерильной инъецируемой среде непосредственно перед применением.
Твердые формы дозировки для перорального введения включают в себя капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых формах дозировки активное соединение может быть смешано с по меньшей мере одним инертным, фармацевтически приемлемым наполнителем или носителем, таким как цитрат натрия или фосфат дикальция, и/или a) наполнителями или сухими разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; b) связующими, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик; c) гигроскопичными веществами, такими как глицерин; d) дезинтенгрирующими агентами, такими как агар-агар, карбонат кальция, помидорный или тапиоковый крахмал, альгиновая кислота, определенные силикаты и карбонат натрия; e) замедляющими растворение агентами, такими как парафин; f) ускорителями абсорбции, такими как четвертичные аммониевые соединения; г) смачивающими агентами, такими как цетиловый спирт и моностеарат глицерина; h) абсорбентами, такими как каолиновая и бентонитовая глина, и i) смазками, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль форма дозировки также может включать в себя буферирующие агенты.
Твердые композиции подобного типа также могут применяться как наполнители в мягких и жестких желатиновых заполненных капсулах, с применением таких наполнителей, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и подобные.
Твердые формы дозировки таблеток, драже, капсул, пилюль и гранул могут быть приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в технике фармацевтического рецептурирования. Они могут необязательно содержать рентгеноконтрастные препараты и также могут представлять собой композицию, так что они высвобождают только активный компонент(ы) или предпочтительно в определенной части кишечного тракта, необязательно, с отсроченным высвобождением. Примеры имплантируемых композиций, которые могут быть применены, включают в себя полимерные вещества и воски.
Активные соединения могут также находиться в микроинкапсулированной форме, если необходимо, с одним или несколькими из упомянутых выше наполнителей.
Жидкие формы дозировки для орального введения включают в себя фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В добавление к активным соединениям жидкие формы дозировки могут содержать инертные разбавители, обычно применяемые в технике, такие как, например, вода или другие растворители, агенты, способствующие растворению, и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и жирнокислотные эфиры сорбита и их смеси.
Кроме инертных разбавителей оральные композиции могут также включать в себя вспомогательные средства, такие как смачивающие агенты, эмульсифицирующие и суспендирующие агенты, подсластители, вкусовые добавки и отдушки.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, который могут быть получены путем смешения соединений по этому изобретению с пригодными нераздражающими наполнителями или носителями, такими как кокосовое масло, полиэтиленгликоль или суппозиторийный воск, которые являются твердыми при комнатной температуре, но жидкими при температуре тела и, таким образом, расплавляются в прямой кишке или вагинальной полости и высвобождают активное соединение.
Соединения по настоящему изобретению также могут быть введены в форме липосом. Как известно в технике, липосомы обычно производятся из фосфолипидов или других липидных веществ. Липосомы образуются моно- или многослойными гидратированными жидкими кристаллами, которые диспергированы в водной среде. Может быть использован любой нетоксичный, физиологически приемлемый и метаболизируемый липид, способный к образованию липосом. Настоящие композиции в липосомной форме могут содержать, в добавление к соединению по настоящему изобретению, стабилизаторы, консерванты, наполнители и подобное. Предпочтительные липиды представляют собой природные и синтетические фосфолипиды и фосфатидилированные холины (лецитины), применяемые отдельно или совместно.
Способы получения липосом известны в технике. Смотри, например, Prescott, Ed., Methods in Cell Biology, v. XIV, Academic Press, New York, N.Y. (1976), p. 33 et seq.
Применяемый здесь термин "фармацевтически приемлемые пролекарства" представляет те пролекарства соединений по настоящему изобретению, которые являются, в пределах объема тщательного медицинского обследования, подходящими для применения при контакте с тканями людей и низших животных без избыточной токсичности, раздражения, аллергического отклика и подобного, соотносятся с приемлемым отношением преимущество/риск и эффективны в отношении их предполагаемого применения соединений по изобретению, также как цвиттер-ионнные формы, где возможно. Пролекарства по настоящему изобретению могут быть быстро трансформированы in vivo в исходное соединение вышеупомянутой формулы, например, путем гидролиза в крови. Исчерпывающее обсуждение проведено в T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Svstems, v. 14 of the A.C.S. Symposium Series, и в Edward B. Roche, ed., Bioreversible Carriers in Drug Design. American Pharmaceutical Association and Pergamon Press (1987), которые включены здесь посредством ссылки.
Соединения по настоящему изобретению могут быть получены с помощью методик, представленных на схемах 1-7. Во всех примерах были использованы представленные после схем общие условия анализа.
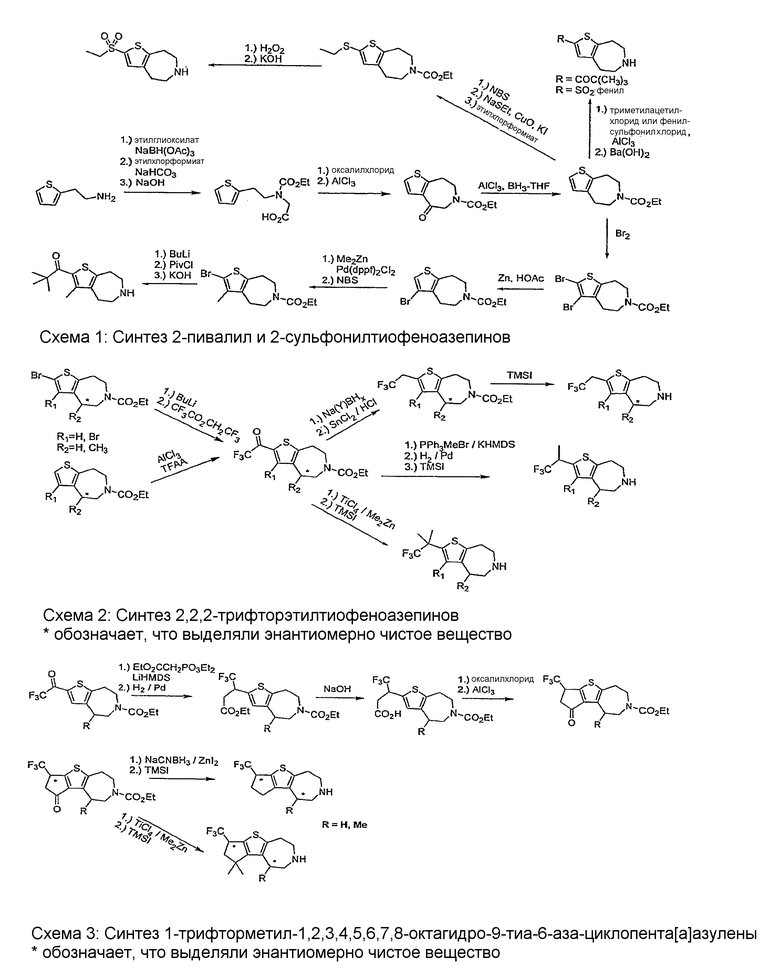
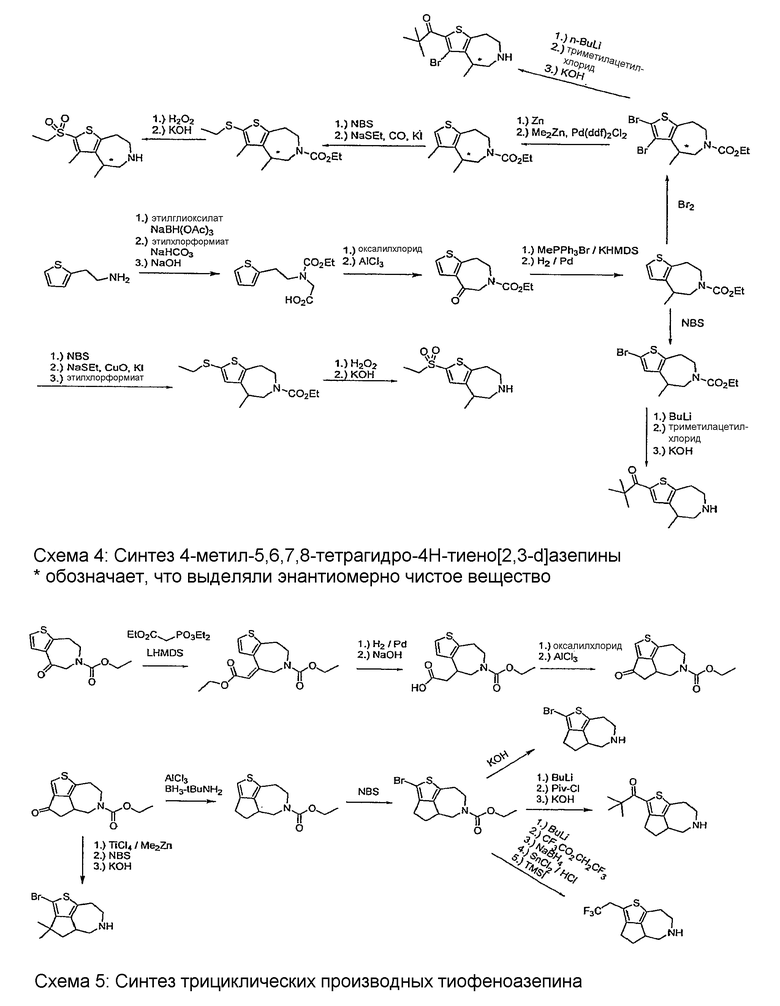
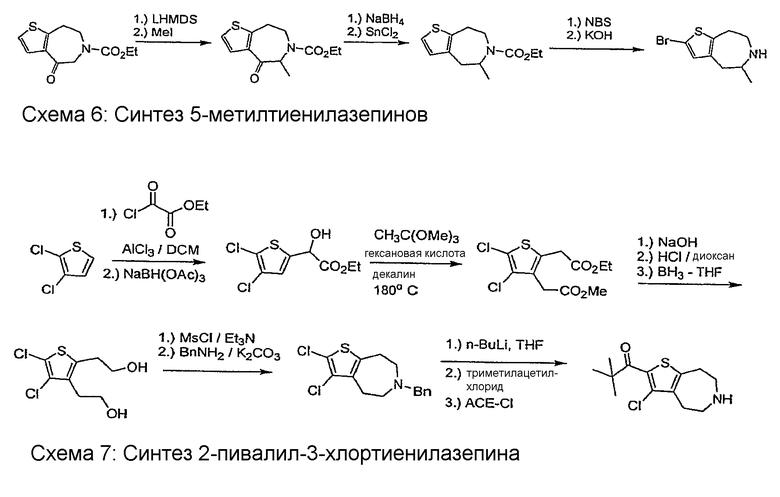
Общие условия анализа
В примерах использовали следующие общие аналитические условия: анализ и очистку методом ВЭЖХ проводили с применением двойного градиентного насоса Waters 2525, пробоотборника Waters 2767, Уф-детектора Waters 2487 (220 и 254 нМ) и электроспрей-масс-спектроскопического детектора Waters Micromass ZQ. Micromass ZQ был настроен как на положительную, так и отрицательную ионизацию (вольтаж конуса =25 и 50, соответственно).
Аналитический ВЭЖХ-анализ проводили, как описано ниже:
50×4,6 мм 3,5 мкм колонка Waters XTerra MS C18;
подвижная фаза: 10 мМ буфер с ацетатом аммония при pH 5,75 и ацетонитрил;
ацетонитрил: 10-75% за 3,5 минут, 75-99% за 3,9 минут, 99% в течение 4,2 минут, от 99% до 10% за 4,5 минут, повторное уравновешивание.
Препаративную ВЭЖХ проводили, как описано ниже:
50×19 мм 5 мкм колонка Waters XTerra Prep MS C18;
подвижная фаза: l0 мМ буфер с ацетатом аммония при pH 5,75 и ацетонитрил;
ацетонитрил: 10-99% за 8 минут, 99% в течение 9 минут, от 99% до 10% за 9,5 минут, повторное уравновешивание.
ЯМР-анализ проводили на приборе Bruker BioSpin UltraShield NMR(300 МГц).
Следующие примеры являются иллюстрациями для получения репрезентативных соединений по настоящему изобретению.
Пример 1. 2,2-Диметил-1-(3-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-пропан-1-он (Схема 1)
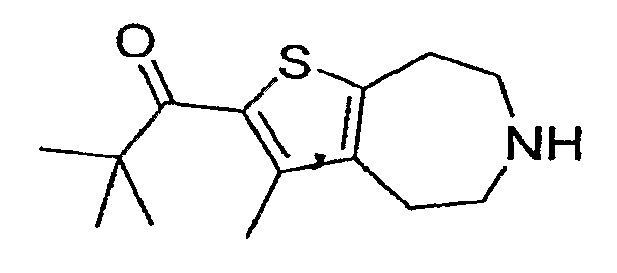
a) Этиловый эфир [этоксикарбонил-(2-тиофен-2-ил-этил)-амино]-уксусной кислоты:
2-Тиофен-2-ил-этиламин (21,0 г, 165 ммоль) перемешивали в 1 литре DCM. Добавляли этил глиоксилат (165 ммоль, 50% в толуоле), затем добавяли 50 мкл HOAc. Реакционную смесь перемешивали в течение 10 минут, после этого времени медленно добавляли NaBH(OAc)3 (214 ммоль, 45 г). Через 15 минут добавляли HOAc (214 ммоль) и реакционную смесь перемешивали в течение 20 минут. Реакционную смесь концентрировали и неочищенный материал повторно растворяли в 500 мл ТГФ и 500 мл воды. Добавляли NaHCO3 (42 г, 500 ммоль), затем добавляли этилхлорформиат (21 мл, 214 ммоль). К реакционной смеси медленно добавляли насыщенный NaHCO3 до момента, когда выделение газа становилось минимальным. После перемешивания в течение ночи реакционную смесь разбавляли EtOAc (400 мл). Продукт экстрагировали 2×EtOAc, высушивали над MgSO4 и концентрировали до продукта, обозначенного в подзаголовке примера, в виде темного масла.
b) [Этоксикарбонил-(2-тиофен-2-ил-этил)-амино]-уксусная кислота:
Неочищенный материал со стадии (a) (165 ммоль, ~47 г) растворяли в EtOH (700 мл) и обрабатывали 600 мл 1М NaOH. После перемешивания в течение ночи реакционную смесь подкисляли концентрированным HCl до pH~1. Неочищенную реакционную смесь разбавляли EtOAc (400 мл) и промывали водой. Воду обратно экстрагировали EtOAc. Объединенные органические экстракты промывали водой (2×) и высушивали над MgSO4. Концентрация и упаривание из толуола (2×) приводила к продукту, обозначенному в подзаголовке примера, в виде твердого вещества.
c) Этиловый эфир 4-оксо-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (b) (-165 ммоль, ~42 г) растворяли в 1 л DCM. Добавляли ДМФА (100 мкл), затем медленно добавляли оксалилхлорид (21,7 мл, 247 ммоль). Через 1 час реакционную смесь концентрировали досуха и неочищенный материал повторно растворяли в DCE (1 л). Аккуратно добавляли AlCl3 (55 г, 410 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ½ часа. Неочищенную реакционную смесь тушили льдом, промывали водой (3×) и высушивали над MgSO4. Продукт, обозначенный в заголовке примера, очищали методом хроматографии на силикагеле (30% EtOAc в гексане), что дало 10,5 граммов соединения, обозначенного в подзаголовке примера, в виде белого твердого вещества. MS: ESI (положительный): 240 (M+H).
d) Этиловый эфир 4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
К 50 мл DCM добавляли AlCl3 (3,95 г, 29,7 ммоль) при 0°C. Добавляли комплекс боран-трет-бутиламин (5,2 г, 59,5 ммоль), затем добавляли продукт со стадии (c) (2,37 г, 9,9 ммоль), растворенный в DCM (50 мл). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, после этого времени добавляли еще 3,95 г (29,7 ммоль) AlCl3. После перемешивания в течение 10 минут реакционную смесь аккуратно тушили 0,1М HCl (~50 мл). После концентрирования органического растворителя неочищенную реакционную смесь распределяли между 1М HCl и EtOAc (70 мл каждого). Водный слой экстрагировали обратно 1×EtOAc. Объединенные органические слои сушили над MgSO4 и концентрировали. Продукт, обозначенный в подзаголовке примера, (1,45 г) получали после очистки методом хроматографии на силикагеле (0-35% EtOAc в гексане). MS: ESI (положительный): 226 (M+H).
e) этиловый эфир 2,3-дибром-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (d) (1,45 г, 6,44 ммоль) и NaHCO3 (3,2 г, 38,6 ммоль) перемешивали в 60 мл циклогексана. Медленно добавляли бром (1,0 мл, 19,3 ммоль) и реакционную смесь перемешивали в темноте в течение 15 минут. Реакцию тушили 5% Na2SO3 и интенсивно перемешивали в течение 15 минут. Соединение, обозначенное в подзаголовке примера, экстрагировали EtOAc (2×). Высушивание над MgSO4 и концентрирование приводило к соединению, обозначенному в подзаголовке примера, (2,6 г) в виде желтого масла.
f) Этиловый эфир 3-бром-4,5,7,8-тетрагидро-тиено|2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (e) (1,3 г, 3,39 ммоль) растворяли в смеси 1:1 HOAc:вода (40 мл), обрабатывали пылью Zn (0,44 г, 6,79 ммоль) и нагревали до кипения с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали, разбавляли водой и экстрагировали 2×EtOAc. Органические экстракты сушили над MgSO4 и концентрировали, что дало 0,76 г соединения, обозначенного в подзаголовке примера.
g) Этиловый эфир 3-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (f) (375 мг, 1,23 ммоль) растворяли в 4 мл диоксана и обрабатывали Me2Zn (1,25 мл 2М в толуоле) и Pd(dppf)2Cl2. После нагревания при 100°C в течение 3 часов реакционную смесь охлаждали, тушили водой, фильтровали через силикагель (промывка EtOAc) и концентрировали, что дало 337 мг соединения, обозначенного в подзаголовке примера, в виде масла.
h) Этиловый эфир 2-бром-3-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (g) (337 мг, 1,4 ммоль) растворяли в 10 мл смеси 1:1 CHCl3:HOAc и обрабатывали NBS (301 мг, 1,7 ммоль). После перемешивания в течение ½ часа реакционную смесь разбавляли DCM и промывали водой (50 мл) и 1М NaOH (2×50 мл). Неочищенный продукт очищали методом хроматографии на силикагеле, что дало 235 мг соединения, обозначенного в подзаголовке примера.
i) 2,2-Диметил-1-(3-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-пропан-1-он:
Продукт со стадии (h) (60 мг, 0,19 ммоль) растворяли в 2 мл ТГФ и охлаждали до -78°C. Добавляли бутиллитий (0,15 мл 1,6М) и реакционную смесь перемешивали в течение 5 минут. Добавляли триметилацетилхлорид (36 мкл, 0,3 ммоль) и реакцию отогревали до комнатной температуры. Реакцию останавливали водой (5 мл) и продукт экстрагировали в DCM (2×). Экстракты концентрировали досуха и обрабатывали 4 мл 1:1 EtOH:40% KOH (водного) и нагревали до 100°C в течение 14 часов. Реакционную смесь охлаждали и разбавляли водой. Продукт экстрагировали в DCM (2×5 мл). Экстракты концентрировали, и соединение, обозначенное в заголовке примера, очищали методом препаративной ВЭЖХ-МС. 1H ЯМР (CD3OD) δ 3,38-3,31 (м, 4H), 3,23 (т, J=5,1 Гц, 2H), 3,04 (т, J=5,2 Гц, 2H), 2,30 (с, 3H), 1,32 (с, 9H); MS: ESI (положительный): 252 (M+H).
Пример 2: 3-Бром-2-(2,2,2-трифтор-этил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин (Схема 2)
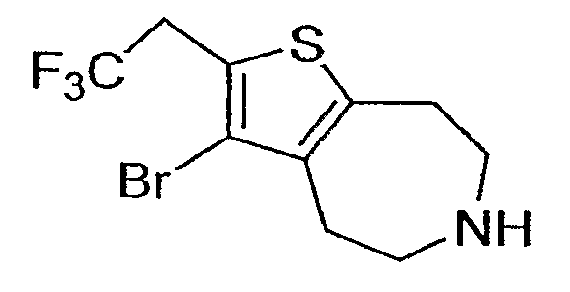
a) Этиловый эфир 3-бром-2-(2,2,2-трифтор-ацетил)-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт примера 1, стадия (e), (200 мг, 0,52 ммоль) растворяли в 5 мл ТГФ, охлаждали до -78°C и обрабатывали BuLi (0,33 мл 1,6М). После перемешивания в течение 15 минут при -78°C добавляли трифторацетил 2,2,2-трифторэтанол (132 мкл, 0,68 ммоль) и реакции позволили отогреться до комнатной температуры. Реакцию тушили водой (50 мл) и концентрировали. Остаток очищали методом хроматографии на силикагеле, что дало 73 мг соединения, обозначенного в подзаголовке примера.
b) Этиловый эфир 3-бром-2-(2,2,2-трифтор-этил-4,5,7,8-тетрагидро-тиено[2,3-диазепин-6-карбоновой кислоты:
Продукт со стадии (a) (73 мг, 0,18 ммоль) перемешивали в 3 мл EtOH и обрабатывали NaBH4 (20 мг, 0,5 ммоль). После перемешивания в течение 20 минут реакцию тушили HOAc до прекращения выделения пузырьков. Реакционную смесь разбавляли водой (5 мл) и неочищенный продукт экстрагировали в DCM (3×5 мл). Объединенные органические экстракты концентрировали, остаток растворяли в HOAc (2 мл) и концентрировали HCl (1 мл). Добавляли SnCl2 (225 мг, 1 ммоль) и реакционную смесь нагревали до 80°C в течение 2 часов. Неочищенную реакционную смесь разбавляли водой (10 мл) и продукт экстрагировали в DCM (3×5 мл). Объединенные органические экстракты концентрировали досуха и применяли без дополнительной очистки.
c) 3-Бром-2-(2,2,2-трифтор-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин:
Продукт со стадии (b) (0,18 ммоль) растворяли в CHCl3 (3 мл) и обрабатывали TMSI (1 ммоль, 200 мкл). После нагревания в течение ½ часа при 70°C добавляли еще 200 мкл TMSI и нагревание продолжали в течение ½ часа. Реакционную смесь охлаждали и тщательно тушили смесью 0,5 мл EtOH и 0,5 мл воды. Реакционную смесь разбавляли 1М NaOH (3 мл) и продукт экстрагировали в DCM (2×5 мл). Органические экстракты концентрировали и остаток очищали методом препаративной ВЭЖХ-МС. 1H-ЯМР (CD3OD) δ 3,75 (кв., J=10,5 Гц, 2H), 3,43-3,34 (м, 4H), 3,26 (т, J=5,4 Гц, 2H), 3,16 (т, J=5,4 Гц, 2H), 3,16 (т, J=5,4 Гц, 2H); MS: ESI (положительный): 316, 314 (M+H).
Пример 3: 2-Бензолсульфонил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин (Схема 1)
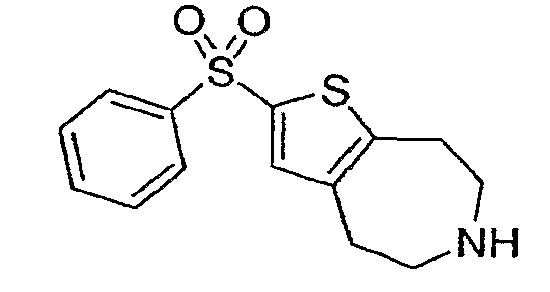
Продукт примера 1, стадия (d) (75 мг, 0,33 ммоль) растворяли в 2 мл DCE и обрабатывали фенилсульфонилхлоридом (83 мкл, 0,66 ммоль), затем добавляли AlCl3 (88 мг, 0,66 ммоль). После нагревания до 80°C в течение ½ часа реакционную смесь охлаждали и аккуратно тушили 1М NaOH. Продукт экстрагировали в DCM (2×). Органические экстракты концентрировали и остаток растворяли в смеси 3 мл EtOH и 3 мл 40% водный KOH. Реакционную смесь нагревали до 100°C в течение 14 часов, охлаждали и разбавляли водой. Соединение, обозначенное в заголовке примера, экстрагировали в DCM (2×5 мл) и очищали методом препаративной ВЭЖХ-МС. 1H-ЯМР (CD3OD) δ 7,96 (д, J=8,1 Гц, 2H), 7,67-7,56 (м, 3H), 5,56 (с, 1H), 3,38-3,32 (м, 4H), 3,27-3,23 (м, 2H), 3,09 (т, J=5,4 Гц, 2H); MS: ESI (положительный): 294 (M+H).
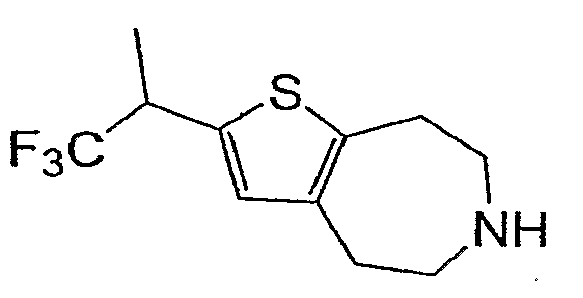
Пример 4: (R,S)-2-(2,2,2-Трифтор-1-метил-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин (Схема 2)
a) Этиловый эфир 2-(2,2,2-трифтор-ацетил)-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт примера 1, стадия (d), (410 мг, 1,82 ммоль) растворяли в 20 мл DCE и обрабатывали трифторуксусным ангидридом (510 мкл, 3,64 ммоль) и AlCl3 (484 мг, 3,64 ммоль). Реакционную смесь нагревали при 50°C в течение 2 часов, затем охлаждали и тушили избытком воды. Продукт экстрагировали в DCM (2×15 мл), высушивали над MgSO4 и концентрировали, что дало 360 мг соединения, обозначенного в подзаголовке примера, в виде полутвердого вещества.
b) Этиловый эфир 2-(1-трифторметил-винил)-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Бромид трифенилфосфония (393 мг, 1,1 ммоль) перемешивали в 7 мл ТГФ. Добавляли KHMDS (199 мг, 1,0 ммоль) и желтый раствор перемешивали в течение 30 минут при комнатной температуре. Продукт стадии (a) (180 мг, 0,56 ммоль) растворяли в 7 мл ТГФ и добавляли к вышеупомянутой реакции. Раствор оставляли перемешиваться в течение 1 часа при комнатной температуре, затем разбавляли EtOAc (25 мл) и промывали водой (2×20 мл). Неочищенный продукт очищали хроматографией на силикагеле (30% EtOAc в гексане), что дало 100 мг соединения, обозначенного в подзаголовке примера.
c) Этиловый эфир (R,S)-2-(2,2,2-трифтор-1-метил-этил)-4,5,7,8-тетрагидро-тиено[2,3-b]азепин-6-карбоновой кислоты:
Продукт со стадии (b) (40 мг, 0,12 ммоль) растворяли в 5 мл EtOH и обрабатывали 10 мг 10% Pd/C (влажный, класс E101 от Degussa). Реакционную смесь интенсивно перемешивали в атмосфере водорода в течение 14 часов. Фильтрование через целит и концентрирование дало 37 мг соединения, обозначенного в подзаголовке примера.
d) (R,S)-2-(2,2,2-Трифтор-1-метил-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин:
Продукт со стадии (c) (37 мг, 0,11 ммоль} растворяли в 1 мл CHCl3 и обрабатывали TMSI (47 мкл, 0,35 ммоль). После нагревания при 60°C в течение 2 часов реакционную смесь охлаждали и концентрировали. Очистка неочищенного остатка методом препаративной ВЭЖХ-МС приводила к соединению, обозначенному в заголовке примера. 1H-ЯМР (CD3OD) δ 6,86 (с, 1H), 3,80 (септ., J=7,9 Гц, 1H), 3,39-3,30 (м, 4H), 3,17 (т, J=5,2 Гц, 2H), 3,05 (т, J=5,2 Гц, 2H), 1,48 (д, J=7,2 Гц, 3H); MS: ESI (положительный): 250 (M+H).
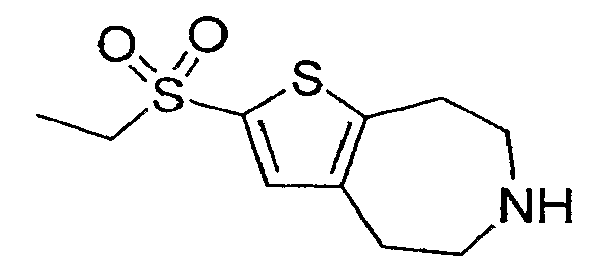
Пример 5: 2-Этансульфонил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин (Схема 1)
a) Этиловый эфир 2-бром-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт примера 1, стадия (d), (80 мг, 0,35 ммоль) растворяли в 2 мл смеси 1:1 CHCl3:HOAc и обрабатывали NBS (62 мг, 035 ммоль). Через 15 минут реакционную смесь концентрировали досуха, растворяли в минимальном количестве EtOAc и фильтровали через слой силикагеля. Фильтрат упаривали, что дало 105 мг соединения, обозначенного в подзаголовке примера.
b) Этиловый эфир 2-этилсульфанил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (a) (105 мг, 0,35 ммоль) растворяли в 1 мл NMP и обрабатывали NaSEt (59 мг, 0,7 ммоль), KI (5 мг, 0,03 ммоль) и CuO (14 мг, 0,18 ммоль). Реакционную смесь нагревали при 120°C в течение 24 часов. Добавляли дополнительный NaSEt (59 мг, 0,7 ммоль), KI (5 мг, 0,03 ммоль), CuO (14 мг, 0,18 ммоль) и нагревание продолжали в течение 24 часов. Реакционную смесь разбавляли водой и DCM (по ~5 мл каждого) и фильтровали для удаления темного порошкообразного осадка. Неочищенный продукт экстрагировали в DCM (3×5 мл) и концентрировали до ~1 мл. Неочищенный остаток разбавляли DCM (2 мл), обрабатывали Et3N (140 мкл, 1,05 ммоль) и этил хлорформиатом (50 мкл, 0,52 ммоль). После перемешивания в течение ночи реакционную смесь разбавляли EtOAc (5 мл) и промывали водой (5×) с целью удаления остаточного NMP. Органический раствор концентрировали, что дало 80 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
c) Этиловый эфир 2-этансульфонил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (b) (80 мг, 0,28 ммоль) растворяли в 3 мл HOAc и обрабатывали H2O2 (300 мкл 30%-ной, ~3 ммоль). После перемешивания при комнатной температуре в течение 3 дней реакционную смесь разбавляли EtOAc и промывали 4× водой. Органический экстракт сушили над MgSO4 и концентрировали, что дало 62 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
d) 2-Этансульфонил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин:
Продукт со стадии (c) (21 мг, 0,066 ммоль) растворяли в 2 мл EtOH и обрабатывали 2 мл 40% KOH, затем нагревали до 100°C в запаянной емкости в течение ночи. Реакционную смесь охлаждали и разбавляли водой. Соединение, обозначенное в заголовке примера, экстрагировали в DCM (3×) и очищали методом препаративной ВЭЖХ-МС. 1H-ЯМР (CD3OD) δ 7,47 (с, 1H), 3,25 (кв, J=7,2 Гц, 2H), 3,24-3,01 (м, 8H), 1,28 (т, J=7,2 Гц, 3H); MS: ESI (положительный): 246 (M+H).
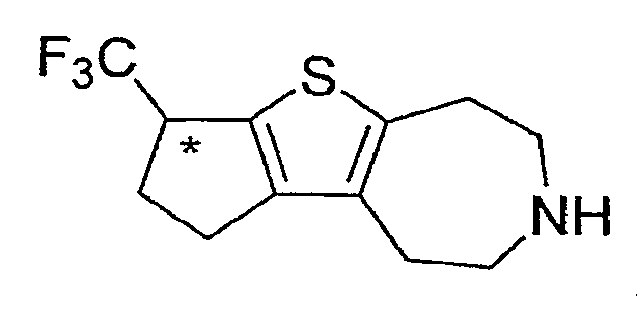
Пример 6: (R,S)-1-Трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен (Схема 3)
a) (R,S)-4,4,4-Трифтор-3-(5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-масляная кислота:
Триэтилфосфоноацетат (56 мг, 0,25 ммоль) и продукт примера 4, стадия (a), (40 мг, 0,12 ммоль) перемешивали в 2 мл ТГФ. Реакцию обрабатывали LiHMDS (0,2 мл 1М) и перемешивали при комнатной температуре в течение 1 часа. Реакцию тушили 5 мл воды, продукт экстрагировали в DCM (2×5 мл) и высушивали над MgSO4. Органический экстракт концентрировали досуха, остаток растворяли в 5 мл EtOH и обрабатывали ~10 мг 10% Pd/C (влажный, от Degussa, класс E101). После перемешивания под атмосферой водорода в течение 3 дней реакционную смесь фильтровали через целит и обрабатывали 1М NaOH (1 мл). После перемешивания в течение 2 часов при 60°C реакционную смесь разбавляли водой и продукт экстрагировали в DCM (3×5 мл), что дало 45 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
b) Этиловый эфир (R,S)-3-оксо-1-трифторметил-2,3,4,5,7,8-гексагидро-1H-9-тиа-6-аза-циклопента[a]азулен-6-карбоновая кислота:
Продукт со стадии (a) (45 мг, 0,12 ммоль) растворяли в DCE (2 мл) и обрабатывали оксалилхлоридом (43 мкл, 0,49 ммоль) и 1 каплей ДМФА. После перемешивания в течение 5 минут при комнатной температуре реакционную смесь концентрировали досуха и растворяли в 2 мл DCM. Добавляли AlCl3 (66 мг, 0,50 ммоль) и реакционную смесь перемешивали в течение 5 минут. Реакцию тушили водой и неочищенный продукт экстрагировали в DCM (2×), что дало 32 мг темного масла, которое применяли без дополнительной очистки. Энантиомеры соединения, обозначенного в подзаголовке примера, могут быть разделены с применением колонки Chiralpak® AD-RH®, 20×250 мм, от Chiral Technologies (подвижная фаза MeOH), что дало энантиомер 1 (время удержания = 9,8 минут) и энантиомер 2 (время удержания = 11,5 минут).
c) (R,S)-1-Трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен:
Продукт со стадии (b) (рацемический, 32 мг, 0,10 ммоль) растворяли в DCE (1 мл) и обрабатывали ZnI2 (64 мг, 0,2 ммоль) и NaCNBH3 (44 мг, 0,7 ммоль). После перемешивания в течение ночи образовывалась густая суспензия. Реакционную смесь фильтровали и твердое вещество промывали DCM. Объединенные фильтраты промывали водой (1×5 мл) и концентрировали досуха. Остаток растворяли в CHCl3 (2 мл) и обрабатывали TMSI (70 мкл, 0,5 ммоль). После нагревания при 60°C в течение 1 часа добавляли еще 50 мкл TMSI и нагревание продолжали в течение 14 часов. Реакционную смесь охлаждали, концентрировали досуха и очищали методом препаративной ВЭЖХ-МС, что дало соединение, обозначенное в заголовке примера. 1H-ЯМР (CD3OD) δ 4,02-3,97 (м, 1H), 3,40-3,33 (м, 4H), 3,18 (т, J=5,2 Гц, 2H), 2,97 (т, J=5,4 Гц, 2H), 2,83-2,65 (м, 3H), 2,57-2,47 (м, 1H); MS: ESI (положительный): 262 (M+H).
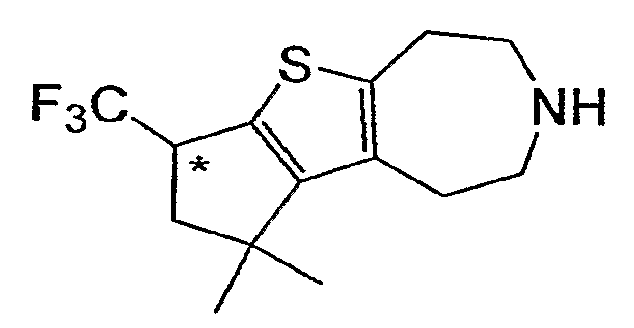
Пример 7: (R,S)-3,3-Диметил-1-трифторметил-1,2,3,4,5,6 , 7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен (Схема 3)
a) Этиловый эфир (R,S)-3,3-диметил-1-трифторметил-2,3,4,5,7,8-гексагидро-1H-9-тиа-6-аза-циклопента[a]азулен-6-карбоновой кислоты:
DCM (3 мл) охлаждали до -78°C и обрабатывали TiCl4 (190 мкл, 1,73 ммоль), затем добавляли Me2Zn (0,86 мл 2М в толуоле). После перемешивания темно-красной суспензии при -78°C в течение 15 минут добавляли продукт примера 6, стадия (b), (рацемический, 100 мг, 0,29 ммоль) в виде раствора в 3 мл DCM. Реакцию отогревали до 0°C и перемешивали в течение 3 часов. Раствор выливали над лед и продукт экстрагировали в DCM (2×10 мл). Органический экстракт сушили над MgSO4 и концентрировали, что дало 84 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
b) (R,S)-3,3-Диметил-1-трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен:
Продукт со стадии (a) (37 мг, 0,10 ммоль) растворяли в CHCl3 (2 мл) и обрабатывали TMSI (1 ммоль, 140 мкл). После нагревания до 60°C в течение 2 часов реакцию тушили MeOH и концентрировали досуха. Соединение, обозначенное в заголовке примера, получали после очистки неочищенного остатка методом препаративной ВЭЖХ-МС. 1H-ЯМР (CD3OD) δ 4,09-3,94 (м, 1H), 3,41-3,32 (м, 4H), 3,17 (т, J=5,2 Гц, 2H), 3,08 (т, J=5,2 Гц, 2H), 2,53 (дд, J=9, 13,5 Гц, 1H), 2,31 (дд, J=6,6, 13,8 Гц, 1H), 1,39 (с, 3H), 1,31 (с, 3H); MS: ESI (положительный): 290 (M+H).
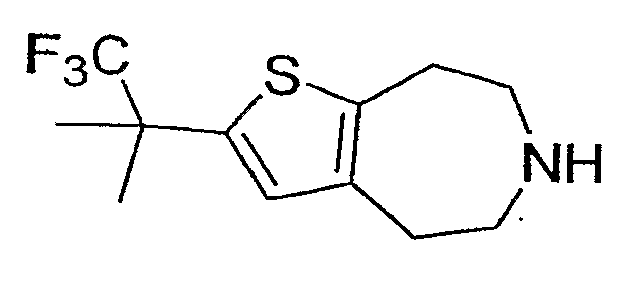
Пример 8: 2-(2,2,2-Трифтор-1,1-диметил-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин (Схема 2)
a) Этиловый эфир 2-(2,2,2-трифтор-1,1-диметил-этил)-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
DCM (2 мл) охлаждали до -78°C и обрабатывали TiCl4 (82 мкл, 0,75 ммоль), затем добавляли Me2Zn (370 мкл до 2М). После перемешивания в течение 15 минут при -78°C, продукт примера 4, стадия (a), (40 мг, 0,125 ммоль) добавляли в виде раствора в 3 мл DCM. Реакцию отогревали до 0°C в течение 1 часа, затем до комнатной температуры в течение 6 часов. Реакцию тушили выливанием на лед и экстрагировали в DCM (2×5 мл). Органические экстракты сушили над MgSO4, концентрировали, что дало 34 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
b) 2-(2,2,2-Трифтор-1,1-диметил-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин:
Продукт со стадии (a) (34 мг, 0,1 ммоль) растворяли в 2 мл CHCl3 и обрабатывали TMSI (140 мкл, 1 ммоль). После нагревания до 60°C в течение 2 часов реакционную смесь концентрировали досуха и соединение, обозначенное в заголовке примера, очищали методом препаративной ВЭЖХ-МС. 1H ЯМР (CD3OD) δ 6,91 (с, 1H), 3,39-3,32 (м, 4H), 3,21-3,17 (м, 2H), 3,09-3,05 (м, 2H), 1,55 (с, 6H); MS: ESI (положительный): 264 (M+H).
Пример 9: 3-Бром-4-метил-2-(2,2,2-трифтор-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин (Схема 2)
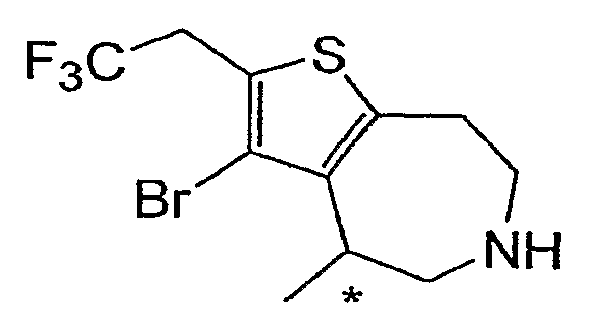
a) Этиловый эфир 4-метилен-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Бромид метилтрифенилфосфония (6,3 г, 17,6 ммоль) растворяли в 150 мл ТГФ и охлаждали до 0°C. Добавляли KHMDS (3,2 г, 16,2 ммоль) частями и реакционную смесь перемешивали в течение ½ часа. Продукт примера 1, стадия (c), (3,0 г, 12,5 ммоль) добавляли в виде раствора в 25 мл ТГФ. Реакцию отогревали до комнатной температуры и перемешивали в течение 1 часа. Смесь концентрировали и продукт, обозначенный в заголовке примера, очищали методом хроматографии на силикагеле (0-40% EtOAc в гексане), что дало 2,6 г соединения, обозначенного в подзаголовке примера.
b) Этиловый эфир (R,S)-4-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (a) растворяли в 100 мл EtOH и обрабатывали 0,5 г 10% Pd/C (влажный, от Degussa, класс E101). После интенсивного перемешивания в течение 14 часов в атмосфере водорода реакционную смесь фильтровали через целит и концентрировали, что дало 2,3 г соединения, обозначенного в подзаголовке примера, в виде прозрачного масла. MS: ESI (положительный): 240 (M+H).
c) Этиловый эфир 2,3-дибром-4-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (b) (5,6 г, 23,4 ммоль) растворяли в 250 мл циклогексана и обрабатывали NaHCO3 (11,8 г, 140 ммоль). Медленно добавляли бром (3,6 мл, 70,3 ммоль) и реакционную смесь перемешивали в течение ½ часа при комнатной температуре, после этого времени реакцию тушили Na2SO3 (180 мл 5% водного раствора). После интенсивного перемешивания в течение 15 минут добавляли EtOAc (~100 мл), органический слой удаляли и высушивали над MgSO4, что дало соединение, обозначенное в подзаголовке примера. Два энантиомера разделяли с применением колонки Chiralpak® AD-RH® 20×250 мм от Chiiral Technologies (подвижная фаза: 10 мл/мин MeOH), что дало энантиомер 1 (время удержания = 9,8 минут) и энантиомер 2 (время удержания = 11,4 минут) соединения, обозначенного в подзаголовке примера.
d) Этиловый эфир 3-бром-4-метил-2-(2,2,2-трифтор-ацетил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (c) (энантиомер 2, 60 мг, 0,15 ммоль) растворяли в 2 мл сухого ТГФ и охлаждали до -78°C. Добавляли бутиллитий (0,11 мл 1,6М) и раствор перемешивали в течение 5 минут, затем тушили трифторацетилированным 2,2,2-трифторэтанолом (50 мкл, 0,24 ммоль). После отогревания до комнатной температуры реакционную смесь фильтровали через слой силикагеля (промывка EtOAc). Фильтрат упаривали, что дало 62 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
e) Этиловый эфир 3-бром-4-метил-2-[2,2,2-трифтор-этил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (d) (62 мг, 0,15 ммоль) растворяли в 2 мл EtOH и обрабатывали NaBH4 (0,6 ммоль). Через 15 минут реакцию тушили HOAc до завершения выделения пузырьков и разбавляли водой. Продукт экстрагировали в DCM (2×5 мл), высушивали над MgSO4 и концентрировали. Неочищенный остаток растворяли в 3 мл смеси 1:1 HOAc:конц. HCl и обрабатывали SnCl2 (225 мг, 1 ммоль). Реакционную смесь нагревали до 70°C в течение 1 часа и затем перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь разбавляли EtOAc (10 мл) и промывали водой (2×) и 1М NaOH (2×). Органический раствор упаривали досуха, что приводило к 40 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
f) 3-Бром-4-метил-2-(2,2,2-трифтор-этил)-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин.
Снимали защиту с продукта со стадии (e) (40 мг, 0,10 ммоль) и очищали согласно методике, описанной для примера 2, стадия (c). 1H-ЯМР (CD3OD) δ 3,75 (кв, J=10,4 Гц, 2H), 3,63-3,55 (м, 3H), 3,42-3,31 (м, 2H), 3,24-3,15 (м, 2H), 1,35 (д, J=7,2 Гц, 3H); MS: ESI (положительный): 328, 330 (M+H).
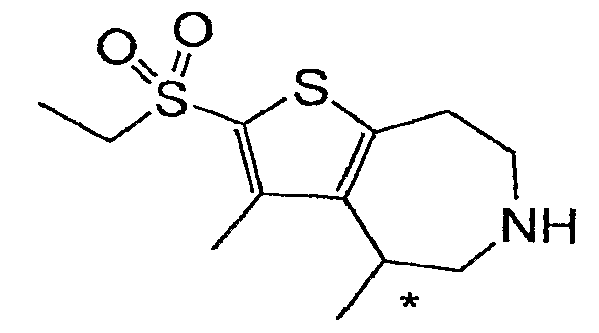
Пример 10: 2-Этансульфонил-3,4-диметил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин (Схема 4)
a) Этиловый эфир 3-бром-4-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт примера 9, стадия (c), (энантиомер 2, 0,75 г, 1,9 ммоль) и Zn (0,25 г, 3,8 ммоль) нагревали до кипения с обратным холодильником в смеси 20 мл воды и 20 мл HOAc. Через ½ часа реакционную смесь охлаждали, разбавляли EtOAc и промывали 2× водой. Органический слой сушили над MgSO4 и концентрировали, что дало 490 мг соединения, обозначенного в подзаголовке примера, в виде масла.
b) Этиловый эфир 3,4-диметил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (a) (150 мг, 0,47 ммоль) растворяли в 3 мл диоксана и обрабатывали Me2Zn (0,47 мл 2М в толуоле) и Pd(ddf)2Cl2 (11 мг, 0,014 ммоль). После нагревания до 100°C в течение 3 часов реакцию тушили водой и фильтровали. Фильтрат распределяли между EtOAc и водой (7 мл каждого). Органический слой сушили над MgSO4 и концентрировали, что дало 92 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
c) Этиловый эфир 2-бром-3,4-диметил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (b) (92 мг, 0,36 ммоль) растворяли в 4 мл смеси 1:1 HOAc/CHCl3 и обрабатывали NBS (67 мг, 0,38 ммоль). После перемешивания в течение ½ часа реакционную смесь разбавляли EtOAc (70 мл), промывали водой (3×30 мл) и 1М NaOH (2×30 мл). Органический раствор сушили над MgSO4 и концентрировали. Неочищенный продукт очищали методом хроматографии на силикагеле (EtOAc/гексан), что дало 90 мг соединения, обозначенного в подзаголовке примера.
d) Этиловый эфир 2-этилсульфанил-3,4-диметил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (c) (60 мг, 0,18 ммоль) растворяли в 2 мл NMP и обрабатывали NaSEt (45 мг, 0,54 ммоль), KI (3 мг, 0,018 ммоль) и CuO (7 мг, 0,09 ммоль). После нагревания в течение 2 дней при 120°C добавляли дополнительное количество NaSEt (45 мг, 0,54 ммоль), KI (3 мг, 0,018 ммоль) и CuO (7 мг, 0,09 ммоль) и нагревание продолжали в течение 3 дней. Реакционную смесь разбавляли 2 мл DCM и 2 мл воды. Полученный черный осадок фильтровали и отбрасывали. Фильтрат упаривали досуха в вакууме, затем разбавляли DCM (4 мл) и обрабатывали Et3N (111 мкл, 0,8 ммоль) и этилхлорформиатом (70 мкл, 0,7 ммоль). После перемешивания в течение ½ часа реакционную смесь разбавляли DCM (5 мл) и промывали водой (2×5 мл). Концентрирование органического слоя дало приблизительно 55 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
e) 2-Этансульфонил-3,4-диметил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин:
Продукт со стадии (d) (55 мг, 0,17 ммоль) растворяли в 2 мл HOAc и обрабатывали 30% H2O2 (220 мкл, 2 ммоль). После нагревания до 70°C в течение 1 часа реакционную смесь разбавляли водой (8 мл) и продукт экстрагировали в DCM (3×5 мл). Неочищенный остаток растворяли в 4 мл смеси 1:1 EtOH:40% KOH (водн.) и нагревали до 100°C в течение 14 часов. Реакционную смесь охлаждали и разбавляли водой. Продукт экстрагировали в DCM (2×5 мл) и очищали методом препаративной ВЭЖХ-МС, что дало соединение, обозначенное в заголовке примера. 1H-ЯМР (CD3OD) δ 3,64-3,19 (м, 7H), 3,25 (кв, J=7,5 Гц, 2H), 2,44 (с, 3H), 1,36 (д, J=7,2 Гц, 3H), 1,27 (т, J=7,4 Гц, 3H); MS: ESI (положительный): 274 (M+H).
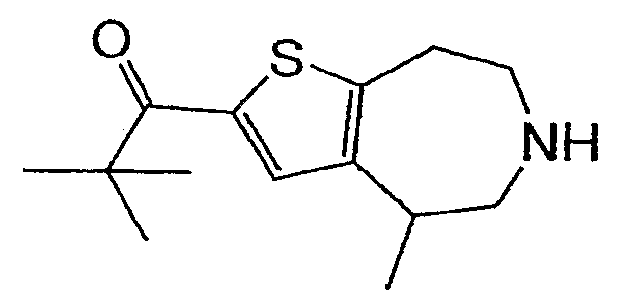
Пример 11: (R,S)-2,2-Диметил-1-(4-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-b]азепин-2-ил)-пропан-1-он (Схема 4)
a) Этиловый эфир (R,S)-2-бром-4-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт примера 9, стадия (b), (рацемический, 80 мг, 0,35 ммоль) растворяли в 2 мл смеси 1:1 CHCl3/HOAc. Добавляли N-бром-сукцинамид (62 мг, 0,35 ммоль) и реакционную смесь перемешивали в течение 15 минут. Концентрирование и очистка методом хроматографии на силикагеле приводили к соединению, обозначенному в подзаголовке примера, в виде желтого масла.
b) (R,S)-2,2-Диметил-1-(4-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-b]азепин-2-ил)-пропан-1-он:
Продукт со стадии (a) (55 мг, 0,17 ммоль) растворяли в ТГФ (2 мл) и охлаждали до -78°C. Добавляли бутиллитий (0,16 мл 1,6М) и реакционную смесь перемешивали в течение 15 минут. После тушения триметилацетилхлоридом (41 мкл, 0,34 ммоль) реакцию отогревали до комнатной температуры и концентрировали. Неочищенный остаток растворяли в 2 мл EtOH и обрабатывали 2 мл 40% KOH (водн.). После нагревания в течение ночи при 100°C реакционную смесь охлаждали и разбавляли водой. Продукт экстрагировали в DCM (2×) и соединение, обозначенное в заголовке примера, очищали методом препаративной ВЭЖХ-МС. 1H-ЯМР (CD3OD) δ 7,69 (с, 1H), 3,51-3,12 (м, 7H), 1,46 (д, J=7,2 Гц, 3H), 1,36 (с, 9H); MS: ESI (положительный): 252 (M+H).
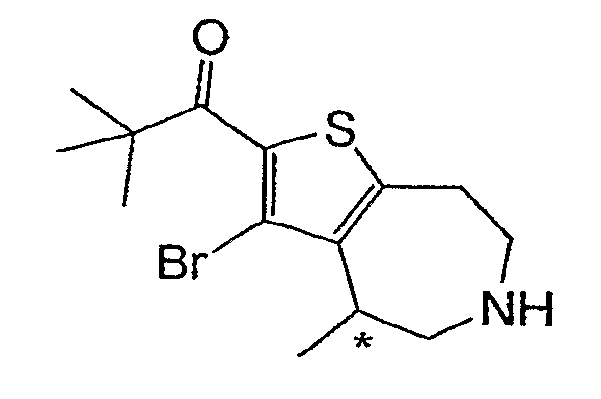
Пример 12: 1-(3-Бром-4-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-он (Схема 4)
a) Этиловый эфир 3-бром-2-(2,2-диметил-пропионил)-4-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт примера 9, стадия (c), (энантиомер 2, 200 мг, 0,50 ммоль) растворяли в 5 мл ТГФ и охлаждали до -78°C. Добавляли бутил литий (0,31 мл 1,6М) и реакционную смесь перемешивали в течение 15 минут. После тушения триметилацетилхлоридом (90 мкл, 0,75 ммоль) реакционную смесь отогревали до комнатной температуры и концентрировали. Очистка сырого остатка методом хроматографии на силикагеле приводила к 170 мг соединения, обозначенного в подзаголовке примера.
b) 1-(3-Бром-4-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-он:
Продукт со стадии (a) (56 мг, 0,14) перемешивали в 2 мл CHCl3 и обрабатывали TMSI (57 мкл, 0,42 ммоль). После нагревания при 70°C в течение 1 часа добавляли еще 0,42 ммоль TMSI и нагревание продолжали в течение 1 часа. Реакционную смесь охлаждали и распределяли между 1М NaOH и CHCl3. Органический слой концентрировали и очищали методом ВЭЖХ-МС, что дало соединение, обозначенное в заголовке примера. 1H ЯМР (CD3OD) δ 3,74-3,56 (м, 4H), 3,43-3,20 (м, 3H), 1,35 (д, J=7,2 Гц, 3H), 1,30 (с, 9H); MS: ESI (положительный): 330, 332 (M+H).
Пример 13: (R,S)-2-(2,2,2-трифтор-этил)-4,4a,5,6,7,8-гексагидро-3H-1-тиа-6-аза-циклопента[cd]азулен (Схема 5)
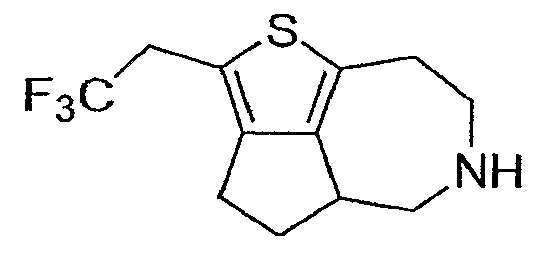
a) Этиловый эфир (E,Z)-4-этоксикарбонилметилен-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
1,6М раствор LHMDS в ТГФ (15 мл) добавляли к продукту примера 1, стадия (c), (2,0 г, 8,37 ммоль) и триэтил фосфоноацетату (4 мл, 16,74 ммоль) в безводном ТГФ (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем обрабатывали дополнительным раствором LHMDS (3,2 мл 1,6М) и триэтилфосфоноацетатом (800 мкл, 3,3 ммоль). После перемешивания в течение 3 часов реакцию тушили водой и разбавляли DCM. Органический слой сушили над MgSO4 и концентрировали, что дало соединение, обозначенное в подзаголовке примера, которое применяли без дополнительной очистки, MS: ESI (положительный): 310 (M+H).
b) Этиловый эфир (R,S)-4-этоксикарбонилметил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (a) (2,47 г, 8 ммоль) перемешивали с 2,0 г 10% Pd/C (влажный, от Degussa, класс El01) в метаноле (8 мл) в атмосфере H2 (1 атм) в течение 72 часов. Реакционную смесь фильтровали через целит, концентрировали досуха, что дало соединение, обозначенное в подзаголовке примера, в виде масла, которое применяли без дополнительной очистки. MS: ESI (положительный): 312 (M+H).
c) Этиловый эфир (R,S)-4-карбоксиметил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (b) (2,47 г, 8 ммоль) перемешивали в этаноле (60 мл) с 1М NaOH (30 мл) при температуре внешней среды в течение ночи. Реакционную смесь подкисляли 1М HCl и распределяли между DCM и водой. Органический слой промывали водой, высушивали над MgSO4, концентрировали досуха, что дало 2,13 г соединения, обозначенного в подзаголовке примера, в виде желтого масла. MS: ESI (положительный): 284 (M+H).
d) Этиловый эфир (R,S)-3-оксо-3,4,4a,5,7,8-гексагидро-1-тиа-6-аза-циклопента[cd]азулен-6-карбоновой кислоты:
Оксалилхлорид (3 мл, 37,7 ммоль) и каталитическое количество ДМФА добавляли к раствору продукта со стадии (c) (2,13 г, 7,54 ммоль) в DCM (40 мл), реакционную смесь перемешивали при температуре внешней среды в течение 1 часа. Реакционную смесь концентрировали досуха и повторно растворяли в дихлорэтане (100 мл). К раствору добавляли AlCl3 (2,0 г, 15,1 ммоль) и реакционную смесь перемешивали при температуре внешней среды в течение ночи. Реакцию тушили льдом и распределяли между DCM и водой. Органический слой концентрировали, что дало соединение, обозначенное в подзаголовке примера, которое очищали методом хроматографии (EtOAc/Hex, выделено 1,02 г) перед применением на последующих стадиях. MS: ESI (положительный): 266 (M+H).
e) Этиловый эфир (R,S)-3,4,4a,5,7,8-гексагидро-1-тиа-6-аза-циклопента[cd]азулен-6-карбоновой кислоты:
A1C13 (627 мг, 4,72 ммоль) добавляли к комплексу BH3-трет-BuNH2 (492 мг, 5,66 ммоль) в DCM (2 мл) при 0°C. Раствор перемешивали в течение 10 минут, затем обрабатывали продуктом со стадии (d) (250 мг, 0,943 ммоль), взятым в виде раствора в DCM (1 мл). После отогревания до комнатной температуры реакцию тушили 0,1М HCl по каплям и концентрировали досуха. Реакционную смесь разбавляли в 1М HCl и экстрагировали в EtOAc. Органический слой концентрировали, что дало соединение, обозначенное в подзаголовке примера, которое очищали методом хроматографии (EtOAc/Hex) перед применением на последующих стадиях. MS: ESI (положительный): 252 (M+H).
f) Этиловый эфир (R,S)-2-бром-3,4,4a,5,7,8-гексагидро-1-тиа-6-аза-циклопента[cd]азулен-6-карбоновой кислоты:
Соединение, обозначенное в подзаголовке примера, получали способом примера 1, стадия (h), с применением продукта со стадии (e) и применяли в неочищенной форме без очистки.
g) Этиловый эфир (R,S)-2-(2,2,2-трифтор-ацетил-3,4,4a,5,7,8-гексагидро-1-тиа-6-аза-циклопента[cd]азулен-6-карбоновой кислоты:
Раствор 1,6М н-BuLi в гексане (130 мкл) добавляли к раствору продукта со стадии (f) (69 мг, 0,208 ммоль) в безводном ТГФ (мл) при -78°C и перемешивали в течение 15 минут. Добавляли 2,2,2-трифтор-этиловый эфир трифторуксусной кислоты (50 мкл, 0,270 ммоль) и реакцию отогревали до температуры внешней среды. Реакцию тушили водой, экстрагировали в этилацетат и органические слои объединяли и концентрировали. Добавляли трифторуксусный ангидрид (750 мкл, 5,36 ммоль) и AlCl3 (600 мг, 4,51 ммоль) к раствору неочищенного продукта в дихлорэтане (10 мл) и реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь охлаждали до температуры внешней среды, тушили водой и экстрагировали в DCM. Органический слой концентрировали, что дало соединение, обозначенное в подзаголовке примера, которое применяли в неочищенной форме без очистки. MS: ESI (положительный): 348 (M+H).
h) Этиловый эфир (R,S)-2-(2,2,2-трифтор-этил)-3,4,4a,5,7,8-гексагидро-1-тиа-6-аза-циклопента[cd]азулен-6-карбоновой кислоты:
NaBH4 (15 мг, 0,208 ммоль) и каталитическое количество уксусной кислоты добавляли к продукту стадии (g) (72 мг, 0,208 ммоль) в этаноле (3 мл) и реакционную смесь перемешивали при температуре внешней среды в течение 20 минут. Реакцию тушили уксусной кислотой (по каплям) и распределяли между водой и DCM. Органический слой концентрировали. Добавляли дигидрат хлорида олова (187 мг, 0,832 ммоль) к неочищенному продукту в уксусной кислоте (2 мл) и реакционную смесь нагревали до 80°C в течение 2 часов. Добавляли дополнительный SnCl2 (150 мг) и реакционную смесь нагревали в течение еще 4 часов. Затем реакционную смесь разбавляли водой и экстрагировали в DCM. Органический слой концентрировали, что дало соединение, обозначенное в подзаголовке примера, которое применяли без дополнительной очистки.
i) (R,S)-2-(2,2,2-Трифтор-этил)-4,4a,5,6,7,8-гексагидро-3H-1-тиа-6-аза-циклопента[cd]азулен:
TMSI (45 мкл) добавляли к продукту со стадии (h) в DCM (2 мл) и нагревали до 50°C в темноте в течение 2 часов. Реакционную смесь охлаждали до температуры внешней среды и тушили метанолом. Реакционную смесь концентрировали и очищали методом препаративной ЖХМС, что дало соединение, обозначенное в заголовке примера. 1H-ЯМР (CD3OD) δ 3,58-3,71 (м,2H), 3,51 (кв, J=10,8 Гц, 2H), 3,07-3,16 (м, 3H), 2,99-3,04 (м, 4H), 1,91-2,07 (м, 2H). MS: ESI (положительный): 262 (M+H).
Пример 14: (R,S)-2-Бром-3,3-диметил-4,4a,5,6,7,8-гексагидро-3H-1-тиа-6-аза-циклопента[cd]азулен (Схема 5)
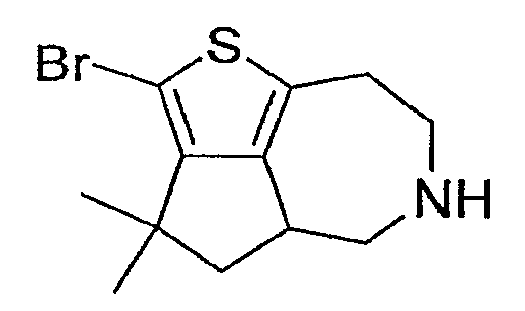
a) Этиловый эфир (R,S)-3,3-диметил-3,4,4a,5,7,8-гексагидро-1-тиа-6-аза-циклопента[cd]азулен-6-карбоновой кислоты:
Раствор 2М Me2Zn в толуоле (1 мл) добавляли к TiCl4 (380 мг, 2,02 ммоль) в DCM при -78°C и перемешивали в течение 10 минут. Добавляли продукт из примера 13, стадия (d), (89 мг, 0,336 ммоль) и реакцию отогревали до 0°C и перемешивали на холоде в течение 1 часа. Реакцию тушили водой и экстрагировали в DCM. Органический слой концентрировали, что дало соединение, обозначенное в подзаголовке примера, которое очищали препаративной ЖХМС перед применением на последующих стадиях. MS: ESI (положительный): 280 (M+H).
b) Этиловый эфир (R,S)-2-Бром-3,3-диметил-3,4,4a,5,7,8-гексагидро-1-тиа-6-аза-циклопента[cd]азулен-6-карбоновой кислоты.
Соединение, обозначенное в подзаголовке примера, получали способом примера 5, стадия (a), с применением продукта со стадии (a) и применяли в неочищенной форме без очистки.
c) (R,S)-2-Бром-3,3-диметил-4,4a,5,6,7,8-гексагидро-3H-1-тиа-6-аза-циклопента[cd]азулен:
Соединение, обозначенное в заголовке примера, получали способом, описанным в примере 5, стадия (d), с применением продукта со стадии (b) и очищали методом препаративной ВЭЖХ-МС. 1H-ЯМР (CD3OD) δ 3,31-3,58 (м, 3H), 2,85-3,07 (м, 3H), 2,63 (т, J=10,5 Гц, 1H), 2,24 (дд, J = 6,9 Гц, J=12,3 Гц, 1H), 1,86-1,95 (м, 1H), 1,44 (с, 3H) 1,26 (с, 3H). MS: ESI (положительный): 288 (М+Н).
Пример 15: (R,S)-2-Бром-5-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин (Схема 6)
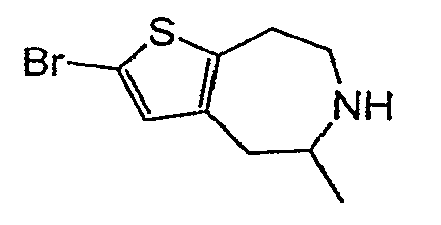
a) Этиловый эфир (R,S)-5-метил-4-оксо-4,5,7,8-тетрагидро-тиено[2,3-b]азепин-6-карбоновой кислоты:
Продукт примера 1, стадия (c), (1,06 г, 4,4 ммоль) растворяли в 20 мл ТГФ и обрабатывали LHMDS (5,3 мл 1М в ТГФ). После перемешивания в течение 1 часа при комнатной температуре добавляли MeI (326 мкл, 5,3 ммоль) и реакционную смесь перемешивали 1 час при комнатной температуре. Реакционную смесь упаривали на силикагеле и очищали методом хроматографии на силикагеле (10% до 30% EtOAc в гексане), что дало 175 мг соединения, обозначенного в подзаголовке примера.
b) Этиловый эфир (R,S)-5-Метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (a) (75 мг, 0,30 ммоль) растворяли в DCE (3 мл) и обрабатывали ZnI2 (143 мг, 0,45 ммоль), затем NaCNBH3 (132 мг, 2,1 ммоль). После перемешивания в течение 3 дней при комнатной температуре реакционную смесь фильтровали, фильтрат промывали DCM и отбрасывали. Органические слои объединяли, промывали водой (2×10 мл) и высушивали над MgSO4. Концентрирование дало 22 мг смеси соединения, обозначенного в подзаголовке примера, и соответствующего спирта, в котором восстановлена одна кетонная группа исходного вещества. Смесь использовали прямо на последующих стадиях.
c) (R,S)-2-Бром-5-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин:
Продукт со стадии (b) (22 мг, 0,092 ммоль) растворяли в 2 мл смеси 1:1 HOAc:CHCl3 и обрабатывали NBS (25 мг, 0,14 ммоль). После перемешивания в течение ½ часа реакционную смесь концентрировали досуха и обрабатывали 2 мл EtOH и 2 мл 40% KOH. После нагревания при 100°C в течение 24 часов и затем при 120°C в течение еще 24 часов реакционную смесь охлаждали, разбавляли водой и экстрагировали 3×DCM. Очистка методом препаративной ВЭЖХ-МС приводила к соединению, обозначенному в заголовке примера. MS: ESI (положительный): 248 (M+H).
Пример 16: 4-Метил-1-трифторметил-l,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен (Схема 3)
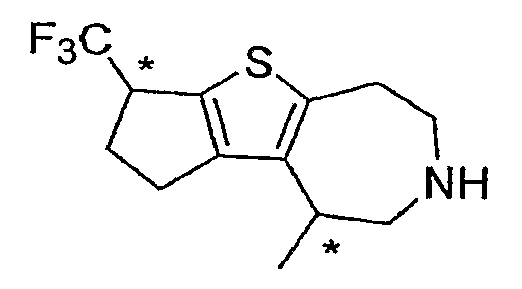
a) Этиловый эфир 4-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт примера 9, стадия (c), (энантиомер 2, 1,0 г, 2,51 ммоль) растворяли в 100 мл EtOH и обрабатывали 1 г Pd/C (влажный, от Degussa, класс E101), и интенсивно перемешивали под 1 атмосферой H2 в течение 2 часов. Реакционную смесь фильтровали через целит и концентрировали, что дало 600 мг соединения, обозначенного в подзаголовке примера.
b) Этиловый эфир 4-метил-2-(2,2,2-трифтор-ацетил)-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (a) растворяли в DCE (100 мл) и обрабатывали A1C13 (3,3 г, 25 ммоль), затем добавляли трифторуксусный ангидрид (3,5 мл, 25 ммоль). После нагревания при 80°C в течение 2 часов добавляли следующую порцию AlCl3 и трифторуксусного ангидрида (25 ммоль каждого). После перемешивания в течение 1 часа при 80°C реакцию тушили выливанием на лед и аккуратно приводили к щелочной реакции среды острожным добавлением Et3N. Густой осадок фильтровали и отбрасывали. Полученную смесь экстрагировали DCM (3×100 мл). После концентрирования экстрактов до приблизительно 100 мл добавляли 0,5 мл этилхлорформиата и 0,5 мл DIEA. После перемешивания в течение ½ часа раствор промывали водой, концентрировали и очищали методом хроматографии на силикагеле (25% EtOAc/гексан), что дало 460 мг соединения, обозначенного в подзаголовке примера.
c) Этиловый эфир 2-(1-карбоксиметил-2,2,2-трифтор-этил)-4-метил-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты:
Продукт со стадии (b) (460 мг, 1,37 ммоль) растворяли в 25 мл ТГФ и обрабатывали триэтилфосфоноацетатом (550 мкл, 2,74 ммоль) и LHMDS (2,2 мл 1М в ТГФ). После перемешивания в течение 15 минут реакцию тушили водой (100 мл) и продукт экстрагировали в DCM (2×50 мл). После высушивания экстрактов над MgSO4 и концентрирования неочищенный остаток растворяли в EtOH (50 мл) и обрабатывали 300 мг Pd/C (10%, влажный, от Degussa, класс E101). После перемешивания под 1 атмосферой H2 в течение 2 часов реакционную смесь фильтровали через целит и обрабатывали 1М NaOH (10 мл). После перемешивания в течение ночи при комнатной температуре реакцию подкисляли 1М HCl и продукт экстрагировали в DCM (2×25 мл). Экстракты сушили над MgSO4 и концентрировали, что дало 0,5 г соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки.
d) Этиловый эфир 4-метил-3-оксо-1-трифторметил-2,3,4,5,7,8-гексагидро-1H-9-тиа-6-аза-циклопента[a]азулен-6-карбоновой кислоты:
Продукт со стадии (c) (500 мг, 1,3 ммоль) растворяли в DCM (20 мл) и обрабатывали ДМФА (50 мкл), затем добавляли оксалилхлорид (240 мкл, 1,45 ммоль). После перемешивания в течение ½ часа добавляли дополнительное количество оксалилхлорида (240 мкл). После перемешивания в течение еще 1 часа реакционную смесь концентрировали досуха и остаток повторно растворяли в DCM (5 мл). Добавляли AlCl3 (360 мг, 2,7 ммоль) и реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Реакцию тушили ледяной водой (10 мл) и продукт экстрагировали в DCM/EtOH (4:1, 2×30 мл). Экстракты сушили над MgSO4 и концентрировали, что дало 468 мг соединения, обозначенного в подзаголовке примера, которое применяли без дополнительной очистки. Диастереомеры могут быть разделены методом хроматографии на силикагеле (25-35% EtOAc в гексане), что давало диастереомер 1 (верхнее пятно, первый продукт элюирования) и диастереомер 2 (нижнее пятно, второй продукт элюирования).
e) Этиловый эфир 4-метил-1-трифторметил-2,3,4,5,7,8-гексагидро-1H-9-тиа-6-аза-циклопента[a]азулен-6-карбоновой кислоты:
Продукт со стадии (d) (смесь диастереомеров, 234 мг, 1,54 ммоль) растворяли в 5 мл EtOH и обрабатывали NaBH4 (117 мг, 3,1 ммоль). После перемешивания в течение 15 минут реакцию тушили HOAc и разбавляли водой (10 мл). Продукт экстрагировали в DCM (2×25 мл) и экстракты концентрировали досуха. Неочищенный остаток растворяли в HOAc (8 мл) и концентрированном HCl (4 мл) и обрабатывали SnCl2 (1,4 г, 6,2 ммоль). После нагревания до 80°C в течение 1 часа реакционную смесь охлаждали и разбавляли водой (15 мл). Продукт экстрагировали в DCM (3×25 мл) и экстракты сушили над MgSO4 и концентрировали досуха. Диастереомеры разделяли методом ВЭЖХ (Novapak C-18, 19×300 мм, 55% CH3CN/вода, 10 мл/мин) для получения диастереомера 1 (время удержания = 23,0 мин) и диастереомер 2 (время удержания = 24,2 мин), что приводило к приблизительно 20 мг каждого из диастереомеров.
f) 4-Метил-1-трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен:
Продукт со стадии (e) (диастереомер 1 и диастереомер 2, по отдельности, 20 мг, 0,06 ммоль) растворяли в 3 мл CHCl3 и обрабатывали TMSI (40 мкл, 0,29 ммоль). После нагревания до 70°C в течение ночи реакцию тушили MeOH (5 мл) и 1М NaOH (2 мл). Соединение, обозначенное в заголовке примера, экстрагировали в CHCl3 (2×10 мл) и затем очищали методом препаративной ВЭЖХ-МС.
Диастереомер 1: 1H-ЯМР (CD3OD) δ 4,01-3,90 (м, 1H), 3,43-2,95 (м, 8H), 2,87-2,64 (м, 3H), 2,56-2,44 (м, 1H), 1,33-1,31 (м, 3H). MS: ESI (положительный): 276 (M+H).
Диастереомер 2: 1H-ЯМР (CD3OD) δ 4,01-3,90 (м, 1H), 3,42-2,94 (м, 8H), 2,84-2,64 (м, 3H), 2,55-2,45 (м, 1H), 1,32-1,29 (м, 3H). MS: ESI (положительный): 276 (M+H).
Пример 17: 3,3,4-Триметил-1-трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[a]азулен (Схема 3)
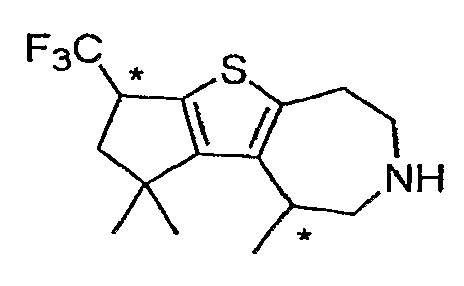
DCM (5 мл) охлаждали до -78°C и обрабатывали TiCl4 (91 мкл, 0,83 ммоль) и Me2Zn (0,4 мл 2М в толуоле). После перемешивания в течение 10 минут продукт примера 16, стадия (d), (диастереомер 1 и диастереомер 2, по отдельности, 50 мг каждого, 0,14 ммоль) растворяли в 5 мл DCM и добавляли к полученной темной суспензии. Реакционную смесь отогревали до комнатной температуры и перемешивали в течение 3 дней. Реакцию тушили выливанием на лед и продукт экстрагировали в DCM (2×15 мл). После высушивания над MgSO4 экстракты концентрировали, повторно растворяли в CHCl3 (10 мл) и обрабатывали TMSI (80 мкл, 0,58 ммоль). Реакционную смесь нагревали в течение ночи до 70°C и затем тушили MeOH (5 мл) и 1М NaOH (2 мл). Соединение, обозначенное в заголовке примера, экстрагировали в CHCl3 (2×10 мл) и очищали методом препаративной ВЭЖХ-МС.
Диастереомер 1: 1H-ЯМР (CD3OD) δ 4,20-3,89 (м, 1H), 3,49-3,34 (м, 3H), 3,28-3,89 (м, 4H), 2,54 (дд, J=9,3, 13,8 Гц, 1H), 2,32 (дд, J=5,4, 13,5 Гц, 1H), 1,39 (с, 3H), 1,35 (с, 3H), 1,34 (д, J=6,3 Гц, 3H). MS: ESI (положительный): 304 (M+H).
Диастереомер 2: 1H-ЯМР (CD3OD) δ 3,98 (секст., J=8,1 Гц, 1H), 3,52-3,34 (м, 3H), 3,27-2,88 (м, 4H), 2,46 (дд, J=8,4, 13,2 Гц, 1H), 2,33 (дд, J=6,0, 13,5 Гц, 1H), 1,42 (с, 3H), 1,38-1,33 (м, 3H), 1,29 (с, 3H). MS; ESI (положительный): 304 (M+H).
Пример 18: 2,2-Диметил-1-(5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-пропан-1-он (Схема 1)
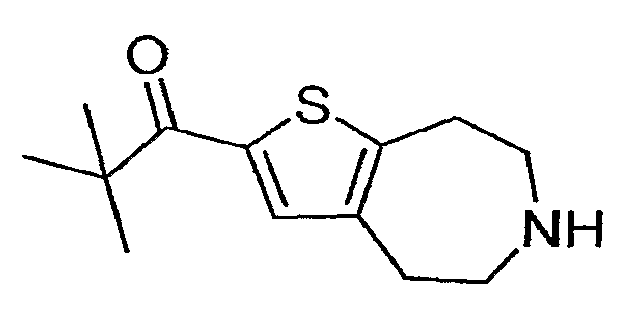
a) Этиловый эфир 2-(2,2-диметил-пропионил)-4,5,7,8-тетрагидро-тиено[2,3-d]азепин-6-карбоновой кислоты
Раствор продукта из Примера 1, стадия (d), (160 мг, 0,71 ммоль) в дихлорэтане (5 мл) при 0°C обрабатывали триметилацетилхлоридом (0,17 мл, 1,42 ммоль) и A1C13 (190 мг, 1,42 ммоль). Реакционную смесь оставляли перемешиваться при 0°C в течение 30 минут и затем позволяли отогреваться до комнатной температуры. Реакционную смесь тушили путем добавления насыщенного раствора NaHCO3 (30 мл) и экстрагировали хлористым метиленом (3×30 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (50 мл), высушивали (MgSO4) и растворитель упаривали в вакууме, что дало желто-коричневое масло. Неочищенное масло очищали методом хроматографии на силикагеле (0-50% EtOAc в гексане), что дало соединение, обозначенное в подзаголовке примера, в виде прозрачного масла (60 мг, 27%). MS: ESI (положительный): 310 (M+H).
b) 2,2-Диметил-1-(5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-пропан-1-он:
Раствор продукта со стадии (a) (50 мг, 0,16 ммоль) в метаноле (5 мл) обрабатывали Ba(OH)2 (200 мг, 1,2 ммоль). Реакционную смесь нагревали в запаянной трубке в течение 30 часов и затем позволяли охладиться до комнатной температуры. Реакционную смесь концентрировали в вакууме и нейтрализовали путем добавления 1Н HCl. Аликвоту этого раствора очищали методом препаративной ВЭЖХ-МС, что дало соединение, обозначенное в заголовке примера, которое конвертировали в его соль с HCl. 1H-ЯМР (300 МГц, ДМСО-d6) δ 9,62 (уш. с, 2H); 7,81 (с, 1H); 3,08-3,23 (м, 8H); 1,30 (с, 9H); MS: ESI (положительный): 238 (M+H).
Пример 19: 1-(3-Хлор-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-он (Схема 7)
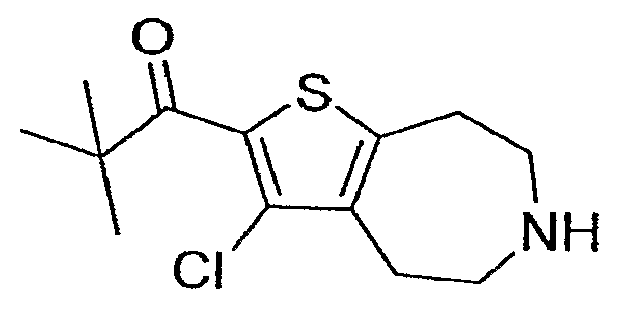
a) Этиловый эфир (4,5-дихлор-тиофен-2-ил)-оксо-уксусной кислоты:
При 5-10°C к 2,3-дихлортиофену (5 г, 32,6 ммоль) добавляли этиловый эфир хлор-оксо-уксусной кислоты (5,43 мл, 48,7 ммоль). Раствор AlCl3 (6,49 г, 48,7 ммоль), растворенный в нитрометане (13 мл), добавляли по каплям таким образом, чтобы внутренняя температура реакции не превышала 10°C. Через 1 час реакционную смесь выливали в ледяную воду и экстрагировали CH2C12 (2×100 мл). Органический слой промывали 10% NaHCO3 (2×50 мл), водой (1×50 мл) и насыщенным солевым раствором (1×50 мл). Высушивание (Na2SO4) и концентрирование приводило к слегка оранжевому твердому веществу, которое очищали методом хроматографии на силикагеле (градиент EtOAc/гексан), что приводило к 6,8 г (82%) соединения, обозначенного в подзаголовке примера.
b) Этиловый эфир (4,5-дихлор-тиофен-2-ил)-гидрокси-уксусной кислоты:
Раствор продукта со стадии (a) (23,0 г, 90,9 ммоль) в ТГФ (500 мл) обрабатывали NaBH(OAc)3 (23,1 г, 109 ммоль) и AcOH (250 мкл) при 60°C в течение 1 часа. Реакцию тушили AcOH (8 мл) и концентрировали до ~250 мл. Содержимое разбавляли H2O (400 мл) и экстрагировали с CH2C12 (1×400 мл; 1×100 мл). Органический слой сушили (MgSO4) и концентрировали, что дало 23 г соединения, обозначенного в подзаголовке примера. 1H-ЯМР (300 МГц, CDC13) δ 6,91 (с, 1H); 5,25 (дд, J1=6 Гц, J2=1 Гц, 1H); 4,22-4,40 (м, 2H); 3,52-3,60 (уш. м, 1H); 1,33 (т, J=7 Гц, 3H).
c) Этиловый эфир (4,5-дихлор-3-метоксикарбонилметил-тиофен-2-ил)-уксусной кислоты:
Раствор продукта со стадии (b) (12,2 г, 48,0 ммоль) в декалине (145 мл) обрабатывали триметилортоацетатом (24,5 мл, 192 ммоль) и гексановой кислотой (0,61 мл). Колбу снабжали колонкой Вигре и нагревали до 180°C. Добавочную гексановую кислоту (3 мл) периодически добавляли в течение 6 часов и реакционную смесь нагревали в течение ночи. Реакционную смесь концентрировали на роторном испарителе и остаток экстрагировали MeOH (2×100 мл). MeOH-экстракты концентрировали и очищали методом хроматографии на силикагеле (градиент EtOAc/Гексан), что приводило к 4,36 г (29%) соединения, обозначенного в подзаголовке примера. MS: ESI (положительный): 311, 313 (M+H).
d) (3-Карбоксиметил-4,5-дихлор-тиофен-2-ил)-уксусная кислота:
Раствор продукта со стадии (c) (1,14 г, 3,66 ммоль) в MeOH (7 мл) при 0°C обрабатывали по каплям с 2М NaOH (3,8 мл). Реакционную смесь отогревали до 22°C и перемешивали в течение ночи. Растворитель упаривали и остаток растворяли в 2М NaOH (50 мл) и экстрагировали эфиром (2×50 мл). Основной слой охлаждали до 0°C и подкисляли до pH1 действием 6М HCl. Кислотный слой обратно экстрагировали EtOAc (4×100 мл), органический слой сушили (MgSO4) и концентрировали. Неочищенное твердое вещество растирали в порошок с гексаном и фильтровали, что приводило к 2,75 г (73%) соединения, обозначенного в подзаголовке примера. MS: ESI (отрицательный): 267, 269 (M-H).
e) 2 [4,5-Дихлор-3-(2-гидрокси-этил)-тиофен-2-ил]-этанол:
Раствор продукта со стадии (d) (2,5 г, 9,33 ммоль) в ТГФ (85 мл) охлаждали до 0°C и по каплям добавляли 1М раствор BH3-ТГФ (46,6 мл, 46,6 ммоль) в течение 10 мин и перемешивали в течение еще 20 минут после завершения добавления. Реакцию отогревали до 22°C и перемешивали в течение 2 часов. Реакционную смесь выливали в ледяной холодный насыщенный NaHCO3 (150 мл) и экстрагировали EtOAc. Неочищенный раствор пропускали через слой силикагеля с промывкой EtOAc. Концентрирование элюента приводило к 1,99 г (88%) соединения, обозначенного в подзаголовке примера.
f) 2-[4,5-дихлор-2-(2-метансульфонилокси-этил)тиофен-3-ил]-этиловый эфир метансульфоновой кислоты:
Раствор продукта со стадии (e) (1,99 г, 8,25 ммоль) в CH2Cl2 (41 мл) охлаждали до 0°C и обрабатывали триэтиламином (3,4 мл, 24,7 ммоль), затем добавляли по каплям метансульфонилхлорид (1,4 мл, 18,1 ммоль) в течение 10 мин. Через 45 минут сырую реакционную смесь разбавляли CH2Cl2 (100 мл) и промывали ледяной водой (25 мл), 10% лимонной кислотой (2×25 мл), насыщенным NaHCO3 (2×25 мл) и насыщенным солевым раствором (1×25 мл). Органический слой сушили (MgSO4), концентрировали до 20 мл и разбавляли безводным диоксаном (76 мл). Эту смесь концентрировали для удаления остатков CH2Cl2 и полученный диоксановый раствор переносили прямо в следующую реакцию.
g) 6-Бензил-2,3-дихлор-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин:
Диоксановый раствор бисмезилата, полученный на стадии (f), переносили в 3-горлую реакционную колбу, снабженную капельной воронкой и обратным холодильником. Добавляли безводный карбонат калия (4,93 г, 35,7 ммоль) и содержимое нагревали до кипения с обратным холодильником. Затем по каплям добавляли раствор бензиламина (2,71 г, 25,3 ммоль) в безводном диоксане (27 мл) в течение 45 минут и нагревание продолжали в течение 16 часов. Соли отфильтровывали и растворитель концентрировали. Сырец очищали хроматографией на силикагеле (градиент EtOAc/гексан), что приводило к 1,43 г (62%) соединения, обозначенного в подзаголовке примера. 1H ЯМР (300 МГц, CDC13) δ 7,20-7,40 (м, 5H); 3,73 (с, 2H); 2,68-2,89 (м, 8H); MS: ESI (положительный): 312, 314 (M+H).
h) 1-(6-Бензил-3-хлор-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-он:
Продукт со стадии (g) (109 мг, 0,35 ммоль) в безводном ТГФ (5 мл) охлаждали до -78°C под атмосферой азота и обрабатывали 1,6М н-бутиллития в гексане (0,25 мл). Реакционную смесь перемешивали при -78°C в течение 5 минут, затем добавляли триметилацетилхлорид (0,22 мл, 1,75 ммоль). После еще 10 минут при -78°C реакционной смеси позволили отогреться до комнатной температуры. Реакцию тушили насыщенным NaHCO3 (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (50 мл), высушивали (MgSO4) и растворитель упаривали в вакууме, что дало соединение, обозначенное в подзаголовке примера, в виде оранжевого масла, которое применяли на следующей стадии без дополнительной очистки. MS: ESI (положительный): 362, 364 (M+H).
i) 1-(3-Хлор-5,6,7,8-тетрагидро-4H-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-он:
Раствор продукта со стадии (h) (предположительно 0,35 ммоль) в безводном дихлорэтане (2 мл) охлаждали до 0°C, обрабатывали K2CO3 (~50 мг) и 1-хлорэтилхлорформиатом (0,38 мл, 3,5 ммоль). Реакция позволили отогреваться до 22°C в течение 18 часов. Реакцию разбавляли CH2Cl2(50 мл) и промытым насыщенным NaHCO3 (2×50 мл), насыщенным солевым раствором (50 мл), высушивали (MgSO4) и растворитель упаривали в вакууме, что приводило к масляному остатку, который растворяли в безводном MeOH (10 мл) и кипятили с обратным холодильником в течение 1,5 часов. MeOH упаривали в вакууме, что приводило к неочищенному продукту. Порцию сырца очищали методом препаративной ВЭЖХ-МС, что дало соединение, обозначенное в заголовке примера. 1H-ЯМР (300 МГц, CDC13) δ 6,25 (уш.с, 1H); 3,05-3,19 (м, 8H); 1,34 (с, 9H); MS: ESI (положительный): 272, 274 (M+H).
Следующая методика использована для исследования репрезентативных соединений по настоящему изобретению в качестве агонистов 5HT2c-рецептора. Результаты этого исследования представлены в таблице 1.
Клеточная культура
HEK 293 EBNA, экспрессирующие 5HT2c-рецептор человека (Изоформа VNV) (Bums et al., NATURE 387: 30308, 1997 Fitzgerald et al., NEUROPSYCHO-PHARMACOLOGY 21: 825-905, 1999), выращивали в среде DMEM, содержащей 10% диализованного FBS, 9 мкг/мл бластицидина при 37°C в атмосфере 5% CO2.
Мобилизация кальция
Клетки HEK 293 EBNA, экспрессирующие 5HT2c-рецептор человека (2×l04/лунку), высевали в черные 384-луночные покрытые коллагеном планшеты и инкубировали в течение ночи при 37°C в атмосфере 5% CO. После удаления среды клетки обрабатывали буфером HBSS (137 мМ NaCl, 5,4 мМ KCl, 5,5 мМ глюкозы, 20 мМ Hepes, pH 7,5, 2,1 мМ MgCl2, 0,3 мМ CaCl2, 0,02 мМ MgSO4, 3,0 мМ NaHCO3 и 0,64 мМ KH2PO4), содержащим краситель Кальций3 (Molecular Device, CA), 2,5 мМ пробенецида и 0,08% плуроновой кислоты, в течение 60 минут согласно указаниям производителя. Соединения разбавляли в буфере Рингера с CsCl (58,3 мМ CsCl, 5,4 мМ KCl, 5,5 мМ глюкозы, 20 мМ Hepes, pH 7,5, 2,1 мМ MgCl2, 1,2 мМ CaCl2). 5-HT применяли в качестве положительного контроля. Индуцируемое лигандом высвобождение кальция и возникающую в процессе флуоресценцию измеряли на ридере планшетов Fluorometric Imaging (FLIPR, Molecular Device, CA).
Анализ данных:
Все данные анализировали в нелинейном среднеквадратичном приближении с применением программного обеспечения Prism 4.0. Стимуляция агонистом индуцируемой кальцием флуоресценции в FLIPR аппроксимировали к сигмоидальной кривой доза-эффект с применением уравнения Y = Фон + (Верхнее Значение - Фон)/(l + 10^((LogEC50-X))), где X представляет собой логарифм концентрации соединений и Y представляет собой флуоресцентный отклик.

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АЗОТИСТЫЕ ПРОИЗВОДНЫЕ ПИРРОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2008 |
|
RU2473543C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ПРОИЗВОДНОЕ п-ФЕНИЛЕНДИАМИНА КАК РЕГУЛЯТОР КАЛИЕВЫХ КАНАЛОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И МЕДИЦИНСКОГО ПРИМЕНЕНИЯ | 2019 |
|
RU2762562C1 |
| ПРОИЗВОДНОЕ НИТРОИМИДАЗОЛА ПРОТИВ ТУБЕРКУЛЕЗА ЛЕГКИХ | 2016 |
|
RU2675622C1 |
| СОЕДИНЕНИЯ ПИПЕРИДИНИЛА, СЕЛЕКТИВНО СВЯЗЫВАЮЩИЕ ИНТЕГРИНЫ | 2003 |
|
RU2333210C2 |
| 6,7,8,9-ТЕТРАГИДРО-5H-ПИРИДО[2,3-D]АЗЕПИНОВЫЕ ЛИГАНДЫ ДОФАМИНОВЫХ РЕЦЕПТОРОВ D3 | 2017 |
|
RU2712455C1 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИЧ | 2011 |
|
RU2564445C2 |
| БИЦИКЛИЧЕСКИЕ 6-АЛКИЛИДЕНПЕНЕМЫ В КАЧЕСТВЕ ИНГИБИТОРОВ β-ЛАКТАМАЗ | 2003 |
|
RU2339640C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИН-4(3H)-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ TRPV4 | 2021 |
|
RU2840769C1 |
Изобретение относится к соединениям формулы
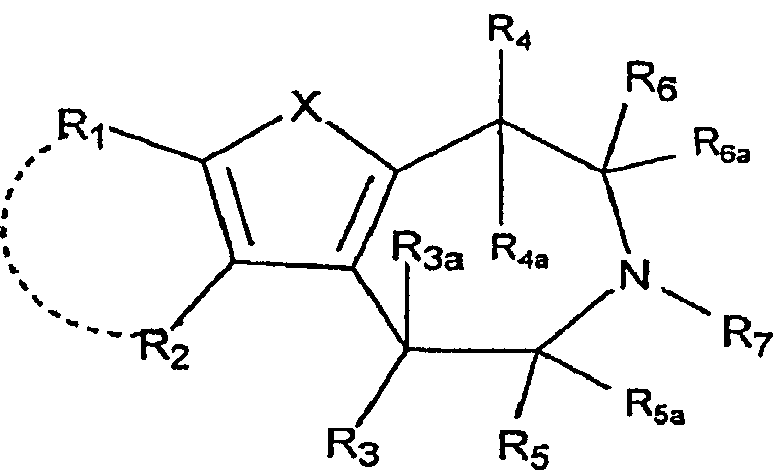 ,
,
где
Х представляет собой S;
R1 и R2, взятые вместе с атомами, к которым они присоединены, образуют 5-членный карбоцикл, замещенный заместителями, вплоть до двух, выбранными из алкила и CF3;
R3 выбран из группы, состоящей из атома водорода и C1-8-алкила;
R3a представляет собой атом водорода;
R4 представляет собой атом водорода;
R4a представляет собой атом водорода;
R5 представляет собой атом водорода;
R5a представляет собой атом водорода;
R6 представляет собой атом водорода;
R6a представляет собой атом водорода;
R7 представляет собой атом водорода;
или к их фармацевтически приемлемым солям. Изобретение также относится к соединениям формулы, к соединениям, выбранным из группы, а также к фармацевтической композиции. Технический результат - получение новых биологически активных соединений, модулирующих активность серотониновых рецепторов (5-HT2C). 5 н. и 1 з.п. ф-лы, 1 табл.
1. Соединение формулы
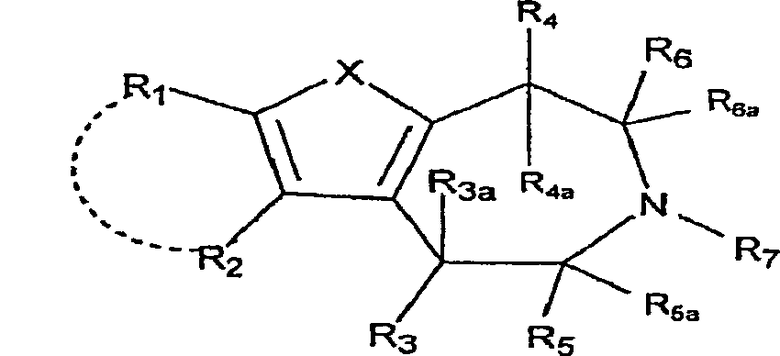 ,
,
где Х представляет собой S;
R1 и R2, взятые вместе с атомами, к которым они присоединены, образуют 5-членный карбоцикл, замещенный заместителями, вплоть до двух, выбранными из алкила и CF3;
R3 выбран из группы, состоящей из атома водорода и C1-8-алкила;
R3a представляет собой атом водорода;
R4 представляет собой атом водорода;
R4a представляет собой атом водорода;
R5 представляет собой атом водорода;
R5a представляет собой атом водорода;
R6 представляет собой атом водорода;
R6a представляет собой атом водорода;
R7 представляет собой атом водорода;
или его фармацевтически приемлемая соль.
2. Соединение формулы
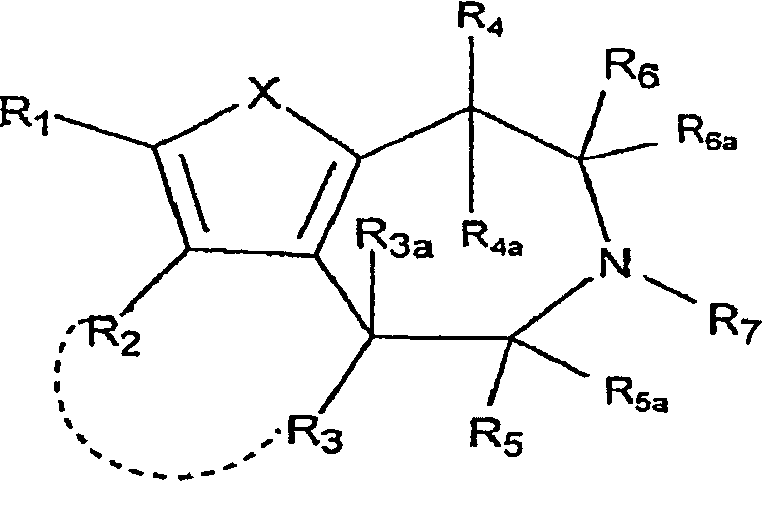 ,
,
где Х представляет собой S;
R1 выбран из группы, состоящей из галогена, C1-8-алкилпергалогеналкила;
R3a представляет собой атом водорода;
R2 и R3, взятые вместе с атомами, к которым они присоединены, образуют 5-членный карбоцикл, замещенный заместителями, вплоть до двух, выбранными из алкила;
R4 представляет собой атом водорода;
R4a представляет собой атом водорода;
R5 представляет собой атом водорода;
R5a представляет собой атом водорода;
R6 представляет собой атом водорода;
R6a представляет собой атом водорода или C1-8-алкил;
R7 представляет собой атом водорода;
или его фармацевтически приемлемая соль.
3. Соединение формулы
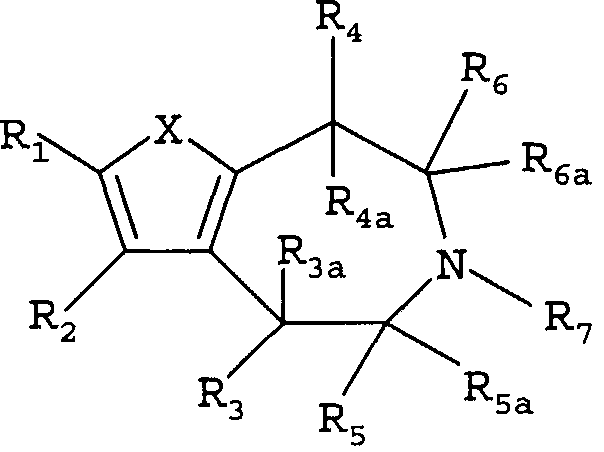
где Х представляет собой S;
R1 выбран из группы, состоящей из -C(=O)R10 и -SO2R10;
R2 выбран из группы, состоящей из атома водорода, галогена и C1-8-алкила;
R3 выбран из группы, состоящей из атома водорода и C1-8-алкила;
R3a представляет собой атом водорода;
R4 представляет собой атом водорода;
R4a представляет собой атом водорода;
R5 представляет собой атом водорода или -C1-8-алкил;
R5a представляет собой атом водорода;
R6 представляет собой атом водорода;
R6a представляет собой атом водорода;
R7 представляет собой атом водорода;
R10 выбран из группы, состоящей из -C1-8-алкила и арила;
или его фармацевтически приемлемая соль.
4. Соединение по п.3, в котором R1 представляет собой C(=O)R10, R2 представляет собой C1-8-алкил, и R3, R3a, R4, R4a, R5 и R5a представляют собой атом водорода.
5. Соединение, выбранное из группы, состоящей из:
2,2-Диметил-1-(3-метил-5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепин-2-ил)-пропан-1-она;
2-Бензолсульфонил-5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепина;
2-Этансульфонил-5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепина;
(R,S)-1-Трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[а]азулена;
(R,S)-3,3-Диметил-1-трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[а]азулена;
2-Этансульфонил-3,4-диметил-5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепина;
(R,S)-2,2-Диметил-1-(4-метил-5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепин-2-ил)-пропан-1-она;
1-(3-Бром-4-метил-5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-она;
(R,S)-2-Бром-5-метил-5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепина;
4-Метил-1-трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[а]азулена;
3,3,4-Триметил-1-трифторметил-1,2,3,4,5,6,7,8-октагидро-9-тиа-6-аза-циклопента[а]азулена;
2,2-Диметил-1-(5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепин-2-ил)-пропан-1-она и
1-(3-Хлор-5,6,7,8-тетрагидро-4Н-тиено[2,3-d]азепин-2-ил)-2,2-диметил-пропан-1-она;
или его фармацевтически приемлемая соль.
6. Фармацевтическая композиция, модулирующая активность серотониновых рецепторов (5-HT2C), включающая, по меньшей мере, одно соединение по любому из пп.1-5 и фармацевтически приемлемый носитель.
| US 2006003990 A1, 05.01.2006 | |||
| US 2002123488 A1, 05.09.2002 | |||
| Способ получения производных азепина или их солей | 1982 |
|
SU1091858A3 |

