Изобретение относится к замещенным бензиламинопиперидиновым соединениям, которые представляют интерес в области медицинской химии и химиотерапии. Более конкретно, изобретение относится к ряду замещенных пиперидиновых соединений, а также к их фармацевтически приемлемым солям, которые имеют особое значение ввиду их способности к антагонизму вещества Р. Эти соединения могут быть использованы при лечении желудочно- кишечного заболевания, заболевания центральной нервной системы (ЦНС), воспалительного заболевания, недержания мочи, боли, мигрени, солнечной эритемы, заболеваний кровеносных сосудов, расстройств и неблагоприятных состояний, вызванных Helicobacter pylori и др. , в частности заболеваний ЦНС млекопитающего, в частности человека.
Вещество P представляет собой природный ундекапептид, принадлежащий к тахикитиновому семейству пептидов, причем последние имеют такое название вследствие их быстрого стимулирующего действия на гладкие мышечные ткани. То есть, вещество P является фармацевтичесм активным нейропептидом, который продуцируется у млекопитающих (первоначально было выделено из кишки) и обладает характерной аминокислотной последовательностью, которая описана D. F. Veber et al. в патенте США 4680283. Широкое вовлечение вещества P и других тахикининов в физиологию многочисленных заболеваний достаточно полно проиллюстрировано в данной области. Например, вещество P, как недавно показано, вовлекается в передачу боли или мигрени, а также в заболевания центральной нервной системы, такие как тревожное состояние и шизофрения, в респираторные и воспалительные заболевания, такие как астма и ревматоидные артриты, соответственно, а также в желудочно-кишечные расстройства и заболевания желудочно-кишечного тракта типа язвенных колитов и болезни Крона, и др. Также сообщается, что тахикининовые антагонисты могут быть полезны при лечении аллергических состояний, при иммунорегуляции, вазодилатации, бронхоспазме, рефлекторном или нейронном контроле внутренних органов и старческого слабоумия типа болезни Альцгеймера, рвоте, солнечной эритеме и инфекциях, вызванных Helicobacter pylori.
В международных публикациях WO 93/01170, WO 93/00331 и WO 93/11110 раскрыт широкий спектр пиперидиновых производных в качестве тахикининовых антагонистов, таких как антагонисты вещества Р.
Настоящее изобретение предлагает замещенные пиперидины следующей общей формулы (1):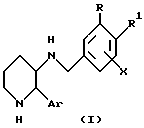
и их фармацевтически приемлемые соли,
где R представляет собой галоген - C1-C8-алкил, галоген - C2-C8-алкенил, галоген - C2-C8-алкинил или галоген - C1-C8 - алкил, замещенный гидрокси - или C1-C8-алкоксигруппой; R1 представляет собой атом водорода, атом галогена или C1-C6-алкоксигруппу; или
R и R1 вместе с двумя атомами углерода, общими для бензольного кольца и заместителей R и R1 образуют конденсированный C4-C6-циклоалкил, где один атом углерода необязательно заменен атомом кислорода, и где один или два атома углерода необязательно имеют до пяти заместителей, выбираемых из числа атома галогена, C1-С6-алкила и галоген -C1-С6-алкила;
X представляет собой C1-С8-алкокси-, галоген -C1-C6- алкокси-, феноксигруппу или атом галогена; и
Ar представляет собой фенильную группу, необязательно замещенную атомом галогена.
Пиперидиновые соединения настоящего изобретения формулы (1) проявляют высокую антагонистическую активность в отношении вещества P, в частности высокую активность против заболеваний ЦНС, и, следовательно, могут быть использованы для лечения желудочно-кишечного расстройства, заболевания центральной нервной системы, воспалительного заболевания, рвоты, недержания мочи, боли, мигрени, солнечной эритемы, развития заболеваний кровеносных сосудов или расстройств и неблагоприятных состояний, вызванных Helicobacter pylori, млекопитающего, в особенности человека.
Таким образом, настоящее изобретение предлагает фармацевтическую композицию для лечения желудочно-кишечного расстройства, расстройства центральной нервной системы, воспалительного заболевания, рвоты, недержания мочи, мигрени, солнечной эритемы, заболеваний кровеносных сосудов, расстройств и неблагоприятных состояний, вызванных Helicobacter pylori и др., в частности заболеваний ЦНС млекопитающего, в особенности человека, которая содержит терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель.
В настоящем изобретении:
термин "галоген -C1-С8- алкил" используется для обозначения линейного, разветвленного или циклического C1 - C8 - алкильного радикала, замещенного одним или более атомами галогена (то есть, Cl, F, I или Br), включая, но не ограничиваясь только ими, трифторметил, дифторэтил, трифторэтил, пентафторэтил, трифторизопропил, тетрафторизопропил, пентафторизопропил, гексафторизопропил и гептафторизопропил и им подобные радикалы;
термин "галоген C2-C8 - алкенил" используется для обозначения линейного, разветвленного или циклического C2-C8- алкенильного радикала, замещенного одним или более атомами галогена (то есть, Cl, F, I или Br), включая, но не ограничиваясь только ими, 3,3,3 - трифторпропенил, 1,1-диметил-4,4,4-трифторбутенил и им подобные радикалы;
термин "галоген -C2-C8- алкинил" используется для обозначения линейного, разветвленного или циклического C2-C8 - алкинильного радикала, замещенного одним или более атомами галогена (то есть, Cl, F, I или Br), включая, но не ограничиваясь только ими, 3,3,3 -трифторпропинил, 1,1-диметил-4,4,4- трифторбутинил и им подобные радикалы; и
термин "галоген -C1-C8-алкокси" используется для обозначения линейного, разветвленного или циклического C1-C8 - алкоксильного радикала, замещенного одним или более атомами галогена (то есть, Cl, F, I или Br), включая, но не ограничиваясь только ими, дифторметокси-, трифторметокси -, 2,2,2-трифторэтоксигруппы и им подобные.
В химической формуле (1):
R предпочтительно представляет собой C1-С6-алкил, гидрокси-C1-С6-алкил, С2 - С6-алкенил или С2-С6-алкинил, в которых алкильный, алкенильный или алкинильный фрагменты замещены двумя - семью атомами галогена.
В предпочтительном варианте осуществления настоящего изобретения заместитель R представляет собой C1-С6-алкил, гидрокси-C1-С6-алкил, С2-С6-алкенил или С2-С6-алкинил, предпочтительно С1-С6-алкил, причем эти группы замещены двумя-тремя атомами фтора. Примерами заместителя R являются трифторметил, дифторэтил, трифторэтил, трифторизопропил, трифтор-трет-бутил, трифтор-1,1-диметилметил-3-бутинил и 2 -хлортрифторизопропил.
В другом предпочтительном варианте осуществления настоящего изобретения R представляет собой C1-С6-алкил, гидрокси-C2-С6-алкил, С2-С6-алкенил или С2-С6-алкинил, причем эти группы замещены четырьмя-семью атомами фтора. Примерами заместителя R являются пентафторэтил, пентафторпропил, пентафторизопропенил, гексафторизопропил, гептафторизопропил, гексафтор - 2 - гидроксиизопропил и гексафтор-третбутил.
Заместитель R1 предпочтительно представляет собой атом водорода или метоксигруппу, более предпочтительно-атом водорода.
В другом предпочтительном варианте осуществления настоящего изобретения заместители R и R1 вместе с двумя атомами углерода, общими для бензольного кольца и заместителей R и R1 образуют конденсированный C4-C6-циклоалкил, где один атом углерода необязательно заменен атомом кислорода. Один или два атома углерода C4-C6-циклоалкильной группы могут необязательно иметь до четырех заместителей, более предпочтительно один - два заместителя, выбираемых из числа атома фтора и трифторметильной группы. Более предпочтительно R и R1 вместе с двумя атомами углерода, общими для бензольного кольца и заместителей R и R1 образуют трифторметилциклопентил, трифторметилциклогексил, дифторциклогексил или дифтордиметилциклогексил.
Заместитель X предпочтительно представляет собой атом галогена, метокси-, дифторметокси, трифторметокси или феноксигруппу, более предпочтительно метокси-, дифторметокси - или трифторметоксигруппу, и наиболее предпочтительно-метоксигруппу. Предпочтительно X находится в положении 2 фенильного кольца.
Ar предпочтительно представляет собой фенильную группу.
Другая предпочтительная группа соединений настоящего изобретения состоит из соединений формулы (Ia):
где R1 представляет собой атом водорода, атом галогена или метоксигруппу; и R2 и R3 независимо друг от друга выбирают из группы, включающей атом галогена, C1-C6- алкил, C2-C6-алкенил и C2-C6-алкинил или R2 и R3 вместе образуют C2-C6-алкилиден, где алкильный, алкенильный, алкинильный и алкилиденовый фрагменты необязательно содержат до семи атомов галогена;
или R1 и R2 вместе образуют конденсированный C4-C6-циклоалкил, в котором один атом углерода необязательно заменен атомом кислорода, причем C4-C8-циклоалкил необязательно замещен заместителями в количестве до четырех, выбираемыми из группы, включающей атом галогена, C1-C4-алкил и галоген-C1-C4- алкил.
В указанных соединениях предпочтительной стереохимической конфигурацией является (2S, 3S).
Предпочтительную группу отдельных соединений настоящего изобретения составляют следующие соединения:
(2S, 3S)-3-(2-Фтор-5-(трифторметил)бензил)амино-2 -фенилпиперидин или его соли:
(2S, 3S)-3-(2-Хлор-5-(трифторметил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(трифторметил)бензил)амино-2 -фенилпиперидин или его соли;
(2S, 3S)-3-(2-Фенокси-5-(трифторметил)бензил)амино-2- фенилпиперидин или его соли;
(2S, 3S)-3-(5-(1,1-Дифторэтил)-2-(трифторметокси)бензил)амино-2- фенилпиперидин или его соли;
(2S, 3S)-3-(5-(1,1-Дифторэтил)-2-метоксибензил)амино-2 фенилпиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(2,2,2-трифторэтил)бензил)амино-2- фенилпиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(1-(трифторметил)этил)бензил)амино-2- фенилпиперидин или его соли;
(2S, 3S)-3-[5-(1,1-Диметил-4,4,4-трифтор-2-бутинил)-2-метоксибензил] амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-[5-(1,1-Диметил-2,2,2-трифторэтил)-2-метоксибензиламино] -2-фенилпиперидин или его соли;
(2S, 3S)-3-(2,4-Диметокси-5-(2,2,2-трифторэтил)бензил)амино-2- фенилпиперидин или его соли; и
(2S, 3S)-3-[5-[1-Хлор-(1-трифторметил)этил]-2-метоксибензиламино]-2 -фенилпиперидин или его соли.
Другая предпочтительная группа отдельных соединений настоящего изобретения включает следующие соединения:
(2S, 3S)-2-Фенил-3-(5-(2,2,2-трифтор-1-(трифторметил)этил)-2- метоксибензил)аминопиперидин или его соли;
(2S, 3S)-2-Фенил-3-(5-(1,2,2,2-тетрафтор-1-(трифторметил)этил)-2- метоксибензил) аминопиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(1,1,2,2,2-пентафторэтил)бензил)амино-2- фенилпиперидин или его соли;
(2S, 3S)-2-Фенил-3-(5-(2,2,2-трифтор-1-метил-1-(трифторметил)этил)-2- метоксибензил)аминопиперидин или его соли;
(2S, 3S)-3-[5-[2,2-Дифтор-1(трифторметил)этенил]-2-метоксибензил] амино-2-фенилпиперидин или его соли; и
(2S, 3S)-3-(2-Метокси-5-(2,2,2-трифтор-1-гидрокси-1- (трифторметил)этил)бензил)амино-2-фенилпиперидин или его соли.
Другой предпочтительной группой отдельных соединений настоящего изобретения является следующая группа:
(2S, 3S)-3-[5-Метокси-1-(трифторметил)индан-6-ил)метиламино] -2- фенилпиперидин или его соли;
(2S, 3S)-3-((6-Метокси-1-(трифторметил)-1,2,3,4 - тетрагидронафталин-7-ил)метил)амино-2-фенилпиперидин или его соли; и
(2S, 3S)-3-((2,2-Дифтор-6-метокси-1,2,3,4-тетрагидронафталин -7-ил)метил)амино-2-фенилпиперидин или его соли.
Пиперидиновые соединения формулы (I) настоящего изобретения могут быть получены в соответствии с приведенными ниже схемами реакций.
Если не отмечено особо, то в схемах реакции заместители R, X и Ar принимают определенные выше значения.
Схема A-I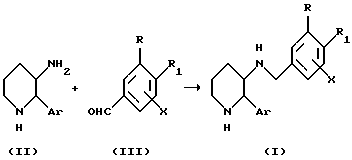
На Схеме A-I показан метод получения соединений формулы (I) путем восстановительного аминирования соединения формулы (III) соединением формулы (II). Восстановление может быть осуществлено с помощью каталитического гидрирования или некоторыми гидридными реагентами в инертном по отношению к реакции растворителе. Каталитическое гидрирование может быть проведено в присутствии металлического катализатора, такого как палладий или никель Ренея. Приемлемые гидридные реагенты включают боргидриды, такие как боргидрид натрия (NaBH4), цианборгидрид натрия (NaBH3CN) и триацетоксиборгидрид натрия (NaB (OAc)3Н), бораны, реагенты на основе алюминия и триалкилсиланы. Приемлемые растворители включают полярные растворители, такие как метанол, этанол, метиленхлорид, тетрагидрофуран (ТГФ), диоксан и этилацетат. Эту реакцию обычно проводят при температуре от - 78oC до температуры кипения растворителя с обратным холодильником, предпочтительно в интервале от 0 до 25oC, в течение от 5 минут до 48 часов, предпочтительно от 0,5 до 12 часов.
С другой стороны, пиперидиновые соединения формулы (I) настоящего изобретения могут быть получены в соответствии со схемой А - II.
Схема A-II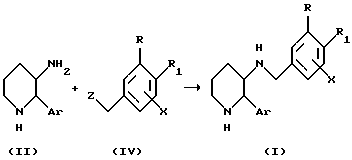
(где Z представляет собой уходящую группу, такую как атом галогена или сульфонатная группа, включая тозилатную и мезилатную).
В соответствии со Схемой A-II соединения формулы (I) настоящего изобретения могут быть получены реакцией соединения формулы (IV) с соединением (II). Соединение (IV) может быть обработано соединением (II) в присутствии основания (например, K2CO3 или Nа2CO3) в полярном растворителе (например, метаноле, этаноле, изопропиловом спирте, ТГФ, диоксане, диметилформамиде (ДМФА) или диметилсульфоксиде (ДМСО). Эту реакцию обычно проводят при температуре от - 78oC до температуры кипения растворителя с обратным холодильником, предпочтительно в интервале от 0 до 25oC, в течение от 5 минут до 48 часов, предпочтительно от 0,5 до 12 часов.
Соединение (IV) может быть получено восстановлением альдегида формулы (III) с последующим превращением гидроксильной группы полученного соединения в уходящую группу Z. Восстановление альдегида (III) может быть осуществлено с использованием различных восстанавливающих агентов в инертном по отношению к реакции растворителе. Приемлемые системы восстанавливающий агент/ растворитель включают боргидрид натрия (NаBH4) в метаноле или этаноле; боргидрид лития (LiBH4) в ТГФ или диэтиловом эфире; литий-алюминий гидрид (LiAlH4), литий- триэтоксиалюминийгидрид (LiAl(OEt)3H), литий-трет- бутоксиалюминийгидрид (LiAl(Obu-t)3Н) или гидрид алюминия (AlH3) в ТГФ или диэтиловом эфире; и изобутилалюминийгидрид (i - BuAlH2) или диизопропилалюминийгидрид (ДИПАЛГ) в дихлорметане, ТГФ или н-гексане. Эту реакцию обычно проводят при температуре от -20 до 25oC в течение от 15 минут до 12 часов. Затем гидроксильную группу полученного соединения превращают в уходящую группу Z (например, в галоген, такой как хлор, бром, иод или фтор, или сульфонатную группу, включая тозилатную или мезилатную). Превращение гидроксильной группы в уходящую группу Z может быть осуществлено в соответствии с методами, известными специалистам в данной области. Например, когда Z представляет собой сульфонатную группу, такую как тозилатная или мезилатная, гидроксильное соединение вводят в реакцию с сульфонатом в присутствии пиридина или триэтиламина в дихлорметане. Если Z представляет собой галоген, такой как хлор или бром, то гидроксильное соединение может быть обработано SOX2 (X представляет собой Cl или Br) в присутствии пиридина.
Соединения формулы (III) могут быть получены в соответствии с приведенной ниже схемой B-I.
Схема B-I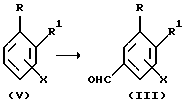
Соединения формулы (III) могут быть получены прямым или непрямым формилированием соединения формулы (V). Для этого может быть использован любой из известных в данной области методов введения формильной группы в бензольное кольцо. Например, прямое формилирование может быть осуществлено обработкой соединения (V) приемлемым формилирующим агентом в присутствии подходящего катализатора. Приемлемыми системами формилирующий агент/катализатор являются дихлорметил - метиловый эфир/хлорид титана (IV) (Cl2CHOCH3/TiCl4), трифторуксусная кислота (CF3CO2H) /гексаметилентетрамин (условия реакции Дуффа) и хлорокись фосфора (POCl3)/ДМФА (условия реакции Вильсмайера). Непрямое формилирование может быть осуществлено путем галогенирования соединения (V), замещением атома галогена на цианогруппу с последующим восстановлением полученного циано-замещенного соединения. Галогенирование может быть осуществлено по методике, описанной в статье G.A.Olah et al. J.:.Org. Chem., 58, 3194(1993). Замещение атома галогена на цианогруппу можно проводить методами, приведенными в публикациях D. М. Tschaem et al. Synth.CommLin. , 24, 887 (1994), K.Takagi et al., Bull.Chem.Soc.Jpn., 64, 1118 (1991). Восстановление может быть осуществлено в присутствии диизопропилалюминийгидрида (ДИПАЛГ) в дихлорметане или никеля Ренея в муравьиной кислоте.
Исходные вещества формулы (V) являются известными и коммерчески доступными соединениями или могут быть синтезированы известными методами. Например, соединение формулы (V), где X представляет собой алкоксигруппу, могут быть получены путем О-алкилирования соответствующих соединений (V), где X представляет собой гидроксигруппу, в присутствии основания (например, NaH или KH) в подходящем растворителе (например, ДМСО, ДМФА и ТГФ).
Соединение (V) также может быть получено другими методами, которые описаны в следующих литературных ссылках:
(А) трифторметилирование, J. Am. Chem. Soc. Ill, 393-395 (1989);
(В) трет-алкилирование, Angew. Chem. lnt. Ed. Engl. , 19, N. 11900-901 (1980);
(С) хемоселективное и позиционно-специфическое метилирование трет-алкилгалогенидов метилтитаном (IV) Angew.Chem.Jnt Ed. Engl., 19, N. 11901-902 (1980), и фторирование кетонов, Organic Reaction (1988), 35.
Кроме того, заместитель R в соединении формулы (III) может быть заменен на любой желаемый заместитель R" (например, CF2CF3 или CF2CH3) по методикам, известным квалифицированным в данной области специалистам, например, в соответствии со следующей схемой B-II.
Схема B-II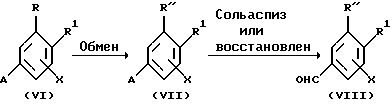
А=ацеталь, СN, еtс...
На схеме B-II исходные соединения формулы (VI) представляют собой известные соединения, которые могут быть получены в соответствии с методиками, описанными, например, в публикации Collect. Czech. Chem. Commun., 52, 980 (1987), или в публикации Bull.Chem.Soc.Jpn., 51, 2435 (1978).
Например, соединение формулы (VI), где А представляет собой CN и заместитель R представляет собой алкилкарбонил (см., (.Collect.Czech.Chem.,Commun. , 52, 980 (1987)), может быть подвергнуто тиокетализации, за которым следует замещение, с получением соединения формулы (VII) (см., J.Org.Chem., 51, 3508 (1986)). Соединение формулы (VI), где А представляет собой ацеталь и заместитель R представляет собой атом галогена (см., Bull.Chem.Soc; Jpn., 51, 2435 (1978)), может быть подвергнуто алкилированию с получением соединения формулы (VII) (см., Synthetic Comm. 18, 965 (1988)).
Затем соединение формулы (VII) может быть подвергнуто сольволизу или восстановлению в подходящих реакционных условиях с получением соединения формулы (VIII), где заместитель R превращен в заместитель R" (например, CF2CF3 CF2CH3) (см., J.Org.Chem., 24, 627 (1959) и Protective group in organic synthesis, John Wiley and sons, inc., 180 и 191 (1991)).
С другой стороны, соединение формулы (1) может быть получено в соответствии с приведенной ниже Схемой A-III.

Схема A-III иллюстрирует получение соединений формулы (I). Из этой схемы видно, что N-защищенное соединение формулы (IX) (Ar представляет собой фенил или аналогичную группу) может быть превращено при обработке (трет-BuOCO)2O(Boc2O) в присутствии основания, такого как бикарбонат натрия (NaHCO3) или триэтиламин (Et3N), в соединение формулы (Х). Соединение (Х) подвергают гидрогенолизу с получением соединения формулы (XI) (где заместитель Ar = фенил). Другой способ введения N - защитной группы в соединение формулы (IX) может быть осуществлен путем обработки карбобензоксихлоридом (Cbz - Cl) в присутствии основания, такого как бикарбонат натрия (NaHCO3) или триэтиламин (Et3N), когда заместитель Ar представляет собой фенил. Гидрогенолиз можно провести при обработке H2 или формиатом аммония (HCO2NH4) в присутствии металлического катализатора, такого как палладий на угле (например, 20%-ный палладий на угле) в подходящем растворителе. Затем соединение (XI) подвергают восстановительному аминированию, как это показано на Схеме A-I. Соединение (XII) может быть превращено в соединение формулы (I) обработкой кислотным катализатором, таким как хлористый водород (HCl) в метаноле, конц.соляная кислота в этилацетате или CF3CO2H в дихлорметане.
Соединения формулы (I) и промежуточные соединения, представленные в приведенных выше реакционных схемах, могут быть выделены и очищены с помощью обычных методик, таких как перекристаллизация или хроматографическое разделение.
Так как пиперидиновые соединения настоящего изобретения имеют по меньшей мере два асимметричных центра, они способны существовать в различных стереоизомерных формах или конфигурациях. Следовательно, соединения могут существовать в разделенных (+)- и (-)- оптически активных формах, а также в виде их смесей. Настоящее изобретение включает все эти формы. Индивидуальные изомеры могут быть получены известными способами, такими как оптическое разделение, оптически селективные реакции или хроматографическое разделение на стадии конечного продукта или его промежуточных соединений.
Поскольку пиперидиновые соединения настоящего изобретения являются основными соединениями, все они обладают способностью образовывать большое число различных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемы для введения животным, часто на практике желательно первоначально выделить пиперидиновое основание из реакционной смеси в виде фармацевтически неприемлемой соли, затем просто превратить в свободное основание путем обработки щелочным реагентом и после этого превратить свободное основание в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли пиперидиновых основных соединений настоящего изобретения легко образуются при обработке основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в водном растворителе или в приемлемом органическом растворителе, таком как метанол или этанол. При осторожном упаривании растворителя легко выделяют желаемую соль. Кислоты, которые используют при получении фармацевтически приемлемых кислотно-аддитивных солей названных выше пиперидиновых основных соединений, представляют собой кислоты, которые дают нетоксичные кислотно-аддитивные соли, например соли, содержащие фармацевтически приемлемые анионы, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат или бисульфат, фосфат или кислый фосфат, ацетат, лактат, цитрат или кислый цитрат, тартрат или би-тартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, п - толуолсульфонат и памоат (то есть, 1,1'-метилен-бис-(2-гидрокси- 3-нафтоат).
Пиперидиновые соединения настоящего изобретения, которые также содержат кислотные группы, способны образовывать основные соли с различными фармацевтически приемлемыми катионами. Примерами таких солей являются соли щелочных или щелочноземельных металлов и в особенности соли натрия и калия. Такие соли получают по обычным методикам.
Химические основания, которые используют в качестве реагентов при получении фармацевтически приемлемых основных солей настоящего изобретения, представляют собой основания, которые дают нетоксичные основные соли с описанными здесь кислыми пиперидиновыми производными. Эти нетоксичные основные соли включают соли, полученные из таких фармацевтически приемлемых катионов, как натрий, калий, кальций, магний и др. Такие соли могут быть получены при обработке упомянутых выше кислых пиперидиновых соединений водными растворами, содержащими необходимый фармацевтически приемлемый катион, с последующим упариванием полученного раствора досуха, предпочтительно при пониженном давлении. С другой стороны, они могут быть приготовлены при смешении растворов в низших спиртах кислых соединений и алкоксида желаемого щелочного металла и упаривании получаемого раствора досуха способом, аналогичным описанному выше. В любом случае предпочтительно использование стехиометрических количеств реагентов для того, чтобы обеспечить полноту протекания реакции и максимальный выход конечного продукта.
Активные пиперидиновые соединения настоящего изобретения проявляют значительную активность в связывании рецептора вещества P и, следовательно, имеют значение при лечении большого спектра клинических состояний, которые характеризуются присутствием избытка указанного вещества Р. Такие состояния включают желудочно-кишечные расстройства, расстройства центральной нервной системы, воспалительные заболевания, рвоту, недержание мочи, боль, мигрень или развитие кровеносных сосудов у млекопитающего, в особенности у человека. Для лечения рвоты такие соединения могут быть предпочтительно использованы в сочетании с антагонистами 5HT3 -рецепторов.
Активные пиперидиновые соединения формулы (1) настоящего изобретения могут быть введены млекопитающему перорально, парентерально или локально. В общем случае эти соединения наиболее предпочтительно вводить человеку в дозах, лежащих в интервале приблизительно от 0,3 до 750 мг в день, хотя обязательно будут существовать варианты в зависимости от веса и состояния пациента и выбранного конкретного способа введения. Однако уровень доз, который находится в интервале приблизительно от 0,06 до 2 мг на кг веса в день, является наиболее желательным для применения. Тем не менее, все равно могут иметь место различные изменения в зависимости от вида животного, нуждающегося в лечении, и его индивидуальной реакции на указанное медицинское средство, а также от типа выбранной фармацевтической рецептуры и временного периода и интервала, в течение которых вводят лекарство. В некоторых случаях могут быть приемлемы уровни доз, ниже нижнего предела названного интервала, хотя в других случаях могут быть использованы еще более высокие дозы без проявления каких - либо побочных эффектов, при условии, что такие более высокие дозы вначале поделены на несколько маленьких доз для применения в течение всего дня.
Соединения настоящего изобретения могут быть введены отдельно или в сочетании с фармацевтически приемлемыми носителями или разбавителями любым из названных выше способов введения, и такое введение может быть осуществлено в виде единственной дозы или в виде кратных доз. Более конкретно, новые терапевтические агенты настоящего изобретения могут быть введены в виде целого ряда различных лекарственных форм, то есть, они могут быть объединены с различными фармацевтически приемлемыми инертными носителями в виде таблеток, капсул, лепешек, пастилок, твердых леденцов, порошков, аэрозолей, кремов, мазей, свечей, желе, гелей, паст, лосьонов, протираний, водных суспензий, растворов для инъекций, эликсиров, сиропов и т.д. К таким носителям относятся твердые разбавители или наполнители, стерильная водная среда и различные нетоксичные органические растворители и т. д. Кроме того, пероральные фармацевтические композиции могут быть соответствующим образом подслащены и/или могут содержать добавки, корригирующие вкус и запах. В общем случае такие лекарственные формы содержат терапевтически эффективные количества соединений настоящего соединения в концентрации приблизительно от 5,0 до 70 мас.%.
Для перорального применения могут быть использованы таблетки, содержащие различные наполнители, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, дикальций фосфат и глицин, вместе с различными диспергаторами, такими как крахмал, предпочтительно кукурузный, картофельный крахмал и крахмал из тапиоки, альгиновая кислота и некоторые комплексные силикаты, а также со связующими веществами для гранулирования типа поливинилпирролидона, сахарозы, желатина и аравийской камеди. Кроме того, очень часто при таблетировании полезно использовать смазывающие агенты, такие как стеарат магния, лаурилсульфонат натрия и тальк. Твердые композиции аналогичного типа могут быть использованы в качестве наполнителей желатиновых капсул; предпочтительные материалы в этом случае содержат лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Когда для перорального применения необходимы водные растворы и/или эликсиры, активный ингредиент может быть смешан с различными подслащивающими или корригирующими вкус и запах агентами, красящими веществами или красителями и, если это желательно, эмульгирующими и/или суспендирующими агентами, а также с такими разбавителями как вода, этанол, пропиленгликоль, глицерин и их различные сочетания.
Для парентерального применения могут быть использованы растворы соединения настоящего изобретения или в кунжутном или арахисовом масле, или в водном пропиленгликоле. Водные растворы должны, если это необходимо, иметь определенное значение pH (предпочтительно pH > 8), а разбавитель должен быть изотоничным. Такие водные растворы приемлемы для внутривенных инъекций. Масляные растворы приемлемы для межсуставных, внутримышечных и подкожных инъекций. Получение всех этих растворов в стерильных условиях может быть легко осуществлено по стандартным фармацевтическим методикам, хорошо известным квалифицированным в данной области специалистам. Кроме того, при лечении воспалительных состояний кожи также возможно местное применение соединений настоящего изобретения, которые могут быть получены в виде кремов, желе, гелей, паст, мазей и других подобных рецептур, по стандартным фармацевтическим технологиям.
Активность соединений настоящего изобретения в качестве антагонистов вещества P определяют по их способности ингибировать связывание вещества P к их рецепторным сайтам в CHO - клетках, которые обнаруживают NK1 рецептор или клетки IM-9, с использованием радиоактивных лигандов. Активность описанных пиперидиновых соединений как антагонистов вещества P оценивают с помощью стандартной методики оценки, описанной М.A.Cascieri et al.(The.Journal of Immunology, 133, 3260 (1984)). Этот метод по существу включает определение концентрации индивидуального соединения, требуемой для уменьшения на 50 % количества лигандов вещества P с радиоактивной меткой у их рецепторных сайтов в указанных изолированных тканях коровы или клетках IM-9, в результате чего получают характеристичные значения IC50 для каждого испытуемого соединения. Если говорить более конкретно, то определяют ингибирование [3H]SP-связывания IM-9 клетками человека соединениями в буфере для оценки [50 мМ Tris-HCl(pH 7,4), 1 мМ MnCl2, 0,02% бычий сывороточный альбумин, бацитрацин (40 мкг/мл), лейпептин (4 мкг/мл), химостатин (2 мкг/мл) и фосфорамидон (30 мкг/мл)] . Реакцию инициируют путем добавления клеток в буфер для оценки, содержащий 0,56 нМ [3H]SP и различные концентрации соединений (общий объем 0,5 мл), и инкубируют в течение 120 минут при 4oC. Инкубирование останавливают путем фильтрации на фильтрах GF/В (предварительно погруженных в 0,1%-ный полиэтиленимин на 2 часа). Неспицифическое связывание определяют по радиоактивности, оставшейся в присутствии 1 мкМ SP. Фильтры помещают в трубки и подсчитывают радиоактивность с помощью жидкостного сцинтилляционного счетчика.
Неблагоприятное влияние на связующее сродство Ca2+ - каналов определяют в исследовании по связыванию верапамила в препаратах сердечных мембран крыс. Более конкретно, связывание верапамила осуществляют в соответствии с описанием Reynolds et al.,(.J.Pharmacol.Exp.Ther. 237, 731, 1986). Инкубацию инициируют добавлением ткани в трубки, содержащие 0,25 нМ [3H] десметоксиверапамила и различные концентрации соединений (общий объем, 1 мл). Неспецифическое связывание определяют по связыванию радиолигандов, оставшихся в присутствии 3-10 мкМ метоксиверапамила. Активность соединений настоящего изобретения в отношении расстройств центральной нервной системы определяют в тесте на поcтукивание лап, индуцированном [SAR9, Met (O2)11] - веществом P, у карликовой песчанки. Карликовых песчанок подвергают легкой анестезии эфиром и раскрывают черепную поверхность. Непосредственно в боковой желудочек иглой 25 размера, введенной на 3,5 см ниже ламбды, вводят [SAR9, Met (O2)11] - вещество P или растворитель (5 мкл). После инъекции песчанок помещают в 2-литровый стакан и наблюдают за повторяющимся постукиванием задних лап. В этом методе испытаний проверены некоторые соединения, полученные в приведенных ниже Примерах. В результате установлено, что соединения настоящего изобретения обладают хорошей антагонистической активностью в отношении вещества P, в частности хорошей активностью против заболеваний ЦНС, при благоприятных метаболитических свойствах. Более конкретно, при сравнении, например, трифторметил - и гексафторизопропилбензил - аминопиперидиновых соединений (Примеры 3 и 5, соответственно) с соответствующими незамещенными галогеном соединениями, найдено, что галогензамещенные соединения проявляют неожиданно повышенную активность против заболеваний ЦНС.
Период полувыведения соединений настоящего изобретения определяют в препаратах микросом печени человека. То есть, соединение (1 мкМ) инкубируют с объединенными микросомами печени человека (2,0 мг/мл), НАДФ (NADP) (1,3 мМ), НАДВ (NADH) (0,93 мМ), глюкоза-6- фосфатом (3,3 мМ), MgCl2 (3,3 мМ) и глюкоза-6-фосфатдегидрогеназой (8 единиц на мл) в суммарном объеме 1,2 мл 100 мМ калийфосфатного буфера, pH 7,4. При различных временных точках (0,5, 10, 30 и 60 минут) 100 мкл-пробы добавляют к ацетонитрильному раствору (1,0 мл), который содержит внутренний стандарт. Осажденный белок отделяют в центрифуге (3000 x g, 5 минут). Надосадочную жидкость анализируют с помощью сочетания ЖХ - МС. Сочетание ЖХ - МС состоит из системы высокоэффективной жидкостной хроматографии Hewlett Packard НР1090 и Sciex API-III. Образцы (10 мкл) впрыскивают с помощью автопробоотборника в колонку Hewlett Packard ODS - Hypersil (2,1 х 20 мм). Подвижная фаза состоит из 80 % ацетонитрила в 10 мМ ацетате аммония. Результаты измерений API - III анализируют с помощью детектора для мониторинга составных реакций (MRM).
Пример 1
Получение дигидрохлорида (2S, 3S)-3-(2-фтор-5-(трифторметил) бензил)амино-2-фенилпиперидина (Соединение 2)
(I) Дигидрохлорид (2S, 3S)-2-фенилпиперидин-3-амина (Соединение 1)
Это соединение получают в соответствии с методиками, описанными в публикации ЕР 558156.
(II) Дигидрохлорид (2S,2S)-3-(2-фтор-5-(трифторметил)бензил)амино- 2-фенилпиперидина (Соединение 2)
К суспензии Соединения 1 (150 мг, 0,60 ммоля) и 2-фтор-5- (трифторметил)бензальдегида (116 мг, 0,60 ммоля) в сухом CH2Cl2 (6 мл) при перемешивании в атмосфере азота и при комнатной температуре добавляют порциями NaBH(OAc)3 (179 мг, 0,84 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение 6,5 часов. Смесь подщелачивают насыщенным водным раствором NaHCO3, экстрагируют CH2Cl2, сушат MgSO4 и концентрируют, получают сырой (2S, 3S)-3-(2-фтор-5- (трифторметил) бензил)амино-2-фенилпиперидин в виде бесцветного масла. Масло очищают с помощью хроматографии, получают чистый (2S, 3S) -3-(2-хлор-5-(трифторметил)бензил)амино-2-фенилпиперидин в виде бесцветного масла (75 мг). Полученный продукт разбавляют этилацетатом, добавляют HCl-МеОН и концентрируют. Белое твердое вещество перекристаллизовывают из смеси МеОН-эфир, получают Соединение 2 в виде белого кристаллического вещества (67 мг, 26,3%).
Т.пл.: 195-203oC.
Спектр 1H-ЯМР (CDCl3, свободное основание): 7,42 - 7,22 (м, 7H); 6,99 (т, J= 8,8 Гц, 1H); 3,90 (д, J= 2,2 Гц, 1H); 3,61 (д, J=15,0 Гц, 1H); 3,46 (д, J = 15,0 Гц, 1H); 3,28 - 3,22 (м, 1H); 2,86 - 2,76 (м, 2H); 2,08-2,03 (м,1H); 1,95-1,78 (м, 1H); 1,69-1,57 (м,1H); 1,48-1,44 (м, 1H).
Пример 2
Получение дигидрохлорида (2S, 3S)-3-(2-хлор-5-(трифторметил)бензил)амино-2-фенилпиперидина (Соединение 3)
К суспензии Соединения 1 (150 мг, 0,60 ммоля) и 2-фтор-5-(трифторметил)бензальдегида (126 мг, 0,60 ммоля) в сухом CH2Cl2 (6 мл) при перемешивании в атмосфере азота и при комнатной температуре добавляют порциями NaBH(OAc)3 (179 мг, 0,84 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение 17 часов. Смесь подщелачивают насыщенным водным раствором NaHCO3, экстрагируют CH2Cl2, сушат MgSO4 и концентрируют, получают сырой (2S, 3S)-3-(2- хлор-5-(трифторметил)бензил) амино-2-фенилпиперидин в виде бесцветного масла. Полученное масло очищают с помощью хроматографии, получают чистый (2S, 3S)-3-(2-хлор-5-(трифторметил)бензил) амино-2-фенилпиперидин в виде бесцветного масла (135 мг). Продукт разбавляют этилацетатом, добавляют HCl-МеОН и концентрируют. Белое твердое вещество перекристаллизовывают из MeOH, получают Соединение 3 в виде белого кристаллического вещества (64 мг, 24,1%).
Т.пл.:200- 210oC.
Спектр 1H-ЯМР (CDCl3, свободное основание): 7,40-7,22 (м, 8H): 3,91 (д, J=2,2 Гц, 1H); 3,66 (д,J= 15,0 Гц, 1H); 3,50 (д,J=15,0 Гц,1H); 3,29-3,23 (м, 1H); 2,86-2,76 (м,2H); 2,10-2,05 (м, 1H); 1,97 -1,80(м,1H); 1,70-1,58(м,1H): 1,51-1,45(м,1H).
Пример 3
Получение дигидрохлорида (2S,3S)-3-(2-метокси-5-(трифторметил)бензил)амино-2-фенилпиперидина (Соединение 5)
(i) 2-Метокси-5-(трифторметил)бензальдегид (Соединение 4)
К охлаждаемому льдом раствору NaOMe (904 мг, 4,68 ммоля) при перемешивании добавляют порциями 2-фтор-5-(трифторметил) бензальдегид (500 мг, 2,60 ммоля). Использованную капельную воронку промывают ТГФ. Полученную суспензию перемешивают при комнатной температуре в течение 5 часов. Реакционную смесь нейтрализуют уксусной кислотой (0,3 мл, 5,0 ммоля), растворитель упаривают. К твердому остатку добавляют воду и смесь экстрагируют CH2Cl2. Объединенные дихлорметановые экстракты промывают насыщенным водным раствором NaHCO3, сушат MgSO4 и концентрируют, получают сырое Соединение 4 в виде белого твердого вещества. Продукт очищают препаративной тонкослойной хроматографией, получают Соединение 4 в виде белого кристаллического вещества (363 мг, 68,4%).
Спектр 1H-ЯМР (CDCl3): 10,47 (с, 1H); 8,11(д, J=2,2Гц, 1H); 7,80 (дд, J= 8,8; 2,2 Гц, 1H): 7,10 (д, J=8,8; 2,2 Гц, 1H); 4,01 (с, 3Н).
(ii) Дигидрохлорид (2S, 3S)-3-(2-метокси-5-(трифторметил) бензил)амино-2-фенилпиперидина (Соединение 5)
К суспензии Соединения 1 (150 мг, 0,60 ммоля) и Соединения 4 (123 мг, 0,60 ммоля) в сухом CH2Cl2 (6 мл) в атмосфере азота при перемешивании и при комнатной температуре добавляют порциями NaBH(OAc)3 (179 мг, 0,84 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение 3,5 часов. Смесь подщелачивают насыщенным водным раствором NaHCO3, экстрагируют CH2Cl2, сушат MgSO4 и концентрируют, получают сырой (2S, 3S)-3-(2- метокси-5-(трифторметил)бензил)амино-2-фенилпиперидин в виде бесцветного масла. Полученное масло очищают хроматографией, выделяют чистый (2S, 3S)-3-(2-метокси-5-(трифторметил)бензил) амино-2-фенилпиперидин в виде бесцветного масла (95 мг). Продукт разбавляют этилацетатом, добавляют HCl-MeOH и концентрируют. Белое твердое вещество перекристаллизовывают из смеси MeOH-эфир, получают Соединение 5 в виде белого кристаллического вещества (85 мг, 32,4 %).
Т.пл.: 228-233oC.
Спектр 1H-ЯМР (CDCl3, свободное основание): 7,40 (т, J = 8,8 Гц, 1H); 7,34-7,23 (м, 6H); 6,71 (д, J= 8,8 Гц, 1H); 3,92 (д, J= 2,2 Гц, 1H); 3,66 (д, J = 14,3 Гц, 1H); 3,52 (с, 3H); 3,42 (д, J = 14,3 Гц, 1H); 3,33 - 3,29 (м, 1H): 2,87 - 2,77 (м, 2H); 2,12-1,90 (м, 2H); 1,66-1,56 (м, 1H); 1,48-1,43 (м, 1H).
Пример 4
Получение дигидрохлорида (2S, 3S)-3-(2-фенокси-5- (трифторметил)бензил)амино-2-фенилпиперидина (Соединение 7)
(i) 2-Фенокси-5-трифторметил)бензальдегид (Соединение 6)
К раствору 2-хлор-5-(трифторметил)бензальдегида (500 мг, 2,40 ммоля) в ДМФА (5 мл) при перемешивании добавляют фенол (226 мг, 2,40 ммоля), K2CO3 (663 мг, 4,79 ммоля). Реакционную суспензию перемешивают при комнатной температуре в течение 1 часа, затем при 80oC в течение 1,5 часов. Реакционную смесь нейтрализуют уксусной кислотой (0,5 мл), растворитель упаривают. К твердому остатку добавляют воду и смесь экстрагируют CH2Cl2. Объединенные дихлорметановые экстракты промывают насыщенным водным раствором NaHCO3, сушат MgSO4 и концентрируют, получают сырое Соединение 6 в виде желтого масла. Масло очищают хроматографией, получают Соединение 6 в виде желтого масла (466 мг, 72,9 %).
Спектр 1H-ЯМР (CDCl3): 10,58 (с, 1H); 8,21(д, J=2,6 Гц,1H): 7,70 (дд, J = 8,8; 2,6 Гц, 1H); 7,50 - 7,42 (м, 2H); 7,31-7,25 (м, 1H); 7,16- 7,10 (м, 2H), 6,93 (д, J=8,8 Гц, 1H).
(II) Дигидрохлорид (2S, 3S)-3-(2-фенокси-5-(трифторметил)бензил)амино-2-фенилпиперидина (Соединение 7)
К суспензии Соединения 1 (150 мг, 0,60 ммоля) и Соединения 6 (160 мг, 0,60 ммоля) в сухом CH2Cl2 (6 мл) в атмосфере азота при перемешивании и при комнатной температуре добавляют порциями NaBH(OAc)3 (179 мг, 0,84 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Смесь подщелачивают насыщенным водным раствором NaHCO3, экстрагируют CH2Cl2, сушат MgSO4 и концентрируют, получают сырой (2S, 3S)-3-(2- фенокси-5-(трифторметил) бензил)амино-2-фенилпиперидин в виде желтого масла. Его очищают хроматографией, получают чистый (2S, 3S)-3-(2-фенокси-5-(трифторметил) бензил) амино-2- фенилпиперидин в виде желтого масла (135 мг). Продукт разбавляют этилацетатом, добавляют HCl-МеОН и концентрируют. Белое твердое вещество перекристаллизовывают из смеси МеОН - эфир, получают Соединение 7 в виде белого кристаллического вещества (108 мг, 36,0%).
Т.пл.: 190-197oC.
Спектр 1-ЯМР (CDCl3, свободное основание): 7,39-7,18 (м, 8H); 7,15- 7,09 (м, 2H); 6,79 - 6,71 (м,3H); 3,90 (д, J = 2,2 Гц, 1H); 3,66 (д, J = 14,7 Гц, 1H), 3,51 (д, J= 14,7 Гц, 1H); 3,28 - 3,23 (м, 1H); 2,90 (д, J = 2,6 Гц, 1H); 2,66 (дт, J = 12,1, 2,9 Гц, 1H); 2,11- 2,06 (м, 1H); 1,96- 1,81 (м, 1H); 1,69-1,56 (м, 1H): 1,46-1,41 (м, 1H).
Пример 5
Получение дигидрохлорида (2S, 3S)-2-фенил-3-(5-(2,2, -2 трифтор-1-(трифторметил)этил)-2-метоксибензил) аминопиперидина(Соединение 15)
(I) 4-(2,2,2-Трифтор-1-(трифторметил)этил)анизол (Соединение 8)
Это соединение получают в соответствии с методиками, описанными в J.Am. Chem.Soc., 820 (1972)
(II) 5-(2,2,2-Трифтор-1-(трифторметил)этил)-2-метоксибензальдегид (Соединение 9)
К охлаждаемому льдом раствору Соединения 8 (650 мг, 2,5 ммоля) в сухом CH2Cl2 (15 мл) при перемешивании добавляют чистый TiCl4 (950 мг, 5,0 ммоля), затем добавляют Cl2CHOMe (600 мг, 5,0 ммоля). По окончании добавления смесь перемешивают при комнатной температуре в течение 5 часов, выливают в H2O (60 мл) и экстрагируют CH2Cl2. Объединенные экстракты сушат (Na2SO4) и концентрируют в вакууме, получают желтое масло, которое очищают колоночной хроматографией на силикагеле, получают Соединение 9 (650 мг, 90%).
Спектр 1H-ЯМР (CDCl3): 10,47(с,1H); 7,86-7,08 (м,3H); 4,05 (геп, J = 8 Гц, 1H); 3,98 (с, 3H).
(III)-3-(2S, 3S)-3-(2-Метоксибензил)амино-2-фенилпиперидин (Соединение 10.)
Это соединение получают в соответствии с методиками, описанными в публикации WO 93-01170.
(IV) (2S, 3S)-1 трет-Бутоксикарбонил-3-(2-метоксибензил) аминопиперидин (Соединение 11)
К охлаждаемой льдом смеси Соединения 10 (10 г, 27 ммолей), 3M водного NaOH (36 мл, 110 ммолей) и трет-BuOH (15 мл) при перемешивании добавляют (т. - BuOCO)2 (7,4 г, 34 ммоль). После перемешивания при комнатной температуре в течение ночи смесь экстрагируют AcOEt. Объединенные этилацетатные слои промывают H2O и насыщенным водным раствором NaCI, сушат (Na2SO4) и концентрируют в вакууме, получают Соединение 11 (11 г, выход количественный) в виде бледно-желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,58 (ушир. д, J = 7,3 Гц, 2H); 7,36 -7,16(м,5H); 6,89 (ддд, J= 7,5, 7,5, 1,1 Гц, 1H); 6,81 (дд, J=8,4, 0,8 Гц, 1H); 5,47 (ушир. с, 1H); 3,96 (дм, J = 13,4 Гц, 1H); 3,87 (д, J = 13,6 Гц, 1H); 3,79 (д, J = 13,6 Гц, 1H); 3,70 (с, 3H); 3,10- 2,99 (м, 1H): 2,94 (дд, J = 12,5, 3,4 Гц, 1H); 1,87-1,74 (м, 2H); 1,74-1,40 (м, 3H); 1,41 (с, 9H).
Это соединение используют на следующей стадии без дополнительной очистки.
(V) (2S, 3S)- 3-Амино-1-трет-бутоксикарбонил-2-фенилпиперидин (Соединение 12)
Смесь Соединения 11 (11 г), 20%- ного Pd (ОН)2/C(3,1 г) и МеОН (90 мл) перемешивают в атмосфере H2 при комнатной температуре в течение ночи. После добавления дополнительного количества 20%-ного Pd (ОН)2/С (0,55 г) перемешивание в атмосфере H2 при комнатной температуре продолжают в течение трех дней. Катализатор отфильтровывают с использованием целита и тщательно промывают МеОН. Объединенные метанольные фильтраты и промывные растворы концентрируют в вакууме, получают сырое Соединение 9 (8,6 г, выход количественный).
Соединение 9 растворяют в EtOH (20 мл) и затем к полученному раствору при комнатной температуре в одну порцию добавляют теплый раствор фумаровой кислоты (1,6 г, 13,5 ммоля) в EtOH (20 мл). Выпадающие в осадок кристаллы отфильтровывают, промывают ледяным EtOH и сушат в вакууме при 50oC, получают полуфумарат (2S, 3S)-3-амино-1-(трет-бутоксикарбонил)-2-фенилпиперидина (6,1 г, 68%) в виде белых коротких иголок.
После охлаждения льдом к суспензии полуфумарата (1,2 г, 3,7 ммоля) в H2O добавляют 20%-ный водный раствор NaOH до получения основной смеси. Затем смесь экстрагируют AcOEt. Объединенные этилацетатные экстракты промывают насыщенным водным раствором NaCl, сушат (Na2SO4) и концентрируют в вакууме, получают Соединение 12 (0,95 г, 93%).
Спектр 1H-ЯМР (CDCl3): 7,47-7,39 (м, 2H); 7,37 -7,23 (м, 5H); 5,19 (ушир. д, J = 6,2 Гц, 1H): 4,00 (дм, J = 13,0 Гц, 1H); 3,25 - 3,05 (м, 2H); 1,94-1,83 (м, 1H); 1,83-1,56 (м,4H); 1,36 (с,9H); 1,32 (ушир.с, 2H).
(VI) (2S, 3S)-1-трет-Бутоксикарбонил-2-фенил-3- (5-(2,2,2-трифтор-1-(трифторметил)этил)-2-метоксибензил) аминопиперидин (Соединение 13)
К охлаждаемому льдом раствору Соединения 12 (100 мг, 0,3 ммоля) и Соединения 9 (100 мг, 0,3 ммоля) в сухом CH2Cl2 (10 мл) при перемешивании в одну порцию добавляют NaBH(ОАс)3 (210 мг, 1 ммоль). Затем смесь перемешивают при комнатной температуре в течение 20 часов. Смесь выливают в водный раствор NaHCO3, экстрагируют CH2Cl2. Объединенные экстракты сушат (Na2SO4) и концентрируют в вакууме, получают Соединение 13 в виде желтого масла (170 мг).
Спектр 1H-ЯМР (CDCl3): 7,61 - 6,82 (м, 8H): 5,45 (ушир., 1H); 4,08 - 3,65 (м, 3H); 3,74 (с, 3H); 3,10-2,96 (м, 6H); 1,90-1,20 (м,4H); 1,39 (с, 9H).
Полученное соединение используют на следующей стадии без дополнительной очистки.
(VII)(2S, 3S)-2-Фенил-3-(5-(2,2,2-трифтор-1-(трифторметил) этил)-2-меnоксибензил)аминопиперидин (Соединение 14)
К раствору Соединения 13 (170 мг) в AcOEt (5 мл) добавляют конц. HCl (1 мл). Смесь перемешивают при комнатной температуре в течение 45 минут, выливают в водный раствор NaHCO3, экстрагируют CH2Cl2. Объединенные экстракты сушат (Na2SO4) и концентрируют в вакууме, получают Соединение 14(160 мг) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,36 - 6,68 (м, 8H); 3,95 - 3,26 (М, 5H); 3,55 (с, 3H); 2,89-1,40 (м, 6H).
Масс - спектр: 446 (M+)
Полученное соединение используют на следующей стадии без дополнительной очистки.
(VIII) Дигидрохлорид (2S, 3S)-2-Фенил-3-(5-(2,2,2-трифтор-1- (трифторметил)этил)-2-метоксибензил) аминопиперидин (Соединение 15)
К раствору соединения 14 (160 мг) с CH2Cl2 (10 мл) добавляют избыток 10%-ной смеси HCl-MeOH (6 мл). После упаривания растворителя в вакууме остаток перекристаллизовывают из изофталевой кислоты, получают Соединение 15 (130 мг, 83%, три ступени) в виде бесцветных кристаллов.
Т.пл.: 290-294oC.
Пример 6
Получение дигидрохлорида (2S, 3S)-2-фенил-3-(5-(1,2,2,2- тетрафтор-1-(трифторметил)этил)-2-метоксибензил)аминопиперидина (Соединение 18)
(I) 4-(1,2,2-Тетрафтор-1-(трифторметил)этил)анизол (Соединение 16)
Это соединение получают в соответствии с методиками, описанными в Nippon Kagaku Kaishi, 2351 (1973)
(II) (5-(1,2,2,2-Тетрафтор-1-(трифторметил)этил)- 2-метоксибензилальдегид (Соединение 17)
Это соединение получают из Соединения 16 по методике, аналогичной методике получения Соединения 9.
Спектр 1H-ЯМР (CDCl3): 10,49 (с, 1H); 8,13- 7,12 (м, ЗН): 4,02 (с, 3H).
(III) Дигидрохлорид (2S, 3S)-2-фенил-3-(5-(1,2,2,2-тетрафтор-1-(трифторметил)этил)-2-метоксибензил) аминопиперидина (Соединение 18)
Это соединение получают из Соединения 1 и Соединения 17 по методике, аналогичной методике получения Соединения 2.
Т.пл.: 265-270oC
Спектр 1H-ЯМР (CDCl3, свободное основание): 7,44-6,72 (м, 8H); 3,96-2,75 (м, 6H); 3,53 (с, 3H); 2,89- 1,40 (м, 4H).
Масс - спектр (свободное основание): 464 (М+).
Пример 7
Получение дигидрохлорида (2S, 3S)-3-(5-(1,1-дифторэтил-2- (трифторметокси)бензил)амино-2-фенилпиперидина (Соединение 28)
(I) 3-Йод-4-(трифторметокси)бензальдегид (Соединение 19)
Трифторметансульфокислоту (18,6 мл, 0,21 ммоля) по каплям при охлаждении льдом в атмосфере N2 добавляют к N - йодсукцинимиду (10,4 г, 46,3 ммоля). К полученной темно-синей смеси по каплям и при охлаждении льдом добавляют 4-(трифторметокси) бензальдегид (4,0 г, 21,0 ммоль). После перемешивания при комнатной температуре в течение 4 часов реакционную смесь выливают в ледяную воду (50 мл). Смесь экстрагируют CH2Cl2. Объединенные растворы промывают водным раствором Na2S2O3, водным раствором Na2CO3 и рассолом, сушат (MgSO4), обрабатывают активированным древесным углем и концентрируют в вакууме, получают сырое Соединение 19 (6,56 г, 99%) в виде бледно-оранжевого масла, которое самопроизвольно затвердевает при стоянии в холодильнике (длинные иголки). Полученное соединение используют на следующей стадии без дополнительной очистки.
Спектр 1H-ЯМР (CDCl3): 9,95 (с, 1H); 8,39 (д, J = 1,9 Гц, 1H): 7,91 (дд, J = 8,5, 1,9 Гц, 1H); 7,41 (дк, J = 8,5, 1,2 Гц, 1H).
(II) 3-Циано-4-(трифторметокси)бензальдегид (Соединение 20)
К суспензии Соединения 19 (6,85 г, 21,7 ммоля) и цианида цинка (4,07 г, 34,7 ммоля) в сухом ДМФА (35 мл) при перемешивании при комнатной температуре порциями добавляют тетракис (трифенилфосфин) палладий (0) (3,00 г, 2,60 ммоля). Реакционную смесь нагревают при 100oC в течение 9 часов. Реакционную смесь разбавляют смесью толуола (100 мл) и 2М водного NH3 (100 мл). Органический слой отделяют. Водный слой разбавляют толуолом (100 мл), фильтруют через слой целита и осадок на фильтре промывают толуолом. Отделяют органический слой и водный слой экстрагируют толуолом. Объединенный раствор промывают 2М водным NH3 (50 мл) и рассолом, сушат (MgSO4) и концентрируют в вакууме, полу чают сырой продукт в виде бледно-рыжего масла.
Сырой продукт очищают колоночной хроматографией на силикагеле (элюент гексан-этилацетат, 10: 1-5: 1), получают Соединение 20 (2,87 г, 62%) в виде бледно-желтого масла.
Спектр 1H-ЯМР (CDCl3): 10,04 (с, 1H); 8,26 (д, J = 1,4 Гц, 1H); 8,19(дд, J=8,4,1,4 Гц, 1H): 7,59 (дк, J = 8,4, 1,8 Гц, 1H).
(III) 5-(-1-Гидроксиэтил)-2-(трифторметокси)бензонитрил (Соединение 21)
К раствору Соединения 20 (2,59 г, 12,0 ммоля) в сухом ТГФ (25 мл) при перемешивании и охлаждении льдом в атмосфере N2 добавляют MeMgBr (4,42 мл, 13,2 ммоля, 3,0М раствор в диэтиловом эфире). Реакционную смесь перемешивают при 0oC в течение 1 часа и затем в течение 2 часов при комнатной температуре. Смесь при охлаждении льдом разбавляют насыщенным раствором NH4Cl (20 мл). Смесь экстрагируют эфиром. Объединенные растворы промывают рассолом, сушат (MgSO4), и концентрируют в вакууме, получают Соединение 21 (2,78 г, выход количественный) в виде желтого масла. Полученное соединение применяют на следующей стадии без дополнительной очистки.
Спектр 1H-ЯМР (CDCl3): 7,75 (д, J = 2,2 Гц, 1H); 7,66 (дд, J = 8,7, 2,2 Гц, 1H); 7,39 (дк, J = 8,7, 1,7 Гц, 1H); 5,03 - 4,90 (м, 1H), 2,02 (ушир. с, 1Н), 1,51 (д, J=6,6 Гц, 3H).
(IV) 3-Циано-4-(трифторметокси)ацетофенон (Соединение 22)
К раствору Соединения 21 (2,78 г, 12,0 ммоля) в сухом CH2Cl2 (100 мл) при перемешивании и при комнатной температуре порциями добавляют оксид марганца (IV) (активированный; 13,9 г). Смесь кипятят с обратным холодильником в течение 2,5 часов. После охлаждения до комнатной температуры смесь фильтруют через слой целита и катализатор промывают CH2Cl2. Фильтрат и промывные растворы концентрируют в вакууме, получают сырое Соединение 22 (2,31 г, 84%) в виде желтого твердого вещества.
Полученное соединение используют на следующей стадии без дополнительной очистки.
Спектр 1H-ЯМР (CDCl3): 8,66 и 8,31 (каждый д, J = 2,2 Гц, всего 1H); 8,24 и 8,17 (каждый дд, J = 8,8, 2,2 Гц, всего 1H); 7,54 - 7,47 и 7,46 - 7,39 (каждый м, всего 1H); 2,66 и 2,65 (каждый с, всего 3H).
(V) 2-Метил-2-(3-циано-4-(трифторметокси)фенил)-1,3-дитиолан (Соединение 23)
К раствору Соединения 22 (2,31 г, 10,1 ммоля) в сухом CH2Cl2 (30 мл) при перемешивании и при комнатной температуре добавляют 1,2-этандитиол (1,42 г, 15,1 ммоля) и эфират трифторида бора (1,14 г, 8,1 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение 15 часов. Смесь разбавляют 5%- ным водным раствором NaOH (40 мл) и отделяют органический слой. Водный слой экстрагируют CH2Cl2. Объединенный раствор промывают рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт в виде пурпурного масла. Сырой продукт очищают колоночной хроматографией на силикагеле (элюент гексан-этилацетат, 50:1-20:1), получают Соединение 23 (2,61 г, 85%) в виде пурпурного масла.
Спектр 1H-ЯМР (CDCl3): 8,15 (д, J = 2,6 Гц, 1H): 8,03 (дд, J = 8,8, 2,6 Гц, 1H); 7,32 (дд, J = 8,8, 1,5 Гц, 1H); 3,58 - 3,44 (м, 2H), 3,43 -3,28(м, 2H), 2,13(с, 3H).
(VI) 5-(1,1-Дифторметокси)бензилонитрил (Соединение 24)
К суспензии 1,3-дибром-5,5-диметилгидантоина (ДБГ (1,07 г, 3,73 ммоля) в сухом CH2Cl2 (8 мл) при перемешивании и при -78oC (ацетон-сухой лед) добавляют HF- пиридин (0,95 мл, 4,11 ммоля). К этой смеси при той же температуре добавляют раствор Соединения 23 (570 мг, 1,87 ммоля) в сухом CH2Cl2 (4 мл). Реакционную смесь перемешивают при -78oC в течение 10 минут и затем в течение 30 мин при комнатной температуре. Полученную смесь выливают в насыщенный водный раствор NaHCO3 (40 мл) и перемешивают при комнатной температуре в течение 15 минут. Смесь фильтруют через слой целита и осадок на фильтре промывают эфиром. Органический слой отделяют и водный слой экстрагируют эфиром. Объединенный раствор промывают 10%-ным водным раствором HCl и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт (670 мг). Сырой продукт очищают с помощью препаративной тонкослойной хроматографии (ТСХ) (элюент гексан- этилацетат, 5:1), получают Соединение 24 (408 мг, 87 %) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3):7,88-7,85 (м, 1H); 7,83-7,76 (м,1H); 7,51 -7,43(м, 1H); 1,94(т,J=18,3 Гц, 3H).
ИК-спектр (пленка): 2245, 1619, 1504, 1417, 1391, 1272, 1265, 1212, 1185, 1120, 924, 844.
(VII) 5-(1,1-Дифторэтил)-2-(трифторметокси)бензальдегид (Соединение 25)
К раствору Соединения 24 (1,31 г, 5,22 ммоля) в сухом CH2Cl2 (20 мл) при перемешивании и при охлаждении льдом добавляют раствор диизобутилалюминийгидрида (ДИПАЛГ) (6,20 мл, 6,26 ммоля, 1,01 М раствор в толуоле). Реакционную смесь перемешивают при температуре 0oC в течение 3 часов. К смеси добавляют H2O (6 мл), и затем 6М водный раствор HCl (20 мл) и перемешивают при комнатной температуре в течение 1,5 часов. Органический слой отделяют и водный слой экстрагируют CH2Cl2. Объединенный раствор промывают насыщенным водным раствором NaHSO4 и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт. Сырой продукт, очищают с помощью колоночной хроматографии на силикагеле (элюент гексан - этилацетат, 50:1-30:1), получают Соединение 25 (1,17 г, 88%) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3): 10,39 (с, 1H); 8,15-8,08 (м,1H); 7,86 -7,78(м, 1H); 7,48- 7,40 (м, 1H); 1,95(т,J=18, 3 Гц, 3H).
ИК-спектр (пленка): 1702, 1618, 1499, 1390, 1269, 1212, 1180, 1115, 923.
(VIII) (2S, 3S)-1-трет-Бутоксикарбонил-3-(5-(1,1-дифторэтил)- 2-(тpифторметокси)бензил)амино-2-фeнилпипеpидин (Соединение 26)
К раствору Соединения 12 (500 мг, 1,81 ммоля) и Соединения 25 (552 мг, 2,17 ммоля) в сухом CH2Cl2 (10 мл) при перемешивании и при комнатной температуре порциями добавляют триацетоксиборгидрид натрия (1,15 г, 5,43 ммоля). К этой смеси при той же температуре добавляют уксусную кислоту (109 мг, 1,81 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение 66 часов и при охлаждении льдом подщелачивают до pH 10-11 с использованием 10%-ного водного раствора NaOH. Органический слой отделяют и водный слой экстрагируют CH2Cl2. Объединенный раствор промывают рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт (1,46 г) в виде бледно-желтого масла. Полученное соединение используют на следующей стадии без дополнительной очистки.
(IX) (2S, 3S)-3-(5-(1,1-Дифторэтил)-2-(трифторметокси)бензил) амино-2-фенилпиперидин (Соединение 27)
К раствору Соединения 26 (660 мг, 1,25 ммоля) в этилацетате (6 мл) при перемешивании и при охлаждении льдом добавляют конц. водную HCl (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение 50 минут. Смесь при охлаждении льдом подщелачивают до pH 10-11 с использованием 10% - ного водного раствора NaOH. Органический слой отделяют и водный слой экстрагируют AcOEt. Объединенный раствор промывают рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт в виде бледно-желтого масла. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле (элюент дихлорметан - метанол, 20: 1), получают Соединение 27 (360 мг, 70 %) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,40-7,30 (м, 7H); 7,17-7,10 (м,1H); 3,91 (д, J = 2,2 Гц, 1H); 3,61 (д, J = 15,0 Гц, 1H); 3,47 (д, J = 15,0 Гц, 1H); 3,30 - 3,20 (м, 1H); 2,89 -2, 73 (м, 2H); 2,13 - 2,00 (м, 1H); 1,97- 1,71 (м, 1H); 1,82 (т, J=18,1 Гц, 3H); 1,70- 1,55 (м,1H); 1,53-1,40 (м, 1H).
ИК-спектр (пленка): 3340, 1605, 1497, 1454, 1419, 1387, 1354, 1309, 1259, 1250, 1221, 1173, 1118, 920, 874, 834, 753, 702.
(X) Дигидрохлорид (2S, 3S)-3-(5-(1,1-дифторэтил)-2-(трифторметокси) бензил)амино-2-фенилпиперидина (Соединение 28)
Соединение 27 (360 мг, 0,87 ммоля) обрабатывают метанолом, насыщенным HCl (20 мл), растворитель упаривают в вакууме, получают сырой продукт в виде белого твердого вещества. Сырой продукт перекристаллизовывают из смеси этанол-диэтиловый эфир, получают Соединение 28 (370 мг, 87%) в виде белого твердого вещества.
Т.пл.: 172-174oC
ИК-спектр (KBr): 3435, 1607, 1573, 1512, 1458, 1303, 1264, 1208, 1173, 1124, 924, 906, 826, 747, 698.
Пример 8
Получение мономанделата (2S, 3S)-3-5(1,1-дифторэтил)- 2-метоксибензил)амино-2-фенилпиперидина (Соединение 37)
(I) 2-Бром-5-(1-гидроксиэтил)анизол (Соединение 29)
Это соединение получают из 3-бром-4-метоксибензальдегида по методике, аналогичной методике получения Соединения 21.
Спектр 1H-ЯМР (CDCl3): 7,57 (д, J = 2,2 Гц, 1H); 7,28 (дд, J = 8,4, 2,2 Гц, 1H); 6,88 (д, J = 8,4 Гц, 1H); 4,84 (кв, J = 6,2 Гц, 1H); 3,89 (с, 1H); 1,78 (ушир, с,1H): 1,47(д,J=6,2 Гц, 3H).
(II) 3-Бром-4-Метоксиацетофенон (Соединение 30)
Это соединение получают из Соединения 29 по методике, аналогичной методике получения Соединения 22.
Спектр 1H-ЯМР (CDCl3): 8,17 (д, J = 2,2 Гц, 1H); 7,92 (дд, J = 8,4, 2,2 Гц, 1H); 6,94 (д, J = 8,4 Гц, 1H): 3,97 (с, 3H); 2,56 (с, 3H).
(III) 3-Циано-4-метоксиацетофенон (Соединение 31).
Это соединение получают из Соединения 30 по методике, аналогичной методике получения Соединения 20.
Спектр 1H-ЯМР (CDCl3): 8,21 - 8,14 (м, 2H); 7,09 - 7,01 (м, 1H); 4,02 (с, 3H): 2,58 (с, 3H).
(IV) 2-Метил-2-(3-циано-4-метоксифенил)-1,3-дитиолан (Соединение 23)
Это соединение получают из Соединения 31 по методике, аналогичной методике получения Соединения 23.
Спектр 1Н-ЯМР (CDCl3): 7,98 (д, J = 2,6 Гц, 1Н); 7,93 (дд, J = 8,8, 2,6 Гц, 1Н); 6,91 (д, J = 8,8 Гц, 1Н); 3,93 (с, 3Н); 3,53 - 3,32 (м, 4Н); 2,11 (с,3Н).
(V) 5-(1,1-Дифторэтил)-2-метоксибензонитрил (Соединение 33)
К суспензии N-йодсукцинимида (12,5 г, 55,7 ммоля) в сухом СН2Cl2 (60 мл) при перемешивании и при -78oC (ацетон - сухой лед) добавляют HF - пиридин (6,81 мл, 30,6 ммоля), а затем при той же температуре раствор Соединения 32 (3,50 г, 13,9 ммоля) в сухом CH2Cl2 (10 мл). Реакционную смесь перемешивают при -78oC в течение 10 минут и затем в течение 30 мин при температуре -10oC (метанол-лед). Смесь выливают в насыщенный водный раствор NaHCO3 (100 мл) и перемешивают при комнатной температуре в течение 2 часов. Смесь фильтруют через слой целита и осадок на фильтре промывают CH2Cl2. Органический слой отделяют и водный слой экстрагируют CH2Cl2. Объединенный раствор промывают насыщенным водным раствором Na2S2O3, 10%-ным водным раствором HCl и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт в виде желтого масла.
Сырой продукт очищают с помощью колоночной хроматографии на силикагеле (элюент гексан-этилацетат, 5:1), получают Соединение 33 (2,67 г, 97%) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,73 - 7,65 (м, 2H); 7,02 (д, J = 8,4 Гц, 1H); 3,97(с, 3H); 1,91(т, J=18,0 Гц, 3Н).
(VI) 5-(1,1-Дифторэтил)-2-метоксибензальдегид (Соединение 34)
Это соединение получают из Соединения 33 по методике, аналогичной методике получения Соединения 25.
Спектр 1H-ЯМР (CDCl3): 10,47 (с, 1H); 7,97 (д, J = 2,6 Гц, 1H); 7,72 (дд, J = 8,8, 2,6 Гц, 1H); 7,05 (д, J = 8,8 Гц, 1H); 3,97 (с, 3H); 1,93 (т, J = 18,0 Гц, 3H).
(VII) (2S,3S)-1-трет-Бутоксикарбонил-3-(5-(1,1-дифторэтил)-2- (метоксибензил)амино-2-фенилпиперидин (Соединение 35)
Это соединение получают из Соединений 12 и 34 по методике, аналогичной методике получения Соединения 26, и используют на следующей стадии без дополнительной очистки.
(VII) (2S, 3S)-3-(5-(1,1-дифторэтил)-2-(метоксибензил)амино-2-фенилпиперидин (Соединение 36)
Это соединение получают из Соединения 35 по методике, аналогичной методике получения Соединения 27.
Спектр 1H-ЯМР (CDCl3): 7,35- 7,18 (м, 6H); 7,15 (д, J= 2,2 Гц, 1H); 6,68 (д, J= 8,4, Гц, 1H); 3,89 (д, J=2,2 Гц, 1H); 3,66 (д, J=13,9 Гц, 1H): 3,49 (с, 3H); 3,41 (д, J = 13,9 Гц, 1H); 3,22 - 3,20 (м, 1 H); 2,86 - 2,72 (м, 2H); 2,18 - 2,05 (м, 1H), 2,02 -1,81 (м, 1H); 1,86 (т, J = 18,0 Гц, 3H); 1,72 (ушир. с, 2H); 1,75-1,52 (м, 1H); 1,47-1,35 (м, 1H).
ИК-спектр (пленка): 3335, 1614, 1502, 1451, 1385, 1308, 1280, 1252, 1174, 1123, 1030, 923, 901, 870, 816, 751, 701.
(IX) Moноманделат (2S, 3S)-3-(5-1,1-Дифторэтил)-2-метоксибензил)амино-2-фенилпиперидина (Соединение 37.)
К раствору Соединения 36 (179 мг, 0,50 ммоля) в этаноле (3 мл) при комнатной температуре добавляют (R)-(-)- миндальную кислоту (75,4 мг, 0,50 ммоля). После упаривания в вакууме растворителя полученный остаток перекристаллизовывают из смеси этанолдиэтиловый эфир, получают Соединение 37 (168 мг, 66%) в виде белого твердого вещества.
Т.пл.: 177-179oC.
ИК-спектр (пленка): 3400, 1615, 1576, 1506, 1473, 1454, 1399, 1384, 1362, 1345, 1318, 1249, 1172, 1115, 1054, 1028, 900, 756, 742, 698.
Пример 9
Получение дигидрохлорида (2S,3S)-3-(2-метокси-5-(1,1,2,2,2-пентафторэтил)бензил)амино-2 -фенилпиперидина (Соединение 43)
(I) 2-(3-Бром-6-метоксифенил)-1,3-диоксан (Соединение 38)
Смесь 5-бром-о-анисового альдегида (10,0 г, 46,5 ммоля), пропан-1,3-диола (3,90 г, 51,2 ммоля) и BF3-Et2O (0,15 мл) в толуоле (50 мл) кипятят с обратным холодильником в приборе Дина-Старка в течение 3 часов. Реакционную смесь охлаждают, разбавляют эфиром. Органический слой промывают последовательно насыщенным водным раствором NaHCO3, водой и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт. Остаток перегоняют, получают Соединение 38 (10,7 г, 84%) в виде бесцветного масла.
Т.пл.: 124-125oC/0,23-0,25 мм рт.ст.
Спектр 1H-ЯМР (CDCl3): 7,74 (д, J = 2,6 Гц, 1H); 7,39 (дд, J = 8,8, 2,6 Гц, 1H); 6,74(д, J=8,8 Гц, 1H); 5,81 (с, 1H); 4,31-4,17 (м, 2H): 4,06 - 3,91 (м, 2H); 3,82 (c, 3H), 2,35 - 2,10 (м, 1H), 1,50- 1,35 (м, 1H).
(II) 2-(2-Метокси-5-(1,1,2,2,2-пентафторэтил)фенил)-1,3-диоксан (Соединение 39)
В круглодонную колбу на 50 мл, снабженную насадкой Дина-Старка и обратным холодильником, помещают Соединение 38 (1,0 r, 3,66 ммоля), пентафторпропионат натрия (1,29 г, 6,95 ммоля), йодид меди (1) (1,46 г, 7,69 ммоля) и ДМФА (15 мл) - толуол (6 мл). Суспензию нагревают до 120-140oC (температура бани) и толуол (6 мл) удаляют дистилляцией. Реакционную смесь нагревают при 140oC (внутренняя температура) в течение 15 часов. Смесь разбавляют смесью вода (40 мл)-толуол (15 мл)-этилацетат (60 мл). Смесь фильтруют через слой целита и осадок на фильтре промывают этилацетатом. Фильтрат промывают водой, рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле (элюент гексан-этилацетат, 10: 1-5: 1), получают Соединение 39 (1,07 r) в виде бледно-желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,87 (д, J = 2,2 Гц, 1H); 7,53 (дд, J = 8,8, 2,2 Гц, 1 Н); 6,96 (д, J=8,8 Гц, 1H); 5,85 (с, 1H); 4,26 (дд, J = 11,0, 4,4 Гц, 2H); 4,00 (тд, J = 12,3, 2,2 Гц, 2H); 3,89 (с, 3H): 2,36 - 2,15 (м, 1H), 1,50- 1,38 (м, 1H).
(III) (2-Метокси-5-(1,1,2,2,2-пентафторэтил)бензальдегид (Соединение 40)
К раствору Соединения 39 (1,0 г) в ацетоне (30 мл) при перемешивании и при комнатной температуре добавляют конц. HCl (4 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 часов. Растворитель упаривают в вакууме и остаток экстрагируют эфиром. Объединенный раствор промывают насыщенным водным раствором NaHSO4 и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт (790 мг) в виде желтого твердого вещества. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле (элюент гексан - изопропиловый эфир, 10:1-3:1), получают Соединение 40 (275 мг) в виде желтого твердого вещества.
Спектр 1H-ЯМР (CDCl3): 10,48 (с, 1H); 8,08 (д, J = 2,2 Гц, 1H); 7,77 (дд, J = 8,8, 2,2 Гц, 1H); 7,13(д, J = 8,8, Гц, 1H): 4,02 (с, 3H).
(IV) (2S, 3S)-1-трет- Бутоксикарбонил-3-(2-метокси-5-(1,1,2,2,2-пентафторэтил)бензил)амино-2- фенилпиперидин (Соединение 41)
Это соединение получают из Соединений 12 и 40 по методике, аналогичной методике получения Соединения 26, и используют на следующей стадии без дополнительной очистки.
(V) (2S,3S)-3-(2-метокси-5-(1,1,2,2,2-пентафторэтил)бензил)амино-2- фенилпиперидин (Соединение 42).
Это соединение получают из Соединения 41 по методике, аналогичной методике получения Соединения 27.
Спектр 1H-ЯМР (CDCl3): 7,38 (дд, J=8,4, 2,2 Гц, 1H); 7,34-7,18 (м, 6H); 6,74 (д, J = 8,4 Гц, 1H); 3,90 (д, J=2,2 Гц, 1H); 3,69 (д, J = 4,3, Гц, 1H); 3,53 (с, 3H): 3,40 (д, J=14,3 Гц, 1H); 3,34 - 3,24 (м, 1H); 2,88 - 2,74 (м, 2H); 2,15 -1,83 (м, 4H); 1,69-1,53 (м,1H); 1,50-1,38 (м,1H).
ИК-спектр (пленка): 3330, 1614, 1501, 1460, 1334, 1304, 1275, 1258, 1203, 1145, 1119, 1096, 1029, 1004, 870, 815, 746, 700.
(VI) Дигидрохлорид (2S, 3S)-3-(2-метокси)-5-(1,1,2,2,2-пентафторэтил)бензил)амино-2-фенилпиперидина (Соединение 43)
Это соединение получают из Соединения 42 по методике, аналогичной методике получения Соединения 28.
Т.пл.: 201-202oC.
ИК-спектр (KBr): 3455, 1617, 1554, 1506, 1453, 1443, 1416, 1337, 1282, 1258, 1221, 1202, 1180, 1148, 1131, 1091, 1010, 744, 693.
Пример 10
Получение дигидрохлорида (2S, 3S)-3-(2-метокси-5-(2,2,2-трифторэтил)бензил)амино-2-фенилпиперидина) (Соединение 49)
(I)-1-(4-Метоксифенил)-2,2,2-трифторэтилбромид (Соединение 44)
Это соединение получают в соответствии с методикой, описанной в J.Am. Chem. Soc., 111, 1455 (1989).
(II) 1-(4-Метоксифенил)-2,2,2-трифторэтан (Соединение 45)
Раствор соединения 44 (1,08 г, 400 ммоль) в этаноле (20 мл) гидрируют на 10%-ном Pd/C (800 мг) при атмосферном давлении в течение 16 часов. Катализатор отфильтровывают через слой целита и осадок на фильтре промывают CH2Cl2. Объединенный раствор промывают разбавленным рассолом и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырое Соединение 45 (760 мг, выход количественный) в виде бледно-желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,24-7,17 (м,2H); 6,93-6,84 (м,2H); 3,81 (с, 3H); 3,30 (кв, J = 10,9, Гц, 2H).
(III) 2-Метокси-5-(2,2,2-трифторэтил)бензальдегид (Соединение 46)
К раствору Соединения 45 (760 мг, 4,00 ммоль) в сухом CH2Cl2 (50 мл) при перемешивании и при охлаждении льдом с помощью шприца добавляют TiCl4 (1,67 г, 8,80 ммоля). Через 15 минут к этой смеси при той же температуре добавляют раствор дихлорметилметилового эфира (920 мг, 8,00 ммолей) в сухом CH2Cl2 (5 мл). Реакционную смесь перемешивают при 0oC в течение 15 минут и затем при комнатной температуре в течение 1,5 часов. Смесь при охлаждении льдом разбавляют водой (20 мл) и перемешивают при комнатной температуре в течение 15 минут. Органический слой отделяют и водный слой экстрагируют CH2Cl2. Объединенный раствор промывают насыщенным водным раствором NaHCO3 и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт в виде желтого масла. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле (элюент гексан - этилацетат, 40:1-20:1), получают Соединение 46 (500 мг, 57 %) в виде бесцветного твердого вещества (в виде игл).
Спектр 1H-ЯМР (CDCl3): 10,46 (с, 1H); 7,76 (д, J = 2,2, Гц, 1H), 7,49 (дд, J = 8,8, 2,2 Гц, 1H), 7,00 (д, J = 8,8, Гц, 1H), 3,95 (с, 3H); 3,34 (кв, J=10,6, Гц,
(IV) (2S, 3S)-1-трет-Бутоксикарбонил-3-(2-метокси-5-(2,2,2-трифторэтил) бензил)амино-2- фенилпиперидин (Соединение 47)
Это соединение получают из Соединений 12 и 46 по методике, аналогичной методике получения Соединения 26, и используют на следующей стадии без дополнительной очистки.
(V) (2S, 3S)-3-(2-Метокси-5-(2,2,2-трифторэтил)бензил)амино -2-фенилпиперидин (Соединение 48)
Это Соединение получают из Соединения 47 по методике, аналогичной методике получения Соединения 27.
Спектр 1H-ЯМР (CDCl3): 7,35 - 7,20 (м, 5H); 7,06 (дд, J = 8,4, 1,8 Гц, 1H); 6,84 (д, J = 1,8 Гц, 1H); 6,65 (д, J = 8,4 Гц, 1H), 3,90 (д, J = 2,2 Гц, 1H); ); 3,68 (д, J = 14,3 Гц, 1H); 3,49 (с, 3H); 3,42 (д, J = 14,3 Гц, 1H); 3,35- 3,24 (м, 1H);); 3,20 (кв, J = 11,0 Гц, 2H); 2,88 - 2,73 (м, 2H): 2,20-1,85 (м,4H); 1,68-1,52 (м, 1H): 1,50-1,37 (м,1H).
ИК-спектр (пленка): 3450, 1614, 1500, 1465, 1445, 1430, 1359, 1328, 1263, 1249, 1237, 1128, 1103, 1074, 1031, 854, 822, 810, 773, 746, 700, 672.
(VI) Дигидрохлорид (2S, 3S)-3-(2-метокси-5-(2,2,2-трифторэтил) бензил)амино-2-фенилпиперидина (Соединение 49)
Это Соединение получают из Соединения 48 по методике, аналогичной методике получения Соединения 28.
Т.пл.: 209-210oC.
ИК-спектр (KBr): 3450, 1552, 1506, 1451, 1441, 1415, 1369, 1333, 1260, 1241, 1170 1132, 1086, 1030, 978, 807, 748, 693.
Пример 11
Получение дигидрохлорида (2S, 3S)-3-(2-метокси-5-(1-трифторметил) бензил)амино-2-фенилпиперидина (Соединение 55)
(I) 1-(4-Метоксифенил)-1-(трифторметил)этилбромид (Соединение 50)
Это Соединение получают в соответствии с методикой, описанной в J.Am. Chem.Soc., 104, 211 (1982).
(II)1-(4-Метоксифенил)-1-(трифторметил)этан (Соединение 51)
Это Соединение получают из Соединения 50 по методике, аналогичной методике получения Соединения 45.
Спектр 1H-ЯМР (CDCl3): 7,28-7,19 (м,2H); 6,93- 6,84 (м,2H); 3,81 (с, 3H); 3,48 - 3,27 (м, 1H); 1,48 (д, J = 7,0 Гц, 3H).
(III) 2-Метокси-5-(1-(трифторметил)этил)бензальдегид (Соединение 52)
Это Соединение получают из Соединения 51 по методике, аналогичной методике получения Соединения 46.
Спектр 1H-ЯМР (CDCl3): 10,46 (с, 1H); 7,79 (д, J = 2,6, Гц, 1H), 7,52 (дд, J = 8,8, 2,6 Гц, 1H), 7,00 (д, J = 8,8, Гц, 1H), 3,94 (с, 3H); 3,53 -3,32 (м,1H); 1,50 (д, J = 7,3, Гц, 3H).
(IV) (2S,3S)-1-трет-Бутоксикарбонил-3-(2-метокси-5-(1- (трифторметил)этил)бензил)амино-2-фенилпиперидин (Соединение 53)
Это соединение получают из Соединений 12 и 52 по методике, аналогичной методике получения Соединения 26, и используют на следующей стадии без дополнительной очистки.
(V)(2S, 3S)-3-(2-Метокси-5-(1-(трифторметил)этил) бензил)амино-2-фенилпиперидин (Соединение 54)
Это Соединение получают из Соединения 53 по методике, аналогичной методике получения Соединения 27.
Спектр 1H-ЯМР (CDCl3): 7,35-7,19 (м,5H); 7,13-7,05 (м,1H); 6,91 - 6,86 (м, 1H); 6,68 - 6,61 (м, 1H); 3,91 (д, J = 2,2 Гц, 1H); 3,75 -3,15(м,7H); 2,88-2,73 (м, 2H); 2,30-1,85 (м,4H): 1,70-1,51 (м, 1H); 1,50-1,3J (м, 1H); 1,42 (д, J = 7,3 Гц, 3H).
ИК-спектр (пленка): 3330, 1612, 1500, 1462, 1385, 1349, 1331, 1295, 1250, 1171, 1157, 1122, 1082, 1049, 1031, 995, 805, 747, 701.
(VI) Дигидрохлорид (2S, 3S)-3-(2-метокси-5-(1-трифторметил)этил)бензил)амино-2- фенилпиперидина (Соединение 55)
Это Соединение получают из Соединения 54 по методике, аналогичной методике получения Соединения 28.
Т.пл.: 217-218oC.
ИК-спектр (KBr): 3450, 1554, 1505, 1465, 1453, 1442, 1417, 1334, 1253, 1169, 1159, 1144, 1120, 1083, 1050, 1030, 748, 693.
Пример 12.
Получение дигидрохлорида (2S, 3S)-3-[(5-(1,1-диметил-4,4,4-трифтор-2-бутинил)-2- метоксибензил)]амино-2-фенилпиперидина (Соединение 60)
(1)4-(1,1-Диметил-2-пропинил)анизол (Соединение 56)
Это соединение получают в соответствии с методикой, описанной в Tetrahedron Lett. 4163 (1977).
(II)4-Диметил-4,4,4-трифтор-2-бутинил)анизол (Соединение 57)
К раствору соединения 56 (0,22 г, 1,26 ммоля) в ТГФ (8 мл) при перемешивании и при - 78oC в атмосфере N2 добавляют н - BuLi (1,69 М раствор в гексане, 0,82 мл, 1,39 ммоля), затем нагревают до 0oC и перемешивают в течение 1 часа. Добавляют S - (трифтор-метил)дибензотиофенилтрифторметансульфонат (1,01 г, 2,52 ммоля) и перемешивают в течение 3 часов при 0oC. Смесь гасят добавлением водного NaHCO3 и экстрагируют CH2Cl2. Объединенные органические слои сушат MgSO4, фильтруют и концентрируют. Полученный остаток очищают хроматографированием на SiO2, получают Соединение 57 (38 мг, 13%) в виде бесцветного масла.
Спектр 1H-ЯМР (CDCl3): 7,37 (д, J = 8,8 Гц, 2H); 6,88 (д, J = 8,8 Гц, 2H); 3,80(с,3H); 1,62(с,6H).
(Ill) 5-(1,1-Диметил-4,4,4-трифтор-2-бутинил)-2- метоксибензальдегид (Соединение 58.)
Это Соединение получают из Соединения 57 по методике, аналогичной методике получения Соединения 9.
Спектр 1H-ЯМР (CDCl3): 10,47 (с, 1H); 7,87 (д, J = 2,9 Гц, 1H); 7,71 (дд, J = 8,8, 2,9 Гц, 1H); 7,01 (д, J = 8,8 Гц, 1H); 3,94 (с, 3H); 1,64 (с, 6H).
(IV)(2S, 3S)-1-трет-Бутоксикарбонил-3-[5-(1,1-Диметил-4,4,4- трифтор-2-бутинил)-2-метоксибензил]амино-2 фенилпиперидин (Соединение 59)
Это соединение получают из Соединения 58 и Соединения 12 по методике, аналогичной методике получения Соединения 13.
Спектр 1H-ЯМР (CDCl3): 7,62 - 7,53 (м, 2H); 7,36 - 7,20 (м, 5H); 6,78 (д, J = 9,2 Гц, 1H); 5,53 - 5,42 (м, 1H); 4,01 - 3,88 (м, 1H); 3,83 (с, 2H); 3,70 (с, 3H); 3,13-2,93 (м, 2H); 1,92-1,35(м,4H); 1,60 (с, 3H); 1,59(с,3H), 1,40(с,9H).
(V) Дигидрохлорид (2S,3S)-3-[5-(1,1-Диметил-4,4,4-трифтор-2- бутил)-2-метоксибензил]амино-2-фенилпиперидин (Соединение 60)
К раствору соединения 59 (34 мг, 0,064 ммоля) в AcOEt (8 мл) добавляют избыток HCl - МeOH. Смесь перемешивают в течение 18 часов и затем упаривают в вакууме, твердый остаток перекристаллизовывают из смеси MeOH - Et2O, получают Соединение 60 (24 мг, 75 %) в виде белого твердого продукта.
Т.пл.: 225-227oC.
ИК-спектр (KBr): 3440, 2980, 2935, 2350, 2275, 1558, 1504, 1455, 1416, 1293, 1130 см-1.
Спектр 1H-ЯМР (свободное основание, CDCl3): 7,38 - 7,25 (м, 6H); 7,10(д, J = 2,6 Гц, 1H); 6,67 (дд, J = 8,4 Гц, 1H); 4,04 - 4,01 (м, 1H); 3,78 (д, J = 13,9 Гц, 1H); 3,53 - 3,38 (м, 2H); 3,45 (с, 3H); 2,96 -2,83 (м, 2H); 2,30-1,60 (м,4H), 1,58 (с, 6H).
Элементный анализ, C25H29F3N2O • 2HCl. Вычислено,%: С 59,65, H 6,21, N 5,56. Найдено,%: С 59,38, H 6,27, N 5,55.
Пример 13
Получение дигидрохлорида (2S, 3S)-3-[(5-Метокси-1-трифторметил)индан-6-ил)метиламино]-2-фенилпиперидина (Соединение 61)
(I) 1-Гидрокси-5-метокси-1-(трифторметил)индан (Соединение 52)
К раствору 5-метокси-1-инданона (1,00 г, 6,17 ммоля) и трифторметилтриметилсилана (1,32 г, 9,26 ммоля) в сухом ТГФ (15 мл) при перемешивании и охлаждении льдом добавляют 1,0 М раствор тетрабутиламмонийфторида в ТГФ (0,05 мл). Реакционную массу перемешивают при комнатной температуре в течение 21 часа. К смеси добавляют 1 н. HCl (20 мл) и перемешивают при комнатной температуре в течение 25 часов. Реакционную массу разбавляют смесью CH2Cl2 - вода. Органический слой отделяют и водный слой экстрагируют CH2Cl2. Объединенный раствор промывают водой и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт в виде темно - желтого масла. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле (элюент гексан: этилацетат, 5:1-3:1), получают Соединение 62 (1,05 г, 73%) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,39 (д, J = 8,4 Гц, 1H); 6,88 - 6,75 (м, 2H); 3,81 (с,3H); 3,17-2,86 (м,2H); 2,74-2,57 (м, 1H); 2,43-2,34 (м, 1H); 2,32-2,15 (м, 1H).
(II) 6-Метокси-3-(трифторметил)индан (Соединение 63)
К соединению 62 (850 мг, 3,66 ммоля) при перемешивании и охлаждении льдом добавляют PBr3 (9,90 г, 36,6 ммоля). Реакционную смесь перемешивают при 80oC в течение 6 часов. Смесь при охлаждении льдом разбавляют водой и экстрагируют CH2Cl2. Объединенный раствор промывают насыщенным раствором NaHCO3 и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт в виде желтого масла. Сырой продукт очищают колоночной хроматографии на силикагеле (элюент гексан - этилацетат, 50:1-40:1). Получают Соединение 63 (727 мг, 93 %) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,46 - 7,38 (м, 1H); 7,10- 7,05 (м, 1H); 6,90 (дд, J = 8,4, 2,6 Гц, 1H); 6,87 - 6,81 (м, 1H); 3,84 (с, 3H); 3,50 - 3,47 (м, 2H).
(III) 5-Метокси-1-(трифторметил)индан (Соединение 64)
Раствор соединения 63 (180 мг, 0,84 ммоля) в этаноле (5 мл) гидрируют на 10%- ном Pd-С (90 мг) при атмосферном давлении в течение 4,5 часов. Катализатор отфильтровывают через слой целита и осадок на фильтре промывают CHCl2. Объединенный раствор промывают разбавленным рассолом и рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырое Соединение 64(147 мг, 81 %) в виде светло - желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,36- 7,20 (м, 1H); 6,88-6,69 (м, 2H); 3,90 - 3,62 (м, 4H); 3,20 - 2,80 (м, 2H); 2,48 -2,19 (м, 2H).
(IV)6-Формил-5-метокси-1-(трифторметил)индан(Соединение 65)
Это соединение получают из Соединения 64 по методике, аналогичной методике получения Соединения 46.
Спектр 1H-ЯМР (CDCl3): 10,41 (с, 1H); 7,85 (с, 1H); 6,90 (с, 1H); 3,93 (с, 3H); 3,90- 3,70 (м, 1 Н); 3,24 - 2,87 (м, 2H); 2,50 - 2,20 (м,2H).
(V) (2S,3S)-1-трет-Бутоксикарбонил-3-[(5-метокси-1-трифторметил)индан-6-ил) метиламино]-2-фенилпиперидин (Соединение 66)
Это соединение получают из Соединений 12 и 65 по методике, аналогичной методике получения Соединения 26. На следующей стадии оно используется без дополнительной очистки.
(VI) (2S, 3S)-3-[(5-Метокси-1-(трифторметил) индан - 6-ил) метиламино] -2-фенилпиперидин (Соединение 67)
Это соединение получают из Соединения 66 по методике, аналогичной методике получения Соединения 27.
Спектр 1H-ЯМР (CDCl3): 7,37 - 7,17 (м, 5H): 6,99 (уш. с, 1H); 6,58 и 6,56 (каждый с, всего 1H); 3,91 (д, J = 2,2 Гц, 1H); 3,82 - 3,60 (м, 2H): 3,44 (с, 3H); 3,39 (д, J = 13,9 Гц, 1H); 3,39 - 3,24 (м, 1H); 3,10 -2,72 (м, 4H); 2,56 (уш. с, 2H); 2,43 -1,85 (м, 4H), 1,71-1,35 (м, 2H).
(VII) Дигидрохлорид (2S, 3S)-3-[(5-метокси-1-(трифторметил)индан-6-ил)метиламино]- 2-фенилпиперидина (Соединение 61)
Это соединение получают из Соединения 67 по методике, аналогичной методике получения Соединения 28.
Т.пл.: 213-214oC.
ИК-спектр (KBr): 3435, 1623, 1579, 1560, 1498, 1464, 1452, 1434, 1421, 1368, 1297, 1271, 1170, 1138, 1103, 1034, 749, 694.
Пример 14
Получение дигидророхлорида (2S, 3S)-3-[5-(1,1-диметил-2,2,2-трифторэтил)-2 -метоксибензиламино]-2-фенилпиперидина (Соединение 68)
(I) 4-(1-Хлор-1-метил-2,2,2-трифторэтил)анизол (Соединение 69)
Это соединение получают в соответствии с методикой, описанной в публикации JP 62634034.
(II) 4-(1,1-Диметил-2,2,2-трифторэтил)анизол (Соединение 70)
К соединению TiCl4 (57 мг, 0,30 ммоля) в сухом CH2Cl2 (5 мл) при перемешивании и -78oC добавляют с помощью шприца раствор (1,05 моля/л) ZnMe2 (0,87 мл, 0,91 ммоля) в толуоле. Через 15 минут к полученной смеси добавляют раствор Соединения 69 (217 мг, 0,91 ммоля) в сухом CH2Cl2 (5 мл) при той же температуре. Реакционную смесь перемешивают при -78oC в течение 1 часа и нагревают до комнатной температуры. Через 2 часа смесь разбавляют водой и перемешивают в течение 10 минут. Объединенный раствор промывают рассолом, сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт (200 мл) (Соединения 69: 70 = 1: 2,4) в виде светло - желтого масла. На следующей стадии этот продукт используют без дополнительной очистки.
(III) 5-(1,1-Диметил-2,2,2-трифторэтил, )-2- метоксибензальдегид (Соединение 71)
Это соединение получают из смеси Соединений 69 и 70 по методике, аналогичной методике получения Соединения 46.
Полученное сырое соединение очищают с помощью препаративной ТХС (элюент гексан: этилацетат, 6: 1), Получают соединение 71 (75 мл) в виде светло-желтого масла.
Спектр 1H-ЯМР (CDCl3): 10,47 (с, 1H); 7,95 (д, J = 2,6 Гц, 1H); 7,74 - 7,64 (м, 1H); 6,99 (д, J = 8,8 Гц, 1H); 3,94 (с, 3H); 1,57 (с, 6H).
(IV)(2S, 3S)-1-трет-Бутоксикарбонил-3-[5-(1,1-диметил-2,2,2- трифторэтил)-2-метоксибензиламино]1-2-фенилпиперидин (Соединение 72)
Это соединение получают из Соединения 12 и Соединения 71 по методике, аналогичной методике получения Соединения 26.
Соединение используют на следующей стадии без дополнительной очистки.
(V) (2S,3S)-3-[5-(1,1-Диметил-2,2,2-трифторэтил)-2- метоксибензиламино] -2-фенилпиперидин (Соединение 73)
Это соединение получают из Соединения 72 по методике, аналогичной методике получения Соединения 27.
Спектр 1H-ЯМР (CDCl3): 7,35-7,16 (м,6H); 7,15-7,08 (м,1H); 6,65 (д, J = 8,8 Гц, 1H); 3,91 (д, J = 2,2 Гц, 1H); 3,69 (д, J =13,9 Гц, 1H); 3,48 (с, 3H); 3,41 (д, J = 13,9 Гц, 1H); 3,36 - 3,22 (м, 1H); 2,90 - 2,71 (м, 2H); 2,44 (уш. с, 2H); 2,20 - 1,85 (м, 2H); 1,70-1,35 (м,2H); 1,49 (с, 6H).
(VI) Дигидрохлорид (2S, 3S)-3-[5-(1,1-диметил-2,2,2- трифторэтил)-2-метоксибензиламино]-2-фенилпиперидин (Соединение 68)
Это соединение получают из Соединения 73 по методике, аналогичной методике получения Соединения 28.
Т.пл.: 220-221oC.
ИК-спектр (KBr): 3425, 1564, 1511, 1469, 1453, 1442, 1420, 1400, 1290, 1255, 1187, 1174, 1131, 1101, 1027, 749, 691.
Пример 15
Получение дигидрохлорида (2S,3S)2-фенил-3-(5-(2,2,2-трифтор -1-метил-1-трифторметил)этил)-2-метоксибензил) аминопиперидина (Соединение 78)
(I) 4-(2,2-Дифтор-1-(трифторметил) этенил) анизол
(Соединение 74)
Это Соединение получают в соответствии с методиками, описанными в публикации J. Am. Chem. Soc. 820 (1972).
(II) -4-(2,2,2-Трифтор-1-метил-1-трифторметил)этил)анизол (Соединение 75)
Смесь соединения 74 (570 мг, 2,4 ммоля), Mel (430 мг, 3,0 ммоля) и CsF (760 мг, 5,0 ммоля) в ДМФА (4 мл) перемешивают при 80oC в течение 3 дней. Смесь разбавляют водой. Органический слой отделяют, а водный слой экстрагируют CH2Cl2. Объединенный раствор сушат (Na2SO4) и концентрируют в вакууме, получают сырой продукт, который очищают колоночной хроматографией на силикагеле. Получают Соединение 75 (70 мг, 10%) в виде желтого масла.
Спектр 1H, Н-ЯМР (CDCl3): 7,53- 6,90 (м,4H); 4,85(с,3H); 3,50-3,42 (м, 3H).
(III) 5-(2,2,2-Трифтор-1-метил -1-(трифторметил)этил)-2-метоксибензальдегид (Соединение 76)
Это соединение получают из Соединения 75 по методике, аналогичной методике получения Соединения 9.
Спектр 1H-ЯМР (CDCl3): 10,49 (с, 1H); 8,09-7,08 (м, 3H); 4,02 (с, 3H); 3,51-3,45 (м, 3H).
(IV) (2S, 3S)-1-трет-Бутоксикарбонил-2-фенил-3-(5 -(2,2,2-трифтор-1-метил-1(трифторметил)этил)-2-метоксибензил)- аминопиперидин (Соединение 77)
Это соединение получают из Соединения 12 и Соединения 76 по методике, аналогичной методике получения Соединения 26.
На следующей стадии это соединение используют без дополнительной очистки.
Спектр 1H-ЯМР (CDCl3): 7,62 - 6,86 (м, 8H), 4,06 - 2,95 (м, 9H); 3,78 (с, 3H); 1,91-1,30 (м,4H); 1,35 (с, 9H).
(V) Дигидрохлорид (2S, 3S)-2-фенил-3-(5-(2,2,2 - трифтор-1-метил-1(трифторметил)этил)-2- метоксибензил)аминопиперидина (Соединение 78)
Это соединение получают из Соединения 77 по методике, аналогичной методике получения Соединения 60.
Т.пл.: 267 - 270oC.
Пример 16.
Получение дигидрохлорида (2S, 3S)-3-[5-[2,2, -дифтор-1- трифторметан)этенил]-2-метоксибензил]амино-2-фенилпиперидина (Соединение 81)
(I) 5-[2,2-Дифтор-1-(трифторметил)этенил 1-2-метокситбензальдегид (Соединение 79)
Это соединение получают из Соединения 74 по методике, аналогичной методике получения Соединения 9.
Спектр 1H-ЯМР (CDCl3): 10,47 (с, 1H); 7,81 (д, J = 2,2 Гц, 1H); 7,51 (дд, J = 8,4, 2,2 Гц, 1H); 7,06 (д, J = 8,4 Гц, 1H); 3,98 (с, 3H).
(II) (2S, 3S)-1-трет-Бутоксикарбонил-3-[5-[2,2-дифтор-1- (трифторметил)этенил]-2-метоксибензил]амино-2-фенилпиперидин (Соединение 80)
Это соединение получают из Соединения 79 по методике, аналогичной методике получения Соединения 13.
Спектр 1H-ЯМР (CDCl3): 7,63-7,55 (м,2H); 7,37-7,15 (м,5H); 6,83 (д, J = 8,4 Гц, 1H); 5,53 - 5,42 (м, 1H); 4,02 - 3,92 (м, 1H); 3,83 (с, 2H); 3,74(с, 3H); 3,12-2,94 (м,2H); 1,94-1,40(м,4H); 1,40(с,9H).
(III) Дигидрохлорид (2S,3S)-3-[5-[2,2-дифтор-1-(трифторметил)этенил]-2- метоксибензил]амино-2-фенилпиперидина (Соединение 81)
Это соединение получают из Соединения 80 по методике, аналогичной методике получения Соединения 60.
Т.пл. 235 - 237oC.
Спектр 1H-ЯМР (свободное основание, CDCl3): 7,35 -7,18(м, 5H); 7,15-7,07 (м, 1H); 6,89-6,86 (м,1H); 6,70 (д,J= 8,4 Гц, 1H); 3,91 (д, J = 2,6 Гц, 1H); 3,69 (д, J = 14,7 Гц, 1H); 3,53 (c, 3H); 3,42 (д, J = 14,7 Гц, 1H); 3,36- 3,24 (м, 1 H); 2,88-2,75 (м, 2H); 2,18- 1,40(м,4H).
Пример 17
Получение дигидрохлорида (2S,3S) -3-(2,4-диметокси-5-(2,2,2,-трифторэтил)бензил)амино-2- фенилпиперидина (Соединение 87)
(I)1-(2,4-Диметоксифенил)-2,2,2-трифторэтанол (Соединение 82)
Это соединение получают из 2,4-диметоксибензальдегида по методике, аналогичной методике получения Соединения 62.
Спектр 1H-ЯМР (CDCl3): 7,32 - 6,48 (м, 3H); 7,81 (квинтет, J = 7 Гц, 1H); 3,85 (с, 3H); 3,82 (с, 3H); 3,42 (д, J = 8 Гц, 1H).
(II) 1-(2,4-Диметоксифенил)-2,2,2-трифторэтилбромид (Соединение 83)
Это соединение получают из Соединения 82 по методике, аналогичной методике получения Соединения 44.
Спектр 1H-ЯМР (CDCl3): 7,58 - 6,42 (м, 3H); 5,81 (кв, J = 11 Гц, 1H); 3,88(с,3H);3,86(с,3H).
(III) 1-(2,4-Диметоксифенил)-2,2,2-трифторэтан (Соединение 84)
Это соединение получают из Соединения 83 по методике, аналогичной методике получения Соединения 45.
Спектр 1H-ЯМР (CDCl3): 7,20-6,45 (м, 3H); 3,82 (с, 3H); 3,81 (с, 3H); 3,37 (кв, J=11 Гц,2H).
(IV) (2,4-Диметоксифенил)-5-(2,2,2-трифторэтил)бензальдегид (Соединение 85)
Это соединение получают из Соединения 84 по методике, аналогичной методике получения Соединения 46.
Спектр 1H-ЯМР (CDCl3): 10,20 (с, 1H); 7,77 (с, 1H); 6,46 (с, 1 Н); 3,94 (с, 3H); 3,38 (кв, J=11 Гц,2H).
(V)(2S, 3S)-1-трет-Бутоксикарбонил-3-(2,4-диметокси-5- (2,2,2,-трифторэтил)бензил)амино-2-фенилпиперидин (Соединение 86)
Это соединение получают из Соединения 12 и Соединения 85 по методике, аналогичной методике получения Соединения 13.
На следующей стадии полученное соединение используют без дополнительной очистки.
Спектр 1H-ЯМР (CDCl3): 7,62- 7,20 (м,5H); 7,04 (с, 1H); 6,42 (с, 1H); 4,00 - 2,92 (м, 8H); 3,83 (с, 3H); 3,71 (с, 3H); 1,90-1,30 (м, 4H): 1,39(с, 9H).
(VI) Дигидрохлорид (2S, 3S)-3-(2,4-диметокси-5- (2,2,2-трифторэтил)бензил)амино-2-фенилпиперидина (Соединение 87)
Это соединение получают из Соединения 86 по методике, аналогичной методике получения Соединения 60.
Спектр 1H-ЯМР (свободное основание CDCl3): 7,40 - 7,00 (м, 5H); 6,80 (с, 1H); 6,27 (с, 1H); 3,90 - 2,72 (м, 8H); 3,80 (с, 3H); 3,49 (с,3H); 2,15-1,20(м,4H).
Пример 18
Получение дигидрохлорида (2S, 3S)-3-((6-метокси-1- (трифторметил)-1,2,3,4-тетрагидронафталин-7-ил)метил)амино - 2-фенилпиперидина (Соединение 94)
(I) 1-Гидрокси-6-метокси-1-(трифторметил)-1,2,3,4- тетрагидронафталин (Соединение 88)
Это соединение получают из 6-метокси-1-тетралона по методике, аналогичной методике получения Соединения 62.
Спектр 1H-ЯМР (CDCl3): 7,61 (д, J = 8,8 Гц, 1H); 6,80 (дд, J = 8,8, 2,6 Гц, 1H); 6,66 (д, J = 2,6 Гц, 1H); 3,80 (с, 3H); 2,90- 2,66 (м, 2H); 2,30 (с, 1H); 2,40- 1,75(м,4H).
(II) 6-Метокси-1-(трифторметил)-3,4- дигидронафталин (Соединение 89)
Это соединение получают из Соединения 88 по методике, аналогичной методике получения Соединения 63.
Спектр 1H-ЯМР (CDCl3): 7,39 - 7,28 (м,1H); 6,80- 6,70 (м, 2H); 6,61 - 6,52 (м, 1H); 3,81 (с, 3H); 2,84- 2,72 (м, 2H); 2,46 - 2,30 (м, 2H).
(III) 6-Метокси-1-(трифторметил)-1,2,3,4-тетрагидронафталин (Соединение 90.)
Это соединение получают из Соединения 89 по методике, аналогичной методике получения Соединения 64.
Спектр 1H-ЯМР (CDCl3): 7,26 (д, J = 8,4 Гц, 1H); 6,74 (дд, J = 8,4, 2,9 Гц, 1H); 6,66 (д, J = 2,9 Гц, 1H); 3,79 (с, 3H): 3,57 - 3,36 (м, 1H); 2,90-2,62 (м, 2H); 2,20 -1,60 (с, 4H).
(IV) 7-Формил-6-метокси-1-(трифторметил)- 1,2,3,4-тетрагидронафталин (Соединение 91)
Это соединение получают из Соединения 90 по методике, аналогичной методике получения Соединения 46.
Спектр 1H-ЯМР (CDCl3): 10,39 (с, 1H); 7,81 (с, 1H); 6,74 (с, 1H); 3,91 (с, 3H); 3,60 - 3,40 (м, 1H); 2,98 - 2,70 (м, 2H); 2,27 - 1,65 (м, 4H).
(V) (2S, 3S)-1-трет-Бутоксикарбонил-3-((6-метокси-1-(трифторметил)- 1,2,3,4-тетрагидронафталин-7-ил)метил)амино-2-фенилпиперидин (Соединение 92)
Это соединение получают из Соединений 12 и 91 по методике, аналогичной методике получения Соединения 26. На следующей стадии его используют без дополнительной очистки.
(VI)(2S, 3S)-3-((6-метокси-1-(трифторметил)-1,2,3,4- тетрагидронафталин-7-ил)метил)амино-2-фенилпиперидин (Соединение 93)
Это соединение получают из Соединения 92 по методике, аналогичной методике получения Соединения 27.
Спектр 1H-ЯМР (CDCl3): 7,38 -7,17 (м, 5H); 6,94 (с, 1H); 6,42 и 6,39 (каждый с, всего 1H); 3,90 (д, J = 1,5 Гц, 1H); 3,73 - 3,58 (м, 1H); 3,50 - 3,23 (м, 6H); 2,90 - 2,60 (м, 4H); 2,39 (уш. с, 2H); 2,20 - 1,52 (м, 7H); 1,50-1,34(м,1H).
(VII) Дигидрохлорид (2S,3S)-3-((6-метокси-1-(трифторметил)- 1,2,3,4-тетрагидронафталин-7-ил)метил)амино-2-фенилпиперидина (Соединение 94)
Это соединение получают из Соединения 93 по методике, аналогичной методике получения Соединения 28.
Т.пл.:227-230oC.
ИК-спектр (KBr): 3435, 1624, 1587, 1561, 1507, 1466, 1452, 1433, 1420, 1336, 1260, 1247, 1171, 1138, 1116, 1106, 1044, 979, 834, 748, 693.
Пример 19
Получение дигидрохлорида (2S,3S)-3-((2,2-дифтор-6-метокси-1,2,3,4-тетрагидронафталин-7-ил) метил)амино-2-фенилпиперидина (Соединение 98).
(I) 6-Метокси-2,2-дифтор-1,2,3,4-тетрагидронафталин (Соединение 95)
К раствору 6-метокси-2-тетралона (352 мг, 2,00 ммоля) в сухом CH2Cl2 (5 мл) при перемешивании и при комнатной температуре добавляют диэтиламинотрифторсульфонат (366 мг, 2,27 ммоля). Реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 7 часов. Смесь разбавляют насыщенным раствором NaHCO3 и экстрагируют CH2Cl2. Объединенный раствор сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт в виде желтого масла. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле (элюент гексан: этилацетат, (25:1), получают Соединение 95 (181 мг, 46%) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3): 7,00 (д, J = 8,1 Гц, 1H); 6,75 (дд, J = 8,4, 2,9 Гц, 1H); 6,68 (R,J= 2,6 Гц, 1H); 3,78(с,3H); 3,18 (т, J=15,0 Гц, 2H); 2,98 (т, J = 7,0 Гц, 2H); 2,27 - 2,11 (м, 2H).
(II) 7-Формил-6-метокси-2,2-дифтор-1,2,3,4-тетрагидронафталин (Соединение 96)
К раствору Соединения 95 (90 мг, 0,45 ммоля) в сухом CH2Cl2 (10 мл) при перемешивании и при температуре -78oC с помощью шприца добавляют TiCl2 (104 мг, 0,55 ммоля). Через 15 минут при той же температуре добавляют дихлорметилметиловый эфир (636 мг, 0,55 ммоля). Реакционную смесь перемешивают при - 78oC в течение 2 часов. Смесь разбавляют водой (10 мл) при охлаждении льдом и перемешивают при комнатной температуре в течение 15 минут. Органический слой отделяют и водный слой экстрагируют CH2Cl2. Объединенный раствор сушат (MgSO4) и концентрируют в вакууме, получают сырой продукт в виде желтого масла. Сырой продукт очищают колоночной хроматографией на силикагеле (элюент гексан: этилацетат, (10: 1). Получают Соединение 96 (44 мг, 43%) в виде желтого масла.
Спектр 1H-ЯМР (CDCl3): 10,39 (с, 1H); 7,56 (с, 1H); 6,75 (с, 1H); 3,91 (с, 3H); 3,21 (т, J = 14,7 Гц, 2H); 3,05 (т, J = 7,0 Гц, 2H); 2,30 -2,15 (м, 2H).
(III) (2S,3S)-1-трет-Бутоксикарбонил-3-((2,2-дифтор-6-метокси- 1,2,3,4-тетрагидронафталин-7-ил)метил)амино-2- фенилпиперидин (Соединение 97)
Это соединение получают из Соединения 96 и Соединения 12 по методике, аналогичной методике получения Соединения 13.
Спектр 1H-ЯМР (CDCl3): 7,58 (д, J = 7,3 Гц, 2H); 7,34 - 7,25 (м, 3H); 6,90 (с, 1H); 6,56 (с, 1H); 5,49 (с, 1H); 3,97 - 3,71 (м, 3H); 3,68 (с, 3H); 3,14 (т, J = 15,0 Гц, 2H); 3,08 - 2,93 (м, 4H); 2,24 - 2,09 (м, 2H); 1,81-1,53(м,4H); 1,42(с,9H).
(IV) Дигидрохлорид (2S, 3S)-3-((2,2-дифтор-6-метокси-1,2,3,4- тетрагидронафталин-7-ил)метил)амино-2-фенилпиперидина (Соединение 98)
Это соединение получают из Соединения 97 по методике, аналогичной методике получения Соединения 60.
Спектр 1H-ЯМР (CDCl3): 7,35-7,29 (м,5H); 6,64(с,1H); 6,43 (с, 1H); 3,94 (с, 1H); 3,71 - 3,27 (м, 3H); 3,44 (с, 3H); 3,07 (т, J = 15,4 Гц, 2H); 2,93 (т, J = 6,9 Гц, 2H); 2,88 - 2,77 (м, 2H); 2,25 -1,90 (м, 4H); 1,75 - 1,43 (м, 2H).
Пример 20
Получение дигидрохлорида (2S, 3S)-3-(2-метокси- 5-(2,2,2-трифтор-1-гидрокси-1-трифторметил)этил)бензил)амино-2- фенилпиперидина (Соединение 101)
(I) 4-(2,2,2-Трифтор-1-гидрокси-1-(трифторметил)этил)анизол (Соединение 99)
Это соединение получают в соответствии с методиками, описанными в журнале "Известия Академии Наук СССР", Сер. Химия, 659 (1979).
(II) 2-Меткси-5-(2,2,2-трифтор-1--гидрокси-1-(трифторметил)этил)бензальдегид (Соединение 100)
Это соединение получают из Соединения 99 по методике, аналогичной методике получения Соединения 9.
Спектр 1H-ЯМР (CDCl3): 10,47 (с, 1H); 8,29-7,03 (м,3H); 4,05 (с, 1H); 3,99(с,3H).
(III) Дигидрохлорид (2S,3S)-2-метокси-3-(5-(2,2,2-трифтор-1 -гидpoкcи-1-(трифторметил)этил)бензил)амино-2-фенилпиперидина (Соединение 101)
Это соединение получают из Соединения 100 и Соединения 1 по методике, аналогичной методике получения Соединения 2.
Спектр 1H-ЯМР (свободное основание, CDCl3): 7,66 - 6,65 (м, 8H); 4,02-2,75 (м,6H); 3,57 (с, 3H); 3,47 (с, 1H); 2,20 -1,25 (м, 4H).
Т.пл.: 299 - 302oC.
Пример 21
Получение дигидрохлорида (2S,3S)-3-[5-[(1-хлор-1-(трифторметил)этил]-2 -метоксибензиламино]-2-фенилпиперидина (Cоединение 105)
(I) 5-[(1-Хлор-1-(трифторметил)этил)-2-метоксибензальдегид (Соединение 102).
Это соединение получают из Соединения 69 по методике, аналогичной методике получения Соединения 46.
Спектр 1H-ЯМР (CDCl3): 10,47 (с, 1H); 8,06 (д, J = 2,9 Гц, 1H); 7,97 - 7,87 (м, 1H); 7,05 (д, J = 8,8 Гц, 1H); 3,98 (с, 3H); 2,15 (с, 3H).
(II) (2S,3S)-1-трет-Бутоксикарбонил-3-[5-[(1-хлор-1- (трифторметил)этил] -2-метоксибензиламино]-2-фенилпиперидин (Соединение 103)
Это соединение получают из Соединения 12 и Соединения 102 по методике, аналогичной методике получения Соединения 26. На следующей стадии его используют без дополнительной очистки.
(III) (2S,3S)-3-[5-[(1-[хор-1-(трифторметил)этил]-2-метоксибензиламино] -2-фенилпиперидин (Соединение 104]
Это соединение получают из Соединения 103 по методике, аналогичной методике получения Соединения 27.
Спектр 1H-ЯМР (CDCl3): 7,50-7,15 (м,7H); 6,72-6,62 (м,1H); 3,89 (д, J = 2,2 Гц, 1H); 3,75 - 3,60 (м, 1H); 3,51 (с, 3H); 3,40 (д, J = 14,3 Гц, 1H); 3,35-3,21 (м, 1H): 2,90-2,71 (м,2H); 2,20-1,80 (м,7H); 1,70-1,35 (м,2H).
(IV) Дигидрохлорид (2s, 3S)-3-[5-[(1-[хлор-1-(трифторметил)этил]-2-метоксибензиламино]-2- фенилпиперидина (Соединение 105)
Это соединение получают из Соединения 104 по методике, аналогичной методике получения Соединения 28.
Химические структуры соединений, полученных в Примерах 1 -21, представлены в Таблице 1.
Соединения, представленные в Таблицах 2 и 3 получены с использованием соответствующих исходных веществ методами, описанными в Примерах 10, 13, 14, 17, 18 или 19.
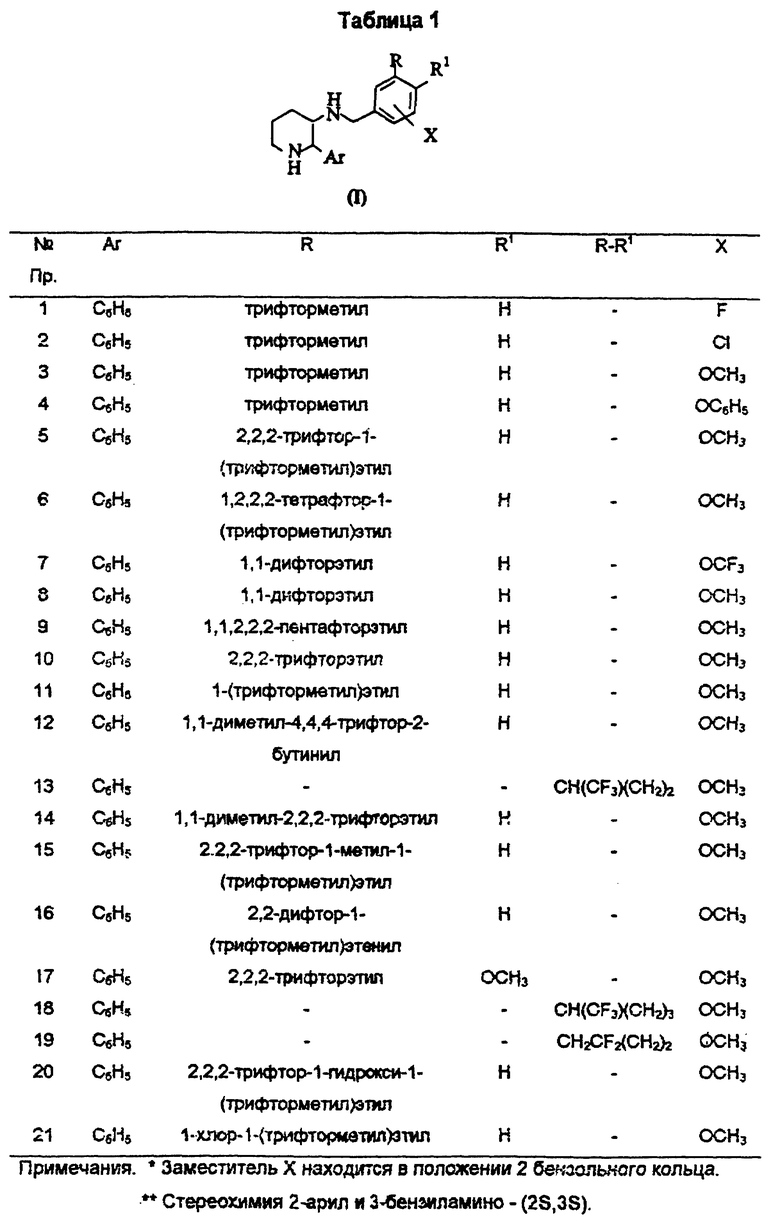




Изобретение относится к соединениям формулы I и их фармацевтически приемлемым солям, где заместитель R представляет собой галоген - C1-C8-алкил, галоген - C2-C8-алкенил, галоген - C2-C8-алкинил или галоген - C1-C8-алкил, замещенный гидрокси- или C1-C8-алкоксигруппой; R1 представляет собой атом водорода, атом галогена или C1-C8-алкоксигруппу; или R и R1 вместе с двумя атомами углерода, общими для бензольного кольца, образуют конденсированный C4-C6-циклоалкил, где один атом углерода необязательно заменен атомом кислорода, и где один или два атома углерода необязательно имеют до пяти заместителей, выбираемых из атома галогена, C1-C6-алкила и галоген - C1-C6-алкила; X представляет собой C1-C6-алкокси-, галоген - C1-C6-алкокси-, феноксигруппу или атом галогена; и Ar представляет собой фенильную группу, необязательно замещенную атомом галогена. Эти соединения могут быть использованы для лечения желудочно-кишечного расстройства, расстройства центральной нервной системы, воспалительного заболевания, рвоты и др. 3 с. и 17 з.п. ф-лы, 4 табл. 
и их фармацевтически приемлемые соли,
где R представляет собой галоген - C1 - C8-алкил, галоген - C2 - C8-алкенил, галоген - C2 - C8-алкинил или галоген - C1 - C8-алкил, замещенный гидрокси- или C1 - C8-алкоксигруппой; R1 представляет собой атом водорода, атом галогена или C1 - C6-алкоксигруппой; или
R и R1 вместе с двумя атомами углерода, общими для бензольного кольца и заместителей R и R1, образуют конденсированный C4 - C6-циклоалкил, где один атом углерода необязательно заменен атомом кислорода, и где один или два атома углерода необязательно имеют до пяти заместителей, выбираемых из числа атома галогена, C1 - C6-алкила и галоген - C1 - C6-алкила;
X представляет собой C1 - C6-алкокси-, галоген - C1 - C6-алкокси-, феноксигруппу или атом галогена; и
Ar представляет собой фенильную группу, необязательно замещенную атомом галогена.
где R1 представляет собой атом водорода, атом галогена или метоксигруппу; R2 и R3 независимо друг от друга выбирают из группы, включающей атом галогена, C1 - C6-алкил, C2 - C6-алкенил и C2 - C6-алкинил, или R2 и R3 вместе образуют C2 - C6-алкилиден, где алкильный, алкенильный, алкинильный и алкилиденовый фрагменты необязательно содержат до семи атомов галогена;
или R1 и R2 вместе образуют конденсированный C4 - C6-циклоалкил, в котором один атом углерода необязательно заменен атомом кислорода, причем C4 - C6-циклоалкил необязательно замещен заместителями в количестве до четырех, выбираемыми из группы, включающей атом галогена, C1 - C4-алкил и галоген - C1 - C4-алкил.
(2S, 3S)-3-(2-Фтор-5-(трифторметил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(2-Хлор-5-(трифторметил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(трифторметил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(2-Фенокси-5-(трифторметил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(5-(1,1-Дифторэтил)-2-(трифторметокси)бензил)амино-2-фенилпиперидин или его соли;
(2S,3S)-3-(5-(1,1-Дифторэтил)-2-метоксибензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(2,2,2-трифторэтил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(1-(трифторметил)этил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-[5-(1,1-Диметил-(4,4,4-трифтор-2-бутинил)-2-метоксибензил] амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-[5-(1,1-Диметил-2,2,2-трифторэтил)-2-метоксибензиламино] -2-фенилпиперидин или его соли;
(2S, 3S)-3-(2,4-Диметокси-5-(2,2,2-трифторэтил)бензил)амино-2-фенилпиперидин или его соли; и
(2S, 3S)-3-[5-[1-Хлор-(1-трифторметил)этил)-2-метоксибензиламино] -2-фенилпиперидин или его соли.
(2S, 3S)-2-Фенил-3-(5-(2,2,2-трифтор-1-трифторметил)этил)-2-метоксибензил)аминопиперидин или его соли;
(2S, 3S)-2-Фенил-3-(5-(1,2,2,2-тетрафтор-1-(трифторметил)этил)-2-метоксибензил)аминопиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(1,1,2,2,2-пентафторэтил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-2-Фенил-3-(5-(2,2,2-трифтор-1-метил-1-(трифторметил)этил)-2-метоксибензил)аминопиперидин или его соли;
(2S, 3S)-3-[5-[2,2-Дифтор-1-(трифторметил)этенил] -2-метоксибензил] амино-2-фенилпиперидин или его соли; и
(2S,3S)-3-(2-Метокси-5-(2,2,2-трифтор-1-гидрокси-1-(трифторметил)этил)бензил)амино-2-фенилпиперидин или его соли.
(2S,3S)-3-[5-Метокси-1-(трифторметил)индан-6-ил)метиламино]-2-фенилпиперидин или его соли;
(2S, 3S)-3-((6-Метокси-1-(трифторметил)-1,2,3,4-тетрагидронафталин-7-ил)метил)амино-2-фенилпиперидин или его соли; и
(2S,3S)-3-((2,2-Дифтор-6-метокси-1,2,3,4-тетрагидронафтали-7-ил)метил)амино-2-фенилпиперидин или его соли.
(2S, 3S)-3-(2-Метокси-5-(трифторметил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(2,2,2-трифторэтил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(2-Метокси-5-(1-(трифторметил)этил)бензил)амино-2-фенилпиперидин или его соли;
(2S, 3S)-3-(5-(1,1-Дифторэти)-2-(трифторметокси)бензил)амино-2-фенилпиперидин или его соли; и
(2S, 3S)-3-[5-(1,1-Диметил-2,2,2-трифторэтил)-2-метоксибензиламино] -2-фенилпиперидин или его соли.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Цифровой измеритель скорости вращения | 1976 |
|
SU610021A1 |
| Грузоподъемный кран | 1977 |
|
SU653208A1 |
| EP 0655246 A1, 31.05.1995 | |||
| Землесос для разработки грунта под водой | 1977 |
|
SU627221A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДПЫХ АМИНОПИПЕРИДИНА | 0 |
|
SU310448A1 |

