Настоящее изобретение относится к способу получения алкена (алкенов) из исходного материала, включающего по меньшей мере один одноатомный алифатический парафиновый спирт.
Олефин (олефины) традиционно получают крекингом с водяным паром или каталитическим крекингом углеводородов. Однако по мере того как запасы нефти уменьшаются, цена нефти неизбежно увеличивается, что делает получение легкого олефина (олефинов) дорогостоящим процессом. Таким образом, существует постоянно растущая потребность в ненефтяных путях получения олефина (олефинов) C2+, практически этилена и пропилена. Такой олефин (олефины) представляет собой эффективный исходный материал для многочисленных химических продуктов, включающих полимерные продукты, такие как полиэтилен.
В последние годы поиск альтернативных материалов для получения олефина (олефинов) C2+ приводит к применению спиртов, таких как метанол, этанол и более высокомолекулярные спирты. Упомянутые спирты могут быть получены ферментацией, например, сахаров и/или целлюлозных материалов.
По другому варианту спирты могут быть получены из синтез-газа. Синтез-газом называют сочетание водорода и оксидов углерода, полученных в установке для синтез-газа из такого источника углерода, как природный газ, нефтяные дистилляты, биомасса и углеродистые материалы, включая уголь, перерабатываемые для вторичного использования пластмассы, муниципальные отходы и любой органический материал. Таким образом, спирт и производные спиртов могут явиться путями для получения олефина (олефинов) и других родственных углеводородов на ненефтяной основе.
Обычно получение оксигенатов, главным образом метанола, проводят осуществлением трех технологических стадий. Этими тремя технологическими стадиями являются получение синтез-газа, синтез метанола и очистка метанола. На стадии получения синтез-газа может быть использована дополнительная стадия, на которой исходный материал обрабатывают, например исходный материал очищают перед превращением в синтез-газ для удаления серы и других потенциальных каталитических ядов. Эта обработка может быть также проведена после получения синтез-газа, например, когда используют уголь или биомассу.
Способы получения смесей оксида (оксидов) углерода и водорода (синтез-газа) известны хорошо. Каждый обладает своими преимуществами и недостатками, и выбор для применения конкретного способа реформинга определяется соображениями экономики и доступности потока исходных материалов, а также целевым мольным соотношением H2:CO в исходном материале, получаемом в результате реакции реформинга. Синтез-газ может быть получен с использованием любых методов, известных в данной области техники, включая частичное окисление углеводородов, реформинг с водяным паром, реформинг с газовым нагревом, микроканальный реформинг (как это изложено, например, в патенте US 6284217, который включен в настоящее описание в качестве ссылки), плазменный реформинг, автотермический реформинг и любое их сочетание. Обсуждение этих технологий получения синтез-газа приведено в журналах "Hydrocarbon Processing", V 78, №4, 87-90, 92-93 (апрель 1999 г.) и "Petrole et Thechniques", №415, 86-93 (июль-август, 1998 г.). Предусмотрена также возможность получения синтез-газа каталитическим частичным окислением углеводородов в микроструктурном реакторе, пример чего представлен в "IMRET 3: Proceedings of the Third International Conference on Microreaction Technology", Editor W Ehrfeld, Springer Verlag, 1999, cc.187-196. По другому варианту синтез-газ может быть получен каталитическим частичным окислением углеводородистых исходных материалов с кратковременным контактированием так, как изложено в ЕР 0303438. В предпочтительном варианте синтез-газ получают посредством способа с "компактной реформинг-установкой" так, как изложено в "Hydrocarbon Engineering", 2000, 5, (5), 67-69; "Hydrocarbon Processing", 79/9, 34 (сентябрь, 2000 г.); "Today's Refinery", 15/8, 9 (август, 2000 г.); WO 99/02254 и WO 200023689.
В случае промышленного получения синтез-газа давление, под которым получают синтез-газ, как правило, находится в интервале от приблизительно 20 до 75 бар, а температура, при которой синтез-газ выходит из реформинг-установки, находится в интервале от приблизительно 700 до 1100°С. Синтез-газ содержит водород и оксид углерода в молярном соотношении, которое в зависимости от исходного материала для синтез-газа находится в интервале от 0,8 до 3.
Получение синтез-газа, также известное как реформинг, можно осуществлять в одну стадию, на которой все потребляющие энергию реакции реформинга протекают в единственной трубной установке реформинга с водяным паром. Применение установки одностадийного реформинга приводит к получению добавочного водорода. В предпочтительном другом варианте получение синтез-газа может происходить в двухстадийном способе реформинга, при осуществлении которого первичный реформинг в трубной установке реформинга с водяным паром совмещают со вторичной стадией реформинга со сжиганием в кислороде, на которой получают синтез-газ с нехваткой водорода. С применением такого сочетания существует возможность регулировать состав синтез-газа с достижением наиболее приемлемого состава для синтеза метанола. В качестве альтернативы автотермический реформинг, в котором автономная установка реформинга со сжиганием в кислороде производит синтез-газ, характеризующийся нехваткой водорода, с последующим в технологической линии удалением диоксида углерода для восстановления целевого отношения водорода к оксиду углерода приводит к упрощенной технологической схеме с уменьшенными капитальными затратами. Важной частью любой стадии со сжиганием в кислороде является конструкция горелки. Горелка смешивает углеводород и кислород и сжиганием в пламени создает тепло для превращения углеводородов.
Реакция превращения синтез-газа в оксигенаты, такие как метанол, является ограниченной экзотермическим равновесием реакцией, протеканию которой содействуют низкие температуры. Над гетерогенным катализатором требуются также высокие давления, поскольку реакции, в которых образуется метанол, проявляют уменьшение объема среды. Как это изложено в US №3326956, синтез метанола под низким давлением основан на катализаторе из оксида меди/оксида цинка/оксида алюминия, который среди разнообразных катализаторов, включая CuO/ZnO/Al2O3, CuO/ZnO/Cr2O3, ZnO/Cr2O3, Fe, Co, Ni, Ru, Os, Pt и Pd, как правило, работает под номинальным давлением от 5 до 10 МПа и при температурах в интервале от приблизительно 150 до 450°С. Для получения метанола и диметилового эфира предпочтительны катализаторы на основе ZnO. Катализатор синтеза метанола под низким давлением на основе меди технически доступен у таких поставщиков, как фирмы BASF, ICI Ltd. of the United Kingdom и Haldor-Topsoe. Значения выхода метанола в случаях катализаторов на основе меди обычно превышают 99,5% от подвергшихся превращению имеющихся СО+СО2. Побочным продуктом превращения синтез-газа в оксигенаты является вода. В статье, озаглавленной "Selection of Technology for Large Methanol Plants", написанной Helge Holm-Larsen, представленной на Всемирной конференции по метанолу 30 ноября -1 декабря 1994 г. в Женеве, Швейцария, которая включена в настоящее описание в качестве ссылки, приведен обзор разработок в производстве метанола и показано, как дальнейшее снижение стоимости получения метанола скажется на конструировании очень больших установок производительностью, приближающейся к 10000 метрических т/день.
В US №4543435 описан способ превращения оксигенатного исходного материала, включающего метанол, диметиловый эфир или т.п., в реакторе превращения оксигената в жидкие углеводороды, включающие С2-С4олефин (олефины) и углеводороды С5+. С2-С4олефин (олефины) сжимают для выделения богатого этиленом газа. Этот богатый этиленом газ возвращают в реактор превращения оксигената. В US №4076761 описан способ превращения оксигенатов в бензин с возвратом богатого водородом газообразного продукта в установку для синтез-газа или реакционную зону превращения оксигената.
В US №5177114 описан способ превращения природного газа в жидкие углеводороды бензинового сорта и/или олефин (олефины) превращением природного газа в синтез-газ, превращением синтез-газа в сырой метанол и/или диметиловый эфир и последующим превращением сырого метанола/диметилового эфира в бензин и олефин (олефины).
Международная заявка на патент №93/13013, поданная Kvisle и др., относится к усовершенствованному способу приготовления кремнеалюмофосфатного катализатора, который более стабилен к дезактивации от закоксовывания. В этой заявке на патент говорится, что по истечении некоторого периода времени все такие катализаторы, используемые для превращения метанола в олефин (олефины) (МвО), утрачивают способность к активному превращению метанола в углеводороды главным образом потому, что микропористая кристаллическая структура закоксовывается, т.е. заполняется низколетучими углеродистыми соединениями, которые блокируют пористую структуру. Эти углеродистые соединения могут быть удалены по обычным методам, таким как сжигание на воздухе.
В ЕРО публикации №0407038 А1 описан способ получения диалкиловых эфиров, включающий подачу потока, содержащего алифатический спирт, в зону подачи ректификационной колонны как реактора, контактирование этого потока с неподвижным слоем твердой кислой каталитической ректификационной структуры с получением соответствующего диалкилового эфира и воды и одновременное отделение ректификацией простого эфира как продукта от воды и непрореагировавших материалов.
В US №5817906 описан способ получения легкого олефина (олефинов) из сырого оксигенатного исходного материала, включающего спирт и воду. В этом способе применяют две реакционные стадии. Во-первых, с использованием реакции с ректификацией спирт превращают в простой эфир. Затем простой эфир в дальнейшем направляют в зону превращения оксигената, содержащую металлалюмосиликатный катализатор, с получением потока легких олефинов.
Процесс превращения метанола в олефин (олефины) (МвО) может быть описан как дегидративное сочетание метанола до олефина (олефинов) и хорошо известен химику, который может его использовать для получения олефина (олефинов) из спирта (спиртов). Полагают, что этот механизм осуществляется посредством сочетания C1-фрагментов, образуемых катализируемой кислотой дегидратацией метанола, возможно через метилоксониевый промежуточный продукт. Однако главный недостаток упомянутого процесса МвО заключается в том, что совместно с ароматическими и алкановыми побочными продуктами образуется ряд олефинов, вследствие чего, в свою очередь, выделение целевого олефина (олефинов) сопряжено с очень большими затруднениями технологического порядка и затратами.
Как содействующие превращению оксигенатов благодаря химическому пути от метанола до олефина (МвО) к углеводородным смесям известны молекулярные сита, такие как микропористый кристаллический цеолит и нецеолитные катализаторы, особенно кремнеалюмофосфаты (SAPO). Такой метод для этих катализаторов различных типов описан в многочисленных патентах: US №№3928483, 4025575, 4252479 (Chang и др.), 4496786 (Santilli и др.), 4547616 (Avidan и др.), 4677243 (Kaiser), 4843183 (Inui), 4499314 (Seddon и др.), 4447669 (Harmon и др.), 5095163 (Barger), 5191141 (Barger), 5126308 (Barger), 4973792 (Lewis) и 4861938 (Lewis).
Однако эта реакция характеризуется стадией высокоэнергетической активации (возможно на стадии получения метанола или диметилового эфира), вследствие чего для достижения высокой степени превращения с целью поступательного ведения реакции существует потребность в высоких температурах, например 450°С. Обычно с целью добиться этих высокотемпературных условий в таких системах предусматривают различные средства, такие как возврат в процесс нагретого катализатора и последующие нагревательные системы. Однако, к сожалению, проведение процесса при этих упомянутых высоких температурах приводит к возникновению таких основных проблем, как дезактивация катализатора, закоксовывание и образование побочных продуктов. С целью избежать этих проблем реакции можно проводить при более низких температурах, но это требует дорогостоящего возврата в процесс промежуточных продуктов и реагентов.
Другой главный недостаток, связанный с этим методом, заключается в том, что совместно с олефином (олефинами) образуются ароматические и алкановые побочные продукты, отделение которых от целевых продуктов сопряжено как с затруднениями технологического порядка, так и с большими затратами; так, например, выделение этилена и этана является дорогостоящим процессом.
Эти и другие недостатки известных технических решений показывают, что существует потребность в разработке усовершенствованного и/или альтернативного способа получения алкена (алкенов) С2+ из спиртов.
Объектом настоящего изобретения конкретно является другой способ получения олефина (олефинов) из спирта (спиртов), отличный от метода МвО. Упомянутый химический способ по настоящему изобретению осуществляют, как полагают, посредством дегидратации спиртов С2+ с получением олефина (олефинов).
Объектом настоящего изобретения является способ получения алкена (алкенов) из исходного материала, включающего по меньшей мере один одноатомный алифатический парафиновый первичный (или вторичный) спирт, содержащий этанол, пропанол (пропанолы) или их смесь, характеризующийся следующими стадиями:
1) одноатомный алифатический парафиновый первичный (или вторичный) спирт (спирты) в реакционно-ректификационной колонне под повышенным давлением и при повышенной температуре превращают в алкен (алкены) с соответствующим аналогичным числом углеродных атомов таким образом, что поток (потоки) головных погонов, отводимый из верхней части упомянутой реакционно-ректификационной колонны, включает, по существу, упомянутый алкен (алкены);
2) затем верхний поток со стадии 1 охлаждают до температуры, достаточной для конденсации по меньшей мере части алкена (алкенов) с самой высокой точкой кипения;
3) далее по меньшей мере часть конденсированного алкена (алкенов) со стадии 2 возвращают назад в упомянутую реакционно-ректификационную колонну в качестве возвращаемой флегмы;
4) одновременно выделяют оставшийся алкен (алкены).
В соответствии с предпочтительным вариантом объектом настоящего изобретения является способ превращения углеводорода в алкен (алкены), включающий следующие стадии:
а) превращение углеводорода в реакторе синтез-газа в смесь оксида (оксидов) углерода и водорода;
б) превращение упомянутой смеси оксида (оксидов) углерода и водорода со стадии а) в присутствии порошкообразного катализатора в реакторе при температуре, находящейся в пределах от 200 до 400°С, и под давлением от 50 до 200 бар в исходный материал, включающий по меньшей мере один первичный (или вторичный) одноатомный алифатический парафиновый спирт, содержащий этанол, пропанол (пропанолы) или их смесь, и
в) проведение процесса в соответствии с вышеуказанными стадиями с 1 по 4 в соответствии с настоящим изобретением с получением вышеупомянутого алкена (алкенов).
В соответствии с настоящим изобретением способ получения алкена (алкенов) из спирта (спиртов) осуществляют посредством дегидратации спиртов С2+; для того чтобы это происходило, должен содержаться один или несколько альфа-водородных атомов, например как у фенола, неопентилгликоля, например 2,2-диметилпропан-1-ола, которые посредством этого механизма не дегидратируются, тогда как в этаноле, н-пропаноле и трет-бутаноле дегидратируются. Эти реакции дегидратации отличаются от вышеупомянутого процесса МвО тем, что хотя в процессе дегидратации никакого сочетания углеродных фрагментов не требуется, во время элиминирования воды образуется двойная связь С-С и в результате может быть достигнута очень высокая селективность. Обычно условия, применяемые в процессе МвО, оказываются намного более жесткими, чем условия, применяемые при дегидратации спиртов.
В целесообразном варианте способ по настоящему изобретению, т.е. превращение исходного материала в простой эфир (эфиры) и/или алкен (алкены) осуществляют в одной реакционно-ректификационной колонне, благодаря чему уменьшаются капитальные и энергетические затраты. Дегидратацию исходного материала, как полагают, проводят любой прямой дегидратацией до алкена (алкенов) и воды:
Уравнение 1
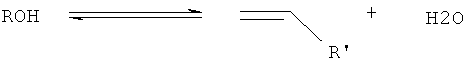 или посредством простого эфирного промежуточного продукта;
или посредством простого эфирного промежуточного продукта;
Уравнение 2

Уравнение 3
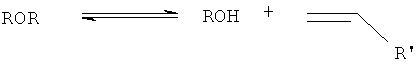
где R и R' обозначают этильную, пропильную, бутильную или пентильную группу.
Уравнение 1 демонстрирует эндотермическое прямое элиминирование спирта до алкена (алкенов) и воды. С уравнением 1 конкурируют уравнения 2 и 3, протекают экзотермическая реакция этерификации (уравнение 2) и эндотермическое элиминирование простого эфира (эфиров) с образованием алкена (алкенов) и спирта (уравнение 3). Однако общая дегидратация спиртов до алкена (алкенов) представляет собой эндотермический процесс.
Все основные представленные выше реакции, протекающие в реакционно-ректификационной колонне, катализируют кислотными катализаторами. Хотя вышеупомянутый механизм, как полагают, является справедливым для первичных и вторичных спиртов, необходимо отметить, что он не является верным для третичных спиртов. Так, например, третичные спирты, такие как трет-бутанол, обычно дегидратируются только непосредственно до изобутена (согласно уравнению 1) и, следовательно, метилтрет-бутиловый эфир в этом процессе не получают. Таким образом, настоящее изобретение обладает дополнительным преимуществом, заключающимся в том, что оно создано с расчетом на получение и возможность располагать не только алкеном (алкенами), но также простым эфиром (эфирами) (образующимися согласно уравнениям 2 и 3).
Все реакции согласно уравнениям 1, 2 и 3 являются ограниченными равновесием. Однако поскольку в соответствии с настоящим изобретением все три реакции протекают в реакционно-ректификационной колонне, при этом благодаря непрерывному удалению продуктов посредством ректификации в ходе протекания ограниченных равновесием реакций достигается увеличенное превращение. Это преимущество является ожидаемым, если основываться на принципе Ле Шателье, который утверждает, что если на систему, находящуюся в равновесии, оказывается какое-либо нарушающее воздействие, то система самостоятельно регулируется для восстановления этого равновесия. Таким образом, при выполнении настоящего изобретения благодаря непрерывному удалению продуктов посредством ректификации скорость протекания ограниченной равновесием реакции повышают за пределы ее термодинамического ограничения, в результате чего существует повышенная концентрация реагентов. Следовательно, олефиновый продукт становится концентрированным в верхней части реакционно-ректификационной колонны и носит название верхнего потока, а вода концентрируется в основании реакционно-ректификационной колонны и известна в качестве нижнего продукта. Спирт (спирты) и простой эфир (эфиры), образующие с водой азеотропы, характеризуются промежуточной точкой кипения и концентрируются в реакционной зоне реакционно-ректификационной колонны.
Хорошо известно, что когда в паровой фазе используют гетерогенный катализатор, этанол ингибирует элиминирование диэтилового эфира благодаря его более сильному каталитическому взаимодействию. Это может привести к последовательности реакций. Так, например, когда этанол направляют в проточный реактор с катализатором дегидратации, уравнения 1 и 2 превалируют до тех пор, пока концентрация этанола не падает до уровня, при котором простой эфир может эффективно конкурировать за каталитические участки. Конкуренция двух реагентов за активный участок может быть описана механизмом Лангмира-Хиншелвуда (например, Chemical Kinetics, издание 3-е, автор K.J.Laidler, сс.249-251, Harper и Row publishers New York). Было установлено, что влияние этого взаимодействия на реакторы периодического действия или проточные уменьшает скорость получения этилена до тех пор, пока не израсходовано основное количество этанола (см., например, Collection of czechoslavak chemical comms, 1986 51 (4), cc. 763-73 V.Moravek и M.Kraus).
Однако в соответствии с настоящим изобретением вследствие сочетания реакции и ректификации это ограничение может быть устранено. Так, например, в реакционно-ректификационной колонне простой эфир (эфиры) и спирт (спирты) разделяют соответственно по их азеотропам и по их точкам кипения. Таким образом, простой эфир (эфиры) концентрируют в катализаторе не в тех местах, где спирт (спирты), и, следовательно, результатом этого является уменьшенное спиртовое ингибирование реакции.
Реакционно-ректификационная колонна, в которой проводят процесс, относится к объединенным ректификационной колонне и реактору. Внутрикорпусные устройства реакционно-ректификационной колонны размещают таким образом, чтобы обеспечить наличие множества "теоретических тарелок", которые содействуют отделению продуктов от реагентов. Этими внутрикорпусными устройствами колонны обычно являются те, которые используют при обычной ректификации, например ситовые тарелки, неструктурированная и структурированная насадки, колпачковые тарелки и их сочетания. Этот конкретный аппарат оказывается очень эффективным для промотирования парожидкостного контактирования и, следовательно, фракционной отгонки продукта (продуктов) из реагентов. Применяемый катализатор (катализаторы) может быть либо гомогенным либо гетерогенным, причем предпочтительным вариантом является гомогенный катализатор (катализаторы).
Когда в соответствии с настоящим изобретением используют гетерогенный катализатор (катализаторы), этот катализатор (катализаторы) размещают таким образом, чтобы он имел возможность для максимального взаимодействия с реагентами и реакционными промежуточными продуктами; это может быть достигнуто с применением в колонне в качестве опор для катализатора (катализаторов) внутрикорпусных устройств; например ионообменные смолы могут опираться на них, находиться в кипах тканей, на ситовых тарелках, пакетах из стекловолокна, в реакционно-ректификационных установках для метилтрет-бутилового эфира (МТБЭ). Катализатор (катализаторы) может также составлять насадку колонны, например он может быть снабжен покрытием, экструдирован, отформован в виде колец Рашига или насадки для колонны любого другого известного типа. Катализатор (катализаторы) может быть также диспергирован внутри немодифицированных насадок колонны. Гетерогенный катализатор (катализаторы) обладает дополнительным преимуществом, заключающимся в том, что разделение реагентов и продуктов достигается тривиальным путем, т.е. его осуществляют физическим разделением, например фильтрованием.
В соответствии с настоящим изобретением приемлемые гетерогенные катализаторы включают, хотя ими их список не ограничен, нерастворимые гетерополикислоты, сульфированные носители (например, продукт Nafion и ионообменные смолы), цеолиты, модифицированные металлами цеолиты, мордениты и их смеси; предпочтительны гетерополикислоты и ионообменные смолы, более предпочтительны гетерополикислоты, а наиболее предпочтительны соли 12-вольфрамокремниевой кислоты и 18-вольфрамофосфорной кислоты.
Гетерополикислоты по настоящему изобретению представляют собой комплексные, высокомолекулярные анионы, включающие связанные атомами кислорода атомы поливалентных металлов. Как правило, каждый анион включает от 12 до 18 связанных атомами кислорода атомов поливалентных металлов. Атомы поливалентных металлов, которые известны как периферические атомы, симметричным образом окружают один или несколько центральных атомов. Периферические атомы обычно представляют собой один или несколько атомов молибдена, вольфрама, ванадия, ниобия, тантала и любого другого поливалентного металла. Центральными атомами в предпочтительном варианте служат атомы кремния или фосфора, но по другому варианту они могут включать любой один из большого разнообразия атомов элементов групп с I по VIII Периодической таблицы элементов. К ним относятся ионы меди, бериллия, цинка, кобальта, никеля, бора, алюминия, галлия, железа, церия, мышьяка, сурьмы, фосфора, висмута, хрома, родия, кремния, германия, олова, титана, циркония, ванадия, серы, теллура, марганца, никеля, платины, тория, гафния, церия, ванадия, теллура и иода. Приемлемые гетерополикислоты включают гетерополикислоты Кеггина (Keggin), Уэллса-Доусона (Wells-Dawson) и Андерсона-Эванса-Перлова (Anderson-Evans-Perloff). Конкретными примерами приемлемых гетерополикислот являются следующие соединения:
и свободная кислота или неполные соли следующих гетерополикислот:
Гетерополикислоты, используемые при выполнении настоящего изобретения, могут обладать молекулярными массами больше 700 и меньше 8500, предпочтительно больше 2800 и меньше 6000. Такие гетерополикислоты также включают димерные комплексы.
Для приготовления катализаторов, которые могут быть целесообразно использованы при выполнении настоящего изобретения, носитель катализатора пропитывают неводным раствором гетерополикислоты и катализатор осаждают получением in situ соли низкой растворимости. Такой раствор готовят растворением гетерополикислоты в неводном растворителе. Приемлемые растворители включают полярные растворители, такие как спирты, кетоны и альдегиды. Приемлемые спирты включают спирты с С1 по C8, предпочтительно спирты с С1 по С4, а наиболее предпочтительно метанол и этанол. Приемлемые кетоны представляют собой кетоны с С2 по С4, например ацетон. Концентрация гетерополикислоты в растворе в предпочтительном варианте составляет от 10 до 80 мас.%, более предпочтительно от 20 до 60 мас.%, а наиболее предпочтительно от 30 до 50 мас.%.
Для приготовления нерастворимого катализатора пропитку можно осуществлять с использованием метода начальной влажности со стадией частичной нейтрализации. Можно использовать любой приемлемый метод сушки, причем предпочтительно выпаривание в стандартном стендовом роторном испарителе.
По другому варианту носитель катализатора может быть погружен в водный раствор и оставлен для пропитки, а затем для осаждения ГПК на носителе добавлен раствор противоиона. Далее пропитанный носитель может быть промыт и высушен. Это может быть достигнуто с использованием любого обычного метода разделения, включая, например, декантацию и/или фильтрование. После выделения пропитанный носитель может быть высушен, предпочтительно помещением носителя в сушильный шкаф. По другому варианту или к тому же можно использовать эксикатор. Количество гетерополикислоты, впитавшейся в носитель, в целесообразном варианте находится в интервале от 10 до 60 мас.%, а предпочтительно от 30 до 50 мас.%, в пересчете на общую массу гетерополикислоты и носителя.
Приемлемые носители катализатора включают кремнеземные носители, такие как силикагелевые носители и носители, полученные гидролизом SiCl4 в пламени. Предпочтительные носители, по существу, свободны от посторонних металлов или элементов, которые могут негативно воздействовать на каталитическую активность системы. Таким образом, приемлемые кремнеземные носители характеризуются чистотой по меньшей мере 99 мас.%. Количество примесей составляет до меньше 1 мас.%, предпочтительно меньше 0,60 мас.%, а более предпочтительно меньше 0,30 мас.%. Удельный объем пор носителя составляет от 0,3 до 1,2 мл/г, предпочтительно от 0,6 до 1,0 мл/г. Средний радиус пор (перед применением) носителя составляет от 10 до 500 Å, предпочтительно от 30 до 100 Å. Носитель обладает сопротивлением раздавливанию по меньшей мере 2 кг силы, целесообразно по меньшей мере 5 кг силы, предпочтительно по меньшей мере 6 кг, а более предпочтительно по меньшей мере 7 кг. Объемная плотность носителя составляет по меньшей мере 380 г/л, предпочтительно по меньшей мере 440 г/л.
Приемлемые силикагелевые носители включают продукты Grace 57 и 1371, причем предпочтителен продукт Grace №1371. Grace №1371 обладает средним размером частиц от 0,1 до 3,5 мм. Однако при необходимости эти частицы могут быть раздавлены и просеяны до меньших размеров, например от 0,5 до 2 мм.
Приемлемые носители, полученные гидролизом SiCl4 в пламени, могут быть приготовлены таблетированием продукта AEROSIL® 200 (фирмы Degussa). Примером такого носителя является продукт Support 350. Приемлемые методы таблетирования описаны в US 5086031, конкретно в примерах. Средний диаметр частиц гранул составляет от 2 до 10 мм, предпочтительно от 4 до 6 мм.
По дополнительному варианту выполнения упомянутого изобретения предусмотрено, чтобы носитель катализатора, используемый при осуществлении настоящего изобретения, был вначале обработан фторирующим агентом; полагают, что благодаря высокоэлектроотрицательной природе атома фтора получаемый эффект проявляется в том, что электронные свойства носителя катализатора, по-видимому, модифицируются, и полагают, что это дает следующие преимущества: инертность носителя и/или улучшенная кислотность, что таким образом улучшает в целом селективность и/или активность катализатора.
Фторирующие агенты, которые можно использовать для обработки носителя, могут включать, хотя ими их список не ограничен, фторид водорода, водные растворы плавиковой кислоты, смеси плавиковой кислоты с меньшими количествами других кислот, таких как соляная и уксусная кислоты, или растворы кислот, в которые предварительно добавляют некоторые алюминиевые соли, или слабые растворы плавиковой кислоты, содержащие алюминиевую соль. Обработку упомянутого носителя катализатора водными растворами плавиковой кислоты можно осуществлять замачиванием частиц катализатора в растворе этой кислоты крепостью в пределах от 1 до 8% в течение периода в пределах от 1 до 24 ч. Затем фторированный носитель может быть пропитан выбранным катализатором.
В соответствии с настоящим изобретением в реакционно-ректификационной колонне может быть также использован гомогенный катализатор (катализаторы). Предпочтительный гомогенный катализатор (катализаторы) обладает более высокой точкой кипения, чем реагенты и продукты, в результате чего он остается по преимуществу в жидкой фазе (фазах) колонны и в конечном счете концентрируется в реакционном сосуде. Взаимодействие между этими упомянутыми катализатором (катализаторами) и реагентами в реакционной зоне можно регулировать варьированием количества катализатора (катализаторов), возвращаемого в реакционно-ректификационную колонну, и заменой внутрикорпусных устройств колонны для увеличения количества удерживаемой жидкости. Отделение гомогенных катализаторов от воды, накапливающейся в ребойлере, может быть достигнуто конденсацией выше ребойлера потока пара по преимуществу чистой воды. Дополнительные преимущества применения гомогенного катализатора (катализаторов) заключаются в том, что может быть свободно изменена концентрация катализатора, и в том, что дезактивированные катализаторы могут быть из системы легко удалены и заменены свежим катализатором. Затем выделенный из ребойлера раствор гомогенного катализатора возвращают в колонну. Для концентрирования катализатора там, где это требуется, может быть использована одна или несколько точек добавления.
Приемлемые гомогенные катализаторы включают, хотя ими их список не ограничен, сульфоновые кислоты, такие как метансульфоновая кислота, пара-толуолсульфоновая кислота, трифторметансульфоновая кислота, серные кислоты, гетерополикислоты и фосфорная кислота; предпочтительны фосфорная кислота и органосульфоновые кислоты.
В соответствии с настоящим изобретением верхний поток алкена (алкенов), который отводят из верхней части реакционно-ректификационной колонны, состоит из одного или нескольких алкенов, по существу, из пропилена и/или этилена, и по преимуществу свободен от оксигенатов (воды, простых эфиров и спиртов). Затем этот упомянутый поток охлаждают до температуры, достаточной для конденсации по меньшей мере части алкена (алкенов) с самой высокой точкой кипения. В виде потока чистого алкена (алкенов) выделяют фракцию неконденсированного пара. Далее по меньшей мере часть конденсированного алкена (алкенов) возвращают назад в реакционно-ректификационную колонну, а оставшийся жидкий алкен (алкены) также выделяют. Возврат по меньшей мере части алкена (алкенов) для выполнения настоящего изобретения имеет существенное значение, поскольку в целесообразном варианте это позволяет удалить простые эфиры, воду и спирты, которые могут содержаться в результате предыдущей реакции дегидратации, и таким образом улучшает селективность в отношении продукта. При создании настоящего изобретения было установлено, что хотя этот механизм реакции внутри реакционно-ректификационной колонны осуществляется посредством простого эфирного промежуточного продукта, выделенный алкен (алкены) никаких примесей не содержит.
Аппарат, применяемый для охлаждения алкена (алкенов) в верхнем потоке, представляет собой один или несколько размещенных последовательно конденсаторов. В целесообразном варианте верхний поток конденсируют только весьма частично с целью как улучшить экономику процесса, так и предотвратить образование льда, который мог бы, в свою очередь, привести к засорению и/или блокированию.
В соответствии с настоящим изобретением коэффициент орошения по массе х/у, где х обозначает интенсивность (кг/ч), с которой алкен (алкены) возвращают из верхнего потока (потоков) назад в реактор, а у обозначает интенсивность (кг/ч), с которой алкен (алкены) (жидкий и газообразный) выделяют из верхнего потока, превышает 0,5, но меньше 20, предпочтительно больше 1, но меньше 10, а наиболее предпочтительно больше 1, но меньше 5.
Простой эфир (эфиры), который образуется внутри реакционно-ректификационной колонны, представляет собой, по существу, дериватизированный из С2-С3спирта простой эфир (эфиры), такой как диэтиловый эфир, н-пропиловый эфир, изопропиловый эфир, бутиловый эфир (эфиры) и смешанные простые эфиры, такие как этилоизопропиловый эфир.
Термодинамические исследования показывают, что выполнение настоящего изобретения позволяет дегидратацию смеси этанола и н-пропанола до соответствующего алкена (алкенов) проводить с намного более высокой селективностью и неожиданно высоким превращением. Это упомянутое более высокое превращение резко улучшает экономику процесса, поскольку благодаря отсутствию побочных продуктов при этом больше не существует потребность в проведении дорогостоящих разделений побочных продуктов и продуктов, как в процессе МвО.
Выпускное приспособление от основания реакционно-ректификационной колонны удаляет по преимуществу избыток воды с целью сохранить внутри колонны сбалансированную среду.
Сырой оксигенатный исходный материал, который вводят в реакционно-ректификационную колонну, включает по меньшей мере один С2-С3спирт, который может представлять собой, например, этанол, н-пропанол, изопропанол и их смеси. Как правило, обычно используют смесь по меньшей мере двух спиртов, которые, по-видимому, выбирают из одноатомных алифатических парафиновых первичных (или вторичных) спиртов, содержащих 2 или 3 углеродных атома, предпочтительно используют, по-видимому, смесь этанола и н-пропанола.
В соответствии с одним из вариантов выполнения настоящего изобретения спирт (спирты), содержащийся в сыром оксигенатном исходном материале, состоит из смеси этанола и пропанола (пропанолов), в которой молярное отношение этанола к пропанолу (пропанолам) находится в пределах от 2 до 5.
В соответствии с настоящим изобретением в сыром оксигенатном исходном материале допустима вода, в предпочтительном варианте проведения процесса сырой оксигенатный исходный материал может включать до 50 мас.% воды. В другом варианте можно применять колонну, где при этом используют способность реакционно-ректификационной колонны эффективно разделять воду, сырой биоэтанол и другой биоспирт (биоспирты), которые могут включать в основном воду.
В соответствии с наиболее предпочтительным вариантом выполнения настоящего изобретения С2-С3спирты совместно с водой составляют по меньшей мере 90 мас.% сырого оксигенатного исходного материала, вводимого в реакционно-ректификационную колонну.
В другом варианте реакционная ректификация в качестве совместного исходного материала может обладать потоком простых эфиров, как он представлен выше.
В соответствии с другим вариантом выполнения настоящего изобретения давление, под которым работает реакционно-ректификационная колонна, превышает 1,5 МПа, но меньше 4,0 МПа, а предпочтительно больше 1,8 МПа, но меньше 2,7 МПа. Температуру, создаваемую в колонне, регулируют точкой кипения указываемых компонентов под данным давлением, в предпочтительном варианте она находится в интервале от 150 до 250°С. Температуры и давления, выходящие за указанные пределы, не исключаются, однако они не охватываются предпочтительными вариантами выполнения настоящего изобретения.
На чертеже представлен один вариант технологической схемы в соответствии с настоящим изобретением. Этот упомянутый вариант включает необязательные и/или предпочтительные технологические стадии в соответствии с настоящим изобретением.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕАКЦИОННАЯ РЕКТИФИКАЦИЯ ДЛЯ ДЕГИДРАТАЦИИ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2419595C2 |
| ДЕГИДРИРОВАНИЕ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2412141C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНОВ ИЗ ОКСИГЕНАТОВ С ИСПОЛЬЗОВАНИЕМ НАНЕСЕННЫХ НА НОСИТЕЛЬ ГЕТЕРОПОЛИКИСЛОТНЫХ КАТАЛИЗАТОРОВ | 2007 |
|
RU2446011C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНА | 2006 |
|
RU2415832C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2006 |
|
RU2419596C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНА | 2006 |
|
RU2415121C2 |
| РЕЦИРКУЛИРОВАНИЕ ДИМЕТИЛОВОГО ЭФИРА В РЕАКЦИОННОЙ СИСТЕМЕ ОКСИГЕНАТ-В-ОЛЕФИН | 2008 |
|
RU2461536C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНА | 2011 |
|
RU2603636C2 |
| СПОСОБ ОТДЕЛЕНИЯ КОНДЕНСИРОВАННОЙ ЖИДКОСТИ ОТ ПОТОКА ОЛЕФИНА | 2008 |
|
RU2460712C2 |
| СПОСОБ СИНТЕЗА УГЛЕВОДОРОДНЫХ КОМПОНЕНТОВ БЕНЗИНА | 2007 |
|
RU2448147C2 |
Изобретение относится к способу получения алкена (алкенов) из исходного материала из одноатомного алифатического парафинового первичного (и/или вторичного) спирта (спиртов), включающего этанол, пропанол (пропанолы) или их смесь, характеризующемуся следующими стадиями: 1) одноатомный алифатический парафиновый первичный (или вторичный) спирт (спирты) в реакционно-ректификационной колонне под повышенным давлением и при повышенной температуре и в присутствии катализатора превращают в алкен (алкены) с соответствующим аналогичным числом углеродных атомов таким образом, что поток головных погонов, отводимый из верхней части упомянутой реакционно-ректификационной колонны, включает, по существу, упомянутый алкен (алкены), 2) затем поток головных погонов со стадии 1 охлаждают до температуры, достаточной для конденсации по меньшей мере части алкена (алкенов) с самой высокой точкой кипения, 3) далее по меньшей мере часть конденсированного алкена (алкенов) со стадии 2 возвращают назад в упомянутую реакционно-ректификационную колонну в качестве возвращаемой флегмы, 4) одновременно выделяют оставшийся алкен (алкены). Также изобретение относится к способу превращения углеводорода в алкен, включающему приведенный выше способ. Настоящий процесс является усовершенствованным процессом получения алкена С2+ из спиртов. 2 н. и 9 з.п. ф-лы, 1 ил.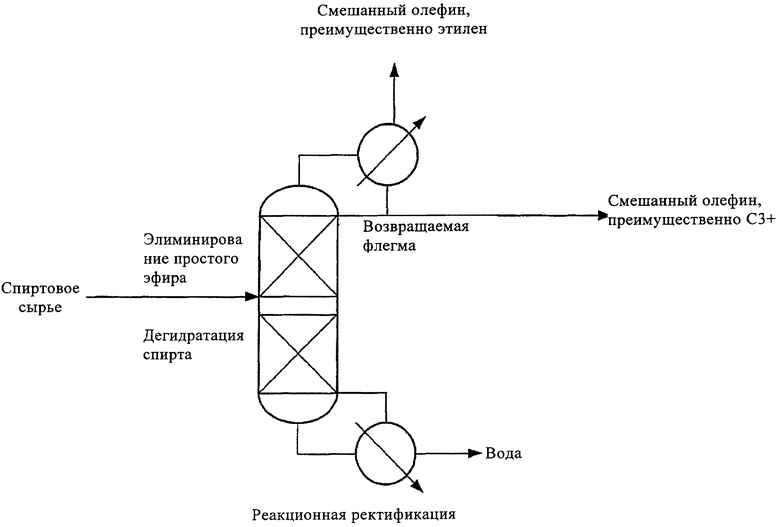
1. Способ получения алкена (алкенов) из исходного материала из одноатомного алифатического парафинового первичного (и/или вторичного) спирта (спиртов), включающего этанол, пропанол (пропанолы) или их смесь, отличающийся следующими стадиями:
1) одноатомный алифатический парафиновый первичный (или вторичный) спирт (спирты) в реакционно-ректификационной колонне под повышенным давлением и при повышенной температуре и в присутствии катализатора превращают в алкен (алкены) с соответствующим аналогичным числом углеродных атомов таким образом, что поток головных погонов, отводимый из верхней части упомянутой реакционно-ректификационной колонны, включает по существу упомянутый алкен (алкены),
2) затем поток головных погонов со стадии 1 охлаждают до температуры, достаточной для конденсации по меньшей мере части алкена (алкенов) с самой высокой точкой кипения,
3) далее по меньшей мере часть конденсированного алкена (алкенов) со стадии 2 возвращают назад в упомянутую реакционно-ректификационную колонну в качестве возвращаемой флегмы,
4) одновременно выделяют оставшийся алкен (алкены).
2. Способ превращения углеводорода в алкен (алкены), включающий следующие стадии:
а) превращение углеводорода в реакторе синтез-газа в смесь оксида (оксидов) углерода и водорода,
б) превращение упомянутой смеси оксида (оксидов) углерода и водорода со стадии а) в присутствии порошкообразного катализатора в реакторе при температуре, находящейся в пределах от 200 до 400°С, и под давлением от 50 до 200 бар в исходный материал, включающий по меньшей мере один одноатомный алифатический парафиновый первичный (или вторичный) спирт (спирты), содержащий 2 или 3 углеродных атома, и
в) одноатомный алифатический парафиновый первичный (или вторичный) спирт (спирты), содержащий 2 или 3 углеродных атома со стадии б) в реакционно-ректификационной колонне под повышенным давлением и при повышенной температуре и в присутствии катализатора превращают в алкен (алкены) с соответствующим аналогичным числом углеродных атомов таким образом, что поток головных погонов, отводимый из верхней части упомянутой реакционно-ректификационной колонны, включает по существу упомянутый алкен (алкены),
г) затем поток головных погонов со стадии в) охлаждают до температуры, достаточной для конденсации по меньшей мере части алкена (алкенов) с самой высокой точкой кипения,
д) далее по меньшей мере часть конденсированного алкена (алкенов) со стадии г) возвращают назад в упомянутую реакционно-ректификационную колонну в качестве возвращаемой флегмы,
е) одновременно выделяют оставшийся алкен (алкены).
3. Способ по п.1, в котором катализатор, используемый в реакционно-ректификационном реакторе, представляет собой гетерогенный катализатор, выбранный среди нерастворимых гетерополикислот, сульфированных носителей (например, Nafion и ионообменных смол), цеолитов, модифицированных металлами цеолитов, морденитов и их смесей, предпочтительно гетерополикислот и ионообменных смол, более предпочтительно гетерополикислот, а наиболее предпочтительно солей 12-вольфрамокремниевой кислоты и 18-вольфрамофосфорной кислоты.
4. Способ по п.3, в котором носитель катализатора вначале обрабатывают фторирующим агентом.
5. Способ по п.1, в котором катализатор, используемый в реакционно-ректификационном реакторе, представляет собой гомогенный катализатор, предпочтительно катализатор с более высокой точкой кипения, чем у реагентов и продуктов.
6. Способ по п.5, в котором катализатор выбирают среди сульфоновых кислот, таких как метансульфоновая кислота, паратолуолсульфоновая кислота, трифторметансульфоновая кислота, серных кислот, гетерополикислот и фосфорной кислоты; предпочтительны фосфорная кислота и органосульфоновые кислоты.
7. Способ по п.1, в котором верхний поток алкена (алкенов), который отводят из верхней части реакционно-ректификационной колонны, состоит из одного или нескольких алкенов, по существу из пропилена и этилена.
8. Способ по п.1, в котором коэффициент орошения х/у, где х обозначает интенсивность, с которой алкен (алкены) возвращают из потока верхних погонов назад в реактор, а у обозначает интенсивность, с которой алкен (алкены) выделяют из потока верхних погонов, превышает 0,5, но меньше 20, предпочтительно больше 1, но меньше 10, а наиболее предпочтительно больше 1, но меньше 5.
9. Способ по п.1, в котором спирт (спирты), содержащийся в сыром оксигенатном исходном материале, который вводят в реакционно-ректификационную колонну, состоит из смеси этанола и пропанола (пропанолов), предпочтительно из этанола и н-пропанола.
10. Способ по п.1, в котором сырой оксигенатный исходный материал включает дополнительно воду, и С2-С3спирты совместно с водой составляют по меньшей мере 90 мас.% сырого оксигенатного исходного материала, вводимого в реакционно-ректификационную колонну.
11. Способ по п.1, в котором в спирт, подаваемый на реакционную ректификацию, добавляют дополнительный исходный простой эфир.
| US 5817906 А, 06.10.1998 | |||
| US 5811620 A, 22.09.1998 | |||
| Плашка для накатывания резьбы | 1961 |
|
SU144145A1 |
| JP 55019247 A, 29.02.1980 | |||
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ И КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2001 |
|
RU2194690C1 |

