Изобретение относится к нанесенному на носитель катализатору на основе гетерополикислоты и/или ее солей и к способу получения упомянутого нанесенного на носитель катализатора на основе гетерополикислоты и/или ее солей.
По настоящему изобретению предлагается, в частности, применение нанесенного на носитель гетерополикислотного катализатора для превращения оксигенатов в алкены, благодаря чему возможен усовершенствованный способ в смысле производительности и селективности по алкенам при одновременном предотвращении образования алканов.
Кроме того, объектом настоящего изобретения является усовершенствованный способ получения алкена (алкенов) из оксигенатного исходного материала с использованием нанесенного на носитель гетерополикислотного катализатора в особых рабочих условиях.
Гетерополикислоты являются ценными химическими соединениями, которые можно использовать в качестве кислотных и/или оксигенатных катализаторов. Упомянутые в дальнейшем способы относятся ко множеству примеров различных гетерополикислот и/или методов их получения.
В US 2006052240 описан нанесенный на носитель катализатор, включающий носитель, обладающий нанесенным на него по меньшей мере одним представителем, выбранным из группы, включающей гетерополикислоты и соли гетерополикислот, где гетерополикислота и/или соль гетерополикислоты содержится по существу в зоне поверхностного слоя носителя до глубины 30% от поверхности носителя.
В US 6624325 описан катализатор получения эфиров низших жирных кислот посредством эстерификации низшей алифатической карбоновой кислоты низшим олефином, включающий гетерополикислоту или ее соль, содержащуюся на носителе, и обладающий удельной площадью поверхности, как это определяют по методу БЭТ, от 65 до 350 м2/г. Предлагаются также способ получения катализатора и способ получения эфира низшей жирной кислоты с использованием этого катализатора.
В US 5227141 описан мембранный каталитический реактор, который включает гетерополикислоту, выбранную из группы, включающей 12-вольфрамофосфорную кислоту, 12-молибдофосфорную кислоту, 12-молибдовольфрамофосфорную кислоту и 12-вольфрамокремниевую кислоту, и предлагается полисульфоновая мембрана. Этот мембранный каталитический реактор применим в отношении парофазной дегидратации, дегидрогенизации, окисления и одновременного разделения органических или неорганических материалов, в особенности парофазной дегидратации этанола.
В US 2004/024918 описаны катализатор для применения при получении эфира низшей алифатической карбоновой кислоты, где катализатор готовят по способу, включающему стадию контактирования катализатора с газом, содержащим по меньшей мере один представитель, выбранный из воды, низших алифатических карбоновых кислот и низших алифатических спиртов; способ получения катализатора и способ получения эфира низшей алифатической карбоновой кислоты с использованием такого катализатора. В этом документе дополнительно описан кремнийсодержащий носитель для применения в катализаторе, который обладает содержанием кремния от 39,7 до 46,3 мас.% или содержанием кремния от 85 до 99 мас.% в виде диоксида кремния или сопротивлением раздавливанию 30 N или больше. С применением катализатора, включающего носитель, эфир низшей алифатической карбоновой кислоты получают из низшего олефина и низшей алифатической карбоновой кислоты в газовой фазе, не вызывая большого снижения каталитической активности или крекинга, или истирания катализатора.
Алкен (алкены) традиционно получают крекингом с водяным паром или каталитическим крекингом углеводородов. Однако по мере того как запасы нефти уменьшаются, цена нефти неизбежно будет увеличиваться, что делает получение легкого олефина (алкенов) дорогостоящим процессом. Таким образом существует постоянно растущая потребность в ненефтяных путях получения алкена (алкенов) С2 и С2+, в особенности этилена и пропилена, поскольку они представляют собой эффективные исходные материалы для ряда химических процессов, включающих получение полимерных продуктов, таких как полиэтилен и полипропилен.
В последние годы поиск альтернативных исходных материалов для получения алкена (алкенов) С2 и С2+ приводит к применению оксигенатов, таких как спирты (например, метанол, этанол и более высокомолекулярные спирты). Эти спирты обеспечивают особенно привлекательный путь для получения алкена (алкенов), поскольку упомянутые спирты могут быть получены ферментацией, например, сахаров и/или целлюлозных материалов.
По другому варианту спирты могут быть получены из синтез-газа. Синтез-газом называют сочетание водорода и оксидов углерода, полученных в установке для синтез-газа из такого источника углерода, как природный газ, нефтяные дистилляты, биомасса и углеродистые материалы, включая уголь, перерабатываемые для вторичного использования пластмассы, муниципальные отходы или любой органический материал. Таким образом, спирт и производные спиртов могут обеспечивать пути для получения алкена (алкенов) и других родственных углеводородов на ненефтяной основе.
Обычно получение спиртов, например метанола, проводят осуществлением трех технологических стадий: получение синтез-газа, синтез метанола и очистка метанола. На стадии получения синтез-газа может быть использована дополнительная стадия, на которой исходный материал вначале обрабатывают, например, исходный материал очищают перед превращением в синтез-газ для удаления серы и других потенциальных каталитических ядов. Эта обработка может быть также осуществлена после получения синтез-газа, например, когда используют уголь или биомассу.
Способы, упоминаемые в дальнейшем, относятся к примерам и альтернативным способам дегидратации оксигенатного исходного материала; в US №4543435 описан способ превращения оксигенатного исходного материала, включающего метанол, диметиловый эфир или т.п., в реакторе превращения оксигената в жидкие углеводороды, включающие С2-С4алкены и углеводороды С5+. С2-С4алкены сжимают для выделения богатого этиленом газа. Этот богатый этиленом газ возвращают в реактор превращения оксигената.
В US №4076761 описан способ превращения оксигенатов в бензин с возвратом богатого водородом газообразного продукта в установку для синтез-газа или реакционную зону превращения оксигената.
В US №5177114 описан способ превращения природного газа в жидкие углеводороды бензинового сорта и/или алкены превращением природного газа в синтез-газ, превращением синтез-газа в сырой метанол и/или диметиловый эфир и последующим превращением сырого метанола/диметилового эфира в бензин и алкены. Международная заявка на патент №93/13013, поданная Kvisle и др., относится к усовершенствованному способу приготовления кремнеалюмофосфатного катализатора, который более стабилен к дезактивации от закоксовывания. В этой заявке на патент говорится, что по истечении некоторого периода времени все такие катализаторы, используемые для превращения метанола в олефин (олефины) (МвО), утрачивают способность к активному превращению метанола в углеводороды главным образом потому, что микропористая кристаллическая структура закоксовывается, т.е. заполняется низколетучими углеродистыми соединениями, которые блокируют пористую структуру. Эти углеродистые соединения могут быть удалены по обычным методам, таким как сжигание на воздухе.
В ЕРО публикации №0407038 А1 описан способ получения диалкиловых эфиров, включающий подачу потока, содержащего алифатический спирт, в зону подачи ректификационной колонны как реактора, контактирование этого потока с неподвижным слоем твердой кислой каталитической ректификационной структуры с получением соответствующего диалкилового эфира и воды и одновременное отделение ректификацией простого эфира как продукта от воды и непрореагировавших материалов.
В US №5817906 описан способ получения легких алкенов из сырого оксигенатного исходного материала, включающего спирт и воду. В этом способе применяют две реакционные стадии. Во-первых, с использованием реакции с ректификацией спирт превращают в простой эфир. Затем простой эфир в дальнейшем направляют в зону превращения оксигената, содержащую металлалюмосиликатный катализатор, с получением потока легких алкенов. Так, например, хорошо известный процесс превращения метанола в олефин (олефины) (МвО) может быть описан как дегидративное сочетание метанола до алкенов и хорошо известен химику, который может его использовать для получения алкенов из спирта (спиртов). Полагают, что этот механизм осуществляется посредством сочетания С1-фрагментов, образуемых катализируемой кислотой дегидратацией метанола, возможно через метилоксониевый промежуточный продукт. Главный недостаток упомянутого процесса МвО также заключается в том, что совместно с рядом ароматических и алкановых побочных продуктов образуется ряд алкенов, вследствие чего, в свою очередь, выделение целевых алкенов сопряжено с очень большими затруднениями технологического порядка и затратами.
Как содействующие превращению оксигенатов благодаря химическому пути от метанола до олефина (МвО) к углеводородным смесям известны молекулярные сита, такие как микропористый кристаллический цеолит и нецеолитные катализаторы, особенно кремнеалюмофосфаты (SAPO). Такой метод для этих катализаторов различных типов описан в многочисленных патентах: US №3928483, 4025575, 4252479 (Chang и др.), 4496786 (Santilli и др.), 4547616 (Avidan и др.), 4677243 (Kaiser), 4843183 (Inui), 4499314 (Seddon и др.), 4447669 (Harmon и др.), 5095163 (Barger), 5191141 (Barger), 5126308 (Barger), 4973792 (Lewis) и 4861938 (Lewis).
Более того, эта реакция характеризуется стадией высокоэнергетической активации (возможно на этапе получения метанола или диметилового эфира), вследствие чего для достижения высокой степени превращения с целью поступательного ведения реакции существует потребность в высоких температурах, например 450°С. Обычно с целью добиться этих высокотемпературных условий в таких системах предусматривают различные средства, такие как возврат в процесс нагретого катализатора и последующие нагревательные системы. К сожалению, проведение процесса при этих упомянутых высоких температурах приводит к возникновению таких основных проблем, как дезактивация катализатора, закоксовывание и образование побочных продуктов. С целью избежать этих проблем реакции можно проводить при более низких температурах, но это требует дорогостоящего возврата в процесс промежуточных продуктов и реагентов.
Эти и другие недостатки известных технических решений показывают, что в данной области техники существует явная потребность в разработке усовершенствованного и/или альтернативного способа получения алкена (алкенов) С2 и С2+ из оксигенатов.
Следовательно, конкретной задачей настоящего изобретения является устранение этих и других недостатков, а его объектом являются:
выбранная группа нанесенного на носитель катализатора на основе гетерополикислоты и/или ее солей, который обладает особыми свойствами, благодаря которым он проявляет улучшенные качества, когда его используют в таких способах, как дегидратация оксигенатов до алкена (алкенов),
получение упомянутой выбранной группы нанесенного на носитель катализатора на основе гетерополикислоты и/или ее солей,
усовершенствованный в смысле селективности, производительности и уменьшенного количества образующегося алкана способ дегидратации оксигенатного исходного материала с целью получить алкен (алкены) и
применение выбранной группы нанесенного на носитель катализатора на основе гетерополикислоты и/или ее солей в способе дегидратации оксигенатов до алкена (алкенов) для улучшения селективности и производительности алкена (алкенов) и уменьшения количества образующегося алкана.
Таким образом, по настоящему изобретению предлагается нанесенный на носитель гетерополикислотный катализатор, который характеризуется тем, что удельный объем его пор удовлетворяет следующей формуле:
ОП>0,6-0,3 [количество ГПК/площадь поверхности высушенного катализатора],
а содержание гетерополикислоты на площадь поверхности нанесенного на носитель гетерополикислотного катализатора в предпочтительном варианте превышает 0,1 (микромоль/м2).
Объектом настоящего изобретения является также применение нанесенного на носитель гетерополикислотного катализатора в способе получения алкена (алкенов) из оксигенатного исходного материала для повышения селективности и производительности по алкенам и уменьшения количества образующихся алканов, где нанесенный на носитель гетерополикислотный катализатор характеризуется тем, что удельный объем его пор удовлетворяет следующей формуле:
ОП>0,6-0,3 [количество ГПК/площадь поверхности высушенного катализатора].
Кроме того, объектом настоящего изобретения является способ получения алкена (алкенов) из оксигенатного исходного материала в реакторе, характеризующийся проведением процесса при температуре, предпочтительно находящейся в пределах от 180 до 250°С, в присутствии нанесенного на носитель гетерополикислотного катализатора, который характеризуется тем, что удельный объем его пор удовлетворяет следующей формуле:
ОП>0,6-0,3 [количество ГПК/площадь поверхности высушенного катализатора],
где, принимая во внимание цели настоящего изобретения и прилагаемой формулы изобретения,
ОП обозначает удельный объем пор высушенного нанесенного на носитель гетерополикислотного катализатора (определен в мл/г катализатора);
количество ГПК представляет собой количество гетерополикислоты, содержащейся в высушенном нанесенном на носитель гетерополикислотном катализаторе (определено в микромолях/г);
площадь поверхности высушенного катализатора является удельной площадью поверхности высушенного нанесенного на носитель гетерополикислотного катализатора (определена в м2/г).
При создании настоящего изобретения было установлено, что получение алкена (алкенов) из оксигенатного исходного материала, как явствует из описания настоящего изобретения, осуществляют посредством сочетания двух разных механизмов дегидратации: первый, являющийся прямой дегидратацией оксигенатов до соответствующего алкена (алкенов) и воды (как проиллюстрировано уравнением 1);
уравнение 1
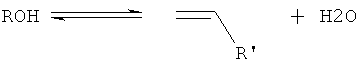
и второй, являющийся двухстадийным механизмом, где оксигенатный исходный материал дегидратируют путем получения простого эфирного промежуточного продукта (как проиллюстрировано уравнениями 2 и 3),
уравнение 2

уравнение 3
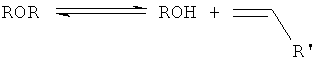
где R обозначает этильную, пропильную, бутильную или пентильную группу, а R' обозначает либо водородный атом, либо метальную, этильную или пропильную группу. В предпочтительном варианте R обозначает либо этильную, либо пропильную группу, a R' обозначает либо водородный атом, либо метильный остаток.
Уравнение 1 демонстрирует эндотермическое прямое элиминирование спирта до олефина (олефинов) и воды; с уравнением 1 конкурируют уравнения 2 и 3, т.е. экзотермическая реакция этерификации (уравнение 2) и эндотермическое элиминирование простого эфира (эфиров) с получением алкена (алкенов) и спирта (уравнение 3). Однако о реакции дегидратации спиртов до алкена (алкенов) в общем говорят как о являющейся эндотермической.
В соответствии с настоящим изобретением и прилагаемой формулой изобретения все основные реакции, которые проводят (т.е. те реакции, которые проиллюстрированы выше) во время осуществления упомянутого способа дегидратации, катализируют применением нанесенного на носитель гетерополикислотного катализатора.
Понятие "гетерополикислота", используемое в данном случае и по всему тексту описания настоящего изобретения, служит как охватывающее, помимо прочего, соли щелочных, щелочно-земельных металлов, аммония, свободных кислот, с объемистыми катионами и/или металлические соли (где соли могут быть либо полными, либо неполными солями) гетерополикислот. Следовательно, гетерополикислоты, используемые при выполнении настоящего изобретения, представляют собой комплексные высокомолекулярные анионы, включающие связанные атомами кислорода атомы поливалентных металлов.
Как правило каждый анион включает от 12 до 18 связанных атомами кислорода атомов поливалентных металлов. Атомы поливалентных металлов, которые известны как периферические атомы, симметричным образом окружают один или несколько центральных атомов. Периферические атомы могут представлять собой один или несколько атомов молибдена, вольфрама, ванадия, ниобия, тантала и любого другого поливалентного металла. Центральными атомами в предпочтительном варианте служат атомы кремния или фосфора, но по другому варианту они могут включать любой один из большого разнообразия атомов элементов групп с I по VIII Периодической таблицы элементов. К ним относятся ионы меди, бериллия, цинка, кобальта, никеля, бора, алюминия, галлия, железа, церия, мышьяка, сурьмы, висмута, хрома, родия, кремния, германия, олова, титана, циркония, ванадия, серы, теллура, марганца, никеля, платины, тория, гафния, церия, ванадия, теллура и иода. Приемлемые гетерополикислоты включают гетерополикислоты Кеггина (Keggin), Уэллса-Доусона (Wells-Dawson) и Андерсона-Эванса-Перлова (Anderson-Evans-Perloff). Конкретными примерами приемлемых гетерополикислот являются следующие соединения:
и свободная кислота или неполные соли следующих гетерополикислот:
Предпочтительными примерами выбранных улучшенных гетерополикислот являются следующие:
и свободная кислота или неполные соли следующих гетерополикислот:
мононатриевая соль 12-вольфрамокремниевой
Кроме того, можно использовать смеси разных гетерополикислот и солей. Предпочтительные для применения в способе, приведенном в описании настоящего изобретения, гетерополикислоты представляет собой любую одну или несколько гетерополикислот, которые основаны на структурах Кеггина или Уэллса-Доусона; более предпочтительная гетерополикислота, выбранная для применения в способе, приведенном в описании настоящего изобретения, представляет собой любую одну или несколько следующих соединений: вольфрамокремневая кислота, фосфовольфрамовая кислота, кремнемолибденовая кислота и фосфомолибденовая кислота, более предпочтительная выбранная для применения в способе, приведенном в описании настоящего изобретения, гетерополикислота представляет собой любую одну или несколько вольфрамокремневых кислот, а наиболее предпочтительная выбранная для применения в способе, приведенном в описании настоящего изобретения, гетерополикислота представляет собой 12-вольфрамокремниевую кислоту H4[SiW12O40]·xH2O.
В предпочтительном варианте гетерополикислоты, используемые в соответствии с настоящим изобретением, обладают молекулярными массами больше 700 и меньше 8500, предпочтительнее больше 2800 и меньше 6000. Такие гетерополикислоты также включают димерные комплексы.
Нанесенный на носитель катализатор может быть эффективно приготовлен растворением выбранной гетерополикислоты в приемлемом растворителе, где приемлемые растворители включают полярные растворители, такие как вода, простые эфиры, спирты, карбоновые кислоты, кетоны и альдегиды и/или их смеси, причем наиболее предпочтительными растворителями являются дистиллированная вода и/или этанол. Полученный кислый раствор характеризуется концентрацией гетерополикислоты, которая в предпочтительном варианте находится в пределах от 10 до 80 мас.%, более предпочтительно от 20 до 70 мас.%, а наиболее предпочтительно от 30 до 60 мас.%. Далее этот упомянутый раствор добавляют к выбранному носителю (или, по другому варианту, носитель погружают в раствор). Фактический объем кислого раствора, добавляемого к носителю, не ограничивают, вследствие чего его может быть достаточно для достижения начальной влажности или мокрой пропитки, где мокрая пропитка (т.е. приготовление с использованием избыточного объема кислого раствора относительно объема пор носителя) является предпочтительным методом, принимая во внимание цели настоящего изобретения.
Получаемая нанесенная на носитель гетерополикислота может быть модифицирована, а затем либо перед, либо во время пропитки кислым раствором носителя в водном растворе различные соли гетерополикислоты могут быть получены введением нанесенной на носитель гетерополикислоты в длительный контакт с раствором приемлемой металлической соли или добавлением фосфорной кислоты и/или других минеральных кислот.
Когда для модификации носителя используют растворимую металлическую соль, эту соль применяют в целевой концентрации с раствором гетерополикислоты. Далее носитель оставляют вымачиваться в упомянутом кислом растворе в течение подходящего времени (например, несколько часов) с периодическим перемешиванием или встряхиванием и по истечении этого времени его отфильтровывают с использованием приемлемого средства с целью удалить весь избыток кислоты.
Когда соль оказывается нерастворимой, в предпочтительном варианте катализатор пропитывают гетерополикислотой, а затем обрабатывают предшественником соли. Этот метод может улучшить диспергирование соли гетерополикислоты. Можно также применять другие методы, такие как вакуумная пропитка.
Затем пропитанный носитель можно промывать и сушить. Это может быть достигнуто с использованием любого обычного метода разделения, включая, например, декантацию и/или фильтрование. После выделения пропитанный носитель может быть высушен, предпочтительно помещением носителя в сушильный шкаф при повышенной температуре. Эту сушку, как правило, проводят при 130°С в токе азота в течение 16 ч, а затем охлаждают до комнатной температуры. Принимая во внимание цель настоящего изобретения и прилагаемую формулу изобретения, понятие "высушенный катализатор" определяют как катализатор, приготовленный по вышеприведенному описанию. По другому варианту или к тому же можно использовать эксикатор. В промышленном масштабе эту стадию сушки часто осуществляют продувкой горячим инертным газом, таким как азот.
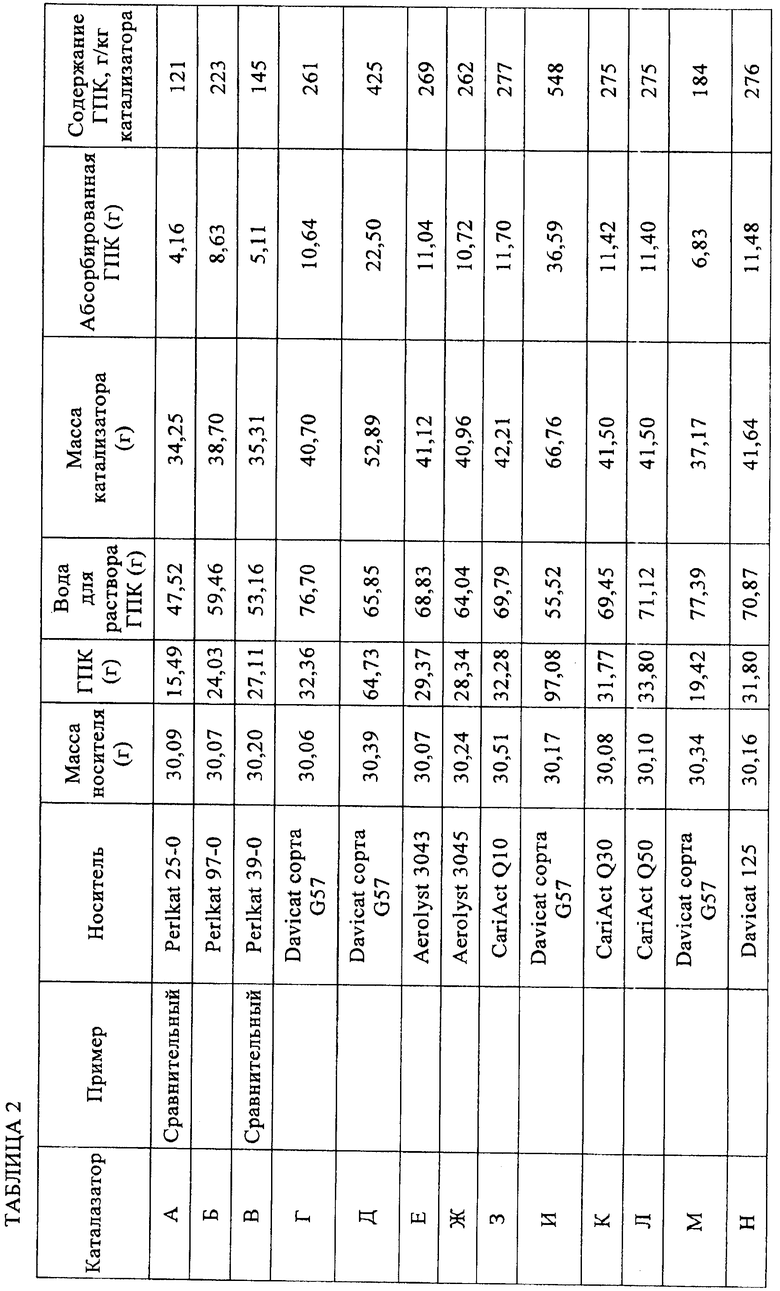
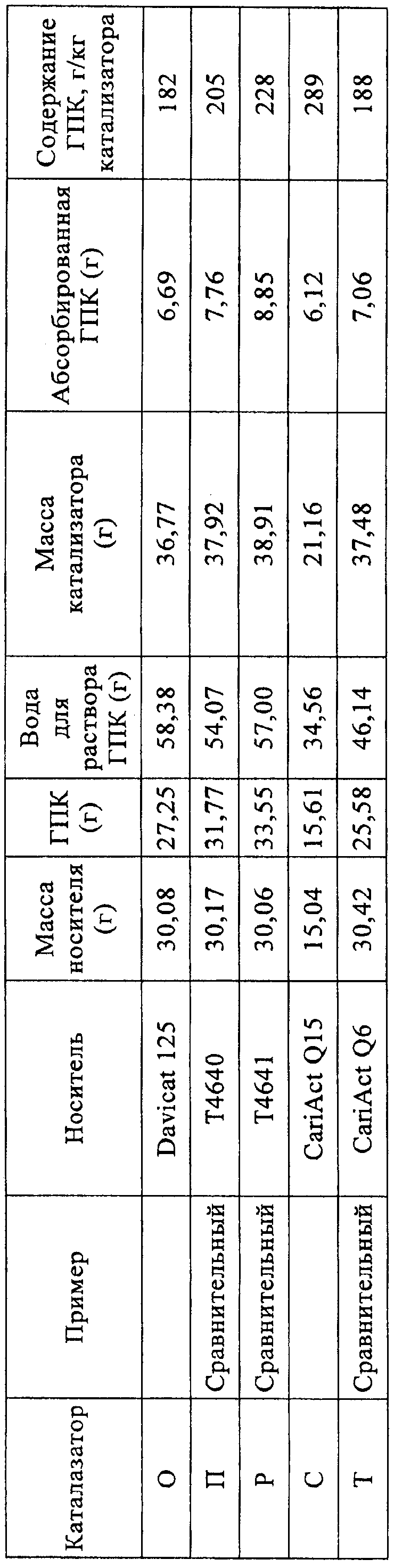
Количество гетерополикислоты, впитавшейся в полученный носитель, в целесообразном варианте находится в интервале от 10 до 80 мас.%, а предпочтительно от 20 до 50 мас.%, в пересчете на общую массу гетерополикислоты и носителя.
Для определения массы кислоты на носителе можно использовать массу катализатора в сухом состоянии и массу применяемого носителя вычитанием этой последней из первой с получением содержания катализатора в граммах гетерополикислоты/килограмм катализатора. С использованием известной или установленной объемной плотности носителя может быть также вычислено содержание катализатора в граммах гетерополикислоты/литр носителя. Предпочтительное содержание каталитической гетерополикислоты составляет от 150 до 600 г ГПК/кг катализатора.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения среднее содержание гетерополикислоты на площадь поверхности высушенного нанесенного на носитель гетерополикислотного катализатора превышает 0,1 микромоля/м2.
Необходимо отметить, что в отношении свежей кислоты, перед тем как ею пропитывают носитель, а преимущественно перед воздействием на нее технологических условий дегидратации по настоящему изобретению, предварительно устанавливают состояния поливалентного окисления и состояния гидратации гетерополикислот и применяют только так, как указано в типичных формулах некоторых конкретных соединений. Степень гидратации гетерополикислоты может повлиять на кислотность нанесенного на носитель катализатора и, следовательно, на его активность и селективность. Следовательно, любое или оба эти действия процесса пропитки и дегидратации могут изменить состояние гидратации и окисления металлов в гетерополикислотах, т.е. фактические используемые каталитические материалы в данных технологических условиях могут не обеспечить достижение состояний гидратации/окисления металлов в гетерополикислотах, используемых для пропитки носителя. Таким образом, следует, естественно, ожидать, что такие состояния гидратации и окисления могут также различаться в отработавших катализаторах после способа дегидратации по настоящему изобретению.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения количество хлорида, содержащегося в упомянутом катализаторе с нанесенной на носитель гетерополикислотой или на нем, составляет меньше 40 част./млн, предпочтительнее меньше 25 част./млн, а наиболее предпочтительно меньше 20 част./млн.
Приемлемые носители катализатора могут находиться в порошкообразной форме или, по другому варианту, могут находиться в форме гранул, в форме пилюль, в форме шариков или в форме экструдатов (включая формованные частицы) и включают, хотя ими их список не ограничен, мордениты, например монтмориллонит, глины, бентонит, диатомовые земли, диоксид титана, активированный уголь, оксид алюминия, диоксид кремния/оксид алюминия, совместные гели диоксида кремния/диоксида титана, совместные гели диоксида кремния/диоксида циркония, оксид алюминия с углеродным покрытием, цеолиты, оксид цинка, полученные пиролизом в пламени оксиды. Носителями могут служить смешанные оксиды, нейтральные или слабоосновные оксиды. В качестве носителей предпочтителен диоксид кремния, такой как силикагелевые носители и носители, полученные гидролизом SiCl4 в пламени. Предпочтительные носители по существу свободны от посторонних металлов или элементов, которые могут негативно воздействовать на каталитическую активность системы. Таким образом, приемлемые кремнеземные носители характеризуются чистотой по меньшей мере 99 мас.%. Количество примесей составляет до меньше 1 мас.%, предпочтительно меньше 0,60 мас.%, а наиболее предпочтительно меньше 0,30 мас.%. Удельный объем пор носителя в предпочтительном варианте превышает 0,50 мл/г, предпочтительнее превышает 0,8 мл/г.
Приемлемые носители из диоксида кремния включают, хотя ими их список не ограничен, продукты Grace Davison сорта 57, GraceDavison Davicat® 1252, Grace Davison Davicat® Si 1254, Fuji Silysia CariAct Q 15, Fuji Silysia CariAct Q10, Degussa Aerolyst 3045 и Degussa Aerolyst 3043. Средний диаметр частиц носителя составляет от 2 до 10 мм, предпочтительно от 3 до 6 мм. Однако при необходимости эти частицы можно раздавливать и просеивать с получением при необходимости частиц меньших размеров, например от 0,5 до 2 мм.
Средний радиус пор (перед пропитыванием гетерополикислотой) носителя составляет от 10 до 500 Å, предпочтительно от 30 до 175 Å, более предпочтительно 50 до 150 Å, а наиболее предпочтительно 60 до 120 Å. Площадь поверхности по БЭТ в предпочтительном варианте находится в пределах от 50 до 600 м2/г, а наиболее предпочтительно находится в пределах от 150 и 400 м2/г. Носитель обладает средним сопротивлением раздавливанию единственной частицы по меньшей мере 1 кг силы, целесообразно по меньшей мере 2 кг силы, предпочтительно по меньшей мере 6 кг силы, а более предпочтительно по меньшей мере 7 кг силы. Объемная плотность носителя составляет по меньшей мере 380 г/л, предпочтительно по меньшей мере 395 г/л.
Сопротивление раздавливанию единственной частицы определяли с использованием динамометра Mecmesin, которым измеряют минимальное усилие, необходимое для раздавливания частицы между параллельными пластинами. Сопротивление раздавливанию основано на среднем значении от того, что устанавливали для группы из 25 каталитических частиц.
Площадь поверхности по БЭТ, объем пор, распределение пор по размерам и средний радиус пор определяли по изотерме адсорбции азота, которую устанавливали при 77К с использованием анализатора статической объемной адсорбции Micromeritics TRISTAR 3000. Применяемый метод является приложением методов по стандартам Великобритании ВS4359:часть 1:1984 "Recommendations for gas absorption (BET) methods" и ВS7591:часть 2:1992, "Porosity and pore size distribution of materials" - Method of evaluation by gas adsorption. Полученные данные уменьшали с использованием метода БЭТ (в диапазоне давлений от 0,05 до 0,20 Р/Po) и по методу Barrett, Joyner & Halenda (BJH) (для диаметров пор от 20 до 1000 Å) с получением соответственно удельной площади поверхности и распределения пор по размерам.
Приемлемыми ссылками на методы уменьшения вышеприведенных данных являются работы Brunauer, S, Emmett, P H, & Teller, E, J. Amer. Chem. Soc. 60, 309 (1938) и Barrett, E.P, Joyner, LG & Halenda P.P, J. Am Chem Soc., 1951, 73, 373-380.
В целях вышеприведенных аналитических определений образцы носителей и "высушенные катализаторы" дегазировали в течение 16 ч при 120°С под вакуумом 5×10-3 Торр.
Дополнительным вариантом выполнения упомянутого изобретения является тот, в котором выбранный носитель катализатора вначале обрабатывают фторирующим агентом; авторы настоящего изобретения полагают, что в результате осуществления упомянутого варианта катализатор становится более инертным и/или кислым, благодаря чему во время вышеупомянутого процесса дегидратации улучшаются селективность и/или эффективность катализатора.
В соответствии с настоящим изобретением исходный материал, который используют в качестве исходного материала для реакции дегидратации (как это изложено в настоящем описании выше), представляет собой "оксигенатный исходный материал", который в предпочтительном варианте может включать либо чистый спирт (спирты), либо чистый простой эфир (эфиры), либо их смеси.
Предпочтительный спирт (спирты), содержащийся в упомянутом оксигенатном исходном материале, представляет собой по меньшей мере один одноатомный алифатический спирт и обладает одним (или несколькими) альфа-водородным атомом (атомами), например этанол, н-пропанол и трет-бутанол, что необходимо для того чтобы находиться в соответствии с предложенным по изобретению механизмом, по которому образуется алкен (алкены) (см. уравнения с 1 по 3). Предпочтительный одноатомный алифатический спирт (спирты) представляет собой этанол и/или пропанол или их смесь, например смесь этанола и н-пропанола и/или изопропанола. В идеальном варианте для дегидратации до соответствующего "целевого алкена (алкенов)" спирты, содержащиеся в упомянутом оксигенатном исходном материале, включают смесь этанола и н-пропанола. Понятие "целевые алкены", используемое в настоящем описании и прилагаемой формуле изобретения до сих пор и в дальнейшем, воспринимают как обозначающее алкены, которые образуются в соответствии со способом, представленным в описании настоящего изобретения, и предпочтительно охватывает либо этен, либо пропен или их смесь.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения оксигенатный исходный материал, который необходимо дегидратировать, обладает содержанием изопропанола меньше 5 мас.%, предпочтительнее меньше 1 мас.%, наиболее предпочтительно меньше 0,1 мас.%, а в идеальном варианте изопропанола не содержит.
Предпочтительная характеризующая особенность в соответствии с настоящим изобретением заключается в том, что оксигенатный исходный материал, который необходимо дегидратировать, обладает содержанием спиртов С3+ (причем в качестве спиртов имеют в виду те, которые содержат по меньшей мере 4 углеродных атома, например н-бутанол, изобутанол и пентанол) меньше 5 мас.%, предпочтительно меньше 1 мас.%, наиболее предпочтительно меньше 0,1 мас.%, а в идеальном варианте спиртов С3+ не содержит.
Другой предпочтительный вариант в соответствии с настоящим изобретением заключается в том, что спирты, содержащиеся в оксигенатном исходном материале, который должен быть дегидратирован, обладают содержанием метанола меньше 5 мас.%, предпочтительно меньше 2 мас.%, наиболее предпочтительно меньше 0,5 мас.%, а в идеальном варианте метанол отсутствует. Удаление метанола может обусловить соответствующие преимущества, т.е.
(I) предотвращение образования диметилового эфира, диметиловый эфир трудно отделить от пропилена и этилена, если сравнивать с диэтиловым эфиром,
(II) предотвращение химизма МвО,
(III) предотвращение алкилирования алкенов, например пропилена до бутена,
(IV) предотвращение образования метилэтилового эфира (который трудно отделить от этилена),
(V) меньше отходов,
(VI) пониженная токсичность,
(VII) более низкое давление пара - легче транспортировать,
(VIII) улучшенное соотношение С:O в исходном материале для транспортировки, т.е. меньше образование воды
В соответствии с настоящим изобретением с целью уменьшить количество/удалить метанол и спирты С3+, содержащиеся в "оксигенатном исходном материале", который должен быть дегидратирован, можно использовать обычную дистилляцию.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения простые эфиры, которые можно использовать в форме смеси со спиртами, содержащимися в упомянутом "оксигенатном исходном материале", который должен быть дегидратирован, включают любой один или несколько из гомо- и смешанных простых эфиров, дериватизированных из спиртов, этанола и пропанола, например диэтиловый эфир, н-пропиловый эфир, этил-н-пропиловый эфир, этилизопропиловый эфир, н-пропилизопропиловый эфир и изопропиловый эфир.
При создании настоящего изобретения было установлено, что с использованием смеси указанных спирта (спиртов) и простого эфира (эфиров) (в смысле концентраций и компонентов) в качестве оксигенатного исходного материала по настоящему изобретению не только существует возможность достижения более высокой селективности в отношении целевых алкенов, но, как при этом дополнительно было установлено, возможность общего улучшения в смысле производительности. Таким образом, по настоящему изобретению предлагается усовершенствованный в смысле уменьшенного количества образующегося алкана и повышенных селективности и производительности по алкену (алкенам) способ дегидратации особого оксигенатного исходного материала до алкенов в присутствии нанесенного на носитель гетерополикислотного катализатора.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения наличие воды в оксигенатном исходном материале, который должен быть дегидратирован, допустимо; упомянутый исходный материал может включать до 50 мас.% воды, но в предпочтительном варианте упомянутый исходный материал включает меньше 25 мас.% воды, а в наиболее предпочтительном варианте исходный материал включает меньше 20 мас.% воды. Однако вследствие технологических затрат, в частности из-за размера реактора, тепла выпаривания и теплоемкости воды, в предпочтительном варианте процесс проводят с исходными материалами, содержащими уменьшенные количества воды, например меньше 10 мас.%, предпочтительнее меньше 5 мас.%.
Поскольку в качестве катализаторов для проведения процесса используют гетерополикислоты, количество воды в контакте с катализатором может повлиять на стабильность и активность катализатора. Так, например, гетерополикислоты демонстрируют пониженную стабильность катализатора при низких концентрациях воды (<1 мас.%) и пониженную активность при высоких концентрациях воды (>50 мас.%). Для специалиста в данной области техники очевидно, что оптимальная концентрация воды обычно зависит от взаимодействия сложной совокупности переменных, включающих состав спиртового сырья, давление, температуру и природу используемой гетерополикислоты. Другими словами, осуществление предлагаемого способа создает хорошую возможность для выделения воды и, следовательно, упрощает применение биоэтанола и другого биоспирта (биоспиртов).
В соответствии с настоящим изобретением способ, благодаря которому дегидратируют оксигенатный исходный материал, в предпочтительном варианте осуществляют в дегидратационном реакторе, причем предпочтителен парофазный дегидратационный реактор. Реактор конструируют для возможности экзотермического получения простых эфиров и эндотермической дегидратации до алкенов. Реакционную температуру в предпочтительном варианте поддерживают в пределах небольшого температурного диапазона, поскольку чрезмерно низкая температура уменьшает скорость получения алкенов и может привести к конденсации реагентов, а чрезмерно высокая температура может привести к загрязнению алкенов неприемлемыми количествами побочных продуктов, таких как алканы с таким же числом углеродных атомов. В предпочтительном варианте температурный профиль слоя катализатора ниже на 30°С, более предпочтительно ниже на 15°С, а наиболее предпочтительно ниже на 10°С. Если общей эндотермической реакции позволяют протекать до термодинамического равновесия, то в случае адиабатического реактора с единственным слоем это могло бы привести к теоретическому падению температуры в 180°С. Очевидной проблемой является проблема теплового регулирования с помощью конструкции реактора. Приемлемые конструкции реактора включают те, которые способны манипулировать тепловыми потоками, в частности реакторы с неподвижным слоем, псевдоожиженным слоем, со множеством трубок и с несколькими неподвижными слоями с межстадийными нагревателями. Тепловое регулирование может быть, что необязательно, улучшено инжектированием предварительно нагретого свежего спиртового сырья в нескольких точках в реакторном слое, причем в такой точке экзотермическая реакция этерификации может частично противодействовать общей эндотермической реакции. Исходный материал может быть также нагрет дополнительно до температуры, превышающей реакционную, с целью обеспечить дополнительный источник тепла.
В соответствии с другим предпочтительным вариантом выполнения настоящего изобретения этанол, пропанол, простые эфиры совместно с водой составляют по меньшей мере 90 мас.% оксигенатного исходного материала, вводимого в парофазный дегидратационный реактор, а предпочтительнее по меньшей мере 99 мас.% оксигенатного исходного материала, вводимого в парофазный дегидратационный реактор.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения рабочие условия, в которых проводят процесс дегидратации, являются такими, при которых процесс дегидратации всегда протекает в парофазном состоянии. В предпочтительном варианте рабочее давление в процессе дегидратации всегда по меньшей мере на 0,1 МПа, предпочтительно на 0,2 МПа, ниже давления точки росы и/или рабочая температура в процессе дегидратации по меньшей мере на 10°С выше точки росы исходного материала, поступающего в парофазный дегидратационный реактор, и получаемой композиции, которая содержится внутри дегидратационного реактора. Эта последняя обычно зависит от таких факторов, как начальный состав исходного материала и степень превращения в реакторе.
Принимая во внимание цели настоящего изобретения, "точку росы" определяют как являющуюся пороговой температурой. Так, например, если для данной смеси под данным давлением температуру системы повышают до уровня выше точки росы, смесь обычно существует в виде сухого газа. Подобным же образом ниже точки росы смесь обычно существует в виде пара, содержащего некоторое количество жидкости. И аналогичным образом "давление точки росы" определяют как являющееся пороговым давлением. Так, например, если для данной смеси при данной температуре давление системы ниже давления точки росы, смесь обычно существует в виде сухого газа; под давлением выше давления точки росы смесь обычно существует в виде пара, содержащего некоторое количество жидкости.
Таким образом, при выполнении настоящего изобретения особая температура, при которой осуществляют способ дегидратации в соответствии с настоящим изобретением, в предпочтительном варианте находится в пределах от 180 до 270°С, более предпочтительно в пределах от 190 до 260°С, а наиболее предпочтительно в пределах от 200 до 250°С. При создании настоящего изобретения было установлено, что благодаря проведению процесса в условиях этих упомянутых температурных диапазонов (которые особенно низки, если сравнивать с типичными температурами, создаваемыми в данной области техники) существует возможность извлечь пользу от обычных преимуществ проведения процесса при пониженной температуре, но, как также было установлено, способ в соответствии с настоящим изобретением обладает дополнительным преимуществом, состоящим в том, что существует возможность достижения значительно уменьшенного количества образующегося алкана, например <1000 част./млн этана в этене.
Парофазный реактор, применяемый для дегидратации оксигенатов, в предпочтительном варианте работает под давлением выше 0,1 МПа, но ниже 4,5 МПа, более предпочтительно под давлением выше 1,5 МПа, но ниже 3,5 МПа, а наиболее предпочтительно под давлением выше 1,8 МПа, но ниже 2,8 МПа.
Предпочтительные реакционные условия внутри парофазного дегидратационного реактора в соответствии с настоящим изобретением являются такими, в которых процесс дегидратации всегда проводят при умеренном превращении оксигенатов в соответствующие алкены.
Принимая во внимание цели настоящего изобретения и прилагаемую формулу изобретения, умеренное превращение "оксигенатного исходного материала" в соответствующие алкены определяют как являющееся превращением спиртов (например, этанола и пропанола) и/или их соответствующих дериватизированных простых эфиров в соответствующие алкены (например, алкены С2 и С3), а это означает, что за проход "превращают" от 10 до 80%, более предпочтительно от 20 до 60%, спиртов и/или простых эфиров. Для реактора с возвратом в процесс непрореагировавшего материала (простого эфира и спирта), работающего таким образом, чтобы достичь стационарного режима, превращение приближается к следующему:
число молей алкенов, полученных за проход, деленное на общее число молей спирта и удвоенное число молей простого эфира в потоке исходных материалов, содержащем возвращаемый в процесс материал с добавленным свежим исходным материалом.
Эта аппроксимация остается справедливой, поскольку в предпочтительных реакционных условиях данная реакция является высокоселективной в отношении алкенов.
Экспериментальные исследования показывают, что выполнение настоящего изобретения дает возможность проводить дегидратацию оксигенатного исходного материала до соответствующих алкенов с гораздо более высокой селективностью, чем ранее достигаемая в данной области техники, например в современной химии МвО. Это упомянутое умеренное превращение, низкая температура, повышенная селективность процесса позволяют извлекать выгоду из улучшенной экономики, а также других преимуществ. Кроме того, в отличие от обычных способов дегидратации в данной области техники осуществление предлагаемого способа, представленного в настоящем описании, не требует осуществления явной и дорогостоящей стадии разделения алканов/алкенов или восстановления катализатора.
Образование простых эфиров во время парофазного процесса дегидратации является термодинамически благоприятным. Это образование простого эфира упрощает выделение воды из потока продуктов, поскольку этанол, н-пропанол и изопропанол - все полностью или в значительной мере смешиваются с водой и, следовательно, во время процесса дегидратации легко образуют водные азеотропы, из-за которых выделение воды (значительного побочного продукта реакции) из потока продуктов является очень трудным процессом. В то же время образование простых эфиров, таких как диэтиловый эфир и ди-н-пропиловый эфир, которые оба обладают ограниченной смешиваемостью с водой и очень низким содержанием воды в азеотропе, дает возможность извлекать воду с применением отстойника даже в присутствии непрореагировавших спиртов.
Необходимо отметить, что осуществлению способа, представленного в описании настоящего изобретения, при среднем превращении с удалением воды во время выделения олефинов свойственно то преимущество, что это позволяет сводить реакционные условия в отношении содержания воды, присутствующей в реакторе, к оптимальным.
Поток жидких продуктов после удаления олефинов включает в основном непрореагировавшие спирты, простые эфиры и воду. При создании настоящего изобретения было установлено, что основную часть спиртов и простых эфиров предпочтительнее возвращать назад в парофазный дегидратационный реактор после удаления воды как побочного продукта. Как указано в настоящем описании выше, пропанол может существовать в виде двух изомеров: н-пропанол и изопропанол; в реакционных условиях эти изомеры способны взаимопревращаться таким образом, что спиртовой рецикловый поток, в дополнение к непрореагировавшим этанолу и н-пропанолу, может включать некоторое количество изопропанола. Эта изомеризация может также затрагивать соединения, содержащиеся в доле простых эфиров предпочтительного рециклового потока. Вышеупомянутые простые эфиры, которые необязательно возвращают назад в парофазный дегидратационный реактор, включают некоторое количество дериватизированного из этанола и пропанола простого эфира (эфиров), такого как диэтиловый эфир, н-пропиловый эфир, этил-н-пропиловый эфир, этилизопропиловый эфир, н-пропилизопропиловый эфир и изопропиловый эфир, которые образуются во время стадии дегидратации.
В соответствии с предпочтительным вариантом выполнения настоящего изобретения в ходе проведения процесса предусмотрена дополнительная стадия разделения, благодаря которой в предпочтительном варианте из алкенов выделяют по меньшей мере 80 мас.%, более предпочтительно по меньшей мере 90 мас.%, наиболее предпочтительно по меньшей мере 99 мас.%, еще более предпочтительно по меньшей мере 99,9 мас.%, простого эфира (эфиров). Таким образом, затем по меньшей мере часть, предпочтительнее все количество полученного вышеупомянутого простого эфира (эфиров), в предпочтительном варианте возвращают в соответствующий парофазный дегидратационный реактор.
Также в соответствии с вариантом выполнения настоящего изобретения по меньшей мере часть, предпочтительно все количество, упомянутого возвращаемого в процесс простого эфира перед поступлением в парофазный дегидратационный реактор по настоящему изобретению предварительно смешивают с оксигенатным исходным материалом, который должен быть дегидратирован.
Объем настоящего изобретения охватывает также вариант, в соответствии с которым предлагается способ превращения углеводорода в алкен (алкены), включающий следующие последовательные стадии:
а) превращение углеводородного исходного материала в реакторе синтез-газа в смесь оксида (оксидов) углерода и водорода,
б) превращение упомянутой смеси оксида (оксидов) углерода и водорода со стадии а) в присутствии порошкообразного катализатора в реакторе при температуре, находящейся в пределах от 200 до 400°С, и под давлением от 50 до 200 бар в исходный материал, включающий по меньшей мере один одноатомный алифатический парафиновый спирт и/или соответствующий простой эфир, содержащий от 2 до 5 углеродных атомов, и
в) продолжение осуществления способа так, как изложено в описании настоящего изобретения, с получением алкенов, благодаря чему оксигенатный исходный материал включает по меньшей мере часть спирта (спиртов) и/или простых эфиров, полученных на стадии б).
С целью осуществления приведенного выше варианта в способах по изобретению может быть использован любой содержащий углеводород поток исходных материалов, который можно превращать в исходный материал, включающий моноксид углерода и водород, наиболее предпочтительно "синтез-газ".
Углеводородный исходный материал, используемый для получения синтез-газа, в предпочтительном варианте представляет собой углеродистый материал, например биомассу, пластик, бензино-лигроиновую фракцию, кубовые остатки нефтепереработки, отходящий из плавильной печи газ, муниципальные отходы, уголь, кокс и/или природный газ, причем предпочтительным углеродистым материалом являются уголь и природный газ, а наиболее предпочтительный углеводородный исходный материал представляет собой природный газ.
Исходные материалы, включающие моноксид углерода и водород, например синтез-газ, перед подачей в любые реакционные зоны можно подвергать очистке. Очистку синтез-газа можно проводить по способам, известным в данной области техники (см., например, Weissermel, К. и Arpe H.-J. в работе Industrial Organic Chemistry, издание второе, переработанное и расширенное, 1993, с.19-21).
Примеры и сравнительные примеры
Материал носителя, используемый в примерах
Гранулы диоксида кремния CariAct® Q6, CariAct® Q10, CariAct® Q15, CariAct® Q30, CariAct® Q50 получали на фирме Fuji Silysia.
Экструдаты диоксида кремния Aerolyst® 3043 и Aerolyst® 3045 получали как экструдаты на фирме Degussa.
Гранулы диоксида кремния Davicat® сорта 57, Davicat® 1252 и Davicat® 1254 получали на фирме Grace Davison. Гранулы диоксида кремния Perlkat® 97-0, Perlkat® 39-3, Perlkat® 25-0 получали на фирме Engelhard.
Гранулы алюмината кремния Т4640 и Т4641 получали на фирме Sud Chemie.
Свойства носителей
Материалы носителя анализировали азотной порозиметрией (таблица 1).
Приготовление катализаторов с использованием в качестве растворителя воды
В широкогорлый пластиковый флакон отвешивали вольфрамокремневую кислоту (H4[SiW12O40]·24H2O, Mw:3310,6) и растворяли в дистиллированной воде. В этот кислый раствор добавляли взвешенное количество материала носителя. Во время этого периода кислый раствор и материал носителя оставляли пропитываться в течение приблизительно 1 ч с периодическим осторожным перемешиванием для удаления всех захваченных воздушных пузырьков.
После пропитки неадсорбировавшийся кислый раствор удаляли сливанием кислого раствора, материал носителя извлекали из пластикового контейнера и помещали на пластиковый фильтр (который содержал фильтровальную бумагу).
Катализатору позволяли освобождаться от жидкости дренированием до истечения приблизительно от 15 до 60 мин, пока жидкость из катализатора больше не сливалась. После того как дренирование завершали, катализатор переносили в керамическую тарелку и сушили в муфельной печи при 130°С в азотной атмосфере.
Высушенный твердый катализатор взвешивали и количество вольфрамокремневой кислоты, адсорбированной на катализаторе, рассчитывали по разнице масс относительно исходного материала, как указано в приведенной ниже таблице 2.
Приготовление катализаторов с использованием в качестве растворителя этанола
Приведенный выше метод повторяли за исключением того, что в качестве растворителя использовали этанол. К этанолу добавляли также небольшое количество н-бутанола (таблица 3).
Свойства катализаторов
Высушенные твердые катализаторы анализировали азотной порозиметрией (таблица 4).
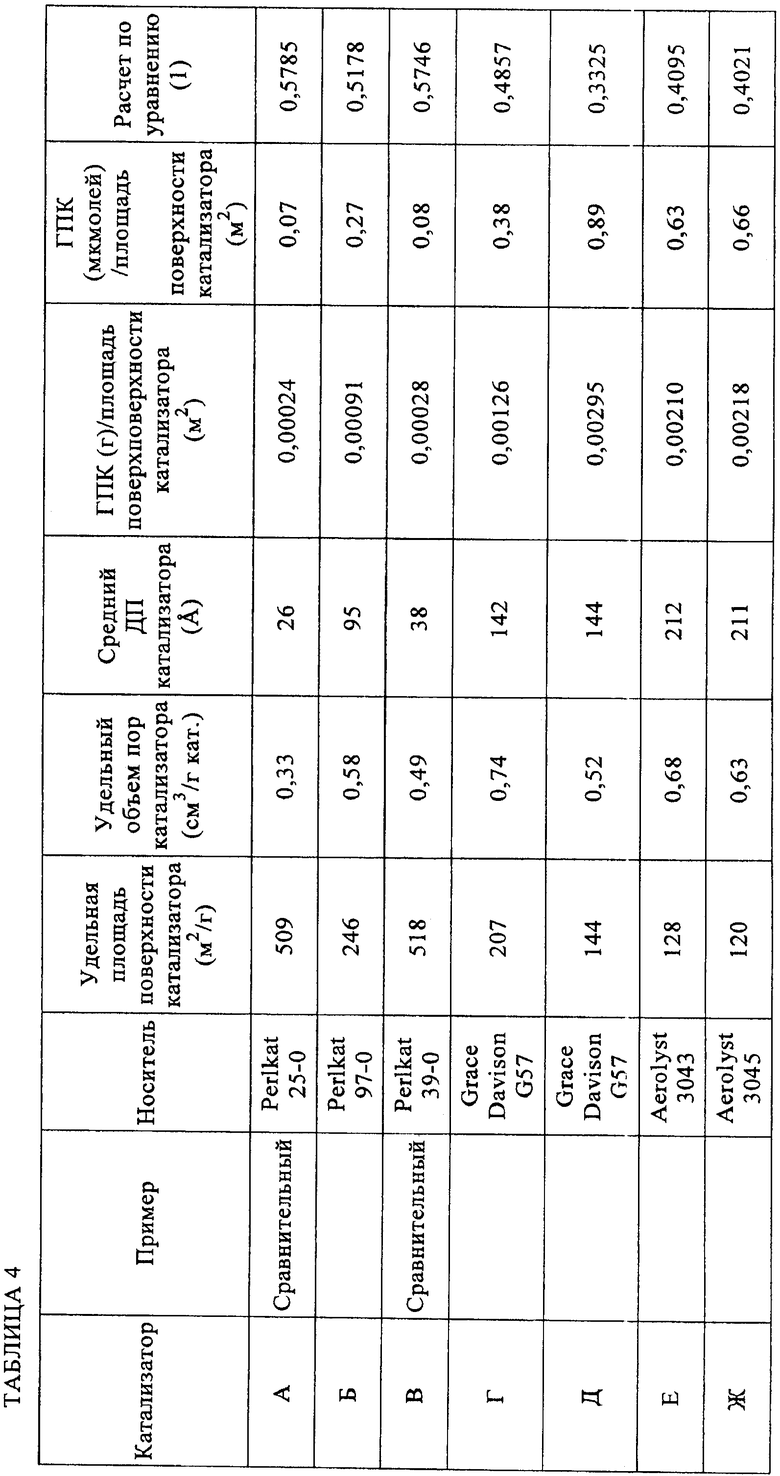
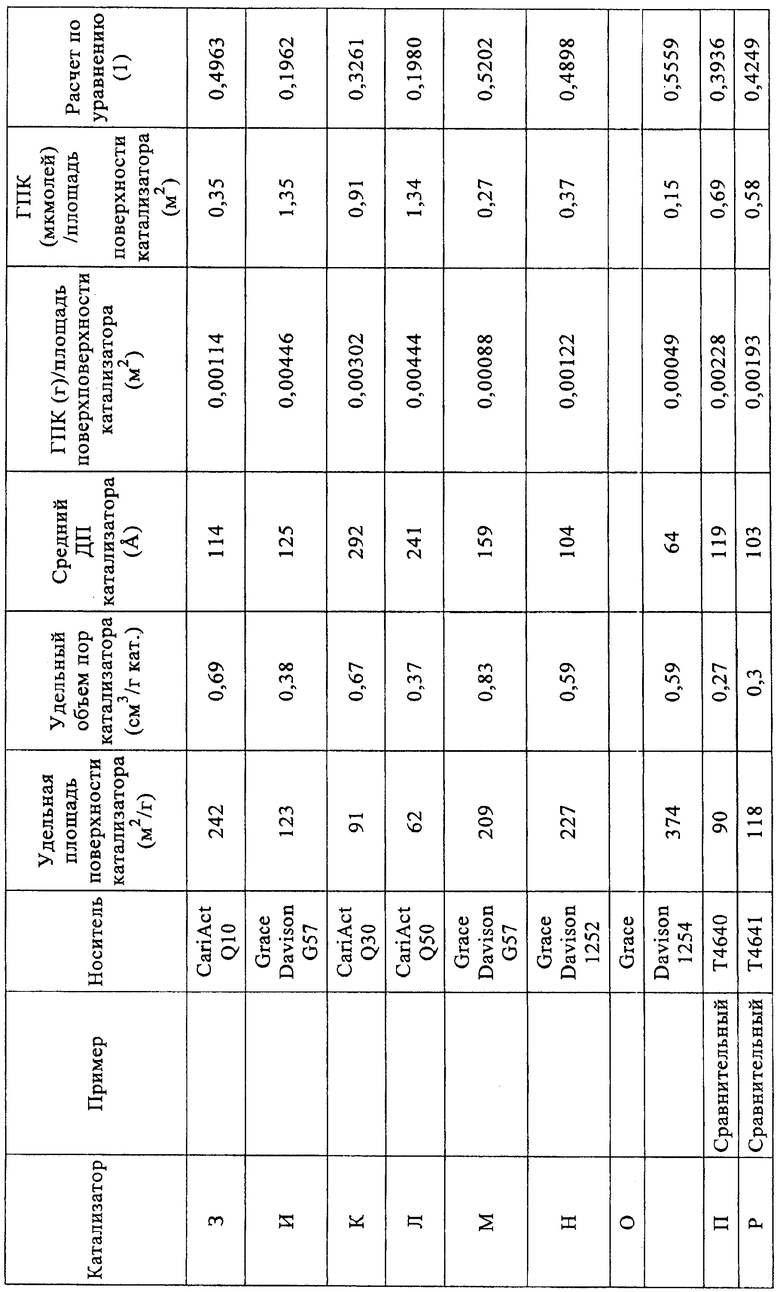
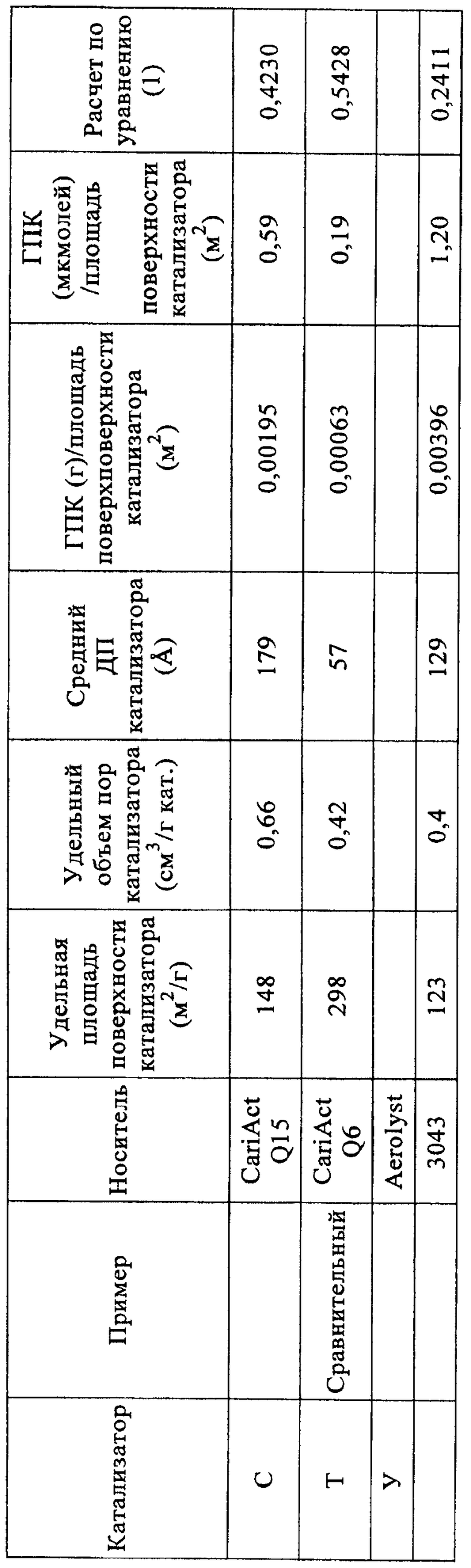
Уравнение (1): 0,6-0,3*[количество ГПК (мкмолей/г)/удельная площадь поверхности катализатора (м2/г)]. Когда рассчитывают количество микромолей гетерополикислоты (ГПК), адсорбированной на катализаторе, полагают, что гетерополикислота полностью гидратирована. Для расчета количества микромолей адсорбированной 12-вольфрамокремневой кислоты (H4[SiW12O40]·24H2O) использовали значение Mw 3310,6.
Испытание катализатора
Катализаторы (от 0,8 до 0,92 мл с размерами частиц от 125 до 180 мкм) загружали в параллельные проточные реакторы. Фиксировали массы и объемы катализаторов.
Реакторы испытывали давлением, а затем в токе азота нагревали до 220°С. Когда температура достигала 220°С, жидкий исходный материал из этанола, диэтилового эфира и воды испаряли и перед подачей в реактор, содержавший катализатор, смешивали с азотом. Реакционные условия были следующими: 20 бар, номинальная ССПГ: 3000 ч-1 для 1-миллилитрового каталитического слоя, EtOH (28 об.%), диэтиловый эфир (34,5 об.%), вода (3,3 об.%) и азот (34,2 об.%).
Когда используют параллельные проточные реакторы небольшого объема, всегда существуют небольшие варьирования фактических массы и объема катализатора (катализаторов), загруженного в индивидуальный реактор. При этом любое варьирование объема загруженных катализаторов приводит к варьированию фактической создаваемой ССПГ, поскольку все реакторы обладают одинаковым общим расходом проходящего через него газа.
Катализаторы испытывали при следующих последовательных температурах: 220°С в течение 24 ч (фаза 1), 210°С в течение 24 ч (фаза 2), 230°С в течение 24 ч (фаза 3), 240°С в течение 24 ч (фаза 4), перед конечным испытанием вновь 220°С в течение 24 ч (фаза 5).
Определяли скорость потока выходившего из реакторов газа и выходивший газ также анализировали ГХ с использованием пламенно-ионизационных детекторов. С помощью ГХ определяли этилен, этан, этанол, диэтиловый эфир, бутаны и ацетальдегид. По газовому потоку и данным анализа состава рассчитывали производительность по продуктам.
Катализаторы проявляли следующие средние катализаторные эксплуатационные свойства в течение периода вышеуказанной фазы с учетом производительности по этилену и селективности в отношении этана (как это определено соотношением этана/этилена (таблица 7).
Уравнение (1): 0,6-0,3*[количество ГПК (мкмолей/г)/удельная площадь поверхности катализатора (м2/г)]. Массовый баланс был существенно выше 95%, определенные побочные продукты приведены в отношениях к этену, использованная аналитическая система позволяла определить углеводороды и оксигенаты с C1 по C6. Как проиллюстрировано выше, катализаторы в соответствии с настоящим изобретением являются высокоселективными.
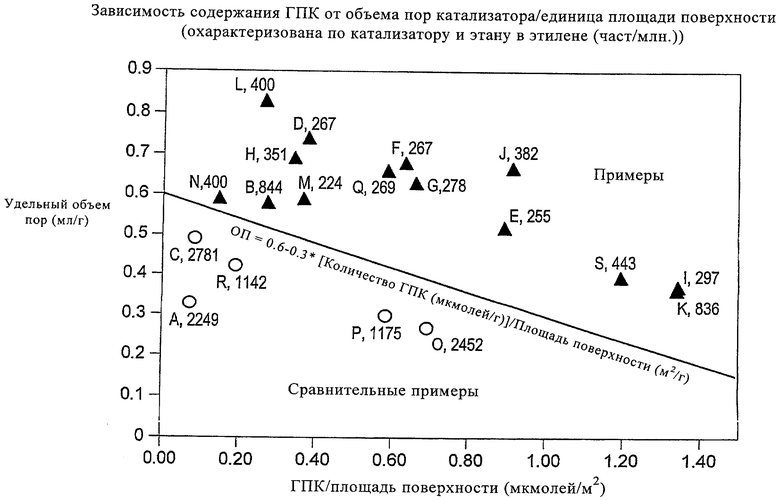
Эти примеры и сравнительные примеры в виде графика представлены на фиг.1, на которой приведен удельный объем пор катализатора (см2/г) в зависимости от отношения количества ГПК (мкмолей/г) к удельной площади поверхности (м2/г) катализатора. Для каждого примера на чертеже указано содержание этана в этене (част./млн).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНА | 2006 |
|
RU2415121C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНА | 2006 |
|
RU2415832C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2006 |
|
RU2419596C2 |
| СПОСОБ ИЗВЛЕЧЕНИЯ КАТАЛИЗАТОРА | 2008 |
|
RU2484900C2 |
| РЕАКЦИОННАЯ РЕКТИФИКАЦИЯ ДЛЯ ДЕГИДРАТАЦИИ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2419595C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНА | 2008 |
|
RU2467992C2 |
| ДЕГИДРИРОВАНИЕ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2412141C2 |
| РЕАКЦИОННАЯ РЕКТИФИКАЦИЯ С ВОЗВРАТОМ В ПРОЦЕСС ОЛЕФИНОВ | 2006 |
|
RU2419597C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНА (ВАРИАНТЫ) | 2011 |
|
RU2593747C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНА | 2011 |
|
RU2603636C2 |
Изобретение относится к использованию гетерополикислотных катализаторов для превращения оксигенатов в алкены. Описан способ получения алкена (алкенов) из оксигенатного исходного материала дегидратацией в реакторе в присутствии нанесенного на носитель гетерополикислотного катализатора, характеризующегося тем, что удельный объем его пор удовлетворяет следующей формуле: ОП>0,6-0,3 [количество ГПК/площадь поверхности высушенного катализатора], где ОП обозначает удельный объем пор высушенного нанесенного на носитель гетерополикислотного катализатора (определен в мл/г катализатора); количество ГПК представляет собой количество гетерополикислоты, содержащейся в высушенном нанесенном на носитель гетерополикислотном катализаторе (определено в микромолях/г); площадь поверхности высушенного катализатора является удельной площадью поверхности высушенного нанесенного на носитель гетерополикислотного катализатора (определена в м2/г). Описан способ превращения углеводорода в алкен (алкены), включающий следующие последовательные стадии: а) превращение углеводородного исходного материала в реакторе синтез-газа в смесь оксида (оксидов) углерода и водорода, б) превращение упомянутой смеси оксида (оксидов) углерода и водорода со стадии а) в присутствии порошкообразного катализатора в реакторе при температуре, находящейся в пределах от 200 до 400°С, и под давлением от 50 до 200 бар в исходный материал, включающий по меньшей мере один одноатомный алифатический парафиновый спирт и/или соответствующий простой эфир, содержащий от 2 до 5 углеродных атомов, и в) продолжение осуществления способа в соответствии с описанным выше способом с получением алкенов, благодаря чему оксигенатный исходный материал включает по меньшей мере часть спирта (спиртов) и/или простых эфиров, полученных на стадии б). Описано применение нанесенного на носитель гетерополикислотного катализатора в способе получения алкена (алкенов) из оксигенатного исходного материала для повышения селективности и производительности по алкенам с одновременным предотвращением образования алканов в присутсвии описанного выше катализатора. Технический результат - увеличение производительности получения алкенов и уменьшение количества образующихся алканов. 4 н. и 16 з.п. ф-лы; 7 табл.; 1 ил.; 19 пр.
1. Способ получения алкена (алкенов) из оксигенатного исходного материала дегидратацией в реакторе в присутствии нанесенного на носитель гетерополикислотного катализатора, отличающийся тем, что удельный объем его пор удовлетворяет следующей формуле
ОП>0,6-0,3 [количество ГПК/площадь поверхности высушенного катализатора],
где ОП обозначает удельный объем пор высушенного нанесенного на носитель гетерополикислотного катализатора (определен в мл/г катализатора); количество ГПК представляет собой количество гетерополикислоты, содержащейся в высушенном нанесенном на носитель гетерополикислотном катализаторе (определено в мкмоль/г); площадь поверхности высушенного катализатора является удельной площадью поверхности высушенного нанесенного на носитель гетерополикислотного катализатора (определена в м2/г).
2. Способ по п.1, в котором количество гетерополикислоты на площадь поверхности нанесенного на носитель гетерополикислотного катализатора превышает 0,1 мкмоль/м2.
3. Способ по п.1, в котором нанесенный на носитель гетерополикислотный катализатор обладает содержанием катализатора в пределах от 150 до 600 г гетерополикислоты/кг катализатора.
4. Способ по п.1, в котором удельный объем пор катализаторного носителя нанесенного на носитель гетерополикислотного катализатора в предпочтительном варианте превышает 0,50 мл/г, а предпочтительнее превышает 0,8 мл/г.
5. Способ по п.1, в котором катализаторный носитель нанесенного на носитель гетерополикислотного катализатора обладает средним радиусом пор в пределах от 10 до 500 Å, предпочтительно в пределах от 30 до 175 Å, более предпочтительно в пределах от 50 до 150 Å, а наиболее предпочтительно в пределах от 60 до 120 Å.
6. Способ по п.1, в котором катализаторный носитель нанесенного на носитель гетерополикислотного катализатора обладает удельной площадью поверхности по БЭТ в пределах от 50 до 600 м2/г, а предпочтительно в пределах от 150 до 400 м2/г.
7. Способ по п.1, в котором катализаторный носитель нанесенного на носитель гетерополикислотного катализатора обладает средним сопротивлением одной частицы раздавливанию по меньшей мере 1 кг силы, предпочтительно по меньшей мере 2 кг силы, более предпочтительно по меньшей мере 6 кг силы, а наиболее предпочтительно по меньшей мере 7 кг силы.
8. Способ по п.1, в котором катализаторный носитель нанесенного на носитель гетерополикислотного катализатора обладает объемной плотностью по меньшей мере 380 г/л, а предпочтительно по меньшей мере 395 г/л.
9. Способ по п.1, в котором катализаторный носитель нанесенного на носитель гетерополикислотного катализатора представляет собой кремнеземный носитель.
10. Способ по п.1, в котором процесс проводят при температуре, находящейся в пределах от 180 до 250°С.
11. Способ по п.1, в котором процесс дегидратации проводят в парофазном состоянии.
12. Способ по п.11, в котором процесс дегидратации проводят под давлением по меньшей мере на 0,1 МПа, предпочтительно по меньшей мере на 0,2 МПа, ниже давления точки росы и/или процесс дегидратации проводят при температуре по меньшей мере на 10°С выше точки росы исходного материала, поступающего в парофазный дегидратационный реактор, и получаемой композиции, которая содержится внутри парофазного дегидратационного реактора.
13. Способ по п.1, в котором процесс дегидратации проводят при температуре в пределах от 180 до 270°С, более предпочтительно в пределах от 190 до 260°С, а наиболее предпочтительно в пределах от 200 до 250°С.
14. Способ по п.1, в котором процесс дегидратации проводят под давлением выше 0,1 МПа, но ниже 4,5 МПа, более предпочтительно под давлением выше 1,0 МПа, но ниже 3,5 МПа, а наиболее предпочтительно под давлением выше 1,0 МПа, но ниже 2,8 МПа.
15. Способ по п.11, в котором основную часть непрореагировавших спиртов и/или простых эфиров, полученных во время процесса дегидратации, возвращают назад в парофазный дегидратационный реактор.
16. Способ по п.15, в котором по меньшей мере часть, предпочтительно все количество, упомянутых возвращаемых в процесс непрореагировавших спиртов и/или простого эфира перед поступлением в парофазный дегидратационный реактор предварительно смешивают с оксигенатным исходным материалом, который должен быть дегидратирован.
17. Способ превращения углеводорода в алкен (алкены), включающий следующие последовательные стадии:
а) превращение углеводородного исходного материала в реакторе синтез-газа в смесь оксида (оксидов) углерода и водорода,
б) превращение упомянутой смеси оксида (оксидов) углерода и водорода со стадии а) в присутствии порошкообразного катализатора в реакторе при температуре, находящейся в пределах от 200 до 400°С, и под давлением от 50 до 200 бар в исходный материал, включающий по меньшей мере один одноатомный алифатический парафиновый спирт и/или соответствующий простой эфир, содержащий от 2 до 5 углеродных атомов, и в) продолжение осуществления способа в соответствии с одним из предыдущих пунктов, с получением алкенов, благодаря чему оксигенатный исходный материал включает по меньшей мере часть спирта (спиртов) и/или простых эфиров, полученных на стадии б).
18. Применение нанесенного на носитель гетерополикислотного катализатора в способе получения алкена (алкенов) из оксигенатного исходного материала для повышения селективности и производительности по алкенам с одновременным предотвращением образования алканов, где удельный объем пор нанесенного на носитель гетерополикислотного катализатора удовлетворяет следующей формуле
ОП>0,6-0,3 [количество ГПК/площадь поверхности высушенного катализатора].
19. Применение по п.18, при котором способ получения алкена (алкенов) осуществляют по одному из пп.2-16.
20. Применение нанесенного на носитель гетерополикислотного катализатора на стадии в) способа по п.17 для повышения селективности и производительности по алкенам с одновременным предотвращением образования алканов, где удельный объем пор нанесенного на носитель гетерополикислотного катализатора удовлетворяет следующей формуле
ОП>0,6-0,3 [количество ГПК/площадь поверхности высушенного катализатора].
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Устройство для вычисления симметричных булевых функций | 1980 |
|
SU959064A1 |
| US 5086031 А, 04.02.1992 | |||
| Способ получения олефинов С @ - С @ | 1987 |
|
SU1657478A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНА | 0 |
|
SU186444A1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ И КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2001 |
|
RU2194690C1 |

