Область изобретения
Настоящее изобретение относится к способу получения изоцианатов.
Предпосылки изобретения
Изоцианаты широко используются в качестве исходных веществ для получения таких продуктов, как пенополиуретан, краски и адгезивы. Основной промышленно используемый способ получения изоцианатов включает взаимодействие аминов с фосгеном (фосгеновый метод) и почти все производимое в мире количество изоцианатов получают в соответствии с фосгеновым методом. Однако фосгеновый метод имеет различные недостатки.
Во-первых, этот метод требует использования большого количества фосгена в качестве исходного материала. Фосген является чрезвычайно токсичным и требует специальных мер защиты при обращении с ним, чтобы предотвратить его воздействие на работающий с ним персонал, и также требуется специальное оборудование для обезвреживания отходов.
Во-вторых, поскольку образуется большое количество коррозионно-агрессивного хлористого водорода в качестве побочного продукта фосгенового метода, помимо того, что требуется способ для обезвреживания хлористого водорода, во многих случаях в получаемых изоцианатах содержится гидролитический хлор, который может неблагоприятно влиять на стойкость к атмосферной коррозии и теплостойкость полиуретановых продуктов в случае использования изоцианатов, получаемых с использованием фосгенового метода.
На основании таких предпосылок предпринимались попытки поиска способа получения изоцианатов, в котором не используют фосген. Один пример способа получения изоцианатных соединений без использования фосгена, который был предложен, включает термическое разложение сложных эфиров карбаминовой кислоты. Давно известно, что изоцианаты и гидроксисоединения получают термическим разложением сложных эфиров карбаминовой кислоты (см., например, Berchte der Deutechen Chemischen Gesellschaft, Vol. 3, p. 653, 1870). Основная реакция иллюстрируется следующей формулой:

(где R представляет собой органический остаток, имеющий валентность a, R' представляет собой одновалентный органический остаток, и a представляет собой целое число, имеющее значение 1 или более).
Реакция термического разложения, представленная указанной выше формулой, является обратимой и, в отличие от ее равновесия, протекает в направлении сложного эфира карбаминовой кислоты с левой стороны при низких температурах, при этом сторона изоцианата и гидроксисоединения становится доминирующей при высоких температурах. Таким образом, необходимо осуществление реакции термического разложения сложного эфира карбаминовой кислоты при высоких температурах. Кроме того, в случае алкилкарбаматов, в частности, поскольку скорость реакции выше для обратной реакции термического разложения, а именно реакции, посредством которой происходит образование алкилкарбамата из изоцианата и спирта, сложный эфир карбаминовой кислоты заканчивается как образуемый до разделения изоцианата и спирта, которые образуются путем термического разложения, часто приводя таким образом к очевидным трудностям дальнейшего прохождения реакции термического разложения.
С другой стороны, термическое разложение алкилкарбаматов связано с одновременным возникновением различных необратимых побочных реакций, таких как реакции термической денатурации, которые являются нежелательными для алкилкарбаматов или конденсации изоцианатов, образуемых путем термического разложения. Примеры таких побочных реакций включают реакцию, в которой образуются мочевинные связи, как представлено следующей формулой (2), реакцию, в которой образуются карбодиимиды, как представлено следующей формулой (3), и реакцию, в которой образуются изоцианураты, как представлено следующей формулой (4) (см. Berchte der Deutechen Chemischen Gesellschaft, Vol. 3, p. 653, 1870 и Journal of American Chemical Society, Vol. 81, p. 2138, 1959):
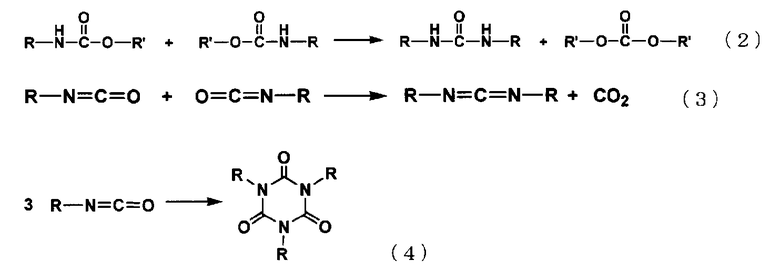
Помимо этих побочных реакций, приводящих к снижению выхода и селективности целевого изоцианата, в способе получения полиизоцианатов, в частности, эти реакции могут сделать долговременную работу способа затруднительной в результате, например, того, что они вызывают осаждение твердых полимерных частиц, что засоряет реактор.
Предлагались различные способы для решения таких проблем. Например, был предложен способ получения полиизоцианата, в котором полиалкилкарбамат, в котором сложноэфирные группы образованы из алкоксигрупп, соответствующих первичному спирту, подвергают реакции переэтерификации с вторичным спиртом с получением полиалкилкарбамата, в котором сложноэфирные группы образованы из алкоксигрупп, соответствующих вторичному спирту, с последующим термическим разложением полиалкилкарбамата (см., например, International Publication No. WO 95/23484). В этом способе описывается, что температура термического разложения полиалкилкарбамата может быть установлена на более низкую температуру, благодаря прохождению через полиалкилкарбамат, в котором сложноэфирные группы образованы из алкоксигрупп, соответствующих вторичному спирту, результатом чего является возможность ингибирования осаждения твердых полимерных частиц. Однако скорость обратной реакции между полиизоцианатом, образованным в результате реакции термического разложения полиалкилкарбамата, и вторичным спиртом все еще остается высокой, оставляя таким образом проблему ингибирования образования полиалкилкарбамата путем обратной реакции нерешенной.
Был раскрыт альтернативный способ, где при получении ароматических изоцианатов, например ароматический полиалкилкарбамат и ароматическое гидроксисоединение подвергают реакции переэтерификации с получением ароматического полиарилкарбамата с последующим термическим разложением ароматического полиарилкарбамата с получением ароматического изоцианата (см., например, Патент США № 3992430). В этом способе описывается эффект получения возможности устанавливать температуру термического разложения на более низкую температуру посредством прохождения через ароматический полиарилкарбамат. Однако, в случае этого ароматического полиарилкарбамата также, при температурах, подобных тем, при которых осуществляют реакцию переэтерификации или реакцию термического разложения, имеется много случаев, в которых все еще возникают побочные реакции, подобные тем, которые описаны выше, приводя к тому, что проблема улучшения выхода изоцианата остается неразрешенной. Более того, известно, что термическое разложение N-замещенных ароматических уретанов в газовой фазе или жидкой фазе часто приводит к возникновению различных нежелательных побочных реакций (см., например, Патент США № 4613466).
Раскрытие изобретения
Задачи, решаемые настоящим изобретением
Как было описано выше, на сегодняшний день вряд ли существуют какие-либо способы промышленного получения полиизоцианатов с хорошим выходом без использования чрезвычайно токсичного фосгена.
Задачей настоящего изобретения является обеспечение способа, который делает возможным стабильное получение изоцианатов в течение длительного периода времени с высоким выходом, не сталкиваясь при этом с различными проблемами, существующими в известном уровне техники при получении изоцианатов без использования фосгена.
Средства решения задач
В свете вышеизложенного, в результате проведения всесторонних исследований, связанных с вышеозначенной проблемой, авторами настоящего изобретения было обнаружено, что способ получения, в котором сложный эфир карбаминовой кислоты и специфическое ароматическое гидроксисоединение подвергают реакции переэтерификации с получением арилкарбамата с последующей реакцией термического разложения арилкарбамата с получением изоцианата, позволяет решить вышеозначенные проблемы, что таким образом привело к созданию настоящего изобретения.
А именно, настоящее изобретение обеспечивает следующее:
[1] способ получения изоцианата, включающий следующие стадии:
взаимодействие сложного эфира карбаминовой кислоты и ароматического гидроксисоединения с получением арилкарбамата, содержащего группу, происходящую из ароматического гидроксисоединения; и
реакцию разложения арилкарбамата,
где ароматическое гидроксисоединение представляет собой ароматическое гидроксисоединение, которое представлено следующей формулой (5) и которое содержит заместитель R1, по меньшей мере, в одном ортоположении гидроксильной группы:

где кольцо A представляет собой ароматическое углеводородное кольцо в форме одного или нескольких колец, которые могут содержать заместитель и, которые содержат от 6 до 20 атомов углерода;
R1 представляет собой группу, отличную от атома водорода, в форме алифатической алкильной группы, содержащей от 1 до 20 атомов углерода, алифатической алкоксигруппы, содержащей от 1 до 20 атомов углерода, арильной группы, содержащей от 6 до 20 атомов углерода, арилоксигруппы, содержащей от 6 до 20 атомов углерода, аралкильной группы, содержащей от 7 до 20 атомов углерода, или аралкиоксигруппы, содержащей от 7 до 20 атомов углерода, при этом такая группа содержит атом, выбранный из атома углерода, атома кислорода и атома азота; и R1 может связываться с A с образованием циклической структуры,
[2] способ в соответствии с пунктом [1], где ароматическое гидроксисоединение представляет собой соединение, представленное следующей формулой (6):

где кольцо A и R1 имеют значения, определенные выше,
R2 представляет собой атом водорода или алифатическую алкильную группу, содержащую от 1 до 20 атомов углерода, алифатическую алкоксигруппу, содержащую от 1 до 20 атомов углерода, арильную группу, содержащую от 6 до 20 атомов углерода, арилоксигруппу, содержащую от 6 до 20 атомов углерода, аралкильную группу, содержащую от 7 до 20 атомов углерода, или аралкиоксигруппу, содержащую от 7 до 20 атомов, при этом алифатическая алкильная, алифатическая алкокси, арильная, арилокси, аралкильная и аралкилокси группы содержат атом, выбранный из атома углерода, атома кислорода и атома азота, и R2 может связываться с A с образованием циклической структуры,
[3] способ в соответствии с пунктом [2], где в формуле (6) общее количество атомов углерода, образующих R1 и R2, составляет от 2 до 20,
[4] способ в соответствии с любым из пунктов [1]-[3], где кольцо A ароматического гидроксисоединения включает структуру, содержащую, по меньшей мере, одну структуру, выбранную из группы, состоящей из бензольного кольца, нафталинового кольца и антраценового кольца,
[5] способ в соответствии с пунктом [4], где ароматическое гидроксисоединение представляет собой соединение, представленное следующей формулой (7):
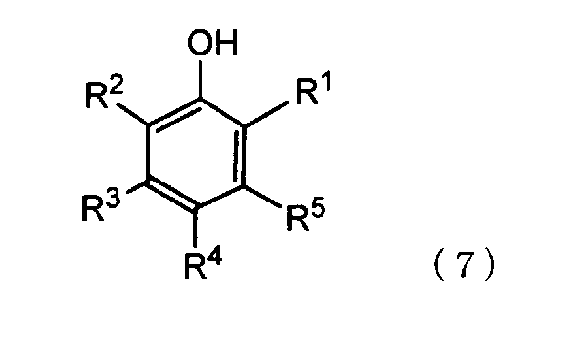
где R1 и R2 имеют значения, определенные выше, и
каждый из R3, R4 и R5 независимо представляет собой атом водорода или алифатическую алкильную группу, содержащую от 1 до 20 атомов углерода, алифатическую алкоксигруппу, содержащую от 1 до 20 атомов углерода, арильную группу, содержащую от 6 до 20 атомов углерода, арилоксигруппу, содержащую от 6 до 20 атомов углерода, аралкильную группу, содержащую от 7 до 20 атомов углерода, или аралкиоксигруппу, содержащую от 7 до 20 атомов углерода, при этом алифатическая алкильная, алифатическая алкокси, арильная, арилокси, аралкильная и аралкилокси группы содержат атом, выбранный из атома углерода, атома кислорода и атома азота,
[6] способ в соответствии с пунктом [5], где ароматическое гидроксисоединение представляет собой такое соединение, где в формуле (7) каждый из R1 и R4 независимо представляет собой группу, представленную следующей формулой (8), и R2, R3 и R5 представляют собой атом водорода:

где X представляет собой разветвленную структуру, выбранную из структур, представленных следующими формулами (9) и (10):
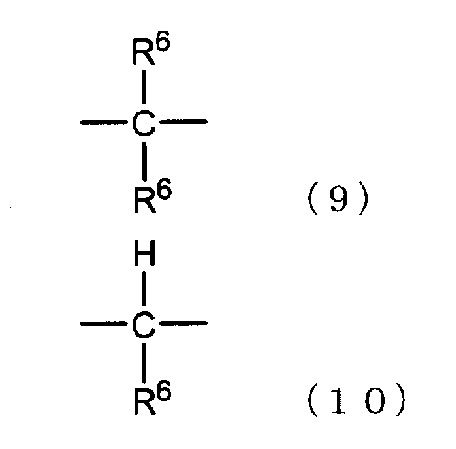
где R6 представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода,
[7] способ в соответствии с пунктом [5], где ароматическое гидроксисоединение представляет собой такое соединение, где в формуле (3) R1 представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 8 атомов углерода, и каждый из R2 и R4 независимо представляет собой атом водорода или линейную или разветвленную алкильную группу, содержащую от 1 до 8 атомов углерода,
[8] способ в соответствии с любым из пунктов [1]-[7], где сложный эфир карбаминовой кислоты представляет собой алифатический сложный эфир карбаминовой кислоты, и низкокипящий компонент, образованный при помощи арилкарбамата, представляет собой алифатический спирт,
[9] способ в соответствии с пунктом [8], где алифатический сложный эфир карбаминовой кислоты представляет собой алифатический сложный эфир поликарбаминовой кислоты,
[10] способ в соответствии с пунктом [8], дополнительно включающий следующие стадии:
непрерывную подачу алифатического сложного эфира карбаминовой кислоты и ароматического гидроксисоединения в реакционный сосуд так, чтобы обеспечить взаимодействие алифатического сложного эфира карбаминовой кислоты и ароматического гидроксисоединения внутри реакционного сосуда;
выделение образованного низкокипящего компонента в форме газообразного компонента; и
непрерывное экстрагирование реакционной жидкости, содержащей арилкарбамат и ароматическое гидроксисоединение, из донной части реакционного сосуда,
[11] способ в соответствии с любым из пунктов [1]-[10], где реакция разложения представляет собой реакцию термического разложения и представляет собой реакцию, в которой соответствующий изоцианат и ароматическое гидроксисоединение образуются из арилкарбамата,
[12] способ в соответствии с пунктом [11], где, по меньшей мере, одно соединение из изоцианата и ароматического гидроксисоединения, которые образуются в результате реакции термического разложения арилкарбамата, выделяют в форме газообразного компонента,
[13] способ в соответствии с пунктом [8], где алифатический сложный эфир карбаминовой кислоты представляет собой соединение, представленное следующей формулой (11):
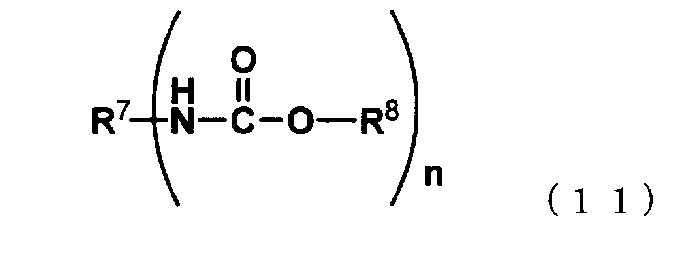
где R7 представляет собой группу, выбранную из группы, состоящей из алифатической группы, содержащей от 1 до 20 атомов углерода, и ароматической группы, содержащей от 6 до 20 атомов углерода, при этом такая группа содержит атом, который выбран из атома углерода, атома кислорода и атома азота и имеет валентность n,
R8 представляет собой алифатическую группу, которая содержит от 1 до 8 атомов углерода и которая содержит атом, выбранный из атома углерода, атома кислорода и атома азота, и
n представляет собой целое число, имеющее значение от 1 до 10,
[14] способ в соответствии с пунктом [13], где алифатический сложный эфир карбаминовой кислоты представляет собой такой, где R8 в соединении, представленном формулой (11), представляет собой группу, выбранную из группы, состоящей из алкильной группы, содержащей от 1 до 20 атомов углерода, и циклоалкильной группы, содержащей от 5 до 20 атомов углерода,
[15] способ в соответствии с пунктом [14], где алифатический сложный эфир карбаминовой кислоты представляет собой, по меньшей мере, одно соединение, выбранное из группы, состоящей из соединений, представленных следующими формулами (12), (13) и (14):

где R8 имеет значения, определенные выше,
[16] полиарилкарбамат, представленный следующими формулами (15), (16) или (17):
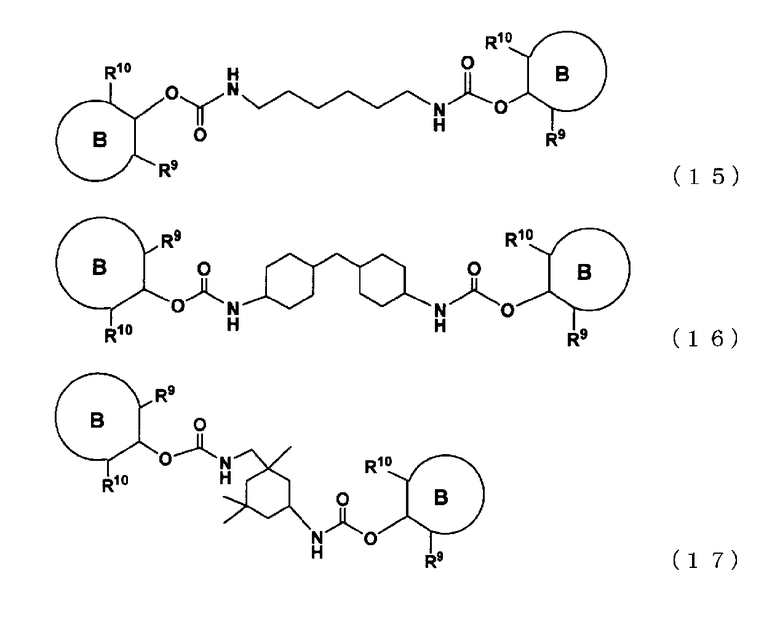
где кольцо B представляет собой структуру, которая может содержать заместитель и которая содержит, по меньшей мере, одну структуру, выбранную из группы, состоящей из бензольного кольца, нафталинового кольца и антраценового кольца,
R9 представляет собой группу, отличную от атома водорода, в форме алифатической алкильной группы, содержащей от 1 до 20 атомов углерода, алифатической алкоксигруппы, содержащей от 1 до 20 атомов углерода, арильной группы, содержащей от 6 до 20 атомов углерода, арилоксигруппы, содержащей от 6 до 20 атомов углерода, аралкильной группы, содержащей от 7 до 20 атомов углерода, или аралкиоксигруппы, содержащей от 7 до 20 атомов углерода, при этом такая группа содержит атом, выбранный из атома углерода, атома кислорода и атома азота, и
R10 представляет собой алифатическую алкильную группу, содержащую от 1 до 20 атомов углерода, алифатическую алкоксигруппу, содержащую от 1 до 20 атомов углерода, арильную группу, содержащую от 6 до 20 атомов углерода, арилоксигруппу, содержащую от 6 до 20 атомов углерода, аралкильную группу, содержащую от 7 до 20 атомов углерода, или аралкиоксигруппу, содержащую от 7 до 20 атомов углерода, при этом алифатическая алкильная, алифатическая алкокси, арильная, арилокси, аралкильная и аралкилокси группа содержит атом, выбранный из атома углерода, атома кислорода и атома азота,
[17] полиарилкарбамат в соответствии с пунктом [16], который представлен следующими формулами (18), (19) или (20):
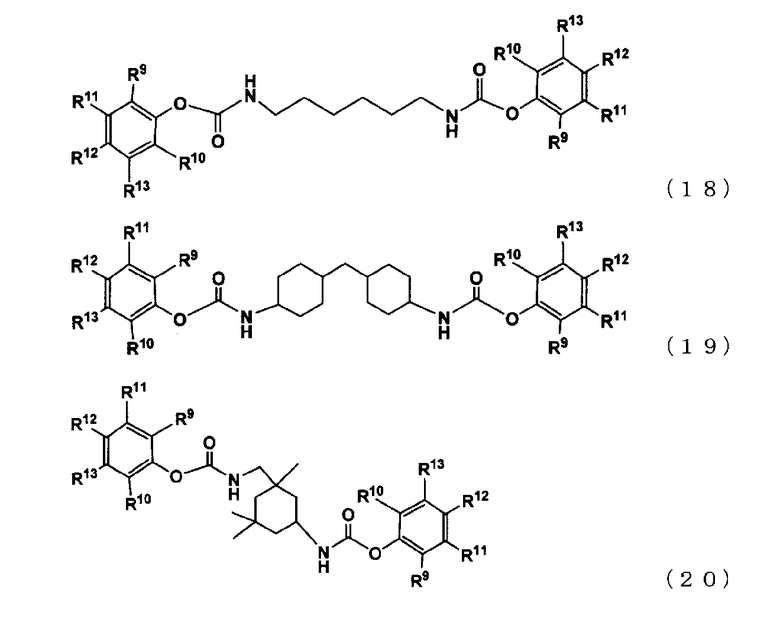
где R9 представляет собой группу, отличную от атома водорода, в форме алифатической алкильной группы, содержащей от 1 до 20 атомов углерода, алифатической алкоксигруппы, содержащей от 1 до 20 атомов углерода, арильной группы, содержащей от 6 до 20 атомов углерода, арилоксигруппы, содержащей от 6 до 20 атомов углерода, аралкильной группы, содержащей от 7 до 20 атомов углерода, или аралкиоксигруппы, содержащей от 7 до 20 атомов углерода, при этом такая группа содержит атом, выбранный из атома углерода, атома кислорода и атома азота, и
каждый из R10, R11, R12 и R13 независимо представляет собой атом водорода или алифатическую алкильную группу, содержащую от 1 до 20 атомов углерода, алифатическую алкоксигруппу, содержащую от 1 до 20 атомов углерода, арильную группу, содержащую от 6 до 20 атомов углерода, арилоксигруппу, содержащую от 6 до 20 атомов углерода, аралкильную группу, содержащую от 7 до 20 атомов углерода, или аралкиоксигруппу, содержащую от 7 до 20 атомов углерода, при этом алифатическая алкильная, алифатическая алкокси, арильная, арилокси, аралкильная и аралкилокси группа содержащит атом, выбранный из атома углерода, атома кислорода и атома азота).
Преимущество настоящего изобретения
В соответствии с настоящим изобретением, изоцианат может быть эффективным образом получен без использования фосгена.
Краткое описание чертежей
Фиг. 1 в общем виде представляет схему устройства для непрерывного процесса получения сложного эфира угольной кислоты в соответствии с одним вариантом воплощения настоящего изобретения;
Фиг. 2 в общем виде представляет схему устройства для реакции переэтерификации в соответствии с одним вариантом воплощения настоящего изобретения;
Фиг. 3 в общем виде представляет схему устройства для реакции термического разложения в соответствии с одним вариантом воплощения настоящего изобретения;
Фиг. 4 в общем виде представляет схему устройства для реакции термического разложения в соответствии с одним вариантом воплощения настоящего изобретения;
Фиг. 5 в общем виде представляет схему устройства для получения изоцианата в соответствии с одним вариантом воплощения настоящего изобретения;
Фиг. 6 представляет 1H-ЯМР спектр ди(2,4-ди-трет-бутилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты, полученного на стадии (3-4) Примера 3 по настоящему изобретению; и,
Фиг. 7 представляет 13C-ЯМР спектр ди(2,4-ди-трет-бутилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты, полученного на стадии (3-4) Примера 3 по настоящему изобретению.
Описание цифровых обозначений:
(Фиг. 1)
101, 107: дистилляционная колонна, 102: реактор колоночного типа, 103, 106: пленочный испаритель, 104: автоклав, 105: резервуар для декарбонизации, 111, 112, 117: ребойлер, 121, 123, 126, 127: конденсатор, 1, 9: линия подачи, 2, 4, 5, 6, 7, 8, 10, 11, 12, 13, 14: передаточная линия, 3, 15: линия выпуска, 16: линия экстракции, 17: питающая линия.
(Фиг. 2)
201: питающий резервуар, 202: пленочный испаритель, 203: дистилляционная колонна, 204: конденсатор, 21, 22, 23, 24, 25, 26, 27, 28, 29: передаточная линия
(Фиг.3)
301: питающий резервуар, 302: пленочный испаритель, 303: дистилляционная колонна, 304: ребойлер, 305: конденсатор, 31, 32, 33, 34, 35, 36, 37, 38, 39, передаточная линия
(Фиг. 4)
401: питающий резервуар, 402: пленочный испаритель, 403, 404: дистилляционная колонна, 405, 407, конденсатор, 406, 408: ребойлер, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53: передаточная линия
(Фиг. 5)
601, 604: смесительный резервуар, 602, 605, 608: резервуар, 603, 606, 609: пленочный испаритель, 607, 610: дистилляционная колонна, 611, 613: конденсатор, 612: ребойлер, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81: передаточная линия
Лучший способ осуществления изобретения
Далее представлено подробное объяснение лучшего способа осуществления настоящего изобретения (далее указан как представленный вариант воплощения настоящего изобретения). Кроме того, настоящее изобретение не ограничивается представленным ниже вариантом воплощения настоящего изобретения, наоборот, его можно модифицировать различными путями, не выходящими за рамки сущности настоящего изобретения.
Способ получения изоцианата согласно представленному варианту воплощения настоящего изобретения представляет собой способ получения изоцианатов, который включает следующие стадии: взаимодействие сложного эфира карбаминовой кислоты и ароматического гидроксисоединения с получением арилкарбамата, содержащего группу, происходящую из ароматического гидроксисоединения; и реакцию разложения арилкарбамата; где в качестве ароматического гидроксисоединения используют ароматическое соединение, имеющее специфическую композицию.
Ниже представлен пример подробной схемы реакций представленного варианта воплощения настоящего изобретения. Однако способ получения изоцианата, заявленный в настоящем изобретении, не ограничивается представленной ниже схемой реакций.
Таблица 1


Для начала представлено разъяснение, касающееся соединений, используемых в способе получения изоцианата представленного варианта воплощения настоящего изобретения.
<Ароматические гидроксисоединения>
Ароматические гидроксисоединения, используемые в способе получения изоцианата представленного варианта воплощения настоящего изобретения, представляют собой ароматические гидроксисоединения, которые представлены следующей формулой (5) и которые содержат заместитель, по меньшей мере, в одном положении орто относительно гидроксильной группы:
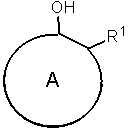
где кольцо A представляет собой ароматическое углеводородное кольцо в форме одного или нескольких колец, которые имеют заместитель и которые содержат от 6 до 20 атомов углерода;
R1 представляет собой группу, отличную от атома водорода, в форме алифатической алкильной группы, содержащей от 1 до 20 атомов углерода, алифатической алкоксигруппы, содержащей от 1 до 20 атомов углерода, арильной группы, содержащей от 6 до 20 атомов углерода, арилоксигруппы, содержащей от 6 до 20 атомов углерода, аралкильной группы, содержащей от 7 до 20 атомов углерода, или аралкиоксигруппы, содержащей от 7 до 20 атомов углерода, при этом такая группа содержит атом, выбранный из атома углерода, атома кислорода и атома азота, и R1 может связываться с A с образованием циклической структуры.
Примеры R1 в формуле (1) выше включают алифатические алкильные группы, в которых количество атомов углерода, образующих такую группу, представляет собой количество, выбранное из целых чисел, имеющих значение от 1 до 20, такие как метильная группа, этильная группа, пропильная группа (изомеры), бутильная группа (изомеры), пентильная группа (изомеры), гексильная группа (изомеры), гептильная группа (изомеры), октильная группа (изомеры), нонильная группа (изомеры), децильная группа (изомеры), додецильная группа (изомеры), октадецильная группа (изомеры) или подобные; алифатические алкоксигруппы, в которых количество атомов углерода, образующих такую группу, представляет собой количество, выбранное из целых чисел, имеющих значение от 1 до 20, такие как метоксигруппа, этоксигруппа, пропоксигруппа (изомеры), бутилоксигруппа (изомеры), пентилоксигруппа (изомеры), гексилоксигруппа (изомеры), гептилоксигруппа (изомеры), октилоксигруппа (изомеры), нонилоксигруппа (изомеры), децилоксигруппа (изомеры), додецилоксигруппа (изомеры), октадецилоксигруппа (включая изомеры) или подобные; арильные группы, в которых количество атомов углерода, образующих такую группу, составляет от 6 до 20, такие как фенильная группа, метилфенильная группа (изомеры), этилфенильная группа (изомеры), пропилфенильная группа (изомеры), бутилфенильная группа (изомеры), пентилфенильная группа (изомеры), гексилфенильная группа (изомеры), гептилфенильная группа (изомеры), октилфенильная группа (изомеры), нонилфенильная группа (изомеры), децилфенильная группа (изомеры), бифенильная группа (изомеры), диметилфенильная группа (изомеры), диэтилфенильная группа (изомеры), дипропилфенильная группа (изомеры), дибутилфенильная группа (изомеры), дипентилфенильная группа (изомеры), дигексилфенильная группа (изомеры), дигептилфенильная группа (изомеры), терфенильная группа (изомеры), триметилфенильная группа (изомеры), триэтилфенильная группа (изомеры), трипропилфенильная группа (изомеры), трибутилфенильная группа (изомеры) или подобные; арилоксигруппы, в которых количество атомов углерода, образующих такую группу, составляет от 6 до 20, такие как феноксигруппа, метилфеноксигруппа (изомеры), этилфеноксигруппа (изомеры), пропилфеноксигруппа (изомеры), бутилфеноксигруппа (изомеры), пентилфеноксигруппа (изомеры), гексилфеноксигруппа (изомеры), гептилфеноксигруппа (изомеры), октилфеноксигруппа (изомеры), нонилфеноксигруппа (изомеры), децилфеноксигруппа (изомеры), фенилфеноксигруппа (изомеры), диметилфеноксигруппа (изомеры), диэтилфеноксигруппа (изомеры), дипропилфеноксигруппа (изомеры), дибутилфеноксигруппа (изомеры), дипентилфеноксигруппа (изомеры), дигексилфеноксигруппа (изомеры), дигептилфеноксигруппа (изомеры), дифенилфеноксигруппа (изомеры), триметилфеноксигруппа (изомеры), триэтилфеноксигруппа (изомеры), трипропилфеноксигруппа (изомеры), трибутилфеноксигруппа (изомеры) или подобные; аралкильные группы, в которых количество атомов углерода, образующих такую группу, составляет от 7 до 20, такие как фенилметильная группа, фенилэтильная группа (изомеры), фенилпропильная группа (изомеры), фенилбутильная группа (изомеры), фенилпентильная группа (изомеры), фенилгексильная группа (изомеры), фенилгептильная группа (изомеры), фенилоктильная группа (изомеры), фенилнонильная группа (изомеры) или подобные; и аралкилоксигруппы, в которых количество атомов углерода, образующих такую группу, составляет от 7 до 20, такие как фенилметоксигруппа, фенилэтоксигруппа (изомеры), фенилпропилоксигруппа (изомеры), фенилбутилоксигруппа (изомеры), фенилпентилоксигруппа (изомеры), фенилгексилоксигруппа (изомеры), фенилгептилоксигруппа (изомеры), фенилоктилоксигруппа (изомеры), фенилнонилоксигруппа (изомеры) или подобные.
Примеры кольца A в формуле (1) выше включают бензольное кольцо, нафталиновое кольцо, антраценовое кольцо, фенантреновое кольцо, нафтаценовое кольцо, хризеновое кольцо, пиреновое кольцо, трифениленовое кольцо, пенталеновое кольцо, азуленовое кольцо, гепталеновое кольцо, андаценовое кольцо, бифениленовое кольцо, аценафтиленовое кольцо, ацеантриленовое кольцо, ацефенантриленовое кольцо или подобные, предпочтительные примеры включают кольца, выбранные из группы, состоящей из бензольного кольца, нафталинового кольца и антраценового кольца. Кроме того, эти кольца могут содержать заместитель, отличный от вышеуказанного R1, примеры которого включают алифатические алкильные группы, в которых количество атомов углерода, образующих такую группу, представляет собой количество, выбранное из целых чисел, имеющих значение от 1 до 20, такие как метильная группа, этильная группа, пропильная группа (изомеры), бутильная группа (изомеры), пентильная группа (изомеры), гексильная группа (изомеры), гептильная группа (изомеры), октильная группа (изомеры), нонильная группа (изомеры), децильная группа (изомеры), додецильная группа (изомеры), октадецильная группа (изомеры) или подобные; алифатические алкоксигруппы, в которых количество атомов углерода, образующих такую группу, представляет собой количество, выбранное из целых чисел, имеющих значение от 1 до 20, так как метоксигруппа, этоксигруппа, пропоксигруппа (изомеры), бутилоксигруппа (изомеры), пентилоксигруппа (изомеры), гексилоксигруппа (изомеры), гептилоксигруппа (изомеры), октилоксигруппа (изомеры), нонилоксигруппа (изомеры), децилоксигруппа (изомеры), додецилоксигруппа (изомеры), октадецилоксигруппа (изомеры) или подобные; арильные группы, в которых количество атомов углерода, образующих такую группу, составляет от 6 до 20, такие как фенильная группа, метилфенильная группа (изомеры), этилфенильная группа (изомеры), пропилфенильная группа (изомеры), бутилфенильная группа (изомеры), пентилфенильная группа (изомеры), гексилфенильная группа (изомеры), гептилфенильная группа (изомеры), октилфенильная группа (изомеры), нонилфенильная группа (изомеры), децилфенильная группа (изомеры), бифенильная группа (изомеры), диметилфенильная группа (изомеры), диэтилфенильная группа (изомеры), дипропилфенильная группа (изомеры), дибутилфенильная группа (изомеры), дипентилфенильная группа (изомеры), дигексилфенильная группа (изомеры), дигептилфенильная группа (изомеры), терфенильная группа (изомеры), триметилфенильная группа (изомеры), триэтилфенильная группа (изомеры), трипропилфенильная группа (изомеры), трибутилфенильная группа (изомеры) или подобные; арилоксигруппы, в которых количество атомов углерода, образующих такую группу, составляет от 6 до 20, такие как феноксигруппа, метилфеноксигруппа (изомеры), этилфеноксигруппа (изомеры), пропилфеноксигруппа (изомеры), бутилфеноксигруппа (изомеры), пентилфеноксигруппа (изомеры), гексилфеноксигруппа (изомеры), гептилфеноксигруппа (изомеры), октилфеноксигруппа (изомеры), нонилфеноксигруппа (изомеры), децилфеноксигруппа (изомеры), фенилфеноксигруппа (изомеры), диметилфеноксигруппа (изомеры), диэтилфеноксигруппа (изомеры), дипропилфеноксигруппа (изомеры), дибутилфеноксигруппа (изомеры), дипентилфеноксигруппа (изомеры), дигексилфеноксигруппа изомеры), дигептилфеноксигруппа (изомеры), дифенилфеноксигруппа (изомеры), триметилфеноксигруппа (изомеры), триэтилфеноксигруппа (изомеры), трипропилфеноксигруппа (изомеры), трибутилфеноксигруппа (изомеры) или подобные; аралкильные группы, в которых количество атомов углерода, образующих такую группу, составляет от 7 до 20, такие как фенилметильная группа, фенилэтильная группа (изомеры), фенилпропильная группа (изомеры), фенилбутильная группа (изомеры), фенилпентильная группа (изомеры), фенилгексильная группа (изомеры), фенилгептильная группа (изомеры), фенилоктильная группа (изомеры), фенилнонильная группа (изомеры) или подобные; и аралкилоксигруппы, в которых количество атомов углерода, образующих такую группу, составляет от 7 до 20, такие как фенилметоксигруппа, фенилэтоксигруппа (изомеры), фенилпропилоксигруппа (изомеры), фенилбутилоксигруппа (изомеры), фенилпентилоксигруппа (изомеры), фенилгексилоксигруппа (изомеры), фенилгептилоксигруппа (изомеры), фенилоктилоксигруппа (изомеры), фенилнонилоксигруппа (изомеры).
Кроме того, ароматические гидроксисоединения можно использовать предпочтительным образом, независимо от того, является ли такое соединение ароматическим гидроксисоединением, содержащим заместитель в одном ортоположении относительно гидроксильной группы, или ароматическим гидроксисоединением, содержащим заместители в двух ортоположениях относительно гидроксильной группы, как в соединениях, представленных следующей формулой (6):

где кольцо A и R1 имеют значения, определенные выше,
R2 представляет собой алифатическую алкильную группу, содержащую от 1 до 20 атомов углерода, алифатическую алкоксигруппу, содержащую от 1 до 20 атомов углерода, арильную группу, содержащую от 6 до 20 атомов углерода, арилоксигруппу, содержащую от 6 до 20 атомов углерода, аралкильную группу, содержащую от 7 до 20 атомов углерода, или аралкиоксигруппу, содержащую от 7 до 20 атомов, при этом алифатическая алкильная, алифатическая алкокси, арильная, арилокси, аралкильная и аралкилокси группы содержат атомы, выбранные из атома углерода, атома кислорода и атома азота, и R2 может связываться с A с образованием циклической структуры.
Примеры R2 в указанной выше формуле (6) включают атом водорода; алифатические алкильные группы, в которых количество атомов углерода, образующих такую группу, представляет собой количество, выбранное из целых чисел, имеющих значение от 1 до 20, такие как метильная группа, этильная группа, пропильная группа (изомеры), бутильная группа (изомеры), пентильная группа (изомеры), гексильная группа (изомеры), гептильная группа (изомеры), октильная группа (изомеры), нонильная группа (изомеры), децильная группа (изомеры), додецильная группа (изомеры), октадецильная группа (изомеры) или подобные; алифатические алкоксигруппы, в которых количество атомов углерода, образующих такую группу, представляет собой количество, выбранное из целых чисел, имеющих значение от 1 до 20, такие как метоксигруппа, этоксигруппа, пропоксигруппа (изомеры), бутилоксигруппа (изомеры), пентилоксигруппа (изомеры), гексилоксигруппа (изомеры), гептилоксигруппа (изомеры), октилоксигруппа (изомеры), нонилоксигруппа (изомеры), децилоксигруппа (изомеры), додецилоксигруппа (изомеры), октадецилоксигруппа (изомеры) или подобные; арильные группы, в которых количество атомов углерода, образующих такую группу, составляет от 6 до 20, такие как фенильная группа, метилфенильная группа (изомеры), этилфенильная группа (изомеры), пропилфенильная группа (изомеры), бутилфенильная группа (изомеры), пентилфенильная группа (изомеры), гексилфенильная группа (изомеры), гептилфенильная группа (изомеры), октилфенильная группа (изомеры), нонилфенильная группа (изомеры), децилфенильная группа (изомеры), бифенильная группа (изомеры), диметилфенильная группа (изомеры), диэтилфенильная группа (изомеры), дипропилфенильная группа (изомеры), дибутилфенильная группа (изомеры), дипентилфенильная группа (изомеры), дигексилфенильная группа (изомеры), дигептилфенильная группа (изомеры), терфенильная группа (изомеры), триметилфенильная группа (изомеры), триэтилфенильная группа (изомеры), трипропилфенильная группа (изомеры), трибутилфенильная группа (изомеры) или подобные; арилоксигруппы, в которых количество атомов углерода, образующих такую группу, составляет от 6 до 20, такие как феноксигруппа, метилфеноксигруппа (изомеры), этилфеноксигруппа (изомеры), пропилфеноксигруппа (изомеры), бутилфеноксигруппа (изомеры), пентилфеноксигруппа (изомеры), гексилфеноксигруппа (изомеры), гептилфеноксигруппа (изомеры), октилфеноксигруппа (изомеры), нонилфеноксигруппа (изомеры), децилфеноксигруппа (изомеры), фенилфеноксигруппа (изомеры), диметилфеноксигруппа (изомеры), диэтилфеноксигруппа (изомеры), дипропилфеноксигруппа (изомеры), дибутилфеноксигруппа (изомеры), дипентилфеноксигруппа (изомеры), дигексилфеноксигруппа (изомеры), дигептилфеноксигруппа (изомеры), дифенилфеноксигруппа (изомеры), триметилфеноксигруппа (изомеры), триэтилфеноксигруппа (изомеры), трипропилфеноксигруппа (изомеры), трибутилфеноксигруппа (изомеры) или подобные; аралкильные группы, в которых количество атомов углерода, образующих такую группу, составляет от 7 до 20, такие как фенилметильная группа, фенилэтильная группа (изомеры), фенилпропильная группа (изомеры), фенилбутильная группа (изомеры), фенилпентильная группа (изомеры), фенилгексильная группа (изомеры), фенилгептильная группа (изомеры), фенилоктильная группа (изомеры), фенилнонильная группа (изомеры) или подобные; и аралкилоксигруппы, в которых количество атомов углерода, образующих такую группу, составляет от 7 до 20, такие как фенилметоксигруппа, фенилэтоксигруппа (изомеры), фенилпропилоксигруппа (изомеры), фенилбутилоксигруппа (изомеры), фенилпентилоксигруппа (изомеры), фенилгексилоксигруппа (изомеры), фенилгептилоксигруппа (изомеры), фенилоктилоксигруппа (изомеры), фенилнонилоксигруппа (изомеры) или подобные.
В случае ароматического гидроксисоединения, используемого в способе получения изоцианата представленного варианта воплощения настоящего изобретения, используют ароматические гидроксисоединения, содержащие заместители в двух орто положениях относительно гидроксильной группы, выбранные из соединений, представленных указанной выше формулой (6), предпочтительно используют ароматические гидроксисоединения, в которых общее количество атомов углерода, образующих R1 и R2, составляет от 2 до 20. Нет никаких конкретных ограничений в том, что касается комбинаций R1 и R2, при условии, что общее количество атомов углерода, образующих R1 и R2, составляет от 2 до 20.
Примеры таких ароматических гидроксисоединений включают соединения, представленные следующей формулой (7):

где R1 и R2 имеют значения, определенные выше, и
каждый из R3, R4 и R5 независимо представляет собой атом водорода или алифатическую алкильную группу, содержащую от 1 до 20 атомов углерода, алифатическую алкоксигруппу, содержащую от 1 до 20 атомов углерода, арильную группу, содержащую от 6 до 20 атомов углерода, арилоксигруппу, содержащую от 6 до 20 атомов углерода, аралкильную группу, содержащую от 7 до 20 атомов углерода, или аралкиоксигруппу, содержащую от 7 до 20 атомов углерода, при этом алифатическая алкильная, алифатическая алкокси, арильная, арилокси, аралкильная и аралкилокси группы содержат атом, выбранный из атома углерода, атома кислорода и атома азота.
В частности, предпочтительно используют ароматические гидроксисоединения, в которых каждый из R1 и R4 в указанной выше формуле (7) независимо представляет собой группу, представленную следующей формулой (8), и R2, R3 и R5 представляют собой атомы водорода, или ароматические гидроксисоединения, в которых R1 в указанной выше формуле (7) представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 8 атомов углерода, и каждый из R2 и R4 независимо представляет собой атом водорода или линейную или разветвленную алкильную группу, содержащую от 1 до 8 атомов углерода:

где X представляет собой разветвленную структуру, выбранную из структур, представленных следующими формулами (9) и (10):
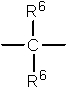

где R6 представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода.
Примеры таких ароматических гидроксисоединений включают 2-этилфенол, 2-пропилфенол (изомеры), 2-бутилфенол (изомеры), 2-пентилфенол (изомеры), 2-гексилфенол (изомеры), 2-гептилфенол (изомеры), 2,6-диметилфенол, 2,4-диэтилфенол, 2,6-диэтилфенол, 2,4-дипропилфенол (изомеры), 2,6-дипропилфенол (изомеры), 2,4-дибутилфенол (изомеры), 2,4-дипентилфенол (изомеры), 2,4-дигексилфенол (изомеры), 2,4-дигептилфенол (изомеры), 2-метил-6-этилфенол, 2-метил-6-пропилфенол (изомеры), 2-метил-6-бутилфенол (изомеры), 2-метил-6-пентилфенол (изомеры), 2-этил-6-пропилфенол (изомеры), 2-этил-6-бутилфенол (изомеры), 2-этил-6-пентилфенол (изомеры), 2-пропил-6-бутилфенол (изомеры), 2-этил-4-метилфенол (изомеры), 2-этил-4-пропилфенол (изомеры), 2-этил-4-бутилфенол (изомеры), 2-этил-4-пентилфенол (изомеры), 2-этил-4-гексилфенол (изомеры), 2-этил-4-гептилфенол (изомеры), 2-этил-4-октилфенол (изомеры), 2-этил-4-фенилфенол (изомеры), 2-этил-4-кумилфенол (изомеры), 2-пропил-4-метилфенол (изомеры), 2-пропил-4-этилфенол (изомеры), 2-пропил-4-бутилфенол (изомеры), 2-пропил-4-пентилфенол (изомеры), 2-пропил-4-гексилфенол (изомеры), 2-пропил-4-гептилфенол (изомеры), 2-пропил-4-октилфенол (изомеры), 2-пропил-4-фенилфенол (изомеры), 2-пропил-4-кумилфенол (изомеры), 2-бутил-4-метилфенол (изомеры), 2-бутил-4-этилфенол (изомеры), 2-бутил-4-пропилфенол (изомеры), 2-бутил-4-пентилфенол (изомеры), 2-бутил-4-гексилфенол (изомеры), 2-бутил-4-гептилфенол (изомеры), 2-бутил-4-октилфенол (изомеры), 2-бутил-4-фенилфенол (изомеры), 2-бутил-4-кумилфенол (изомеры), 2-пентил-4-метилфенол (изомеры), 2-пентил-4-этилфенол (изомеры), 2-пентил-4-пропилфенол (изомеры), 2-пентил-4-бутилфенол (изомеры), 2-пентил-4-гексилфенол (изомеры), 2-пентил-4-гептилфенол (изомеры), 2-пентил-4-октилфенол (изомеры), 2-пентил-4-фенилфенол (изомеры), 2-пентил-4-кумилфенол (изомеры), 2-гексил-4-метилфенол (изомеры), 2-гексил-4-этилфенол (изомеры), 2-гексил-4-пропилфенол (изомеры), 2-гексил-4-бутилфенол (изомеры), 2-гексил-4-пентилфенол (изомеры), 2-гексил-4-гептилфенол (изомеры), 2-гексил-4-октилфенол (изомеры), 2-гексил-4-фенилфенол (изомеры), 2-гексил-4-кумилфенол (изомеры), 2-гептил-4-метилфенол (изомеры), 2-гептил-4-этилфенол (изомеры), 2-гептил-4-пропилфенол (изомеры), 2-гептил-4-бутилфенол (изомеры), 2-гептил-4-пентилфенол (изомеры), 2-гептил-4-гексилфенол (изомеры), 2-гептил-4-октилфенол (изомеры), 2-гептил-4-фенилфенол (изомеры), 2-гептил-4-кумилфенол (изомеры), 2,4,6-триметилфенол, 2,6-диметил-4-этилфенол, 2,6-диметил-4-пропилфенол (изомеры), 2,6-диметил-4-бутилфенол (изомеры), 2,6-диметил-4-пентилфенол (изомеры), 2,6-диметил-4-гексилфенол (изомеры), 2,6-диметил-4-фенилфенол (изомеры), 2,6-диметил-4-кумилфенол (изомеры), 2,4,6-триэтилфенол, 2,6-диэтил-4-метилфенол, 2,6-диэтил-4-пропилфенол (изомеры), 2,6-диэтил-4-бутилфенол (изомеры), 2,6-диэтил-4-пентилфенол (изомеры), 2,6-диэтил-4-гексилфенол (изомеры), 2,6-диэтил-4-фенилфенол (изомеры), 2,6-диэтил-4-кумилфенол (изомеры), 2,4,6,-трипропилфенол (изомеры), 2,6-дипропил-4-этилфенол (изомеры), 2,6-дипропил-4-метилфенол (изомеры), 2,6-дипропил-4-бутилфенол (изомеры), 2,6-дипропил-4-пентилфенол (изомеры), 2,6-дипропил-4-гексилфенол (изомеры), 2,6-дипропил-4-фенилфенол (изомеры), 2,6-дипропил-4-кумилфенол (изомеры), 2,4-диметил-6-этилфенол, 2-метил-4,6-диэтилфенол, 2-метил-4-пропил-6-этилфенол (изомеры), 2-метил-4-бутил-6-этилфенол (изомеры), 2-метил-4-пентил-6-этилфенол (изомеры), 2-метил-4-гексил-6-этилфенол (изомеры), 2-метил-4-фенил-6-этилфенол (изомеры), 2-метил-4-кумил-6-этилфенол (изомеры), 2,4-диметил-6-пропилфенол (изомеры), 2-метил-4,6-дипропилфенол (изомеры), 2-метил-4-этил-6-пропилфенол (изомеры), 2-метил-4-бутил-6-пропилфенол (изомеры), 2-метил-4-пентил-6-пропилфенол (изомеры), 2-метил-4-гексил-6-пропилфенол (изомеры), 2-метил-4-фенил-6-пропилфенол (изомеры), 2-метил-4-кумил-6-пропилфенол (изомеры), 2,4-диметил-6-бутилфенол (изомеры), 2-метил-4,6-дибутилфенол (изомеры), 2-метил-4-пропил-6-бутилфенол (изомеры), 2-метил-4-этил-6-бутилфенол (изомеры), 2-метил-4-пентил-6-бутилфенол (изомеры), 2-метил-4-гексил-6-бутилфенол (изомеры), 2-метил-4-фенил-6-бутилфенол (изомеры), 2-метил-4-кумил-6-бутилфенол (изомеры), 2,4-диметил-6-пентилфенол, 2-метил-4,6-дипентилфенол, 2-метил-4-пропил-6-пентилфенол (изомеры), 2-метил-4-бутил-6-пентилфенол (изомеры), 2-метил-4-этил-6-пентилфенол (изомеры), 2-метил-4-гексил-6-пентилфенол (изомеры), 2-метил-4-фенил-6-пентилфенол (изомеры), 2-метил-4-кумил-6-пентилфенол (изомеры), 2,4-диметил-6-гексилфенол, 2-метил-4,6-дигексилфенол, 2-метил-4-пропил-6-гексилфенол (изомеры), 2-метил-4-бутил-6-гексилфенол (изомеры), 2-метил-4-пентил-6-гексилфенол (изомеры), 2-метил-4-этил-6-гексилфенол (изомеры), 2-метил-4-фенил-6-гексилфенол (изомеры), 2-метил-4-кумил-6-гексилфенол (изомеры), 2-этил-4-метил-6-пропилфенол (изомеры), 2,4-диэтил-6-пропилфенол (изомеры), 2-этил-4,6-дипропилфенол (изомеры), 2-этил-4-бутил-6-пропилфенол (изомеры), 2-этил-4-пентил-6-пропилфенол (изомеры), 2-этил-4-гексил-6-пропилфенол (изомеры), 2-этил-4-гептил-6-пропилфенол (изомеры), 2-этил-4-октил-6-пропилфенол (изомеры), 2-этил-4-фенил-6-пропилфенол (изомеры), 2-этил-4-кумил-6-пропилфенол (изомеры), 2-этил-4-метил-6-бутилфенол (изомеры), 2,4-диэтил-6-бутилфенол (изомеры), 2-этил-4,6-дибутилфенол (изомеры), 2-этил-4-пропил-6-бутилфенол (изомеры), 2-этил-4-пентил-6-бутилфенол (изомеры), 2-этил-4-гексил-6-бутилфенол (изомеры), 2-этил-4-гептил-6-бутилфенол (изомеры), 2-этил-4-октил-6-бутилфенол (изомеры), 2-этил-4-фенил-6-бутилфенол (изомеры), 2-этил-4-кумил-6-бутилфенол (изомеры), 2-этил-4-метил-6-пентилфенол (изомеры), 2,4-диэтил-6-пентилфенол (изомеры), 2-этил-4,6-дипентилфенол (изомеры), 2-этил-4-бутил-6-пентилфенол (изомеры), 2-этил-4-пропил-6-пентилфенол (изомеры), 2-этил-4-гексил-6-пентилфенол (изомеры), 2-этил-4-гептил-6-пентилфенол (изомеры), 2-этил-4-октил-6-пентилфенол (изомеры), 2-этил-4-фенил-6-пентилфенол (изомеры), 2-этил-4-кумил-6-пентилфенол (изомеры), 2-этил-4-метил-6-гексилфенол (изомеры), 2,4-диэтил-6-гексилфенол (изомеры), 2-этил-4,6-дигексилфенол (изомеры), 2-этил-4-пропил-6-гексилфенол (изомеры), 2-этил-4-пентил-6-гексилфенол (изомеры), 2-этил-4-бутил-6-гексилфенол изомеры), 2-этил-4-гептил-6-гексилфенол (изомеры), 2-этил-4-октил-6-гексилфенол (изомеры), 2-этил-4-фенил-6-гексилфенол (изомеры), 2-этил-4-кумил-6-гексилфенол (изомеры), 2-пропил-4-метил-6-бутилфенол (изомеры), 2,4-дипропил-6-бутилфенол (изомеры), 2-пропил-4,6-дибутилфенол (изомеры), 2-пропил-4-этил-6-бутилфенол (изомеры), 2-пропил-4-пентил-6-бутилфенол (изомеры), 2-пропил-4-гексил-6-бутилфенол (изомеры), 2-пропил-4-гептил-6-бутилфенол (изомеры), 2-пропил-4-октил-6-бутилфенол (изомеры), 2-пропил-4-фенил-6-бутилфенол (изомеры) и 2-пропил-4-кумил-6-бутилфенол (изомеры) или подобные соединения.
Авторы настоящего изобретения к удивлению обнаружили, что с использованием конкретных ароматических гидроксисоединений, описанных выше, ароматические сложные эфиры карбаминовой кислоты, которые традиционно считали нестабильными, способны ингибировать побочные реакции, подобные тем, которые описаны выше в реакции переэтерификации и/или реакции термического разложения, которая будет описана ниже. Хотя механизм, по которому ароматическое гидроксисоединение ингибирует побочные реакции, остается неясным, авторы настоящего изобретения предположили, что в случае, например, когда R' представляет собой группу, происходящую из ароматического гидроксисоединения, в реакции, посредством которой образуется мочевинная связь, представленная указанной выше формулой (2), заместитель в ортоположении относительно гидроксильной группы стерически защищает уретановую связь, препятствуя таким образом реакции между другим сложным эфиром карбаминовой кислоты и уретановой связью.
Ароматические гидроксисоединения, предпочтительно, представляют собой ароматические гидроксисоединения, имеющие стандартную температуру кипения, которая выше, чем стандартная температура кипения гидроксисоединения, соответствующего алифатической алкоксигруппе, арилоксигруппе или аралкилоксигруппе, которая образует сложноэфирную группу сложного эфира карбаминовой кислоты, описанного ниже. Термин "стандартная температура кипения", как он используется в настоящем изобретении, указывает температуру кипения при давлении 1 атмосфера.
<Сложные эфиры карбаминовой кислоты>
Нет никаких конкретных ограничений, касающихся сложного эфира карбаминовой кислоты, используемого в способе получения изоцианата представленного варианта воплощения настоящего изобретения, и предпочтительно используют алифатический сложный эфир карбаминовой кислоты. Примеры алифатических сложных эфиров карбаминовой кислоты включают соединения, представленные следующей формулой (11):
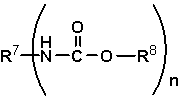
где R7 представляет собой группу, выбранную из группы, состоящей из алифатической группы, содержащей от 1 до 20 атомов углерода, и ароматической группы, содержащей от 6 до 20 атомов углерода, при этом такая группа содержит атом, выбранный из атома углерода и атома кислорода, и имеет количество атомов равное n,
R8 представляет собой алифатическую группу, содержащую от 1 до 8 атомов углерода, содержащую атом, выбранный из атома углерода и атома кислорода, и
n представляет собой целое число, имеющее значение от 1 до 10.
В формуле (11) выше, n, предпочтительно, представляет собой число, выбранное из целых чисел, имеющих значение 2 или больше, и более предпочтителен алифатический сложный эфир поликарбаминовой кислоты, в котором n имеет значение 2.
Примеры R7 в формуле (11) включают линейные углеводородные группы, такие как метилен, диметилен, триметилен, тетраметилен, пентаметилен, гексаметилен, октаметилен или подобные; незамещенные ациклические углеводородные группы, такие как циклопентан, циклогексан, циклогептан, циклооктан, бис(циклогексил)алкан или подобные; алкил-замещенные циклогексаны, такие как метилциклопентан, этилциклопентан, метилциклогексан (изомеры), этилциклогексан (изомеры), пропилциклогексан (изомеры), бутилциклогексан (изомеры), пентилциклогексан (изомеры), гексилциклогексан (изомеры) или подобные; диалкил-замещенные циклогексаны, такие как диметилциклогексан (изомеры), диэтилциклогексан (изомеры), дибутилциклогексан (изомеры) или подобные; триалкил-замещенные циклогексана, такие как 1,5,5-триметилциклогексан, 1,5,5-триэтилциклогексан, 1,5,5-трипропилциклогексан (изомеры), 1,5,5-трибутилциклогексан (изомеры) или подобные; моноалкил-замещенн бензолы, такие как толуол, этилбензол, пропилбензол или подобные; диалкил-замещенные бензолы, такие как ксилол, диэтилбензол, дипропилбензол или подобные; и ароматические углеводороды, такие как дифенилалкан, бензол или подобные. В частности, предпочтительно используют гексаметилен, фенилен, дифенилметан, толуол, циклогексан, ксилол, метилциклогексан, изофорон и дициклогексилметан.
Примеры R8 включают алкильные группы, в которых количество атомов углерода, образующих такую группу, выбрано из целого числа, имеющего значение от 1 до 8, такие как метильная группа, этильная группа, пропильная группа (изомеры), бутильная группа (изомеры), пентильная группа (изомеры), гексильная группа (изомеры), гептильная группа (изомеры), октильная группа (изомеры) или подобные; и циклоалкильные группы, в которых количество атомов углерода, образующих такую группу, выбрано из целого числа, имеющего значение от 5 до 14, такие как циклопентильная группа, циклогексильная группа, циклогептильная группа, циклооктильная группа, дициклопентильная группа (изомеры), дициклогексильная группа (изомеры), циклогексил-циклопентильная группа или подобные.
Примеры полиалкилкарбаматов, представленных указанной выше формулой (11), включают алкилкарбаматы, такие как диметиловый эфир N,N'-гександиил-бис-карбаминовой кислоты, диэтиловый эфир N,N'-гександиил-бис-карбаминовой кислоты, дибутиловый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), дипентиловый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), дигексиловый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), диоктиловый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), диметил-4,4'-метилен-дициклогексилкарбамат, диэтил-4,4'-метилен-дициклогексилкарбамат, дипропил-4,4'-метилен-дициклогексилкарбамат (изомеры), дибутил-4,4'-метилен-дициклогексилкарбамат (изомеры), дипентил-4,4'-метилен-дициклогексилкарбамат (изомеры), дигексил-4,4'-метилен-дициклогексилкарбамат (изомеры), дигептил-4,4'-метилен-дициклогексилкарбамат (изомеры), диоктил-4,4'-метилен-дициклогексилкарбамат (изомеры), метиловый эфир 3-(метоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, этиловый эфир 3-(этоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, пропиловый эфир 3-(пропилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), бутиловый эфир 3-(бутилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), пентиловый эфир 3-(пентилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), гексиловый эфир 3-(гексилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), гептиловый эфир 3-(гептилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), октиловый эфир 3-(октилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), диметиловый эфир толуол-дикарбаминовой кислоты (изомеры), диэтиловый эфир толуол-дикарбаминовой кислоты (изомеры), дипропиловый эфир толуол-дикарбаминовой кислоты (изомеры), дибутиловый эфир толуол-дикарбаминовой кислоты (изомеры), дипентиловый эфир толуол-дикарбаминовой кислоты (изомеры), дигексиловый эфир толуол-дикарбаминовой кислоты (изомеры), дигептиловый эфир толуол-дикарбаминовой кислоты (изомеры), диоктиловый эфир толуол-дикарбаминовой кислоты (изомеры), диметиловый эфир N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты, диэтиловый эфир N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты, дипропиловый эфир N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты, N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты дибутиловый эфир, дипентиловый эфир N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты, дигексиловый эфир N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты, дигептиловый эфир N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты, диоктиловый эфир N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты или подобные.
Из них предпочтительно используют алкилкарбаматы, в которых R7 в формуле (11) выше представляет собой группу, выбранную из группы, состоящей из алкильной группы, содержащей от 1 до 20 атомов углерода, и циклоалкильной группы, содержащей от 5 до 20 атомов углерода, при этом алкилкарбаматы, представленные любой из следующих формул (12)-(14), являются особенно предпочтительными для использования:
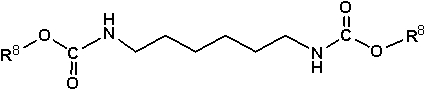


где R8 имеет значения, определенные выше.
Примеры полиалкилкарбаматов, представленных формулой (12), включают диметиловый эфир N,N'-гександиил-бис-карбаминовой кислоты, диэтиловый эфир N,N'-гександиил-бис-карбаминовой кислоты, дибутиловый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), дипентиловый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), дигексиловый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры) и диоктиловый эфир N,N'-гександиил-бис-карбаминовой кислоты (включая изомеры). Кроме того, примеры полиалкилкарбаматов, представленных формулой (13), включают диметил-4,4'-метилен-дициклогексилкарбамат, диэтил-4,4'-метилен-дициклогексилкарбамат, дипропил-4,4'-метилен-дициклогексилкарбамат (изомеры), дибутил-4,4'-метилен-дициклогексилкарбамат (изомеры), дипентил-4,4'-метилен-дициклогексилкарбамат (изомеры), дигексил-4,4'-метилен-дициклогексилкарбамат (изомеры), дигептил-4,4'-метилен-дициклогексилкарбамат (изомеры) и диоктил-4,4'-метилен-дициклогексилкарбамат (изомеры). Более того, примеры полиалкилкарбаматов, представленных формулой (14), включают полиалкилкарбаматы, такие как метиловый эфир 3-(метоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, этиловый эфир3-(этоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, пропиловый эфир 3-(пропилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), бутиловый эфир 3-(бутилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), пентиловый эфир 3-(пентилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), гексиловый эфир 3-(гексилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), гептиловый эфир 3-(гептилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), октиловый эфир 3-(октилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры) или подобные.
Можно использовать известный способ для получения сложных эфиров карбаминовой кислоты. Например, сложные эфиры карбаминовой кислоты можно получить путем взаимодействия аминовых соединений с оксидом углерода, кислородом и алифатическими спиртами или ароматическими гидроксисоединениями, или путем взаимодействия аминовых соединений с мочевиной и алифатическими спиртами или ароматическими гидроксисоединениями. В представленном варианте воплощения настоящего изобретения сложные эфиры карбаминовой кислоты, предпочтительно, получают путем взаимодействия сложных эфиров угольной кислоты и аминовых соединений.
Далее объясняется получение алкилкарбаматов путем взаимодействия диалкилкарбонатов и аминовых соединений.
В качестве диалкилкарбонатов можно использовать диалкилкарбонаты, представленные следующей формулой (21):
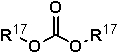
где R17 представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 8 атомов углерода.
Примеры R17 включают алкильные группы в форме алифатических углеводородных групп, в которых количество атомов углерода, образующих такую группу, представляет собой количество, выбранное из целого числа, имеющего значение от 1 до 8, такие как метильная группа, этильная группа, пропильная группа (изомеры), бутильная группа (изомеры), пентильная группа (изомеры), гексильная группа (изомеры), гептильная группа (изомеры), октильная группа (изомеры) или подобные. Примеры таких диалкилкарбонатов включают диметилкарбонат, диэтилкарбонат, дипропилкарбонат (изомеры), дибутилкарбонат (изомеры), дипентилкарбонат (изомеры), дигексилкарбонат (изомеры), дигептилкарбонат (изомеры) и диоктилкарбонат (изомеры). Из них диалкилкарбонат, в котором количество атомов углерода, образующих алкильные группы, представляет собой количество, выбранное из целого числа, имеющего значение от 4 до 6, является особенно предпочтительным для использования.
В качестве аминовых соединений предпочтительно используют аминовые соединения, представленные следующей формулой (22):

где R7 представляет собой группу, выбранную из группы, состоящей из алифатической группы, содержащей от 1 до 20 атомов углерода, и ароматической группы, содержащей от 6 до 20 атомов углерода, при этом такая группа содержит атом, выбранный из атома углерода и атома кислорода, и имеет валентность n, и n представляет собой целое число, имеющее значение от 1 до 10.
В формуле (22) выше используют полиаминовое соединение, в котором n предпочтительно имеет значение от 1 до 3, и более предпочтительно n имеет значение 2.
Примеры таких полиаминовых соединений включают алифатические диамины, такие как гексаметилендиамин, 4,4'-метиленбис(циклогексиламин) (изомеры), циклогександиамин (изомеры), 3-аминометил-3,5,5-триметилциклогексиламин (изомеры) или подобные; и ароматические диамины, такие как фенилендиамин (изомеры), толуолдиамин (изомеры), 4,4'-метилендианилин (изомеры) или подобные. Из них предпочтительно используют алифатические диамины, такие как гексаметилендиамин, 4,4'-метиленбис(циклогексиламин) (изомеры), циклогександиамин (изомеры), 3-аминометил-3,5,5-триметилциклогексиламин (изомеры) или подобные, при этом более предпочтительно использование гексаметилендиамина, 4,4'-метиленбис(циклогексиламин) и 3-аминометил-3,5,5-триметилциклогексиламина.
Условия реакции варьируются в зависимости от взаимодействующих соединений, и, хотя диалкилкарбонат предпочтительно используют в избыточном количестве в расчете на аминогруппы аминового соединения для повышения скорости реакции и быстрого завершения реакции, при стехиометрическом отношении диалкилкарбоната к аминогруппам аминового соединения в пределах от 2 до 1000 раз, этот диапазон, предпочтительно, составляет от 2 до 100 раз, и более предпочтительно от 2,5 до 30 раз, с учетом размера реакционного сосуда. Температура реакции обычно находится в пределах от нормальной температуры (20°C) до 300°C, и хотя более высокие температуры являются предпочтительными для повышения скорости реакции, поскольку при высоких температурах могут иметь место обратные нежелательные реакции, температура реакции, предпочтительно, находится в пределах от 50 до 150°C. Реактор может быть снабжен известным охлаждающим устройством или нагревательным устройством для поддержания постоянной температуры реакции. Кроме того, хотя его можно варьировать в соответствии с типами используемых соединений и температурой реакции, реакционное давление может представлять собой пониженное давление, нормальное давление или повышенное давление, и реакцию обычно осуществляют при давлении в пределах от 20 до 1×106 Па. Нет никаких конкретных ограничений в том, что касается времени реакции (время нахождения в реакторе в случае непрерывного способа) и оно обычно составляет от 0,001 до 50 часов, предпочтительно от 0,01 до 10 часов, и более предпочтительно от 0,1 до 5 часов. Кроме того, реакция также может быть завершена путем подтверждения, что желаемое количество алкилкарбамата уже образовано, например, при помощи жидкостной хроматографии после отбора проб реакционной жидкости. В представленном варианте воплощения настоящего изобретения можно использовать катализатор, если это необходимо, и примеры катализаторов, которые можно использовать, включают металлоорганические соединения и неорганические соединения металлов олова, свинца, меди или титана, и щелочные катализаторы, такие как алкилаты щелочных металлов или щелочно-земельных металлов в форме метилатов, этилатов и бутиратов (изомеры) лития, натрия, калия, кальция или бария. Хотя в представленном варианте воплощения настоящего изобретения необязательно требуется использование реакционного растворителя, предпочтительно, когда используют подходящий инертный растворитель в качестве реакционного растворителя для облегчения процедуры реакции, примеры которого включают алканы, такие как гексан (изомеры), гептан (изомеры), октан (изомеры), нонан (изомеры), декан (изомеры) или подобные; ароматические углеводороды и алкил-замещенные ароматические углеводороды, так как бензол, толуол, ксилол (изомеры), этилбензол, диизопропилбензол (изомеры), дибутилбензол (изомеры), нафталин или подобные; ароматические соединения, замещенные галогеном или нитрогруппой, такие как хлорбензол, дихлорбензол (изомеры), бромбензол, дибромбензол (изомеры), хлорнафталин, бромнафталин, нитробензол, нитронафталин или подобные; полициклические углеводородные соединения, такие как дифенил, замещенный дифенил, дифенилметан, терфенил, антрацен, дибензилтолуол или подобные; алифатические углеводороды, такие как циклогексан, циклопентан, циклооктан, этилциклогексан или подобные; кетоны, такие как метилэтилкетон, ацетофенон или подобные; сложные эфиры, такие как дибутилфталат, дигексилфталат, диоктилфталат, бензилбутилфталат или подобные; простые эфиры и тиоэфиры, такие как дифениловый эфир, дифенилсульфид или подобные; и сульфоксиды, такие как диметилсульфоксид, дифенилсульфоксид или подобные. Эти растворители можно использовать отдельно или два или более таких видов можно использовать в виде смеси. Кроме того, диалкилкарбонат, используемый в избыточном количестве в расчете на аминогруппы аминового соединения, также предпочтительно используют в качестве растворителя в реакции.
В качестве реакционного сосуда можно использовать известный реактор в форме бака, реактор колоночного типа или дистилляционную колонну, и хотя можно использовать известные материалы для реакционного сосуда и линий, которые используют, при условии, что они не оказывают вредного действия на исходные вещества или реагенты, предпочтительно, можно использовать SUS304, SUS316 или SUS316L и подобные, поскольку они являются дешевыми.
<Реакция переэтерификации>
В способе получения изоцианата представленного варианта воплощения настоящего изобретения сложные эфиры карбаминовой кислоты и ароматические гидроксисоединения сначала подвергают взаимодействию для получения арилкарбаматов, содержащих группу, происходящую из ароматических гидроксисоединений. Эта реакция включает обмен между алифатической алкоксигруппой или аралкилоксигруппой, образующей сложноэфирную группу сложного эфира карбаминовой кислоты, и арилоксигруппой, происходящей из ароматических гидроксисоединений, приводя к образованию соответствующего арилкарбамата и гидроксисоединения, происходящего из сложного эфира карбаминовой кислоты (и в настоящем описании эта реакция указана как реакция переэтерификации).
Хотя возможно варьирование в соответствии с взаимодействующими соединениями, реакционные условия этой реакции переэтерификации таковы, что ароматическое гидроксисоединение используют в количестве, от 2 до 1000 раз превышающим количество сложноэфирной группы сложного эфира карбаминовой кислоты, которое выражают как стехиометрическое отношение. В результате проведения всесторонних исследований авторы настоящего изобретения к удивлению обнаружили, что при использовании ароматических гидроксисоединений, содержащих заместитель, по меньшей мере, в одном ортоположении относительно гидроксильной группы, в этой реакции переэтерификации, описанной выше, побочные реакции, описанные выше как относимые за счет сложного эфира карбаминовой кислоты и/или продукта в форме арилкарбамата, могут ингибироваться в реакции переэтерификации. В реакции переэтерификации, хотя ароматическое гидроксисоединение предпочтительно используют в избыточном количестве, в расчете на сложноэфирную группу сложного эфира карбаминовой кислоты, для ингибирования побочных реакций, относимые за счет сложного эфира карбаминовой кислоты и/или продукта в форме арилкарбамата, а также для обеспечения быстрого завершения реакции, ароматическое гидроксисоединение предпочтительно используют в количестве, в 2 до 100 раз большем, и предпочтительно в 5 до 50 раз большем, с учетом размеров реакционного сосуда. Температура реакции обычно находится в пределах от 100 до 300°C, и хотя высокие температуры и являются предпочтительными для повышения скорости реакции, но поскольку при высоких температурах, наоборот, может быть бόльшая вероятность возникновения побочных реакций, температура реакции, предпочтительно, находится в пределах от 150 до 250°C. Реактор может быть снабжен известным охлаждающим устройством или нагревательным устройством для поддержания постоянной температуры реакции. Кроме того, хотя его можно варьировать в соответствии с типами используемых соединений и температурой реакции, реакционное давление может представлять собой пониженное давление, нормальное давление или повышенное давление, и реакцию обычно осуществляют при давлении в пределах от 20 до 1×106 Па. Нет никаких конкретных ограничений в том, что касается времени реакции (время нахождения в реакторе в случае непрерывного способа), и оно обычно составляет от 0,001 до 100 часов, предпочтительно от 0,01 до 50 часов, и более предпочтительно от 0, 1 до 30 часов. Кроме того, реакция также может быть завершена путем подтверждения, что желаемое количество арилкарбамата уже образовано, например, при помощи жидкостной хроматографии после отбора проб реакционной жидкости. В представленном варианте воплощения настоящего изобретения катализатор используют в количестве от 0,01 до 30% мас., и предпочтительно в количестве от 0,5 до 20% мас., в расчете на массу сложного эфира карбаминовой кислоты. Например, подходящими для использования являются металлоорганические катализаторы, такие как дилаурат дибутилолова, октоат железа или октоат олова, или амины, такие как 1,4-диазабицикло[2,2,2]октан, триэтилендиамин или триэтиламин, при этом металлоорганические катализаторы, такие как дилаурат дибутилолова, октоат железа или октоат олова, являются особенно предпочтительными. Эти соединения можно использовать отдельно или два или более таких видов можно использовать в виде смеси. Хотя в представленном варианте воплощения настоящего изобретения необязательно требуется использование реакционного растворителя, предпочтительно, когда используют подходящий инертный растворитель в качестве реакционного растворителя для облегчения процедуры реакции, примеры которого включают алканы, такие как гексан (изомеры), гептан (изомеры), октан (изомеры), нонан (изомеры), декан (изомеры) или подобные; ароматические углеводороды и алкил-замещенные ароматические углеводороды, такие как бензол, толуол, ксилол (изомеры), этилбензол, диизопропилбензол (изомеры), дибутилбензол (изомеры), нафталин или подобные; ароматические соединения, замещенные галогеном или нитрогруппой, такие как хлорбензол, дихлорбензол (изомеры), бромбензол, дибромбензол (изомеры), хлорнафталин, бромнафталин, нитробензол, нитронафталин или подобные; полициклические углеводородные соединения, такие как дифенил, замещенный дифенил, дифенилметан, терфенил, антрацен, дибензилтолуол (изомеры) или подобные; алифатические углеводороды, такие как циклогексан, циклопентан, циклооктан, этилциклогексан или подобные; кетоны, такие как метилэтилкетон, ацетофенон или подобные; сложные эфиры, такие как дибутилфталат, дигексилфталат, диоктилфталат, бензилбутилфталат или подобные; простые эфиры и тиоэфиры, такие как дифениловый эфир, дифенилсульфид или подобные; и сульфоксиды, такие как диметилсульфоксид, дифенилсульфоксид или подобные; и силиконовое масло. Эти растворители можно использовать отдельно или два или более таких видов можно использовать в виде смеси.
Как было описано выше, хотя реакция переэтерификациии в представленном варианте воплощения настоящего изобретения включает обмен между алифатической алкоксигруппой, образующей сложноэфирную группу сложного эфира карбаминовой кислоты, и арилоксигруппой, происходящей из ароматического гидроксисоединения, приводя к образованию соответствующих арилкарбаматов и спиртов, реакция переэтерификации представляет собой равновесную реакцию. Таким образом, для эффективного получения арилкарбаматов посредством этой реакции переэтерификации, предпочтительно удаление продуктов из этой реакционной системы. Поскольку соединениями, имеющими самую низкую стандартную температуру кипения в реакционной системе, являются спирты, образованные реакцией переэтерификации, эти спирты, предпочтительно, удаляют из реакционной системы таким способом, как дистилляционное разделение.
Кроме того, реакцию переэтерификации предпочтительно осуществляют непрерывным способом для эффективного прохождения реакции переэтерификации. А именно, предпочтительно используют способ, в котором сложные эфиры карбаминовой кислоты и ароматические гидроксисоединения непрерывно подают в реакционный сосуд для осуществления реакции переэтерификации, образуемые спирты удаляют из реакционного сосуда в форме газообразных компонентов, и реакционные жидкости, содержащие образованные арилкарбаматы и ароматические гидроксисоединения, непрерывно извлекают из донной части реакционного сосуда. В случае осуществления реакции переэтерификации в соответствии с этим способом, помимо промотирования реакции переэтерификации, также имеет место удивительный эффект возможности улучшения конечного выхода изоцианатов путем ингибирования побочных реакций, как описано выше.
Хотя можно использовать известные материалы для реакционного сосуда и линий, используемых для осуществления реакции переэтерификации, при условии, что они не оказывают вредного действия на исходные вещества или реагенты, предпочтительно, можно использовать SUS304, SUS316 или SUS316L и подобные, поскольку они являются дешевыми. Нет никаких конкретных ограничений в отношении типа реакционного сосуда, и можно использовать известный реактор в форме бака или реактор колоночного типа. Предпочтительно, используют реакционный сосуд, который обеспечивается вместе с линиями для экстракции низкокипящей реакционной смеси, содержащей спирт, образованный в реакции переэтерификации, из реакционного сосуда в форме газообразных компонентов, и для удаления жидкой смеси, содержащей образованные арилкарбаматы и ароматические гидроксисоединения, из нижней части реакционного сосуда в форме жидкости. Для такого реакционного сосуда используют различные известные способы, примеры которых включают типы с использованием реакционных сосудов, содержащих смесительный резервуар, многоступенчатый смесительный резервуар, дистилляционную колонну, многоступенчатую дистилляционную колонну, многотрубный реактор, многоступенчатую дистилляционную колонну непрерывного действия, насадочную колонну, пленочный испаритель, реактор, снабженный находящимся внутри него носителем, реактор с принудительной циркуляцией, испаритель с падающей пленкой, испаритель с падающими каплями, реактор с медленным протеканием жидкости через неподвижный слой, колпачковую колонну и типы с использованием их комбинаций. Способы с использованием пленочного испарителя или реактора колоночного типа являются предпочтительными с точки зрения эффективности сдвига равновесия в сторону продуктов, при этом структура, имеющая большую площадь контакта газ-жидкость, является предпочтительной, поскольку способна обеспечивать быстрый перенос образуемого спирта в газовую фазу.
Многоступенчатая дистилляционная колонна относится к дистилляционной колонне с несколькими ступенями, в которой количество теоретических тарелок дистилляции составляет 2 или более, и можно использовать любую многоступенчатую дистилляционную колонну, при условии, что она обеспечивает возможность непрерывной дистилляции. В качестве многоступенчатой дистилляционной колонны можно использовать любую многоступенчатую дистилляционную колонну, при условии, что ее обычно используют в качестве многоступенчатой дистилляционной колонны, примеры которой включают типы тарельчатых колонн с использованием колпачковой тарелки, пористой пластинчатой тарелки, клапанной тарелки, противоточной тарелки или т.п., и типы насадочных колонн, заполненных насадочными материалами различного типа, такими как кольцо Рашига, кольцо Лессинга, кольцо Полла, седловидная насадка Берля, седло “Интерлок”, насадка Диксона, седловидная сетчатая насадка Мак Магона, Helipack, насадка Сульцера, Mellapak или подобные. Можно использовать любую насадочную колонну, при условии, что такая колонна заполнена известными насадочными материалами, как описано выше. Более того, также предпочтительно, когда используют колонну комбинированного тарелочно-насадочного типа, которая объединяет тарелочную часть с частью, заполненной насадочными материалами. Предпочтительно, когда реакционный сосуд снабжен линией для подачи смеси, содержащей сложные эфиры карбаминовой кислоты и ароматические гидроксисоединения, линией для удаления газофазных компонентов, содержащих спирты, образованные реакцией переэтерификации, и линией для экстракции смеси жидкостей, содержащей сложные эфиры карбаминовой кислоты и ароматические гидроксисоединения, и предпочтительно, когда указанная линия для удаления газофазных компонентов, содержащих спирты, расположена таким образом, чтобы обеспечить возможность удаления газофазных компонентов из реакционного сосуда, и особенно предпочтительно, чтобы указанная линия для экстракции смеси жидкостей, содержащих арилкарбаматы и ароматические гидроксисоединения, была расположена ниже.
Линия для подачи инертного газа и/или жидкого инертного растворителя из нижней части реакционного сосуда может быть отдельно подсоединена, и в случае, когда смесь жидкостей, содержащих образованные арилкарбаматы и ароматические гидроксисоединения, содержит непрореагировавшие сложные эфиры карбаминовой кислоты, линия может быть подсоединена для рециркулирования всей или части смеси жидкостей в реакционный сосуд. Следует отметить, что в случае использования указанного выше инертного растворителя такой инертный растворитель может быть в форме газа и/или жидкости.
Газообразные компоненты, содержащие спирты, экстрагированные из реакционного сосуда, могут быть очищены известным способом, таким как с использованием дистилляционной колонны, и азеотропное и/или сопутствующее ароматическое гидроксисоединение и т.п. можно рециркулировать. К каждой линии может быть добавлено оборудование для подогрева, охлаждения или нагревания, учитывая проблемы засорения и подобные.
<Арилкарбаматы>
Арилкарбаматы, предпочтительно полученные путем реакции переэтерификации, представляют собой арилкарбаматы, представленные любой из следующих формул (15)-(17):

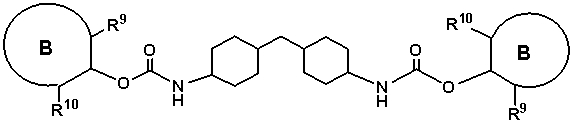

где кольцо B представляет собой структуру, которая может содержать заместитель и которая содержит, по меньшей мере, одну структуру, выбранную из группы, состоящей из бензольного кольца, нафталинового кольца и антраценового кольца,
R9 представляет собой группу, отличную от атома водорода, в форме алифатической алкильной группы, содержащей от 1 до 20 атомов углерода, алифатической алкоксигруппы, содержащей от 1 до 20 атомов углерода, арильной группы, содержащей от 6 до 20 атомов углерода, арилоксигруппы, содержащей от 6 до 20 атомов углерода, аралкильной группы, содержащей от 7 до 20 атомов углерода или аралкиоксигруппы, содержащей от 7 до 20 атомов углерода, при этом такая группа содержит атом, выбранный из атома углерода, атома кислорода и атома азота, и
R10 представляет собой алифатическую алкильную группу, содержащую от 1 до 20 атомов углерода, алифатическую алкоксигруппу, содержащую от 1 до 20 атомов углерода, арильную группу, содержащую от 6 до 20 атомов углерода, арилоксигруппу, содержащую от 6 до 20 атомов углерода, аралкильную группу, содержащую от 7 до 20 атомов углерода, или аралкиоксигруппу, содержащую от 7 до 20 атомов углерода, при этом алифатическая алкильная, алифатическая алкокси, арильная, арилокси, аралкильная и аралкилокси группы содержат атом, выбранный из атома углерода, атома кислорода и атома азота.
Из них более предпочтительные полученные арилкарбаматы представляют собой арилкарбаматы, представленные любой из следующих формул (18)-(20):
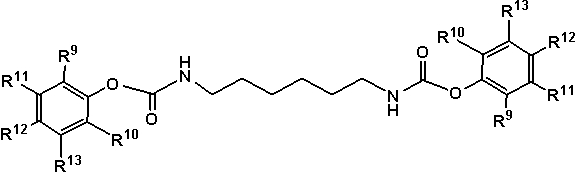


где R9 представляет собой группу, отличную от атома водорода, в форме алкильной группы, содержащей от 1 до 20 атомов углерода, алкоксигруппы, содержащей от 1 до 20 атомов углерода, арильной группы, содержащей от 6 до 20 атомов углерода, арилоксигруппы, содержащей от 6 до 20 атомов углерода, аралкильной группы, содержащей от 7 до 20 атомов углерода, или аралкиоксигруппы, содержащей от 7 до 20 атомов углерода, при этом такая группа содержит атом, выбранный из атома углерода, атома кислорода и атома азота, и
каждый из R10, R11, R12 и R13 независимо представляет собой атом водорода или алифатическую алкильную группу, содержащую от 1 до 20 атомов углерода, алифатическую алкоксигруппу, содержащую от 1 до 20 атомов углерода, арильную группу, содержащую от 6 до 20 атомов углерода, арилоксигруппу, содержащую от 6 до 20 атомов углерода, аралкильную группу, содержащую от 7 до 20 атомов углерода, или аралкиоксигруппу, содержащую от 7 до 20 атомов углерода, при этом алифатическая алкильная, алифатическая алкокси, арильная, арилокси, аралкильная и аралкилокси группы содержат атом, выбранный из атома углерода, атома кислорода и атома азота.
Примеры полиарилкарбаматов, представленных формулой (18), включают бис(2-этилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты, бис(2-пропилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2-бутилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты(изомеры), бис(2-пентилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2-гексилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2-октилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2-кумилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты, бис(2,4-диэтилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты, бис(2,4-дипропилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2,4-дибутилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), кислоты бис(2,4-дипентилфенил)овый эфир N,N'-гександиил-бис-карбаминовой (изомеры), бис(2,4-дигексилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2,4-диоктилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2,4-дикумилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2,6-диметилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты, бис(2,6-диэтилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2,6-дипропилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры), бис(2,4,6-триметилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты, бис(2,3,6-триметилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты, бис(2,4,6-триэтилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты и бис(2,4,6-трипропилфенил)овый эфир N,N'-гександиил-бис-карбаминовой кислоты (изомеры). Кроме того, примеры полиалкилкарбаматов, представленных формулой (19), включают бис(2-этилфенил)-4,4'-метилен-дициклогексилкарбамат, бис(2-пропилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2-бутилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2-пентилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2-гексилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2-гептилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2-октилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2-кумилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4-диэтилфенил)-4,4'-метилен-дициклогексилкарбамат, бис(2,4-дипропилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4-дибутилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4-дипентилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4-дигексилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4-дигептилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4-диоктилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4-дикумилфенил)-4,4'-метилен-дициклогексилкарбамат, бис(2,6-диметилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,6-диэтилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,6-дипропилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4,6-триметилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры), бис(2,4,6-триэтилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры) и бис(2,4,6-трипропилфенил)-4,4'-метилен-дициклогексилкарбамат (изомеры). Более того, примеры полиалкилкарбаматов, представленных формулой (20), включают (2-этилфенил)овый эфир 3-((2-этилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, (2-пропилфенил)овый эфир 3-((2-пропилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2-бутилфенил)овый эфир 3-((2-бутилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2-пентилфенил)овый эфир 3-((2-пентилфенокси)карбониламинометил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2-гексилфенил)овый эфир 3-((2-гексилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2-гептилфенил)овый эфир 3-((2-гептилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2-октилфенил)овый эфир 3-((2-октилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2-кумилфенил)овый эфир 3-((2-кумилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2,4-диэтилфенил)овый эфир 3-((2,4-диэтилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, (2,4-дипропилфенил)овый эфир 3-((2,4-дипропилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2,4-дибутилфенил)овый эфир 3-((2,4-дибутилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2,4-дипентилфенил)овый эфир 3-((2,4-дипентилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2,4-дигексилфенил)овый эфир 3-((2,4-дигексилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2,4-дигептилфенил)овый эфир 3-((2,4-дигептилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2,4-диоктилфенил)овый эфир 3-((2,4-диоктилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2,4-дикумилфенил)овый эфир 3-((2,4-дикумилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, (2,6-диметилфенил)овый эфир 3-((2,6-диметилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, (2,6-диэтилфенил)овый эфир 3-((2,6-диэтилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, (2,6-дипропилфенил)овый эфир 3-((2,6-дипропилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры), (2,4,6-триметилфенил)овый эфир 3-((2,4,6-триметилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, (2,4,6-триэтилфенил)овый эфир 3-((2,4,6-триэтилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты и (2,4,6-трипропилфенил)овый эфир 3-((2,4,6-трипропилфенокси)карбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты (изомеры).
Арилкарбаматы, полученные в реакции переэтерификации, можно подвергнуть следующей реакции термического разложения пока они еще находятся в виде жидкой смеси, содержащей арилкарбаматы и ароматические гидроксисоединения, которую извлекают из реактора, или арилкарбаматы можно подвергнуть реакции термического разложения после очистки из жидкой смеси. Для очистки арилкарбамата из реакционной жидкости можно использовать известный способ, примеры которого включают удаление ароматических гидроксисоединений путем дистилляции, промывку растворителями и очистку арилкарбаматов при помощи кристаллизации.
Поскольку арилкарбаматы представленного варианта воплощения настоящего изобретения представляют собой сложные эфиры карбаминовой кислоты, образованные из ароматических гидроксисоединений и изоцианатов, температура термического разложения низкая, как это общеизвестно. Кроме того, арилкарбаматы представленного варианта воплощения настоящего изобретения, к удивлению, являются чрезвычайно стойкими к возникновению побочных реакций (таких как реакция, приводящая к образованию мочевинной связи, как описано выше) при высоких температурах (таких как 180°C), при которых осуществляют термическое разложение. Хотя механизм, по которому происходит ингибирование побочных реакций, остается неясным, как было описано выше, предполагается, что заместитель в орто положении относительно гидроксильной группы стерически защищает уретановую связь, препятствуя, таким образом, реакции между другим сложным эфиром карбаминовой кислоты и уретановой связью. Более того, хотя ароматические гидроксисоединения, образованные в результате реакции термического разложения арилкарбаматов, согласно представленному варианту воплощения настоящего изобретения представляют собой ароматические гидроксисоединения, содержащие заместитель в ортоположении относительно гидроксильной группы, поскольку скорость реакции между ароматическими гидроксисоединениями и изоцианатами является удивительно нихкой, а именно скорость обратной реакции в реакци термического разложения является медленной, при осуществлении реакции термического разложения арилкарбаматов существует преимущество, позволяющее легко разделять ароматические гидроксисоединения и изоцианаты.
<Реакция термического разложения арилкарбаматов>
Далее представлено разъяснение, касающееся реакции разложения арилкарбамата представленного варианта воплощения настоящего изобретения.
Реакция разложения представленного варианта воплощения настоящего изобретения представляет собой реакцию термического разложения, посредством которой образуются соответствующие изоцианаты и ароматические гидроксисоединения из арилкарбаматов.
Температура реакции обычно находится в пределах от 100 до 300°C и хотя высокая температура является предпочтительной для повышения скорости реакции, но, поскольку побочные реакции, как описано выше, могут быть вызваны обратным образом арилкарбаматами и/или продуктами реакции в форме изоцианатов, температура реакции, предпочтительно, находится в пределах от 150 до 250°C. Реактор может быть снабжен известным охлаждающим устройством или нагревательным устройством для поддержания постоянной температуры реакции. Кроме того, хотя его можно варьировать в соответствии с типами используемых соединений и температурой реакции, реакционное давление может представлять собой пониженное давление, нормальное давление или повышенное давление, и реакцию обычно осуществляют при давлении в пределах от 20 до 1×106 Па. Нет никаких конкретных ограничений в том, что касается времени реакции (время нахождения в реакторе в случае непрерывного способа), и оно обычно составляет от 0,01 до 50 часов, и более предпочтительно от 0,1 до 30 часов. В представленном варианте воплощения настоящего изобретения можно использовать катализатор, и катализатор используют в количестве от 0,0 1 до 30% мас., и предпочтительно в количестве от 0,5 до 20% мас., в расчете на массу арилкарбаматов. Например, металлоорганические катализаторы, такие как дилаурат дибутилолова, октоат железа, октоат олова или подобные, или амины, такие как 1,4-диазабицикло[2,2,2]октан, триэтилендиамин, триэтиламин или подобные, являются подходящими для использования в качестве катализаторов, при этом металлоорганические катализаторы, такие как дилаурат дибутилолова, октоат железа, октоат олова или подобные, являются особенно предпочтительными. Эти соединения можно использовать отдельно или два или более таких видов можно использовать в виде смеси. В случае использования катализаторов в указанной выше реакции переэтерификации, катализаторы, содержащиеся в жидкой смеси после реакции переэтерификации, можно использовать в качестве катализатора в реакции термического разложения, или катализаторы могут быть свежедобавленными к арилкарбаматам при осуществлении реакции термического разложения. Хотя в представленном варианте воплощения настоящего изобретения необязательно требуется использование реакционного растворителя, предпочтительно, когда используют подходящий инертный растворитель в качестве реакционного растворителя для облегчения процедуры реакции, примеры которого включают алканы, такие как гексан (изомеры), гептан (изомеры), октан (изомеры), нонан (изомеры), декан (изомеры) или подобные; ароматические углеводороды и алкил-замещенные ароматические углеводороды, такиекак бензол, толуол, ксилол (изомеры), этилбензол, диизопропилбензол (изомеры), дибутилбензол (изомеры), нафталин или подобные; ароматические соединения, замещенные галогеном или нитрогруппой, такие как хлорбензол, дихлорбензол (изомеры), бромбензол, дибромбензол (изомеры), хлорнафталин, бромнафталин, нитробензол, нитронафталин или подобные; полициклические углеводородные соединения, такие как дифенил, замещенный дифенил, дифенилметан, терфенил, антрацен, дибензилтолуол (изомеры) или подобные; алифатические углеводороды, такие как циклогексан, циклопентан, циклооктан, этилциклогексан или подобные; кетоны, такие как метилэтилкетон, ацетофенон или подобные; сложные эфиры, такие как дибутилфталат, дигексилфталат, диоктилфталат, бензилбутилфталат или подобные; простые эфиры и тиоэфиры, такие как дифениловый эфир, дифенилсульфид или подобные; и сульфоксиды, такие как диметилсульфоксид, дифенилсульфоксид или подобные; и силиконовое масло. Эти растворители можно использовать отдельно или два или более таких видов можно использовать в виде смеси.
Как было описано выше, хотя реакция термического разложения представленного варианта воплощения настоящего изобретения представляет собой реакцию, посредством которой образуются соответствующие изоцианаты и ароматические гидроксисоединения из арилкарбаматов, реакция термического разложения является равновесной реакцией. Таким образом, для эффективного получения изоцианатов в этой реакции термического разложения, предпочтительно, когда осуществляют удаление, по меньшей мере, одного из продуктов этой реакции термического разложения в форме изоцианатов и ароматических гидроксисоединений из системы реакции термического разложения в виде газообразного компонента таким способом, как дистилляция. Какой продукт в виде газообразных компонентов следует удалять: изоцианаты или ароматические гидроксисоединения - произвольно определяют в соответствии с используемыми соединениями, и, например, сравнивают соответствующие стандартные температуры кипения изоцианатов и ароматических гидроксисоединений с последующим удалением соединений, имеющих более низкую стандартную температуру кипения, в форме газообразных компонентов.
Арилкарбаматы также подвержены побочным реакциям, как описано выше, в случае, когда их выдерживают при высокой температуре в течение длительного времени, хотя и в значительно меньшей степени, чем сложные эфиры карбаминовой кислоты. Кроме того, указанные выше побочные реакции также могут быть индуцированы изоцианатами, образованными реакцией термического разложения. Таким образом, период времени, в течение которого арилкарбаматы и изоцианаты выдерживают при высокой температуре, предпочтительно, должен быть как можно более коротким, и реакцию термического разложения предпочтительно осуществляют непрерывным способом. Непрерывный способ относится к способу, в котором арилкарбаматы непрерывно подают в реакционный сосуд, где их подвергают реакции термического разложения, и, по меньшей мере, либо образованные изоцианаты, либо ароматические гидроксисоединения удаляют из реакционного сосуда в форме газообразного компонента.
Хотя можно использовать известные материалы для используемых реакционного сосуда и линий, при условии, что они не оказывают вредного действия на арилкарбамат или продукты в форме ароматических гидроксисоединений и изоцианатов, предпочтительно, можно использовать SUS304, SUS316 или SUS316L и подобные, поскольку они являются дешевыми. Нет никаких конкретных ограничений в отношении типа реакционного сосуда, и можно использовать известный реактор в форме бака или реактор колоночного типа. Предпочтительно, используют реакционный сосуд, который обеспечивается вместе с линиями для экстракции низкокипящей смеси, содержащей, по меньшей мере, либо изоцианаты, либо ароматические гидроксисоединения, образованные в реакции термического разложения, из реакционного сосуда в форме газообразных компонентов, и для удаления смеси жидкостей, содержащей непрореагировавшие арилкарбаматы и соединения, которые не были экстрагированы в форме газообразных компонентов из нижней части реакционного сосуда. Используют известные способы различного типа для таких реакторов, примеры которых включают типы, в которых используют реакторы, содержащие смесительный резервуар, многоступенчатый смесительный резервуар, дистилляционную колонну, многоступенчатую дистилляционную колонну, многотрубный реактор, многоступенчатую дистилляционную колонну непрерывного действия, насадочную колонну, пленочный испаритель, реактор, снабженный находящимся внутри него носителем, реактор с принудительной циркуляцией, испаритель с падающей пленкой, испаритель с падающими каплями, реактор с медленным протеканием жидкости через неподвижный слой или колпачковую колонну, и типы с использованием сочетаний таких реакторов. Способы с использованием пленочного испарителя или реактора колоночного типа являются предпочтительными с точки зрения возможности быстрого удаления низкокипящих компонентов из реакционной системы, при этом структура, имеющая большую площадь контакта газ-жидкость, является предпочтительной, поскольку способна обеспечивать быстрый перенос образуемого спирта в газовую фазу.
Реакционный сосуд предпочтительно обеспечивается вместе линией для подачи арилкарбаматов, линией для удаления газообразного компонента, содержащего, по меньшей мере, либо изоцианаты, либо ароматические гидроксисоединения, образованные реакцией термического разложения, и линией для удаления смеси жидкостей, содержащей соединения, которые не были удалены в виде газообразного компонента, и непрореагировавшие арилкарбаматы, при этом линия для удаления газообразных компонентов, содержащая, по меньшей мере, либо изоцианаты, либо ароматические гидроксисоединения, предпочтительно расположена в таком месте, которое позволяет удалять находящиеся в реакционном сосуде газообразные компоненты, и особенно предпочтительно, когда линия для экстракции смеси жидкостей, содержащей соединения, которые не были удалены в виде газообразного компонента, и непрореагировавшие арилкарбаматы, расположена ниже.
Кроме того, линия для подачи инертного газа и/или жидкого инертного растворителя из нижней части реакционного сосуда может быть подсоединена отдельно, и также может быть подсоединена линия для рециркулирования всей или части смеси жидкостей, содержащей непрореагировавшие арилкарбаматы и соединения, которые не были удалены в виде газообразных компонентов, в реакционный сосуд. К каждой линии может быть добавлено оборудование для подогрева, охлаждения или нагревания, учитывая проблемы засорения и подобные. Кроме того, в случае использования указанного выше инертного растворителя, этот инертный растворитель может быть в форме газа и/или жидкости.
Изоцианат, полученный указанным выше способом получения, можно предпочтительным образом использовать в качестве исходного сырья для получения пенополиуретана, красок, адгезивов и т.п. Поскольку данный способ обеспечивает эффективное получение изоцианатов без использования чрезвычайно токсичного фосгена, настоящее изобретение имеет чрезвычайно важное промышленное значение.
ПРИМЕРЫ
Хотя далее представлено подробное разъяснение настоящего изобретения на базе его примеров, объем настоящего изобретения не ограничивается этими примерами.
<Аналитические методы>
1) Анализ ЯМР
Устройство: система JNM-A400 FT-ЯМР, JEOL Ltd., Japan
(1) Получение образцов для анализа 1H и 13C-ЯМР
Отвешивали около 0,3 г образца раствора с последующим добавлением около 0,7 г тяжелого хлороформа (99,8%, Aldrich Corp., USA) и около 0,05 г внутреннего стандарта в форме тетраметилолова (гарантированный реагент, Wako Pure Chemical Industries, Ltd., Japan) и смешиванием до однородности с получением растворов, которые использовали в качестве образцов для анализа ЯМР.
(2) Количественный анализ
Осуществляли анализы для каждого стандарта и количественные анализы осуществляли на аналитических образцах растворов на основании полученной калибровочной кривой.
2) Жидкостная хроматографиия
Устройство: система LC-10AT, Shimadzu Corp., Japan
Колонка: колонка Silica-60, Tosoh Corp., Japan, две колонки, соединенные в ряд
Проявляющий растворитель: Смесь жидкостей гексан/тетрагидрофуран (80/20) (v/v)
Скорость потока растворителя: 2 мл/мин
Температура колонки: 35°C
Детектор: R.I. (рефрактометр)
(1) Образцы для анализа методом жидкостной хроматографии
Отвешивали около 0,1 г образца с последующим добавлением около 1 г тетрагидрофурана (dehydrated, Wako Pure Chemical Industries, Ltd., Japan) и около 0,02 г внутреннего стандарта в форме бисфенола A (гарантированный реагент, Wako Pure Chemical Industries, Ltd., Japan) и смешиванием до однородности с получением растворов, которые использовали как образцы для анализа методом жидкостной хроматографии.
(2) Количественный анализ
Осуществляли анализы для каждого стандарта и количественные анализы осуществляли на аналитических образцах растворов на основании полученной калибровочной кривой.
3) Газовая хроматография
Устройство: GC-2010, Shimadzu Corp., Japan
Колонка: колонка DB-1, Agilent Technologies Corp., USA, length: 30 м, внутренний диаметр: 0,250 мм, толщина пленки: 1,00 мкм
Температура колонки: выдерживали при 50°C в течение 5 минут с последующим повышением со скоростью 10°C/мин до 200°C; выдерживали при 200°C в течение 5 минут с последующим повышением со скоростью 10°C/мин до 300°C
Детектор: FID
(1) Образцы для анализа методом газовой хроматографии
Отвешивали около 0,05 г образца с последующим добавлением около 1 г ацетона (дегидрированный, Wako Pure Chemical Industries, Ltd., Japan) и около 0,02 г внутреннего стандарта в виде толуола (дегидрированный, Wako Pure Chemical Industries, Ltd., Japan) и смешиванием до однородности с получением растворов, которые использовали как образцы для анализ методом газовой хроматографии.
(2) Количественный анализ
Осуществляли анализы для каждого стандарта и количественные анализы осуществляли на аналитических образцах растворов на основании полученной калибровочной кривой.
[Ссылочный Пример 1] Получение бис(3-метилбутил)карбоната
Стадия (I-1): Получение катализатора на основе диалкилолова
625 г (2,7 моль) оксида ди-н-бутилолова (Sankyo Organic Chemicals Co., Ltd., Japan) и 2020 г (22,7 моль) 3-метил-1-бутанола (Kuraray Co., Ltd., Japan) помещали в 5000-мл мерную грушевидную колбу. Колбу соединяли с испарителем (R-144, Shibata Co., Ltd., Japan), к которому была подсоединена масляная баня (OBH-24, Masuda Corp., Japan), снабженным автоматическим терморегулятором, вакуумным насосом (G-50A, Ulvac Inc., Japan) и регулятором вакуума (VC-10S, Okano Seisakusho Co., Ltd., Japan). Выходное отверстие продувочного клапана этого испарителя соединяли с трубопроводом, содержащим газообразный азот, протекающий при нормальном давлении. После закрытия продувочного клапана испарителя для снижения давления внутри системы, продувочный клапан постепенно окрывали, обеспечивая возможность протекания азота в систему и возвращения к нормальному давлению. Температуру масляной бани устанавливали около 145°C, колбу погружали в масляную баню и начинали вращение испарителя. После нагревания в течение около 40 минут в присутствии азота при атмосферном давлении, оставляя при этом продувочный клапан испарителя открытым, начиналась дистилляция 3-метил-1-бутанола, содержащего воду. После поддержания его в таком состоянии в течение 7 часов, продувочный клапан закрывали, давление внутри системы постепенно уменьшали и избыточное количество 3-метил-1-бутанола отгоняли при давлении внутри системы от 74 до 35 кПа. После того, как фракция больше не появлялась, колбу извлекали из масляной бани. После того, как колбе давали охладиться приблизительно до комнатной температуры (25°C), колбу извлекали из масляной бани, продувочный клапан постепенно окрывали и давление внутри системы снова возвращали к атмосферному давлению. Получали 1173 г реакционной жидкости в колбе. На основании результатов 119Sn-, 1H- и 13C-ЯМР анализов было подтверждено, что получен 1,1,3,3-тетра-н-бутил-1,3-бис(3-метилбутилокси)дистанноксан с выходом 99% в расчете на оксид ди-н-бутилолова. Эту же процедуру затем повторяли 12 раз с получением в общем 10335 г 1,1,3,3-тетра-е-бутил-1,3-бис(3-метилбутилокси) дистанноксана.
Стадия (I-2): Получение бис(3-метилбутил)карбоната
Бис(3-метилбутил)карбонат получали в устройстве непрерывного действия, аналогичном тому, которое показано на Фиг.1. 1,1,3,3-тетрабутил-1,3-бис(3-метилбутилокси) дистанноксан, полученный способом, описанным выше, подавали со скоростью 4388 г/час из передаточной линии 4 в реактор колоночного типа 102, имеющий насадку Metal Gauze CY Packing (Sulzer Chemtech Ltd., Switzerland) и имеющий внутренний диаметр 151 мм и эффективную длину 5040 мм, и 3-метил-1-бутанол, очищенный при помощи дистилляционной колонны 101, подавали со скоростью 14953 г/час из передаточной линии 2. Температуру жидкости внутри реакционного сосуда 102 регулировали до 160°C при помощи нагревателя и ребойлера 112, и давление регулировали до около 120 кПа(изб.) при помощи уравнительного клапана. Время нахождения в реакционном сосуде составляло около 17 минут. 3-Метил-1-бутанол, содержащий воду, при скорости 15037 г/час из верхней части реакционного сосуда через передаточную линию 6 и 3-метил-1-бутанол при скорости 825 г/час через питающую линию 1 подавали при помощи насоса в дистилляционную колонну 101, имеющую насадку Metal Gauze CY Packing и снабженную ребойлером 111 и конденсатором 121, для осуществления дистилляционной очистки. В верхней части дистилляционной колонны 101 фракцию, имеющую высокую концентрацию воды, конденсировали при помощи конденсатора 121 и извлекали из линии выпуска 3. Очищенный 3-метил-1-бутанол подавали при помощи насоса в реактор колоночного типа 102 через передаточную линию 2, расположенную в нижней части дистилляционной колонны 101. Композицию катализатора на основе алкоксида алкилолова, содержащую ди-н-бутил-бис(3-метилбутилокси)олово и 1,1,3,3-тетра-н-бутил-1,3-бис(3-метилбутилокси)дистанноксан, получали из донной части реактора колоночного типа 102 и подавали в пленочный испаритель 103 (Kobelco Eco-Solutions Co., Ltd., Japan) через передаточную линию 5. 3-Метил-1-бутанол отгоняли в пленочном испарителе 103 и возвращали в реактор колоночного типа 102 через конденсатор 123, передаточную линию 8 и передаточную линию 4. Композицию катализатора на основе алкоксида алкилолова откачивали из донной части пленочного испарителя 103 через передаточную линию 7 и подавали в автоклав 104, регулируя скорость потока ди-н-бутил-бис(3-метилбутилокси)олова и 1,1,3,3-тетра-н-бутил-1,3-бис(3-метилбутилокси)дистанноксана, доводя ее до около 5130 г/час. Диоксид углерода подавали в автоклав по передаточной линии 9 при скорости 973 г/час, и давление внутри автоклава поддерживали при 4 МПа(изб.). Температуру внутри автоклава устанавливали на 120°C, время нахождения в реакторе регулировали до около 4 часов и осуществляли реакцию между диоксидом углерода и композицией катализатора на основе алкоксида алкилолова с получением реакционной жидкости, содержащей бис(3-метилбутил)карбонат. Эту реакционную жидкость передавали в резервуар для декарбонизации 105 через передаточную линию 10 и регулировочный клапан для удаления остаточного диоксида углерода, и диоксид углерода извлекали из передаточной линии 11. Затем реакционную жидкость перекачивали в пленочный испаритель (Kobelco Eco-Solutions Co., Ltd., Japan) 106, установленный на около 142°C и около 0,5 кПа, через передаточную линию 12 и подавали, регулируя при этом скорость потока 1,1,3,3-тетра-н-бутил-1,3-бис(3-метилбутилокси)дистанноксана до около 4388 г/час, с получением фракции, содержащей бис(3-метилбутил)карбонат. С другой стороны, осуществляли циркуляцию остатка, полученного в результате выпаривания, в реактор колоночного типа 102 через передаточную линию 13 и передаточную линию 4, регулируя при этом скорость потока 1,1,3,3-тетра-н-бутил-1,3-бис(3-метилбутилокси)дистанноксана до около 4388 г/час. Фракцию, содержащую бис(3-метилбутил)карбонат, подавали в дистилляционную колонну 107, имеющую насадку Metal Gauze CY и снабженную ребойлером 117 и конденсатором 127, через конденсатор 126 и передаточную линию 14 при скорости 959 г/час с последующей дистилляционной очисткой, с получением 99% мас. бис(3-метилбутил)карбоната из линии выпуска 16 при скорости 944 г/час. Когда композицию катализатора на основе алкоксида алкилолова из передаточной линии 13 анализировали методом 119Sn-, 1H- и 13C-ЯМР, было обнаружено, что она содержит 1,1,3,3-тетра-н-бутил-1,3-бис(3-метилбутилокси)дистанноксан, но не содержит ди-н-бутил-бис(3-метилбутилокси)олова. После осуществления указанной выше непрерывной работы процесса в течение около 240 часов композицию катализатора на основе алкоксида алкилолова экстрагировали из линии экстракции 16 при скорости 18 г/час, при этом 1,1,3,3-тетра-н-бутил-1,3-бис(3-метилбутилокси)дистанноксан, полученный в соответствии с описанным выше способом, подавали из питающей линии 17 при скорости 18 г/час.
[Ссылочный Пример 2] Получение дибутилкарбоната
Стадия (II-1): Получение катализатора на основе диалкилолова
692 г (2,78 моль) оксида ди-н-бутилолова и 2000 г (27 моль) 1-бутанола (Wako Pure Chemical Industries, Ltd., Japan) помещали в 3000-мл мерную грушевидную колбу. Колбу, содержащую белую похожую на суспензию смесь, соединяли с испарителем, к которому была подсоединенена масляная баня, снабженным автоматическим терморегулятором, вакуумным насосом и регулятором вакуума. Выходное отверстие продувочного клапана этого испарителя соединяли с трубопроводом, содержащим газообразный азот, протекающий при нормальном давлении. После закрытия продувочного клапана испарителя для снижения давления внутри системы продувочный клапан окрывали постепенно, обеспечивая возможность протекания азота в систему и возвращения к нормальному давлению. Температуру масляной бани устанавливали на около 126°C, колбу погружали в масляную баню и начинали вращение испарителя. После вращения и нагревания в течение около 30 минут при нормальном давлении, оставляя при этом продувочный клапан испарителя открытым, происходило кипение смеси, и начиналась дистилляция низкокипящего компонента. После поддержания в таком состоянии в течение 8 часов продувочный клапан закрывали, давление внутри системы постепенно уменьшали и остаточный низкокипящий компонент отгоняли при давлении внутри системы от 76 до 54 кПа. После того, как уже не оставалось никакого низкокипящего компонента, колбу извлекали из масляной бани. Реакционная жидкость была в форме прозрачной жидкости. Колбу затем извлекали из масляной бани, окрывали постепенно продувочный клапан и давление внутри системы возвращали к нормальному давлению. Получали 952 г реакционной жидкости в колбе. На основании результатов 119Sn-, 1H- и 13C-ЯМР анализов был получен продукт в форме 1,1,3,3-тетра-н-бутил-1,3-ди(н-бутилокси)дистанноксана с выходом 99% в расчете на оксид ди-н-бутилолова. Эту же процедуру затем повторяли 12 раз с получением в общем 11480 г 1,1,3,3-тетра-н-бутил-1,3-ди(бутилокси)дистанноксана.
Стадия (II-2): Получение дибутилкарбоната
Сложный эфир угольной кислоты получали в устройстве для непрерывного процесса получения аналогично показанному на Фиг. 1. 1,1,3,3-Тетра-н-бутил-1,3-ди(н-бутилокси)дистанноксан, полученный на стадии (II-1), подавали со скоростью 4201 г/час из передаточной линии 4 в реактор колоночного типа, имеющий насадку Mellapak 750Y (Sulzer Chemtech Ltd., Switzerland) и имеющий внутренний диаметр 151 мм и эффективную длину 5040 мм, и 1-бутанол, очищенный при помощи дистилляционной колонны 101, подавали в реактор колоночного типа 102 при скорости 24717 г/час из питающей линии 2. Температуру жидкости внутри реакционного сосуда регулировали до 160°C при помощи нагревателя и ребойлера 112 и давление регулировали до около 250 кПа(изб.) при помощи уравнительного клапана. Время нахождения в реакционном сосуде составляло около 10 минут. 1-Бутанол, содержащий воду, при скорости 24715 г/час из верхней части реакционного сосуда через передаточную линию 6 и 1-бутанол при скорости 824 г/час через питающую линию 1 подавали при помощи насоса в дистилляционную колонну 101, имеющую насадку Metal Gauze CY (Sulzer Chemtech Ltd., Switzerland) и снабженную ребойлером 111 и конденсатором 121, для осуществления дистилляционной очистки. В верхней части дистилляционной колонны 101 фракцию, содержащую высокую концентрацию воды, конденсировали при помощи конденсатора 121 и извлекали из передаточной линии 3. Очищенный 1-бутанол откачивали через передаточную линию 2, расположенную в нижней части дистилляционной колонны 101. Композицию катализатора на основе алкоксида алкилолова, содержащую ди-н-бутоксид ди-н-бутилолова и 1,1,3,3-тетра-н-бутил-1,3-ди(н-бутилокси)дистанноксан, получали из донной части реактора колоночного типа 102 и подавали в пленочный испаритель 103 (Kobelco Eco-Solutions Co., Ltd., Japan) через передаточную линию 5. 1-Бутанол отгоняли в пленочном испарителе 103 и возвращали в реактор колоночного типа 102 через конденсатор 123, передаточную линию 8 и передаточную линию 4. Композицию катализатора на основе алкоксида алкилолова откачивали из донной части пленочного испарителя 103 через передаточную линию 7 и подавали в автоклав 104, регулируя скорость потока активных компонентов в форме дибутоксида дибутилолова и 1,1,3,3-тетра-н-бутил-1,3-ди(н-бутилокси)дистанноксана до около 4812 г/час. Диоксид углерода подавали в автоклав через питающую линию 9 при скорости 973 г/час и давление внутри автоклава поддерживали при 4 МПа(изб.). Температуру внутри автоклава устанавливали на 120°C, время нахождения в реакторе регулировали до около 4 часов и осуществляли реакцию между диоксидом углерода и композицией катализатора на основе алкоксида алкилолова с получением реакционной жидкости, содержащей дибутилкарбонат. Эту реакционную жидкость передавали в резервуар для декарбонизации 105 через передаточную линию 10 и регулировочный клапан для удаления остаточного диоксида углерода и диоксид углерода извлекали из передаточной линии 11. Затем реакционную жидкость перекачивали при помощи насоса в пленочный испаритель 106 (Kobelco Eco-Solutions Co., Ltd., Japan), устанавленный на около 140°C и около 1,4 кПа, через передаточную линию 12 и подавали, регулируя при этом скорость потока 1,1,3,3-тетра-н-бутил-1,3-ди(н-бутилокси)дистанноксана до около 4201 г/час, с получением фракции, содержащей дибутилкарбонат. С другой стороны, осуществляли циркуляцию остатка, полученного в результате выпаривания, в реактор колоночного типа 102 через передаточную линию 13 и передаточную линию 4, регулируя при этом скорость потока 1,1,3,3-тетра-н-бутил-1,3-ди(н-бутилокси)дистанноксана до около 4201 г/час. Фракцию, содержащую дибутилкарбонат, подавали в дистилляционную колонну 107, имеющую насадку Metal Gauze CY (Sulzer Chemtech Ltd., Switzerland) и снабженную ребойлером 117 и конденсатором 127, через конденсатор 126 и передаточную линию14 при скорости 830 г/час с последующей дистилляционной очисткой с получением 99% мас. бис(3-метилбутил)карбоната из линии выпуска 16 при скорости 814 г/час. Когда композицию катализатора на основе алкоксида алкилолова из передаточной линии 13 анализировали методом 119Sn-, 1H- и 13C-ЯМР, было обнаружено, что она содержат 1,1,3,3-тетра-н-бутил-1,3-ди(н-бутилокси)дистанноксан, но не содержит ди-н-бутоксид ди-н-бутилолова. После осуществления указанной выше непрерывной работы процесса в течение около 600 часов композицию катализатора на основе алкоксида алкилолова экстрагировали из линии экстракции 16 при скорости 16 г/час, при этом 1,1,3,3-тетра-н-бутил-1,3-ди(н-бутилокси)дистанноксан, полученный на стадии (II-1), подавали из питающей линии 17 при скорости 16 г/час.
[Пример 1]
Стадия (1-1): Получение ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
1818 г (9,0 моль) бис(3-метилбутил)карбоната и 208,8 г (1,8 моль) гексаметилендиамина (Aldrich Corp., USA) помещали в 5-л мерную четырехгорлую колбу, в колбу помещали мешалку и колбу соединяли с конденсатором Dimroth и трехходовым клапаном. После замещения атмосферы внутри системы азотом четырехгорлую колбу погружали в масляную баню (OBH-24, Masuda Corp., Japan), нагретую до 80°C, с последующим добавлением 3,5 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd., Japan) для запуска реакции. Образцы реакционной жидкости подходящим образом собирали и подвергали анализу ЯМР, и реакцию останавливали в тот момент, когда больше не определяли присутствия гексаметилендиамина. В результате анализа полученного раствора методом жидкостной хроматографии было обнаружено, что раствор содержит 29,9% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (1-2): Отгонка низкокипящего компонента
Раствор, полученный на стадии (1-1), помещали в 5-л мерную колбу, снабженную трехходовым клапаном, конденсатором, сборником дистиллята и термометром, и атмосферу внутри колбы замещали азотом в вакууме. Колбу погружали в масляную баню, нагретую до около 130°C. Дистилляцию осуществляли при постепенном снижении давления в колбе до конечного давления 0,02 кПа. Получали 1410 г дистиллята. В результате анализа методом газовой хроматографии было обнаружено, что дистиллят представляет собой раствор, содержащий 78,3% мас. бис(3-метилбутил)карбоната и 21,4% мас. изоамилового спирта. Кроме того, в результате анализа полученного остатка в колбе после дистилляции методом жидкостной хроматографии, было обнаружено, что остаток после дистилляции содержит 98,0% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (1-3): Получение ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
Реакцию переэтерификации осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 2.
111 г дилаурата дибутилолова (химической чистоты, Wako Pure Chemical Industries, Ltd., Japan) и 4119 г 2,4-ди-трет-амилфенола (Tokyo Chemical Industry Co., Ltd., Japan) добавляли к 618 г остатка после дистилляции, полученного на стадии (1-2), и перемешивали с получением гомогенного раствора, который затем помещали в питающий резервуар 201. Аппарат для пленочной дистилляции 202 (Kobelco Eco-Solutions Co., Ltd., Japan), имеющий теплопроводящую площадь поверхности 0,2 м2, нагревали до 240°C и атмосферу внутри аппарата для пленочной дистилляции заменяли на атмосферу азота при атмосферном давлении. Раствор подавали в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час. Газообразную смесь, содержащую 3-метил-1-бутанол и 2,4-ди-трет-амилфенол, экстрагировали из линии 25, обеспечиваемой в верхней части аппарата для пленочной дистилляции 202, и подавали в дистилляционную колонну 203, содержащую насадку Metal Gauze CY Packing (Sulzer Chemtech Ltd., Switzerland). 3-метил-1-бутанол и 2,4-ди-трет-амилфенол разделяли в дистилляционной колонне 203 и 2,4-ди-трет-амилфенол возвращали в верхнюю часть аппарата для пленочной дистилляции 202 по линии 26, обеспечиваемой в нижней части дистилляционной колонны 203. Реакционную жидкость экстрагировали из линии 22, обеспечиваемой внижней части аппарата для пленочной дистилляции 202, и возвращали в питающий резервуар 201 через линию 23.
После осуществления этой стадии в течение 62 часов реакционную жидкость экстрагировали из линии 24. Экстрагировали 4532 г экстрагированной жидкостости и 304 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203.
В результате анализа экстрагированной реакционной жидкости методом жидкостной хроматографии было обнаружено, что реакционную жидкость содержит 24,2% мас. ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты. Кроме того, в результате анализа раствора, извлеченного из линии 27, методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 98% мас. 3-метил-1-бутанола.
Стадия (1-4): Получение гексаметилендиизоцианата путем термического разложения ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Реакцию термического разложения осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 2.
Аппарат для пленочной дистилляции 302 (Kobelco Eco-Solutions Co., Ltd., Japan), имеющий теплопроводящую площадь поверхности 0,2 м2, нагревали до 200°C и давление внутри аппарата для пленочной дистилляции устанавливали на около 1,3 кПа. Раствор, полученный на стадии (1-3), помещали в питающий резервуар 301 и подавали в аппарат для пленочной дистилляции при скорости около 980 г/час через линию 31. Жидкий компонент экстрагировали из линии 33, обеспечиваемой в нижней части аппарата для пленочной дистилляции 302, и возвращали в питающий резервуар 301 через линию 34. Газообразный компонент, содержащий гексаметилендиизоцианат и 2,4-ди-трет-амилфенол, экстрагировали из линии 32, обеспечиваемой в верхней части аппарата для пленочной дистилляции 302. Газообразный компонент вводили в дистилляционную колонну 303 с последующим разделением на гексаметилендиизоцианат и 2,4-ди-трет-амилфенол, и часть 2,4-ди-трет-амилфенола возвращали в питающий резервуар 301 по линии 34 через посредство линии 36, обеспечиваемой в нижней части дистилляционной колонны 303. По прошествии 13 часов осуществления этой реакции 266 г раствора извлекали из линии 35 и в результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 88%.
[Пример 2]
Стадия (2-1): Получение ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Раствор, содержащий 12,8% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что добавляли 2230 г 3-метил-1-бутанола в 5-л мерную четырехгорлую колбу с последующим добавлением в колбу 1515 г (7,5 моль) бис(3-метилбутил)карбоната и 174 г (1,5 моль) гексаметилендиамина и использовали 28,9 г метоксида натрия. Раствор пропускали через колонну, содержащую в качестве насадки ионообменную смолу (Amberlyst 15 Dry, Rohm и Haas Co., USA), и извлекали 3919 г раствора.
Стадия (2-2): Отгонка низкокипящего компонента
3373 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (2-1) вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 27,0% мас. бис(3-метилбутил)карбоната и 72,9% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 92,1% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (2-3): Получение ди(2,4-ди-трет-бутилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
3351 г реакционной жидкости экстрагировали из линии 24 путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 544 г остатка после дистилляци, полученного на стадии (2-2), вместо остатка после дистилляци, полученного на стадии (1-2), использовали 92 г дилаурата дибутилолова, использовали 2999 г 2,4-ди-трет-бутилфенола (Wako Pure Chemical Industries, Ltd., Japan) вместо 2,4-ди-трет-амилфенола и осуществляли реакцию в течение 70 часов. Кроме того, 246 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203. Экстрагированная реакционная жидкость содержала 24,2% мас. ди(2,4-ди-трет-бутилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (2-4): Получение гексаметилендиизоцианата путем термического разложения ди(2,4-ди-трет-бутилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
225 г раствора извлекали из линии 35 путем осуществления такого же способа, как на стадии (1-4) Примера 1, в реакционном устройстве, аналогичном показанному на Фиг. 2, за исключением того, что нагревали аппарат 302 для пленочной дистилляции до 200°C, устанавливая давление внутри аппарата для пленочной дистилляции до около 1,3 кПа, подавая раствор, полученный на стадии (2-3), в аппарат для пленочной дистилляции при скорости около 980 г/час через линию 31 и осуществляли реакцию в течение 11 часов. В результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 89%.
[Пример 3]
Стадия (3-1): Получение ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
2754 г раствора, содержащего 21,6% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 2545 г (12,6 моль) бис(3-метилбутил)карбоната и 209 г (1,8 моль) гексаметилендиамина, наряду с использованием мешалки, и использовали 3,5 г метоксида натрия.
Стадия (3-2): Отгонка низкокипящего компонента
2150 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (3-1), вместо раствора, полученного на стадии (1-1). В результате анализа дистиллята методом газовой хроматографии было обнаружено, что дистиллят содержит 85,6% мас. бис(3-метилбутил)карбоната и 14,0% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии, было обнаружено, что остаток в колбе после дистилляции содержит 98,4% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (3-3): Получение ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
4834 г реакционной жидкости экстрагировали из линии 24 путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 602 г остатка после дистилляции, полученного на стадии (3-2), вместо остатка после дистилляции, полученного на стадии (1-2), использовали 109 г дилаурата дибутилолова, использовали 4431 г 2,4-ди-трет-амилфенола и осуществляли реакцию в течение 70 часов. Кроме того, 297 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203. Экстрагированная реакционная жидкость содержала 22,2% мас. ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (3-4): Дистилляция 2,4-ди-трет-амилфенола
Вакуумный насос и регулятор вакуума подсоединяли к устройству для молекулярной дистилляции, снабженному нагревательным устройством с двойными стенками, работающим при циркуляции масла (ϕ80 устройство для молекулярной дистилляции, Asahi Seisakusho Co., Ltd., Japan), и линию продувки регулятора вакуума соединяли с трубопроводом для газообразного азота. Воздух внутри устройства для молекулярной дистилляции заменяли азотом и нагревательное устройство нагревали до 150°C при помощи масляного циркуляционного насоса. Давление в устройстве для молекулярной дистилляции снижали до 0,3 кПа, и раствор, полученный на стадии (3-3), подавали в устройство для молекулярной дистилляции при скорости около 5 г/мин при вращении движка устройства для молекулярной дистилляции при скорости около 300 об/мин для отгонки 2,4-ди-трет-амилфенола. 1291 г высококипящего вещества извлекали в сборник образцов с высокой температурой кипения, поддерживаемый при температуре 120°C, и в результате анализа методом жидкостной хроматографии было обнаружено, что это вещество содержит 83,1% мас. ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (3-5): Получение гексаметилендиизоцианата путем термического разложения ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Реакцию термического разложения осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 2.
290 г раствора извлекали из линии 35 путем осуществления такого же способа, как на стадии (1-4) Примера 1, за исключением того, что подавали жидкую смесь в форме суспензии, включающей смесь 1289 г вещества, содержащего сложный эфир карбаминовой кислоты, полученного на стадии (3-4), и 3422 г бензилбутилфталата (гарантированный реагент, Wako Pure Chemical Industries, Ltd., Japan), в питающий резервуар 301, с подачей в аппарат для пленочной дистилляции при скорости около 980 г/час и взаимодействием в течение 14 часов. В результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 83%.
[Пример 4]
Стадия (4-1): Получение дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты
Раствор, содержащий 30,8% мас. дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 1479 г (8,5 моль) дибутилкарбоната, полученного в соответствии со способом Ссылочного Примера 2, и 197 г (1,7 моль) гексаметилендиамина (Aldrich Corp., USA), и использовали 3,3 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd., Japan) в 5-л мерной четырехгорлой колбе.
Стадия (4-2): Отгонка низкокипящего компонента
1156 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (4-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 78,5% мас. дибутилкарбоната и 20,8% мас. н-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии, было обнаружено, что остаток в колбе после дистилляции содержит 98,8% мас. дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (4-3): Получение ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
4929 г реакционной жидкости экстрагировали из линии 24 путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали остаток после дистилляции, полученный на стадии (4-2), вместо остатка после дистилляции, полученного на стадии (1-2), использовали 82 г дилаурата дибутилолова, добавляли 4566 г 2,4-ди-трет-амилфенола с последующим перемешиванием с получением гомогенного раствора, который затем подавали в питающий резервуар 201, нагревали аппарат для пленочной дистилляции 202, имеющий теплопроводящую площадь поверхности 0,2 м2, до 240°C и осуществляли взаимодействие в течение 86 часов. 233 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203.
Когда экстрагированную реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 20,4% мас. ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99,9% мас. 1-бутанола.
Стадия (4-4): Получение гексаметилендиизоцианата путем термического разложения ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Реакцию термического разложения осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 2.
248 г раствора извлекали из линии 35 путем осуществления такого же способа, как на стадии (1-4) Примера 1, за исключением того, что использовали раствор, который экстрагировали из линии 24 на стадии (4-3), вместо раствора, который экстрагировали из линии 24 на стадии (1-3), и осуществляли взаимодействие в течение 14 часов. В результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 87%.
[Пример 5]
Стадия (5-1): Получение дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты
2091 г раствора, содержащего 26,2% мас. дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 1879 г (10,8 моль) дибутилкарбоната и 209 г (1,8 моль) гексаметилендиамина, добавляли мешалку и добавляли 3,5 г метоксида натрия.
Стадия (5-2): Отгонка низкокипящего компонента
1537 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (5-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 82,9% мас. дибутилкарбоната и 16,6% мас. н-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 99,0% мас. дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (5-3): Получение ди(2,6-диметилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
3554 г реакционной жидкости экстрагировали из линии 24 путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 548 г остатка после дистилляции, полученного на стадии (5-2), вместо остатка после дистилляции, полученного на стадии (1-2), использовали 3142 г 2,6-диметилфенола (Aldrich Corp., USA) вместо 2,4-ди-трет-амилфенола, использовали 109 г дилаурата дибутилолова, обеспечивали температуру аппарата 202 для пленочной дистилляции 200°C и осуществляли реакцию в течение 225 часов. Кроме того, 239 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203. Экстрагированная реакционная жидкость содержала 18,7% мас. ди(2,6-диметилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (5-4): Получение гексаметилендиизоцианата путем термического разложения ди(2,6-диметилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Реакцию термического разложения осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 4.
Аппарат 402 для пленочной дистилляции (Kobelco Eco-Solutions Co., Ltd., Japan), имеющий теплопроводящую площадь поверхности 0,2 м2, нагревали до 200°C и давление внутри аппарата для пленочной дистилляции устанавливали на около 1,3 кПа. Раствор, полученный на стадии (5-3), помещали в питающий резервуар 401 и подавали в аппарат для пленочной дистилляции при скорости около 680 г/час через линию 41. Жидкий компонент экстрагировали из линии 43, обеспечиваемой в нижней части аппарата для пленочной дистилляции 402, и возвращали в питающий резервуар 401 через линию 44. Газообразный компонент, содержащий гексаметилендиизоцианат и 2,6-диметилфенол, экстрагировали из линии 42, обеспечиваемой в верхней части аппарата для пленочной дистилляции 402. Газообразный компонент вводили в дистилляционную колонну 403 с последующим разделением на гексаметилендиизоцианат и 2,6-диметилфенол, при этом 2,6-диметилфенол экстрагировали из линии 45 через верхнюю часть дистилляционной колонны 403, и газообразный компонент, содержащий гексаметилендиизоцианат, экстрагировали из линии 47, обеспечиваемой в дистилляционной колонне 403. С другой стороны, высококипящее вещество экстрагировали из линии 46, обеспечиваемой в нижней части дистилляционной колонны, и часть возвращали в питающий резервуар 401 по линии 44. Газообразный компонент, содержащий гексаметилендиизоцианат, который экстрагировали из линии 47, подавали при помощи насоса в дистилляционную колонну 404, и гексаметилендиизоцианат отгоняли и выделяли в дистилляционной колонне 404. Высококипящее вещество экстрагировали из линии 48, обеспечиваемой вдистилляционной колонне 404, и часть возвращали в питающий резервуар 401 по линии 44. С другой стороны, газообразный компонент экстрагировали из линии 49, и гексаметилендиизоцианат экстрагировали из линии 52 через конденсатор. После взаимодействия в течение 11 часов 249 г раствора, содержащего 99% мас. гексаметилендиизоцианата, извлекали из линии 47. Выход в расчете на гексаметилендиамин составил 82%.
[Пример 6]
Стадия (6-1): Получение диметилового эфира N,N'-гександиил-бис-карбаминовой кислоты
Раствор, содержащий 39,0% мас. диметилового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 765 г (8,5 моль) диметилкарбоната (Aldrich Corp., USA) и 197 г (1,7 моль) гексаметилендиамина и использовали 3,3 г метоксида натрия в 2-л мерной четырехгорлой колбе.
Стадия (6-2): Отгонка низкокипящего компонента
582 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (6-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 80,8% мас. диметилкарбоната и 17,9% мас. метанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 98,9% мас. диметилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (6-3): Получение ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
4517 г реакционной жидкости экстрагировали из линии 24 путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 376 г остатка в колбе после дистилляции, полученного на стадии (6-2), вместо остатка в колбе после дистилляции, полученного на стадии (1-2), использовали 82 г дилаурата дибутилолова, использовали 4161 г 2,4-ди-трет-амилфенола и осуществляли реакцию в течение 86 часов. 100 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203.
Когда экстрагированную реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 22,1% мас. ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99,4% мас. метанола.
Стадия (6-4): Получение гексаметилендиизоцианата путем термического разложения ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
242 г раствора извлекали из линии 35 путем осуществления такого же способа, как на стадии (1-4) Примера 1, за исключением того, что использовали раствор, который экстрагировали из линии 24 на стадии (6-3), вместо раствора, который экстрагировали из линии 24 на стадии (1-3), и осуществляли реакцию в течение 13 часов. В результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 85%.
[Пример 7]
Стадия (7-1): Получение диметилового эфира N,N'-гександиил-бис-карбаминовой кислоты
934 г раствора, содержащего 42,4% мас. диметилового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 729 г (8,1 моль) диметилкарбоната и 209 г (1,8 моль) гексаметилендиамина и использовали 0,35 г метоксида натрия.
Стадия (7-2): Отгонка низкокипящего компонента
533 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (7-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 77,8% мас. диметилкарбоната и 20,6% мас. метанола. Кроме того, в результате анализа методом жидкостной хроматографии, было обнаружено, что остаток в колбе после дистилляции содержит 99,7% мас. диметилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (7-3): Получение ди(2,6-диметилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
3537 г реакционной жидкости экстрагировали из линии 24 путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 395 г остатка после дистилляции, полученного на стадии (7-2), вместо остатка после дистилляции, полученного на стадии (1-2), использовали 108 г дилаурата дибутилолова, использовали 3133 г 2,6-диметилфенола вместо 2,4-ди-трет-амилфенола, нагревали аппарат 202 для пленочной дистилляции до 200°C и осуществляли реакцию в течение 250 часов. Кроме того, 100 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203. Экстрагированная реакционная жидкость содержала 18,3% мас. ди(2,6-диметилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (7-4): Получение гексаметилендиизоцианата путем термического разложения ди(2,6-диметилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
243 г раствора, содержащего 99% мас. гексаметилендиизоцианата, извлекали из линии 52 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что использовали реакционную жидкость, полученную из линии 24 на стадии (7-3), вместо раствора, полученного на стадии (5-3), и осуществляли реакцию в течение 16 часов. Выход в расчете на гексаметилендиамин составил 81%.
[Пример 8]
Стадия (8-1): Получение (3-метилбутил)ового эфира 3-((3-метилбутил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты
1980 г (9,8 моль) бис(3-метилбутил)карбоната и 239 г (1,8 моль) 3-аминометил-3,5,5-триметилциклогексиламина (Aldrich Corp., USA) помещали в 5-л мерную четырехгорлую колбу, в колбу помещали мешалку и подсоединяли к колбе конденсатор Dimroth и трехходовый клапан. После замещения атмосферы внутри системы азотом, четырехгорлую колбу погружали в масляную баню (OBH-24, Masuda Corp., Japan), нагретую до 100°C, с последующим добавлением 2,7 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd., Japan) для запуска реакции. Образцы реакционной жидкости подходящим образом собирали и подвергали анализу ЯМР, и реакцию останавливали в тот момент, когда 3-аминометил-3,5,5-триметилциклогексиламин больше нельзя было обнаружить. В результате анализа полученного раствора методом жидкостной хроматографии было обнаружено, что раствор содержит 23,9% мас. (3-метилбутил)ового эфира 3-((3-метилбутил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты.
Стадия (8-2): Отгонка низкокипящего компонента
1683 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (8-1) вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 85,4% мас. бис(3-метилбутил)карбоната и 13,8% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 99,4% мас. (3-метилбутил)ового эфира 3-((3-метилбутил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты.
Стадия (8-3): Получение (2,4-ди-трет-амилфенил)ового эфира 3-((2,4-ди-трет-амилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты при помощи переэтерификации
5034 г реакционной жидкости экстрагировали из линии 24 и 221 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 530 г остатка после дистилляции, полученного на стадии (8-2), вместо остатка после дистилляции, полученного на стадии (1-2), использовали 84 г дилаурата дибутилолова, использовали 4645 г 2,4-ди-трет-амилфенола, нагревали аппарат 202 для пленочной дистилляции до 240°C, подавали раствор в аппарат для пленочной дистилляции через питающую линию 21 при скорости около 1200 г/час и осуществляли реакцию в течение 75 часов.
Когда экстрагированную реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционную жидкость содержит 17,2% мас. (2,4-ди-трет- амилфенил)ового эфира 3-((2,4-ди-трет-амилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 3-метил-1-бутанола.
Стадия (8-4): Получение изофорондиизоцианата путем термического разложения (2,4-ди-трет-амилфенил)пентилового эфира 3-((2,4-ди-трет-амилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты
257 г раствора, содержащего 99% мас. изофорондиизоцианата, извлекали из линии 52 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 200°C, создавали давление в аппарате для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (8-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час, и осуществляли реакцию в течение 11 часов. Выход в расчете на 3-аминометил-3,5,5-триметилциклогексиламин составил 83%.
[Пример 9]
Стадия (9-1): Получение бутилового эфира 3-(бутилоксикарбониламино-метил)-3,5,5-триметилциклогексил-карбаминовой кислоты
Осуществляли такой же способ, как на стадии (8-1) Примера 8, за исключением того, что использовали 2349 г (13,5 моль) дибутилкарбоната, 255 г (1,5 моль) 3-аминометил-3,5,5-триметилциклогексиламина и 2,9 г метоксида натрия. В результате анализа полученного раствора методом жидкостной хроматографии было обнаружено, что раствор содержит 20,0% мас. бутилового эфира 3-(бутилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты.
Стадия (9-2): Отгонка низкокипящего компонента
2075 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (9-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 89,2% мас. дибутилкарбоната и 10,0% мас. н-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 98,7% мас. бутилового эфира 3-(бутилоксикарбониламино-метил)-3,5,5-триметилциклогексил-карбаминовой кислоты.
Стадия (9-3): Получение (2,4-ди-трет-бутилфенил)ового эфира 3-((2,4-ди-трет-бутилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты при помощи переэтерификации
4077 г реакционной жидкости экстрагировали из линии 24 и 197 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 525 г остатка после дистилляции, полученного на стадии (9-2), использовали 89 г дилаурата дибутилолова, использовали 3751 г 2,4-ди-трет-бутилфенола с получением гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 240°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавали раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час и осуществляли реакцию в течение 84 часов.
Когда извлеченную реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 20,7% мас. (2,4-ди-трет-бутилфенил)ового эфира 3-((2,4-ди-трет-бутилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 98% мас. 1-бутанола.
Стадия (9-4): Получение изофорондиизоцианата путем термического разложения (2,4-ди-трет-бутилфенил)ового эфира 3-((2,4-ди-трет-бутилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты
271 г раствора, содержащего 99% мас. изофорондиизоцианата, извлекали из линии 52 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 200°C, создавали давление в аппарате для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (9-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час, и осуществляли реакцию в течение 11 часов. Выход в расчете на 3-аминометил-3,5,5-триметилциклогексиламин составил 82%.
[Пример 10]
Стадия (10-1): Получение бутилового эфира 3-(бутилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты
Раствор, содержащий 25,1% мас. бутилового эфира 3-(бутилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (8-1) Примера 8, за исключением того, что использовали 1949 г (11,2 моль) дибутилкарбоната, 272 г (1,6 моль) 3-аминометил-3,5,5-триметилциклогексиламин и 3,1 г метоксида натрия.
Стадия (10-2): Отгонка низкокипящего компонента
1657 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (10-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 85,7% мас. дибутилкарбоната и 13,4% мас. н-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии, было обнаружено, что остаток в колбе после дистилляции содержит 98,9% мас. бутилового эфира 3-(бутилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты.
Стадия (10-3): Получение 3-((2,4,6-три-метилфенил)оксикарбониламино-метил)-3,(2,4,6-три-метилфенил)ового эфира 3-((2,4,6-три-метилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты при помощи переэтерификации
3067 г реакционной жидкости экстрагировали из линии 24 и 208 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 76 г дилаурата дибутилолова к 560 г остатка после дистилляции, полученного на стадии (10-2), добавляли 2645 г 2,4,6-триметилфенола (Aldrich Corp., USA) и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 220°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 120 г/час, и осуществляли реакцию в течение 180 часов.
Когда экстрагированную реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 22,7% мас. (2,4,6-триметилфенил)ового эфира 3-((2,4,6-три-метилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 1-бутанола.
Стадия (10-4): Получение изофорондиизоцианата путем термического разложения (2,4,6-триметилфенил)ового эфира 3-((2,4,6-триметилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты
286 г раствора, содержащего 99% мас. изофорондиизоцианата, извлекали из линии 52 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 200°C, создавали давление в аппарате для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (10-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час, и осуществляли реакцию в течение 14 часов. Выход в расчете на 3-аминометил-3,5,5-триметилциклогексиламин составил 81%.
[Пример 11]
Стадия (11-1): Получение метилового эфира 3-(метилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты
Раствор, содержащий 33,6% мас. метилового эфира 3-(метилоксикарбониламино-метил)-3,5,5-триметилциклогексил-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (8-1) Примера 8, за исключением того, что использовали 1323 г (14,7 моль) диметилкарбоната, 357 г (2,1 моль) 3-аминометил-3,5,5-триметилциклогексиламина и 4,1 г метоксида натрия.
Стадия (11-2): Отгонка низкокипящего компонента
1111 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (11-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 86,7% мас. диметилкарбоната и 11,3% мас. метанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 99,1% мас. метилового эфира 3-(метилоксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты.
Стадия (11-3): Получение (2,4,6-три-метилфенил)ового эфира 3-((2,4,6-три-метилфенил)оксикарбониламинометил)-3,5,5-триметилциклогексилкарбаминовой кислоты при помощи переэтерификации
Реакцию переэтерификации осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 2.
4457 г реакционной жидкости экстрагировали из линии 24 и 118 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 99 г дилаурата дибутилолова и 4006 г 2,4,6-триметилфенола к 567 г остатка после дистилляции, полученного на стадии (11-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 220°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 90 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 20,5% мас. (2,4,6-триметилфенил)ового эфира 3-((2,4,6-три-метилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. метанола.
Стадия (11-4): Получение изофорондиизоцианата путем термического разложения (2,4,6-триметилфенил)ового эфира 3-((2,4,6-триметилфенил)оксикарбониламино-метил)-3,5,5-триметилциклогексилкарбаминовой кислоты
368 г раствора, содержащего 99% мас. изофорондиизоцианата, извлекали из линии 47 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 200°C, создавали давление в аппарате для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (11-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 900 г/час, и осуществляли реакцию в течение 13 часов. Выход в расчете на 3-аминометил-3,5,5-триметилциклогексиламин составил 79%.
[Пример 12]
Стадия (12-1): Получение бис(3-метилбутил)-4,4'-метилен-дициклогексилкарбамата
1313 г (6,5 моль) бис(3-метилбутил)карбоната и 273 г (1,3 моль) 4,4'-метиленбис(циклогексиламин) (Aldrich Corp., USA) помещали в 5-л мерную четырехгорлую колбу, в колбу помещали мешалку и подсоединяли к колбе конденсатор Dimroth и трехходовый клапан. После замещения атмосферы внутри системы азотом, четырехгорлую колбу погружали в масляную баню (OBH-24, Masuda Corp., Japan), нагретую до 100°C, с последующим добавлением 2,5 г метоксида натрия для запуска реакции. Образцы реакционной жидкости подходящим образом собирали и подвергали анализу ЯМР и реакцию останавливали в тот момент, когда 4,4'-метиленбис(циклогексиламин) больше нельзя было обнаружить. В результате анализа полученного раствора методом жидкостной хроматографии было обнаружено, что раствор содержит 34,3% мас. бис(3-метилбутил)-4,4'-метилен-дициклогексилкарбамата.
Стадия (12-2): Отгонка низкокипящего компонента
1034 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (12-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 77,8% мас. бис(3-метилбутил)карбоната и 21,2% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 99,0% мас. бис(3-метилбутил)-4,4'-метилендициклогексилкарбамата.
Стадия (12-3): Получение бис(2,4-ди-трет-амилфенил)-4,4'-метилендициклогексилкарбамата при помощи переэтерификации
4702 г реакционной жидкости экстрагировали из линии 24 и 210 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 79 г дилаурата дибутилолова и 4358 г 2,4-ди-трет-амилфенола к 547 г остатка после дистилляции, полученного на стадии (12-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 260°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 58 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 18,2% мас. бис(2,4-ди-трет-амилфенил)-4,4'-метилен-дициклогексилкарбамата. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 3-метил-1-бутанола.
Стадия (12-4): Получение гексаметилендиизоцианата путем термического разложения бис(2,4-ди-трет-амилфенил)-4,4'-метилендициклогексилкарбамата
287 г вещества, содержащего 99% мас. 4,4'-метилен- ди(циклогексилизоцианат), извлекали из линии 47 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 210°C, создавали давление в аппарате для пленочной дистилляции около 0,13 кПа, подавали раствор, полученный на стадии (12-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час, и осуществляли реакцию в течение 11 часов. Выход в расчете на 4,4'-метиленбис(циклогексиламин) составил 84%.
[Пример 13]
Стадия (13-1): Получение бис(3-метилбутил)-4,4'-метилен-дициклогексилкарбамата
Раствор, содержащий 29,4% мас. бис(3-метилбутил)-4,4'-метилендициклогексилкарбамата, получали путем осуществления такого же способа, как на стадии (12-1) Примера 12, за исключением того, что использовали 1818 г (9,0 моль) бис(3-метилбутил)карбоната, 315 г (1,5 моль) 4,4'-метиленбис(циклогексиламин) и 2,9 г метоксида натрия.
Стадия (13-2): Отгонка низкокипящего компонента
1490 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (13-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 82,6% мас. бис(3-метилбутил)карбоната и 16,8% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 98,0% мас. бис(3-метилбутил)-4,4'-метилендициклогексилкарбамата.
Стадия (13-3): Получение бис(2,4-ди-трет-бутилфенил)-4,4'-метилендициклогексилкарбамата при помощи переэтерификации
4987 г реакционной жидкости экстрагировали из линии 24 и 238 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 90 г дилаурата дибутилолова и 4511 г 2,4-ди-трет-бутилфенола к 633 г остатка после дистилляции, полученного на стадии (13-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 240°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 78 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 18,3% мас. бис(2,4-ди-трет-бутилфенил)-4,4'-метилендициклогексилкарбамата. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 3-метил-1-бутанола.
Стадия (13-4): Получение 4,4'-метилен-ди(циклогексилизоцианат) путем термического разложения бис(2,4-ди-трет-бутилфенил)-4,4'-метилен-дициклогексилкарбамата
325 г вещества, содержащего 99% мас. 4,4'-метилен- ди(циклогексилизоцианат), извлекали из линии 52 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 210°C, создавали давление в аппарате для пленочной дистилляции около 0,13 кПа, подавали раствор, полученный на стадии (13-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час, и осуществляли реакцию в течение 14 часов. Выход в расчете на 4,4'-метиленбис(циклогексиламин) составил 83%.
[Пример 14]
Стадия (14-1): Получение дибутил-4,4'-метилен-дициклогексилкарбамата
Раствор, содержащий 29,0% мас. дибутил-4,4'-метилен-дициклогексилкарбамата, получали путем осуществления такого же способа, как на стадии (12-1) Примера 12, за исключением того, что использовали 1696 г (9,8 моль) дибутилкарбоната, 315 г (1,5 моль) 4,4'-метиленбис(циклогексиламин) и 2,9 г метоксида натрия.
Стадия (14-2): Отгонка низкокипящего компонента
1409 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (14-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 84,0% мас. дибутилкарбоната и 14,9% мас. н-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии, было обнаружено, что остаток в колбе после дистилляции содержит 97,3% мас. дибутил-4,4'-метилен-дициклогексилкарбамата.
Стадия (14-3): Получение бис(2,6-диметилфенил)-4,4'-метилендициклогексилкарбамата при помощи переэтерификации
3923 г реакционной жидкости экстрагировали из линии 24 и 194 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 89 г дилаурата дибутилолова и 3447 г 2,6-диметилфенола к 595 г остатка после дистилляции, полученного на стадии (14-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 200°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 350 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 16,8% мас. бис(2,6-диметилфенил)-4,4'- метилендициклогексилкарбамата. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 97% мас. 1-бутанола.
Стадия (14-4): Получение 4,4'-метилен-ди(циклогексилизоцианат) путем термического разложения бис(2,6-диметилфенил)-4,4'-метилендициклогексилкарбамата
313 г раствора, содержащего 99% мас. 4,4'-метилен- ди(циклогексилизоцианат), извлекали из линии 52 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 210°C, создавали давление в аппарате для пленочной дистилляции около 0,13 кПа, подавали раствор, полученный на стадии (14-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 700 г/час, и осуществляли реакцию в течение 15 часов. Выход в расчете на 4,4'-метиленбис(циклогексиламин) составил 80%.
[Пример 15]
Стадия (15-1): Получение дибутил-4,4'-метилен-дициклогексилкарбамата
Раствор, содержащий 27,0% мас. дибутил-4,4'-метилен-дициклогексилкарбамата, получали путем осуществления такого же способа, как на стадии (12-1) Примера 12, за исключением того, что использовали 1705 г (9,8 моль) дибутилкарбоната, 294 г (1,4 моль) 4,4'-метиленбис(циклогексиламин) и 0,27 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.).
Стадия (15-2): Отгонка низкокипящего компонента
1643 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (15-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 87,7% мас. дибутилкарбоната и 11,7% мас. н-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 99% мас. дибутил-4,4'-метилен-дициклогексилкарбамата.
Стадия (15-3): Получение бис(2-трет-бутилфенил)-4,4'-метилендициклогексилкарбамата при помощи переэтерификации
4256 г реакционной жидкости экстрагировали из линии 24 и 181 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 84 г дилаурата дибутилолова и 3980 г 2-трет-бутилфенола к 562 г остатка после дистилляции, полученного на стадии (15-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 220°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 180 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 15,5% мас. бис(2-трет-бутилфенил)-4,4'-метилендициклогексилкарбамата. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 1-бутанола.
Стадия (15-4): Получение 4,4'-метилен-ди(циклогексилизоцианат) путем термического разложения бис(2-трет-бутилфенил)-4,4'-метилендициклогексилкарбамата
287 г раствора, содержащего 99% мас. 4,4'-метилен- ди(циклогексилизоцианат), извлекали из линии 52 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 210°C, создавали давление в аппарате для пленочной дистилляции около 0,13 кПа, подавали раствор, полученный на стадии (15-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 710 г/час, и осуществляли реакцию в течение 14 часов. Выход в расчете на 4,4'-метиленбис(циклогексиламин) составил 78%.
[Пример 16]
Стадия (16-1): Получение диметил-4,4'-метилен-дициклогексилкарбамата
Раствор, содержащий 28,2% мас. диметил-4,4'-метилен-дициклогексилкарбамата, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 1440 г (16,0 моль) диметилкарбоната, 336 г (1,6 моль) 4,4'-метиленбис(циклогексиламин) и 1,5 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.).
Стадия (16-2): Отгонка низкокипящего компонента
1271 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (16-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 91,3% мас. диметилкарбоната и 7,7% мас. метанола. Кроме того, в результате анализа методом жидкостной хроматографии, было обнаружено, что остаток в колбе после дистилляции содержит 99,4% мас. дибутил-4,4'-метилен-дициклогексилкарбамата.
Стадия (16-3): Получение бис(2,4-ди-трет-амилфенил)-4,4'-метилендициклогексилкарбамата при помощи переэтерификации
5151 г реакционной жидкости экстрагировали из линии 24 и 88 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 97 г дилаурата дибутилолова и 4645 г 2,4-ди-трет-амилфенола к 501 г остатка после дистилляции, полученного на стадии (16-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 240°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 80 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 19,5% мас. бис(2,4-ди-трет-амилфенил)-4,4'-метилен-дициклогексилкарбамата. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. метанола.
Стадия (16-4): Получение гексаметилендиизоцианата путем термического разложения бис(2,4-ди-трет-амилфенил)-4,4'-метилендициклогексилкарбамата
Осуществляли такой же способ, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 210°C, создавали давление в аппарате для пленочной дистилляции около 0,13 кПа, подавали раствор, полученный на стадии (16-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час, и осуществляли реакцию в течение 16 часов. 323 г раствора, содержащего 99% мас. 4,4'-метилен-ди(циклогексилизоцианат), извлекали из линии 52. Выход в расчете на 4,4'-метиленбис(циклогексиламин) составил 77%.
[Пример 17]
Стадия (17-1): Получение бис(3-метилбутил)ового эфира толуол-2,4-дикарбаминовой кислоты
1818 г (9,0 моль) бис(3-метилбутил)карбоната и 220 г (1,8 моль) 2,4-толуолдиамина (Aldrich Corp., USA) помещали в 5-л мерную четырехгорлую колбу, в колбу помещали мешалку и подсоединяли к колбе конденсатор Dimroth и трехходовый клапан. После замещения атмосферы внутри системы азотом четырехгорлую колбу погружали в масляную баню (OBH-24, Masuda Corp., Japan), нагретую до 80°C с последующим добавлением 0,35 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.) для запуска реакции. Образцы реакционной жидкости подходящим образом собирали и подвергали анализу ЯМР и реакцию останавливали в тот момент, когда 2,4-толуолдиамин больше нельзя было обнаружить. В результате анализа полученного раствора методом жидкостной хроматографии было обнаружено, что раствор содержит 29,7% мас. бис(3-метилбутил)ового эфира толуол-2,4-дикарбаминовой кислоты.
Стадия (17-2): Отгонка низкокипящего компонента
1422 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (17-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 78,2% мас. бис(3-метилбутил)карбоната и 21,2% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 98,0% мас. бис(3-метилбутил)ового эфира толуол-2,4-дикарбаминовой кислоты.
Стадия (17-3): Получение бис(2,4-ди-трет-амилфенил)ового эфира толуол-2,4-дикарбаминовой кислоты при помощи переэтерификации
5258 г реакционной жидкости экстрагировали из линии 24 и 289 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 109 г дилаурата дибутилолова и 4835 г 2,4-ди-трет-амилфенола к 615 г остатка после дистилляции, полученного на стадии (17-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 240°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 70 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 20,0% мас. бис(2,4-ди-трет-амилфенил)ового эфира толуол-2,4-дикарбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 98% мас. 3-метил-1-бутанола.
Стадия (17-4): Получение толуол-2,4-диизоцианата путем термического разложения бис(2,4-ди-трет-амилфенил)ового эфира толуол-2,4-дикарбаминовой кислоты
Осуществляли такой же способ, как на стадии (1-4) Примера 1, за исключением того, что нагревали аппарат 302 для пленочной дистилляции до 200°C, создавали давление в аппарате для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (17-3), в питающий резервуар 301, подавая в аппарат для пленочной дистилляции через линию 31 при скорости около 1000 г/час, и осуществляли реакцию в течение 15 часов. 267 г раствора извлекали из линии 35 и в результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. толуол-2,4-диизоцианата. Выход в расчете на 2,4-толуолдиамин составил 85%.
[Пример 18]
Стадия (18-1): Получение дибутилового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты
1774 г (10,2 моль) дибутилкарбоната и 336 г (1,7 моль) 4,4'-метилендианилина (Aldrich Corp., USA) помещали в 5-л мерную четырехгорлую колбу, в колбу помещали мешалку и подсоединяли к колбе конденсатор Dimroth и трехходовый клапан. После замещения атмосферы внутри системы азотом четырехгорлую колбу погружали в масляную баню (OBH-24, Masuda Corp., Japan), нагретую до 80°C, с последующим добавлением 3,3 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.) для запуска реакции. Образцы реакционной жидкости подходящим образом собирали и подвергали анализу ЯМР и реакцию останавливали в тот момент, когда 4,4'-метилендианилин больше нельзя было обнаружить. В результате анализа полученного раствора методом жидкостной хроматографии было обнаружено, что раствор содержит 30,8% мас. дибутилового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты.
Стадия (18-2): Отгонка низкокипящего компонента
1452 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (18-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 82,7% мас. дибутилкарбоната и 16,6% мас. 1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 98,5% мас. дибутилового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты.
Стадия (18-3): Получение бис(2,4-ди-трет-амилфенил)ового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты при помощи переэтерификации
4322 г реакционной жидкости экстрагировали из линии 24 и 226 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 103 г дилаурата дибутилолова и 3799 г 2,4-ди-трет-амилфенола к 656 г остатка после дистилляции, полученного на стадии (18-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 240°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 62 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 25,3% мас. бис(2,4-ди-трет-амилфенил)ового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 1-бутанола.
Стадия (18-4): Получение 4,4'-дифенилметандиизоцианата путем термического разложения бис(2,4-ди-трет-амилфенил)ового эфира N,N'-(4,4'-метандиилдифенил)-бискарбаминовой кислоты
Когда аппарат для пленочной дистилляции 402 нагревали до 210°C, создавали давление в аппарате для пленочной дистилляции составил около 0,1 кПа, раствор, полученный на стадии (18-3), подавали в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час, и реакцию осуществляли в течение 11 часов, 351 г раствора, содержащего 99% мас. 4,4'-дифенилметандиизоцианата, извлекали из линии 52. Выход в расчете на 4,4'-метилендианилин составил 83%.
[Пример 19]
Стадия (19-1): Получение дибутилового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты
Раствор, содержащий 26,7% мас. дибутилового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (18-1) Примера 18, за исключением того, что использовали 1583 г (9,1 моль) дибутилкарбоната, 257 г (1,3 моль) 4,4'-метилендианилина и 2,5 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.).
Стадия (19-2): Отгонка низкокипящего компонента
1342 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (19-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 85,5% мас. дибутилкарбоната и 13,6% мас. 1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 98,6% мас. дибутилового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты.
Стадия (19-3): Получение бис(2,6-диметилфенил)ового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты при помощи переэтерификации
2824 г реакционной жидкости экстрагировали из линии 24 и 160 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 74 г дилаурата дибутилолова и 2441 г 2,6-диметилфенола к 475 г остатка после дистилляции, полученного на стадии (19-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 200°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 662 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 18,9% мас. бис(2,6-диметилфенил)ового эфира толуол-2,4-дикарбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 1-бутанола.
Стадия (19-4): Получение 4,4'-дифенилметандиизоцианата путем термического разложения бис(2,6-диметилфенил)ового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты
244 г раствора, содержащего 99% мас. 4,4'-дифенилметандиизоцианата, извлекали из линии 52 путем осуществления такого же способа, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 210°C, создавали давление в аппарате для пленочной дистилляции около 0,1 кПа, подавали раствор, полученный на стадии (19-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 700 г/час, и осуществляли реакцию в течение 13 часов. Выход в расчете на 4,4'-метилендианилин составил 75%.
[Пример 20]
Стадия (20-1): Получение дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты
Раствор, содержащий 22,7% мас. дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 2192 г (12,6 моль) дибутилкарбонатаа, 209 г (1,8 моль) гексаметилендиамина и 3,5 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.).
Стадия (20-2): Отгонка низкокипящего компонента
1845 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (20-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии, было обнаружено, что дистиллят содержит 85,9% мас. дибутилкарбоната и 13,6% мас. 1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии, было обнаружено, что остаток в колбе после дистилляции содержит 98,6% мас. дибутилового эфира N,N'-(4,4'-метандиил-дифенил)-бискарбаминовой кислоты.
Стадия (20-3): Получение ди(2,6-ди-трет-бутилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
5395 г реакционной жидкости экстрагировали из линии 24 и 206 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что получали гомогенный раствор 550 г остатка после дистилляции, полученного на стадии (20-3), 109 г дилаурата дибутилолова и 4950 г 2,6-ди-трет-бутилфенола, нагревали аппарат 202 для пленочной дистилляции до 240°C и осуществляли реакцию в течение 86 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 14,9% мас. ди(2,6-ди-трет-бутилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 1-бутанола.
Стадия (20-4): Получение гексаметилендиизоцианата путем термического разложения ди(2,6-ди-трет-бутилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Осуществляли такой же способ, как на стадии (1-4) Примера 1, за исключением того, что нагревали аппарат 302 для пленочной дистилляции до 200°C, создавали давление в аппарате для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (20-3) в питающий резервуар 301, подавая в аппарат для пленочной дистилляции через линию 31 при скорости около 980 г/час, и осуществляли реакцию в течение 13 часов. 210 г раствора извлекали из линии 35. В результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 70%.
[Пример 21]
Стадия (21-1): Получение ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Раствор, содержащий 24,0% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 2290 г (11,3 моль) бис(3-метилбутил)карбоната, 208,8 г (1,8 моль) гексаметилендиамина и 3,5 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.).
Стадия (21-2): Отгонка низкокипящего компонента
1891 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (21-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 83,6% мас. бис(3-метилбутил)карбонат и 16,1% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 98,6% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (21-3): Получение ди(2-фенилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
3977 г реакционной жидкости экстрагировали из линии 24 и 276 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 110 г дилаурата дибутилолова и 3545 г 2-фенилфенола к 606 г остатка после дистилляции, полученного на стадии (21-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 240°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 80 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 20,0% мас. ди(2-фенилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 3-метил-1-бутанола.
Стадия (21-4): Получение гексаметилендиизоцианата путем термического разложения ди(2-фенилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
241 г раствора извлекали из линии 35 путем осуществления такого же способа, как на стадии (1-4) Примера 1, за исключением того, что нагревали аппарат 302 для пленочной дистилляции до 200°C, создавали давление в аппарате для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (21-3), в питающий резервуар 301, подавая в аппарат для пленочной дистилляции через линию 31 при скорости около 980 г/час, и осуществляли реакцию в течение 13 часов. В результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 80%.
[Пример 22]
Стадия (22-1): Получение ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Раствор, содержащий 23,8% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 2163 г (10,7 моль) бис(3-метилбутил)карбоната, 197 г (1,7 моль) гексаметилендиамина и 3,3 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.).
Стадия (22-2): Отгонка низкокипящего компонента
1783 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (22-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 83,5% мас. бис(3-метилбутил)карбоната и 16,0% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 97,1% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (22-3): Получение ди(2,4-бис(α,α-диметилбензил)фенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
4689 г реакционной жидкости экстрагировали из линии 24 и 259 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что добавляли 103 г дилаурата дибутилолова и 4385 г 2,4-бис(α,α-диметилбензил)фенола (Aldrich Corp., USA) к 575 г остатка после дистилляции, полученного на стадии (22-2), и использовали в форме гомогенного раствора, нагревали аппарат 202 для пленочной дистилляции до 240°C, заменяя атмосферу внутри аппарата для пленочной дистилляции азотом при атмосферном давлении, подавая раствор в аппарат для пленочной дистилляции через линию подачи 21 при скорости около 1200 г/час, и осуществляли реакцию в течение 80 часов.
Когда реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 25,8% мас. ди(2,4-бис(α,α-диметилбензил)фенилового эфира N,N'-гександиил-бис-карбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. 3-метил-1-бутанола.
Стадия (22-4): Получение гексаметилендиизоцианата путем термического разложения ди(2,4-бис(αα-диметилбензил)фенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
220 г раствора извлекали из линии 35 путем осуществления такого же способа, как на стадии (1-4) Примера 1, за исключением того, что нагревали аппарат 302 для пленочной дистилляции до 200°C, создавали давление в аппарате для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (22-3), в питающий резервуар 301, подавая в аппарат для пленочной дистилляции через линию 31 при скорости около 980 г/час, и осуществляли реакцию в течение 18 часов. В результате анализа раствора методом 1H- и 13C-ЯМР было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 77%.
[Пример 23]
Гексаметилендиизоцианат получали в реакционном устройстве, аналогичном показанному на Фиг. 5.
Стадия (23-1): Способ получения ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Смесительный резервуар 601 (внутренний объем: 5 л) нагревали до 80°C. Бис(3-метилбутил)карбонат переносили в смесительный резервуар 601 из линии 60 при скорости 678 г/час, при этом линия 62 была закрыта, и смешанный раствор гексаметилендиамина, 3-метил-1-бутанола и метоксида натрия (28% раствор в метаноле) (смешиваемое отношение: гексаметилендиамин 50 частей/3-метил-1-бутанол 50 частей/метоксид натрия 0,42 части) одновременно переносили из линии 61 при скорости 112 г/час. Через 4 часа линию 62 окрывали при закрытой линии 63 и начинали перенос реакционной жидкости в резервуар 602 при скорости 790 г/час. Температуру в линии 62 поддерживали при 80°C для предотвращения осаждения твердых частиц из реакционной жидкости.
Когда реакционную жидкость, перенесенную на линию 602, анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 20,3% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (23-2): Способ отгонки низкокипящего компонента
Аппарат для пленочной дистилляции 603 (теплопроводящая площадь поверхности 0,2 м2, Kobelco Eco-Solutions Co., Ltd., Japan) нагревали до 150°C и создавали давление внутри устройство около 0,02 кПа.
Раствор, который хранили в резервуаре 602, передавали в аппарат для пленочной дистилляции 603 из линии 63 при скорости 790 г/час, где низкокипящий компонент, содержащийся в растворе, отгоняли. Низкокипящий компонент, который был отогнан, экстрагировали из аппарата 603 для пленочной дистилляции через линию 64. С другой стороны, высококипящий компонент экстрагировали из аппарата 603 для пленочной дистилляции через линию 65, где поддерживали температуру 150°C, и передавали в смесительный резервуар 604, где поддерживали температуру 120°C. В то же время 2,4-ди-трет-амилфенол переносили через линию 66 в смесительный резервуар 604 при скорости 1306 г/час и дилаурат дибутилолова передавали в смесительный резервуар 604 через линию 67 при скорости 29 г/час.
Жидкую смесь, полученную в смесительном резервуаре 604, переносили в резервуар 605 через линию 68, при этом линия 69 была закрыта, и хранили в резервуаре 605. Когда раствор, который хранили в резервуаре 605, анализировали методом жидкостной хроматографии, было обнаружено, что раствор содержит 10,7% мас. ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (23-3): Способ получения ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
Аппарат для пленочной дистилляции 606 (теплопроводящая площадь поверхности 0,2 м2, Kobelco Eco-Solutions Co., Ltd., Japan) нагревали до 240°C.
Реакцию переэтерификации осуществляли путем переноса жидкой смеси ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты, 2,4-ди-трет-амилфенола и дилаурата дибутилолова, которую хранили в резервуаре 605, в аппарат 606 для пленочной дистилляции через линию 69 при скорости 1496 г/час, при этом линия 72 была закрыта. Газообразную смесь, содержащую 3-метил-1-бутанол и 2,4-ди-трет-амилфенол, экстрагировали из линии 73, обеспечиваемой в верхней части аппарата 606 для пленочной дистилляции, и подавали в дистилляционную колонну 607. 3-Метил-1-бутанол и 2,4-ди-трет-амилфенол разделяли в дистилляционной колонне 607 и 2,4-ди-трет-амилфенол возвращали в верхнюю часть аппарата 606 для пленочной дистилляции через линию 74, обеспечиваемую в нижней части дистилляционной колонны 607. Реакционную жидкость экстрагировали из линии 70, обеспечиваемой в нижней части аппарата 606 для пленочной дистилляции, и подавали в аппарат 606 для пленочной дистилляции через линию 71. Когда содержание ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты в реакционной жидкости, экстрагируемой из линии 70, достигало 20,3% мас., линию 72 окрывали, при закрытии линии 75, и реакционную жидкость переносили в резервуар 608.
Стадия (23-4): Способ получения гексаметилендиизоцианата путем термического разложения ди(2,4-ди-трет-амилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Раствор, который хранили в резервуаре 608, подавали в аппарат 609 для пленочной дистилляции (теплопроводящая площадь поверхности 0,2 м2, Kobelco Eco-Solutions Co., Ltd., Japan), нагретый до 200°C и установленный на внутреннее давление около 1,3 кПа, через линию 75 при скорости 1395 г/час. Газообразный компонент, содержащий гексаметилендиизоцианат, экстрагировали из линии 77, обеспечиваемой в верхней части аппарата 609 для пленочной дистилляции, и подавали в дистилляционную колонну 610. Дистилляционное разделение осуществляли в дистилляционной колонне 610 и гексаметилендиизоцианат извлекали из линии 79 при скорости 72 г/час.
[Сравнительный Пример 1]
Стадия (A-1): Получение диметилового эфира N,N'-гександиил-бис-карбаминовой кислоты
1044 г раствора, содержащего 29,6% мас. метилового эфира N,N-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 882 г (9,8 моль) диметилкарбоната и 162 г (1,4 моль) гексаметилендиамина, добавляли мешалку и использовали 2,7 г метоксида натрия.
Стадия (A-2): Отгонка низкокипящего компонента
729 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (A-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 87,5% мас. диметилкарбоната и 11,7% мас. метанола. В результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 98,2% мас. диметилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (A-3): Получение дифенилового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
4101 г реакционной жидкости экстрагировали из линии 24 и 65 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 316 г остатка после дистилляции, полученного на стадии (A-2), вместо остатка после дистилляции, полученного на стадии (1-2), использовали 85 г дилаурата дибутилолова и 3770 г фенола (для нуклеиновокислотной экстракции, Wako Pure Chemical Industries, Ltd., Japan), устанавливая нагревательное устройство аппарата для пленочной дистилляции на 180°C, и осуществляли реакцию в течение 430 часов. Экстрагированная реакционная жидкость содержала 8,7% мас. дифенилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (A-4): Получение путем термического разложения дифенилового эфира N,N'-гександиил-бис-карбаминовой кислоты
Осуществляли такой же способ, как на стадии (5-4) Примера 5, за исключением того, что нагревали аппарат 402 для пленочной дистилляции до 200°C, создавали давление внутри аппарата для пленочной дистилляции около 1,3 кПа, подавали раствор, полученный на стадии (A-3), в питающий резервуар 401, подавая в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час, и осуществляли реакцию в течение 11 часов. 134 г раствора, содержащего 99% мас. гексаметилендиизоцианата, извлекали из линии 47. Выход в расчете на гексаметилендиамин составил 57%. Кроме того, после завершения стадии (A-4) наблюдали прилипание черного твердого вещества к боковым стенкам аппарата 402 для пленочной дистилляции.
[Сравнительный Пример 2]
Стадия (B-1): Получение ди(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
2146 г раствора, содержащего 23,1% мас. ди(3-метилбутил)ового эфира N,N-гександиил-бис-карбаминовой кислоты, получали путем осуществления такого же способа, как на стадии (1-1) Примера 1, за исключением того, что использовали 1970 г (9,8 моль) бис(3-метилбутил)карбоната и 174 г (1,5 моль) гексаметилендиамина, добавляли мешалку и использовали 2,9 г метоксида натрия.
Стадия (B-2): Отгонка низкокипящего компонента
1631 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (B-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 84,2% мас. бис(3-метилбутил)карбоната и 15,4% мас. 3-метил-1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 96,7% мас. бис(3-метилбутил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (B-3): Получение ди(4-метилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
4978 г реакционной жидкости экстрагировали из линии 24 и 185 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 510 г остатка после дистилляции, полученного на стадии (B-2), вместо остатка после дистилляции, полученного на стадии (1-2), использовали 91 г дилаурата дибутилолова и 4645 г 4-метилфенола (Aldrich Corp., USA), и осуществляли реакцию в течение 58 часов. Экстрагированная реакционная жидкость содержала 8,1% мас. ди(4-метилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (B-4): Получение гексаметилендиизоцианата путем термического разложения ди(4-метилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Реакцию термического разложения осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 4.
Аппарат для пленочной дистилляции 402 (Kobelco Eco-Solutions Co., Ltd., Japan), имеющий теплопроводящую площадь поверхности 0,2 м2, нагревали до 200°C и создавали давление внутри аппарата для пленочной дистилляции около 1,3 кПа. Раствор, полученный на стадии (B-3), подавали в питающий резервуар 401 и подавали в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час. Жидкий компонент экстрагировали из линии 43, обеспечиваемой в нижней части аппарата для пленочной дистилляции 402, и возвращали в питающий резервуар 401 через линию 44. Газообразный компонент, содержащий гексаметилендиизоцианат и 4-метилфенол, экстрагировали из линии 42, обеспечиваемой в верхней части аппарата для пленочной дистилляции 402. Газообразный компонент подавали в дистилляционную колонну 403, где осуществляли разделение гексаметилендиизоцианата и 4-метилфенола, 4-метилфенол экстрагировали из линии 45, соединенной с верхней частью дистилляционной колонны 403, гексаметилендиизоцианат экстрагировали из линии 47, обеспечиваемой в средней части дистилляционной колонны 403, высококипящее вещество экстрагировали из линии 46, обеспечиваемой в нижней части дистилляционной колонны 403, и часть возвращали в питающий резервуар 401 через линию 44. Когда реакцию осуществляли в течение 11 часов, 114 г раствора, содержащего 99% мас. гексаметилендиизоцианата, извлекали из линии 47. Выход в расчете на гексаметилендиамин составил 57%. Кроме того, после завершения стадии (В-4) наблюдали прилипание черного твердого вещества к боковым стенкам аппарата 402 для пленочной дистилляции.
[Сравнительный Пример 3]
Стадия (C-1): Получение дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты
1818 г (10,5 моль) дибутилкарбоната, полученного с использованием способа Ссылочного Примера 2, и 220 г (1,9 моль) гексаметилендиамина помещали в 5-л мерную четырехгорлую колбу, в колбу помещали мешалку и подсоединяли к колбе конденсатор Dimroth и трехходовый клапан. После замещения атмосферы внутри системы азотом четырехгорлую колбу погружали в масляную баню (OBH-24, Masuda Corp.), нагретую до 80°C, с последующим добавлением 3,7 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.) для запуска реакции. Образцы реакционной жидкости подходящим образом собирали и подвергали анализу ЯМР, и реакцию останавливали в тот момент, когда больше не определяли присутствия гексаметилендиамина. В результате анализа полученного раствора методом жидкостной хроматографии было обнаружено, что раствор содержит 28,3% мас. дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (C-2): Отгонка низкокипящего компонента
1444 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (C-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 80,9% мас. дибутилкарбоната и 18,6% мас. 1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии, было обнаружено, что остаток в колбе после дистилляции содержит 98,0% мас. дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (C-3): Получение ди(4-октилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
6122 г реакционной жидкости экстрагировали из линии 24 и 182 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 594 г остатка после дистилляции, полученного на стадии (6-3), вместо остатка после дистилляции, полученного на стадии (1-2), добавляли 114 г дилаурата дибутилолова и 5611 г 4-октилфенола и использовали в форме гомогенного раствора, подавали в питающий резервуар 201, нагревали аппарат для пленочной дистилляции 202, имеющий теплопроводящую площадь поверхности 0,2 м2, до 240°C и осуществляли реакцию в течение 86 часов.
Когда экстрагированную реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 11,5% мас. ди(4-октилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99,0% мас. 1-бутанола.
Стадия (C-4): Получение гексаметилендиизоцианата путем термического разложения ди(4-октилфенил)ового эфира N,N'-гександиил-бис-карбаминовой кислоты
Реакцию термического разложения осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 3.
Аппарат 302 для пленочной дистилляции, имеющий теплопроводящую площадь поверхности of 0,2 м2, нагревали до 200°C и создавали давление внутри аппарата для пленочной дистилляции около 1,3 кПа. Раствор, полученный на стадии (C-3), подавали в питающий резервуар 301 и подавали в аппарат для пленочной дистилляции через линию 31 при скорости около 980 г/час. Жидкий компонент экстрагировали из линии 33, обеспечиваемой в нижней части аппарата 302 для пленочной дистилляции, и возвращали в питающий резервуар 301 через линию 34. Газообразный компонент, содержащий гексаметилендиизоцианат и 4-октилфенол, экстрагировали из линии 32, обеспечиваемой в верхней части аппарата 302 для пленочной дистилляции. Газообразный компонент подавали в дистилляционную колонну 303, где осуществляли разделение гексаметилендиизоцианата и 4-октилфенола, и часть 4-октилфенола возвращали в питающий резервуар 301 по линии 34 через линию 36, обеспечиваемую в нижней части дистилляционной колонны 303. Когда реакцию осуществляли в течение 13 часов, 167 г раствора извлекали из линии 35, и в результате анализа раствора методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99% мас. гексаметилендиизоцианата. Выход в расчете на гексаметилендиамин составил 53%. Кроме того, после завершения стадии (С-4) наблюдали прилипание черного твердого вещества к боковым стенкам аппарата 302 для пленочной дистилляции.
[Сравнительный Пример 4]
Стадия (D-1): Получение дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты
1914 г (11,0 моль) дибутилкарбоната, полученного с использованием способа Ссылочного Примера 2, и 232 г (2,0 моль) гексаметилендиамина помещали в 5-л мерную четырехгорлую колбу, в колбу помещали мешалку и подсоединяли к колбе конденсатор Dimroth и трехходовый клапан. После замещения атмосферы внутри системы азотом четырехгорлую колбу погружали в масляную баню, нагретую до 80°C, с последующим добавлением 0,37 г метоксида натрия (28% раствор в метаноле, Wako Pure Chemical Industries, Ltd.) для запуска реакции. Образцы реакционной жидкости подходящим образом собирали и подвергали анализу ЯМР, и реакцию останавливали в тот момент, когда больше не определяли присутствия гексаметилендиамина. В результате анализа полученного раствора методом жидкостной хроматографии было обнаружено, что раствор содержит 28,3% мас. дибутилового эфира N,N'- гександиил-бис-карбаминовой кислоты.
Стадия (D-2): Отгонка низкокипящего компонента
1532 г дистиллята получали путем осуществления такого же способа, как на стадии (1-2) Примера 1, за исключением того, что использовали раствор, полученный на стадии (D-1), вместо раствора, полученного на стадии (1-1). В результате анализа методом газовой хроматографии было обнаружено, что дистиллят содержит 80,9% мас. дибутилкарбоната и 18,5% мас. 1-бутанола. Кроме того, в результате анализа методом жидкостной хроматографии было обнаружено, что остаток в колбе после дистилляции содержит 99,5% мас. дибутилового эфира N,N'-гександиил-бис-карбаминовой кислоты.
Стадия (D-3): Получение ди(3-октилокси)эфира N,N'-гександиил-бис-карбаминовой кислоты при помощи переэтерификации
4203 г реакционной жидкости экстрагировали из линии 24 и 250 г раствора извлекали из линии 27, обеспечиваемой в верхней части дистилляционной колонны 203, путем осуществления такого же способа, как на стадии (1-3) Примера 1, за исключением того, что использовали 605 г остатка после дистилляции, полученного на стадии (6-3), вместо остатка после дистилляции, полученного на стадии (1-2), добавляли 120 г дилаурата дибутилолова и 3727 г 3-октанола (Aldrich Corp., USA) и использовали в форме гомогенного раствора, подавали в питающий резервуар 201, нагревали аппарат 202 для пленочной дистилляции, имеющий теплопроводящую площадь поверхности 0,2 м2, до 175°C и осуществляли реакцию в течение 180 часов.
Когда экстрагированную реакционную жидкость анализировали методом жидкостной хроматографии, было обнаружено, что реакционная жидкость содержит 17,1% мас. ди(3-октилокси)эфира N,N'-гександиил-бис-карбаминовой кислоты. Кроме того, когда раствор, извлеченный из линии 27, анализировали методом 1H- и 13C-ЯМР, было обнаружено, что раствор содержит 99,0% мас. 1-бутанола.
Стадия (D-4): Получение гексаметилендиизоцианата путем термического разложения ди(3-октилокси)эфира N,N'-гександиил-бис-карбаминовой кислоты
Реакцию термического разложения осуществляли в реакционном устройстве, аналогичном показанному на Фиг. 4.
Аппарат 402 для пленочной дистилляции (Kobelco Eco-Solutions Co., Ltd., Japan), имеющий теплопроводящую площадь поверхности 0,2 м2, нагревали до 200°C и создавали давление внутри аппарата для пленочной дистилляции около 1,3 кПа. Раствор, полученный на стадии (D-3), подавали в питающий резервуар 401 и подавали в аппарат для пленочной дистилляции через линию 41 при скорости около 680 г/час. Жидкий компонент экстрагировали из линии 43, обеспечиваемой в нижней части аппарата для пленочной дистилляции 402, и возвращали в питающий резервуар 401 через линию 44. Газообразный компонент, содержащий гексаметилендиизоцианат и 3-октанол, экстрагировали из линии 42, обеспечиваемой в верхней части аппарата 402 для пленочной дистилляции. Газообразный компонент подавали в дистилляционную колонну 403, где осуществляли разделение гексаметилендиизоцианата и 3-октанола, 3-октанол экстрагировали из линии 45, соединенной с верней частью дистилляционной колонны 403, гексаметилендиизоцианат экстрагировали из линии 47, обеспечиваемой в средней части дистилляционной колонны 403, высококипящее вещество экстрагировали из линии 46, обеспечиваемой в нижней части дистилляционной колонны 403, и часть возвращали в питающий резервуар 401 через линию 44. Когда реакцию осуществляли в течение 11 часов, 149 г раствора, содержащего 99% мас. гексаметилендиизоцианата, извлекали из линии 47. Выход в расчете на гексаметилендиамин составил 45%. Кроме того, после завершения стадии (D-4) наблюдали прилипание черного твердого вещества к боковым стенкам аппарата 402 для пленочной дистилляции.
Промышленная применимость
Поскольку способ получения изоцианата в соответствии с настоящим изобретением делает возможным эффективное получение изоцианатов без использования чрезвычайно токсичного фосгена, способ получения по настоящему изобретению является чрезвычайно промышленно полезным и обладает высокой коммерческой ценностью.
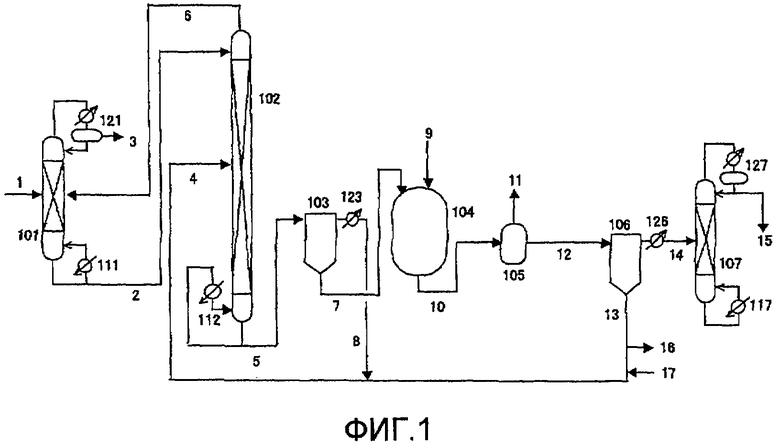
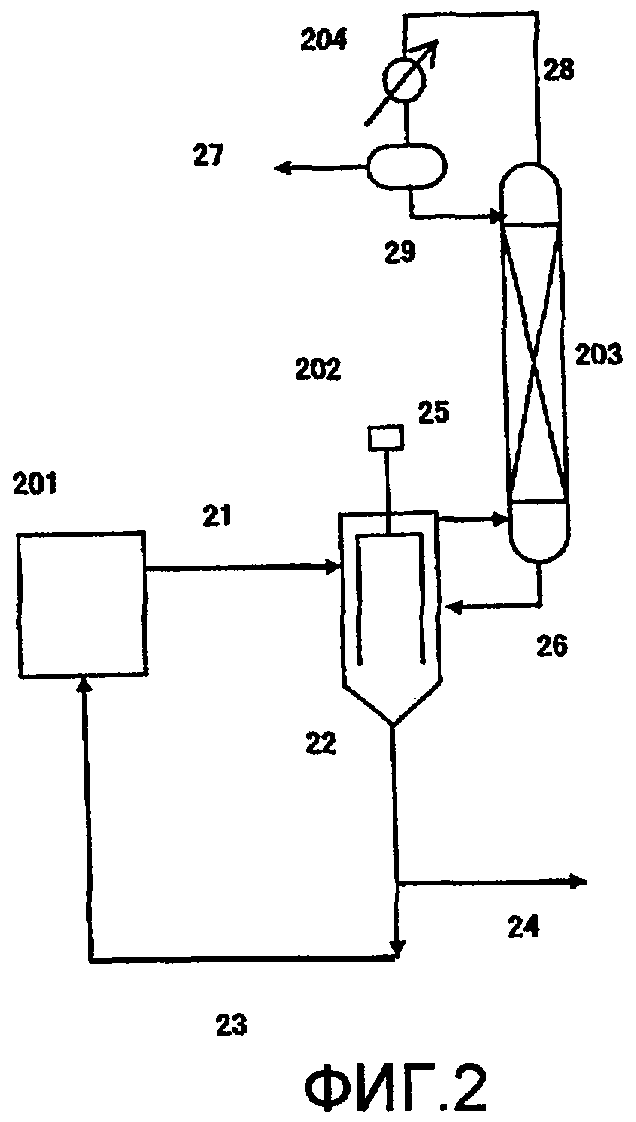
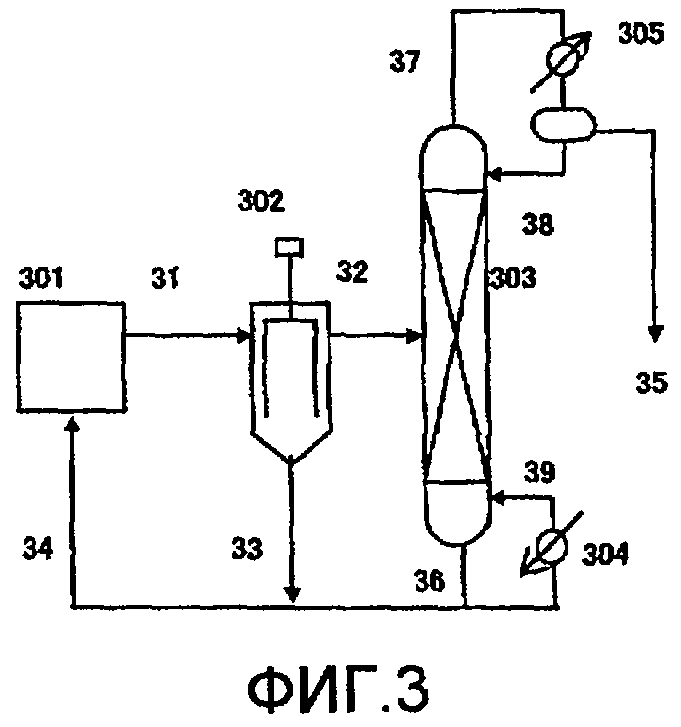
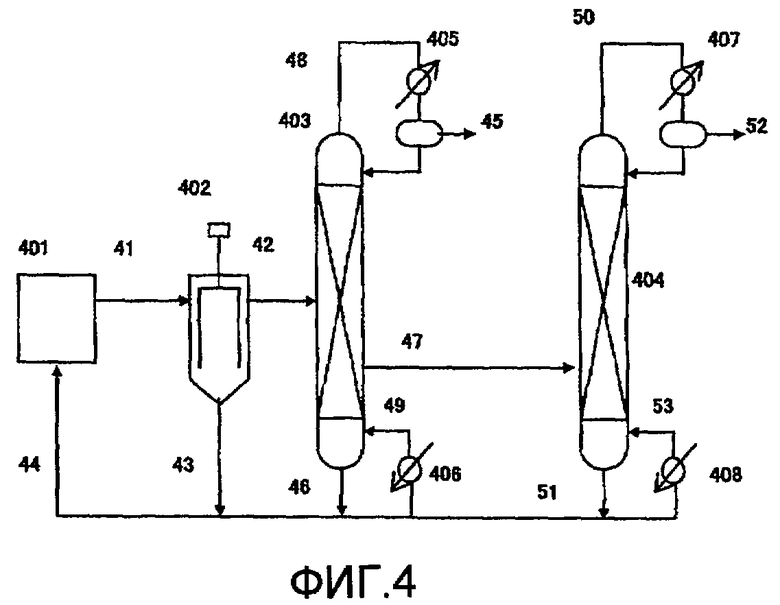
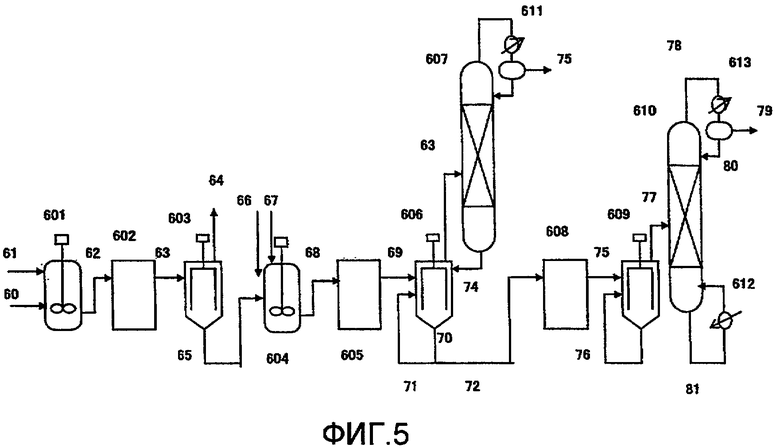
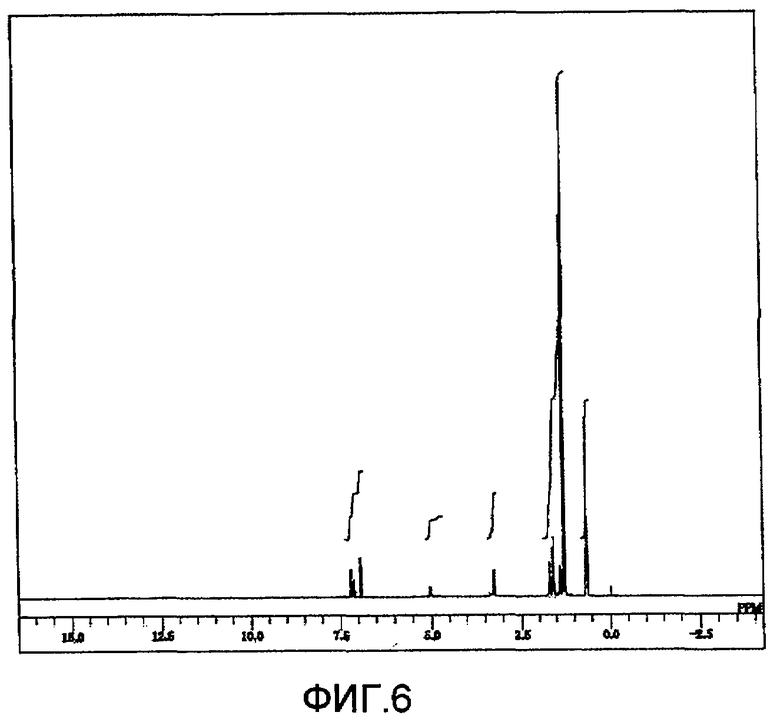
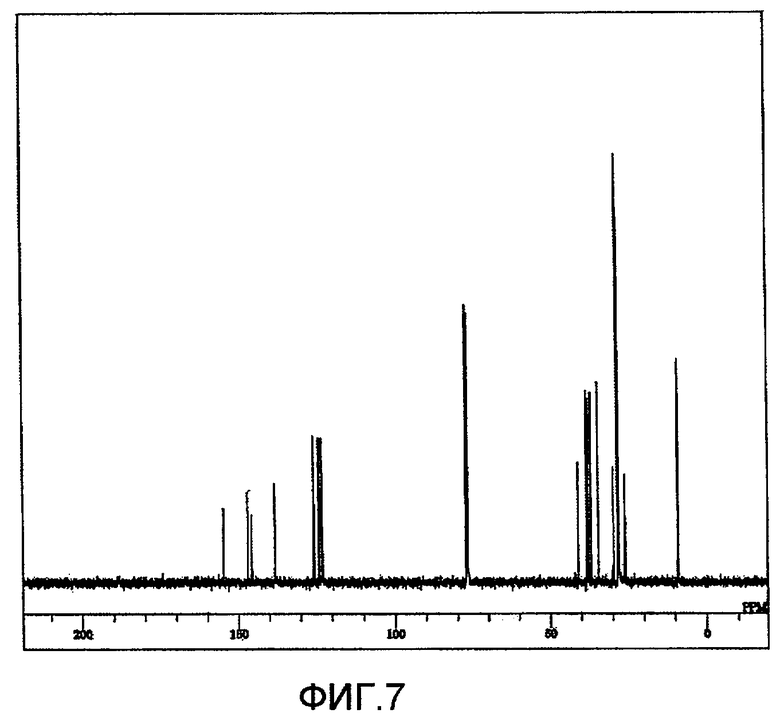
Изобретение относится к способу получения изоцианатов без использования токсичного фосгена. Способ включает следующие стадии: взаимодействие алифатического сложного эфира карбаминовой кислоты формулы
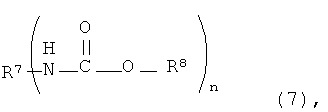
где R7 представляет собой группу, выбранную из линейной алкильной группы, содержащей от 1 до 20 атомов углерода, бис(С5-С6-циклоалкил)алкильной группы, содержащей от 11 до 20 атомов углерода, С5-С6-циклоалкилалкильной группы, содержащей от 6 до 20 атомов углерода, причем циклоалкильное кольцо может быть замещено 1-3 заместителями, выбранными из алкила, содержащего от 1 до 8 атомов углерода; и ароматической группы, содержащей от 6 до 20 атомов углерода и имеющей 1 или 2 бензольных кольца; при этом R7 имеет валентность n, R8 представляет собой алкильную группу, содержащую от 1 до 8 атомов углерода, n равно 2, с ароматическим гидроксисоединением формулы
 ,
,
которое содержит заместитель R1, по меньшей мере, в одном ортоположении гидроксильной группы, где кольцо А представляет собой бензольное кольцо, которое может содержать дополнительный заместитель R2 в другом ортоположении по отношению к гидроксигруппе; R1 представляет собой группу, отличную от атома водорода, в форме алкильной группы, содержащей от 1 до 20 атомов углерода; фенильной или фенилалкильной группы, содержащей от 7 до 20 атомов углерода, при этом дополнительный заместитель R2 выбирается из водорода, алкильной группы, содержащей от 1 до 20 атомов углерода; фенильной группы или фенилалкильной группы, содержащей от 7 до 20 атомов углерода; с получением соответствующего арилкарбамата, содержащего группу, происходящую из ароматического гидроксисоединения; и соответствующего алифатического спирта; и реакцию термического разложения арилкарбамата с образованием соответствующего изоцианата и ароматического гидроксисоединения. Способ позволяет увеличить выход изоцианатов и снизить количество побочных продуктов, накапливающихся внутри технологического оборудования. 8 з.п. ф-лы, 1 табл., 7 ил.
1. Способ получения изоцианата, включающий следующие стадии: взаимодействие алифатического сложного эфира карбаминовой кислоты формулы (7):
где R7 представляет собой группу, выбранную из линейной алкильной группы, содержащей от 1 до 20 атомов углерода, бис(С5-С6-циклоалкил)алкильной группы, содержащей от 11 до 20 атомов углерода, С5-С6-циклоалкилалкильной группы, содержащей от 6 до 20 атомов углерода, причем циклоалкильное кольцо может быть замещено 1-3 заместителями, выбранными из алкила, содержащего от 1 до 8 атомов углерода; и ароматической группы, содержащей от 6 до 20 атомов углерода и имеющей 1 или 2 бензольных кольца, причем бензольное кольцо может быть замещено 1-3 заместителями, выбранными из алкила, содержащего от 1 до 8 атомов углерода; при этом R7 имеет валентность n,
R8 представляет собой алкильную группу, содержащую от 1 до 8 атомов углерода, и
n представляет собой целое число, имеющее значение 2,
с ароматическим гидроксисоединением, где ароматическое гидроксисоединение представляет собой ароматическое гидроксисоединение, которое представлено следующей формулой (I):
и которое содержит заместитель R1, по меньшей мере, в одном орто положении гидроксильной группы,
где кольцо А представляет собой бензольное кольцо, которое может содержать дополнительный заместитель R2 в другом орто положении по отношению к гидрокси группе;
R1 представляет собой группу, отличную от атома водорода, в форме алкильной группы, содержащей от 1 до 20 атомов углерода; фенильной или фенилалкильной группы, содержащей от 7 до 20 атомов углерода, при этом дополнительный заместитель R2 выбирается из водорода, алкильной группы, содержащей от 1 до 20 атомов углерода; фенильной группы или фенилалкильной группы, содержащей от 7 до 20 атомов углерода; при этом кольцо А также может быть замещено другими дополнительными заместителями, выбранными из водорода, алкильной группы, содержащей от 1 до 20 атомов углерода; фенильной группы или фенилалкильной группы, содержащей от 7 до 20 атомов углерода;
с получением соответствующего арилкарбамата, содержащего группу, происходящую из ароматического гидроксисоединения; и соответствующего алифатического спирта; и
реакцию термического разложения арилкарбамата с образованием соответствующего изоцианата и ароматического гидрокси соединения.
2. Способ по п.1, где ароматическое гидроксисоединение представляет собой соединение, представленное следующей формулой (2):

где кольцо A, R1 и R2 имеют значения, определенные выше.
3. Способ по п.2, где в формуле (2), общее количество атомов углерода, образующих R1 и R2, составляет от 2 до 20.
4. Способ по п.1, где ароматическое гидроксисоединение представляет собой соединение, представленное следующей формулой (3):

где R1 и R2 имеют значения, определенные выше,
R3 и R5 представляют собой атом водорода,
R4 представляет собой атом водорода, алкильную группу, содержащую от 1 до 20 атомов углерода, фенильную или фенилалкильную группу, содержащую от 7 до 20 атомов углерода.
5. Способ по п.4, где ароматическое гидроксисоединение представляет собой такое соединение, где в формуле (3) каждый из R1 и R4 независимо представляет собой группу, представленную следующей формулой (4):
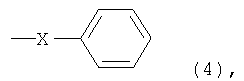
и R2, R3 и R5 представляют собой атом водорода,
Х представляет собой разветвленную структуру, выбранную из структур, представленных следующими формулами (5) и (6):
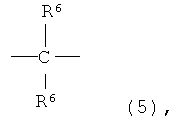
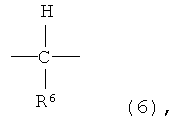
где R6 представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода.
6. Способ по п.4, где ароматическое гидроксисоединение представляет собой такое соединение, где в формуле (3) R1 представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 8 атомов углерода, и каждый из R2 и R4 независимо представляет собой атом водорода или линейную или разветвленную алкильную группу, содержащую от 1 до 8 атомов углерода.
7. Способ по п.1, отличающийся тем, что в результате реакции алифатического сложного эфира карбаминовой кислоты и ароматического гидроксисоединения получают низкокипящий алифатический спирт.
8. Способ по п.1, где по меньшей мере одно соединение из изоцианата и ароматического гидроксисоединения, которые образуются в результате реакции термического разложения арилкарбамата, выделяют в форме газообразного компонента.
9. Способ по п.1, где алифатический сложный эфир карбаминовой кислоты представляет собой, по меньшей мере, одно соединение, выбранное из группы, состоящей из соединений, представленных следующими формулами (8), (9) и (10):

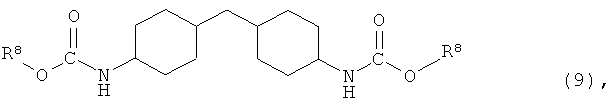
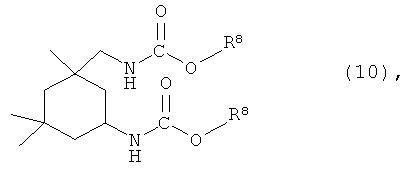
где R8 имеет значения, определенные выше.
| US 3992430, 16.11.1976 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Cs | |||
| KOSA et al, New combined phenol-hindered amine stabilizers for polymers based on diphenylmethane-4,4'-diisocyanate and dicyclohexylmethane-4,4'-diisocyanate, POLYMER DEGRADATION AND STABILITY, 2004, 86(3), 391-400 | |||
| W.D.HABICHER et al, Synthesis and antioxidative | |||

