ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к гетероциклическому соединению, содержащему бета-аминогруппу, которое обладает превосходной ингибиторной активностью в отношении дипептидилпептидазы-IV (в дальнейшем в настоящем документе обозначаемой как "DPP-IV") и высокой биодоступностью, и к фармацевтической композиции, содержащей указанное гетероциклическое соединение или его фармацевтически приемлемую соль в качестве активного ингредиента.
УРОВЕНЬ ТЕХНИКИ
Фермент дипептидилпептидаза IV, сокращаемый в настоящем документе как DPP-IV (и в других документах как DP-IV, DP-4 или DAP-IV) а также по классификации известный как EC. 3.4.14.5, представляет собой сериновую протеазу (Barrett A. J. et al., Arch. Biochem. Biophys., 1995, 247-250), которая отщепляет N-концевой дипептид от пептидов, которые начинаются с последовательности H-Xaa-Pro-Y или H-Xaa-Ala-Y, где Xaa представляет собой любую липофильную аминокислоту, Pro представляет собой пролин и Ala представляет собой аланин (Heins J., et al., Biochim. et Biophys. Acta 1988, 161). DPP-IV широко распространен и встречается в различных тканях млекопитающих, таких как почка, печень и тонкий кишечник (Hegen M. et al., J. Immunol, 1990, 2908-2914). Впервые DPP-IV был идентифицирован как мембраносвязанный белок. Позднее была идентифицирована растворимая форма (Duke-Cohan J. S. et al., J. Biol. Chem., 1995, 14107-14114). Согласно недавно опубликованному исследованию и отчету, было выявлено, что такая растворимая форма DPP-IV обладает той же структурой и функцией, что и мембраносвязанная форма фермента, и она находится в крови без определенного мембраносвязанного домена (Christine D. et al., Eur. J. Biochem., 2000, 5608-5613).
Первоначальный интерес к DPP-IV был сфокусирован на его роли в активации T-лимфоцитов. DPP-IV, ответственный за активацию T-лимфоцитов, был конкретно обозначен как CD26. В отчете, в котором показано, что CD26 связывается или взаимодействует с вирусом иммунодефицита человека (HIV) (Guteil W. G. et al., Proc. Natl. Acad. Sci., 1994, 6594-6598), было сделано предположение, что ингибиторы DPP-IV могут быть пригодны для лечения СПИД (Doreen M. A. et al., Bioorg. Med. Chem. Lett., 1996, 2745-2748).
В дополнение к ключевой роли участия в иммунной системе основная функция DPP-IV связана с его пептидолитической активностью, как описано выше. В частности, роли DPP-IV уделяется внимание, поскольку было выявлено, что DPP-IV является ключевым ферментом, вовлеченным в деградацию глюкагон-подобного белка-1 (в дальнейшем в настоящем документе обозначаемого как "GLP-1") в тонком кишечнике (Mentlein R. et al., Eur. J. Biochem., 1993, 829-835). GLP-1 представляет собой пептидный гормон из 30 аминокислот, который секретируется L-клетками кишечника в качестве ответа тонкого кишечника на прием пищи (Goke R. et al., J. Biol. Chem., 1993, 19650-19655). Поскольку известно, что GLP-1 оказывает усиливающие эффекты на действие инсулина в отношении контроля уровней глюкозы в крови после приема пищи (Hoist J. J. et al., Diabetes Care, 1996, 580-586), было предположено, что ингибиторы DPP-IV также могут быть подходящим образом использованы для лечения диабета 2 типа. Исходя из этого предположения, была разработана ранняя форма ингибитора DPP-IV, и в некоторых отчетах была продемонстрирована терапевтическая эффективность лекарственного средства в экспериментах на животных (Pauly R. P. et al., Metabolism, 1999, 385-389). Кроме того, у дефицитных по DPP-IV мышей или крыс сохранялась активность GLP-1 и высокие уровни инсулина, что приводило к снижению уровней глюкозы в крови, и такое генетическое нарушение или мутация гена DPP-IV не оказывала существенного эффекта на выживаемость отдельных животных (Marguet D. et al., Proc. Natl. Acad. Sci., 2000, 6874-6879). Следовательно, было предположено, что DPP-IV является возможным сильнодействующим лекарственным средством для лечения диабета 2 типа, что привело к ускоренному исследованию и разработке ингибитора DPP-IV.
Связывание GLP-1 с рецептором в различных тканях приводит к сытости (ощущению наполненности), замедленному опорожнению желудка и ускоренному росту бета-клеток поджелудочной железы. Таким образом, постепенно увеличивается количество клинических испытаний лечения диабета 2 посредством внутривенного введения непосредственно GLP-1 (Verdich C. et al., J. Clin. Endocrinol. Metab., 2001, 4382-4389). Время полужизни GLP-1 составляет только 2 мин (Kieffer T. J., et al., Endocrinology, 1995, 3585-3596), так что короткое время полужизни является основным препятствием для прямого применения GLP-1 в качестве лекарственного средства. После этого большим количеством исследовательских групп и институтов было предпринято множество попыток получения производных GLP-1, что привело к разработке и коммерциализации пептида, который способен удлинять короткое время полужизни in vivo (Deacon C. F., Diabetes, 2004, 2181-2189). Однако такое производное GLP-1 все еще обладает существенным ограничением, состоящим в том, что оно представляет собой инъецируемый состав. Кроме того, значительный интерес все больше и больше фокусируется на разработке эффективного ингибитора DPP-IV, поскольку активный GLP-1 (7-36) деградируется посредством DPP-IV, а затем превращается в неактивный GLP-1 (9-36) только в течение короткого периода времени, например 2 мин.
Начало разработки ингибиторов DPP-IV было сходным с путем развития других ингибиторов. Это значит, что большинство результатов исследований было получено для аналогов субстратов. Репрезентативным аналогом субстрата является дипептидное производное, которое было получено в качестве результата раннего исследования, которое проводили на исходном ядре, имеющем структуру, сходную со структурой пролина (Pro), на основании того факта, что DPP-IV обладает выраженной аффинностью к пептиду, содержащему конкретную аминокислоту - пролин (Chinnaswamy T. et al., J. Biol. Chem., 1990, 1476-1483). Типичные примеры пролиноподобных структур включают пирролидид и тиазолидид, и производные, содержащие эти соединения с исходными ядрами, обладают обратимой и конкурентной ингибиторной активностью в отношении фермента DPP-IV (Augustyns KJL., et al., Eur. J. Med. Chem., 1997, 301-309).
В число результатов таких обширных исследований и разработки входят продолжающиеся эксперименты по механизму действия и эффективности определенных соединений, конкретно Val-Pyr (валин-пирролидид), Ile-Thia (изолейцин-тиазолидид) и т.п. В частности, большое внимание уделено Ile-Thia, поскольку структура Val-Pyr оказывает относительно слабую ингибиторную активность на DPP-IV (Hanne B. R., et al., Nat. Struct. Biol., 2003, 19-25), что, по существу, вызвало интенсивное исследование и изучение производных соединения Ile-Thia.
Из производных соединений Ile-Thia, на которых было сосредоточено указанное выше изучение и исследование, и которые были получены, соединение, обладающее наиболее выраженной активностью, представляло собой соединение серии тиазолидидов бета-аминокислот, которое попытались получить в Merck & Co., Inc. Однако, согласно результатам фармакодинамических и фармакокинетических экспериментов, проведенных у крыс, полученное соединение обладало в значительной степени низкой биодоступностью в сочетании с очевидным ограничением в ингибировании ферментативной активности (Jinyou Xu, et al., Bioorg. Med. Chem. Lett., 2004, 4759-4762). Следовательно, последующая разработка соединений этого класса была прервана вследствие существенных недостатков.
В ходе указанного выше исследования в Merck было отмечено, что бета-аминокислота, в дополнение к тиазолидидному исходному ядру, также является ключевым фактором, оказывающим выраженные эффекты на ингибиторную активность DPP-IV. Это открытие было применено в подходе с заменой тиазолидидного исходного ядра отличающимся соединением исходного ядра (Linda L. B., et al., Bioorg. Med. Chem. Lett., 2004, 4763-4766). В таком последовательном исследовании было синтезировано множество производных, имеющих замену тиазолидидного исходного ядра пиперазиновым исходным ядром, с тестированием эффективности лекарственных средств и фармакодинамическими исследованиями. К сожалению, пиперазиновые производные Merck все еще обладали в значительной степени недостаточной биодоступностью. На основании оптимизации соединения для устранения такого недостатка был разработан продукт MK-0431 (торговая марка: JANUVIA) с модификацией пиперазиновой группы до триазолопиперазиновой группы. Этот продукт в настоящее время доступен в рамках одобрения новых лекарственных средств US FDA в 2006. Кроме того, после MK-0431, в настоящее время разрабатывается соединение с включением группы диазепанона (семичленное кольцо) (WO 2004037169; WO 2005011581; WO 2006104997; и Bioorg. Med. Chem. Lett, 2007, 49-52). В частности, согласно статье, опубликованной в журнале (Bioorg. Med. Chem. Lett., 2007, 49-52), было показано, что имидазолон (пятичленное кольцо) и пиперазинон (шестичленное кольцо) обладают значительно более низкой активностью in vitro по сравнению с диазепаноном, что, таким образом, привело к усиленному сосредоточению внимания на оптимизации диазепанона.
[MK-0431]
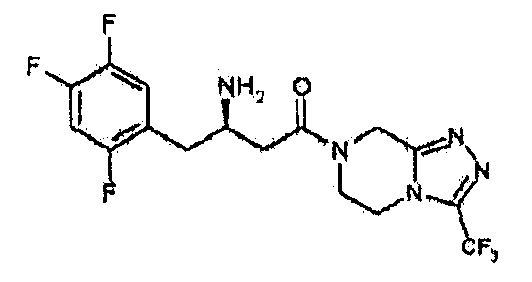
В результате множества обширных и интенсивных исследований и экспериментов для решения проблем, описанных выше, и для достижения оптимизации представляющего интерес соединения авторы настоящего изобретения открыли, что когда в группе пиперазинона сделана замена, включающая гетероатом, модифицированное таким образом соединение не только обладает превосходной ингибиторной активностью в отношении DPP-IV, но также способно достигать значительно повышенной биодоступности по сравнению с общепринятым ингибитором DPP-IV, а затем успешно провели синтез нового гетероциклического соединения, содержащего бета-аминогруппу. Настоящее изобретение было сделано на основании этих открытий.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ПРОБЛЕМА
Задачей настоящего изобретения является предоставление гетероциклического соединения, содержащего бета-аминогруппу и обладающего ингибиторной активностью в отношении DPP-IV, или его фармацевтически приемлемой соли, гидрата или сольвата.
Другой задачей настоящего изобретения является предоставление фармацевтической композиции для предупреждения и лечения диабета или ожирения, содержащей в качестве активного ингредиента указанное выше гетероциклическое соединение или его фармацевтически приемлемую соль, гидрат или сольват.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
Далее настоящее изобретение описано более подробно. Настоящее изобретение относится к гетерциклическим соединениям с бета-аминогруппой, представленным формулой 1:
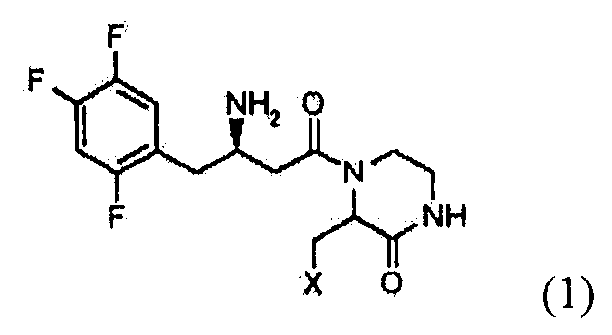
где X представляет собой OR1, SR1 или NR1R2, где R1 и R2 независимо представляют собой C1-C5 низший алкил, и R1 и R2 в NR1R2 могут образовывать 5-7-членное кольцо с включением гетероатома O; или к их фармацевтически приемлемой соли.
Предпочтительно, соединение формулы 1 в соответствии с настоящим изобретением включает соединение формулы 2, которое представляет собой стереоизомер, индуцирующий оптическую активность на атоме углерода в 3 положении кольца пиперазинона, и соответствует формуле 2, ниже.
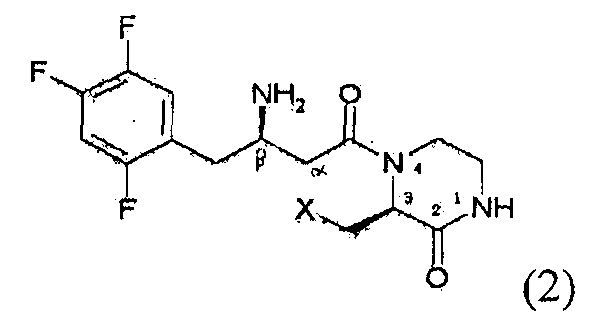
где X является таким, как определено для формулы 1.
Следовательно, соединение формулы 1 может иметь два асимметричных центра. Конкретно, соединение формулы 1, как показано на формуле 2, может иметь асимметричные центры на бета-углероде и на углероде в 3 положении кольца пиперазинона, так что оно может быть представлено в форме одного диастереоизомера, рацемата, рацемической смеси или диастереоизомерной смеси, все из которых относятся к соединению формулы 1 в соответствии с настоящим изобретением.
Кроме того, соединение формулы 1 может частично присутствовать в качестве таутомера. Также в соединение формулы 1 включены отдельные таутомеры, а также их смеси.
Стереоизомерную форму соединения формулы 1 можно получать стереоселективным синтезом в соответствии с общепринятым способом, известным в данной области, с использованием оптически чистого исходного материала или известного реагента.
Предпочтительные примеры содержащего бета-аминогруппу гетероциклического соединения формулы 1 в соответствии с настоящим изобретением могут включать следующие соединения:
1) гидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
2) гидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(метоксиметил)пиперазин-2-она;
3) гидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(этоксиметил)пиперазин-2-она;
4) гидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(изопропоксиметил)пиперазин-2-она;
5) гидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(циклопентилоксиметил)пиперазин-2-она;
6) дигидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-[(диэтиламино)метил]пиперазин-2-она;
7) дигидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-[(этилметиламино)метил]пиперазин-2-она;
8) дигидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(морфолинометил)пиперазин-2-она;
9) гидрохлорид (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутилтиометил)пиперазин-2-она;
10) гидрохлорид (S)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
11) (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-он;
12) тартрат (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
13) цитрат (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
14) фосфат (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
15) ацетат (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
16) малат (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
17) сукцинат (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она; и
18) адипат (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она.
Содержащее бета-аминогруппу гетероциклическое соединение формулы 1 в соответствии с настоящим изобретением включает его фармацевтически приемлемую соль, а также гидрат и сольват, которые можно получать из него.
Фармацевтически приемлемую соль гетероциклического соединения формулы 1 можно получать любым общепринятым способом получения солей, известным в данной области.
Как используют в настоящем документе, термин "фармацевтически приемлемая соль" относится к соли, полученной из фармацевтически приемлемого нетоксичного основания или кислоты, включающих неорганическое или органическое основание и неорганическую или органическую кислоту. Примеры фармацевтически приемлемой соли могут включать соли соединения 1 с неорганическим основанием, например с ионом алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, манганата, марганца, калия, натрия или цинка. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Твердая соль может иметь одну или несколько кристаллических структур, или в ином случае она может иметь форму гидрата. Примеры фармацевтически приемлемой нетоксичной органической соли могут включать соли соединения 1 с первичным, вторичным или третичным амином, замещенным амином, таким как встречающийся в природе замещенный амин, циклическим амином, или основной ионообменной смолой, такой как аргининовая, бетаиновая, кофеиновая, холиновая, N,N'-дибензилэтилендиаминовая, диэтиламиновая, 2-диэтиламиноэтаноловая, 2-диметиламиноэтаноловая, этаноламиновая, этилендиаминовая, N-этилморфолиновая, N-этилпиперидиновая, глюкаминовая, глюкозаминовая, гистидиновая, гидрабаминовая, изопропиламиновая, лизиновая, метилглюкаминовая, морфолиновая, пиперазиновая, пиперидиновая, полиаминовая смола, прокаином, пурином, теобромином, триэтиламином, триметиламином, трипропиламином и трометамином.
Когда соединение по настоящему изобретению является основанием, его соль можно получать из фармацевтически приемлемых нетоксичных кислот, включающих неорганические и органические кислоты. Примеры кислоты могут включать уксусную кислоту, бензолсульфоновую кислоту, бензойную кислоту, камфорсульфоновую кислоту, лимонную кислоту, этансульфоновую кислоту, фумаровую кислоту, глюконовую кислоту, глутаминовую кислоту, бромисто-водородную кислоту, хлористо-водородную кислоту, изетионовую кислоту, молочную кислоту, малеиновую кислоту, яблочную кислоту, миндальную кислоту, метансульфоновую кислоту, муциновую кислоту, азотную кислоту, памовую кислоту, пантотеновую кислоту, фосфорную кислоту, янтарную кислоту, серную кислоту, винно-каменную кислоту, п-толуолсульфоновую кислоту и адипиновую кислоту. Особенно предпочтительными являются уксусная, лимонная, хлористо-водородная, яблочная, фосфорная, янтарная, винно-каменная и адипиновая кислоты.
Когда в настоящем документе представлено соединение формулы 1, этот термин включает его фармацевтически приемлемую соль.
Как используют в настоящем документе, термин "гидрат" означает соединение формулы 1 или его фармацевтически приемлемую соль, которые дополнительно включают стехиометрическое или нестехиометрическое количество воды, связанной с ними посредством нековалентных межмолекулярных сил. Гидрат может содержать более 1 эквивалента воды, как правило, от 1 до 5 эквивалентов воды. Гидрат можно получать кристаллизацией соединения формулы 1 или его фармацевтически приемлемой соли в воде или содержащем воду растворителе.
Как используют в настоящем документе, термин "сольват" означает соединение формулы 1 или его соль, которые дополнительно включают стехиометрическое или нестехиометрическое количество растворителя, связанного с ними посредством нековалентных межмолекулярных сил. Предпочтительные растворители являются летучими, нетоксичными и/или приемлемыми для введения человеку. Например, могут быть упомянуты этанол, метанол, пропанол, метиленхлорид и т.д.
В соответствии с другим аспектом настоящего изобретения предусмотрен способ получения гетероциклического соединения с бета-аминогруппой, представленного формулой 1, или его фармацевтически приемлемой соли.
Настоящее изобретение, как показано на схеме реакции 1, ниже, включает способ получения гетероциклического соединения, представленного формулой 2, включающий: 1) реакцию соединения формулы 3, имеющего бета-аминогруппу, с замещенным гетероциклическим соединением формулы 4 в присутствии 1-гидроксибензотриазола (HOBT), 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC) и третичного амина с получением, таким образом, соединения формулы 5, имеющего пептидную связь, и 2) реакцию соединения формулы 5 в присутствии кислоты с получением гетероциклического соединения формулы 2, имеющего бета-аминогруппу.
[Схема реакции 1]
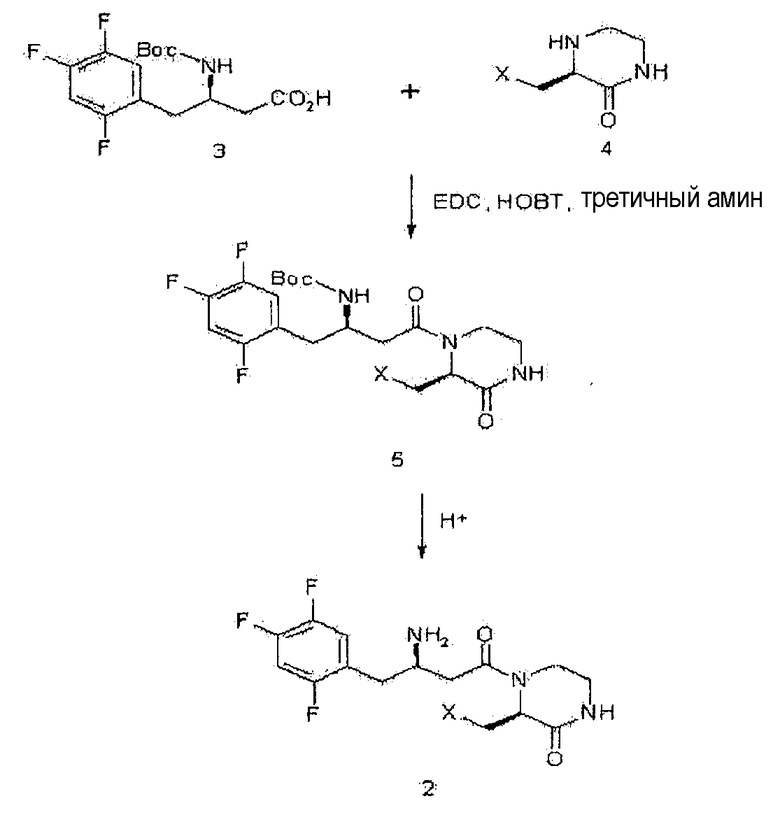
где X является таким, как определено для формулы 1.
Например, промежуточное соединение формулы 5 можно получать реакцией соединения формулы 3 и соединения формулы 4 обычным путем в растворителе, таком как N,N-диметилформамид (DMF) или дихлорметан, в присутствии связывающего реагента, такого как 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC) или 1-гидроксибензотриазол (HOBT), и основания, такого как диизопропилэтиламин или триэтиламин, при температуре от 0°С до комнатной температуры в течение от 3 до 48 часов.
Для предотвращения участия соединения в пептидизации атом азота промежуточного соединения формулы 5, которое получено пептидизацией, защищают защитной группой. Требуемое гетероциклическое соединение формулы 2, имеющее бета-аминогруппу, можно получить удалением защитной группы посредством реакции снятия защитной группы. Следовательно, поскольку защитная группа представляет собой Boc, удаление защитной группы можно проводить в кислых условиях, как правило, с использованием смеси трифторуксусная кислота/дихлорметан, смеси этилацетат/хлористо-водородная кислота, смеси хлористо-водородная кислота/дихлорметан или смеси метанол/хлористо-водородная кислота, при температуре от 0°С до комнатной температуры в течение от 1 до 24 часов.
Если необходимо, соединение формулы 2, полученное реакцией пептидной связи и удалением защитной группы, можно очищать от нежелательных побочных продуктов любым общепринятым способом, таким как перекристаллизация, порошкование, препаративная тонкослойная хроматография, флэш-хроматография на силикагеле (см. W.C. Still et al., J. Org. Chem., 43, 2923 (1978)) или ВЭЖХ. Соединение, очищенное посредством ВЭЖХ, можно выделять в качестве соответствующей его соли. Соединение формулы 5 также можно очищать аналогичным образом.
В настоящем изобретении смесь стереоизомеров соединения формулы 1 получают с использованием смеси стереоизомеров в качестве исходного материала, и полученную смесь разделяют на отдельные стереоизомеры с получением, таким образом, соединения формулы 1. Кроме того, каждый стереоизомер соединения формулы 1 можно получать с использованием каждого стереоизомера в качестве исходного материала. Выделение стереоизомера можно проводить общепринятой колоночной хроматографией или перекристаллизацией.
При получении соединения формулы 2 соединение формулы 3, используемое в схеме реакции 1, является коммерчески доступным, или его можно легко получать любым способом, известным в данной области.
При получении соединения формулы 2 соединение формулы 4, используемое в схеме реакции 1, можно получать в соответствии с синтетическим каскадом схемы реакции 2 и схемы реакции 3.
В схеме реакции 2 соединение 6 может быть коммерчески доступным, или оно может не быть коммерчески доступным, в зависимости от заместителя X, так что соединение 6 является коммерчески доступным, или его можно легко получить любым способом, известным в данной области, например способом, представленным на схеме реакции 3, ниже.
На схеме реакции 2 соединение 4, используемое для получения соединения по настоящему изобретению, можно получать из соединения 6. Конкретно, соединение 6 подвергают реакции с N-бутилоксикарбонил-2-аминоацетальдегидом в присутствии восстановителя с получением соединения 7, из которого затем получают соединение 8, имеющее вторичный амин, защищенный бензилоксикарбонилом (Cbz), с последующим удалением защитной группы с получением, таким образом, соединения 9, где бутилоксикарбонил (Boc) удален. Затем соединение 9 подвергают циклизации с использованием триметилалюминия (или смеси диизопропилэтиламин/этанол, смеси гидрокарбонат натрия/метанол и т.д.) с получением соединения 10, за которой следует удаление защитной группы Cbz с получением соединения 4. Примеры восстановителя, который можно использовать для получения соединения 7 из соединения 6, могут включать цианоборгидрид натрия, триацетоксиборгидрид натрия, боргидрид натрия и т.п.
[Схема реакции 2]
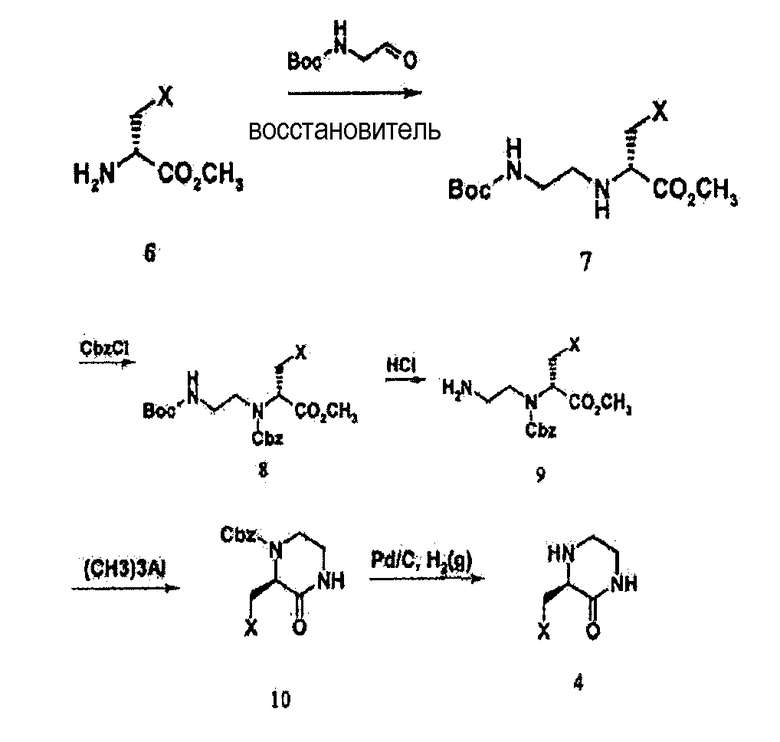
где X является таким, как определено в формуле 1.
Когда соединение 6 на схеме реакции 2 не является коммерчески доступным, его можно получать аналогично схеме реакции 3, ниже. Соединение 6, имеющее множество заместителей R1 на схеме реакции 3, получают замещением метилового сложного эфира D-серина тритилхлоридом с получением соединения 11 и заменой гидроксильной группы соединения 11 мезильной группой, с последующим кипячением с обратным холодильником с получением, таким образом, соединения азиридина 12. Затем тритильную группу соединения 12 удаляют с использованием трифторуксусной кислоты, а затем защищают бензилоксикарбонилом (Cbz) с получением соединения 13. Затем соединение 13 подвергают реакции с HX, имеющим множество заместителей R1, с получением соединения 14, с последующим удалением Cbz с получением соединения 6.
[Схема реакции 3]
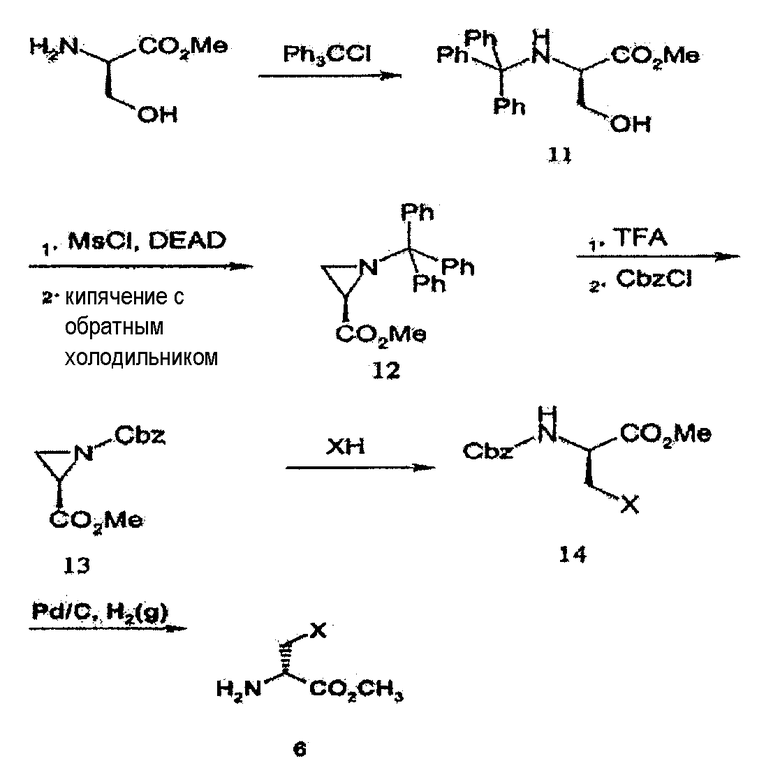
где X является таким, как определено в формуле 1.
Для облегчения представляющей интерес реакции или избежания образования нежелательного продукта реакции для некоторых из соединений формулы 1 по настоящему изобретению указанные выше условия реакции и последовательности реакции можно варьировать, если желательно.
Как описано выше, соединения формулы 1 по настоящему изобретению, исходные материалы и промежуточные соединения можно синтезировать различными способами, известными в данной области.
В соответствии со следующим аспектом настоящего изобретения предусмотрена фармацевтическая композиция для предупреждения и лечения диабета или ожирения, содержащая соединение формулы 1 или его фармацевтически приемлемую соль в качестве активного ингредиента.
Соединение формулы 1 в соответствии с настоящим изобретением обладает превосходной ингибиторной активностью в отношении DPP-IV. Когда определяли ингибиторную способность соединения формулы 1 в отношении фермента DPP-IV, IC50, концентрация лекарственного средства, которая требуется для ингибирования ферментативной реакции DPP-IV на 50%, была практически равна диапазону от 0,5 до 20 нМ, что соответствует более высокой ингибиторной активности в отношении DPP-IV по сравнению с общепринятым ингибитором DPP-IV, для которого описана IC50 от нескольких сотен нМ до нескольких тысяч нМ или даже до нескольких десятков тысяч нМ (Jinyou Xu, et al., Bioorg. Med. Chem. Lett., 2004, 4759-4762; и Linda L. B., et al., Bioorg. Med. Chem. Lett., 2004, 4763-4766).
Кроме того, соединение формулы 1 в соответствии с настоящим изобретением обладает высокой пероральной толерантностью к глюкозе. Таким образом, в тесте пероральной толерантности к глюкозе (OGTT) было измерено, что соединение формулы 1 обладает эффектами снижения глюкозы в крови более чем на 35%, предпочтительно более чем на 50%, таким образом демонстрируя, что оно обладает более высокой биодоступностью по сравнению с общепринятыми ингибиторами DPP-IV. Кроме того, результаты экспериментов in vivo, включающие фармакокинетические/фармакодинамические корреляции, измерение длительности периода ингибиторной активности DPP-IV и эксперименты кинетики in vivo, демонстрируют, что соединение по настоящему изобретению обладает более высокой ингибиторной активностью в отношении DPP-IV и биодоступностью.
Таким образом, фармацевтическую композицию, содержащую в качестве активного ингредиента соединение формулы 1, можно эффективно использовать для лечения и предупреждения диабета и ожирения, которые являются репрезентативными заболеваниями, вызываемыми с помощью DPP-IV.
В соответствии с другим аспектом настоящего изобретения предусмотрены применение указанной выше композиции для предупреждения и лечения диабета или ожирения и способ предупреждения и лечения диабета или ожирения, включающий введение эффективного количества указанной выше композиции млекопитающему (включая человека).
Фармацевтическую композицию, содержащую в качестве активного ингредиента соединение формулы 1 или его стереоизомер, фармацевтически приемлемую соль, гидрат или сольват, можно изготавливать в виде следующих пероральных или парентеральных лекарственных форм, не ограничиваясь ими.
Примеры лекарственной формы для перорального введения могут включать таблетки, пилюли, мягкие и твердые капсулы, растворы, суспензии, эмульсии, сиропы, гранулы, эликсиры и т.п. Эти фармацевтические составы могут содержать, в дополнение к указанному выше активному ингредиенту, один или несколько общепринятых разбавителей или эксципиентов, таких как наполнители, разбавители, смачивающие вещества, дезинтегрирующие вещества, вещества, способствующие скольжению, связующие вещества и поверхностно-активные вещества. Примеры дезинтегрирующих веществ могут включать агар, крахмал, альгиновую кислоту или ее натриевую соль, безводный моногидрофосфат кальция и т.п. Примеры веществ, способствующих скольжению, могут включать диоксид кремния, тальк, стеариновую кислоту или их магниевую или кальциевую соль, полиэтиленгликоль и т.п. Примеры связующего вещества могут включать алюмосиликат магния, крахмальную пасту, желатин, трагакант, метилцеллюлозу, карбоксиметилцеллюлозу натрия, поливинилпирролидон, низкозамещенную гидроксипропилцеллюлозу и т.п. Кроме того, фармацевтический состав может содержать разбавители, например лактозу, декстрозу, сахарозу, маннит, сорбит, целлюлозу и/или глицин. Если желательно, состав может дополнительно содержать широко известные шипучие смеси, абсорбенты, красители, вкусовые добавки и подсластители.
Фармацевтическую композицию, содержащую в качестве активного ингредиента соединение формулы 1 или его фармацевтически приемлемую соль, можно вводить парентеральным путем, например посредством суппозитория, подкожной инъекции, внутривенной инъекции, внутримышечной инъекции или внутригрудной инъекции. Для приготовления композиции по настоящему изобретению в виде препарата для парентерального введения соединение формулы 1 или его фармацевтически приемлемую соль смешивают со стабилизатором или буфером в присутствии воды с получением раствора или суспензии, из которых затем изготавливают единичные лекарственные формы в виде ампул или флаконов.
Композиция может быть стерилизованной и/или она может содержать адъюванты, такие как консерванты, стабилизаторы, гидратирующие вещества, эмульгаторы, соли для контроля осмотического давления и/или буферы, и терапевтически пригодные вещества, и их можно изготавливать в соответствии с общепринятыми способами, такими как смешивание, грануляция и нанесение покрытия.
Если желательно, соединение формулы 1 или фармацевтическую композицию, содержащую его в качестве активного ингредиента, можно вводить в сочетании с другими лекарственными средствами, например противодиабетическими лекарственными средствами.
Когда соединение формулы 1 или фармацевтическую композицию, содержащую его в качестве активного ингредиента, изготавливают в виде стандартной лекарственной формы, соединение формулы 1 применяют предпочтительно в единичной дозе приблизительно от 0,1 до 1,500 мг при пересчете на активный ингредиент. Как будет очевидно специалистам в данной области, эффективную дозу активного соединения в соответствии с настоящим изобретением можно определять в соответствии с назначением врача, в зависимости от различных факторов, таких как масса тела и возраст пациентов, тип и тяжесть заболевания и т.п. Для взрослых эффективная доза активного соединения, как правило, находится в диапазоне приблизительно от 1 до 500 мг/сутки, с учетом частоты и интенсивности введения. В случае внутримышечной или внутривенной инъекции взрослым могут быть пригодными от 5 до 300 мг общей дозы, разделенной на несколько единичных доз, хотя для некоторых пациентов может потребоваться даже более высокая суточная доза.
ПРЕИМУЩЕСТВЕННЫЕ ЭФФЕКТЫ
Как конкретно проиллюстрировано в дальнейшем в данном документе, настоящее изобретение относится к гетероциклическому соединению, содержащему бета-аминогруппу и обладающему превосходными ингибиторными эффектами в отношении ферментативной активности DPP-IV. Фармацевтическая композиция, содержащая указанное соединение по настоящему изобретению в качестве активного ингредиента, обладает превосходной ингибиторной активностью в отношении DPP-IV и биодоступностью, и поэтому она может быть пригодной для профилактики или лечения различных заболеваний, считающихся вызываемыми посредством DPP-IV, таких как диабет и ожирение.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
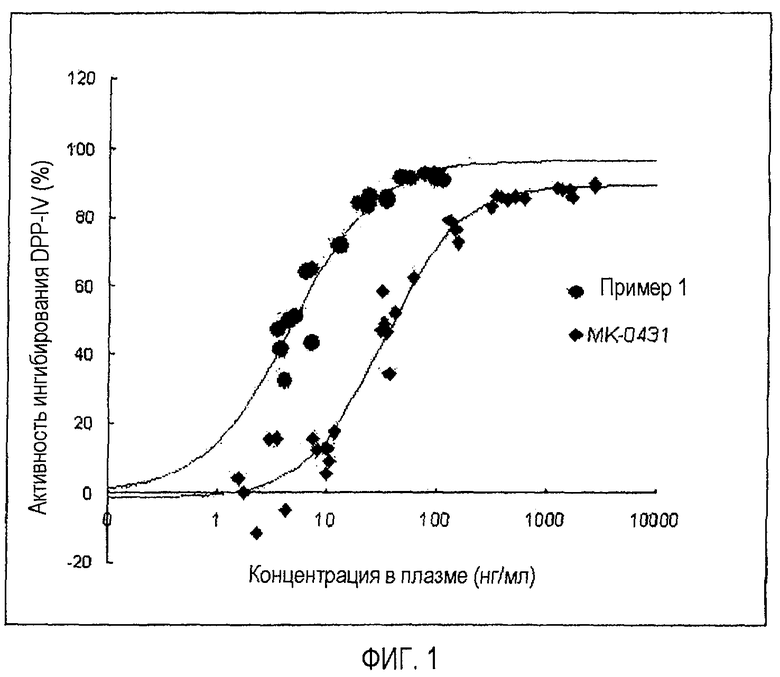
На фиг.1 представлена корреляция между активностью DPP-IV в плазме и дозой лекарственного средства, полученная для MK-0431 и соединения примера 1; и
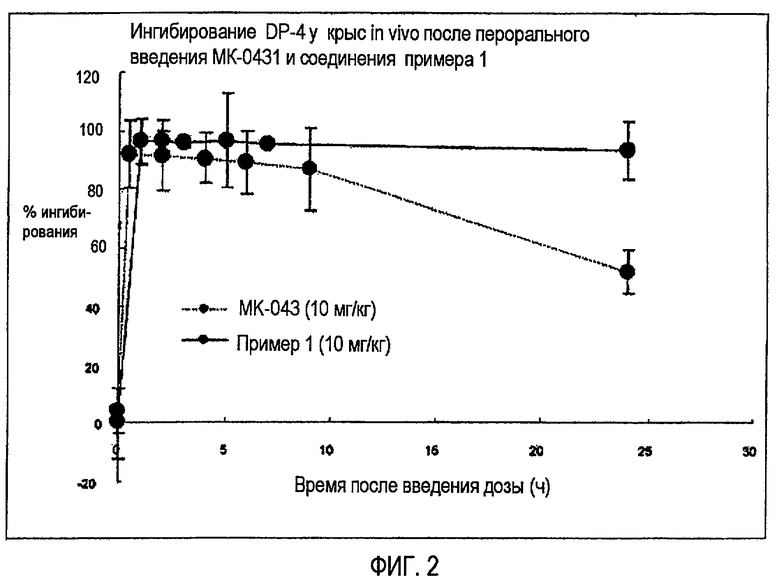
на фиг.2 показаны результаты измерения и сравнения длительности ингибиторной активности DPP-IV, полученной для MK-0431, и соединения примера 1 у лабораторных крыс.
СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение описано более подробно с помощью следующих примеров. Эти примеры предоставлены только для иллюстрации настоящего изобретения, и их не следует истолковывать как ограничивающие объем и сущность настоящего изобретения.
Пример 1: получение гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенилбутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
Стадия 1: получение (R)-метил-1-тритилазиридин-2-карбоксилата
200 г гидрохлорида метилового сложного эфира D-серина добавляли к 1,8 л хлороформа, и реакционный раствор охлаждали до 0°С, и затем к нему медленно добавляли 448 мл триэтиламина. В реакционную смесь медленно добавляли 358,4 г тритилхлорида и затем ее перемешивали в течение 1 часа. Реакционную смесь нагревали до комнатной температуры, и к ней добавляли 1 л хлороформа, а затем промывали 2,5 л воды. Органический слой сушили над сульфатом магния и охлаждали до 0°С, а затем к нему последовательно медленно добавляли 484 мл триэтиламина и 15,7 г 4-метиламинопиридина. Реакционную смесь перемешивали в течение 5 мин и к ней медленно добавляли 139 мл сульфонилхлорида метана. Реакционную смесь нагревали до комнатной температуры, перемешивали еще в течение 4 часов, а затем кипятили с обратным холодильником в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и промывали 4 л воды, а затем 3 л рассола. Органический слой сушили над сульфатом магния и концентрировали до высушивания при пониженном давлении. К полученному осадку добавляли 3 л этанола, а затем его перемешивали. Полученные твердые вещества отфильтровывали с получением 329 г указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): 7,42-7,49 (м, 6H), 7,18-7,32 (м, 9H), 7,68 (с, 1H), 3,74 (с, 3H), 2,24 (м, 1H), 1,87 (м, 1H) и 1,40 (м, 1H).
Стадия 2: Получение (R)-1-бензил-2-метилазиридин-1,2-дикарбоксилата
328,4 г (R)-метил-1-тритилазиридин-2-карбоксилата растворяли в 1,4 л хлороформа и реакционный раствор охлаждали до 0°С, а затем к нему медленно добавляли 462 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 1 часа, и затем к ней добавляли 2 л воды, и затем перемешивали в течение 10 мин и удаляли органический слой. Водный слой нейтрализовывали гидрокарбонатом натрия и использовали в последующих реакциях без дальнейшей очистки.
К водному слою добавляли 2 л диэтилового эфира и 120,5 г гидрокарбоната натрия, и реакционный раствор охлаждали до 0°С, а затем к нему медленно по каплям добавляли 165 мл бензилхлорформиата. Реакционную смесь перемешивали в течение еще 2 часов и водный слой удаляли. Органический слой сушили над сульфатом магния, концентрировали и сушили при пониженном давлении, и очищали колоночной хроматографией, с получением, таким образом, 108,5 г указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO): 7,32-7,36 (м, 5H), 5,13 (с, 2H), 3,09 (д.д. J=3,2, 5,4 Гц, 1H), 2,58 (д.д. J=1,2, 3,2 Гц, 1H) и 2,47 (д.д. J=1,2, 5,4 Гц, 1H).
Стадия 3: получение метилового сложного эфира (R)-2-амино-3-трет-бутоксипропана
1,1 г (R)-1-бензил-2-метилазиридин-1,2-дикарбоксилата растворяли в 11 мл хлороформа и затем к нему добавляли 18 мл трет-бутанола. К реакционной смеси медленно по каплям добавляли 1,2 мл BF3OEt2, а затем перемешивали в течение 12 часов. Реакцию завершали добавлением в реакционную смесь 2 л воды. Затем органический слой отделяли и сушили над сульфатом магния, концентрировали и сушили при пониженном давлении, а затем использовали в последующих реакциях без дальнейшей очистки.
Полученный осадок растворяли в 10 мл метанола, и затем к нему добавляли 740 мг смеси палладий/углерод в 2 мл этилацетата, после этого барботировали водородом в течение 1 часа при внешнем атмосферном давлении. Реакционную смесь фильтровали и сушили при пониженном давлении с получением 736 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, CD3OD): 4,21 (м, 1H), 3,82 (с, 3H), 3,74-3,88 (м, 2H) и 1,20 (с, 9H)
Стадия 4: получение метилового сложного эфира (R)-3-трет-бутокси-2-(2-(трет-бутоксикарбониламино)этиламино)пропионовой кислоты
736 мг метилового сложного эфира (R)-2-амино-3-трет-бутоксипропана, полученного на стадии 3, растворяли в 14 мл дихлорметана, и к нему медленно добавляли 6335 мг N-трет-бутоксикарбонил-2-аминоацетальдегида метанола. Реакционную смесь охлаждали до 0°С, а затем постепенно добавляли 1,2 мл триэтиламина и 1,78 г триацетоксиборгидрида натрия. Реакционную смесь нагревали до комнатной температуры, а затем перемешивали в течение 12 часов. Для завершения реакции добавляли насыщенный раствор гидрокарбоната натрия и органический слой промывали 10 мл воды и рассола, концентрировали и сушили при пониженном давлении. Полученный осадок очищали колоночной хроматографией с получением, таким образом, 355 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): 5,10 (м, 1H), 3,71 (с, 3H), 3,56 (м, 2H), 3,40 (м, 1H), 3,15-3,28 (м, 2H), 2,81 (м, 1H), 2,67 (м, 1H), 1,42 (с, 9H) и 1,13 (с, 9H)
Стадия 5: получение метилового сложного эфира (R)-2-((бензилоксикарбонил)(2-трет-бутоксикарбониламино)этил)амино)-3-трет-бутоксипропионовой кислоты
355 мг метилового сложного эфира (R)-3-трет-бутокси-2-(2-(трет-бутоксикарбониламино)этиламино)пропионовой кислоты, полученного на стадии 4, растворяли в 11 мл тетрагидрофурана, и реакционную смесь охлаждали до 0°С и затем к ней добавляли 187 мг гидрокарбоната натрия. К этой смеси медленно по каплям добавляли 192 мкл бензилхлорформиата и реакционную смесь нагревали до комнатной температуры. Через 12 часов реакционную смесь сушили при пониженном давлении, а затем добавляли 10 мл этилацетата и органический слой промывали 10 мл воды. Органический слой сушили над сульфатом магния, сушили при пониженном давлении и очищали колоночной хроматографией, с получением, таким образом, 410 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): 7,36-7,25 (м, 5H), 5,82-5,72 (м, 1H), 5,17-5,03 (м, 2H), 4,15 (м, 1H), 3,98 (м, 1H), 3,81 (м, 1H), 3,73 (с, 3H), 3,60 (м, 1H), 3,42-3,28 (м, 3H), 1,40 (с, 9H) и 1,l4 (с, 9H).
Стадия 6: получение (R)-бензил 2-(трет-бутоксиметил)-3-оксопиперазин-1-карбоксилата
410 мг метилового сложного эфира (R)-2-((бензилоксикарбонил)(2-трет-бутоксикарбониламино)этил)амино)-3-трет-бутоксипропионовой кислоты, полученного на стадии 5, растворяли в 10 мл метанола и реакционную смесь охлаждали до 0°С, и к ней медленно добавляли 4 мл смеси 2-N-хлористо-водородная кислота/диэтиловый эфир, а затем перемешивали в течение 3 часов. Реакционную смесь сушили при пониженном давлении и использовали в последующих реакциях без дальнейшей очистки.
Полученный осадок растворяли в 10 мл дихлорметана и реакционную смесь охлаждали до 0°С, а затем к ней медленно добавляли 152 мкл триэтиламина. К этой смеси медленно добавляли 1,1 мл триметилалюминия (2,0 M раствор в толуоле), и реакционную смесь нагревали до комнатной температуры, а затем перемешивали в течение 12 часов. Реакционную смесь охлаждали до 0°С и для завершения реакции добавляли насыщенный водный раствор хлорида аммония. К реакционной смеси добавляли 10 мл этилацетата, и затем ее промывали 10 мл рассола. Органический слой сушили над сульфатом магния и сушили при пониженном давлении. Полученный осадок очищали колоночной хроматографией с получением 103 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): 7,34-7,25 (м, 5H), 6,27 (м, 1H), 5,14 (м, 2H), 4,57 (м, 1H), 4,19 (м, 1H), 4,08 (м, 1H), 3,94 (м, 1H), 3,74 (м, 1H), 3,64 (м, 1H), 3,42 (м, 1H), 3,29 (м, 1H) и 1,09 (с, 9H)
Стадия 7: получение (R)-(3-трет-бутоксиметил)пиперазин-2-она
103 мг (R)-бензил-2-(трет-бутоксиметил)-3-оксопиперазин-1-карбоксилата, полученного на стадии 6, растворяли в 2 мл метанола и затем к нему добавляли 50 мг смеси палладий/углерод в 1 мл этилацетата, а затем барботировали водородом в течение 1 часа при обычном атмосферном давлении. Реакционную смесь фильтровали и сушили при пониженном давлении с получением 58 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): 6,41 (шир.с, 1H), 3,76 (м, 3H), 3,63 (м, 1H), 3,52 (м, 1H), 3,42 (м, 1H), 3,28 (м, 1H), 3,16 (м, 1H), 2,95 (м, 1H), 2,45 (шир.с, 1H) и 1,17 (с, 9H).
Стадия 8: получение трет-бутил-(R)-4-[(R)-2-(трет-бутоксметил)-3-оксопиперазин-1-ил]-4-оксо-1-(2,4,5-трифторфенил)бутан-2-илкарбамата
104 мг (3R)-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)бутановой кислоты и 58 мг (R)-(3-трет-бутоксиметил)пиперазин-2-она добавляли к 4 мл N,N-диметилформамида, и затем к нему добавляли 63 мг 1-гидроксибензотриазола (HOBT) и 217 мкл диизопропилэтиламина. Реакционную смесь охлаждали до 0°С и к ней добавляли 78 мг 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC), а затем перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь разбавляли 10 мл этилацетата и промывали два раза рассолом. Органический слой сушили над сульфатом магния и концентрировали. Полученный осадок очищали колоночной хроматографией с получением 97 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): 7,03 (м, 1H), 6,88 (м, 1H), 5,97 (м, 1H), 5,48 (м, 1H), 4,16-4,07 (м, 1H), 4,02-3,91 (м, 1H), 3,74 (м, 2H), 3,37 (м, 2H), 3,24 (м, 1H), 2,92 (м, 2H), 2,80 (м, 1H), 2,59 (м, 2H), 1,34 (д, 9H) и 1,13 (с, 9H)
Стадия 9: получение гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
97 мг трет-бутил-(R)-4-[(R)-2-(трет-бутоксиметил)-3-оксопиперазин-1-ил]-4-оксо-1-(2,4,5-трифторфенил)бутан-2-илкарбамата, полученного на стадии 8, растворяли в 3 мл метанола, а затем добавляли 2 мл смеси 2Н хлористо-водородная кислота/диэтиловый эфир и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали и сушили при пониженном давлении с получением 64 мг указанного в заголовке соединения в виде пенистого твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,37 (м, 1H), 7,23 (м, 1H), 4,80 (м, 1H), 4,59-4,40 (м, 1H), 3,93 (м, 1H), 3,90-3,83 (м, 2H), 3,70 (м, 1H), 3,38 (м, 2H), 3,27 (м, 1H), 3,07 (м, 2H), 2,89-2,66 (м, 2H), 1,18 (с, 3H), и 1,11 (с, 6H)
Mass (M+1): 402
Пример 2: получение гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(метоксиметил)пиперазин-2-она
Вместо трет-бутанола стадии 3 примера 1 использовали метанол, и затем синтезировали 40 мг указанного в заголовке соединения аналогично стадиям с 4 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,34 (м, 1H), 7,23 (м, 1H), 4,82 (м, 1H), 4,62-4,46 (м, 1H), 3,92 (м, 1H), 3,87-3,82 (м, 2H), 3,66 (м, 1H), 3,35 (м, 2H), 3,24 (м, 1H), 3,04 (м, 2H), 2,94-2,72 (м, 2H) и 3,27 (с, 3H)
Mass (M+1): 360
Пример 3: получение гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(этоксиметил)пиперазин-2-она
Вместо трет-бутанола стадии 3 примера 1 использовали этанол, и затем синтезировали 66 мг указанного в заголовке соединения аналогично стадиям с 4 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,38 (м, 1H), 7,23 (м, 1H), 4,83 (м, 1H), 4,54-4,44 (м, 1H), 3,98 (м, 1H), 3,93-3,82 (м, 2H), 3,71 (м, 1H), 3,53 (м, 2H), 3,36 (м, 2H), 3,26 (м, 1H), 3,07 (м, 2H), 2,90-2,70 (м, 2H) и 1,11 (т, 3H)
Mass (M+1): 374
Пример 4: получение гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(изопропоксиметил)пиперазин-2-она
Вместо трет-бутанола стадии 3 примера 1 использовали изопропанол, и синтезировали 69 мг указанного в заголовке соединения аналогично стадиям с 4 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,38 (м, 1H), 7,23 (м, 1H), 4,86 (м, 1H), 4,62-4,43 (м, 1H), 3,96 (м, 1H), 3,90-3,87 (м, 2H), 3,77 (м, 1H), 3,69 (м, 1H), 3,44 (м, 2H), 3,26 (м, 1H), 3,08 (м, 2H), 2,95-2,69 (м, 2H) и 1,15 (м, 6H)
Mass (M+1): 388
Пример 5: получение гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(циклопентилоксиметил)пиперазин-2-она
Вместо трет-бутанола стадии 3 примера 1 использовали циклопентанол, и синтезировали 51 мг указанного в заголовке соединения аналогично стадиям с 4 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,38 (м, 1H), 7,23 (м, 1H), 4,82 (м, 1H), 4,61-4,42 (м, 1H), 3,93 (м, 1H), 3,90-3,82 (м, 2H), 3,67 (м, 1H), 3,40 (м, 1H), 3,36 (м, 2H), 3,25 (м, 1H), 3,08 (м, 2H), 3,01-2,62 (м, 2H) и 1,67-1,50 (м, 8H)
Mass (M+1): 414
Пример 6: получение дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-[(диэтиламино)метил]пиперазин-2-она
Вместо трет-бутанола добавляли диэтиламин и вместо добавления BF3OEt2 проводили кипячение с обратным холодильником на стадии 3 примера 1, и синтезировали 68 мг указанного в заголовке соединения аналогично стадиям с 4 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,41 (м, 1H), 7,24 (м, 1H), 5,21 (м, 1H), 3,59-3,53 (м, 2H), 3,53-3,50 (м, 4H), 3,43-3,37 (м, 4H), 3,35 (м, 2H), 3,09 (м, 2H), 2,97-2,81 (м, 2H) и 1,37 (м, 6H)
Mass (M+1): 401
Пример 7: получение дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноин-3-[(этилметиламино)метил]пиперазин-2-она
Вместо трет-бутанола добавляли этилметиламин и вместо добавления BF3OEt2 проводили кипячение с обратным холодильником на стадии 3 примера 1, и затем синтезировали 67 мг указанного в заголовке соединения аналогично стадиям с 4 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,42 (м, 1H), 7,24 (м, 1H), 5,22 (м, 1H), 4,08-3,87 (м, 2H), 3,86-3,75 (м, 2H), 3,68-3,57 (м, 2H), 3,56-3,33 (м, 4H), 3,09 (м, 2H), 3,02-2,81 (м, 5H), и 1,38 (м, 3H)
Mass (M+1): 387
Пример 8: получение дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(морфолинометил)пиперазин-2-она
Вместо трет-бутанола добавляли морфолин и вместо добавления BF3OEt2 проводили кипячение с обратным холодильником на стадии 3 примера 1, и затем синтезировали 27 мг указанного в заголовке соединения аналогично стадиям с 4 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,37 (м, 1H), 7,23 (м, 1H), 5,32 (м, 1H), 4,12-3,98 (м, 4H), 3,97-3,77 (м, 4H), 3,74-3,52 (м, 4H), 3,48-3,39 (м, 2H), 3,14-2,91 (м, 4H) и 2,86-2,72 (м, 2H)
Mass (M+1): 415
Пример 9: получение гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутилтиометил)пиперазин-2-она
Вместо трет-бутанола использовали трет-бутилтиол на стадии 3 примера 1, и затем синтезировали 25 мг указанного в заголовке соединения аналогично стадиям с 4 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,34 (м, 1H), 7,25 (м, 1H), 5,04 (м, 1H), 4,60 (с, 1H), 4,60-4,41 (м, 1H), 3,86 (м, 2H), 3,70 (м, 1H), 3,40 (м, 2H), 3,25 (м, 1H), 3,05 (м, 2H), 2,95 (м, 1H), 2,81 (м, 2H), и 1,26 (с, 9H)
Mass (M+1): 418
Пример 10: Получение гидрохлорида (S)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
Вместо гидрохлорида метилового сложного эфира D-серина использовали гидрохлорид метилового сложного эфира L-серина на стадии 1 примера 1, и затем синтезировали 31 мг указанного в заголовке соединения аналогично стадиям с 2 по 9 примера 1.
1H ЯМР (400 МГц, CD3OD): 7,34 (м, 1H), 7,24 (м, 1H), 4,79 (м, 1H), 4,580-4,40 (м, 1H), 3,96 (м, 1H), 3,86-3,74 (м, 2H), 3,70 (м, 1H), 3,36 (м, 2H), 3,19 (м, 1H), 3,05-2,86 (м, 3H), 2,67 (м, 1H), 1,15 (с, 4H) и 1,03 (с, 5H)
Mass (M+1): 402
Пример 11: получение (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
60 мг соединения, полученного по примеру 1, добавляли к 10 мл 5% водного раствора гидрокарбоната натрия, и смесь экстрагировали два раза 10 мл смешанного раствора дихлорметан/2-пропанол (4/1 (об./об.)). Органический слой сушили при пониженном давлении с получением 55 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,27 (м, 1H), 7,14 (м, 1H), 4,56-4,39 (м, 1H), 3,96-3,81 (м, 3H), 3,70 (м, 1H), 3,46 (м, 1H), 3,43-3,32 (м, 1H), 2,83-2,65 (м, 3H), 2,58-2,40 (м, 2H), 1,16 (с, 3H) и 1,11 (с, 6H)
Mass (M+1): 402
Пример 12: получение тартрата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
55 мг соединения, полученного по примеру 11, растворяли в 0,56 мл ацетона и затем к нему медленно добавляли раствор 26 мг L-винно-каменной кислоты в 0,35 мл смеси этанол/вода (9/1 (об./об.)), а затем перемешивали в течение 30 мин. К этой смеси добавляли 0,56 мл 2-пропанола, а затем перемешивали в течение 10 мин и фильтровали с получением 77 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,38 (м, 1H), 7,22 (м, 1H), 4,80 (м, 1H), 4,59-4,40 (м, 1H), 4,40 (с, 2H), 3,93 (м, 1H), 3,90-3,83 (м, 2H), 3,70 (м, 1H), 3,38 (м, 2H), 3,27 (м, 1H), 3,07 (м, 2H), 2,89-2,66 (м, 2H), 1,15 (с, 3H) и 1,11 (с, 6H)
Mass (M+1): 402
Пример 13: получение цитрата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
496 мг соединения, полученного по примеру 11, растворяли в 2 мл этанола и затем к нему медленно добавляли раствор 273 мг безводной лимонной кислоты в 1 мл воды, а затем перемешивали в течение 30 мин. Реакционную смесь концентрировали и затем к ней добавляли 2 мл этилацетата и 1 мл 2-пропанола при перемешивании. К этой смеси добавляли 15 мл гексана, а затем перемешивали в течение 10 мин и фильтровали с получением 637 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,34 (м, 1H), 7,22 (м, 1H), 4,81 (м, 1H), 4,58-4,40 (м, 1H), 3,94 (м, 1H), 3,87 (м, 2H), 3,70 (м, 1H), 3,36 (м, 2H), 3,25 (м, 1H), 3,03 (м, 2H), 2,94-2,70 (м, 4H), 1,18 (с, 3H) и 1,12 (с, 6H)
Mass (M+1): 402
Пример 14: получение фосфата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
501 мг соединения, полученного по примеру 11, растворяли в 3 мл 2-пропанола и затем к нему медленно добавляли 84 мкл 85% водного раствора фосфорной кислоты при перемешивании в течение 30 мин. К нему добавляли 3 мл 2-пропанола, и полученную смесь перемешивали в течение 10 мин и фильтровали с получением 100 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,33 (м, 1H), 7,19 (м, 1H), 4,81 (м, 1H), 4,58-4,41 (м, 1H), 3,94 (м, 1H), 3,85 (м, 2H), 3,65 (м, 1H), 3,37 (м, 2H), 3,22 (м, 1H), 2,95 (м, 2H), 2,69 (м, 2H), 1,17 (с, 3H), и 1,12 (с, 6H)
Mass (M+1): 402
Пример 15: получение ацетата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
500 мг соединения, полученного по примеру 11, растворяли в 3 мл этилацетата и к нему медленно добавляли раствор 74,5 мг уксусной кислоты в 1 мл этилацетата, а затем перемешивали в течение 30 мин. Реакционную смесь концентрировали и затем к ней добавляли 2 мл этилацетата и 1 мл 2-пропанола, а затем перемешивали. К этой смеси добавляли 15 мл гексана, и полученную смесь перемешивали в течение 10 мин и фильтровали с получением 495 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,32 (м, 1H), 7,20 (м, 1H), 4,79 (м, 1H), 4,60-4,40 (м, 1H), 3,94 (м, 1H), 3,87 (м, 2H), 3,70 (м, 1H), 3,34 (м, 2H), 3,24 (м, 1H), 2,90 (м, 2H), 2,76-2,58 (м, 2H), 1,94 (с, 3H), 1,17 (с, 3H) и 1,12 (с, 6H)
Mass (M+1): 402
Пример 16: получение малата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
498 мг соединения, полученного по примеру 11, растворяли в 4 мл ацетона и к нему медленно добавляли раствор 166 мг L-яблочной кислоты в 1 мл ацетона, а затем перемешивали в течение 30 мин. Реакционную смесь концентрировали и затем к ней добавляли 2 мл этилацетата и 1 мл 2-пропанола, а затем перемешивали. К этой смеси добавляли 15 мл гексана, и полученную смесь перемешивали в течение 10 мин и фильтровали с получением 506 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,34 (м, 1H), 7,21 (м, 1H), 4,80 (м, 1H), 4,58-4,39 (м, 1H), 4,26 (м, 1H), 3,94 (м, 1H), 3,84 (м, 2H), 3,71 (м, 1H), 3,36 (м, 2H), 3,22 (м, 1H), 3,02 (м, 2H), 2,82-2,63 (м, 3H), 2,50 (м, 1H), 1,17 (с, 3H) и 1,12 (с, 6H)
Mass (M+1): 402
Пример 17: получение сукцината (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
498 мг соединения, полученного по примеру 11, растворяли в 3 мл ацетона и затем к нему медленно добавляли раствор 147 мг янтарной кислоты в 2 мл смеси ацетон/вода (20/1 (об./об.)), а затем перемешивали в течение 30 мин. Реакционную смесь концентрировали до высушивания при пониженном давлении с получением 596 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,34 (м, 1H), 7,21 (м, 1H), 4,81 (м, 1H), 4,58-4,40 (м, 1H), 3,95 (м, 1H), 3,85 (м, 2H), 3,70 (м, 1H), 3,36 (м, 2H), 3,25 (м, 1H), 2,92 (м, 2H), 2,81-2,64 (м, 2H), 2,51 (с, 4H), 1,18 (с, 3H) и 1,12 (с, 6H)
Mass (M+1): 402
Пример 18: Получение адипата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она
503 мг соединения, полученного по примеру 11, растворяли в 4 мл этилацетата и к затем к нему медленно добавляли раствор 183 мг адипиновой кислоты в 3 мл смеси ацетон/вода (30/1 (об./об.)), а затем перемешивали в течение 30 мин. Реакционную смесь концентрировали и к ней добавляли 2 мл этилацетата и 1 мл 2-пропанола, а затем перемешивали. К этой смеси добавляли 15 мл гексана, и полученную смесь перемешивали в течение 10 мин и фильтровали с получением 336 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD): 7,32 (м, 1H), 7,19 (м, 1H), 4,80 (м, 1H), 4,56-4,40 (м, 1H), 3,94 (м, 1H), 3,87 (м, 2H), 3,70 (м, 1H), 3,35 (м, 2H), 3,25 (м, 1H), 2,92 (м, 2H), 2,83-2,58 (м, 2H), 2,25 (м, 4H), 1,63 (м, 4H), 1,21 (с, 3H) и 1,12 (с, 6H)
Mass (M+1): 402
Экспериментальный пример 1: анализ ингибиторной активности DPP-IV
Для исследования ингибиторной способности DPP-IV соединений формулы 1 согласно изобретению, полученных по примерам 1-18, проводили следующие эксперименты.
DPP-IV, известную как сериновая протеаза, приобретали от R & D Systems. MK-0431 в качестве контроля получали в соответствии со способом, описанным в J. Med. Chem., 2005, 48, 141-151. Для оценки лекарственной эффективности соединений формулы 1 согласно изобретению измеряли связывающую активность синтетических ингибиторов DPP-IV с использованием флуорогенного субстрата Gly-Pro-AMC. Ферментативную реакцию проводили при 25°С в буферном растворе, содержащем 25 мМ Tris/HCl (pH 8,0) с использованием 50 мкМ Gly-Pro-AMC относительно 100 нг/мл DPP-IV с различными концентрациями ингибитора. IC50, которая представляет собой константу ингибирования для ингибитора, получали измерением флуоресценции спектрофотометром после ферментативной реакции в течение 1 часа с последующим вычислением концентрации ингибитора, которая требуется для ингибирования ферментативной реакции DPP-IV на 50%. Спектрофотометр представлял собой спектрофотометр Tecan SpectraFluor с длиной волны возбуждения 360 нм и длиной волны испускания 465 нм. В результате IC50, измеренная в качестве способности соединения формулы 1 ингибировать активность DPP-IV, находилась в диапазоне от 0,5 до 20 нМ (таблица 1: ингибиторная активность в отношении DPP-IV человека in vitro). Из этого результата можно видеть, что соединение по изобретению формулы 1 обладает превосходной ингибиторной активностью в отношении DPP-IV, по сравнению со значением IC50, описанным для коммерчески доступного JANUVIA или общепринятых ингибирующих DPP-IV соединений (находящимся в диапазоне от нескольких сотен нМ до нескольких тысяч нМ).
Экспериментальный пример 2: пероральный тест толерантности к глюкозе (OGTT)
Для исследования противодиабетических эффектов фармацевтической композиции, содержащей в качестве активного ингредиента соединение по изобретению формулы 1, проводили пероральный тест толерантности к глюкозе (OGTT), посредством которого измеряют способность организма метаболизировать глюкозу в течение данного периода времени.
Для этой цели лабораторных животных (мышей C57BL/6) подвергали голоданию в течение от 16 до 17 часов перед экспериментами. Кровь собирали из хвостовых вен животных утром в день эксперимента и измеряли уровень глюкозы в крови с помощью Accu-Chek Active Blood Glucose Meter (Roche Diagnostics). Фармацевтическую композицию с носителем вводили перорально за 30 мин до введения глюкозы (-30 мин), а затем перорально вводили раствор глюкозы (2 г/кг/10 мл) через 30 мин (0 мин). Сбор крови проводили в указанные моменты времени - непосредственно перед введением лекарственного средства, непосредственно перед введением глюкозы и через 5, 15, 30, 60 и 90 мин после введения глюкозы.
В результате примеры 1, 3 и 12 обладали наиболее высокими эффектами снижения глюкозы в крови, составляющими 54%, 52% и 62%, соответственно, в дозе 1 мг/кг, по сравнению с контрольной группой (без введения композиции с носителем). Из этих результатов можно видеть, что соединение по изобретению формулы 1 может быть подходящим для лечения связанных с DPP-IV заболеваний, включающих диабет и ожирение, вследствие высокой биодоступности.
Экспериментальный пример 3: фармакокинетическая/фармакодинамическая корреляция ингибитора DPP-IV (активность DPP-IV в плазме относительно дозы лекарственного средства)
Для выявления противодиабетических эффектов соединения по изобретению формулы 1 проводили сравнительную оценку ингибиторной активности в отношении DPP-IV между соединениями по изобретению и MK-0431. Мышам в возрасте 8 недель C57BL6 перорально вводили MK-0431 и соединение по изобретению (соединение примера 1) в индивидуальных дозах, а затем вводили глюкозу в дозе 2 г/кг через 1 час. Через 10 мин кровь собирали через глаза животных. Из собранной крови получали плазму крови и измеряли активность DPP-IV в плазме и концентрацию лекарственного средства в плазме.
Активность DPP-IV в плазме определяли посредством измерения количества флуоресцентного AMC (7-амино-4-метилкумарина), высвобожденного под действием DPP-IV при использовании в качестве субстрата Gly-Pro-AMC (Bachem, Switzerland). Для этой цели к реакционному раствору добавляли 50 мкл плазмы (100 мМ HEPES, pH 7,6, 0,1 мг/мл, 50 мкМ Gly-Pro-AMC) и вычисляли скорость высвобождения AMC при 25°С в течение 5 мин.
В результате соединение формулы 1 (пример 1) обладало от 4 до 5 раз более высокой ингибиторной активностью при концентрации в плазме 10 нг/мл и от 8 до 9 раз превышающей EC50 (50% эффективной концентрацией) и EC80 (80% эффективной концентрацией), по сравнению с MK-0431 (см. фиг.1).
Экспериментальный пример 4: анализ DPP-IV in vivo (длительность ингибиторной активности в отношении DPP-IV)
Для исследования противодиабетических эффектов соединения по изобретению формулы 1 проводили сравнительную оценку ингибиторной активности в отношении DPP-IV в плазме и ее длительности между MK-0431 и соединением по изобретению (пример 1), после введения лекарственных соединений нормальным крысам SD.
Для этой цели лабораторных животных (крыс SD) подвергали голоданию в течение от 16 до 17 часов перед экспериментами. В день эксперимента голодным животным проводили анестезию эфиром, а затем канюлирование абдоминального отдела аорты. Затем MK-0431 и соединение по изобретению (пример 1) разбавляли до 0,5% MC и вводили животным. Перед введением лекарственного средства (0 ч) и через указанные периоды времени после введения лекарственного средства кровь собирали в предварительно подготовленные 500-мкл пробирки с гепарином и отделяли плазму. В каждый реакционный раствор добавляли 50 мкл плазмы (0,1 M HEPES, pH 7,6, 0,1 мг/мл, 50 мкМ Gly-Pro-AMC) и проводили исследование кинетики в течение 5 мин для вычисления скорости реакции.
В результате было выявлено, что соединение по изобретению в дозе 10 мг/кг сохраняет 90% или более ингибиторной активности в отношении DPP-IV до 24 часов после введения, что является значительно более высокой активностью с учетом того факта, что MK-0431 сохранял только 50% ингибиторной активности в отношении DPP-IV после того же периода, составляющего 24 часа (см. фиг.2).
Экспериментальный пример 5: кинетические эксперименты in vivo
Для измерения времени полужизни in vivo соединения по изобретению формулы 1 нормальным крысам SD (в возрасте 8 недель) перорально вводили MK-0431 и соединение по изобретению (примеры 1, 3 и 12) в дозе 10 мг/кг. Периодически проводили забор крови из бедренного отдела аорты и измеряли время удерживания исходного соединения in vivo. В результате соединение по изобретение обладало более высоким временем полужизни in vivo (T1/2) относительно времени полужизни MK-0431.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Как очевидно из приведенного выше описания, настоящее изобретение обеспечивает продукцию гетероциклического соединения, содержащего бета-аминогруппу, которое обладает превосходными ингибиторными эффектами на активность DPP-IV. Кроме того, фармацевтическая композиция, содержащая указанное соединение по настоящему изобретению в качестве активного ингредиента, обладает превосходной ингибиторной активностью в отношении DPP-IV и биодоступностью, и поэтому она может быть пригодна для предупреждения или лечения различных заболеваний, считающихся вызываемыми посредством DPP-IV, таких как диабет и ожирение.
Изобретение относится к соединению формулы 1

где Х представляет собой OR1, SR1 или NR1R2, где R1 и R2 независимо представляют собой С1-С5 низший алкил, и R1 и R2 в NR1R2 могут образовывать 5-7-членное кольцо, включающее гетероатом О; или к его стереоизомеру, фармацевтически приемлемой соли, гидрату или сольвату. Изобретение также относится к способу его получения и к фармацевтической композиции на его основе, обладающей ингибиторной активностью в отношении DPP-IV. Технический результат - получены новые соединения, которые могут найти свое применение в медицине для предупреждения или лечения связанных с DPP-IV заболеваний, таких как диабет или ожирение. 6 н. и 6 з.п. ф-лы, 1 табл., 2 ил.
1. Соединение, представленное формулой 1
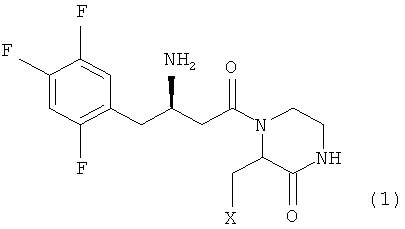
где X представляет собой OR1, SRI или NR1R2, где R1 и R2 независимо представляют собой C1-С5 низший алкил, и R1 и R2 в NR1R2 могут образовывать 5-7-членное кольцо, включающее гетероатом О; или его стереоизомер, фармацевтически приемлемая соль, гидрат или сольват.
2. Соединение по п.1, где соединение представлено формулой 2
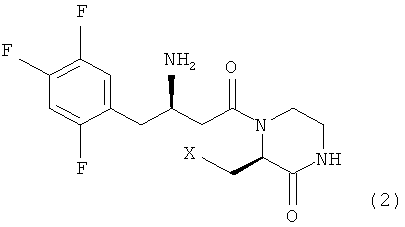
где X является таким, как определено в п.1, или его стереоизомер, фармацевтически приемлемая соль, гидрат или сольват.
3. Соединение по п.1, где соединение выбрано из группы, состоящей из:
1) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(метоксиметил)пиперазин-2-она;
2) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(этоксиметил)пиперазин-2-она;
3) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(изопропоксиметил)пиперазин-2-она;
4) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
5) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(циклопентилоксиметил)пиперазин-2-она;
6) дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-[(диэтиламино)метил]пиперазин-2-она;
7) дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-[(этилметиламино)метил]пиперазин-2-она;
8) дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(морфолинометил)пиперазин-2-она;
9) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутилтиометил)пиперазин-2-она;
10) гидрохлорида (S)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
11) (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-он;
12) тартрата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
13) цитрата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
14) фосфата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
15) ацетата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
16) малата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
17) сукцината (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она и
18) адипата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она.
4. Способ получения соединения, представленного формулой 2 по п.2, включающий:
1) реакцию соединения формулы 3, имеющего бета-аминогруппу с замещенным гетероциклическим соединением формулы 4, в присутствии 1-гидроксибензотриазола (НОВТ), 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC) и третичного амина с получением таким образом соединения формулы 5, и
2) обработку соединения формулы 5, полученного на стадии (1), кислотой с получением соединения формулы 2
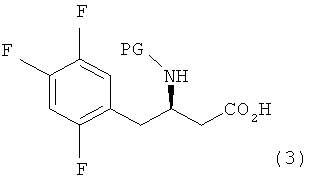
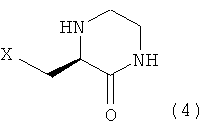
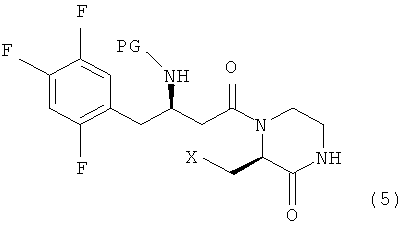
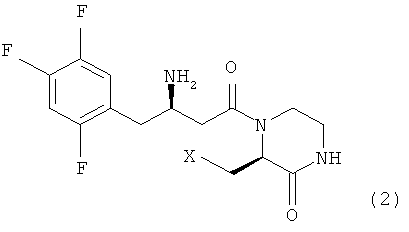
где X представляет собой OR1, SR1 или NR1R2, где R1 и R2 независимо представляют собой C1-C5 низший алкил, и R1 и R2 в NR1R2 могут образовывать 5-7-членное кольцо, включающее гетероатом О.
5. Фармацевтическая композиция для предупреждения и лечения диабета или ожирения, содержащая соединение по п.1, представленное формулой 1, или его стереоизомер, фармацевтически приемлемую соль, гидрат или сольват в качестве активного ингредиента.
6. Композиция по п.5, где соединение выбрано из группы, состоящей из:
1) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
2) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(этоксиметил)пиперазин-2-она;
3) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(изопропоксиметил)пиперазин-2-она;
4) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(метоксиметил)пиперазин-2-она;
5) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(циклопентилоксиметил)пиперазин-2-она;
6) дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-[(диэтиламино)метил]пиперазин-2-она;
7) дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-[(этилметиламино)метил]пиперазин-2-она;
8) дигидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(морфолинометил)пиперазин-2-она;
9) гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутилтиометил)пиперазин-2-она;
10) гидрохлорида (S)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
11) (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-он;
12) тартрата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
13) цитрата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
14) фосфата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
15) ацетата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
16) малата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она;
17) сукцината (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она и
18) адипата (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она.
7. Способ предупреждения и лечения диабета или ожирения, включающий введение эффективного количества композиции, содержащей соединение по любому из пп.1-3, млекопитающему, нуждающемуся в этом.
8. Применение фармацевтической композиции, содержащей соединение по любому из пп.1-3, для получения лекарственного средства для предупреждения и лечения диабета или ожирения.
9. Способ по п.4, где PG означает Воc.
10. Соединение, представленное формулой 4, предназначенное для получения соединения формулы 2 по п.2
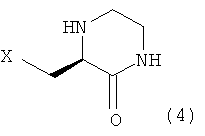
где Х означает OR1, SR1 или NR1R2, где R1 и R2 независимо представляют собой С1-С5 низший алкил, и R1 и R2 в NR1R2 могут образовывать 5-7-членное кольцо, включающее гетероатом О.
11. Соединение по п.10, где Х выбирают из группы, состоящей из трет-бутокси, метокси, этокси, изопропокси, циклопентилокси, диэтиламино, этилметиламино, морфолино и трет-бутилтио.
12. Соединение по п.10, где Х означает трет-бутокси.
| WO 2005123685 A1, 29.12.2005 | |||
| WO 2005056003 A1, 23.06.2005 | |||
| WO 2005121131 A1, 22.12.2005 | |||
| WO 2005095343 A1, 13.10.2005 | |||
| US 20070049619 A1, 01.03.2007 | |||
| ЗАМЕЩЕННЫЕ 1-АМИНОАЛКИЛЛАКТАМЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2243222C2 |
| ЗАМЕЩЕННЫЕ 1-АМИНОАЛКИЛЛАКТАМЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2241702C2 |

