Предпосылки изобретения
Диабет относится к патологическому процессу, вызванному многочисленными этиологическими факторами, и характеризуется повышенными уровнями глюкозы в плазме либо гипергликемией натощак или после введения глюкозы при проведении теста на толерантность к глюкозе при пероральном приеме. Стойкая или неконтролируемая гипергликемия связана с повышенным числом заболеваний и преждевременной смертностью. Часто аномальный гомеостаз глюкозы связан как с прямыми, так и опосредованными изменениями метаболизма липидов, липопротеинов и аполипопротеинов и другим метаболическим и гемодинамическим нарушением. Следовательно, пациенты, страдающие сахарным диабетом типа 2, имеют особенно повышенный риск макрососудистых и микрососудистых осложнений, включающих коронарную болезнь сердца, инсульт, заболевание периферических сосудов, гипертензию, нефропатию, нейропатию и ретинопатию. Поэтому терапевтическое регулирование гомеостаза глюкозы, метаболизма липидов и гипертензии является крайне важным в клинической терапии и при лечении сахарного диабета.
Существуют две обычно определяемые формы диабета. При диабете типа 1 или инсулинзависимом сахарном диабете (ИЗСД) у пациентов вырабатывается небольшое количество инсулина либо не вырабатывается инсулин, гормон, который регулирует утилизацию глюкозы. При диабете типа 2 или инсулиннезависимом сахарном диабете (ИНЗСД) содержание инсулина в плазме пациентов часто такое же или даже повышенное по сравнению с таковым у людей, которые не страдают диабетом; однако у таких пациентов развивается резистентность к инсулинстимулирующему действию на глюкозу и метаболизм липидов в основных тканях, чувствительных к инсулину, таких как мышцы, печень и жировые ткани, и содержание инсулина в плазме, хотя и повышенное, является недостаточным для компенсации четко выраженной резистентности к инсулину.
Резистентность к инсулину, по существу, не является следствием уменьшенного количества инсулиновых рецепторов, а следствием дефекта связывания пост-инсулинового рецептора, который еще не изучен. Такая резистентность к инсулиновой восприимчивости приводит в результате к недостаточной активации инсулина при накоплении глюкозы, окислению и сохранению глюкозы в мышцах, и недостаточному подавлению инсулина при липолизисе в жировых тканях и продуцированию и секреции глюкозы в печени.
Доступные способы лечения диабета типа 2, которые не изменились существенным образом за многие годы, выявили ряд ограничений. Несмотря на то, что физические упражнения и снижение калорий в пищевом рационе может эффективно улучшить диабетическое состояние, соблюдение такого лечения является очень сложным из-за прочно укрепившегося малоподвижного образа жизни и избытка потребления пищи, особенно пищи, содержащей высокое количество насыщенных жиров. Увеличение уровня инсулина в плазме путем введения сульфонилмочевины (например, толбутамида и глипизида) или меглитинида, которые стимулируют β-клетки поджелудочной железы для секреции большего количества инсулина, и/или путем инъекции инсулина, если сульфонилмочевина или меглитинид становятся неэффективными, могут привести к концентрациям инсулина выше достаточной для стимуляции особенно инсулин-резистентных тканей. Однако опасно низкие уровни глюкозы в плазме могут быть результатом введения инсулина или стимуляторов секреции инсулина (сульфонилмочевины или меглитинида) и могут привести к повышенному уровню резистентности к инсулину вследствие даже более высоких уровней инсулина в плазме. Бигуаниды увеличивают чувствительность к инсулину, приводящую к некоторой коррекции гипергликемии. Однако два бигуанидина, фенформин и метформин могут индуцировать лактоцидоз и тошноту/диарею. Метформин имеет меньше побочных эффектов, чем фенформин, и часто назначается при лечении диабета типа 2.
Глитазоны (т.е. 5-бензилтиазолидин-2,4-дионы) представляют собой недавно описанный класс соединений с потенциалом улучшения многих симптомов диабета типа 2. Такие агенты, по существу, увеличивают чувствительность к инсулину в мышцах, печени и жировой ткани в некоторых моделях животных диабета типа 2, что приводит в результате к частичной или полной коррекции повышенных уровней глюкозы в плазме без случаев гипогликемии. Глитазоны, которые продаются в настоящее время, являются агонистами рецептора, активируемыми пролифераторами пероксисом (PPAR), главным образом подтипа PPAR-гамма. Обычно предполагается, что PPAR-гамма агонизм является ответственным за улучшение сенсибилизации к инсулину, которая обнаружена с помощью глитазонов. Более новые агонисты PPAR, которые начали тестировать для лечения диабета типа 2, являются агонистами подтипа альфа, гамма или дельта, или их комбинацией и во многих случаях химически отличаются от глитазонов (т.е. они не являются тиазолидиндионами). Серьезные побочные эффекты (например, гепатотоксичность) возникали с некоторыми глитазонами, такими как троглитазон.
Другие способы лечения заболевания все еще находятся на стадии исследования. Новые биохимические достижения, которые введены недавно или все еще находятся на стадии разработки, включают в себя лечение ингибиторами альфа-глюкозидов (например, акарбоз) и ингибиторами протеина тирозин-фосфатазы-1B (PTP-1B).
Соединения, которые являются ингибиторами фермента дипептидилпептидазы-IV ("DP-IV" или "DPP-IV"), также находятся на стадии исследования как лекарственные средства, которые могут быть пригодными при лечении диабета, и более конкретно диабета типа 2. См., например, WO 97/40832, WO 98/19998, патент США №. 5939560, Bioorg. Med. Chem. Lett., 6: 1163-1166 (1996); и Bioorg. Med. Chem. Lett. 6: 2745-2748 (1996). Применимость ингибиторов DP-IV при лечении диабета типа 2 основана на том факте, что DP-IV in vivo легко инактивирует глюкагон-подобный пептид-1 (GLP-1) и гастроингибирующий кишечный пептид (GIP). GLP-1 и GDP являются инкретинами и продуцируются при потреблении пищи. Инкретины стимулируют продуцирование инсулина. Ингибирование DP-IV приводит к снижению инактивации инкретинов и это, в свою очередь, приводит к увеличению эффективности инкретинов при стимуляции продуцирования инсулина поджелудочной железой. Таким образом, ингибирование DP-IV приводит в результате к повышенному уровню инсулина в сыворотке. Преимущественно, поскольку инкретины продуцируются телом только при приеме пищи, предполагается, что ингибирование DP-IV не увеличивает уровень инсулина в несоответствующие моменты времени, такие как между приемами пищи, что может привести к чересчур низкому уровню сахара в крови (гипогликемии). Следовательно, предполагается, что ингибирование DP-IV увеличивает инсулин без увеличения риска гипогликемии, которая представляет собой опасный побочный эффект, связанный с использованием стимуляторов секреции инсулина.
Ингибиторы DP-IV также имеют другие терапевтические применения, как изложено в настоящем описании. Ингибиторы DP-IV в настоящее время еще не изучены в достаточной степени, особенно с точки зрения их применимости в отношении не только диабета, но и других заболеваний. Для того чтобы можно было обнаружить улучшенные DP-IV для лечения диабета и потенциально других заболеваний и состояний, необходимы новые соединения. Терапевтический потенциал ингибиторов DP-IV для лечения диабетов типа 2 обсуждается D. J. Drucker в Exp. Opin. Invest. Drugs, 12: 87-100 (2003) и K. Augustyns, и др., в Exp. Opin. Ther. Patents. 13: 499-510 (2003).
Сущность изобретения
Настоящее изобретение относится к соединениям, которые являются ингибиторами фермента дипептидилпептидазы-IV ("DP-IV inhibitors") и могут использоваться при лечении или предупреждении заболеваний, в которые вовлечен фермент дипептидилпептидаза-IV, таких как диабет и более конкретно диабет типа 2. Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие соединения, и применению таких соединений и композиций для предупреждения или лечения таких заболеваний, в которые вовлечен фермент дипептидилпептидаза-IV.
Подробное описание изобретения
Настоящее изобретение относится к производным гексагидродиазепинона, которые могут использоваться в качестве ингибитора дипептидилпептидазы-IV. Соединения по настоящему изобретению описываются структурной формулой I:
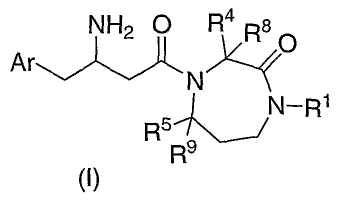
или ее фармацевтически приемлемой солью; где каждый n, независимо, равен 0, 1 или 2;
Ar представляет собой фенил, замещенный от одного до пяти заместителями R3;
R1 выбран из группы, состоящей из
водорода,
C1-10алкила, где алкил является незамещенным или замещенным от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкокси, карбокси, C1-6алкилоксикарбонила и фенил-C1-3алкокси, где алкокси является незамещенным или замещенным от одного до пяти атомами галогена,
(CH2)n-арила, где арил является незамещенным или замещенным от одного до пяти заместителями, независимо выбранными из галогена, CN, гидрокси, R2, OR2, NHSO2R2, NR2SO2R2, SO2R2, CO2H, и C1-6алкилоксикарбонила,
(CH2)n-гетероарила, где гетероарил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из гидрокси, галогена, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-гетероциклила, где гетероциклил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из оксо, гидрокси, галогена, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-C3-6циклоалкила, где циклоалкил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена; и
где любой атом углерода метилена (CH2) в R1 является незамещенным или замещенным одной или двумя группами, независимо выбранными из галогена, гидрокси и C1-4алкила, незамещенного или замещенного от одного до пяти атомами галогена;
каждый R3 независимо выбран из группы, состоящей из
водорода,
галогена,
циано,
гидрокси,
C1-6алкила, незамещенного или замещенного от одного до пяти атомами галогена,
C1-6алкокси, незамещенного или замещенного от одного до пяти атомами галогена,
карбокси,
алкоксикарбонила,
амино,
NHR2,
NR2R2,
NHSO2R2,
NR2SO2R2,
NHCOR2,
NR2COR2,
NHCO2R2,
NR2CO2R2,
SO2R2,
SO2NH2,
SO2NHR2 и
SO2NR2R2;
каждый R2 независимо представляет собой C1-6алкил, незамещенный или замещенный от одного до пяти заместителями, независимо выбранными из галогена, CO2H и C1-6алкилоксикарбонила;
R4 и R5 независимо выбраны из группы, состоящей из:
водорода,
циано,
карбокси,
C1-6алкилоксикарбонила,
C1-10алкила, незамещенного или замещенного от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкокси, карбокси, C1-6алкилоксикарбонила, и фенил-C1-3алкокси, где алкокси является незамещенным или замещенным от одного до пяти атомами галогена,
(CH2)n-арила, где арил является незамещенным или замещенным от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-гетероарила, где гетероарил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из гидрокси, галогена, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-гетероциклила, где гетероциклил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из оксо, гидрокси, галогена, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-C3-6циклоалкила, где циклоалкил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)nCONR6R7, где R6 и R7 независимо выбраны из группы, состоящей из водорода, тетразолила, тиазолила, (CH2)n-фенила, (CH2)n-C3-6циклоалкила и C1-6алкила, где алкил является незамещенным или замещенным от одного до пяти атомами галогена, и где фенил и циклоалкил являются незамещенными или замещенными от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена и;
или где R6 и R7 вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо, выбранное из азетидина, пирролидина, пиперидина, пиперазина и морфолина; и где указанное гетероциклическое кольцо является незамещенным или замещенным от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена; и где любой атом углерода метилена (CH2) в R4 или R5 является незамещенным или замещенным одной или двумя группами, независимо выбранными из галогена, гидрокси и C1-4алкила, незамещенного или замещенного от одного до пяти атомами галогена; и
R8 и R9, каждый, независимо, представляют собой водород или C1-6алкил.
В одном из вариантов осуществления соединений по настоящему изобретению атом углерода, помеченный *, имеет Rконфигурацию, как изображено на формуле Ia:
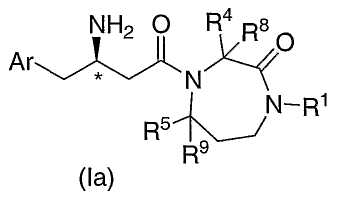
где Ar, R1, R4, R5, R8 и R9 являются такими, как определено в настоящем описании.
Во втором варианте осуществления соединений по настоящему изобретению R3 выбран из группы, состоящей из
водорода,
галогена,
циано,
гидрокси,
C1-6алкила, незамещенного или замещенного от одного до пяти атомами галогена, и
C1-6алкокси, незамещенного или замещенного от одного до пяти атомами галогена.
В классе таких вариантов осуществления R3 выбран из группы, состоящей из водорода, фтора, хлора, брома, трифторметила и метила. В подклассе указанного класса R3 выбран из группы, состоящей из водорода, фтора и хлора.
В третьем варианте осуществления соединений по настоящему изобретения R1 выбран из группы, состоящей из:
водорода,
C1-6алкила, где является незамещенным или замещенным от одного до пяти заместителями, выбранными из галогена, гидрокси, C1-6алкокси, карбокси, C1-6алкилоксикарбонила и фенил-C1-3алкокси, где алкокси является незамещенным или замещенным от одного до пяти атомами галогена,
(CH2)n-C3-6циклоалкила, где циклоалкил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена; и
где любой атом углерода метилена (CH2) в R1 является незамещенным или замещенным одной или двумя группами, независимо выбранными из галогена, гидрокси и C1-4алкила, незамещенного или замещенного от одного до пяти атомами галогена.
В классе такого варианта осуществления соединений по настоящему изобретению R1 выбран из группы, состоящей из
водорода,
C1-4алкила,
2,2,2-трифторэтила,
метоксикарбонилметила,
карбоксиметила,
гидроксиэтила,
бензилоксиметила,
бензилоксиэтила и
циклопропила.
В подклассе указанного класса R1 выбран из группы, состоящей из водорода, метила, трет-бутила и циклопропила.
В четвертом варианте осуществления соединений по настоящему изобретению R4 и R5 независимо выбраны из группы, состоящей из:
водорода,
С1-10алкила, незамещенного или замещенного от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкокси, карбокси, C1-6алкилоксикарбонила и фенил-C1-3алкокси, где алкокси является незамещенным или замещенным от одного до пяти атомами галогена,
(CH2)n-арила, где арил является незамещенным или замещенным от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-гетероарила, где гетероарил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из гидрокси, галогена, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-гетероциклила, где гетероциклил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из оксо, гидрокси, галогена, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-C3-6циклоалкила, где циклоалкил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
где любой атом углерода метилена (CH2) в R4 или R5 является незамещенным или замещенным одной или двумя группами, независимо выбранными из галогена, гидрокси и C1-4алкила, незамещенного или замещенного от одного до пяти атомами галогена.
В классе такого варианта осуществления соединений по настоящему изобретению R4 и R5 независимо выбраны из группы, состоящей из:
водорода,
C1-6алкила, незамещенного или замещенного от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкокси, карбокси, C1-6алкилоксикарбонила и фенил-C1-3алкокси, где алкокси является незамещенным или замещенным от одного до пяти атомами галогена,
(CH2)n-арила, где арил является незамещенным или замещенным от одного до пяти заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-гетероарила, где гетероарил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из гидрокси, галогена, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена,
(CH2)n-C3-6циклоалкила, где циклоалкил является незамещенным или замещенным от одного до трех заместителями, независимо выбранными из галогена, гидрокси, C1-6алкила и C1-6алкокси, где алкил и алкокси являются незамещенными или замещенными от одного до пяти атомами галогена, и
где любой атом углерода метилена (CH2) в R4 или R5 является незамещенным или замещенным одной или двумя группами, независимо выбранными из галогена, гидрокси и C1-4алкила, незамещенного или замещенного от одного до пяти атомами галогена.
В подклассе указанного класса R4 и R5 независимо выбраны из группы, состоящей из:
водорода,
CH3,
CH2CH3,
CH2CH(CH3)2,
CH2-циклопропила,
CH2-циклогексила,
CH2OCH2Ph,
CH2OH
CH2Ph,
CH2(3-OCF3-Ph),
CH2(4-OCF3-Ph),
CH2(3-CF3,5-CF3-Ph),
CH2(2-CF3-Ph),
CH2(2-Cl-Ph),
CH2(2-Me-Ph),
CH2(2-Me,5-Me-Ph),
CH2(2-Ph-Ph),
CH2(2-F,5-F-Ph),
CH2(2-F-Ph),
CH2(2-F,3-F-Ph),
CH2(2-пиридинил),
CH2(3-пиридинил),
CH2(4-пиридинил),
CH2(1-оксидопиридин-2-ил),
CH2(1-оксидопиридин-3-ил),
CH2(1H-пиразол-1-ил),
CH2(2-F,6-F-Ph) и
CH2CF3.
В дополнительном подклассе указанного класса R5 представляет собой водород.
В пятом варианте осуществления соединений по настоящему изобретению R8 и R9 независимо выбираны из водорода и метила.
В классе такого варианта осуществления R8 и R9 представляют собой водород.
В шестом варианте осуществления по настоящему изобретению представлены соединения формулы Ia, где R1 выбран из группы, состоящей из
водорода,
C1-4алкила,
2,2,2-трифторэтила,
метоксикарбонилметила,
карбоксиметила,
гидроксиэтила,
бензилоксиметила,
бензилоксиэтила и
циклопропила;
R3 представляет собой водород, хлор или фтор;
R4 выбран из группы, состоящей из:
водорода,
CH3,
CH2CH3,
CH2CH(CH3)2,
CH2-циклопропила,
CH2-циклогексила,
CH2OCH2Ph,
CH2OH
CH2Ph,
CH2(3-OCF3-Ph),
CH2(4-OCF3-Ph), и
CH2(3-CF3,5-CF3-Ph),
CH2(2-CF3-Ph),
CH2(2-Cl-Ph),
CH2(2-Me-Ph),
CH2(2-Me,5-Me-Ph),
CH2(2-Ph-Ph),
CH2(2-F,5-F-Ph),
CH2(2-F-Ph),
CH2(2-F,3-F-Ph),
CH2(2-пиридинил),
CH2(3-пиридинил),
CH2(4-пиридинил),
CH2(1-оксидопиридин-2-ил),
CH2(1-оксидопиридин-3-ил),
CH2(1H-пиразол-1-ил),
CH2(2-F,6-F-Ph) и
CH2CF3; и
R8 и R9 представляют собой водород.
В классе такого варианта осуществления R5 представляет собой водород.
Иллюстративные, но не ограничивающие примеры соединений по настоящему изобретению, которые могут быть использованы в качестве ингибиторов дипептидилпиптедазы-IV являются следующими:
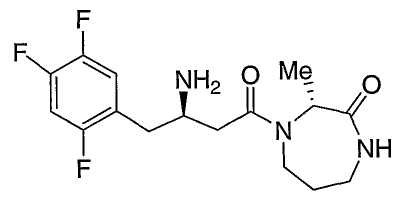
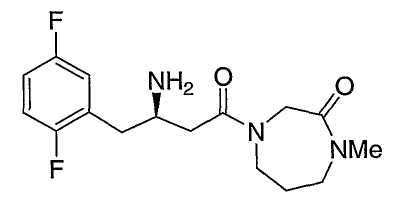
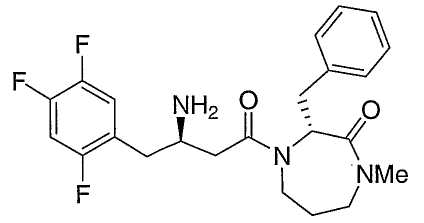
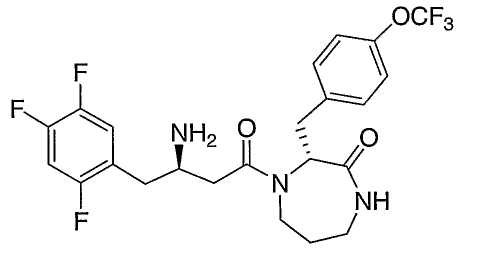
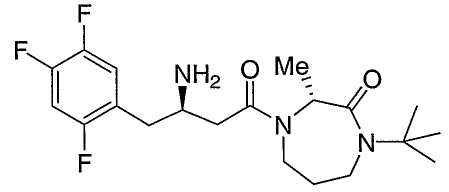
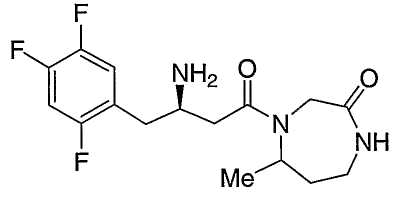
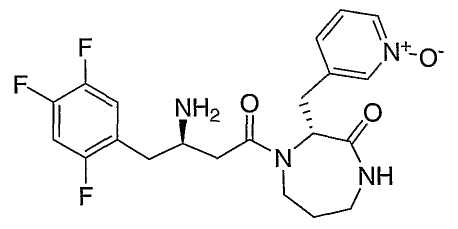
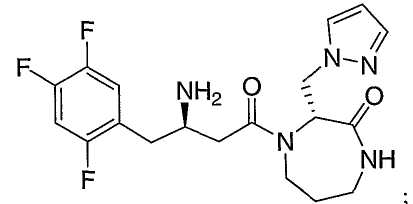
или их фармацевтически приемлемые соли.
Как используется в настоящем описании, применяются нижеследующие определения:
"Алкил", а также другие группы, имеющие приставку "алк", такие как алкокси и алканоил, обозначают углеродную цепь, которая может быть линейной или разветвленной, и их сочетания, кроме углеродной цепи, определенной иным способом. Примеры алкильных групп включают в себя метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и т.п. Там, где допускается определенное количество атомов углерода, например из C3-10, термин алкил также включает в себя циклоалкильные группы и сочетания линейных или разветвленных алкильных цепей, объединенных с циклоалкильными структурами. Если количество атомов углерода не указано, подразумевается C1-6.
"Циклоалкил" представляет собой подкласс алкила и означает насыщенное углеводородное кольцо, имеющее определенное количество атомов углерода. Примеры циклоалкила включают в себя циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и т.п. Обычно циклоалкильная группа представляет собой моноциклическую, кроме установленных иным способом. Группы циклоалкила являются насыщенными, кроме определенных иным способом.
Термин "алкокси" относится к алкоксидам линейной или разветвленной цепи с определенным количеством атомов углерода (например, C1-6алкокси) или любым количеством внутри данного диапазона [т.е. метокси (MeO-), этокси, изопропокси и т.д.].
Термин "алкилтио" относится к алкилсульфидам линейной или разветвленной цепи с определенным количеством атомов углерода (например, C1-6алкилтио) или любым количеством внутри данного диапазона [т.е. метилтио (MeS-), этилтио, изопропилтио и т.д.].
Термин "алкиламино" относится к линейным или разветвленным алкиламинам с определенным количеством атомов углерода (например, C1-6алкиламино) или любым количеством внутри данного диапазона [т.е. метиламино, этиламино, изопропиламино, трет-бутиламино, и т.д.].
Термин "алкилсульфонил" относится к алкилсульфонилам линейной или разветвленной цепи с определенным количеством атомов углерода (например, C1-6алкилсульфонил) или любым количеством внутри данного диапазона [т.е. метилсульфонил (MeSO2-), этилсульфонил, изопропилсульфонил, и т.д.].
Термин "алкилоксикарбонил" относится к сложным эфирам, линейной или разветвленной цепи, производным карбоновых кислот по настоящему изобретению с определенным количеством атомов углерода (например, C1-6 алкилоксикарбонил), или любым количеством внутри данного диапазона [т.е. метилоксикарбонил (MeOCO-), этилоксикарбонил или бутилоксикарбонил].
"Арил" означает моно- или систему полициклических ароматических колец, содержащих атомы углерода. Предпочтительными арилами являются моноциклические или системы бициклических 6-10-членных ароматических колец. Фенил и нафтил являются предпочтительными арилами. Наиболее предпочтительным арилом является фенил.
"Гетероцикл" и "гетероциклил" относятся к насыщенным или ненасыщенным неароматическим кольцам или системам колец, содержащим, по меньшей мере, один гетероатом, выбранный из O, S и N, дополнительно включающим в себя окисленные формы серы, а именно SO и SO2. Примеры гетероциклов включают в себя тетрагидрофуран (THF), дигидрофуран, 1,4-диоксан, морфолин, 1,4-дитиан, пиперазин, пиперидин, 1,3-диоксолан, имидазолидин, имидазолин, пирролин, пирролидин, тетрагидропиран, дигидропиран, оксатиолан, дитиолан, 1,3-диоксан, 1,3-дитиан, оксатиан, тиоморфолин и т.п.
"Гетероарил" означает ароматический или частично ароматический гетероцикл, который содержит, по меньшей мере, один гетероатом в кольце, выбранный из O, S и N. Таким образом, гетероарилы включают в себя гетероарилы, слитые с другими видами колец, такими как арилы, циклоалкилы и гетероциклы, которые не являются ароматическими. Примеры гетероарильных групп включают в себя: пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фурил, триазинил, тиенил, пиримидил, бензизоксазолил, бензоксазолил, бензотиазолил, бензотиадиазолил, дигидробензофуранил, индолинил, пиридазинил, индазолил, изоиндолил, дигидробензотиенил, индолизинил, циннолинил, фталазинил, хиназолинил, нафтиридинил, карбазолил, бензодиоксолил, хиноксалинил, пуринил, фуразанил, изобензилфуранил, бензимидазолил, бензофуранил, бензотиенил, хинолил, индолил, изихинолил, дибензофуранил и т.п. Для гетероциклильных и гетероарильных групп включаются кольца и системы колец, содержащие от 3-15 атомов, образующие 1-3 кольца.
"Галоген" относится к фтору, хлору, брому и йоду. В общем случае хлор и фтор являются предпочтительными. Фтор является наиболее предпочтительным, если галогены являются замещенными на алкильной или алкокси группе (например, CF3O и CF3CH2O).
Соединения по настоящему изобретению могут содержать один или несколько асимметричных центров и таким образом могут встречаться в виде рацематов и рацемических смесей, одних энантиомеров, смесей диастереомеров и отдельных диастереомеров. Соединения по настоящему изобретению имеют один асимметрический центр на атоме углерода, помеченном * в формуле Ia. Дополнительные асимметрические центры могут присутствовать в молекуле в зависимости от природы различных заместителей. Каждый такой асимметрический центр может независимо создавать два оптических изомера и предполагается, что все возможные оптические изомеры и диастереомеры в смесях и в виде чистых или частично очищенных соединений включены в объем по настоящему изобретению. Подразумевается, что настоящее изобретение охватывает все такие изомерные формы указанных соединений.
Некоторые соединения, представленные в настоящем описании, содержат олефиновые двойные связи, и, если только не указано противное, подразумевается, что они включают в себя как E, так и Z геометрические изомеры.
Некоторые соединения, представленные в настоящем описании, могут существовать в виде таутомеров, которые имеют разные точки присоединения водорода, сопровождаемые одним или несколькими перемещениями двойных связей. Например, кетон и его енольная форма представляют собой кето-енольный таутомер. Отдельные таутомеры, а также их смеси охватываются соединениями по настоящему изобретению.
Формула I показывает структуры класса соединений без предпочтительной стереохимии. Формула Ia показывает предпочтительную стереохимию атома углерода, к которому присоединена аминогруппа бета-аминокислоты, из которой эти соединения получают.
Независимый синтез этих диастереомеров или их хроматографическое разделение могут быть осуществлены, как известно, в данной области техники, при помощи подходящей модификации методов, описанных в настоящем описании. Их абсолютная стереохимия может быть определена при помощи рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных продуктов, которые преобразуют, если необходимо, с помощью реагентов, содержащих асимметричный центр известной абсолютной конфигурации.
При желании, рацемические смеси соединений могут быть разделены для того, чтобы выделить отдельные энантиомеры. Разделение может быть осуществлено способами, хорошо известными в данной области техники, такими как связывание рацемической смеси соединений с энантиомерно чистым соединением для образования смеси диастереомеров, с последующим разделением отдельных диастереомеров стандартными способами, такими как фракционная кристаллизация или хроматография. Реакция присоединения часто представляет собой образование солей с использованием энантиомерно чистых кислоты или основания. Затем производные диастереомеров могут быть преобразованы в чистые энантиомеры расщеплением дополнительных хиральных остатков. Рацемические смеси таких соединений также могут быть разделены непосредственно хроматографическими способами, использующими хиральные неподвижные фазы, способами, которые хорошо известны в данной области техники.
В качестве альтернативы любой энантиомер соединения может быть получен стереоселективным синтезом, используя оптически чистые исходные вещества или реагенты известной конфигурации, способами, хорошо известными в данной области техники.
Должно быть понятно, как используется в настоящем описании, что ссылки на соединения структурной формулы I также означают включение фармацевтически приемлемых солей, а также солей, которые не являются фармацевтически приемлемыми, если они использованы в качестве предшественников для свободных соединений или их фармацевтически приемлемых солей, или при других синтетических манипуляциях.
Соединения по настоящему изобретению могут быть введены в виде фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли основных соединений, охваченные термином "фармацевтически приемлемая соль", относятся к нетоксичным солям соединений по настоящему изобретению, которые обычно получают при взаимодействии реакции свободного основания с подходящей органической или неорганической кислотой. Характерные соли основных соединений по настоящему изобретению включают в себя, без ограничения, следующие: ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, камзилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глутамат, глюколлиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, соль N-метилглюкамин аммония, олеат, оксалат, памоат (эмбонат), палмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат. Кроме того, если соединения по настоящему изобретению несут кислую составляющую, их подходящие фармацевтически приемлемые соли включают в себя, без ограничения, соли, полученные из неорганических оснований, включающих в себя алюминий, аммоний, кальций, медь, железо (III), железо (II), литий, магний, марганец (III), марганец (II), калий, натрий, цинк и т.п. Более конкретно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксических оснований, включают в себя соли первичных, вторичных и третичных аминов, циклических аминов и основных неионнообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Также в случае присутствия в соединениях по настоящему изобретению группы карбоновой кислоты (-COOH) или спирта могут использоваться фармацевтически приемлемые сложные эфиры, производные карбоновых кислот, такие как метиловый, этиловый, или пивалоилоксиметиловый, или ацильные производные спиртов, такие как ацетат или малеат. Включены эфиры и ацильные группы, известные в данной области техники для модификации характеристик растворимости или гидролиза для использования в качестве составов с замедленным высвобождением или пролекарства.
Сольваты и более конкретно гидраты соединений структурной формулы I также включены в настоящее изобретение.
Иллюстрацией по настоящему изобретению является применение соединений, приведенное в примерах и настоящем описании.
Целевые соединения пригодны в способе ингибирования фермента дипептидилпептидазы-IV у пациента, такого как млекопитающее, при необходимости такого ингибирования, путем введения эффективного количества соединения. Настоящее изобретение относится к применению соединений, приведенных в настоящем описании, в качестве ингибиторов активности фермента дипептидилпептидазы-IV.
Помимо приматов, например людей, согласно способу по настоящему изобретению, может быть излечено множество других млекопитающих. Например, могут быть излечены млекопитающие, включающие в себя, без ограничения, коров, овец, коз, лошадей, собак, кошек, морских свинок, крыс или другие виды крупного рогатого скота, мелкого рогатого скота, лошадиных, псовых, кошачьих, грызунов или мышевидных. Однако способ также может быть применен к другим видам, таким как виды птиц (например, цыплята).
Настоящее изобретение дополнительно относится к способу получения лекарственного средства для ингибирования активности фермента дипептидилпептидазы-IV у людей и животных, включающего объединение соединения по настоящему изобретению с фармацевтически приемлемым носителем или разбавителем.
Объектом, излечиваемым настоящими способами, обычно является млекопитающее, предпочтительно человек, мужчина или женщина, у которого желательно ингибировать активность фермента дипептидилпептидазу-IV. Термин "терапевтически эффективное количество" означает количество целевого соединения, которое может вызвать биологический или медицинский ответ ткани, системы, животного или человека, который установлен исследователем, ветеринаром, врачом или другим клиницистом.
Термин "композиция", как используется в настоящем описании, обозначает продукт, содержащий определенные ингредиенты в определенных количествах, а также любой продукт, который получается, прямо или опосредованно, из комбинации определенных ингредиентов в определенных количествах. Такой термин относительно фармацевтической композиции, предназначен для охвата продукта, содержащего активный ингредиент(ингредиенты), и инертный ингредиент(ингредиенты), который составляет носитель, а также любой продукт, который получается прямо или опосредованно при сочетании комплексообразования или агрегации любых двух или нескольких ингредиентов, или диссоциации одного или нескольких из ингредиентов, или других типов реакций или взаимодействия одного или нескольких ингредиентов. Таким образом, фармацевтические композиции по настоящему изобретению охватывают любую композицию, полученную смешиванием соединения по настоящему изобретению и фармацевтически приемлемого носителя. Под "фармацевтически приемлемым" подразумевается носитель, разбавитель или наполнитель, который должен быть совместимым с другими ингредиентами состава и не опасен для его реципиента.
Термины "применение" и или "введение" соединения следует понимать как означающие предоставление соединения по настоящему изобретению или пролекарства соединения по настоящему изобретению индивиду, нуждающемуся в лечении.
Полезность соединений согласно настоящему изобретению в качестве ингибиторов активности фермента дипептидилпептидазы-IV может быть продемонстрирована при помощи методов, известных в данной области техники. Константы ингибирования определяют следующим образом. Применяют непрерывный флуоресцентный анализ с субстратом Gly-Pro-AMC, который очищают при помощи DP-IV для освобождения флуоресцентной AMC выходящей группы. Кинетические параметры, которые описывают такую реакцию, являются следующими: Km=50 мкМ; kcat=75 s-1; kcat/Km=1,5 x 106 M-1s-1. Обычно реакция содержит приблизительно 50 пМ фермента, 50 мкМ Gly-Pro-AMC, и буфер (100 мМ HEPES, pH 7,5, 0,1 мг/мл BSA) в общем реакционном объеме 100 мкл. Выделение AMC постоянно контролируют в флуорометре с 96-луночным планшетом, с использованием длины волны возбуждения 360 нм и длины волны излучения 460 нм. В таких условиях, приблизительно 0,8 мкМ AMC продуцируется за 30 минут при 25°C. Фермент, использованный в таких исследованиях, являлся растворимым (трансмембранный домен и цитоплазматическое расширение исключили) человеческим белком, продуцированным в системе экспрессии бакуловируса (Bac-To-Bac, Gibco BRL). Было установлено, что кинетические константы гидролиза Gly-Pro-AMC и GLP-1 соответствуют литературным значениям нативного фермента. Для измерения констант диссоциации соединений растворы ингибитора в ДМСО добавляли в реакционные смеси, содержащие фермент и субстрат (конечная концентрация ДМСО составила 1%). Все эксперименты проводили при комнатной температуре, используя стандартные реакционные условия, описанные выше. Для определения констант диссоциации (Ki) установили скорости реакции при помощи нелинейной регрессии по уравнению Михаэлиса-Ментона для конкурентного ингибирования. Ошибки при воспроизведении констант диссоциации обычно не превышали двух.
В частности, соединения по следующим примерам имели активность при ингибировании фермента дипептидилпептидазы-IV в вышеуказанном анализе, обычно с IC50 меньше, чем примерно 1 мкМ. Такой результат служит признаком природной активности соединений при использовании в качестве ингибиторов активности фермента дипептидилпептидазы-IV.
Фермент дипептидилпептидазы-IV (DP-IV) является белком клеточной поверхности, который вовлечен в широкий спектр биологических функций. Он имеет широкое распространение в тканях (кишечника, почек, печени, поджелудочной железы, плаценты, тимуса, селезенки, эпителиальных клетках, эндотелии сосудов, лимфоидных и миелоидных клетках, сыворотке), и уровни экспрессии в различных тканях и типах клеток. DP-IV является идентичным CD26 маркеру активации T клеток и он может расщеплять некоторое количество пептидов иммунорегуляции, эндокринных и нейрологических пептидов in vitro. Это подтверждает потенциальную роль указанной пептидазы в ряде процессов заболеваний у человека или других видов.
Следовательно, целевые соединения могут быть использованы при предупреждении или лечении следующих заболеваний, нарушений и состояний.
Диабет типа 2 и связанные заболевания: Действительно установлено, что инкретины GLP-1 и GIP легко инактивируются in vivo при помощи DP-IV. Исследования на мышах с дефицитом DP-IV(-/-) и предварительные клинические испытания показывают, что ингибирование DP-IV увеличивает устойчивые концентрации GLP-1 и GIP, являющиеся результатом улучшенной толерантности к глюкозе. По аналогии с GLP-1 и GIP, возможно, что другие пептиды семейства глюкагонов, которые вовлечены в регуляцию глюкозы, также инактивируются при помощи DP-IV (например, PACAP). Инактивация таких пептидов при помощи DP-IV также может играть роль в гомеостазе глюкозы. Следовательно, ингибиторы DP-IV по настоящему изобретению могут быть использованы при лечении диабета типа II и для лечения и предупреждения многочисленных состояний, которые часто сопровождают диабет типа 2, включая синдром X (также известный как метаболический синдром), реактивную гипоглекемию и диабетическую дислипидемию. Ожирение, обсуждаемое ниже, является другим состоянием, которое часто обнаруживают при диабете типа 2, и оно может быть подвержено лечению соединениями по настоящему изобретению.
Нижеследующие заболевания, нарушения и состояния связаны с диабетом типа 2, и, следовательно, могут быть излечены, контролируемы или в некоторых случаях предупреждены при помощи лечения соединениями по настоящему изобретению: (1) гипергликемия, (2) низкая толерантность к глюкозе, (3) резистентность к инсулину, (4) ожирение, (5) липидные нарушения, (6) дислипидемия, (7) гиперлипидемия, (8) гипертриглицеридемия, (9) гиперхолестеролемия, (10) низкие уровни HDL, (11) высокие уровни LDL, (12) атеросклероз и его последствия, (13) сосудистый рестеноз, (14) синдром раздраженного кишечника, (15) воспалительное заболевание кишечника, включая болезнь Крона и язвенные колиты, (16) другие воспалительные состояния, (17) панкреатит, (18) абдоминальное ожирение, (19) нейродегенеративное заболевание, (20) ретинопатия, (21) нефропатия, (22) нейропатия, (23) синдром X, (24) овариальный гиперандрогенизм (синдром поликистоза яичников), и другие заболевания, при которых резистентность к инсулину является составляющей. При синдроме X, также известном как метаболический синдром, предполагается, что ожирение способствует резистентности к инсулину, диабету, дислипидемии, гипертензии и к увеличенному риску сердечно-сосудистых заболеваний. Следовательно, ингибиторы DP-IV также могут быть использованы для лечения гипертензии, связанной с таким состоянием.
Ожирение: ингибиторы DP-IV могут быть использованы для лечения ожирения. Это предположение основано на наблюдаемом ингибирующем действии GLP-1 и GLP-2 на прием пищи и опорожнение желудка. Экзогенное введение GLP-1 людям существенно увеличивает прием пищи и замедляет опорожнение желудка (Am. J. Physiol., 277: R910-R916 (1999)). Введение ICV (интрацервикально) GLP-1 крысам и мышам также имеет выраженное действие на прием пищи (Nature Medicine, 2: 1254-1258 (1996)). Это подавление приема пищи не наблюдается у GLP-1R(-/-) мышей, что указывает на то, что такие эффекты опосредованы рецепторами GLP-1 мозга. По аналогии с GLP-1, возможно, что GLP-2 также регулируется при помощи DP-IV. ICV введение GLP-2 также подавляет прием пищи аналогично эффектам, наблюдаемым при GLP-1 (Nature Medicine, 6: 802-807 (2000)). Дополнительно исследования мышей с дефицитом DP-IV подтвердило, что такие животные являются резистентными к ожирению, вызываемому питанием, и связанной патологии (например, гиперинсулинонемии).
Дефицит гормона роста: Ингибирование DP-IV может быть пригодным для лечения дефицита гормона роста, что базируется на гипотезе, что фактор (GRF), высвобождающий гормон роста, белок который стимулирует высвобождение гормона роста из передней доли гипофиза, расщепляется ферментом DP-IV in vivo (WO 00/56297). Следующие данные предоставляют доказательство того, что GRF является эндогенным субстратом: (1) GRF эффективно расщепляется in vitro, создавая неактивный продукт GRF[3-44] (BBA 1122: 147-153 (1992)); (2) GRF легко разрушается в плазме до GRF[3-44]; это предупреждается дипротином A ингибитором DP-IV; и (3) GRF[3-44] обнаружен в плазме трансгенной свиньи с человеческим GRF (J. Clin. Invest., 83: 1533-1540 (1989)). Такие ингибиторы DP-IV могут быть использованы для такого же спектра признаков, который был обсужден для средств, усиливающих секрецию гормона роста.
Повреждения кишечника: Возможность использования ингибиторов DP-IV для лечения повреждений кишечника подтверждена результатами исследований, указывающих, что глюкагон-подобные пептиды-2 (GLP-2), подобно эндогенному субстрату для DP-IV, могут проявлять трофические эффекты на эпителий кишечника (Regulatory Peptides, 90: 27-32 (2000)). Введение GLP-2 приводит к некоторому увеличению массы кишечника у грызунов и ослабляет внутреннее повреждение в моделях колита и энтерита грызунов.
Иммуносупрессия: ингибирование DP-IV может быть использовано для моделирования иммунного ответа, основанного на исследованиях, подразумевающих фермент DP-IV при активации T клеток и при обработке хемокином, и эффективность ингибиторов DP-IV в моделях заболеваний in vivo. Показано, что DP-IV является идентичным CD26, маркером клеточной поверхности активированных иммунных клеток. Экспрессия CD26 регулируется состоянием дифференциации и активации иммунных клеток. Общепризнано, что CD26 функционирует как ко-стимуляторная молекула в моделях активации T клеток in vitro. Некоторые хемокины содержат пролин в предпоследнем положении, по-видимому, для защиты от разрушения неспецифическими аминопептидазами. Показано, что многие из них обрабатываются in vitro при помощи DP-IV. В некоторых случаях (RANTES, LD78-бета, MDC, эотаксин, SDF-1альфа) расщепление дает измененную активность в исследованиях хемотаксиса и передачи сигналов. Также показано, что селективность рецепторов в некоторых случаях модифицируется (RANTES). Множество N-терминально усеченных форм некоторых хемокинов идентифицировано в системах клеточных культур in vitro, включающих в себя предсказанные продукты гидролиза DP-IV.
Показано, что ингибиторы DP-IV являются эффективными иммуносупрессантами в животных моделях трансплантации и артрита. Продипин (Pro-Pro-дифенил-фосфонат), неизменяемый ингибитор DP-IV, показал увеличение вдвое выживания сердечного аллотрансплантанта у крыс с 7 дня по 14 день (Transplantation, 63:1495-1500 (1997)). Ингибиторы DP-IV протестированы в коллагене и при артритах, индуцированных алкилдиамином, у крыс, и показали статистически значимое смягчение задней лапы, опухшей в такой модели [Int. J. Immunopharmacology, 19:15-24 (1997) и Immunopharmacology, 40: 21-26 (1998)]. DP-IV является завышенно регулируемым в некоторых аутоимунных заболеваниях, включающих в себя ревматоидные артриты, рассеянный склероз, болезнь Грейвса, и тиреоидит Хашимото (Immunology Today, 20: 367-375 (1999)).
ВИЧ инфекция: Ингибирование DP-IV может быть использовано для лечения или предупреждения ВИЧ инфекции или СПИДа, поскольку некоторые хемокины, которые ингибируют проникновение ВИЧ клеток, являются возможными субстратами для DP-IV (Immunology Today 20: 367-375 (1999)). В случае SDF-1альфа расщепление усиливает антивирусную активность (PNAS, 95: 6331-6 (1998)). Таким образом, ожидается, что стабилизация SDF-1альфа путем ингибирования DP-IV может увеличить ВИЧ инфективность.
Гемопоэз: Ингибирование DP-IV может быть использовано для лечения или предупреждения гемопоэза, поскольку DP-IV может быть вовлечен в гемопоэз. Ингибитор DP-IV, Val-Boro-Pro стимулировал гемопоэз в мышиной модели нейтропении, индуцированной циклофосфамидом (WO 99/56753).
Нейронные нарушения: Ингибирование DP-IV может быть использовано для лечения или предупреждения различных нейронных или психиатрических нарушений, поскольку некоторые пептиды, вовлеченные в различные нейронные процессы, расщепляются in vitro при помощи DP-IV. Ингибитор DP-IV таким образом может иметь терапевтическое преимущество при лечении нейронных нарушений. Показано, что эндоморфин-2, бета-казоморфин и субстанция Р - все являются in vitro субстратами для DP-IV. Во всех случаях расщепление in vitro высокоэффективно, с kcat/Km примерно 106 M-1s-1 или больше. В тестовой модели анальгезии прыжка при электрошоке у крыс, ингибитор DP-IV показал значимый эффект, который не зависел от присутствия экзогенного эндоморфина-2 (Brain Research, 815: 278-286 (1999)).
Нейропротективный и нейрорегенеративный эффекты ингибиторов DP-IV были также доказаны способностью ингибиторов защищать моторные нейроны от эксцитотоксической гибели клеток, защищать стриарную иннервацию додаминергических нейронов при введении конкурентно с MPTP и защищать восстановление плотности стриарной иннервации, если дается, при терапевтическом лечении MPTP нижеследующим способом [см. Yong-Q. Wu, и др., "Neuroprotective Effects of Inhibitors of Dipeptidyl Peptidase-IV In Vitro and In Vivo," Int. Conf. On Dipeptidyl Aminopeptidases: Basic Science and Clinical Applications, September 26-29, 2002 (Berlin, Germany)].
Инвазия опухоли и метастазов: Ингибирование DP-IV может быть использовано для лечения или предупреждения инвазии опухоли и метастазов, поскольку увеличение или уменьшение в экспрессии некоторых эктопептидаз, включающих в себя DP-IV, обнаружено в процессе трансформации нормальных клеток в злокачественный фенотип (J. Exp. Med., 190: 301-305 (1999)). Обнаружено, что увеличение или уменьшение регуляции таких белков является специфичным для типа клеток и тканей. Например, увеличенная CD26/DP-IV экспрессия наблюдавалась в лимфоме T клеток, острой лимфобластной лейкемии T клеток, клетках, производных карциномы щитовидной железы, базальной клеточной карциноме и карциноме молочной железы. Таким образом, ингибиторы DP-IV могут быть использованы при лечении таких карцином.
Доброкачественная гипертрофия простаты: Ингибирование DP-IV может быть использовано для лечения или предупреждения доброкачественной гипертрофии простаты, поскольку была отмечена увеличенная активность DP-IV в ткани простаты пациентов с ДГП (Eur. J. Clin. Chem. Clin. Biochem. 30: 333-338 (1992)).
Подвижность сперматозоидов/женская контрацепция: Ингибирование DP-IV может быть использовано для изменения подвижности сперматозоидов и для контрацепции женщин, поскольку семенная жидкость, простатосома, органеллы, выделенные из простаты, важные для подвижности сперматозоидов, обладают очень высокими уровнями активности DP-IV (Eur. J. Clin. Chem. Clin. Biochem.. 30: 333-338 (1992)).
Гингивит: Ингибирование DP-IV может быть использовано для лечения гингивита, поскольку активность DP-IV обнаружена в десенной жидкости в десневой борозде и в некоторых исследованиях, коррелированных с тяжестью периодонтального заболевания (Arch. Oral Biol., 37: 167-173 (1992)).
Остеопороз: Ингибирование DP-IV может быть использовано для лечения или предупреждения остеопороза, поскольку в остеобластах присутствуют GIP рецепторы.
Соединения по настоящему изобретению могут быть использованы при лечении или предупреждении одного или нескольких из нижеследующих состояний или заболеваний: (1) гипергликемия, (2) низкая толерантность к глюкозе, (3) резистентность к инсулину, (4) ожирение, (5) липидные нарушения, (6) дислипидемия, (7) гиперлипидемия, (8) гипертриглицеридемия, (9) гиперхолестеролемия, (10) низкие уровни HDL, (11) высокие уровни LDL, (12) атеросклероз и его последствия, (13) сосудистый рестеноз, (14) синдром раздражения кишечника, (15) воспалительное заболевание кишечника, включая болезнь Крона и язвенные колиты, (16) другие воспалительные состояния, (17) панкреатит, (18) абдоминальное ожирение, (19) нейродегенеративное заболевание, (20) ретинопатия, (21) нефропатия, (22) нейропатия, (23) синдром X, (24) овариальный гиперандрогенизм (синдром поликистоза яичников), (25) диабет типа II, (26) дефицит гормона роста, (27) нейтропения, (28) нейронные нарушения, (29) метастазы рака, (30) доброкачественная гипертрофия простаты, (32) гингивит, (33) гипертензия, (34) остеопороз и другие состояния, которые могут быть излечены или предупреждены ингибированием DP-IV.
Целевые соединения, кроме того, могут быть использованы для предупреждения и лечения вышеуказанных заболеваний, нарушений и состояний в сочетании с другими агентами.
Соединение по настоящему изобретению может быть использовано в сочетании с одним или несколькими другими лекарственными средствами при лечении, предупреждении, подавлении или облегчении заболеваний или состояний, для которых соединения формулы I или другие лекарственные средства могут быть использованы, причем совместная комбинация лекарственных средств является более безопасной и более эффективной, чем какое-либо одно лекарственное средство. Такое другое лекарственное средство(средства) могут быть введены способом и в количестве, обычно используемом в этом случае, одновременно или последовательно с соединением формулы I. Если соединение формулы I используется одновременно с одним или несколькими другими лекарственными средствами, готовят фармацевтическую композицию в виде стандартной лекарственной дозы, содержащей эти другие лекарственные средства и соединение формулы I. Однако комбинированное лечение также может включать в себя также виды лечения, при которых соединение формулы I и одно или несколько других лекарственных средств вводят при разных перекрывающихся режимах. Также предполагается, что при использовании в сочетании с одним или несколькими другими активными ингредиентами соединения по настоящему изобретению и другие активные ингредиенты могли быть использованы в более низких дозах, чем когда каждое используется отдельно. Таким образом, фармацевтические композиции по настоящему изобретению включают в себя такие, которые содержат одно или несколько других активных ингредиентов дополнительно к соединению формулы I.
Примеры других активных ингредиентов, которые могут быть введены в сочетании с соединением формулы I, и либо введены отдельно, либо в той же фармацевтической композиции, включают в себя, без ограничений:
(a) другой ингибитор дипептидилпептидазы IV (DP-IV);
(b) инсулиновые сенсибилизаторы, включая (i) агонист PPARγ, такой как глитазоны (например, троглитазон, пиоглитазон, энглитазон, МСС-555, розиглитазон и т.п.) и другие PPAR лиганды, включающие в себя двойной агонист PPARα/γ, такой как KRP-297, и агонист PPARα, такой как производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и безафибрат), (ii) бигуаниды, такие как метформин и фенформин, и (iii) ингибитора белка тирозин-фосфатазы-1B (PTP-1B);
(с) инсулин или миметик инсулина;
(d) сульфонилмочевину и вещества, повышающие секрецию инсулина, такие как толбутамид, глибенкламид, глипизид, глимепирид и меглитиниды, такие как репаглинид;
(е) ингибиторы α-глюкозидазы (такие как акарбоз и миглитол);
(f) антагонисты глюкагонового рецептора, такие как те, которые, раскрыты в WO 98/04528, WO 99/01423, WO 00/39088, WO 00/69810;
(g) GLP-1, миметик GLP-1 или агонисты GLP-1 рецептора, такие как те, которые раскрыты в WO 00/42026 и WO 00/59887;
(h) GIP, миметик GIP, такой как тот, который раскрыт в WO 00/58360, и агонист GIP рецептора;
(i) PACAP, миметик PACAP и агонисты PACAP рецептора, такие как те, которые раскрыты в WO 01/23420;
(j) агенты, понижающие уровень холестерина, такие как (i) ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин, правастатин, церивастатин, флувастатин, аторвастатин, итавастатин, розувастатин и др.), (ii) секвестранты (холестирамин, колестипол и производные диалкиламиноалкила декстрана с поперечными связями), (iii) никотиниловый спирт, никотиновая кислота или ее соли, (iv) агонисты PPARα, такие как производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и безафибрат), (v) двойной агонист PPARα/γ, такой как KRP-297, (vi) ингибитор абсорбции холестерина, такой как бета-ситостерол и эзетимиб, (vii) ингибиторы ацил CoA:холестерин ацилтрансферазы, такие как авасимиб, и (viii) антиоксиданты, такие как пробукол;
(k) агонисты PPARδ, такие как те, которые раскрыты в WO 97/28149;
(1) соединения против ожирения, такие как фенфлурамин, дексфенфлурамин, фентермин, сибутрамин, орлистат, антагонисты нейропептида Y1 или Y5, инверсивные агонисты и антагонисты рецептора СВ-1, агонисты β3 адренергического рецептора, агонисты рецептора меланокортина, более конкретно агонисты рецептора меланокортина-4, антагонисты грелина и антагонисты рецептора меланин-концентрирующего гормона (MCH);
(m) ингибиторы илеального транспортера для желчных кислот;
(n) агенты, предназначенные для использования при воспалительных состояниях, такие как аспирин, нестероидные противовоспалительные лекарственные средства, глюкокортикоиды, азулфидин и ингибиторы селективной циклооксигеназы-2;
(o) антигипертензивные средства, такие как ингибиторы АСЕ (эналаприл, лизиноприл, каптоприл, хинаприл, тандолаприл), блокаторы рецептора А-II (лозартан, кандезартан, ирбезартан, валзартан, телмизартан, эпрозартан), бета блокаторы и блокаторы кальциевого канала; и
(р) активаторы глюкокиназы (GKA).
Ингибиторы дипептидилпептидазы-IV, которые могут быть объединены с соединениями структурной формулы I, включают в себя такие, которые описаны в WO 03/004498 (16 января 2003); WO 03/004496 (16 января 2003); EP 1 258 476 (20 ноября 2002); WO 02/083128 (24 октября 2002); WO 02/062764 (15 августа 2002); WO 03/000250 (3 января 2003); WO 03/002530 (9 января 2003); WO 03/002531 (9 января 2003); WO 03/002553 (9 января 2003); WO 03/002593 (9 января 2003); WO 03/000180 (3 января 2003); и WO 03/000181 (3 января 2003). Некоторые соединения ингибиторов DP-IV включают в себя тиазолидид изолейцина; NVP-DPP728; P32/98; P93/01; и LAF 237.
Соединения против ожирения, которые могут быть объединены с соединениями структурной формулы I, включают в себя фенфлурамин, дексфенфлурамин, фентермин, сибутрамин, орлистат, атагонисты нейропептида Y1 или Y5, атагонисты или инверсивные агонисты каннабиноидного CB1 рецептора, агонисты рецепторов меланокортина, более конкретно агонисты рецепторов меланокортина-4, антагонисты грелина и антагонисты рецептора меланин-концентрирующего гормона (MCH). Для обзора соединений против ожирения, которые могут быть объединены с соединениями структурной формулы I, см. S. Chaki и др., "Recent advances in feeding suppressing agents: potential therapeutic strategy for the treatment of obesity," Expert Opin. Ther. Patents. 11: 1677-1692 (2001) and D. Spanswick and K. Lee, "Emerging antiobesity drugs," Expert Opin. Emerging Drugs, 8: 217-237 (2003).
Антагонисты нейропептида Y5, которые могут быть объединены с соединениями структурной формулы I, включают в себя такие, которые описаны в патенте США No. 6 335 345 (1 января 2002) и WO 01/14376 (1 марта 2001); некоторые соединения, идентифицированные как GW 59884A; GW 569180A; LY366377; и CGP-71683A.
Антагонисты каннабиноидного CB1 рецептора, которые могут быть объединены с соединениями структурной формулы I, включают в себя такие, которые описаны в заявке PCT WO 03/007887; патенте США No. 5 624 941, такие как римонабант; заявке PCT WO 02/076949, такие как SLV-319; патенте США No. 6 028 084; заявке PCT WO 98/41519; заявке PCT WO 00/10968; заявке PCT WO 99/02499; патенте США No. 5 532 237; и патенте США No. 5 292 736.
Агонисты рецептора меланокортина, которые могут быть объединены с соединениями структурной формулы I, включают в себя такие, которые описаны в WO 03/009847 (6 февраля 2003); WO 02/068388 (6 сентября 2002); WO 99/64002 (16 декабря 1999); WO 00/74679 (14 декабря 2000); WO 01/70708 (27 сентября 2001); и WO 01/70337 (27 сентября 2001), а также описанные в J.D. Speake и др., "Recent advances in the development of melanocortin-4 receptor agonists," Expert Opin. Ther. Patents, 12: 1631-1638 (2002).
Потенциальная полезность безопасных и эффективных активаторов глюкокиназы (GKA) для лечения диабета описана в J. Grimsby и др., "Allosteric Activators of Glucokinase: Potential Role in Diabetes Therapy," Science, 301: 370-373 (2003).
Вышеуказанные комбинации включают в себя комбинации соединения по настоящему изобретению не только с каким-либо одним активным соединением, но также с двумя или несколькими другими активными соединениями. Неограничивающие примеры включают в себя комбинации соединений, имеющих формулу I, с двумя или несколькими активными соединениями, выбранными из бигуанидинов, сульфонилмочевины, ингибиторов редуктазы HMG-CoA, агонистов PPAR, ингибиторов PTP-1B, других ингибиторов DP-IV и соединений против ожирения.
Более того, соединения по настоящему изобретению могут быть использованы в сочетании с другими лекарственными средствами, которые используются для лечения/предупреждения/подавления или облегчения заболеваний или состояний, для которых пригодны соединения по настоящему изобретению. Эти другие лекарственные средства могут быть введены способом и в количестве, обычно используемом в таких случаях, одновременно или последовательно с соединением по настоящему изобретению. Если соединение по настоящему изобретению используется одновременно с одним или несколькими другими лекарственными средствами, предпочтительна фармацевтическая композиция, содержащая такие другие лекарственные средства дополнительно к соединению по настоящему изобретению. Следовательно, фармацевтические композиции по настоящему изобретению включают в себя такие, которые также содержат один или несколько других активных ингредиентов, дополнительно к соединению по настоящему изобретению.
Массовое соотношение соединения по настоящему изобретению со вторым активным ингредиентом может быть различным и будет зависеть от эффективной дозы каждого ингредиента. В общем случае может быть использована эффективная доза каждого. Таким образом, например, если соединение по настоящему изобретению объединено с другим агентом, массовое соотношение соединения по настоящему изобретению с другим агентом будет в общем случае находиться в пределах от примерно 1000:1 до примерно 1:1000, предпочтительно от примерно 200:1 до примерно 1:200. Комбинации соединения по настоящему изобретению и других активных ингредиентов будет в общем случае также находиться в вышеуказанном диапазоне, но в каждом случае должна быть использована эффективная доза каждого активного ингредиента.
В таких комбинациях соединение по настоящему изобретению и другие активные агенты могут вводиться отдельно или в комбинации. Дополнительно один элемент может быть введен перед введением другого агента(агентов), одновременно или после него.
Соединение по настоящему изобретению может вводиться орально, парентерально (например, внутримышечно, интраперитонеально, внутривенно, ICV, интрацистернально инъекцией или вливанием, подкожной инъекцией или в виде имплантанта), при помощи ингаляционного спрея, назальным, вагинальным, ректальным, сублингвальным или местным способами введения, и может быть разработана рецептура одного или совместно в подходящем составе единичной дозы, содержащей обычные нетоксические фармацевтически приемлемые носители, добавки и наполнители, подходящие для каждого способа введения. Кроме лечения теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, кошки, обезьяны, и т.д., соединения по настоящему изобретению эффективны для использования людьми.
Фармацевтические композиции для введения соединений по настоящему изобретению могут обычно находиться в виде единичной дозы и могут быть приготовлены любым способом, хорошо известным в области фармацевтики. Все способы включают в себя стадию объединения активного ингредиента с носителем, который составляет один или несколько дополнительных ингредиентов. В общем случае фармацевтические композиции готовят путем однородного и тщательного смешивания активного ингредиента с жидким носителем или тонко измельченным твердым носителем, или с обоими, а затем, если необходимо, формование продукта в желаемый состав. В фармацевтической композиции активное целевое соединение включено в количестве, эффективном для создания желаемого эффекта в процессе или состоянии заболевания. Как используется в настоящем описании, термин "композиция" предназначен для охвата продукта, содержащего определенные ингредиенты в определенных количествах, а также любой продукт, который получают в результате, прямо или опосредованно из объединения определенных ингредиентов в определенных количествах.
Фармацевтические композиции, содержащие активный ингредиент, могут быть в виде, подходящем для перорального применения, например в виде пилюль, таблеток, лепешек, водных или масляных суспензий, дисперсных порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального использования, могут быть приготовлены согласно любому способу, известному в области получения фармацевтических композиций, и такие композиции могут содержать один или несколько агентов, выбранных из группы, включающей подсластители, вкусовые добавки, красители и консерванты для получения фармацевтически удобного и приятного лекарства. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые подходят для производства таблеток. Такие наполнители могут быть, например, инертными разбавителями, такими как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие агенты, например кукурузный крахмал или альгиновая кислота; связывающие агенты, например крахмал или гуммиарабик, и смазывающие агенты, например стеарат магния, стериновая кислота или тальк. Таблетки могут быть непокрытыми, или они могут быть покрыты хорошо известными в данной области способами для замедления распада и абсорбции в пищеварительном тракте и таким образом достижения продолжительного воздействия в течение более длительного периода. Например, может быть использовано вещество, задерживающее высвобождение, такое как глицерилмоностеарат или глицерилдистеарат. Также они могут быть покрыты способами, описанными в патенте США 4 256 108; 4 166 452; и 4 265 874, с получением осмотических терапевтических таблеток для регулируемого освобождения.
Составы для перорального применения также могут быть представлены в виде желатиновых капсул, где активный ингредиент смешивают с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешивают с водной или масляной средой, например с арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии содержат активные вещества в смеси с наполнителями, подходящими для получения водных суспензий. Такие наполнители представляют собой суспендирующие агенты, например карбоксиметилцеллюлозу натрия, метилцеллюлозу, гидроксипропилметилцеллюлозу, алгинат натрия, поливинилпирролидон, трагакантовую камедь и Аравийскую камедь смолу; диспергирующие или смачивающие агенты могут быть фосфатидами природного происхождения, например лецитин, или продуктами конденсации оксида алкилена с жирными кислотами, например полиоксиэтилена стеарат, или продуктами конденсации оксида этилена со спиртами с длинной алифатической цепью, например гептадекаэтиленоксицетанол, или продуктами конденсации оксида этилена с неполными эфирами, полученными из жирных кислот и гекситола, такие как полиоксиэтиленмоноолеатсорбитола, или продуктами конденсации оксида этилена с неполными эфирами, полученными из жирных кислот и ангидридов гекситола, например полиэтиленмоноолеатсорбитана. Водные суспензии также могут содержать один или несколько консервантов, например этил или н-пропил, п-гидроксибензоат, один или несколько красителей, и одну или несколько вкусовых добавок, и один или несколько подсластителей, таких как сахароза или сахарин.
Масляные суспензии могут быть получены суспендированием активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загустители, например воск, твердые парафины или цетиловый спирт. Могут быть добавлены подсластители, такие как указанные выше, и вкусовые добавки для придания приятного вкуса пероральным препаратам. Такие композиции могут быть защищены путем добавления антиоксидантов, таких как аскорбиновая кислота.
Дисперсные порошки и гранулы, подходящие для приготовления водных суспензий путем добавления воды, представляют собой активные ингредиенты в смеси с диспергирующими или смачивающими агентами, суспендирующими агентами и одним или несколькими консервантами. Подходящие диспергирующие или смачивающие агенты и суспендирующие агенты проиллюстрированы таковыми уже упомянутыми выше. Дополнительные наполнители, например подсластители, вкусовые добавки и красители, также могут присутствовать.
Фармацевтические композиции по настоящему изобретению также могут быть в виде эмульсий масло-в-воде. Масляная фаза может быть растительным маслом, например оливковым маслом или арахисовым маслом, или минеральным маслом, например жидкими парафинами или их смесью. Подходящие эмульгаторы могут быть смолами природного происхождения, например акароидной смолой или трагакантовой смолой, фосфатидами природного происхождения, например соя, лецитин, и эфирами или неполными эфирами, полученными из жирных кислот и ангидридами гекситола, например моноолеат сорбитана, и продуктами конденсации указанных неполных эфиров с оксидом этилена, например полиоксиэтилен моноолеат сорбитана. Эмульсии также могут содержать подсластители и вкусовые добавки.
Сиропы и эликсиры могут быть приготовлены вместе с подсластителями, например глицерином, пропиленгликолем, сорбитолом или сахарозой. Такие составы могут также содержать успокоительное средство, консерванты, вкусовые добавки и красители.
Фармацевтические композиции могут быть в виде стерильных инъецируемых водных или масляных суспензий. Такая суспензия может быть приготовлена, как известно, из уровня техники с использованием таких подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые были указаны выше. Стерильные инъецируемые лекарства также могут быть стерильными инъецируемыми растворами или суспензиями в нетоксичном разбавителе или растворителе, приемлемыми для парентерального применения, например, в виде раствора в 1,3-бутандиоле. Среди допустимых разбавителей и растворителей, которые могут быть использованы, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителей или в качестве суспендирующей среды обычно используются стерильные жирные масла. Для этой цели может использоваться любое мягкое жирное масло, включая синтетические моно- или диглицириды. Дополнительно жирные кислоты, такие как олеиновая кислота, находят применение в инъецируемых лекарствах.
Соединения по настоящему изобретению также могут быть введены в виде свечей для ректального введения лекарственного средства. Такие композиции могут быть приготовлены смешиванием лекарственного средства с подходящим нераздражающим наполнителем, который является твердым при обычных температурах, но жидким при температуре прямой кишки, и, следовательно, может таять в прямой кишке, высвобождая лекарственное вещество. Такими веществами являются масло какао и полиэтиленгликоли.
Для местного применения используются кремы, мази, желе, растворы или суспензии и т.д., содержащие соединение по настоящему изобретению. (В целях такого использования местное использование должно включать жидкости для полоскания рта и растворы для полоскания горла.)
Фармацевтическая композиция и способ по настоящему изобретению могут дополнительно содержать другие терапевтические активные соединения, как указано в настоящем описании, которые обычно применяются при лечении вышеупомянутых патологических состояний.
При лечении или предупреждении состояний, при которых требуется ингибирование активности фермента дипептидилпептидазы-IV, подходящая доза обычно может составлять примерно 0,01-500 мг на 1 кг массы тела пациента в сутки, которая может быть введена в виде единичной дозы или большим количеством доз.
Предпочтительно доза может составлять от примерно 0,1 до примерно 250 мг/кг в сутки; более предпочтительно от примерно 0,5 до примерно 100 мг/кг в сутки. Подходящая доза может составлять от примерно 0,01 до 250 мг/кг в сутки, от примерно 0,05 до 100 мг/кг в сутки, или от примерно 0,1 до 50 мг/кг в сутки. Внутри такого диапазона доза может составлять 0,05-0,5, 0,5-5 или 5-50 мг/кг в сутки. Для перорального применения композиции предпочтительно представлены в виде таблеток, содержащих 1,0-1000 мг активного ингредиента, более конкретно 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 и 1000,0 мг активного ингредиента для симптоматического регулирования дозой для пациента, которого необходимо вылечить. Соединения могут вводиться в режиме от 1 до 4 раз в сутки, предпочтительно один или два раза в сутки.
При лечении или предупреждении сахарного диабета и/или гипергликемии, или гипертриглицеридемии или других заболеваний, для которых предназначены соединения по настоящему изобретению, обычно удовлетворительные результаты получают, когда соединения по настоящему изобретению вводят суточной дозой от примерно 0,1 мг до примерно 100 мг на килограмм массы тела животного, предпочтительно давая в виде единичной суточной дозы или разделенных доз от двух до шести раз в сутки, или в виде препарата замедленного освобождения. Для большинства млекопитающих общая суточная доза составляет от примерно 1,0 мг до примерно 1000 мг, предпочтительно от примерно 1 мг до примерно 50 мг. В случае взрослого человека массой 70 кг общая суточная доза может обычно составлять от примерно 7 мг до примерно 350 мг. Такой режим дозирования может быть установлен для обеспечения оптимального терапевтического ответа.
Однако очевидно, что конкретный уровень доз и частота доз для любого конкретного пациента могут быть различными и зависеть от множества факторов, включающих активность конкретного используемого соединения, метаболическую стабильность и длительность воздействия такого соединения, возраст, массу тела, общее состояние здоровья, пол, диету, режим и время приема, скорость экскреции, сочетание лекарственных средств, тяжесть конкретного состояния и лечение, которому подвергают пациента.
Некоторые способы получения соединений по настоящему изобретению проиллюстрированы нижеследующими схемами и примерами. Исходные вещества получены согласно способам, известным в данной области техники, или как проиллюстрировано в настоящем описании.
Соединения по настоящему изобретению могут быть получены из промежуточных соединений с получением бета-аминокислоты, таких как соединения формулы II, и замещенных промежуточных соединений с получением гексагидродиазепинона, таких как соединения формулы III, в стандартных условиях связывания пептидов с последующим снятием защиты. Получение таких интермедиатов описано на следующих схемах.
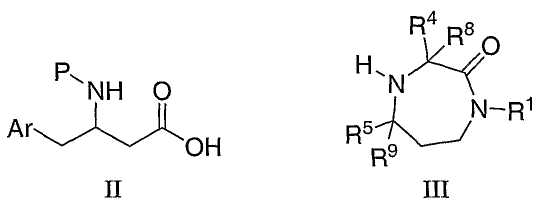
где Ar, R1, R4, R5, R8, и R9 являются такими, как определено выше, и P представляет собой подходящую группу, защищающую азот, такую как трет-бутоксикарбонил (BOC), бензилоксикарбонил (Cbz) или 9-флуоренилметоксикарбонил (Fmoc).
Схема 1
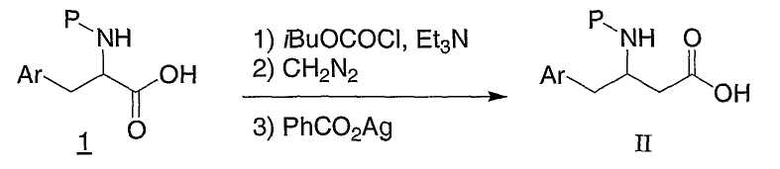
Соединения формулы II являются коммерчески доступными, известными из литературы, или могут быть получены обычным образом многими способами, хорошо известными специалистам в данной области техники. Один из обычных путей проиллюстрирован на схеме 1. Защищенную альфа-аминокислоту 1, которая может быть коммерчески доступной или легко получена из соответствующей аминокислоты путем использования защиты, например ди-трет-бутилдикарбоната (для P=BOC), карбобензилоксихлорида (для P=Cbz), или N-(9-флуоренилметоксикарбонилокси)сукцинимида (для P=Fmoc), обрабатывают изобутилхлорформаитом и основанием, таким как триэтиламин или N,N-диизопропилэтиламин, далее диазометаном. Полученный в результате диазокетон затем обрабатывают бензоатом серебра в растворе, таком как метанол или водный раствор диоксана, и можно подвергнуть обработке ультразвуком, следуя процедуре по Sewald и др., Synthesis, 837 (1997), для предоставления бета-аминокислоты II. Как должно быть понятно специалистам в данной области техники, для получения энантиомерно чистых бета-аминокислот II могут быть использованы энантиомерно чистые альфа-аминокислоты I. Альтернативные способы получения защищенных интермедиатов бета-аминокислоты II могут быть найдены в следующих обзорах: E. Juaristi, Enantioselective Synthesis of β-Amino Acids, Ed., Wiley-VCH, New York: 1997; Juaristi et al., Aldrichimica Acta, 27: 3 (1994); and Cole et al., Tetrahedron, 32: 9517 (1994).
Схема 2
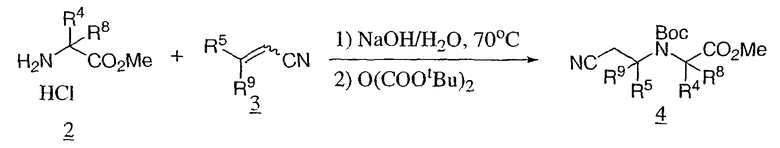
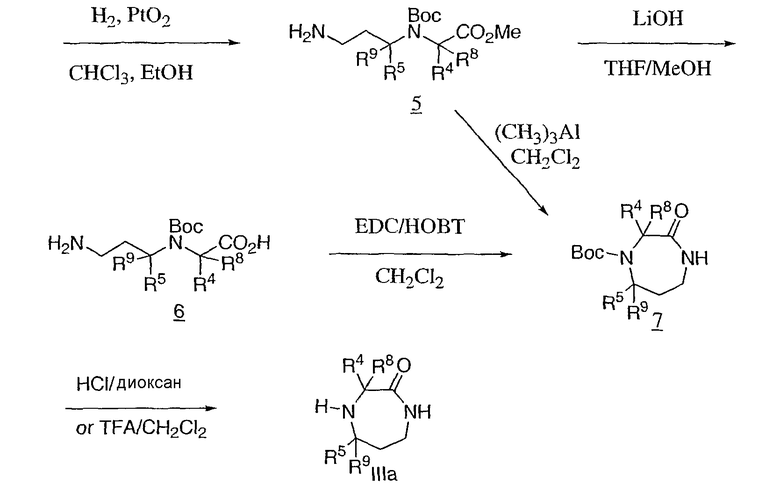
Соединения формулы III являются коммерчески доступными, известными из литературы, или могут быть получены обычным образом многими способами, хорошо известными специалистам в данной области техники. Один из обычных способов, в котором R1 является водородом, показан на схеме 2. Аминоэфир 2, обычно используемый в виде его гидрохлоридной соли, конденсируют с акрилонитрилом 3 и защищают образованную аминогруппу продукта, например, в виде его трет-бутоксикарбонильного (Boc) производного, для предоставления 4, который восстанавливают до первичного амина 5. Циклизацию 5 в N-защищенный гексагидродиазепинон 7 можно проводить с использованием триэтилалюминия. В качестве альтернативы аминоэфир 5 может быть гидролизован с кислотой 6 и циклизован с использованием реагентов для связывания аминокислот, таких как EDC, для получения промежуточного соединения с получением 7. Снятие защиты, например в случае Boc, путем обработки кислотой, такой как соляная в диоксане, или трифторуксусной кислотой в дихлорметане, дает промежуточное соединение IIIa.
Схема 3
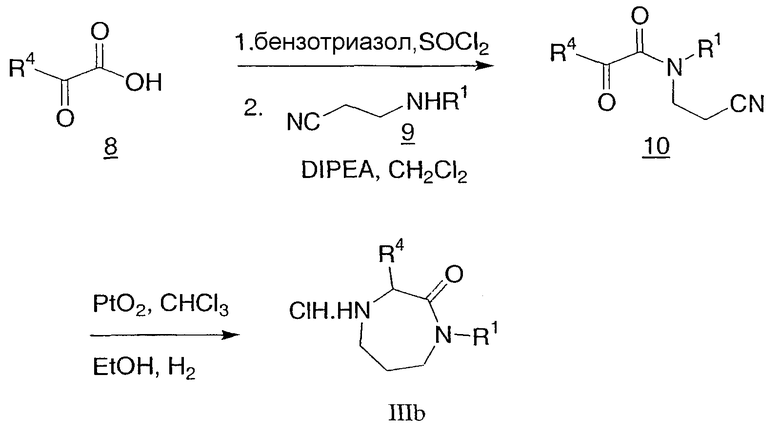
Альтернативный способ получения гексагидродиазепинона IIIb (в котором R5, R8 и R9 представляют собой водород) показан на схеме 3. α-Кетокислоты 8, такие как пировиноградная кислота, могут быть конденсированы с аминопропионитрилом 9 для получения цианоэтилоксопропанамидов 10, которые могут подвергаться восстановительной циклизации до гексагидродиазепинона IIIb восстановителем, таким как оксид платины и водород.
Схема 4
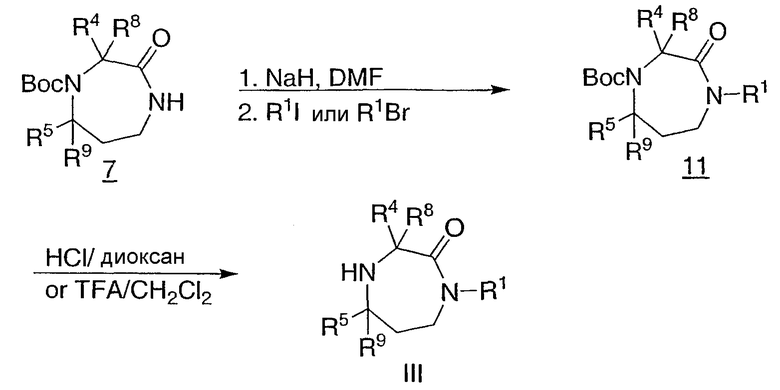
Промежуточные соединения для синтеза гексагидродиазепинона III и промежуточные соединения для их синтеза могут быть преобразованы различными способами. Например, азот амида промежуточного соединения 7, полученный, как показано на схеме 2, может быть алкилирован депротонированием с основанием, таким как гидрид натрия, с последующей обработкой галогеналкилом, как показано на схеме 4. Снятие защиты полученного промежуточного соединения 11 дает промежуточное соединение III.
Схема 5
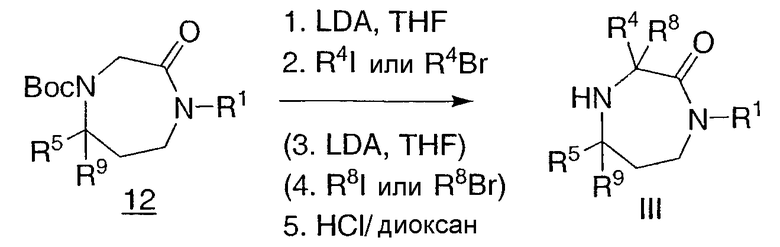
Другой такой пример проиллюстрирован на схеме 5. Защищенный гексагидродиазепинон 12, который может быть получен, как описано для промежуточного соединения 11 на схеме 4, где R4 и R8 представляют собой водород, или при помощи защиты промежуточного соединения IIIa из схемы 3, где R5 представляет собой водород, может быть алкилирован с использованием основания, такого как LDA, с последующей обработкой различными галогеналкилами. Способ может быть повторен для введения второй алкильной группы R8. Снятие защиты дает промежуточное соединение III.
Схема 6
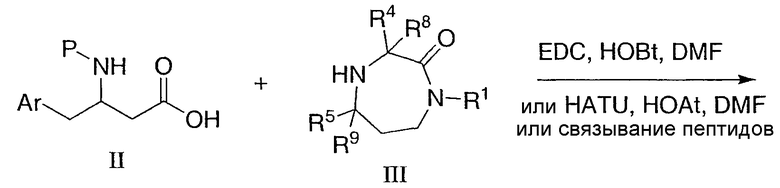
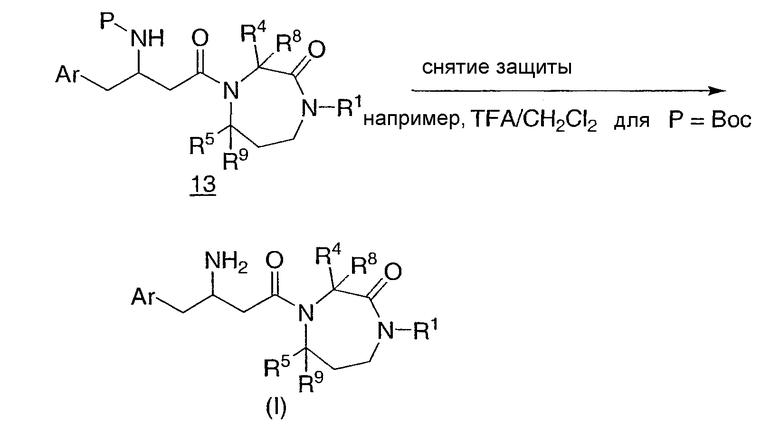
Промежуточные соединения II и III связывают в стандартных условиях связывания пептидов, например, используя 1-этил-3-(3-диметиламинопропил)карбодиимид и 1-гидроксибензотриазол (EDC/HOBT) или O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфат и 1-гидрокси-7-азабензотриазол (HATU/HOAT) в растворителе, таком как N,N-диметилформамид (ДМФ) или дихлорметан, в течение от 3 до 48 часов при температуре окружающей среды с получением промежуточного соединения 13, как показано на схеме 6. В некоторых случаях промежуточное соединение III может быть солью, такой как соль гидрохлорида или трифторуксусной кислоты, и в этом случае удобно добавлять основание, в общем случае N,N-диизопропилэтиламин, для реакции связывания. Затем удаляют защитную группу, например, трифторуксусной кислотой или метанольным раствором хлористоводородной кислоты в случае Boc с получением желаемого амина I. Продукт очищают, если необходимо, перекристаллизацией, растиранием в порошок, препаративной тонкослойной хроматографией, флэш-хроматографией на селикагеле, такой как с устройством Biotage®, или ВЭЖХ. Соединения, которые очищены ВЭЖХ, могут быть выделены в виде соответствующих солей. Очистку промежуточных соединений выполняют таким же образом.
В некоторых случаях продукт I или синтетические промежуточные соединения, проиллюстрированные в вышеприведенных схемах, могут быть дополнительно модифицированы, например, при помощи введения заместителей на Ar, R1, R4 или R5. Такие введения могут включать в себя, без ограничений, реакции восстановления, окисления, алкилирования, ацилирования и гидролиз, которые обычно известны специалистам в данной области техники.
В некоторых случаях для осуществления вышеуказанного взаимодействия схемы могут быть изменены для облегчения реакции или во избежание нежелательных продуктов реакции. Следующие примеры предоставлены для того, чтобы настоящее изобретение могло быть понято более полно. Такие примеры являются только иллюстративными и не должны быть истолкованы как ограничивающие настоящее изобретение каким-либо способом.
Промежуточное соединение 1
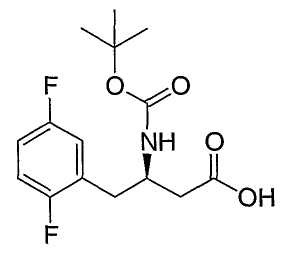
(3R)-3-[(трет-Бутоксикарбонил)амино]-4-(2,5-дифторфенил)бутановая кислота
Стадия A: (R,S)-N-(трет-Бутоксикарбонил)-2,5-дифторфенилаланин
К раствору 0,5 г (2,49 мМ) 2,5-дифтор-DL-фенилаланина в 5 мл трет-бутанола добавляли последовательно 1,5 мл 2н. водного раствора гидроксида натрия и 543 мг ди-трет-бутилдикарбоната. Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч и разбавляли этилацетатом. Органическую фазу промывали последовательно 1н. соляной кислотой и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией (силикагель, 97:2:1 дихлорметан:метанол:уксусная кислота) и получали указанное в заголовке соединение.
MS 302 (M+1).
Стадия B: (R,S)-3-[(трет-Бутоксикарбонил)амино]-1-диазо-4-(2,5-дифторфенил)бутан-2-он
К раствору 2,23 г (7,4 мМ) (R,S)-N-(трет-бутоксикарбонил)-2,5-дифторфенилаланина в 100 мл диэтилового эфира при 0°C последовательно добавляли 1,37 мл (8,1 мМ) триэтиламина и 0,931 мл (7,5 мМ) изобутилхлорформиата, и реакционную смесь перемешивали при данной температуре в течение 15 мин. Затем добавляли охлажденный раствор диазометана в эфире до тех пор, пока сохранялся желтый цвет, и перемешивание продолжали еще в течение 16 ч. Избыток диазометана удаляли прибавлением по каплям уксусной кислоты, и реакционную смесь разбавляли этилацетатом и промывали последовательно 5% соляной кислотой, насыщенным раствором бикарбоната натрия и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме.
Очистка флэш-хроматографией (силикагель, 4:1 гексан:этилацетат) давала диазокетон.
1ЯМР (500 МГц, CDCl3) δ 7,03-6,95 (м, 1H), 6,95-6,88 (м, 2H), 5,43 (ушир.с, 1H), 5,18 (ушир.с, 1H), 4,45 (ушир.с, 1H), 3,19-3,12 (м, 1H), 2,97-2,80 (м, 1H), 1,38 (с, 9H).
Стадия C: (3R)-3-[(трет-Бутоксикарбонил)амино]-4-(2,5-дифторфенил)бутановая кислота
В раствор 2,14 г (6,58 мМ) (R,S)-3-[(трет-бутоксикарбонил)амино]-1-диазо-4-(2,5-дифторфенил)бутан-2-она, растворенного в 100 мл метанола при -30°C, последовательно добавляли 3,3 мл (19 мМ) диизопропилэтиламина и 302 мг (1,32 мМ) бензоата серебра. Реакционную смесь перемешивали в течение 90 мин, разбавляли этилацетатом и промывали последовательно 2н. соляной кислотой, насыщенным водным раствором бикарбоната натрия и солевым раствором. Органическую фазу сушили над сульфатом магния, концентрировали в вакууме и разделяли энантиомеры препаративной хиральной ВЭЖХ (колонка Chiralpak AD, 5% этанол в гексане) с получением 550 мг желаемого (R)-энантиомера, который элюировали первым. Указанное вещество растворяли в 50 мл смеси тетрагидрофуран:метанол:1н. водный раствор гидроксида лития (3:1:1) и перемешивали при 50°C в течение 4 ч. Реакционную смесь охлаждали, подкисляли 5% разбавленной соляной кислотой и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого пенообразного твердого вещества.
1ЯМР (500 МГц, CDCl3) δ 7,21 (м, 1H), 6,98 (м, 2H), 6,10 (ушир.с, 1H), 5,05 (м, 1H), 4,21 (м, 1H), 2,98 (м, 2H), 2,60 (м, 2H), 1,38 (с, 9H).
Промежуточное соединение 2
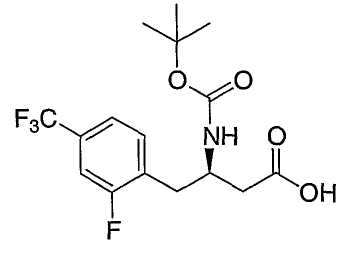
(3R)-3-[(трет-бутоксикарбонил)амино]-4-[2-фтор-4-(трифторметил)фенил]-бутановая кислота
Стадия A: (2R,5S)-2,5-Дигидро-3,6-диметокси-2-(2'-фтор-4'-(трифторметил)бензил)-5-изопропилпиразин
К раствору 3,32 г (18 мМ) коммерчески доступного (2S)-2,5-дигидро-3,6-диметокси-2-изопропилпиразина в 100 мл тетрагидрофурана при -70°C добавляли 12 мл (19 мМ) 1,6 M раствора бутиллития в гексане. После перемешивания при указанной температуре в течение 20 мин добавляли 5 г (19,5 мМ) 2-фтор-4-трифторметилбензилбромида в 20 мл тетрагидрофурана и продолжали перемешивать в течение 3 ч, затем нагревали реакционную смесь до температуры окружающей среды. Реакционную смесь гасили водой, концентрировали в вакууме и экстрагировали этилацетатом. Объединенные органические фазы промывали солевым раствором, сушили и концентрировали в вакууме. Очистка флэш-хроматографией (силикагель, 0-5% этилацетат в гексане) давала указанное в заголовке соединение.
1ЯМР (500 МГц, CDCl3) δ 7,33-7,25 (м, 3H), 4,35-4,31 (м, 1H), 3,75 (с, 3H), 3,65 (с, 3H), 3,60 (т, 1H, J=3,4 Гц), 3,33 (дд, 1H, J=4,6, 13,5 Гц), 3,03 (дд, 1H, J=7, 13,5 Гц), 2,25-2,15 (м, 1H), 1,0 (д, 3H, J=7 Гц), 0,66 (д, 3H, J=7 Гц).
Стадия B: (R)-N-(трет-бутоксикарбонил)-2-фтор-4-трифторметил-фенилаланинметиловый эфир
К раствору 5,5 г (15 мМ) (2R,5S)-2,5-дигидро-3,6-диметокси-2-(2'-фтор-4'-(трифторметил)бензил)-5-изопропилпиразина в 50 мл смеси ацетонитрил:дихлорметан (10:1) добавляли 80 мл 1н. водного раствора трифторуксусной кислоты. Реакционную смесь перемешивали в течение 6 ч и органические растворители отгоняли в вакууме. Добавляли карбонат натрия до тех пор, пока раствор не становился щелочным (>pH 8), и затем реакционную смесь разбавляли 100 мл тетрагидрофурана и добавляли 10 г (46 мМ) ди-трет-бутилдикарбоната. Полученную взвесь перемешивали в течение 16 ч, концентрировали в вакууме и экстрагировали этилацетатом. Объединенную органическую фазу промывали солевым раствором, сушили и концентрировали в вакууме. Очистка флэш-хроматографией (силикагель, 20% этилацетат в гексане) давала указанное в заголовке соединение.
1ЯМР (500 МГц, CDCl3) δ 7,38-7,28 (м, 3H), 5,10 (шир.д, 1H), 4,65-3,98 (м, 1H), 3,76 (с, 3H), 3,32-3,25 (м, 1H), 3,13-3,05 (м, 1H), 1,40 (с, 9H).
Стадия C: (R)-N-(трет-Бутоксикарбонил)-2-фтор-4-трифторметил)фенилаланин
Раствор 5,1 г (14 мМ) (R,S)-N-(трет-бутоксикарбонил)-2-фтор-4-трифторметил)фенилаланинметилового эфира в 350 мл смеси тетрагидрофуран:метанол:1н. гидроксид лития (3:1:1) перемешивали при 50°C в течение 4 ч. Реакционную смесь охлаждали, подкисляли 5% соляной кислотой и экстрагировали этилацетатом. Объединенные органические фазы промывали солевым раствором, сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения.
1ЯМР (500 МГц, CD3OD) δ 7,45-7,38 (м, 3H), 4,44-4,40 (м, 1H), 3,38-3,33 (м, 1H), 2,98 (дд, 1H, J=9,6, 13,5 Гц), 1,44 (с, 9H).
Стадия D: (3R)-3-[(трет-Бутоксикарбонил)амино]-4-[2-фтор-4-(трифторметил)фенил]бутановая кислота
К раствору 3,4 г (9,7 мМ) продукта со стадии C в 60 мл тетрагидрофурана при 0°C добавляли последовательно 2,3 мл (13 мМ) диизопропилметиламина и 1,7 мл (13 мМ) изобутилхлорформиата, и реакционную смесь перемешивали при указанной температуре в течение 30 мин. Затем добавляли охлажденный эфирный раствор диазометана до тех пор, пока сохранялся желтый цвет, и перемешивание продолжили далее в течение 16 ч. Избыток диазометана гасили прибавлением по каплям уксусной кислоты, и реакционную смесь разбавляли этилацетатом и последовательно промывали 5% соляной кислотой, насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Очистка флэш-хроматографией (силикагель, 9:1 гексан:этилацетат) давала 0,5 г диазокетона. К раствору 0,5 г (1,33 мМ) диазокетона, растворенного в 100 мл метанола, при 0°C последовательно добавляли 0,7 мл (4 мМ) диизопропилэтиламина и 32 мг (0,13 мМ) бензоата серебра. Реакционную смесь перемешивали в течение 2 ч, затем разбавляли этилацетатом и промывали последовательно 2н. соляной кислотой, насыщенным водным раствором бикарбоната натрия и солевым раствором. Органическую фазу сушили над сульфатом магния, концентрировали в вакууме и растворяли в 50 мл смеси тетрагидрофуран:метанол:1н. водный раствор гидроксида лития (3:1:1) и перемешивали при 50°C в течение 3 ч. Реакционную смесь охлаждали, подкисляли 5% соляной кислотой и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого пенообразного твердого вещества.
1ЯМР (500 МГц, CD3OD) δ 7,47-7,33 (м, 3H), 4,88 (ушир.с, 1H), 4,26-3,98 (м, 1H), 3,06-3,01 (м, 1H), 2,83-2,77 (м, 1H), 2,58-2,50 (м, 2H), 1,29 (с, 9H).
Промежуточное соединение 3
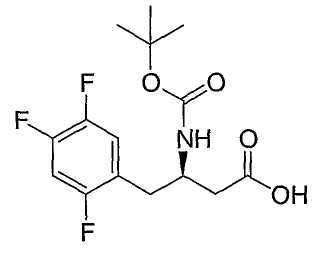
(3R)-3[(трет-Бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутановая кислота
Стадия A: (2S,5R)-2,5-Дигидро-3,6-диметокси-2-изопропил-5-(2',4',5'трифторбензил)пиразин
Указанное в заголовке соединение (3,81 г) получали из 3,42 г (18,5 мМ) (2S)-2,5-дигидро-3,6-диметокси-2-изопропилпиразина и 5 г (22,3 мМ) 2,4,5-трифторбензилбромида, используя способ, описанный для промежуточного соединения 2, стадия A.
1ЯМР (500 МГц, CD3OD) δ 7,01 (м, 1H), 6,85 (м, 1H), 4,22 (м, 1H), 3,78 (м, 3H), 3,64 (м, 3H), 3,61 (м, 1H), 3,20 (м, 1H), 2,98 (м, 1H), 2,20 (м, 1H), 0,99 (д, 3H, J=8 Гц), 0,62 (д, 3H, J=8 Гц).
Стадия В: (R)-N-(трет-Бутоксикарбонил)-2,4,5-трифторфенилаланин метиловый эфир
К раствору 3,81 г (11,6 мМ) (2S,5R)-2,5-дигидро-3,6-диметокси-2-изопропил-5-(2',4',5'трифторбензил)пиразина в 20 мл ацетонитрила добавляли 20 мл 2н. соляной кислоты. Реакционную смесь перемешивали в течение 72 ч и концентрировали в вакууме. Реакционную смесь растворили в 30 мл дихлорметана и добавляли 10 мл (72 мМ) триэтиламина и 9,68 г (44,8 мМ) ди-трет-бутилдикарбоната. Реакционную смесь перемешивали в течение 16 ч, разбавляли этилацетатом и промывали последовательно 1н. соляной кислотой и солевым раствором. Органическую фазу сушили над сульфатом магния, концентрировали в вакууме и очищали флэш-хроматографией (силикагель 9:1 гексан:этилацетат) с получением указанного в заголовке соединения.
1ЯМР (500 МГц, CDCl3) δ 6,99 (м, 1H), 6,94 (м, 1H), 5,08 (м, 1H), 4,58 (м, 1H), 3,78 (м, 3H), 3,19 (м, 1H), 3,01 (м, 1H), 1,41 (с, 9H).
Стадия C: (R)-N-(трет-Бутоксикарбонил)-2,4,5-трифторфенилаланин
Указанное в заголовке соединение (2,01 г) получали из 2,41 г (7,5 М) (R)-N-(трет-бутоксикарбонил)-2,4,5-трифторфенилаланин метилового эфира, используя способ, описанный для промежуточного соединения 2, стадия C.
LC-MS 220,9 (M+1-BOC).
Стадия D: (3R)-3-[(трет-Бутоксикарбонил)амино]-4-(2,4,5-трифторфенилбутановая кислота
К раствору 0,37 г (1,16 мМ) (R)-N-(1,1-диметилэтоксикарбонил)-2,4,5-трифторфенилаланина в 10 мл диэтилового эфира при -20°C последовательно добавляли 0,193 мл (1,3 мМ) триэтиламина и 0,18 мл (1,3 мМ) изобутилхлорформиата, и реакционную смесь перемешивали при указанной температуре в течение 15 мин. Затем добавляли охлажденный эфирный раствор диазометана до тех пор, пока сохранялся желтый цвет, и перемешивание продолжили далее в течение 1 ч. Избыток диазометана гасили прикапыванием уксусной кислоты, и реакционную смесь разбавляли этилацетатом, и последовательно промывали насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Очистка флэш-хроматографией (силикагель, 3:1 гексан:этилацетат) давала 0,36 г диазокетона. К раствору 0,35 г (1,15 мМ) диазокетона, растворенного в 12 мл смеси 1,4 диоксан:вода (5:1), добавляли 26 мг (0,113 мМ) бензоата серебра. Полученный раствор обрабатывали ультразвуком в течение 2 ч, затем разбавляли этилацетатом и промывали последовательно 1н. соляной кислотой и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Очистка флэш-хроматографией (силикагель, 97:2:1 дислорметан:метанол:уксусная кислота) давала указанное в заголовке соединение.
1ЯМР (500 МГц, CDCl3) δ 7,06 (м, 1H), 6,95 (м, 1H), 5,06 (ушир.с, 1H), 4,18 (м, 1H), 2,98 (м, 2H), 2,61 (м, 2H), 1,39 (с, 9H).
Промежуточное соединение 4
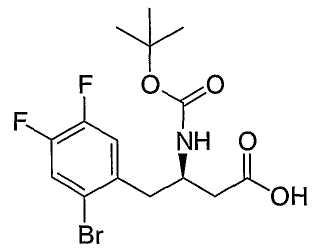
(3R)-4-(2-Бром-4,5-дифторфенил)-3-[(трет-бутоксикарбонил)амино]-бутаноая кислота
К раствору 2,4 г (10 мМ) 2-бром-4,5-дифторбензойной кислоты [приготовленной согласно процедуре по Braish и др., Syn. Comm., 3067-3074 (1992)] в 75 мл тетрагидрофурана добавляли 2,43 г (15 мМ) карбонилдиимидазола. Раствор кипятили с обратным холодильником в течение 3,5 ч, охлаждали до температуры окружающей среды и добавляли 0,38 г (10 мМ) боргидрида натрия в 15 мл воды. Реакционную смесь перемешивали в течение 10 мин и разделяли между этилацетатом и 10% водным раствором бикарбоната натрия. Органический слой промывали дважды теплой водой, солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Очистка флэш-хроматографией (силикагель, 4:1 гексан:этилацетат) давала 1,9 г 2-бром-4,5-дифторбензиловый спирт. К раствору 1,9 г (8,4 мМ) 2-бром-4,5-дифторбензилового спирта в 30 мл дихлорметана при 0°C добавляли 3,4 г (10 мМ) тетрабромида углерода и 2,7 г (10 мМ) трифенилфосфина. Реакционную смесь перемешивали в течение 2 ч при указанной температуре, растворитель отгоняли в вакууме и остаток перемешивали с 100 мл диэтилового эфира. Раствор фильтровали, концентрировали в вакууме и очищали флэш-хроматографией (силикагель, 20:1 гексан:этилацетат) с получением 2,9 г 2-бром-4,5-дифторбензилбромида, с примесью тетрабромида углерода, который затем использовали без дополнительной очистки. Использованием процедуры, описанной с получением промежуточного соединения 2-4, преобразовывали производное бензилбромида в указанное в заголовке соединение.
LC-MS 394 и 396 (M+1).
По существу следуя процедурам, описанным для получения промежуточных соединений 1-4, были получения промежуточных соединений, приведенные в таблице 1.
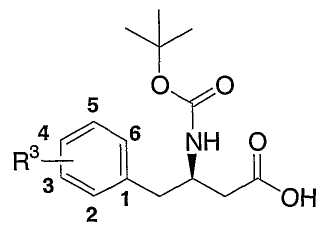
Пример 1
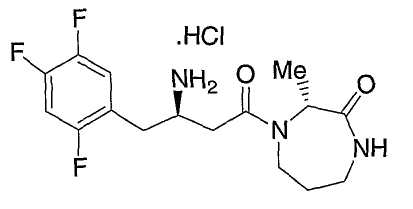
(3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-метил-2Н-1,4-диазепин-2-она гидрохлорид
Стадия A: Метил N-(трет-бутоксикарбонил)-N-(2-цианоэтил)-D-аланинат
К перемешиваемой суспензии гидрохлорида D-аланинметилового эфира (2,0 г) и 5н. водного раствора гидроксида натрия (2,9 мл) в воде (15 мл) при 0°C добавляли акрилонитрил (1,1 мл). Полученную в результате смесь перемешивали при 70°C в течение 3,5 ч и охлаждали до комнатной температуры. Добавляли ди-трет-бутилдикарбонат (30 мл), и реакционную смесь перемешивали в течение двух дней. Реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Органический слой отделяли, промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной флэш-хроматографией (силикагель, этилацетат/гексан 2:3) с получением метил-N-(трет-бутоксикарбонил)-N-(2-цианоэтил)-D-аланината.
Стадия B: Метил N-(3-аминопропил)-N-(трет-бутоксикарбонил)-D-аланинат
К раствору метил N-(трет-бутоксикарбонил)-N-(2-цианоэтил)-D-аланината (1,5 г) в этаноле (80 мл) и хлороформе (1,4 мл) добавляли оксид платины (350 мг), и реакционную смесь перемешивали в атмосфере водорода в течение 16 ч. Смесь фильтровали через целит, и целит промывали метанолом и дихлорметаном. Фильтрат концентрировали с получением N-(3-аминопропил)-N-(трет-бутоксикарбонил)-D-аланината в виде масляного остатка.
Стадия C: трет-Бутил-(2R)-гексагидро-2-метил-3-оксо-1Н-1,4-диазепин-1-карбоксилат
К 2M раствору триметилалюминия в дихлорметане (30 мл) медленно добавляли раствор метил-N-(3-аминопропил)-N-(трет-бутоксикарбонил)-D-аланината (11,5 г) в дихлорметане. Реакционную смесь перемешивали при комнатной температуре в течение четырех дней и затем выливали в колбу, содержащую 30 г целита. Смесь перемешивали и гасили медленным добавлением ˜10 мл насыщенного водного раствора хлорида аммония. Добавляли сульфат натрия (20 г) и метанол (50 мл). Смесь перемешивали в течение 1 ч, затем фильтровали. Твердое вещество промывали 5% смесью метанол/дихлорметан. Фильтрат концентрировали. Остаток очищали флэш-хроматографией (силикагель, элюированием последовательно с 4, 6, 7 и 12% смесью 10:1 метанол/водный раствор концентрированного гидроксида аммония в дихлорметане) с получением указанного в заголовке соединения, содержащего менее 3% (3S)-изомера.
LC/MS 228.9 (M+1).
Стадия D: (3R)-гексагидро-3-метил-2Н-1,4-диазепин-2-она гидрохлорид
Трет-бутил-(2R)-гексагидро-2-метил-3-оксо-1Н-1,4-диазепин-1-карбоксилат, полученный на предыдущей стадии, растворяли в 4М соляной кислоты в диоксане и спустя 2,5 ч упаривали с получением гидрохлоридной соли желаемого соединения.
Стадия E: (3R)-4-[(3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-метил-2H-1,4-диазепин-2-он
К раствору N-метилморфолина (0,38 мл) и (3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутановой кислоты (1,0 г) в 20 мл дихлорметана при -20°C добавляли изобутилхлорформиат (0,39 мл). Полученную в результате смесь перемешивали в течение 1 ч. Добавляли (3R)-гексагидро-3-метил-2Н-1,4-диазепин-2-она гидрохлорид (500 мг) и N-метилморфолин (0,40 мл) в дихлорметане (5 мл) и DMF (8 мл). Смесь перемешивали в течение 28 ч, сначала при -20°C и затем постепенно нагревали до температуры окружающей среды. Реакционную смесь гасили добавлением насыщенного раствора хлорида аммония и экстрагировали последовательно дихлорметаном и этилацетатом. Объединенный органический слой промывали последовательно водой и солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток очищали хроматографией (силикагель, от 3 до 7% 10:1 метанол/концентрированный гидроксид аммония в дихлорметане) с получением двойного продукта. Он был дополнительно очищен путем растворения продукта в смеси этанол (7,5 мл) и гексан (16 мл) при 50°C. Раствор оставляли остывать до комнатной температуры на всю ночь и затем помещали в холодильник на 3 ч. Твердое вещество охладили и промывали, охлаждали смесью 5%этанол/гексан с получением указанного в заголовке соединения.
Стадия F: (3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-метил-2H-1,4-диазепин-2-она гидрохлорид
(3R)-4-[(3R)-3-[(трет-Бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-метил-2Н-1,4-диазепин-2-он из стадии E обрабатывали 4н. соляной кислотой в диоксане, перемешивали в течение 2,5 ч и упаривали с получением указанного в заголовке соединения.
LC/MS 344,1 (M+1).
Пример 2
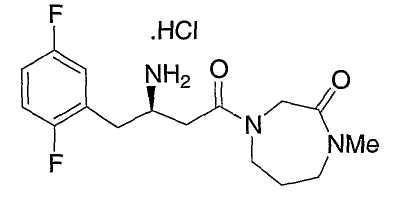
4[(3R)-3-Амино-4-(2,5-дифторфенил)бутаноил]гексагидро-1-метил-2Н-1,4-диазепин-2-она гидрохлорид
Стадия A: Метил-N-(3-аминопропил)-N-(трет-бутоксикарбонил)глицинат
Указанное в заголовке соединение получали из гидрохлорида глицин метилового эфира, следуя способам, описанным в вышеупомянутом примере 1, стадия A-B.
LC/MS 241,0 (M+1).
Стадия В: трет-Бутилгексагидро-3-оксо-Н-1,4-диазепин-1-карбоксилат
К раствору метил-N-(3-аминопропил)-N-(трет-бутоксикарбонил)глицината (10,2 г) в тетрагидрофуране (THF)/метанол (2/1, 300 мл) добавляли 1M водный раствор гидроксида лития (60 мл). Полученную смесь перемешивали при комнатной температуре всю ночь. Добавляли дополнительные 20 мл 1М водного раствора гидроксида лития и смесь перемешивали в течение 6 ч. Растворитель отгоняли при пониженном давлении, и остаток растворяли в 50 мл метанола и 200 мл толуола и концентрировали в вакууме. К остатку в дихлорметане (300 мл) добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC, 9,6 г) и 1-гидроксибензотриазол (HOBT, 6,8 г). Смесь перемешивали при комнатной температуре в течение трех дней, затем обрабатывали насыщенным водным раствором хлорида аммония и экстрагировали тремя частями этилацетата. Объединенные органические фазы промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток очищали хроматографией (силикагель, от 4 до 5% метанол/водный раствор гидроксида аммония (10:1) в дихлорметане) с получением указанного в заголовке соединения.
LS/MS 215,0 (M+1).
Стадия C: трет-Бутилгексагидро-4-метил-3-оксо-1Н-1,4-диазепин-1-карбоксилат
Гидрид натрия (103 мг) добавляли к перемешиваемой смеси трет-бутилгексагидро-3-оксо-1Н-1,4-диазепин-1-карбоксилата в DMF (5 мл) при 0°C. Спустя 1 ч добавляли йодметан (0,15 мл) и полученную в результате смесь перемешивали при 0°C, затем всю ночь при комнатной температуре. Разбавляли насыщенным водным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой отделяли, промывали последовательно насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над сульфатом натрия и концентрировали с получением продукта, который использовали без дополнительной очистки.
Стадия D: Гексагидро-1-метил-2Н-1,4-диазепин-2-она гидрохлорид
Трет-бутилгексагидро-4-метил-3-оксо-1Н-1,4-диазепин-1-карбоксилат, полученный в примере 2, Стадия C, растворяли в 4М соляной кислоты в диоксане и спустя 2,5 ч упаривали с получением указанного в заголовке соединения.
Стадия E: 4-[(3R)-3-[(трет-бутоксикарбонил)амино]-4-2,5-дифторфенил)бутаноил]гексагидро-1-метил-2Н-1,4-диазепин-2-он
К перемешиваемому раствору (3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,5-дифторфенил)бутановой кислоты (40 мг), EDC (29 мг), и HOBT (21 мг) в дихлорметане добавляли триэтиламин (0,042 мл) и гексагидро-1-метил-2Н-1,4-диазепин-2-она гидрохлорид (33 мг). Полученную в результате смесь перемешивали при температуре окружающей среды всю ночь и затем концентрировали. Остаток очищали препаративной ТСХ (силикагель, 8% 10:1 метанол/концентрированный гидроксид аммония в дихлорметане) с получением указанного в заголовке соединения.
Стадия D: 4-[(3R)-3-Амино-4-(2,5-дифторфенил)бутаноил]гексагидро-1-метил-2Н-1,4-диазепин-2-он
(3R)-4-[(3R)-3-[(трет-Бутоксикарбонил)амино]-4-(2,5-трифторфенил)бутаноил]гексагидро-3-метил-2Н-1,4-диазепин-2-он из стадии E обрабатывали 4н.соляной кислотой в диоксане, перемешивали в течение 4 ч и упаривали с получением указанного в заголовке соединения.
LC/MS 326,0 (M+1).
Пример 3
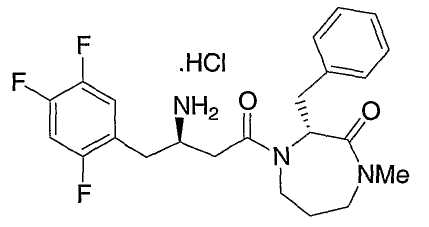
(3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-бензилгексагидро-1-метил-2Н-1,4-диазепин-2-она гидрохлорид
Стадия A: трет-Бутил-2-бензилгексагидро-4-метил-3-оксо-1Н-1,4-диазепин-1-карбоксилат
К перемешиваемому раствору трет-бутилгексагидро-4-метил-3-оксо-1Н-1,4-диазепин-1-карбоксилата, приготовленного как описано в примере 2, стадия C (180 мг), в THF (8 мл) при -78°C добавляли раствор диизопропиламид лития (LDA) (1,5Mв циклогексане, 0,53 мл). После перемешивания смеси в течение 40 мин добавляли бензилбромид (0,28 мл). Полученную в результате смесь продолжали перемешивать при -78°C в течение 6 ч. Затем реакционную смесь разбавляли насыщенным водным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой промывали последовательно насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над безводным сульфатом натрия и концентрировали. Остаток очищали хроматографией (силикагель, 2% метанол/дихлометан) с получением указанного в заголовке соединения.
Стадия B: 3-Бензилгексагидро-1-метил-2Н-1,4-диазепин-2-она гидрохлорид
Трет-бутил-2-бензилгексагидро-4-метил-3-оксо-1Н-1,4-диазепин-1-карбоксилат, полученный на стадии A, растворяли в 4Мсоляной кислоты в диоксане и спустя 2,5 ч упаривали с получением гидрохлоридной соли желаемого соединения.
Стадия C: (3R)-4-[(3R)-3-[(трет-Бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]-3-бензилгексагидро-1-метил-2Н-1,4-диазепин-2-он
N-метилморфолин (0,048 мл) и изобутилхлорформиат (0,026 мл) добавляли к перемешиваемому раствору (3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутановой кислоты (67 мг) в THF (1 мл) при -20°C и полученную в результате смесь перемешивали в течение 1 ч. Добавляли 3-бензилгексагидро-1-метил-2Н-1,4-диазепин-2-она гидрохлорид, полученный на стадии B выше (48 мг), и N-метилмморфолин (0,024 мл) в DMF (1 мл). Смесь перемешивали в течение 30 мин при -20°C и в течение 36 ч при температуре окружающей среды и затем упаривали. Остаток обрабатывали насыщенным водным раствором хлорида аммония, экстрагировали этилацетатом и органический экстракт упаривали. Полученный продукт очищали препаративной ТСХ (силикагель, метанол/концентрированный гидроксид аммония/дихлорметан 4,4:0,1:95,5) с получением двойного продукта в виде смеси диастереомеров. Изомеры разделяли ВЭЖХ (ChiralPAK AD, 14% этанол/гексан) и собирали быстрее элюируемый (R,S)-изомер и медленнее элюируемый (R,R)-изомер.
Стадия D: Гидрохлорид (3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-бензилгексагидро-1-метил-2Н-1,4-диазепин-2-она
Указанное в заголовке соединение со стадии C растворяли в 4М соляной кислоте в диоксане и спустя 2,5 ч упаривали с получением желаемого продукта.
LC/MS 434,1 (M+1).
Пример 4
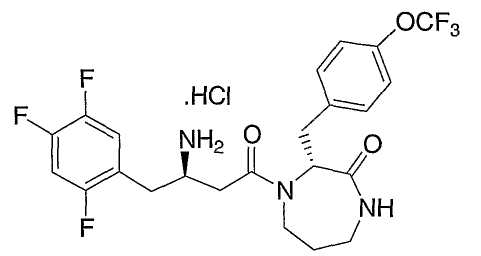
(R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-[4-(трифторметокси)бензил]-2Н-1,4-диазепин-2-он
Стадия A: трет-Бутил-4-[(бензилокси)метил]гексагидро-3-оксо-1Н-1,4-диазепин-1-карбоксилат
Указанное в заголовке соединение получали из трет-бутилгексагидро-3-оксо-1Н-1,4-диазепин-1-карбоксилата (пример 2, стадия B) бензилхлорметилового эфира, по существу, следуя способу, описанному в примере 2, стадия C.
Стадия В: трет-Бутил-4-[(бензилокси)метил]гексагидро-3-оксо-2-[4-(трифторметокси)бензил]-1H-1,4-диазепин-1-карбоксилат
Указанное в заголовке соединение получали из трет-бутил-4-[(бензилокси)метил]гексагидро-3-оксо-1H-1,4-диазепин-1-карбоксилата и 4-(трифторметокси)бензилбромида, по существу, следуя способу, описанному в примере 3, стадия 1.
Стадия C: трет-Бутилгексагидро-3-оксо-2-[4-(трифторметокси)бензил]-1Н-1,4-диазепин-1-карбоксилат
К раствору трет-бутил-4-[(бензилокси)метил]гексагидро-3-оксо-2-[4-(трифторметокси)бензил]-1Н-1,4-диазепин-1-карбоксилата (520 мг) в этаноле (17 мл) добавляли 10% палладий на угле (300 мг). Реакционную смесь перемешивали в течение 22 ч в атмосфере водорода. Добавляли несколько капель воды и продолжали перемешивание в течение дополнительных 20 ч. Смесь фильтровали через целит, и целит промывали этилацетатом. Фильтрат упаривали при пониженном давлении. Остаток очищали флэш-хроматографией на устройстве Biotage® (силикагель; метанол/концентрированный водный раствор гидроксида аммония/дихлорметан 1,5:0,1:98,4). Полученный продукт растворяли в толуоле и кипятили с обратным холодильником в течение 3 ч. Упаривание в вакууме дало указанное в заголовке соединение, которое использовали на следующих стадиях без дополнительной очистки.
Стадия D: Гидрохлорид гексагидро-3-[4-(трифторметокси)бензил]-2Н-1,4-диазепин-2-она
Трет-бутилгексагидро-3-оксо-2-[4-(трифторметокси)бензил]-1H-1,4-диазепин-1-карбоксилат растворяли в 4М соляной кислоте в диоксане и спустя 2,5 ч упаривали с получением желаемого продукта.
Стадия E: (3R)-4-[(3R)-3-[(трет-Бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-[4-трифторметокси)бензил]-2Н-1,4-диазепин-2-он
N-метилморфолин (0,072 мл) и изобутилхлорформиат (0,039 мл) добавляли к перемешиваемому раствору (3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутановой кислоты (95 мг) в дихлорметане (5 мл) при -20°C и полученную в результате смесь перемешивали в течение 30 мин. Добавляли гексагидро-3-[4-(трифторметокси)бензил]-2Н-1,4-диазепин-2-она гидрохлорид, полученный на стадии D (91 мг), и N-метилморфолин (0,036 мл). Реакционную смесь перемешивали в течение 30 мин при -20°C и 2 ч при температуре окружающей среды и затем упаривали. Остаток очищали препаративной ТСХ (силикагель, метанол/насыщенный водный раствор гидроксида аммония/дихлорметан 4,4:0,1:95,5), и изомеры последовательно разделяли ВЭЖХ (ChiralPAK OD, 7% этанол/гексан) с получением более медлено элюируемого (R,R)-изомера.
Стадия F: Гидрохлорид (3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-[4-(трифторметокси)бензил]-2Н-1,4-диазепин-2-она
(3R)-4-[(3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-[4-(трифторметокси)бензил]-2H-1,4-диазепин-2-он растворяли в 4М соляной кислоте в диоксане и спустя 2,5 ч упаривали с получением желаемого продукта.
LC/MS 504,2 (M+1).
Пример 5
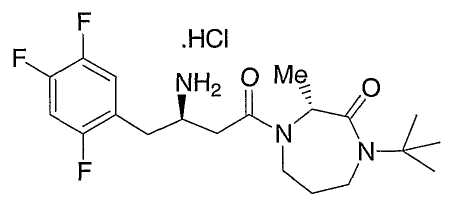
(3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]-1-трет-бутилгексагидро-3-метил-2Н-1,4-диазепин-2-она гидрохлорид
Стадия A: N-(трет-Бутил)-N-(2-цианоэтил)-2-оксопропанамид
Раствор бензотриазола (2,4 г, 20 мМ) и тионилхлорида (1,5 мл) в дихлорметане (10 мл) добавляли по каплям к перемешиваемому раствору пировиноградной кислоты в дихлорметане (10 мл) и смесь перемешивали в течение 10 мин. Образованный остаток фильтровали и промывали дихлорметаном. Фильтрат обрабатывали сульфатом магния, фильтровали и фильтрат перемешивали всю ночь с раствором 3-(трет-бутиламино)пропионитрила, растворенным в дихлорметане (10 мл). Реакционную смесь обрабатывали насыщенным водным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния, фильтровали, упарили и очищали при помощи Biotage® флэш-хроматографией (силикагель, этилацетат/гексан 1:1) с получением желаемого продукта в виде масла.
1H ЯМР (400 МГц, CDCl3) δ 1,49 (с, 9H), 2,45 (с, 3H), 2,74 (т, J=7,4 Гц, 2H), 3,6 (ушир., 2H).
Стадия B: 1-трет-Бутилгексагидро-3-метил-2H-1,4-диазепин-2-он
N-(трет-бутил)-N-(2-цианоэтил)-2-оксопропанамид, полученный на стадии A (440 мг), и оксид платины (60 мг) суспендировали в этаноле (40 мл), содержащем хлороформ (0,3 мл), и перемешивали в шейкере Parr в атмосфере водорода при 40 фунт/дюйм2 в течение 16 ч. Смесь фильтровали, катализатор промывали смесью метанол/дихлорметан (10:90) и объединенный фильтрат упаривали и очищали при помощи Biotage® флэш-хроматографии (силикагель, 5-10% метанол/дихлорметан) с получением желаемого продукта.
1H ЯМР (400 МГц, CDCl3) δ 1,23 (д, J=6,6, 3H), 1,43 (с, 9H), 1,5 (м, 1H), 1,7 (м, 1H), 2,9 (м, 1H), 3,2 (м, 1H), 3,4 (м, 1H.), 3,6 (м, 2H). LC/MS 185,2 (M+1).
Стадия C: (3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]-1-трет-бутил-гексагидро-3-метил-2H-1,4-диазепин-2-он
Указанное в заголовке соединение получали из (3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутановой кислоты и 1-трет-бутилгексагидро-3-метил-2Н-1,4-диазепин-2-она способом, описанным в примере 3, стадии C и D. LC/MS 400,1 (M+1).
Пример 6
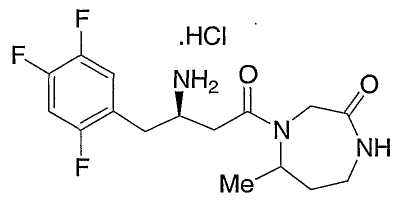
4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-5-метил-2Н-1,4-диазепин-2-она гидрохлорид
Стадия A: Гексагидро-5-метил-2Н-1,4-диазепин-2-он
Указанное в заголовке соединение получали из кротононитрила, по существу, следуя процедурам, описанным в примере 2, стадии A и B.
Стадия В: 4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил] гексагидро-5-метил-2H-1,4-диазепин-2-он
Указанное в заголовке соединение получали из (3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутановой кислоты гексагидро-5-метил-2Н-1,4-диазепин-2-она способом, описанным в примере 3, стадии C и D. LC/MS 344,1 (M+1).
Пример 7
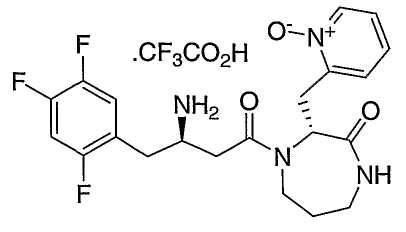
(3R)-4[(3R)-3-Амино-4(2,4,5-трифторфенил)бутаноил]гексагидро-3[(1-оксидопиридин-2-ил)метил]-2Н-1,4-диазепин-2-он, соль трифторуксусной кислоты
Стадия A: (3R)-4-[(3R)-3-[трет-Бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-[(1-оксидопиридин-2-ил)метил]-2Н-1,4-диазепин-2-он
Указанное в заголовке соединение получали, по существу, следуя процедурам, описанным в примере 4, стадии от A до E.
Стадия B: (3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-[(1-оксидопиридин-2-ил)метил]-2Н-1,4-диазепин-2-он, соль трифторуксусной кислоты
К раствору (3R)-4-[(3R)-3-[трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-[(1-оксидопиридин-2-ил)метил]-2Н-1,4-диазепин-2-она (20 мг, 0,038 мМ) в дихлорметане (2,5 мл) при 0°C добавляли MCPBA (28 мг, 0,096 мМ) и реакционную смесь перемешивали всю ночь при температуре окружающей среды. Раствор обрабатывали насыщенным водным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток очищали препаративной ТСХ (силикагель, 11:89 10% аммоний в метанол/дихлорметан) с получением N-BOC защищенного N-оксида пиридина. Снятие защиты смесью трифторуксусная кислота-дихлорметан (1:1) при температуре окружающей среды в течение 1 ч с последующей концентрацией давало желаемый продукт. MS 437,2 (M+1).
Пример 8
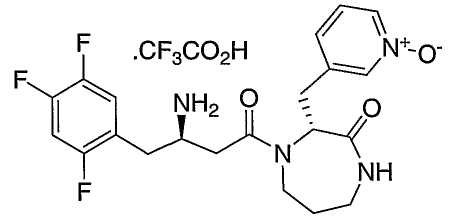
(3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-[(1-оксидопиридин-3-ил)метил]-2Н-1,4-диазепин-2-он, соль трифторуксусной кислоты
Указанное в заголовке соединение получали, по существу, следуя процедурам, описанным в примере 7. MS 437,2 (M+1).
Пример 9
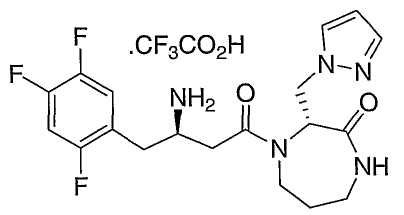
(3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-(1Н-пиразол-1-илметил)-2Н-1,4-диазепин-2-он, соль трифторуксусной кислоты
Стадия A: трет-Бутил-4-[(бензилокси)метил]гексагидро-3-оксо-1Н-1,4-диазепин-1-карбоксилат
Указанное в заголовке соединение получали из трет-бутилгексагидро-3-оксо-1Н-1,4-диазепин-1-карбоксилат (пример 2, стадия B) и бензилхлорметилового эфира, по существу следуя способу, описанному в примере 2, стадия C.
Стадия В: трет-Бутил-4-[(бензилокси)метил]гексагидро-2-метилен-3-оксо-1,4-диазепин-1-карбоксилат
Указанное в заголовке соединение получали из трет-бутил-4-[(бензилокси)метил]гексагидро-3-оксо-1Н-1,4-диазепин-1-карбоксилата и бензилхлорметилового эфира, по существу, следуя способу, описанному в примере 3, стадия 1.
Стадия C: трет-Бутил-4-[(бензилокси)метил]гексагидро-2-(1Н-пиразол-1-илметил)-3-оксо-1,4-диазепин-1-карбоксилат
К раствору пиразола (258 мг, 3,78 мМ) в 10 мл DMF при 0°C добавляли гидрид натрия (60%, 91 мг). Полученную в результате смесь перемешивали в течение 30 мин и затем добавляли продукт со стадии B (655,4, 1,89 мМ). Реакционную смесь перемешивали при температуре окружающей среды всю ночь и гасили добавлением воды. Водную смесь экстрагировали тремя частями этилацетата. Объединенные органические фазы концентрировали. Очистка флэш-хроматографией на приборе Biotage® (силикагель, градиент 40-80% этилацетат/гексан) давала указанное в заголовке соединение.
Стадия D: 3-(Н-Пиразол-1-илметил)-1,4-диазепин-2-он
Продукт со стадии C обрабатывали трифторуксусной кислотой. Реакционную смесь перемешивали при температуре окружающей среды всю ночь и затем концентрировали. Остаток растворяли в толуоле и кипятили с обратным холодильником в течение 3 ч. Очистка флэш-хроматографией на приборе Biotage® (силикагель, 5-15% смеси 10:1 метанол/хлорид аммония в дихлорметане) давала указанное в заголовке соединение.
Стадия E: (3R)-4-[(3R)-3-Амино-4-(2,4,5-трифторфенил)бутаноил]гексагидро-3-(1Н-пиразол-1-илметил)-2Н-1,4-диазепин-2-он, соль трифторуксусной кислоты
Указанное в заголовке соединение получали из продукта со стадии D и (3R)-3-[(трет-бутоксикарбонил)амино]-4-(2,4,5-трифторфенил)бутановой кислоты, по существу, следуя способу связывания, описанному в примере 4, стадия E. Очистка препаративной ТСХ (силикагель, 1:9 этанол/дихлорметан) давала N-BOC продукт в виде смеси диастереомеров. ВЭЖХ (колонка Chiralcell OJ, 7% этанол/гексан) давала отдельные диастереомеры. Снятие защиты смесью 1:1 трифторуксусная кислота/дихлорметан в течение 1 ч при температуре окружающей среды с последующей концентрацией давала отдельные диастереомеры указанного в заголовке соединения. MS 410,2 (M+1).
По существу, следуя способам, описанным в примерах 1-9, получали соединения таблицы 2.
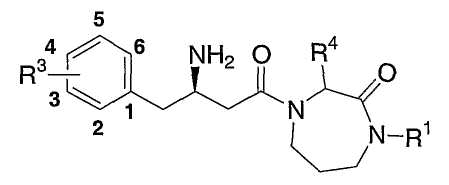
C6H5
Пример фармацевтического препарата
В качестве конкретного варианта воплощений фармацевтической композиции для перорального применения готовили таблетку с содержанием действующего вещества 100 мг из 100 мг любого соединения по настоящему изобретению, 268 мг микрокристаллической целлюлозы, 20 мг натрийкроскармелозы и 4 мг стеарата магния. Действующее вещество, микрокристаллическую целлюлозу и кроскармелозу смешивали первыми. Затем в смесь вводили смазывающий агент в виде стеарата магния и прессовали в таблетку.
Хотя настоящее изобретение описано и проиллюстрировано со ссылками на основные конкретные варианты его осуществления, специалисты в данной области техники признают, что различные доработки, изменения, модификации, замещения, сокращения или дополнения процедур и протоколов могут быть сделаны без отступления от сути и объема по настоящему изобретению. Например, могут быть пригодными эффективные дозы, отличающиеся от конкретных доз, приведенных ранее в настоящем описании, как следствие различий реактивности млекопитающего, которое находится на лечении с любым из указанных симптомов соединениями по настоящему изобретению, указанными выше. Определенные наблюдаемые фармакологические ответы могут отличаться согласно и в зависимости от конкретных выбранных активных соединений или присутствия фармакологических носителей, а также от типа состава и используемого режима введения, и такие ожидаемые вариации или различия в результатах предполагаются соответствующими задачам и видам практического применения по настоящему изобретению. Следовательно, подразумевается, что настоящее изобретение определено объемом нижеследующей формулы изобретения, и что указанная формула изобретения должна быть интерпретирована максимально широко.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ [(3-ГИДРОКСИПИРИДИН-2-КАРБОНИЛ)АМИНО]АЛКАНОВЫХ КИСЛОТ, СЛОЖНЫХ ЭФИРОВ И АМИДОВ | 2012 |
|
RU2764667C2 |
| ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ-4 ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2-ГО ТИПА, СОЕДИНЕНИЯ (ВАРИАНТЫ) | 2018 |
|
RU2712097C1 |
| ИНГИБИТОР DPP-IV, ВКЛЮЧАЮЩИЙ БЕТА-АМИНОГРУППУ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ И ЛЕЧЕНИЯ ДИАБЕТА ИЛИ ОЖИРЕНИЯ | 2008 |
|
RU2419615C1 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА ДИПЕПТИДИЛПЕПТИДАЗЫ-IV И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2010 |
|
RU2499792C2 |
| ПРОИЗВОДНЫЕ ДИАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ АРИТМИИ | 1994 |
|
RU2155587C2 |
| СПОСОБ ПОЛУЧЕНИЯ [(3-ГИДРОКСИПИРИДИН-2-КАРБОНИЛ)АМИНО]АЛКАНОВЫХ КИСЛОТ, СЛОЖНЫХ ЭФИРОВ И АМИДОВ | 2012 |
|
RU2602083C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА ДИПЕПТИДИЛПЕПТИДАЗЫ-IV И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2010 |
|
RU2498976C9 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ДИАБЕТА ИЛИ ОЖИРЕНИЯ, СОДЕРЖАЩАЯ СОЕДИНЕНИЕ, ИНГИБИРУЮЩЕЕ АКТИВНОСТЬ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV, И ДРУГИЕ ПРОТИВОДИАБЕТИЧЕСКИЕ СРЕДСТВА ИЛИ СРЕДСТВА ПРОТИВ ОЖИРЕНИЯ В КАЧЕСТВЕ АКТИВНЫХ ИНГРЕДИЕНТОВ | 2009 |
|
RU2466727C1 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
Описываются новые производные гексагидродиазепинона общей формулы I
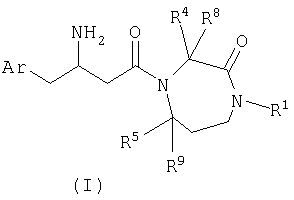
или их фармацевтически приемлемые соли; где Ar представляет собой фенил, замещенный одним-тремя атомами галогена;
R1 означает водород, C3-6циклоалкил, C1-10алкил, возможно замещенный гидрокси, галогеном C1-10алкоксикарбонилом или фенил-С1-3алкокси;
R4 - означает
водород, C1-10алкил, необязательно замещенный
(a) фенилом, который может быть замещен галогеном, C1-3алкилом, галоген-С1-3алкилом, галоген-С1-3алкокси,
(b)гидрокси,
(c) C3-6циклоалкилом,
(d) фенил-С1-3алкокси,
(e) бифенилом,
(f) пиридилом,
(g) 1-оксидо-пиридинилом,
(i) пиразолилом;
R5 означает водород или C1-10алкил;
R8 и R9 означает водород, фармацевтическая композиция, их содержащая, и применение новых соединений для получения лекарственного средства для лечения инсулиннезависимого диабета. 7 н. и 9 з.п. ф-лы, 2 табл.
или их фармацевтически приемлемые соли;
где Ar представляет собой фенил, замещенный одним-тремя атомами галогена;
R1 означает водород, C1-10алкил, необезательно замещенный галогеном, гидрокси, С1-10алкоксикарбонилом или фенил-С1-3алкокси, или С3-6циклоалкил;
R4 выбран из группы, состоящей из:
водорода, C1-10алкила, необязательно замещенного
(a) фенилом, который может быть замещен галогеном, C1-3алкилом, галоген-C1-3алкилом, галоген-C1-3алкокси,
(b)гидрокси,
(c) C3-6циклоалкилом,
(d) фенил-C1-3алкокси,
(e) бифенилом,
(f) пиридилом,
(g) 1-оксидопиридинилом,
(h) галоген-C1-3алкилом,
(i) пиразолилом;
R5 представляет собой водород или C1-10алкил;
R8 и R9, каждый, независимо, представляют собой водород.
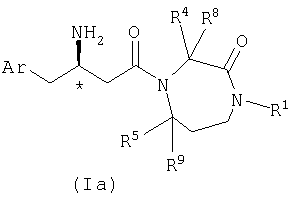
где атом углерода, помеченный *, имеет R конфигурацию и Ar, R1, R4, R5, R8 и R9 являются такими, как определено в п.1.
водорода,
C1-4алкила,
2,2,2-трифторэтила,
метоксикарбонилметила,
гидроксиэтила,
бензилоксиметила,
бензилоксиэтила и
циклопропила.
водорода,
CH3,
CH2CH3,
CH2CH(CH3)2,
CH2-циклопропила,
CH2-циклогексила,
CH2OCH2Ph,
CH2OH,
CH2Ph,
CH2(3-OCF3-Ph),
CH2(4-OCF3-Ph),
CH2(3-CF3,5-CF3-Ph),
CH2(2-CF3-Ph),
CH2(2-Cl-Ph),
CH2(2-Me-Ph),
CH2(2-Me,5-Me-Ph),
CH2(2-Ph-Ph),
CH2(2-F,5-F-Ph),
CH2(2-F-Ph),
CH2(2-F,3-F-Ph),
CH2(2-пиридинил),
CH2(3-пиридинил),
CH2(4-пиридинил),
CH2(1-оксидопиридин-2-ил),
CH2(1-оксидопиридин-3-ил),
CH2(1Н-пиразол-1-ил),
CH2(2-F,6-F-Ph) и
CH2CF3.
водорода,
C1-4алкила,
2,2,2-трифторэтила,
метоксикарбонилметила,
гидроксиэтила,
бензилоксиметила,
бензилоксиэтила и
циклопропила;
Ar представляет собой фенил, замещенный атомами хлора или фтора;
R4 выбран из группы, состоящей из
водорода,
CH3,
CH2CH3,
CH2CH(CH3)2,
CH2-циклопропила,
CH2-циклогексила,
CH2OCH2Ph,
CH2OH,
CH2Ph,
CH2(3-OCF3-Ph),
CH2(4-OCF3-Ph), и
CH2(3-CF3,5-CF3-Ph),
CH2(2-CF3-Ph),
CH2(2-Cl-Ph),
CH2(2-Me-Ph),
CH2(2-Me,5-Me-Ph),
CH2(2-Ph-Ph),
CH2(2-F,5-F-Ph),
CH2(2-F-Ph),
CH2(2-F,3-F-Ph),
CH2(2-пиридинил),
CH2(3-пиридинил),
CH2(4-пиридинил),
CH2(1-оксидопиридин-2-ил),
CH2(1-оксидопиридин-3-ил),
CH2(1Н-пиразол-1-ил),
CH2(2-F,6-F-Ph) и
CH2CF3.
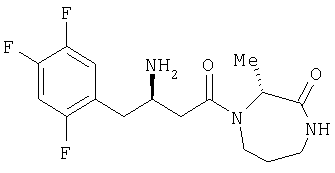
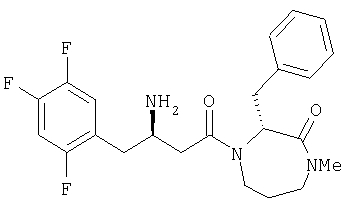
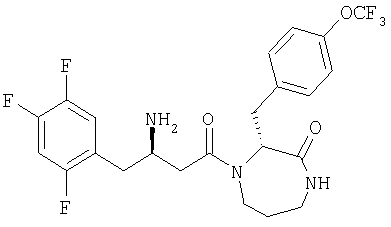
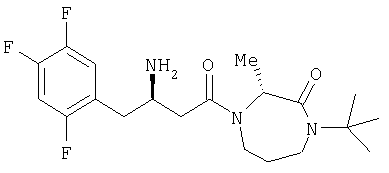
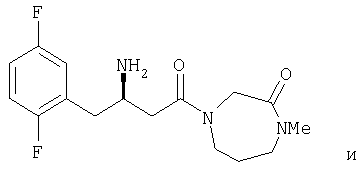
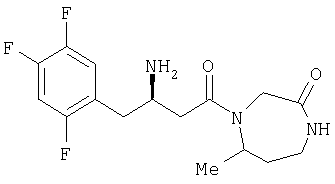
или их фармацевтически приемлемые соли.
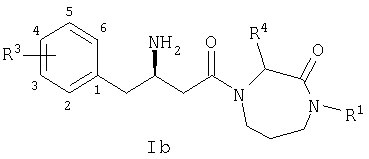
или их фармацевтически приемлемые соли.
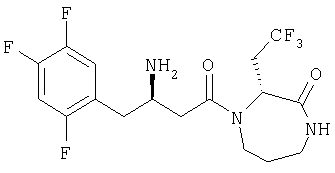
или его фармацевтически приемлемая соль.
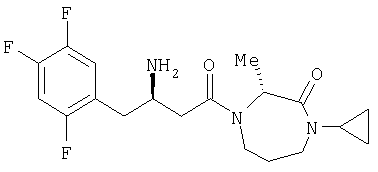
или его фармацевтически приемлемая соль.
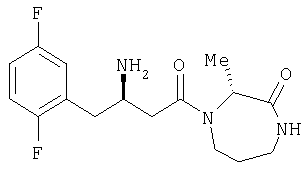
или его фармацевтически приемлемая соль.
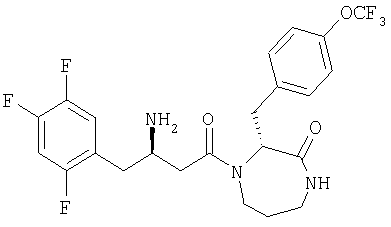
или его фармацевтически приемлемая соль.
| Камера разделения электрофоретического сепаратора | 1985 |
|
SU1364647A1 |
| ДИАЗЕПАНОВОЕ ПРОИЗВОДНОЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ЕГО СОДЕРЖАЩАЯ, И ИНГИБИТОР АКТИВИРОВАННОГО ФАКТОРА X КОАГУЛЯЦИИ КРОВИ | 2001 |
|
RU2257381C2 |

