Настоящее изобретение относится к способу получения дигидрохиназолинов, которые применяются для приготовления лекарственных средств.
Соединения, полученные способом, предлагаемым в настоящем изобретении, пригодны для применения в качестве противовирусных средств, в частности для борьбы с цитомегаловирусами, как это описано в WO 04/072048 и WO 04/096778.
Синтез дигидрохиназолинов, описанный в этих документах, начинают с 2-галогензамещенного анилина (А), который путем сочетания по Хеку превращают в производное 2-аминокоричной кислоты (В). По реакции с трифенилфосфином в тетрахлориде углерода получают фосфинимид (С), который затем вводят в реакцию с изоцианатом с выделением трифенилфосфиноксида и с получением карбодиимида (D). По реакции карбодиимида (D) с амином образуется дигидрохиназолинметиловый эфир (Е), который разделяют на энантиомеры с помощью хроматографии на хиральной фазе. Затем при стандартных условиях проводят гидролиз в дигидрохиназолинкарбоновую кислоту (F1). Синтез иллюстрируют приведенные ниже схемы 1 и 2.
Схема 1:
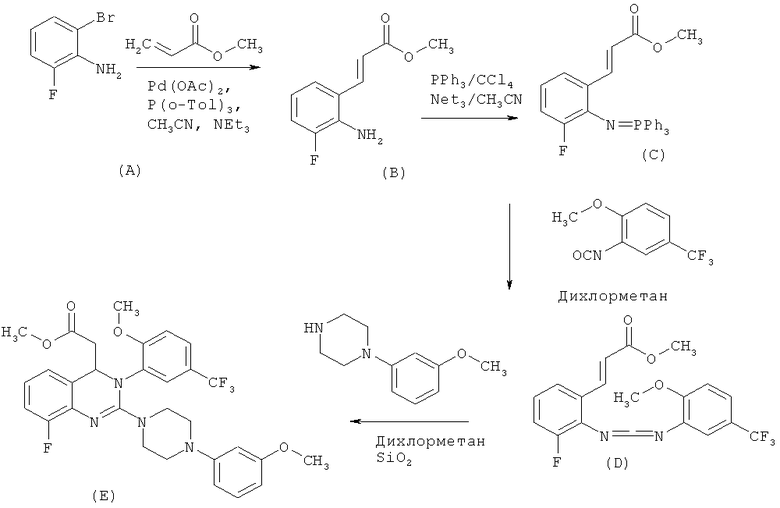
Схема 2
Описанные выше стадии реакции при проведении в промышленном масштабе сопряжены со значительным риском и образованием побочных продуктов, а также стехиометрических количеств органических отходов. При использовании фосфинимида (С) и карбодиимида (D) во время реакции образуются промежуточные продукты, содержащие высокореакционноспособные функциональные группы, что приводит к значительному количеству побочных продуктов. Эти побочные продукты можно отделить только с помощью весьма трудоемкой хроматографической очистки или трудоемкой процедуры экстракции.
Кроме того, во время реакции соединения формулы (С) с образованием соединения формулы (D) в стехиометрических количествах образуется трифенилфосфиноксид, хроматографическое отделение которого от искомого продукта является трудоемкой процедурой. Хроматография является особенно неподходящей для синтеза соединений в промышленном масштабе, поскольку она требует много времени и является трудоемкой и при этом используется большое количество растворителей.
Разделение энантиомеров соединения формулы (Е) проводят с помощью трудоемкой хроматографии на хиральной фазе и при этом в качестве отхода выделяется нежелательный R-энантиомер.
В основу настоящего изобретения была положена задача разработки применимого в промышленности способа получения дигидрохиназолина формулы (I), в частности, (S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусной кислоты, в котором устранены недостатки указанных выше стадий предшествующего уровня техники и в котором не образуется нежелательный отход - R-энантиомер.
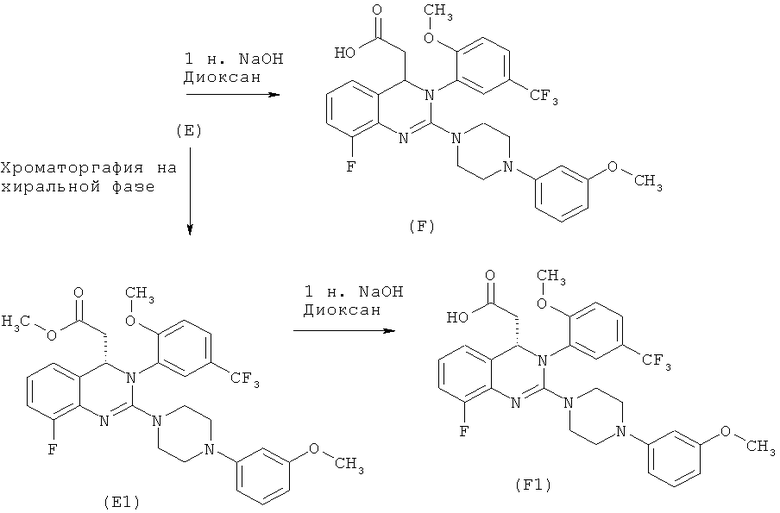
В контексте настоящего изобретения эта задача решена описанным ниже образом. Приведенные ниже схемы 3 и 4 иллюстрируют отдельные стадии реакции.
Схема 3:
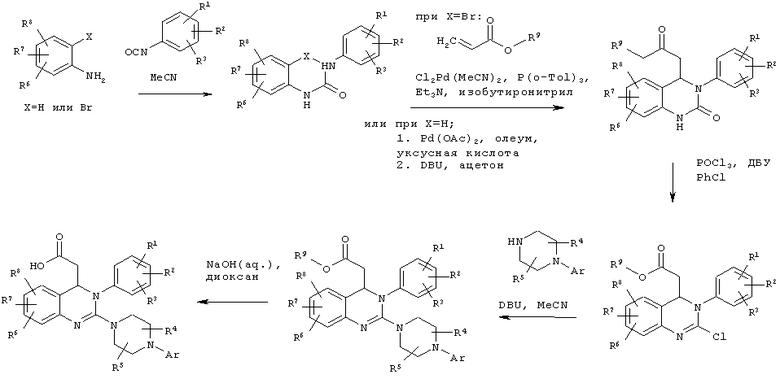
Схема 4:
Согласно изобретению неожиданно было установлено, что соединения формулы (I) можно получить способом, предлагаемым в настоящем изобретении, т.е. по реакции 2-галогензамещенного анилина с изоцианатом с последующей реакцией Хека с алкилакрилатом, предпочтительно метилакрилатом, и при этом можно избежать образования фосфинимида и карбодиимида в качестве реакционноспособных промежуточных продуктов и образования стехиометрических количеств трифенилфосфиноксида соответственно.
Кроме того, согласно изобретению неожиданно было установлено, что соединения промежуточных стадий образуют кристаллы и их можно очистить с помощью кристаллизации без хроматографии или экстракции и таким образом возможно промышленное применение этих стадий способа.
Кроме того, согласно изобретению неожиданно было установлено, что структурный блок метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил} ацетат можно эффективно синтезировать с помощью орто-палладирования. В этом случае N-(2-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевина вводят в реакцию с метилакрилат и окислительным реагентом в присутствии кислоты и получают метил-(2Е)-3-{3-фтор-2-[({[2-метокси-5-(трифторметил)фенил]амино}карбонил)амино]фенил}акрилат. Затем замывание цикла тетрагидрохиназолина проводят в щелочной среде.
Кроме того, согласно изобретению неожиданно было установлено, что алкил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетаты, предпочтительно метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат, можно разделить на энантиомеры путем кристаллизации с (28,38)-2,3-бис[(4-метилбензоил)окси]янтарной кислотой.
Кроме того, согласно изобретению неожиданно было установлено, что R-энантиомер алкил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата, предпочтительно -метил- {8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата можно рацемизировать в щелочной среде на стадии образования кислоты после гидролиза алкилового или метилового эфира и после повторной этерификации можно разделить с помощью кристаллизации с (2S,3S)-2,3-бис[(4-метилбензоил)-окси]янтарной кислотой и при этом повышается полный выход S-энантиомера.
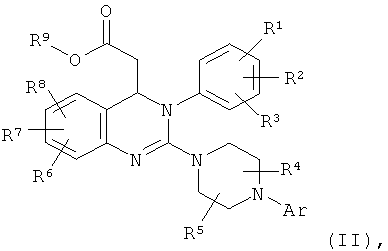
При более подробном описании предлагаемый в настоящем изобретении способ получения соединения формулы (I)
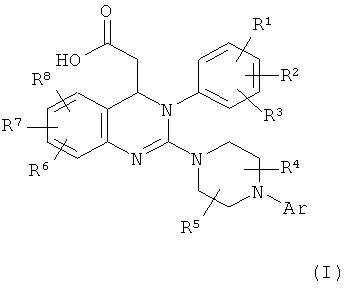
в которой
Аr обозначает арил, где арил может содержать от 1 до 3 заместителей и заместители независимо друг от друга выбраны из группы, включающей алкил, алкоксигруппу, формил, карбоксигруппу, алкилкарбонил, алкоксикарбонил, трифторметил, галоген, цианогруппу, гидроксигруппу, аминогруппу, алкиламиногруппу, аминокарбонил и нитрогруппу, где алкил может содержать от 1 до 3 заместителей и заместители независимо друг от друга выбраны из группы, включающей галоген, аминогруппу, алкиламиногруппу, гидроксигруппу и арил, или два из заместителей арила вместе с атомами углерода, с которыми они связаны, образуют 1,3-диоксолановое кольцо, циклопентановое кольцо или циклогексановое кольцо и необязательно содержащийся третий заместитель независимо выбран из указанной группы,
R1 обозначает водород, аминогруппу, алкил, алкоксигруппу, алкиламиногруппу, алкилтиогруппу, цианогруппу, галоген, нитрогруппу или трифторметил,
R2 обозначает водород, алкил, алкоксигруппу, алкилтиогруппу, цианогруппу, галоген, нитрогруппу или трифторметил,
R3 обозначает аминогруппу, алкил, алкоксигруппу, алкиламиногруппу, алкилтиогруппу, цианогруппу, галоген, нитрогруппу, трифторметил, алкилсульфонил или алкиламиносульфонил
или
один из радикалов R1, R2 и R3 обозначает водород, алкил, алкоксигруппу, цианогруппу, галоген, нитрогруппу или трифторметил и два других вместе с атомами углерода, с которыми они связаны, образуют 1,3-диоксолановое кольцо, циклопентановое кольцо или циклогексановое кольцо,
R4 обозначает водород или алкил,
R5 обозначает водород или алкил
или
радикалы R4 и R5 в пиперазиновом кольце связаны с расположенными друг напротив друга атомами углерода и образуют метиленовый мостик, который необязательно замещен 1 или 2 метальными группами,
R6 обозначает водород, алкил, алкоксигруппу, алкилтиогруппу, формил, карбоксигруппу, аминокарбонил, алкилкарбонил, алкоксикарбонил, трифторметил, галоген, цианогруппу, гидроксигруппу или нитрогруппу,
R7 обозначает водород, алкил, алкоксигруппу, алкилтиогруппу, формил, карбоксигруппу, алкилкарбонил, алкоксикарбонил, трифторметил, галоген, цианогруппу, гидроксигруппу или нитрогруппу
и
R8 обозначает водород, алкил, алкоксигруппу, алкилтиогруппу, формил, карбоксигруппу, алкилкарбонил, алкоксикарбонил, трифторметил, галоген, цианогруппу, гидроксигруппу или нитрогруппу,
включает гидролиз сложного эфира соединения формулы (II)
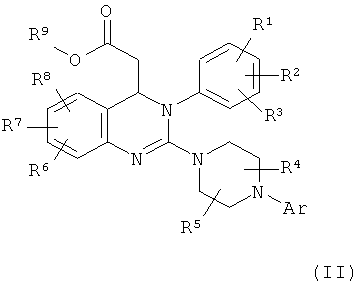
в которой
Аr, R1, R2, R3, R4, R5, R6, R7 и R8 обладают указанными выше значениями и
R9 обозначает С1-С4-алкил,
с использованием основания или кислоты.
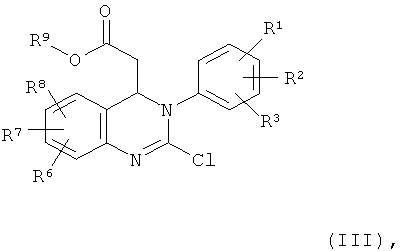
Соединение формулы (II) можно получить по реакции соединения формулы (III)
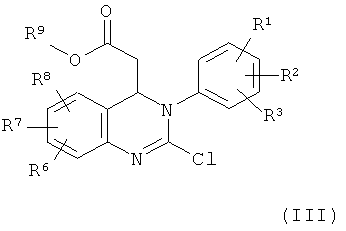
в которой
R1, R2, R3, R6, R7 и R8 обладают указанными выше значениями и
R9 обозначает С1-С4-алкил,
в присутствии основания
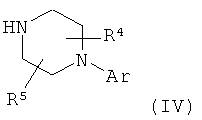
с соединением формулы (IV)
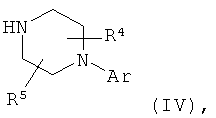
в которой
Аr, R4 и R5 обладают указанными выше значениями.
Соединение формулы (III) можно получить по реакции соединения формулы (V)
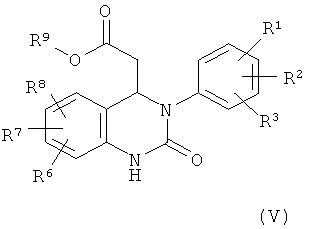 ,
,
в которой
R1, R2, R3, R6, R7 и R8 обладают указанными выше значениями и
R9 обозначает С1-С4-алкил,
с оксихлоридом фосфора, трихлоридом фосфора или пентахлоридом фосфора в присутствии основания.
Соединение формулы (V) можно получить по реакции соединения формулы (VI)
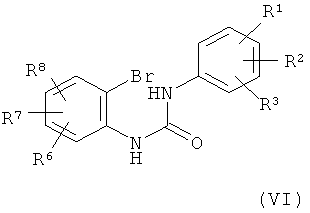
в которой
R1, R2, R3, R6, R7 и R8 обладают указанными выше значениями, с соединением формулы
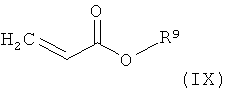
в которой
R9 обозначает С1-С4-алкил,
в присутствии палладиевого катализатора и основания.
Соединения формул (IV), (VI) и (IX) известны специалисту в данной области техники или можно получить по обычным методикам, известным из литературы.
В альтернативной методике соединение формулы (V) можно получить по реакции соединения формулы (VII)
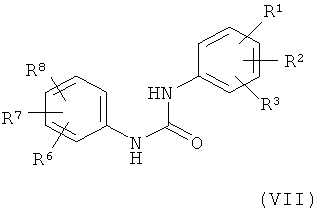
в которой
R1, R2, R3, R6, R7 и R8 обладают указанными выше значениями,
на первой стадии с соединением формулы (IX) в уксусной кислоте в присутствии палладиевого катализатора, окислительного реагента и кислоты с получением соединения формулы (VIII)
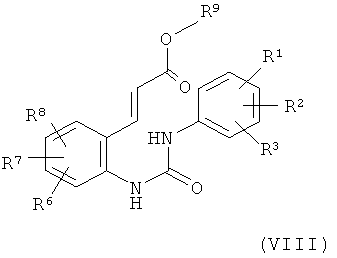
в которой
R1, R2, R3, R6, R7 и R8 обладают указанными выше значениями и
R9 обозначает С1-С4-алкил,
и на второй стадии с основанием с получением соединения формулы (V).
Соединения формулы (VII) известны специалисту в данной области техники или можно получить по обычным методикам, известным из литературы.
В предпочтительном варианте осуществления настоящего изобретения в методике синтеза радикал R9 в соединениях формул (II), (III), (V), (VIII) и (IX) обозначает метил.
Кроме того, настоящее изобретение относится к соединениям формулы (III)
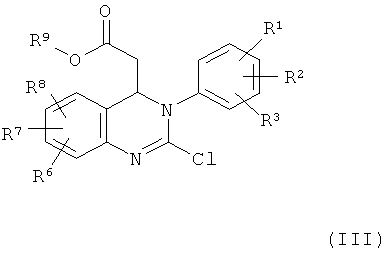
в которой
R1, R2, R3, R6, R7 и R8 обладают указанными выше значениями и
R9 обозначает С1-С4-алкил.
Соединения формулы (III) в которой R9 обозначает метил, являются предпочтительными.
Кроме того, настоящее изобретение относится к соединениям формулы (V)
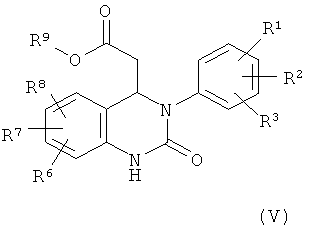
в которой
R1, R2, R3, R6, R7 и R8 обладают указанными выше значениями и
R9 обозначает С1-С4-алкил.
Соединения формулы (V), в которой R9 обозначает метил, являются предпочтительными.
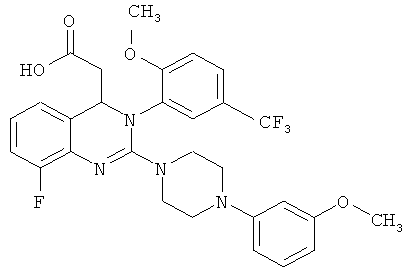
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) представляет собой следующее соединение:
{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}уксусная кислота
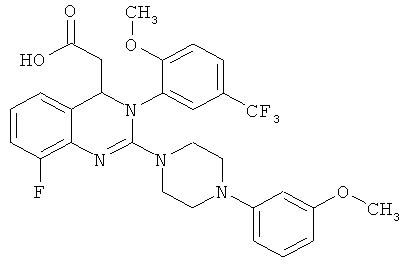
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (II) представляет собой следующее соединение:
метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат
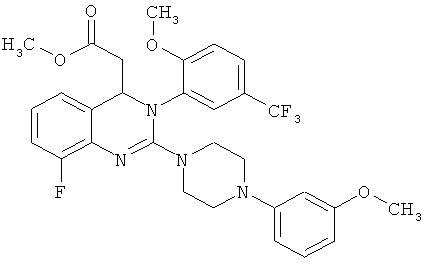
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (III) представляет собой следующее соединение:
метил-2-хлор-8-фтор-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}-ацетат
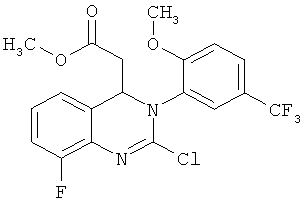
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (IV) представляет собой следующее соединение:
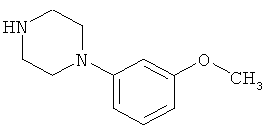
1-(3-метоксифенил)пиперазин
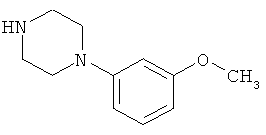
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (V) представляет собой следующее соединение:
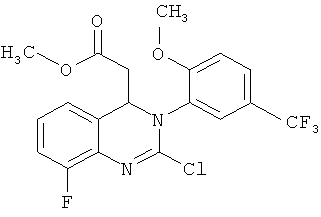
метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил}ацетат
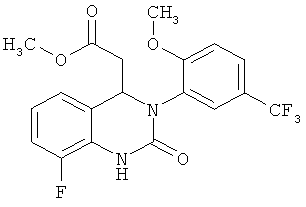
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (VI) представляет собой следующее соединение:
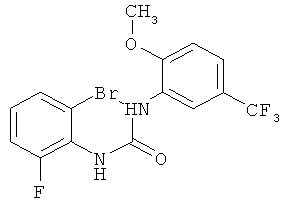
N-(2-бром-6-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевина
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (VII) представляет собой следующее соединение:
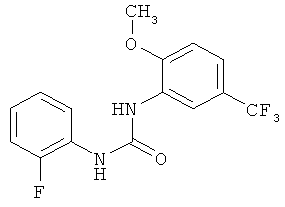
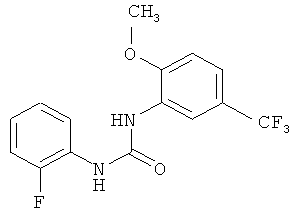
N-(2-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевина
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (VIII) представляет собой следующее соединение:
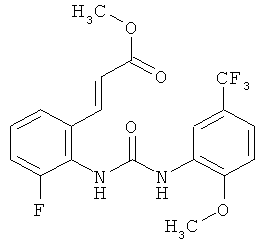
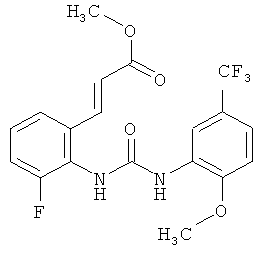
метил-(2Е)-3-{3-фтор-2-[({[2-метокси-5-(трифторметил)фенил]амино}карбонил)амино]фенил}акрилат
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (IX) представляет собой следующее соединение:
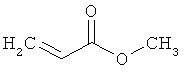
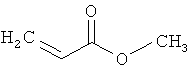
метилакрилат
Гидролиз сложного эфира соединения формулы (II) с образованием соединения формулы (I) проводят по реакции соединения формулы (II) с основанием в инертном растворителе, в температурном диапазоне от 18 до температуры кипения растворителя, предпочтительно от 18 до 50°С, особенно предпочтительно - от 20 до 30°С, при атмосферном давлении в течение, например, от 0,5 до 10 ч, предпочтительно - в течение от 1 до 5 ч.
Основаниями являются, например, гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид лития или гидроксид калия, или карбонаты щелочных металлов, такие как карбонат цезия, карбонат натрия или карбонат калия, или алкоксиды, такие как метоксид натрия или метоксид калия или этоксид натрия или этоксид калия, и основание необязательно содержится в водном растворе.
Инертными растворителями являются, например, простые эфиры, такие как 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол или трет-бутанол, или вода, или смеси растворителей.
Гидроксид натрия в воде и диоксане является предпочтительным.
Гидролиз сложного эфира соединения формулы (II) в соединение формулы (I) проводят по реакции соединения формулы (II) с кислотой в растворителе в присутствии воды, в температурном диапазоне от 18°С до температуры кипения растворителя, предпочтительно - от 18 до 50°С, особенно предпочтительно - от 20 до 30°С, при атмосферном давлении, в течение, например, от 0,5 до 48 ч, предпочтительно - в течение от 5 до 24 ч.
Кислотами в растворителе являются, например, хлористоводородная кислота, серная кислота или фосфорная кислота в диоксане или тетрагидрофуране.
Хлористоводородная кислота в диоксане является предпочтительной.
Синтез соединения формулы (II) из соединения формулы (III) и соединения формулы (IV) в присутствии основания проводят в инертном растворителе, в температурном диапазоне от 40°С до температуры кипения растворителя, предпочтительно - при температуре кипения растворителя, при атмосферном давлении, в течение, например, от 5 до 48 ч, предпочтительно - в течение от 10 до 24 ч.
Основаниями являются, например, амиды, такие как амид натрия, бис(триметилсилил)амид лития или диизопропиламид лития, или амины, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), 1-(3-метоксифенил)пиперазин или триэтиламин, или другие основания, такие как трет-бутоксид калия или гидрид натрия.
Инертными растворителями являются, например, хлорбензол или простые эфиры, такие как 1,2-диметоксиэтан, диоксан, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля.
ДБУ в диоксане является предпочтительным.
Реакцию соединения формулы (V) с получением соединения формулы (III) проводят по реакции соединения формулы (V) с оксихлоридом фосфора, трихлоридом фосфора или пентахлоридом фосфора, причем реакция с оксихлоридом фосфора является предпочтительной, в присутствии основания в инертном растворителе, в температурном диапазоне от 40°С до температуры кипения растворителя, предпочтительно - при температуре кипения растворителя, при атмосферном давлении, в течение, например, от 5 до 48 ч, предпочтительно - в течение от 10 до 24 ч.
Основаниями являются, например, амины, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), пиридин или триэтиламин, или амиды, такие как амид натрия, бис(триметилсилил)амид лития или диизопропиламид лития, или другие основания, такие как трет-бутоксид калия.
Инертными растворителями являются, например, углеводороды, такие как бензол, ксилол, толуол или хлорбензол.
ДБУ в хлорбензоле является предпочтительным.
Реакцию соединения формулы (VI) с получением соединения формулы (V) проводят по реакции соединения формулы (VI) с соединением формулы (IX) в присутствии палладиевого катализатора и основания в инертном растворителе, в температурном диапазоне от 40°С до температуры кипения растворителя, предпочтительно - при температуре кипения растворителя, при атмосферном давлении, в течение, например, от 5 до 48 ч, предпочтительно - в течение от 10 до 24 ч.
Палладиевыми катализаторами являются, например, бис(трифенилфосфин)палладий(II)хлорид, тетракис(трифенилфосфин)палладий(0), бис(трис(о-толил)фосфино)палладий(II)хлорид или палладиевый катализатор, полученный из бис(ацетонитрил)дихлорпалладия или ацетата палладий(II) и лиганда, например, трис(о-толил)фосфин, трифенилфосфин или дифенилфосфиноферроцен.
Основаниями являются, например, 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), триэтиламин или диизопропилэтиламин.
Инертными растворителями являются, например, простые эфиры, такие как 1,2-диметоксиэтан, диоксан, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, углеводороды, такие как бензол, ксилол или толуол, или другие растворители, такие как изобутиронитрил, ацетонитрил, нитробензол, диметилформамид, диметилацетамид, диметилсульфоксид или N-метилпирролидон.
Палладиевые катализаторы, полученные из бис(ацетонитрил)дихлорпалладия и трис(о-толил)фосфина и триэтиламина в изобутиронитриле являются предпочтительными.
Реакцию соединения формулы (VII) с получением соединения формулы (VIII) проводят по реакции соединения формулы (VII) с соединением формулы (IX) в уксусной кислоте в присутствии палладиевого катализатора, окислительного реагента и кислоты, в температурном диапазоне от 0 до 50°С, предпочтительно - при комнатной температуре, при атмосферном давлении, в течение, например, от 5 до 48 ч, предпочтительно - в течение от 10 до 24 ч.
Палладиевыми катализаторами являются, например, соли палладия, такие как хлорид палладия(II), ацетилацетонат палладия(II), ацетат палладия(II) или тетрахлорпалладат натрия; ацетат палладия(II) является предпочтительным.
Окислительными реагентами являются, например, п-бензохинон или пероксиды, такие как, например, пероксид водорода, трет-бутилгидропероксид или перборат натрия, или комплекс триоксида серы с пиридином, диоксид марганца, 2,3-дихлор-5,6-дициано-п-бензохинон (DDQ), пероксодисульфат натрия или олеум (дымящая серная кислота); п-бензохинон, пероксодисульфат натрия или олеум (дымящая серная кислота) является предпочтительным и олеум является особенно предпочтительным.
Кислотами являются, например, метансульфоновая кислота, трифторметансульфоновая кислота или замещенные бензолсульфоновые кислоты, такие как, например, 4-метилбензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота или 4-нитробензолсульфоновая кислота, или концентрированная серная кислота в форме олеума; трифторметансульфоновая кислота или серная кислота в форме олеума являются предпочтительным, олеум является особенно предпочтительным.
Реакцию соединения формулы (VIII) с получением соединения формулы (V) проводят по реакции соединения формулы (VIII) с основанием в инертном растворителе, в температурном диапазоне от 40°С до температуры кипения растворителя, предпочтительно - при температуре кипения растворителя, при атмосферном давлении, в течение, например, от 1 до 48 ч, предпочтительно - в течение от 2 до 14 ч.
Основаниями являются, например, карбонаты щелочных металлов, такие как карбонат цезия, карбонат натрия или карбонат калия, 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), триэтиламин или диизопропилэтиламин; карбонат калия или ДБУ является предпочтительным.
Инертными растворителями являются, например, простые эфиры, такие как 1,2-диметоксиэтан, диоксан, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, или углеводороды, такие как бензол, ксилол или толуол, или кетоны, такие как ацетон или метилизобутилкетон (МИБК), или другие растворители, такие как изобутиронитрил, ацетонитрил, хлорбензол, нитробензол, диметилформамид, диметилацетамид, диметилсульфоксид, N-метилпирролидон или тетрагидротиофен-1,1-диоксид (сульфолан); ацетон является предпочтительным.
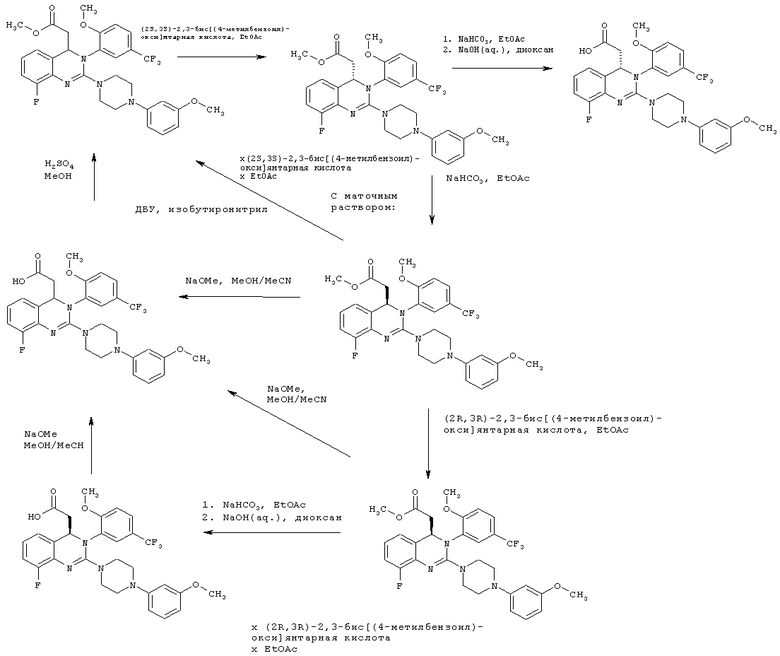
Кроме того, настоящее изобретение относится к способу разделения энантиомеров (С1-С4)-алкил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата и выделения (С1-С4)-алкил-(S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата, отличающемуся тем, что рацемический сложный эфир кристаллизуют с (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислотой. Кристаллизацию проводят в температурном диапазоне от 0 до 25°С в этилацетате. Соль S-энантиомера осаждается из раствора первой.
Способ разделения энантиомеров метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата и выделения метил-(S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата, отличающийся тем, что рацемический сложный эфир кристаллизуют с (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислотой является предпочтительным. Кристаллизацию проводят в температурном диапазоне от 0 до 25°С в этилацетате. Соль S-энантиомера осаждается из раствора первой.
Кроме того, настоящее изобретение относится к солям (С1-С4)-алкил-(S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты.
Кроме того, настоящее изобретение относится к соли метил-(S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат (2S,3S)-2,3 бис[(4-метилбензоил)окси]янтарной кислоты.
Кроме того, настоящее изобретение относится к способу рацемизации (C1-С4)-алкил-(R)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата, отличающемуся тем, что
на первой стадии алкиловый сложный эфир гидролизуют в кислоту,
на второй стадии кислоту рацемизуют с помощью метоксида натрия или этоксида натрия и на третьей стадии кислоту повторно превращают в алкиловый сложный эфир.
Способ рацемизации метил-(R)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата, отличающийся тем, что
на первой стадии метиловый сложный эфир гидролизуют в кислоту, на второй стадии кислоту рацемизуют с помощью метоксида натрия или этоксида натрия и
на третьей стадии кислоту повторно превращают в метиловый сложный эфир, является предпочтительным.
Гидролиз на первой стадии с помощью кислоты или основания проводят при тех же условиях проведения реакции, как и при гидролизе сложного эфира соединения формулы (II), с получением соединения формулы (I).
Рацемизацию на второй стадии проводят путем нагревания кислоты с метоксидом натрия или этоксидом натрия, и предпочтительно используют не менее 2 эквивалентов основания и метоксид натрия или этоксид натрия необязательно используют в спиртовом растворе, в растворителе при кипячении с обратным холодильником, при атмосферном давлении, в течение, например, от 36 до 72 ч, предпочтительно - в течение от 50 до 70 ч.
Растворителями являются, например, простые эфиры, такие как 1,2-диметоксиэтан, диоксан, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, углеводороды, такие как бензол, ксилол или толуол, или другие растворители, такие как изобутиронитрил, ацетонитрил или диметилсульфоксид; ацетонитрил является предпочтительным.
Этерификацию на третьей стадии проводят, например, по реакции кислоты с серной кислотой в метаноле или другом спирте при кипячении с обратным холодильником, при атмосферном давлении, в течение, например, от 12 до 48 ч, предпочтительно - в течение от 20 до 30 ч.
Кроме того, настоящее изобретение относится к способу рацемизации (С1-С4)-алкил-(R)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата, отличающемуся тем, что
алкиловый сложный эфир вводят в реакцию с основанием в инертном растворителе.
Способ рацемизации метил-(R)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5 -(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат, отличающийся тем, что
метиловый сложный эфир вводят в реакцию с основанием в инертном растворителе является предпочтительным.
Реакцию проводят в температурном диапазоне от 40°С до температуры кипения растворителя, предпочтительно - при температуре кипения растворителя, при атмосферном давлении, в течение, например, от 5 до 48 ч, предпочтительно - в течение от 12 до 24 ч.
Основаниями являются, например, органические азотистые основания, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ) или тетраметилгуанидин; ДБУ является предпочтительным.
Инертными растворителями являются, например, углеводороды, такие как бензол, ксилол или толуол, или другие растворители, такие как изобутиронитрил; изобутиронитрил является предпочтительным.
Соединения, описанные в контексте способа, предлагаемого в настоящем изобретении, также могут находиться в форме своих солей, сольватов или сольватов солей.
Соединения, описанные в контексте способа, предлагаемого в настоящем изобретении, в зависимости от своей структуры могут находиться в стереоизомерных формах (энантиомеры, диастереоизомеры). Поэтому способ, предлагаемый в настоящем изобретении, также включает получение и применение энантиомеров или диастереоизомеров и соответствующих их смесей. Стереоизомерно однородные компоненты можно выделить из таких смесей энантиомеров и/или диастереоизомеров по методикам, известным специалисту данной области техники.
Соединения, описанные в контексте способа, предлагаемого в настоящем изобретении, в зависимости от своей структуры также могут находиться в форме своих таутомеров.
Солями, предпочтительными в контексте настоящего изобретения, являются физиологически приемлемые соли применяющихся соединений, полученные способом, предлагаемым в настоящем изобретении.
Физиологически приемлемые соли соединений, применяющиеся в способе, предлагаемом в настоящем изобретении, и получаемые этим способом, включают соли присоединения неорганических кислот, карбоновых кислот и сульфоновых кислот, например, соли хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, нафталиндисульфоновой кислоты, уксусной кислоты, пропионовой кислоты, молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты и бензойной кислоты. Физиологически приемлемые соли соединений, применяющиеся в способе, предлагаемом в настоящем изобретении, и получаемые этим способом, также включают соли обычных оснований, такие как, например и предпочтительно, соли щелочных металлов (например, соли натрия и калия), соли щелочноземельных металлов (например, соли кальция и магния) и соли аммония, полученные из аммиака или органических аминов, содержащих от 1 до 16 атомов С, такие как, например и предпочтительно, этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дихлоргексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метилморфолин, дигидроабиетиламин, аргинин, лизин, этилендиамин и метилпиперидин.
Сольваты в контексте настоящего изобретения означают такие формы соединений, применяющиеся в способе, предлагаемом в настоящем изобретении, и получаемые этим способом, которые в твердом или жидком состоянии образуют комплекс путем координации с молекулами растворителя. Гидраты являются особой формой сольватов, в которых координация происходит с водой.
В контексте настоящего изобретения "рацемические" означает, что соединения не содержатся в энантиомерно чистой форме, т.е. соединения содержатся в виде смесей (S)- и (R)-энантиомеров. Поэтому отношение количества (S)-энантиомера к количеству (R)-энантиомера не является постоянным. Смесь (S)-энантиомера с (R)-энантиомера в соотношении 1:1 является предпочтительной.
В контексте настоящего изобретения, если не указано иное, заместители обладают следующими значениями.
Алкил сам по себе и "алкил" в алкоксигруппе, алкиламиногруппе, алкилтиогруппе, алкилкарбониле, алкилсульфониле, алкоксикарбониле и алкиламиносульфониле означает линейный или разветвленный алкильный радикал, обычно содержащий от 1 до 6 ("C1-С6-алкил"), предпочтительно - от 1 до 4, более предпочтительно - от 1 до 3 атомов углерода, например и предпочтительно метил, этил, н-пропил, изопропил, трет-бутил, н-пентил и н-гексил.
Алкоксигруппа например и предпочтительно означает метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, трет-бутоксигруппу, н-пентоксигруппу или н-гексоксигруппу.
Алкиламиногруппа означает алкиламиновый радикал, содержащий 1 или 2 алкильных заместителя (выбранных независимо друг от друга), например и предпочтительно метиламиногруппу, этиламиногруппу, н-пропиламиногруппу, изопропиламиногруппу, трет-бутиламиногруппу, н-пентиламиногруппу, н-гексиламиногруппу, N,N-диметиламиногруппу, N,N-диэтиламиногруппу, N-этил-N-метиламиногруппу, "N-метил-N-н-пропиламиногруппу, N-изопропил-N-н-пропиламиногруппу, N-трет-бутил-N-метиламиногруппу, N,N-этил-N-н-пентиламиногруппу или N-н-гексил-N-метиламиногруппу. C1-С3-Алкиламиногруппа означает, например, моноалкиламиновый радикал, содержащий от 1 до 3 атомов углерода, или диалкиламиновый радикал, содержащий от 1 до 3 атомов углерода в алкильном заместителе.
Алкилтиогруппа, например и предпочтительно, означает метилтиогруппу, этилтиогруппу, н-пропилтиогруппу, изопропилтиогруппу, трет-бутилтиогруппу, н-пентилтиогруппу или н-гексилтиогруппу.
Алкилкарбонил, например и предпочтительно, означает метилкарбонил, этилкарбонил, н-пропилкарбонил, изопропилкарбонил, трет-бутилкарбонил, н-пентилкарбонил или н-гексилкарбонил.
Алкилсульфонил, например и предпочтительно, означает метилсульфонил, этилсульфонил, н-пропилсульфонил, изопропилсульфонил, трет-бутилсульфонил, н-пентилсульфонил или н-гексилсульфонил.
Алкоксикарбонил, например и предпочтительно, означает метоксикарбонил, этоксикарбонил, н-пропоксикарбонил, изопропоксикарбонил, трет-бутоксикарбонил, н-пентоксикарбонил или н-гексоксикарбонил.
Алкиламиносульфонил означает алкиламиносульфонильный радикал, содержащий 1 или 2 алкильных заместителя (выбранных независимо друг от друга), например и предпочтительно метиламиносульфонил, этиламиносульфонил, н-пропиламиносульфонил, изопропиламиносульфонил, трет-бутиламиносульфонил, н-пентиламиносульфонил, н-гексиламиносульфонил, N,N-диметиламиносульфонил, N,N-диэтиламиносульфонил, N-этил-N-метиламиносульфонил, N-метил-N-н-пропиламиносульфонил, N-изопропил-N-н-пропиламиносульфонил, N-трет-бутил-N-метиламиносульфонил, N-этил-N-н-пентиламиносульфонил или N-н-гексил-N-метиламиносульфонил. С1-С3-Алкиламиносульфонил, например, означает моноалкиламиносульфонил, содержащий от 1 до 3 атомов углерода, или диалкиламиносульфонильный радикал, содержащий от 1 до 3 атомов углерода в алкильном заместителе.
Арил означает моно- или бициклический ароматический карбоциклический радикал, обычно содержащий от 6 до 10 атомов углерода; например и предпочтительно фенил или нафтил.
Галоген означает фтор, хлор, бром или йод,
Настоящее изобретение описано ниже с помощью неограничивающих предпочтительных примеров и сравнительных примеров. Если не указано иное, то все количественные значения приведены в массовых процентах.
Типичные варианты осуществления
Перечень аббревиатур:
АЦН ацетонитрил
ИАД-ЭР-отр. ионизация электрораспылением при атмосферном давлении, в режиме отрицательных ионов (в МС)
ХИ, NН3 химическая ионизация (с использованием аммиака)
ДБУ 1,8-диазабицикло[5.4.0]ундец-7-ен
ДМАП 4-(диметиламино)пиридин
ДМСО диметилсульфоксид
ВНСТ внешная стандартизация
ч час(ы)
ВЭЖХ высокоэффективная жидкостная хроматография
МИБК метилизобутилкетон
мин минуты
МС масс-спектроскопия
ЯМР спектроскопия ядерного магнитного резонанса
RT время удерживания (в ВЭЖХ)
Общие методики ВЭЖХ:
Методика 1 (ВЭЖХ): Прибор: HP 1050 с детектированием при нескольких длинах волн; колонка: Phenomenex-Prodigy ODS (3) 100А, 150 мм×3 мм, 3 мкм; элюент А: (1,0 г КН2РO4+1,0 мл Н3РO4) / л воды, элюент В: ацетонитрил; градиентный режим: 0 мин 10% В, 25 мин 80% В, 35 мин 80% В; скорость потока: 0,5 мл/мин; температура: 45°С; УФ-детектирование: 210 нм.
Методика 2 (ВЭЖХ): Прибор: HP 1050 с детектированием при нескольких длинах волн; колонка: хиральная AD-H, 250 мм×4,6 мм, 5 мкм; элюент А: н-гептан+0,2% диэтиламин, элюент В: изопропанол+0,2% диэтиламин; градиентный режим: 0 мин 12,5% В, 30 мин 12,5% В; скорость потока: 1 мл/мин; температура: 25°С; УФ-детектирование: 250 нм.
Для S-энантиомера приведены положительные значения ЭИ, (энантиомерный избыток), для R-энантиомера приведены отрицательные значения ЭИ.
Методика 3 (ВЭЖХ): Прибор: HP 1050 с детектированием при нескольких длинах волн; колонка: хиральная AD-H, 250 мм×4,6 мм, 5 мкм; элюент А: н-гептан + 0,2% диэтиламин, элюент В: изопропанол + 0,2% диэтиламин; градиентный режим: 0 мин 25% В, 15 мин 25% В; скорость потока: 1 мл/мин; температура: 30°С; УФ-детектирование: 250 нм.
Для S-энантиомера приведены положительные значения ЭИ, для R-энантиомера приведены отрицательные значения ЭИ.
Методика 4 (ВЭЖХ): Прибор: HP 1100 с детектированием при нескольких длинах волн; колонка: Phenomenex-Prodigy C8, 150 мм×3 мм, 5 мкм; элюент А: (1,36 г КН2РO4+1,15 г 85% Н2РO4)/л воды, элюент В: ацетонитрил; градиентный режим: 0 мин 10% В, 20 мин 80% В, 30 мин 80% В; скорость потока: 0,5 мл/мин; температура: 40°С; УФ-детектирование: 210 нм.
Приведенные выходы не скорректированы на содержание.
Схема 5:
Синтез {8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}уксусной кислоты
Пример 1
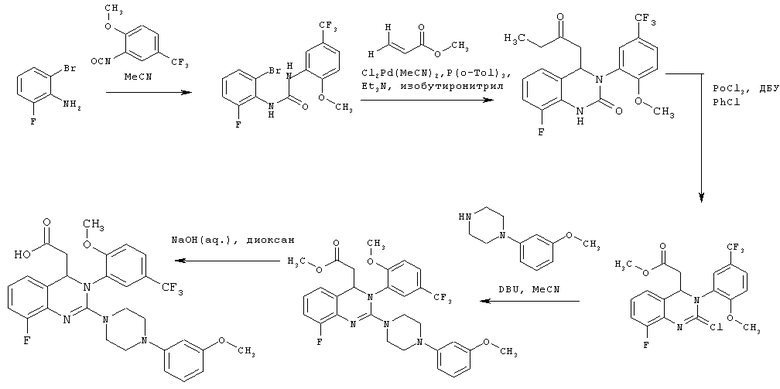
N-(2-Бром-6-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевина
2-Метокси-5-трифторметилфенилизоцианат (274,3 г) растворяют в ацетонитриле (1 л), затем прибавляют 2-бром-6-фторанилин (200 г) и промывают ацетонитрилом (50 мл). Полученный прозрачный раствор перемешивают при кипячении с обратным холодильником (примерно 85°С) в течение 38 ч, затем концентрируют в вакууме при 40°С и получают вязкую взвесь. Твердое вещество собирают фильтрованием с отсасыванием, промывают ацетонитрилом (260 мл, охлажденным до 0-5°С) и сушат в течение ночи при 45°С в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 424,3 г N-(2-бром-6-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевины в виде твердого вещества, что соответствует 99,2% от теоретического выхода.
1H ЯМР (300 МГц, d6-ДМСО): δ = 8,93 (s, 1Н), 8,84 (s, 1H), 8,52 (d, 3J=2,3, 2H), 7,55 (d, 2J=7,7, 1H), 7,38-7,26 (m, 3Н), 7,22 (d, 2J=8,5, 1H), 4,00 (s, 3Н) част./млн;
МС (ИАД-ЭР-пол.): m/z=409 [(М+Н)+, 100%];
ВЭЖХ (методика 1): RT=22,4 и 30,6 мин.
Пример 2
N-(2-Бром-6-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевина (альтернативный синтез)
2-Метокси-5-трифторметилфенилизоцианат (1,19 кг) расплавляют примерно при 35°С и растворяют в ацетонитриле (4,2 л), затем прибавляют 2-бром-6-фторанилин (870 г) и промывают ацетонитрилом (380 мл). Полученный прозрачный раствор перемешивают при 74-88°С в течение 45 ч, затем концентрируют в вакууме (200 мбар) при 50°С и получают вязкую взвесь (количество дистиллята равно 4,4 л). Ее разбавляют при комнатной температуре диизопропиловым эфиром (1,5 л), твердое вещество собирают фильтрованием с отсасыванием, промывают диизопропиловым эфиром (1,15 л) и сушат до постоянной массы (24 ч) при 45°С в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 1,63 кг N-(2-бром-6-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевины в виде твердого вещества, что соответствует 87,5% от теоретического выхода.
ВЭЖХ (методика 1): RT=22,6 и 30,8 мин.
Пример 3
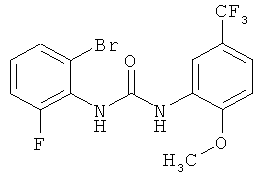
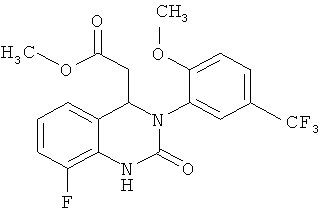
Метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил} ацетат
N-(2-Бром-6-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевину (300 г) суспендируют в изобутиронитриле (1,2 л) в атмосфере азота, затем в указанном порядке прибавляют триэтиламин (210 мл), бис(ацетонитрил)дихлорпалладий (7,5 г), трис-(о-толил)фосфин (18,0 г) и метилакрилат (210 мл). Полученную суспензию перемешивают при кипячении с обратным холодильником (примерно 102°С) в течение 16 ч и затем охлаждают до комнатной температуры. Прибавляют воду (1,2 л) и смесь перемешивают при комнатной температуре в течение 1 ч, затем твердое вещество собирают фильтрованием с отсасыванием и промывают смесью вода/метанол (1:1, 300 мл) и ацетонитрилом (100 мл). Остаток сушат в течение ночи при 45°С в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 208 г метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил} ацетата в виде твердого вещества, что соответствует 68,5% от теоретического выхода.
1Н ЯМР (300 МГц, d6-ДМСО): δ=9,73 (s, 1Н), 7,72 (d, 2J=7,3, 1H), 7,71 (s, 1H), 7,33 (d, 2J=9,3, 1H), 7,15 (dd, 2J=9,6, 2;=8,6, 1H), 7,01 (d, 2J=7,3, 1H), 6,99-6,94 (m, 1H), 5,16 (t, 2;=5,9, 1H), 3,84 (s, 3Н), 3,41 (s, 3H), 2,81 (dd, 2J=15,4, 2]=5,8, 1H), 2,62 (dd, 2J - 15,4, 2J=6,3, 1H) част./млн;
MC (ИАД-ЭР-пол.): m/z=413 [(М+Н)+, 100%], 825 [(2М+Н)+ 14%];
ВЭЖХ (методика 1): RT=19,3 мин; Pd (ICP): 16000 част./млн.
Пример 4
Метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил} ацетат (альтернативный синтез)
N-(2-Бром-6-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевину (2,5 кг) суспендируют в изобутиронитриле (9 л) в атмосфере азота, затем в указанном порядке прибавляют триэтиламин (1,31 кг), бис(ацетонитрил)дихлорпалладий (64,9 г), трис(о-толил)фосфин (149 г) и метилакрилат (1,59 кг). Полученную суспензию перемешивают при 90-100°С в течение 22 ч, затем охлаждают до комнатной температуры. Прибавляют воду (9 л) и смесь перемешивают при комнатной температуре в течение 1 ч, затем твердое вещество собирают фильтрованием с отсасыванием и промывают смесью вода/метанол (1:1, 2,5 л) и ацетонитрилом (850 мл). Остаток сушат в течение ночи при 45°С в вакуумном сушильном шкафу до постоянной массы (21 ч) с подачей азота. Таким образом получают всего 1,90 кг метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил}ацетата в виде твердого вещества, что соответствует 74,9% от теоретического выхода.
ВЭЖХ (методика 1): RT=19,4 мин.
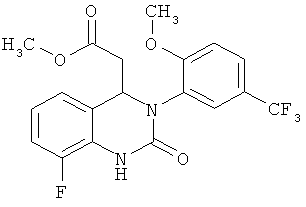
Пример 5
Метил-{2-хлор-8-фтор-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат/хлорирование
Раствор 2,84 кг метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил}ацетата в 14,8 л хлорбензола кипятят с обратным холодильником и растворитель отгоняют до прекращения отделения воды. Смесь охлаждают до 120°С. В течение 10 мин прибавляют 3,17 кг оксихлорида фосфора, и затем в течение еще 10 мин прибавляют 2,10 кг ДБУ. Смесь кипятят с обратным холодильником в течение 9 ч.
Для проведения обработки смесь охлаждают до 40°С, перемешивают в течение ночи и содержимое сосуда прибавляют к 11,4 л воды, температуру которой предварительно устанавливают равной 40°С. Во время прибавления необходимо поддерживать внутреннюю температуру, равную 40-45°С. Смеси дают охладиться до комнатной температуры, прибавляют 11,4 л дихлорметана, смесь фильтруют через фильтровальную чашку Seitz и фазы разделяют. Органическую фазу промывают с помощью 11,4 л воды, 11,4 л насыщенного водного раствора гидрокарбоната натрия и повторно с помощью 11,4 л воды. Органическую фазу концентрируют в вакууме в роторном испарителе и полученный остаток (2,90 кг) используют на следующей стадии без дополнительной обработки.
1Н ЯМР (300 МГц, d6-ДМСО): δ=7,93-7,82 (m, 2Н), 7,38 (d, 2J=8,9, 1Н), 7,17 (m, 2Н), 6,97-6,91 (m, 1Н), 5,45 и 5,29 (m и t, 2J=5,4, 1Н), 3,91 и 3,84 (2s, 3H), 3,48 (s, 3H), 3,0-2,6 (m, 2Н) част./млн;
МС (ХИ, NН3): m/z=431 [(М+Н)+, 100%];
ВЭЖХ (методика 1): RT=23,5 мин; типичное значение для Pd (ICP): 170 част./млн.
Пример 6
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат/аминирование
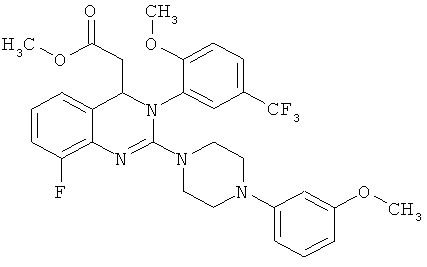
Метил-{2-хлор-8-фтор-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат (52,5 г) растворяют в 1,4-диоксане (100 мл), затем при комнатной температуре прибавляют 3-метоксифенилпиперазин (25,8 г) и ДБУ (20,4 г) и при этом температура повышается. Смесь перемешивают при кипячении с обратным холодильником в течение 22 ч, затем охлаждают до комнатной температуры, разбавляют этилацетатом (500 мл) и водой (200 мл) и фазы разделяют. Органическую фазу промывают с помощью 0,2 н. хлористоводородной кислоты (три раза по 100 мл) и водой (200 мл), сушат над сульфатом натрия и концентрируют в роторном испарителе. Таким образом получают всего 62,5 г метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетатав виде твердого вспененного вещества, которое вводят в реакцию в виде неочищенного продукта без дополнительной очистки. ВЭЖХ (методика 1): RT=16,6 мин.
Пример 7
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат/однореакторное хлорирование+аминирование
Метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил} ацетат (50,0 г) растворяют в хлорбензоле (300 мл), затем хлорбензол частично отгоняют (50 мл). Смесь охлаждают до 120°С, прибавляют ДБУ (36,9 г), затем при 120-128°С в течение 10 мин прибавляют оксихлорид фосфора (33,4 мл). Смесь перемешивают при кипячении с обратным холодильником (примерно 130°С) в течение 9 ч. Затем смесь охлаждают до 40°С, медленно при 40-45°С прибавляют воду (200 мл), смесь охлаждают до комнатной температуры и разбавляют дихлорметаном (200 мл), экстрагируют при перемешивании и затем фазы разделяют. Органическую фазу промывают водой (200 мл), насыщенным водным раствором гидрокарбоната натрия (200 мл) и повторно водой (200 мл), сушат над сульфатом натрия, концентрируют в роторном испарителе и затем сушат при 50°С в высоком вакууме. Остаток (48,1 г) растворяют в хлорбензоле (20 мл), затем раствор разбавляют 1,4-диоксаном (80 мл) и при комнатной температуре прибавляют 3-метоксифенилпиперазин (23,6 г) и ДБУ (18,7 г) и при этом температура повышается. Смесь перемешивают при кипячении с обратным холодильником в течение 22 ч, затем охлаждают до комнатной температуры, разбавляют этилацетатом (500 мл) и водой (200 мл) и фазы разделяют. Органическую фазу промывают с помощью 0,2 н. хлористоводородной кислоты (три раза по 100 мл) и водой (200 мл), сушат над сульфатом натрия и концентрируют в роторном испарителе. Таким образом получают всего 55,6 г метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетатав виде твердого вспененного вещества, которое вводят в реакцию в виде неочищенного продукта без дополнительной очистки.
ВЭЖХ (методика 1): RT=16,2 мин.
Пример 8
(±)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил} уксусная кислота/гидролиз рацемата
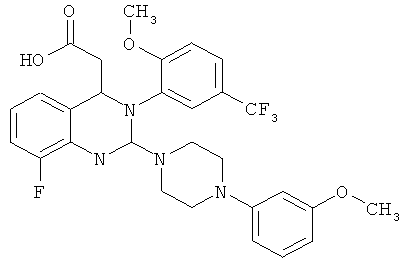
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат (64 г) растворяют в 1,4-диоксане (450 мл) и 1 н. растворе гидроксид натрия (325 мл) и перемешивают при комнатной температуре в течение 2 ч, затем часть растворителя (400 мл) отгоняют при 30°С в вакууме. Затем прибавляют толуол (300 мл) и фазы разделяют. Водную фазу промывают толуолом (дважды по 150 мл), затем объединенные органические фазы экстрагируют повторно 1 н. раствором гидроксид натрия (50 мл). Значение рН объединенной водной фазы доводят до 7,5 с помощью 2 н. хлористоводородной кислоты (примерно 150 мл), затем прибавляют МИБК (150 мл). Фазы разделяют, водную фазу повторно экстрагируют с помощью МИБК (150 мл), затем объединенные фазы в МИБК сушат над сульфатом натрия и концентрируют при 45°С. Таким образом получают всего 64 г (±)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусной кислоты с количественным выходом в виде аморфного твердого вещества.
ВЭЖХ (методика 1): RT=14,9 мин.
Схема 6:
Разделение энантиомеров метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата
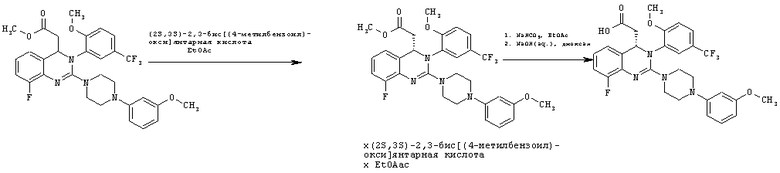
Пример 9
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат (28,38)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1)/кристаллизация
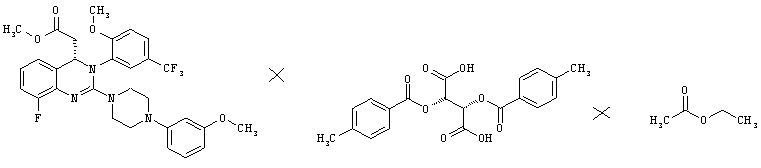
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат (62,5 г, неочищенный продукт) растворяют в этилацетате (495 мл) и фильтруют. К фильтрату прибавляют (28,38)-2,3-бис[(4-метилбензоил)окси]янтарную кислоту (42,0 г), смесь перемешивают при комнатной температуре в течение 30 мин, затем вносят затравку метил- {8-фтор-2- [4-(3 -метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата (28,38)-2,3-бис[(4-метилбензоил)окси]-янтарной кислоты (соль состава 1:1) (165 мг) и перемешивают при комнатной температуре в течение 3 дней, затем охлаждают до 0-3°С и перемешивают в течение еще 3 ч. Суспензию собирают фильтрованием с отсасыванием и промывают холодным этилацетатом (0-10°С, 35 мл). Кристаллы сушат при 40°С в течение 18 ч в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 37,1 г соли в виде твердого вещества, что соответствует 30,4% от теоретического выхода за три стадии (хлорирование, аминирование и кристаллизация) в пересчете на рацемат или 60,8% в пересчете на полученный S-энантиомер.
1H ЯМР (300 МГц, d6-ДМСО): δ=7,90 (d, 2J=7,8, 4Н), 7,56 (d, 2J=8,3, 1H), 7,40 (d, 2J=7,8, 4Н), 7,28-7,05 (m, 4Н), 6,91-6,86 (m, 2H). 6,45 (d, 2J=8,3, 1H), 6,39-6,36 (m, 2H), 5,82 (s, 2H), 4,94 (m, 1H), 4,03 (q, 2J=7,1, 2H), 3,83 (brs, 3H), 3,69 (s, 3H), 3,64 (s, 3H), 3,47-3,36 (m, 8H и вода, 2H), 2,98-2,81 (m, 5H), 2,58-2,52 (m, 1H), 2,41 (s, 6H), 1,99 (s, 3H), 1,18 (t, 2J=7,2, 3H) част./млн;
ВЭЖХ (методика 1): RT=16,6 и 18,5 мин.
Пример 10
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат (28,38)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1)/перекристаллизация
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат (28,38)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (36,8 г) суспендируют в этилацетате (370 мл) и растворяют при кипячении с обратным холодильником (77°С). Смесь медленно охлаждают до комнатной температуры. В это время происходит самопроизвольная кристаллизация. Суспензию перемешивают при комнатной температуре в течение 16 ч, затем охлаждают до 0-5°С и перемешивают в течение еще 3 ч. Суспензию собирают фильтрованием с отсасыванием и промывают холодным этилацетатом (0-10°С, два раза по 15 мл). Кристаллы сушат при 45°С в течение 18 ч в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 33,6 г соли в виде твердого вещества, что соответствует 91,3% от теоретического выхода. ВЭЖХ (методика 1): RT=16,9 и 18,8 мин;
ВЭЖХ (методика 3): 99,9% ЭИ.
Пример 11
(S)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусная кислота
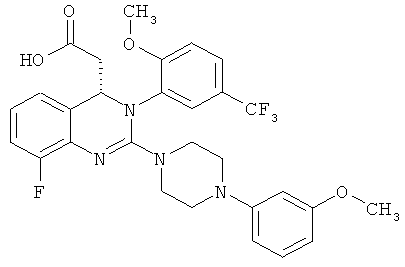
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат (28,38)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (10,1 г, содержащий 14 част./млн Pd) суспендируют в этилацетате (100 мл) и встряхивают с насыщенным водным раствором бикарбоната натрия (100 мл), пока обе фазы не станут прозрачными. Фазы разделяют и органическую фазу концентрируют в роторном испарителе. Остаток растворяют в 1,4-диоксане (100 мл) и 1 н. растворе гидроксида натрия (31,2 мл) и перемешивают при комнатной температуре в течение 3 ч. Затем значение рН доводят до 7,5 с помощью 1 н. хлористоводородной кислоты (примерно 17 мл), прибавляют МИБК (80 мл) и затем значение рН повторно доводят до 7,0 с помощью 1 н. хлористоводородной кислоты (примерно 2 мл). Фазы разделяют и органическую фазу сушат над сульфатом натрия и концентрируют. Остаток растворяют в этаноле (40 мл) и концентрируют, затем повторно растворяют в этаноле (40 мл) и концентрируют и сушат при 50°С в высоком вакууме. Затвердевшее вспененное вещество сушат при 45°С в течение 18 ч в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 5,05 г (S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусной кислоты в виде аморфного твердого вещества, что соответствует 85,0% от теоретического выхода.
1Н ЯМР (300 МГц, d6-ДМСО): δ=7,53 (d, 2J=8,4, 1Н), 7,41 (brs, 1H), 7,22 (d, 2J=8,5, 1H), 7,09-7,01 (m, 2H), 6,86 (m, 2H), 6,45 (dd, 2J=8,2, 3J=1,8, 1H), 6,39-6,34 (m, 2H), 4,87 (t, 2J=7,3, 1H), 3,79 (brs, 3Н), 3,68 (s, 3H), 3,50-3,38 (m, 4H), 2,96-2,75 (m, 5H), 2,45-2,40 (m, 1H) част./млн;
MC (ИАД-ЭР-отр.): m/z=571 [(М-Н), 100%];
ВЭЖХ (методика 1); RT=15,1 мин;
ВЭЖХ (методика 2): 99,8% ЭИ; Pd (ICP): <1 част./млн.
Пример 12
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат(2R,3R)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1)/кристаллизация R-изомера из маточного раствора
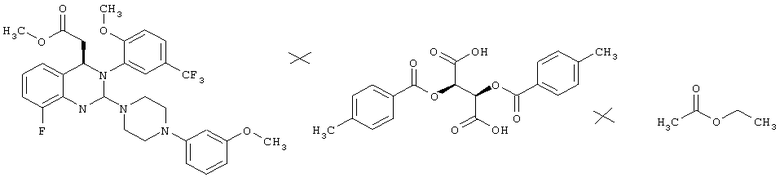
Маточный раствор после кристаллизации метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1 -ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата (28,38)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) встряхивают с насыщенным водным раствором бикарбоната натрия (1,5 л), фазы разделяют и органическую фазу встряхивают с разбавленным вдвое насыщенным водным раствором бикарбоната натрия (1,5 л). Фазы разделяют и органическую фазу сушат над сульфатом натрия и концентрируют в роторном испарителе. Остаток (188,4 г) растворяют в этилацетате (1,57 л), затем прибавляют (2R,3R)-2,3-бис[(4-метилбензоил)окси]янтарную кислоту (121,7 г) и смесь перемешивают при комнатной температуре в течение 10 мин. Затем в смесь вносят затравку метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата (2R,3R)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (0,38 г) и перемешивают при комнатной температуре в течение 18 ч, затем охлаждают до 0-3°С и перемешивают в течение еще 3 ч. Суспензию собирают фильтрованием с отсасыванием и промывают холодным этилацетатом (0-10°С, 500 мл). Кристаллы сушат при 40°С в течение 18 ч в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 160 г соли в виде твердого вещества.
ВЭЖХ (методика 1): RT=16,6 и 18,5 мин.;
ВЭЖХ (методика 3): -99,0% ЭИ.
Пример 13
(R)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусная кислота/получение R-изомера
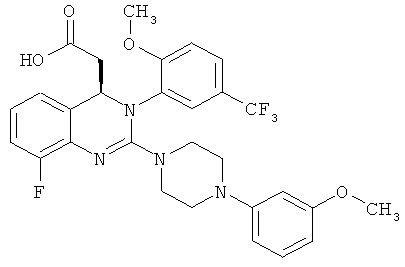
Метил{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат (2R,3R)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (170 г) суспендируют в этилацетате (850 мл) и встряхивают с насыщенным водным раствором бикарбоната натрия (850 мл), пока обе фазы не станут прозрачными (примерно 5 мин). Фазы разделяют и растворитель органической фазы при атмосферном давлении заменяют 1,4-диоксаном до конечной температуры, равной 99°С (порциями отгоняют всего 2,55 л растворителя и используют 2,55 л 1,4-диоксана). Смесь охлаждают до комнатной температуры и перемешивают при комнатной температуре в течение 18 ч с 1 н. раствором гидроксид натрия (525 мл). Затем значение рН доводят до 7,5 с помощью концентрированной хлористоводородной кислоты (примерно 35 мл), прибавляют МИБК (850 мл) и затем значение рН повторно доводят до 7,0 с помощью концентрированной хлористоводородной кислоты (примерно 10 мл). Фазы разделяют и органическую фазу сушат над сульфатом натрия и концентрируют. Остаток растворяют в этаноле (350 мл) и концентрируют, затем повторно растворяют в этаноле (350 мл) и концентрируют при 50°С. Таким образом получают всего 91,6 г (R)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусной кислоты в виде аморфного твердого вещества, что соответствует 91,6% от теоретического выхода.
ВЭЖХ (методика 1): RT=14,8 мин.
Пример 14
{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусная кислота/рацемизация R-энантиомера
(R)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусную кислоту (50 г) растворяют в ацетонитриле (500 мл) и прибавляют метоксид натрия (30% в метаноле, 32,4 мл) и затем смесь перемешивают при кипячении с обратным холодильником в течение 60 ч. После охлаждения до комнатной температуры смесь концентрируют до половины объема в вакууме, затем значение рН доводят до 7,5 с помощью хлористоводородной кислоты (20%, примерно 20 мл) прибавляют, МИБК (200 мл) и значение рН смеси повторно доводят до 7 с помощью хлористоводородной кислоты (20%). Фазы разделяют и органическую фазу сушат над сульфатом натрия и концентрируют в роторном испарителе и получают твердое вспененное вещество. Остаток растворяют в этаноле (150 мл) и концентрируют, затем повторно растворяют в этаноле (150 мл) и концентрируют.Таким образом получают всего 54,2 г (±)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусной кислоты в виде аморфного твердого вещества с количественным выходом.
ВЭЖХ (методика 1): RT=14,9 мин;
ВЭЖХ (методика 4): 80,8 мас.%.
Пример 15
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат/этерификация рацемата
(±)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусную кислоту (54 г) растворяют в метаноле (540 г), затем прибавляют концентрированную серную кислоту (7,85 мл). Смесь перемешивают при кипячении с обратным холодильником в течение 26 ч, затем охлаждают и концентрируют в вакууме примерно до трети исходного объема. Прибавляют, воду (150 мл) и дихлорметан (150 мл), затем фазы разделяют. Органическую фазу экстрагируют насыщенным раствором гидрокарбоната натрия (два раза по 140 мл), сушат над сульфатом натрия и концентрируют и получают вспененный остаток. Его дважды последовательно растворяют в этаноле (по 150 мл) и концентрируют и затем сушат в течение 18 ч в вакууме с подачей азота. Таким образом получают всего 41,6 г метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата в виде аморфного твердого вещества, что соответствует 75,2% от теоретического выхода.
ВЭЖХ (методика 1): RT=16,8 мин;
ВЭЖХ (методика 4): 85,3 мас.%;
ВЭЖХ (методика 3): -8,5% ЭИ.
Пример 16
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1)/кристаллизация этерифицированного рацемата
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат (41,0 г) суспендируют в этилацетате (287 мл), затем прибавляют (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарную кислоту (27,5 г). Смесь перемешивают при комнатной температуре в течение 30 мин, затем вносят затравку метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (0,08 г). Суспензию перемешивают при комнатной температуре в течение 16 ч, затем охлаждают до 0-5°С и перемешивают в течение еще 3 ч, затем твердое вещество собирают фильтрованием с отсасыванием и промывают холодным этилацетатом (0-10°С, четыре раза по 16 мл). Кристаллы сушат при 45°С в течение 18 ч в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 25,4 г соли в виде твердого вещества, что соответствует 37,4% от теоретического выхода.
ВЭЖХ (методика 1): RT=16,9 и 18,8 мин;
ВЭЖХ (методика 4): 99,5 мас.%;
ВЭЖХ (методика 3): 99,3% ЭИ.
Пример 17
(S)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил} уксусная кислота/гидролиз кристаллического продукта
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (25,1 г) суспендируют в этилацетате (250 мл) и встряхивают с насыщенным водным раствором бикарбоната натрия (250 мл), пока обе фазы не станут прозрачными. Фазы разделяют и органическую фазу концентрируют в роторном испарителе. Остаток растворяют в 1,4-диоксане (250 мл) и 1 н. растворе гидроксида натрия (77,4 мл) и перемешивают при комнатной температуре в течение 18 ч. Затем значение рН доводят до 7,5 с помощью 1 н. хлористоводородной кислоты (примерно 50 мл), МИБК (240 мл) прибавляют и затем значение рН повторно доводят до 7,0 с помощью 1 н. хлористоводородной кислоты (примерно 15 мл). Фазы разделяют и органическую фазу сушат над сульфатом натрия и концентрируют. Остаток растворяют в этаноле (90 мл) и концентрируют, затем повторно растворяют в этаноле (90 мл) и концентрируют. Затвердевшее вспененное вещество сушат при 45°С в течение 18 ч в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 12 г (S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусной кислоты в виде аморфного твердого вещества, что соответствует 81,2% от теоретического выхода.
ВЭЖХ (методика 1): RT=15,1 мин;
ВЭЖХ (методика 2): 97,5% ЭИ.; Pd (ICP): <20 част./млн.
Альтернативная методика рацемизации:
Пример 18
(±)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусная кислота/гидролиз концентрированного R-изомера, полученного из маточного раствора после кристаллизации
Маточный раствор после кристаллизации 207 г метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата(2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) встряхивают с насыщенным водным раствором бикарбоната натрия (500 мл), фазы разделяют и органическую фазу встряхивают с разбавленным вдвое насыщенным водным раствором бикарбоната натрия (500 мл). Фазы разделяют и органическую фазу сушат над сульфатом натрия и концентрируют в роторном испарителе. Остаток растворяют в этаноле (500 мл) и концентрируют в роторном испарителе и получают твердое вспененное вещество. Его растворяют в 1,4-диоксане (1,6 л) и 1 н. растворе гидроксида натрия (1,04 л) и перемешивают при комнатной температуре в течение 18 ч, затем прибавляют толуол (1,5 л) и фазы разделяют. Значение рН водной фазы доводят равного от 14 до 8 с помощью хлористоводородной кислоты (20%, примерно 155 мл), затем прибавляют МИБК (1,25 л) и значение рН смеси повторно доводят до 7 с помощью хлористоводородной кислоты (20% примерно 25 мл). Фазы разделяют и органическую фазу сушат над сульфатом натрия и концентрируют в роторном испарителе и получают твердое вспененное вещество. Его сушат при 45°С в течение 18 ч в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 150 г {8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил} уксусной кислоты в виде (R/S) смеси в виде аморфного твердого вещества.
ВЭЖХ (методика 2): -14,6% ЭИ.
Пример 19
(±)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусная кислота/рацемизация
{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусную кислоту (150 г, R/S-смеси с 14,6% ЭИ) растворяют в ацетонитриле (1,5 л) и прибавляют метоксид натрия (30% в метаноле, 97,2 мл) и затем смесь перемешивают при кипячении с обратным холодильником в течение 77 ч. После охлаждения до комнатной температуры смесь концентрируют до половины объема в вакууме и затем значение рН доводят равного от 13 до 7,5 с помощью хлористоводородной кислоты (20%, примерно 80 мл), прибавляют МИБК (0,6 л) и значение рН смеси повторно доводят до 7 с помощью хлористоводородной кислоты (20%, примерно 3 мл). Фазы разделяют и органическую фазу сушат над сульфатом натрия и концентрируют в роторном испарителе и получают твердое вспененное вещество. Остаток растворяют в этаноле (500 мл) и концентрируют, затем повторно растворяют в этаноле (500 мл) и концентрируют, затем сушат в течение 18 ч при 45°С в вакуумном сушильном шкафу с подачей азота. Всего получают 148 г (±)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил} уксусной кислоты в виде аморфного твердого вещества, что соответствует 98,7% от теоретического выхода.
ВЭЖХ (методика 2): 1,5% ЭИ.
Пример 20
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат (этерификация)
(±)-{8-Фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-(2-метокси-5-трифторметилфенил)-3,4-дигидрохиназолин-4-ил}уксусную кислоту (148 г) растворяют в метаноле (1480 г), затем прибавляют концентрированную серную кислоту (21,5 мл). Смесь перемешивают при кипячении с обратным холодильником в течение 6 ч, затем охлаждают и концентрируют в вакууме примерно до трети исходного объема. Прибавляют воду (400 мл) и дихлорметан (400 мл), затем фазы разделяют. Органическую фазу экстрагируют насыщенным раствором гидрокарбоната натрия (два раза по 375 мл, разбавленного с помощью 300 мл воды), сушат над сульфатом натрия и концентрируют и получают вспененный остаток. Его дважды последовательно растворяют в этаноле (по 400 мл) и концентрируют и затем сушат в вакууме в течение 18 ч с подачей азота. Таким образом получают всего 124 г метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата в виде аморфного твердого вещества, что соответствует 81,9% от теоретического выхода.
ВЭЖХ (методика 1): RT=16,9 мин.;
Пример 21
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1)/кристаллизация этерифицированного рацемата
Метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетат (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (123 г, 14,4% ЭИ) суспендируют в этилацетате (861 мл) и фильтруют, затем прибавляют (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарную кислоту (82,5 г). Смесь перемешивают при комнатной температуре в течение 30 мин, затем вносят затравку метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата (28,38)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (0,24 г). Суспензию перемешивают при комнатной температуре в течение 4 дней, затем концентрируют примерно до 600 мл и повторно вносят затравку метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил} ацетата (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты (соль состава 1:1) (0,24 г). Суспензию перемешивают при комнатной температуре в течение 1 недели, охлаждают до 0-5°С и перемешивают в течение еще 3 ч, затем твердое вещество собирают фильтрованием с отсасыванием и промывают холодным этилацетатом (0-10°С, 4×40 мл). Кристаллы сушат при 45°С в течение 18 ч в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 11,8 г соли в виде твердого вещества, что соответствует 5,8% от теоретического выхода.
Схема 7:
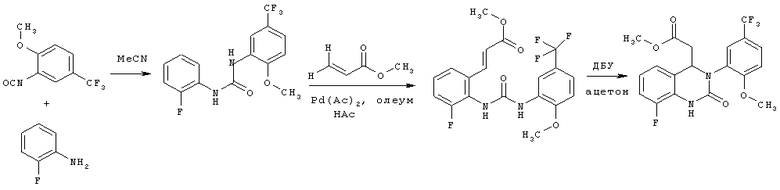
Пример 22
N-(2-Фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевина
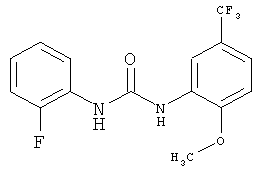
2-Метокси-5-трифторметилфенилизоцианат (1057,8 г) растворяют в ацетонитриле (4240 мл), затем прибавляют 2-фторанилин (540,8 г) и промывают ацетонитрилом (50 мл). Полученный прозрачный раствор перемешивают при кипячении с обратным холодильником (примерно 82°С) в течение 4 ч, затем вносят затравку примерно при 78°С и перемешивают в течение примерно 15 мин. Суспензию охлаждают до 0°С и продукт собирают фильтрованием с отсасыванием и промывают ацетонитрилом (950 мл, охлажденным до 0-5°С). Продукт сушат в течение ночи при 45°С в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 1380,8 г N-(2-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевины в виде твердого вещества, что соответствует 86,4% от теоретического выхода.
1Н ЯМР (500 МГц, d6-ДМСО): δ=9,36 (s, 1Н), 9,04 (s, 1H), 8,55 (d, l,7 H2, 1H), 8,17 (t, 8,2Hz, 1H), 7,33 (d, 8,5Hz, 1H), 7,20-7,26 (m, 2H), 7,14 (t, 7,6Hz, 1H), 7,02 (m, 1H), 3,97 (s, 3Н) част./млн;
MC (ИАД-ЭР-пол.): m/z - 329 [(M+H)+, 100%];
ВЭЖХ: RT=48,7 мин.
Прибор: HP 1100 с детектированием при нескольких длинах волн; колонка:
Phenomenex-Prodigy ODS (3) 100А, 150 мм×3 мм, 3 мкм; элюент А: (1,36 г КН2РO4+0,7 мл Н3РO4)/л воды, элюент В: ацетонитрил; градиентный режим: 0 мин 20% В, 40 мин 45% В, 50 мин 80% В, 65 мин 80% В; скорость потока: 0,5 мл/мин; температура: 55°С; УФ-детектирование: 210 нм.
Пример 23
Метил-(2Е)-3-{3-фтор-2-[({[2-метокси-5-(трифторметил)фенил]амино}карбонил)амино]фенил}акрилат
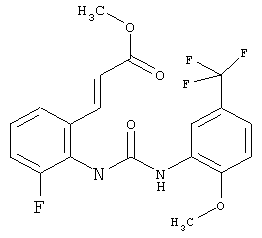
N-(2-Фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевину (0,225 кг) растворяют в уксусной кислоте (6,75 л) и прибавляют ацетат палладия (30,3 г). Затем прибавляют 65% олеум (247,5 г) и метилакрилат (90 г) затем прибавляют. Раствор перемешивают при комнатной температуре в течение ночи. Затем уксусную кислоту (3740 г) отгоняют примерно при 30°С и примерно 30 мбар. К суспензии прибавляют воду (2,25 л) и смесь перемешивают в течение примерно 1 ч. Продукт собирают фильтрованием с отсасыванием, дважды промывают водой (0,5 л) и сушат при 50°С в течение ночи в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 210,3 г метил-(2Е)-3-{3-фтор-2-[({[2-метокси-5-(трифторметил)фенил]амино}карбонил)амино]фенил}акрилата в виде твердого вещества, что соответствует 72,2% от теоретического выхода.
1Н ЯМР (300 МГц, d6-ДМСО): δ=9,16 (s, 1Н), 8,84 (s, 1H), 8,45 (d, l,7Hz, 1H), 7,73 (m, 2H), 7,33 (m, 3H), 7,22 (d, 8,6Hz, 1H), 6,70 (d, 16Hz, 1H), 3,99 (s, 3H), 3,71 (s, 3H) част./млн;
MC (ИАД-ЭР-пол.): m/z=429,9 [(M+NH4)+]; 412,9 [(M+H)+]
ВЭЖХ: RT=46,4 мин.
Прибор: HP 1100 с детектированием при нескольких длинах волн; колонка: Phenomenex-Prodigy ODS (3) 100А, 150 мм×3 мм, 3 мкм; элюент А: (1,36 г КН2РO4+0,7 мл Н3РO4)/л воды, элюент В: ацетонитрил; градиентный режим: 0 мин 20% В, 40 мин 45% В, 50 мин 80% В, 65 мин 80% В; скорость потока: 0,5 мл/мин; температура: 55°С; УФ-детектирование: 210 нм.
Пример 24
Метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил} ацетат
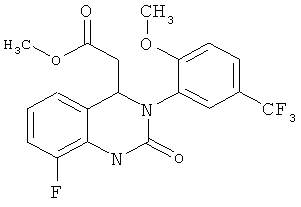
Метил-(2Е)-3-{3-фтор-2-[({[2-метокси-5-(трифторметил)фенил]амино}карбонил)амино] фенил }акрилат (50 г) суспендируют в ацетоне (1,2 л) и прибавляют 1,8-диазабицикло[5.4.0]ундец-7-ен (3,7 г). Суспензию кипятят с обратным холодильником (примерно 56°С) и перемешивают в течение 4 ч. Полученный прозрачный раствор фильтруют теплым через кизельгур (5 г). Кизельгур промывают теплым ацетоном (100 мл). Затем ацетон (550 г) отгоняют. Полученную суспензию охлаждают до 0°С в течение 3 ч и перемешивают. Продукт собирают фильтрованием с отсасыванием, дважды промывают холодным ацетоном (50 мл) и сушат при 45°С в течение ночи в вакуумном сушильном шкафу с подачей азота. Таким образом получают всего 44,5 г метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил}ацетата в виде твердого вещества, что соответствует 89% от теоретического выхода.
1Н ЯМР (300 МГц, d6-ДМСО): δ=9,73 (s, 1Н), 7,72 (d, 2J=7,3, 1H), 7,71 (s, 1H), 7,33 (d, 2J=9,3, 1H), 7,15 (dd, 2J=9,6, 2J=8,6, 1H), 7,01 (d, 1J=7,3, 1H), 6,99-6,94 (m, 1H), 5,16 (t, 2J=5,9, 1H), 3,84 (s, 3Н), 3,41 (s, 3H), 2,81 (dd, 2J=15,4, 2J=5,8, 1H), 2,62 (dd, 2J=15,4, 2J=6,3, 1H) част./млн;
MC (ИАД-ЭР-пол.): m/z=413 [(M+H)+, 100%], 825 [(2М+Н)+, 14%];
ВЭЖХ: RT=37,1 мин.
Прибор: HP 1100 с детектированием при нескольких длинах волн; колонка: Phenomenex-Prodigy ODS (3) 100А, 150 мм×3 мм, 3 мкм; элюент А: (1,36 г КН2РO4+0,7 мл Н3РO4)/л воды, элюент В: ацетонитрил; градиентный режим: 0 мин 20% В, 40 мин 45% В, 50 мин 80% В, 65 мин 80% В; скорость потока: 0,5 мл/мин; температура: 55°С; УФ-детектирование: 210 нм.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ СОМТ | 2013 |
|
RU2642779C2 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ | 2019 |
|
RU2783414C2 |
| СОЕДИНЕНИЯ 2,3-ДИГИДРОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NaV1.8 | 2020 |
|
RU2833870C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА | 2015 |
|
RU2709810C2 |
| СОЕДИНЕНИЯ КАРБОНОВЫХ КИСЛОТ И МЕДИЦИНСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2004 |
|
RU2375353C2 |
| ОКСОХИНАЗОЛИНИЛБУТАНАМИДНЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2669393C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ АЗОЛОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1994 |
|
RU2128659C1 |
| З-АМИНОПИРИДИНЫ В КАЧЕСТВЕ АГОНИСТОВ GPBAR1 | 2012 |
|
RU2594886C2 |
| АЗАБЕНЗИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ И ФАРМКОМПОЗИЦИЯ | 2019 |
|
RU2804485C2 |
| ИНГИБИТОРЫ СЕТР | 2006 |
|
RU2513107C2 |
Изобретение относится к улучшенному способу получения дигидрохиназолинов формулы (I), которые применяют для приготовления лекарственных средств. В формуле (I)
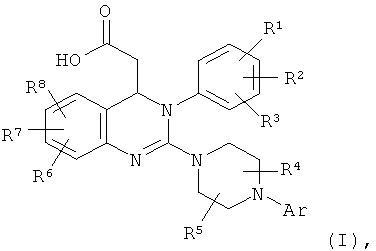
Аr обозначает фенил, возможно замещенный С1-С4алкоксигруппой, R1 и R2 выбраны из водорода, С1-С4алкоксигруппы и трифторметила, R3 выбран из С1-С4алкоксигруппы и трифторметила, R4 обозначает водород или С1-С4алкил, R5 обозначает водород или C1-С4алкил, каждый из R6 R7 и R8 обозначает водород или галоген. Способ заключается в гидролизе сложного эфира соединения формулы (II), в которой Ar, R1, R2, R3, R4, R5, R6, R7 и R8 имеют указанные выше значения и R9 обозначает С1-С4-алкил, с основанием или кислотой, при этом используют соединение формулы (II) полученное в результате реакции соединения
формулы (III), в которой R1, R2, R3, R6, R7 и R8 имеют указанные выше значения и R9 обозначает С1-С4-алкил, в присутствии основания с соединением формулы (IV), в которой Ar, R4 и R5 имеют указанные выше значения. Способ позволяет упростить выделение продуктов. Изобретение также относится к новым промежуточным соединениям и их получению. 5 н. и 6 з.п. ф-лы.
1. Способ получения соединения формулы (I)
в которой Аr обозначает арил, где арил представляет собой фенил, необязательно замещенный C1-С4алкоксигруппой,
R1 и R2 выбраны из водорода, С1-С4алкоксигруппы и трифторметила,
R3 выбран из С1-С4алкоксигруппы и трифторметила,
R4 обозначает водород или С1-С4алкил,
R5 обозначает водород или С1-С4алкил,
R6 обозначает водород или галоген,
R7 обозначает водород или галоген и
R8 обозначает водород или галоген,
путем гидролиза сложного эфира соединения формулы (II)
в которой Ar, R1, R2, R3, R4, R5, R6, R7 и R8 имеют указанные выше значения и
R9 обозначает С1-С4-алкил,
с основанием или кислотой, при этом используют соединение формулы (II), полученное в результате реакции соединения формулы (III) ,
в которой R1, R2, R3, R6, R7 и R8 имеют указанные выше значения и
R9 обозначает С1-С4-алкил,
в присутствии основания с соединением формулы (IV)
 ,
,
в которой Ar, R4 и R5 имеют указанные выше значения.
2. Способ по п.1, отличающийся тем, что соединение формулы (III) по п.1 получают по реакции соединения формулы (V)
,
в которой R1, R2, R3, R6, R7 и R8 имеют значения, указанные в п.1, и
R9 обозначает С1-С4-алкил,
с оксихлоридом фосфора, трихлоридом фосфора или пентахлоридом фосфора в присутствии основания.
3. Способ по п.2, отличающийся тем, что соединение формулы (V) по п.2 получают по реакции соединения формулы (VI)
,
в которой R1, R2, R3, R6, R7 и R8 имеют значения, указанные в п.2,
с соединением формулы
,
в которой R9 обозначает С1-С4-алкил,
в присутствии палладиевого катализатора и основания.
4. Способ по п.2, отличающийся тем, что соединение формулы (V) по п.2 получают по реакции соединения формулы (VII)
,
в которой R1, R2, R3, R6, R7 и R8 имеют значения, указанные в п.2,
на первой стадии с соединением формулы (IX) в уксусной кислоте в присутствии палладиевого катализатора, окислительного реагента и кислоты с получением соединения формулы (VIII)
,
в которой R1, R2, R3, R6, R7 и R8 имеют значения, указанные в п.2, и
R9 обозначает С1-С4-алкил,
и на второй стадии с основанием с получением соединения формулы (V).
5. Способ по одному из пп.1-4, отличающийся тем, что соединение формулы (I) представляет собой
{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}уксусную кислоту
соединение формулы (II) представляет собой метил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат
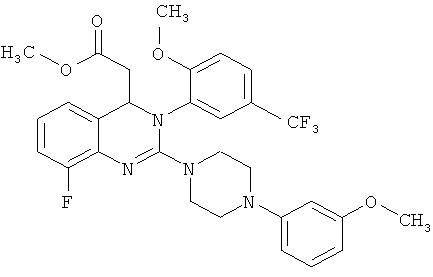
соединение формулы (III) представляет собой метил-2-хлор-8-фтор-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат
соединение формулы (IV) представляет собой 1-(3-метоксифенил)пиперазин
соединение формулы (V) представляет собой
метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил}ацетат
соединение формулы (VI) представляет собой N-(2-бром-6-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевину
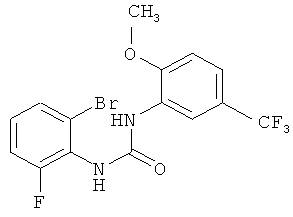
соединение формулы (VII) представляет собой N-(2-фторфенил)-N'-[2-метокси-5-(трифторметил)фенил]мочевину
соединение формулы (VIII) представляет собой метил-(2Е)-3-{3-фтор-2-[({[2-метокси-5-(трифторметил)фенил]амино}карбонил)амино]фенил} акрилат
соединение формулы (IX) представляет собой метилакрилат
 .
.
6. Способ разделения энантиомеров (С1-С4)-алкил-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата и выделения (С1-С4)-алкил-(S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5-(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетата, отличающийся тем, что рацемический сложный эфир кристаллизуют с (2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислотой.
7. Метил-(S)-{8-фтор-2-[4-(3-метоксифенил)пиперазин-1-ил]-3-[2-метокси-5(трифторметил)фенил]-3,4-дигидрохиназолин-4-ил}ацетат(2S,3S)-2,3-бис[(4-метилбензоил)окси]янтарной кислоты.
8. Соединение формулы (III)
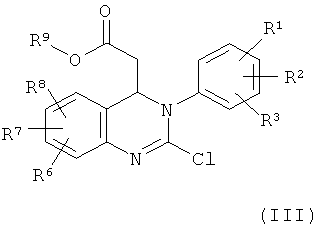 ,
,
в которой R1, R2, R3, R6, R7 и R8 имеют значения, указанные в п.1, и
R9 обозначает С1-С4-алкил.
9. Соединение по п.8, отличающееся тем, что оно представляет собой метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-3,4-дигидрохиназолин-4-ил}ацетат
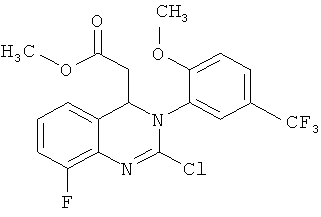
10. Соединение формулы (V)
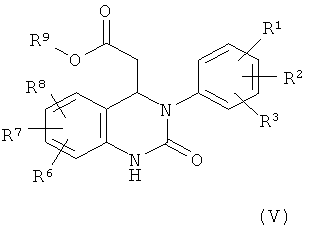 ,
,
в которой R1, R2, R3, R6, R7 и R8 имеют значения, указанные в п.1, и
R9 обозначает С1-С4-алкил.
11. Соединение по п.10, отличающееся тем, что оно представляет собой метил-{8-фтор-3-[2-метокси-5-(трифторметил)фенил]-2-оксо-1,2,3,4-тетрагидрохиназолин-4-ил}ацетат
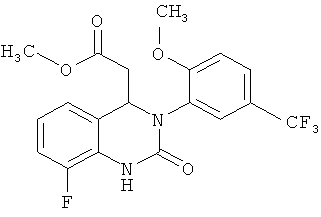 .
.
| P.MOLINS et al "Tetrabutylammonium fluoride promoted intramolecular nucleophilic attack of a cardbodiimide group on an α,β-unsaturated ester group" Synthesis, 1998, 283-287 | |||
| ZHILI XIN et al | |||
| "A practical and efficient intramolecular Michael addition of ureas to α,β-unsaturated esters" Tetrahedron Letters, 2000, 41, 1147-1150 | |||
| WO |

