Изобретение относится к замещенным производным азолонам, являющимися эффективными противо-Helicobacter средствами, которые могут быть использованы в монотерапии с целью уничтожения Helicobacter pylori и родственных видов.
Известны (патент США 4,791,111) 4-(4-фенил-1-пиперазинил)фенолы, являющиеся промежуточными соединениями в синтезе [[4-[4-(4-фенил-1-пиперазинил)феноксиметил] -1,3-диоксолин-2- ил]метил]-1H-имидазолов и -1H-1,2,4-триазолов.
Описаны также 4-(4-фенил-1-пиперазинил)фенолы, обладающие ингибирующей 5-липоксигеназу активностью (патент США 4.931.444). Соединения настоящего изобретения отличаются от указанных соединений своей полезной против-Helicobacter активностью.
Болезни желудочно-кишечного тракта широко распространены. Современная медицина все еще бессильна в изучении целого ряда таких заболеваний, в частности, заболеваний, связанных с присутствием в слизистой желудка бактерии Helicobacter, например: хронического гастрита, язвы двенадцатиперстной кишки и обострения язвы двенадцатиперстной кишки. Двойственная терапия для ликвидации Helicobacter, состоящая до введения по отдельности двух лекарств-антибиотиков до настоящего времени не давала удовлетворительных результатов по одной или нескольким следующим причинам: низкая степень ликвидации Helicobacter, многочисленные побочные эффекты и создание устойчивости у Helicobacter.
Тройственная терапия, состоящая во введении двух антибиотиков и соединения висмута, как показано, эффективна, но потребность в них у больных очень велика и их применение также осложняется побочными эффектами.
Задачей настоящего изобретения являются соединения, обладающие противо-Helicobacter активностью, пригодные для получения лекарственного средства для лечения заболеваний, вызываемых бактериями Helicobacter, а также создание способа лечения указанных заболеваний с помощью нового лекарственного средства на основе данных соединений.
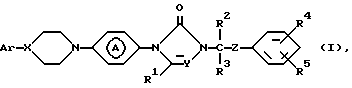
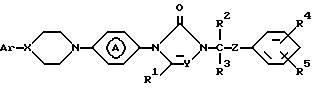
Поставленная задача достигается применением замещенными производными азолона формулы: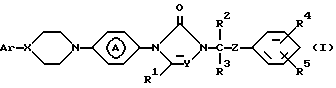
его фармацевтически приемлемых солей с кислотами и его стереохимических изомеров:
где X и Y независимо представляют CH или N;
R1, R2 и R3 каждый независимо представляет водород или C1-C4-алкил;
R4 и R5 каждый независимо представляет водород, галоген, C1-C4-алкил, C1-C4-алкилоксигруппу, гидроксигруппу, трифторметил, трифторметилоксигруппу или дифторметилоксигруппу;
Z представляет C=O или CHOH;
Ar представляет фенил, возможно замещенный вплоть до трех заместителями, выбранными из гидроксигруппы, C1-C4-алкила, C1-C4-алкилоксигруппы, галогена, трифторметила, три(C1-C4-алкил)силилоксигруппы, нитрогруппы, аминогруппы и цианогруппы или пиридинила, замещенного гидроксигруппой или C1-C4-алкилоксигруппой; группа
представляет радикал формул: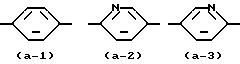
Настоящее изобретение относится также к способу лечения пациентов, страдающих от связанных с Helicobacter заболеваний, включающий введение субъекту эффективного противо-Helicobacter количества соединения формулы (I).
Кроме того, настоящее изобретение относится к фармацевтическим препаратам, содержащим фармацевтически приемлемый носитель и в качестве активного компонента соединение формулы (I), его фармацевтически приемлемую соль с кислотой или его стереохимический изомер, где X, Y, R1-R5, Z, Ar и группа
Принимают вышеуказанные значения, при условии, что Ar отличен от 4-гидроксифенила, 3-(C1-C4-алкил)-4-гидроксифенила или 3,5-ди(C1-C4-алкил)-4-гидроксифенила, когда X = N и группа
представляет радикал формулы (a-I).
Настоящее изобретение относится также к соединению формулы (I), его фармацевтически приемлемой соли с кислотой или его стереохимическому изомеру, где X, Y, R1-R5, Z, Ar и группа
принимают вышеуказанные значения, при условии, что Ar отличен от 4-гидроксифенила, 3-(C1-C4-алкил)-4-гидроксифенила, 3,5-ди(C1-C4-алкил)-4-гидроксифенила или 4-метоксифенила, когда X = N и группа
представляет радикал формулы (a-I).
Вышеприведенные определения в применяемом здесь значении означают: галоген включает хлор, фтор, бром и йод; C1-C4-алкил определяет насыщенный углеводородный радикал с прямой или разветвленной цепью и 1-4 атомами углерода, например: метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил и 1,1-диметилэтил.
Термин "фармацевтически приемлемая соль с кислотой" в применяемом значении определяет неядовитые терапевтически активные соли, которые соединения формулы (I) способны образовать при добавлении кислот. Соединения формулы (I), обладающие основными свойствами, могут быть превращены в соответствующие терапевтически активные соли с кислотами обработкой свободного основания по обычной методике приемлемым количеством соответствующей кислоты. Примеры приемлемых кислот включают неорганические кислоты, такие как гидрогалогеновые, например: хлористоводородную, бромистоводородную и аналогичные кислоты, серную кислоту, азотную кислоту, фосфорную кислоту и т.п. или органические кислоты, такие как, например: уксусную, пропионовую, гидроксиуксусную, 2-гидроксипропионовую, 2-оксопропионовую, этандикислоту, пропандикислоту, бутандикислоту, Z-2-бутендикислоту, (E)-бутендикислоту, 2-гидроксибутендикислоту, 2,3-дигидроксибутандикислоту, 2-гидрокси-1,2,3-пропантрикарбоновую, метансульфоновую, этансульфоновую, бензолсульфоновую, 4-метилбензолсульфоновую, циклогексансульфаминовую, 2-гидроксибензойную, 4-амино-2-гидроксибензойную и аналогичные кислоты. Термин "фармацевтически приемлемые соли с кислотами" включает также сольфаты, которые способны образовывать соединения формулы (I), например: гидраты, алкаголяты и т.п.
Термин "стереохимические изомеры" в применяемом здесь значении определяет различные изомерные, а также конформационные формы, присущие соединениям формулы (I). Если нет других упоминаний или указаний, химическое обозначение соединения означает смесь всех возможных стереохимических и конформационных изомерных форм, причем такие смеси содержат все диастереомеры, энантиомеры и/или конформеры базовой молекулярной структуры.
Абсолютная конфигурация каждого хирального центра может быть указана стереохимическими обозначениями R и S.
Отдельные соединения настоящего изобретения могут существовать в различных таутомерных формах и такие таутомерные формы охватываются объемом настоящего изобретения.
К конкретным соединениям изобретения относятся те соединения формулы (I), в которой R4 и R5 каждый независимо представляет водород или галоген и Ar представляет фенил, замещенный вплоть до трех заместителями, выбранными из гидроксигруппы, C1-C4-алкила и C1-C4-алкилоксигруппы, или пиридинил, замещенный гидроксигруппой или C1-C4-алкилоксигруппой.
Первую группу представляющих интерес соединений образуют те соединения формулы (I), в которой Ar - радикал формулы: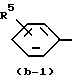
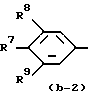
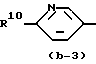
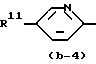
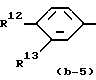
где R6, R7, R10, R11 и R12 каждый независимо представляет гидроксигруппу или C1-C4-алкилоксигруппу; R8 и R9 - C1-C4-алкил.
Вторая группа представляющих интерес соединений включает те соединения формулы (I), в которой X = N.
К третьей группе представляющих интерес соединений относятся те соединения формулы (I), в которой группа
представляет радикал формул (a-1), (a-2) или (a-3).
Четвертую группу представляющих интерес соединений образуют те соединения формулы (I), в которой Y = N и R1 - водород.
Пятая группа представляющих интерес соединений включает те соединения формулы (I), в которой R2 - этил и R3 - водород.
К шестой группе представляющих интерес соединений относятся те соединения формулы (I), в которой R4 - галоген, замещенный в пара-положении и R5 - водород.
Предпочтительные соединения формулы (I), в которой R2 - C1-C4-алкил,
R4 - галоген, замещенный в пара-положении и R1, R3 и R5 - водород.
Более предпочтительны те из рекомендуемых соединений формулы (I), в которой Ar представляет гидроксифенил, метоксифенил, диметоксифенил, C1-C4-алкилфенил, ди(C1-C4-алкил)фенил или метоксипиридинил; группа
представляет радикал формул (a-1), (a-2) или (a-3).
К наиболее предпочтительным относятся следующие соединения:
[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(4- метоксифенил)-1-пиперазинил]-3-пиридинил]-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(3- гидроксифенил)-1-пиперазинил]-3-пиридинил]-3H-1,2,4-триазол-3-он;
2-[1-[(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4- [6-[4-(3-гидроксифенил)-1-пиперазинил]-3-пиридинил]- 3H-1,2,4-триазол-3-он;
2-[1-[(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4- [4-[4-(6-метокси-3-пиридинил)-1-пиперазинил]фенил]- 3H-1,2,4-триазол-3-он;
2-[1-(4-хлорбензоил)пропил] -4-[6-[4-(3-метоксифенил)-1- пиперазинил]-3-пиридинил]-2,4-дигидро- 3H-1,2,4-триазол-3-он;
2-[1-[(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4-[6-[4-(3-метоксифенил)-1-пиперазинил]-3-пиридинил]- 3H-1,2,4-триазол-3-он;
2-[1-(4-хлорбензоил)пропил]-4-[6-[4-(3,4-диметоксифенил)-1- пиперидинил] -3-пиридинил]-2,4-дигидро-3H-1,2,4-триазол-3-он;
2-[1-[(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4- [4-[4-(6-метокси-2-пиридинил)-1-пиперазинил]фенил]- 3H-1,2,4-триазол-3-он;
2-[1-[(4-хлорфенил)гидроксиметил] пропил] -4-[6-[4-(3,4- диметоксифенил)-1-пиперазинил]-3-пиридинил]-2,4-дигидро- 3H-1,2,4-триазол-3-он и
2-[1-[(4-хлорбензоил)пропил] -4-[6-[4-(3,4-диметоксифенил)-1- пиперазинил] -3-пиридинил] -2,4-дигидро-3H-1,2,4-триазол-3-он, их фармацевтически приемлемые соли с кислотами и их стереохимические изомеры.
Методики получения соединений настоящего изобретения формулы (I) приведены, например, в патенте США 4,791,111 и патенте США 4,931,444.
В частности, соединения формулы (I) могут быть получены N-алкилированием промежуточного соединения формулы (II) реактивом формулы (III):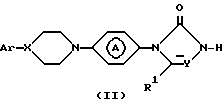
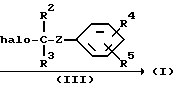
Реакцию N-алкилирования соединения формулы (II) с соединением формулы (III) удобно проводить при перемешивании и нагревании смеси реагентов в приемлемом растворителе в присутствии соответствующего основания. Приемлемые растворители включают, например, диполярные апротонные растворители, например: N, N-диметилформамид, N,N-диметилацетамид, 1,3-диметил-2-имидазолидинон; ароматические растворители, например: бензол, метилбензол; простые эфиры, например: 1,1'-оксибисэтан, тетрагидрофуран; галогенированные углеводороды, например: дихлорметан, тетрагидрофуран или смеси указанных растворителей.
Приемлемые основания включают, например: бис(триметилсилил) амид натрия, карбонаты и гидрокарбонаты щелочных и щелочноземельных металлов, например: карбонат натрия или калия, или органические основания, например: триэтиламин и аналогичные основания.
Соединения формулы (I) могут быть также превращены друг в друга известными способами превращения функциональных групп.
К примеру соединения формулы (I), где Z представляет C=O, могут быть превращены в соединения формулы (I), где Z представляет CHOH, известными специалистам методами восстановления. К примеру, восстановление проводят с гидридом металла или комплексом гидрида металла, например: боргидридом натрия, цианоборгидридом натрия и т.п. в воде, 1-метилпирролидоне, в спиртовой среде, например, метаноле, этаноле или простом эфире, например: тетрагидрофуране, 1,4-диоксане или в смеси указанных растворителей.
Или же восстановление может быть проведено реакцией с гидроборатом трис(1-метилэтокси)калия в инертном, в условиях реакции, растворителе, например: тетрагидрофуране.
Кроме того, соединения формулы (I), в которой Ar замещен, по меньшей мере одной гидроксигруппой, могут быть получены из соответствующих C1-C4-алкилоксипроизводных в приемлемой реакции дезалкилирования, например, применением трифторуксусной кислоты, или, в частности, минеральной кислоты, такой как концентрированная гидрогалогеновая кислота, например: бромистоводородная кислота, йодистоводородная кислота, возможно в смеси с насыщенным раствором йодистоводородной кислоты в ледяной уксусной кислоты; кислоты Льюиса, например: трехбромистого бора в инертном, в условиях реакции, растворителе, например: дихлорметане. В случае применения бромистоводородной кислоты рекомендуется реакцию дезалкилирования проводить в присутствии связывающего бромвещества, такого как, например: сульфит или гидросульфит натрия.
Напротив, соединения формулы (I), в которой Ar замещен, по меньшей мере, C1-C4-алкилоксигруппой, могут быть получены алкилированием соответствующего гидроксипроизводного приемлемым алкилирующим средством, например: диметилсульфатом и т.п. Возможно проведение реакции алкилирования в известных специалистам условиях каталитической реакции с переносом фаз. Эти условия включают перемешивание реагентов с приемлемым основанием, например: гидроксидом натрия в инертных в условиях реакции растворителе, например: дихлорметане в присутствии приемлемого катализатора переноса фаз, например: бензилтриэтиламмонийхлоридом и т.п.
Соединения формулы (I), в которой Ar замещен гидроксигруппой, могут быть превращены в соответствующие три(C1-C4-алкил)силилоксипроизводные реакцией с три (C1-C4-алкил)-Si-L7, где L7 представляет реакционноспособную отходящую группу, например, галоген в инертном условиях реакции растворителе, например, пиридине, дихлорметане.
И напротив, соединения формулы (I), в которой Ar замещен три (C1-C4-алкил)силилоксигруппой, могут быть превращены в соответствующие гидроксипроизводные реакцией с фторидом, например, (н-C4H9)4 N+F- в инертном в условиях реакции растворителе, например: тетрагидрофуране, дихлорметане.
И наконец, чистые изомерные формы соединений формулы (I) могут быть выделены из смеси обычными методами разделения. В частности, энантиомеры могут быть разделены колоночной хроматографией с применением хиральной неподвижной фазы, например, дериватизированной целлюлозы, такой как три(диметилкарбамоил)целлюлоза (Хиральцель, OD®) и аналогичных хиральных неподвижных фаз.
Во всех предшествующих и последующих методах получения продукты реакции могут быть выделены из реакционной и при необходимости подвергнуты дополнительной очистке по общим известным специалистам методикам.
Некоторые промежуточные и исходные соединения, используемые в вышеприведенных способах получения, являются известными соединениями, которые могут быть синтезированы по известным специалистам методикам получения таких или аналогичных соединений.
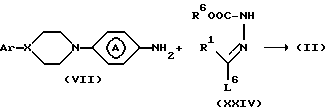
Промежуточные соединения формулы (II) могут быть синтезированы циклизацией промежуточного соединения формулы (IV) с реагентом формулы (V) или его производным.
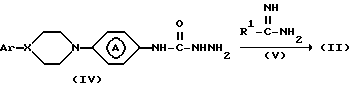
Приемлемым для указанной реакции растворителем является, например, инертный в условиях реакции диполярный апротонный растворитель, например: N, N-диметилформамид, диметилсульфоксид и т. п. или спирт, например: этанол, 1-бутанол и т.п.
Промежуточное соединение формулы (IV) может быть синтезировано реакцией промежуточного соединения формулы (VI) с гидразином или его производным в инертном в условиях реакции растворителе, например: 1,4-диоксане и т.п. (формулу (IV) см. ниже).
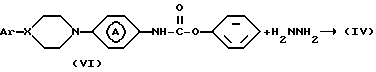
Промежуточное соединение формулы (VI) может быть синтезировано реакцией промежуточного соединения формулы (VII) с фенилхлорформатом в инертном в условиях реакции растворителе, таком как, например: N,N-диметилформамид, N, N-диметилацетамид, ароматическом растворителе, например: пиридине или галогенированном углеводороде, например: дихлорметане и т.п. или смеси указанных растворителей.
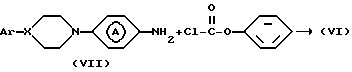
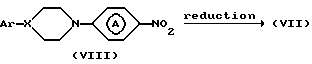
Промежуточные соединения формулы (VII) могут быть получены восстановлением по известным специалистам методикам соответствующих нитропроизводных формулы (VIII).
Приемлемые известные методики восстановления включают, например: каталитическое гидрирование в подходящем растворителе, например: метаноле, тетрагидрофуране, N, N-диметилформамиде в присутствии соответствующего катализатора, например: палладия на угле, никеля Ренея и т.п. под давлением водорода, возможно в присутствии тиофена.
Или же промежуточное соединение формулы (VII) может быть получено реакцией соединения формулы (VIII) с гидразином или его производным в инертном в условиях реакции растворителе, например: метаноле в присутствии катализатора, например: никеля Ренея.
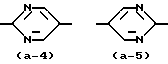
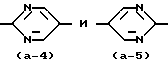
Кроме того, промежуточные соединения формулы (VII), в которой группа  представляет радикал формулы (a-4), могут быть получен реакцией промежуточного соединения формулы (IX) или его производного согласно следующей схеме реакции:
представляет радикал формулы (a-4), могут быть получен реакцией промежуточного соединения формулы (IX) или его производного согласно следующей схеме реакции: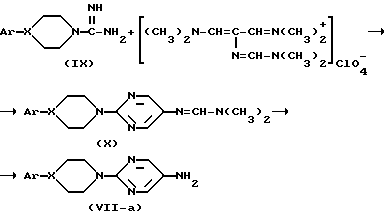
Получение промежуточных соединений вышеприведенной формулы (X) обычно проводят в инертных условиях реакции растворителе, например: 1-пропаноле, метаноле или их смеси, предпочтительно в присутствии основания, например: метоксида натрия. Синтез промежуточных соединений формулы (VII-a) из промежуточных соединений формулы (X) может быть осуществлен в присутствии основания, например: гидроксида натрия и т.п.
Промежуточные соединения формулы (VIII) могут быть получены реакцией промежуточных соединений формулы (XI) с реагентом формулы (XII), где L - реакционноспособная отходящая группа, например: галоген в инертном в условиях реакции растворителе, например: N,N-диметилацетамиде, диметилсульфоксид, N, N-диметилформамид, бис(2-метоксиэтиловый) эфир, 3-метокси-1-пропанол и т.п., предпочтительно в присутствии основания, например: карбоната калия и т.п.
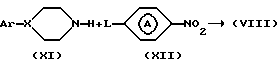
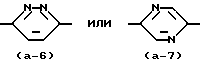
Промежуточное соединения формулы (XI), в которой X = N, то есть промежуточные соединения, представленные формулой (XI-a), могут быть получены реакцией промежуточных соединений формулы (XIII) с реагентом формулы (XIV), где L1 и L2 представляют реакционноспособные отходящие группы, например: галоген, в инертном в условиях реакции растворителе, например: 1-бутаноле, гексаноле и т.п., предпочтительно в присутствии основания, например: карбоната калия и возможно в присутствии небольшого количества йодида калия.
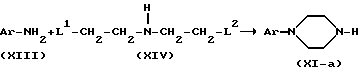
Возможно проведение указанных реакций применением N-защищенного промежуточного соединения формулы (XIV), например, тозильного производного. После реакции с промежуточным соединением формулы (XIII) защитные группы могут быть удалены действием разбавленной кислоты, например, серной кислоты.
Или же промежуточные соединения формулы (XI-a), в которой Ar замещен метоксигруппой могут быть получены согласно следующей схеме реакции.
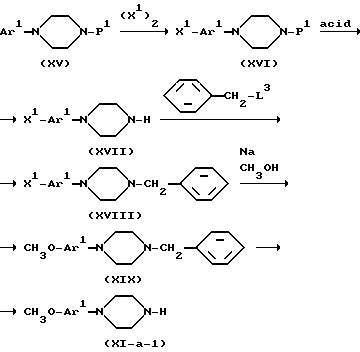
Ar1 в применяемом здесь значении представляет фенил или пиридинил, R1 представляет защитную группу, например, C1-C4-алкилоксикарбонил, X1 представляет галоген, в частности, бром или йод и L3 представляет реакционноспособную отходящую группу, например, галоген. Синтез промежуточных соединений формулы (XVI) рекомендуют проводить в инертном в условиях реакции растворителе, например, сероуглероде.
Защитная группа в промежуточных соединениях формулы (XVI) может быть удалена действием кислоты, например, бромистоводородной кислоты. Получение промежуточных соединений формулы (XVIII) рекомендуют проводить в инертном в условиях реакции растворителе, например, диметилбензоле, возможно в присутствии основания, например, гидрокарбоната натрия. Кроме того, получение вышеуказанных промежуточных соединений формулы (XIX) возможно в присутствии катализатора, например, йодида меди (I) и т.п., возможно в атмосфере азота. И наконец, бензильная группа в промежуточных соединениях формулы (XIX) может быть удалена каталитическим гидрированием по известным методикам. Или же промежуточное соединение формулы (XVI) может быть получено из соответствующего анилинопроизводного применением известных методик.
Для получения промежуточных соединений формулы (II) промежуточное соединение формулы (VI) может быть введено в реакцию с реактивом формулы (XX), где L4 и L5 представляют реакционноспособную отходящую группу, например C1-C4-алкилоксигруппу в инертном в условиях реакции растворителе, например, 1,4-диоксане и т. п. с получением промежуточного соединения формулы (XXI). Последнее промежуточное соединение затем циклизуют действием кислоты, например, хлористоводородной кислоты.
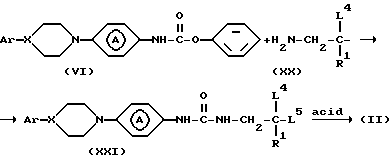
В другом варианте промежуточные соединения формулы (II) могут быть синтезированы реакцией промежуточного соединения формулы (VI) с реактивом формулы (XXII) или его производным в инертном в условиях реакции растворителе, например, 1,4-диоксане, возможно в присутствии основания, например, N, N-диметил-4-пиридинамина с образованием промежуточного соединения формулы (XXIII). Полученное промежуточное соединение может быть циклизовано действием кислоты, например, муравьиной кислоты.
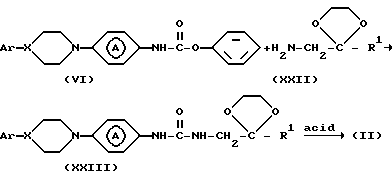
Промежуточные соединения формулы (II) могут быть также получены реакцией промежуточного соединения формулы (VII) с реагентом формулы (XXIV), где R6-C1-C6-алкил, например: метил или этил и L6 представляет реакционноспособную отходящую группу, например: C1-C6-алкилоксигруппу или ди(C1-C6-алкил)аминогруппу, например: метокси-, этокси- или диметиламиногруппу в инертном в условиях реакции растворителе, например, тетрагидротиофен-1,1-диоксиде и т.п.
Соединения формулы (I), их фармацевтически приемлемые соли с кислотами и их стереохимические изомеры проявляют полезную фармакологическую активность против бактерий вида Helicobacter, например: Helicobacter pylori, Helicobacter mustelae, Helicobacter felis и т.п., в частности Helicobacter pylori.
В этой связи особенно важным является открытие того, что рассматриваемые соединения проявляют ингибирующую активность по отношению к росту Helicobacter, а также бактерицидную активность против указанной бактерии. Бактерицидное действие на Helicobacter определяют в суспендированных культурах с помощью методики, приведенной в издании "Химиотерапия противомикробными средствами", 1991, т. 35, стр. 869-872.
Интересным признаком соединений настоящего изобретения является их высокая специфическая активность против Helicobacter. Соединения формулы (I) не проявляют ингибирующей активности против каких-либо следующих видов: Campylobacter jejuni, Campylobacter coli, Campylobacter fetus, Campolybacter sputorum, Vibrio spp., Staphylococcus aureus и Escherichial coli при испытании в концентрации вплоть до 10-5 М.
Важной характеристикой соединений настоящего изобретения является их устойчивая активность против H. pylori при pH ниже нормальных нейтральных значений pH. Активность при низких pH in vitro может служить указанием на то, что на соединения не будет оказывать неблагоприятное воздействие кислотная среда желудка in vivo.
Следовательно указанные соединения можно считать представляющими ценность лечебными средствами для теплокровных животных, в частности, человека, страдающих от заболеваний и поражений, возбудителем которых является Helicobacter. Примеры подобных заболеваний или недугов включают гастрит, язву желудка, язву двенадцатиперстной кишки и рак желудка.
Обладающие полезными противо- Helicobacter свойствами данные соединения могут быть введены в состав разнообразных фармацевтических форм для введения в лечебных целях. Для получения фармацевтических препаратов, согласно настоящему изобретению, эффективное количество конкретного соединения (в виде основания или в виде соли с кислотой) в качестве активного компонента смешивают в однородной с фармацевтически приемлемым носителем, который может иметь вид самых разнообразных форм в зависимости от целевой формы предназначенного для введения препарата. Такие фармацевтические препараты рекомендуются в единичных дозированных формах, пригодных предпочтительно для введения перорально, ректально или парентеральными инъекциями. К примеру, при получении препаратов в пероральной дозированной форме может быть использована любая обычная фармацевтическая среда, такая как, например: вода, гликоли, масла, спирты и т.п. в случае жидких пероральных препаратов, например: суспензий, сиропов, эликсиров или растворов, или твердые носители, например: крахмал, сахар, каолин, смазки, связующие вещества, структураторы и т.п. в случае порошков, пилюль и таблеток. Вследствие легкости их введения таблетки и капсулы представляют собой наиболее рекомендуемые пероральные дозированные формы, и в этом случае, очевидно, применяют твердые фармацевтические носители. Для парентеральных препаратов в качестве носителя обычно применяют стерильную воду, составляющую, по меньшей мере, большую часть препарата, хотя препарат может включать и другие компоненты, например, способствующие растворимости вещества. Могут быть приготовлены, например, инъектированные растворы, в которых носителем является солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Могут быть также приготовлены инъектируемые суспензии, и в этом случае применяют соответствующие жидкие носители, суспендирующие средства и т.п.
В случае, если фармацевтический препарат имеет вид водного раствора, те соединения формулы (I), которые отличаются низкой растворимостью, могут быть введены в препарат в виде соли или может быть добавлен смешивающийся с водой и физиологически приемлемый сорастворитель, например: диметилсульксид и т.п. , или же соединение формулы (I) может быть солюбилизировано приемлемым носителем, например, циклодекстрином (ЦД) или, в частности, производным циклодекстрина, например, производными циклодекстрина, раскрытыми в патенте США 3,459,731, Европейском патенте EP-A-149,197 (июль 24, 1985), Европейском патенте EP-A-197,571 (октябрь 15, 1986), патенте США 4,535,152 или WO 90/12035 (октябрь 18, 1990). Обычно такие производные состоят из α-, β- или γ-ЦД, в котором одна или несколько гидроксильных групп замещены C1-C6-алкилом, в частности, метилом, этилом или изопропилом; гидрокси-C1-C6-алкилом, в частности, гидроксиэтилом, гидроксипропилом или гидроксибутилом; карбокси-C1-C6-алкилом, в частности, карбоксиметилом или карбоксиэтилом; C1-C6-алкилкарбонилом, в частности, ацетилом; C1-C6-алкилоксикарбонил-C1-C6-алкилом; карбокси-C1-C6-алкилокси-C1-C6-алкилом, в частности, карбоксиметоксипропилом или карбоксиэтоксипропилом, или C1-C6-алкилкарбонилокси-C1-C6-алкилом, в частности, 2-ацетилпропилом. Особого внимания в качестве комплексообразующих и/или солюбилизирующих средств заслуживают β-ЦД, 2,6-диметил-β-ЦД, и особенно 2-гидроксипропил -β-ЦД,2-гидроксиэтил-β-ЦД, 2-гидроксиэтил-γ-ЦД, 2-гидроксипропил -γ-ЦД и (2-карбоксиметокси)пропил-β-ЦД. Для вышеупомянутых производных циклодекстрина С3 (степень замещения, то есть среднее число замещенных гидроксифункций на звено глюкозы) находится в интервале 0,125-3, в частности, 0,2-2 или 0,2-1,5. Более предпочтительные значения С3 находятся в интервале 0,2-0,7, в частности 0,35-5 и особенно около 0,4. МС3 (молярная степень замещения, то есть среднее число молей замещающего вещества на звено глюкозы) находится в интервале 0,125-10, в частности, 0,3-3 или 0,3-1,5. Более предпочтительные значения МС3 находятся в интервале 0.3-0,8, в частности, 0,35-0,5 и особенно около 0,4. Наиболее рекомендуемыми для использования в препаратах настоящего изобретения производными циклодекстрина являются гидроксипропил -β-циклодекстрин с МС3 в интервале 0,35-0,5 и содержащий менее 1,5% незамещенного β-циклодекстрина. Количество циклодекстрина или его производного в конечном препарате, как правило, находится в интервале 1-40%, в частности, 2,5-25% и особенно 5-20%.
Вышеупомянутые фармацевтические препараты особенно рекомендуется изготовлять в дозированных единичных формах, облегчающих введение и обеспечивающих одинаковость доз. В данном описании и в формуле изобретения термин "дозированная единичная форма" относится к физически дискретной единице, применяемой в виде разовой дозы, при этом каждая единица содержит заданное количество активного компонента, рассчитанное на создание нужного терапевтического эффекта, в смеси с необходимым фармацевтическим носителем. Примеры подобных дозированных единичных форм включают: таблетки (в том числе рифленые и имеющие оболочку таблетки), капсулы, пилюли, порошки в пакетиках, вафли, инъектируемые растворы или суспензии и т.п., и такие единичные формы могут быть объединены в одной упаковке.
Ввиду применимости рассматриваемых соединений для лечения связанных с Helicobacter заболеваний очевидно, что настоящим изобретением дается способ лечения теплокровных животных, в том числе и человека, страдающих связанным с Helicobacter заболеванием. Способ состоит в системном введении фармацевтически эффективного количества соединения формулы (I), его фармацевтически приемлемой соли с кислотой или его стереохимического изомера в смеси с фармацевтическим носителем. Как правило, считают, что ежедневно эффективное количество находится в интервале 0,05-20 мг/кг массы тела, предпочтительно 0,1-10 мкг/кг массы тела и более предпочтительно 0,5-5 мг/кг массы тела. Очевидно, что эффективная ежедневная доза может быть уменьшена или увеличена в зависимости от реакции подвергаемого лечению субъекта и/или в зависимости от наблюдений лечащего врача, прописывающего соединения настоящего изобретения. Вышеупомянутые интервалы эффективных ежедневных количеств, таким образом, являются всего лишь ориентировочными, и не предназначены ни в коей мере для ограничения объема изобретения. В основном пригодная фармацевтическая композиция включает соединение формулы 1 в количестве 3,5-1400 мг, в частности 7-700 мг, более предпочтительно 35-350 мг.
В комбинации с соединениями настоящего изобретения возможно также введение и других активных соединений, применяемых для подавления Helicobacter. Введение может быть осуществлено отдельно (например: одновременно, совместно или последовательно) или различные лекарственные средства могут быть объединены в одной дозированной форме. Рекомендуемыми для комбинированного лечения соединения являются соединения висмута, например: субцитрат висмута, субсалицилат висмута и т.п. и ингибиторы перекачки протонов, например: смепразол, ланзодразол и т.п.
Экспериментальная часть
Получение промежуточных соединений
Пример 1.
а) смесь 2-(4-метоксифенил)пиперазина дигидрохлорида (0,05 моля), 1-хлор-4-нитробензола (0,05 моля) и карбоната калия (10 г) в N,N-диметилформамиде (100 мл) кипятят с перемешиванием примерно сутки. Реакционную смесь разбавляют водой и экстрагируют дважды дихлорметаном. Органические слои объединяют, сушат (MgSO4), фильтруют и испаряют в вакууме. Остаток последовательно ополаскивают 4-метил-2-пентаноном и перекристаллизовывают из 1,4-диоксана. Фильтрованием и высушиванием продукта получают 10,5 г (67%) 1-(4-метоксифенил)-4-(4-нитрофенил)пиперазина, т.пл. 195,1oC (промеж. соед. I).
b) смесь промежуточного соединения (1) (0,038 моля) в метаноле (250 мл) и тетрагидрофуране (250 мл) гидрируют под нормальным давлением и при комнатной температуре с применением в качестве катализатора 10% палладия на активированном угле (2 г). После поглощения водорода (3 экв.) катализатор отфильтровывают и ополаскивают N,N-диметилацетамидом. Объединенные фильтраты переносят в воду. Осадок отфильтровывают и перекристаллизовывают из 1-бутанола. Фильтрованием и сушкой продукта получают 8 г (74%) 4-[4-(4-метоксифенил)-1-пиперазинил]бензоламина, т.пл. 191,8oC (промеж. соед. 2).
c) Смесь промежуточного соединения (2) (0,021 моля) и фенилхлорформата (0,023 моля) в пиридине (75 мл) и дихлорметане (75 мл) перемешивают и нагревают до полного растворения реагентов. Перемешивание продолжают 30 минут при комнатной температуре. Реакционную смесь переносят в смесь 500 мл воды и 300 мл 2,2'-оксибиспропана. Осадок после перемешивания отфильтровывают, сушат и перекристаллизовывают из 1-бутанола. Фильтрованием и высушиванием продукта получают 5,2 г (61%) фенил [4-[4-(4-метоксифенил)-1-пиперазинил]фенил] карбамата, т.пл. 204,5oC (промеж. соед. 3).
d) Смесь промежуточного соединения (3) (0,008 моля) с моногидратом гидразина (50 мл) в 1,4-диоксане (100 мл) кипятят с перемешиванием 3 часа. После охлаждения реакционную смесь переносят в воду. Осадок отфильтровывают и перекристаллизовывают из N,N-диметилформамида. Фильтрованием и высушиванием продукта получают 1,7 г (62%) N-[4-[4-метоксифенил)-1-пиперазинил]фенил]гидравзинкарбоксамида, т.пл. > 300oC (промеж. соед. 4).
e) Смесь промежуточного соединения (4) (0,001 моля) и ацетата метанимидамида (0,029 моля) в диметилсульфоксиде (10 мл) нагревают 2 часа при 160oC. После охлаждения реакционную смесь переносят в смесь 4-метил-2-пентанона и 2,2'-оксибиспропана. Осадок отфильтровывают и обрабатывают активированным углем в N,N-диметилформамиде. После фильтрования продукт оставляют для кристаллизации. Фильтрованием и сушкой продукта получают 1 г (28%) 2,4-дигидро-4-[4-[4-(4-метоксифенил)-1-пиперазинил]фенил]- 3H-1,2,4-триазол-3-она, т. пл. > 300oC (промеж. соед. 5).
Пример 2.
Смесь промежуточного соединения (4) (0,015 моля) и гидрохлорида этанимидамида (0,56 моля) в N,N-диметилформамиде (150 мл) перемешивают 3 часа при 130oC. После охлаждения реакционную смесь переносят в воду. Осадок отфильтровывают, промывают водой и CH3OH и кристаллизуют из N,N-диметилформамида. Фильтрованием продукта и его перекристаллизацией из 1,4-диоксана получают 19,5 г (33,3%) 2,4-дигидро-4-[4-[4-(4-метоксифенил)-1-пиперазинил]фенил]-5-метил- 3H-1,2,4-триазол-4-она, т.пл. 298.4oC (промеж. соед. 6).
Пример 3.
a) Смесь 10 г промежуточного соединения (3), 3 г 2,2-диметоксиэтанамина и 100 мл 1,4-диоксана кипятят с перемешиванием 6 часов, после чего реакционную смесь охлаждают. Осадившийся продукт отфильтровывают, промывают 1,4-диоксаном и очищают колоночной хроматографией на силикагеле с применением в качестве элюента смеси трихлорметана с метанолом (99:1). Собирают чистые фракции и элюент испаряют. Кристаллизацией остатка из 1,4-диоксана получают 3,9 г N-(2,2-диметоксиэтил)-N'-[4-[4-метоксифенил)-1-пиперазинил] фенил]-мочевины, т.пл. 225oC (промеж. соед. 7).
b) Смесь 70 г промежуточного соединения (7), 84 г концентрированной соляной кислоты, 300 мл воды и 350 мл метанола нагревают с перемешиванием 30 минут при 80oC. Реакционную смесь оставляют нагреваться до комнатной температуры с одновременной кристаллизацией продукта. Продукт фильтруют, промывают водой и после сушки получают 24,5 г (37%) моногидрата моногидрохлорида 1,3-дигидро-1-[4-[4-(4-метоксифенил)-1-пиперазинил]фенил]-2H-имидазол-2-она, т.пл. 256,2oC (промеж. соед. 8).
Пример 4.
a) Смесь 4,4 г промежуточного соединения (3), 1,6 г 2-аминометил-2-метил-1,3-диоксалана моногидрата, 100 мл 1,4-диоксана, 1 г N,N-диметил-4-пиридинамина и 4 г N,N-диэтилэтанамина кипятят с перемешиванием 3 часа. Добавляют воду и смесь охлаждают с кристаллизацией продукта. Продукт отфильтровывают, промывают водой и 2-пропанолом и после сушки получают 3,8 г (89,1%) N-[4-[4-(4-метоксифенил)-1-пиперазинил] фенил]-N'-[(2-метил-1,3- диоксалан-2-ил)метил]мочевины (промеж. соед. 9).
b) Смесь 3,3 г промежуточного соединения (9) и 100 мл муравьиной кислоты перемешивают 1 час при 70oC. Реакционную смесь испаряют и остаток нейтрализуют раствором гидрокарбоната натрия. Добавляют 4-метил-2-пентанон, смесь перемешивают и продукт отфильтровывают. Осадок промывают водой и 4-метил-2-пентаноном. Перекристаллизацией продукта из N,N-диметилформамида получают 2 г (71,3%) 1,3-дигидро-1-[4-[4-(4-метоксифенил)-1-пиреразинил]фенил]-5-метил -2H-имидазол-2-она, т.пл. > 300oC (промеж. соед. 10).
Пример 5.
a) К перемешиваемой и охлаждаемой смеси ( < 10oC) 150 г этилового эфира 4-(2-пиридинил)-1-пиперазинкарбоновой кислоты и 1535 мл сероуглерода прибавляют по каплям 32,8 мл брома. По окончании прикапывания перемешивание продолжают 18 часов с повышением температуры до комнатной. При температуре ниже 20oC добавляют раствор 70 г 10 н. гидроксида натрия в 300 мл воды. После перемешивания 3 часа при комнатной температуре слои разделяют. Водную фазу экстрагируют дважды трихлорметаном. Объединенные органические фазы промывают водой, сушат, фильтруют и испаряют. К остатку добавляют 46 мл бензола и смесь вновь испаряют. После стояния 48 часов продукт затвердевает. Маслянистую фазу декантируют и твердый продукт кристаллизуют дважды из 2,2'-оксибиспропана при 10oC. Фильтрованием и сушкой получают 100 г этилового эфира 4-(5-бром-2-пиридинил)-1-пиперазинкарбоновой кислоты, т.пл. 68oC (промеж. соед. 11).
b) Смесь 18 г промежуточного соединения (11) и 50 мл 48%-ой водной бромистоводородной кислоты кипятят с перемешиванием 2 часа. Реакционную смесь охлаждают и испаряют. Твердый остаток растворяют в воде и при температуре ниже 20oC раствор подщелачивают 10 н. раствором гидроксида натрия. Продукт дважды экстрагируют трихлорметаном. Объединенные экстракты промывают водой, сушат, фильтруют и испаряют. Сушкой твердого продукта на воздухе при комнатной температуре получают 12,3 г 1-(5-бром-2-пиридинил)пиперазина, т.пл. 70,6oC (промеж. соед. 12).
c) Смесь 9,7 г промежуточного соединения (12) 3,7 г гидрокарбоната натрия и 40 мл диметилбензола кипятят с перемешиванием. Затем в кипящую смесь по каплям прибавляют раствор 4,8 мл (бромметил) бензола в 10 мл диметилбензола. По окончании прибавления кипячение с перемешиванием продолжают 4 часа. Реакционную смесь охлаждают до комнатной температуры, добавляют 75 мл воды и слои разделяют. Водную фазу экстрагируют трихлорметаном. Объединенные органические фазы сушат, фильтруют и испаряют в вакууме. Остаток кристаллизуют при 4oC из 2,2'-оксибиспропана. Продукт отсасывают и после сушки получают 6,5 г 1-(5-бром-2-пиридинил)-4-(фенилметил)-пиперазина, т.пл. 100oC (промеж. соед. 13).
d) К 500 мл метанола при перемешивании добавляют порциями натрий (0,6 моля) и смесь перемешивают до полного вступления в реакцию натрия. Затем добавляют промежуточное соединение (13) (0,15 моля), йодид меди (1) (0,15 моля) и N, N-диметилформамид (500 мл) и смесь кипятят с перемешиванием 48 часов. Затем смесь фильтруют и испаряют. Остаток очищают колоночной хроматографией на силикагеле (элюент: EtOAc-гексан-CH2Cl2, 1 : 1 : 2). Чистые фракции собирают и упаривают, получая 33,5 г (79%) 1-(5-метокси-2-пиридинил)-4-(фенилметил)-пиперазина (промеж. соед. 14).
e) Промежуточное соединение (14) (0,11 моля) в метаноле гидрируют при 50oC в присутствии в качестве катализатора 10% палладия на активированном угле (7 г). После поглощения 2 экв. водорода катализатор отфильтровывают и испарением фильтрата получают 19,3 г (88%) 1-(5-метокси-2-пиридинил)пиперазина (промеж. соед. 15).
f) Смесь промежуточного соединения (15) (0,094 моля) и N,N-диметилацетамида (0,15 моля) в растворе карбоната калия (100 мл) перемешивают при комнатной температуре. По каплям прибавляют 1-фтор-4-нитробензол (0,12 моля) и смесь перемешивают примерно сутки при комнатной температуре. Смесь переносят в воду, осадок отфильтровывают и после сушки получают 20,6 г (70%) продукта. Образец продукта (1 г) кристаллизован из 2-пропанола и очищен колоночной хроматографией на силикагеле (элюент: CH2Cl2-CH3CN-CH3OH, 97 : 2 : 0,25). Чистые фракции собирают и испаряют. Промывкой остатка (0,8 г) 2,2'-оксибиспропаном получают 0,8 г 1-(5-метокси-2-пиридинил)-4-(4-нитрофенил)пиперазина, т. пл. 160,2oC (промеж. соед. 16).
g) Промежуточное соединение (16) (0,061 моля) в N,N-диметилформамиде (500 мл) гидрируют примерно сутки при 50oC с использованием в качестве катализатора никеля Ренея. После поглощения 3 экв. водорода катализатор отфильтровывают и фильтрат испаряют. Остаток используют без дополнительной очистки. Получают 17,6 г (100 %) 4-[4-(5-метокси-2-пиридинил)-1-пиперазинил] бензоламина (промеж. соед. 17).
h) К перемешиваемой на водяной бане смеси промежуточного соединения (17) (0,062 моля) и N,N-диметилформамида (100 мл) прибавляют по каплям фенилхлорформат (0,11 моля) и смесь перемешивают примерно сутки. Затем добавляют воду, осадок отфильтровывают и после сушки получают 25 г (99%) фенил[4-[4-(5-метокси-2-пиридинил)-1- пиперазинил]фенил]карбамата (промеж. соед. 18).
i) Смесь промежуточного соединения (18) (0,062 моля) и моногидрата гидразина (0,62 моля) в 1,4-диоксане (500 мл) перемешивают 48 часов пр 50oC. Смесь переносят в воду, осадок отфильтровывают и после сушки получают 15 г (71%) N-[4-[4-(5-метокси-2-пиридинил)-1-пиперазинил] фенил] гидразинкарбоксамида (промеж. соед. 19).
j) Смесь промежуточного соединения (19) (0,044 моля) и ацетата метанимидамида (0,22 моля) в 1-бутаноле (300 мл) кипятят с перемешиванием примерно сутки. Смесь переносят в воду и экстрагируют CH2Cl2. Органический слой сушат, фильтруют и испаряют. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2-CH3OH, 95 : 5). Чистые фракции собирают и после испарения получают 3,2 г 2,4-дигидро-4-[4-[4-(5-метокси-2-пиридинил)-1-пиперазинил]фенил]-3H-1,2,4-триазол-3-она (промеж. соед. 20).
Пример 6.
a) Смесь 36 г дигидрохлорида 1-(4-метоксифенил)пиперазина, 22 г 2-хлор-5-нитропиридина, 58 г карбоната калия и 227 мл диметилсульфоксида перемешивают примерно сутки при 140oC. Реакционную смесь охлаждают и переносят в воду. Осадившийся продукт отфильтровывают, промывают водой и растворяют в дихлорметане. Раствор обрабатывают активированным углем. Уголь отфильтровывают и фильтрат испаряют. Остаток ополаскивают 2-пропанолом. Фильтрованием продукта и его кристаллизацией из 1-бутанола получают 24,5 г 1-(4-метоксифенил)-4-(5-нитро-2-пиридинил)пиперазина, т. пл. 170oC (промеж. соед. 21).
b) К кипящей смеси 36 мл моногидрата гидразина, 4 г никеля Ренея и 1000 мл метанола добавляют порциями в течение 25 минут 35 г промежуточного соединения (21). После кипячения 35 минут с перемешиванием добавляют еще 11 мл моногидрата гидразина и кипячение продолжают 10 минут. Добавляют дополнительно 0,5 г никеля Ренея и смесь кипятят 15 минут. Реакционную смесь охлаждают, катализатор отфильтровывают и испарением фильтрата получают 33,57 г 6-[4-(4-метоксифенил)-1-пиперазинил] -3-пиридинамина в виде сырого продукта, т. пл. 144 - 147oC (промеж. соед. 22).
c) Смесь 31,21 г промежуточного соединения (22), 38,4 г этилового эфира (1-этоксиэтилден)гидразинкарбоновой кислоты и 10 мл тетрагидротиофен-1,1-диоксида нагревают 17 часов на масляной бане при 154 - 158oC. После охлаждения добавляют 150 мл 2-пропанола и смесь фильтруют. Остаток перекристаллизовывают из смеси ацетонитрила с N,N-диметилформамидом (90 : 10). Остаток очищают вытеснительной хроматографией (элюент: CH2Cl2-CH3OH(NH4OH 10%), 98 : 2 ---> 96 : 4). Испарением элюента в целевых фракциях и перекристаллизацией остатка из смеси N,N-диметилформамида с ацетонитрилом получают 2,4-дигидро-4-[6-[4-(4-метоксифенил)-1-пиперазинил] -3-пиридинил] -5-метил-3H-1,2,4-триазол-3-она, т. пл. 251 - 252oC (промеж. соед. 23).
Пример 7.
a) К смеси 20 г 4-(4-метоксифенил)-1-пиперазинкарбоксидамида и 21 г N-[3-(диметиламино)метилен] амино] -2-пропенилиден]-N- метилметанаминийперхлората в 200 мл 1-пропанола прибавляют по каплям 70 мл раствора метоксида натрия в метаноле (1М). После перемешивания 2 часа вновь по каплям прибавляют 70 мл раствора метоксида натрия в метаноле (1М). Затем смесь кипятят с перемешиванием 2 часа. После охлаждения реакционную смесь испаряют и остаток очищают колоночной хроматографией (элюент: CH2Cl2-CH3OH, 97 : 3). Испарением элюента в целевых фракциях получают 15 г (62,5%) N-[(диметиламино)метилен] -2-[4-(4-метоксифенил)-1-пиперазинил]- 5-пиримидинамина (промеж. соед. 24).
b) Смесь 10 г промежуточного соединения (24) и 150 мл серной кислоты (0,2 М) кипятят с перемешиванием 4 часа. После охлаждения смесь нейтрализуют водным раствором карбоната калия. Продукт экстрагируют дихлорметаном, экстракт промывают водой, сушат, фильтруют и испаряют. Остаток очищают колоночной хроматографией (элюент: CH2Cl2-CH3OH, 98 : 2). Элюент в целевых фракциях испаряют и остаток кристаллизуют из этилацетата. Продукт фильтруют и после сушки в вакууме при 50oC получают 3,5 г (40,9%) 2-[4-(4-метоксифенил)-1-пиперазинил]-5-пиримидинамина, т. пл. 157,1oC (промеж. соед. 25).
c) К перемешиваемой в бане со льдом смеси 10 г промежуточного соединения (25) и 100 мл N.N-диметилацетамида прибавляют по каплям 4,8 мл фенилхлорформата. После перемешивания 2 часа при комнатной температуре реакционную смесь переносят в ледяную воду. Осадок отфильтровывают и после сушки в вакууме при 50oC получают 7,7 г (54,3,%) фенил[2-[4-(4-метоксифенил)-1-пиперазинил]-5-пиримидинил]карбамата (промеж. соед. 26).
d) Смесь 7,7 г промежуточного соединения (26), 10 мл моногидрата гидразина и 60 мл 1,4-диоксана перемешивают при комнатной температуре примерно сутки. Реакционную смесь переносят в воду, осадок отфильтровывают и промывают метанолом. Продукт очищают колоночной хроматографией (элюент: CH2Cl2-CH3OH, 96 : 4). Элюент в целевых фракциях испаряют, остаток перемешивают в метаноле, фильтруют и после сушки в вакууме при 75oC получают 1,5 г (23%) N-[2-[4-(4-метоксифенил)- 1-пиперазинил]-5-пиридинил]гидразин-карбоксамида, т. пл. > 300oC (разл.) (промеж. соед. 27).
e) Смесь 3 г промежуточного соединения (27), 3,7 г ацетата метанимидамида и 35 мл 1-бутанола кипятят с перемешиванием примерно сутки. Реакционную смесь охлаждают, осадок отфильтровывают, промывают 2,2'-оксибиспропаном и после сушки в вакууме при 70oC получают 2 г (62,9%) 2,4-дигидро-4-[2-[4-(4-метоксифенил)-1-пиперазинил] -5- пиримидинил]-3H-1,2,4-триазол-3-она, т. пл. 272,5oC (промеж. соед. 28).
Пример 8.
a) Смесь 6-метокси-3-пиримидинамида (0,2 моля) и 2-хлор-N-(2-хлорэтил)этанамина гидрохлорида (0,3 моля) в 2-бутанола (500 мл) кипятят с перемешиванием. После добавления порциями (20 г/ч) карбоната калия (0,7 моля) смесь кипятят 48 часов, добавляют дополнительное количество карбоната калия (30 г) и смесь перемешивают 48 часов. Смесь переносят в воду, экстрагируют CH2Cl2 и разделяют. Органический слой сушат, фильтруют и после испарения получают 38 г (98%) 1-(6-метокси-3-пиридинил)пиперазина (промеж. соед. 29).
b) Смесь промежуточного соединения (29) (0,2 моля) и карбоната калия (0,5 моля) в N,N-диметилацетамиде (500 мл) перемешивают при комнатной температуре. По каплям прибавляют 1-фтор-4-нитробензол (0,24 моля) и смесь перемешивают примерно сутки. Смесь переносят в воду и фильтрованием осадка получают 16,3 г (30 %) 1-(6-метокси-3-пиридинил)-4-(4-нитрофенил)пиперазина (промеж. соед. 30).
Аналогичным путем, но используя в качестве растворителя бис(2-метоксиэтиловый) эфир синтезированы:
1-(3-метоксифенил)-4-(5-нитро-2-пиридинил)пиперазин (промеж. соед. 31);
2-[4-(3-метоксифенил)-1-пиперидинил] -5-нитропиридин, т. пл. 147,3oC (промеж. соед. 32) и
1-(2-метоксифенил)-4-(5-нитро-2-пиридинил)пиперазин (промеж. соед. 33).
c) Промежуточное соединение (30) (0,042 моля) в N,N-диметилформамиде (500 мл) гидрируют примерно сутки при 60oC с применением в качестве катализатора никеля Ренея (6 г). После поглощения 3 экв. водорода катализатор отфильтровывают и фильтрат испаряют. Ополаскиванием остатка 2,2'-оксибиспропаном получают 11,5 г (96%) продукта. Перекристаллизацией образца продукта (0,5 г) из 2,2'-оксибиспропана получают 0,3 г 4-[4-(6-метокси-3-пиридинил)-1- пиперазинил]бензоламина, т. пл. 150oC (промеж. соед. 34).
Аналогичным путем синтезированы:
6-[4-(3-метоксифенил)-1-пиперазинил] -3-пиридинамин, т. пл. 110,6oC (промеж. соед. 35);
6-[4-(3-метоксифенил)-1-пиперидинил] -3-пиридинамин, т. пл. 124,9oC (промеж. соед. 36) и
6-[4-(2-метоксифенил)-1-пиперазинил]-3-пиридинамин (промеж. соед. 37).
d) Смесь промежуточного соединения (34) (0,025 моля) и этил-2-[(диметиламино)метилен] гидразинкарбоксилата (0,075 моля) в тетрагидротиофен-1,1-диоксиде (10 мл) перемешивают 5 часов при 150oC. Добавляют смесь 2-пропанола с 2,2'-оксибиспропаном (50 : 50), осадок отфильтровывают, промывают и после сушки получают 6,5 г (74%) 2,4-дигидро-4-[4-[4-(6-метокси-3-пиридинил)-1-пиперазинил]фенил]-3H-1,2,4-триазол-3-она (промеж. соед. 38).
Аналогичным путем синтезированы:
2,4-дигидро-4-[6-[4-(3-метоксифенил)-1-пиперазинил] -3-пиридинил] -3H-1,2,4-триазол-3-он, т. пл. 242,6oC (промеж. соед. 39);
2,4-дигидро-4-[6-[4-(3-метоксифенил)-1-пиперидинил] -3- пиридинил]-3H-1,2,4-триазол-3-он (промеж. соед. 40) и
2,4-дигидро-4-[6-[4-(2-метоксифенил)-1-пиперазинил] -3- пиридинил]-3H-1,2,4-триазол-3-он (промеж. соед. 41).
Пример 9.
a) Смесь 10 г гидрохлорида 4-метокси-3,5-диметилбензоламина, 19,9 г N, N-бис(2-хлорэтил)-4-метилбензолсульфонамида, 16,8 г карбоната натрия. 0,5 г йодида калия и 100 мл циклогексанола перемешивают примерно сутки при 150 - 160oC. После охлаждения реакционную смесь переносят в воду и продукт экстрагируют дихлорметаном. Экстракт промывают водой, сушат, фильтруют и испаряют. Остаток очищают колоночной хроматографией на силикагеле с применением в качестве элюента трихлорметана. Чистые фракции собирают и растворитель испаряют. Кристаллизацией остатка из 1-бутанола получают 10 г (50,4%) 4-(4-метокси-3,5-диметилфенил)-1-[(4-метилфенил) сульфонил] пиперазина, т. пл. 174,2oC (промеж. соед. 42).
b) Смесь 77,6 г промежуточного соединения (42), 121,2 мл концентрированной серной кислоты и 140 мл воды кипятят с перемешиванием примерно сутки. Еще одну порцию в 30,4 г концентрированной серной кислоты добавляют и кипячение с перемешиванием продолжают еще примерно сутки. Реакционную смесь охлаждают и обрабатывают гидроксидом натрия. Продукт экстрагируют дихлорметаном. Экстракт промывают водой, сушат, фильтруют и испаряют. Остаток превращают в соль с хлористоводородной кислотой в 2-пропаноле и 2,2'-оксибиспропане. Соль фильтруют и кристаллизацией из 2-пропанола получают 18,5 г 1-(4-метокси-3,5-диметилфенил)пиперазина дигидрохлорида (промеж. соед. 43).
c) Смесь 14,1 г 1-фтор-4-нитробензола, 26 г промежуточного соединения (43), 15 г карбоната натрия и 272,7 мл диметилсульфоксида перемешивают 4 часа при 60oC. Реакционную смесь охлаждают и переносят в воду. Продукты экстрагируют метилбензолом. Экстракт промывают водой, сушат, фильтруют и испаряют. Остаток ополаскивают 2,2-оксибиспропаном. Продукт фильтруют и кристаллизацией из 2-пропанола получают 17,5 г (64,1%) 1-(4-метокси-3,5-диметилфенил)-4-(4-нитрофенил)пиперазина, т. пл. 135,3oC (промеж. соед. 44).
d) Смесь 15 г промежуточного соединения (44), 1 мл 4%-го раствор тиофена в метаноле и 202,5 мл метанола гидрируют под нормальным давлением и при 50oC в присутствии в качестве катализатора 10% палладия на угле (2 г). После поглощения расчетного количества водорода катализатор фильтруют и фильтрат испаряют. Остаток превращают в 2-пропаноле в гидрохлорид. Соль фильтруют и после сушки получают 15,7 г (92,8%) дигидрохлорида 4-[4-(4-метокси-3,5-диметилфенил)-1-пиперазинил]бензоламина, т. пл. 289,5oC (промеж. соед. 45).
e) Смесь 65 г промежуточного соединения (45), 39,3 г (1-этоксиэтилиденгидразинкарбоновой кислоты этилового эфира и 100 мл тетрагидротиофен-1,1-диоксида перемешивают 3 часа при 120oC в атмосфере азота. Смесь охлаждают и добавляют 2-пропанол. Образовавшийся осадок отфильтровывают и сушат. Остаток очищают колоночной хроматографией (элюент: CHCl3-CH3OH, 99 : 1). Элюент в целевых фракциях испаряют и кристаллизацией остатка из 1,4-диоксана получают 36,75 г (44,9%) 2,4-дигидро-4-[4-[4-(4-метокси-3,5-диметилфенил)-1-пиперазинил] фенил] -5-метил-3H-1,2,4-триазол-3-она, т. пл. 280,1oC (промеж. соед. 46).
B. Получение целевых соединений
Пример 10.
Смесь 2,4-дигидро-4-[6-[4-(4-метоксифенил)-1-пиперазинил]-3- пиридинил] -3H-1,2,4-триазол-3-она (0,014 моля) и карбоната натрия (0,06 моля) в метилбензоле (30 мл) и N,N-диметилформамиде (70 мл) кипятят с перемешиванием. По каплям прибавляют 2-бром-1-(4-хлорфенил)-1-бутанон (0,015 моля) в трихлорметане (20 мл) и смесь кипятят с перемешиванием 1 час с ловушкой для воды. Смесь фильтруют в теплом состоянии и фильтрат испаряют. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2-CH3OH, 98 : 2). Чистые фракции собирают и испаряют. Кристаллизацией остатка из 2-пропанола получают 6 г (80%) (±)-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[6-[4-(4-метоксифенил)-1-пиперазинил] -3-пиридинил] -3H-1,2,4-триазол-3-она, т. пл. 142,4oC (соед. 1).
Аналогичным путем синтезированы:
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[5-[4-(4- метоксифенил)-1-пиперазинил]-2-пиридинил]-3H-1,2,4-триазол-3-он, т. пл. 127,2oC (соед 2);
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[4-[4- (5-метокси-2-пиридинил)-1-пиперазинил] фенил-3H-1,2,4-триазол-3-он, т. пл. 165,1oC (соед. 3);
(±)-2-[1-(4-хлорбензоил)пропил]-4-[6-[4-(3-метоксифенил)-1- пиперазинил] -3-пиридинил]-2,4-дигидро-3H-1,2,4-триазол-3-он, т. пл. 128,3oC (соед. 4);
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(2- метоксифенил)-1-пиперазинил]-3-пиридинил]-3H-1,2,4-триазол-3-он, т. пл. 195,6oC (соед. 5);
(±)-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[4-[4-(6- метокси-3-пиридинил)-1-пиперазинил]фенил]-3H-1,2,4-триазол-3-он, т. пл. 188,4oC (соед. 6);
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4[6-[4-(3- метоксифенил)-1-пиперидинил]-3-пиридинил]-3H-1,2,4-триазол-3-он, т. пл. 125,7oC (соед. 7);
(±)-2-[1-(4-хлорбензоил)пропил] -4-[6-[4-(5-метокси-2- пиридинил)-1-пиперазинил] -3-пиридинил] -2,4-дигидро -3H-1,2,4-триазол-3-он, т. пл. 145,9oC (соед. 37);
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[4-(4- фенил-1-пиперазинил)фенил]-3H-1,2,4-триазол-3-он, т. пл. 180,7oC (соед. 39);
(±)-2-[1-(4-хлорбензоил)пропил] -4-[6-[4-(2-хлорфенил)-1- пиперазинил]-3-пиридинил]-2,4-дигидро-3H-1,2,4-триазол-3-он, (соед. 41);
±-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[6-[4-[3- (трифторметил)фенил] -1-пиперазинил]-3-пиридинил]-3H-1,2,4- триазол-3-он, т. пл. 126,7oC (соед. 42);
(±)-2-[1-(4-хлорбензоил)пропил-4-[6-[4-(3,5-диметилфенил)- 1-пиперазинил] -3-пиридинил] -2,4-дигидро-3H-1,2,4-триазол-3-она дигидрохлорид (соед. 43);
(±)-2-[1-(4-хлорбензоил)пропил-4-[6-[4-(3-этилфенил)-1- пиперазинил] -3-пиридинил]-2,4-дигидро-3H-1,2,4-триазол-3-она дигидрохлорид (соед. 44);
(±)-2-[1-(4-хлорбензоил)пропил-4-[6-[4-(3,4- диметоксифенил)-1-пиперидинил] -3-пиридинил] -2,4-дигидро- 3H-1,2,4-триазол-3-он, т. пл. 107,7oC (соед. 45) и
(±)-2-[1-(4-хлорбензоил)пропил-4-[6-[4-(3,4- диметоксифенил)-1-пиперазинил] -3-пиридинил] -2,4-дигидро- 3H-1,2,4-триазол-3-она дигидрохлорид, т. пл. 197,3oC (соед. 48).
Пример 11.
Перемешивают 2,4-дигидро-4-[2-[4-(4-метоксифенил)-1-пиперазинил] -5-пиримидинил] -3H-1,2,4-триазол-3-он (0,019 моля) и N,N-диметилформамид (200 мл). По каплям прибавляют под N2 раствор бис(триметилсилил)амида натрия в тетрагидрофуране (1 М, 21 мл) и смесь перемешивают 30 минут при комнатной температуре. По каплям прибавляют 2-бром-1-(4-хлорфенил)-1-бутанон (0,021 моля), растворенный в небольшом количестве N,N-диметилформамида, и смесь перемешивают 3 часа при комнатной температуре. Смесь переносят в воду и экстрагируют дихлорметаном. Органический слой промывают водой, сушат и испаряют. Остаток кристаллизуют из 1-пропанола. Осадок отфильтровывают и после сушки в вакууме при 75oC получают 6 г (60%) (±)-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[2-[4-(4- метоксифенил)-1-пиперазинил] -5-пиримидинил] -3H-1,2,4-триазол-3-она, т. пл. 184,6oC (соед. 8).
Пример 12.
К охлажденному раствору (t = -5oC) 5,4 г 1-[1-(4-нитробензоил)этил]-1,3-дигидро-3-[4-[4-(4-гидроксифенил)-1- пиперазинил]фенил]-2H-имидазол-2-она в 500 мл тетрагидрофурана по каплям прибавляют 20 мл 1 М раствора трис(1-метилэтокси)калийгидробората в тетрагидрофуране. После перемешивания 2 часа при комнатной температуре реакционную смесь разбавляют 1500 мл воды и перемешивание продолжают еще 2 час. Осадок отфильтровывают, промывают водой и сушат. Продукт перекристаллизовывают из 1,4-диоксана. Продукт отфильтровывают и после сушки в вакууме получают 3,4 части (64,5%) [A] + [B]-1-[2-(4-бромфенил)-2-гидрокси-1-метилэтил] -1,3-дигидро-3- [4-[4-(4-гидроксифкенил)-1-пиперазинил]фенил-2H-имидазол-2-она, т. пл. 253,8oC (соед. 9).
Аналогичным путем синтезированы:
±-(R*, R*)-2-[2-(3,4-дихлорфенил)-2-гидрокси-1- метилэтил]-2,4-дигидро-4-[4-[4-(4-гидрокси-3,5-диметилфенил)-1- пиперазинил] фенил] -5-метил-3H-1,2,4-триазол-3-она полугидрат, т. пл. 184,9oC (соед. 10);
1-[2-(4-хлорфенил)-2-гидрокси-1-метилэтил] -1,3-дигидро-3- [4-[4-(4-гидроксифенил)-1-пиперазинил]фенил]-4-метил-2H-имидазол-2-он, т. пл. 245,7oC (соед. 11);
1-[2-(4-хлорфенил)-2-гидрокси-1-метилэтил] -2,3-дигидро-3-[4-[4- (4-гидроксифенил)-1-пиперазинил] фенил]-2H-имидазол-2-он, т. пл. 252,8oC (соед. 12);
(±)-2-[1-[(4-бромфенил)гидроксиметил] -1-метилпропил] - 2,4-дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил]фенил]-5- метил-3H-1,2,4-триазол-3-он, т. пл. 215,1oC (соед. 13);
(±)-2-[1-[(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро- 4-[6-[4-(4-гидроксифенил)-1-пиперазинил] -3-пиридинил] - 3H-1,2,4-триазол-3-он, т. пл. 231,7oC (соед. 14);
(±)-(RR, SS)-2-[1-[(4-хлорфенил)гидроксиметил] пропил] - 2,4-дигидро-4-[4-[4-(5-метокси-2-пиридинил)-1-пиперазинил] фенил] - 3H-1,2,4-триазол-3-он, т. пл. 208,5oC (соед. 15);
(±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил]-2,4-дигидро-4-[6-[4-(3-метоксифенил)-1-пиперазинил] -3- пиридинил]-3H-1,2,4-триазол-3-он, т. пл. 203,8oC (соед. 16);
(±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил-2,4-дигидро-4-[6-[4-(2-метоксифенил)-1-пиперазинил] -3- пиридинил]-3H-1,2,4-триазол-3-он, т. пл. 199,3oC (соед. 17);
(±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил]-2,4-дигидро-4-[6-[4-(3-гидроксифенил)-1-пиперазинил] -3- пиридинил]-3H-1,2,4-триазол-3-он, т. пл. 240,8oC (соед. 18);
(±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил-2,4-дигидро-4-[6-[4-(2-гидроксифенил)-1-пиперазинил] -3- пиридинил]-3H-1,2,4-триазол-3-он, т. пл. 221,6oC (соед. 19);
(±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил]-2,4-дигидро-4-[4-[4-(6-метокси-3-пиридинил)-1-пиперазинил] фенил]-3H-1,2,4-триазол-3-он, т. пл. 209,5oC (соед. 20);
(±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил]-1,3-дигидро-3-[6-[4-(4-метоксифенил)-1-пиперазинил]-3- пиридинил]-2H-имидазол-2-он, т. пл. 192oC (соед. 38) и
(±)-(R*, R*)-2-[1-[(4-хлорфенил) гидроксиметил]пропил]-2,4-дигидро-4-[4-[4-(6-метокси-2- пиридинил)-1-пиперазинил]фенил-3H-1,2,4-триазол-3-он, т. пл. 180,8oC (соед. 46).
Пример 13.
Смесь 2-[2-(4-бромфенил)-1,1-диметил-2-оксоэтил]-2,4- дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил]-3H- 1,2,4-триазол-3-она (0,009 моля) тетрагидробората натрия (0,025 моля) в 1,4 диоксане (100 мл), метаноле (30 мл) и воде (50 мл) перемешивают примерно сутки. После добавления уксусной кислоты (5 мл) и затем воды (500 мл) происходит кристаллизация. Реакционную смесь перемешивают 4 часа при комнатной температуре, осадок отфильтровывают с отсасыванием на воронке Бюхнера и твердое вещество кристаллизуют из 1-бутанола. Осадок отфильтровывают и после сушки получают 4,4 г (87%) (±)-2-[2-(4-бромфенил-2-гидрокси-1,1-диметилэтил] -2,4- дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил] -3H-1,2,4- триазол-3-она, т.пл. 224,5oC (соед. 21).
Аналогичным путем синтезированы:
(±)-2-[2-(4-бромфенил)-2-гидрокси-1,1-диметилэтил] -2,4-дигидро-4- [4-[4-(4-гидроксифенил)-1-пиперазинил]фенил]-5-метил-3H-1,2,4- триазол-3-он, т. пл. > 300oC (соед. 22);
2,4-дигидро-2-[2-гидрокси-1-метил-2-(4-метилфенил)этил] -4-[4-[4- (4-гидроксифенил)-1-пиперазинил] фенил] -3H-1,2,4-триазол-3-он, т. пл. 259,8oC (соед. 33);
(±)-(R*, R*)-2-[2-(4-бромфенил)-2-гидрокси-1-метилэтил] -4-[4-[4- [4-[[(1,1-диметилэтил)диметилсилил] окси] фенил] -1-пиперазинил] фенил]-2,4-дигидро-3H-1,2,4-триазол-3-он, т.пл. 241,3oC (соед. 34);
(±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] бутил]-2,4-дигидро- 4-[6-[4-(3-метоксифенил)-1-пиперазинил] -3-пиридинил] -3H-1,2,4- триазол-3-он, т.пл. 202,4oC (соед. 40) и
(±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил]пропил]-4-[6-[4- (3,4-диметоксифенил)-1-пиперазинил]-3-пиридинил]-2,4-дигидро-3H- 1,2,4-триазол-3-он (соед. 49).
Аналогичным путем, но в присутствии 1-метил-2-пирролидинона вместо 1,4-диоксана синтезирован:
2-[2-(4-бромфенил)-2-гидрокси-1-метилэтил] -2,4-дигидро-4-[6-[4- (4-гидроксифенил)-1-пиперазинил] -3-пиридинил] -5-метил-3H-1,2,4- триазол-3-он, т.пл. 195,1oC (соед. 23).
Пример 14.
К 48%-ной водной бромистоводородной кислоты (100 мл) добавляют несколько кристалликов сульфата натрия с целью обесцвечивания кислоты. Затем добавляют (±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[5-[4-(4-метоксифенил)-1-пиперазинил] -2-пиридинил]-3H-1,2,4-триазол-3-он (0,0047 моля) и смесь кипятят с перемешиванием 2 часа. Смесь переносят в воду и нейтрализуют NH4OH. Осадок отфильтровывают и сушат. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2-Cl2-CH3OH, 98:2). Чистые фракции собирают и после испарения получают 2,2 г (0%) продукта. Кристаллизацией образца продукта (1 г) из C4H9OH получают 0,7 г (±)-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4- [5-[4-(4-гидроксифенил)-1-пиперазинил] -2-пиридинил]-3H-1,2,4-триазол -3-он, т.пл. 185,6oC (соед. 24).
Аналогичным путем синтезированы:
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(4-гидроксифенил) -1-пиперазинил] -3-пиридинил] -3H-1,2,4-триазол-3-он, т. пл. 185,2oC (соед. 25);
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[2-[4-(4-гидроксифенил) -1-пиперазинил] -5-пиримидинил] -3H-1,2,4-триазол-3-он. т.пл. 195,3oC (соед. 26);
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[4-[4-(5- гидрокси)-2-пиридинил)-1-пиперазинил] фенил]-3H-1,2,4-триазол-3-он, т.пл. 193,3oC (соед. 27);
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(3- гидроксифенил)-1-пиперазинил]-3-пиридинил]-3H-1,2,4-триазол-3-он, т.пл. 103,1oC (соед. 28) и
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(2- гидроксифенил)-1-пиперазинил]-3-пиридинил]-3H-1,2,4-триазол-3-он (соед. 29).
Пример 15.
К смеси 25 г трехбромистого бора и 100 мл дихлорметана прибавляют по каплям раствор 6,1 г 1-[1-(4-хлорбензоил)этил]- 1,3-дигидро-3-[4-[4-(4-метоксифенил)-1-пиперазинил]фенил]-4-метил- 2H-имидазол-2-она в 200 мл дихлорметана (t < 10oC). После перемешивания 2 часа реакционную смесь переносят в смесь NH4OH со льдом. Затем добавляют 300 мл дихлорметана и все перемешивают 1 час. Органический слой отделяют, сушат, фильтруют и испаряют. Остаток очищают колоночной хроматографией (силикагель; CH2Cl2-CH3OH, 99:1). Элюент в целевых фракциях испаряют, остаток ополаскивают метанолом, затем фильтруют и после сушки получают 4,3 г (72,3%) 1-[1-(4-хлорбензоил)этил]-1,3-дигидро- 3-[4-[4-(4-гидроксифенил)-1-пиперазинил]фенил]-4-метил-2H-имидазол -2-он, т. пл. 229,7oC (соед. 30).
Аналогичным путем синтезирован:
2,4-дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил]фенил]-2-[1- (4-метоксибензоил)этил]-3H-1,2,4-триазол-3-он, т.пл. 204,5oC (соед. 35).
Пример 16.
Смесь 2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[4-[4-(4- гидроксифенил)-1-пиперазинил] фенил] -3H-1,2,4-триазол-3-она (0,009) и бензилтриэтиламмонийхлорида (0,25) перемешивают в дихлорметане (150 мл). Добавляют 25%-ный раствор гидроксида натрия (50 мл) и смесь перемешивают 10 минут. После добавления диметилсульфата (0,015 моля) смесь перемешивают 1 час при комнатной температуре. Смесь разбавляют водой (100 мл) и слои разделяют. Органический слой промывают водой и сушат. Остаток очищают хроматографией на силикагеле (элюент: CH2Cl2-CH3OH, 98: 2). Чистые фракции собирают и после испарения получают 4,3 г продукта. Продукт дополнительно очищают на колонке, заполненной 1 кг ХИРАЦЕЛЯ OD® (элюент: C2H5OH). Чистые фракции собирают и после испарения получают 0,9 г (+)-(A)-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[4-[4-(4-метоксифенил)-1-пиперазинил] фенил]-3H-1,2,4-триазол-3-она, т.пл. 199,6oC, [α]
Пример 17.
Моногидрат (±)-(R*, R*)-2-[2-(2,4-дихлорфенил)-2-гидрокси-1-метилэтил] -2,4-дигидро- 4-[4-[4-(4-гидрокси-3,5-диметилфенил)-1-пиперазинил] фенил]-5-метил-3H -1,2,4-триазол-3-он (1,5 г) очищают колоночной хроматографией на ХИРАЦЕЛЕ OD® (1 кг) с элюированием смесью н-гексана с 2-пропанолом (80:20). Чистые фракции собирают и испаряют. Кристаллизацией остатка из 2-пропанола получают 0,5 г (+)-(R*, R*)-2-[2-(2,4-дихлорфенил)-2-гидрокси-1-метилэтил] -2,4-дигидро-4- [4-[4-(4-гидрокси-3,5-диметилфенил)-1-пиперазинил] фенил]-5-метил-3H- 1,2,4-триазол-3-она дигидрата, т. пл. 123,5oC, [α]
Пример 18.
В 100 мл дихлорметана при перемешивании растворяют (+)-4-[4-[4- [4-[[(1,1-диметилэтил)диметилсилил] окси] фенил] -1-пиперазинил] фенил]-2,4-дигидро-2-[2-гидрокси-2-[4-(трифторметокси)фенил] этил] -5-метил-3H-1,2,4-триазол-3-он (0,0043 моля). Добавляют раствор тетрабутиламмонийфторида (0,0045 моля) в тетрагидрофуране и смесь перемешивают 15 минут. Затем добавляют воду и смесь перемешивают 30 минут Осадок отфильтровывают, сушат и после кристаллизации из н-бутанола получают 1,1 г (58,2%) (±)-2,4-дигидро-4-[4-[4-(4- гидроксифенил)-1-пиперазинил] фенил] -2-[2-гидрокси-2-[4-(трифторметокси) фенил]этил]-5-метил-3H-1,2,4-триазол-3-она, т.пл. 230,6oC (соед. 36).
Пример 19.
К перемешиваемому раствору 2-[1-(4-бромбензоил)этил]-2,4- дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил]-3H-1,2,4- триазол-3-она (0,009 моля) в пиридине (20 мл) в атмосфере азота добавляют раствор 2-(хлордиметилсилил)-2-метилпропана (0,011 моля) в дихлорметане (20 мл). Перемешивание продолжают 1 неделю при комнатной температуре. Реакционную смесь испаряют и остаток растворяют в дихлорметане. Полученный раствор очищают на силикагеле со стеклянным фильтром. Чистые фракции собирают и растворитель испаряют. Остаток (3,5 г) перекристаллизовывают из этилацетата. Продукт фильтруют и после сушки получают 2,7 г (45%) 2-[1-(4-бромфенил)этил]-4-[4-[4-[4- [[(1,1-диметилэтил)диметилсилил] окси] фенил] -1-пиперазинил]фенил]- 2,4-дигидро-3H-1,2,4-триазол-3-она, т.пл. 177,8oC (соед. 47).
С. Фармакологический пример
Активность соединений изобретения против Helicobacter выявлена применением следующей методики испытаний in vitro.
Активность испытуемых соединений относительно Helicobacter
Активность испытуемых соединений против Helicobacter pylori определялась применением стандартного выбора из 5 штаммов H. pylori, полученного на основе клинического материала. Минимальную ингибирующую концентрацию (МИК) определяют путем измерения активности уреазы H. pylori после обработки растущих культур бактерии противомикробными средствами.
Испытуемые соединений растворяют в концентрации 10-3М в ДМСО. Кроме того, готовят разбавление до 10-4М в ДМСО. Полученные растворы пипеткой переносят в объеме 10 мкл в лунки планшета Репли-Диш (® Стерилин). В каждом планшете в качестве контрольных предусмотрены лунки, содержащие только ДМСО. В каждой серии опытов в качестве ссылочных соединений применяют ампицилин (Тригидрат (+)-6-[(2-амино-2-фенилацетил)амино[-3,3-диметил-7- оксо-4-тиа-1-азабицикло [3.2.0] гептан-2-карбоновой кислоты) и метронидазол (2-метил-5-нитро-1H-имидазол-1-этанол). Указанные соединения применяют в конечных концентрациях в 10-5, 10-6, 10-7 и 10-8М. Планшеты для испытаний хранят при 4oC до момента применения. Пять изолятов H. pylori поддерживают субкультивированием на 10%-ом кровяном агаре каждые 2 или 3 дня. Бактерии выращивают при 37oC в атмосфере, содержащей 5% кислорода, 10% CO2 и 85% азота. Суспензии Helicobacter pylori для инокулума готовят сердечно-мозговом инфузионном бульоне, и суспензии доводят до поглощения 1,5±0,3 при 530 нМ.
Свежеприготовленный 10%-ый кровяной агар, нагретый до 45oC, добавляют в объеме 1 мл в лунки планшетов для испытаний с разбавлением тем самым испытуемые соединения до 10-5 и 10-6М. Среду оставляют охлаждаться, после чего на поверхность агара пипеткой наносят 10 мкл бактериальной суспензии. Планшеты инкубируют 48 часов при 37oC в вышеприведенной микроаэрофильной атмосфере. Для облегчения считывания планшетов и для гарантирования того, что рост на среде связан только с H. pylori, используют характерную для того вида бактерий чрезвычайно высокую уреазную активность. После 48 часов инкубирования в каждую лунку планшета Репли-Диш осторожно вносят по 1 мл бульона уреазы и планшеты инкубируют 2 часа. Затем из каждой лунки пипеткой отбирают 100 мкл жидкого образца и переносят в лунки планшета для микроразбавлений на 96 мест. Пурпурная окраска считается свидетельством роста бактерий, в то время как желто-оранжевая окраска свидетельствует об отсутствии роста H. pylori. Этим методом получают четкую конечную точку, на основании которой может быть определено ингибирующее действие соединений. Все соединения, для которых было показано проявление активности в любой их двух концентраций, подвергнуты дополнительными испытаниями в еще большем разбавлении с определением МИК и с еще более широким спектром видом бактерий в качестве целевых микроорганизмов.
В таблице 1 суммированы данные для величин МИК, определенных по вышеприведенной методике для целого ряда соединений изобретения.
D. Примеры препаратов
Термин "активный компонент" (АК) в значении, применяемом во всех последующих примерах, относится к соединениям формулы (I), к их фармацевтически приемлемым солям с кислотами или к их стереохимическим изомерам.
Пример 20. Пероральные капли
В 0,5 л 2-гидроксипропановой кислоты и 1,5 л полиэтиленгликоля растворяют при 60 - 80oC 500 г А.К. После охлаждения до 30 - 40oC добавляют 35 л полиэтиленгликоля и смесь тщательно перемешивают. Затем добавляют раствор 1750 г сахарина натрия в 2,5 л очищенной воды и при перемешивании добавляют 2,5 л вкусового коррегента какао и полиэтилен в количестве, необходимом для доведения общего объема до 50 л, с получением раствора пероральных капель, содержащего 10 мг/мл А. К. Полученным раствором заполняют соответствующие контейнеры.
Пример 21. Капсулы
Интенсивным перемешиванием образуют смесь 20 г А.К., 6 г лаурилсульфата натрия, 56 г крахмала, 56 г лактозы, 0,8 г коллоидального диоксида кремния и 1,2 г стеарата магния. Полученной смесью заполняют соответствующие твердые желатиновые капсулы (1000 штук), каждая из которых содержит 20 мг активного компонента.
Пример 22. Таблетки с пленочным покрытием.
Получение центральной части таблеток
Смесь 100 г А.К., 570 г лактозы и 200 г крахмала тщательно смешивают, после чего увлажняют раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона примерно в 200 мл воды.
Увлажненную порошковую смесь просеивают, сушат и вновь просеивают. Затем добавляют 100 г микрокристаллической целлюлозы и 15 г гидрированного растительного масла. Все тщательно смешивают и прессуют в таблетки с получением 10000 таблеток, каждая из которых содержит 10 мг активного компонента.
Оболочка
К раствору 10 г метиленцеллюлозы в 75 мл денатурированного этанола добавляют раствор 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. Полиэтиленгликоль (10 г) переводят в расплавленное состояние и растворяют в 75 мл дихлорметана. Последний раствор добавляют к ранее полученному раствору, после чего добавляют 2,5 г октадеканоата магния, 5 г поливинилпирролидона, 30 мл концентрированного раствора красителя и все доводят до гомогенного состояния. В аппаратах для нанесения покрытия на центральных частях таблеток создают оболочку из полученной в результате смеси.
Пример 23. Инъектируемый раствор
Примерно в 0,5 о кипящей воды для инъекций растворяют 1,8 г метилового эфира 4-гидроксибензойной кислоты и 0,2 г пропилового эфира той же кислоты. После охлаждения примерно до 50oC при перемешивании добавляют 4 г молочной кислоты, 0,05 г пропиленгликоля и 4 г А.К. Раствор охлаждают до комнатной температуры и к нему добавляют воду для инъекций в количестве, необходимом для доведения общего объема до 1 л, с получением раствора, содержащего 4 мг/мл А.К. Раствор стерилизуют фильтрованием и им заполняют стерильные контейнеры.
Пример 24. Суппозиторий
В растворе 3 г 2,3-дигидроксибутандиовой кислоты в 25 мл полиэтиленгликоля 400 растворяют 3 г А.К. Совместно плавят 12 г поверхностно-активного вещества и триглицерид в количестве, необходимом для получения в общей сложности 300 г. Полученную в результате смесь разливают в формы при температуре 37 - 38oC с приготовлением 100 суппозиториев, каждый из которых содержит 30 мг/мл А.К.
Пример 25. Содержащий циклодекстрин препарат
Пропиленгликоль (100 мл) обрабатывают 3,76 концентрированной HCl, перемешивают и слегка нагревают. Добавляют 10 г А.К. и перемешивание продолжают до гомогенного состояния. В отдельных сосуде в 400 мл дистиллированной воды растворяют 400 г гидроксипропил -β- циклодекстрина. К перемешиваемому раствору активного компонента медленно прибавляют раствор циклодекстрина. Добавляют раствор сорбита (190 мл) и перемешивают до гомогенного состояния. В 50 мл дистиллированной воды растворяют сахарин натрия (0,6 г) и полученный раствор добавляют к ранее приготовленной смеси. Добавляют корррегенты и pH смеси (примерно 1,7) добавленным 10 н. NaOH устанавливают равным 2,0±0,1. Полученный раствор разбавляют дистиллированной водой до конечного объема в 1 литр. Фармацевтические дозированные формы получают фильтрованием полученного раствора и заполнением им приемлемых контейнеров, например, стеклянных бутылочек на 100 мл с завинчивающимися крышками.

Для получения лекарственных средств, предназначенных для лечения связанных с Helicobacter заболеваний, применяют соединения формулы (I), их фармацевтически приемлемые соли с кислотами или их стереохимические изомеры, где Х и У каждый независимо представляет СH или N; R1; R2 и R3 каждый независимо представляет водород или С1-С4-алкил; R4 и R5 каждый независимо представляет водород, галоген, С1-С4-алкил, С1-С4-алкилоксигруппу, гидроксигруппу, трифторметил, трифторметилоксигруппу или дифторметилоксигруппу; Z - представляет С=О или СНОН и Ar представляет фенил, возможно замещенный вплоть до трех заместителями выбранными из гидроксигруппы, С1-С4-алкила, С1-С4-алкилоксигруппы, галогена, трифторметила, три(С1-С4-алкил)силилоксигруппы, или пиридинил, замещенный гидроксигруппой и С1-С4-алкилоксигруппой, и (А) представляет радикал формул (а-1), (а-2), (а-3), (а-4), (а-5). Описывается способ их получения, а также фармацевтический препарат, содержащий в качестве активного компонента замещенные производные азолона формулы (I) и способ его получения. 4 c. и 1 з.п. ф-лы, 1 табл.
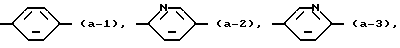
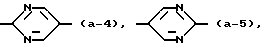
их фармацевтически приемлемые соли с кислотой или стереохимические изомеры,
где X и Y каждый независимо представляет CH или N;
R1, R2 и R3 каждый независимо представляет водород или C1-C4-алкил;
R4 и R5 каждый независимо представляет водород, галоген, C1-C4-алкил, C1-C4-алкилоксигруппу, гидроксигруппу, трифторметил, трифторметилоксигруппу или дифторметилоксигруппу;
Z представляет C=O или CHOH;
Ar представляет фенил, возможно замещенный вплоть до трех заместителями, выбранными из гидроксигруппы, C1-C4-алкила, C1-C4-алкилоксигруппы, галогена, трифторметила, три(C1-C4-алкил) силилоксигруппы, или пиридинил, замещенный гидроксигруппой или C1-C4-алкилоксигруппой,
группа,
представляет радикал формулы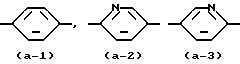
при условии, что Ar отличен от 4-гидроксифенила, три(C1-C4-алкил)-4-гидроксифенила, 3,5-ди(C1-C4-алкил)-4-гидроксифенила или 4-метоксифенила, когда X = N и группа
представляет радикал формулы (а-1).
[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(4-метоксифенил)-1-пиперизинил]-3-пиридинил]-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[6-[4-(3-гидроксифенил)-1-пиперазинил]-3-пиридинил]-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4-[6-[4-(3-гидроксифенил)-1-пиперазинил]-3-пиридинил]-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4-[4-[4-(6-метокси-3-пиридинил)-1-пиперазинил]-фенил]-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорбензоил)пропил]дигидро-4-[6-[4-(3-метоксифенил)-1-пиперазинил]-3-пиридинл]-2,4-дигидро-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4-[6-[4-(3-метоксифенил)-1-пиперазинил]-3-пиридинил]-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорбензоил)пропил] -4-[6-[4-(3,4-диметоксифенил)-1-пиперидинил] -3-пиридинил]-2,4-дигидро-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4-[4-[4-(6-метокси-2-пиридинил)-1- пиперазинил]-фенил]-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорфенил)гидроксиметил] пропил] -4-[6-[4-(3,4-диметоксифенил)-1-пиперазинил]-3-пиридинил]-2,4-дигидро-3H-1,2,4-триазол-3-он;
2-[1-(4-хлорбензоил)пропил] -4-[6-[4-(3,4-диметоксифенил)-1-пиперазинил] -3-пиридинил]-2,4-дигидро-3H-1,2,4-триазол-3-он,
их фармацевтически приемлемые соли с кислотами или их стереохимические изомеры.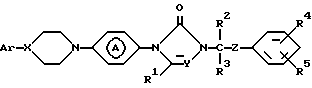
где Ar, X, R1, R2, R3, R4, R5, Z и группа
принимают значения по п.1, отличающийся тем, что промежуточное соединение формулы (II) N-алкилируют реагентом формулы (III)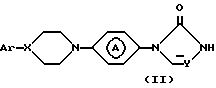
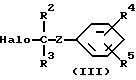
в инертном в условиях реакции растворителе и в присутствии основания с последующим при желании превращением соединений формулы (I) друг в друга путем превращения функциональных групп, превращением соединений формулы (I) в их соли с кислотами обработкой фармацевтически приемлемыми кислотами или наоборот превращением солей в свободные основания обработкой щелочью и/или получением стереохимических изомеров.
| ХОЛОДИЛЬНАЯ УСТАНОВКАВСЕСОЮЗНАЯS •(• ,ч,г-.,-!--';;л ^TVIIi ii:-' «;asin;«-lt-.sHi. | 0 |
|
SU331232A1 |
| EP 0228125 А, 1987 | |||
| SU, 604496, 1978 | |||
| SU, 492085, 1976 | |||
| SU, 503516, 1977 | |||
| H | |||
| Ravtelin, clu vitro activity of autifungal azolos against heliobacter pylori | |||
| EVR | |||
| J | |||
| Clin | |||
| Microbiol | |||
| Jnfect | |||
| dis, 1992, 11(3), 273-4. | |||
