Настоящее изобретение относится к новому применению макролидных соединений, таких как санглиферины, которые связываются с циклофилинами.
Санглиферины представляют собой класс полициклических макролидных соединений, которые выделяют из ферментационной культуральной среды актиномицетов. Установлено, что санглиферины характеризуются активностью при связывании с циклофилинами, а санглиферины A-D проявляют иммунодепрессантную и ингибирующую активность в отношении пролиферации В- и Т-клеток. Однако в отличие от известных иммунодепрессантантов и противовоспалительных соединений, таких как циклоспорин А и FK506, санглиферины не обладают активностью в отношении связывания FKBP или ингибирования кальциневрина. Таким образом, санглиферины относятся к новой категории лекарственных средств как с точки зрения химической структуры, так и различных профилей активности.
Санглиферины и способы их получения описаны в документах WO 97/02285, WO 98/07743 и US 006124453. Ряд производных санглиферина, минисанглиферины, описаны в статьях J.Am.Chem.Soc., т.125, №13, сс.3849-3859 (2003), J.Org.Chem., т.65, сс.9255-9260 (2000), и Angew. Chem. Int. Ed., т.38, №16, сс.2443-2446 (1999).
Неожиданно было установлено, что санглиферины, связывающиеся с циклофилином, проявляют ингибирующую активность в отношении вируса гепатита С (ВГС).
Соответственно, в настоящем изобретении предлагается применение соединения, связывающегося с циклофилином, такого как санглиферин, для профилактики или лечения инфекций, вызванных вирусом гепатита С, нарушений, вызванных ВГС, или ингибиторов пептидилпролил-цис-транс-изомеразы, или состояний, связанных с заболеваниями печени. Агенты можно использовать также, например, для профилактического лечения новорожденных, родившихся у матерей, инфицированных ВГС, или для профилактики персонала, подвергающегося опасности заражения вирусом, или для лечения пациентов после пересадки органов, например пациентов с трансплантом печени, чтобы исключить возможностьповторного заражения ВГС после трансплантации.
Настоящее изобретение относится к применению макролидов (так называемые санглиферины) для профилактики или лечения инфекций, вызванных вирусом гепатита С. Санглиферины выделяют из ферментационной культуральной среды актиномицетов. Стандартные санглиферины включают санглиферины А-D, например, соединение формулы
Макроциклическое кольцо в составе саглиферинов А-D характеризуется тем, что 1) в положениях 1-6 находится остаток 3-карбоксипиперидазинилкарбоновой кислоты, 2) в положениях 7-9 находится остаток ароматической α-аминокислоты и 3) в положениях 10-12 находится остаток алифатической α-аминокислоты. Остальная часть макроциклического кольца содержит остаток гидроксикарбоновой кислоты при условии, что в случае санглиферинов А-D в исходном макроциклическом кольце содержится 11 дополнительных атомов углерода.
Стандартная нумерация макроциклического кольца, принятая в химии макролидов, показана выше для санглиферина А, причем нумерация начинается с атома углерода в карбонильной группе макроциклического лактона в мостиковой связи (положение 1).
Санглиферины А-D содержат также новую бициклическую спиросистему, присоединенную в положении 23 макроциклического кольца через гидрокарбильную связующую группу.
Для получения других макролидов класса санглиферинов санглиферины А-D модифицируют в различных условиях модификации. Такие химические реакции включают отщепление макроциклического кольца, прежде всего по оксигруппе в составе лактона, отщепление связующей группы между макролидом и спироциклической системой, а также модификацию, например введение защитных групп, или другую химическую модификацию заместителей, например, как описано в данном контексте. Другие способы модификации известны специалистам в данной области техники.
В настоящем изобретении установлено, что санглиферины обладают специфической и абсолютно новой биологической активностью в отношении инфекций, вызванных ВГС. Прежде всего, было установлено, что они одновременно проявляют комбинацию различных видов активности, как описано в данном контексте.
Санглиферины характеризуются профилем активности, который отличается от профилей активностей известных иммунодепрессантов и противовоспалительных соединений, таких как циклоспорины и макролиды, например рапамицин и NIM811, что свидетельствует о том, что механизм действия санглиферинов отличается от механизма действия указанных соединений. Таким образом, санглиферины являются новым классом лекарственных средств как с точки зрения структуры, так и с точки зрения активности, что позволяет значительно расширить спектр действия иммунотерапии, противовоспалительной или противовирусной терапии, например исключить или снизить нежелательные побочные эффекты при лечении иммунодепрессантами или противовоспалительными агентами, описанными в предшествующем уровне техники, и/или улучшить или расширить применение такой терапии для лечения новых видов заболевания или новых категорий пациентов.
Санглиферины, в которых, например, макролидное кольцо является незамкнутым, и в положениях 26 и 27 углеводородной связующеей группы между макролидом и спироциклической системой находятся гидроксигруппы, или в котором спироостаток, присоединенный к макролидному циклу, отщеплен или укорочен, в основном не проявляют некоторых или всех видов активности, характерных для санглиферина. Например, санглиферины с отщепленным спироостатком связываются с циклофилином, но не проявляют важную иммунодепрессантную активность. Однако, как известно специалистам в данной области техники, такие соединения можно использовать в качестве ценных компонентов, промежуточных соединений или главных фрагментов, необходимых для получения новых санглиферинов, они, следовательно, могут найти применение при разработке лекарственных средств класса санглиферинов.
Таким образом, наличие такой структуры обеспечивает биологическую активность материала, например санглиферинов А-D, а бициклическую спиросистему можно рассматривать как важный структурный компонент, который можно использовать для дальнейшей модификации или для получения других производных санглиферинов или для модификации других лекарственных средств, например для модификации активности других иммунодепрессантов класса макролидов.
В первом объекте настоящего изобретения предлагается макролид, в котором 1) в положениях 2-6 макролидного цикла находятся остатки пиперидазинилкарбоновой кислоты и/или 2) в положениях 7-9 макролидного кольца находится остаток ароматической α-аминокислоты и/или 3) в положениях 10-12 макролидного цикла находится остаток алифатической α-аминокислоты, в свободной или защищенной форме или в форме соли.
Пригодные макролиды по настоящему изобретению характеризуются двумя, прежде всего всеми тремя структурными признаками 1), 2) и 3).
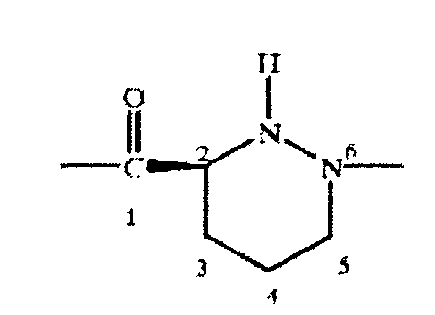
Пригодным остатком пиперидазинилкарбоновой кислоты является остаток 1,2-пиперидазин-3-карбокси-1-ил, карбоксильная группа в котором расположена в положении 1, а первый атом азота в положении 6 макролидного цикла, т.е. остаток формулы I

где указанные номера означают положения атомов в макроциклическом кольце. Указанный остаток может содержать или не содержать заместители в цикле. Пригодным является незамещенный остаток.
Карбоксильная группа в составе остатка ароматической α-аминокислоты обычно расположена в положении 7 макроциклического кольца, а α-аминогруппа - в положении 9. Пригодной ароматической α-аминокислотой являются замещенный или незамещенный фенилаланин, прежде всего содержащий заместители гидрокси, метокси, этокси, OnPr, OiPr, в свободной, защищенной или активированной форме.
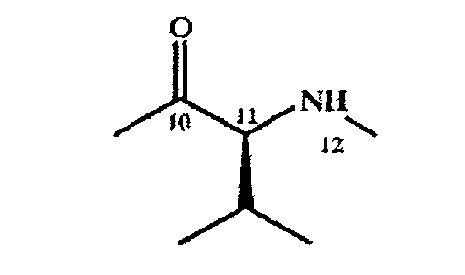
Карбоксильная группа в составе остатка алифатической α-аминокислоты обычно расположена в положении 10 макроциклического кольца, а α-аминогруппа- в положении 12. Пригодной ароматической α-аминокислотой является остаток валина в свободной, защищенной или активированной форме.
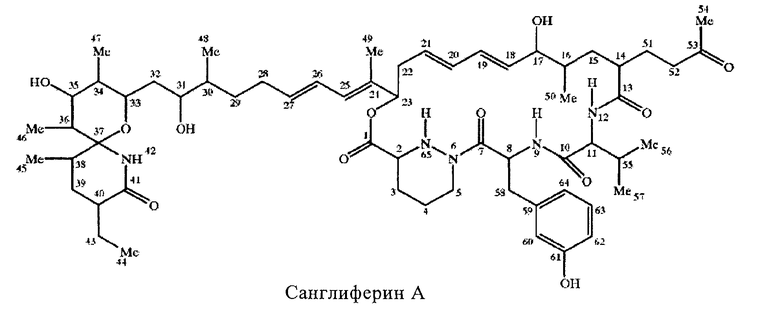
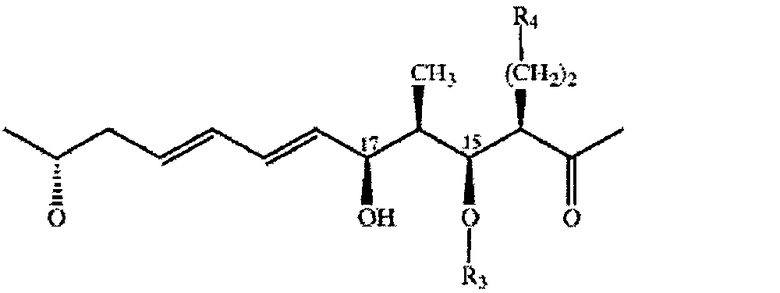
Остальная часть макроциклического кольца прежде всего включает остаток гидроксикарбоновой кислоты, оксигруппу, расположенную а концевой части макроциклической лактонной группы, и карбонильную группу, которая образует амидную связь с α-аминогруппой в положении 12 макроциклического кольца. Длина цепи указанного остатка гидроксикарбоновой кислоты прежде всего составляет от 6 до 20, более предпочтительно 11, атомов углерода. Указанный остаток является замещенным или незамещенным и/или содержит одну или более ненасыщенных связей прежде всего в виде чередующихся двойных связей вдоль цепи. Предпочтительная остальная часть макроциклического кольца включает остатки 11-оксиундеканоил-11-ил, прежде всего 11-оксиундека-6,8-диеноил-11-ил, необязательно замещенные, например, в положениях 2, 3, 4 и/или 5. Более пригодным остатком указанной гидроксикарбоновой кислоты является остаток формулы II
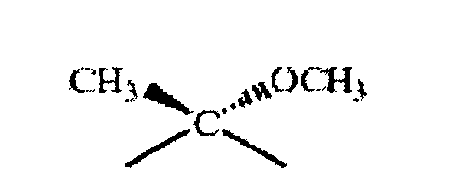
где R1 означает Н, ОН или дополнительную связь, R2 означает Н или дополнительную связь, R3 означает Н, R4 означает -СО-СН3 или -СН(ОН)- СН3 или R3 и R4 вместе означают остаток формулы III

в свободной или защищенной форме или в форме соли. Предпочтительными макролидами по настоящему изобретению являются макролиды, содержащие макроциклическое кольцо формулы IV

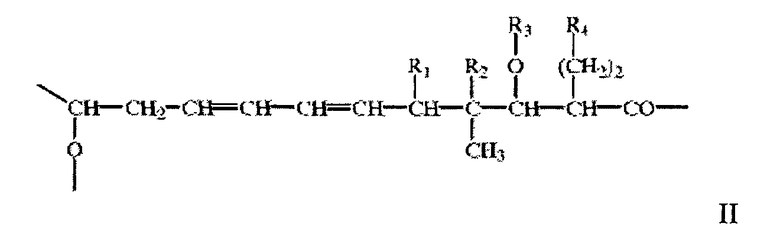
где X, Y и Z означают остатки 1), 2) и 3), значения которых описаны выше, а А означает остаток гидроксикарбоновой кислоты, описанный выше, в свободной или защищенной форме или в форме соли, прежде всего макроциклическое кольцо формулы V
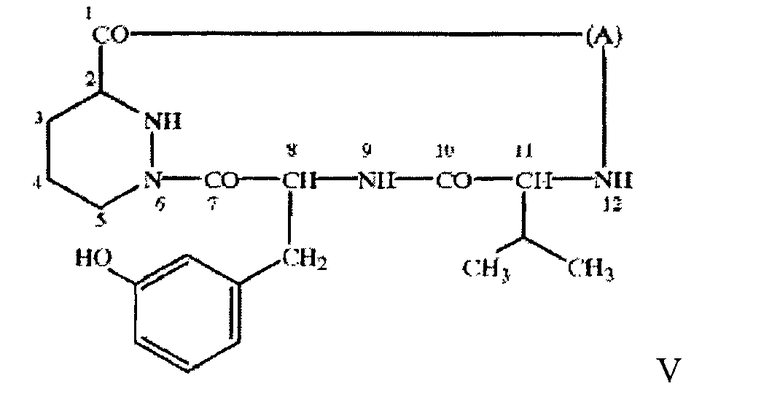
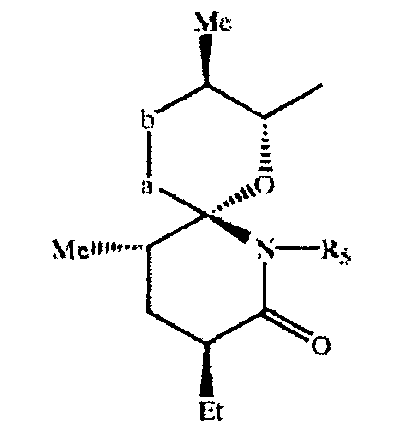
в свободной или защищенной форме или в форме соли. В большинстве случаев в санглиферинах макроциклическое кольцо является замещенным по атому углерода, соседнему с оксигруппой в составе лактонной мостиковой связи. В большинстве случаев таким заместителем является остаток 1-оксо-7-азаспиро-{5.5}-ундекан-8-он-2-ил, например, формулы VI
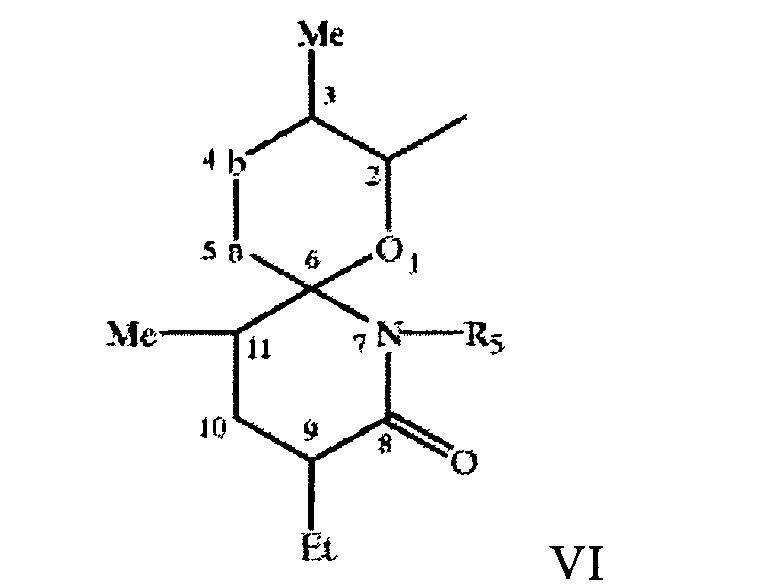
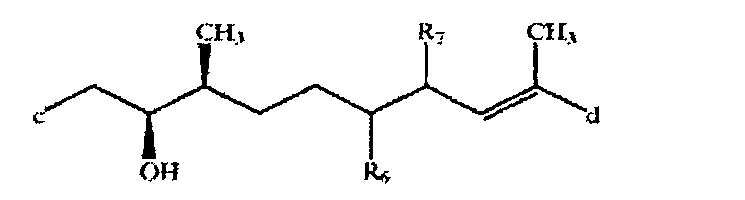
где -а-b- означает - (Ме)С=СН- или -(Ме)СН-СН(ОН)-, a R означает Н или Me (где Me и Et означают метил и этил соответственно) в свободной или защищенной форме, или соли указанных соединений, причем указанный остаток связан с макролидным кольцом через связующую группу, включающую линейную цепь из 6-11, прежде всего 9, атомов углерода между спироостатком и макролидным кольцом.
Связующая группа является замещенной или незамещенной и/или содержит одну или более ненасыщенных связей прежде всего в виде чередующихся двойных связей вдоль цепи. Пригодная связующая группа замещена метильной группой, например двумя метильными группами. Пригодная связующая группа замещена гидроксигруппами, например тремя гидроксигруппами, и/или является этиленненасыщенной, например, содержит 2 углерод-углерод двойные связи. Более пригодная связующая группа содержит остаток 1,7-диметилнонан-9-ил, прежде всего 1,7-диметилнон-1-ен-9-ил или 1,7-диметилнона-1,3-диен-9-ил, необязательно замещенный, например, в положениях 3, 4 и/или 8. Предпочтительная связующая группа является группой формулы VII

где с означает связь с спироостатком, d означает связь с макроциклическим кольцом, a R6 и R7 каждый означает ОН или вместе означают дополнительную связь, в свободной или защищенной форме.
Связующая группа в большинстве случаев присоединена к макроциклическому кольцу через атом углерода, соседний с лактонной оксигруппой, то есть, если макроциклическое кольцо содержит остаток 11-оксиундеканоил-11-ил, то в положении 11 указанного цикла.
В настоящем изобретении предлагаются соединения формулы VIII
S--L-M VIII
где S означает спиробициклогруппу, как описано выше, L означает связующую группу, как описано выше, а М означает макролидное кольцо, значение которого описано выше, в свободной или защищенной форме, или соли указанных соединений.
Предпочтительными соединениями по настоящему изобретению являются соединения формул IХа и IX
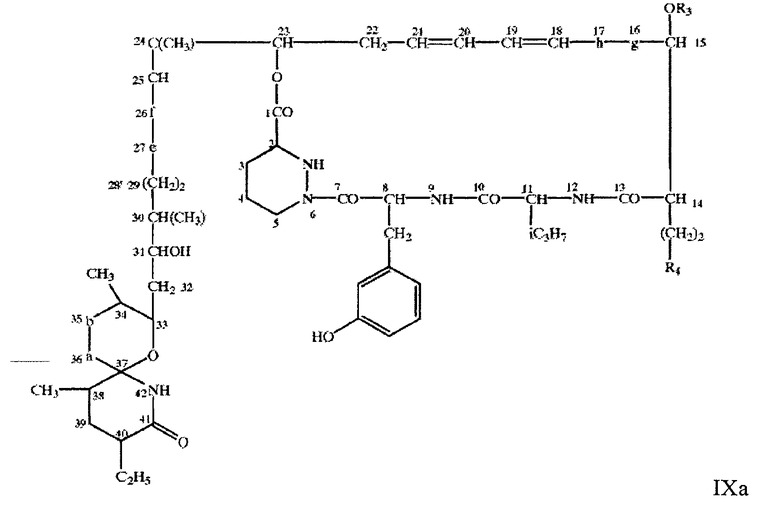
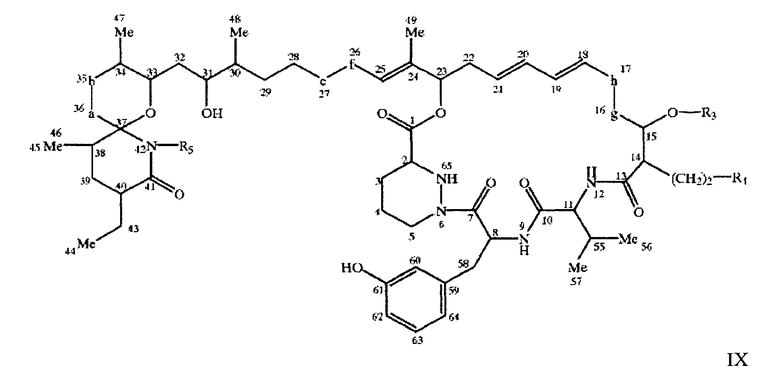
где -a-b- имеет значение, описанное выше, -e-f- означает -СН(ОН)- СН(ОН)- или -СН=СН-, -g-h-имеет значение, описанное выше для -a-b-, a R3, R4 и R5 имеют значения, описанные выше, в свободной или защищенной форме, или соли указанных выше соединений.
Соединения формул I - IX содержат асимметричные атомы углерода и поэтому могут существовать в виде различных эпимерных форм. Все возможные эпимеры, а также их диастереомерные смеси включены в объем настоящего изобретения. Однако соединения формул VIII и IX, в которых макролидное кольцо замкнуто и которые характеризуются соответствующей стереохимией, проявляют активность, характерную для санглиферинов, как описано в данном контексте. Предпочтительными являются эпимеры, проявляющие активность, характерную для санглиферина. В большинстве случаев предпочтительными, например, для применения в фармацевтике по настоящему изобретению, являются эпимеры, проявляющие виды активности, характерные для санглиферинов, в основном содержащие индивидуальный изомер или в форме частично индивидуальных изомеров (т.е. не содержащие или в основном не содержащие эпимеры, не обладающие характерной для санглиферинов активностью), например, содержащие по крайней мере 90%, например, по крайней мере 95% активного эпимера (т.е. содержащие менее 10%, например менее 5% неактивного эпимера).
Предпочтительный остаток 3-карбоксипиперидазинилкарбоновой кислоты 1) в положениях 1-6 макроциклического кольца характеризуется следующей конформацией
Предпочтительная ароматическая аминокислота 2) в положениях 7-9 макроциклического кольца характеризуется конфигурацией L, например следующей конфигурацией
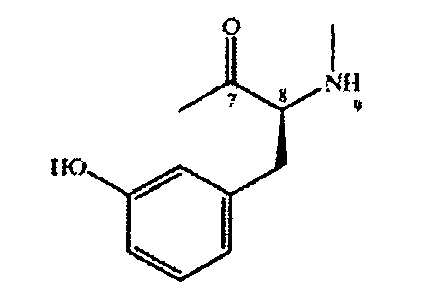
Предпочтительная алифатическая аминокислота 3) в положениях 10-12 макроциклического кольца характеризуется конфигурацией L, например следующей конфигурацией
Если остальной фрагмент макроциклического кольца содержит остаток формулы II, то он предпочтительно характеризуется конфигурацией
или

если R3 и R4 вместе характеризуются конфигурацией
предпочтительный остаток 1 -оксо-7-азаспиро-{5.5}-ундекан-8-он-2-ила характеризуется конфигурацией
где если -а-b- означает -(Ме)СН-СН(ОН)-, то он предпочтительно характеризуется конфигурацией
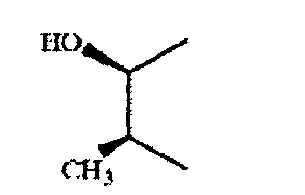
Если связующая группа характеризуется формулой VII, то соединение предпочтительно имеет конфигурацию
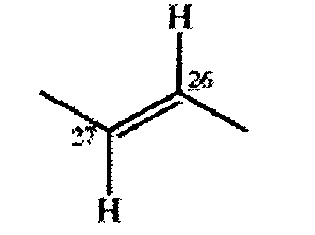
Если R6 и R7 каждый означает ОН, то конфигурация в положениях 26 и 27 предпочтительно означает 26(S), 27(S) или 26(R), 27(R). Если R6 и R7 вместе образуют дополнительную связь, то конфигурация в положениях 26 и 27 предпочтительно означает следующую конфигурацию
Соединения по настоящему изобретению формулы IX предпочтительно характеризуются следующей конформацией
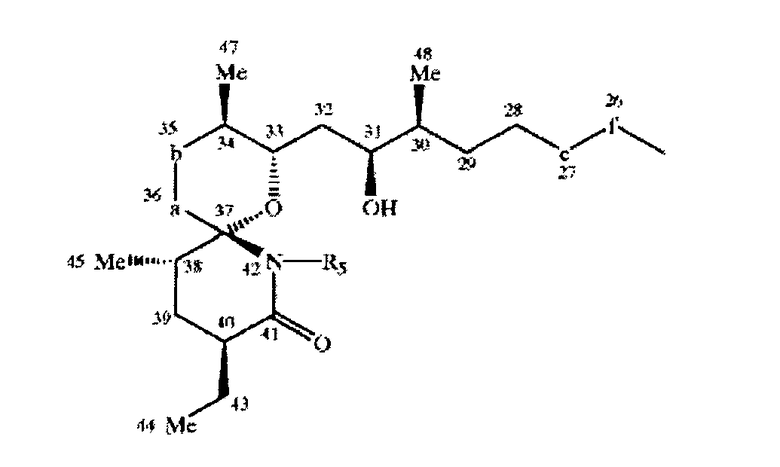

где если -a-b- означает -(Ме)СН-СН(ОН)-, то он предпочтительно характеризуется следующей конформацией
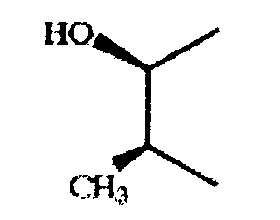
если -e-f- означает -СН(ОН)-СН(ОН)-, то соединение предпочтительно характеризуется конфигурацией (S), (S) или (R), (R), если -g-h- означает -(Me) СН-СН(ОН)-, то он предпочтительно характеризуется следующей конфигурацией
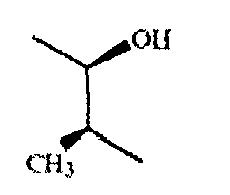
если -g-h- означает - (Ме)С=СН-, то он предпочтительно характеризуется конфигурацией
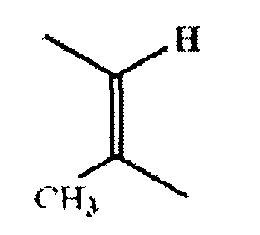
и если R3 и R4 объединены, то они характеризуются конфигурацией
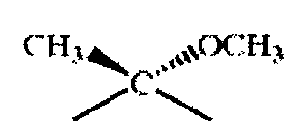
Соединения по настоящему изобретению применяют в свободной или защищенной форме, например в защищенной форме, как описано в книге «Protective Groups in Organic Synthesis», Т.W.Greene и P.G.M.Wuts, 2oe изд., John Wiley & Sons Inc., New York (1991). Прежде всего группы ОН присутствуют в защищенной форме, например, в форме силильных эфиров (например, как описано на стр.68-86 книги Greene и Wuts), сложных эфиров (см., например, стр.87-103 книги Greene и Wuts) и карбонатов (см., например, стр.104-111 книги Greene и Wuts). Такие защищенные формы включают также внутренние защищенные формы, например, в случае макролидов формулы IX, где -g-h- означает -СН(СН3)-СН(ОН)-, защищенную форму, в которой в положениях 14-17 макроциклического кольца находится остаток формулы X
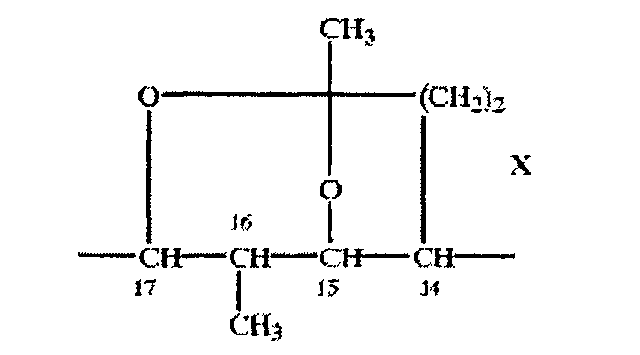
Например, 1,3-диолы, присутствующие в санглиферинах, можно защитить за счет образования соответствующих циклических структур, например, как описано на стр.118-142 книги Greene и Wuts.
Соединения по настоящему изобретению существуют также в форме солей. Примеры фармацевтически приемлемых солей по настоящему изобретению включают кислотно- и основно-аддитивные соли в зависимости от соответствующих заместителей в составе соединения, например гидрохлориды.
Как указано выше, макроциклическое кольцо в составе соединений по настоящему изобретению можно отщеплять, прежде всего по лактонной оксигруппе, при этом получают соединения, в которых макроциклическое кольцо не замкнуто. В большинстве случаев отщепление лактонной оксигруппы осуществляют при гидролизе (сольволизе), например, для получения соединений с открытым макроциклическим кольцом формулы XI
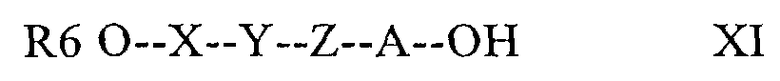
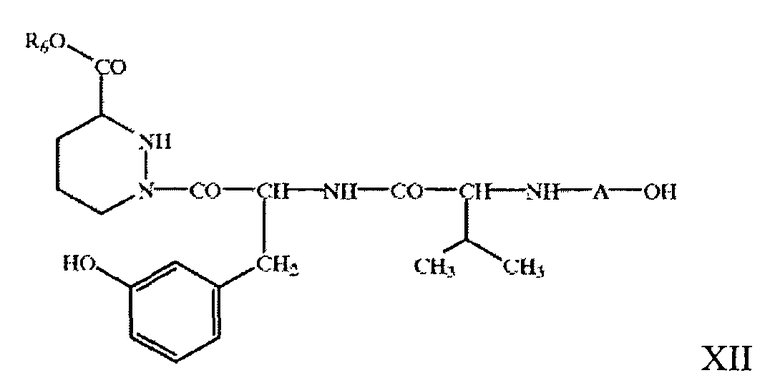
например, формулы XII
например, соединение формулы IX'
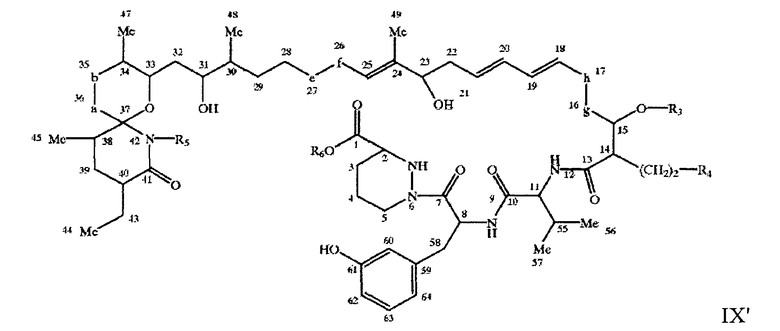
где X, Y, Z, A, -a-b-, -e-f-, -g-h-, R3, R4 и R5 имеют значения, описанные выше, a R6 означает Н или С1-С4алкил, например метил.
Такие незамкнутые формы можно использовать в качестве промежуточных соединений для модификации основной макроциклической системы санглиферинов и такие формы являются частью настоящего изобретения.
В другом объекте настоящего изобретения предлагаются макролид, описанный выше, в форме незамкнутого цикла в свободной или защищенной форме или его соль, соединение R6 O-X-Y-Z-A-ОН, описанное выше, в свободной или защищенной форме или его соль, соединение R6 O-X-Y-Z-A'--CH(OH)-L-S, где -А'- СН(ОН)- означает остаток гидроксикарбоновой кислоты, например остаток формулы II, как описано выше, а значения других символов описаны выше, в свободной или защищенной форме, или его соль,
соединение формулы XII'
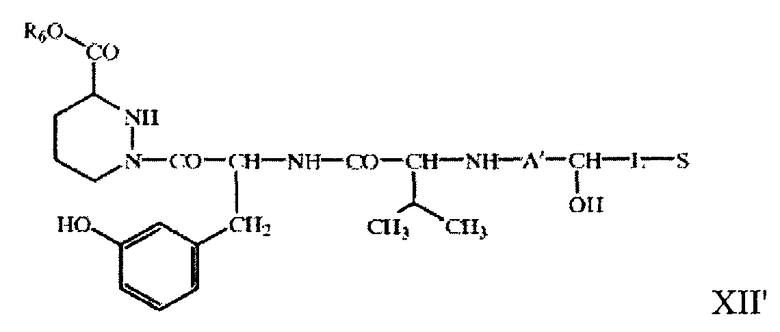
в свободной или защищенной форме или его соль, соединение формулы IX'
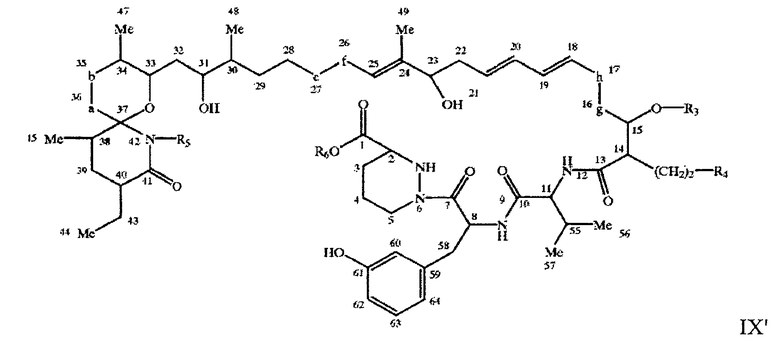
в свободной или защищенной форме или его соль.
В предпочтительных вариантах осуществления настоящего изобретения (указанного объекта) предлагается соединение формулы IX"
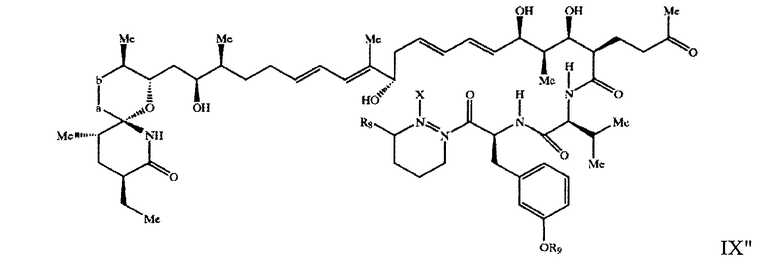
где -а-b- означает -(Ме)С=СН- или -(Me) CH-CH(OH)-, R8 означает Н или -C(O)-OR10, где R10 означает Н или Me, R9 означает Н или, при условии, что -а-b- означает -(Ме)СН-СН(ОН)- и R означает -С(O)- ОМе, R9 означает Me, а Х означает Н, если -N-N- означает простую связь между двумя атомами азота, или Х вместе с -N-N- означает двойную связь между двумя атомами азота, в свободной или защищенной форме, или его соль.
В более предпочтительных вариантах настоящего изобретения предлагаются соединения формул W, Y (секосанглиферин A), S (секосанглиферин В) и Т
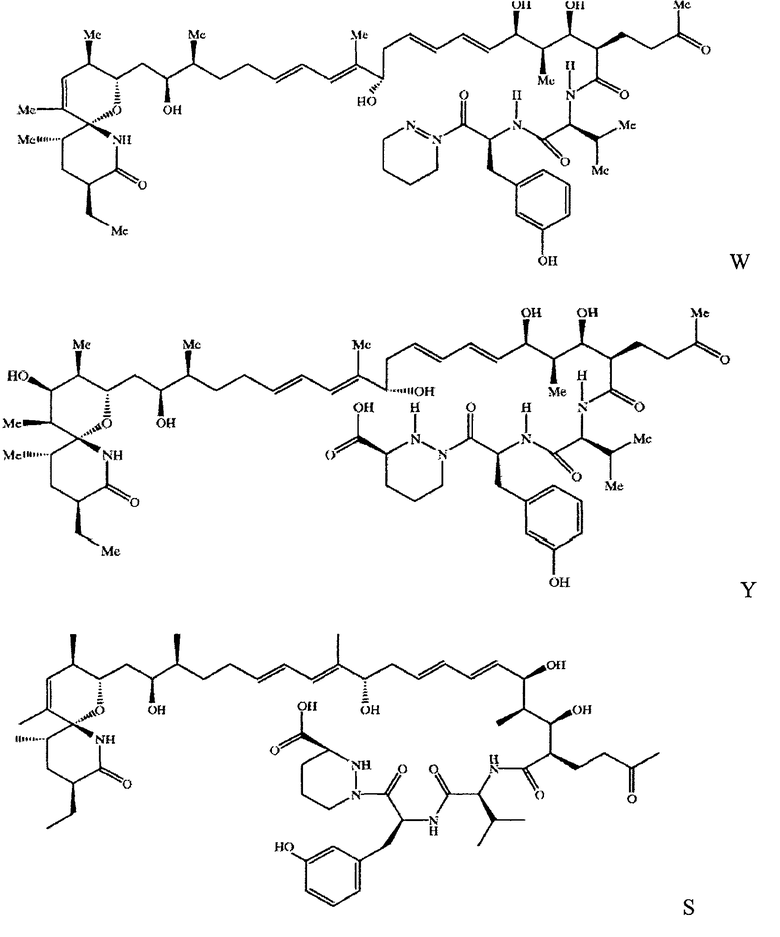
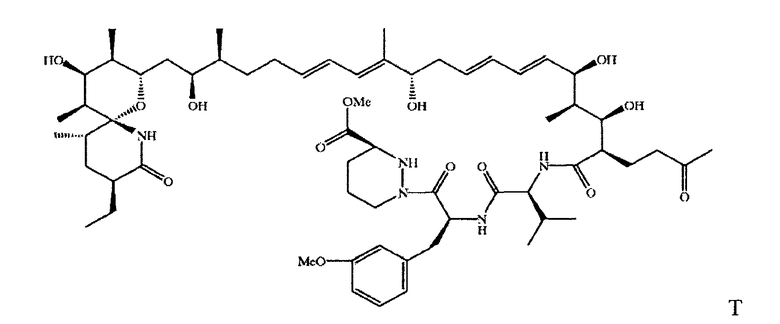
В объем настоящего изобретения включены соединения, в которых циклическая система 1-оксо-7-азаспиро-{5.5}-ундекан-8-он-2-ил находится в виде незамкнутого цикла, например соединение формулы XIII
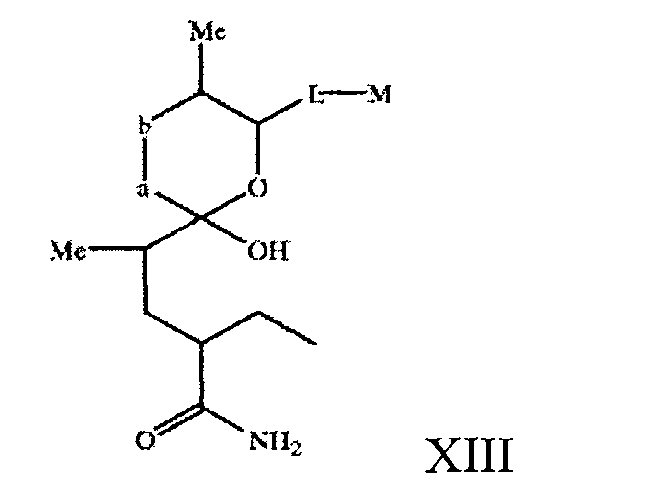
где a, b, L и М имеют значения, описанные выше, в свободной или защищенной форме, или его соль.
Соединения в виде незамкнутого цикла по настоящему изобретению предпочтительно характеризуются конформацией, описанной выше, в качестве предпочтительной для соединений с замкнутым циклом. Спиробициклоциклическая система соединений формулы XIII в виде незамкнутого цикла предпочтительно характеризуется конформацией
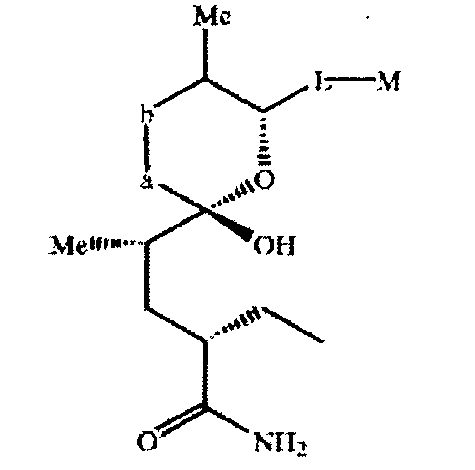
где -а-b- означает -(Me)CH-CH(OH)-.
Прежде всего в настоящем изобретении предлагается соединение формулы Z (санглиферин Е)

в свободной или защищенной форме, или его соль.
Соединения формул IX", S, Т, W, Y, XIII и Z можно использовать в качестве промежуточных соединений для получения санглиферинов, в основном содержащих замкнутые циклы и расширенные замкнутые циклы.
Соединения формулы IX" получают отщеплением макролидного цикла в составе санглиферина с замкнутым циклом, например санглиферина А или В, по связи лактонной оксигруппы или по соседней связи. Таким образом, например, соединения формул Y и Т получают при обработке санглиферина А в присутствии основания, например, в случае соединения формулы III в присутствии гидроксида щелочного металла, и, например, в случае соединения формулы IV в присутствии метанола и карбоната щелочного металла.
В другом варианте соединения формулы IX" получают выделением из культуральной среды микроорганизмов, продуцирующих санглиферин. Например, соединения формул Y и S выделяют из культуральной среды Streptomyces sp.A92-308110, как описано в данном контексте.
Выделение соединений формул Y и S из культуральной среды Streptomyces sp.A92-308110 описано в патенте US 6124453.
Соединение формулы Z получают отщеплением спироциклической системы между атомом азота и центральным атомом в спиросистеме. В другом варианте соединение формулы Z получают выделением из культуральной среды микроорганизмов, продуцирующих санглиферин. Например, выделение соединения формулы Z из культуральной среды Streptomyces sp.A92-308110 описано в патенте US 6124453.
Макролиды по настоящему изобретению, содержащие спиробициклический остаток, присоединенный к макроциклическому кольцу, обрабатывают в условиях отщепления промежуточной связующей группы, например, в случае соединения формулы IX, прежде всего по связи между остатками 26 и 27, при этом получают новые спиробициклические соединения и другие макролиды. Как указано выше, такие соединения можно использовать в качестве промежуточных соединений, при этом спиробициклический остаток санглиферинов вносит основной вклад в биологическую активность класса санглиферинов.
В настоящем изобретении предлагается 1-оксо-7-азаспиро-{5.5}-ундекан-8-он-2-ил в свободной или защищенной форме, его соль прежде всего соединение формулы VI'

где R7 означает Н, необязательно защищенную ОН-группу, реакционноспособную функциональную группу или a CH2-СН(ОН)-СН(СН3)-СН2-СН2-СНО группу или их аналог δ-лактона, в свободной или защищенной форме, или его соль.
Предпочтительно соединение формулы VI' характеризуется следующей конфигурацией
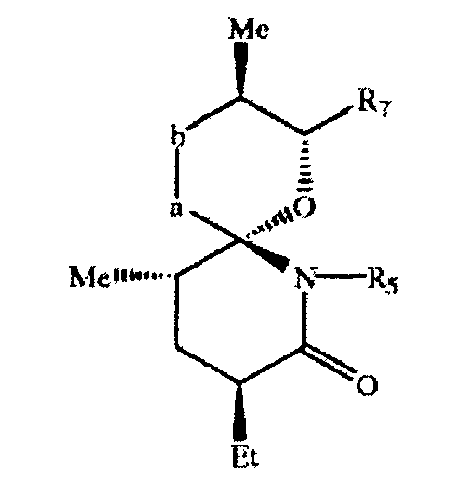
где если -а-b- означает -(Ме)СН-СН(ОН)-, то соединение предпочтительно характеризуется конфигурацией, как описано выше.
В настоящем изобретении предлагается 1-оксо-7-азаспиро-{5.5}-ундекан-8-он-2-ил в виде незамкнутого цикла в свободной или защищенной форме, его соль, прежде всего соединение формулы XIII'
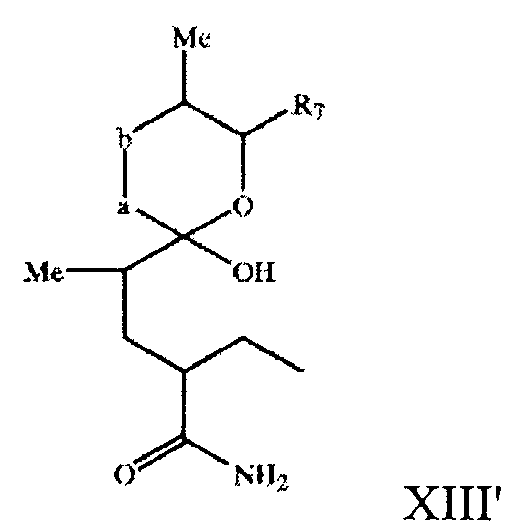
где -a-b- и R7 имеют значения, описанные выше, в свободной или защищенной форме или его соль. Спиробициклосистема соединений формулы XIII' с незамкнутым циклом предпочтительно характеризуется конфигурацией

где если -а-b- означает -(Ме)СН-СН(ОН)-, то соединение предпочтительно характеризуется конфигурацией, как описано выше.
В настоящем изобретении предлагается также макролид формулы XIV

где М означает макролидное кольцо, описанное выше, прежде всего макролид формулы XV

который предпочтительно характеризуется конформацией
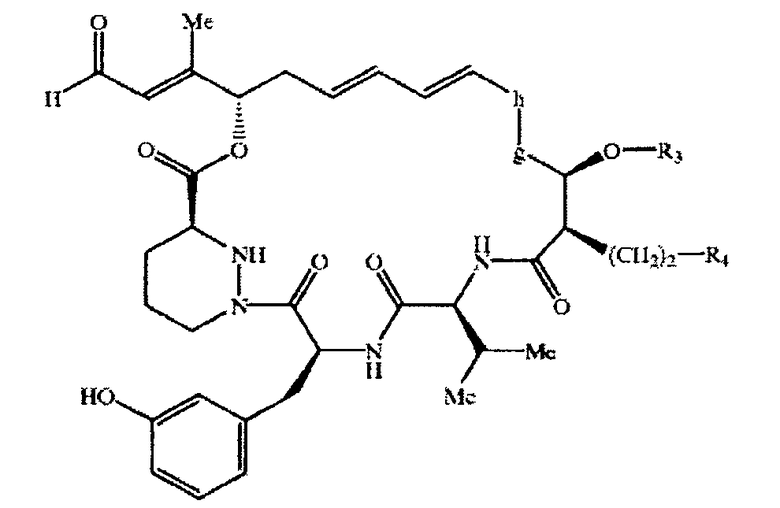
где если -g-h- означает -(Me)CH-CH(OH)-, то соединение предпочтительно характеризуется конфигурацией, как описано выше, и если -g-h- означает -(Ме)С=СН-, то оно предпочтительно характеризуется конфигурацией, как описано выше, и если R3 и R4 объединены, то они предпочтительно характеризуется конфигурацией, как описано выше, в свободной или защищенной форме, в форме системы с незамкнутым циклом, или его соль.
В настоящем изобретении предлагается также макролид формулы XVI

где Х означает Н, метокси, этокси, OnPr, OiPr,
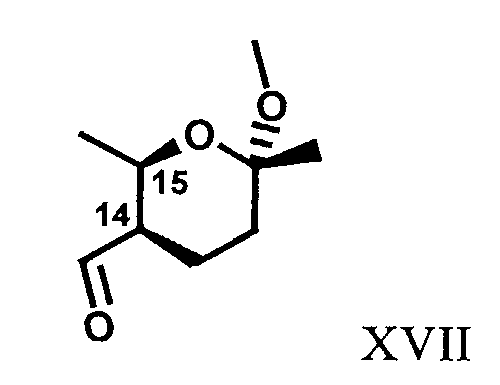
R14 и R15 независимо означают Н, метил, алкил, С1-С4алкил, C1-C4-CO-СН3 или R14 и R15 вместе образуют 5-7-членный замещенный или незамещенный циклоалкил, который выбирают, например, из соединения формулы XVII
R16 означает Н, или Me, или С1-С4алкил;
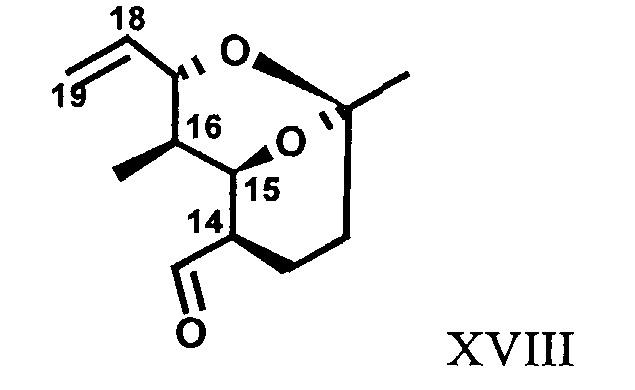
R17 означает Н, ОН, -O-С1-С4алкил или R14, R15 и R17 вместе образуют циклоалкил формулы XVIII
R23 означает Н, С1-С10алкил с прямой или разветвленной цепью и/или содержит одну или более ненасыщенных связей, прежде всего чередующихся двойных или тройных связей вдоль цепи, предпочтительно -СН=СН2, -СН2-ОН, -СН2-ОСН3, -СНО, -СООН или формулы XIX'
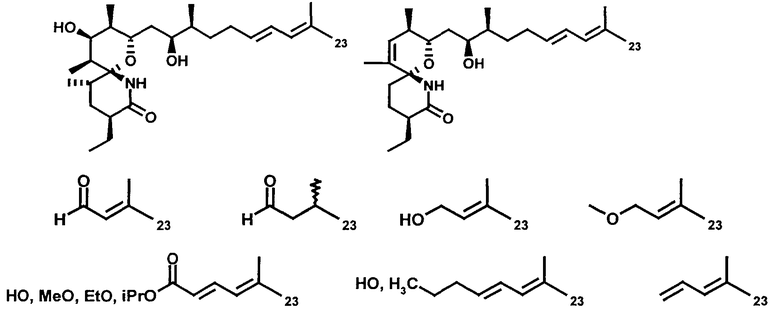
Пригодный R23 является замещенным, как описано в данном контексте.
В другом объекте настоящего изобретения предпочтительные макролиды выбирают из соединений формулы XX
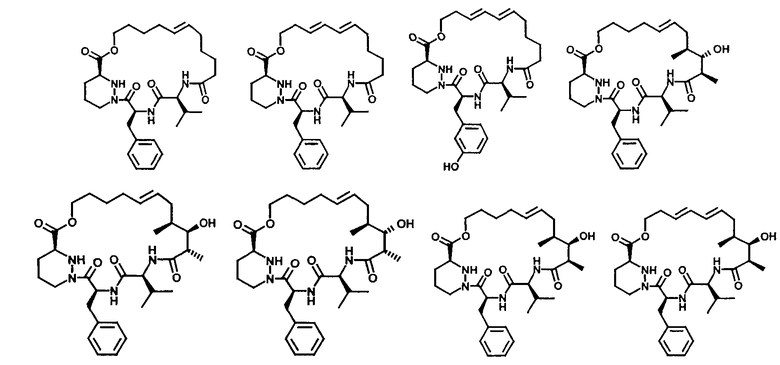
В другом объекте настоящего изобретения предлагаются макролиды и соединения по настоящему изобретению, прежде всего полученные из природных продуктов в основном в очищенной форме, например, содержащие по крайней мере 90%, предпочтительно 95%, прежде всего по крайней мере 98% очищенной формы.
В предпочтительном варианте осуществления настоящего изобретения санглиферины А, В, С и D, кроме других, выделяют из новых штаммов микроорганизмов Streptomyces sp.А92- 308110. Образцы штаммов Streptomyces sp.A92-308110 хранятся в банке микроорганизмов фирмы Deutsche Sammlung von Mikroorganismen und Zelikulturen GmbH (Mascheroder Weg Ib, D-38124 Braunschweig, Германия) и зарегестрированы 3 мая 1995 г. согласно Будапештскому договору под номером DSM 9954. Выделение санглиферинов А, В, С и D из культуральной среды Streptomyces sp.A92-30810 описано в патенте US 6124453.
Макролиды по настоящему изобретению, например соединения формулы IX, например санглиферины А, В, С и D и их фармацевтические соли (в данном контексте «агенты по настоящему изобретению»), характеризуются видами активности, харктерными для санглиферинов, т.е. следующей комбинацией активностей:
связывание с циклоспорином,
ингибирующая активность в отношении пептидилпролил-цис-транс-изомеразы,
иммунодепрессантная активность,
подавление пролиферации В- и Т-клеток,
но не связываются с белком FK и не ингибируют активность кальциневрина.
Биологическую активность макролидов по настоящему изобретению, например, формулы XVI можно оценивать стандартными методами анализа in vitro и in vivo, например, как описано ниже.
Изучение пролиферативной ответной реакции лимфоцитов на аллогенную стимуляцию проводили следующим образом. Клетки селезенки (2×10)5 мыши Balb/c (самка, возраст 8-10 недель) инкубировали в течение 4 суток в смеси с клетками селезенки (2×10) мыши СВА (самка, возраст 8-10 недель). Аллогенные клетки индуцировали пролиферативную ответную реакцию в популяции клеток-респондеров селезенки, которую оценивали по включению меченного предшественника в ДНК. Величина IC50 для макролидов по настоящему изобретению, например соединений формулы IX и их фармацевтически приемлемых солей, например санглиферинов А, В, С и D, составляла приблизительно от 30 до приблизительно 200 нМ по сравнению с величиной IC50 20 нМ для циклоспорина А, определенной по данным указанного анализа (см. статью Т. Мео, The MLR in the mouse, в книге "Immunological Methods", под ред. L.Lefkovits и В.Pernis, Academic Press, N.Y. cc.227-239 (1979)).
Клетки селезенки (2×10)5 мыши СВА инкубировали в течение 48 ч в смеси с LPS (мЕд..г/мл) и исследуемым соединением. Пролиферативную ответную реакцию оценивали по включению меченого предшественника в ДНК. Макролиды по настоящему изобретению, например соединения формулы IX и их фармацевтически приемлемые соли, например санглиферины А, В, С и D, ингибировали пролиферацию В-клеток, а величина IC50 составляла приблизительно от 40 до приблизительно 100 мкМ (см. статьи Greaves M. и J.Janossy, Elicitation of selective T and В lymphocyte response by cell surface binding ligands. Transplant Rev., т.11, с.87 (1972), Janossy G. и M.F.Greaves, Lymphocyte activation, I, Response of T and В lymphocytes to phytomitogens, Clin. Exp. Immunol, т.9, cc.483-498 (1971).
Цитотоксичность определяли с использованием клеточной культуры моноцитов человека ТНР1 (5×104 клеток/лунка), которые инкубировали в присутствии IFNγ (100 Ед./мл), LPS (5 мкг/мл) и исследуемого соединения (не более 10 мкМ) в течение от 24 до 72 ч при 37°С. Количество выживших клеток определяли с использованием колориметрического метода МТТ, который позволяет определять ферментативную акивность митохондриальной дегидрогеназы в живых клетках (см. статью Mossman (1983)). Величина IC50 для макролидов по настоящему изобретению, например соединений формулы IX и их фармацевтически приемлемых солей, например санглиферинов А, В, С и D, составляла приблизительно от 1000 до 5000 нМ после инкубирования в течение 24 ч (см. статью Mossman Т.J., Rapid calorimetric assay for cellular growth and survival: application to proliferation and cytotoxic assays, J. Imm. Methods., т.65, cc.55-63 (1983)).
Моноядерные клетки получали из периферической крови здоровых добровольцев в градиенте Ficoll-Hypaque по методике, как описано в статье Hansell и др. (1991). Клетки (105 клеток/лунка в 200 мкл среды RPMI, содержащей 10 об.% ЭТС) перед добавлением стимулирующего соединения инкубировали с серийными разбавлениями исследуемых соединений в течение 30 мин при 37°С. В качестве стимулирующего соединения для индуцирования высвобождения α-фактора некроза опухолей (TNF) моноядерными клетками периферической крови использовали γ-интерферон (100 Ед./мл) и LPS (5 мкг/мл). Через 3 ч инкубирования клетки центрифугировали (1200 об/мин в течение 10 мин) и собирали супернатанты. Количество TNF в супернатантах клеток определяли с использованием коммерческого набора для анализа методом ИФА. Величина IС50 для макролидов по настоящему изобретению, например соединений формулы IX и их фармацевтически приемлемых солей, например санглиферинов А, В, С и D, составляла приблизительно от 200 до приблизительно 1000 нМ.
Пригодным методом анализа связывания с циклофилином является конкурентный метод ИФА, описанный в статье Quesniaux, Eur. J. Immunol, т.17, cc.1359-1365 (1987). Исследуемое соединение добавляли в процессе инкубирования циклофилина (рекомбинантный циклофилин А человека) с комплексом БСА-циклоспорин А в составе покрытия планшета и затем рассчитывали концентрацию, необходимую для обеспечения 50% ингибирования контрольной реакции в отсутствие конкурентного агента (IС50). Другим методом анализа является конкурентное связывание, описанное в статье Schneider и др., Biochemistry, т.33, cc.8218-8224 (1994), который заключается в добавлении исследуемого соединения в процессе инкубирования биотинилированного циклофилина (рекомбинантный циклофилин А человека) с комплексом БСА-циклоспорин А в составе покрытия. Количество биотинилированного циклофилина, связывающегося в присутствии и отсутствие исследуемого соединения, определяли при инкубировании с конъюгатом стрептавидина и щелочной фосфатазы. Величина IС50 для макролидов по настоящему изобретению, например соединений формулы IX и их фармацевтически приемлемых солей, например санглиферинов А, В, С и D, составляла приблизительно от 10 до приблизительно 100 нМ по сравнению с величиной IС50 приблизительно 80 нМ для циклоспорина А, определенной указанным методом анализа.
Другой метод анализа in vitro, который можно использовать для определения биологической активности санглиферинов, включает анализ с использованием репортерного гена IL-2 и клеток селезенки, стимулированных СоnА (оценка действия на активацию Т-клеток).
С использованием стандартных методов анализа установлено, что макролиды по настоящему изобретению, например соединения формулы IX, например санглиферины А, В, С и D, не проявляют активности в отношении связывания белков FK и не ингибируют активность кальциневрина.
Агенты по настоящему изобретению можно использовать в качестве лекарственных средств, например в качестве иммунодепрессантов, противовоспалительных и/или противовирусных агентов.
Они являются пригодными, прежде всего, для профилактики нарушений, связанных с инфекциями вируса гепатита, прежде всего гепатита С. Кроме того, агенты по настоящему изобретению можно использовать для подавления нарушений, связанных с активностью пептидилпролил-цис-транс-изомеразы (PPI). PPI представляют собой вид циклофилина, ускоряющего укладку белковых цепей за счет катализа цис-транс-изомеризации имидных пептидных связей пролина в олигопептидах. Макролиды по настоящему изобретению связываются с циклофилинами и ингибируют активность PPI.
Агенты по настоящему изобретению можно использовать также для лечения аутоиммунных заболеваний и воспалительных состояний, прежде всего воспалительных состояний с этиологией, включающей аутоиммунную компоненту, таких как артрит (например, ревматоидный артрит, хронический прогрессирующий артрит и деформирующий артрит) и ревматоидные заболевания.
Ряд других вариантов настоящего изобретения включает также:
1.1 Способ профилактики или лечения инфекций, вызванных вирусом гепатита С, или нарушений, вызванных ВГС, таких как фиброз печени, цирроз печени или гепатоцеллюлярная карцинома, у пациентов, нуждающихся в таком лечении, который заключается во введении указанному пациенту терапевтически эффективного количества соединения, связывающегося с циклофилином, такого как санглиферин, например соединения формулы, в отдельности или в комбинации с одним или более ко-агентов.
1.2 Способ подавления репликации ВГС в среде, который заключаются в добавлении в среду эффективного количества соединения, связывающегося с циклофилином, такого как санглиферин, например соединения формулы.
1.3 Способ подавления репликации ВГС в организме пациента, нуждающегося в таком ингибировании, который заключаются во введении указанному пациенту терапевтически эффективного количества соединения, связывающегося с циклофилином, такого как санглиферин, например соединения формулы.
1.4 Способ профилактики рецидива инфекции ВГС у пациента после трансплантации, нуждающегося в такой профилактике, который заключается во введении указанному пациенту терапевтически эффективного количества соединения, связывающегося с циклофилином, такого как санглиферин, например соединения формулы.
2. Применение соединения, связывающегося с циклофилином, такого как санглиферин, для получения фармацевтической композиции, предназначенной для применения согласно любому методу, описанному выше.
3. Фармацевтическя композиция, предназначенная для применения согласно любому способу, описаному выше, включающая соединение, связывающееся с циклофилином, такое как санглиферин, в комбинации с одним или более фармацевтически приемлемых разбавителей или носителей.
Кроме того, санглиферины по настоящему изобретению, обладающие активностью в отношении связывания циклофилина, можно использовать в качестве реагентов в заместительных иммунных анализах циклоспоринов и других соединений, связывающихся с циклофилином, например, в анализе, описанном в заявке на выдачу патента WO 95/07468. В указанной заявке описан метод анализа для определения концентрации фармацевтического средства, связывающего иммунофилин, например, циклоспорина, в крови; и указанный метод анализа заключается в добавлении в кровь связывающего конкурентного агента, который вытесняет фармацевтическое средство из комплексов иммунодепрессант-иммунофилин в крови; в добавлении рецептора, который связывается с фармацевтическим средством, но незначительно связывается с конкурентным агентом; в выделении комплекса рецептор-фармацевтическое средство из образца и в определении количества фармацевтического средства. Санглиферины можно использовать в качестве связывающего конкурирующего агента в таких методах анализах, например, для замещения циклоспоринов из циклофилинов, при этом происходит высвобождение циклоспорина, который определяют количественно, например, с использованием моноклональных антител, которые являются специфичными к циклоспорину.
В соответствии с описанным выше в настоящем изобретении предлагаются также следующие объекты:
4. Фармацевтическая комбинация, включающая а) первый агент, который является соединением, связывающимся с циклофилином, таким как санглиферин, например соединение формулы I, и б) ко-агент, например одно или более лекарственных средств, как определено в данном контексте.
5. Способ, описанный выше, который заключается в совместном введении, например, одновременно или последовательно, терапевтически эффективного количества соединения, связывающегося с циклофилином, такого как санглиферин, например соединения формулы I, и ко-агента, например одного или более лекарственных средств, как определено в данном контексте.
Соединение по настоящему изобретению можно вводить любым стандартным способом, например энтеральным способом, например, пероральным способом, например, в виде растворов, таблеток или капсул, или парентеральным способом, например, в форме растворов или суспензий для инъекций. Предпочтительными фармацевтическими композициями являются, например, композиции на основе микроэмульсий, как описано в патенте UK 2222770 А.
В другом варианте пригодные ко-агенты вводят в комбинации или согласно другому курсу лечения. Термин «совместное введение» или т.п., использованный в данном контексте, означает введение определенных лекарственных средств пациенту, и включает способы лечения, при которых агенты необязательно вводят одинаковым способом или одновременно. Фиксированные комбинации также включены в объем настоящего изобретения. При введении фармацевтической композиции по настоящему изобретению наблюдается благоприятный эффект например, синергетическое или аддитивноое терапевтическое действие, по сравнению с монотерапией, при которой вводят только один из фармацевтически активных компонентов. Ко-агенты, пригодные для совместного введения с соединением по настоящему изобретению, включают, без ограничения перечисленным:
(1) Интерфероны или конъюгаты интерферонов, такие как:
(а) продукт Intron-A®, интерферон α-2b (фирмы Sobering Corporation, Kenilworth, NJ),
(б) продукт PEG-Intron®, пегинтерферон α-2b (фирмы Schering Corporation, Kenilworth, NJ),
(в) продукт Roferon®, рекомбинантный интерферон α-2а (фирмы Hoffmann-La Roche, Nutley, NJ),
(г) продукт Pegasys®, пегинтерферон α-2а (фирмы Hoffmann-La Roche, Nutley, NJ),
(д) продукт Berefor®, интерферон α2 (фирмы Boehringer Ingelheim Pharmaceutical, Inc., Ridgefield, CT),
(е) продукт Sumiferon®, очищенная смесь природных α- интерферонов (фирмы Sumitomo, Japan),
(ж) продукт Wellferon®, лимфобластоидный интерферон αn1 (фирмы GlaxoSmithKline),
(з) продукт Infergen®, консенсусный α-интерферон (фирм InterMune Pharmaceuticals, Inc., Brisbane, CA и Amgen, Inc., Newbury Park, CA),
(и) продукт Alferon®, смесь природных α-интерферонов (фирм Interferon Sciences и Purdue Frederick Co., CT),
(к) продукт Viraferon®;
(л) конъюгированные интерфероны включают, например, продукт Albuferon (фирмы Human Genome Science), то есть конъюгат с альбумином человека, а также интерферон, конъюгат с водорастворимым полимером или гомополимерами полиалкиленоксида, такими как полиэтиленгликоль (ПЭГ) или полипропиленгликоли, поликосиэтилированные полиолы, их сополимеры и блок-сополимеры. В качестве альтернативы полимерам на основе полиалкиленоксида эффективно используют неантигенные материалы, такие как декстран, поливинилпирролидон, полиакриламиды, поливиниловые спирты, полимеры на основе углеводов и т.п. Конъюгаты интерферона с полимерами описаны в документах US 4766106, US 4917888, EPA 0236987, EPA 0510356 и WO 95/13090. Поскольку полимерная модификация значительно снижает антигенную ответную реакцию, интерфероны из внешних источников не обязательно должны быть аутологичными. Интерферон, который используют для получения конъюгатов с полимерами, получают из экстрактов тканей млекопитающих, таких как человек, жвачные животные или крупный рогатый скот, или рекомбинантным способом.
Другие формы интерферонов включают интерфероны β, γ, τ и ω, такие как продукт Rebif (Interferon β 1a) фирмы Serono, продукт Omniferon (природный интерферон) фирмы Viragen или интерферон ω фирмы Boehringer Ingelheim;
Пероральные интерфероны, такие как пероральный интерферон α фирмы Amarillo Biosciences.
В другом объекте примеры интерферонов включают пегилированный интерферон α, например пегилированный интерферон α-2а, пегилированный интерферон α-2b, пегилированный консенсусный интерферон или очищенный пегилированный интерферон α. Пегилированный интерферон α-2а описан в Европейском патенте 593868 (полностью включенном в настоящее изобретение в качестве ссылки), который является коммерческим препаратом, например, под торговым названием PEGASYS® (фирмы Hoffmann-La Roche). Пегилированный интерферон α-2b описан, например, в Европейском патенте 975369 (полностью включенном в настоящее изобретение в качестве ссылки), который является коммерческим препаратом, например, под торговым названием PEG-INTRON А® (фирмы Schering Plough). Пегилированный консенсусный интерферон описан в заявке на выдачу патента WO 96/11953 (полностью включенной в настоящее изобретение в качестве ссылки).
(2) Противовирусные агенты
Противовирусными агентами являются химические соединения или природные вещества, которые эффективно подавляют образование и/или репликацию вируса в организме млекопитающих. Они включают агенты, которые подавляют явления, наблюдаемые в организме хозяина или в частицах вируса, и которые необходимы для образования и/или репликации вируса в организме млекопитающих, такие как рибавирин (1-β-D-рибофуранозил-1Н-1,2,4-триазол-3-карбоксамид) фирмы Valeant Pharmaceuticals, Inc., Costa Mesa, CA, продукт Rebetol® фирмы Schering-Plough Corporation, Kenilworth, NJ, и Copegus® фирмы Hoffmann-La Roche, Nutley, NJ, аналоги рибавирина, такие как продукты Levovirin и Viramidine фирмы Valeant, и монофосфат мизорибина, находящиеся в стадии разработки;
(3) Ингибиторы протеазы
Ингибиторы сериновой протеазы NS3-4A вируса гепатита В, такие как ингибиторы субстратной протеазы (см. документы Attwood и др., Antiviral peptide derivatives, PCT WO 98/22496 (1998); Attwood и др., Antiviral Chemistry and Chemotherapy, т.10, cc.259-273 (1999); Attwood и др., Preparation and use of amino acid derivatives as anti-viral agents, публикация патента Германии DE 19914474; Tung и др., Inhibitors of serine proteases, particularly hepatitis С virus NS3 protease; PCT WO 98/17679), включающие α-кетоамиды и гидразиномочевины, и ингибиторы, содержащие в качестве концевой группы электрофильную группу, такую как бороновая кислота или фосфонат (см. Llinas-Brunet и др., Hepatitis С inhibitor peptide analogues, PCT WO 99/07734); ингибиторы протеазы, описанные в патентах США, предназначенные для лечения ВГС, например, в патентах US №6004933, Spruce и др., в котором описан класс ингибиторов цистеинпротеаз для ингибирования эндопептидазы 2 ВГС; US №5990276, Zhang и др., в котором описаны синтетические ингибиторы протеазы NS3 вируса гепатита С; US №5538865, Reyes и др.; пептиды в качестве ингибиторов сериновой протеазы NS3 ВГС, которые описаны в заявке WO 02/008251, Corvas International, Inc., и в заявках WO 02/08187 и WO 02/008256, Schering Corporation, трипептиды-ингибиторы ВГС, которые описаны в патентах US №6534523, 6410531 и 6420380, Boehringer Ingelheim, и в заявке WO 02/060926, Bristol Myers Squibb (которые полностью включены в объем настоящего изобретения в качестве ссылок); диарилпептиды в качестве ингибиторов сериновых протеаз NS3 ВГС, которые описаны в заявке WO 02/48172, Schering Corporation; имидазолидиноны в качестве ингибиторов сериновых протеаз NS3 ВГС, которые описаны в заявках WO 02/18198, Schering Corporation, и WO 02/48157, Bristol Myers Squibb (которые полностью включены в объем настоящего изобретения в качестве ссылок); в заявках WO 98/17679, Vertex Pharmaceuticals, и WO 02/48116, Bristol Myers Squibb, также описаны ингибиторы протеаз ВГС (которые полностью включены в объем настоящего изобретения в качестве ссылок).
(4) Ингибиторы несубстратных протеаз NS3, такие как производные 2,4,6-тригидрокси-3-нитробензамида (см. статьи Sudo K. и др., Biochemiscal and Biophysical Research Communications, т.238, cc.643-647 (1997), Sudo K. и др., Antiviral Chemistry and Chemotherapy, т.9, с.186 (1998)), включая RD3-4082 и RD3-4078;
(5) Фенантренхинон, обладающий активностью в отношении протеазы по данным анализа электрофорезов в ДСН-ПААГ и авторадиографии, который выделяют из ферментационной культуральной среды штамма Streptomyces sp., Sch 68631 (см. статьи Chu М. и др., Tetrahedron Letters, т.37, cc.7229-7232 (1996), Chu М. и др., Tetrahedron Letters т.37, cc.7229-7232 (1996)); Sch 351633, который выделяют из грибов Penicillium grieofulvum (см. статью Chu М. и др., Bioorganic and Medicinal Chemistry Letters, т.9, cc.1949-1952); селективные ингибиторы, разработанные на основе макромолекул eglin с., которые выделяют из пиявок и которые являются эффективными ингибиторами некоторых сериновых протеаз, таких как протеазы S. griseus А и В, α-химотрипсин, химаза и субтилизин (см. статью Qasim М.А. и др., Biochemistry, т.36, cc.1598-1607 (1997)).
(6) Тиазолидины и бензалидины, описанные в статьях Kakiuchi N. и др., J. FEBS Letters, т.421, cc.217-220, Takeshita N. и др., Analytical Biochemistry, т.247, cc. 242-246 (1997), Производные тиазолидина, характеризующиеся значительным ингибированием (по данным анализа ОФ-ЖХВР) гибридного белка NS3/4A и субстрата NS5A/5B (см. статью Sudo K. и др., Antiviral Research, т.32, cc.9-18 (1996)), прежде всего соединение RD-16250, содержащее конденсированное циннамоилпроизводное, замещенное длинной алкильной цепью, соединения RD4 6205 и RD4 6193.
(7) Ингибиторы сериновой протеазы NS3-4A ВГС, включающие продукт BILN 2061 фирмы Boehringer Ingelheim, продукт VX-950 фирмы Vеrtех, продукты SCH-503034 и SCH-351633 фирмы Schering-Plough, а также другие ингибиторы серинпротеаз ВГС, находящиеся на стадии доклинических и клинических испытаний, фирм GlaxoSmithKline, Bristol Myers Squibb, Abbot, Roche, Merck, Pfizer и Gilead;
(8) Аналоги нуклеозидов
Продукт Telbivudine фирмы Idenix (см. патенты US 6444652, US6596700 и заявку WO 0196353). Нуклеозидные и ненуклеозидные ингибиторы РНК-зависимой РНК-полимеразы NS5B ВГС, такие как сложный эфир 2'-С-метил-3'-O-L-валина и рибофуранозилцитидина (продукт NM283 фирмы Idenix), как описано в заявке WO 2004/002422.
Разветвленные нуклеозиды описаны фирмой Idenix Pharmaceuticals для лечения флавирусов (включая ВГС) и пестивирусов, в публикациях заявок на выдачу международных патентов WO 01/90121 и WO 01/92282 (полностью включенных в объем настоящего изобретения в качестве ссылок). Способ лечения инфекции, вызванной вирусом гепатита С (а также флавирусами и пестивирусами), у человека и других животных, описан в публикациях фирмы Idenix, который заключается во введении эффективного количества биологически активных 1', 2', 3' или 4'-разветвленных B-D или B-L нуклеозидов или их фармацевтически приемлемых солей или пролекарств, которые вводят в отдельности или в комбинации с другим противовирусным агентом, необязательно в фармацевтически приемлемом носителе.
Другие заявки на выдачу патента, в которых описано применение аналогов нуклеозидов для лечения вируса гепатита С, включают РСТСА00/01316 (WO 01/32153, поданная 3 ноября 2000 г.) и РСТ/СА 01/00197 (WO 01/60315, поданная 19 февраля 2001 г.), поданная фирмой BioChem Pharma, Inc. (новое название Shire Biochem, Inc.); PCT/US 02/01531 (WO 02/057425, поданная 18 января 2002 г.) и PCT/US 02/03086 (WO 02/057287; поданная 18 января 2002 г), поданная фирмой Merck & Co., Inc., PCT/EP 01/09633 (WO 02/18404, поданная 21 августа 2001 г.), поданная фирмой Roche, и публикация заявки РСТ WO 01/79246 (поданная 13 апреля 2001 г.), WO 02/32920 (поданная 18 октября 2001 г.) и WO 02/48165, поданная фирмой Pharmasset, Ltd. (содержание которых полностью включено в объем настоящего изобретения в качестве ссылок);
2'-фторнуклеозиды, описанные в публикации заявки РСТ WO 99/43691 "2'-Fluoronucleosides", Emory University (содержание которой полностью включено в объем настоящего изобретения в качестве ссылки);
2'-модифицированные нуклеозиды, описанные в публикации Eldrup и др. (Oral Session V, Hepatitis С Virus, Flaviviridae; 16th International Conference on Antiviral Research (April 27, 2003, Savannah, GA));
Аналоги нуклеозидов и 2'-модифицированные нуклеозиды, описанные в публикации Bhat и др. (Oral Session V, Hepatitis С Virus, Flaviviridae, 2003 (Oral Session V, Hepatitis С Virus, Flaviviridae; 16th International conference on Antiviral Research (April 27, 2003, Savannah, Ga); p A75);
2'-модифицированные нуклеозиды, описанные в публикации Olsen и др. (Oral Session V, Hepatitis С Virus, Flaviviridae; 16th International Conference on Antiviral Research (April 27, 2003, Savannah, Ga)p A76);
Соединения, находящиеся на стадии доклинических испытаний, включают продукт R803 (фирмы Rigel), продукт JTK-003 (фирмы Japan Tabacco), продукт HCV-086 (фирмы ViroPharma/Wyeth), продукт R-1479 (фирмы Roche);
(9) Ингибиторы нуклеотидполимеразы и глиотоксин (см. статью Ferrari R. и др., Journal of Virology, т.73, cc.1649-1654 (1999)), а также природный продукт церуленин (см. статью Lohmann V. и др., Virology, т.249, cc.108-118 (1998));
(10) Ингибиторы геликазы NS3 ВГС, такие как продукт VP_50406 фирмы ViroPhama и другие соединения фирмы Vertex, другие ингибиторы геликазы, такие как описанные в патенте US №5633388 и заявке на выдачу патента РСТ WO 97/36554;
(11) Антисмысловые молекулы против генома ВГС или против любых компонентов клетки, необходимых для репликации вируса.
Фосфоротиоаты олигонуклеотидов (S-ODN), комплементарные цепям в 5'-некодирующем фрагменте (NCR) вируса (см. статью Alt M. и др., Hepatology, т.22, cc.707-717 (1995)), или нуклеотиды 326-348, содержащие 3'-фрагмент NCR, и нуклеотиды 371-388, расположенные в середине кодирующего фрагмента РНК ВГС (см. статьи Alt M. и др., Archives of Virology, т.142, cc.589-599 (1997), Galderisi U. и др., Journal of Cellular Physiology, т.181, cc.251-257 (199)), такие как продукт ISIS 14803 фирмы Isis Pharm/Elan, антисмысловые олигонуклеотиды фирм Hybridon, AVI bioPharma и Chugai, продукт AVI-4065 (фирмы AVI BioPharma) или антисмысловая последовательность, комплементарная любой части генома ВГС и повышающая эффективность терапии;
(12) Ингибиторы IRES-зависимой трансляции (см. Ikeda N и др.. Agent for the prevention and treatment of hepatitis С, публикация патента Японии JP-08268890, Kai Y и др., Prevention and treatment of viral diseases, публикация патента Японии JP-10101591), такие как продукт ISIS 14803 фирмы Isis Pharm/Elan, ингибиторы IRES фирм РТС Therapeutics, Anadys, Immusol, RiboTargets и SomaGenics;
(13) Рибозимы, такие как рибозимы, резистентные к нуклеазам (см. статью Maccjak D.J. и др., Hepatology, т.30, абстракт 995 (1999)), и описанные в патентах US №6043077, Barber и др., и US №№5869253 и 5610054, Draper и др. (полностью включенные в объем настоящего изобретения в качестве ссылок), например, продукт HEPTAZYME фирм RPI и Sirna Therapeutics Inc.;
(14) Ингибитор других мишеней жизненного цикла ВГС, включающего проникновение, сборку и созревание вируса, такие как продукт Celgosivir (MBI 3253), ингибитор процессинга гликопротеинов фирмы Migenix, гибридный ингибитор фирмы Trimeris, продукт АСН-0137171 фирмы Achillion;
(15) Иммуномодулирующие агенты
Агонисты рецепторов, такие как контрольные рецепторы (TLR), включающие продукты ANA245, ANA971, ANA975 (см. патенты US 5041426, 4880784) фирмы Anadys; агонист TLR-9 (продукт CpG-10101 фирмы Coley Pharmaceuticals); ингибитор IMPDH, микофенольная кислота, ее соли и пролекарства, микофенолят натрия и микофенолят мофетила или меримебодиб (продукт VX-497 фирмы Vertex); тимозин α-1 (продукт Zadaxin или комбинации на его основе фирмы SciClone); продукт SCV-07 (фирмы SciClone), продукт Belerofon (модифицированный интерферон-α фирмы Nautilus); продукт CIVACIR (иммуноглобулин вируса гепатита С) фирмы NABI, или агонист рецептора S1P, например продукт FTY720 или его аналоги, необязательно фосфорилированные, например, как описано в документах ЕР 627406А1, ЕР 778263А1, ЕР 1002792А1, WO 02/18395, WO 02/76995, WO 02/06268, JP 2002316985, WO 03/29184, WO 03/29205, WO 03/62252 и WO 03/62248, содержание которых полностью включено в настоящее изобретение в качестве ссылок; резихимод (продукт VML 600) фирмы 3М Pharmaceuticals, аналог имихимода, который является эффективным индуктором интерферона-α и других цитокинов;
(16) Противофиброзный агент, такой как производное N-фенил-2-пиримидинамина иматиниб (фирмы Gleevac), продукт IP-501 фирмы Indevus, и интерферон-γ 1b фирмы InterMune, продукт Pirfenidone (многокомпонентный продукт: агонист TGF Beta, антагонист FGF, антагонист PDGF) фирм Intermune, Marnac, Shionogi;
(17) Терапевтические вакцины фирмы Intercell (терапевтическая пептидная вакцина IC41 HCV), продукты Epimmune/Genecor, Merix, Tripep (фирмы Chiron-VacC), иммунотерапевтический агент (продукт Therapore) фирмы Avant, агент для обработки Т клеток фирмы CellExSys, моноклональные антитела XTL-002 фирмы STL, продукты ANA 246 и ANA 246 фирмы Anadys, терапевтическая вакцина против Е2 фирмы Innogenetics, моноклональные антитела mAb против белка оболочки Е2 фирмы XTL Bio, продукт GI-5005 (фирмы Globelmmune Inc), продукт InnoVac-C (WO 9967285, фирмы Innogenetics), продукт IC-41 (фирмы Intercell), интерферон α-n3 (фирмы Interferon Sciences), продукт Engerix В (фирмы SmithKline Beecham);
(18) Другие соединения включают продукт Ursodiol (EP00269516, фирмы Axcan Pharma), продукт HE-2000 (аналог DHE, фирмы Colthurst Ltd), продукт ЕНС-18 (иммуномодулятор фирмы Enzo biochem), дигидрохлорид гистамина (агонист Н2, WO 09104037, фирмы Estero-Anstalt), 1-аминоалкилциклогексаны (патент US №6034134), алкилированные липиды (патент US №5922757), витамин Е и другие антиоксиданты (патент US №5922757), желчные кислоты (патент US №584699964), N-(фосфоноацетил)-L-аспарагиновая кислота (патент US №5830905), бензолдикарбоксамиды (патент US №5633388), производные полиадениловой кислоты (патент US №5496546), 2'3'-дидезоксиинозин (патент US №5026687), бензимидазолы (патент US №5891874), экстракты растений (патенты US №5837257, 5725859 и 6056961) и пиперидины (патент US №5830905); N-(фосфоноацетил)-L-аспарагиновая кислота, бензолдикарбоксамиды, производные полиадениловой кислоты, ингибиторы гликозилирования и неспецифичные цитозащитные агенты, которые блокируют повреждение клеток, вызванное вирусной инфекцией;
(19) Любые другие соединения, находящиеся на стадии доклинических и клинических испытаний, предназначенные для лечения ВГС, включая интерлейкин-10 (фирмы Schering-Plough), AMANTADINE (Symmetrel) фирмы Endo Labs Solvay, ингибитор каспазы IDN-6556 фирмы Idun Pharma, продукт HCV/MF59 фирмы Chiron, продукт CEPLENE (дихлорид гистамина) фирмы Maxim, продукт IDN-6556 фирмы Idun PHARM, продукт Т67, ингибитор β-тубулина фирмы Tularik, продукт FK788 фирмы Fujisawa Healthcare, продукт IdB1016 (фирмы Siliphos, пероральная силибинфосфатидилхолинфитосома), проудкт Dication фирмы Immtech, очиститель крови фирмы Aethlon Medical, продукт UT 231 В фирмы United Therapeutics; продукты НереХ-С SM1, PPVO-Вау55-8800 (Parapoxvirus ovis) фирмы Bayer; продукт Refanalin (модель HGF) фирмы Angion, продукт R803 (фирмы Rigel), продукты JTK-003, JTK-002 и JTK-109 (фирмы Japan Tobacco), продукт HCV-086 (фирмы ViroPharma/Wyeth), продукт ISIS-14803 (фирмы ISIS Pharmaceuticals), продукт GS-9132 (ингибитор полимеразы фирмы Achillion Pharmaceuticals), продукт HCV-793 (фирм Pharmasset и Roche), продукт R1626 (фирмы Roche).
Специалисты в данной области техники могут найти соединения или лекарственные средства на стадии доклинических испытаний при поиске информации о клинических испытаниях в Интернете, например, пресс-релизов соответствующих компаний.
(20) Соединения, повышающие эффективность комбинированной терапии, включающие антифолат, а 5-фторпиримидин (включая 5-форурацил), аналог цитидина, такой как β-L-1,3-диоксоланилцитидин или β-L-1,3-диоксоланил-5-фторцитидин, антиметаболиты (включая антиметаболиты пурина, циатрабин, фударабин, флоксуридин, 6-меркаптопурин, метотрексат и 6-тиогуанин), гидроксимочевину, грибковые ингибиторы (включая продукты СРТ-11, Etoposide (VP-21), таксол и алкалоиды барвинка, такие как винкристин и винбластин, алкилирующий агент (включая, без ограничения перечисленным, бусульфан, хлорамбуцил, циклофосфамид, ифофамид, мехлоретамин, мелфалан и тиотепа), неклассические алкилирующие агенты, соединения, содержащие платину, блеомицин, противораковый антибиотик, антрациклин, такой как доксорубицин и дауномицин, антрацендион, ингибиторы топоизомеразы II, гормональные агенты (включая, без ограничения перечисленным, кортикостероиды (дексаметазон, преднизон и метилпреднизон), андрогены, такие как флуоксиместрон и метилтестостерон, эстрогены, такие как диэтилстилбестрол, антиэстрогены, такие как тамоксифен, аналоги LHRH, такие как лейпролид, антиандрогены, такие как флутамид, аминоглутетимид, ацетат мегестрола и медроксипрогестерон), аспарагиназа, кармустин, ломустин, гексаметилмеламин, дакарбазид, метотан, стрептозоцин, цисплатин, карбоплатин, левамазол и лейковорин. Соединения по настоящему изобретению можно использовать также в комбинации с агентами ферментативной терапии и модуляторами иммунной системы, такими как интерлейкин, фактор некроза опухолей, колониестимулирующим фактором макрофагов и колониестимулирующим фактором.
Применение соединения, связывающегося с циклофилином, или лекарственного средства, такого как санглиферин, для лечения заболеваний или состояний, указанных выше, можно оценивать с использованием стандартных методов испытаний in vitro или клинических испытаний, например, с использованием методик, описанных в данном контексте.
А. Анализ in vitro
Влияние различных санглиферинов по настоящему изобретению на репликацию генома ВГС исследовали с использованием клеток репликона ВГС.
Для анализа используют клеточную культуру репликона ВГС, клон А, фирмы Apath, LLC. Клетки культивировали в среде Игла, модифицированной Дульбекко (DMEM, фирмы Gibco), содержащей 10% эмбриональной телячьей сыворотки, инактивированной нагреванием (ЭТС, фирмы Gibco), 2 мМ L-глутамина, заменимые аминокислоты 1х (фирмы Gibco) и 1 мг/мл продукта G418 (фирмы Invitrogen, Carlsbad, CA).
Для определения противовирусной активности соединений клеточную культуру репликона ВГС (клон А) обрабатывали серийными разведениями соединений в DMEM, содержащей 2% ЭТС и 0,5% ДМСО, в течение 48 ч, затем экстрагировали общую внутриклеточную РНК и определяли уровень РНК ВГС методом ОТ-ПЦР в реальное время (фирмы Taqman) с использованием праймеров, специфичных к ВГС (5'-ТСТ ТСА CGC AGA AAG CGT CTA-3' и 5'-CTG GCA АТТ CCG GTG ТАС Т-3), последовательность ID №1, и зонда (5'-6-FAM-TCC TGG AGG CTG САС GAC ACT CAT A-TAMRA-3'), последовательность ID №2. Для каждой обработки количество РНК ВГС нормализовали по общему количеству экстрагированной общей РНК, которое определяли методом Quant-iT (фирмы Molecular Probe). Каждый полученный результат означает среднюю величину 6 повторных анализов в клеточной культуре. IС50 означает концентрацию соединения, при которой уровень РНК ВГС в клетках репликона снижается на 50%. Для оценки цитотоксичности оценивали выживаемость клеток репликона после обработки соединением в течение 48 ч при окрашивании клеток производным тетразолия (MTS) (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega, Madison, WI). CC50 означает концентрацию соединения, при которой выживаемость клеток снижается на 50%.
В. Клинические испытания
15-30 пациентов, страдающих от инфекции ВГС, обследовали в течение 2-12 недель. Каждый пациент получал терапевтически эффективную дозу соединения, связывающегося с циклофилином, такого как санглиферин. В основном для всех пациентов определяли уровень РНК в сыворотке в течение курса лечения и в течение последующих 2 недель.
Продолжительность курса лечения и дозы, требуемые для обеспечения устойчивой ответной реакции на подавление вируса у пациентов, страдающих ВГС, будут определены в следующих клинических испытаниях, включающих большее число пациентов и более продолжительный период лечения вплоть до 12 месяцев с последующим периодом 6 месяцев.
Суточные дозы для введения согласно способу по настоящему изобретению зависят от, например, природы соединения, связывающегося с циклофилином, организма хозяина, способа введения и тяжести заболевания, предназначенного для лечения.
Санглиферин вводят в отдельности или в комбинации с другими лекарственными средствами, например лекарственными средствами с активностью против ВГС, таким как интерферон-α, пегилированный интерферон-α, рибавирин, ингибитор полимеразы ВГС, такой как NM283, или ингибитор протеазы ВГС, такой как SCH503034 и VX-950. Суточные дозы ко-агентов зависят от, например, синергетических эффектов с соединениями, организма хозяина, способа введения и тяжести заболевания, предназначенного для лечения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ МОДИФИЦИРОВАННЫХ ЦИКЛОСПОРИНОВ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ HCV | 2004 |
|
RU2389501C2 |
| ПРИМЕНЕНИЕ МОДИФИЦИРОВАННЫХ ЦИКЛОСПОРИНОВ | 2007 |
|
RU2463071C2 |
| ПРОГНОЗ КИНЕТИКИ ВИРУСА ГЕПАТИТА С ПРИ ЛЕЧЕНИИ, ИСКЛЮЧАЮЩЕМ ИНТЕРФЕРОН | 2011 |
|
RU2590691C2 |
| ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2544010C2 |
| ПРИМЕНЕНИЕ СРЕДСТВ, СВЯЗЫВАЮЩИХ EDG-РЕЦЕПТОР, В ЛЕЧЕНИИ РАКОВОГО ЗАБОЛЕВАНИЯ | 2008 |
|
RU2426555C2 |
| ПРИМЕНЕНИЕ СРЕДСТВ, СВЯЗЫВАЮЩИХ EDG-РЕЦЕПТОР, В ЛЕЧЕНИИ РАКОВОГО ЗАБОЛЕВАНИЯ | 2003 |
|
RU2358717C2 |
| Пангенотипичный ингибитор белка NS5A вируса гепатита С, фармацевтическая композиция и способы их получения и применения | 2019 |
|
RU2723482C1 |
| ПРИМЕНЕНИЕ ЦИТОКИНА ИЗ СЕМЕЙСТВА ИНТЕРЛЕЙКИНА-6 ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИИ ДЛЯ КОМБИНИРОВАННОГО ВВЕДЕНИЯ С ИНТЕРФЕРОНОМ-АЛЬФА | 2006 |
|
RU2413529C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2355418C2 |
| МОЛЕКУЛЫ АНАЛОГОВ ЦИКЛОСПОРИНА, МОДИФИЦИРОВАННЫЕ ПО 1 и 3 АМИНОКИСЛОТЕ | 2011 |
|
RU2630690C9 |
Группа изобретений относится к медицине, а именно к инфекционным болезням, и может быть использована для получения лекарственного средства для лечения и профилактики гепатита С. Предложено применение санглиферина, связывающегося с циклофилином, формулы
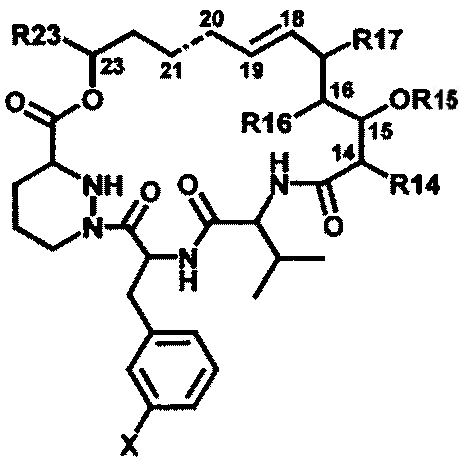
в получении лекарственного средства для лечения и профилактики гепатита С и связанных с ним заболеваний, ингибирования репликации вируса гепатита С, профилактики рецидива гепатита С. Использование изобретений позволяет расширить арсенал средств для профилактики и лечения гепатита С. 3 н.п. ф-лы.
1. Применение терапевтически эффективного количества соединения, связывающегося с циклофилином, для получения лекарственного средства для профилактики или лечения инфекции вируса гепатита С (HCV) и связанных с ним заболеваний, таких как фиброз печени, цирроз печени и гепатоцеллюлярная карцинома, при котором соединением, связывающимся с циклофилином, является соединение формулы
где Х означает Н, метокси, этокси, OnPr, OiPr,
R14 и R15 независимо означают Н, метил, алкил, С1-С4алкил, C1-C4-CO-СН3, или R14 и R15 вместе образуют 5-7-членный замещенный или незамещенный циклоалкил, который выбирают из соединения формулы
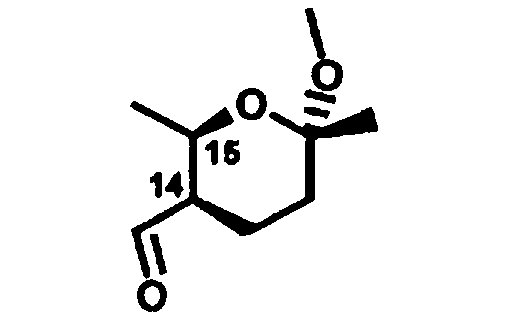
R16 означает Н, или Me или С1-С4алкил;
R17 означает Н, ОН, -O-С1-С4алкил или R14, R15 и R17 вместе образуют циклоалкил формулы

R23 означает -СН=СН2, -СН2-ОН, -СН2-ОСН3, -СНО, -СООН или группу формулы XIX',

2. Применение терапевтически эффективного количества соединения, связывающегося с циклофилином, такого как санглиферин, для получения лекарственного средства для ингибирования репликации HCV.
3. Применение терапевтически эффективного количества соединения, связывающегося с циклофилином, такого как санглиферин, для получения лекарственного средства для профилактики рецидива инфекции HCV у пациента после трансплантации, нуждающегося в ней.
| СПОСОБ ЛЕЧЕНИЯ ХРОНИЧЕСКИХ ВИРУСНЫХ ГЕПАТИТОВ | 1999 |
|
RU2180862C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ПРОГРЕССИРУЮЩЕГО ФИБРОЗА И ЦИРРОЗА ПЕЧЕНИ | 2001 |
|
RU2250768C2 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| INOUE K, et al Interferon combined with cyclosporine treatment as an effective countermeasure against hepatitis С virus recurrence in liver transplant patients with end-stage hepatitis С virus related disease // | |||

