Настоящее изобретение относится к замещенным гетероциклиламидом тиазолам, пирролам и тиофенам, к способу их получения, к их применению для лечения и/или профилактики определенных заболеваний, а также к их применению для приготовления лекарственных средств, предназначенных для лечения и/или профилактики определенных заболеваний, прежде всего для применения в качестве противовирусных средств, главным образом против цитомегаловирусов.
В WO 99/23091 описаны ароматические гетероциклические соединения в качестве противовоспалительных средств, которые помимо прочего могут применяться и для лечения вирусных инфекций, а в WO 04/052852 описаны производные 3-пирролилмочевины в качестве противовирусных средств, несущие карбоцикл в качестве заместителя у мочевины.
Несмотря на наличие на рынке обладающих противовирусным действием средств структурно иного типа используемые в современной терапии препараты, такие как ганцикловир, валганцикловир, фоскарнет и цидофовир, обладают серьезными побочными действиями, например, вызывают нефротоксикоз, нейтропению или тромбоцитопению. Помимо этого к подобным препаратам часто может развиваться резистентность. Поэтому для эффективной терапии требуются новые средства.
Исходя из вышеизложенного, в основу настоящего изобретения была положена задача - предложить новые соединения, которые обладали бы таким же, что и известные средства, или лучшим противовирусным действием для лечения вирусных инфекционных заболеваний человека и животных.
При создании изобретения неожиданно было установлено, что предлагаемые в нем и более подробно рассмотренные в последующем описании замещенные гетероциклы обладают высокой противовирусной эффективностью.
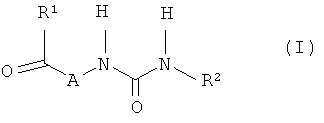
В соответствии с этим объектом настоящего изобретения являются соединения формулы (I)

в которой
R1 представляет собой группу формулы
 или
или  , где
, где
* обозначает место присоединения к карбонильной группе,
R3 обозначает фенил или 5- либо 6-членный гетероарил, каждый из которых может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, гидроксигруппу, оксогруппу, нитрогруппу, цианогруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, трифторметилтиогруппу, C1-С6алкил, C1-С6алкоксигруппу,гидроксикарбонил, C1-С6алкоксикарбонил, аминогруппу, C1-С6алкиламиногруппу, аминокарбонил и C1-С6алкиламинокарбонил,
R4 обозначает фенил или 5- либо 6-членный гетероарил, каждый из
которых может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, гидроксигруппу, оксогруппу, нитрогруппу, цианогруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, трифторметилтиогруппу, C1-С6алкил, C1-С6алкоксигруппу, гидроксикарбонил, C1-С6алкоксикарбонил, аминогруппу, C1-С6алкиламиногруппу, аминокарбонил и C1-С6алкиламинокарбонил, а
R5 и R6 независимо друг от друга обозначают водород, метил или этил,
R2 представляет собой фенил, который может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, гидроксигруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, трифторметилтиогруппу, C1-С6алкил и C1-С6алкоксигруппу,
A представляет собой группу формулы
 ,
,  ,
, 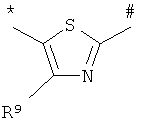 или
или  , где
, где
* обозначает место присоединения к карбонильной группе,
# обозначает место присоединения к атому азота мочевины,
R7 обозначает C1-С6алкил, который может быть замещен заместителем, выбранным из группы, включающей C3-С6циклоалкил, C6-С10арил и 5- либо 6-членный гетероарил, каждый из которых, в свою очередь, может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, гидроксигруппу, оксогруппу, нитрогруппу, цианогруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, трифторметилтиогруппу, C1-С6алкил, C1-С6алкоксигруппу, гидроксикарбонил, C1-С6алкоксикарбонил, аминогруппу, C1-С6алкиламиногруппу, аминокарбонил и C1-С6алкиламинокарбонил, а
R8 и R9 независимо друг от друга обозначают водород, галоген или C1-С6алкил, который может быть замещен заместителем, выбранным из группы, включающей C3-С6циклоалкил, C6-С10арил и 5- либо 6-членный гетероарил, каждый из которых, в свою очередь, может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, гидроксигруппу, оксогруппу, нитрогруппу, цианогруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, трифторметилтиогруппу, C1-С6алкил, C1-С6алкоксигруппу, гидроксикарбонил, C1-С6алкоксикарбонил, аминогруппу, C1-С6алкиламиногруппу, аминокарбонил и C1-С6алкиламинокарбонил,
а также их соли, их сольваты и сольваты их солей.
Предлагаемые в изобретении соединения представляют собой соединения формулы (I) и их соли, сольваты и сольваты этих солей, подпадающие под формулу (I) соединения приведенных ниже формул и их соли, сольваты и сольваты этих солей, а также подпадающие под формулу (I), указанные далее в качестве примеров соединения, их соли, сольваты и сольваты этих солей, при условии, что под подпадающими под формулу (I) указанными ниже соединениями уже не имеются в виду соли, сольваты и сольваты солей.
Предлагаемые в изобретении соединения в зависимости от их структуры могут существовать в стереохимических формах (энантиомеров, диастереомеров). В соответствии с этим в объем изобретения включены также соответствующие энантиомеры и диастереомеры и их соответствующие смеси. Из таких смесей энантиомеров и/или диастереомеров можно известным путем выделять их компоненты в виде индивидуальных стереизомеров.
В объем настоящего изобретения включены также все возможные таутомерные формы предлагаемых в изобретении соединений.
В качестве солей согласно настоящему изобретению предпочтительны физиологически безвредные соли предлагаемых в нем соединений. Вместе с тем в объем изобретения включены и те соли, которые как таковые не пригодны для фармацевтического применения, но могут использоваться, например, для выделения или очистки предлагаемых в изобретении соединений.
К физиологически безвредным солям предлагаемых в изобретении соединений относятся кислотно-аддитивные соли, образованные с минеральными кислотами, карбоновыми кислотами и сульфоновыми кислотами, например, соли, образованные с хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, толуолсульфоновой кислотой, бензолсульфоновой кислотой, нафталиндисульфоновой кислотой, уксусной кислотой, трифторуксусной кислотой, пропионовой кислотой, молочной кислотой, винной кислотой, яблочной кислотой, лимонной кислотой, фумаровой кислотой, малеиновой кислотой и бензойной кислотой.
К физиологически безвредным солям предлагаемых в изобретении соединений относятся также соли, образованные с обычными основаниями, предпочтительно такие, например, как соли с щелочными металлами (например, натриевые и калиевые соли), соли с щелочноземельными металлами (например, кальциевые и магниевые соли) и аммониевые соли, представляющие собой производные аммиака или органических аминов с 1-16 атомами углерода, предпочтительно таких, например, как этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метилморфолин, аргинин, лизин, этилендиамин и N-метилпиперидин.
Под сольватами согласно изобретению подразумеваются такие формы предлагаемых в нем соединений, которые в твердом или жидком состоянии образуют комплекс в результате образования координационной связи с молекулами растворителя. Гидраты представляют собой особую форму сольватов, образующихся в результате образования координационной связи с молекулами воды.
В объем настоящего изобретения включены также пролекарства предлагаемых в нем соединений. Под термином "пролекарства" подразумеваются такие соединения, которые сами могут обладать или не обладать биологической активностью, но при своем нахождении в организме превращаются в предлагаемые в изобретении соединения (например, метаболическим либо гидролитическим путем).
Согласно изобретению используемые для обозначения заместителей понятия имеют, если не указано иное, следующие значения.
Алкил как таковой и фрагменты "алк" и "алкил" в составе алкоксигруппы, алкиламиногруппы, алкоксикарбонила и алкиламинокарбонила представляют собой линейный либо разветвленный алкильные остаток обычно с 1-6 атомами углерода (C1-С6алкил), предпочтительно с 1-4 атомами углерода, особенно предпочтительно с 1-3 атомами углерода, предпочтительно, например, метил, этил, н-пропил, изопропил, трет-бутил, н-пентил и н-гексил.
Алкоксигруппа предпочтительно представляет собой, например, метокси-, этокси-, н-пропокси-, изопропокси-, трет-бутокси-, н-пентокси- и н-гексоксигруппу.
Алкиламиногруппа представляет собой алкиламиноостаток с одним либо двумя (выбранными независимо друг от друга) алкильными заместителями, предпочтительно, например, метиламино-, этиламино-, н-пропиламино-, изопропиламино-, трет-бутиламино-, н-пентиламино-, н-гексиламино-, N,N-диметиламино-, N,N-диэтиламино-, N-этил-N-метиламино-, N-метил-N-н-пропиламино-, N-изопропил-N-н-пропиламино-, N-трет-бутил-N-метиламино-, N-этил-N-н-пентиламино- и N-н-гексил-N-метиламиногруппу. C1-С3алкиламиногруппа представляет собой, например, моноалкиламиноостаток с 1-3 атомами углерода или диалкиламиноостаток, имеющий по 1-3 атома углерода в каждом алкильном заместителе.
Алкоксикарбонил предпочтительно представляет собой, например, метоксикарбонил, этоксикарбонил, н-пропоксикарбонил, изопропоксикарбонил, трет-бутоксикарбонил, н-пентоксикарбонил и н-гексоксикарбонил.
Алкиламинокарбонил представляет собой алкиламинокарбонильный остаток с одним либо двумя (выбранными независимо друг от друга) алкильными заместителями, предпочтительно, например, метиламинокарбонил, этиламинокарбонил, н-пропиламинокарбонил, изопропиламинокарбонил, трет-бутиламинокарбонил, н-пентиламинокарбонил, н-гексиламинокарбонил, N,N-диметиламинокарбонил, N,N-диэтиламинокарбонил, N-этил-N-метиламинокарбонил, N-метил-N-н-пропиламинокарбонил, N-изопропил-N-н-пропиламинокарбонил, N-трет-бутил-N-метиламинокарбонил, N-этил-N-н-пентиламинокарбонил и N-н-гексил-N-метиламинокарбонил. С1-С3алкиламинокарбонил представляет собой, например, моноалкиламинокарбонильный остаток с 1-3 атомами углерода или диалкиламинокарбонильный остаток, имеющий по 1-3 атома углерода в каждом алкильном заместителе.
Арил представляет собой моно- либо бициклический ароматический карбоциклический остаток обычно с 6-10 атомами углерода, предпочтительно, например, фенил и нафтил.
5- либо 6-членный гетероарил согласно изобретению в целом представляет собой ароматический моноциклический остаток с 5 либо 6 кольцевыми атомами и с 1-4 гетероатомами из группы, включающей S, О и/или N. Гетероарильный остаток может быть присоединен через атом углерода либо гетероатом. В качестве предпочтительных примеров гетероарила можно назвать тиенил, фурил, пирролил, тиазолил, оксазолил, пиразолил, имидазолил, пиридил, пиримидил и пиридазинил.
Циклоалкил представляет собой циклоалкильную группу обычно с 3-6 атомами углерода, в качестве предпочтительных примеров которой можно назвать циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Галоген может представлять собой фтор, хлор, бром или иод.
К предпочтительным согласно настоящему изобретению относятся предлагаемые в нем соединения формулы (I), в которой
R1 представляет собой группу формулы
 , где
, где
* обозначает место присоединения к карбонильной группе,
R3 обозначает фенил или 5- либо 6-членный гетероарил, каждый из которых может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, гидроксигруппу, оксогруппу, нитрогруппу, цианогруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, трифторметилтиогруппу, C1-С6алкил, C1-С6алкоксигруппу, гидроксикарбонил, C1-С6алкоксикарбонил, аминогруппу, C1-С6алкиламиногруппу, аминокарбонил и C1-С6алкиламинокарбонил,
R2 представляет собой фенил, который может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, гидроксигруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, трифторметилтиогруппу, C1-С6алкил и C1-С6алкоксигруппу,
А представляет собой группу формулы
, , или , где
* обозначает место присоединения к карбонильной группе,
# обозначает место присоединения к атому азота мочевины,
R7 обозначает C1-С6алкил, который может быть замещен заместителем, выбранным из группы, включающей C3-С6циклоалкил, C6-С10арил и 5- либо 6-членный гетероарил, каждый из которых, в свою очередь, может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, гидроксигруппу, оксогруппу, нитрогруппу, цианогруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, трифторметилтиогруппу, C1-С6алкил, C1-С6алкоксигруппу, гидроксикарбонил, C1-С6алкоксикарбонил, аминогруппу, C1-С6алкиламиногруппу, аминокарбонил и C1-С6алкиламинокарбонил, а
R8 и R9 независимо друг от друга обозначают водород, галоген или C1-С6алкил,
а также их соли, их сольваты и сольваты их солей.
К предпочтительным согласно изобретению относятся также предлагаемые в нем соединения формулы (I), в которой
R1 представляет собой группу формулы
, где
* обозначает место присоединения к карбонильной группе,
R3 обозначает фенил или пиридил, каждый из которых может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, нитрогруппу, цианогруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, C1-С4алкил и C1-С4алкоксигруппу,
R2 представляет собой фенил, который может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей фтор, хлор, трифторметоксигруппу, дифторметоксигруппу, трифторметилтиогруппу и метил,
А представляет собой группу формулы
, , или , где
* обозначает место присоединения к карбонильной группе,
# обозначает место присоединения к атому азота мочевины,
R7 обозначает метил, этил или н-бутил, каждый из которых может быть замещен заместителем, выбранным из группы, включающей циклопропил и фенил, который, в свою очередь, может быть замещен трифторметилом, а
R8 и R9 независимо друг от друга обозначают водород, бром, хлор, метил или этил,
а также их соли, их сольваты и сольваты их солей.
К предпочтительным согласно изобретению относятся далее предлагаемые в нем соединения формулы (I), в которой
R1 представляет собой группу формулы
, где
* обозначает место присоединения к карбонильной группе, а
R3 обозначает фенил или пиридил, каждый из которых может быть
замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей галоген, нитрогруппу, цианогруппу, трифторметил, дифторметил, трифторметоксигруппу, дифторметоксигруппу, монофторметоксигруппу, C1-С4алкил и C1-С4алкоксигруппу.
К предпочтительным согласно изобретению относятся также предлагаемые в нем соединения формулы (I), в которой R2 представляет собой фенил, который может быть замещен 1-3 заместителями, независимо друг от друга выбранными из группы, включающей фтор, хлор, трифторметоксигруппу, дифторметоксигруппу, трифторметилтиогруппу и метил.
К предпочтительным согласно изобретению относятся далее предлагаемые в нем соединения формулы (I), в которой
А представляет собой группу формулы
, , или , где
* обозначает место присоединения к карбонильной группе,
# обозначает место присоединения к атому азота мочевины,
R7 обозначает метил, этил или н-бутил, каждый из которых может быть замещен заместителем, выбранным из группы, включающей циклопропил и фенил, который, в свою очередь, может быть замещен трифторметилом,
R8 обозначает водород, бром, хлор или метил, а
R9 обозначает водород.
Объектом изобретения является также способ получения соединений формулы (I), заключающийся в том, что
[А] соединения формулы (II)
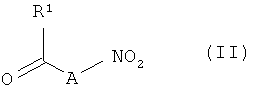 ,
,
в которой R1 имеет указанные выше значения, на первой стадии подвергают взаимодействию с восстановителем, а на второй стадии подвергают в присутствии производного угольной кислоты взаимодействию с соединениями формулы (III)
 ,
,
в которой R2 имеет указанные выше значения, или
[Б] соединения формулы (II) на первой стадии подвергают взаимодействию с восстановителем, а на второй стадии подвергают взаимодействию с соединениями формулы (IV)
 ,
,
в которой R2 имеет указанные выше значения, или
[В] соединения формулы (V)
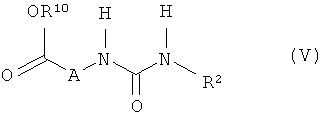 ,
,
в которой R2 имеет указанные выше значения, a R10 обозначает метил или этил, на первой стадии подвергают взаимодействию с соответствующим основанием, а на второй стадии подвергают в присутствии дегидратирующих агентов взаимодействию с соединениями формулы (VI)
 ,
,
в которой R1 имеет указанные выше значения.
Соединения формул (III), (IV) и (VI) в принципе известны или же их можно получать известными способами из соответствующих эдуктов.
Пояснения к вариантам [А] и [Б], стадия 1
Реакцию проводят обычно в инертных растворителях, предпочтительно при температуре в интервале от 0°С до температуры отгонки растворителей и при давлении в интервале от нормального до 3 бар.
В качестве восстановителей можно использовать, например, палладий на активированном угле и водород, смесь муравьиной кислоты, триэтиламина и палладия на активированном угле, цинк, смесь цинка и соляной кислоты, железо, смесь железа и соляной кислоты, смесь сульфата железа (II) и соляной кислоты, сульфид натрия, дисульфид натрия, дитионит натрия, полисульфид аммония, смесь борогидрида натрия и хлорида никеля, дихлорид олова, трихлорид титана или никель Ренея и водный раствор гидразина, предпочтительны из которых никель Ренея, водный раствор гидразина, палладий на активированном угле и водород или смесь муравьиной кислоты, триэтиламина и палладия на активированном угле.
В качестве инертных растворителей можно использовать, например, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол или трет-бутанол, углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или нефтяные фракции, или же другие растворители, такие как диметилформамид, диметилацетамид, ацетонитрил или пиридин, а в случае смешивающихся с водой растворителей можно также использовать их смеси с водой, при этом предпочтительны в качестве растворителей метанол, этанол, изопропанол, а в случае никеля Ренея и водного раствора гидразина - тетрагидрофуран.
Пояснения к варианту [А], стадия 2
Реакцию проводят обычно в инертных растворителях, предпочтительно при температуре в интервале от комнатной до 40°С и при нормальном давлении.
В качестве примера производных угольной кислоты можно назвать N,N-карбонилдиимидазол, фосген, дифосген, трифосген, фениловый эфир хлормуравьиной кислоты или 4-нитрофениловый эфир хлормуравьиной кислоты, предпочтителен среди которых N,N-карбонилдиимидазол.
В качестве инертных растворителей можно использовать, например, галогенированные углеводороды, такие как метиленхлорид, трихлорметан, тетрахлорметан, трихлорэтан, тетрахлорэтан, 1,2-дихлорэтан или трихлорэтилен, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или нефтяные фракции, или же другие растворители, такие как этилацетат, ацетон, диметилформамид, диметилацетамид, 2-бутанон, диметилсульфоксид, ацетонитрил или пиридин, а в случае смешивающихся с водой растворителей можно использовать также их смеси с водой, при этом предпочтителен среди указанных растворителей диметилсульфоксид.
Пояснения к варианту [Б], стадия 2
Реакцию проводят обычно в инертных растворителях, необязательно в присутствии основания, предпочтительно при температуре в интервале от комнатной до температуры отгонки растворителей и при нормальном давлении.
В качестве инертных растворителей можно использовать, например, галогенированные углеводороды, такие как метиленхлорид, трихлорметан, тетрахлорметан, трихлорэтан, тетрахлорэтан, 1,2-дихлорэтан или трихлорэтилен, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или нефтяные фракции, или же другие растворители, такие как этилацетат, ацетон, диметилформамид, диметилацетамид, 2-бутанон, диметилсульфоксид, ацетонитрил или пиридин, при этом предпочтителен среди указанных растворителей тетрагидрофуран или метиленхлорид.
В качестве оснований можно использовать, например, карбонаты щелочных металлов, такие как карбонат цезия, карбонат натрия либо калия или трет-бутилат калия, а также иные основания, такие как гидрид натрия, ДБУ (диазабициклоундецен), триэтиламин или диизопропилэтиламин, предпочтителен среди которых триэтиламин.
Пояснения к варианту [В], стадия 1
Реакцию проводят обычно в инертных растворителях, предпочтительно при температуре в интервале от 0°С до температуры отгонки растворителей и при нормальном давлении.
В качестве оснований можно использовать, например, гидроксиды щелочных металлов, такие как гидроксид натрия, лития либо калия, или карбонаты щелочных металлов, такие как карбонат цезия, карбонат натрия либо калия, предпочтителен среди которых гидроксид натрия.
В качестве инертных растворителей можно использовать, например, галогенированные углеводороды, такие как метиленхлорид, трихлорметан, тетрахлорметан, трихлорэтан, тетрахлорэтан, 1,2-дихлорэтан или трихлорэтилен, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол или трет-бутанол, углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или нефтяные фракции, или же другие растворители, такие как диметилформамид, диметилацетамид, диметилсульфоксид, ацетонитрил или пиридин, либо смеси таких растворителей с водой, при этом предпочтительна в качестве растворителя смесь этанола и воды.
Пояснения к варианту [В], стадия 2
Реакцию проводят обычно в инертных растворителях, необязательно в присутствии основания, предпочтительно при температуре в интервале от -70 до 40°С и при нормальном давлении.
В качестве дегидратирующих агентов на этой стадии можно использовать, например, карбодиимиды, такие, например, как N,N′-диэтил-, N,N′-дипропил-, N,N′-диизопропил-, N,N′-дициклогексилкарбодиимид, гидрохлорид N-(3-диметиламиноизопропил)-N′-этилкарбодиимида (ГДЭК), N-циклогексилкарбодиимид-N′-пропилоксиметил-полистирол (ПС-карбодиимид), карбонильные соединения, такие как карбонилдиимидазол, 1,2-оксазолиевые соединения, такие как 3-сульфат 2-этил-5-фенил-1,2-оксазолия или перхлорат 2-тред-бутил-5-метилизоксазолия, ациламиносоединения, такие как 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин, пропанфосфоновый ангидрид, изобутилхлорформиат, бис-(2-оксо-3-оксазолидинил)фосфорилхлорид либо гексафторфосфат бензотриазолилокситри(диметиламино)фосфония, гексафторфосфат O-(бензотриазол-1-ил)-N,N,N′,N′-тетраметилурония (ГБТУ), тетрафторборат 2-(2-оксо-1-(2H)-пиридил)-1,1,3,3-тетраметилурония (ТПТУ) либо гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилурония (ГАТУ), 1-гидроксибензотриазол (ГОБТ), гексафторфосфат бензотриазол-1-илокси-трис-(диметиламино)фосфония (ГБФ) или смеси указанных соединений с основаниями.
В качестве оснований можно использовать, например, карбонаты щелочных металлов, такие как карбонат или гидрокарбонат натрия либо калия, органические основания, такие как триалкиламины, например, триэтиламин, N-метилморфолин, N-метилпиперидин, 4-диметиламинопиридин (ДМАП) или диизопропилэтиламин, либо ДБУ (1,8-диазабицикло[5.4.0]ундец-7-ен), ДБН (1,5-диазабицикло[4.3.0]нон-5-ен) или пиридин, предпочтителен среди которых триэтиламин.
Конденсацию предпочтительно проводить с использованием ТБТУ (тетрафторбората O-бензотриазол-N,N,N′,N′-тетраметилурония) и ДМАП.
В качестве инертных растворителей можно использовать, например, галогенированные углеводороды, такие как метиленхлорид, трихлорметан, тетрахлорметан, трихлорэтан, тетрахлорэтан, 1,2-дихлорэтан или трихлорэтилен, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля, углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или нефтяные фракции, или же другие растворители, такие как этилацетат, ацетон, диметилформамид, диметилацетамид, 2-бутанон, диметилсульфоксид, ацетонитрил или пиридин, а в случае смешивающихся с водой растворителей можно также использовать их смеси с водой, при этом предпочтителен среди указанных растворителей диметилформамид.
В другом варианте полученные на первой стадии варианта [В] карбоновые кислоты можно на второй стадии сначала взаимодействием с соответствующим хлорирующим агентом, таким, например, как тионилхлорид, превращать в хлорангидрид карбоновой кислоты, а затем взаимодействием с соединениями формулы (VI) в присутствии соответствующего основания превращать в соединения формулы (I).
Соединения формулы (II) известны или же их можно получать из соединений формулы
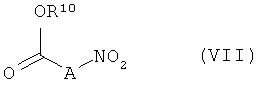
в которой R10 имеет указанное выше значение, взаимодействием на первой стадии с соответствующим основанием и взаимодействием на второй стадии с соединениями формулы (VI) в присутствии дегидратирующих агентов.
Такую реакцию проводят аналогично варианту [В].
Соединения формулы (VII) известны или же их можно получать взаимодействием соединений формулы

в которой R10 имеет указанное выше значение, с дымящей азотной кислотой, концентрированной азотной кислотой, нитрующей кислотой или смесью серной кислоты и азотной кислоты в другом соотношении между ними, необязательно в уксусном ангидриде в качестве растворителя, предпочтительно при температуре в интервале от комнатной до 60°С и при нормальном давлении.
Соединения формулы (VIII) известны или же их можно синтезировать известными способами из соответствующих эдуктов.
Соединения формулы (V) известны или же их можно получать из соединений формулы (VIII) взаимодействием на первой стадии с соответствующим восстановителем и взаимодействием на второй стадии в присутствии соответствующего производного угольной кислоты с соединениями формулы (III) либо взаимодействием на второй стадии с соединениями формулы (IV).
Такую реакцию проводят аналогично вариантам [А] и [Б].
Схема синтеза 1

Схема синтеза 2
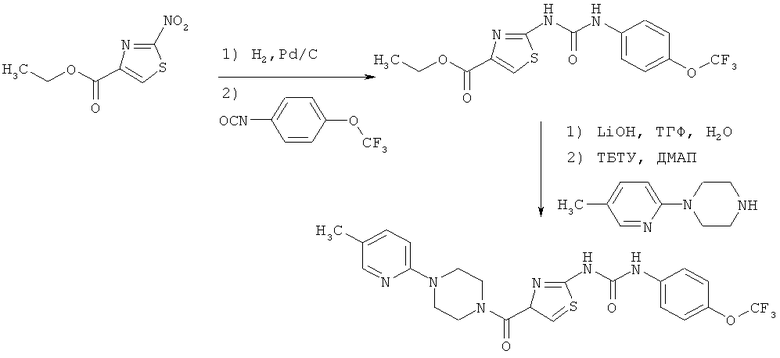
Предлагаемые в изобретении соединения общей формулы (I) обладают неожиданным спектром действия, который невозможно было предсказать заранее. Предлагаемые в изобретении соединения проявляют противовирусную активность в отношении представителей группы вирусов Herpes viridae (вирусов герпеса), прежде всего в отношении цитомегаловируса (CMV); главным образом в отношении человеческого цитомегаловируса (HCMV). Тем самым предлагаемые в изобретении соединения пригодны для лечения и профилактики определенных заболеваний, в первую очередь вирусных инфекций, прежде всего инфекций, возбудителями которых являются вышеуказанные вирусы, а также обусловленных ими инфекционных заболеваний. Под понятием "вирусная инфекция" в последующем описании подразумеваются и сама возбуждаемая определенным вирусом инфекция, и обусловленное такой вирусной инфекцией заболевание.
Соединения общей формулы (I) благодаря их особым свойствам можно использовать для приготовления лекарственных средств, пригодных для профилактики и/или лечения соответствующих заболеваний, прежде всего вирусных инфекций.
В качестве примера показаний к применению предлагаемых в изобретении соединений можно назвать следующие:
1) лечение и профилактика HCMV-инфекций у больных СПИДом (ретинит, пневмонит, желудочно-кишечные инфекции);
2) лечение и профилактика вызываемых цитомегаловирусом инфекций у пациентов с трансплантированным костным мозгом и трансплантированными органами, подверженных часто угрожающим их жизни таким заболеваниям, как HCMV-пневмонит, -энцефалит, а также желудочно-кишечные и системные HCMV-инфекции;
3) лечение и профилактика HCMV-инфекций у новорожденных и детей младшего возраста;
4) лечение острой HCMV-инфекции у беременных женщин;
5) лечение HCMV-инфекции у пациентов с подавленными иммунными реакциями, страдающих раком и проходящих курс противораковой терапии;
6) лечение HCMV-положительных онкологических больных с целью уменьшить опосредуемое HCMV-вирусом прогрессирование опухоли (см. J. Cinatl и др., FEMS Microbiology Reviews, 28, 2004, сс.59-77).
Предлагаемые в изобретении соединения предпочтительно использовать для приготовления лекарственных средств, предназначенных для профилактики и/или лечения инфекций, возбуждаемых одним из представителей группы вирусов герпеса, особенно цитомегаловирусом, прежде всего человеческим цитомегаловирусом.
Предлагаемые в изобретении соединения благодаря их фармакологическим свойствам можно использовать для лечения и/или предупреждения вирусных инфекций, прежде всего HCMV-инфекций, индивидуально, а при необходимости - и в комбинации с действующими веществами других типов, прежде всего с противовирусными действующими веществами, такими, например, как ганцикловир, валганцикловир или ацикловир.
Еще одним объектом настоящего изобретения является применение предлагаемых в нем соединений для лечения и/или профилактики определенных заболеваний, предпочтительно вирусных инфекций, прежде всего инфекций, вызываемых человеческим цитомегаловирусом или иным представителем из группы вирусов герпеса.
Еще одним объектом настоящего изобретения является применение предлагаемых в нем соединений для лечения и/или профилактики определенных заболеваний, прежде всего вышеуказанных заболеваний.
Еще одним объектом настоящего изобретения является применение предлагаемых в нем соединений для приготовления лекарственного средства, предназначенного для лечения и/или профилактики определенных заболеваний, прежде всего вышеуказанных заболеваний.
Еще одним объектом настоящего изобретения является способ лечения и/или профилактики определенных заболеваний, прежде всего вышеуказанных заболеваний, с применением предлагаемых в изобретении соединений в противовирусно эффективном количестве.
Предлагаемые в изобретении соединения могут проявлять свое действие системно и/или местно. С этой целью их можно применять соответствующим образом, например, перорально, парентерально, пульмонально, интраназально, сублингвально, лингвально, буккально, ректально, дермально, чрескожно, конъюктивально, в уши или в виде имплантата, соответственно стента.
Для подобного применения предлагаемых в изобретении соединений их можно вводить в организм в составе соответствующих лекарственных форм.
Для перорального применения пригодны лекарственные формы с известным из уровня техники принципом действия и с быстрым и/или модифицированным высвобождением из них предлагаемых в изобретении соединений, которые содержатся в таких лекарственных формах в кристаллическом и/или аморфном и/или растворенном виде. В качестве примера подобных лекарственных форм можно назвать таблетки (без покрытия или с покрытием, например, с покрытием, устойчивым к действию желудочного сока, или с замедленно растворяющимся или нерастворимым покрытием, регулирующим или контролирующим высвобождение предлагаемого в изобретении соединения), быстро распадающиеся в ротовой полости таблетки, облатки с пленочным покрытием, лиофилизаты с пленочным покрытием, капсулы (например, твердо- или мягкожелатиновые капсулы), драже, грануляты, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное применение предлагаемых в изобретении соединений предполагает возможность их введения в организм без всасывания (например, при внутривенном, внутриартериальном, внутрисердечном, интраспинальном или интралюмбальном введении) или же со всасыванием (например, при внутримышечном, подкожном, внутрикожном, чрескожном или внутрибрюшинном введении). В качестве пригодных для парентерального применения лекарственных форм можно назвать помимо прочего лекарственные формы для инъекций и инфузий в виде растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для применения предлагаемых в изобретении соединений остальными методами пригодны, например, ингаляционные лекарственные формы (в частности, порошковые ингаляторы, небулайзеры), назальные капли, растворы и спреи, лингвальные и сублингвальные таблетки или таблетки для медленного растворения в щечном кармане, облатки с пленочным покрытием или капсулы, суппозитории, ушные и глазные препараты, вагинальные капсулы, водные суспензии (примочки, болтушки), липофильные суспензии, мази, кремы, трансдермальные терапевтические системы, молочко, пасты, пены, присыпки, имплантаты или стенты.
Предлагаемые в изобретении соединения можно переводить в указанные выше формы применения. Для этого предлагаемые в изобретении соединения можно известным образом смешивать с инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами. К таким вспомогательным веществам относятся, в частности, носители (например, микрокристаллическая целлюлоза, лактоза, маннит), растворители (например, жидкие полиэтиленгликоли), эмульгаторы, диспергаторы или смачиватели (например, додецилсульфат натрия, полиоксисорбитанолеат), связующие (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (например, антиокислители, такие как аскорбиновая кислота), красители (например, неорганические пигменты, такие как оксиды железа) и корригенты вкуса и/или запаха.
Еще одним объектом настоящего изобретения являются лекарственные средства, содержащие по меньшей мере одно предлагаемое в изобретении соединение, обычно совместно с одним либо несколькими инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами, а также их применение в указанных выше целях.
В целом для достижения эффективных результатов предлагаемые в изобретении соединения предпочтительно вводить в организм в дозе, которая при внутривенном применении составляет примерно от 0,001 до 10 мг/кг веса тела, предпочтительно примерно от 0,01 до 5 мг/кг веса тела, а при пероральном применении составляет примерно от 0,01 до 25 мг/кг веса тела, предпочтительно от 0,1 до 10 мг/кг веса тела. Однако в некоторых случаях, а именно: в зависимости от веса тела пациента, пути введения действующего вещества в организм, индивидуальной реакции на действующее вещество, типа содержащей его лекарственной формы и времени, соответственно интервала времени введения лекарственного средства в организм может потребоваться применять предлагаемые в изобретении соединения в дозах, отличных от указанных выше количеств. Так, в частности, иногда может оказаться достаточным использовать предлагаемые в изобретении соединения в дозе, меньшей указанного выше нижнего предела, тогда как в других случаях может потребоваться использовать предлагаемые в изобретении соединения в дозе, превышающей вышеуказанный верхний предел. При необходимости применения предлагаемых в изобретении соединений в больших дозах может оказаться целесообразным дробить их на несколько более мелких доз из расчета на несколько приемов в сутки.
Приведенные в последующей экспериментальной части описания и в примерах процентные данные представляют собой, если не указано иное, массовые проценты, части представляют собой массовые части, а соотношения между растворителями, пропорции разбавления или разведения и концентрации жидкостно-жидкостных растворов в каждом случае указаны в пересчете на объем.
А. Примеры
Используемые сокращения
Методы ЖХВД- и ЖХ-МС-анализа
Метод 1 (ЖХ-МС): тип масс-спектрометра: Micromass ZQ; тип ЖХВД-хроматографа: Waters Alliance 2795; колонка: Phenomenex Synergi 2µ Hydro-RP Mercury, 20×4 мм; элюент А: 1 л воды+0,5 мл 50%-ной муравьиной кислоты, элюент Б: 1 л ацетонитрила+0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% элюента А→2,5 мин 30% элюента А→3,0 мин 5% элюента А→4,5 мин 5% элюента А; скорость потока: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; печь: 50°С; УФ-обнаружение: 210 нм.
Метод 2 (ЖХ-МС): прибор: масс-спектрометр Quattro LCZ с ЖХВД-хроматографом Agilent Serie 1100; колонка: Phenomenex Synergi 2µ Hydro-RP Mercury, 20×4 мм; элюент A: 1 л воды+0,5 мл 50%-ной муравьиной кислоты, элюент Б: 1 л ацетонитрила+0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% элюента А →2,5 мин 30% элюента А→3,0 мин 5% элюента А→4,5 мин 5% элюента А; скорость потока: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; печь: 50°С; УФ-обнаружение: 208-400 нм.
Метод 3 (ЖХ-MС): тип масс-спектрометра: Micromass ZQ; тип ЖХВД-хроматографа: HP 1100 Series с УФ-детектором на диодной матрице; колонка: Phenomenex Synergi 2µ Hydro-RP Mercury, 20x4 мм; элюент А: 1 л воды+0,5 мл 50%-ной муравьиной кислоты, элюент Б: 1 л ацетонитрила+0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% элюента А→2,5 мин 30% элюента А→3,0 мин 5% элюента А→4,5 мин 5% элюента А; скорость потока: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; печь: 50°С; УФ-обнаружение: 210 нм.
Исходные соединения
Пример 1А
1-метил-2-трихлорацетил-1H-пиррол

1,09 мл (12,3 ммоля) трихлорацетилхлорида добавляют в атмосфере аргона в 5 мл ДХМ, после чего при КТ в течение 30 мин по каплям добавляют раствор N-метилимидазола в 3 мл ДХМ. Затем смесь оставляют перемешиваться на ночь при КТ, после чего раствор концентрируют и остаток очищают фильтрованием через флеш-фритту (циклогексан, циклогексан/этилацетат в соотношении 40:1). Таким путем получают требуемый продукт в виде жидкости.
Выход: 2,12 г (76% от теории).
ЖХ-МС (метод 1): Rt=2,34 мин.
МС (EI+): m/z=225 (M+).
Пример 2А
1-метил-4-нитро-2-трихлорацетил-1Н-пиррол

2,12 г (9,34 ммоля) 1-метил-2-трихлорацетил-1Н-пиррола растворяют в 9,5 л уксусного ангидрида, охлаждают до -20°С и смешивают с 0,43 мл (9,34 моля) азотной кислоты. Затем смеси дают медленно нагреться до КТ и перемешивают еще в течение 1 ч при КТ. После этого реакционную смесь сливают на 95 г льда и в течение 2,5 ч интенсивно перемешивают (сначала образуется маслянистый осадок, а затем происходит кристаллизация). Осадок отделяют вакуум-фильтрацией, размешивают с 20 мл метанола, фильтруют и оставляют сушиться на ночь в вакууме. Для удаления одновременно образовавшегося региоизомера смесь в течение 2 ч размешивают с 10 мл смеси уксусной кислоты и воды в их соотношении 1:1, затем твердое вещество отделяют вакуум-фильтрацией и сушат в вакууме. Таким путем получают требуемый продукт в виде твердого вещества.
Выход: 1,71 г (67% от теории).
ЖХ-МС (метод 2): Rt=2,51 мин.
1H-ЯМР (400 МГц, ДМСО-d6):δ=8,58 (d, 1Н), 7,80 (d, 1H), 4,00 (s, 3H).
Пример 3А
Этиловый эфир 1-метил-4-нитро-1Н-пиррол-2-карбоновой кислоты

0,50 г (1,84 ммоля) 1-метил-4-нитро-2-трихлорацетил-1Н-пиррола добавляют в 5 мл этанола, после чего смешивают с 0,26 мл (1,84 ммоля) триэтиламина и в течение 2 ч перемешивают при КТ. Затем реакционную смесь смешивают с 5 мл воды, перемешивают в течение 30 мин при 0°С и в завершение осадок отделяют вакуум-фильтрацией и сушат в вакууме.
Выход: 321 мг (88% от теории).
ЖХ-МС (метод 3): Rt=2,25 мин.
МС (ESI+): m/z=199 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6):δ=8,29 (d, 1H), 7,31 (d, 1H), 4,27 (q, 2H), 3,92 (s, 3H), 1,30 (t, 3H).
Пример 4А
Этиловый эфир 1-метил-4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1H-пиррол-2-карбоновой кислоты
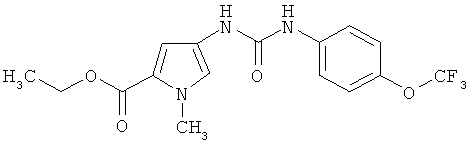
304 мг (1,53 ммоля) этилового эфира 1-метил-4-нитро-1H-пиррол-2-карбоновой кислоты добавляют в 6 мл смеси этилацетата и этанола в их соотношении 1:1, после чего смешивают с 163 мг (0,15 ммоля) палладия (10%-ного на активированном угле) и 580 мг (9,20 ммоля) формиата аммония и в течение 1 ч перемешивают при 80°С. После охлаждения смесь фильтруют через кизельгур, промывают этанолом и из фильтрата в вакууме удаляют растворитель. Остаток растворяют в 6 мл ТГФ, смешивают с 374 мг (1,84 ммоля) 4-трифторметоксифенилизоцианата и в течение 1 ч перемешивают при КТ. Затем раствор концентрируют и остаток очищают посредством ОФ-ЖХВД (ацетонитрил/вода). Таким путем получают требуемый продукт в виде твердого вещества.
Выход: 486 мг (85%» от теории).
ЖХ-МС (метод 3): Rt=2,61 мин.
МС (ESI+): m/z=372 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6):δ=8,80 (s, 1H), 8,39 (s, 1H), 7,53 (m, 2H), 7,26 (d, 2H), 7,20 (d, 1H), 6,72 (d, 1H), 4,20 (q, 2H), 3,82 (s, 3H), 1,28 (t, 3H).
Пример 5А
1-метил-4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1H-пиррол-2-карбоновая кислота

470 мг (1,27 ммоля) этилового эфира 1-метил-4-[({[4-(трифторметокси)-фенил]амино}карбонил)амино]-1H-пиррол-2-карбоновой кислоты добавляют 5 мл ТГФ, после чего добавляют 152 мг (6,33 ммоля) гидроксида лития в 1 мл воды и оставляют перемешиваться на ночь при нагревании с обратным холодильником. Затем реакционную смесь концентрируют, остаток подкисляют 2-молярной соляной кислотой, образовавшийся осадок отделяют вакуум-фильтрацией и сушат в вакууме. Таким путем получают требуемый продукт в виде твердого вещества.
Выход: 429 мг (98% от теории).
ЖХ-МС (метод 2): Rt=2,09 мин.
МС (ESI+): m/z=344 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6):δ=12,16 (шир. s, 1H), 9,01 (s, 1H), 8,58 (s; 1H), 7,53 (m, 2H), 7,25 (d, 2H), 7,18 (d, 1H), 6,63 (d, 1H), 3,80 (s, 3H).
Пример 6А
1-(5-метилпиридин-2-ил)пиперазин

Стадия 1
1-(трет-бутилоксикарбонил)-4-(5-метилпиридин-2-ил)пиперазин
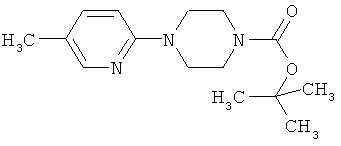
В атмосфере аргона 2,50 г (19,6 ммоля) 2-метил-5-хлорпиридина и 4,38 г (23,5 ммоля) N-(трет-бутилоксикарбонил)пиперазина растворяют в 50 мл абсолютного толуола. Затем добавляют 2,26 г (23,5 ммоля) трет-бутилата натрия, 0,37 г (0,59 ммоля) БИНАФ и 0,36 г (0,39 ммоля) трис-(дибензилиденацетон)дипалладия и нагревают до 70°С с выдержкой при этой температуре в течение 12 ч. После охлаждения реакционную смесь смешивают с диэтиловым эфиром, трижды промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и в вакууме удаляют растворитель. Остаток очищают экспресс-хроматографией (смесь циклогексана и этилацетата в соотношении 9:1).
Выход: 5,27 г (97% от теории).
ЖХ-МС (метод 1): Rt=1,26 мин.
МС (ESI+): m/z=278 (М+Н)+.
1H-ЯМР (300 МГц, CDCl3):δ=8,02 (d, 1H), 7,34 (dd, 1H), 6,59 (d, 1H), 3,55 (m, 4H), 3,45 (m, 4H), 2,21 (s, 3H), 1,49 (s, 9H).
Стадия 2
1-(5-метилпиридин-2-ил)пиперазин
3,47 г (12,5 ммоля) 1-(трет-бутилоксикарбонил)-4-(5-метилпиридин-2-ил)пиперазина растворяют в 10 мл диоксана и смешивают с 31 мл (125 ммолей) хлористого водорода в диоксане (4-молярном). Затем смесь перемешивают в течение 2 ч при КТ. Далее смесь концентрируют, остаток подщелачивают 1-молярным раствором едкого натра и несколько раз экстрагируют дихлорметаном. Объединенные органические фазы сушат над сульфатом натрия, концентрируют и сушат в вакууме.
Выход: 2,18 г (98% от теории).
ЖХ-МС (метод 3): Rt=0,38 мин.
МС (ESI+): m/z=177 (М+Н)+.
1H-ЯМР (300 МГц, CDCl3): δ=8,02 (d, 1H), 7,32 (dd, 1H), 6,59 (d, 1H), 3,45 (m, 4H), 3,00 (m, 4H), 2,20 (s, 3H).
Пример 7А
1-этил-4-[({[4-(трифторметокси)фенил]амино)карбонил)амино]-1H-пиррол-2-карбоновая кислота

Указанное соединение получают аналогично примеру 5А.
ЖХ-МС (метод 2): Rt=2,10 мин.
МС (ESI+): m/z=358 (М+Н)+.
Пример 8А
1-бутил-4-[({[4-(трифторметокси)фенил]амино)карбонил)амино]-1H-пиррол-2-карбоновая кислота

Указанное соединение получают аналогично примеру 5А.
ЖХ-МС (метод 3): Rt=2,47 мин.
МС (ESI+): m/z=386 (М+Н)+.
1H-ЯМР (300 МГц, ДМОС-d6): δ=8,90 (шир. s, 1H), 8,48 (шир. s, 1H), 7,54 (d, 2H), 7,26 (d, 2H), 7,24 (d, 1H), 6,73 (d, 1H), 4,21 (t, 2H), 1,62 (quint., 2H), 1,25 (sext., 2H), 0,88 (t, 3H).
Пример 9А
1-(циклопропилметил)-4-[({[4-трифторметокси)фенил]амино}карбонил)-амино]-1H-пиррол-2-карбоновая кислота
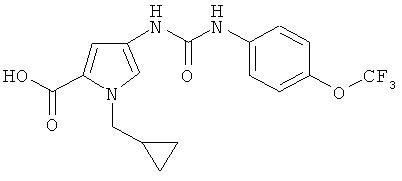
Указанное соединение получают аналогично примеру 5А.
ЖХ-МС (метод 2): Rt=2,33 мин.
МС (ESI+): m/z=384 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,86 (шир. s, 1H), 8,45 (шир. s, 1H), 7,55 (m, 2H), 7,23-7,32 (m, 3H), 6,68 (d, 1H), 4,11 (d, 2H), 1,21 (m, 1H), 0,45 (m, 2H), 0,32 (m, 2H).
Пример 10А
4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]тиофен-2-карбоновая кислота

Указанное соединение получают аналогично примерам 4А и 5А исходя из метилового эфира 4-аминотиофен-2-карбоновой кислоты (синтезированного по методу, описанному у A.A.Kiryano и др. в Tetrahedron Lett., 42, 2001, сс.8797-8800).
Выход: 72 мг (27% от теории, 2 стадии).
ЖХ-МС (метод 2): Rt=2,25 мин.
МС (ESI+): m/z=347 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ=13,1 (шир. s, 1H), 9,10 (шир. s, 1H), 8,98 (шир. s, 1H), 7,69 (s, 1H), 7,53-7,60 (m, 3H), 7,29 (d, 2H).
Пример 11А
2-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1,3-тиазол-4-карбоновая кислота

Указанное соединение получают аналогично примерам 4А и 5А исходя из этилового эфира 2-амино-1,3-тиазол-4-карбоновой кислоты (коммерчески доступный продукт, выпускаемый фирмой ACROS).
Выход: 290 мг (61% от теории, по 2 стадиями).
ЖХ-МС (метод 2): Rt=2,05 мин.
МС (ESI+): m/z=348 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=12,8 (шир. s, 1H), 10,9 (шир. s, 1H), 9,22 (шир. s, 1H), 7,91 (s, 1H), 7,60 (d, 2H), 7,32 (d, 2H).
Пример 12А
5-метил-2-[({[4-(трифторметокси)фенил]амино1карбонил)амино]-1,3-тиазол-4-карбоновая кислота

Указанное соединение получают аналогично примерам 4А и 5А исходя из метилового эфира 2-амино-5-метил-1,3-тиазол-4-карбоновой кислоты (коммерчески доступный продукт, поставляемый фирмой Tyger Scientific).
Выход: 148 мг (40% от теории, 2 стадии).
ЖХ-МС (метод 3): Rt=2,47 мин.
МС (ESI+): m/z=362 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=12,7 (шир. s, 1H), 10,7 (шир. s, 1H), 9,18 (шир. s, 1H), 7,59 (d, 2H), 7,32 (d, 2H), 3,34 (s, 3H).
Пример 13А
5-хлор-2-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1,3-тиазол-4-карбоновая кислота

Указанное соединение получают аналогично примерам 4А и 5А исходя из этилового эфира 2-амино-5-хлор-1,3-тиазол-4-карбоновой кислоты (синтез этого соединения описан у K.J.Hodgetts и др. в Org. Lett., 4, 2002, сс.1363-1366).
Выход: 365 мг (89% от теории, по 2-м стадиям).
ЖХ-МС (метод 3): Rt=2,51 мин.
МС (ESI+): m/z=382 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=13,2 (шир. s, 1Н), 11,2 (шир. s, 1Н), 9,28 (шир. s, 1H), 7,58 (m, 2H), 7,33 (m, 2H).
Пример 14А
5-бром-2-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1,3-тиазол-4-карбоновая кислота
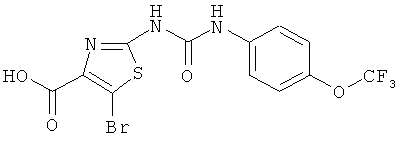
Указанное соединение получают аналогично примерам 4А и 5А исходя из этилового эфира 2-амино-5-бром-1,3-тиазол-4-карбоновой кислоты (синтез этого соединения описан у J.F. Okonya и др. в Tetrahedron Lett., 43, 2002, сс.7051-7054).
Выход: 343 мг (74% от теории, по 2 стадиям).
ЖХ-МС (метод 2): Rt=2,22 мин.
МС (ESI+): m/z=426 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=13,1 (шир. s, 1H), 11,2 (шир. s, 1H), 9,28 (шир. s, 1H), 7,58 (m, 2H), 7,32 (m, 2H).
Пример 15А
2-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1,3-тиазол-5-карбоновая кислота

Указанное соединение получают аналогично примерам 4А и 5А исходя из этилового эфира 2-амино-1,3-тиазол-5-карбоновой кислоты (коммерчески доступный продукт, выпускаемый фирмой RareChem).
Выход: 200 мг (55% от теории, по 2-м стадиям).
ЖХ-МС (метод 3): Rt=2,36 мин.
МС (ESI+): m/z=348 (М+Н)+.
1H-ЯМР (300 МГц, ДМСО-d6): δ=12,8 (шир. s, 1H), 10,9 (шир. s, 1H), 9,21 (шир. s, 1H), 7,92 (s, 1H), 7,60 (d, 2H), 7,33 (d, 2H).
Конечные соединения
Пример 1
N-{1-метил-5-[(4-пиридин-2-илпиперазин-1-ил)карбонил]-1H-пиррол-3-ил}-N′-[4-(трифторметокси)фенил]мочевина

50 мг (0,15 ммоля) 1-метил-4-[({[4-(трифторметокси)фенил]амино}-карбонил)амино]-1Н-пиррол-2-карбоновой кислоты (пример 5А) добавляют в 1 мл ДМФ, после чего смешивают с 56 мг (0,18 ммоля) тетрафторбората О-(бензотриазол-1-ил)-N,N,N′,N′-тетраметилурония (ТБТУ) и с 8,9 мг (0,07 ммоля) 4-(диметиламино)пиридина (ДМАП). Затем добавляют 29 мг (0,18 ммоля) 1-(2-пиридил)пиперазина и перемешивают в течение 8 ч при КТ. В завершение реакционную смесь очищают посредством ОФ-ЖХВД (ацетонитрил/вода). Таким путем получают указанное в заголовке соединение в виде твердого вещества.
Выход: 54 мг (76% от теории).
ЖХ-МС (метод 1): Rt=1,61 мин.
МС (ESI+): m/z=489 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,75 (s, 1H), 8,32 (s, 1H), 8,13 (dd, 1H), 7,50-7,59 (m, 3H), 7,25 (d, 2H), 7,02 (d, 1H), 6,86 (d, 1H), 6,68 (dd, 1H), 6,28 (d, 1H), 3,72 (m, 4H), 3,64 (s, 3H), 3,55 (m, 4H).
Пример 2
N-(1-метил-5-([4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил) -1H-пиррол-3-ил)-N′-[4-(трифторметокси)фенил]мочевина
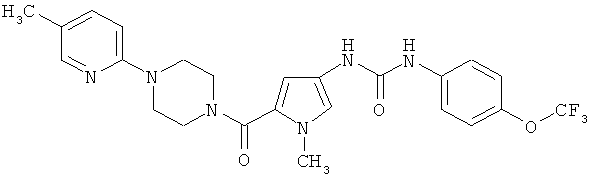
Указанное соединение получают аналогично примеру 1 исходя из соединений из примеров 5А и 6А.
Выход: 50 мг (68% от теории).
ЖХ-МС (метод 1): Rt=1,66 мин.
МС (ESI+): m/z=503 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,75 (шир. s, 1H), 8,32 (шир. s, 1H), 7,98 (d, 1H), 7,53 (m, 2H), 7,42 (dd, 1H), 7,25 (d, 2H), 7,02 (d, 1H), 6,80 (d, 1H), 6,28 (d, 1H), 3,68-3,76 (m, 4H), 3,63 (s, 3H), 3,45-3,53 (m, 4H), 2,17 (s, 3H).
Пример 3
N-{1-этил-5-[(4-пиридин-2-илпиперазин-1-ил)карбонил]-1H-пиррол-3-ил}-N′-[4-(трифторметокси)фенил] мочевина

Указанное соединение получают аналогично примеру 1 исходя из соединений из примеров 7А и 6А.
Выход: 29 мг (43% от теории).
ЖХ-МС (метод 2): Rt=1,79 мин.
МС (ESI+): m/z=503 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,74 (шир. s, 1H), 8,32 (шир. s, 1H), 8,13 (d, 1H), 7,49-7,60 (m, 3H), 7,22-7,28 (m, 2H), 7,08 (s, 1H), 6,87 (d, 1H), 6,68 (dd, 1H), 6,26 (s, 1H), 4,04 (q, 2H), 3,68-3,77 (m, 4H), 3,50-3,58 (m, 4H), 1,26 (t, 3H).
Пример 4
N-(1-этил-5-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил)-1H-пиррол-3-ил)-N′-[4-(трифторметокси)фенил] мочевина
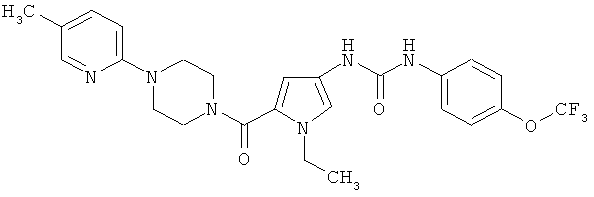
Указанное соединение получают аналогично примеру 1 исходя из соединений из примеров 7А и 6А.
Выход: 26 мг (39% от теории).
ЖХ-МС (метод 2): Rt=1,80 мин.
МС (ESI+): m/z=517 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,75 (шир. s, 1H), 8,32 (шир. s, 1H), 7,98 (m, 1H), 7,54 (m, 2H), 7,42 (m, 1H), 7,26 (m, 2H), 7,08 (m, 1H), 6,80 (d, 1H), 6,25 (s, 1H), 4,03 (q, 2H), 3,68-3,76 (m, 4H), 3,44-3,52 (m, 4H), 2,18 (s, 3H), 1,25 (t, 3H).
N-{1-бутил-5-[(4-пиридин-2-илпиперазин-1-ил)карбонил]-1H-пиррол-3-ил}-N′-[4-(трифторметокси)фенил] мочевина

Указанное соединение получают аналогично примеру 1 исходя из соединения из примера 8А.
Выход: 41 мг (69% от теории).
ЖХ-МС (метод 2): Rt=2,11 мин.
МС (ESI+): m/z=531 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ=8,75 (шир. s, 1H), 8,33 (шир. s, 1H), 8,12 (d, 1H), 7,58 (m, H), 7,54 (d, 2H), 7,26 (d, 2H), 7,07 (d, 1H), 6,87 (d, 1H), 6,68 (dd, 1H), 6,26 (d, 1H), 4,02 (t, 2H), 3,72 (m, 4H), 3,53 (m, 4H), 1,59 (quint., 2H), 1,18 (sext, 2H), 0,84 (t, 3H).
Пример 6
N-(1-бутил-5-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил] карбонил}-1H-пиррол-3-ил)-N′-[4-(трифторметокси)фенил]мочевина
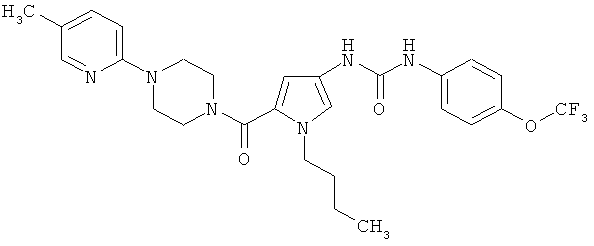
Указанное соединение получают аналогично примеру 1 исходя из соединений из примеров 8А и 6А.
Выход: 19 мг (20% от теории).
ЖХ-МС (метод 2): Rt=1,97 мин.
МС (ESI+): m/z=545 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,74 (шир. s, 1H), 8,32 (шир. s, 1H), 7,98 (d, 1H), 7,53 (d, 2H), 7,41 (dd, 1H), 7,25 (d, 2H), 7,07 (d, 1H), 6,80 (d, 1H), 6,25 (d, 1H), 4,02 (t, 2H), 3,72 (m, 4H), 3,47 (m, 4H), 2,16 (s, 3H), 1,59 (quint., 2H), 1,18 (sext., 2H), 0,83 (t, 3H).
Пример 7
N-{1-(циклопропилметил)-5-[(4-пиридин-2-илпиперазин-1-ил)карбонил]-1H-пиррол-3-ил)-N′-[4-(трифторметокси)фенил] мочевина

Указанное соединение получают аналогично примеру 1 исходя из соединения из примера 9А.
Выход: 51 мг (86% от теории).
ЖХ-МС (метод 1): Rt=1,84 мин.
МС (ESI+): m/z=529 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,75 (шир. s, 1Н), 8,33 (шир. s, 1H), 8,13 (dt, 1H), 7,51-7,61 (m, 3H), 7,27 (d, 2H), 7,13 (d, 1H), 6,88 (d, 1H), 6,69 (dd, 1H), 6,28 (d, 1H), 3,90 (d, 2H), 3,73 (m, 4H), 3,53 (m, 4H), 1,12 (m, 1H), 0,45 (m, 2H), 0,28 (m, 2H).
Пример 8
N-(1-(циклопропилметил)-5-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1H-пиопол-3-ил)-N′-[4-(трифторметокси)фенил]мочевина

Указанное соединение получают аналогично примеру 1 исходя из соединений из примеров 9А и 6А.
Выход: 52 мг (85% от теории).
ЖХ-МС (метод 1): Rt=1,86 мин.
МС (ESI+): m/z=543 (М+Н)+.
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,77 (шир. s, 1H), 8,34 (шир. s, 1H), 7,98 (d, 1H), 7,54 (d, 2H), 7,41 (dd, 1H), 7,25 (d, 2H), 7,12 (d, 1H), 6,80 (d, 1Н), 6,27 (d, 1H), 3,89 (d, 2Н), 3,72 (m, 4Н), 3,48 (m, 4Н), 2,16 (s, 3Н), 1,12 (m, 1H), 0,45 (m, 2Н), 0,28 (m, 2Н).
Пример 9
N-(5-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-3-тиенил)-N′-[4-(трифторметокси)фенил]мочевина
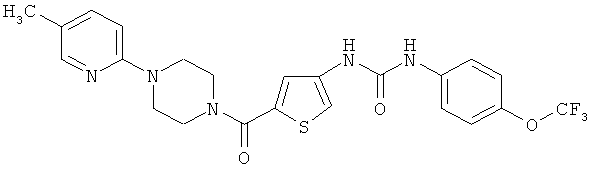
Указанное соединение получают аналогично примеру 1 исходя из соединений из примеров 10А и 6А.
Выход: 60 мг (63%) от теории).
ЖХ-МС (метод 3): Rt=2,05 мин.
МС (ESI+): m/z=506 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=9,05 (шир. s, 1H), 8,95 (шир. s, 1H), 7,98 (d, 1H), 7,56 (d, 2Н), 7,47 (s, 1Н), 7,39-7,44 (m, 2Н), 7,29 (d, 2Н), 6,80 (d, 1H), 3,75 (m, 4Н), 3,51 (m, 4Н), 2,16 (s, 3Н).
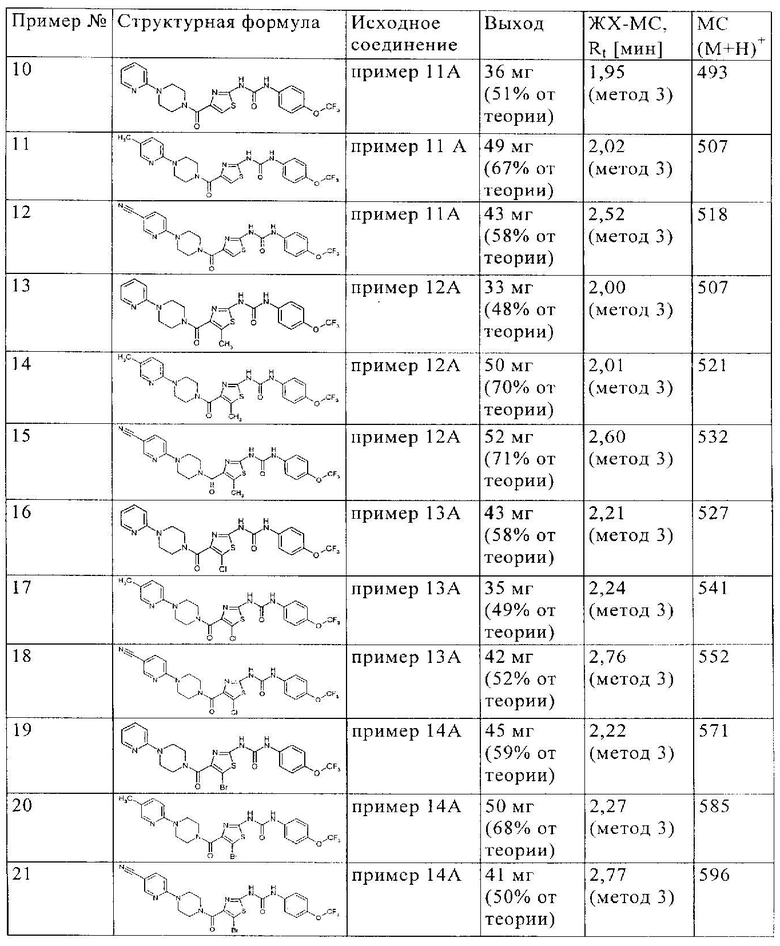
Представленные ниже в таблице соединения синтезируют аналогично примеру 1 из соответствующих исходных соединений:

Б. Оценка физиологической эффективности предлагаемых в изобретении соединений
Действие предлагаемых в изобретении соединений in vitro можно продемонстрировать на примере следующих опытов.
Опыты по подавлению цитопатогенности HCMV-вируса (человеческого цитомегаловируса) предлагаемыми в изобретении соединениями
Тестируемые соединения используют в виде 50-миллимолярных (50 мМ) растворов в диметилсульфоксиде (ДМСО). В качестве сравнительных соединений используют ганцикловир, фоскарнет и цидофовир. После добавления по 2 мкл 50-, 5-, 0,5- и 0,05-миллимолярных исходных растворов в ДМСО к каждым 98 мкл культуральной среды для выращивания клеток в ряду 2 А-Н в двух повторностях проводят разведения в пропорции 1:2 культуральной средой, по 50 мкл которой добавляют вплоть до ряда 11 96-луночного планшета. Лунки в рядах 1 и 12 содержат только по 50 мкл культуральной среды. Затем в лунки ряда 1 пипеткой добавляют по 150 мкл суспензии 1×104 клеток (человеческих фибробластов крайней плоти [NHDF]) (клетки в лунках ряда 1 служат контрольными), соответственно в лунки рядов с 2 по 12 пипеткой добавляют по 150 мкл суспензии 1×104 клеток в виде смеси из инфицированных HCMV-вирусом и неинфицированных клеток (множественность заражения равняется 0,001-0,002, т.е. на каждые 1000 неинфицированных клеток приходится 1-2 инфицированные клетки). Лунки ряда 12 (без тестируемых соединений) содержат только вирус, который служит контрольным. Конечная концентрация тестируемых соединений составляет от 250 до 0,0005 мкМ. Планшеты инкубируют в течение 6 дней при 37°С в атмосфере с 5%-ным содержанием CO2, т.е. все клетки, включая в лунках с контрольным вирусом, оказываются инфицированы им (100%-ный цитопатогенный эффект [ЦПЭ]). После этого клетки в лунках фиксируют и окрашивают добавлением смеси из формалина и красителя Гимза (30 мин), затем промывают дважды дистиллированной водой и сушат в сушильном шкафу при 50°С. Далее клетки в планшетах визуально оценивают с помощью микроскопа типа "overhead" (Plaque multiplier фирмы Technomara).
При анализе опытных планшетов можно получить следующие данные:
показатель CC50 (NHDF): максимальная концентрация тестируемого соединения в мкМ, при которой оно не оказывает на клетки никакого заметного цитостатического действия в сравнении с необработанными контрольными клетками;
показатель EC50 (HCMV): концентрация тестируемого соединения в мкМ, при которой оно подавляет цитопатический эффект на 50% в сравнении с необработанным контрольным вирусом;
SI (индекс избирательности): CC50 (NHDF)/EC50 (HCMV).
Репрезентативные данные о действии предлагаемых в изобретении соединений in vitro приведены ниже в таблице А.
Пригодность предлагаемых в изобретении соединений для лечения вызываемых HCMV-вирусом инфекций можно продемонстрировать в следующем опыте на животных-моделях.
Модель с ксенотрансплантатом Gelfoam®, инфицированным HCMV-вирусом
Животные
Для опытов используют 3-4-недельных самок иммунодефицитных мышей (16-18 г) линии Fox Chase SCID, Fox Chase SCID-NOD или SCID-beige, которых приобретают у коммерческих, разводящих их поставщиков (Bomholtgaard, Jackson). Животных содержат в изоляторах в стерильных условиях (включая использование стерильных подстилки и корма).
Размножение вируса
Человеческий цитомегаловирус (HCMV), штамм Davis, размножают in vitro на человеческих эмбриональных фибробластах крайней плоти (NHDF-клетках). После инфицирования NHDF-клеток со множественностью заражения, равной 0,01, инфицированные вирусом клетки собирают по истечении 5-7 дней и хранят при -40°С в присутствии минимальной поддерживающей среды (MEM), дополненной 10% фетальной телячьей сыворотки (ФТС) с 10% ДМСО. После последовательного разведения инфицированных вирусом клеток с десятичным шагом определяют титр на 24-луночных планшетах с конфлюэнтными NHDF-клетками после их предварительного витального окрашивания нейтральным красным или фиксации и окрашивания смесью формалина с красителем Гимза (см. выше).
Подготовка губок, трансплантация, обработка и анализ результатов
Коллагеновые губки размером 1×1×1 см (Gelfoam®, фирма Peasel & Lorey, кат. №407534; К.Т.Chong и др., Abstracts of 39th Interscience Conference on Antimicrobial Agents and Chemotherapy, 1999, c. 439; P.M.Kraemer и др., Cancer Research, 43, 1983, cc. 4822-4827) сначала смачивают забуференным фосфатом физиологическим раствором (ЗФР), путем вакуумирования удаляют включенные воздушные пузырьки и затем хранят в MEM-среде, дополненной 10% ФТС. Далее 1×106 инфицированных вирусом NHDF-клеток (инфицирование штаммом Davis HCMV-вируса со множественностью заражения, равной 0,01) отделяют через 3 ч после инфицирования и в 20 мкл MEM-среды с 10% ФТС накапывают на влажную губку. При необходимости через 12-13 ч на инфицированные губки наносят основной фактор роста фибробластов (bFGF) в концентрации 5 нг/мкл в 25 мкл ЗФР с 0,1% БСА и 1 мМ ДТТ (дитиотреитол) и инкубируют в течение 1 ч. Для трансплантации губок иммунодефицитным мышам их наркотизируют авертином или смесью из ацепромазина с ксилазином и кетамина, шерсть на спине удаляют электробритвой, в эпидермисе делают надрез длиной 1-2 см, ослабляют и под кожу спины трансплантируют влажные губки. Операционную рану закрывают клеем для склеивания тканей. Через 24 ч после трансплантации мышам на протяжении 8 дней трижды в день (в 7.00, 14.00 и 19.00 часов), дважды в день (в 8.00 и 17.00 часов) либо однократно в день (в 14.00 часов) перорально вводят тестируемое соединение. Тестируемое соединение применяют в дозе 3, 10, 30 или 100 мг/кг веса тела при вводимом объеме 10 мл/кг веса тела. Тестируемые соединения вводят мышам в виде 0,5%-ной суспензии в тилозе, необязательно с добавкой 2% ДМСО. Через 9 дней после трансплантации и через 16 ч после последнего введения тестируемого соединения животных подвергают безболезненному умерщвлению и извлекают губку. Инфицированные вирусом клетки выделяют из губки путем ее разложения коллагеназой (330 ед./1,5 мл) и хранят при -140°С в присутствии MEM-среды, дополненной 10% ФТС с 10% ДМСО. Эффективность применения тестируемых соединений оценивают после последовательного разведения инфицированных вирусом клеток с десятичным шагом, определяя титр на 24-луночных планшетах с конфлюэнтными NHDF-клетками после их предварительного витального окрашивания нейтральным красным или фиксации и окрашивания смесью формалина с красителем Гимза (см. выше). При этом определяют количество инфекционных вирусных частиц после применения тестируемых соединений в сравнении с контрольной группой животных, которым вводили плацебо.
В. Примеры фармацевтических композиций
Предлагаемые в изобретении соединения можно перерабатывать в следующие фармацевтические композиции.
Таблетка
Состав: 100 мг соединения из примера 1,50 мг лактозы (в виде моногидрата), 50 мг кукурузного крахмала (природного), 10 мг поливинилпирролидона (PVP 25, продукт фирмы BASF, Людвигсхафен, Германия) и 2 мг стеарата магния.
Масса одной таблетки 212 мг, диаметр 8 мм, радиус выпуклости 12 мм.
Получение
Смесь действующего вещества с лактозой и крахмалом гранулируют в воде с использованием 5%-ного по массе раствора поливинилпирролидона. После сушки гранулят в течение 5 мин смешивают со стеаратом магния. Из этой смеси на обычном таблетировочном прессе прессуют таблетки (параметры таблетки см. выше). Ориентировочное значение усилия прессования составляет 15 кН.
Суспензия для перорального применения
Состав: 1000 мг соединения из примера 1, 1000 мг этанола (96%-ного), 400 мг ксантановой камеди Rhodigel (продукт фирмы FMC, шт.Пенсильвания, США) и 99 г воды.
Разовой дозе, содержащей 100 мг предлагаемого в изобретении соединения, соответствуют 10 мл пероральной суспензии.
Получение
Камедь Rhodigel суспендируют в этаноле и затем к полученной суспензии добавляют действующее вещество. Далее при перемешивании добавляют воду. Смесь перемешивают в течение примерно 6 ч до завершения набухания камеди Rhodigel.
Раствор для внутривенного введения
Состав: 1 мг соединения из примера 1,15 г полиэтиленгликоля 400 и 250 г воды для инъекций.
Получение
Предлагаемое в изобретении соединение совместно с полиэтиленгликолем 400 при перемешивании растворяют в воде. Полученный раствор стерилизуют фильтрацией (через фильтр с диаметром пор 0,22 мкм) и в асептических условиях разливают в подвергнутые тепловой стерилизации бутылки для инфузий. В завершение их укупоривают прокалываемыми пробками и отрывными колпачками.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЛАМИДОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 2006 |
|
RU2415851C2 |
| ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИНГИБИТОРЫ JAK | 2012 |
|
RU2632870C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СПОСОБЫ, ОТНОСЯЩИЕСЯ К НИМ | 2012 |
|
RU2596946C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИН-4(3H)-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ TRPV4 | 2021 |
|
RU2840769C1 |
| СОЕДИНЕНИЕ ТРИАЗИНОНА И ИНГИБИТОР КАЛЬЦИЕВЫХ КАНАЛОВ Т-ТИПА | 2013 |
|
RU2645158C2 |
| ТРИЦИКЛИЧЕСКИЕ ПИРРОЛО ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2591191C2 |
| АЗОТСОДЕРЖАЩИЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ ВИЧ | 2016 |
|
RU2720145C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2005 |
|
RU2384562C2 |
| КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛЬПАИНА | 2010 |
|
RU2540856C2 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
Настоящее изобретение относится к соединению формулы (I) и одной из его фармацевтически активных солей. Соединение формулы (I) обладает противовирусной активностью в отношении человеческого цитомегаловируса (HCMV) или иного представителя группы Herpes virida. В формуле (I)
 ,
,
R1 представляет собой группу формулы , где * обозначает место присоединения к карбонильной группе, R3 обозначает пиридил, который может быть замещен заместителем, независимо выбранным из группы, включающей C1-С6алкил и цианогруппу, R5 и R6 независимо друг от друга обозначают водород, R2 представляет собой фенил, который может быть замещен заместителем, выбранным из группы, включающей трифторметоксигруппу, дифторметоксигруппу и монофторметоксигруппу, А представляет собой группу формулы  ,
,  ,
,  или , где * обозначает место присоединения к карбонильной группе, # обозначает место присоединения к атому азота мочевины, R7 обозначает C1-С6алкил, который может быть замещен заместителем, выбранным из группы, включающей С3-С6циклоалкил, R8 и R9 независимо друг от друга обозначают водород, галоген или C1-С6алкил. Изобретение также относится к способу получения соединения формулы (I) из соединения формулы (V), к способу получения соединения формулы (V), к лекарственному средству, содержащему соединение изобретения, к применению соединения для приготовления лекарственного средства и к способу борьбы с вирусными инфекциями, выбранными из человеческого цитомегаловируса (HCMV) или иного представителя группы Herpes viridae. 6 н. и 3 з. п. ф-лы, 2 табл.
или , где * обозначает место присоединения к карбонильной группе, # обозначает место присоединения к атому азота мочевины, R7 обозначает C1-С6алкил, который может быть замещен заместителем, выбранным из группы, включающей С3-С6циклоалкил, R8 и R9 независимо друг от друга обозначают водород, галоген или C1-С6алкил. Изобретение также относится к способу получения соединения формулы (I) из соединения формулы (V), к способу получения соединения формулы (V), к лекарственному средству, содержащему соединение изобретения, к применению соединения для приготовления лекарственного средства и к способу борьбы с вирусными инфекциями, выбранными из человеческого цитомегаловируса (HCMV) или иного представителя группы Herpes viridae. 6 н. и 3 з. п. ф-лы, 2 табл.
1. Соединение формулы (I)
в которой R1 представляет собой группу формулы
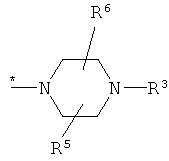
где * обозначает место присоединения к карбонильной группе,
R3 обозначает пиридил, который может быть замещен заместителем, независимо выбранным из группы, включающей C1-С6алкил и цианогруппу,
R5 и R6 независимо друг от друга обозначают водород,
R2 представляет собой фенил, который может быть замещен заместителем, выбранным из группы, включающей трифторметоксигруппу, дифторметоксигруппу и монофторметоксигруппу,
А представляет собой группу формулы
,  ,
, 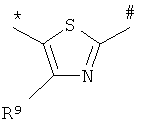 или
или 
где * обозначает место присоединения к карбонильной группе,
# обозначает место присоединения к атому азота мочевины,
R7 обозначает C1-С6алкил, который может быть замещен заместителем, выбранным из группы, включающей С3-С6циклоалкил,
R8 и R9 независимо друг от друга обозначают водород, галоген или C1-С6алкил, или одна из его фармацевтически активных солей.
2. Соединение по п.1, отличающееся тем, что
R1 представляет собой группу формулы
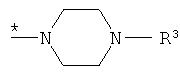
где * обозначает место присоединения к карбонильной группе,
R3 обозначает пиридил, который может быть замещен заместителем, независимо выбранным из группы, включающей С1-С4алкил и цианогруппу,
R2 представляет собой фенил, который может быть замещен заместителем, выбранным из группы, включающей трифторметоксигруппу, дифторметоксигруппу и монофторметоксигруппу,
А представляет собой группу формулы
,  ,
,  или
или 
где * обозначает место присоединения к карбонильной группе,
# обозначает место присоединения к атому азота мочевины,
R7 обозначает метил, этил или н-бутил, каждый из которых может быть замещен заместителем, выбранным из группы, включающей циклопропил, а
R8 и R9 независимо друг от друга обозначают водород, бром, хлор, метил или этил,
или одна из его фармацевтически активных солей.
3. Способ получения соединения формулы (I) по п.1, отличающийся тем, что соединение формулы (V)
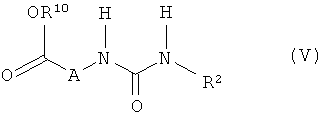
в которой R2 имеет указанные в п.1 значения, a R10 обозначает метил или этил, на первой стадии подвергают взаимодействию с соответствующим основанием, а на второй стадии в присутствии дегидратирующих агентов подвергают взаимодействию с соединением формулы (VI)

в которой R1 имеет указанные в п.1 значения.
4. Способ получения соединения формулы (V), определенного в п.3, отличающийся тем, что соединение формулы (II)

в которой R1 имеет указанные в п.1 значения, на первой стадии подвергают взаимодействию с восстановителем, а на второй стадии подвергают взаимодействию с соединением формулы (IV)
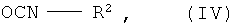
в которой R2 имеет указанные в п.1 значения.
5. Соединение по одному из пп.1 или 2, обладающее противовирусной активностью в отношении человеческого цитомегаловируса (HCMV) или иного представителя группы Herpes viridae.
6. Лекарственное средство, обладающее противовирусной активностью в отношении человеческого цитомегаловируса (HCMV) или иного представителя группы Herpes viridae, содержащее соединение по одному из пп.1 или 2 в сочетании с по меньшей мере одним инертным, нетоксичным, фармацевтически приемлемым вспомогательным веществом.
7. Лекарственное средство по п.6, предназначенное для лечения и/или профилактики указанных вирусных инфекций.
8. Применение соединения по одному из пп.1 или 2 для приготовления лекарственного средства, обладающего противовирусной активностью в отношении человеческого цитомегаловируса (HCMV) или иного представителя группы Herpes viridae.
9. Способ борьбы с вирусными инфекциями, выбранными из человеческого цитомегаловируса (HCMV) или иного представителя группы Herpes viridae, заключающийся в том, что вводят в противовирусно эффективном количестве по меньшей мере одно соединение по одному из пп.1 или 2, либо лекарственное средство по п.6.
| АРОМАТИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ПРОТИВОВОСПАЛИТЕЛЬНЫЕ СРЕДСТВА | 1999 |
|
RU2220142C2 |
| WO 2004002481 А1, 08.01.2004 | |||
| WO 2004052852 А1, 24.06.2004. | |||

