Настоящее изобретение относится к трициклическим производным пиррола, к способу их получения, к фармацевтическим композициям, содержащим их, и к их применению в качестве терапевтических средств, в частности, при лечении рака и расстройств, связанных с клеточной пролиферацией.
Поэтому соединения по изобретению могут применяться при лечении заболеваний, вызванных нарушениями регуляции активности протеинкиназы. Настоящее изобретение также относится к способам получения этих соединений, фармацевтическим композициям, содержащим эти соединения, и к способам лечения заболеваний с использованием фармацевтических композиций, содержащих эти соединения.
Нарушение функции протеинкиназ (PK) является «краеугольным камнем» многочисленных заболеваний. Большой вклад онкогенов и протоонкогенов, вовлеченных в патологические процессы при злокачественных заболеваниях у людей, связан с кодированием PK. Повышенная активность PK также задействована при многих не злокачественных заболеваниях, таких как доброкачественная гиперплазия предстательной железы, семейный аденоматоз, полипоз, нейрофиброматоз, псориаз, пролиферация сосудистых гладкомышечных клеток, связанная с атеросклерозом, легочный фиброз, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз.
PK также задействованы при воспалительных состояниях и в размножении вирусов и паразитов. PK могут также играть основную роль в патогенезе и развитии нейродегенеративных расстройств.
Общую ссылку на нарушение функций или нарушение регуляции PK можно найти, например, в публикациях Chemical Biology 1999, 3, 459-465 и Carcinogenesis 2008, 29, 1087-1091.
Применение митотических ингибиторов при лечении рака является широко принятой клинической стратегией для лечения широкого диапазона злокачественных заболеваний людей. Таксаны (паклитаксел и доцетаксел) и алкалоиды винки (винкристин и винбластин) оказывают действие или стабилизацией, или дестабилизацией микротрубочек с катастрофическими последствиями для клеток, проходящих стадии митоза. Они представляют собой терапевтические средства первой линии по поводу нескольких типов опухолей и второй линии при устойчивом к цисплатину раке яичников, молочных желез, легких, мочевого пузыря и пищевода (таксаны). Однако ввиду роли микротрубочек в таких процессах, как движение клеток, фагоцитоз и аксональный транспорт, при применении этих средств часто наблюдаются определенные виды токсичности, такие как периферическая нейропатия. Прохождение стадий митоза является требованием всех пролиферирующих клеток, и, следовательно, способы лечения рака, имеющие мишени в митозе, в целом могут применяться при широком диапазоне типов опухолей.
Несколько протеинкиназ играют ключевые роли в координации клеточного цикла, и некоторые из них уже применяются в способах прицельной терапии в онкологии, включая Cdk-2 и Aurora-A. Точность воспроизведения митоза имеет первостепенное значение, и в нормальных клетках существуют несколько «контрольных пунктов» для поддержании целостности хромосом во время клеточного цикла. Контрольный пункт сборки веретена (SAC), в частности, требуется для должной хромосомной сегрегации в две дочерние клетки после клеточного деления. Он обеспечивает то, что сестринские хроматиды, совмещенные в метафазной пластинке, не разделяются перед биполярным прикреплением всех удвоенных хромосом к митотическому веретену (Обзор см. в публикации Musacchio A. and Salmon D. Nat Rev Mol Cell Biol, May; 8(5): 379-93, 2007).
Даже одна не совмещенная хромосома достаточна для запуска сигнала SAC, это жестко регулируемый путь, который в конечном счете приводит к ингибированию опосредованного стимулирующим анафазу комплексом/цитохромом (APC/C) полиубиквитилирования и разрушению двух ключевых митотических компонентов: циклина B1 и секурина. Секурин, в частности, требуется для получения разделения сестринских хроматид и перехода в анафазу, а вместо этого, циклин B1 инактивирует основную митотическую киназу CDK1, стимулируя выход из митоза. (Обзор см. в публикации Musacchio A. and Salmon D. Nat Rev Mol Cell Biol, May; 8(5): 379-93, 2007).
Уже была идентифицирована большая группа белков, играющих роль в функциях SAC: человеческая MPS1-киназа (монополярная веретенная 1) (также известная как TTK) действительно играет основную роль. MPS1 представляет собой двойную тирозин и серин/треонинкиназу, высоко сохранную от дрожжей до млекопитающих. Человеческий геном кодирует лишь один член семейства генов MPS1, который не имеет высокого подобия последовательностей с другими протеинкиназами.
MPS1 представляет собой фермент, регулирующий клеточный цикл, который подвергается стимулирующей регуляции и активации при митозе после фосфорилирования (Stucke V.M., et al., Embo J. 21 (7): 1723, 2002).
У Saccharomyces cerevisiae MPS1 регулирует дупликацию тела полюса веретена (Winey M. et al., J. Cell Biol 114: 745, 1991), сборку челнока (Jones, M.H. et al., Curr. Biol. 15: 160, 2005) и контрольный пункт сборки веретена (Weiss and Winey, J. Cell. Biol 132: 111, 1996). Вместо этого, у высших эукариот активность киназы MPS1 главным образом вовлечена в регуляцию и функции SAC (Jelluma, N. et al., Cell 132: 233, 2008).
Эксперименты интерференции РНК указывают на то, что в отсутствие MPS1 функции SAC нарушены: длительность митоза уменьшается, и клетки быстро делятся без совмещения метафазной пластинки, что, в конечном счете, вызывает аберрантную анеуплоидизацию, митотическую катастрофу и больше несовместимо с клеточным выживанием (Jelluma N. et al., Cell 132: 233, 2008; Tighe A. et al., J Cell Biol 2008; Jelluma N. et al., Plos ONE 3(6): e2415, 2008). Кроме того, для подтверждения этих результатов, был описан мелкомолекулярный АТФ-конкурентный ингибитор MPS1, и, несмотря на его не чистый профиль селективности, было показано, что он способен инактивировать функции SAC, инактивировать опосредованную нокодазолом и таксолом остановку митоза и стимулировать гибель клеток главным образом в линиях туморигенных клеток (Schmidt et al., EMBO Rep, 6(9): 866, 2005).
Несмотря на то, что большинство опухолей являются анеуплоидными, при раке никогда не обнаруживали мутированный MPS1, вместо этого была обнаружена его стимулирующая регуляция при ряде опухолей различных происхождений, подобных раку мочевого пузыря, анапластической щитовидной железы, молочных желез и предстательной железы (Yuan B. et al., Clin Cancer Res, 12(4): 1121, 2006). Кроме того, он был обнаружен в характерных признаках верхних 25 генов при CIN (внутриэпителиальной неоплазии шейки матки) и при анеуплоидных опухолях, что прогнозирует клинический исход при раке молочных желез и легких, медуллобластоме, глиоме, мезотелиоме и лимфоме (Carter S.L. et al., Nat Genet. 38(9): 1043, 2006). Наконец, его уровень очень повышен при метастатических опухолях, и была обнаружена его сверхэкспрессия при раковых опухолях молочных желез с мутированным p53 (Bertheau P. et al., Plos Med 4(3): e90, 2007).
Вместе с тем фактом, что была обнаружена стимулирующая регуляция других компонентов SAC, подобных MAD2, BUBR1 или BUB1, при различных опухолях (deCarcer G. et al., Curr Med Chem 14(9): 969, 2007), представляется, что функции SAC могут требоваться и являются существенными для поддержания способности сегрегироваться опухолевых клеток с высокой частотой анеуплоидии, и опухолевая селективность ингибиторов SAC предвидится, в частности, для опухолей с высокой частотой анеуплоидии, подобных карциномам ободочной кишки, легких и молочных желез (Kops G.J. et al., Nat. Rev Cancer, 5: 773, 2005).
Наконец, было показано, что индукция массивной анеуплоидии и нарушение регуляции SAC снижают канцерогенез у склонных к образованию опухолей мышей, подтверждая гипотезу, что ингибирование SAC может обеспечивать ингибирование роста опухоли (Weaver et al., Cancer Cell 11(1): 25, 2007). Таким образом, по этим причинам фармакологическое ослабление функции MPS1 может оказывать благоприятное терапевтическое воздействие при лечении нескольких разнообразных видов рака.
Первоначально идентифицированные в качестве активированных генов провирусным мутагенезом на мышиной модели лимфомы, PIM (PIM1, PIM2 и/или PIM-3 по всей настоящей заявке) представляют собой протеин-серин/треонинкиназы. PIM-киназы слабо экспрессированы в нормальных тканях и сверхэкспрессированы или даже мутированы при дискретном числе различных видов рака у людей, включая лимфому, лейкоз, рак предстательной железы и рак желудка [Shah et al. Eur. J. Cancer, 44, 2144-51, (2008)].
PIM-киназы являются конститутивно активными, и их активность поддерживает рост и выживание опухолевых клеток in vitro и in vivo посредством модификации увеличивающегося числа обычных, а также специфических по изоформе субстратов, включая несколько регуляторов клеточного цикла и медиаторы апоптоза. Представляется, что PIM1, но не PIM2, также опосредует хоминг и миграцию нормальных и злокачественных гематопоэтических клеток регуляцией поверхностной экспрессии хемокиновых рецепторов [Brault et al. Haematologica 95: 1004-1015 (2010)].
Имеется все больше доказательств того, что PIM1 и PIM2 киназы могут быть вовлечены в опосредование онкогенных эффектов некоторых онкогенов, связанных с острыми миелогенными лейкозами (AML). В частности, онкогенной роли FLT3-мутаций (мутаций ITD и KD, присутствующих при 30% случаев AML) и/или транслокаций, вовлекающих ген MLL (встречающийся в 20% случаев AML), [Kumar, et al. J. Mol. Biol. 348, 183-193, (2005)]. PIM1 больше экспрессирован в FLT3-ITD-трансформированных клетках AML, чем в клетках WT костного мозга. Данные свидетельствуют о том, что ингибирование PIM1, а также PIM2, может опосредовать FLT3ITD-зависимую гибель клеток AML. Представляет интерес, что клетки, трансформированные мутациями FLT3, которые придают устойчивость к ингибиторам мелкомолекулярной тирозинкиназы, все еще были чувствительны к нокауту PIM2 или PIM-1 и PIM-2 РНКи, [Kim et al., Blood 105, 1759-67, (2005)].
Кроме того, сообщалось, что PIM2 сверхэкспрессирован и связан с прогрессированием нескольких злокачественных заболеваний, которые происходят из B-клеточной линии дифференциации, таких как хронический лимфоцитарный лейкоз (CLL), диффузная B-крупноклеточная лимфома (DLBCL), мантийноклеточная лимфома (MCL) или миелома [Cohen et al. Leuk. Lymphoma 94 51 (2004), Huttmann et al. Leukemia 20 1774 (2006)].
Представляет интерес, что PIM AKT/PKB, как представляется, играют частично чрезмерные роли в опосредовании роста и выживания гематопоэтических клеток, наиболее вероятно, вследствие перекрывающих субстратов, подобных BAD, p21WAF1/CIP1, p27KIP1 или Cot/Tpl-2 [Choudhary et al., Mol Cell. 36: 326-39 (2009)].
Было показано, что PIM-киназы регулируют ингибирование резистентности (рапамицин), пролиферации и выживания mTOR. Поэтому, комбинация мелкомолекулярных ингибиторов, мишенью которых являются несколько киназ выживания, может быть существенна для мощной терапевтической платформы при раке [Amaravadi R., et al. J. Clin. Invect. 2005, 115(10): 2618-24]. Представляется, что синтез онкогенного белка посредством белка 1 (4E-BP1), связывающего elF4E, является независимым от mTOR и регулируемым PIM-2. Эти наблюдения свидетельствуют о том, что комплекс, инициирующий онкогенную трансформацию elF4F, может блокироваться мелкомолекулярными ингибиторами PIM-2 [Tamburini J. et al. Blood 2009, 114(8), 1718-27 и Brault L. et al. Haematologica 2010, 95(6) 1004-1015].
Производные тетрагидробензоциклогептена, известные в данной области в качестве иммуносупрессивных средств и средств для лечения и предотвращения воспалительных состояний, аллергических расстройств и иммунных расстройств, описаны в заявке на международный патент WO2009/089305. Производные тетрагидробензоциклогептена, известные в данной области в качестве ингибиторов протеинкиназы, описаны в заявке на международный патент WO2005/037843.
Производные трициклициндола, обладающие ингибирующей киназу активностью, были описаны в заявке на международный патент WO2008/065054, поданной от имени самого заявителя настоящего изобретения; некоторые определенные соединения указанной выше заявки на международный патент WO2008/065054 исключены из общей формулы по настоящему изобретению.
Несмотря на эти разработки, все еще сохраняется потребность в эффективных средствах для лечения указанных заболеваний.
Заявители в настоящее время обнаружили, что соединения формулы (I), описанные ниже, являются ингибиторами киназы и, таким образом, могут применяться при лечении в качестве противоопухолевых средств, и лишены, с точки зрения и токсичности, и побочных эффектов, указанных выше недостатков, связанных с применением имеющихся в настоящее время противоопухолевых средств.
Соответственно, первой целью настоящего изобретения является получение замещенного трициклического соединения формулы (I):
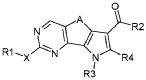 ,
,
где
R1 обозначает водород, галоген или необязательно замещенную группу, выбранную из амино, прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила;
R2 обозначает группу, выбранную из -NR"R"', -N(OR"')R" и OR", где R" и R'" обозначают, каждый независимо, водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или вместе с атомом азота, с которым они связаны, R" и R'" могут образовывать 5-6-членную гетероарильную или гетероциклильную группу, необязательно содержащую один дополнительный гетероатом, выбранный среди N, O и S;
R3 обозначает водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила;
R4 обозначает водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила;
X обозначает одиночную связь или двухвалентный радикал, выбранный из -NR'-, -CONR'-, -NH-CO-NH-, -O-, -S-, -SO2- и -OSO2-, где R' обозначает водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или вместе с атомом азота, с которым они связаны, R1 и R' могут образовывать 5-6-членную гетероарильную или гетероциклильную группу, необязательно содержащую один дополнительный гетероатом, выбранный среди N, O и S;
A обозначает группу, выбранную из -CH2-, -(CH2)2-, -(CH2)3-, -CH=CH-, -C(CH3)2-CH2- и -CH2-C(CH3)2;
или его фармацевтически приемлемой соли,
при условии, что исключены следующие соединения:
этил-2-амино-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат,
2-амино-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота,
2-амино-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
этил-2-амино-8-фенил-9H-пирроло[3,2-h]хиназолин-7-карбоксилат,
2-амино-8-фенил-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
2-амино-9-метил-8-фенил-9H-пирроло[3,2-h]хиназолин-7-карбоксамид и
2-амино-9-метил-8-фенил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид.
Настоящее изобретение также относится к способам синтеза замещенного трициклического соединения, представленного формулой (I), полученного посредством способа, состоящего из стандартных синтетических трансформаций, и изомеров, таутомеров, гидратов, сольватов, комплексов, метаболитов, пролекарств, носителей, N-оксидов.
Настоящее изобретение также относится к способу лечения заболеваний, вызванных и/или связанных с нарушенной регуляцией активности протеинкиназы, в частности, ABL, ACK1, AKT1, ALK, AUR1, AUR2, BRK, BUB1, CDC7/DBF4, CDK2/CYCA, CHK1, CK2, EEF2K, EGFR1, EphA2, EphB4, ERK2, FAK, FGFR1, FLT3, GSK3бета, Haspin, IGFR1, IKK2, IR, JAK1, JAK2, JAK3, KIT, LCK, LYN, MAPKAPK2, MELK, MET, MNK2, MPS1, MST4, NEK6, NIM1, P38альфа, PAK4, PDGFR, PDK1, PERK, PIM1, PIM2, PIM3, РКАальфа, РКСбета, PLK1, RET, ROS1, SULU1, Syk, TLK2, TRKA, TYK, VEGFR2, VEGFR3, ZAP70, конкретнее, MPS1, PIM1, PIM2, PIM3.
Предпочтительный способ по настоящему изобретению представляет собой лечение заболевания, вызванного и/или связанного с нарушением регуляции активности протеинкиназы, выбранного из группы, состоящей из рака, клеточных пролиферативных расстройств, вирусных инфекций, аутоиммунных и нейродегенеративных расстройств.
Другой предпочтительный способ по настоящему изобретению относится к лечению определенных типов рака, включая без ограничения: карциному, такую как карцинома мочевого пузыря, молочных желез, ободочной кишки, почек, печени, легких, включая мелкоклеточный рак легких, пищевода, желчного пузыря, яичников, поджелудочной железы, желудка, шейки матки, щитовидной железы, предстательной железы и кожи, включая плоскоклеточную карциному; гематопоэтические опухоли лимфоидной линии дифференциации, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластический лейкоз, B-клеточную лимфому, T-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому и лимфому Буркетта; гематопоэтические опухоли миелоидной линии дифференцировки, включая острый и хронический миелогенные лейкозы, миелодиспластический синдром и промиелоцитарный лейкоз; опухоли мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включая астроцитому, нейробластому, глиому и шванному; другие опухоли, включая меланому, семиному, тератокарциному, остеосаркому, пигментозную ксеродермию, кератоксантому, фолликулярный рак щитовидной железы, саркому Капоши и мезотелиому, опухоли с высокой частотой анеуплоидии и опухоли, которые сверхэкспрессируют митотический контрольный пункт.
Другой предпочтительный способ по настоящему изобретению относится к лечению определенных клеточных пролиферативных расстройств, таких как, например, доброкачественная гиперплазия предстательной железы, семейный аденоматозный полипоз, нейрофиброматоз, псориаз, пролиферация сосудистых гладкомышечных клеток, связанная с атеросклерозом, легочный фиброз, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз.
Другой предпочтительный способ по настоящему изобретению относится к лечению заболеваний и расстройств, связанных с иммунными клетками, таких как воспалительные и аутоиммунные заболевания, например, рассеянный склероз, системная красная волчанка, воспалительные кишечные заболевания (IBD), болезнь Крона, синдром раздраженного кишечника, панкреатит, язвенный колит, дивертикулез, генерализованная миастения, васкулит, псориаз, склеродермия, астма, аллергия, системный склероз, витилиго, артрит, такой как остеоартрит, ювенильный ревматоидный артрит, анкилозирующий спондилит.
Другой предпочтительный способ по настоящему изобретению относится к лечению вирусных инфекций, в частности, предотвращению развития СПИДа у индивидов, инфицированных ВИЧ.
Другой предпочтительный способ по настоящему изобретению относится к лечению нейродегенеративных расстройств, таких как болезнь Альцгеймера, болезнь Паркинсона и болезнь Хантингтона.
Кроме того, способ по настоящему изобретению также относится к ингибированию ангиогенеза и метастазирования опухолей, а также к лечению отторжения трансплантированных органов и болезни хозяин против трансплантата.
Настоящее изобретение также относится к фармацевтической композиции, содержащей одно или более соединений формулы (I) или их фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент, носитель и/или разбавитель.
Настоящее изобретение, кроме того, относится к фармацевтической композиции, содержащей соединение формулы (I), в комбинации с известными противораковыми способами лечения, такими как лучевая терапия или схема химиотерапии, в комбинации с цитостатическими или цитотоксическими средствами, средствами типа антибиотиков, алкилирующими средствами, антиметаболитными средствами, гормональными средствами, иммунологическими средствами, средствами типа интерферона, ингибиторами циклооксигеназы (например, ингибиторами COX-2), ингибиторами матричной металлопротеазы, ингибиторами теломеразы, ингибиторами тирозинкиназы, средствами против рецепторов фактора роста, средствами против HER, средствами против EGFR (рецепторов эндотелиального фактора роста), средствами против ангиогенеза (например, ингибиторов ангиогенеза), ингибиторами фарнезилтрансферазы, ингибиторами пути передачи сигналов ras-raf, ингибиторами клеточного цикла, другими ингибиторами cdks, средствами, связывающими тубулин, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II и тому подобными.
Настоящее изобретение, кроме того, относится к способу ингибирования активности протеинкиназы in vitro, который включает обеспечение контакта киназы с эффективным количеством соединения формулы (I), как определено выше.
Кроме того, изобретение относится к продукту или набору, включающему соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, или его фармацевтические композиции и одно или более химиотерапевтических средств, в качестве комбинированного препарата для одновременного, отдельного или последовательного применения при лечении злокачественных заболеваний.
В другом аспекте изобретение относится к соединению формулы I) или его фармацевтически приемлемой соли, как определено выше, для применения в качестве лекарственного препарата.
Кроме того, изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, при получении лекарственного препарата с противораковой активностью.
Наконец, изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, как определено выше, для применения в способе лечения рака.
Пока нет иных определений, при ссылке на сами соединения формулы (I), а также любую их фармацевтическую композицию или любое терапевтическое лечение, включающее их, настоящее изобретение включает все из гидратов, сольватов, комплексов, метаболитов, пролекарств, носителей, N-оксидов и фармацевтически приемлемых солей соединений по настоящему изобретению.
Другими словами, в случае возможного легкого получения из соединений формулы (I), как определено выше, их изомеры, таутомеры, гидраты, сольваты, комплексы, метаболиты, пролекарства, носители и N-оксиды являются целью настоящего изобретения.
Метаболит соединения формулы (I) представляет собой любое соединение, в которое именно это соединение формулы (I) превращается in vivo, например, после введения нуждающемуся в нем млекопитающему. Обычно, однако без представления ограничивающего примера, после введения соединения формулы (I), указанное производное может быть превращено в разнообразные соединения, например, включая более растворимые производные, подобные гидроксилированным производным, которые легко выделяются. Следовательно, в зависимости от конкретного метаболического пути, любые из этих гидроксилированных производных могут расцениваться как метаболит соединения формулы (I).
Пролекарства представляют собой любые ковалентно связанные соединения, которые высвобождают in vivo активное материнское лекарственное средство в соответствии с формулой (I).
N-оксиды представляют собой соединения формулы (I), где азот и кислород связаны посредством дативной связи.
Если стереогенный центр или другая форма изомерного центра присутствует в соединении по настоящему изобретению, то подразумевается, что настоящее изобретение охватывает все формы такого изомера или изомеров, включая энантиомеры и диастереомеры. Соединения, содержащие стереогенный центр, могут применяться в виде рацемической смеси, энантиомерно обогащенной смеси, или рацемическая смесь может быть разделена с использованием хорошо известных технологий, и отдельный энантиомер может применяться отдельно. В случаях, при которых соединения имеют ненасыщенные межуглеродные двойные связи, и цис (Z), и транс (E) изомеры входят в объем настоящего изобретения.
В случаях, когда соединения могут существовать в таутомерных формах, таких как кето-енольные таутомеры, каждая таутомерная форма предусматривается как включенная в настоящее изобретение, существуя или в равновесии, или преимущественно в одной форме.
Термин «арил» включает карбоциклические или гетероциклические углеводороды с 1-2 кольцевыми составляющими, или конденсированными, или связанными друг с другом одиночными связями, причем по меньшей мере одно из колец является ароматическим; в случае присутствия, любой ароматический гетероциклический углеводород, также именуемый гетероарильной группой, содержит 5-6-членное кольцо с 1-3 гетероатомами, выбранными из N, O и S.
Примерами арильных групп в соответствии с изобретением являются, например, фенил, бифенил, α- или β-нафтил, дигидронафтил, тиенил, бензотиенил, фурил, бензофуранил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолил, изоиндолил, пуринил, хинолил, изохинолил, дигидрохинолинил, хиноксалинил, бензодиоксолил, инданил, инденил, триазолил и тому подобные.
Под термином «гетероциклил» (также известном как «гетероциклоалкил») подразумевается 3-7-членное, насыщенное или частично ненасыщенное карбоциклическое кольцо, где один или более атомов углерода заменены гетоатомами, такими как азот, кислород или сера. Неограничивающими примерами гетероциклильных групп являются, например, пиран, пирролидин, пирролин, имидазолин, имидазолидин, пиразолидин, пиразолин, тиазолин, тиазолидин, дигидрофуран, тетрагидрофуран, 1,3-диоксолан, пиперидин, пиперазин, морфолин и тому подобные.
Пока нет других указаний, под термином «C3-C7 циклоалкил» подразумевается 3-7-членное, полностью углеродное моноциклическое кольцо, которое может содержать одну или более двойных связей, но не имеет полностью конъюгированную π-электронную систему. Примерами циклоалкильных групп без ограничения являются циклопропан, циклобутан, циклопентан, циклопентен, циклогексан, циклогексен, циклогексадиен, циклоептан, циклоептен, циклоептадиен.
Под термином «прямой или разветвленный C1-C6 алкил», следовательно, охватывающим C1-C4 алкил, подразумевается любая из групп, такая как, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, н-гексил и тому подобные.
Под термином «прямой или разветвленный C2-C6 алкенил» подразумевается любая из групп, такая как, например, винил, аллил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-пентенил, 1-гексенил и тому подобные.
Под термином «прямой или разветвленный C2-C6 алкинил» подразумевается любая из групп, такая как, например, этинил, 2-пропинил, 4-пентинил и тому подобные.
В соответствии с настоящим изобретением и пока нет иных указаний, любая из указанных выше групп R1, R2, R3, R4, R', R" и R'" может быть необязательно замещена в любом из их свободных положений одной или более группами, например 1-6 группами, независимо выбранными из атома галогена, нитро, оксогрупп (=О), циано, C1-C6 алкила, полифторированного алкила, полифторированной алкоксигруппы, алкенила, алкинила, гидроксиалкила, арила, арилалкила, гетероциклила, C3-C7 циклоалкила, гидрокси, алкокси, арилокси, гетероциклилокси, метилендиокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси, гетероциклилкарбонилокси, алкилиденаминоокси, карбокси, алкоксикарбонила, арилоксикарбонила, циклоалкилоксикарбонила, гетероциклилоксикарбонила, амино, уреидо, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонила, алкиламинокарбонила, диалкиламинокарбонила, ариламинокарбонила, гетероциклиламинокарбонила, алкоксикарбониламино, гидроксиаминокарбонила, алкоксиимино, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, формила, алкилкарбонила, арилкарбонила, циклоалкилкарбонила, гетероциклилкарбонила, алкилсульфонила, арилсульфонила, аминосульфонила, алкиламиносульфонила, диалкиламиносульфонила, ариламиносульфонила, гетероциклиламиносульфонила, арилтио, алкилтио, фосфоната и алкилфосфоната.
В свою очередь, при целесообразности, каждый из указанных выше заместителей может быть дополнительно замещен одной или более из указанных выше групп.
В этом отношении, под термином «атом галогена» подразумевается атом фтора, хлора, брома или йода.
Под термином «циано» подразумевается остаток -CN.
Под термином «нитро» подразумевается группа -NO2.
Под термином «алкенил» или «алкинил» подразумевается любая из указанных выше прямых или разветвленных C2-C6 алкильных групп, дополнительно несущих двойную или тройную связь. Неограничивающими примерами алкенильных или алкинильных групп являются, например, винил, аллил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-пентенил, 1-гексенил, этинил, 2-пропинил, 4-пентинил и тому подобные.
Под термином «полифторированный алкил или алкокси» подразумевается любая из указанных выше прямых или разветвленных C1-C6 алкильных или алкокси групп, которые замещены более чем одним атомом фтора, таких как, например, трифторметил, трифторэтил, 1,1,1,3,3,3-гексафторпропил, трифторметокси и тому подобные.
Под термином «алкокси», «арилокси», «гетероциклилокси» и их производными подразумевается любая из указанных выше C1-C6 алкильной, арильной или гетероциклильной групп, связанных с остальной молекулой через атом кислорода (-O-).
Из всего сказанного выше, специалисту в данной области ясно, что любая группа, название которой представляет собой составное название, такое как, например, ариламино, должно подразумевать обычное трактование частями, из которых она происходит, например, аминогруппой, которая дополнительно замещена арилом, где арил представляет, как определено выше.
Аналогичным образом, любой из терминов, таких как, например, алкилтио, алкиламино, диалкиламино, алкоксикарбонил, алкоксикарбониламино, гетероциклилкарбонил, гетероциклилкарбониламино, циклоалкилоксикарбонил и тому подобные, включает группы, где алкильная, алкокси, арильная, C3-C7 циклоалкильная и гетероциклильная части представляют, как определено выше.
Фармацевтически приемлемые соли соединений формулы (I) включают кислотно-аддитивные соли с неорганическими и органическими кислотами, например, азотной, хлористоводородной, бромистоводородной, серной, перхлорной, фосфорной, уксусной, трифторуксусной, пропионовой, гликолевой, фумаровой, молочной, щавелевой, малоновой, яблочной, малеиновой, винной, лимонной, бензойной, коричной, миндальной, метансульфоновой, изетионовой и салициловой кислотой. Предпочтительно, кислотно-аддитивная соль соединений по изобретению выбрана между гидрохлоридом и мезилатом.
Фармацевтически приемлемые соли соединений формулы (I) также включают соли с неорганическими и органическими основаниями, например, щелочных и щелочноземельных металлов, в частности, гидроксиды, карбонаты или бикарбонаты натрия, калия, кальция, аммония или магния, ациклические или циклические амины, предпочтительно метиламин, этиламин, диэтиламин, триэтиламин, пиперидин и тому подобные.
Предпочтительные соединения формулы (I) представляют собой соединения, где X обозначает группу -NR'- и R2 обозначает группу, выбранную из -NHR" и -N(OR"')R", где R" обозначает водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила; и R', R'", R1, R3, R4 и A обозначают, как определено выше.
Другие предпочтительные соединения представляют собой соединения формулы (I), где X обозначает группу -O- и R2 обозначает группу, выбранную из -NHR" и -N(OR"')R", где R" обозначает водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила; и R'", R1, R3, R4 и A обозначают, как определено выше.
Другие предпочтительные соединения представляют собой соединения формулы (I), где X обозначает группу -S- и R2 обозначает группу, выбранную из -NHR" и -N(OR"')R", где R" обозначает водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила; и R'", R1, R3, R4 и A обозначают, как определено выше.
Другие предпочтительные соединения представляют собой соединения формулы (I), где X обозначает одиночную связь и R2 обозначает группу, выбранную из -NHR" и -N(OR"')R", где R" обозначает водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила; и R'", R1, R3, R4 и A обозначают, как определено выше.
Другие предпочтительные соединения представляют собой соединения формулы (I), где X обозначает группу -NR'-; R2 обозначает группу -NHR" или -N(OR"')R", где R" обозначает водород или необязательно замещенную группу, выбранную из прямой или разветвленной C1-C4 алкильной группы и арила; и R1 обозначает необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила; и R', R'", R3, R4 и A обозначают, как определено выше.
Другие предпочтительные соединения представляют собой соединения формулы (I), где X обозначает группу -O-; R2 обозначает группу -NHR" или -N(OR"')R", где R" обозначает водород или необязательно замещенную группу, выбранную из прямой или разветвленной C1-C4 алкильной группы и арила; и R1 обозначает необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила; и R'", R3, R4 и A обозначают, как определено выше.
Другие предпочтительные соединения представляют собой соединения формулы (I), где X обозначает группу -S-; R2 обозначает группу -NHR" или -N(OR"')R", где R" обозначает водород или необязательно замещенную группу, выбранную из прямой или разветвленной C1-C4 алкильной группы и арила; и R1 обозначает необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила; и R'", R3, R4 и A обозначают, как определено выше.
Другие предпочтительные соединения представляют собой соединения формулы (I), где X обозначает одиночную связь; R2 обозначает группу -NHR" или -N(OR"')R", где R" обозначает водород или необязательно замещенную группу, выбранную из прямой или разветвленной C1-C4 алкильной группы и арила; и R1 обозначает необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, C3-C7 циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила; и R'", R3, R4 и A обозначают, как определено выше.
Предпочтительными определенными соединениями формулы (I) или их солью являются соединения, перечисленные ниже:
1) N-(2,6-диэтилфенил)-9-(метоксиметил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
2) 2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хинозолин-7-карбоксамид,
3) N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
4) N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил}амино)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
5) N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
6) N-(2,6-диэтилфенил)-2-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метоксифенил}амино)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
7) 2-{[2-метокси-(4-метилпиперазин-1-ил)фенил]амино}-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
8) 2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
9) N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
10) N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил}амино)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
11) N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
12) N-(2,6-диэтилфенил)-2-[(4-{[3-(диметиламино)пропил](метил)амино}-2-метоксифенил)амино]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
13) N-(2,6-диэтилфенил)-2-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метоксифенил}амино)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
14) 2-[(4-бром-2-метоксифенил)амино]-N-[(1S)-2-(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)-1-фенилэтил]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
15) 2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-10-метил-5,6,7,10-тетрагидропирроло[3',2',6,7]циклогепта[1,2-d]пиримидин-8-карбоксамид,
16) N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-10-метил-5,6,7,10-тетрагидропирроло[3',2',6,7]циклогепта[1,2-d]пиримидин-8-карбоксамид,
17) N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил}амино)-10-метил-5,6,7,10-тетрагидропирроло[3',2',6,7]циклогепта[1,2-d]пиримидин-8-карбоксамид и
18) N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-10-метил-5,6,7,10-тетрагидропирроло[3',2',6,7]циклогепта[1,2-d]пиримидин-8-карбоксамид,
19) 8-метил-2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
20) 8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
21) 2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
22) 2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
23) 2-(диметиламино)-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
24) 9-(2-гидроксиэтил)-8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
25) 9-(2-гидроксиэтил)-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
26) 2-(диметиламино)-8-метил-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
27) 9-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
28) 8-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
29) 9-(2-гидроксиэтил)-8-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
30) 8-метил-2-(метилсульфанил)-9-(пропан-2-ил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
31) 9-этил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
32) 2-(метилсульфанил)-9-(пиперидин-4-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
33) 9-(цис-4-аминоциклогексил)-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
34) 9-(цис-4-аминоциклогексил)-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
35) 2-(метилсульфанил)-9-(пиперидин-4-ил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
36) 2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид,
37) 2-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид,
38) 9-(3-амино-2,2-диметилпропил)-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид гидрохлорид,
39) 9-(азепан-3-ил)-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид гидрохлорид.
Ссылка на любое определенное соединение формулы (I) по изобретению, необязательно, в форме фармацевтически приемлемой соли, имеется в экспериментальном разделе и формуле изобретения.
Настоящее изобретение также относится к способу получения соединения формулы (I), как определено выше, использованием описанных ниже путей реакций и синтетических схем, с использованием технологий, доступных в данной области, и общедоступных исходных материалов. Получение по определенным вариантам осуществления описано в следующих примерах, но средним специалистам в данной области понятно, что описанные способы получения могут быть легко адаптированы для получения других вариантов осуществления настоящего изобретения. Например, синтез не иллюстрируемых соединений в соответствии с изобретением может быть выполнен модификациями, очевидными для специалистов в данной области, например, соответствующей защитой интерферирующих групп, переключением на другие подходящие реагенты, известные в данной области, или внесением обычных модификаций в условия реакций. Альтернативно, другие реакции, указанные в настоящем описании, или известные в данной области, будут признаны как адаптируемые для получения других соединений по изобретению.
На представленной Схеме 1 показано получение соединения формулы (I).
Схема 1
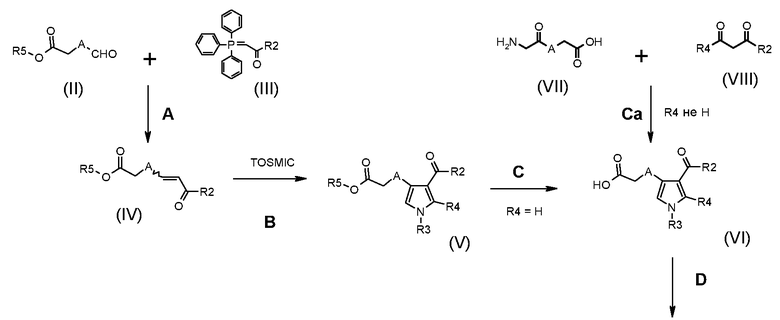
В приведенной выше схеме 1 X обозначает одиночную связь или двухвалентный радикал, выбранный из -NR', -O- и -S-; R2 обозначает необязательно замещенную алкоксигруппу; A обозначает, как определено в формуле (I), за исключением -CH=CH-; R1, R3, R4 и R' обозначают, как определено в формуле (I), и R5 обозначает необязательно замещенный C1-C6 алкил.
Всем специалистам в данной области понятно, что любая трансформация, выполняемая в соответствии с указанными способами, может требовать стандартных модификаций, таких как, например, защита интерферирующих групп, замена другими подходящими реагентами, известными в данной области, или внесение обычных модификаций условий реакций.
Соответственно, способ по настоящему изобретению включает следующие стадии:
Стадия A) взаимодействие соединения формулы (II):
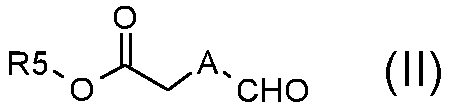 ,
,
где A обозначает, как определено в формуле (I), за исключением -CH=CH-, и R5 обозначает необязательно замещенный C1-C6 алкил (например, метил, этил или трет-бутил), с соединением формулы (III):
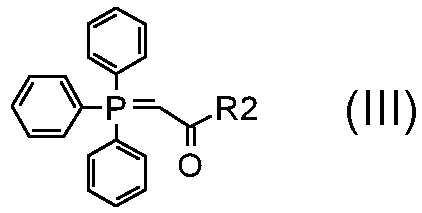 ,
,
где R2 обозначает необязательно замещенный алкокси (например, метокси, этокси или трет-бутокси);
стадия B) взаимодействие полученного соединения формулы (IV):
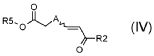 ,
,
где R2 обозначает необязательно замещенный алкокси, R5 обозначает необязательно замещенный C1-C6 алкил, и A обозначает, как определено в формуле (I), за исключением -CH=CH-, с толуолсульфонилметилизоцианидом в присутствии сильного основания;
стадия C) селективный гидролиз в кислотных или основных условиях полученного соединения формулы (V):
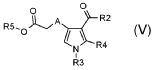 ,
,
где R3 и R4 обозначают водород, A обозначает, как определено в формуле (I), за исключением -CH=CH-, R2 обозначает необязательно замещенный алкокси, и R5 обозначает необязательно замещенный C1-C6 алкил, с тем, чтобы получить соединение формулы (VI):
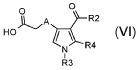 ,
,
где R3 и R4 обозначают водород, A обозначает, как определено в формуле (I), за исключением -CH=CH-, и R2 обозначает необязательно замещенный алкокси;
альтернативно,
стадия Ca) соединение формулы (VI), где R3 обозначает водород, R4 обозначает, как определено в формуле (I), за исключением водорода, A обозначает, как определено в формуле (I), за исключением -CH=CH-, и R2 обозначает необязательно замещенный алкокси, может быть получено взаимодействием соединения формулы (VII):
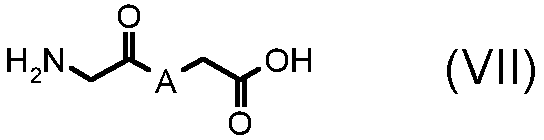 ,
,
где A обозначает, как определено выше, с соединением формулы (VIII):
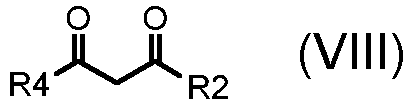 ,
,
где R2 и R4 обозначают, как определено выше;
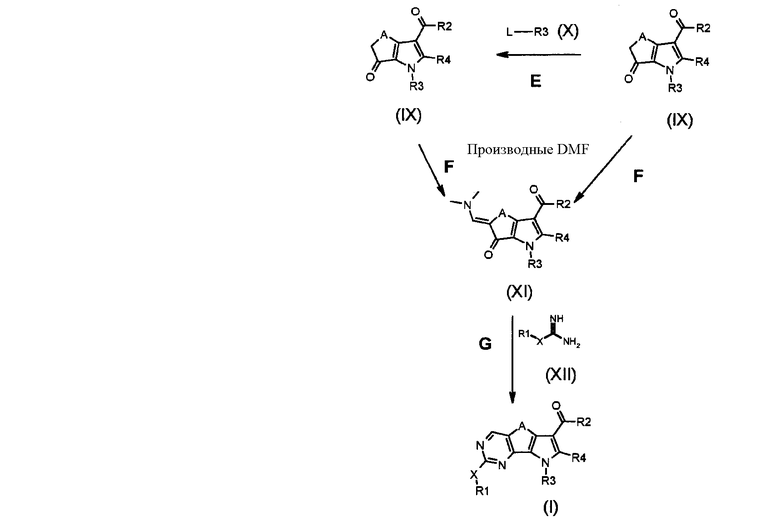
стадия D) циклизация полученного соединения формулы (VI), где R2 обозначает необязательно замещенный алкокси, R3 обозначает водород, R4 обозначает, как определено в формуле (I), и A обозначает, как определено в формуле (I), за исключением -CH=CH-, в кислотных условиях, с тем, чтобы получить соединение формулы (IX):
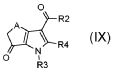 ,
,
где R2, R3, R4 и A обозначают, как определено выше;
если необходимо или желательно,
стадия E) алкилирование соединения формулы (IX), где R3 обозначает водород, соединением формулы (X):
R3-L (X),
где L обозначает OH или группу, которая необязательно после активации может работать как подходящая уходящая группа, такая как йод-, бром-, хлор- или сульфонатная группа (например -OS(О)2CF3, -OS(О)2CH3 или -OS(О)2PhMe), и R3 обозначает, как определено в формуле (I), за исключением водорода;
стадия F) взаимодействие полученного соединения формулы (IX):
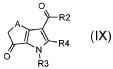 ,
,
где R2 обозначает необязательно замещенный алкокси, R3 и R4 обозначают, как определено в формуле (I), и A обозначает, как определено в формуле (I), за исключением -CH=CH-, с производным Ν,Ν-диметилформамида;
стадия G) взаимодействие полученного соединения формулы (XI):
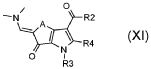 ,
,
где R2 обозначает необязательно замещенный алкокси, R3 и R4 обозначают, как определено в формуле (I), и A обозначает, как определено в формуле (I), за исключением -CH=CH-, с соединением формулы (XII):
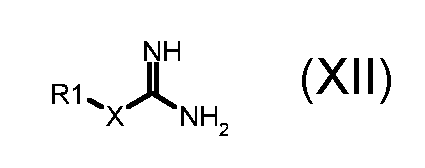 ,
,
где X обозначает одиночную связь или двухвалентный радикал, выбранный из -NR', -O- и -S-; и R1 и R' обозначают, как определено в формуле (I), с тем, чтобы получить соединение формулы (I):
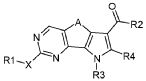 (I),
(I),
где X обозначает одиночную связь или двухвалентный радикал, выбранный из -NR', -O- и -S-; R2 обозначает необязательно замещенный алкокси; A обозначает, как определено в формуле (I), за исключением -CH=CH-; и R1, R3, R4 и R' обозначают, как определено в формуле (I); необязательно, превращение соединения формулы (I) в другое отличающееся соединение формулы (I), и, при желании, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
Как указано выше, соединения формулы (I), которые получены в соответствии со способом, являющимся целью изобретения, могут быть подходящим образом превращены в другие соединения формулы (I) действиями в соответствии с хорошо известными условиями синтеза, причем далее приведены примеры возможных превращений:
Превращение 1) превращение соединение формулы (I), где R3 обозначает защитную группу P, такую как метоксиметил или п-метоксибензил, в соответствующее соединение формулы (I), где R3 обозначает атом водорода, в кислотных или основных условиях:
 ;
;
Превращение 2) превращение соединения формулы (I), где R3 обозначает водород, в соответствующее соединение формулы (I), где R3 обозначает, как определено в формуле (I), но не водород, посредством взаимодействия с соединением формулы R3-L (X), где L обозначает OH или группу, которая необязательно после активации может работать как подходящая уходящая группа, такая как йод-, бром-, хлор- или сульфонатная группа (например, -OS(О)2CF3, -OS(О)2CH3 или -OS(О)2PhMe), и R3 обозначает, как определено выше, но не атом водорода:
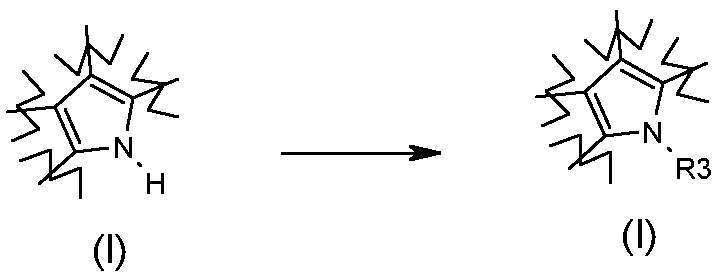 ;
;
Превращение 3) превращение соединения формулы (I), где R2 обозначает OR5, где R5 обозначает необязательно замещенный C1-C6 алкил, в соответствующее соединение формулы (I), где R2 обозначает гидрокси, или или его соответствующую соль, посредством кислотного или основного гидролиза:
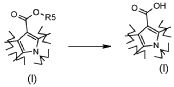 ;
;
Превращение 4) превращение соединения формулы (I), где R2 обозначает гидрокси, или его соответствующей соли в соответствующее соединение формулы (I), где R2 обозначает группу -NR"R"' или -N(OR"')R", где R" и R'" обозначают, как определено в формуле (I), посредством взаимодействия с производным формулы R"R"'NH (XIII) или R"NHOR"' (XIV), где R" и R'" обозначают, как определено выше, в основных условиях и в присутствии подходящего конденсирующего агента;
альтернативно, соединение формулы (I), где R2 обозначает гидрокси, может быть сначала превращено в соответствующее хлоридное производное с использованием хлорирующего агента, затем полученное соединение подвергается взаимодействию с производным формулы R"R"'NH (XIII) или R"NHOR"' (XIV), где R" и R'" обозначают, как определено выше, в основных условиях, с тем, чтобы получить соединение формулы (I), где R2 обозначает группу -NR"R"' или -N(OR"')R":
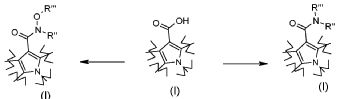 ;
;
Превращение 5) превращение соединения формулы (I), где R2 обозначает OR5, где R5 обозначает необязательно замещенный C1-C6 алкил, в соответствующее соединение формулы (I), где R2 обозначает группу -NR"R"' или -N(OR"')R", где R" и R'" обозначают, как определено в формуле (I), посредством взаимодействия с производным формулы R"R"'NH (XIII) или R"NHOR"' (XIV), где R" и R'" обозначают, как определено выше:
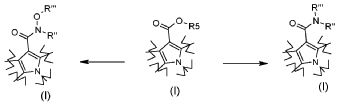 ;
;
Превращение 6) превращение соединения формулы (I), где X обозначает, как определено в формуле (I), за исключением SO2 и -OSO2-, и R1 обозначает арил, т.е. фенил, замещенный бромом, в соответствующее соединение формулы (I), где R1 обозначает арил, т.е. фенил, замещенный NR"R"', обработкой амином формулы R"R"'-NH (XIII):
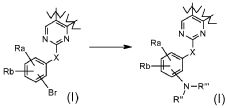 ,
,
где Ra и Rb независимо обозначают атом галогена, за исключением брома, водород, нитро, циано, C1-C6 алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, C3-C7 циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, метилендиокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси, гетероциклилкарбонилокси, алкилиденаминоокси, карбокси, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, гетероциклилоксикарбонил, амино, уреидо, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, ариламинокарбонил, гетероциклиламинокарбонил, алкоксикарбониламино, гидроксиаминокарбонил, алкоксиимино, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, формил, алкилкарбонил, арилкарбонил, циклоалкилкарбонил, гетероциклилкарбонил, алкилсульфонил, арилсульфонил, аминосульфонил, алкиламиносульфонил, диалкиламиносульфонил, ариламиносульфонил, гетероциклиламиносульфонил, арилтио, алкилтио, фосфонат или алкилфосфонат;
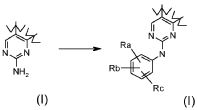
Превращение 7) превращение соединения формулы (I), где X обозначает -NH- и R1 обозначает водород, в соответствующее соединение формулы (I), где R1 обозначает арил, т.е. фенил, замещенный Ra, Rb, Rc:
 ,
,
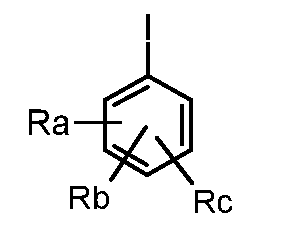
где Ra, Rb и Rc обозначают независимо водород, нитро, циано, C1-C6 алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, C3-C7 циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, метилендиокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси, гетероциклилкарбонилокси, алкилиденаминоокси, карбокси, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, гетероциклилоксикарбонил, амино, уреидо, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, ариламинокарбонил, гетероциклиламинокарбонил, алкоксикарбониламино, гидроксиаминокарбонил, алкоксиимино, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, формил, алкилкарбонил, арилкарбонил, циклоалкилкарбонил, гетероциклилкарбонил, алкилсульфонил, арилсульфонил, аминосульфонил, алкиламиносульфонил, диалкиламиносульфонил, ариламиносульфонил, гетероциклиламиносульфонил, арилтио, алкилтио, фосфонат или алкилфосфонат,
обработкой йодпроизводным формулы (XV):
 ,
,
где Ra, Rb и Rc обозначают, как определено выше, в присутствии палладия;
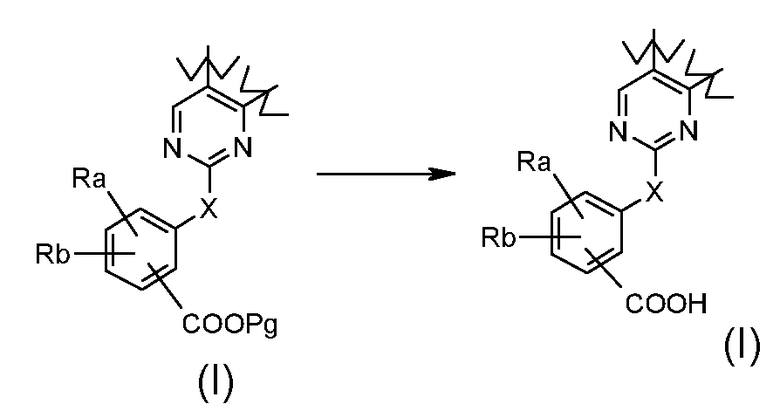
Превращение 8) превращение соединения формулы (I), где X обозначает, как определено в формуле (I), за исключением SO2 и -OSO2-, и R1 обозначает арил, т.е. фенил, замещенный -COOPg, где Pg обозначает подходящую защитную группу, в соответствующее соединение формулы (I), где R1 обозначает арил, т.е. фенил, замещенный -COOH, путем использования условий, хорошо известных в литературе (см. Teodora W. Green PereG. M.Wuts):
 ,
,
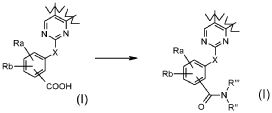
где Ra и Rb обозначают независимо атом галогена, водород, нитро, циано, C1-C6 алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, C3-C7 циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, метилендиокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси, гетероциклилкарбонилокси, алкилиденаминоокси, карбокси, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, гетероциклилоксикарбонил, амино, уреидо, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, ариламинокарбонил, гетероциклиламинокарбонил, алкоксикарбониламино, гидроксиаминокарбонил, алкоксиимино, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, формил, алкилкарбонил, арилкарбонил, циклоалкилкарбонил, гетероциклилкарбонил, алкилсульфонил, арилсульфонил, аминосульфонил, алкиламиносульфонил, диалкиламиносульфонил, ариламиносульфонил, гетероциклиламиносульфонил, арилтио, алкилтио, фосфонат или алкилфосфонат;
Превращение 9) превращение соединения формулы (I), где X обозначает, как определено в формуле (I), за исключением SO2 и -OSO2-, и R1 обозначает арил, т.е. фенил, замещенный -COOH, в соответствующее соединение формулы (I), где R1 обозначает арил, т.е. фенил, замещенный -CONR"R"', где R" и R'" обозначают, как определено выше, обработкой амином формулы R"R"'-NH (XIII), в присутствии подходящих конденсирующих агентов:
 ,
,
где Ra и Rb обозначают независимо атом галогена, водород, нитро, циано, C1-C6 алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, C3-C7 циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, метилендиокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси, гетероциклилкарбонилокси, алкилиденаминоокси, карбокси, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, гетероциклилоксикарбонил, амино, уреидо, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, ариламинокарбонил, гетероциклиламинокарбонил, алкоксикарбониламино, гидроксиаминокарбонил, алкоксиимино, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, формил, алкилкарбонил, арилкарбонил, циклоалкилкарбонил, гетероциклилкарбонил, алкилсульфонил, арилсульфонил, аминосульфонил, алкиламиносульфонил, диалкиламиносульфонил, ариламиносульфонил, гетероциклиламиносульфонил, арилтио, алкилтио, фосфонат или алкилфосфонат;
Превращение 10) превращение соединения формулы (I), где R1 обозначает водород и X обозначает -NH-, в соответствующее соединение формулы (I), где R1 обозначает йод и X обозначает одиночную связь, взаимодействием с изоамилнитритом и дийодметаном или йодидом цезия в присутствии йода и CuI:
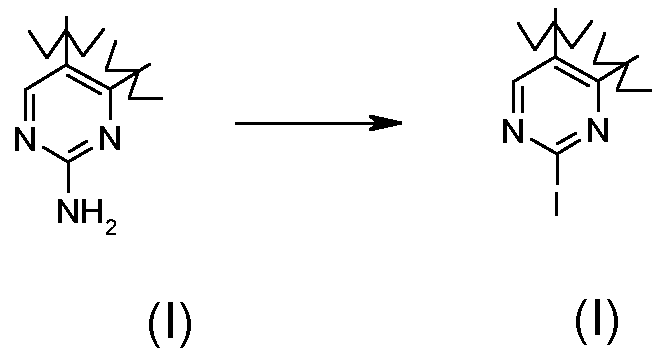 ;
;
Превращение 11) превращение соединения формулы (I), где R1 обозначает йод и X обозначает одиночную связь, в соответствующее соединение формулы (I), где X обозначает -NH- и R1 обозначает необязательно замещенный арил, взаимодействием с необязательно замещенным ариламином формулы RI-NH2 (XVI), где R1 обозначает, как определено выше, в присутствии Pd(OAc)2 и BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил):
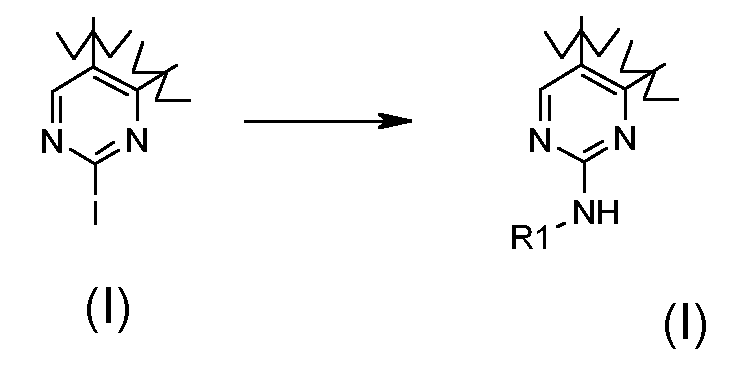 ,
,
Превращение 12) превращение соединения формулы (I), где R1 обозначает йод и X обозначает одиночную связь, в соответствующее соединение формулы (I), где X обозначает одиночную связь и R1 обозначает, как определено в формуле (I), взаимодействием с соединением формулы (XVII):
R1-Q (XVII),
где R1 обозначает, как определено выше, и Q обозначает подходящую группу, такую как -B(OH)2, -B(OAlk)2, -Sn(Alk)4, ZnHal или MgHal, которое может быть подвергнуто опосредованному палладием образованию углеродной связи:
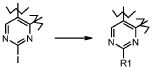 ;
;
Превращение 13) превращение соединения формулы (I), где R1 обозначает, как определено в формуле (I), и X обозначает -S-, в соответствующее соединение формулы (I), где X обозначает -SO2-, в окислительных условиях:
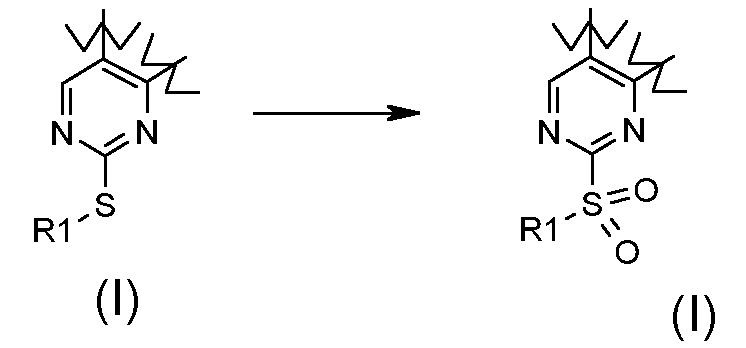 ;
;
Превращение 14) превращение соединения формулы (I), где R1 обозначает, как определено в формуле (I), и X обозначает -SO2-, в соответствующее соединение формулы (I), где X обозначает -NR'-, взаимодействием сульфонильной группы с амином формулы R1-NHR' (XVIa), где R1 и R' обозначают, как определено в формуле (I):
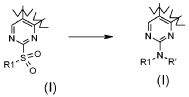 ;
;
Превращение 15) превращение соединения формулы (I), где R1 обозначает, как определено в формуле (I), и X обозначает -SO2-, в соответствующее соединение формулы (I), где X обозначает -O-, взаимодействием сульфонильной группы с соединением формулы R1-OH (XVIII), где R1 обозначает, как определено в формуле (I), за исключением водорода:
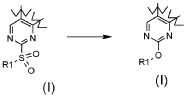 ,
,
Превращение 16) превращение соединения формулы (I), где R1 обозначает метил и X обозначает -O-, в соответствующее соединение формулы (I), где R1 обозначает водород и X обозначает -O-:
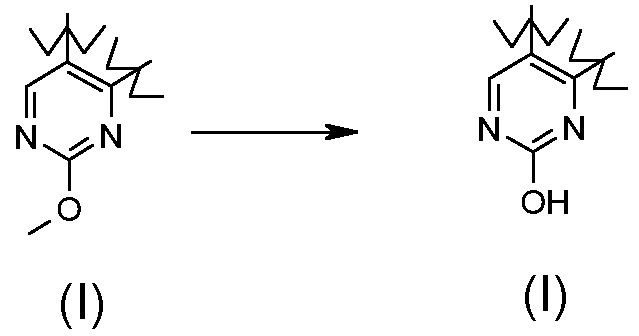 ,
,
Превращение 17) превращение соединения формулы (I), где R1 обозначает водород и X обозначает -O-, в соединение формулы (I), где R1 обозначает трифторметил и X обозначает -OSO2-, взаимодействием с трифлатирующим агентом:
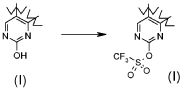 ;
;
Превращение 18) превращение соединение формулы (I), где R1 обозначает трифторметил и X обозначает -OSO2-, в соответствующее соединение формулы (I), где X обозначает -O- и R1 обозначает, как определено в формуле (I), взаимодействием с соединением формулы R1-OH (XVIII), где R1 обозначает, как определено выше, за исключением водорода:
 ;
;
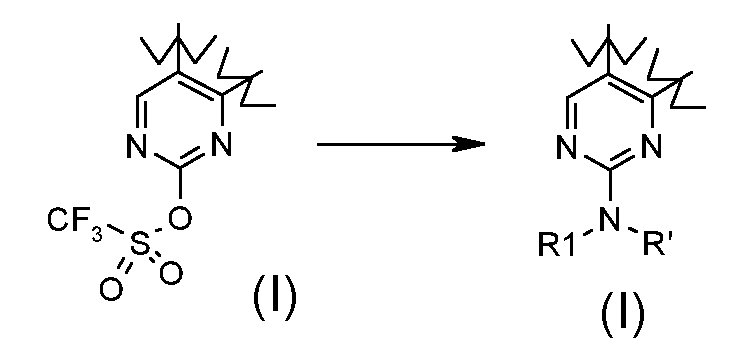
Превращение 19) превращение соединения формулы (I), где R1 обозначает трифторметил и X обозначает -OSO2-, в соответствующее соединение формулы (I), где X обозначает -NR'- и R1 обозначает, как определено в формуле (I), за исключением водорода, взаимодействием с соединением формулы R1-NHR' (XVIa), где R1 обозначает, как определено выше:
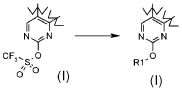
 ;
;
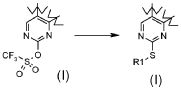
Превращение 20) превращение соединения формулы (I), где R1 обозначает трифторметил и X обозначает -OSO2-, в соответствующее соединение формулы (I), где X обозначает -S- и R1 обозначает, как определено в формуле (I), за исключением водорода, взаимодействием с тиолом формулы R1-SH (XIX), где R1 обозначает, как определено выше, за исключением водорода:
 ;
;
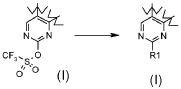
Превращение 21) превращение соединения формулы (I), где R1 обозначает трифторметил и X обозначает -OSO2-, в соответствующее соединение формулы (I), где X обозначает одиночную связь и R1 обозначает, как определено в формуле (I), за исключением водорода, взаимодействием с соединением формулы R1-Q (XVII), где R1 обозначает, как определено выше, за исключением водорода, и Q обозначает подходящую группу, такую как -B(OH)2, -B(OAlk)2, -Sn(Alk)4, ZnHal или MgHal, которое может подвергаться опосредованному палладием образованию углеродной связи:
 ;
;
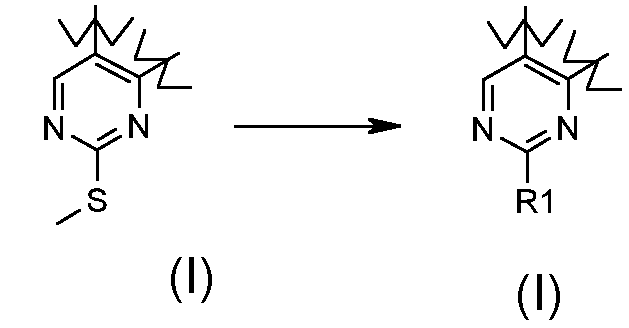
Превращение 22) превращение соединения формулы (I), где R1 обозначает метил и X обозначает -S-, в соответствующие соединения формулы (I), где R1 обозначает необязательно замещенный арил и X обозначает одиночную связь, взаимодействием его с арилборной кислотой формулы R1-B(OH)2 (XVIIa), где R1 обозначает необязательно замещенный арил, в присутствии палладиевого производного:
 ;
;
Превращение 23) превращение соединения формулы (I), где A обозначает двухвалентную группу, такую как -CH2-CH2-, в соответствующее соединение формулы (I), где A обозначает группу -CH=CH-, обработкой окисляющим агентом, или в дегидрирующих рабочих условиях в присутствии Pd или Pt катализатора:
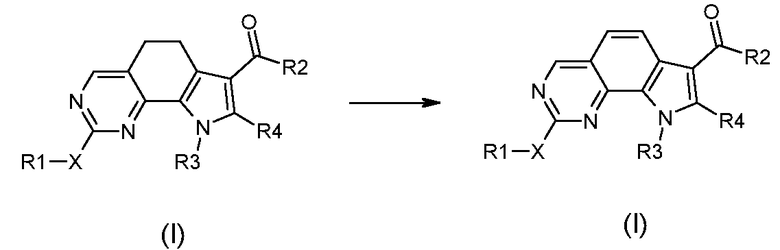 ,
,
где X, R1, R2, R3 и R4 обозначают, как определено в формуле (I);
Превращение 24) превращение соединения формулы (I), где R4 обозначает водород и A обозначает двухвалентную группу, такую как -CH2-CH2-, в соответствующее соединение формулы (I), где R4 обозначает водород и A обозначает группу -CH=CH-, сначала превращением в соединение формулы (XX) избытком N-йодсукцинимида и последующим удалением йода, в присутствии палладиевого производного:
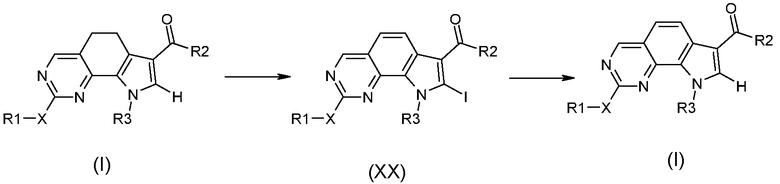 ,
,
где X, R1, R2 и R3 обозначают, как определено в формуле (I), и R4 обозначает водород;
Превращение 25) удаление любой защитной группы или групп и, при желании, образование соли.
В соответствии со стадиями (стадией A) способа, альдегид формулы (II) взаимодействует с фосфораном формулы (III) в подходящем растворителе, таком как, например, толуол, ксилол, THF или Et2O, при температуре в диапазоне от комнатной до дефлегмации, и в течение времени в диапазоне от 1 до примерно 12 часов. Предпочтительно, указанная выше реакция проводится в толуоле при кипячении в сосуде с обратным холодильником, с тем, чтобы получить соединение формулы (IV).
В соответствии со стадией (стадией B), соединение формулы (IV) взаимодействует с TOSMIC (толуолсульфонилметилизоцианидом), в присутствии основания, такого как KOH, NaH, LiN(TMS)2, в подходящем растворителе, таком как, например, толуол, THF или Et2O, при температуре в диапазоне от -78°C до комнатной температуры, и в течение времени в диапазоне от 1 до примерно 12 часов. Предпочтительно, указанная выше реакция проводится в присутствии LiN(TMS)2 в THF при -78°C, с тем, чтобы получить соединение формулы (V).
В соответствии со стадией (стадией C), соединение формулы (V), где R3 обозначает водород, R4 обозначает водород и A обозначает, как определено в формуле (I), за исключением -CH=CH-, R2 обозначает необязательно замещенную алкоксигруппу, и R5 обозначает необязательно замещенный алкил, превращается в производное монокарбоновой кислоты (VI), в присутствии основания, такого как KOH, NaOH, LiOH или Na2CO3, в подходящем растворителе, таком как, например, H2O, диоксан или их смеси, при температуре в диапазоне от 0°C до комнатной температуры, и в течение времени в диапазоне от 1 до примерно 24 часов. Предпочтительно, указанная выше реакция проводится в присутствии LiOH в смеси диоксан/H2O при комнатной температуре, с тем, чтобы получить соединение формулы (VI).
Альтернативно, в соответствии со стадией (стадией Ca), соединение формулы (VII) взаимодействует с соединением формулы (VIII), в присутствии AcONa или этилата натрия, в подходящем растворителе, таком как, например, H2O, EtOH или AcOH, при температуре в диапазоне от комнатной до дефлегмации, и в течение времени в диапазоне от 1 до примерно 24 часов. Предпочтительно, указанная выше реакция проводится в присутствии AcONa в H2O при кипячении в сосуде с обратным холодильником, с тем, чтобы получить соединение формулы (VI), где R3 обозначает водород, R4 обозначает, как определено выше, за исключением водорода, и R2 и A обозначают, как определено выше.
В соответствии со стадией (стадией D) способа, соединение формулы (VI), где R4 обозначает, как определено в формуле (I), может быть превращено в соединение формулы (IX) в присутствии TFAA (трифторуксусного ангидрида) или PPA (полифосфоновой кислоты) в подходящем растворителе, таком как TFA, при температуре в диапазоне от комнатной до дефлегмации, и в течение времени в диапазоне от 1 до примерно 8 часов. Предпочтительно, указанная выше реакция проводится в присутствии TFAA в TFA при комнатной температуре, с тем, чтобы получить соединение формулы (IX).
В соответствии со стадией (стадией E) способа, соединение формулы (IX), где R3 обозначает атом водорода, взаимодействует с соединением формулы (X), как определено выше, где L обозначает OH, и в этом случае могут использоваться условия Митсунобу, или группу, которая необязательно после активации может работать как уходящая группа, такая как атом галогена, тозилат, мезилат или трифлат.
В первом случае, то есть, когда используется протокол Митсунобу, реакция может осуществляться с использованием диалкилазодикарбоксилата, такого как диэтилазодикарбоксилат (DEAD), диизопропилазодикарбоксилат (DIAD) или тому подобное, в присутствии триалкила или триарилфосфина, предпочтительно трифенилфосфина, в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, ацетонитрил.
Когда L обозначает галоген или группу, такую как тозилат, мезилат или трифлат или тому подобную, превращение может быть осуществлено с использованием подходящего основания, такого как, например, NaH, K2CO3, Cs2CO3, DBU, KO-t-Bu и тому подобное, в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил, N,N-диметилформамид, Ν,Ν-диметилацетамид и тому подобные. Указанные реакции могут проводиться при температуре в диапазоне от 0°C до дефлегмации и в течение времени в диапазоне от 30 минут до примерно 48 часов.
В соответствии со стадией (стадией F) способа, соединение формулы (IX) взаимодействует с производным N,N-диметилформамида, например, с производным N,N-диметилформамида, таким как N,N-диметилформамид-ди-трет-бутилацеталь, N,N-диметилформамид-диизопропилацеталь, N,N-диметилформамид-диметилацеталь, N,N-диметилформамид-диэтилацеталь или трис(диметиламино)метан, в подходящем растворителе, таком как, например, DMF или толуол, при температуре в диапазоне от комнатной до дефлегмации, и в течение времени в диапазоне от 1 до примерно 48 часов. Предпочтительно, реакция проводится в присутствии чистого трис(диметиламино)метана, чистого или в DMF при 90°C, для получения соединения формулы (XI).
В соответствии со стадией (стадией G) способа, соединение формулы (XI) взаимодействует с производным формулы (XII), где X обозначает одиночную связь или двухвалентный радикал, выбранный из -NR'-, -O- и -S-, где R' обозначает, как определено в формуле (I), R1 обозначает, как определено в формуле (I); с тем, чтобы получить соединение формулы (I), как определено выше, где X и R1 обозначают, как определено выше, посредством образования пиримидинового кольца в присутствии, в конечном счете, основания, такого как AcOK, K2CO3 или Na2CO3, в подходящем растворителе, таком как, например, DMF, EtOH или толуол, при температуре в диапазоне от комнатной до дефлегмации, и в течение времени в диапазоне от примерно 1 до примерно 48 часов. Предпочтительно, реакция проводится в присутствии DMF при 120°C. Альтернативно, микроволновое облучение может использоваться вместо нагревания.
В соответствии с превращением (превращение 1) способа, соединение формулы (I), где R3 обозначает группу, выбранную из метоксиметила или п-метоксибензила, может быть превращено в другое соединение формулы (I), где R3 обозначает атом водорода, взаимодействием в кислотных условиях, например, с AcOH, TFA или HCl, или в основных условиях, например, с NaOH, и в присутствии подходящего растворителя, такого как MeOH, DCM или диоксан, при температуре в диапазоне от комнатной до дефлегмации и в течение времени в диапазоне от 1 до примерно 12 часов.
В соответствии с превращением (превращение 2) способа, соединение формулы (I), где R3 обозначает атом водорода, может быть превращено в соединение формулы (I), где R2 обозначает, как определено выше, за исключением атома водорода, взаимодействием с подходящим соединением формулы (X), как определено выше, когда L обозначает OH, и в этом случае могут использоваться условия Митсунобу, или группу, которая необязательно после активации может работать как уходящая группа, такая как атом галогена, тозилат, мезилат или трифлат.
В первом случае, то есть, когда используется протокол Митсунобу, реакция может осуществляться с использованием диалкилазодикарбоксилата, такого как диэтилазодикарбоксилат (DEAD), диизопропилазодикарбоксилат (DIAD) или тому подобное, в присутствии триалкила или триарилфосфина, предпочтительно, трифенилфосфина, в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, ацетонитрил.
Когда L обозначает галоген или группу, такую как тозилат, мезилат или трифлат или тому подобное, превращение может быть осуществлено с использованием подходящего основания, такого как, например, NaH, K2CO3, Cs2CO3, DBU, KO-t-Bu и тому подобное, в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил, N,N-диметилформамид, Ν,Ν-диметилацетамид и тому подобное. Указанные реакции могут проводиться при температуре в диапазоне от 0°C до дефлегмации и в течение времени в диапазоне от 30 минут до примерно 48 часов.
В соответствии с превращением (превращение 3) способа, соединение формулы (I), где R2 обозначает OR5, где R5 обозначает необязательно замещенный алкил, может быть превращено в соответствующее производное карбоновой кислоты формулы (I), где R2 обозначает гидроксил или его соответствующие соли, посредством условий основного или кислотного гидролиза, широко известных в данной области. Предпочтительно, реакция проводится в присутствии NaOH в диоксане/H2O при кипячении в сосуде с обратным холодильником.
В соответствии с превращением (превращение 4) способа, соединение формулы (I), где R2 обозначает гидрокси или соответствующую соль, может быть превращено в производное формулы (I), где R2 обозначает группу NR"R"' или N(OR"')R", где R" и R'" обозначают, как определено в формуле (I). Реакция проводится в присутствии соединения формулы или (XIII), или (XIV), как определено выше, в присутствии основания, например, с DIPEA (N,N-диизопропилэтиламином) или TEA (триэтиламином), в подходящем растворителе, таком как DCM (дихлорметан), DMF (диметилформамид), THF (тетрагидрофуран) или диоксан, и в присутствии подходящего конденсирующего агента, такого как DCC (N,N-дициклогексилкарбодиимид), EDCI (1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид) или TBTU (1-(1H-бензотиазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторборат); могут также потребоваться каталитические количества PyBOP (бензотриазол-1-ил-окситрипирролидинфосфония гексафторфосфата) или HOBt (1-гидроксибензотриазола). Предпочтительно, реакция проводится в присутствии DIPEA и TBTU в DMF при комнатной температуре.
Альтернативно, такое же превращение может быть получено сначала взаимодействием соединения формулы (I), где R2 обозначает гидрокси, или соответствующей соли с хлорирующим агентом, например, оксалилдихлоридом или SOCl2, в подходящем растворителе, например, DCM (дихлорметане), толуоле, THF, диоксане или DMF, при температуре в диапазоне от комнатной температуры до 100°C, с тем, чтобы получить соответствующее хлоридное производное. Предпочтительно, реакция проводится в присутствии SOCl2 в THF при кипячении в сосуде с обратным холодильником.
В соответствии с превращением (превращение 5) способа, соединение формулы (I), где R2 обозначает OR5, где R5 обозначает необязательно замещенный алкил, взаимодействует с подходящим соединением формулы (XIII) или (XIV), как определено выше, в присутствии основания, такого как NaH, NaN(TMS)2 или LiN(TMS)2, в подходящем растворителе, например, Et2O, THF или диоксане, при температуре в диапазоне от -10°C до 40°C, и в течение времени в диапазоне от примерно 10 минут до примерно 12 часов, с тем, чтобы получить другое соединение формулы (I), где R2 обозначает аминогруппу формулы -NR"R"' или -N(OR"')R". Предпочтительно, реакция проводится в присутствии LiN(TMS)2 в THF при 0°C.
В соответствии с превращением (превращение 6) способа, замещение брома составляющей -NR"R"' достигали взаимодействием исходного материала с амином формулы (XIII), как определено выше, в подходящем растворителе, таком как THF или диоксан, и в присутствии каталитических количеств Pd2(dba)3, 2-дициклогексилфосфино-2'-(N,N-диметиламино)-бифенила и основания, такого как LiN(TMS)2, при температуре в диапазоне от комнатной до дефлегмации и в течение времени в диапазоне от 1 до примерно 24 часов.
В соответствии с превращением (превращение 7) способа, соединение формулы (I), как определено выше, взаимодействует с соединениями формулы (XV), как определено выше, в соответствии с обычными способами. В качестве примера, реакция может проводиться в подходящем растворителе, таком как DMF, DME, диоксан или CH3CN, и в присутствии необязательно замещенного арилйода формулы (XV), как определено выше, кататических количеств Pd2(dba)3, BINAP или 2-(дициклогексилфосфино)-2',4',6'-триизопропил-1,1'-бифенила (X-фос) и основания, такого как K2CO3, фосфат калия или Cs2CO3, при температуре в диапазоне от комнатной температуры до 110°C и в течение времени в диапазоне от 2 до примерно 24 часов.
В соответствии с превращением (превращение 8) способа, снятие защиты карбоксильного остатка в соответствующую кислоту может быть достигнуто с использованием процедуры, хорошо известной в данной области, в кислотных условиях, например с HCl или TFA, в подходящем растворителе, например, THF или диоксане, при температуре в диапазоне от комнатной температуры до 60°C и в течение времени в диапазоне от примерно 1 до примерно 12 часов.
В соответствии с превращением (превращение 9) способа, трансформация кислотного остатка в соответствующее амидное производное -CONR"R"', где R" и R'" обозначают, как определено выше, может быть получена взаимодействием кислотных производных с амином формулы (XIII), как определено выше, в основных условиях, предпочтительно с DIPEA или TEA, в подходящем растворителе, таком как DCM, DMF, THF или диоксан, и в присутствии подходящего конденсирующего агента, такого как DCC, EDCI или TBTU; могут также потребоваться каталитические количества PyBOP или HOBt, при температуре в диапазоне от комнатной температуры до 60°C и в течение времени в диапазоне от примерно 1 до примерно 24 часов.
В соответствии с превращением (превращение 10) способа, соединения формулы (I), где R1 обозначает йод и X обозначает одиночную связь, могут быть получены соответствующими соединениями формулы (I), где R1 обозначает водород и X обозначает -NH-; реакция проводится с использованием изоамилнитрита и дийодметана или йодида цезия в присутствии йода и CuI в подходящем растворителе, таком как THF, Et2O или DME, при температуре в диапазоне от комнатной температуры до примерно 70°C, и в течение времени от примерно 8 часов до примерно 48 часов.
В соответствии с превращением (превращение 11) способа, замещение йода ариламином формулы R1-NH2 (XVI) может проводиться в подходящем растворителе, таком как DMF, DME или CH3CN, и в присутствии каталитических количеств Pd(OAc)2, BINAP или Xantphos и основания, такого как K2CO3, фосфат калия или Cs2CO3, при температуре в диапазоне от комнатной температуры до 110°C и в течение времени в диапазоне от примерно 2 до примерно 24 часов.
В соответствии с превращением (превращение 12) способа, замещение йода группой формулы R1 может проводиться путем использования любой из реакций поперечной сшивки, подходящих для образования межуглеродных связей. Указанные реакции, которые хорошо известны в данной области, предусматривают соединение с подходящим металлоорганическим реагентом, таким как, например, борорганическое соединение (реакция Сузуки), органоолово (реакция Штилле), органомагний (реакция Кумады) или органоцинк (реакция Негиши) и тому подобные. Предпочтительной реакцией является реакция Сузуки, где соответствующее арильное или гетероарильное производное используется в присутствии катализатора на основе палладия, такого как PdCl2(dppf)2CH2Cl2 или Pd2(dba)3 или Pd(PPh3)4, в подходящем растворителе, таком как DMF, DCM, MeOH, CH3CN, или в смеси растворителей, таких как диметоксиэтан и вода, необязательно в присутствии основания, такого как карбонат натрия, цезия или фторид цезия, при температуре в диапазоне от комнатной температуры до 100°C.
В соответствии с превращением (превращение 13) способа, трансформация тиогруппы в сульфонильную группу может быть получена взаимодействием с окисляющим агентом, хорошо известным специалистам в данной области, таким как, например, оксон, в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, ацетон, необязательно в присутствии воды в качестве совместного растворителя или м-хлорпербензойной кислоты, и в присутствии подходящего растворителя, предпочтительно DCM, при комнатной температуре.
В соответствии с превращением (превращение 14) способа, замещение сульфонильной группы подходящим амино производным предпочтительно проводится амином формулы R1-NHR' (XVIa) в присутствии DMF, DME, диоксана, CH3CN, N-метилпирролидона или диглима, при температуре в диапазоне от комнатной температуры до примерно 100°C.
В соответствии с превращением (превращение 15) способа, замещение сульфонильной группы может быть легко получено взаимодействием со спиртом или фенольным производным формулы (XVIII). Реакция может проводиться в присутствии основания, такого как K2CO3 или Na2CO3, бутиллитий, LiN(TMS)2, NaH или тому подобного, в подходящем растворителе, таком как DMF или THF, и обработкой при температуре в диапазоне от комнатной температуры до примерно 100°C.
В соответствии с превращением (превращение 16) способа, удаление метильного остатка может быть получено в присутствии триметилсилилхлорида и йодида натрия. Реакция может проводиться в подходящем растворителе, таком как CH3CN, и обработкой при температуре в диапазоне от комнатной температуры до примерно дефлегмации.
В соответствии с превращением (превращение 17) способа, соединения с трифторметансульфонильной группой могут быть получены взаимодействием соединений формулы (I), где X обозначает -O- и R1 обозначает водород, с трифлатирующим агентом, таким как трифторметансульфоновый ангидрид, трифторметансульфонилхлорид или N-фенил-бис(трифторметансульфонимид), необязательно в присутствии основания, такого как TEA или DIPEA, в подходящем растворителе, таком как CM, THF или диоксан, при температуре в диапазоне от -78°C до комнатной температуры.
В соответствии с превращением (превращение 18) способа, реакция может проводиться со спиртом формулы (XVIII) операцией в подходящем растворителе, таком как диоксан, THF, DME, CH3CN, DMF или DMSO, при температуре в диапазоне от комнатной температуры до примерно 90°C, необязательно в присутствии основания, такого как K2CO3, трет-бутоксид калия или NaH.
Альтернативно, реакция может проводиться в подходящем растворителе, таком как толуол, DMF, DME или CH3CN, в присутствии Pd(OAc)2, (±)-BINAP и основания, такого как фосфат калия или K2CO3 или CsCO3, при температуре в диапазоне от 0°C до 100°C.
В соответствии с превращением (превращение 19) способа, соединения формулы (I), где R1 обозначает, как определено в формуле (I), за исключением водорода, и X обозначает -NR'-, могут быть получены из соответствующих трифторметансульфонильных соединений амином формулы R1-NHR' (XVIa). Взаимодействие обычно получается операцией в подходящем растворителе, таком как диоксан, THF, DME, CH3CN, DMF или DMSO, при температуре в диапазоне от комнатной температуры до 90°C, необязательно в присутствии основания, такого как K2CO3 или TEA.
В соответствии с превращением (превращение 20) способа, соединения формулы (I), где R1 обозначает, как определено в формуле (I), за исключением водорода, и X обозначает -S-, могут быть получены из соответствующих трифторметансульфонильных соединениий. Превращение проводится взаимодействием с тиолом формулы R1-SH (XIX), где R1 обозначает, как определено выше, в подходящем растворителе, таком как THF, DMF, DCM, MeOH, DME или CH3CN, при температуре в диапазоне от комнатной температуры до 100°C.
В соответствии с превращением (превращение 21) способа, соединения формулы (I), где R1 обозначает, как определено выше, могут быть получены соответствующим трифторметансульфонилом. Превращение проводится взаимодействием с производными формулы (XVII) в подходящем растворителе, таком как DMF, DCM, MeOH, DME или CH3CN, в присутствии Pd2(dba)3, PdCl2(dppf) или Pd(PPh3)4, необязательно в присутствии фторида цезия, при температуре в диапазоне от комнатной температуры до 100°C.
В соответствии с превращением (превращение 22) способа, соединения формулы (I), где R1 обозначает необязательно замещенный арил и X обозначает одиночную связь, могут быть получены соответствующим соединением формулы (I), где X обозначает -S- и R1 обозначает метил. Превращение проводится взаимодействием с бороновыми кислотами формулы (XVIIa) в подходящем растворителе, таком как DMF, THF, DCM, MeOH, DME или CH3CN, в присутствии CuTC и Pd2(dba)3 или Pd(PPh3)4, необязательно в присутствии фторида цезия, при температуре в диапазоне от комнатной до дефлегмации.
В соответствии с превращением (превращение 23) способа, соединение формулы (I), где A обозначает -(CH2)2-, может подвергаться дегидрогенизации в присутствии палладия, необязательно в носителе, палладия или платины или 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ), с тем, чтобы получить соответствующее ароматическое производное формулы (I), операцией в подходящем растворителе, таком как толуол, 1,4-диоксан, хлорбензол, дихлорбензол, при температуре в диапазоне от 90°C до дефлегмации, в течение времени, варьирующегося от 2 часов до 8 часов.
В соответствии с превращением (превращение 24) способа, соединение формулы (I), где R4 обозначает водород и A обозначает -(CH2)2-, может взаимодействовать с избытком N-йодсукцинимида в DMF при комнатной температуре с тем, чтобы получить соединение формулы (XX), которое в последующем дегалогенируется в присутствии палладиевого катализатора, например, тетракис(трифенилфосфин)палладия, и формиата натрия, с тем, чтобы получить соответствующее ароматическое производное формулы (I) операцией в подходящем растворителе, таком как N,N-диметилформамид, при температуре в диапазоне от 90°C до дефлегмации в течение времени, варьирующегося от 2 часов до 8 часов.
В соответствии с превращением (превращение 25) способа, снятие защиты атома азота соединения формулы (I), где R' обозначает защитную группу, сможет осуществляться в соответствии с обычными способами, обеспечивающими возможность селективного гидролиза трет-бутоксикарбонильной, бензильной, 4-метоксибензильной, 2,4-диметоксибензильной и трифенилметильной защитных групп. Предпочтительно, эта реакция протекает в кислотных условиях, например, в присутствии неорганической или органической кислоты, такой как хлористоводородная, трифторуксусная или метансульфоновая, в подходящем растворителе, таком как DCM, 1,4-диоксан, низший спирт, такой как метанол или этанол, при температуре в диапазоне от комнатной до дефлегмации и в течение периода времени в диапазоне от примерно 1 часа до примерно 48 часов.
В соответствии с любым вариантом способа получения соединений формулы (I), исходный материал и любой другой реагент известен или легко получается в соответствии с известными способами.
Соединение формулы (II), где A обозначает -CH2- и R5 обозначает метил, имеется в продаже.
Соединение формулы (II), где A обозначает -(CH2)2- и R5 обозначает метил, может быть получено, как описано в публикации в J. Org. Chem., 1998, 63(5), 1668.
Соединение формулы (II), где A обозначает -(СΗ2)3- и R5 обозначает метил, может быть получено, как описано в публикации в European Journal of Organic Chemistry, 2008, 23, 3917.
Соединение формулы (II), где A обозначает -C(CH3)2-CH2- и R5 обозначает метил, может быть получено, как описано в патенте США № 5750769.
Соединение формулы (II), где A обозначает -CH2-C(CH3)2- и R5 обозначает метил, может быть получено, как описано в публикации в J. Org. Chem., 1964, 29, 801.
Соединения формулы (III), где R2 обозначает метил, этил и трет-бутил, имеются в продаже.
Соединение формулы (VII), где A обозначает -CH2CH2-, может быть получено, как описано в заявке на патент США US2010/160318.
Соединение формулы (VIII) где R2 обозначает этокси и R4 обозначает метил, имеется в продаже.
Соединения формул (X), (XII), (XIII), (XIV), (XV), (XVI), (XVIa), (XVII) и (XVIIa) или имеются в продаже, или могут быть получены известными способами.
ПРИМЕРЫ
Синтетическое получение некоторых соединений формулы (I) по изобретению описано в следующих примерах.
Соединения по настоящему изобретению, полученные в соответствии со следующими примерами, были также охарактеризованы аналитическими данными 1H ЯМР или ВЭЖХ/МС (ВЭЖХ/МС, высокоэффективной жидкостной хроматографией/масс-спектроскопией); данные ВЭЖХ/МС получали, соблюдая любой из способов 1, 2, 3 и 4.
Аналитический способ ВЭЖХ/МС 1
Оборудование ВЭЖХ состояло из системы Waters Acquity™ UPLC, оборудованной детектором Waters 2996 PDA, детектором Waters Acquity ELSD™ и одиночного квадрупольного масс-спектрометра Waters модели SQD, оборудованного электрораспылительным (ESI) источником ионов. Регулировка приборов, получение данных и обработку данных обеспечивали программными обеспечениями Empower 2 и MassLynx 4.1.
ВЭЖХ проводили при 45°C при скорости потока 0,7 мл/мин, используя колонку Waters Acquity™ BEH C18, 1,7 мкм, 50×2,1 мм. Подвижная фаза A представляла собой 0,1% трифторуксусную кислоту в H2O/CH3CN (95:5), а подвижная фаза B представляла собой H2O/CH3CN (5:95); градиент составлял от 5 до 95% B за 2 минуты, затем удерживался 95% B 0,1 минуты. Объем инжекции составил 0,8 мкл. Масс-спектрометр работал в положительном и отрицательном ионном режиме, капиллярное напряжение было установлено на 3 кВ (ES+ и ES-); конус составил 30 В (ES+ и ES-); температура источника составила 120°C; полный скан, был установлен диапазон массы от 100 до 800 amu (единиц атомной массы).
Аналитический способ ВЭЖХ/МС 2
Оборудование ВЭЖХ состояло из системы Waters Alliance™ HT 2795, оборудованной детектором Waters 2996 PDA, и одиночного квадрупольного масс-спектрометра Waters модели ZQ, оборудованного электрораспылительным (ESI) источником ионов. Регулировка приборов, получение данных и обработку данных обеспечивали программными обеспечениями Empower 2 и MassLynx 4.1. ВЭЖХ проводили при 25°C при скорости потока 1,0 мл/мин, используя колонку Phenomenex Gemini C18, 3 мкм, 50×4,6 мм. Подвижная фаза A представляла собой ацетат аммония 5 мМ pH=5,2 буфер с CH3CN (95:5), а подвижная фаза B представляла собой H2O/CH3CN (5:95); градиент составил от 10 до 90% B за 8 минут, затем постепенное повышение до 100% B за 0,1 минуты. Объем инжекции составлял 10 мкл. Масс-спектрометр работал в положительном и отрицательном ионном режиме, капиллярное напряжение было установлено на 3,5 кВ (ES+) и 2,8 кВ (ES-); конус составил 14 В (ES+) и 28 В (ES-); температура источника составила 120°C; полный скан, был установлен диапазон массы от 100 до 800 amu.
Аналитический способ ВЭЖХ/МС 3
Оборудование ВЭЖХ состояло из системы Waters 2790, оборудованной детектором Waters 996 PDA, и одиночного квадрупольного масс-спектрометра Waters модели ZQ 2000, оборудованного электрораспылительным (ESI) источником ионов. Регулировка приборов, получение данных и обработку данных обеспечивали программными обеспечениями Empower 2 и MassLynx 4.1. ВЭЖХ проводили при 25°C при скорости потока 1 мл/мин, используя колонку RP18 Waters X Terra 3 мкм (3,0×20 мм). Подвижная фаза A представляла собой гидроксид аммония 0,05 мМ pH=10 буфер с CH3CN (95:5), а подвижная фаза B представляла собой H2O/CH3CN (5:95); градиент составил от 10 до 90% B за 4 минуты, затем удерживали 90% B 1 минуту. Объем инжекции составлял 10 мкл. Масс-спектрометр работал в положительном и отрицательном ионном режиме, капиллярное напряжение было установлено на 2,5 кВ; температура источника составила 120°C; конус составил 10 В; полный скан, был установлен диапазон массы от 100 до 800 amu.
Несколько соединений по изобретению формулы (I), полученных в соответствии со следующими примерами, очищали препаративной ВЭЖХ.
Рабочие условия определены ниже:
Способ 1 препаративной ВЭЖХ/МС
Оборудование ВЭЖХ состояло из системы Waters FractionLynx™, оборудованной детектором 2996 Waters PDA, и одиночного квадрупольного масс-спектрометра Waters модели ZQ 2000, оборудованного электрораспылительным (ESI) источником ионов. Регулировку приборов, получение данных и обработку данных обеспечивали программными обеспечениями Empower 2 и MassLynx 4.1. ВЭЖХ проводили при 25°C при скорости потока 20 мл/мин, используя колонку RP18 Waters X Terra 10 мкм (19×250 мм). Подвижная фаза A представляла собой гидроксид аммония 0,05% pH=10 буфер с CH3CN (95:5), и подвижная фаза B представляла собой CH3CN; градиент составил от 10 до 90% B за 15 минут, затем удерживали 90% B 3 минуты. Объем инжекции составил 200 мкл.
Масс-спектрометр работал в положительном и отрицательном ионном режиме, капиллярное напряжение было установлено на 2,5 кВ; температура источника составила 120°C; конус составил 10 В; полный скан, был установлен диапазон массы от 100 до 800 amu.
Способ 2 препаративной ВЭЖХ/МС
Оборудование ВЭЖХ состояло из системы FractionLynx™, оборудованной детектором 2996 Waters PDA, и одиночного квадрупольного масс-спектрометра Waters модели ZQ 2000, оборудованного электрораспылительным (ESI) источником ионов. Регулировку приборов, получение данных и обработку данных обеспечивали программными обеспечениями Empower 2 и MassLynx 4.1. ВЭЖХ проводили при 25°C при скорости потока 20 мл/мин, используя колонку RP18 Waters X Terra 10 мкм (19×250 мм). Подвижная фаза A представляла собой 0,1% TFA в H2O/CH3CN (95:5), и подвижная фаза B представляла собой CH3CN; градиент составил от 10 до 90% B за 15 минут, затем удерживали 90% B 3 минуты. Объем инжекции составил 200 мкл.
Масс-спектрометр работал в положительном и отрицательном ионном режиме, капиллярное напряжение было установлено на 2,5 кВ; температура источника составила 120°C; конус составил 10В; полный скан, был установлен диапазон мыссы от 100 до 800 amu.
Точная масс-спектроскопия
Точные данные масс-спектроскопии ESI(+) получали на приборе Waters Q-Tof Ultima, прямо соединенном с микроанализатором ВЭЖХ 1100 Agilent, как описано ранее (M. Colombo, F. Riccardi-Sirtori, V. Rizzo, Rapid Commun. Mass Spectrom. 2004, 18, 511-517).
ЯМР
1H-ЯМР спектры регистрировали при постоянной температуре 28°C на спектрометре Varian INOVA 400, работающем при 400,50 МГц и оборудованном 5-мм z-осевым датчиком непрямого выявления PFG (прецизионного функционального генератора) (1H{15N-31P}).
Химические сдвиги указывались в отношении сигналов остаточных растворителей (ДМСО-d6: 2,50 м.д. для 1H, при отсутствии иных определений). Данные представлены следующим образом: химический сдвиг (δ), множественность (с=синглет, д=дублет, т=триплет, кв=квартет, шир.с=широкий синглет, тд=триплет дублетов, дд=дублет дублетов, ддд=дублет дублетов дублетов, м=мультиплет, септ=септет), константы соединения (J, Гц), и число протонов.
В примерах ниже, а также по всей заявке следующие аббревиатуры имеют следующие значения.
При отсутствии определений, термины имеют их общепринятые значения.
Получение A (стадия A)
диметил-(2E)-гепт-2-ендиоат
Раствор метил-5-оксопентаноата (1,9 г, 14,6 ммоль) и (карбоэтоксиметилена) трифенилфосфорана (5,0 г, 14,9 ммоль) в толуоле (50 мл) кипятили в сосуде с обратным холодильником в течение 8 часов. Растворитель удаляли в вакууме, и неочищенный продукт очищали флэш-хроматографией на силикагеле (элюент: AcOEt/гексан 2/8) для получения 1,32 г (выход 48%) указанного в заголовке соединения в виде бесцветного масла.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,61-1,75 (м, 2H) 2,16-2,27 (м, 2H) 2,27-2,35 (м, 2H) 3,58 (с, 3H) 3,64 (с, 3H) 5,87 (дт, J=15,65, 1,56 Гц, 1H) 6,87 (дт, J=15,65, 6,94 Гц, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
диметил-(2Z)-5,5-диметилгепт-2-ендиоат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 0,98 (с, 6H) 2,24 (с, 2H) 2,63 (дт, J=7,75, 1,65 Гц, 2H) 3,57 (м, 3H) 3,62 (с, 3H) 5,90 (дт, J=11,60, 1,65 Гц, 1H) 6,38 (дт, J=11,60, 7,75 Гц, 1H).
диметил-(2E)-5,5-диметилгепт-2-ендиоат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 0,96 (с, 6H) 2,21 (с, 2H) 2,22-2,24 (м, 2H) 3,58 (с, 3H) 3,65 (с, 3H) 5,90 (дт, J=15,50, 5,85 Гц, 1H) 6,88 (дт, J=15,50, 7,80 Гц, 1H).
Диметил-(2E)-гекс-2-ендиоат
Диметил-(2E)-окт-2-ендиоат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,36-1,47 (м, 4H) 1,37-1,57 (м, 2H) 2,20 (кв.д, J=7,05, 1,60 Гц, 2H) 2,31 (т, J=7,32 Гц, 2H) 3,58 (с, 3H) 3,64 (с, 3H) 5,88 (дт, J=15,65, 1,60 Гц, 1H) 6,87 (дт, J=15,65, 7,05 Гц, 1H).
1-этил-8-метил-(2E)-окт-2-ендиоат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,20 (т, J=7,16 Гц, 3H) 1,36-1,46 (м, 2H) 1,47-1,57 (м, 2H) 2,20 (кв.д, J=7,10, 1,46 Гц, 2H) 2,31 (т, J=7,14 Гц, 2H) 3,58 (с, 3H) 4,10 (кв, J=7,16 Гц, 2H) 5,86 (дт, J=15,56, 1,46 Гц, 1H) 6,86 (дт, J=15,56, 7,10 Гц, 1H).
1-этил-7-метил-(2E)-5,5-диметилгепт-2-ендиоат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 0,96 (с, 6H) 1,21 (т, J=7,08 Гц, 3H) 2,21 (с, 2H) 2,22 (дд, J=7,87, 1,30 Гц, 2H) 3,58 (с, 3H) 4,11 (кв, J=7,08 Гц, 2H) 5,88 (дт, J=15,47, 1,30 Гц, 1H) 6,86 (дт, J=15,47, 7,87 Гц, 1H).
1-этил-7-метил-(2E)-гепт-2-ендиоат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,20 (т, J=7,14 Гц, 3H) 1,68 (квин, J=7,30 Гц, 2H) 2,15-2,27 (м, 2H) 2,31 (т, J=7,30 Гц, 2H) 3,58 (с, 3H) 4,11 (кв, J=7,14 Гц, 2H) 5,85 (дт, J=15,65, 1,59 Гц, 1H) 6,86 (дт, J=15,65, 6,94 Гц, 1H).
Получение B (стадия B)
метил-4-(4-метокси-4-оксобутил)-1H-пиррол-3-карбоксилат
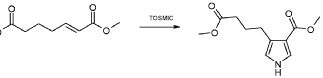
К 1M раствору LiN(TMS)2 в THF (5,9 мл, 5,9 ммоль), охлажденному при -78°C, в атмосфере аргона по каплям добавляли раствор TOSMIC (1,15 г, 5,9 ммоль) в THF (15 мл). Через 40 мин при -78°C медленно добавляли раствор диметил-(2E)-гепт-2-ендиоата (1,1 г, 5,9 ммоль) в THF (15 мл) при -78°C. Раствор перемешивали в течение 10 мин, затем холодную баню удаляли, и реакционной смеси давали возможность согреться при комнатной температуре. THF выпаривали, и остаток разделяли между H2O (200 мл) и DCM (200 мл). Водный слой экстрагировали DCM, и объединенные органические слои обезвоживали над Na2SO4, фильтровали и концентрировали для получения остатка, который хроматографировали на силикагеле (элюент: AcOEt/гексан 3/7) для получения 633 мг (выход: 40%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,77 (квин, J=7,45 Гц, 2H) 2,28 (т, J=7,45 Гц, 2H) 2,62 (т, J=7,45 Гц, 2H) 3,57 (с, 3H) 3,66 (с, 3H) 6,60 (т, J=2,20 Гц, 1H) 7,32 (дд, J=3,17, 2,20 Гц, 1H) 11,15 (шир.с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
метил-4-(4-метокси-2,2-диметил-4-оксобутил)-1H-пиррол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 0,90 (с, 6H) 2,06 (с, 2H) 2,74 (с, 2H) 3,65 (с, 3H) 6,60 (с, 1H) 7,33 (т, J=2,56 Гц, 1H) 11,21 (шир.с, 1H) 11,84 (шир.с, 1H).
метил-4-(3-метокси-3-охопропил)-1H-пиррол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,52-2,58 (м, 2H) 2,83-2,90 (м, 2H) 3,58 (с, 3H) 3,67 (с, 3H) 6,60 (т, J=2,25 Гц, 1H) 7,33(дд, J=3,17, 2,25 Гц, 1H) 11,16 (шир.с, 1H).
метил-4-(5-метокси-5-оксопентил)-1H-пиррол-3-карбоксилат
MS рассчитанный: 240,1231; MS найденный: 240,1226.
этил-4-(4-метокси-2,2-диметил-4-оксобутил)-1H-пиррол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 0,89 (с, 6H) 1,24 (т, J=7,08 Гц, 3H) 2,15 (с, 2H) 2,73 (с, 2H) 3,56 (с, 3H) 4,12 (кв, J=7,08 Гц, 2H) 6,59 (т, J=2,30 Гц, 1H) 7,31 (дд, J=3,11, 2,30 Гц, 1H) 11,21 (шир.с, 1H).
этил-4-(4-метокси-4-оксобутил)-1H-пиррол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,24 (т, J=7,14 Гц, 3H) 1,70-1,83 (м, 2H) 2,28 (т, J=7,51 Гц, 2H) 2,61 (т, J=7,51 Гц, 2H) 3,57 (с, 3H) 4,13 (кв, J=7,14 Гц, 1H) 6,59 (т, J=2,20 Гц, 1H) 7,31 (дд, J=3,17, 2,20 Гц, 1H) 11,13 (шир.с, 1H).
Получение C (стадия C)
4-[4-(метоксикарбонил)-1H-пиррол-3-ил]бутановая кислота

Метил-4-(4-метокси-4-оксобутил)-1H-пиррол-3-карбоксилат (50 мг, 0,220 ммоль) суспендировали в безводном диоксане (2 мл) и добавляли H2O (0,5 мл) и LiOH (5,3 мг, 0,220 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор подкисляли 1н HCl и AcOEt (50 мл) и добавляли H2O (20 мл). Водный слой экстрагировали AcOEt, и объединенные органические слои обезвоживали над Na2SO4, фильтровали и концентрировали для получения 46 мг (количественный выход) указанного в заголовке соединения в виде белого твердого вещества.
MS рассчитанный: 212,0918; MS найденный: 212,0917.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,74 (квин, J=7,50 Гц, 2H) 2,19 (т, J=7,50 Гц, 2H) 2,62 (т, J=7,50 Гц, 2H) 3,66 (с, 3H) 6,60 (т, J=2,05 Гц, 1H) 7,32 (дд, J=3,10, 2,05 Гц, 1H) 11,14 (шир.с, 1H) 11,93 (шир.с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
4-[4-(метоксикарбонил)-1H-пиррол-3-ил]-3,3-диметилбутановая кислота
MS рассчитанный: 240,1230; MS найденный: 240,1229.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 0,90 (с, 6H) 2,06 (с, 2H) 2,74 (с, 2H) 3,65 (с, 3H) 6,60 (с, 1H) 7,33 (т, J=2,56 Гц, 1H) 11,21 (шир.с, 1H) 11,84 (шир.с, 1H).
3-[4-(метоксикарбонил)-1H-пиррол-3-ил]пропановая кислота
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,45 (т, J=7,50 Гц, 2H) 2,84 (т, J=7,50 Гц, 2H) 3,67 (с, 3H) 6,60 (т, J=2,20 Гц, 1H) 7,33 (дд, J=3,17, 2,20 Гц, 1H) 11,15 (шир.с, 1H) 11,96 (шир.с, 1H).
5-[4-(метоксикарбонил)-1H-пиррол-3-ил]пентановая кислота
MS рассчитанный: 248,0893; MS найденный: 248,0896.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,45-1,56 (м, 4H) 2,20 (т, J=6,90 Гц, 2H) 2,60 (т, J=6,90 Гц, 2H) 3,66 (с, 3H) 6,59 (т, J=2,20 Гц, 1H) 7,31 (дд, J=3,17, 2,20 Гц, 1H) 11,11 (шир.с, 1H) 11,92 (шир.с, 1H).
4-[4-(этоксикарбонил)-1H-пиррол-3-ил]-3,3-диметилбутановая кислота
4-[4-(этоксикарбонил)-1H-пиррол-3-ил]бутановая кислота
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,24 (т, J=7,08 Гц, 3H) 1,74 (квин, J=7,50 Гц, 2H) 2,19 (т, J=7,50 Гц, 2H) 2,61 (т, J=7,50 Гц, 2H) 4,14 (кв, J=7,08 Гц, 2H) 6,59 (т, J=2,20 Гц, 1H) 7,31 (дд, J=3,17, 2,20 Гц, 1H) 11,12 (шир.с, 1H) 11,92 (шир.с, 1H).
Получение D (стадия Ca)
4-[4-(этоксикарбонил)-5-метил-1H-пиррол-3-ил]бутановая кислота
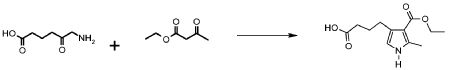
Гидрохлорид 6-амино-5-оксогексановой кислоты (9,73 г, 49,8 ммоль) растворяли в H2O (35 мл), и к указанному выше раствору добавляли этилацетоцетат (5,51 г, 42,34 ммоль) и AcONa (20,3 г, 14,95 ммоль). Реакционный раствор кипятили в сосуде с обратным холодильником в течение 1 ч, охлаждали до комнатной температуры и добавляли 0,5н HCl до тех пор, пока ни достигался pH примерно 5. Объединенные органические слои обезвоживали над Na2SO4, фильтровали и концентрировали для получения 7,15 г (выход: 60%) указанного в заголовке соединения в виде коричневого твердого вещества.
MS рассчитанный: 240,1231; MS найденный: 240,1225.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,25 (т, J=7,14 Гц, 3H) 1,71 (квин, J=7,50 Гц, 2H) 2,18 (т, J=7,50 Гц, 2H) 2,36 (с, 3H) 2,56 (т, J=7,50 Гц, 2H) 4,13 (кв, J=7,14 Гц, 2H) 6,39 (д, J=2,32 Гц, 1H) 10,91 (шир.с, 1H) 11,90 (шир.с, 1H).
Получение E (стадия D)
метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
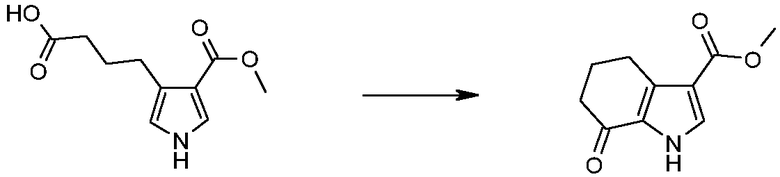
4-[4-(Метоксикарбонил)-1H-пиррол-3-ил]бутановую кислоту (500 мг, 2,36 ммоль) растворяли в TFA (3 мл). Добавляли TFAA (0,329 мл, 2,36 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Органический растворитель выпаривали, и остаток суспендировали в Et2O (15 мл) и фильтровали для получения 320 мг (выход: 64%) указанного в заголовке соединения в виде светло-желтого твердого вещества.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,02 (квин, J=6,30 Гц, 2H) 2,40 (т, J=6,30 Гц, 2H) 2,91 (т, J=6,30 Гц, 2H) 3,72 (с, 3H) 7,58 (д, J=3,42 Гц, 1H) 12,38 (шир.с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
метил-5,5-диметил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 0,97 (с, 6H) 2,30 (с, 2H) 2,82 (с, 2H) 3,72 (с, 3H) 7,60 (d, J=3,42 Гц, 1H) 12,38 (шир.с, 1H).
метил-6-оксо-1,4,5,6-тетрагидроциклопента[b]пиррол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,71-2,80 (м, 2H) 2,87-2,93 (м, 2H) 3,74 (с, 3H) 7,85 (с, 1H) 12,37 (шир.с, 1H).
метил-8-оксо-1,4,5,6,7,8-гексагидроциклогепта[b]пиррол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,70-1,80 (м, 2H) 1,81 -1,89 (м, 2H) 2,59-2,65 (м, 2H) 3,06-3,15 (м, 2H) 3,70 (с, 3H) 7,51 (д, J=3,66 Гц, 1H) 12,01 (шир.с, 1H).
этил-5,5-диметил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,03 (с, 6H) 1,26 (т, J=7,08 Гц, 3H) 2,30 (с, 2H) 2,83 (с, 2H) 4,19 (кв, J=7,08 Гц, 2H) 7,57 (д, J=3,42 Гц, 1H) 12,36 (шир.с, 1H).
этил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,24 (т, J=7,08 Гц, 3H) 1,74 (квин, J=7,50 Гц, 2H) 2,19 (т, J=7,50 Гц, 2H) 2,61 (т, J=7,50 Гц, 2H) 4,14 (кв, J=7,08 Гц, 2H) 6,59 (т, J=2,20 Гц, 1H) 7,31 (дд, J=3,17, 2,20 Гц, 1H) 11,12 (шир.с, 1H) 11,92 (шир.с, 1H).
этил-2-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
MS рассчитанный: 222,1125; MS найденный: 222,1136.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,27 (т, J=7,08 Гц, 3H) 1,99 (квин, J=6,30 Гц, 2H) 2,36 (т, J=6,30 Гц, 2H) 2,43 (с, 3H) 2,87 (т, J=6,30 Гц, 2H) 4,18 (кв, J=7,08 Гц, 2H) 12,15 (шир.с, 1H).
Получение F (стадия E)
метил-1-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат

К раствору метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилата (300 мг, 1,55 ммоль) в сухом DMF (5 мл) добавляли K2CO3 (429 мг, 3,10 ммоль) и метилйодид (0,193 мл, 3,10 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, затем добавляли H2O (100 мл), и продукт экстрагировали DCM (330 мл). Органические фракции сушили над Na2SO4, фильтровали и концентрировали в вакууме для получения 305 мг (выход: 95%) в виде светло-желтого твердого вещества.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,95-2,03 (м, 2H) 2,40 (т, J=6,15 Гц, 2H) 2,91 (т, J=6,04 Гц, 2H) 3,72 (с, 3H) 3,85 (с, 3H) 7,71 (с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
метил-1-метил-6-оксо-1,4,5,6-тетрагидроциклопента[b]пиррол-3-карбоксилат
MS рассчитанный: 194,0812; MS найденный: 194,0810.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,72-2,81 (м, 2H) 2,83-2,92 (м, 2H) 3,73 (с, 3H) 3,74 (с, 3H) 7,81-7,91 (м, 1H).
метил-1,5,5-триметил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
MS рассчитанный: 236,1281; MS найденный: 236,1281.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,02 (с, 6H) 2,30 (с, 2H) 2,83 (с, 2H) 3,72 (с, 3H) 3,85 (с, 3H) 7,72 (с, 1H).
метил-1-метил-8-оксо-1,4,5,6,7,8-гексагидроциклогепта[b]пиррол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,60-1,74 (м, 4H) 2,53-2,62 (м, 2H) 3,13 (т, J=6,04 Гц, 2H) 3,68 (с, 3H) 3,78 (с, 3H) 7,66 (с, 1H).
этил-1,5,5-триметил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
MS рассчитанный: 250,1438; MS найденный: 250,1444.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,02 (с, 6H) 1,26 (т, J=7,08 Гц, 3H) 2,30 (с, 2H) 2,83 (с, 2H) 3,85 (с, 3H) 4,19 (кв, J=7,08 Гц, 2H) 7,70 (с, 1H).
этил-1-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
MS рассчитанный: 222,1125; MS найденный: 222,1134.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,26 (т, J=7,16 Гц, 3H) 1,93-2,05 (м, 2H) 2,40 (т, J=6,10 Гц, 2H) 2,92 (т, J=6,10 Гц, 2H) 3,86 (с, 3H) 4,19 (кв, J=7,16 Гц, 2H) 7,70 (с, 1H).
этил-1-(метоксиметил)-2-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
MS рассчитанный: 266,1387; MS найденный: 266,1376.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,29 (т, J=7,12 Гц, 3H) 1,92-2,03 (м, 2H) 2,39-2,45 (м, 2H) 2,53 (с, 3H) 2,93 (т, J=6,16 Гц, 2H) 3,19 (с, 3H) 4,22 (кв, J=7,12 Гц, 2H) 5,75 (с, 2H).
этил-1,2-диметил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
MS рассчитанный: 236,1281; MS найденный: 236,1283.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,28 (т, J=7,08 Гц, 3H) 1,95 (квин, J=6,25 Гц, 2H) 2,38 (т, J=6,25 Гц, 2H) 2,48 (с, 3H) 2,90 (т, J=6,25 Гц, 2H) 3,83 (с, 3H) 4,20 (кв, J=7,08 Гц, 2H).
Получение G (стадия F)
Метил-(6E)-6-[(диметиламино)метилиден]-1-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
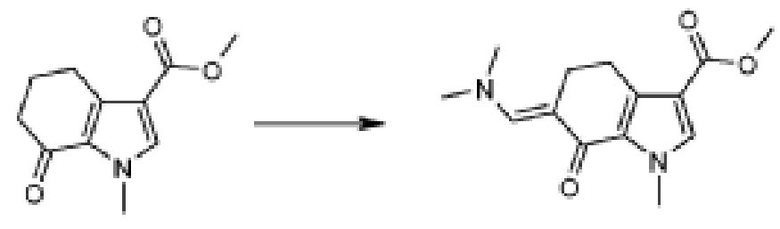
Метил-1-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат (288 мг, 1,39 ммоль) обрабатывали трис(диметиламино)метаном (2,4 мл, 13,9 ммоль), и реакционную смесь перемешивали при 90°C в течение 10 ч. Летучие вещества удаляли под пониженным давлением, и остаток использовали без дополнительной очистки.
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
метил-(6E)-5,5-диметил-6-[(метиламино)метилиден]-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
MS рассчитанный: 263,1390; MS найденный: 263,1384.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,15 (с, 6H) 2,74 (с, 2H) 2,97 (д, J=5,05 Гц, 3H) 3,64-3,73 (м, 3H) 6,91 (д, J=12,30 Гц, 1H) 7,40 (д, J=3,30 Гц, 1H) 9,58 (дд, J=12,30, 5,05 Гц, 1H) 12,00 (шир.с, 1H).
метил-(7E)-7-[(диметиламино)метилиден]-1-метил-8-оксо-1,4,5,6,7,8-гексагидроциклогепта[b]пиррол-3-карбоксилат
MS рассчитанный: 277,1547; MS найденный: 277,1554.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,76 (квин, J=6,80 Гц, 2H) 2,32 (т, J=6,80 Гц, 2H) 2,89 (т, J=6,80 Гц, 2H) 3,07 (с, 6H) 3,69 (с, 3H) 3,74 (с, 3H) 7,36 (с, 1H) 7,51 (с, 1H).
этил-(6E)-6-[(диметиламино)метилиден]-1-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,25 (т, J=7,14 Гц, 3H) 2,76-2,89 (м, 4H) 3,05 (с, 6H) 3,87 (с, 3H) 4,17 (кв, J=7,14 Гц, 2H) 7,29 (с, 1H) 7,53 (с, 1H).
этил-(6E)-6-[(диметиламино)метилиден]-2-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
MS рассчитанный: 277,1547; MS найденный: 277,1544.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,26 (т, J=7,08 Гц, 3H) 2,41 (с, 3H) 2,77-2,83 (м, 2H) 2,85-2,91 (м, 2H) 3,04 (с, 6H) 4,16 (кв, J=7,08 Гц, 2H) 7,26 (с, 1H) 11,86 (шир.с, 1H).
этил-(6E)-6-[(диметиламино)метилиден]-1-(метоксиметил)-2-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
метил-(6E)-6-[(диметиламино)метилиден]-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
LC/MS (254 нм) ВЭЖХ способ 2 Время удерживания 4,5 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 2,80-2,84 (м, 2H) 2,88-2,92 (м, 2H) 3,06 (с, 6H) 3,70 (с, 3H) 7,31 (с, 1H) 7,41 (д, J=2,75 Гц, 1H) 12,06 (шир.с, 1H).
метил-(6E)-1-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-6-[(диметиламино)метилиден]-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
Получение H (стадия E)
метил-1-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилат
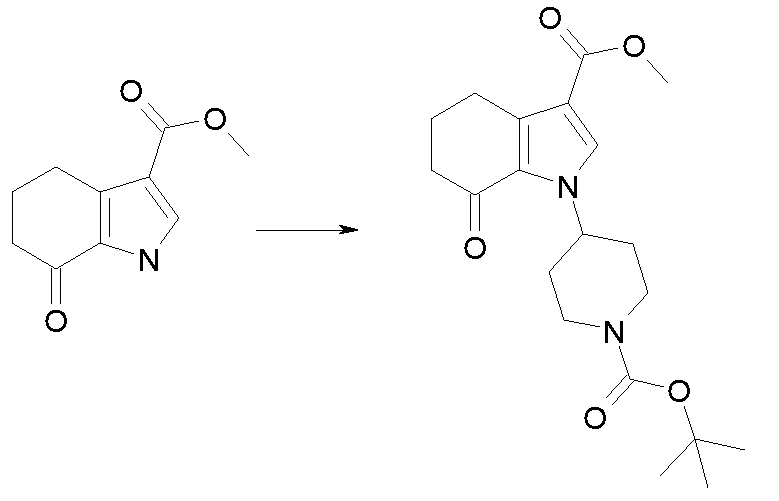
К смеси метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилата (100 мг, 0,52 ммоль), трет-бутил-4-гидроксипиперидин-1-карбоксилата (105 мг, 0,52 ммоль) и трифенилфосфина (136 мг, 0,52 ммоль) в безводном THF (5 мл) при комнатной температуре добавляли ди-трет-бутил-диазадикарбоксилат (DTAD) (120 мг, 0,52 ммоль). Смесь перемешивали при комнатной температуре в течение 8 ч. ВЭЖХ/МС свидетельствовали о 40% превращении, и оставались 60% SM (исходных соединений). Добавляли реагенты, трифенилфосфин (136 мг, 0,52 ммоль) и DTAD (120 мг, 0,52 ммоль), смесь перемешивали в течение 4 часов. ВЭЖХ/МС показали 80% превращение, оставались 20% SM. В реагенты снова добавляли TPP (тетрафенилфосфония бромид) (136 мг, 0,52 ммоль) и DTAD (120 мг, 0,526 ммоль), и раствор перемешивали в течение еще 4 часов. Летучие вещества удаляли в вакууме, неочищенное твердое вещество очищали флэш-хроматографией на силикагеле (гексан/EtOAc 7/3) для получения 140 мг (выход 70%) указанного в заголовке соединения в виде белого твердого вещества.
Пример 1 (стадия G)
метил-2-[(4-бром-2-метоксифенил)амино]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-O-метил, R3=метил, R4=H, A=-CH2CH2-]
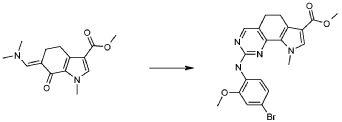
К суспензии метил-(6E)-6-[(диметиламино)метилиден]-1-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилата (365 мг, 1,39 ммоль) в DMF (5 мл) добавляли N-(4-бром-2-метоксифенил)гуанидин (340 мг, 1,39 ммоль). Смесь перемешивали при 120°C в течение 3 часов. Полученную смесь охлаждали при комнатной температуре и выпаривали до сухости. Неочищенное твердое вещество очищали флэш-хроматографией на силикагеле (элюент: AcOEt/гексан 4/6) для получения 306 мг (выход: 50%) указанного в заголовке соединения в виде светло-оранжевого твердого вещества.
MS рассчитанный: 443,0714; MS найденный: 443,0704.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,76 (т, J=7,81 Гц, 2H) 2,93 (т, J=7,81 Гц, 2H) 3,72 (с, 3H) 3,88 (с, 3H) 4,03 (с, 3H) 7,12 (дд, J=8,60, 2,20 Гц, 1H) 7,20 (д, J=2,20 Гц, 1H) 7,65 (д, J=0,49 Гц, 1H) 7,91 (с, 1H) 8,07 (д, J=8,60 Гц, 1H) 8,21 (с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
этил-2-[(4-бром-2-метоксифенил)амино]-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-O-этил, R3=H, R4=метил, A=-CH2CH2-]
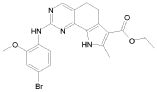
MS рассчитанный: 457,087; MS найденный: 457,0868.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,28 (т, J=7,08 Гц, 3H) 2,51 (шир.с, 3H) 2,75-2,83 (м, 2H) 2,91-2,98 (м, 2H) 3,92 (с, 3H) 4,19 (кв, J=7,08 Гц, 2H) 7,13 (дд, J=8,65, 2,20 Гц, 1H) 7,20 (д, J=2,20 Гц, 1H) 7,60 (с, 1H) 8,17 (с, 1H) 8,50 (д, J=8,65 Гц, 1H) 12,07 (с, 1H).
этил-9-метил-2-{[4-(4-метилпиперазин-1-ил)фенил]амино}-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=4-(4-метилпиперазин-1-ил)фенил, X=-NH-, R2=-O-этил, R3=метил, R4=H, A=-CH2CH2-]
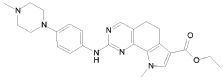
MS рассчитанный: 447,2503; MS найденный: 447,2485.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,27 (т, J=7,08 Гц, 3H) 2,22 (с, 3H) 2,42-2,47 (м, 4H) 2,70-2,77 (м, 2H) 2,89-2,95 (м, 2H) 3,02-3,09 (м, 4H) 4,08 (с, 3H) 4,19 (кв, J=7,08 Гц, 2H) 6,84-6,90 (м, 2H) 7,45-7,53 (м, 2H) 7,61 (с, 1H) 8,14 (с, 1H) 8,97 (с, 1H).
этил-2-[(4-бром-2-метоксифенил)амино]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-O-этил, R3=метил, R4=H, A=-CH2CH2-]
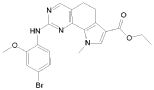
MS рассчитанный: 457,0870; MS найденный: 457,0876.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,27 (т, J=7,08 Гц, 3H) 2,71-2,83 (м, 2H) 2,89-2,98 (м, 2H) 3,88 (с, 3H) 4,03 (с, 3H) 4,20 (кв, J=7,08 Гц, 2H) 7,12 (дд, J=8,60, 2,20 Гц, 1H) 7,20 (д, J=2,20 Гц, 1H) 7,63 (с, 1H) 7,92 (с, 1H) 8,06 (д, J=8,60 Гц, 1H) 8,20 (с, 1H).
метил-2-[(4-бром-2-метоксифенил)амино]-10-метил-5,6,7,10-тетрагидропирроло[3',2':6,7]циклогепта[1,2-d]пиримидин-8-карбоксилат [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-O-метил, R3=метил, R4=H, A=-(CH2)3-]
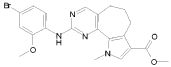
MS рассчитанный: 457,0870; MS найденный: 457,0851.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,92-2,07 (м, 2H) 2,91 (т, J=7,08 Гц, 2H) 3,71 (с, 3H) 3,85 (с, 3H) 3,86 (с, 3H) 7,13 (дд, J=8,61, 2,20 Гц, 1H) 7,20 (д, J=2,20 Гц, 1H) 7,66 (с, 1H) 7,93-8,08 (м, 2H) 8,30 (с, 1H).
этил-2-амино-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(l), R1=H, X=-NH-, R2=-O-этил, R3=H, R4=метил, A=-CH2CH2-]
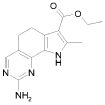
этил-9-(метоксиметил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=2-метокси-4-(4-метилпиперазин-1-ил)фенил, X=-NH-, R2=-O-этил, R3=метоксиметил, R4=метил, A=-CH2CH2-]
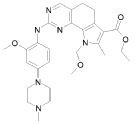
Пример 2 (превращение 3)
2-[(4-бром-2-метоксифенил)амино]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-OH, R3=метил, R4=H, A=-CH2CH2-]
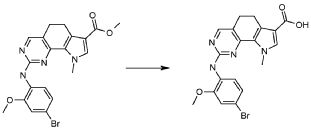
Метил-2-[(4-бром-2-метоксифенил)амино]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат (300 мг, 0,68 ммоль) суспендировали в диоксане (10 мл) и обрабатывали 2н раствором NaOH (5,1 мл, 10,2 ммоль) при температуре дефлегмации в течение 3 ч. Добавляли H2O (50 мл), и раствор подкисляли 2н HCl. Полученный осадок собирали фильтрацией для получения 211 мг (выход 72%) указанного в заголовке соединения в виде белого твердого вещества.
MS рассчитанный: 429,0557; MS найденный: 429,0566.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,73 (т, J=7,93 Гц, 2H) 2,93 (т, J=7,93 Гц, 2H) 3,88 (с, 3H) 4,03 (с, 3H) 7,12 (дд, J=8,67, 2,20 Гц, 1H) 7,20 (д, J=2,20 Гц, 1H) 7,57 (с, 1H) 7,88 (с, 1H) 8,08 (д, J=8,67 Гц, 1H) 8,20 (с, 1H) 12,00 (шир.с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
2-[(4-бром-2-метоксифенил)амино]-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-OH, R3=метил, R4=метил, A=-CH2CH2-]
MS рассчитанный: 443,0714; MS найденный: 443,0703.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,53 (с, 3H) 2,67-2,74 (м, 2H) 2,87-2,97 (м, 2H) 3,88 (с, 3H) 4,00 (с, 3H) 7,12 (дд, J=8,54, 2,20 Гц, 1H) 7,20 (д, J=2,20 Гц, 1H) 7,86 (с, 1H) 8,09 (д, J=8,54 Гц, 1H) 8,16 (с, 1H) 12,02 (шир.с, 1H).
2-амино-9-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(l), R1=H, X=-NH-, R2=-OH, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=H, A=-CH2CH2-]
Пример 3 (превращение 4)
2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-CH2CH2- (соединение 8)
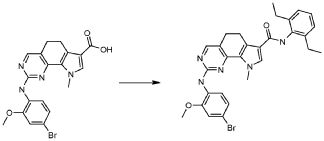
2-[(4-Бром-2-метоксифенил)амино]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновую кислоту (206 мг, 0,48 ммоль) суспендировали в сухом THF (10 мл) и добавляли SOCl2 (0,7 мл, 9,6 ммоль) в атмосфере аргона. Реакционную смесь кипятили в сосуде с обратным холодильником в течение 2 ч, затем все летучие вещества удаляли при пониженном давлении. Неочищенный остаток растворяли в сухом DCM (10 мл), затем добавляли DIPEA (0,43 мл, 2,4 ммоль) и 2,6-диэтиланилин (143 мг, 0,96 ммоль) и реакционную смесь кипятили в сосуде с обратным холодильником в течение 2 ч. Добавляли DCM (100 мл), и органическую фазу экстрагировали H2O (3×25 мл). Органический слой сушили над безводным Na2SO4, и растворитель выпаривали до сухости для получения 190 мг (выход: 70%) указанного в заголовке соединения в виде бледно-желтого твердого вещества.
MS рассчитанный: 560,1656; MS найденный: 560,1655.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,57 Гц, 6H) 2,55 (кв, J=7,57 Гц, 4H) 2,72-2,79 (м, 2H) 2,95-3,02 (м, 2H) 3,89 (с, 4H) 4,08 (с, 3H) 7,08-7,16 (м, 3H) 7,16-7,24 (м, 2H) 7,74 (с, 1H) 7,88 (с, 1H) 8,12 (д, J=8,70 Гц, 1H) 8,20 (с, 1H) 9,05 (с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
2-[(4-бром-2-метоксифенил)амино]-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-NH2, R3=метил, R4=метил, A=-CH2CH2-]
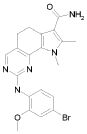
Пример 4 (превращение 5)
2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=метил, A=-CH2CH2-] (соединение 2)
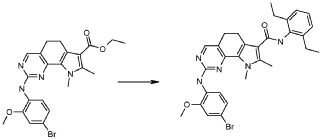
К раствору 2,6-диэтиланилина (300 мг, 2,01 ммоль) в сухом THF (10 мл) в атмосфере аргона по каплям добавляли 1M раствор LiN(TMS)2 (4,02 мл, 4,02 ммоль) в THF при 0°C. Смесь перемешивали при 0°C в течение 10 минут, затем по каплям добавляли этил-2-[(4-бром-2-метоксифенил)амино]-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат (0,315 г, 0,67 ммоль) в сухом THF (10 мл) при 0°C. Ледяную баню удаляли, и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли H2O (20 мл) и смесь экстрагировали AcOEt (2×30мл). Органический слой сушили над безводным Na2SO4, и растворитель выпаривали до сухости. Неочищенное твердое вещество очищали флэш-хроматографией на силикагеле (элюент: AcOEt/циклогексан 1/1) для получения 355 мг (выход 92%) указанного в заголовке соединения в виде светло-желтого твердого вещества.
MS рассчитанный: 574,1812; MS найденный: 574,1818.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,14 (т, J=7,57 Гц, 6H) 2,44 (с, 3H) 2,58 (кв, J=7,57 Гц, 4H) 2,71-2,81 (м, 2H) 2,85-2,94 (м, 2H) 3,89 (с, 3H) 4,02 (с, 3H) 7,10-7,16 (м, 3H) 7,18-7,23 (м, 2H) 7,85 (с, 1H) 8,13 (д, J=8,67 Гц, 1H) 8,16 (с, 1H) 8,84 (с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
N-(2,6-диэтилфенил)-9-(метоксиметил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=2-метокси-4-(4-метилпиперазин-1-ил)фенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метоксиметил, R4=метил, A=-CH2CH2-] (соединение 1)
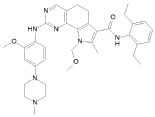
MS рассчитанный: 624,3657; MS найденный: 624,3660.
2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-10-метил-5,6,7,10-тетрагидропирроло[3',2':6,7]циклогепта[1,2-d]пиримидин-8-карбоксамид [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-(CH2)3-] (соединение 15)
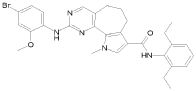
MS рассчитанный: 574,1812; MS найденный: 574,1797.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,12 (т, J=7,57 Гц, 6H) 1,94-2,05 (м, 2H) 2,55 (кв, J=7,57 Гц, 4H) 2,92 (т, J=7,14 Гц, 2H) 3,88 (с, 3H) 3,89 (с, 3H) 7,09-7,20 (м, 4H) 7,21 (д, J=2,32 Гц, 1H) 7,69 (с, 1H) 7,98 (с, 1H) 8,10 (д, J=8,54 Гц, 1H) 8,30 (с, 1H) 9,05 (с, 1H).
Пример 5 (превращение 6)
N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=2-метокси-4-(4-метилпиперазин-1-ил)фенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-CH2CH2-] (соединение 9)
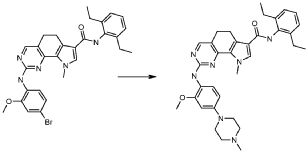
Pd2(dba)3, (10 мг, 0,010 ммоль), 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил (10 мг, 0,025 ммоль) и 2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид (100 мг, 0,178 ммоль) в сухом THF (5 мл) загружали в круглодонную колбу, продутую аргоном. Содержимое колбы эвакуировали и снова заполняли аргоном. Добавляли раствор LiN(TMS)2 (1M в THF, 1,39 мл) и N-метилпиперазина (0,058 мл, 0,522 ммоль), реакционную смесь нагревали при 85°C в течение 0,5 ч. Затем реакционной смеси давали возможность охладиться до комнатной температуры, и растворитель выпаривали до сухости. Неочищенное твердое вещество очищали флэш-хроматографией на силикагеле (элюент: DCM/MeOH 95/5) для получения 72 мг (выход 70%) указанного в заголовке соединения в виде желтого твердого вещества.
MS рассчитанный: 580,3395; MS найденный: 580,3373.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,57 Гц, 6H) 2,25 (шир.с, 3H) 2,54 (кв, J=7,57 Гц, 4H) 2,69-2,76 (м, 2H) 2,91-2,99 (м, 2H) 3,05-3,19 (м, 4H) 3,82 (с, 3H) 4,04 (с, 3H) 6,49 (дд, J=8,67, J=2,56 Гц, 1H) 6,63 (д, J=2,56 Гц, 1H) 7,08-7,14 (м, 2H) 7,16-7,23 (м, 1H) 7,66 (с, 1H) 7,69 (с, 1H) 7,76 (д, J=8,67 Гц, 1H) 8,10 (с, 1H) 9,03 (с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=метил, A=-CH2CH2-] (соединение 3)
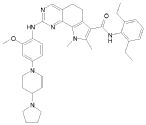
MS рассчитанный: 648,4021; MS найденный: 648,4026.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,13 (т, J=7,57 Гц, 6H) 1,47-1,61 (м, 2H) 1,64-1,76 (м, 4H) 1,88-1,98 (м, 2H) 2,42 (с, 3H) 2,52-2,63 (м, 8H) 2,64-2,75 (м, 4H) 2,83-2,92 (м, 2H) 3,26-3,29 (м, 1H) 3,54-3,65 (м, 2H) 3,82 (с, 3H) 3,98 (с, 3H) 6,49 (дд, J=8,65, 2,50 Гц, 1H) 6,63 (д, J=2,40 Гц, 1H) 7,08-7,15 (м, 2H) 7,17-7,24 (м, 1H) 7,62 (с, 1H) 7,74 (д, J=8,65 Гц, 1H) 8,06 (с, 1H) 8,81 (с, 1H).
N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил}амино)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=метил, A=-CH2CH2-] (соединение 4)
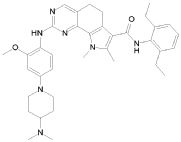
MS рассчитанный: 622,3864; MS найденный: 622,3868.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,13 (т, J=7,57 Гц, 6H) 1,44-1,60 (м, 2H) 1,68-1,90 (м, 2H) 2,23 (шир.с, 6H) 2,42 (с, 3H) 2,58 (кв, J=7,57 Гц, 4H) 2,60-2,69 (м, 2H) 2,69-2,75 (м, 2H) 2,83-2,93 (м, 2H) 3,61-3,70 (м, 2H) 3,82 (с, 3H) 3,98 (с, 3H) 6,49 (дд, J=8,70, 2,50 Гц, 1H) 6,63 (д, J=2,50 Гц, 1H) 7,06-7,16 (м, 2H) 7,17-7,24 (м, 1H) 7,62 (с, 1H) 7,74 (д, J=8,70 Гц, 1H) 8,06 (с, 1H) 8,81 (с, 1H).
N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=2-метокси-4-(4-метилпиперазин-1-ил)фенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=метил, A=-CH2CH2-] (соединение 5)
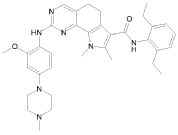
MS рассчитанный: 594,3551; MS найденный: 594,3554.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,13 (т, J=7,57 Гц, 6H) 2,25 (с, 3H) 2,42 (с, 3H) 2,58 (кв, J=7,57 Гц, 4H) 2,68-2,79 (м, 2H) 2,84-2,91 (м, 2H) 3,07-3,17 (м, 4H) 3,82 (с, 3H) 3,99 (с, 3H) 6,49 (дд, J=8,80, 2,45 Гц, 1H) 6,63 (д, J=2,45 Гц, 1H) 7,06-7,17 (м, 2H) 7,17-7,24 (м, 1H) 7,62 (с, 1H) 7,77 (д, J=8,80 Гц, 1H) 8,07 (с, 1H) 8,82 (с, 1H).
N-(2,6-диэтилфенил)-2-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метоксифенил}амино)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=метил, A=-CH2CH2-] (соединение 6)
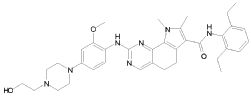
MS рассчитанный: 624,3657; MS найденный: 624,3643.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,13 (т, J=7,57 Гц, 6H) 2,42 (с, 3H) 2,44-2,49 (м, 6H) 2,58 (кв, J=7,57 Гц, 4H) 2,73 (м, 2H) 2,88 (м, 2H) 3,06-3,20 (м, 4H) 3,49-3,61 (м, 2H) 3,82 (с, 3H) 3,99 (с, 3H) 4,41 (шир.с, 1H) 6,48 (дд, J=8,70, 2,35 Гц, 1H) 6,63 (д, J=2,35 Гц, 1H) 7,04-7,16 (м, 2H) 7,17-7,23 (м, 1H) 7,63 (с, 1H) 7,76 (д, J=8,70 Гц, 1H) 8,06 (с, 1H) 8,82 (с, 1H).
2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=2-метокси-4-(4-метилпиперазин-1-ил)фенил, X=-NH-, R2=-NH2, R3=метил, R4=метил, A=-CH2CH2-] (соединение 7)
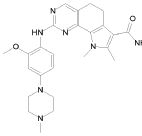
MS рассчитанный: 462,2612; MS найденный: 462,2595.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 2,22 (с, 3H) 2,38 (с, 3H) 2,43-2,47 (м, 4H) 2,61-2,69 (м, 2H) 2,73-2,81 (м, 2H) 3,06-3,13 (м, 4H) 3,80 (с, 3H) 3,93 (с, 3H) 6,47 (дд, J=8,70, 2,56 Гц, 1H) 6,61 (д, J=2,56 Гц, 1H) 6,89 (шир.с, 2H) 7,58 (с, 1H) 7,74 (д, J=8,70 Гц, 1H) 8,04 (с, 1H).
N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил}амино)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-CH2CH2-] (соединение 10)
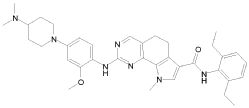
MS рассчитанный: 608,3708; MS найденный: 608,3712.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,57 Гц, 6H) 1,41-1,61 (м, 2H) 1,77-1,92 (м, 2H) 2,17-2,32 (м, 7H) 2,54 (кв, J=7,57 Гц, 46H) 2,60-2,68 (м, 2H) 2,69-2,77 (м, 2H) 2,90-3,01 (м, 2H) 3,59-3,64 (м, 2H) 3,82 (с, 3H) 4,03 (с, 3H) 6,49 (дд, J=8,80, 2,56 Гц, 1H) 6,63 (д, J=2,56 Гц, 1H) 7,05-7,16 (м, 2H) 7,16-7,23 (м, 1H) 7,66 (с, 1H) 7,69 (с, 1H) 7,74 (д, J=8,80 Гц, 1H) 8,10 (с, 1H) 9,03 (с, 1H).
N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-CH2CH2-] (соединение 11)
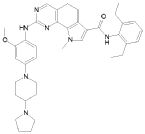
MS рассчитанный: 634,3864; MS найденный: 634,3874.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,57 Гц, 6H) 1,45-1,60 (м, 2H) 1,64-1,74 (м, 4H) 1,87-1,99 (м, 2H) 2,04-2,16 (м, 1H) 2,54 (кв, J=7,57 Гц, 4H) 2,65-2,81 (м, 4H) 2,90-3,03 (м, 2H) 3,50-3,66 (м, 2H) 3,82 (с, 3H) 4,04 (с, 3H) 6,49 (дд, J=8,70, 2,50 Гц, 1H) 6,63 (д, J=2,50 Гц, 1H) 7,04-7,15 (м, 2H) 7,15-7,29 (м, 1H) 7,65 (с, 1H) 7,69 (с, 1H) 7,74 (д, J=8,70 Гц, 1H) 8,10 (с, 1H) 9,03 (с, 1H).
N-(2,6-диэтилфенил)-2-[(4-{[3-(диметиламино)пропил](метил)амино}-2-метоксифенил)амино]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-{[3-(диметиламино)пропил]метиламино}-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-CH2CH2-] (соединение 12)
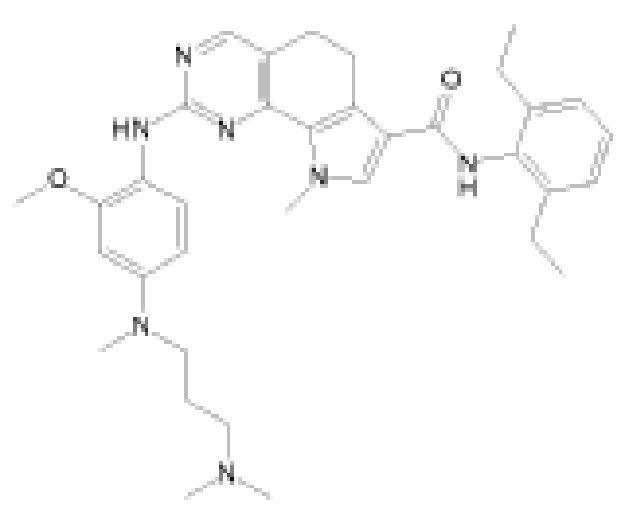
MS рассчитанный: 596,3708; MS найденный: 596,3782.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,51 Гц, 6H) 1,64 (квин, J=6,80 Гц, 2H) 2,15 (с, 6H) 2,24 (т, J=6,80 Гц, 2H) 2,54 (кв, J=7,51 Гц, 4H) 2,65-2,75 (м, 2H) 2,87 (с, 3H) 2,90-2,97 (м, 2H) 3,79 (с, 3H) 4,01 (с, 3H) 6,27 (дд, J=8,80, 2,50 Гц, 1H) 6,39 (д, J=2,50 Гц, 1H) 7,09-7,14 (м, 2H) 7,16-7,23 (м, 1H) 7,57 (д, J=8,80 Гц, 1H) 7,62 (с, 1H) 7,67 (с, 1H) 8,06 (с, 1H) 9,02 (с, 1H).
N-(2,6-диэтилфенил)-2-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метоксифенил}амино)-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-CH2CH2-] (соединение 13)
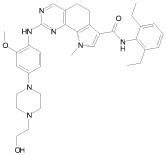
MS рассчитанный: 610,3500; MS найденный: 610,3498.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,51 Гц, 6H) 2,45 (т, J=6,10 Гц, 2H) 2,52-2,61 (м, 8H) 2,68-2,76 (м, 2H) 2,91-3,00 (м, 2H) 3,09-3,16 (м, 4H) 3,54 (кв, J=6,10 Гц, 2H) 3,82 (с, 2H) 4,04 (с, 2H) 4,37-4,45 (м, 1H) 6,48 (дд, J=8,79, 2,44 Гц, 1H) 6,62 (д, J=2,44 Гц, 1H) 7,09-7,13 (м, 2H) 7,15-7,23 (м, 1H) 7,66 (с, 1H) 7,69 (с, 1H) 7,75 (д, J=8,79 Гц, 1H) 8,10 (с, 1H) 9,03 (с, 1H).
N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-10-метил-5,6,7,10-тетрагидропирроло[3',2':6,7]циклогепта[1,2-d]пиримидин-8-карбоксамид [(I), R1=2-метокси-4-(4-метилпиперазин-1-ил)фенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-(CH2)3-] (соединение 16)
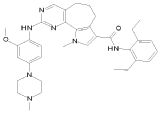
MS рассчитанный: 594,3551; MS найденный: 594,3524.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,57 Гц, 6H) 1,89-2,03 (м, 2H) 2,23 (с, 3H) 2,43-2,48 (м, 4H) 2,55 (кв, J=7,57 Гц, 4H) 2,91 (т, J=7,14 Гц, 2H) 3,09-3,15 (м, 4H) 3,80 (с, 3H) 3,81 (с, 3H) 6,49 (дд, J=8,70, 2,50 Гц, 1H) 6,63 (д, J=2,50 Гц, 1H) 7,08-7,15 (м, 2H) 7,15-7,23 (м, 1H) 7,61-7,68 (м, 2H) 7,79 (с, 1H) 8,19 (с, 1H) 9,01 (с, 1H).
N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил}амино)-10-метил-5,6,7,10-тетрагидропирроло[3',2':6,7]циклогепта[1,2-d]пиримидин-8-карбоксамид [(I), R1=4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-(CH2)3-] (соединение 17)
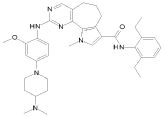
MS рассчитанный: 622,3864; MS найденный: 622,3876.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,57 Гц, 6H) 1,42-1,58 (м, 2H) 1,76-1,89 (м, 2H) 1,92-2,02 (м, 2H) 2,13-2,19 (м, 1H) 2,18-2,23 (м, 6H) 2,43-2,48 (м, 2H) 2,55 (кв, J=7,57 Гц, 4H) 2,60-2,70 (м, 2H) 2,92 (т, J=7,08 Гц, 2H) 3,62-3,72 (м, 2H) 3,79 (с, 3H) 3,80 (с, 3H) 6,50 (дд, J=8,75, 2,44 Гц, 1H) 6,62 (д, J=2,44 Гц, 1H) 7,05-7,14 (м, 2H) 7,13-7,24 (м, 1H) 7,58-7,68 (м, 2H) 7,79 (с, 1H) 8,19 (с, 1H) 9,01 (с, 1H).
N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-10-метил-5,6,7,10-тетрагидропирроло[3',2':6,7]циклогепта[1,2-d]пиримидин-8-карбоксамид [(I), R1=2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил, X=-NH-, R2=-N-(2,6-диэтилфенил), R3=метил, R4=H, A=-(CH2)3-] (соединение 18)
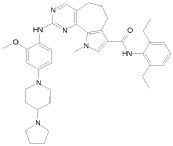
MS рассчитанный: 648,4021; MS найденный: 648,4023.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,11 (т, J=7,57 Гц, 6H) 1,42-1,59 (м, 2H) 1,63-1,76 (м, 4H) 1,83-2,04 (м, 4H) 2,04-2,18 (м, 1H) 2,42-2,50 (м, 6H) 2,54 (кв, J=7,57 Гц, 4H) 2,65-2,77 (м, 2H) 2,92 (т, J=7,14 Гц, 2H) 3,51-3,65 (м, 2H) 3,79 (с, 3H) 3,80 (с, 3H) 6,50 (дд, J=8,85, 2,50 Гц, 1H) 6,62 (д, J=2,45 Гц, 1H) 7,03-7,15 (м, 2H) 7,15-7,24 (м, 1H) 7,58-7,68 (м, 2H) 7,78 (с, 1H) 8,19 (с, 1H) 9,01 (с, 1H).
Пример 6 (превращение 2)
этил-2-[(4-бром-2-метоксифенил)амино]-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=-бром-2-метоксифенил, X=-NH-, R2=-O-этил, R3=метил, R4=метил, A=-CH2CH2-]
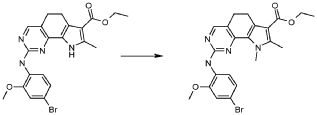
К раствору этил-2-[(4-бром-2-метоксифенил)амино]-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата (50 мг, 0,11 ммоль) в DMF (1 мл) добавляли Cs2CO3 (73 мг, 0,22 ммоль) и метилйодид (0,007 мл, 0,11 ммоль). Смесь перемешивали при комнатной температуре в течение 8 ч, растворитель удаляли в вакууме, затем добавляли DCM (10 мл), и органическую фазу промывали водой (2×15 мл). Органическую фракцию сушили над Na2SO4, фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией на силикагеле (элюент: AcOEt/гексан 4/6) давала 40 мг (выход: 80%) указанного в заголовке соединения в виде бледно-желтого твердого вещества.
MS рассчитанный: 471,1027; MS найденный: 471,1031.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,28 (т, J=7,08 Гц, 3H) 2,53 (с, 3H) 2,72 (т, J=7,63 Гц, 2H) 2,91 (т, J=7,63 Гц, 2H) 3,88 (с, 3H) 4,00 (с, 3H) 4,20 (кв, J=7,08 Гц, 2H) 7,12 (дд, J=8,55, 2,14 Гц, 1H) 7,20 (д, J=2,14 Гц, 1H) 7,89 (с, 1H) 8,07 (д, J=8,55 Гц, 1H) 8,17 (с, 1H).
В соответствии с такой же методологией, но используя подходящие замещенные производные, получали следующие соединения:
этил-2-амино-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=-H, X=-NH-, R2=-O-этил, R3=метил, R4=метил, A=-CH2CH2-]
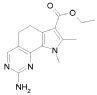
Пример 7 (превращение 7)
этил-2-({2-метокси-4-[(1-метилпиперидин-4-ил)карбамоил]фенил}амино)-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=2-метокси-4-[(1-метилпиперидин-4-ил)карбамоил]фенил, X=NH-, R2=-O-этил, R3=метил, R4=метил, A=-CH2CH2-]
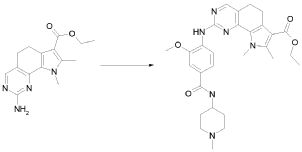
К раствору этил-2-амино-8,9-диметил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата (87 мг, 0,280 ммоль) в диоксане (2 мл) добавляли 4-йод-3-метокси-N-(1-метилпиперидин-ил)бензамид (112 мг, 0,254 ммоль) и Cs2CO3 (92 мг, 0,280 ммоль), содержимое колбы эвакуировали и снова заполняли аргоном. Затем загружали Pd2(dba)3 (4,7 мг, 0,005 ммоль) и Xantphos (6,5 мг, 0,011 ммоль) и смесь нагревали при 80°C в атмосфере аргона в течение 8 часов. После охлаждения до комнатной температуры, реакционную смесь концентрировали, суспендировали в H2O (10 мл) и экстрагировали AcOEt (3×15 мл). Органическую фазу обезвоживали над Na2SO4, фильтровали и выпаривали до сухости, неочищенное твердое вещество очищали флэш-хроматографией на силикагеле (элюент: DCM/MeOH 9/2) для получения 100 мг (выход: 70%) указанного соединения в виде желтого твердого вещества.
1H ЯМР (500 МГц, ДМСО-d6) δ м.д. 1,29 (т, J=7,00 Гц, 3H) 1,54-1,67 (м, 2H) 1,72-1,83 (м, 2H) 1,91-2,08 (м, 2H) 2,20 (шир.с, 3H) 2,55 (с, 3H) 2,70-2,76 (м, 2H) 2,77-2,86 (м, 2H) 2,89-2,98 (м, 2H) 3,69-3,81 (м, 1H) 3,94 (с, 2H) 4,05 (с, 2H) 4,20 (кв, J=7,00 Гц, 2H) 7,40-7,54 (м, 2H) 7,97 (с, 1H) 8,12 (д, J=7,69 Гц, 1H) 8,22 (с, 1H) 8,29 (д, J=8,24 Гц, 1H).
Пример 8 (превращение 4)
2-[(4-бром-2-метоксифенил)амино]-N-[(1S)-2-(1,3-диохо-1,3-дигидро-2H-изоиндол-2-ил)-1-фенилэтил]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=4-бром-2-метоксифенил, X=-NH-, R2=N-[(1S)-2-(1,3-диохо-1,3-дигидро-2H-изоиндол-2-ил)-1-фенилэтил], R3=метил, R4=H, A=-CH2CH2-] (соединение 14)
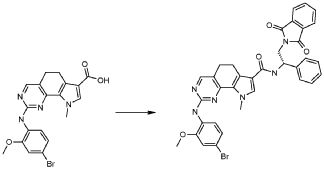
2-[(4-Бром-2-метоксифенил)амино]-9-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновую кислоту (50 мг, 0,116 ммоль) в сухом DMF (5,0 мл) обрабатывали DIPEA (0,056 мл, 0,033 ммоль) и TBTU (65 мг, 0,200 ммоль). Затем смесь обрабатывали 2-[(2S)-2-амино-2-фенилэтил]-1H-изоиндол-1,3(2H)-дионом (3 мг, 0,011 ммоль).
Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь разбавляли водой, и полученный осадок собирали фильтрацией для получения 35 мг (выход: 45%) указанного в заголовке соединения в виде желтого твердого вещества.
MS рассчитанный: 677,1507; MS найденный: 677,1521.
Пример 9 (стадия G)
этил-8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-этил, R3=H, R4=метил, A=-CH2CH2-] и этил-2-(диметиламино)-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-N(Me)-, R2=-O-этил, R3=H, R4=метил, A=-CH2CH2-]
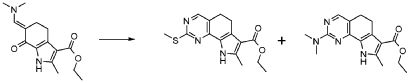
К раствору этил-(6E)-6-[(диметиламино)метилиден]-2-метил-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилата 2 г (7,22 ммоль) в 20 мл безводного DMF добавляли 1,41 г (14,4 ммоль) безводного ацетата калия и 4,0 г (14,4 ммоль) сульфата метилизотиомочевины. Реакционную смесь перемешивали при 100°C в течение 3 часов. Смесь разбавляли этилацетатом, промывали H2O, сушили над Na2SO4, фильтровали и выпаривали. Неочищенный продукт очищали хроматографией на силикагеле (этилацетат:гексан 4:6) для получения в качестве основного соединения 0,8 г этил-8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата (40%).
ЖХ/МС (254 нм) Способ ВЭЖХ 2. Время удерживания 5,79 мин.
1H ЯМР (500 МГц, ДМСО-d6) δ 1,28 (т, J=7,05 Гц, 3H) 2,52 (с, 3H) 2,82 (т, J=8,05 Гц, 2H) 2,95 (т, J=8,05 Гц, 2H) 3,33 (с, 3H) 4,18 (кв, J=7,05 Гц, 2H) 8,25 (с, 1H) 12,13 (шир.с, 1H).
HRMS (масс-спектроскопия высокого разрешения) (ESI) рассчитанный для C15H18N3O2S [M+H]+ 304,1114, найденный 304,1120;
и в качестве второстепенного продукта 0,2 г этил-2-(диметиламино)-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата (10%).
ЖХ/МС (254 нм) Способ ВЭЖХ 2 Время удерживания 5,44 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 1,27 (т, J=7,08 Гц, 3H) 2,50 (с, 3H) 2,70 (т, J=7,88 Гц, 2H) 2,89 (т, J=7,88 Гц, 2H) 3,13 (с, 6H) 4,18 (кв, J=7,08 Гц, 2H) 7,99 (с, 1H) 11,78 (шир.с, 1H).
HRMS (ESI) рассчитанный для C16H21N4O [M+H]+ 301,1659 найденный 301,1655.
Используя такой же способ, было получено следующее соединение:
метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-метил, R3=R4=H, A=-CH2CH2-]
ЖХ/МС (254 нм) Способ ВЭЖХ 2 Время удерживания 5,02 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,53 (с, 3H) 2,86 (т, J=8,06 Гц, 2H) 2,99 (т, J=8,06 Гц, 2H) 3,73 (с, 3H) 7,57 (с, 1H) 8,31 (с, 1H) 12,42 (шир.с, 1H).
HRMS (ESI) рассчитанный для C13H14N3O2S [M+H]+ 276,0801, найденный 276,0799.
Пример 10 (превращение 2)
этил-8-метил-2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=O-этил, R3=H, R4=метил, A=-CH2CH2-]
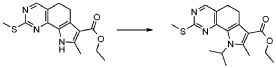
К раствору этил-8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата (100 мг, 0,33 ммоль) в сухом DMF (2 мл) добавляли Cs2CO3 (160 мг, 0,495 ммоль) и 2-йод-пропан (0,08 мл, 0,825 ммоль). Реакционную смесь перемешивали при 80°C в течение 8 ч. Анализ ВЭЖХ/МС показал 50% превращения, поэтому в котел добавляли дополнительное количество реагентов и перемешивали при той же температуре в течение еще 8 ч. Смесь выливали в H2O (100 мл), и продукт экстрагировали EtOAc (3×30 мл). Органические фракции сушили над Na2SO4, фильтровали и концентрировали в вакууме. Неочищенное твердое вещество очищали флэш-хроматографией на силикагеле (элюент: AcOEt/гексан 1/9) для получения 75 мг (выход: 66%) указанного в заголовке соединения в виде не совсем белого твердого вещества.
ЖХ/МС (254 нм) Способ ВЭЖХ 2. Время удерживания 7,34 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 1,28 (т, J=7,08 Гц, 3H) 1,55 (д, J=7,14 Гц, 6H) 2,51 (с, 3H) 2,66 (с, 3H) 2,70-2,74 (м, 2H) 2,87-2,91 (м, 2H) 4,20 (кв, J=7,08 Гц, 2H) 5,90 (шир.с, 1H) 8,28 (с, 1H).
HRMS (ESI) рассчитанный для C18H24N3O2S [M+H]+ 346,1584, найденный 346,1595.
Пример 11 (превращение 2)
метил-9-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-метил, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=H, A=-CH2CH2-]
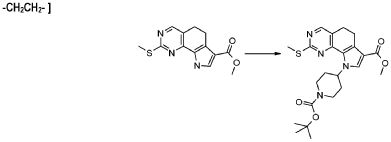
К смеси метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата (60 мг, 0,218 ммоль), трет-бутил-4-гидроксипиперидин-1-карбоксилата (88 мг, 0,436 ммоль) и трифенилфосфина (120 мг, 0,436 ммоль) в безводном THF (5 мл) при комнатной температуре добавляли ди-трет-бутилдиазадикарбоксилат (DTAD) (100 мг, 0,436 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч. ВЭЖХ/МС свидетельствовали о 30% превращении и оставались 70% SM, добавляли реагенты, трифенилфосфин (120 мг, 0,436 ммоль) и DTAD (100 мг, 0,436 ммоль) в 5 мл THF, смесь перемешивали в течение 6 часов. ВЭЖХ/МС показали 70% превращения и оставались 30% SM. В реагенты повторно добавляли TPP (120 мг, 0,436 ммоль) и DTAD (100 мг, 0,436 ммоль) в 5 мл THF, раствор дополнительно перемешивали в течение 18 часов. Летучие вещества удаляли в вакууме, неочищенное твердое вещество очищали флэш-хроматографией на силикагеле (гексан/EtOAc 7/3) для получения 69 мг (70% выход) указанного в заголовке соединения.
ЖХ/МС (254 нм) Способ ВЭЖХ 2. Время удерживания 8,10 мин.
Применяя такой же способ, получали следующее соединение:
метил-9-[1-(трет-бутоксикарбонил)пиперидин-4-yl]-8-йод-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-этил, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=I, A=-CH=CH-]
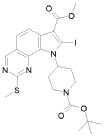
Пример 12 (превращение 2)
метил-9-{цис-4-[(трет-бутоксикарбонил)амино]циклогексил}-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=O-метил, R3=цис-4-[(трет-бутоксикарбонил)амино]циклогексил, R4=H, A=-CH2CH2-]
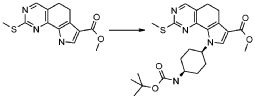
К раствору метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата (100 мг, 0,363 ммоль) в THF (5 мл) добавляли транс-трет-бутил-4-гидроксициклогексилкарбамат (156 мг, 0,727 ммоль), Ph3P (190 мг, 0,727 ммоль) и DEAD (113 мкл, 0,727 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, ВЭЖХ/МС свидетельствовали о наличии не вступившего в реакцию материала (80%) и желательного продукта (20%), затем добавляли дополнительные 190 мг Ph3P и 113 мкл DEAD. Через 5 ч растворитель удаляли роторным выпариванием для получения вязкого оранжевого масла. Смесь SM (60%) и желательного продукта (40%) разделяли флэш-хроматографией на силикагеле, используя 20:80 EtOAc-гексан в качестве элюента. Смесь, растворенную в THF (5 мл), снова обрабатывали транс-трет-бутил-4-гидроксициклогексилкарбаматом (156 мг, 0,727 ммоль), Ph3P (190 мг, 0,727 ммоль), DEAD (113 мкл, 0,727 ммоль) и перемешивали при комнатной температуре в течение 16 ч. ВЭЖХ/МС свидетельствовали о наличии не вступившего в реакцию исходного материал (40%) и желательного продукта (60%), затем добавляли еще 95 мг Ph3P и 56 мкл DEAD и перемешивали в течение 4 ч. Требовалось еще пять дополнительных добавлений свежих реагентов, перед тем как реакция протекала до завершения. Летучие вещества удаляли в вакууме, и неочищенный продукт очищали хроматографией на силикагеле (гексан/EtOAc 8/2) для получения указанного в заголовке соединения в виде желтого твердого вещества (160 мг, выход 93%).
ЖХ/МС (254 нм) Способ ВЭЖХ 2. Время удерживания 8,11 мин.
Пример 13 (превращение 3)
8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-S-, R2=OH, R3=H, R4=метил, A=-CH2CH2-]

Этил-8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат (110 мг, 0,36 ммоль) суспендировали в диоксане (10 мл) и обрабатывали 2н раствором NaOH (4,0 мл, 8 ммоль) при 95°C в течение 18 ч. Добавляли H20 (20 мл), и раствор подкисляли 2н HCl. Смесь разделяли между этилацетатом и водой, органический слой сушили над Na2SO4, фильтровали и концентрировали для получения 95 мг (95%) указанного в заголовке соединения в виде не совсем белого твердого вещества.
ЖХ/МС (254 нм) Способ ВЭЖХ 2 Время удерживания 4,13 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,48 (с, 3H) 2,52 (с, 3H) 2,77-2,83 (м, 2H) 2,92-2,96 (м, 2H) 8,24 (с, 1H) 12,05 (шир.с, 1H).
HRMS (ESI), рассчитанный для C13H14N3O2S [M+H]+ 276,0801, найденный 276,0804.
Действуя аналогичным образом, получали следующие соединения:
2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-S-, R2=OH, R3=R4=H, A=-CH2CH2-]
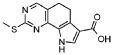
ЖХ/МС (254 нм) Способ ВЭЖХ 2 Время удерживания 3,61 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,53 (с, 3H) 2,85 (т, J=7,75 Гц, 2H), 2,98 (т, J=7,75 Гц, 2H) 7,48-7,52 (м, 1H) 8,30 (с, 1H) 12,32 (шир.с, 1H).
HRMS (ESI), рассчитанный для C12H12N3O2S [M+H]+ 262,0645, найденный 262,0649.
8-метил-2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X= -S-, R2=OH, R3=изопропил, R4=метил, A=-CH2CH2-]
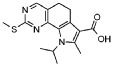
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 5,43 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 1,55 (д, J=6,96 Гц, 6H) 2,48 (с, 3H) 2,66 (с, 3H) 2,71 (т, J=7,78 Гц, 2H) 2,89 (т, J=7,75 Гц, 2H) 8,27 (с, 1H) 12,20 (шир.с, 1H).
HRMS (ESI) рассчитанный для C16H20N3O2S [M+H]+ 318,1271, найденный 318,1263.
2-(диметиламино)-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-N(Me)-, R2=OH, R3=H, R4=метил, A=-CH2CH2-]
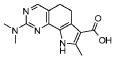
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,17 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,54 (с, 3H) 2,79 (т, J=7,20 Гц, 2H) 2,98 (т, J=7,20 Гц, 2H) 3,23 (с, 6H) 7,49 (с, 1H) 7,91 (с, 1H) 12,20 (шир.с, 1H).
HRMS (ESI) рассчитанный для C14H17N4O2 [M+H]+ 273,1346, найденный 273,1346.
9-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-S-, R2=OH, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=H, A=-CH2CH2-]

ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 6,13 мин.
9-{цис-4-[(трет-бутоксикарбонил)амино]циклогексил}-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-S-, R2=OH, R3=цис-4-[(трет-бутоксикарбонил)амино]циклогексил, R4=H, A=-CH2CH2-]
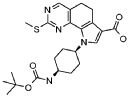
ЖХ/МС (254 нм) Способ ВЭЖХ 2. Время удерживания 6,38 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 12,13 (шир.с, 1H), 8,31 (с, 1H), 8,04 (с, 1H), 7,23 (д, J=8,97 Гц, 1H), 5,45 (ддд, J=4,03, 8,33, 12,00 Гц, 1H), 3,81 (шир.с, 1H), 2,91-2,96 (м, 2H), 2,72-2,80 (м, 2H), 2,00-2,12 (м, 2H), 1,77-1,86 (м, 2H), 1,67-1,75 (м, 2H), 1,59 (м, 2H), 1,42 (с, 9H).
HRMS (ESI) рассчитанный для C23H31N4O4S [M+H]+ 459,2061, найденный 459,2066.
2-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=одиночная связь, R2=OH, R3=H, R4=H, A=-CH2CH2-]
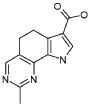
Пример 14 (превращение 4)
8-метил-2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=изопропил, R4=метил, A=-CH2CH2-] (Соединение 19)
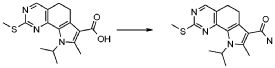
8-Метил-2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоновая кислота (100 мг, 0,363 ммоль) в сухом DMA (диметиламине) (2,0 мл) обрабатывали NH4Cl (0,062 г, 0,108 ммоль), DIPEA (0,253 мл, 0,14 ммоль) и TBTU (175 мг, 0,544 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли насыщенным NaHCO3, и полученный осадок собирали фильтрацией, промывали простым диэтиловым эфиром для получения 90 мг (выход: 90%) указанного в заголовке соединения в виде желтого тведого вещества.
ЖХ/МС (254 нм) Способ ВЭЖХ 2 Время удерживания 4,63 мин.
HRMS (ESI) рассчитанный для C16H21N4OS [M+H]+ 317,1431, найденный 317,1435.
В результате работы в соответствии с этим способом, были получены следующие соединения:
8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=H, R4=метил, A=-CH2CH2-] (соединение 20)
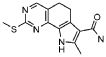
ЖХ/МС (254 нм) Способ ВЭЖХ 3, время удерживания 4,63 мин.
1H ЯМР (500 МГц, ДМСО-d6) δ 2,42 (с, 3H) 2,52 (с, 3H) 2,79 (т, J=8,05 Гц, 2H) 2,87 (т, J=8,05 Гц, 2H) 6,53-7,04 (м, 2H) 8,21 (с, 1H) 11,79 (шир.с, 1H).
HRMS (ESI) рассчитанный для C13H15N4OS [M+H]+ 275,0961, найденный 275,0968.
2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=R4=H, A=-CH2CH2-] (соединение 21)
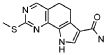
ЖХ/МС (254 нм) Способ ВЭЖХ 3, время удерживания 5,43 мин.
1H ЯМР (500 МГц, ДМСО-d6) δ 2,52 (с, 3H) 2,81 (т, J=7,78 Гц, 2H) 3,00 (т, J=7,78 Гц, 2H) 6,79 (шир.с, 1H) 7,31 (шир.с, 1H) 7,61 (д, J=3,11 Гц, 1H) 8,27 (с, 1H) 12,02 (шир.с, 1H).
HRMS (ESI) рассчитанный для C12H13N4OS [M+H]+ 261,0805, найденный 261,0814.
2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=изопропил, R4=H, A=-CH2CH2-] (соединение 22)
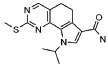
ЖХ/МС (254 нм) Способ ВЭЖХ 3 Время удерживания 4,48 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 1,44 (д, J=6,59 Гц, 6H) 2,75 (т, J=7,78 Гц, 2H) 2,98 (т, J=7,78 Гц, 2H) 5,63-5,74 (м, 1H) 6,81 (шир.с, 1H) 7,30 (шир.с, 1H) 7,88 (с, 1H) 8,28 (с, 1H).
HRMS (ESI) рассчитанный для C15H19N4OS [M+H]+ 303,1274, найденный 303,1277.
2-(диметиламино)-8-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-N(Me)-, R2=NH2, R3=H, R4=метил, A=-CH2CH2- (соединение 23)
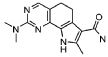
ЖХ/МС (254 нм) Способ ВЭЖХ 3, время удерживания 2,65 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,43 (с, 3H) 2,67 (т, J=7,69 Гц, 2H) 2,81 (т, J=7,69 Гц, 2H) 3,13 (с, 6H) 6,50-6,95 (м, 2H) 7,96 (с, 1H) 11,45 (шир.с, 1H).
HRMS (ESI) рассчитанный для C14H18N50 [M+H]+ 272,1506, найденный 272,1509.
трет-бутил-4-[7-карбамоил-2-(метилсульфанил)-5,6-дигидро-9H-пирроло[3,2-h]хиназолин-9-ил]пиперидин-1-карбоксилат [(I), R1=метил, X=-S-, R2=NH2, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=H, A=-CH2CH2-]
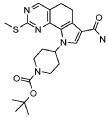
ЖХ/МС (254 нм) Способ ВЭЖХ 1. Время удерживания 1,473 мин.
трет-бутил-{цис-4-[7-карбамоил-2-(метилсульфанил)-5,6-дигидро-9H-пирроло[3,2-h]хиназолин-9-ил]циклогексил}карбамат [(I), R1=метил, X=-S-, R2=NH2, R3=цис-4-[(трет-бутоксикарбонил)амино]циклогексил, R4=H, A=-CH2CH2-]
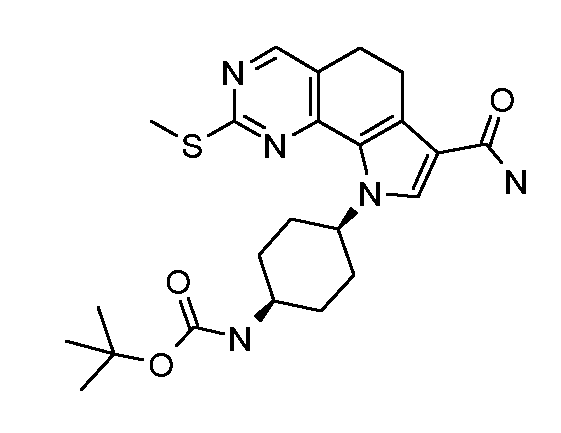
ЖХ/МС (254 нм) Способ ВЭЖХ 1, время удерживания 1,501 мин.
2-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=одиночная связь, R2=NH2, R3=H, R4=H, A=-CH2CH2-] (соединение 37)
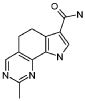
HRMS (ESI) рассчитанный для C14H18N50 [M+H]+ 229,1084, найденный 229,1085.
трет-бутил-4-(2-амино-7-карбамоил-5,6-дигидро-9H-пирроло[3,2-h]хиназолин-9-ил)пиперидин-1-карбоксилат [(I), R1=H, X=-NH-, R2=NH2, R3=4-[(трет-бутоксикарбонил)амино]циклогексил, R4=H, A=-CH2CH2-]
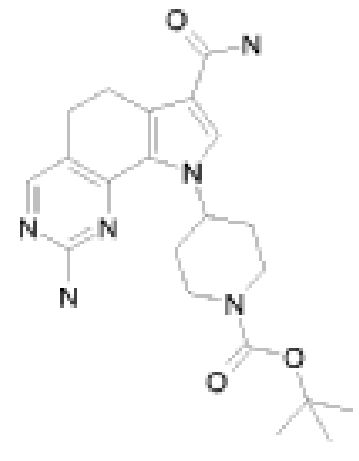
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,43 (с, 9H) 1,56-1,67 (м, 2H) 1,98-2,05 (м, 2H) 2,61 (т, J=7,69 Гц, 2H) 2,92 (т, J=7,69 Гц, 2H) 2,95-3,05 (м, 2H) 4,01-4,16 (м, 2H) 5,57-5,68 (м, 1H) 6,37 (шир.с, 2H) 6,75 (шир.с, 2H) 7,80 (с, 1H) 7,95 (с, 1H).
HRMS (ESI) рассчитанный для C15H18N4O2S [M+H]+ 413,2296; найденный 413,2296.
Пример 15 (превращение 2)
9-(2-гидроксиэтил)-8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=2-гидроксиэтил, R4=метил, A=-CH2CH2-] (соединение 24)
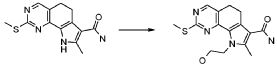
К раствору 8-метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамида (30 мг, 0,109 ммоль) в сухом диметилформамиде (2 мл) добавляли 2-йодэтанол (37 мкл, 0,437 ммоль) и карбонат цезия (106 мг, 0,327 ммоль). Полученную смесь нагревали при 90°C в течение 8 часов. После охлаждения до комнатной температуры, смесь выливали в воду и экстрагировали EtOAc. Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле, элюируя DCM/MeOH 95/5, для получения указанного в заголовке соединения 8 мг (25%).
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,32 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 2,45 (с, 3H) 2,47 (с, 3H) 2,74 (т, J=8,05 Гц, 2H) 2,83 (т, J=8,05 Гц, 2H) 3,68 (кв, J=5,90 Гц, 2H) 4,54 (т, J=5,90 Гц, 2H) 4,86 (т, J=5,90 Гц, 1H) 6,70-7,09 (м, 2H) 8,22 (с, 1H).
HRMS (ESI) рассчитанный для C15H18N4O2S [M+H]+ 319,1223, найденный 319,1215.
Работая в соответствии этим способом, получали следующие соединения:
9-(2-гидроксиэтил)-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=2-гидроксиэтил, R4=H, A=-CH2CH2-] (соединение 25)
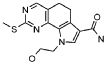
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 4,21 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 2,47 (с, 3H) 2,77 (т, J=7,80 Гц, 2H) 2,99 (т, J=7,80 Гц, 2H) 3,71 (кв, J=5,50 Гц, 2H) 4,52 (т, J=5,50 Гц, 2H) 4,90 (т, J=5,50 Гц, 1H) 6,81 (шир.с, 1H) 7,30 (шир.с, 1H) 7,63 (с, 1H) 8,27 (с, 1H).
HRMS (ESI), рассчитанный для C14H17N4O2S [M+H]+ 305,1067, найденный 305,1062.
Пример 16 (превращение 23)
метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-метил, R3=R4=H, A=-CH=CH-]

Раствор метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата 250 мг (0,91 ммоль) и 330 мг (1,82 ммоль) DDQ в хлорбензоле нагревали при 140°C в течение 2 часов. Летучие вещества удаляли в вакууме, остаток растворяли этилацетатом и промывали насыщенным водным раствором NaHCO3. Органическую фазу сушили Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали колоночной хроматографией на силикагеле, элюируя этилацетатом и гексаном (1:4), обеспечивая выход указанного в заголовке соединения 180 мг (90%).
ЖХ/МС (254 нм) Способ ВЭЖХ 2. Время удерживания 5,72 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 2,73 (с, 3H) 3,86 (с, 3H) 7,74 (д, J=8,61 Гц, 1H) 8,20 (д, J=8,61 Гц, 1H) 8,26 (д, J=3,11 Гц, 1H) 9,37 (с, 1H) 13,23 (шир.с, 1H).
HRMS (ESI) рассчитанный для C13H12N3O2S [M+H]+ 274,0645, найденный 274,065.
Используя такой же способ как в описанном выше примере, также синтезировали следующие аналоги:
этил-8-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-метил, R3=H, R4=метил, A=-CH=CH-]
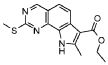
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 5,95 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,38 (т, J=7,14 Гц, 3H) 2,73 (с, 3H) 2,77 (с, 3H) 4,33 (кв, J=7,14 Гц, 2H) 7,68 (д, J=8,61 Гц, 1H) 8,15 (д, J=8,61 Гц, 1H) 9,32 (с, 1H) 12,93 (шир.с, 1H).
HRMS (ESI), рассчитанный для C15H16N3O2S [M+H]+ 302,0958, найденный 302,0957.
2-(диметиламино)-8-метил-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=H, R4=метил, A=-CH=CH-] (соединение 26)
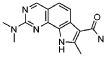
ЖХ/МС (254 нм) Способ ВЭЖХ 2. Время удерживания 3,52 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 2,67 (с, 3H) 3,30 (с, 6H) 7,01 (с, 2H) 7,35 (д, J=8,61 Гц, 1H) 7,67 (д, J=8,61 Гц, 1H) 9,04 (с, 1H) 12,08 (шир.с, 1H).
HRMS (ESI), рассчитанный для C14H16N5OS [M+H]+ 270,1350, найденный 270,1352.
Пример 17 (превращение 2)
метил-9-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-метил, R3=метил, R4=H, A=-CH=CH-]

К раствору метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилата (80 мг, 0,29 ммоль) в DMF (1,5 мл) добавляли Cs2CO3 (191 мг, 0,58 ммоль) и метилйодид (18 мкл, 0,29 ммоль). Смесь перемешивали при комнатной температуре в течение 8 ч, растворитель удаляли в вакууме, затем добавляли DCM (10 мл), и органическую фазу промывали водой (2×15 мл). Органическую фракцию сушили над Na2SO4, фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией на силакагеле (элюент: AcOEt/гексан 4/6) давала 58 мг (выход: 70%) указанного в заголовке соединения в виде бледно-желтого твердого вещества.
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 6,4 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 2,68 (с, 3H) 3,86 (с, 3H) 4,49 (с, 3H) 7,75 (д, J=8,67 Гц, 1H) 8,21 (д, J=8,67 Гц, 1H) 8,35 (с, 1H) 9,35 (с, 1H).
HRMS (ESI) рассчитанный для C14H14N3O2S [M+H]+ 288,0801, найденный 288,0802.
Пример 18 (превращение 3)
9-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-S-, R2=-OH, R3=R4=H, A=-CH=CH-]

Метил-9-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилат (50 мг, 0,174 ммоль) суспендировали в диоксане (5 мл) и обрабатывали 2н раствором NaOH (2,0 мл, 4 ммоль) при 95°C в течение 2 ч. Добавляли H2O (20 мл), и раствор подкисляли (pH~6) 2н HCl. Твердое вещество отфильтровывали и промывали водой и простым диэтиловым эфиром для получения 40 мг (85%) указанного в заголовке соединения в виде не совсем белого твердого вещества.
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 4,13 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,68 (с, 3H) 4,48 (с, 3H) 7,71 (д, J=8,61 Гц, 1H) 8,23 (д, J=8,61 Гц, 1H) 8,26 (с, 1H) 9,34 (с, 1H) 12,35 (шир.с, 1H).
HRMS (ESI) рассчитанный для C13H12N3O2S [M+H]+ 274,0645, найденный 274,064.
Работая в соответствии с тем же способом, получали следующие соединения:
8-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-S-, R2=-OH, R3=H, R4=метил, A=-CH=CH-]
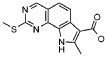
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 4,21 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,73 (с, 3H) 2,76 (с, 3H) 7,65 (д, J=8,61 Гц, 1H) 8,18 (д, J=8,61 Гц, 1H) 9,31 (с, 1H) 12,23 (шир.с, 1H) 12,83 (шир.с, 1H).
HRMS (ESI) рассчитанный для C13H12N3O2S [M+H]+ 274,0645, найденный 274,065.
9-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-S-, R2=-OH, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=H, A=-CH=CH-]
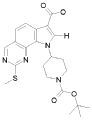
2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоновая кислота [(I), R1=метил, X=-S-, R2=-OH, R3=H, R4=метил, A=-CH=CH-]
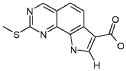
Пример 19 (превращение 4)
9-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=Метил, R4=H, A=-CH=CH- (соединение 27)

9-Метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоновую кислоту (30 мг, 0,109 ммоль) в сухом DMA (2,0 мл) обрабатывали NH4Cl (0,040 г, 0,74 ммоль), DIPEA (0,120 мл, 0,68 ммоль) и TBTU (70 мг, 0,218 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли насыщенным NaHCO3, и продукт экстрагировали EtOAc (3×30 мл). Органические фракции сушили над Na2SO4, фильтровали и выпаривали в вакууме для получения 27 мг (выход: 93%) указанного в заголовке соединения в виде не совсем белого твердого вещества.
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,84 мин.
1H ЯМР (500 МГц, ДМСО-d6) δ 2,68 (с, 3H) 4,46 (с, 3H) 7,01 (шир.с, 1H) 7,58 (шир.с, 1H) 7,64 (д, J=8,61 Гц, 1H) 8,19 (с, 1H) 8,39 (д, J=8,61 Гц, 1H) 9,31 (с, 1H).
HRMS (ESI) рассчитанный для C13H13N4OS [M+H]+ 273,0805, найденный 273,0814.
Работая в соответствии с тем же способом, получали следующие соединения:
8-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=H, R4=метил, A=-CH=CH-] (соединение 28)
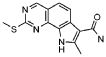
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,54 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,71 (с, 3H) 2,72 (с, 3H) 7,14 (с, 2H) 7,58 (д, J=8,61 Гц, 1H) 8,04 (д, J=8,61 Гц, 1H) 9,29 (с, 1H) 12,57 (с, 1H).
HRMS (ESI) рассчитанный для C13H13N3O2S [M+H]+ 273,0805, найденный 273,0807.
трет-бутил-4-[7-карбамоил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-9-yl]пиперидин-1-карбоксилат [(I), R1=метил, X=-S-, R2=-NH2, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=H, A=-CH=CH-]
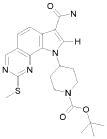
2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=-NH2, R3=H, R4=H, A=-CH=CH-] (соединение 36)
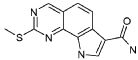
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 2,72 (с, 3H) 6,98 (шир.с, 1H) 7,62 (д, J=8,61 Гц, 1H) 7,64 (шир.с, 1H) 8,28 (с, 1H) 8,37 (д, J=8,61 Гц, 1H) 9,33 (с, 1H) 12,80 (шир.с, 1H).
HRMS (ESI) рассчитанный для C13H13N3O2S [M+H]+ 259,0648; найденный 259,0646.
Пример 20 (превращение 2)
9-(2-гидроксиэтил)-8-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=2-гидроксиэтил, R4=метил, A=-CH=CH-] (соединение 29)

К раствору 8-метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамида (20 мг, 0,072 ммоль) в DMF (1,5 мл) добавляли 2-йодэтанол (24 мкл, 0,288 ммоль) и карбонат цезия (70 мг, 0,216 ммоль). Полученную смесь нагревали при 80°C в течение 8 часов. После охлаждения до комнатной температуры, реакционную смесь выливали в воду (10 мл) и разделяли этилацетатом. Органические слои промывали рассолом, сушили над Na2SO4 и концентрировали. Неочищенные продукт очищали хроматографией на силикагеле (DCM/MeOH/ацетон 85/0,5/1) для получения 10 мг (45%) указанного в заголовке соединения в виде белого твердого вещества.
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 4,31 мин.
1H ЯМР (400 МГц, ДМСО-d6) 2,62 (с, 3H) 2,73 (с, 3H) 3,85 (кв, J=5,50 Гц, 2H) 4,92 (т, J=5,50 Гц, 1H) 4,98 (т, J=5,50 Гц, 2H) 7,29 (шир.с, 2H) 7,62 (д, J=8,61 Гц, 1H) 8,01 (д, J=8,61 Гц, 1H) 9,28 (с, 1H).
HRMS (ESI) рассчитанный для C15H17N4O2S [M+H]+ 317,1067; найденный 317,1064.
Работая аналогичным образом, получали следующие соединения:
8-метил-2-(метилсульфанил)-9-(пропан-2-ил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=изопропил, R4=метил, A=-CH=CH- (соединение 30)
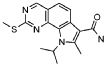
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 6,1 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,54-1,93 (м, 6H) 2,63 (с, 3H) 2,79 (с, 3H) 4,96-5,10 (м, 1H) 7,35 (шир.с, 2H) 7,57-7,68 (м, 1H) 7,90-8,04 (м, 1H) 9,28 (с, 1H).
HRMS (ESI) рассчитанный для C16H19N4OS [M+H]+ 315,1274; найденный 315,1281.
9-этил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=этил, R4=H, A=-CH=CH-] (соединение 31)
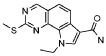
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 4,13 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 1,51 (т, J=7,14 Гц, 3H) 2,66 (с, 3H) 4,94 (кв, J=7,14 Гц, 2H) 7,02 (шир.с, 1H) 7,56 (шир.с, 1H) 7,65 (д, J=8,61 Гц, 1H) 8,28 (с, 1H) 8,41 (д, J=8,61 Гц, 1H) 9,32 (с, 1H).
HRMS (ESI) рассчитанный для C14H15N4OS [M+H]+ 287,0961; 287,0961.
Пример 21 (превращение 23)
трет-бутил-{цис-4-[7-карбамоил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-9-yl]циклогексил}карбамат [(I), R1=метил, X=-S-, R2=NH2; R3=цис-4-[(трет-бутоксикарбонил)амино]циклогексил, R4=H, A=-CH=CH-]
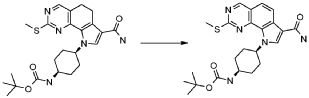
Раствор трет-бутил-{цис-4-[7-карбамоил-2-(метилсульфанил)-5,6-дигидро-9H-пирроло[3,2-h]хиназолин-9-ил]циклогексил}карбамата 15 мг (0,032 ммоль) и 15 мг (0,064 ммоль) DDQ в хлорбензоле нагревали при 140°C в течение 2 часов. Летучие вещества удаляли в вакууме, остаток растворяли этилацетатом и промывали насыщенным водным раствором NaHCO3. Органическую фазу сушили Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали колоночной хроматографией на силикагеле, элюируя DCM/MeOH (97:3), с выходом указанного в заголовке соединения 10 мг (71%).
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 5,72 мин.
Пример 22 (превращение 24)
метил-2-(метилсульфанил)-9-(пиперидин-4-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат гидрохлорид [(I), R1=метил, X=-S-, R2=-O-метил, R3=пиперидин-4-ил, R4=H, A=-CH2CH2-]
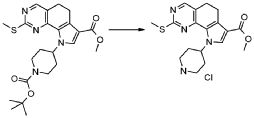
Метил-9-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат 10 мг (0,021 ммоль) растворяли в 1,4-диоксане (2 мл) и добавляли 4M HCl в 1,4-диоксане 3 мл (3 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Летучие вещества удаляли в вакууме, и полученный остаток истирали в порошок с простым диэтиловым эфиром, фильтровали и сушили для получения 8 мг (97%) указанного в заголовке соединения.
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,64 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 2,08-2,19 (м, 2H) 2,22-2,29 (м, 2H) 2,54 (с, 3H) 2,81 (т, J=7,88 Гц, 2H) 2,97 (т, J=7,88 Гц, 2H) 2,99-3,03 (м, 2H) 3,44-3,51 (м, 2H) 3,75 (с, 3H) 5,51-5,59 (м, 1H) 7,71 (с, 1H) 8,38 (с, 1H) 8,78 (шир.с, 1H) 8,83 (шир.с, 1H).
HRMS (ESI), рассчитанный для C18H24N4O2S [M+H]+ 359,1536; найденный 359,1531.
Действуя аналогичным образом, получали следующие соединения:
2-(метилсульфанил)-9-(пиперидин-4-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид гидрохлорид [(I), R1=метил, X=-S-, R2=NH2, R3=пиперидин-4-ил, R4=H, A=-CH2CH2-] (соединение 32)
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,61 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 1,91-2,11 (м, 2H) 2,23-2,32 (м, 2H) 2,53 (с, 3H) 2,76 (т, J=7,60 Гц, 2H) 2,89-3,06 (м, 4H) 3,45-3,56 (м, 2H) 5,40-5,50 (м, 1H) 6,89 (шир.с, 1H) 7,45 (шир.с, 1H) 7,83 (с, 1H) 8,33 (с, 1H) 8,85 (шир.с, 2H).
HRMS (ESI), рассчитанный для C17H23N5OS [M+H]+ 344,154; найденный 344,1544.
9-(цис-4-аминоциклогексил)-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид гидрохлорид [(I), R1=метил, X=-S-, R2=NH2, R3=цис-4-аминоциклогексил, R4=H, A=-CH2CH2-] (соединение 33)
ЖХ/МС (254 нм) Способ ВЭЖХ 2. Время удерживания 3,91 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 1,72-1,86 (м, 2H) 1,89-2,01 (м, 4H) 2,01-2,13 (м, 2H) 2,76 (т, J=7,70 Гц, 2H) 2,94 (т, J=7,70 Гц, 2H) 3,43-3,54 (м, 1H) 5,31-5,44 (м, 1H) 6,93 (шир.с, 1H) 7,16 (шир.с, 1H) 8,00 (с, 1H) 8,08 (шир.с, 3H) 8,30 (с, 1H).
HRMS (ESI), рассчитанный для C18H24N5OS [M+H]+ 358,1696; найденный 358,1694.
9-(цис-4-аминоциклогексил)-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=цис-4-аминоциклогексил, R4=H, A=-CH=CH-] (соединение 34)
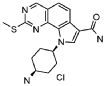
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,15 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 1,85-1,96 (м, 2H) 2,00-2,07 (м, 2H) 2,07-2,14 (м, 2H) 2,17-2,29 (м, 2H) 2,68 (с, 3H) 3,51-3,58 (м, 1H) 6,01-6,11 (м, 1H) 7,11 (шир.с, 1H) 7,54 (шир.с, 1H) 7,67 (д, J=8,61 Гц, 1H) 8,13 (шир.с, 3H) 8,38 (д, J=8,61 Гц, 1H) 8,64 (с, 1H) 9,33 (с, 1H).
HRMS (ESI) рассчитанный для C18H22N5OS [M+H]+ 356,1696; найденный 356,1694.
9-(3-амино-2,2-диметилпропил)-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид гидрохлорид [(I), R1=метил, X=-S-, R2=NH2, R3=3-амино-2,2-диметилпропил, R4=H, A=-CH=CH-] (соединение 38)
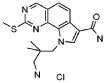
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,89 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 9,35 (с, 1H), 8,47 (д, J=8,61 Гц, 1H), 8,28 (с, 1H), 7,83 (шир.с, 4H), 7,69 (д, J=8,79 Гц, 1H), 7,08 (шир.с, 1H), 5,11 (с, 2H), 2,76 (д, J=5,86 Гц, 2H), 2,71 (с, 3H), 1,02-1,07 (м, 6H).
HRMS (ESI) рассчитанный для C17H22N5OS [M+H]+ 344,1540; найденный 344,1544.
9-(азепан-3-ил)-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид гидрохлорид [(I), R1=метил, X=-S-, R2=NH2, R3=азепан-3-ил, R4=H, A=-CH=CH-] (соединение 39)
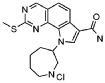
ЖХ/МС (254 нм) Способ ВЭЖХ 2, время удерживания 3,82 мин.
1H ЯМР (600 МГц, ДМСО-d6) δ 9,34 (с, 1H), 8,80-9,09 (м, 2H), 8,50 (с, 1H), 8,43 (д, J=8,61 Гц, 1H), 7,72-7,75 (м, 1H), 7,67 (д, J=8,61 Гц, 1H), 6,90-7,17 (м, 1H), 6,26 (шир.с, 1H), 3,21 (м, 2H), 2,69 (с, 3H), 2,32-2,46 (м, 3H), 2,07-2,11 (м, 2H), 1,91 (м, 1H).
HRMS (ESI), рассчитанный для C18H22N5OS [M+H]+ 356,1540; найденный 356,1538.
2-амино-9-(пиперидин-4-ил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксамид гидрохлорид [(I), R1=H, X=-NH-, R2=NH2, R3=пиперидин-4-ил, R4=H, A=-CH2CH2-]
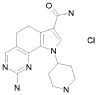
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,95-2,11 (м, 2H) 2,22-2,33 (м, 2H) 2,53 (с, 3H) 2,76 (т, J=7,60 Гц, 1H) 2,89-3,06 (м, 4H) 3,45-3,55 (м, 2H) 5,40-5,50 (м, 1H) 6,89 (шир.с, 1H) 7,45 (шир.с, 1H) 7,83 (с, 1H) 8,33 (с, 1H) 8,85 (шир.с, 2H).
HRMS (ESI) рассчитанный для C18H22N5O [M+H]+ 344,1540; найденный 344,1544.
2-(метилсульфанил)-9-(пиперидин-4-ил)-9H-пирроло[3,2-h]хиназолин-7-карбоксамид [(I), R1=метил, X=-S-, R2=NH2, R3=пиперидин-4-ил, R4=H, A=-CH=CH-] (соединение 35)
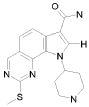
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,80-1,94 (м, 2H) 2,11-2,19 (м, 2H) 2,71 (с, 3H) 2,72-2-80 (м, 2H) 3,15-3,24 (м, 2H) 6,07-6,17 (м, 1H) 7,01 (шир.с, 1H) 7,63 (шир.с, 1H) 7,66 (д, J=8,61 Гц, 1H) 8,45 (д, J=8,61 Гц, 1H) 8,55 (с, 1H) 9,32 (с, 1H).
HRMS (ESI), рассчитанный для C18H22N5O [M+H]+ 245,1033; найденный 245,1041.
Пример 23 (стадия G)
метил-2-метил-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=одиночная связь, R2=-O-метил, R3=H, R4=H, A=-CH2CH2-]
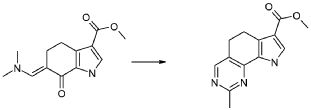
К раствору метил-(6E)-6-[(диметиламино)метилиден]-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилата (50 мг, 0,2 ммоль) в DMF (4 мл) добавляли ацетамидин гидрохлорид (190 мг, 2,0 ммоль) и K2CO3 (275 мг, 2,0 ммоль) и смесь нагревали при 180°C в течение 1 ч в условиях микроволнового облучения. Летучие вещества удаляли в вакууме, остаток растворяли DCM и промывали H2O. Органическую фазу сушили Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали колоночной хроматографией на силикагеле, элюируя DCM/MeOH (10:1), обеспечивая выход указанного в заголовке соединения 20 мг (40%) в виде темно-желтого твердого вещества.
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 2,55 (с, 3H) 2,85-2,90 (м, 2H) 2,96-2,99 (м, 2H) 3,72 (с, 3H) 7,50 (с, 1H) 8,35 (с, 1H) 12,40 (шир.с, 1H).
HRMS (ESI), рассчитанный для C18H22N5O [M+H]+ 244,1081; найденный 244,1087.
Пример 24 (стадия G)
метил-2-амино-9-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=H, X=-NH-, R2=-O-метил, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=H, A=-CH2CH2-]
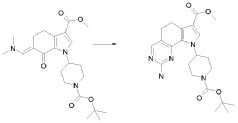
К раствору метил-(6E)-1-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-6-[(диметиламино)метилиден]-7-оксо-4,5,6,7-тетрагидро-1H-индол-3-карбоксилата (10 мг, 0,023 ммоль) в DMF (1 мл) добавляли гуанидинкарбонат (10 мг, 0,055 ммоль). Смесь перемешивали при 110°C в течение 8 часов. Полученную смесь охлаждали при комнатной температуре и выпаривали до сухости. Неочищенное твердое вещество очищали флэш-хроматографией на силикагеле (элюент: AcOEt) для получения 8 мг (выход: 80%) указанного в заголовке соединения в виде коричневого твердого вещества.
МС, рассчитанный: 428,2293; МС найденный: 428,2292.
1H ЯМР (401 МГц, ДМСО-d6) δ м.д. 1,43 (с, 9H) 1,66-1,84 (м, 2H) 1,92-2,04 (м, 2H) 2,62-2,69 (м, 2H) 2,86-2,92 (м, 2H) 2,92-3,10 (м, 2H) 3,71 (с, 3H) 3,98-4,07 (м, 2H) 5,59-5,72 (м, 1H) 6,28 (с, 2H) 7,72 (с, 1H) 7,99 (с, 1H).
Действуя аналогичным образом, получали следующие соединения:
метил-2-амино-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=H, X=-NH-, R2=-O-метил, R3=H, R4=H, A=-CH2CH2-]
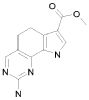
МС, рассчитанный: 245,1033; МС найденный: 245,1041.
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 2,71 (т, J=7,78 Гц, 2H) 2,91 (т, J=7,78 Гц, 2H) 3,71 (с, 3H) 6,08 (шир.с, 2H) 7,44 (д, J=2,93 Гц, 1H) 7,98 (с, 1H) 12,09 (шир.с, 1H).
Получение I
метил-2-(метилсульфанил)-8-йод-9H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-метил, R3=H, R4=I, A=-CH=CH-]

К раствору метил-2-(метилсульфанил)-6,9-дигидро-5H-пирроло[3,2-h]хиназолин-7-карбоксилата (100 мг, 0,363 ммоль) в DMF (5 мл) добавляли N-йодсукцинимид (3,25 мг, 1,44 ммоль) и смесь перемешивали при комнатной температуре в течение 24 ч. Летучие вещества удаляли в вакууме, остаток растворяли в DCM и промывали H2O. Органическую фазу сушили Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали колоночной хроматографией на силикагеле, элюируя гексаном/AcOEt (4:2), при выходе указанного в заголовке соединения 85 мг (60%) в виде желтого твердого вещества.
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 2,75 (с, 3H) 3,89 (с, 3H) 7,69 (д, J=8,79 Гц, 1H) 8,15 (д, J=8,79 Гц, 1H) 9,35 (с, 1H) 13,75 (с, 1H).
МС, рассчитанный: 399,9611; МС найденный: 399,9610.
Пример 25 (превращение 23)
метил-9-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-метил, R3=1-(трет-бутоксикарбонил)пиперидин-4-ил, R4=H, A=-CH=CH-]
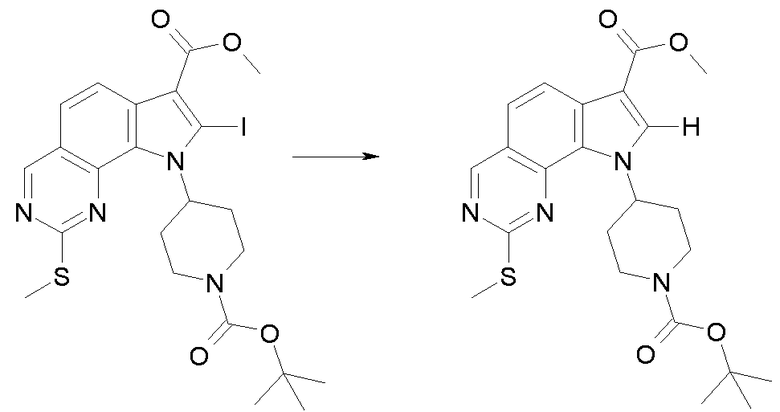
К раствору метил-9-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-8-йод-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилата (30 мг, 0,05 ммоль) в DMF (2 мл) добавляли формиат натрия (7 мг, 10 ммоль) и Pd(PPh3)4 (50 мг, 0,004 ммоль) и смесь нагревали при 120°C в течение 3 часов. Летучие вещества удаляли в вакууме. Неочищенный материал очищали колоночной хроматографией на силикагеле, элюируя DCM/MeOH (95:5) при выходе указанного в заголовке соединения 15 мг (65%) в виде белого твердого вещества.
Действуя аналогичным образом, получали следующее соединение:
метил-2-(метилсульфанил)-9H-пирроло[3,2-h]хиназолин-7-карбоксилат [(I), R1=метил, X=-S-, R2=-O-метил, R3=H, R4=H, A=-CH=CH-]
ФАРМАКОЛОГИЯ
Соединения формулы (I) активны в качестве ингибиторов протеинкиназы и поэтому могут применяться, например, для ограничения неконтролируемой пролиферации опухолевых клеток.
В терапии они могут применяться при лечении различных опухолей, таких как ранее определенные опухоли, а также при лечении других клеточных пролиферативных расстройств, таких как доброкачественная гиперплазия предстательной железы, семейный аденоматоз полипоз, нейрофиброматоз, псориаз, пролиферация сосудистых гладкомышечных клеток, связанная с атеросклерозом, легочный фиброз, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз.
Ингибирующую активность предполагаемых ингибиторов MPS1 и активность выбранных соединений определяли посредством описанных ниже анализов.
Краткие формы и аббревиатуры, используемые в настоящем описании, имеют следующее значение:
Клонирование, экспрессия и очистка белка полноразмерного рекомбинантного MPS1
MPS1 полной длины (соответствующий остаткам 2-857 полноразмерной последовательности, см. номер доступа в депозитарии Swiss-Prot P33981) был амплифицирован PCR (ПЦР, полимеразной цепной реакцией) из человеческого гена MPS1 полной длины, присутствующего в депозитарии в виде клона pGEX4t_MPS1. Амплификацию выполняли с использованием прямого олигонуклеотида:
5'ggggacaagtttgtacaaaaaagcaggcttactggaagttctgttccaggggcccgaatccgaggatttaagtggcagag3';
и обратного олигонуклеотида: 5'ggggaccactttgtacaagaaagctgggttttatttttttcccctttttttttcaaaagtcttggaggatgaag3'.
Оба олигонуклеотида описаны в заявке на международный патент WO2009/156315, опубликованной 30 декабря 2009 г.
В целях клонирования олигонуклеотиды включали сайты attB для получения attB-фланкирующего продукта ПЦР, подходящего для клонирования с использованием технологии Gateway® (Invitrogen). Кроме того, в целях очистки, прямой праймер включал сайт расщепления протеазой. Полученный продукт ПЦР клонировали в плазмиде pDONR201 и затем переносили в экспрессионный вектор бакуловируса pVL1393GST (Invitrogen), модифицированный технологией Gateway®. Клонирование выполняли в соответствии с последовательностями операций, описанными в руководстве по технологии Gateway®.
Бакуловирусы получали совместной трансфекцией клеток насекомых Sf9 экспрессионным вектором и вирусной ДНК, используя набор для трансфекции BaculoGold® (Pharmingen). Вирусный супернатант извлекали через 5 дней и подвергали 3 циклам амплификации для увеличения вирусного титра. Рекомбинантный белок получали инфекцией клеток насекомых High5. Через 72 часа после инфекции при 21°C, клетки извлекали, осаждали центрифугированием и замораживали при -80°C. Для очистки рекомбинантного белка клеточный осадок после центрифугирования оттаивали, ресуспендировали в лизисном буфере (PBS (солевой раствор с фосфатным буфером), NaCl 150 мМ, 10% глицерин, 0,1% CHAPS 3-((3-холамидопропил)диметиламмонио)-1-пропансульфоновой кислотой), 20 мМ DTT (дитиотреитола), ингибиторы протеазы и фосфатазы) и лизировали гаулином. Лизат очищали центрифугированием и загружали на аффинную колонку GST. После обильного промывания рекомбинантный белок расщепляли специфической протеазой и элюировали инкубацией.
Для получения полностью активированного фермента белок затем подвергали автофосфорилированию в присутствии 1 мМ АТФ при 25°C в течение 2 часов в киназном буфере (Hepes pH 7,5 50 мМ, MgCl2 2,5 мМ, MnCl2 1 мМ, DTT 1 мМ, ингибиторы фосфатазы); затем АТФ удаляли обессоливающей колонкой.
Биохимический анализ для выявления активности ингибиторов MPS1 киназы
Ингибиторную активность предполагаемых ингибиторов киназы и силу действия выбранных соединений определяли, используя анализ трансфосфорилирования.
Определенные пептидные или белковые субстраты трансфосфорилировали их специфической ser-thr или tyr киназой в присутствии АТФ, меченной 33Ρ-γ-АТФ, и в присутствии их собственного оптимального буфера и кофакторов.
В конце реакции фосфорилирования более чем 98% немеченого АТФ и радиоактивного АТФ захватывается избытком ионообменной смолы dowex; затем смола осаждается на дно реакционного планшета под действием гравитации. Супернатант в последующем удаляют и переносят в планшет для подсчета, затем оценивают расчетом коэффициента β.
Реагенты/условия анализа
i. Получение смолы Dowex
Отвешивают 500 г влажной смолы (SIGMA, полученная на заказ смола DOWEX 1×8 200-400 меш, 2,5 кг) и разбавляют до 2 л в 150 мМ формиата натрия, pH 3,00.
Смоле дают возможность осесть (несколько часов), и затем супернатант удаляли.
После трех промываний, как указано выше, в течение пары дней смоле дают возможность осесть, и добавляют два объема (на основании объема смолы) буфера формиата натрия 150 мМ.
Затем измеряют pH, и он должен составлять около 3,00.
Промытая смола устойчива в течение более одной недели; исходную смолу держат при 4°C перед применением.
ii. Киназный буфер (KB)
Буфер для анализа MPS1 был составлен из 50 мМ HEPES при pH 7,5, с 2,5 мМ MgCl2, 1 мМ MnCl2, 1 мМ DTT, 3 мкМ Na3V04, 2 мМ β-глицерофосфата и 0,2 мг/мл BSA (бычьего сывороточного альбумина).
iii. Условия анализа
Анализ проводили при конечной концентрации MPS1 5 нМ, в присутствии 15 мкМ АТФ и 1,5 нМ 33Ρ-γ-АТФ; субстрат представлял собой Ρ38-βtide, используемый в концентрации 200 мкМ.
Роботизированный анализ смолы dowex
Тестируемая смесь состояла из:
1) 3x Ферментной смеси (приготовленной в киназном буфере 3X), 5 мкл/лунку;
2) 3x смеси субстрата и АТФ (приготовленной в ddH2O (дважды деионизированной H2O)), вместе с 33Ρ-γ-АТФ, 5 мкл/лунку;
3) 3x тестируемых соединений (разведенных в ddH2O - 3% DMSO) - 5 мкл/лунку.
Разведение соединений и схема анализа представлены ниже
i. Разведение соединений
Тестируемые соединения получают в виде раствора 1 мМ в 100% DMSO, распределяют в 96- или 384-луночные планшеты:
a) для исследования ингибирования в процентах (HTS) отдельные планшеты разведения в концентрации 1 мМ разбавляют в 3X концентрации (30 мкМ) в ddH20 (3% DMSO=конечная концентрация), используя автоматизированную пипетирующую станцию Beckman NX. Тот же прибор используют для распределения разбавленных материнских планшетов в тестируемые планшеты;
b) для определения IC50 (станция KSS) 100 мкл каждого соединения в концентрации 1 мМ в 100% DMSO переносят из первоначального планшета в первую колонку другого 96-луночного планшета (из A1 в G1); лунку H1 оставляют пустой для ингибитора внутреннего стандарта, обычно стауроспорина.
Автоматизированную установку для серийных разведений (Biomek FX, Beckman) используют для получения разведений 1:3 в 100% DMSO, из линии A1 в линию A10, и для всех семи соединений в колонке. Кроме того, получают 4-5 экземпляров дочерних планшетов переформатированием 5 мкл этого первого набора планшетов разведения в 100% DMSO в 384-глубоколуночные планшеты: один экземпляр дочерних планшетов с серийными разведениями тестируемых соединений оттаивают в день экспериментов, восстанавливают влагосодержание в 3X концентрации водой и используют в анализах определения IC50. В стандартном эксперименте самая высокая концентрация (3X) всех соединений составляет 30 мкМ, тогда как самая низкая концентрация составляет 1,5 нМ.
Каждый 384-луночный планшет содержит эталонные лунки (полная ферментная активность в сравнении с отсутствием ферментной активности) для оценки Z' и отношения сигнала к фону.
ii. Схема анализа
384-луночные планшеты с V-образным дном (тестируемые планшеты) получают с 5 мкл разбавленного соединения (3X) и затем помещали на роботизированную установку PlateTrak 12 (Perkin Elmer; робот имеет пипетирующую головку с 384 наконечниками для начала анализа плюс одну головку с 96 наконечниками для подачи смолы) вместе с одним резервуаром для ферментной смеси (3X) и одним для смеси АТФ (3X).
В начале прогона робот аспирирует 5 мкл смеси АТФ, делает воздушный зазор внутри наконечников (2 мкл) и аспирирует 5 мкл смеси MPS1. Следующая подача в планшеты обеспечивает возможность начала реакции киназы после 3 циклов смешивания, совершаемых самим роботом.
В этот момент восстанавливается должная концентрация для всех реагентов.
Робот инкубирует планшеты в течение 60 минут при комнатной температуре и затем останавливает реакцию пипетированием 70 мкл суспензии смолы dowex в реакционную смесь. Три цикла смешивания выполняются непосредственно после добавления смолы.
Суспензия смолы очень плотная; во избежание окклюзии наконечника, для ее подачи используют наконечники с широкими внутренними диаметрами.
Другой цикл смешивания выполняют после остановки всех планшетов, причем на этот раз используются обычные наконечники: затем планшетам дают возможность находиться в покое в течение примерно одного часа для максимизации захвата АТФ. В этот момент 22 мкл супернатанта переносят в 384-ленточные планшеты 384-Optiplates (Perkin-Elmer) с 50 мкл Microscint 40 (Perkin-Elmer); через 5 мин орбитального встряхивания планшеты считывают на счетчике радиоактивности Perkin-Elmer Top Count.
iii. Анализ данных
Данные анализируют в соответствии с внутренней стандартной версией программного обеспечения «Assay Explorer», которая обеспечивает получение или % ингибирования для первичных анализов, или подгонки к сигмоидальным кривым десяти разведений для определения IC50 при вторичных анализах/обычных анализах, подтверждающих совпадение.
Анализ клеточной пролиферации in vitro
Клетки рака яичников человека A2780, клетки рака молочной железы человека MCF7 и клетки MV-4-11 (бифенотипического B миеломоноцитарного лейкоза) (1250 клеток/лунку) высевали в белые 384-луночные планшеты в полную среду (RPMI 1640 или EMEM плюс 10% фетальная телячья сыворотка) и обрабатывали соединениями, растворенными в 0,1% DMSO, через 24 ч после посева. Клетки инкубировали при 37°C и 5% CO2, и через 72 часа планшеты обрабатывали, используя анализ CellTiter-Glo (Promega), соблюдая инструкции производителя.
CellTiter-Glo представляет собой гомогенный способ, основанный на количественном определении присутствующего АТФ, индикатора метаболически активных клеток. АТФ количественно определяют, используя систему на основе люциферазы и D-люциферина, что приводит к генерированию света. Люминесцентный сигнал пропорционален числу клеток, присутствующих в культуре.
Вкратце, 25 мкл/лунку раствора реагента добавляли в каждую лунку, и после 5 минут встряхивания микропланшеты считывали люминометром Envision (PerkinElmer). Люминесцентный сигнал был пропорционален числу клеток, присутствующих в культуре.
Ингибиторную активность оценивали, сравнивая данные при обработке в сравнении с контролем, используя программу Assay Explorer (MDL). IC50 рассчитывали, используя сигмоидальную интерполяционную кривую.
Учитывая результаты представленных выше анализов ингибирования, можно утверждать, что соединения формулы (I) по изобретению обладают достаточной ингибиторной активностью в отношении MPS1, обычно с IC50 в диапазоне от 0,001 до 5 мкМ.
Кроме того, соединения формулы (I) по изобретению проявляют достаточную ингибиторную активность в отношении клеточной пролиферации, обычно при IC50 в диапазоне от 0,010 до 5 мкМ для клеток A2780.
Биохимический анализ для выявления ингибиторов активности PIM-1-киназы
Ингибиторную активность предполагаемых ингибиторов киназы и силу действия выбранных соединений определяли, используя анализ трансфосфорилирования.
Определенные пептидные или белковые субстраты трансфосфорилируют их специфической серин-треонин- или тирозин-киназой в присутствии АТФ, меченного 33Ρ-γ-ΑΤΡ, и в присутствии их собственного оптимального буфера и кофакторов.
В конце реакции фосфорилирования более чем 98% немеченого АТФ и радиоактивного АТФ захватывается избытком ионообменной смолы dowex; затем смола осаждается на дно реакционного планшета под действием гравитации. Супернатант в последующем удаляют и переносят в планшет для подсчета, затем оценивают расчетом коэффициента β.
Реагенты/условия анализа
Получение смолы Dowex
Отвешивают 500 г влажной смолы (SIGMA, полученная на заказ смола DOWEX 1×8 200-400 меш, 2,5 кг) и разбавляют до 2 л в 150 мМ формиата натрия, pH 3,00.
Смоле дают возможность осесть (несколько часов), и затем супернатант удаляют.
После трех промываний, как указано выше, в течение пары дней смоле дают возможность осесть, и добавляют два объема (на основании объема смолы) буфера формиата натрия 150 мМ.
Затем измеряют pH, и он должен составлять около 3,00.
Промытая смола устойчива в течение более одной недели; исходную смолу держат при 4°C перед применением.
Киназный буфер (KB)
Буфер для анализа MPS1 был составлен из 50 мМ HEPES при pH 7,5, с 10 мМ MgCl2, 1 мМ DTT, 3 мкМ Na3V04 и 0,2 мг/мл BSA.
Человеческий PIM-2 полной длины экспрессировали и очищали, как описано в публикации Bullock AN, et al., J. Biol. Chem. 2005, 280, 41675-82.
Фермент проявил линейную кинетику поле стадии преактивации автофосфорилированием в следующих условиях: 1,7 мкМ PIM1 инкубировали 1 час при RT=28°C в присутствии 125 мкМ АТФ.
Условия анализа
Концентрация АТФ: 200 мкМ.
33P-γ-АТФ: 6 нМ.
Концентрация фермента: 1 нМ.
Концентрация субстрата Актид (Aktide) (Номер в Реестре Химической Реферативной Службы 324029-01-8): 25 мкМ.
Роботизированный анализ смолы dowex.
Тестируемая смесь состояла из:
1) 3x ферментной смеси (приготовленной в Киназном Буфере 3X), 5 мкл/лунку;
2) 3x смеси субстрата и АТФ (приготовленной в ddH2O), вместе с 33Ρ-γ-АТФ, 5 мкл/лунку;
3) 3x тестируемых соединений (разбавленных в ddH2O - 3% DMSO -) 5 мкл/лунку.
Разведение соединений и схема анализа представлены ниже.
Разведение соединений
Для определения IC50 тестируемые соединения получают в виде раствора 1 мМ в 100% DMSO и распределяли в 96-луночные планшеты; затем соединения высевают в первую колонку нового 96-луночного планшета (с A1 по G1), 100 мкл/лунку.
Автоматизированную установку для серийных разведений (Biomek FX, Beckman) используют для получения разведений 1:3 в 100% DMSO, от линии A1 до линии A10, для всех семи соединений в колонке. Кроме того, получают 4-5 экземпляров дочерних планшетов переформатированием 5 мкл этого первого набора планшетов разведения в 100% DMSO в 384-глубоколуночные планшеты: один экземпляр дочерних этих планшетов с серийными разведениями тестируемых соединений оттаивают в день исследования, восстанавливают влагосодержание в рабочей концентрации (в 3 раза превышающей конечную концентрацию) с 162 мкл/лунку воды и используют для анализов определения IC50. В стандартном эксперименте самая высокая концентрация (3X) всех соединений обычно составляет 30 мкМ, тогда как самая низкая концентрация составляет 1,5 нМ.
Каждый 384-луночный планшет используют для построения по меньшей мере одной кривой стандартного ингибитора стауроспорина и эталонные лунки (полная ферментная активность в сравнении с отсутствием ферментной активности) для оценки Z' и отношения сигнала к фону (S/B).
Схема анализа
384-луночные планшеты с V-образным дном (тестируемые планшеты) получают с 5 мкл разбавленного соединения, как описано ранее (3X), и затем помещают на роботизированную установку PlateTrak 12 (Perkin Elmer; робот имеет пипетирующую головку с 384 наконечниками для начала анализа плюс одну головку с 384 наконечниками для начала анализа плюс одну головку с 96 наконечниками для подачи смолы) вместе с одним резервуаром для ферментной смеси (3X) и одним для смеси АТФ (3X).
Данные анализируют принятой для внутреннего использования версией программного обеспечения «Assay Explorer», которая обеспечивает получение подгонки к сигмоидальным кривым десяти разведений для определения IC50 при вторичных анализах/обычных анализах, подтверждающих совпадение.
Способ анализа ингибирования PIM-2 киназы: методика Dowex
Киназный буфер (KB).
Буфер для анализа PIM-2 был составлен из 50 мМ HEPES при pH 7,5, с 1 мМ MgCl2, 1 мМ DTT, 3 мкМ Na3V04 и 0,2 мг/мл BSA.
Человеческий PIM-2 полной длины был экспрессирован и очищен, как описано в публикации Fedorov O, et al., PNAS 2007 104, 51, 20523-28.
Условия анализа (конечные концентрации)
Концентрация фермента=1,5 нМ.
Актид (Aktide) (Номер в Реестре Химической Реферативной Службы 324029-01-8): 5 мкМ.
АТФ=4 мкМ.
33P-γ-ATP=1 нМ.
Роботизированный анализ Dowex
См. выше: такая же процедура, как описана для PIM-1.
В следующей таблице A представлены экспериментальные данные по некоторым репрезентативным соединениям формулы (I) по изобретению, тестируемым по ферментам MPS1, PIM-1 и PIM-2 в описанных выше специфических анализах киназы in vitro (IC50 мкМ).
В следующей таблице A также представлена ингибиторная активность некоторых из ближайших соединений уровня техники.
Эталонные соединения 1, 2, 3 и 4 соответствуют соединениям, закодированным под обозначениями M3, N9, N4 и N10, соответственно, в приведенной выше заявке на международный патент WO2008/065054; эти соединения соответствуют третьему, пятому, седьмому и шестому отвергнутым соединениям по настоящему изобретению.
Дополнительные экспериментальные данные
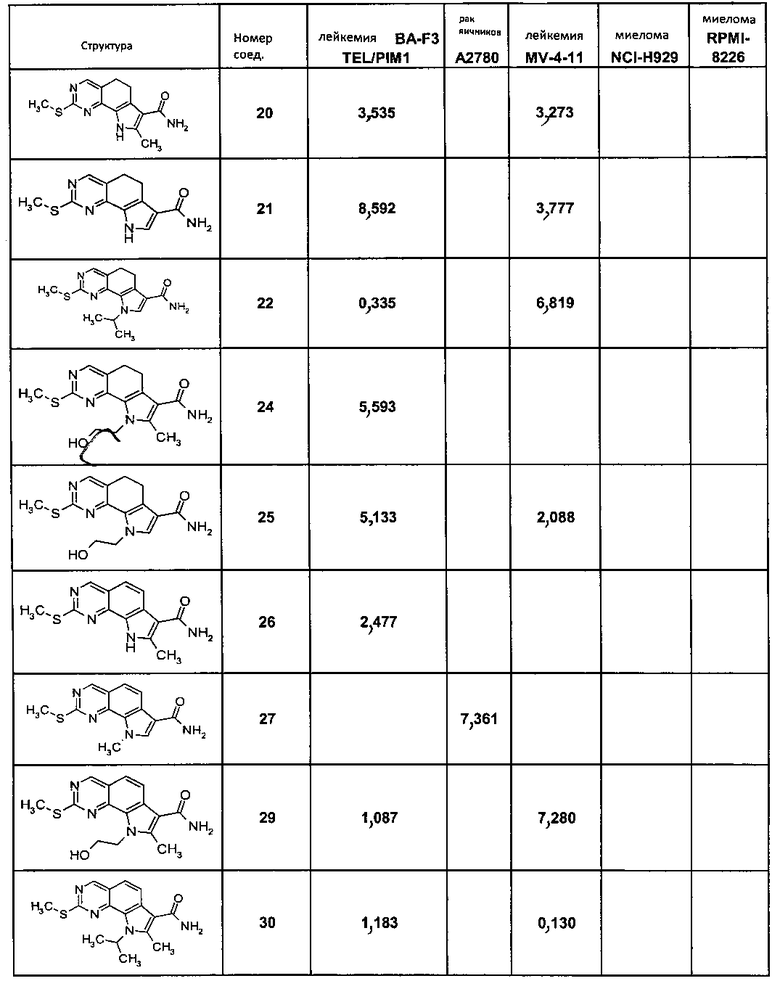
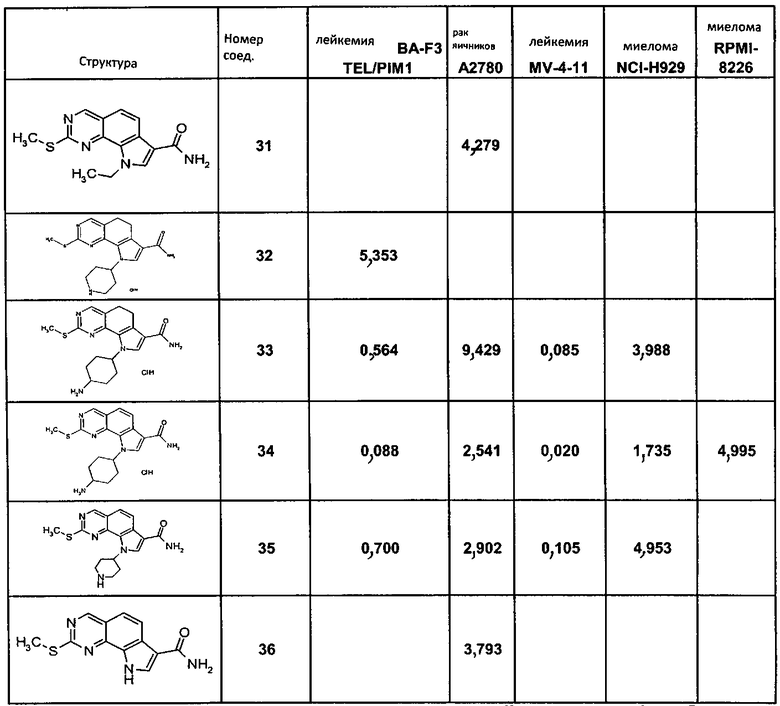
Ингибирующая активность, выраженная как IC50 (микромоль) на выбранные раковые линии клеток.
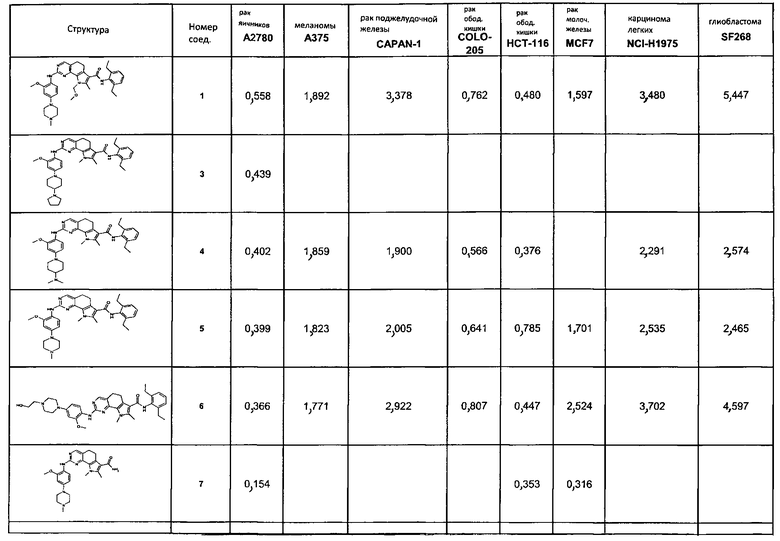
Ингибирующая активность, выраженная как IC50 (микромоль) на выбранные раковые линии клеток.
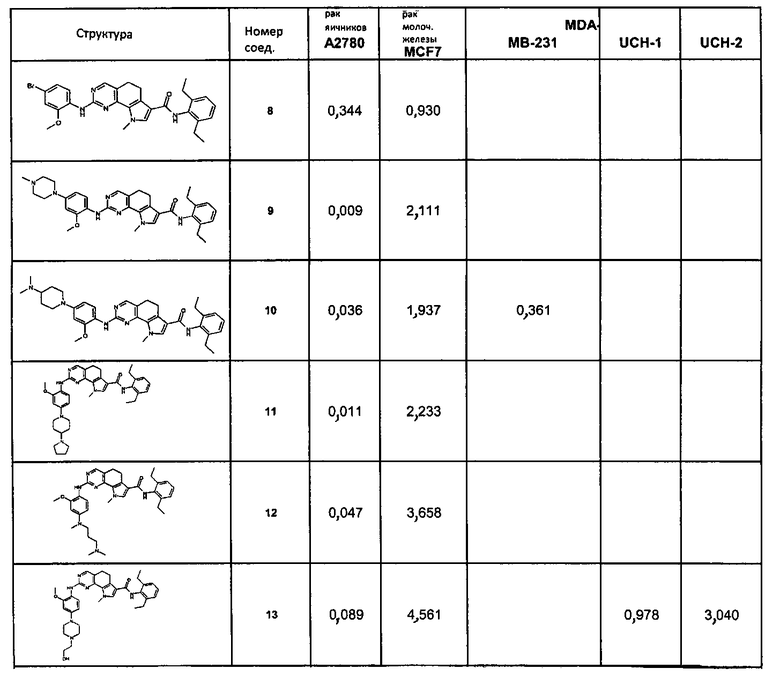
Ингибирующая активность, выраженная как IC50 (микромоль) на выбранные раковые линии клеток.
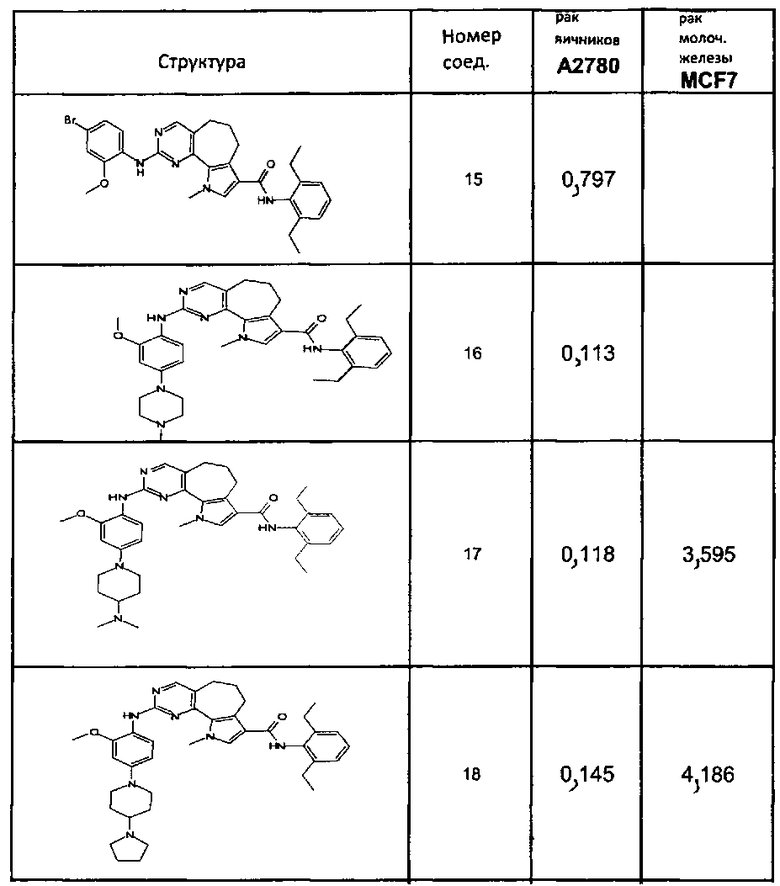
Ингибирующая активность, выраженная как IC50 (микромоль) на выбранные раковые линии клеток.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2666538C2 |
| ФОСФОИНДОЛЫ КАК ИНГИБИТОРЫ ВИЧ | 2005 |
|
RU2393163C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛПИРИМИДИНОНА, СПОСОБ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2263676C2 |
| ИНДОЛОПИРРОЛОКАРБАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ САХАРОВ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РОСТА ОПУХОЛЕЙ | 1997 |
|
RU2167880C2 |
| ГЕТЕРОЦИКЛО-ЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ АМИНОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ХОЛИНЭСТЕРАЗЫ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1992 |
|
RU2119920C1 |
| ПИРАЗОЛБЕНЗОДИАЗЕПИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ CDK2, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ | 2000 |
|
RU2249593C2 |
| ПРОИЗВОДНЫЕ ДИАМИНОВ | 2002 |
|
RU2319699C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНИЛПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2621732C2 |
Настоящее изобретение относится к новым трициклическим производным пиррола формулы (I) или их фармацевтически приемлемым солям, которые модулируют активность протеинкиназ, выбранных из MPS1, PIM-1 и PIM-2. Соединения могут применяться при лечении заболеваний, вызванных нарушением регуляции протеинкиназной активности. Такие заболевания могут быть выбраны из рака яичников, рака молочной железы, меланомы, рака ободочной кишки, лейкемии, рака поджелудочной железы и миеломы. В формуле (I)
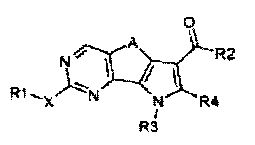 ,
,
где R1 обозначает водород, прямой или разветвленный C1-C6алкил, или арил, выбранный из фенила, содержащего заместители, выбранные из атомов галогена, C1-C6алкокси, C1-C6алкила, диС1-C6алкиламино, 6-членный гетероциклил, содержащий один или два атома азота в цикле, и необязательно замещенный в свою очередь C1-C6алкилом, 5-членный гетероциклил, содержащий один атом азота, ди(C1-C6алкил)амино, [ди(C1-C6алкил)аминоС1-C6алкил](C1-C6алкил)амино, гидроксиС1-C6алкил; R2 обозначает группу -NR″R′″, где R″ и R′″ оба обозначают водород или прямой или разветвленный C1-C6 алкил, или один из R″ и R′″ обозначает водород, а другой выбирают из C1-С6алкила, замещенного фенилом и 2,3-диоксо-1,3-дигидро-2Н-изоиндол-2-илом; и арила, представляющего собой фенил, замещенный C1-С6алкилом и С1-С6алкокси; R3 выбирают из водорода; прямого или разветвленного C1-C6алкила, необязательно замещенного амино; гидроксиС1-C6алкила; C1-C6алкоксиС1-C6алкила; 6-7-членного гетероциклила, содержащего атом азота в качестве гетероатома и необязательно замещенного C1-C6алкоксикарбонилом; и C3-C6циклоалкила, необязательно замещенного амино; R4 обозначает водород или прямой или разветвленный C1-C6алкил; X обозначает группу, выбранную из -NR′- и -S-, где R′ обозначает водород; А обозначает группу, выбранную из -(CH2)3-, -(CH2)2- и -CH=CH-. Изобретение также описывает способ получения соединений. 6 н. и 2 з.п. ф-лы, 4 табл., 32 пр.
1. Соединение формулы (I):
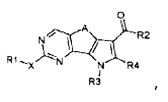
где R1 обозначает водород, прямой или разветвленный C1-C6 алкил, или арил, выбранный из фенила, содержащего заместители, выбранные из атомов галогена, C1-С6алкокси, C1-С6алкила, диС1-С6алкиламино, 6-членный гетероциклил, содержащий один или два атома азота в цикле, и необязательно замещенный в свою очередь C1-С6алкилом 5-членный гетероциклил, содержащий один атом азота, ди(C1-С6алкил)амино, [ди(C1-С6алкил)аминоС1-С6алкил](C1-С6алкил)амино, гидроксиС1-С6алкил;
R2 обозначает группу -NR″R′″, где R″ и R′″ оба обозначают водород или прямой или разветвленный C1-C6 алкил, или один из R″ и R′″ обозначает водород, а другой выбирают из C1-С6алкила, замещенного фенилом и 2,3-диоксо-1,3-дигидро-2Н-изоиндол-2-илом; и арила, представляющего собой фенил, замещенный C1-С6алкилом и С1-С6алкокси;
R3 выбирают из водорода; прямого или разветвленного C1-C6 алкила, необязательно замещенного амино; гидроксиС1-С6алкила; C1-С6алкоксиС1-С6алкила; 6-7-членного гетероциклила, содержащего атом азота в качестве гетероатома и необязательно замещенного C1-С6алкоксикарбонилом; и С3-С6циклоалкила, необязательно замещенного амино;
R4 обозначает водород или прямой или разветвленный C1-C6 алкил;
X обозначает группу, выбранную из -NR′- и -S-, где R′ обозначает водород;
А обозначает группу, выбранную из -(СН2)3-, -(СН2)2- и -СН=СН-;
или его фармацевтически приемлемая соль.
2. Соединение формулы (I) по п. 1,
где X обозначает группу -NR′-; R2 обозначает группу -NR″R′″, где R″ и R′″ оба обозначают водород или один из R″ и R′″ обозначает водород, а другой обозначает фенил, замещенный C1-С6алкилом; и R1 обозначает фенил, содержащий заместители, определенные в п. 1.
3. Соединение формулы (I) по п. 1,
где X обозначает группу -S-; R2 обозначает группу -NR″R′″, где R″ и R′″ оба обозначают водород; и R1 обозначает прямой или разветвленный C1-C6 алкил.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, которое выбрано из группы, состоящей из:
N-(2,6-диэтилфенил)-9-(метоксиметил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-8,9-диметил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-8,9-диметил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-
2-метоксифенил}амино)-8,9-диметил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8,9-диметил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метоксифенил}амино)-8,9-диметил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-8,9-диметил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-9-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-9-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил}амино)-9-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-9-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-[(4-{[3-(диметиламино)пропил](метил)амино}-2-метоксифенил)амино]-9-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
N-(2,6-диэтилфенил)-2-({4-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метоксифенил}амино)-9-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-[(4-бром-2-метоксифенил)амино]-N-[(1S)-2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)-1-фенилэтил]-9-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-[(4-бром-2-метоксифенил)амино]-N-(2,6-диэтилфенил)-10-метил-5,6,7,10-тетрагидропирроло[3′,2′:6,7]циклогепта[1,2-d]пиримидин-8-карбоксамида,
N-(2,6-диэтилфенил)-2-{[2-метокси-4-(4-метилпиперазин-1-ил)фенил]амино}-10-метил-5,6,7,10-тетрагидропирроло[3′,2′:6,7]циклогепта[1,2-d]пиримидин-8-карбоксамида,
N-(2,6-диэтилфенил)-2-({4-[4-(диметиламино)пиперидин-1-ил]-2-метоксифенил}амино)-10-метил-5,6,7,10-тетрагидропирроло[3′,2′:6,7]циклогепта[1,2-d]пиримидин-8-карбоксамида,
N-(2,6-диэтилфенил)-2-({2-метокси-4-[4-(пирролидин-1-ил)пиперидин-1-ил]фенил}амино)-10-метил-5,6,7,10-тетрагидропирроло[3′,2′:6,7]циклогепта[1,2-d]пиримидин-8-карбоксамида,
8-метил-2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
8-метил-2-(метилсульфанил)-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-(метилсульфанил)-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-(метилсульфанил)-9-(пропан-2-ил)-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-(диметиламино)-8-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
9-(2-гидроксиэтил)-8-метил-2-(метилсульфанил)-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
9-(2-гидроксиэтил)-2-(метилсульфанил)-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-(диметиламино)-8-метил-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
9-метил-2-(метилсульфанил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
8-метил-2-(метилсульфанил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
9-(2-гидроксиэтил)-8-метил-2-(метилсульфанил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
8-метил-2-(метилсульфанил)-9-(пропан-2-ил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
9-этил-2-(метилсульфанил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-(метилсульфанил)-9-(пиперидин-4-ил)-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
9-(цис-4-аминоциклогексил)-2-(метилсульфанил)-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
9-(цис-4-аминоциклогексил)-2-(метилсульфанил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-(метилсульфанил)-9-(пиперидин-4-ил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-(метилсульфанил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
2-метил-6,9-дигидро-5Н-пирроло[3,2-h]хиназолин-7-карбоксамида,
9-(3-амино-2,2-диметилпропил)-2-(метилсульфанил)-9Н-пирроло[3,2-h]хиназолин-7-кабоксамида гидрохлорида и
9-(азепан-3-ил)-2-(метилсульфанил)-9Н-пирроло[3,2-h]хиназолин-7-карбоксамида гидрохлорида.
5. Способ получения соединения формулы (I) по п. 1 или его фармацевтически приемлемой соли, включающий следующие стадии:
стадия А) взаимодействие соединения формулы (II):
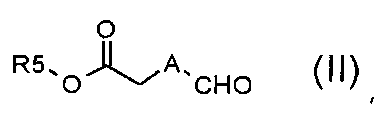
где А обозначает, как определено в п. 1, за исключением -СН=СН-, и R5 обозначает необязательно замещенный C1-C6 алкил, с соединением формулы (III):
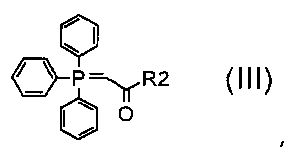
где R2 обозначает необязательно замещенный алкокси;
стадия В) взаимодействие полученного соединения формулы (IV):
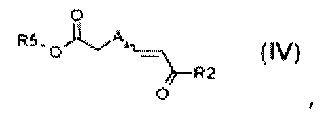
где R2 обозначает необязательно замещенный алкокси, R5 обозначает необязательно замещенный C1-C6 алкил, и А обозначает, как определено в п. 1, за исключением -СН=СН-, с толуолсульфонилметилизоцианидом в присутствии сильного основания;
стадия С) селективный гидролиз в кислотных или основных условиях полученного соединения формулы (V):
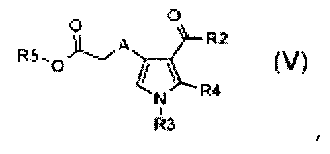
где R3 и R4 обозначают водород, А обозначает, как определено в п. 1, за исключением -СН=СН-, R2 обозначает необязательно замещенный алкокси, и R5 обозначает необязательно замещенный C1-C6 алкил, с тем, чтобы получить соединение формулы (VI):
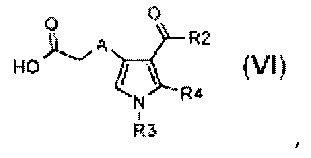
где R3 и R4 обозначают водород, А обозначает, как определено в п. 1, за исключением -СН=СН-, и R2 обозначает необязательно замещенный алкокси;
альтернативно,
стадия Са) соединение формулы (VI), где R3 обозначает водород, R4 обозначает, как определено в п. 1, за исключением водорода, А обозначает, как определено в п. 1, за исключением -СН=СН-, и R2 обозначает необязательно замещенный алкокси, может быть получено взаимодействием соединения формулы (VII):
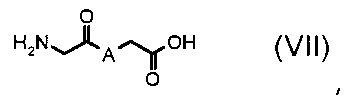
где А обозначает, как определено в п. 1, за исключением
-СН=СН-, с соединением формулы (VIII):
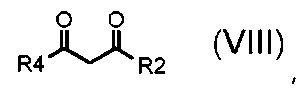
где R2 обозначает необязательно замещенный алкокси и R4 обозначает, как определено в п. 1, за исключением водорода;
стадия D) циклизация полученного соединения формулы (VI), где R2 обозначает необязательно замещенный алкокси, R3 обозначает водород, R4 обозначает, как определено в п. 1, и А обозначает, как определено в п. 1, за исключением -СН=СН-, в кислотных условиях, с тем, чтобы получить соединение формулы (IX):
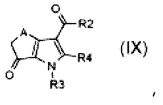
где R2 обозначает необязательно замещенный алкокси, R3 обозначает водород, R4 обозначает, как определено в п. 1, и А обозначает, как определено в п. 11, за исключением -СН=СН-;
если требуется или желательно,
стадия Е) алкилирование соединения формулы (IX), где R3 обозначает водород, соединением формулы (X):
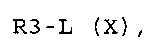
где L обозначает подходящую уходящую группу, такую как мезил, тозил, атом галогена, и R3 обозначает, как определено в п. 1, за исключением водорода;
стадия F) взаимодействие полученного соединения формулы (IX):
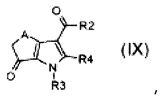
где R2 обозначает необязательно замещенный алкокси, R3 и R4 обозначают, как определено в п. 1, и А обозначает, как определено в п. 1, за исключением -СН=СН-, с производным N,N-диметилформамида;
стадия G) взаимодействие полученного соединения формулы (XI):
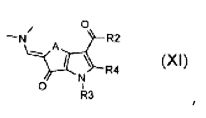
где R2 обозначает необязательно замещенный алкокси, R3 и R4 обозначают, как определено в п. 1, и А обозначает, как определено в п. 1, за исключением -СН=СН-, с соединением формулы (XII)
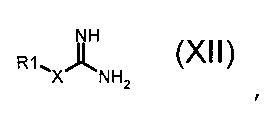
где X обозначает двухвалентный радикал, выбранный из -NR′ и -S-; и R1 и R′ обозначают, как определено в п. 1, с тем, чтобы получить соединение формулы (Ia):
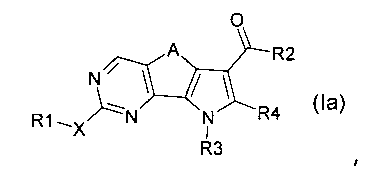
где X обозначает двухвалентный радикал, выбранный из -NR′ и -S-; R2 обозначает необязательно замещенный алкокси; А обозначает, как определено в п. 1, за исключением -СН=СН-; и R1,
R3, R4 и R′ обозначают, как определено в п. 1;
превращение соединения формулы (Ia), где R2 обозначает необязательно замещенный алкокси, в соответствующее соединение формулы (I), где R2 обозначает, как определено в п. 1, посредством взаимодействия с производным формулы R″R′″NH (XIII), где R″ и R′″ обозначают, как определено в п. 1, в присутствии основания в подходящем растворителе:
и, при желании, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
6. Способ лечения заболевания, вызванного и/или связанного с нарушением регуляции активности протеинкиназ MPS1, PIM-1 и PIM-2, где заболевание выбирают из группы, состоящей из рака яичников, рака молочной железы, меланомы, рака ободочной кишки, лейкемии, рака поджелудочной железы и миеломы, включающий введение эффективного количества соединения формулы (I) по п. 1.
7. Способ ингибирования активности протеинкиназы in vitro, включающий контакт указанного белка с эффективным количеством соединения формулы (I) по п. 1, где протеинкиназу выбирают из группы, состоящей из MPS1, PIM-1 и PIM-2.
8. Фармацевтическая композиция, обладающая свойствами ингибитора активности протеинкиназ MPS1, PIM-1 и PIM-2, содержащая эффективное количество одного или более соединений формулы (I) или их фармацевтически приемлемой соли по п. 1 и по меньшей мере один фармацевтически приемлемый эксципиент, носитель и/или разбавитель.
| WO 2008065054 A1, 05.06.2008 | |||
| WO 2009156315 A1, 30.12.2009 | |||
| ГЕТЕРОЦИКЛИЧЕСКИЕ ДИГИДРОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2296766C2 |
| Весы | 1928 |
|
SU10904A1 |

