Настоящее изобретение относится к новым замещенным N-(3-бензоиламинофенил)-4-пиридил-2-пиримидинаминовым производным, способам их получения, содержащим их фармацевтическим композициям, их применению необязательно в комбинации с одним или несколькими другими фармацевтически активными соединениями для лечения заболевания, которое связано с ингибированием активности протеинкиназы, особенно пролиферативного заболевания, и к способам лечения такого заболевания.
Протеинкиназы (РК) являются ферментами, которые катализируют фосфорилирование остатков серина, треонина или тирозина в клеточных белках. Эти посттрансляционные модификации субстратных белков выступают в качестве молекулярных регуляторов клеточной пролиферации, активации и/или дифференциации. Ненормальная или избыточная активность РК наблюдается в многочисленных заболеваниях, включая доброкачественные и злокачественные пролиферативные заболевания. В ряде случаев лечение заболеваний, таких как пролиферативные заболевания, возможно с помощью применения РК ингибиторов in vitro и in vivo.
Ввиду большого количества ингибиторов протеинкиназы и множества пролиферативных и других заболеваний, связанных с РК, все еще существует потребность в получении новых классов соединений, которые были бы полезными в качестве ингибиторов РК, и таким образом для лечения этих заболеваний, связанных с РК, необходимы новые классы фармацевтически приемлемых соединений, ингибирующих РК.
Филадельфийская хромосома является признаком хронической миеломной лейкемии (CML) и несет гибридный ген, который содержит N-терминальные экзоны bcr гена и основную С-терминальую часть (экзоны 2-11) с-abl гена. Ген кодирует химерный белок 190 кДа, 210 кДа или 230 кДа в зависимости от того, какие из трех альтернативных точек остановки включены в bcr. Abl-часть Bcr-Abl белка содержит Abl-тирозинкиназу, которая слабо регулируется в диком виде с-Abl, но сильно активируется в Bcr-Abl белке слияния. Это неодинаковое регулирование тирозинкиназы соотносится с многочисленными путями клеточных сигналов, приводящих к трансформации и неодинаковому регулированию пролиферации клеток (Lugo и др., Science 247, 1079 [1990]).
р210 Bcr-Abl экспрессируется у 95% пациентов с CML и приблизительно у 33% пациентов с острой лимфобластомной лейкемией (ALL). Экспрессия меньшего белка р190 кДа чаще всего происходит при ALL, но иногда при CML, и характеризуется клинически усиленным моноцитозом. Белок слияния 230 кДа связан с редкой хронической нейтрофильной лейкемией, чья прогрессия в преодолении кризиса является медленной. На развитой стадии CML и особенно ALL часто появляются клоны, в которых мутирует киназный домен Bcr-Abl белка. Такие мутанты включают, например, E225V и М351Т трансформации (Shah и др., Cancer Research 2, 117-225 [2002]).
Мутантные ras онкогены часто связаны с ростом опухоли. Белки Ras экспрессируются из трех различных генов, а именно Neuroblastoma (N)-ras, Harvey (Ha)-ras и Kirsten (К)-ras. K-ras мутирует чаще всего в твердых опухолях, таких как рак толстой кишки, легких и особенно поджелудочной железы, и N-ras мутирует в гематопоэтических опухолях, преимущественно в острой миеломной лейкемии (Lyons и др., Endocrine-Related Cancer 8, 219 [2001]). Было показано, что Ras регулирует некоторые пути клеточной трансформации, включая, например, Raf/MEK пути посредством связывания и активации Raf киназы.
N-(3-бензоиламинофенил)-4-пиридил-2-пиримидинаминовые производные формулы 1, описанные далее более подробно, проявляют отличное ингибирование активности протеинкиназ, особенно ингибирование одной или нескольких тирозинкиназ, таких как Bcr-Abl, мутантная Bcr-Abl, с-Abl, Raf, рецептор тирозинкиназ PDGF-R, Flt3, VEGF-R, EGF-R и c-Kit, а также комбинации двух или нескольких из них. В частности, соединения по изобретению проявляют хорошую активность в отношении некоторых мутированных форм Bcr-Abl, которые наблюдались у пациентов, резистентных к лекарственным средствам. Благодаря этим активностям соединения могут использоваться для лечения заболеваний, связанных особенно с неправильной или избыточной активностью таких типов киназ, например, для лечения некоторых видов лейкемий и твердых опухолей, таких как рак толстой кишки, легких и поджелудочной железы.
Данное изобретение относится к соединению формулы 1,

где
R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой низший алкил, фторалкил, гидроксиалкил или карбамоил;
R4 представляет собой водород, низший алкил или галоген; и
R5 и R6 представляют собой, независимо друг от друга, водород, низший алкил, гидрокси-низший алкил, низший алкокси-низший алкил, низший асилокси-низший алкил, карбокси-низший алкил, низший алкоксикарбонил-низший алкил, амино-низший алкил, низший алкиламино-низший алкил, ди(низший алкил)амино-низший алкил, N-низший алкилпиперидинил, N-низший алкилпирролидинил или низший ацил, или R5R6 вместе представляют собой алкилен с четырьмя, пятью или шестью атомами углерода, окса-низший алкилен с одним атомом кислорода и тремя или четырьмя атомами углерода, или азанизший алкилен с одним атомом азота и тремя или четырьмя атомами углерода, где атом азот является незамещенным или замещенным низшим алкилом, гидрокси-низшим алкилом или низшим алкокси-низшим алкилом, и где низший алкилен в каждом случае может быть частично или полностью ненасыщенным и/или атомы углерода низшего алкилена могут быть замещены низшим алкилом, гидрокси или низшим алкокси;
и N-оксиду или фармацевтически приемлемой соли такого соединения.
Общие термины, которые используются здесь и далее, предпочтительно имеют следующие значения в контексте описания, если конкретно не указано иное:
"Низший" обозначает радикал, имеющий до 7 включительно, особенно до 4 включительно атомов углерода, радикалы являются либо линейными, либо разветвленными с одним или несколькими ответвлениями.
В случае если для соединений, солей и им подобных используется множественное число, предполагается, что одно соединение, соль или им подобные также включены.
В (R)-, (S)- или (R,S)-конфигурации, предпочтительно в (R)- или (S)-конфигурации могут присутствовать асимметричные атомы углерода. Соединения могут присутствовать в виде смесей изомеров или в виде чистых изомеров, предпочтительно в виде энантиомерно чистых диастереомеров.
Настоящее изобретение также относится к возможным таутомерам соединений формулы 1.
Низший алкил предпочтительно представляет собой алкил с 1-7, предпочтительно с 1-4 атомами углерода, и является линейным или разветвленным; предпочтительно, низший алкил представляет собой бутил, такой как н-бутил, втор-бутил, изобутил, трет-бутил, пропил, такой как н-пропил или изопропил, этил или метил. Предпочтительно низший алкил представляет собой метил, пропил или трет-бутил.
Низший ацил предпочтительно представляет собой формил или низший алкилкарбонил, особенно ацетил.
Гидроксиалкил представляет собой особенно гидрокси-низший алкил, предпочтительно гидроксиметил, 2-гидроксиэтил или 2-гидрокси-2-пропил.
Фторалкил представляет собой особенно фтор-низший алкил, предпочтительно трифторметил или пентафторэтил.
Галоген представляет собой особенно фтор, хлор, бром или йод, особенно фтор, хлор или бром.
Низший алкокси представляет собой особенно метокси, этокси, изопропилокси или трет-бутилокси.
Низший алкоксикарбонил представляет собой особенно трет-бутоксикарбонил, изопропоксикарбонил, метоксикарбонил или этоксикарбонил.
Соли особенно представляют собой фармацевтически приемлемые соли соединений формулы 1.
Такие соли получают, например, в виде кислотных аддитивных солей, предпочтительно с органическими или неорганическими кислотами, из соединений формулы 1 с основным атомом азота, особенно в виде фармацевтически приемлемых солей. Подходящими неорганическими кислотами являются, например, галогенсодержащие кислоты, такие как соляная кислота, серная кислота или фосфорная кислота. Подходящими органическими кислотами являются, например, карбоновые, фосфорные, сульфоновые или сульфаминовые кислоты, например, уксусная кислота, пропановая кислота, октановая кислота, декановая кислота, додекановая кислота, гликолевая кислота, молочная кислота, фумаровая кислота, янтарная кислота, адипиновая кислота, пимелиновая кислота, субериновая кислота, азелаиновая кислота, яблочная кислота, винная кислота, лимонная кислота, аминокислоты, такие как глутаминовая кислота или аспарагиновая кислота, малеиновая кислота, гидроксималеиновая кислота, метилмалеиновая кислота, циклогексанкарбоновая кислота, адамантанкарбоновая кислота, бензойная кислота, салициловая кислота, 4-аминосалициловая кислота, фталевая кислота, фенилуксусная кислота, манделиновая кислота, коричная кислота, метан- или этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, этан-1,2-дисульфоновая кислота, бензолсульфоновая кислота, 2-нафталенсульфоновая кислота, 1,5-нафталендисульфоновая кислота, 2-, 3- или 4-метилбензолсульфоновая кислота, метилсерная кислота, этилсерная кислота, додецилсерная кислота, N-циклогексилсерная кислота, N-метил-, N-этил- или N-пропилсульфаминовая кислота, или другие органические протонные кислоты, такие как аскорбиновая кислота.
Для выделения или очистки также возможно применение фармацевтически неприемлемых солей, например, пикратов или перхлоратов. Для терапевтического применения используются только фармацевтически приемлемые соли или свободные формы соединений (где применимо в форме фармацевтических составов), и, следовательно, они являются предпочтительными.
Ввиду сродства между новыми соединениями в свободной форме и соединениями в форме их солей, включая те соли, которые могут использоваться в качестве промежуточных соединений, например, для очистки или идентификации новых соединений, любое указание на свободную форму соединений здесь и далее должно относиться также к соответствующим солям, которые являются подходящими и приемлемыми.
Соединения формулы 1 и их N-оксиды имеют ценные фармакологические свойства, как описано здесь и далее.
Эффективность соединений по изобретению в качестве ингибиторов активности с-Abl, Bcr-Abl, Raf и VEGF-рецептора тирозинкиназы может быть определена следующим образом.
Тест на активность в отношении с-Abl протеинтирозинкиназы. Тест проводили как фильтровальный анализ на связывание следующим образом: домен His-меченой киназы с-Abl клонировали и экспрессировали в системе бакуловирус/Sf9, как описано Bhat и др., J. Biol. Chem. 272, 16170-5 (1997). Белок 37 кДа (с-Abl киназа) очищали двухстадийной методикой на колонке с хелатом металлического кобальта с последующей анионообменной колонкой с выходом 1-2 мг/л Sf9 клеток. Чистота с-Abl киназы составляла >90%, что определяли SDS-PAGE после подкрашивания кумачевым синим. Анализ включает: с-Abl киназу (50 нг), 20 мМ Tris·HCl, рН 7,5, 10 мМ MgCl2, 10 мкМ Na3VO4, 1 мМ DTT и 0,06 мк Ci/анализ [γ33Р]-АТФ (5 мкМ АТФ), используя 30 мкг/мл поли-Ala, Glu, Lys, Tyr-6:2:5:1 (Poly-AEKY, Sigma P1152) в присутствии 1% ДМСО, общий объем 30 мкл. Реакции обрывали добавлением 10 мкл 250 мМ EDTA, и 30 мкл реакционной смеси переносили на мембрану Immobilon-PVDF (Millipore, Bedford, MA, USA), предварительно смоченную в течение 5 мин метанолом, промывали водой, затем промывали 5 мин 0,5% H3PO4 и загружали в вакуумную трубку с неподсоединенным вакуумным источником. После загрузки всех образцов подсоединяли вакуум и каждую ячейку промывали 200 мкл 0,5% H3PO4. Мембраны удаляли и промывали на вибраторе 0,5% H3PO4 (4 раза) и один раз этанолом. Мембраны исследовали после высушивания при температуре окружающей среды, загружали в 96-ячеечную подложку Packard TopCount и добавляли 10 мкл/ячейку Microscint TM (Packard).
Тест на активность в отношении Bcr-Abl. Миелоидную прародительскую клеточную линию мыши 32Dcl3, трансфецированную р210 Bcr-Abl вектором экспрессии pGDp210Bcr/Abl (32D-bcr/abl), получали от J.Griffin (Dana Faber Cancer Institute, Boston, MA, USA). Клетки экспрессировали белок слияния Bcr-Abl с изначально активной Abl киназой и независимо пролиферировали фактор роста. Клетки обрабатывали RPMI 1640 (AMIMED), 10% эмбриональной телячьей сывороткой, 2 мМ глутамина (Gibco) („полная среда”) и рабочий раствор получали замораживанием аликвот 2×106 клеток на сосуд в среде для замораживания (95% FCS, 5% ДМСО-d6 (SIGMA)). После размораживания клетки использовали максимально 10-12 раза в экспериментах.
Для клеточного испытания соединения растворяли в ДМСО и разбавляли полной средой с получением начальной концентрации 10 мкМ, затем получали 3-кратно разбавленные растворы в полной среде. 200'000 32D-Bcr/Abl клетки в 50 мкл полной среды высаживали в каждую ячейку 96-ячейчных круглодонных подложек для культуры тканей. К клеткам трижды добавляли 50 мкл на ячейку серийных 3-кратных разбавлений тестируемого соединения. Необработанные клетки использовали в качестве контроля. Соединение инкубировали вместе с клетками в течение 90 мин при 37°С, 5% CO2, затем центрифугировали подложки с культурой тканей при 1300 об/мин (центрифуга Beckman GPR) и супернатанты удаляли, сливая осторожно, чтобы не удалить остаток клеток. Остатки клеток лизировали добавлением 150 мкл лизисного буфера (50 мМ Tris/HCl, рН 7,4, 150 мМ хлорида натрия, 5 мМ EDTA, 1 мМ EGTA, 1% NP-40, 2 мМ орто-ванадата натрия, 1 мМ PMSF, 50 мкг/мл апротинина и 80 мкг/мл леупептина) и непосредственно использовали в ELISA или хранили замороженными в подложках при -20°С до использования.
Черные подложки ELISA (черные подложки Packard HTRF-96) предварительно обрабатывали в течение ночи при 4°С 50 нг/ячейку поликлональным анти-abl-SH3 доменом Ab 06-466 кролика от Upstate в 50 мкл PBS. После промывания 3 раза 200 мкл/ячейку PBS, содержащим 0,05% Tween20 (PBST) и 0,5% TopBlock (Juro), оставшиеся сайты связывания белка блокировали 200 мкл/ячейку PBST, 3% TopBlock в течение 4 ч при комнатной температуре, затем инкубировали с 50 мкл лизатов необработанных или обработанных соединением клеток (20 мкг всего белка на ячейку) в течение 3-4 ч при 4°С. После 3 промываний добавляли 50 мкл/ячейку анти-фосфотирозина Ab PY20(AP), меченого щелочной фосфатазой (Zymed), разбавленного до 0,2 мкг/мл в блокирующем буфере и инкубировали в течение ночи (4°С). На всех стадиях инкубации подложки покрывали крышками (Costar). Наконец, подложки промывали еще три раза промывочным буфером и один раз деионизированной водой перед добавлением 90 мкл/ячейку АР-субстрата CDPStar RTU с Emerald II. Подложки, закрытые крышками Packard TopSeal™-А, инкубировали в течение 45 мин при комнатной температуре в темноте и количественно определяли люминесценцию измерением показателей в секунду (CPS) с помощью Packard Top Count Microplate Scintillation Counter (Top Count).
Определяли разницу между показаниями ELISA (CPS), полученными для лизатов необработанных 32D-Bcr/Abl клеток, и показаниями базового анализа (все компоненты, но без клеточного лизата) и за 100% брали отражение изначально фосфорилированного Bcr-Abl белка, присутствующего в этих клетках. Влияние соединения на активность Bcr-Abl киназы выражали как процентное снижение фосфорилирования Bcr-Abl. Значения IC50 и IC90 определяли по кривым зависимостей от доз графическим экстраполированием.
Тест на активность в отношении мутантной Bcr-Abl: активность соединений в отношении активности М351Т мутантной Bcr-Abl киназы осуществляли, как описано выше, за исключением того, что использовали 32Dcl3 клетки, трансфецированные мутантным Bcr-Abl вместо р210 Bcr-Abl.
Анализ с c-Raf-1 протеинкиназой: рекомбинантный c-Raf-1 белок получали тройной обработкой Sf21 клеток GST-c-Raf-1 рекомбинантным бакуловирусом вместе с v-Src и v-Ras рекомбинантными бакуловирусами, что требуется для активации продуцирования c-Raf-1 киназы (Williams и др., PNAS 1992; 89:2922-6). Активный Ras (v-Ras) необходим для проникновения c-Raf-1 в клеточную мембрану и v-Src для фосфорилирования c-Raf-1 для полного его активирования. Клетки высаживали в количестве 2,5×107 клеток в 150 мм плашку и оставляли связываться в плашках на 150 в течение 1 час при комнатной температуре. Среду (SF900II, содержащий 10% FBS) удаляли и добавляли рекомбинантный бакуловирус GST-c-Raf-1, v-Ras и v-Src в количестве MOI 3,0, 2,5 и 2,5 соответственно, общим объемом 4-5 мл. Клетки инкубировали в течение 1 часа при комнатной температуре и затем добавляли 15 мл среды. Обработанные клетки инкубировали в течение 48-72 ч при 27°С. Обработанные Sf21 клетки выскабливали и собирали в пробирки на 50 мл и центрифугировали в течение 10 мин при 4°С в количестве 1100 г на центрифуге Sorvall. Остаток клеток промывали один раз ледяным PBS и лизировали 0,6 мл лизисного буфера в количестве 2,5×107 клеток. Полный лизис клеток достигался через 10 мин во льду при редком прикапывании. Клеточные лизаты центрифугировали в течение 10 мин при 4°С в количестве 14,500 г на центрифуге Sorvall с SS-34 ротором и супернатант переносили в чистую пробирку и хранили при -80°С. c-Raf-1 очищали от клеточных лизатов с помощью 100 мкл расфасованных гранул глутатион-сефарозы 4В, уравновешенных в охлажденном льдом PBS, в количестве 2,5×107 клеток. GST-c-Raf-1 оставляли связываться с гранулами при 4°С в течение 1 часа при покачивании. Связавшийся с гранулами GST-c-Raf-1 переносили на колонку. Колонку промывали один раз лизисным буфером и два раза охлажденным льдом TRis буферным соляным раствором. Добавляли охлажденный льдом буфер для промывки и поток колонки останавливали для того, чтобы позволить свободному глутатиону разорвать взаимодействие GST-c-Raf-1 с глутатион-сефарозными гранулами. Фракцию (1 мл) собирали в предварительно охлажденные пробирки. Каждая пробирка содержит 10% глицерина (конечная концентрация) для сохранения активности киназы в процессе циклов замораживания-оттаивания. Очищенные фракции белка GST-c-Raf-1 киназы хранили при -80°С.
IκВ использовали в качестве субстрата для c-Raf-1 киназы. IκВ экспрессировался в бактериях в виде His-меченого белка BL21. LysS бактерии, содержащие плазмиду IκВ, высаживали в OD600 0,6 в среде LB, затем индуцировали экспрессию IκВ с помощью IPTG (конечная концентрация 1 мМ) в течение 3 часов при 37°С и затем бактерии подвергали лизису озвучиванием (microtip ограничение, установленное 3 раза на 1 мин), в буфере для озвучивания [50 мМ Tris рН 8,0, 1 мМ DTT, 1 мМ EDTA] и центрифугировали при 10,000 об/мин в течение 15 мин. Супернатант смешивали с сульфатом аммония с получением конечной концентрации 30%. Эту смесь встряхивали в течение 15 мин при 4°С, затем раскручивали при 10,000 об/мин в течение 15 мин. Остаток повторно суспендировали в связующем буфере (Novagen), содержащем 10 мМ БСА. Этот раствор наносили на Ni-агарозу (Novagen) и промывали в соответствии с инструкцией Novagen. IκВ смывали с колонки буфером для промывки (0,4 М имидазола, 0,2 М NaCl, 8 мМ Tris рН 7,9). Фракции, содержащие белок, подвергали диализу в 50 мМ Tris рН 8,1 мМ DTT.
Активность c-Raf-1 протеинкиназы измеряли в присутствии или отсутствии ингибиторов, измерением проникновения 33Р из [γ33Р] АТФ в IκВ. Анализ осуществляли в 96-ячеечных подложках при температуре окружающей среды в течение 60 мин. Они содержали (общий объем 30 мкл): c-Raf-1 киназу (400 нг), 25 мМ Tris·HCl, рН 7,5, 5 мМ MgCl2, 5 мМ MnCl2, 10 мкМ Na3VO4, 1 мМ DTT и 0,3 мк Ci/анализ [γ33Р]-АТФ (10 мкМ АТФ), используя 600 нг IκВ в присутствии 1% ДМСО. Реакции обрывали добавлением 10 мкл 250 мМ EDTA и 30 мкл реакционной смеси переносили на мембрану Immobilon-PVDF (Millipore, Bedford, MA, USA), предварительно намоченную в течение 5 мин метанолом, промытую водой, затем намоченную в течение 5 мин 0,5% H3PO4 и помещенную на вакуумный сборник с отсоединенным источником вакуума. После загрузки всех образцов присоединяли вакуум и каждую ячейку промывали 200 мкл 0,5% H3PO4. Мембраны удаляли и промывали 4 раза на вибраторе с 0,5% H3PO4, один раз с этанолом. Мембраны количественно измеряли после высушивания при температуре окружающей среды, устанавливая на Packard TopCount 96-ячеечную рамку и добавляя 10 мкл/ячейку Microscint TM (Packard).
Тест на активность в отношении тирозинкиназы VEGF-рецептора. Тест проводили с помощью тирозинкиназы Flt-1 VEGF-рецептора. Подробная методика приведена далее: раствор 30 мкл киназы (10 нг киназного домена Flt-1, Shibuya и др., Oncogene 5, 519-24 [1990]) в 20 мМ Tris·HCl pH 7,5, 3 мМ дихлорида марганца (MnCl2), 3 мМ хлорида магния (MgCl2), 10 мкМ ванадата натрия, 0,25 мг/мл полиэтиленгликоля (PEG) 20000, 1 мМ дитиотреитола и 3 мкг/мкл поли(Glu, Tyr) 4:1 (Sigma, Buchs, Switzerland), 8 мкМ [33P]-АТФ (0,2 мк Ci), 1% ДМСО и 0-100 мкМ тестируемого соединения инкубировали вместе в течение 10 минут при комнатной температуре. Реакцию затем останавливали добавлением 10 мкл 0,25 М этилендиаминтетраацетата (EDTA) pH 7. Используя многоканальный передатчик (LAB SYSTEMS, USA), аликвоты 20 мкл переносили на PVDF (= поливинилдифторид) Immobilon P мембраны (Millipore, Bedford, USA), на Gibco-BRL микротитровальный фильтр и подсоединяли к вакууму. После полного расщепления жидкости мембрану промывали 4 раза последовательно в бане, содержащей 0,5% фосфорной кислоты (H3PO4), и один раз этанолом, инкубировали в течение 10 минут все, время встряхивая, затем загружали на Hewlett Packard TopCount Manifold и измеряли радиоактивность добавлением 10 мкл Microscint® (β-стинтилляционный счетчик для жидкостей). IC50-значения определяли линейным регрессионным анализом процентного соотношения ингибирования каждого соединения по крайней мере в четырех концентрациях (как правило, 0,01, 0,1, 1,0 и 10 мкмоль). IC50-значения для соединений формулы 1 находятся в пределах 1-10'000 нМ, предпочтительно в пределах 1-100 нМ.
Ингибирование VEGF-индуцированного автофосфорилирования KDR-рецептора может быть подтверждено следующим экспериментом in vitro на клетках: трансфецированные СНО клетки, которые постоянно экспрессируют клетки VEGF рецептора (KDR), высаживали в полную культуральную среду с 10% эмбриональной телячьей сывороткой (FCS) в 6-ячеечные подложки для клеток и инкубировали при 37°С под 5% СО2 приблизительно до 80% слияния. Затем тестируемые соединения разбавляли культуральной средой (без FCS, с 0,1% бычьим сывороточным альбумином) и добавляли к клеткам (контроли включали среду без тестируемых соединений). Через два часа инкубирования при 37°С добавляли рекомбинантный VEGF; конечная концентрация VEGF составляет 20 нг/мл. Еще через пять минут инкубирования при 37°С клетки промывали два раза охлажденным льдом PBS (фосфат-буферный соляной раствор) и немедленно подвергали лизису в 100 мкл лизисного буфера на ячейку. Затем лизаты центрифугировали для удаления клеточных ядер и концентрации белка супернатантов измеряли с помощью коммерчески доступного белкового анализа (BIORAD). Лизаты затем могут использоваться немедленно или, при необходимости, храниться при -20°С.
Для измерения фосфорилирования KDR-рецептора проводили сэндвич ELISA: моноклональное антитело к KDR (например, Mab 1495, 12, 14) иммобилизировали на черных ELISA подложках (OptiPlate™ HTRF-96 от Packard). Затем подложки промывали и оставшиеся свободные белок-связывающие сайты насыщали 1% БСА в PBS. Клеточные лизаты (20 мкг белка на ячейку) затем инкубировали в этих же подложках в течение ночи при 4°С вместе с антителом к фосфотирозину, связанным с щелочной фосфатазой (PY20:AP от Transduction Laboratories). Подложки промывали опять и связывание антитела к фосфотирозину с захваченным фосфорилированным рецептором, затем определяли с помощью люминесцентного АР субстрата (CDP-Star, готовый к применению, с Emerald II; TROPIX). Люминесценцию измеряли на Packard Top Count Microplate Scintillation Counter (Top Count). Разница между сигналом положительного контроля (стимулируемого VEGF) и сигналом отрицательного контроля (не стимулируемого VEGF) соответствует VEGF-индуцированному фосфорилированию KDR-рецептора (=100%). Активность тестируемых веществ рассчитывали как % ингибирование VEGF-индуцированного фосфорилирования KDR-рецептора, где концентрацию вещества, которая вызывает половину максимального ингибирования, определяли как ED50 (эффективная доза для 50% ингибирования). Соединения формулы 1 предпочтительно имеют значения ED50 в области от 0,25 нМ до 1000 нМ, предпочтительно от 0,25 до 250 нМ.
Соединение формулы 1 или его N-оксид в различной степени ингибирует также другие тирозинкиназы, вовлеченные в сигнальную трансдукцию, которые опосредуются трофными факторами, например, Raf, Bcr-Abl и Abl киназу, Arg, киназы из семейства Src, особенно c-Src киназу, Lck и Fyn; также киназы семейства EGF, например, с-erbB2 киназу (HER-2), с-erbB3 киназу, с-erbB4 киназу; киназу инсулиноподобного рецептора фактора роста (IGF-1 киназа), особенно членов семейства тирозинкиназы PDGF-рецептора, таких как киназа PDGF-рецептора, киназа CSF-1-рецептора, киназу Kit-рецептора и киназу VEGF-рецептора; а также серин/треонинкиназы, каждая из которых играет важную роль для регулирования и трансформации клеток млекопитающих, включая клетки человека.
Ингибирование с-erbB2 тирозинкиназы (HER-2) может быть измерено, например, тем же способом, что и ингибирование EGF-R протеинкиназы, используя известные методики.
На основе этих исследований соединение формулы 1 по изобретению проявляет терапевтическую эффективность, особенно в отношении заболеваний, зависящих от протеинкиназы, особенно пролиферативных заболеваний.
На основе этой эффективности в качестве ингибиторов активности тирозинкиназы VEGF-рецептора соединения формулы 1 ингибируют главным образом рост сосудов крови и таким образом являются, например, эффективными против ряда заболеваний, связанных с нерегулируемым ангиогенезом, особенно заболеваний, вызванных глазной неоваскуляризацией, особенно ретинопатиями, такими как диабетическая ретинопатия или возрастная дегенерация сетчатки, псориазом, гемангиобластомой, такой как гемангиома, мезанглиальных клеточных пролиферативных заболеваний, таких как хронических или острых заболеваний почек, например, диабетическая нефропатия, злокачественный нефросклероз, тромботические микроангиопатические синдромы или отторжение трансплантата, или особенно воспалительное заболевание почек, такое как гломерулонефрит, особенно мезангиопролиферативный гломерулонефрит, гемолитического-уремического синдрома, диабетической нефропатии, гипертензивного нефросклероза, атеромы, артериального рестеноза, аутоиммунных заболеваний, диабетов, эндометриоза, хронической астмы и особенно неопластических заболеваний (твердые опухоли, а также лейкемии и другие "жидкие опухоли", особенно опухоли, экспрессирующие c-kit, KDR, Flt-1 или Flt-3), таких как особенно рак груди, рак толстой кишки, рак легких (особенно рак легких малых клеток), рак простаты или саркома Капоши. Соединение формулы 1 (или его N-оксид) ингибирует рост опухолей и особенно полезно для предотвращения распространения метастаз опухолей и роста микрометастаз.
Соединение формулы 1 может вводиться отдельно или в комбинации с одним или несколькими другими терапевтическими агентами, возможна комбинированная терапия в форме фиксированных комбинаций или введением соединения по изобретению и одного или нескольких терапевтических агентов последовательно или независимо друг от друга, или объединенным введением фиксированных комбинаций и одного или нескольких других терапевтических агентов. Соединение формулы 1 может, помимо этого, использоваться особенно при терапии рака, такой как терапия лейкемии, в комбинации с химиотерапией, радиотерапией, иммунотерапией, хирургическим вмешательством или их комбинацией. Длительная терапия также возможна в качестве вспомогательной терапии при других методиках лечения, как описано выше. Другими возможными лечениями являются терапия для поддержания состояния пациента во время роста опухоли или даже химиопредотвращающей терапии, например, у пациентов группы риска.
Терапевтическими агентами для возможной комбинации являются особенно одно или несколько антипролиферативных, цитостатических или цитотоксических соединений, например, химиотерапевтический агент или несколько агентов, выбранных из группы, включающей, но не ограничиваясь ими, ингибитор биосинтеза полиаминов, ингибитор протеинкиназы, особенно серин/треонин протеинкиназы, такой как протеинкиназа С, или тирозинпротеинкиназы, такой как тирозинкиназа EGF рецептора, например, PKI166, тирозинкиназы VEGF рецептора, например, РТК787, или тирозинкиназы PDGF рецептора, например, STI571, цитокин, регулятор отрицательного роста, такой как TGF-β или IFN-β, ингибитор ароматазы, например, летрозол или анастрозол, ингибитор взаимодействия SH2 домена с фосфорилированным белком, антиэстрогены, ингибиторы топоизомеразы I, такие как иринотекан, ингибиторы топоизомеразы II, микротубулярные активные агенты, например, паклитаксел, дискодермолид или эпотилон, алкилирующие агенты, антинеопластические антиметаболиты, такие как гемцитабин или капецитабин, платиновые соединения, такие как карбоплатин или цисплатин, антиангиогенные соединения, агонисты гонадорелина, антиандрогены, бисфосфонаты, например, AREDIA® или ZOMETA®, и трастузумаб. Предпочтительные терапевтические агенты для комбинации особенно предпочтительно выбраны из группы, включающей индарубицин, цитарабин, интерферон, гидроксимочевина и бисульфан. Структура активных агентов, обозначенных кодом nos., дженерики или товарные знаки могут быть взяты из текущего стандартного издания "The Merck Index" или из баз данных, например, Patents International (например, IMS World Publications). Их соответствующее содержание включено здесь в качестве ссылки.
Соединение по изобретению предназначено не только для (профилактического и предпочтительно терапевтического) введения людям, а также для лечения других теплокровных животных, например, промышленных животных, например, грызунов, таких как мыши, кролики или крысы, или морских свинок. Такие соединения могут также использоваться в качестве ссылочного стандарта в описанных выше тестовых системах для осуществления сравнения с другими соединениями.
Вообще изобретение также относится к применению соединения формулы 1 или его N-оксида для ингибирования активности тирозинкиназы, in vitro или in vivo.
Для групп предпочтительных соединений формулы 1 и их N-оксидов, упомянутых далее, могут использоваться определения заместителей из общих упомянутых ранее определений, например, для замены общих определений на более конкретные определения или особенно для определений, являющихся наиболее предпочтительными.
В частности, изобретение относится к соединениям формулы 1, где R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой низший алкил, фторалкил, гидроксиалкил или карбамоил;
R4 представляет собой низший алкил; и
R5 и R6 представляют собой, независимо друг от друга, водород, низший алкил, гидрокси-низший алкил, низший алкокси-низший алкил, низший асилокси-низший алкил, карбокси-низший алкил, низший алкоксикарбонил-низший алкил, амино-низший алкил, низший алкиламино-низший алкил, ди(низший алкил)амино-низший алкил, N-низший алкилпиперидинил, N-низший алкилпирролидинил, или низший ацил, или R5R6 вместе представляют собой алкилен с четырьмя, пятью или шестью атомами углерода, окса-низший алкилен с одним атомом кислорода и тремя или четырьмя атомами углерода, или азанизший алкилен с одним атомом азота и тремя или четырьмя атомами углерода, где атом азота является незамещенным или замещенным низшим алкилом, гидрокси-низшим алкилом или низшим алкокси-низшим алкилом, и где низший алкилен в каждом случае может быть частично или полностью ненасыщенным и/или атомы углерода низшего алкилена могут быть замещены низшим алкилом, гидрокси или низшим алкокси;
и к N-оксиду или фармацевтически приемлемой соли такого соединения.
Более конкретно, настоящее изобретение относится к соединениям формулы 1, где
R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой метил; и
R5 и R6 представляют собой, независимо друг от друга, водород, низший алкил, гидрокси-низший алкил, низший алкокси-низший алкил, низший асилокси-низший алкил, карбокси-низший алкил, низший алкоксикарбонил-низший алкил, амино-низший алкил, низший алкиламино-низший алкил, ди(низший алкил)амино-низший алкил, N-низший алкилпиперидинил, N-низший алкилпирролидинил, или ацетил, или R5R6 вместе представляют собой алкилен с четырьмя, пятью или шестью атомами углерода, окса-низший алкилен с одним атомом кислорода и тремя или четырьмя атомами углерода, или азанизший алкилен с одним атомом азота и тремя или четырьмя атомами углерода, где атом азота является незамещенным или замещенным низшим алкилом, гидрокси-низшим алкилом или низшим алкокси-низшим алкилом, и где низший алкилен в каждом случае может быть частично или полностью ненасыщенным и/или атомы углерода низшего алкилена могут быть замещены низшим алкилом, гидрокси или низшим алкокси;
и N-оксиду или фармацевтически приемлемой соли такого соединения.
Более конкретно, настоящее изобретение относится к соединениям формулы 1, где
R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой метил; и
R5 и R6 представляют собой, независимо друг от друга, водород, низший алкил, гидрокси-низший алкил, амино-низший алкил, низший алкиламино-низший алкил, ди(низший алкил)амино-низший алкил, N-низший алкилпиперидинил, или низший ацил, или R5R6 вместе представляют собой алкилен с четырьмя или пятью атомами углерода, окса-низший алкилен с одним атомом кислорода и тремя или четырьмя атомами углерода, или азанизший алкилен с одним атомом азота и тремя или четырьмя атомами углерода, где атом азота является незамещенным или замещенным низшим алкилом, гидрокси-низшим алкилом или низшим алкокси-низшим алкилом, и где низший алкилен в каждом случае может быть частично или полностью ненасыщенным и/или атомы углерода низшего алкилена могут быть замещены низшим алкилом;
и N-оксиду или фармацевтически приемлемой соли такого соединения.
Предпочтительными являются соединения формулы 1, где
R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой метил; и
R5 и R6 представляют собой, независимо друг от друга, водород, низший алкил, ди(низший алкил)амино-низший алкил, N-низший алкилпиперидинил, или низший ацетил, или R5R6 вместе представляют собой алкилен с четырьмя или пятью атомами углерода, окса-низший алкилен с одним атомом кислорода и четырьмя атомами углерода, или азанизший алкилен с одним атомом азота и тремя или четырьмя атомами углерода, где атом азота является незамещенным или замещенным низшим алкилом, и где азанизший алкилен может быть ненасыщенным и/или атомы углерода азанизшего алкилена могут быть замещены низшим алкилом;
и N-оксид или фармацевтически приемлемая соль такого соединения.
Особенно предпочтительными являются соединения формулы 1, где
R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой метил; и
R5 и R6 представляют собой, независимо друг от друга, водород, метил, этил, 2-диметиламиноэтил, 4-метил-1-пиперидинил или ацетил, или NR5R6 вместе представляют собой пирролидино, пиперидино, морфолино, N-метилпиперазино, 1H-имидазолил, 1H-2-метилимидазолил, 1Н-4-метилимидазолил или 1Н-2,4-диметилимидазолил;
и N-оксид или фармацевтически приемлемая соль такого соединения.
Особенно предпочтительными являются соединения, описанные в примерах.
Особенно настоящее изобретение относится к применению соединения формулы 1 или его N-оксида или возможного таутомера; или фармацевтически приемлемой соли такого соединения для изготовления фармацевтической композиции для лечения заболевания, которое связано с ингибированием активности протеинкиназы, где заболевание представляет собой неопластическое заболевание.
Более конкретно, настоящее изобретение относится к применению соединения формулы 1 или его N-оксида или возможного таутомера; или фармацевтически приемлемой соли такого соединения для изготовления фармацевтической композиции для лечения лейкемии, которая связана с ингибированием активности Raf и/или Abl тирозинкиназы.
Кроме того, настоящее изобретение относится к применению соединения формулы 1 или его N-оксида или возможного таутомера или фармацевтически приемлемой соли такого соединения для лечения заболевания, которое связано с ингибированием активности протеинкиназы.
Кроме того, настоящее изобретение относится к способу лечения заболевания, которое связано с ингибированием активности протеинкиназы, который включает введение соединения формулы 1 или его N-оксида или фармацевтически приемлемой соли, где радикалы и символы имеют указанные выше значения, в количестве, эффективном против указанного заболевания, теплокровному животному, нуждающемуся в таком лечении.
Соединение настоящего изобретения может быть получено способами, которые, хотя и не применялись ранее для новых соединений настоящего изобретения, являются известными, особенно способом, заключающимся в том, что для синтеза соединения формулы 1, где символы R1, R2, R3 и R4 являются такими, как определено для соединения формулы 1, замещенную бензойную кислоту формулы 2

где R1 R2 и R3 являются такими, как определено для соединения формулы 1, или ее производное, где карбоксигруппа -СООН находится в активированной форме, подвергают реакции с 3-(4-(3-пиридил)-2-пиримидинамино)анилином формулы 3

где R4 является таким, как определено для соединения формулы 1, необязательно в присутствии дегидрирующего агента и инертного основания и/или подходящего катализатора, и необязательно в присутствии инертного растворителя;
где указанные выше исходные соединения формулы 2 и 3 могут также присутствовать, при необходимости, с функциональными группами в защищенной форме и/или в форме солей, при условии, что присутствует солеобразующая группа и реакция в форме соли является возможной;
любая защитная группа в защищенном производном соединения формулы 1 является удаляемой;
и, при необходимости, полученное соединение формулы 1 превращают в другое соединение формулы 1 или его N-оксид, свободное соединение формулы 1 превращают в соль, полученную соль соединения формулы 1 превращают в свободное соединение или другую соль, и/или смесь изомерных соединений формулы 1 разделяют на индивидуальные изомеры.
Производное соединения формулы 2, где карбоксигруппа находится в активированной форме, представляет собой особенно активированный эфир, активированный ангидрид или активированный циклический амид.
Активированные эфиры кислот формулы представляют собой особенно эфиры, ненасыщенные на атоме углерода, связанном с эфирным радикалом, например, эфиры винилового типа, такие как виниловые эфиры (полученные, например, трансэтерификацией соответствующего эфира винилацетатом; метод активированного винилового эфира), карбамоилвиниловые эфиры (полученные, например, обработкой соответствующей кислоты реагентом изоксазола; 1,2-оксазольный метод или метод Woodward) или 1-низший алкоксивиниловые эфиры (полученные, например, обработкой соответствующей кислоты низшим алкоксиацетиленом; этоксиацетиленовый метод) или эфиры амидинового типа, такие как N,N'-дизамещенные амидиноэфиры (полученные, например, обработкой соответствующей кислоты подходящим N,N'-дизамещенным карбодиимидом, например, N,N'-дициклогексилкарбодиимидом; карбодиимидный метод) или N,N'-дизамещенные амидиноэфиры (полученные, например, обработкой соответствующей кислоты N,N'-дизамещенным цианамидом; цианамидный метод), подходящие арильные эфиры, особенно фениловые эфиры, подходящим образом замещенные электроноакцепторными заместителями (полученные, например, обработкой соответствующей кислоты подходящим образом замещенным фенолом, например, 4-нитрофенолом, 4-метилсульфонил-фенолом, 2,4,5-трихлорфенолом, 2,3,4,5,6-пентахлор-фенолом или 4-фенилдиазофенолом, в присутствии конденсирующего агента, такого как N,N'-дициклогексилкарбодиимида; метод активированных ариловых эфиров), цианометиловые эфиры (полученные, например, обработкой соответствующей кислоты хлорацетонитрилом в присутствии основания; метод цианометиловых эфиров), тиоэфиры, особенно незамещенные или замещенные, например, нитрозамещенные, фенилтиоэфиры (полученные, например, обработкой соответствующей кислоты незамещенными или замещенными, например, нитро-замещенными, тиофенолами, собственно ангидридным или карбодиимидным методом; метод активированных тиольных эфиров), амино или амидоэфиры (полученные, например, обработкой соответствующей кислоты N-гидрокси-амино или N-гидрокси-амидосоединением, например, N-гидрокси-сукцинимидом, N-гидрокси-пиперидином, N-гидрокси-фталимидом или 1-гидрокси-бензотриазолом, например, ангидридным или карбодиимидным методом; метод активированных N-гидроксиэфиров), или силильные эфиры (полученные, например, обработкой соответствующей кислоты силирующим агентом, например, гексаметилдисилазаном и быстрой реакцией с гидроксигруппами, но не с аминогруппами).
Ангидриды кислоты формулы 2 могут быть симметричными или предпочтительно смешанными ангидридами этой кислоты, например, ангидридами с неорганическими кислотами, такими как галогениды кислоты, особенно хлориды кислоты (полученными, например, обработкой соответствующей кислоты тионилхлоридом, пентахлоридом фосфора или оксалилхлоридом; метод кислотных хлоридов), азидами (полученными, например, из соответствующего кислотного эфира через соответствующий гидразид и обработкой его азотной кислотой; азидный метод), ангидридами с полупроизводными карбоновой кислоты, такими как соответствующие эфиры, например, низший алкильный полуэфир карбоновой кислоты (полученными, например, обработкой соответствующей кислоты низшим алкиловым эфиром галогенмуравьиной, такой как хлормуравьиной, кислоты или 1-низшим алкоксикарбонил-2-низшим алкокси-1,2-дигидрохинолином, например, 1-низшим алкоксикарбонил-2-этокси-1,2-дигидрохинолином; метод смешанных O-алкилкарбонильных кислотных ангидридов), или ангидридами с дигалогенированной, особенно дихлорированной, фосфорной кислотой (полученные, например, обработкой соответствующей кислоты оксихлоридом фосфора; метод оксихлорида фосфора), или ангидридами с органическими кислотами, такими как смешанные ангидриды с органическими карбоновыми кислотами (полученные, например, обработкой соответствующей кислоты с незамещенным или замещенным галогенидом низший алкан- или фенилалкан-карбоновой кислоты, например, хлоридом фенилуксусной кислоты, хлоридом триметилуксусной кислоты или хлоридом трифторуксусной кислоты; метод смешанных ангидридов карбоновой кислоты), с органическими сульфоновыми кислотами (полученными, например, обработкой соли, такой как соль щелочного металла, соответствующей кислоты, подходящим галогенидом органической сульфоновой кислоты, таким как хлорид низший алкан- или арил-, например, метан- или п-толуол-сульфоновой кислоты; метод смешанных ангидридов сульфоновой кислоты), или органическими фосфоновыми кислотами (полученными, например, обработкой соответствующей кислоты подходящим ангидридом органического фосфонового или фосфониевого цианида; метод смешанных ангидридов фосфоновой кислоты), и симметричными ангидридами (полученными, например, конденсацией соответствующей кислоты в присутствии карбодиимида или 1-диэтиламинопропина; метод симметричных ангидридов).
Подходящими циклическими амидами являются особенно амиды с пятичленными диазациклами ароматического характера, такие как амиды с имидазолами, например, имидазол (полученные, например, обработкой соответствующей кислоты N,N'-карбонилдиимидазолом; имидазолидиновый метод), или пиразолы, например, 3,5-диметил-пиразол (полученный, например, из гидразида кислоты обработкой ацетилацетоном; пиразолидиновый метод).
Производные кислоты формулы 2, где карбоксигруппа находится в активированной форме, предпочтительно получают in situ. Например, N,N'-дизамещенные амидиноэфиры могут быть получены in situ реакцией смеси кислоты формулы 2 и амина формулы 3 в присутствии подходящего N,N-дизамещенного карбодиимида, например, N,N'-дициклогексилкарбодиимида. Активированные смешанные ангидриды кислоты формулы 2 с органической фосфоновой кислотой могут быть получены in situ реакцией, например, с пропилфосфоновым ангидридом или диэтилцианофосфонатом в присутствии подходящего основания, предпочтительно третичного амина, например, триэтиламина или диметиламинопиридина.
Реакция может осуществляться известным способом, в котором условия реакции зависят главным образом от того, является ли активированной карбоксигруппа карбоновой кислоты формулы 2, и если да то как, обычно в присутствии подходящего растворителя или разбавителя или их смеси и, при необходимости, в присутствии конденсирующего агента, который, например, когда карбоксигруппа в реакции находится в форме ангидрида, может также выступать агентом для связывания кислоты, при охлаждении или нагревании, например, в температурной области от приблизительно -30°С приблизительно до +150°С, особенно от приблизительно 0°С до +100°С, предпочтительно от комнатной температуры (приблизительно +20°С) до +70°С, в открытом или закрытом реакционном сосуде и/или в атмосфере инертного газа, например, азота. Обычными конденсирующими агентами являются, например, карбодиимиды, например, N,N'-диэтил-, N,N'-дипропил-, N,N'-дициклогексил- или N-этил-N'-(3-диметиламинопропил)-карбодиимид, подходящие карбонильные соединения, например, карбонилдиимидазол, или соединения 1,2-оксазола, например, 3'-сульфонат 2-этил-5-фенил-1,2-оксазола и перхлорат 2-трет-бутил-5-метил-изоксазола, или подходящее ациламиносоединение, например, 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин. Обычными связывающими кислоту конденсирующими агентами являются, например, карбонаты щелочных металлов или гидрокарбонаты, например, карбонат натрия или калия или гидрокарбонат (обычно вместе с сульфатом), или органические основания, такие как, обычно, пиридин или триэтиламин, или стерически затрудненные тринизшие алкиламины, например, N,N-диизопропил-N-этиламин.
В предпочтительном варианте карбоновую кислоту формулы 2 подвергают реакции с амином формулы 3 в подходящем растворителе, таком как, например, N,N-диметилформамид, в присутствии пропилфосфонового ангидрида или диэтилцианофосфаната и триэтиламина, от 1 до 48 часов при от 0°С и около 50°С, предпочтительно при комнатной температуре.
Если одна или несколько функциональных групп, например карбокси, гидрокси или амино, являются защищенными или нуждаются в защите в соединении формулы 2 или 3, из-за того, что они не должны участвовать в реакции, они являются такими группами, которые обычно используются в синтезе амидов, в частности, пептидных соединений, а также цефалоспоринов и пенициллинов, а также производных нуклеиновых кислот и сахаров.
Защитные группы могут уже присутствовать в предшественниках и должны защищать функциональные группы от нежелательных побочных реакций, таких как ацилирование, получение простого эфира, этерификация, окисление, сольволиз и аналогичные реакции. Характеристикой защитных групп является то, что они сами легко подходят, т.е. без нежелательных побочных реакций, для удаления, обычно сольволизом, восстановлением, фотолизом или также ферментативной активностью, например, в условиях, аналогичных физиологическим условиям, и что они не присутствуют в конечных продуктах. Специалисту известны или могут быть легко установлены, какие защитные группы являются подходящими для реакций, указанных выше и далее.
Защита таких функциональных групп такими защитными группами, защита самих групп и реакции для их удаления описаны, например, в указанных ранее стандартных источниках пептидного синтеза, и в специальных источниках по защитным группам, таких как J.F.W.McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973, в "Methoden der organischen Chemie" (Methods of organic chemistry), Houben-Weyl, 4th edition. Volume 15/1, Georg Thieme Verlag, Stuttgart 1974, и in Т.W.Greene, "Protective Groups in Organic Synthesis", Wiley, New York.
На дополнительных стадиях способа, проводимых при необходимости, функциональные группы исходных соединений, которые не должны участвовать в реакции, могут присутствовать в незащищенной форме или могут быть защищены, например, одной или несколькими защитными группами, указанными выше для "защитных групп". Защитные группы затем полностью или частично удаляются в соответствии с одним из описанных здесь способов.
Соли соединения формулы 1 с солеобразующей группой могут быть получены известным способом. Кислотные аддитивные соли соединений формулы 1 могут быть таким образом получены обработкой кислотой или подходящим анионообменным реагентом.
Соли могут обычным образом превращаться в свободные соединения, например, обработкой подходящими основными агентами, например, карбонатами щелочных металлов, гидрокарбонатами щелочных металлов или гидроксидами щелочных металлов, обычно карбонатом калия или гидроксидом натрия.
Стереоизомерные смеси, например, смеси диастереомеров, могут быть разделены на их соответствующие изомеры известными способами подходящего разделения. Диастереомерные смеси, например, могут быть разделены на их индивидуальные диастереомеры с помощью фракционной кристаллизации, хроматографии, выбора растворителя и аналогичных методик. Это разделение может также производиться либо на стадии исходного соединения, либо в самом соединении формулы 1. Энантиомеры могут быть разделены путем образования диастереомерных солей, например, образованием соли с энантиомерно-чистой хиральной кислотой, или с помощью хроматографии, например, ВЭЖХ, используя хроматографические субстраты с хиральными лигандами.
В соединении формулы 1, где в группе R1 или R2 водород присоединен к атому азота или кислорода, и должно быть превращено в соответствующее соединение, где водород замещен низшим алкилом, это может осуществляться реакцией, например, с соединением диазонизшего алкила, особенно диазометаном, в инертном растворителе, предпочтительно в присутствии катализатора благородного металла, особенно в диспергированной форме, например меди, или соли благородного металла, например хлорида меди(I) или сульфата меди(II). Также возможна реакция с низшими алкилгалогенидами или с другой уходящей группой, переносящей низшие алканы, например, низшими алкиловыми спиртами, этерифицированными сильной органической сульфоновой кислотой, такой как низший алкансульфоновая кислота (необязательно замещенная галогеном, таким как фтор), ароматической сульфоновой кислотой, например, незамещенной или замещенной бензолсульфоновой кислотой, где заместители предпочтительно выбраны из низшего алкила, такого как метил, галогена, такого как бром, и/или нитро, например, этерифицированы метансульфоновой кислотой или п-толуолсульфоновой кислотой. Алкилирование производят в обычных условиях алкилирования амидов, особенно в водном растворе и/или в присутствии полярных растворителей, обычно спиртов, например, метанола, этанола, изопропанола, или этиленгликоля, или диполярных апротонных растворителей, например, тетрагидрофурана, диоксана или диметилформамида, где приемлемо в присутствии кислотных или основных катализаторов, обычно при температурах приблизительно от 0°С до температуры кипения соответствующей реакционной смеси, предпочтительно между 20°С и температурой кипения, при необходимости при повышенном давлении, например, в запаянной пробирке, и/или в атмосфере инертного газа, обычно азота или аргона.
Следует отметить, что реакции, аналогичные превращениям, описанным в этом разделе, могут также осуществляться для соответствующих промежуточных соединений.
Все стадии описанного здесь способа могут осуществляться в известных реакционных условиях, предпочтительно в конкретно указанных условиях, без растворителя или обычно в присутствии растворителей или разбавителей, предпочтительно таких, которые являются инертными в отношении используемых реагентов и способными их растворять, без катализатора или в присутствии катализаторов, конденсирующих агентов или нейтрализующих агентов, например, ионообменников, обычно катионообменников, например, в Н+ форме, в зависимости от типа реакции и/или реагентов при пониженной, нормальной или повышенной температуре, например, в области от -100°С приблизительно до 190°С, предпочтительно приблизительно от -80°С приблизительно до 150°С, например, при от -80 до -60°С, при комнатной температуре, при от -20 до 40°С или при температуре кипения используемого растворителя, при атмосферном давлении или в закрытом сосуде, при необходимости под давлением и/или в инертной атмосфере, например, в атмосфере аргона или азота.
Соли могут присутствовать во всех исходных соединениях и промежуточных веществах, если они имеют солеобразующие группы. Соли могут также присутствовать в процессе реакции таких соединений, при условии, что реакция при этом не затрудняется.
На всех стадиях реакции изомерные смеси могут быть разделены на их индивидуальные изомеры, например, диастереомеры или энантиомеры, или на любые смеси изомеров, например, рацематы или диастереомерные смеси.
Данное изобретение также относится к тем формам способа, в которых способ начинается из соединения, полученного на любой стадии в качестве промежуточного соединения, и осуществляются пропущенные стадии, или прекращению способа на любой стадии, или формам исходного сырья в реакционных условиях, или к применению указанного исходного сырья в форме его активированного производного или соли, или соединению, полученному способом по изобретению и способом получения указанного соединения in situ. В предпочтительном варианте осуществления способ начинают с тех исходных реагентов, которые приводят к получению указанных выше предпочтительных соединений, особенно самых предпочтительных, очень предпочтительных и/или предпочтительных из всех.
В предпочтительном варианте осуществления соединение формулы 1 получают в соответствии со способами и стадиями способа, описанными в примерах, или аналогично им.
Соединения формулы 1, включая их соли, также получают в форме гидратов, или их кристаллы могут включать, например, растворитель, используемый для кристаллизации (представленные в виде сольватов).
Настоящее изобретение, кроме того, относится к способу лечения неопластического заболевания, которое связано с ингибированием активности протеинкиназы, который включает введение соединения формулы 1 или его N-оксида или фармацевтически приемлемой соли, где радикалы и символы имеют значения, определенные выше для формулы 1, в количестве, эффективном против указанного заболевания, теплокровному животному, нуждающемуся в таком лечении.
В частности, изобретение относится к способу лечения лейкемии, которая связана с ингибированием активности Raf и/или Abl тирозинкиназы, который включает введение соединения формулы 1 или его N-оксида или фармацевтически приемлемой соли, где радикалы и символы имеют значения, определенные выше для формулы 1, в количестве, эффективном против указанной лейкемии, теплокровному животному, нуждающемуся в таком лечении.
Настоящее изобретение относится также к фармацевтическим композициям, которые включают соединение формулы 1 или его N-оксид в качестве активного ингредиента и которые могут использоваться особенно для лечения заболеваний, упомянутых ранее. Композиции для энтерального введения, такого как назальное, буккальное, ректальное или, особенно, оральное введение, и для парентерального введения, такого как внутривенное, внутримышечного или подкожного введения теплокровному животному, особенно человеку, являются особенно предпочтительными. Композиции включают активный ингредиент отдельно или, предпочтительно, вместе с фармацевтически приемлемым носителем. Дозировка активного ингредиента зависит от излечиваемого заболевания и от типа, возраста, веса и индивидуальных условий, индивидуальных фармакокинетических показателей и от способа введения.
Настоящее изобретение особенно относится к фармацевтическим композициям, которые включают соединение формулы 1, его таутомер, N-оксид или фармацевтически приемлемую соль, или гидрат или сольват, и по крайней мере один фармацевтически приемлемый носитель.
Данное изобретение также относится к фармацевтическим композициям для применения в способе для профилактики или особенно терапевтического лечения человека или животного, к способу ее получения (особенно в форме композиций для лечения опухолей) и к способу лечения опухолевых заболеваний, особенно тех, которые указаны выше.
Данное изобретение относится также к способам и к применению соединений формулы 1 или их N-оксидов для изготовления фармацевтических составов, которые включают соединения формулы 1 или их N-оксиды в качестве активного компонента (активного ингредиента).
В предпочтительном варианте осуществления фармацевтический состав подходит для введения теплокровному животному, особенно человеку или промышленным млекопитающим, страдающим заболеванием, связанным с ингибированием Abl тирозинкиназы, например, хронической миелогенной лейкемией (CML), острой лимфобластомной лейкемией (ALL) и им подобных, и включает эффективное количество соединения формулы 1 или их N-оксидов для ингибирования белка слияния Bcr-Abl, а также ингибирования мутированного белка слияния Bcr-Abl, такого как Е255К, E225V, F317L или М351Т мутированного Bcr-Abl, или его фармацевтически приемлемую соль, если присутствуют солеобразующие группы, вместе с по крайней мере одним фармацевтически приемлемым носителем. В предпочтительном варианте осуществления соединения формулы 1 или их N-оксиды полезны для лечения лейкемий, резистентных к лечению STI571. Соединения формулы 1 или их N-оксиды особенно полезны для подавления резистентности в отношении лечения STI571. Пациенты, страдающие лейкемиями, резистентными к лечению STI571, описаны в многочисленных публикациях, таких как Susan Brandford и др. (Blood. 2002 May 1; 99(9): 3472-5), Christophe Barthe и др. или Andreas Hochhaus и др. (Science. 2001 Sep 21; 293(5538): 2163). Предпочтительно, термин "резистентный" обозначает, что STI571 ингибирует соответствующий домен функциональной Abl киназы с IC50 большей, чем IC50 природного домена Abl киназы человека, т.е. большей приблизительно 0,025 мкМ, предпочтительно большей приблизительно 0,15 мкМ, более предпочтительно большей приблизительно 0,25 мкМ, наиболее предпочтительно большей приблизительно 5 мкМ.
В другом предпочтительном варианте осуществления фармацевтический состав подходит для введения теплокровному животному, особенно людям или промышленным млекопитающим, страдающим заболеванием, связанным с ингибированием Raf киназы, например, острой миелогенной лейкемией или твердыми опухолями, такими как опухоль толстой кишки, легкого или поджелудочной железы, и включает эффективное количество соединения формулы 1 или его N-оксидов для ингибирования Raf киназы, или его фармацевтически приемлемую соль, если присутствует солеобразующие группы, вместе с по крайней мере одним фармацевтически приемлемым носителем.
Фармацевтическая композиция для профилактики или особенно терапевтического лечения неопластических и других пролиферативных заболеваний теплокровного животного, особенно человека или промышленного млекопитающего, нуждающегося в таком лечении, особенно страдающего от такого заболевания, включающая в качестве активного ингредиента в количестве, которое является профилактическим или особенно терапевтически активным против указанных заболеваний, новое соединение формулы 1 или его N-оксиды, является таким образом предпочтительной.
Фармацевтические композиции включают приблизительно от 1% приблизительно до 95% активного ингредиента, формы для единичного введения включают в предпочтительном варианте осуществления приблизительно от 20% приблизительно до 90% активного ингредиента и формы, которые не являются формами для единичного введения, включают в предпочтительном варианте осуществления приблизительно от 5% приблизительно до 20% активного ингредиента. Отдельными дозированными формами являются, например, покрытые и непокрытые таблетки, ампулы, пузырьки, свечи или капсулы. Другими дозированными формами являются, например, масла, кремы, пасты, пены, настойки, помады для губ, гранулы, спреи, дисперсии и т.д. Примерами являются капсулы, содержащие приблизительно от 0,05 г приблизительно до 1,0 г активного ингредиента.
Фармацевтические композиции настоящего изобретения получают известным способом, например, с помощью обычных способов смешения, гранулирования, покрытия, растворения или лиофилизации.
Предпочтительным является применение растворов активного ингредиента, а также суспензий или дисперсий, особенно изотонических водных растворов, дисперсий или суспензий, которые, например, в случае лиофильных композиций, включающих активный ингредиент отдельно или вместе с носителем, например, маннитом, могут быть получены перед применением. Фармацевтические композиции могут быть стерилизованы и/или могут включать эксципиенты, например, консерванты, стабилизаторы, увлажняющие агенты и/или эмульсификаторы, солюбилизаторы, соли для регулирования осмотического давления и/или буферы и получаются известными способами, например, обычными способами растворения и лиофилизации. Указанные растворы или суспензии могут включать агенты, повышающие вязкость, обычно карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу, декстран, поливинилпирролидон или желатины, или также солюбилизаторы, например, Tween 80® [моноолеат полиоксиэтилен(20)сорбита; товарный знак ICI Americas, Inc, USA].
Суспензии в масле включают в качестве масляного компонента растительные, синтетические или полусинтетические масла, обычно используемые в инъекционных целях. В качестве таковых могут быть особо упомянуты жидкие эфиры жирных кислот, которые содержат в качестве кислотного компонента длинноцепочечные жирные кислоты, имеющие от 8 до 22, особенно от 12 до 22 атомов углерода, например, лауриновая кислота, тридецильная кислота, миристиновая кислота, пентадецильная кислота, пальмитиновая кислота, маргариновая кислота, стеариновая кислота, арахидоновая кислота, бегеновая кислота или соответствующие ненасыщенные кислоты, например, олеиновая кислота, элаидиновая кислота, эруковая кислота, бразидиновая кислота или линолевая кислота, при необходимости с дополнительными антиоксидантами, например, витамином Е, β-каротином или 3,5-ди-трет-бутил-4-гидрокситолуолом. Спиртовой компонент этих эфиров жирных кислот имеет максимум 6 атомов углерода и является моновалентным или поливалентным, например, моно-, ди- или тривалентный спирт, например, метанол, этанол, пропанол, бутанол или пентанол или их изомеры, но особенно гликоль и глицерин. В качестве эфиров жирных кислот, следовательно, могут быть упомянуты следующие: этилолеат, изопропилмиристат, изопропилпальмитат, "Labrafil M 2375" (полиоксиэтиленглицерина триолеат от Gattefossé, Paris), "Labrafil M 1944 CS" (ненасыщенные полигликолированные глицериды, полученные алкоголизом масла абрикосовых зернышек и состоящие из глицеридов и полиэтиленгликолевого эфира; Gattefossé, France), "Labrasol" (насыщенные полигликолированные глицериды, полученные алкоголизом ТСМ и состоящие из глицеридов и полиэтиленгликолевого эфира; Gattefossé, France) и/или "Miglyol 812" (триглицериднасыщенных жирных кислот цепи длиной C8-C12 от Hüls AG, Germany), но особенно предпочтительными являются растительные масла, такие как хлопковое масло, миндальное масло, оливковое масло, касторовое масло, кунжутное масло, соевое масло и особенно арахисовое масло.
Изготовление инъекционных составов обычно производится в стерильных условиях, например, наполнением в ампулы или пробирки, и запаиванием емкостей.
Фармацевтические композиции для орального введения могут быть получены, например, объединением активного ингредиента с одним или несколькими твердыми носителями, при необходимости гранулированием полученной смеси и включением в смеси или гранулы, если это желательно или необходимо, дополнительных эксципиентов, в форму таблеток или ядер для таблеток.
Подходящие носители особенно являются наполнителями, такие как сахара, например, лактоза, сахароза, маннит или сорбит, соединения целлюлозы и/или фосфаты кальция, например, трикальцийфосфат или гидрофосфат кальция, а также связующими веществами, такими как крахмалы, например, кукурузный, пшеничный, рисовый или картофельный крахмал, метилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза натрия и/или поливинилпирролидон, и/или, при необходимости, разрыхлителями, такими как указанные выше крахмалы, также карбоксиметилированный крахмал, поперечно сшитый поливинилпирролидон, альгиновая кислота или ее соль, такая как альгинат натрия. Дополнительные эксципиенты особенно являются поточными кондиционерами и лубрикантами, например, кремниевая кислота, тальк, стеариновая кислота или ее соли, такие как стеарат магния или кальция, и/или полиэтиленгликоль или его производные.
Ядра для таблеток могут быть при необходимости покрыты подходящими, необязательно энтеральными покрытиями в процессе применения, inter alia, концентрированными растворами сахаров, которые могут включать гуммиарабик, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, или растворами для покрытий в подходящих органических растворителях или смесях растворителей, или для состава энтеральных покрытий растворами подходящих производных целлюлозы, таких как фталат ацетилцеллюлозы или фталат гидроксипропилметилцеллюлозы. К таблеткам или покрытиям для таблеток могут быть добавлены красители или пигменты, например, в целях идентификации или для указания различных доз активного ингредиента.
Фармацевтические композиции для орального введения также включают твердые капсулы, состоящие из желатина, а также мягкие запаянные капсулы, состоящие из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы могут содержать активный ингредиент в форме гранул, например, в смеси с наполнителями, такими как кукурузный крахмал, связующими веществами и/или глидантами, такими как тальк или стеарат магния, и необязательно стабилизаторами. В мягких капсулах активный ингредиент предпочтительно растворен или суспендирован в подходящих жидких эксципиентах, таких как жирные масла, парафиновое масло или жидкие полиэтиленгликоли или эфиры жирных кислот этилен- или пропиленгликоля, к которым могут быть также добавлены стабилизаторы и детергенты, например, типа эфиров жирных кислот полиоксиэтиленсорбита.
Фармацевтическими композициями, подходящими для ректального введения, являются, например, свечи, которые содержат комбинацию активного ингредиента и основы для свечи. Подходящими основами для свечей являются, например, природные или синтетические триглицериды, парафиновые углеводороды, полиэтиленгликоли или высшие алканолы.
Для парентерального введения особенно подходящими являются водные растворы активного ингредиента в водорастворимой форме, например, водорастворимой соли, или в форме водных инъекционных суспензий, которые содержат вещества, повышающие вязкость, например, карбоксиметилцеллюлозу натрия, сорбит и/или декстран, и при необходимости стабилизаторы. Активный ингредиент, необязательно вместе с эксципиентами, может также присутствовать в форме лиофилизата и могут быть введены в раствор перед парентеральным введением добавлением подходящих растворителей.
Растворы, которые используются, например, для парентерального введения, могут также применяться в качестве инфузионных растворов.
Предпочтительными консервантами являются, например, антиоксиданты, такие как аскорбиновая кислота, или микробициды, такие как сорбиновая кислота или бензойная кислота.
Данное изобретение относится также к способу или методу лечения одного из патологических состояний, указанных выше, особенно заболевания, которое связано с ингибированием тирозинкиназы, особенно соответствующего неопластического заболевания. Соединения формулы 1 или их N-оксиды могут вводиться непосредственно или предпочтительно в форме фармацевтических композиций, профилактически или терапевтически, предпочтительно в количестве, эффективном против указанных заболеваний, теплокровному животному, например, человеку, нуждающемуся в таком лечении. Для пациента с весом тела приблизительно 70 кг ежедневно вводимая доза составляет приблизительно от 0,05 г приблизительно до 5 г, предпочтительно приблизительно от 0,25 г приблизительно до 1,5 г соединения настоящего изобретения.
Настоящее изобретение также особенно относится к применению соединения формулы 1 или его N-оксидов, или его фармацевтически приемлемой соли, особенно соединения формулы 1, которое указано как предпочтительное, или его фармацевтически приемлемой соли, непосредственно или в форме фармацевтического состава по крайней мере с одним фармацевтически приемлемым носителем для лечения и также профилактики одного или нескольких заболеваний, указанных выше, предпочтительно заболевания, которое связано с ингибированием протеинкиназы, особенно неопластического заболевания, более предпочтительно лейкемии, которая связана с ингибированием тирозинкиназы Abl, или опухоли, которая связана с ингибированием Raf киназы.
Предпочтительные количества доз, композиция и состав фармацевтических составов (лекарственных средств), которые должны использоваться в каждом случае, описаны выше.
Новые исходные реагенты и/или промежуточные соединения, а также способы их получения являются также объектами изобретения. В предпочтительном варианте осуществления используются такие исходные реагенты и условия реакции, которые приведут к получению предпочтительных соединений.
Исходные реагенты формулы 2 и 3 являются известными, коммерчески доступными или могут быть получены аналогично или в соответствии со способами, которые являются известными из уровня техники.
Следующие примеры иллюстрируют настоящее изобретение без ограничения его объема.
Примеры
Пример 1
4-Диэтиламино-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-3-(трифторметил)-бензамид
Раствор, содержащий приблизительно 50% пропилфосфонового ангидрида в N,N-диметилформамиде (Fluka, Buchs, Switzerland; 1,14 мл, ~1,8 ммоль) добавляли к перемешиваемой смеси 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамина (277,3 мг, 1 ммоль), 4-диэтиламино-3-(трифторметил)-бензойной кислоты (261,3 мг, 1 ммоль) и триэтиламина (1,33 мл, 9,6 ммоль) в 3 мл N,N-диметилформамида. После перемешивания в течение 24 часов при комнатной температуре смесь обрабатывали полунасыщенным водным раствором гидрокарбоната натрия и экстрагировали три раза этилацетатом. Объединенные органические экстракты сушили (Na2SO4) и растворитель упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле, элюент дихлорметан/метанол. Чистые фракции объединяли, упаривали и остаток перекристаллизовывали из ацетона с получением указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 0,96 (t, 6H); 2,23 (s, 3Н); 3,02 (q, 4H); 7,23 (d, 1H); 7,44 (d, 1H); 7,48 (dd, 1H); 7,51-7,54 (m, 1H); 7,66 (d, 1H); 8,06 (d, 1H); 8,21 (dd, 1H); 8,24 (m, 1H); 8,48 (dt, 1H); 8,52 (d, 1H); 8,68 (dd, 1H); 9,0 (s, 1H); 9,28 (d, 1H); 10,34 (s, 1H).
Пример 1,1: 4-Диэтиламино-3-(трифторметил)-бензонитрил
Смесь 4-бром-3-(трифторметил)-бензонитрила (Yonezawa и др., Synthetic Communications (1996), 26, 1575-1578; 6,0 г, 24 ммоль), диэтиламина (8,3 мл, 80 ммоль) и 25 мл N,N-диметилацетамида перемешивали в водонепроницаемом закрытом сосуде в течение 16 часов при 135°С. После охлаждения реакционную смесь обрабатывали полунасыщенным водным раствором гидрокарбоната натрия и экстрагировали три раза этилацетатом. Объединенные органические экстракты сушили (Na2SO4) и растворитель упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле, элюент гексан/этилацетат с получением указанного в заголовке соединения в виде оранжевого масла.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 096 (t, 6Н); 3,08 (q, 4H); 7,61 (d, 1H); 8,04 (dd, 1H); 8,16 (d, 1H).
Пример 1,2: 4-Диэтиламино-3-(трифторметил)-бензойная кислота
Смесь 4-диэтиламино-3-(трифторметил)-бензонитрила (1,21 г, 5 ммоль), 12 мл уксусной кислоты и 8 мл дымящейся соляной кислоты (37%) перемешивали в течение 20 часов при 95°С. После охлаждения реакционную смесь упаривали досуха при пониженном давлении. Твердый остаток растворяли в теплом полунасыщенном водном растворе карбоната натрия и рН доводили до ~5-6 добавлением по каплям 2 М соляной кислоты. Полученный осадок отфильтровывали, промывали водой и сушили в вакууме с получением белого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 0,94 (t, 6Н); 3,02 (q, 4H); 7,58 (d, 1H); 8,11-8,16 (m, 2H); 13,35 (br., 1H).
Пример 2
N-[4-Метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-4-(1-пирролидинил)-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогично способу, описанному в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-(1-пирролидинил)-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 1,91-1,96 (m, 4H); 2,22 (s, 3H); 3,38-3,46 (m, 4H); 7,06 (d, 1H); 7,20 (d, 1H); 7,43 (d, 1H); 7,48 (dd, 1H); 7,50-7,54 (m, 1H); 8,05-8,07 (m, 2Н); 8,24 (d, 1H); 8,48 (dt, 1H); 8,51 (d, 1H); 8,68 (dd, 1H); 8,97 (s, 1H); 9,28 (m, 1H); 10,08 (s, 1H).
Пример 2,1: 4-(1-Пирролидинил)-3-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 4-бром-3-(трифторметил)-бензонитрил и пирролидин (Fluka, Buchs, Switzerland), при температуре реакции 95°С.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 1,90-1,96 (m, 4H); 3,39-3,47 (m, 4H); 7,03 (d, 1H); 7,75 (dd, 1H); 7,99 (d, 1H).
Пример 2,2: 4-(1-Пирролидинил)-3-(трифторметил)-бензойная кислота
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,2, используя 4-(1-пирролидинил)-3-(трифторметил)-бензонитрил. Сырой продукт перекристаллизовывали из смеси метиленхлорид/метанол.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 1,90-1,97 (m, 4H); 3,38-3,45 (m, 4H); 7,01 (d, 1H); 7,90 (dd, 1H); 8,10 (d, 1H); 12,65 (br., 1H).
Пример 3
N-[4-Метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-4-(4-морфолинил)-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-(4-морфолинил)-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1Н-ЯМР (400 МГц, ДМСО-d6, δ).: 2,23 (s, 3Н); 2,96 (m, 4H); 3,74 (m, 4H); 7,23 (d, 1H); 7,44 (d, 1H); 7,48 (dd, 1H); 7,52 (ddd, 1H); 7,66 (d, 1H); 8,07 (d, 1H); 8,23-8,25 (m, 2H); 8,48 (dt, 1H); 8,52 (d, 1H); 8,69 (dd, 1H); 8,99 (s, 1H); 9,28 (m, 1H); 10,34 (s, 1H).
Пример 3,1: 4-(4-Морфолинил)-3-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 4-бром-3-(трифторметил)-бензонитрил и морфолин (Fluka, Buchs, Switzerland), при температуре реакции 95°С.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 3,00 (m, 4H); 3,72 (m, 4H); 7,60 (d, 1H); 8,09 (dd, 1H); 8,19 (d, 1H).
Пример 3,2: 4-(4-Морфолинил)-3-(трифторметил)-бензойная кислота
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,2, используя 4-(4-морфолинил)-3-(трифторметил)-бензонитрил.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,92-3,01 (m, 4H); 3,68-3,76 (m, 4H); 7,58 (d, 1H); 8,12-8,19 (m, 2H); 13,25 (br., 1H).
Пример 4
N-[4-Метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-4-(1-пиперидинил)-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-(1-пиперидинил)-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 1,51-1,70 (m, 6H); 2,23 (s, 3H); 2,89-2,95 (m, 4H); 7,22 (d, 1H); 7,44 (d, 1H); 7,48 (dd, 1H); 7,52 (ddd, 1H); 7,57 (d,1H); 8,06 (d, 1H); 8,18-8,23 (m, 2H); 8,48 (dt, 1H); 8,51 (d, 1H); 8,68 (dd, 1H); 8,99 (s, 1H); 9,28 (d, 1H); 10,30 (s, 1H).
Пример 4,1: 4-(1-Пиперидинил)-3-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 4-бром-3-(трифторметил)-бензонитрил и пиперидин (Fluka, Buchs, Switzerland), при температуре реакции 95°С.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 1,51-1,59 (m, 2H); 1,59-1,68 (m, 4H); 2,93-3,00 (m, 4H); 7,51 (d, 1H); 8,03 (dd, 1H); 8,14 (d, 1H).
Пример 4,2: 4-(1-Пиперидинил)-3-(трифторметил)-бензойная кислота
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,2, используя 4-(1-пиперидинил)-3-(трифторметил)-бензонитрил.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 1,51-1,59 (m, 2H); 1,59-1,69 (m, 4H); 2,89-2,97 (m, 4H); 7,49 (m, 1H); 8,10-8,15 (m, 2H); 13,19 (br., 1H).
Пример 5
4-(4-Метил-1-пиперазинил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил1амино]фенил]-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-(4-метил-1-пиперазинил)-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,23 (s, 3Н); 2,39-2,48 (br.s, 3H); 2,63-2,85 (br., 4H); 3,00-3,09 (br.m, 4H); 7,23 (d, 1H); 7,44 (d, 1H); 7,49 (dd, 1H); 7,52 (ddd, 1H); 7,64 (d, 1H); 8,07 (d, 1H); 8,23-8,25 (m, 2H); 8,48 (dt, 1H); 8,52 (d, 1H); 8,69 (dd, 1H); 9,0 (s, 1H); 9,28 (m, 1H); 10,35 (s, 1H)
Пример 5,1: 4-(4-Метил-1-пиперазинил)-3-(трифторметил)-бензойная кислота
Смесь 4-бром-3-(трифторметил)-бензонитрила (Yonezawa и др.. Synthetic Communications (1996) 26, 1575-8; 2,47 г, 12 ммоль), 1-метилпиперазина (Fluka, Buchs, Switzerland, 5,33 мл, 48 ммоль) и 15 мл N,N-диметилацетамида перемешивали в водонепроницаемом закрытом сосуде в течение 14 часов при 95°С. После охлаждения реакционную смесь упаривали досуха при пониженном давлении и остаток обрабатывали полунасыщенным водным раствором карбоната натрия и экстрагировали этилацетатом. Объединенные экстракты сушили (Na2SO4) и растворитель упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле, элюент метиленхлорид/метанол с получением 4-(4-метил-1-пиперазинил)-3-(трифторметил)-бензонитрила в виде светло-желтого масла.
К 4-(4-метил-1-пиперазинил)-3-(трифторметил)-бензонитрилу добавляли смесь, состоящую из 30 мл диоксана, 15 мл воды и 11,25 мл 2 М водного раствора гидроксида натрия и реакционную смесь перемешивали в течение 16 часов при 95°С. После охлаждения смесь упаривали. Полученный остаток обрабатывали водой, рН доводили до ~5-6 с помощью 1 М соляной кислоты и растворитель упаривали при пониженном давлении. Остаток обрабатывали горячим метанолом, нерастворившуюся соль отфильтровывали и фильтрат упаривали с получением сырого указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,28 (s, 3Н); 2,50-2,58 (m, 4H); 2,94-3,02 (m, 4H); 7,52 (m, 1H); 8,11-8,17 (m, 2H); 13,19 (br., 1H).
Пример 6
4-(1Н-Имидазол-1-ил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил1-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-(1Н-имидазол-1-ил)-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,25 (s, 3Н); 7,12-7,15 (m, 1H); 7,26 (d, 1H); 7,43-7,55 (m, 4H); 7,78 (d, 1H); 7,91 (s, 1H); 8,12 (br. 1H); 8,38-8,42 (m, 1H); 8,46-8,54 (m, 3H); 8,67-8,70 (m, 1H); 9,01 (s, 1H); 9,27-9,30 (m, 1H); 10,57 (br.s, 1H).
Пример 6,1: 4-(1Н-Имидазол-1-ил)-3-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 4-хлор-3-(трифторметил)-бензонитрил (Lancaster Syhthesis, GmbH) и имидазол (Fluka, Buchs, Switzerland), при температуре реакции 110°С.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 7,13 (m, 1H); 7,47 (s, 1H); 7,85 (d, 1H); 7,91 (s, 1H); 8,37 (dd, 1H); 8,57 (m, 1H).
Пример 6,2: 4-(1Н-Имидазол-1-ил)-3-(трифторметил)-бензойная кислота
Смесь 4-(1Н-имидазол-1-ил)-3-(трифторметил)-бензонитрила (1,99 г, 8,4 ммоль), 12 мл уксусной кислоты и 6 мл 12 М соляной кислоты (37%) перемешивали в течение 16 часов при 95°С. После охлаждения реакционную смесь упаривали при пониженном давлении. Полученный остаток растворяли в воде и рН доводили до ~5-6 добавлением по каплям 1 М раствора гидроксида натрия. Осадок отфильтровывали, промывали водой и сушили в вакууме с получением указанного в заголовке соединения в виде твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 7,13 (s, 1H); 7,47 (s, 1H); 7,75 (d, 1H); 7,91 (s, 1H); 8,31-8,39 (m, 2H); 13,84 (br., 1H).
Пример 7
4-(2-Метил-1Н-имидазол-1-ил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-(2-метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,09 (s, 3Н); 2,26 (s, 3Н); 6,96 (d, 1H); 7,24-7,28 (m, 2H); 7,45 (d, 1H); 7,50-7,55 (m, 2H); 7,78 (d, 1H); 8,12 (d, 1H); 8,40 (m, 1H); 8,46-8,51 (m, 2H); 8,53 (d, 1H); 8,69 (dd, 1H); 9,03 (s, 1H); 9,30 (d, 1H); 10,59 (s, 1H).
Пример 7,1: 4-(2-Метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 4-хлор-3-(трифторметил)-бензонитрил (Lancaster Synthesis GmbH) и 2-метил-имидазол (Fluka, Buchs, Switzerland), при температуре реакции 145°С в течение 38 часов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,06 (s, 3Н); 6,95 (m, 1H); 7,25 (m, 1H); 7,86 (d, 1H); 8,39 (dd, 1H); 8,58 (m, 1H).
Пример 7,2: 4-(2-Метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензойная кислота
Смесь 4-(2-метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензонитрила (1,01 г, 4 ммоль), 6 мл уксусной кислоты и 3 мл 12 М соляной кислоты (37%) перемешивали в течение 16 часов при 95°С. После охлаждения реакционную смесь упаривали досуха при пониженном давлении. Полученный остаток упаривали дважды с толуолом, растворяли в воде и рН доводили до ~5-6 добавлением по каплям 1 М раствора гидроксида натрия. Водную фазу экстрагировали дважды н-бутанолом и органическую фазу упаривали с получением указанного в заголовке соединения в виде бежевого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,06 (s, 3Н); 6,98 (d, 1H); 7,28 (br., 1H); 7,75 (m, 1H); 8,34-8,38 (m, 2H).
Пример 8
4-(4-Метил-1Н-имидазол-1-ил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,19 (s, 3Н); 2,25 (s, 3Н); 7,16 (s, 1H); 7,26 (d, 1H); 7,45 (d, 1H); 7,49-7,56 (m, 2H); 7,72-7,77 (m, 2H); 8,12 (br, 1H); 8,38 (br.d, 1H); 8,45-8,51 (m, 2H); 8,53 (d, 1H); 8,69 (dd, 1H); 9,01 (s, 1H); 9,29 (m, 1H); 10,55 (s, 1H).
Пример 8,1: 4-(4-Метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 4-хлор-3-(трифторметил)бензонитрил (Lancaster Synthesis GmbH) и 4(5)-метил-имидазол (Fluka, Buchs, Switzerland), при температуре реакции 145°С в течение 14 часов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,17 (s, 3Н); 7,16 (br.s, 1H); 7,76 (br.s, 1H); 7,81 (d, 1H); 8,34 (dd, 1H); 8,53-8,57 (m, 1H).
Пример 8,2: 4-(4-Метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензойная кислота
Смесь 4-(4-метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензонитрила (1,01 г, 4 ммоль), 6 мл уксусной кислоты и 3 мл 12 М соляной кислоты (37%) перемешивали в течение 16 часов при 95°С. После охлаждения реакционную смесь упаривали досуха при пониженном давлении. Полученный остаток упаривали дважды с толуолом, растворяли в воде и рН доводили до ~5-6 добавлением по каплям 1 М раствора гидроксида натрия. Водную фазу экстрагировали дважды этилацетатом. Органическую фазу сушили (Na2SO4) и упаривали с получением указанного в заголовке соединения в виде светло-желтого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,18 (s, 3Н); 7,16 (br.s, 1H); 7,69-7,77 (m, 2H); 8,30-8,37 (m, 2H).
Пример 9
4-(2,4-Диметил-1Н-имидазол-1-ил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-(2,4-диметил-1Н-имидазол-1-ил)-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,03 (s, 3Н); 2,11 (s, 3Н); 2,25 (s, 3Н); 6,94 (s, 1H); 7,26 (d, 1H); 7,45 (d, 1H); 7,49-7,55 (m, 2H); 7,74 (d, 1H); 8,11 (d, 1H); 8,38 (dd, 1H); 8,45 (d, 1H); 8,49 (dt, 1H); 8,53 (d, 1H); 8,69 (dd, 1H); 9,02 (s, 1H); 9,29 (d, 1H); 10,57 (s, 1H).
Пример 9,1: 4-(2,4-Диметил-1Н-имидазол-1-ил)-3-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 4-хлор-3-(трифторметил)-бензонитрил (Lancaster Synthesis GmbH) и 2,4-диметил-имидазол (Trans World Chemicals), при температуре реакции 145°С в течение 20 часов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,01 (s, 3H); 2,09 (s, 3H); 6,93 (s, 1H); 7,81 (d, 1H); 8,36 (dd, 1H); 8,54 (d, 1H).
Пример 9,2: 4-(2,4-Диметил-1Н-имидазол-1-ил)-3-(трифторметил)-бензойная кислота
К 4-(2,4-метил-1Н-имидазол-1-ил)-3-(трифторметил)-бензонитрилу (0,65 г, 2,45 ммоль) добавляли смесь, состоящую из 11 мл диоксана, 5,5 мл воды и 4,9 мл 2 М водного раствора гидроксида натрия, и реакционную смесь перемешивали в течение 16 часов при 95°С. После охлаждения смесь упаривали досуха при пониженном давлении. Полученный остаток обрабатывали водой, рН доводили до ~5-6 с помощью 2 М соляной кислоты и водную фазу экстрагировали дважды н-бутанолом. Объединенные органические экстракты упаривали с получением указанного в заголовке соединения в виде твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,14 (s, 3H); 2,18 (s, 3H); 7,18 (br.s, 1H); 7,81 (d, 1H); 8,31-8,44 (m,2H).
Пример 10
3-(1Н-Имидазол-1-ил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-5-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 3-(1Н-имидазол-1-ил)-5-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,26 (s, 3H); 7,19 (s, 1H); 7,27 (d, 1H); 7,45 (d, 1H); 7,49-7,56 (m, 2H); 8,02 (br, 1H); 8,11 (br.s, 1H); 8,21 (s, 1H); 8,30 (s, 1H); 8,45-8,54 (m, 4Н); 8,69 (dd, 1H); 9,01 (s, 1H); 9,30 (m, 1H); 10,50 (br.s, 1H).
Пример 10,1: 3-(1Н-имидазол-1-ил)-5-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 3-фтор-5-(трифторметил)-бензонитрил (Lancaster Synthesis GmbH) и имидазол (Fluka, Buchs, Switzerland), при температуре реакции 110°С в течение 24 часов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 7,17 (s, 1H); 8,03 (m, 1H); 8,32 (s, 1H); 8,46 (br.s, 1H); 8,54 (d, 1H); 8,62 (m, 1H).
Пример 10,2: 3-(1Н-имидазол-1-ил)-5-(трифторметил)-бензойная кислота
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 6,2, используя 3-(1Н-имидазол-1-ил)-5-(трифторметил)-бензонитрил.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 7,17 (s, 1H); 8,03 (s, 1H); 8,12 (s, 1H); 8,35 (s, 1H); 8,41 (s, 1H); 8,53 (s, 1H); 13,90 (br., 1H).
Пример 11
3-(2-Метил-1Н-имидазол-1-ил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-5-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 3-(2-метил-1Н-имидазол-1-ил)-5-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,25 (s, 3Н); 2,37 (s, 3H); 6,99 (d, 1H); 7,26 (d, 1H); 7,45 (d, 1H); 7,49-7,54 (m, 3H); 8,10-8,15 (m, 2H); 8,35 (m, 2H); 8,48 (dt, 1H); 8,53 (d, 1H); 8,68 (dd, 1H); 9,01 (s, 1H); 9,29 (m, 1H); 10,49 (s, 1H).
Пример 11,1: 3-(2-Метил-1Н-имидазол-1-ил)-5-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 3-фтор-5-(трифторметил)-бензонитрил (Lancaster Synthesis GmbH) и 2-метил-имидазол (Fluka, Buchs, Switzerland), при температуре реакции 145°С в течение 24 часов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,36 (s, 3H); 6,97 (d, 1H); 7,48 (d, 1H); 8,26 (br.s, 1H); 8,41 (m, 1H); 8,46 (br.s, 1H).
Пример 11,2: 3-(2-Метил-1Н-имидазол-1-ил)-5-(трифторметил)-бензойная кислота
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 9,2, используя 3-(2-метил-1Н-имидазол-1-ил)-5-(трифторметил)-бензонитрил.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,33 (s, 3Н); 6,97 (d, 1H); 7,48 (d, 1H); 8,10 (br., 1H); 8,15 (br., 1H); 8,22 (br., 1H).
Пример 12
3-(4-Метил-1Н-имидазол-1-ил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-5-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 3-(4-метил-1Н-имидазол-1-ил)-5-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,20 (s, 3Н); 2,26 (s, 3Н); 7,27 (d, 1H); 7,45 (d, 1H); 7,49-7,56 (m, 2H); 7,72 (s, 1H); 8,12 (br., 1H); 8,18 (s, 1H); 8,25 (s, 1H); 8,39-8,55 (m, 4Н); 8,69 (m, 1H); 9,01 (s, 1H); 9,31 (m, 1H); 10,48 (s, 1H).
Пример 12,1: 3-(4-Метил-1Н-имидазол-1-ил)-5-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 3-фтор-5-(трифторметил)-бензонитрил (Lancaster Sinthesis GmbH) и 4(5)-метил-имидазол (Fluka, Buchs, Switzerland), при температуре реакции 145°С в течение 24 часов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,18 (s, 3Н); 7,74 (m, 1H); 8,27 (br.s, 1H); 8,39 (br.s, 1H); 8,43 (d, 1H); 8,56 (br.s, 1H).
Пример 12,2: 3-(4-Метил-1Н-имидазол-1-ил)-5-(трифторметил)-бензойная кислота
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 9,2, используя 3-(4-метил-1Н-имидазол-1-ил)-5-(трифторметил)-бензонитрил.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 2,27 (s, 3Н); 8,00 (s, 1H); 8,18 (s, 1H); 8,40 (m); 8,47 (br., 1H).
Пример 13
N-[4-Метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-3-(4-морфолинил)-5-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 3-(4-морфолинил)-5-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 2,24 (s, 3Н); 3,28-3,32 (m, 4H); 3,75-3,79 (m, 4H); 7,23 (d, 1H); 7,39 (br., 1H); 7,44 (d, 1H); 7,48 (dd, 1H); 7,51 (ddd, 1H); 7,65 (br., 1H); 7,73 (br., 1H); 8,07 (d, 1H); 8,47 (dt, 1H); 8,52 (d, 1H); 8,68 (dd, 1H); 8,98 (s, 1H); 9,29 (m, 1H); 10,32 (s, 1H).
Пример 13,1: 3-(4-Морфолинил)-5-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 3-фтор-5-(трифторметил)-бензонитрил (Lancaster Synthesis GmbH) и морфолин (Fluka, Buchs, Switzerland), при температуре реакции 105°С в течение 14 часов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 3,25-3,35 (m, 4H); 3,69-3,77 (m, 4H); 7,49 (br.s, 1H); 7,56 (br.s, 1H); 7,66 (br.s, 1H).
Пример 13,2: 3-(4-Морфолинил)-5-(трифторметил)-бензойная кислота
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 7,2, используя 3-(4-морфолинил)-5-(трифторметил)-бензонитрил.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 3,20-3,28 (m, 4H); 3,69-3,77 (m, 4H); 7,21 (br.s, 1H); 7,33 (br.s, 1H); 7,43 (br.s, 1H).
Пример 14
3-(4-Метил-1-пиперазинил)-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-5-(трифторметил)-бензамид.
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 3-(4-метил-1-пиперазинил)-5-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,24 (s, 6H); 2,46-2,50 (m, 4H); 3,30-3,36 (m, 4H); 7,24 (d, 1H); 7,37 (br.s, 1H); 7,44 (d, 1H); 7,49 (dd, 1H); 7,52 (dd, 1H); 7,62 (br.s, 1H); 7,72 (br.s, 1H); 8,08 (d, 1H); 8,47 (dt, 1H); 8,52 (d, 1H); 8,70 (dd, 1H); 8,99 (s, 1H); 9,30 (d, 1H); 10,31 (s, 1H).
Пример 14,1: 3-(4-Метил-1-пиперазинил)-5-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 3-фтор-5-(трифторметил)-бензонитрил (Lancaster Synthesis GmbH) и 1-метилпиперазин (Fluka, Buchs, Switzerland).
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,22 (s, 3Н); 2,41-2,46 (m, 4H); 3,31-3,37 (m, 4H); 7,48 (br.s, 1H); 7,52 (br.s, 1H); 7,65 (br.s, 1H).
Пример 14,2: 3-(4-Метил-1-пиперазинил)-5-(трифторметил)-бензойная кислота
К 3-(4-метил-1-пиперазинил)-5-(трифторметил)-бензонитрилу (2,69 г, 10 ммоль) добавляли смесь, состоящую из 50 мл диоксана, 25 мл воды и 18,75 мл 2 М водного раствора гидроксида натрия, и реакционную смесь перемешивали в течение 16 часов при 95°С. После охлаждения смесь упаривали досуха при пониженном давлении. Полученный остаток обрабатывали водой, рН доводили до ~5-6 с помощью 2 М соляной кислоты. Осадок отфильтровывали и фильтрат экстрагировали дважды н-бутанолом. Объединенные органические экстракты упаривали с получением указанного в заголовке соединения в виде твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,41 (s, 3Н); 2,69-2,76 (m, 4H); 3,37-3,42 (m, 4H); 7,45 (br.s, 1H); 7,55 (br.s, 1H); 7,70 (br.s, 1H).
Пример 15
4-[[2-(Диметиламино)этил]метиламино]-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-[[2-(диметиламино)этил]метиламино]-3-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,10 (s, 6H); 2,23 (s, 3Н); 2,35 (m, 2H); 2,78 (s, 3Н); 3,14 (m, 2H); 7,22 (d, 1H); 7,43 (d, 1H); 7,48 (dd, 1H); 7,51 (ddd, 1H); 7,59 (d, 1H); 8,07 (d, 1H); 8,16-8,23 (m, 2H); 8,48 (dt, 1H); 8,51 (d, 1H); 8,68 (dd, 1H); 8,99 (s, 1H); 9,28 (m, 1H); 10,28 (s, 1H).
Пример 15,1: 4-[[2-(Диметиламино)этил]метиламино]-3-(трифторметил)-бензонитрил
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1,1, используя 4-хлор-3-(трифторметил)-бензонитрил (Lancaster Synthesis GmbH) и N,N,N'-триметил-1,2-этандиамин (Fluka, Buchs, Switzerland).
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,09 (s, 6H); 2,38 (t, 2H); 2,86 (s, 3H); 3,24 (t, 2H); 7,45 (d, 1H); 7,94 (dd, 1H); 8,09 (d, 1H).
Пример 15.2: 4-[[2-(Диметиламино)этил]метиламино]-3-(трифторметил)-бензойная кислота
К 4-[[2-(диметиламино)этил]метиламино]-3-(трифторметил)-бензонитрилу (1,35 г, 5 ммоль) добавляли смесь, состоящую из 25 мл диоксана, 12,5 мл воды и 9,4 мл 2 М водного раствора гидроксида натрия и реакционную смесь перемешивали в течение 16 часов при 95°С. После охлаждения смесь упаривали досуха при пониженном давлении. Полученный остаток обрабатывали водой, рН доводили до ~5 с помощью 1 М соляной кислоты и смесь упаривали досуха при пониженном давлении. Остаток твердого вещества обрабатывали метанолом, суспензию отфильтровывали и фильтрат упаривали с получением указанного в заголовке соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,57 (s, 6H); 2,76 (s, 3H); 2,96 (m, 2H); 3,38 (m, 2H); 7,62 (d, 1H); 8,11-8,16 (m, 2H).
Пример 16
4-[Метил-(1-метил-4-пиперидинил)амино]-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-3-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 4-[метил(1-метил-4-пиперидинил)амино]-5-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 1,46-1,57 (m, 2H); 1,62-1,68 (m, 2H); 1,79-1,88 (m, 2H); 2,13 (s, 3H); 2,23 (s, 3H); 2,64 (s, 3H); 2,73-2,80 (m, 2H); 2,87-2,97 (m, 1H); 7,22 (d, 1H); 7,43 (d, 1H); 7,48 (dd, 1H); 7,51 (ddd, 1H); 7,66 (d, 1H); 8,06 (d, 1H); 8,17-8,24 (m, 2H); 8,48 (dt, 1H); 8,51 (d, 1H); 8,68 (dd, 1H); 8,99 (s, 1H); 9,28 (m, 1H); 10,32 (s, 1H)
Пример 16,1: 4-[Метил-(1-метил-4-пиперидинил)амино]-3-(трифторметил)-бензойная кислота
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 5,1, используя 4-хлор-3-(трифторметил)-бензонитрил (Lancaster Synthesis GmbH) и 1-метил-4-(метиламино)-пиперидин (Aldrich, Buchs, Switzerland). Последующий гидролиз нитрила осуществляли с помощью гидроксида натрия в смеси диоксана и воды как описано в примере 5,1.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 1,77-1,86 (m, 4Н); 2,54 (s, 3H); 2,63 (s, 3Н); 2,65-2,74 (m); 3,13-3,23 (m); 7,63 (d, 1H); 8,12-8,17 (m, 2H).
Пример 17
3-Этиламино-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-5-(трифторметил)-бензамид
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 1, используя 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамин и 3-этиламино-5-(трифторметил)-бензойную кислоту в качестве исходных реагентов.
1H-ЯМР (400 МГц, ДМСО-d6, δ): 1,19 (t, 3Н); 2,23 (s, 3Н); 3,14 (m, 2H); 6,36 (t, 1H); 6,98 (br.s, 1H); 7,22 (d, 1H); 7,32 (br.s, 1H); 7,37 (br.s, 1H); 7,43 (d, 1H); 7,48 (dd, 1H); 7,51 (dd, 1H); 8,06 (d, 1H); 8,48 (dt, 1H); 8,51 (d, 1H); 8,68 (dd, 1H); 9,00 (s, 1H); 9,28 (m, 1H); 10,25 (s, 1H).
Пример 17,1: Метиловый эфир 3-этиламино-5-(трифторметил)-бензойной кислоты
Смесь метилового эфира 3-амино-5-(трифторметил)-бензойной кислоты (J. Med. Chem. (1969) 12, 299-303; 4,23 г, 19,3 ммоль), карбоната калия (8,0 г, 57,9 ммоль) и йодэтана (3,12 мл, 38,6 ммоль) в 20 мл N,N-диметилформамида перемешивали при 65°С в течение 14 часов в водонепроницаемом закрытом сосуде. После охлаждения реакционную смесь отфильтровывали и фильтрат упаривали досуха при пониженном давлении. Остаток обрабатывали водой и экстрагировали три раза этилацетатом. Объединенные экстракты сушили (Na2SO4) и растворитель упаривали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, элюент гексан/метиленхлорид (1:1).
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 1,18 (t, 3Н); 3,10 (m, 2H); 3,85 (s, 3H); 6,46 (t, 1H); 7,02 (br. 1H); 7,29 (br.s, 1H); 7,37 (br., 1H).
Пример 17,2: 3-Этиламино-5-(трифторметил)-бензойная кислота
Смесь метилового эфира 3-этиламино-5-(трифторметил)-бензойной кислоты (1,38 г, 5,6 ммоль), 5,5 мл 1 М водного раствора гидроксида натрия в 12 мл этанола перемешивали в течение 4 часов при 70°С. После охлаждения смесь упаривали досуха при пониженном давлении. Полученный остаток растворяли в воде, рН доводили до 5 с помощью 1 М соляной кислоты. Осадок отфильтровывали, промывали водой и сушили в вакууме с получением указанного в заголовке соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 1,18 (t, 3H); 3,10 (m, 2Н); 6,39 (m, 1H); 6,99 (br.s, 1H); 7,29 (br.s, 1H); 7,36 (br.s, 1H); 13,15 (br., 1H).
Пример 18
3-Ацетиламино-N-[4-метил-3-[[4-(3-пиридинил)-2-пиримидинил]амино]фенил]-5-(трифторметил)-бензамид
Диэтилцианофосфонат (Aldrich, Buchs, Switzerland; 0,66 мл, 4,0 ммоль) добавляли к перемешиваемой смеси 4-метил-N-[4-(3-пиридинил)-2-пиримидинил]-1,3-бензолдиамина (554 мг, 2,0 ммоль), 3-ацетиламино-5-(трифторметил)-бензойной кислоты (495 мг, 2,0 ммоль) и триэтиламина (1,12 мг, 8,0 ммоль) в 10 мл N,N-диметилформамида при 20°С в атмосфере аргона. После перемешивания в течение 18 часов при 20°С смесь обрабатывали насыщенным водным раствором гидрокарбоната натрия и экстрагировали дважды этилацетатом. Объединенные экстракты сушили (MgSO4), отфильтровывали и растворитель упаривали при пониженном давлении с получением сырого продукта. Сырой продукт очищали колоночной хроматографией на силикагеле, элюент дихлорметан/метанол/водный аммиак. Чистые фракции объединяли, растворитель упаривали при пониженном давлении и остаток перекристаллизовывали из смеси этилацетат/гексан с получением указанного в заголовке соединения в виде кремового кристаллического твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 2,10 (s, 3H); 2,23 (s, 3H); 7,22 (dd, 1H); 7,43 (dd, 1H); 7,45-7,50 (m, 1H); 7,51-7,54 (m, 1H); 7,97 (d, 1H); 8,04 (d, 1H); 8,24 (dd, 1H); 8,28 (m, 1H); 8,49 (dt, 1H); 8,50 (dd, 1H); 8,68 (dd, 1H); 8,99 (s, 1H); 9,25 (d, 1H); 10,43 (dd, 1H).
Пример 18,1: 3-Ацетиламино-5-(трифторметил)-бензойная кислота
Смесь 3-нитро-5-(трифторметил)бензойной кислоты (5,10 г, 20 ммоль) и уксусного ангидрида (2,1 мл, 22 ммоль) в 50 мл пиридина перемешивали при 22°С в течение 14 часов. Смесь затем упаривали досуха при пониженном давлении с получением остатка, который обрабатывали 2 М соляной кислотой и экстрагировали три раза этилацетатом. Объединенные экстракты промывали водой, сушили (MgSO4) и растворитель упаривали при пониженном давлении с получением сырого продукта, который очищали перекристаллизацией из смеси этилацетат/гексан с получением указанного в заголовке соединения в виде бежевого кристаллического твердого вещества, tпл 194-220°С.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 7,80 (d, 1H); 8,27 (d, 1H); 8,35 (d, 1H); 10,46 (s, 1H); 13,50 (br.s, 1H).
Пример 18,2: 3-Амино-5-(трифторметил)-бензойная кислота
Раствор 3-нитро-5-(трифторметил)бензойной кислоты (Lancaster Synthesis GmbH; 11,75 г, 50 ммоль) в этаноле (300 мл) гидрировали при атмосферном давлении над никелем Ренея (1 г) при 40°С. Рассчитанное количество водорода подавали через 8 часов. Смесь затем отфильтровывали и растворитель упаривали при пониженном давлении с получением сырого продукта, который очищали перекристаллизацией из смеси диэтиловый эфир/гексан с получением указанного в заголовке соединения в виде бежевого кристаллического твердого вещества, tпл 134-139°С.
1H-ЯМР (400 МГц, ДМСО-d6, δ).: 5,86 (br.s, 2H); 7,02 (d, 1H); 7,24 (d, 1H); 7,38 (d, 1H); 13,11 (br.s, 1H).
Пример 19
Мягкие капсулы
5000 мягких желатиновых капсул, каждая из которых включает в качестве активного ингредиента 0,05 г одного из соединений формулы 1, указанного в предшествующих примерах, получали следующим образом:
250 г измельченного активного ингредиента суспендировали в 2 л Lawoglykol® (пропиленгликольлаурат, Gattefossé S.A., Saint Priest, France) и загружали во влажный пульверизатор для получения частиц размером приблизительно от 1 до 3 мкм. Порции 0,419 г смеси затем вводили в мягкие желатиновые капсулы с помощью устройства для наполнения капсул.
Пример 20
Фармакокинетические данные
Соединение формулы 1 тестировали в составе для введения самкам OF1 мышей от IFACREDO, France, сначала растворением N-метилпирролидона (NMP), а затем разбавлением PEG300 до конечной концентрации 10% об/об NMP: 90% об/об PEG300, получая чистый раствор соединения. Концентрации доводили до постоянного объема 10 мл/кг веса тела. Соединение получали непосредственно перед применением. Составы соединения вводили перорально через катетер дозировками 50 мг/кг. В определенное время (4 в каждый момент времени) анестезировали 3% изофлураном в медицинском кислороде и получали образцы крови сердечной пункцией в гепаринизированные пробирки (приблизительно 30 IU/мл). Животных последовательно умерщвляли до окончания действия анестетика. Из крови получали плазму центрифугированием (10,000 г, 5 мин) и анализировали немедленно или хранили замороженной при -70°С.
В образцы плазмы (10-250 мкл), например, добавляли 5 мкл внутреннего стандарта, смешивали с 200 мкл 0,1 М NaOH и 500 мкл хлороформа в пробирке Эппендорфа на 1,5 мл и тщательно перемешивали 10 минут на смесителе Эппендорфа. Затем смесь центрифугировали (3 мин при 10'000 об/мин), органическую фазу переносили во вторую пробирку Эппендорфа и упаривали досуха на вакуумной центрифуге (Speedvac 5301). Сухой остаток, например, растворяли в 250 мкл 10% об/об ацетонитрила в воде, содержащей 0,1% муравьиной кислоты. Последующий анализ осуществляли, например, жидкостной хроматографией высокого давления/тандемной масс-спектрометрией (ВЭЖХ/MS-MS) с помощью системы для ВЭЖХ Agilent 1100 Series (Agilent, Palo Alto, CA, USA) с вакуумным дегазатором, бинарным насосом и термостатическим колоночным отделением, объединенным с автоохлаждающей системой образцов (HTS PAL, CTC Analytics, Zwingen, Switzerland). Образец (5-15 мкл) вводили, например, в колонку Ultra Phenyl (размер частиц 3 мкм, 50×1 мм; Restek, Bellefonte, USA) с защитой колонки (4×2 мм) из того же материала (Phenomenex, Torrance, USA). После уравновешивания, например, с водой и периода выдерживания 1 мин образцы разбавляли, например, линейным градиентом 0-100% ацетонитрилом в воде, содержащей 0,2% об/об муравьиной кислоты, в течение 11 мин при скорости потока 60 мкл/мин. Колонку готовили для следующего образца, например, повторным уравновешиванием в течение 3 мин с 100% водой до исходных условий. Выделение осуществляли, например, при температуре колонки 40°С. Поток колонки вводили, например, непосредственно в ионный источник трехстадийного квадропольного масс-спектрометра (Quattro Ultima™, Micromass, Manchester, UK), контролируя устройством Masslynx™ 3,5 (Micromass, Manchester, UK), используя в качестве ионизирующего устройства электроспрей-ионизацию положительного заряда (ESI+). Соединение определяли с помощью MS/MS с последующей фрагментацией родственных ионов. Предел квантования определяется, например, 0,002 нмоль/л. Калибрационные кривые строили по известным количествам соединения, включающего фиксированное количество внутреннего стандарта в плазме, что осуществляется, как описано выше. Концентрации неизвестных образцов рассчитывали из графика из соотношения площади пика выбранного дочернего иона к продукту его внутреннего стандарта (ордината) относительно номинальной концентрации (абсцисса). Регрессионный анализ осуществляли с помощью Quanlynx™, Masslynx™ оборудования 3,5 (Micromass, Manchester, UK).
Пример 21
Данные ингибирования in vitro
Данные ферментативного ингибирования (с-Abl, Bcr-Abl) in vitro показаны в сопроводительной таблице. Значения IC50 (в нМ) представлены в области, в которую попадали конкретные значения IC50. Соответствующие значения (±SEM) для соединения, известного как STI571, составляют 170±23 нМ (с-Abl, IC50; 23 определения) и 198±7 нМ (Bcr-Abl, IC50; 71 определения).
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗ | 2008 |
|
RU2445309C2 |
| ДИФЕНИЛЬНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2175319C2 |
| ПРОИЗВОДНЫЕ ТИЕНО[3,2-d]ПИРИМИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ПРОТЕИНКИНАЗЫ | 2011 |
|
RU2524210C2 |
| МОРФОЛИНИЛПИРИДОНЫ | 2018 |
|
RU2803158C2 |
| ПРОИЗВОДНЫЕ 1-(4-БЕНЗИЛПИПЕРАЗИН-1-ИЛ)-3-ФЕНИЛПРОПЕНОНА И ИХ ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РЕЦЕПТОРОВ ХЕМОКИНА (CCR-1) | 2003 |
|
RU2347782C2 |
| НОВЫЕ 4-АМИНО-N-ГИДРОКСИБЕНЗАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDAC ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2591190C2 |
| МОДУЛЯТОРЫ ИНТЕГРИРОВАННОГО СИГНАЛЬНОГО ПУТИ СТРЕССА | 2017 |
|
RU2769327C2 |
| ПРОИЗВОДНЫЕ 2-ОКСОХИНАЗОЛИНА КАК ИНГИБИТОРЫ МЕТИОНИН АДЕНОЗИЛТРАНСФЕРАЗЫ 2A | 2019 |
|
RU2830169C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2015 |
|
RU2691389C2 |
| 2-ЦИАНОПИРИМИДИН-4-ИЛКАРБАМАТ, ИЛИ ПРОИЗВОДНОЕ МОЧЕВИНЫ, ИЛИ ЕГО СОЛЬ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2788740C2 |
Настоящее изобретение относится к новым замещенным производным N-(3-бензоиламинофенил)-4-пиридил-2-пиримидинамина общей формулы (1), обладающим ингибирующей активностью в отношении протеинкиназы, способу их получения и фармацевтическим композициям на их основе. Соединения могут найти применение для лечения заболевания, которое связано с ингибированием активности протеинкиназы, такого как неопластического заболевания или лейкемии. В соединении формулы 1

R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород; R3 представляет собой трифторметил; R4 представляет собой низший алкил; и R5 и R6 представляют собой, независимо друг от друга, водород, низший алкил, ди(низший алкил)аминонизший алкил, N-низший алкилпиперидинил, N-низший алкилпирролидинил, или низший ацил, или
NR5R6 вместе представляют собой пирролидино, пиперидино, морфолино, N-низший алкилпиперазино, 1H-имидазолил, 1Н-2-низший алкилимидазолил, 1H-4-низший алкилимидазолил или 1Н-2,4-ди-низший алкилимидазолил, или фармацевтически приемлемая соль такого соединения. 6 н. и 7 з.п. ф-лы, 1 табл.
1. Соединение формулы 1
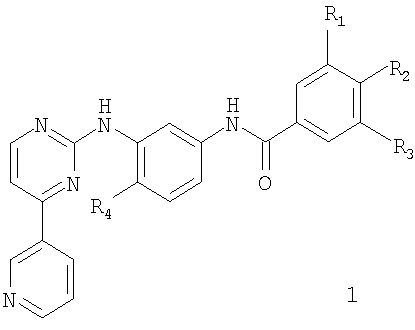
где R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой низший алкил; и
R5 и R6 представляют собой, независимо друг от друга, водород, низший алкил, ди(низший алкил)амино-низший алкил, N-низший алкилпиперидинил, N-низший алкилпирролидинил, или низший ацил, или NR5R6 вместе представляют собой пирролидино, пиперидино, морфолино, N-низший алкилпиперазино, 1H-имидазолил, 1H-2-низший алкилимидазолил, 1Н-4-низший алкилимидазолил или 1Н-2,4-ди-низший алкилимидазолил, или фармацевтически приемлемая соль такого соединения.
2. Соединение формулы 1 по п.1, где
R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой метил; и
R5 и R6 представляют собой, независимо друг от друга водород, низший алкил, ди(низший алкил)амино-низший алкил, N-низший алкилпиперидинил, N-низший алкилпирролидинил или низший ацил, или R5R6 вместе представляют собой пирролидино, пиперидино, морфолино, N-низший алкилпиперазино, 1H-имидазолил, 1Н-2-низший алкилимидазолил, 1H-4-низший алкилимидазолил или 1Н-2,4-ди(низший алкил)имидазолил;
или фармацевтически приемлемая соль такого соединения.
3. Соединение формулы 1 по п.1, где
R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой метил; и
R5 и R6 представляют собой, независимо друг от друга водород, низший алкил, ди(низший алкил)амино-низший алкил, N-низший алкилпиперидинил или низший ацил, или R5R6 вместе представляют собой пирролидино, пиперидино, морфолино, N-низший алкилпиперазино, 1H-имидазолил, 1H-2-низший алкилимидазолил, 1H-4-низший алкилимидазолил или 1H-2,4-ди(низший алкил)имидазолил;
или фармацевтически приемлемая соль такого соединения.
4. Соединение формулы 1 по п.1, где
R1 представляет собой водород и R2 представляет собой NR5R6, или R1 представляет собой NR5R6 и R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой метил; и
R5 и R6 представляют собой, независимо друг от друга водород, метил, этил, 2-диметиламиноэтил, 4-метил-1-пиперидинил или ацетил, или NR5R6 вместе представляют собой пирролидино, пиперидино, морфолино, N-метилпиперазино, 1H-имидазолил, 1H-2-метилимидазолил, 1H-4-метилимидазолил или 1Н-2,4-диметилимидазолил;
или фармацевтически приемлемая соль такого соединения.
5. Способ получения соединения формулы 1 по п.1

или его фармацевтически приемлемой соли, где символы R1, R2, R3 и R4 являются такими, как определено в п.1, заключающийся в том, что соединение формулы 2
где R1, R2 и R3 являются такими, как определено для соединения формулы 1, или его производное, где карбоксигруппа -СООН находится в активированной форме, подвергают реакции с амином формулы 3
где R4 является таким, как определено для соединения формулы 1, необязательно в присутствии дегидрирующего агента и инертного основания и необязательно в присутствии инертного растворителя;
где указанные выше исходные соединения формулы 2 и 3, при необходимости, могут также присутствовать с функциональными группами в защищенной форме и/или в форме солей, при условии, что присутствует солеобразующая группа и реакция в форме соли является возможной;
любые защитные группы в защищенном производном соединения формулы 1 удаляют; и,
при необходимости, полученное свободное соединение формулы 1 превращают в фармацевтически приемлемую соль, полученную соль соединения формулы 1 превращают в свободное соединение или фармацевтически приемлемую другую соль.
6. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении протеинкиназы, включающая в качестве активного ингредиента соединение формулы 1 по п.1 или фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
7. Применение соединения формулы 1 по любому из пп.1-4 или фармацевтически приемлемой соли такого соединения для получения фармацевтической композиции, предназначенной для лечения пролиферативного заболевания, связанного с ингибированием активности протеинкиназы.
8. Применение соединения формулы 1 по любому из пп.1-4 или фармацевтически приемлемой соли такого соединения, обладающего ингибирующей активностью в отношении протеинкиназы для лечения пролиферативного заболевания.
9. Применение по любому из пп.7 и 8, где заболевание представляет собой неопластическое заболевание.
10. Применение по любому из пп.7 и 8, где заболевание представляет собой лейкемию, которая связана с ингибированием активности тирозинкиназы Raf и/или Abl.
11. Способ лечения пролиферативного заболевания, которое связано с ингибированием активности протеинкиназы, который включает введение соединения формулы 1 по п.1 или его фармацевтически приемлемой соли.
12. Способ по п.11, где заболевание представляет собой неопластическое заболевание.
13. Способ по п.11, где заболевание представляет собой лейкемию, которая связана с ингибированием активности тирозинкиназы Raf и/или Abl.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Гидроабразивный перфоратор | 1975 |
|
SU564409A1 |

