Настоящая заявка испрашивает приоритет на основании заявки США № 60/791327, поданной 12 апреля 2006 г., и заявки США № 60/838720, поданной 18 августа 2006 г., каждая из которых включена в данное описание посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, пригодным в качестве ингибиторов протеинкиназ. Изобретение также обеспечивает фармацевтически приемлемые композиции, содержащие соединения по изобретению, и способы применения композиций при лечении различных заболеваний. Изобретение также обеспечивает способ получения соединений изобретения.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
В последние годы поиску новых терапевтических агентов значительно способствовало лучшее понимание структуры ферментов и других биомолекул, связанных с заболеваниями. Одним важным классом ферментов, который был объектом интенсивных исследований, являются протеинкиназы.
Протеинкиназы представляют собой большое семейство структурно-родственных ферментов, которые ответственны за контроль различных процессов трансдукции сигнала в клетке (см. Hardie, G. и Hanks, S. The Protein Kinase Fact Book, I и II, Academic Press, San Diego, CA: 1995). Считают, что протеинкиназы происходят от общего наследуемого гена вследствие сохранения их структуры и каталитической функции. Почти все киназы содержат схожий 250-300 аминокислотный каталитический домен. Киназы могут быть классифицированы в семейства в зависимости от субстратов, которые они фосфорилируют (например, протеин-тирозин, протеин-серин/треонин, липиды и т.д.). Были определены мотивы последовательностей, которые, в большинстве случаев, соответствуют каждому из этих семейств киназ (например, см. Hanks, S.K., Hunter, T., FASEB J. 1995, 9, 576-596; Knighton и др., Science. 1991, 253, 407-414; Hiles и др., Cell. 1992, 70, 419-429; Kunz и др., Cell. 1993, 73, 585-596; Garcia-Bustos и др., EMBO J. 1994, 13, 2352-2361).
В основном, протеинкиназы опосредуют внутриклеточную передачу сигнала посредством переноса фосфорила от нуклеозидтрифосфата к белковому акцептору, который включен в пути передачи сигнала. Данный процесс фосфорилирования выступает в качестве молекулярного переключателя, который может модулировать или регулировать биологическую функцию белка мишени. В конечном счете, данный процесс фосфорилирования запускается в ответ на различные внеклеточные и другие стимулы. Примеры такого рода стимулов включают стрессовые сигналы окружающей среды и химические стрессовые сигналы (например, шок, тепловой шок, ультрафиолетовое излучение, бактериальные эндотоксины и Н2О2), цитокины (например, интерлейкин-1 (IL-1), и фактор некроза опухолей альфа (TNF-a), и факторы роста (например, гранулоцит макрофаг-колониестимулирующий фактор (GM-CSF), и фактор роста фибробластов (FGF)). Внеклеточные стимулы могут влиять на один или несколько клеточных ответов, связанных с ростом клеток, миграцией, дифференцировкой, секрецией гормонов, активацией факторов транскрипции, мышечным сокращением, метаболизмом глюкозы, контролем синтеза белка, выживанием и регуляцией клеточного цикла.
Множество заболеваний связано с аномальным клеточным ответом, вызванным процессом, опосредованным протеинкиназой, как описано выше. Данные заболевания включают, но не ограничиваются этим, рак, аутоиммунные заболевания, воспалительные заболевания, болезни костей, метаболические заболевания, неврологические и нейродегенеративные заболевания, сердечно-сосудистые заболевания, аллергии и астму, болезнь Альцгеймера и гормонозависимые заболевания. В связи с этим, в медицинской химии были предприняты значительные попытки по поиску ингибиторов киназ, которые являются эффективными терапевтическими агентами.
Киназы типа Поло (Plk) относятся к семейству серин/треонин киназ, которые являются высококонсервативными у представителей разных видов, от дрожжей до человека (описано Lowery DM и др., Oncogene. 2005, 24; 248-259). Plk киназы играют множественные роли в клеточном цикле, включая контроль за началом и развитием митоза.
Plk1 являются наиболее изученным из членов семейства Plk. Plk1 широко экспрессирован и наиболее часто встречается в тканях с высокими митотическим индексом. Белковые уровни Plk1 возрастают и достигают пика во время митоза (Hamanaka, R и др., J Biol Chem. 1995, 270, 21086-21091). Описанными субстратами Plk1 являются все молекулы, которые известны как регуляторы при вхождении в митоз и его развитии, и включают CDC25C, циклин В, р53, АРС, BRCA2 и протеасому. Plk1 активируется при различных видах рака, и уровни экспрессии коррелируют с тяжестью заболевания. (Macmillan, JC и др., Ann Surg Oncol. 2001, 8, 729-740). Plk1 является онкогеном и может трансформировать NIH-3Т3 клетки (Smith, MR и др., Biochem Biophys Res Commun. 1997, 234, 397-405). Элиминация или ингибирование Plk1 посредством siRNA, антисмысловой конструкции, микроинъекции антител или трансфекции доминантной негативной конструкции Plk1 в клетки уменьшает пролиферацию и выживаемость опухолевых клеток in vitro (Guan, R и др., Cancer Res. 2005, 65, 2698-2704; Liu, X и др., Proc Natl Acad Sci USA 2003, 100, 5789-5794, Fan, Y и др., Worl J Gastroenterol. 2005, 11, 4596-4599l; Lane, HA и др., J Cell Biol. 1996, 135, 1701-1713). Опухолевые клетки, у которых был элиминирован Plk1, проявляют активированное состояние перехода от метафазы к анафазе и имеют дефекты при образовании веретена, в расположении и разделении хромосом и в цитокинезе. Описанное понижение выживаемости является результатом индукции апоптоза. С другой стороны показано, что нормальные клетки сохраняют уровень выживаемости при элиминации Plk1. Понижение уровня Plk1 in vivo посредством siRNA или при использовании доминантных негативных конструкций приводит к ингибированию роста или к регрессии опухолей в ксенотрансплантатных моделях.
Plk2 главным образом экспрессируется во время фазы G1 клеточного цикла и локализуется в центросоме интерфазных клеток. Plk2 нокаутированные мыши нормально развиваются, способны к деторождению и имеют нормальный уровень выживаемости, однако примерно на 20% меньше по сравнению с диким типом. Клетки нокаутированных животных медленнее проходят через клеточный цикл по сравнению с обычными мышами. (Ma, S и др., Mol Cell Biol. 2003, 23, 6936-6943). Элиминация Plk2 посредством siRNA или трансфекции мутантных неактивных киназ в клетки приводит к блокированию дупликации центриоли. Деактивация Plk2 также делает чувствительными опухолевые клетки к таксолу и стимулирует митотическую катастрофу, в частичности посредством супрессии р53 ответа (Burns TF и др., Moll Cell Biol. 2003, 23, 5556-5571).
Plk3 экспрессируется в ходе клеточного цикла, и его концентрация увеличивается от стадии G1 до митоза. Экспрессия активируется в высокопролиферирующих опухолях яичников и клетках рака груди, и это можно рассматривать как прогноз ухудшения состояния (Weichert, W и др. Br J Cancer. 2004, 90, 815-821; Weichert, W и др., Vichows Arch. 2005, 446, 442-450). Считается, что кроме регуляции митоза Plk3 включен в процесс фрагментации аппарата Гольджи в ходе клеточного цикла и в ходе формирования ответа на повреждение ДНК. Описано, что ингибирование Plk3 посредством доминантной негативной экспрессии вызывает р53-независимый апоптоз после повреждения ДНК и подавляет образование колоний опухолевых клеток (Li, Z и др., J Biol Chem. 2005, 280, 16843-16850).
Plk4 структурно сильнее отличается от других членов семейства Plk. Элиминация данной киназы вызывает апоптоз в раковых клетках (Li, J и др., Neoplasia. 2005, 7, 312-323). Plk4 нокаутированные мыши приостанавливаются в развитии при Е7.5 с высокой фракцией клеток в стадии митоза и частично сегрегированными хромосомами (Hudson, JW и др., Current Biology. 2001, 11, 441-446).
Молекулы семейства протеинкиназ вовлечены в процесс роста раковых клеток, пролиферации и выживаемости. В связи с этим существует большая потребность в разработке соединений, пригодных в качестве ингибиторов протеинкиназ. Доказательство того, что Plk киназы вовлечены в процесс деления клетки в качестве неотъемлемых составляющих, является убедительным. Блокирование клеточного цикла является клинически признанным подходом для ингибирования пролиферации и выживаемости опухолевых клеток. Следовательно, было бы желательно разработать соединения, пригодные в качестве ингибиторов протеинкиназ семейства Plk (например, Plk1, Plk2, Plk3 и Plk4), которые будут ингибировать пролиферацию и уменьшать выживаемость опухолевых клеток, в особенности для важных медицинских целей разработки новых методов лечения рака.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы I

где:
Кольцо А является 5-членным гетероарильным кольцом, где кольцо, необязательно, замещено С1-6 галогеналкилом, галогеном, NO2, -ОН, -CN или, необязательно, замещенным С1-6 алкилом;
Х1 является связью, -О-, NR8-, -S-, -S(O)- или -S(O)2-;
R1 является Н, С1-10 алифатической, С3-10 циклоалифатической, С6-10 арильной, 5-10-членной гетероарильной группой или 3-10-членным гетероциклилом, где R1, необязательно, замещен 0-5 J1;
каждый R2 и R3 независимо является Н, C1-10 алифатической или C3-10 циклоалифатической группой, где каждый R2 и R3 независимо и необязательно замещен 0-5 J2 и J3 соответственно, или
R2 и R3 вместе с атомом углерода, к которому они присоединены, образуют 3-8-членное насыщенное или частично ненасыщенное моноциклическое кольцо, содержащее 0-4 гетероатомов, независимо выбранных из O, N и S, где указанное моноциклическое кольцо, образованное R2 и R3, необязательно замещено 0-4 J23;
R4 является Н, -C(O)R, -C(O)OR, -C(O)NRR`, C1-10 алифатической, C3-10 циклоалифатической группой, С6-10 арилом, С5-10 гетероарилом, 3-10-членным гетероциклилом, (С1-6 алифатической)-(С3-10 циклоалифатической) группой или (С1-6 алифатической)-(5-10-членной гетероарильной) группой, где указанный R4, необязательно, замещен 0-5 J4;
R8 является Н, C1-6 алифатической, C3-8 циклоалифатической группой, -C(O)R, -C(O)OR, -C(O)NRR`;
каждый J1 независимо является С1-6 галогеналкилом, галогеном, NO2, CN, Q или -Z-Q, или две J1, взятые вместе, необязательно, могут образовывать =О;
каждый Z независимо является С1-6 алифатической группой, в которой 0-3 -СН2- звеньев в указанной С1-6 алифатической группе необязательно заменены на -NR-, -O-, -S-, -C(O)-, -C(=NR)-, -C(=NOR)-, -S(O)- или -S(O)2-, где любое из незамененных -СН2- звеньев в указанной С1-6 алифатической группе необязательно замещено 0-2 Jz;
каждый Q независимо является Н, С1-6 алифатической группой, 3-8-членным ароматическим или неароматическим моноциклическим кольцом, имеющим 0-3 гетероатомов, независимо выбранных из O, N и S, или 8-12-членной ароматической или неароматической бициклической системой, имеющей 0-5 гетероатомов, независимо выбранных из O, N и S, где каждый Q необязательно замещен 0-5 JQ;
каждый J2 и J3 независимо является С1-6 алифатической группой, С3-6 циклоалифатической группой или -(С1-4 алкил)n- V1, где
n является 0 или 1,
каждый V1 независимо является галоген(С1-4 алифатической группой), -О(галогенС1-4 алифатической группой), галогеном, NO2, CN, OH, OR``, SH, SR``, NH2, NHR``, N(R``)2, COH, COR``, CO2H, CO2R``, CONH2, CONHR``, CONR``2, OCOR``, OCONH2, OCONHR``, OCON(R``)2, NHCOR``, NR``COR``, NHCO2R``, NR``CO2R``, NHCO2H, NR``CO2H, NHCONH2, NHCONHR``, NHCON(R``)2, SO2NH2, SO2NHR``, SO2N(R``)2, NHSO2R``, NR``SO2R``, или
V1 является циклической группой, выбранной из С3-6 циклоалифатической группы, фенила, 5-6-членного гетероарила или 3-6-членного гетероциклила, где указанная циклическая группа необязательно замещена 0-3 JV;
каждый R`` независимо является незамещенной С1-4 алифатической группой, или две одинаковые J2 и J3, присоединенные к одному и тому же атому, вместе могут необязательно образовывать =О;
каждый JZ и JV независимо является галогеном, С1-6 алифатической группой, С3-6 циклоалифатической группой, NO2, CN, OH, NH2, NH(С1-4 алифатической группой), N(С1-4 алифатической группой)2, -О(С1-4 алифатической группой), -CO2H, -СО2(С1-4 алифатической группой), -О(галоген С1-4 алифатической группой) или галоген(С1-4 алифатической группой);
каждый JQ, J4 и J23 независимо является -M или -Y-M;
каждый Y независимо является ненасыщенной С1-6 алифатической группой, в которой 0-3 -СН2- звеньев в указанной С1-6 алифатической группе необязательно заменены на -NR-, -O-, -S-, -C(O)-, -S(O)- или -S(O)2-;
каждый М независимо является Н, С1-6 алифатической группой, С3-6 циклоалифатической группой, галоген(С1-4 алифатической группой), О(галоген С1-4 алифатической группой), 3-6-членным гетероциклилом, галогеном, NO2, CN, OH, OR`, SH, SR`, NH2, NHR`, N(R`)2, COH, COR`, CO2H, CO2R`, CONH2, CONHR`, CONR2`, OCOR`, OCONH2, OCONHR`, OCON(R`)2, NHCOR`, NR`COR`, NHCO2R`, NR`CO2R`, NHCO2H, NR`CO2H, NHCONH2, NHCONHR`, NHCON(R`)2, SO2NH2, SO2NHR`, SO2N(R`)2, NHSO2R` или NR`SO2R`;
каждый R независимо является Н или ненасыщенной С1-6 алифатической группой; и
каждый R` независимо является С1-6 алифатической группой, или две R` группы вместе с атомом, к которому они присоединены, образуют незамещенное 3-8-членное насыщенное или частично ненасыщенное моноциклическое кольцо, имеющее 0-1 гетероатомов, независимо выбранных из O, N и S.
Соединения по настоящему изобретению и их фармацевтически приемлемые композиции являются эффективными ингибиторами протеинкиназ. В некоторых вариантах, данные соединения являются эффективными ингибиторами PLK протеинкиназ; в некоторых вариантах являются ингибиторами PLK1 протеинкиназ. Как определено в данном описании, данные соединения обладают формулой I или являются ее фармацевтически приемлемыми солями.
Данные соединения и их фармацевтически приемлемые композиции являются пригодными для лечения или предупреждения различных заболеваний, расстройств или состояний, включая, но не ограничиваясь ими, аутоиммунные, воспалительные, пролиферативные или гиперпролиферативные заболевания, нейродегенеративные заболевания или заболевания, опосредованные расстройством иммунитета. Также, соединения по настоящему изобретению являются пригодными для изучения киназ в биологических и патологических процессах; изучения внутриклеточных путей трансдукции сигнала, опосредованного такими киназами; и сравнительного исследования новых ингибиторов киназ.
Соединения по настоящему изобретению включают такие, которые описаны в данном описании, и далее представлены раскрываемыми в данном описании классами, подклассами и конкретными представителями (см., например, Варианты 1-22). Используемые в данном описании следующие определения следует применять, если не указано иное. Для целей настоящего изобретения, химические элементы называются в соответствии с Периодической Таблицей Элементов, CAS версия, Handbook of Chemistry and physics, 75th Ed. Кроме этого общие принципы органической химии описаны в “Organic Chemistry”, Tomas Sorrell, University Science Books, Sauslito: 1999 и “March`s Advanced Organic Chemistry” 5th Ed., под редакцией: Smith, M.B. и March, J., John Wiley & Sons, New York: 2001, содержание которых таким образом включено посредством ссылки.
Используемые в данном описании определенные числовые диапазоны атомов включают любое целое число в этом диапазоне. Например, группа, имеющая от 1-4 атомов, может иметь 1, 2, 3 или 4 атома.
Как описывается в данном описании, соединения настоящего изобретения необязательно могут быть замещены одним или несколькими заместителями, например, такими как в общем приведено выше, или могут быть представлены определенным классом, подклассом и конкретными представителями соединений по изобретению. Следует понимать, что фраза «необязательно замещен» используется в том же смысле, что и фраза «замещен или незамещен». В основном, термин «замещенный», предшествует ли ему термин «необязательно» или нет, относится к замещению водородного радикала в данной структуре на радикал определенного заместителя. Если не указано иное, необязательно замещенная группа может иметь заместитель при каждом положении группы, способном к замещению, и если более чем одно положение в любой данной структуре может быть замещено более чем одним заместителем, выбранным из определенной группы, то при каждом положении, заместители могут быть как одинаковыми, так и различными. Предусматриваемые настоящим изобретением комбинации заместителей предпочтительно являются такими, что приводят к образованию стабильных или практически осуществимых с химической точки зрения соединений.
Используемый в данном описании термин «стабильные» относится к соединениям, которые в значительной степени не изменяются при условиях, необходимых для их получения, определения, выделения, очистки и применения для одной или нескольких описанных здесь целей. В некоторых вариантах, стабильными соединениями или практически осуществимыми соединениями с химической точки зрения являются такие, которые в значительной степени не изменяются при хранении при 40°С или ниже, в отсутствие влаги или других химически активных условий в течение, по крайней мере, недели.
Используемые в данном описании термины «алифатический» или «алифатическая группа» означают прямолинейную (т.е. неразветвленную) или разветвленную, замещенную или незамещенную углеводородную цепь, которая полностью насыщена или которая содержит одно или несколько звеньев ненасыщенности и которая имеет одно место присоединения к остальной молекуле.
Если не определено иначе, алифатические группы содержат от 1-20 алифатических атомов углерода. В некоторых вариантах, алифатическая группа содержит 1-10 алифатических атомов углерода. В других вариантах, алифатическая группа содержит 1-8 алифатических атомов углерода. В некоторых других вариантах, алифатическая группа содержит 1-6 алифатических атомов углерода, и в других вариантах, алифатическая группа содержит 1-4 алифатических атомов углерода. Подходящие алифатические группы включают, но не ограничиваются ими, линейные или разветвленные, насыщенные или ненасыщенные алкильные, алкенильные или алкинильные группы. Конкретные примеры включают, но не ограничиваются ими, метил, этил, изопропил, н-пропил, втор-бутил, винил, н-бутенил, этинил и трет-бутил.
Термины «циклоалифатический» (или «карбоцикл», или «карбоциклил», или «циклоалкил») относятся к моноциклическому С3-8 углеводороду или бициклическому С8-12 углеводороду, который является полностью насыщенным или содержит одно или несколько звеньев ненасыщенности, но не является ароматическим, который имеет одно место присоединения к остальной молекуле, где любое индивидуальное кольцо в указанной бициклической системе содержит 3-7 членов в кольце. Подходящие циклоалифатические группы включают, но не ограничиваются ими, циклоалкильные и циклоалкенильные группы. Конкретные примеры включают, но не ограничиваются ими, циклогексил, циклопентил и циклобутил.
Используемые в данном описании термины «гетероцикл», «гетероциклил» или «гетероциклический» означают неароматическую, моноциклическую, бициклическую или трициклическую систему колец, в которой один или несколько членов колец являются независимо выбранным гетероатомом. В некоторых вариантах «гетероцикл», «гетероциклил» или «гетероциклическая группа» имеют 3=14 членов кольца, где одним или более членов кольца является гетероатом, независимо выбранный из кислорода, серы, азота или фосфора, и каждое кольцо в системе содержит 3-7 членов кольца.
Подходящие гетероциклы включают, но не ограничиваются ими, 3-1Н-бензимидазол-2-он, 3-(1-алкил)-бензимидазол-2-он, 2-тетрагидрофуранил, 3-тетрагидрофуранил, 2-тетрагидротиофенил, 3- тетрагидротиофенил, 2-морфолино, 3-морфолино, 4-морфолино, 2-тиоморфолино, 3-тиоморфолино, 4-тиоморфолино, 1-пирролидинил, 2- пирролидинил, 3-пирролидинил, 1-тетрагидропиперазинил, 2- тетрагидропиперазинил, 3-тетрагидропиперазинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-пиперидинил, 2-тиазолидинил, 3-тиазолидинил, 4-тиазолидинил, 1-имидазолидинил, 2-имидазолидинил, 4-имидазолидинил, 5-имидазолидинил, индолинил, тетрагидрохинолинил, тетрагидроизохинолинил, бензотиолан, бензодитиан и 1,3-дигидроимидазол-2-он.
Циклические группы (например, циклоалифатические и гетероциклические) могут быть линейно конденсированными, связанными через мостик или спироциклическими.
Термин «гетероатом» означает один или несколько атомов кислорода, серы, азота, фосфора или кремния (включая любую окисленную форму азота, серы, фосфора или кремния; кватернизированную форму любого основного азота или способного к замещению азота гетероциклического кольца, например N (как в 3,4-дигидро-2Н-пирролиле), NH (как в пирролидиниле) или NR+ (как в N-замещенном пирролидиниле)).
Используемый в данном описании термин «ненасыщенный» означает, что остаток имеет одно или несколько звеньев ненасыщенности.
Используемый в данном описании термин «неароматический» описывает кольца, которые являются либо насыщенными, либо частично ненасыщенными.
Используемые в данном описании термины «алкокси» или «тиоалкил» относятся к алкильной группе, как определено ранее, присоединенной через атом кислорода («алкокси») или атом серы («тиоалкил»).
Термины «галогеналкил», «галогеналкенил», «галогеналифатическая группа» и «галогеналкокси» означают алкил, алкенил или алкоксил соответственно, замещенные одним или несколькими атомами галогена.
Термины «галоген», «гало» и «гал» означают F, Cl, Br или I.
Термин «арил», используемый самостоятельно или как часть большего остатка, как, например, «аралкил», «аралкокси» или «арилоксиалкил», относится к моноциклической, бициклической и трициклической системе колец, имеющей всего от пяти до четырнадцати членов колец, где, по крайней мере, одно кольцо в системе является ароматическим и где каждое кольцо в системе содержит от 3 до 7 членов кольца. Термин «арил» может быть использован наряду с термином «арильное кольцо».
Термин «гетероарил», используемый самостоятельно или как часть большего остатка, как, например, «гетероаралкил» или «гетероарилалкокси», относится к моноциклической, бициклической и трициклической системе колец, имеющей всего от пяти до четырнадцати членов колец, где, по крайней мере, одно кольцо в системе является ароматическим, по крайней мере, одно кольцо в системе содержит один или несколько гетероатомов и где каждое кольцо в системе содержит от 3 до 7 членов кольца. Термин «гетероарил» может быть использован наряду с термином «гетероарильное кольцо» или термином «гетероароматическая группа». Подходящие гетероароматические кольца включают, но не ограничиваются ими, 2-фуранил, 3-фуранил, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, бензимидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, N-пирролил, 2-пирролил, 3-пирролил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пиридазинил (например, 3-пиридазинил), 2-тиазолил, 4-тиазолил, 5-тиазолил, тетразолил (например, 5-тетразолил), триазолил (например, 2-триазолил и 5-триазолил), 2-тиенил, 3-тиенил, бензофурил, бензотиофенил, индолил, (например, 2-индолил), пиразолил (например, 2-пиразолил), изотиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-триазолил, 1,2,3-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил, пуринил, пиразинил, 1,3,5-триазинил, хинолинил (например, 2-хинолинил, 3-хинолинил, 4-хинолинил) и изохинолинил (например, 1-изохинолинил, 3-изохинолинил или 4-изохинолинил).
Используемые в данном описании термины «блокирующая группа» или «защитная группа» являются взаимозаменяемыми и относятся к временно используемым агентам для блокирования одной или нескольких необходимых функциональных групп в соединении с несколькими реакционноспособными центрами. В некоторых вариантах, защитные группы обладают одной, или несколькими, или, предпочтительно, всеми следующими характеристиками: а) селективно вводятся по функциональной группе, с хорошим выходом давая защищенный субстрат, который является b) стабильным к реакциям, протекающим по одному или нескольким другим реакционноспособным центрам; и с) селективно удаляются с хорошим выходом под действием реагентов, которые не атакуют регенерированную, деблокированную функциональную группу. Для специалиста в области техники будет ясно, что в некоторых случаях реагенты не воздействуют на другие реакционноспособные группы в соединении. В других случаях, реагенты также могут реагировать с другими реакционноспособными группами в соединении. Примеры защитных групп подробно описаны в Greene, T. W., Wuts, P. G в “Protective Groups in Organic Synthesis”, Third Edition, John Wiley & Sons, New York: 1999 (и другие издания книги), которая включена в данное описание посредством ссылки. Используемый в данном описании термин «защитная группа атома азота» относится к агентам, используемым для временной защиты одного или нескольких требуемых азотных реакционных центров в многофункциональном соединении. Предпочтительные защитные группы атома азота также обладают указанными выше характеристиками, и некоторые примеры защитных групп атома азота также подробно описаны в Главе 7 в Greene, T. W., Wuts, P. G в “Protective Groups in Organic Synthesis”, Third Edition, John Wiley & Sons, New York: 1999, которая включена в данное описание посредством ссылки.
В некоторых вариантах алкильная или алифатическая цепь необязательно может быть заменена на другой атом или группу. Это означает, что метиленовое звено алкила или алифатической цепи необязательно заменено на указанный другой атом или группу. Примеры таких атомов или групп могут включать, но не ограничиваться ими, -NR-, -O-, -C(O)-, -C(=N-CN), -C(=NR)-, -C(=NOR)-, -S-, -SO- -или -SO2-. Данные атомы или группы могут быть объединены для образования больших групп. Примеры таких групп включают, но не ограничиваются, -OC(O)-, -C(O)CO-, -CO2-, -C(O)NR-, -C(=N-CN), -NRCO-, -NRC(O)O-, -SO2NR-, -NRSO2-, NRC(O)NR-, -OC(O)NR- или -NRSO2NR-, где R является таким, как определено здесь.
Если специально не указанно, необязательные замены приводят к химически стабильному соединению. Необязательные замены могут быть как внутри цепи, так и в конце цепи; например, как в месте присоединения и/или также в конце цепи. Две необязательные замены могут быть рядом друг с другом в цепи, если это приводит к химически стабильному соединению. Необязательные замены могут полностью заменить все атомы углерода в цепи. Например, С3 алифатическая группа может быть необязательно заменена группами -NR-, -C(O)- и -NR-, давая -NRC(O)NR- (мочевину).
Если не указано иное, при замене в концевом положении, замененный атом присоединен к Н в конце цепи. Например, если -СН2-СН2-СН3 необязательно заменена на -О-, то результирующим соединением может быть -ОСН2СН3, -СН2ОСН3 или -СН2СН2ОН.
Если не указано иное, то подразумевается, что приведенные в данном описании структуры также включают все изомеры (например, энантиомерные, диастереомерные, геометрические, конформационные и ротационные формы структуры). Например, настоящее изобретение включает R и S конфигурации для каждого асимметрического центра, (Z) и (E) изомеры при двойной связи и (Z) и (E) конформационные изомеры. Как будет понятно специалисту в данной области техники, заместитель может свободно вращаться вокруг любой способной к вращению связи. Например, заместитель, показанный как 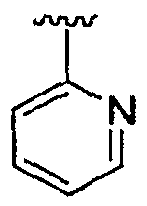 , также представляет и
, также представляет и 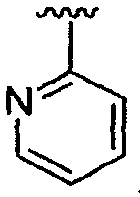 .
.
Следовательно, отдельные стереохимические изомеры, а также энантиомерные, диастереомерные, геометрические, конформационные или ротационные смеси настоящих соединений включены в объем изобретения.
Если не указано иное, все таутомерные формы соединений изобретения включены в объем изобретения.
Кроме этого, если не указано иначе, подразумевается, что приведенные здесь структуры также включают соединения, которые отличаются лишь присутствием одного или нескольких изотопно обогащенных атомов. Например, соединения, имеющие данную структуру за исключением того, что водород заменен на дейтерий или тритий или углерод заменен на обогащенные углеродные атомы 13С- или 14С-, включены в объем изобретения. Например, такие соединения являются пригодными в качестве аналитических инструментов или зондов в биологических анализах.
Соединения по настоящему изобретению могут быть в свободной форме для лечения или, при необходимости, в виде фармацевтически приемлемой соли.
Используемый в данном описании термин «фармацевтически приемлемая соль» относится к соединению, которое является пригодным для применения по предлагаемому назначению. В некоторых вариантах соли являются пригодными для применения в контакте с тканями человека и низших животных без чрезмерной токсичности, раздражения, аллергического ответа и т.п. и являются сопоставимыми с приемлемым соотношением преимущество/риск. В других вариантах, соли могут быть пригодными для применений в анализах in vitro, кинетических исследованиях, кристаллографических исследованиях и т.п.
Фармацевтически приемлемые соли хорошо известны в области техники. Например, S. M. Berge и др. подробно описывают пригодные соли в J. Pharmaceutical Sciences, 1977, 66, 1-19, включенной в данное описание посредством ссылки. Фармацевтически приемлемые соли соединений настоящего изобретения включают такие, которые получены из подходящих неорганических и органических кислот и оснований. Данные соли могут быть получены in situ во время конечного выделения и очистки соединений. Соли, образующиеся при добавлении кислоты, могут быть получены посредством 1) реакции очищенного соединения в форме его свободного основания с подходящей органической или неорганической кислотой и 2) выделения образовавшейся таким образом соли.
Примерами фармацевтически приемлемых, нетоксичных кислотно-аддитивных солей являются соли аминогруппы, полученные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с помощью других способов, используемых в данной области техники, например с помощью ионного обмена. Другие фармацевтически приемлемые соли включают адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, глюколат, глюконат, гемисульфат, гептаноат, гексаноат, гидроиодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, стеарат, сукцинат, сульфат, тартрат, тиоционат, п-толуолсульфонат, ундеканоат, соли валериановой кислоты и т.п. Соли, полученные из соответствующих оснований, включают соли щелочных металлов, щелочноземельных металлов, аммония и N+(C1-4алкил)4 соли. Данное изобретение также предусматривает кватернизацию любых основных азотсодержащих групп описанных в данном описании соединений. С помощью такой кватернизации могут быть получены водо- или маслорастворимые или диспергируемые продукты.
Основно-аддитивные соли могут быть получены посредством 1) реакции очищенного соединения в форме кислоты с подходящим органическим или неорганическим основанием и 2) выделения образовавшейся таким образом соли. Основно-аддитивные соли включают соли щелочных или щелочноземельных металлов. Типичные соли щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция, магния и т.п. Кроме того, фармацевтически приемлемые соли включают соответствующие нетоксичные аммониевые, четвертичные аммониевые и амино катионы, образованные с использованием противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, сульфонат низшего алкила и арилсульфонат. При получении солей, пригодных в качестве промежуточных соединений для получения соединений по изобретению и их фармацевтически приемлемых солей или основно- или кислотно-аддитивных солей, могут применяться другие кислоты и основания, которые сами по себе не являются фармацевтически приемлемыми.
Используются следующие аббревиатуры:
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В некоторых аспектах, изобретение обеспечивает соединения формулы I или фармацевтически приемлемые соли, пригодные в качестве ингибиторов протеинкиназ
где:
Кольцо А является 5-членным гетероарильным кольцом, где кольцо необязательно замещено С1-6 галогеналкилом, галогеном, NO2, -ОН, -CN или необязательно замещенным С1-6 алкилом;
Х1 является связью, -О-, NR8-, -S-, -S(O)- или -S(O)2-;
R1 является Н, С1-10 алифатической, С3-10 циклоалифатической, С6-10 арильной, 5-10-членной гетероарильной группой или 3-10-членным гетероциклилом, где R1 необязательно замещен 0-5 J1;
Каждый R2 и R3 независимо является Н, C1-10 алифатической или C3-10 циклоалифатической группой, где каждый R2 и R3 независимо необязательно замещен 0-5 J2 и J3 соответственно, или
R2 и R3 вместе с атомом углерода, к которому они присоединены, образуют 3-8-членное насыщенное или частично ненасыщенное моноциклическое кольцо, содержащее 0-4 гетероатомов, независимо выбранных из O, N и S, где указанное моноциклическое кольцо, образованное R2 и R3, необязательно замещено 0-4 J23;
R4 является Н, -C(O)R, -C(O)OR, -C(O)NRR`, C1-10 алифатической, C3-10 циклоалифатической группой, С6-10 арилом, С5-10 гетероарилом, 3-10-членным гетероциклилом, (С1-6 алифатической)-(С3-10 циклоалифатической) группой или (С1-6 алифатической)-(5-10-членной гетероарильной) группой, где указанный R4 необязательно замещен 0-5 J4;
R8 является Н, C1-6 алифатической, C3-8 циклоалифатической группой, -C(O)R, -C(O)OR, -C(O)NRR`;
каждый J1 независимо является С1-6 галогеналкилом, галогеном, NO2, CN, Q или -Z-Q, или две J1, взятые вместе, необязательно могут образовывать =О;
каждый Z независимо является С1-6 алифатической группой, в которой 0-3 -СН2- звеньев в указанной С1-6 алифатической группе необязательно заменены на -NR-, -O-, -S-, -C(O)-, -C(=NR)-, -C(=NOR)-, -S(O)- или -S(O)2-, где любое из незамененных -СН2- звеньев в указанной С1-6 алифатической группе необязательно замещено 0-2 Jz;
каждый Q независимо является Н, С1-6 алифатической группой, 3-8-членным ароматическим или неароматическим моноциклическим кольцом, имеющим 0-3 гетероатомов, независимо выбранных из O, N и S, или 8-12-членной ароматической или неароматической бициклической системой, имеющей 0-5 гетероатомов, независимо выбранных из O, N и S, где каждый Q необязательно замещен 0-5 JQ;
каждый J2 и J3 независимо является С1-6 алифатической группой, С3-6 циклоалифатической группой или -(С1-4алкил)n- V1, где
n является 0 или 1,
каждый V1 независимо является галоген(С1-4 алифатической группой), -О(галогенС1-4 алифатической группой), галогеном, NO2, CN, OH, OR``, SH, SR``, NH2, NHR``, N(R``)2, COH, COR``, CO2H, CO2R``, CONH2, CONHR``, CONR``2, OCOR``, OCONH2, OCONHR``, OCON(R``)2, NHCOR``, NR``COR``, NHCO2R``, NR``CO2R``, NHCO2H, NR``CO2H, NHCONH2, NHCONHR``, NHCON(R``)2, SO2NH2, SO2NHR``, SO2N(R``)2, NHSO2R``, NR``SO2R``, или
V1 является циклической группой, выбранной из С3-6 циклоалифатической группы, фенила, 5-6-членного гетероарила или 3-6-членного гетероциклила, где указанная циклическая группа необязательно замещена 0-3 JV;
каждый R`` независимо является незамещенной С1-4 алифатической группой, или две одинаковые J2 и J3, присоединенные к одному и тому же атому, вместе могут необязательно образовывать =О;
каждый JZ и JV независимо является галогеном, С1-6 алифатической группой, С3-6 циклоалифатической группой, NO2, CN, OH, NH2, NH(С1-4 алифатической группой), N(С1-4 алифатической группой)2, -О(С1-4 алифатической группой), -CO2H, -СО2(С1-4 алифатической группой), -О(галоген С1-4 алифатической группой) или галоген(С1-4 алифатической группой);
каждый JQ, J4 и J23 независимо является -M или -Y-M;
каждый Y независимо является ненасыщенной С1-6 алифатической группой, где 0-3 -СН2- звеньев в указанной С1-6 алифатической группе необязательно заменены на -NR-, -O-, -S-, -C(O)-, -S(O)- или -S(O)2-;
каждый М независимо является Н, С1-6 алифатической группой, С3-6 циклоалифатической группой, галоген(С1-4 алифатической группой), О(галоген С1-4 алифатической группой), 3-6-членным гетероциклилом, галогеном, NO2, CN, OH, OR`, SH, SR`, NH2, NHR`, N(R`)2, COH, COR`, CO2H, CO2R`, CONH2, CONHR`, CONR2`, OCOR`, OCONH2, OCONHR`, OCON(R`)2, NHCOR`, NR`COR`, NHCO2R`, NR`CO2R`, NHCO2H, NR`CO2H, NHCONH2, NHCONHR`, NHCON(R`)2, SO2NH2, SO2NHR`, SO2N(R`)2, NHSO2R` или NR`SO2R`;
каждый R независимо является Н или ненасыщенной С1-6 алифатической группой; и
каждый R` независимо является С1-6 алифатической группой, или две R` группы вместе с атомом, к которому они присоединены, образуют незамещенное 3-8-членное насыщенное или частично ненасыщенное моноциклическое кольцо, имеющие 0-1 гетероатомов, независимо выбранных из O, N и S.
В некоторых вариантах, кольцо А является триазольным кольцом, необязательно замещенным С1-6 галогеналкилом, галогеном, NO2, -ОН, -CN или необязательно замещенным С1-6 алкилом.
В некоторых вариантах, кольцо А является имидазольным кольцом, необязательно замещенным С1-6 галогеналкилом, галогеном, NO2, -ОН, -CN или необязательно замещенным С1-6 алкилом.
В некоторых вариантах, Х1 является -О-, -NR8- или -S-.
В некоторых вариантах, Х1 является -NR8-.
В некоторых вариантах, R8 является Н или -С(О)OR, где R является С1-6 алкилом; например R8 является -С(О)ОСН3.
В некоторых вариантах, R1 является Н, необязательно замещенным С6-10 арилом, необязательно замещенным аралкилом или необязательно замещенным С5-10 гетероарилом.
В некоторых вариантах, R1 необязательно замещен группой -O-Q, галогеном, -C(O)N(R)-Q или Q, в которых каждый Q в Q, -C(O)N(R)-Q, -O-Q независимо необязательно замещен 0-5 JQ.
В некоторых вариантах, R1 является фенилом, необязательно замещенным в пара-положении группой -C(O)N(R)-Q и в любом другом положении группой -O-Q, галогеном, Q, где каждый Q в Q, -C(O)N(R)-Q, -O-Q независимо необязательно замещен 0-5 JQ.
В некоторых вариантах, JQ является С1-6 алифатической группой, необязательно замещенной С3-6 циклоалифатической группой, галоген(С1-4 алифатической) группой, О(галоген С1-4 алифатической) группой или 3-6-членным гетероциклилом.
В некоторых вариантах, R1 является гетероарилом, замещенным группой -C(O)N(R)-Q, и в любом другом положении - группой -O-Q или Q, в которых каждый Q в Q, -C(O)N(R)-Q, -O-Q независимо необязательно замещен 0-5 JQ.
В некоторых вариантах, Q в -C(O)N(R)-Q является Н, С1-4 алифатической группой, С1-4 галогеналифатической группой, С3-7 циклоалифатической группой, С3-7 гетероциклоалифатической группой, С1-6 алкокси группой, (С1-6 алкокси)С1-6 алкильной группой или С1-6 галогеналкоксигруппой.
В некоторых вариантах, где R1 является фенильной группой, замещенной группой Q в пара-положении, фенильная группа необязательно замещена в любом другом свободном положении галогеном, С1-4 алифатической группой, С1-4 галогеналифатической группой, С3-7 гетероциклоалифатической группой, С1-6 алкоксигруппой, (С1-6 алкокси) С1-6 алкильной или С1-6 галогеналкоксигруппой.
В некоторых вариантах, Q в -C(O)N(R)-Q является метилом, этилом, 1-метилпиперидин-4-илом, циклопропилом, циклопентилом, 3-фуранилом, 3-фторпирролидин-1-илом или 3,3-дифторциклобутилом.
В некоторых вариантах, R1 необязательно замещен С1-6 алкилом или С3-7 циклоалкилом.
В некоторых вариантах, R1 является фенилом, необязательно замещенным, по крайней мере, одним Q в пара-положении фенильного кольца, и Q является фтором, карбокси, трифторметилом, 4-метилпиперазин-1-илом, дифторметокси, морфолин-1-илом, пиразол-1-илом или пирролидин-1-илом.
В некоторых случаях, R1 является гетероарилом, который замещен группой -C(O)N(R)-Q, и в любом другом положении - группой -O-Q или Q, в которых каждый Q в Q, -C(O)N(R)-Q, -O-Q независимо необязательно замещен 0-5 JQ.
В некоторых вариантах, Q в -C(O)N(R)-Q является Н, С1-4 алифатической группой, С1-4 галогеналифатической группой, С3-7 циклоалифатической группой, С3-7 гетероциклоалифатической группой, С1-6 алкоксигруппой, (С1-6 алкокси)С1-6 алкильной группой или С1-6 галогеналкоксигруппой.
В некоторых других вариантах, Q в -C(O)N(R)-Q является метилом, этилом, 1-метилпиперидин-4-илом, циклопропилом, циклопентилом, 3-фуранилом, 3-фторпирролидин-1-илом или 3,3-дифторциклобутилом.
В некоторых случаях, R1 является необязательно замещенным С1-6 алкилом или С3-7 циклоалкилом. В некоторых других случаях, R1 является Н, этилом, циклопропилом или циклопентилом.
В некоторых вариантах, R1 является фенилом, замещенным в пара-положении группой Q или -ZQ. В некоторых случаях, заместителем в пара-положении является фтор, карбокси, трифторметил, 4-метилпиперазин-1-ил, дифторметокси, морфолин-1-ил, пиразол-1-ил или пирролидин-1-ил.
В некоторых вариантах, R1 является тиофен-2-илом, пиридин-3-илом, пиридин-4-илом или 6-трифторметилпиридин-3-илом.
В некоторых вариантах, каждый R2 и R3 независимо представляет H или C1-3 алкил, необязательно и независимо замещенный 0-5 J2 и J3. В некоторых случаях R2 представляет H и R3 представляет C1-3 алкил, такой как этил.
В некоторых вариантах, каждый из R2 и R3, вместе с атомом углерода, к которому они присоединены, образуют 3-8-членное насыщенное или частично ненасыщенное моноциклическое кольцо, содержащее 0-4 гетероатомов, независимо выбранных из O, N и S, где указанное моноциклическое кольцо, образованное R2 и R3, необязательно замещено 0-4 J23.
В некоторых случаях, J23 является Н, галогеном, С1-4 алкильной группой, ОН, С1-4 алкоксигруппой, С1-4 галогеналкоксигруппой или аминогруппой.
В некоторых вариантах, каждый из J2 и J3 независимо является С1-6 алифатической группой, С3-6 циклоалифатической группой или -(С1-4алкил)n- V1, где
n является 0 или 1,
каждый V1 независимо является галоген(С1-4 алифатической группой), -О(галогенС1-4 алифатической группой), галогеном, NO2, CN, OH, OR``, SH, SR``, NH2, NHR``, N(R``)2, COH, COR``, CO2H, CO2R``, CONH2, CONHR``, CONR``2, OCOR``, OCONH2, OCONHR``, OCON(R``)2, NHCOR``, NR``COR``, NHCO2R``, NR``CO2R``, NHCO2H, NR``CO2H, NHCONH2, NHCONHR``, NHCON(R``)2, SO2NH2, SO2NHR``, SO2N(R``)2, NHSO2R``, NR``SO2R``, или
V1 является циклической группой, выбранной из С3-6 циклоалифатической группы, фенила, 5-6-членного гетероарила или 3-6-членного гетероциклила, где указанная циклическая группа необязательно замещена 0-3 JV; и
каждый R`` независимо является незамещенной С1-4 алифатической группой или две одинаковые J2 и J3, присоединенные к одному и тому же атому, вместе могут необязательно образовывать =О.
В некоторых случаях, каждый J2 и J3 независимо являются С1-6 алифатической группой, С3-6 циклоалифатической группой или -(С1-4алкил)n- V1, где
n является 0 или 1,
каждый V1 независимо является галоген(С1-4 алифатической группой), -О(галогенС1-4 алифатической группой), галогеном, NO2, CN, OH, SH, NH2, COH, CO2H, CONH2, OCONH2, NHCO2H, NHCONH2, SO2NH2 или V1 является циклической группой, выбранной из С3-6 циклоалифатической группы, фенила, 5-6-членного гетероарила или 3-6-членного гетероциклила, где указанная циклическая группа необязательно замещена 0-3 Jv; и
каждый R`` независимо является незамещенной С1-4 алифатической группой, или две одинаковые J2 и J3, присоединенные к одному и тому же атому, вместе могут необязательно образовывать =О.
В других вариантах, каждая J2 и J3 независимо является аминогруппой, амидной группой, CN, OH, С1-4 алкоксигруппой или С1-4 галогеналкоксигруппой и V1 является Н, галогеном, С1-4 алкильной группой, ОН, С1-4 алкоксигруппой, С1-4 галогеналкоксигруппой или аминогруппой.
В некоторых вариантах, R4 является С1-6 алифатической группой, С3-10 циклоалифатической группой, С3-10 гетероциклоалифатической группой, С6-14 арильной группой или С5-14 гетероарильной группой, каждая из которых необязательно замещена 0-5 J4.
В некоторых вариантах, R4 является циклопентилом.
В некоторых вариантах, каждый JQ, J4 и J23 независимо являются -М или -Y-M.
В некоторых вариантах, каждый Y независимо является ненасыщенной С1-6 алифатической группой, где 0-3 -СН2- звеньев в указанной С1-6 алифатической группе необязательно заменены на -NR-, -O-, -S-, -C(O)-, -S(O)- или -S(O)2-.
В другом варианте, каждый М независимо является Н, С1-6 алифатической группой, С3-6 циклоалифатической группой, галоген(С1-4 алифатической группой), О(галоген С1-4 алифатической группой), 3-6-членным гетероциклилом, галогеном, NO2, CN, OH, OR`, SH, SR`, NH2, NHR`, N(R`)2, COH, COR`, CO2H, CO2R`, CONH2, CONHR`, CONR2`, OCOR`, OCONH2, OCONHR`, OCON(R`)2, NHCOR`, NR`COR`, NHCO2R`, NR`CO2R`, NHCO2H, NR`CO2H, NHCONH2, NHCONHR`, NHCON(R`)2, SO2NH2, SO2NHR`, SO2N(R`)2, NHSO2R` или NR`SO2R`.
В других случаях, каждый М независимо является Н, С1-6 алифатической группой, С3-6 циклоалифатической группой, галоген(С1-4 алифатической группой), О(галоген С1-4 алифатической группой), 3-6-членным гетероциклилом.
В некоторых вариантах, соединения по настоящему изобретению могут быть представлены формулой II;

где X1, R1, R2, R3 и R4 описаны раннее, и JA является Н, С1-4 алкильной группой или ОН.
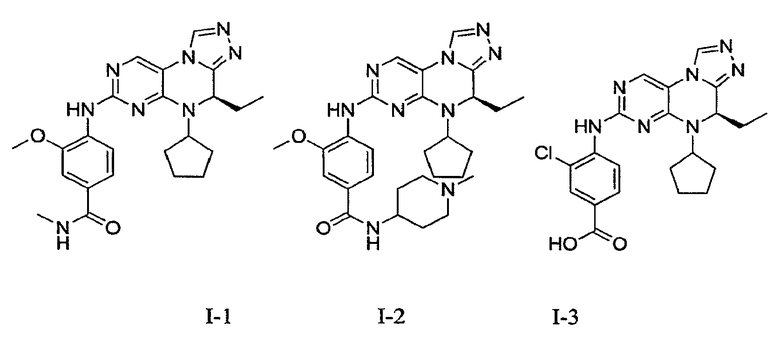
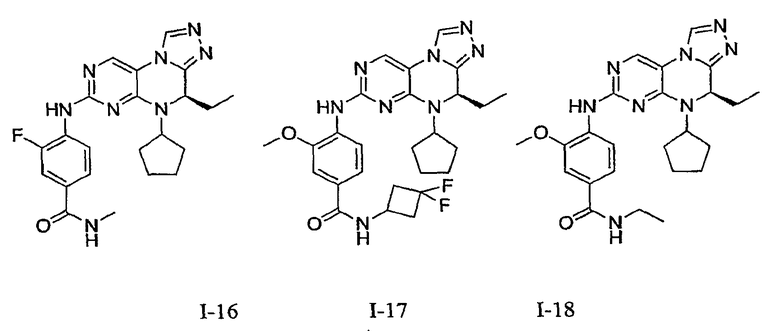
В некоторых вариантах, соединения настоящего изобретения представлены в Таблице 1
Таблица 1





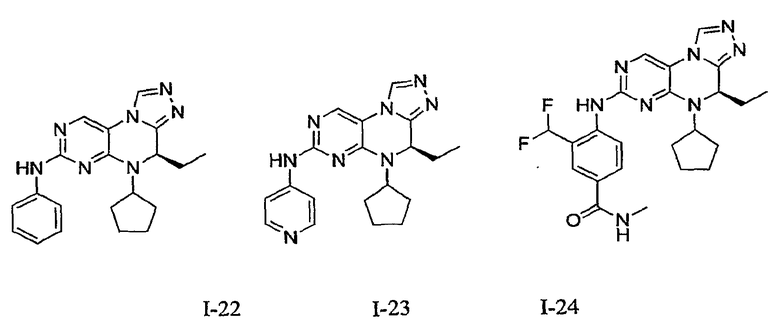


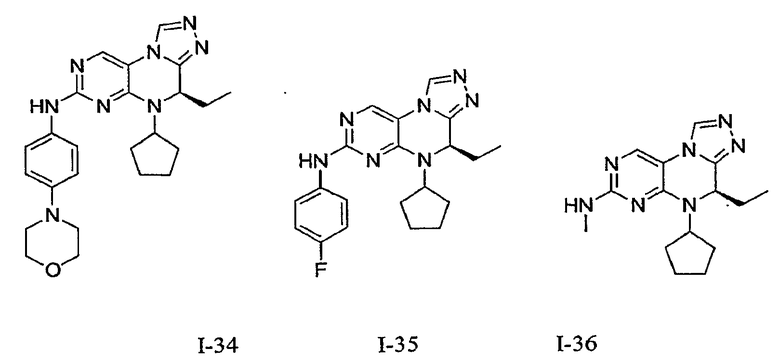
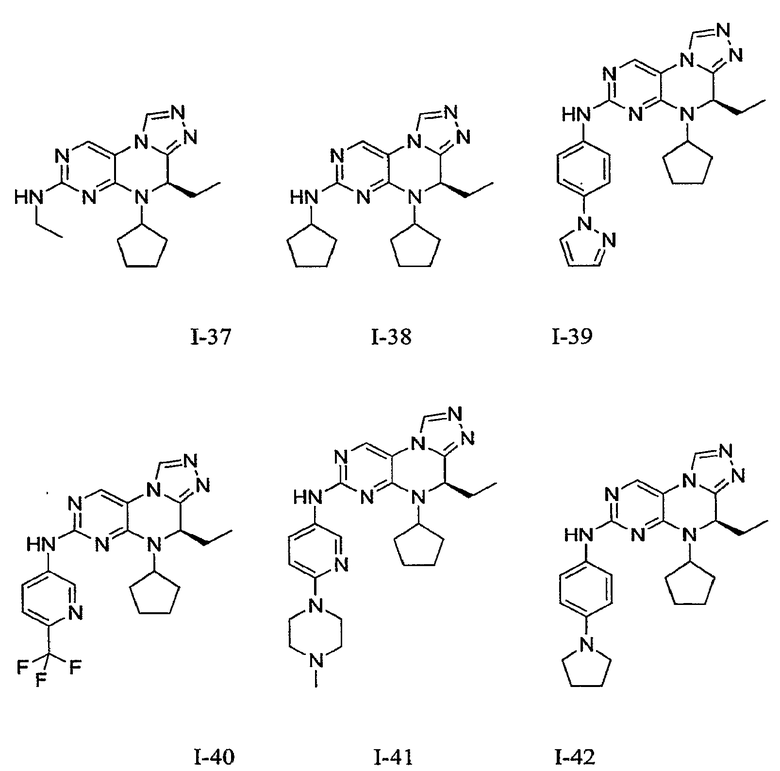
Общая синтетическая методология
В основном, соединения по настоящему изобретению, могут быть получены с помощью способов, которые описаны далее в общих схемах и в последующих препаративных примерах.
Схема 1
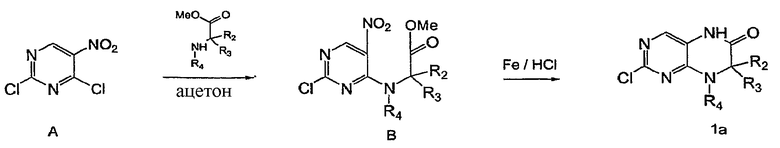
На приведенной выше Схеме 1 показан синтетический путь получения исходного соединения 1а для приведенной далее последовательности (см. US 20040176380). Хлор в положении 4 соединения А замещается аминоэфиром в ацетоне (или гексане), давая В. Восстановление нитрогруппы с последующей внутримолекулярной циклизацией in situ дает соединение 1а.
Схема 2

На приведенной выше Схеме 2 показан общий синтетический путь получения соединений настоящего изобретения. Лактамная функциональная группа в 1 (см. US 20040176380), где LG является подходящей уходящей группой, активируется при известных условиях, давая соединение 2 (Х может быть, но не ограничивается, галогеном, алкоксигруппой и фосфатом). Затем соединение 2 вводят в 1-2-стадийную последовательность (в зависимости от кольца А), с образованием соединений 4. В ином случае, карбониламид в 1 может быть превращен в тиокарбонил, давая тиолактам 3. Затем соединение 3 превращается в соединение 2 (Х=алкилтио) или вводится непосредственно в 1-2-стадийную последовательность (в зависимости от кольца А), с образованием соединений 4. На последней стадии LG может быть использована в качестве основы для получения соединений формулы I. На данной последней стадии LG, например, может быть замещена аминами или введена в известные палладий-катализируемые реакции сочетания, например реакции Сузуки, Стиле или Бухвальда.
Схема 3

На приведенной выше Схеме 3 показан общий синтетический путь получения соединений настоящего изобретения, где кольцо А является триазолом. Хлоримидат в 2А (Х=Cl) или фосфат (X=OP(O)OEt)2) в 2А реагирует с гидразином, давая промежуточное соединение 5. Реакция 5 с ортоэфирами JaC(OR)3 приводит к соединениям 4А, в которых кольцо А является триазолом. Альтернативно, хлоримидиат или фосфат (Х=ОР(О)(OEt)2)в 2А может взаимодействовать с ацилгидразинами JaC(O)NHNH2, непосредственно давая соединение 4А, где кольцо А является триазолом. На последней стадии LG может быть использована в качестве основы для получения соединений формулы IA. На данной последней стадии LG, например, может быть замещена аминами или введена в палладий-катализируемые реакции сочетания, известные в данной области техники (например, Сузуки, Стиле или Бухвальда).
Аналогичные подходы для превращения амидов R1-NH-CO-R2 в R1-триазол-R2 описаны в литературе, например:
- Trends в Het Chem, 8, 79-60, 2002,
- J Org Chem, 70(7), 2878-2880, 2005,
- Biorg Med Chem Lett, 15(19), 4359-4362, 2005.
Схема 4
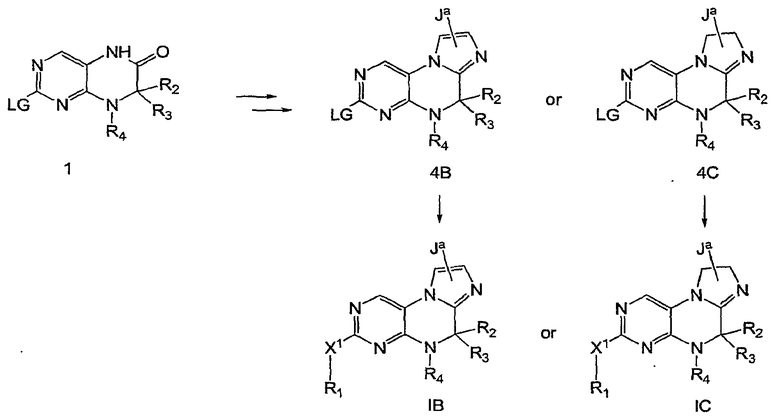
На приведенной выше Схеме 4 показан общий синтетический путь получения соединений настоящего изобретения, где кольцо А может быть, но не ограничено этим, имидазолом 1В или имидазолином 1С.
Аналогичные подходы для превращения амидов R1-NH-CO-R2 в R1-имидазол-R2 описаны в литературе, например:
- Afinidad, 45 (417), 443-446, 1988,
- J Org Chem, 59(7), 5084-5087, 1994,
- Hev. Chim. Acta, 80(3), 979-987, 1997,
- US 2004132708,
- Biorg Med Chem Lett, 12(21), 3219-3222, 2002.
Аналогичные подходы для превращения амидов R1-NH-CO-R2 в R1-имидазолин-R2 описаны в литературе, например:
- Afinidad, 45 (417), 443-446, 1988,
- J Het Chem, 19(1), 193-200, 1982,
- J Org Chem, 50(13), 2220-2224, 1985,
- Indian J Chem, 12(3), 263-269, 1974,
- Heterocecles, 60 (6), 1425-1432, 2003.
Аналогичные подходы для превращения амидов R1-NH-CO-R2 в R1-тетрагидропиримидины-R2 описаны в литературе, например:
J Am Chem Soc, 126(7), 1971-1979, 2004,
Angew Chemie, 43(4), 478-782, 2004,
J Am Chem Soc, 103(14), 4186-4194, 1981,
Indian J Chem, 12(3), 263-269, 1974.
В одном из вариантов, настоящее изобретение обеспечивает способ получения соединения формулы I:
где
X1, R1, R2, R3, R4 и кольцо А являются такими, как определено здесь.
Используемый в данном описании термин «реакция сочетания» относится к реакциям, в которых образуется связь углерод-углерод с помощью металлического катализатора. Обычно, один из атомов углерода связан с функциональной группой («группа кросс-сочетания»), тогда как другой атом углерода связан с галогеном. Примеры реакций кросс-сочетания включают, но не ограничиваются ими, кросс-сочетание Сузуки, кросс-сочетание Стиле, кросс-сочетание Негиши и кросс-сочетание Бухвальда.
Используемый в данном описании термин «группа сочетания» относится к функциональной группе, реагирующей с другой функциональной группой (например, галогеном) в реакции сочетания с образованием связи углерод-углерод («С-С») или связи углерод-азот («С-N»). В некоторых вариантах, С-С связь образуется между двумя ароматическими группами.
Используемый в данном описании термин «условия сочетания» относится к химическим условиям (например, температуре, продолжительности времени реакции, необходимому объему растворителя), требуемым для протекания реакции сочетания.
Примеры групп сочетания и их соответствующих условий сочетания включают, но не ограничиваются ими, бороновые кислоты и бороновые сложные эфиры в условиях сочетания Сузуки, SnBu3 при условиях сочетания Стиле и ZnX при условиях сочетания Негиши. Все эти три условия сочетания, обычно, включают использование катализатора, подходящего растворителя и необязательно основания. Условия сочетания Сузуки включают использование палладиевого катализатора, подходящего основания и подходящего растворителя. Примеры подходящих палладиевых катализаторов включают, но не ограничиваются ими, PdCl2(PPh3)2, Pd(PPh3)4 и PdCl2(dppf). Подходящие основания включают, но не ограничиваются ими, K2CO3 и Na2CO3. Подходящие растворители включают, но не ограничиваются ими, тетрагидрофуран, толуол и этанол.
Условия сочетания Стиле включают использование катализатора (обычно палладиевого, но иногда и никелевого), подходящего растворителя и других подходящих реагентов. Примеры подходящих катализаторов включают, но не ограничиваются ими, PdCl2(PPh3)2, Pd(PPh3)4 и PdCl2(dppf). Подходящие растворители включают, но не ограничиваются ими, тетрагидрофуран, толуол и диметилформамид.
Условия сочетания Негиши включают использование катализатора (палладиевого или никелевого) и подходящего растворителя. Примеры подходящих катализаторов включают, но не ограничиваются ими, Pd2(dba)3, Ni(PPh3)2Cl2, PdCl2(PPh3)2 и Pd(PPh3)4. Подходящие растворители включают, но не ограничиваются ими, тетрагидрофуран, толуол и диметилформамид.
Условия реакций Сузуки, Стиле и Негиши известны специалисту в области техники и более подробно описаны в различных источниках, включая “March`s Advanced Organic Chemistry”.
Условия сочетания Бухвальда включают использование палладиевого катализатора, подходящего основания и подходящего растворителя. Примеры подходящих палладиевых катализаторов включают, но не ограничиваются ими, Pd(OAc)2 c Xanthphos, PdCl2(PPh3)2, Pd(PPh3)4 и PdCl2(dppf). Подходящие основания включают, но не ограничиваются ими, Cs2CO3, K2CO3 и Na2CO3. Подходящие растворители включают, но не ограничиваются ими, диоксан, тетрагидрофуран, толуол и этанол.
Как будет ясно специалисту в данной области техники, группы сочетания образуются из предшественников групп сочетания. Предшественник группы сочетания является реагентом или группой реагентов, используемых для получения группы кросс-сочетания. Примеры включают, но не ограничиваются ими, бис(пинаколато)диборан для получения бороновых эфиров, триметилбораты для получения бороновых кислот, Bu3SnCl для получения станнанов и ZnCl2 для получения цинкатов в реакциях сочетания Негиши. Примеры подходящих условий образования групп сочетания включают, но не ограничиваются ими, создание бороновых эфиров через палладий-катализируемые реакции; создание бороновых кислот с помощью гидролиза бороновых эфиров; создание станнатов в ходе двухстадийного процесса: 1) обмена галогена на металл с последующим 2) трансметаллированием с помощью Bu3SnCl; и создание цинкатов в ходе двухстадийного процесса: 1) обмена галогена на металл с последующим 2) добавлением ZnCl2.
В другом аспекте, настоящее изобретение обеспечивает соединения, которые являются ингибиторами протеинкиназ и, таким образом, которые являются пригодными для лечения заболеваний, расстройств и патологических состояний, наряду с другими применениями, которые описаны в данном описании. В другом аспекте, настоящее изобретение обеспечивает фармацевтически приемлемые композиции, где данные композиции содержат любые описанные в данном описании соединения и необязательно содержат фармацевтически приемлемый носитель, вспомогательное вещество и наполнитель. В некоторых вариантах, данные композиции необязательно также содержат один или несколько дополнительных терапевтических агентов.
Наносящее изобретение обеспечивает соединения и композиции, которые являются пригодными в качестве ингибиторов протеинкиназ. В некоторых вариантах, протеинкиназами являются PLK. В некоторых вариантах - PLK1.
В качестве ингибиторов протеинкиназ, соединения и композиции по настоящему изобретению, в частности, являются пригодными для лечения или уменьшения тяжести заболевания, состояния или расстройства, где протеинкиназа участвует в заболевании, состоянии или расстройстве. В одном из аспектов, настоящее изобретение обеспечивает способы лечения или уменьшения тяжести заболевания, состояния или расстройства, где протеинкиназа принимает участие в болезненном состоянии. В другом аспекте, настоящее изобретение обеспечивает способ лечения или уменьшения тяжести киназного заболевания, состояния или расстройства, где ингибирование ферментной активности участвует в лечении заболевания. В другом аспекте, настоящее изобретение обеспечивает способ лечения или уменьшения тяжести заболевания, состояния или расстройства соединениями, которые ингибируют ферментную активность, связываясь с протеинкиназой. Другой аспект обеспечивает способ лечения или уменьшения тяжести киназного заболевания, состояния или расстройства посредством ингибирования ферментной активности киназы ингибитором протеинкиназы.
В некоторых вариантах, указанным ингибитором протеинкиназы является ингибитор PLK.
Один из аспектов настоящего изобретения относится к способу ингибирования активности протеинкиназы у пациента, причем способ включает введение пациенту соединения формулы I или композиции, содержащей указанное соединение.
В некоторых вариантах, указанный способ применяется для лечения или профилактики состояния, выбранного из аутоиммунных заболеваний, воспалительных заболеваний, пролиферативных заболеваний и гиперпролиферативных заболеваний, заболеваний, опосредованных иммунной системой, болезни костей, метаболических заболеваний, неврологических и нейродегенеративных заболеваний, сердечно-сосудистых заболеваний, гормональных заболеваний, аллергии и астмы, болезни Альцгеймера. В некоторых вариантах, указанные протеинкиназы входят в семейства PLK. В других вариантах, указанные состояния выбраны из пролиферативных заболеваний и нейродегенеративных заболеваний.
В зависимости от конкретных опосредованных протеинкиназой состояний, которые необходимо лечить или предотвратить, вместе с ингибиторами по настоящему изобретению могут быть введены дополнительные лекарства, которые обычно применяются для лечения или профилактики данных состояний. Например, для лечения пролиферативных заболеваний, хемотерапевтические агенты или другие антипролиферативные агенты могут быть объединены с ингибиторами протеинкиназ настоящего изобретения.
Указанные дополнительные агенты могут быть введены отдельно от соединения или композиции, содержащей ингибитор протеинкиназы, как часть многократной схемы приема лекарственного средства. В ином случае, данные агенты могут быть частью одной дозированной формы, смешанной с ингибитором протеинкиназы в одной композиции.
Соединения настоящего изобретения также пригодны в качестве ингибиторов протеинкиназ в биологических образцах. Один из аспектов изобретения относится к способу ингибирования активности протеинкиназы в биологических образцах, который включает контактирование указанного биологического образца с соединением формулы I или содержащей указанное соединение композиции. Используемый в данном описании термин «биологический образец» означает in vitro или ex vivo образец, включающий, не ограничиваясь этим, клеточные культуры или их экстракты, материал для биопсии, полученный из млекопитающего, или их экстракты; кровь, слюну, мочу, кал, семенную жидкость, слезы или другие жидкости организма или его экстракты.
Специалистам в данной области техники известно, что ингибирование активности протеинкиназы в биологических образцах является полезным для различных целей. Примеры таких целей включают, но не ограничиваются ими, переливание крови, пересадка органов и хранение биологических образцов.
Другой аспект настоящего изобретения относится к изучению протеинкиназ в биологических и патологических процессах; изучению путей внутриклеточного преобразования сигнала, опосредованного такими протеинкиназами; и сравнительной оценке новых ингибиторов протеинкиназ. Примеры таких применений включают, но не ограничиваются ими, биологические анализы, такие как анализы ферментов и анализы на основе клеток.
Активность соединений в качестве ингибиторов протеинкиназ может быть исследована in vitro, in vivo или на линии клеток. Анализы in vitro включают анализы, которые определяют ингибирование как киназной активности, так и АТФазной активности активированной киназы. Чередование тестов in vitro позволяет произвести количественный анализ способности связывания ингибитора с протеинкиназой, что может быть измерено с помощью введения радиоактивных меток в ингибитор перед связыванием, выделением ингибитор/киназного комплекса и определением количества связанного радиомеченного вещества или проведением конкурентного эксперимента, в котором новые ингибиторы инкубируются с киназой, присоединенной к известным радиолигандам. Подробные условия для проведения анализов соединений, используемых в настоящем изобретении в качестве ингибиторов PLK1, PLK2, PLK3 и PLK4, приведены в Примерах далее.
Один аспект настоящего изобретения обеспечивает соединения, которые пригодны для лечения заболеваний, расстройств и состояний, характеризующихся чрезмерной или аномальной клеточной пролиферацией. Такие заболевания включают пролиферативные или гиперпролиферативные заболевания и нейродегенеративные заболевания.
Примеры пролиферативных и гиперпролиферативных заболеваний, без ограничения, включают рак.
Термин «рак» включает, но не ограничивается ниже перечисленными видами рака, рак груди; яичников; шейки матки; простаты; яичка; мочеполового тракта; пищевода; гортани; глиобластомы; нейробластомы; желудка; кожи; кератоакантомы; легкого; эпидермойдной карциномы; крупноклеточный рак; мелкоклеточный рак; аденокарциному легкого; костей; кишечника; колоректальный рак; аденому; поджелудочной железы; аденокарциному; щитовидной железы; фолликулярную карциному; недифференцированную карциному; папиллярный рак; семиному; меланому; саркому; карциному мочевого пузыря; карциному печени и желчного протока; почечную карциному; миелоидные расстройства; лимфоидные расстройства; ворсистых клеток Ходжкина; полости рта и носоглотки (рот); губ; языка; рта; глотки; тонкой кишки; ободочной кишки; толстого кишечника; прямой кишки; мозга и центральной нервной системы; хронический миелолейкоз (CML) и лейкемию.
Примеры нейродегенеративных заболеваний, без ограничений, включают болезнь Альцгеймера.
Другой аспект настоящего изобретения обеспечивает способ лечения или уменьшения тяжести заболевания, выбранного из пролиферативного или гиперпролиферативного заболевания или нейродегенеративного заболевания, включающий введение эффективного количества соединения или фармацевтически приемлемой композиции, содержащей соединение, нуждающемуся в этом субъекту.
В некоторых вариантах, «эффективным количеством» соединения или фармацевтически приемлемой композиции является такое количество, которое эффективно для лечения указанного заболевания. Соединения или композиции согласно способам настоящего изобретения могут быть введены с использованием любого количества и посредством любого пути введения, эффективного для лечения или уменьшения тяжести указанного заболевания.
В некоторых вариантах, указанное заболевание является состоянием, опосредованным протеинкиназой. В некоторых вариантах, указанное заболевание является заболеванием, опосредованным PLK.
Используемый здесь термин «состояние, опосредованное протеинкиназой» означает любое заболевание или другое пагубное состояние, в котором участвует протеинкиназа. Такие состояния включают, но не ограничиваются, аутоиммунные заболевания, воспалительные заболевания, пролиферативные и гиперпролиферативные заболевания, заболевания опосредованные иммунной системой, болезни костей, метаболические нарушения, неврологические и нейродегенеративные заболевания, сердечно-сосудистые заболевания, гормональные заболевания, аллергии и астму, болезнь Альцгеймера.
Используемый в данном описании термин «патологическое состояние, опосредованное PLK» означает любое заболевание или другое пагубное состояние, в котором участвует PLK. Такие патологические состояния включают, без ограничения, пролиферативные или гиперпролиферативные заболевания или нейродегенеративные заболевания.
В другом аспекте настоящего изобретения обеспечиваются фармацевтически приемлемые композиции, где данные композиции содержат любые соединения, как описано в настоящем описании, и необязательно содержат фармацевтически приемлемый носитель, адъювант и наполнитель.
В некоторых вариантах, данные композиции также необязательно содержат один или несколько дополнительных терапевтических агентов.
Например, с соединениями настоящего изобретения могут быть объединены хемотерапевтические агенты или другие антипролиферативные агенты для лечения пролиферативных заболеваний и рака.
Примеры известных хемотерапевтических агентов включают, но не ограничиваются ими, GleevecTM, адриамицин, дексаметазон, винкристин, циклофосфамид, фторурацил, топотекан, таксол, интерфероны и производные платины.
Другие примеры агентов, с которыми также могут быть объединены ингибиторы настоящего изобретения, включают без ограничения: лекарственные средства от болезни Альцгеймера, такие как Aricept® и Excelon®; лекарственные средства от болезни Паркинсона, такие как L-ДОФА/карбидопа, энтакапон, ропинрол, прамипексол, бромокриптин, перголид, тригексефендил и амантадин; агенты для лечения рассеянного склероза (MS), такие как бетааинтерферон (например, Avonex® и Rebif®), Copaxone® и митксантрон; для лечения астмы, такие как альбутерол и Singular®; агенты для лечения шизофрении, такие как зипрекса, риспердал, серокуэль и галоперидол; противовоспалительные агенты, такие как кортикостероиды, блокаторы фактора некроза опухоли, IL-1 RA, азатиоприн, циклофосфамид и сульфасалазин; иммуномодулирующие и иммунодепрессантные агенты, такие как циклоспорин, такролимус, рапамицин, мофетила микофенолят, интерфероны, кортикостероиды, циклофосфамид, азатиоприн и сульфасалазин; нейротропные факторы, такие как ингибиторы ацетилхолинэстеразы, ингибиторы МАО, интерфероны, противосудорожные средства, блокаторы ионных каналов и статины; агенты для лечения заболеваний печени, такие как кортикостероиды, холестирамины, интерфероны и противовирусные агенты; агенты для лечения заболеваний крови, такие как кортикостероиды, противолейкозные средства и факторы роста; и агенты для лечения иммунодефицита, такие как гаммаглобулин.
Как описано в данном описании, фармацевтически приемлемые композиции по настоящему изобретению дополнительно содержат фармацевтически приемлемый носитель, адъювант или наполнитель, которые, как используется в данном описании, включают любые и все растворители, разбавители или другие жидкие среды, диспергирующие или суспендирующие вспомогательные средства, поверхностно-активные агенты, изотонические агенты, загустители или эмульгаторы, консерванты, твердые связующие агенты, лубриканты и т.п., подходящие для конкретной требуемой дозируемой формы. Различные носители, используемые для получения фармацевтически приемлемых композиций, и известные способы их получения описаны в Remington`s Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Eatson, Pa., 1980). За исключением любых обычных сред носителя, которые являются несовместимыми с соединениями настоящего изобретения, например, давая нежелательный биологический эффект или, иначе говоря, пагубно взаимодействуют с любым другим компонентом(и) фармацевтически приемлемой композиции, предусматривается, что их применение находится в объеме настоящего изобретения.
Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничиваются ими, ионообменное вещество, окись алюминия, стеарат алюминия, лектин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота или сорбат калия, частичные смеси глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный оксид кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, блок-сополимеры полиэтилена и полиоксипропилена, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлозу и стеарат целлюлозы; порошковый трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и суппозиторные воски; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль или полиэтиленгликол; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксические совместимые лубриканты, такие как лаурилсульфат натрия и стеарат магния, а также, по усмотрению разработчика рецептур, в композиции могут присутствовать красящие агенты, высвобождающие агенты, покрывающие агенты, подсластители, вкусовые и ароматизирующие добавки, консерванты и антиоксиданты.
Ингибиторы протеинкиназы или их фармацевтически приемлемые соли могут быть включены в фармацевтические композиции для введения животным или людям. В другом варианте настоящего изобретения, данные фармацевтические композиции содержат количество ингибитора белка, которое эффективно для лечения или профилактики состояния, опосредованного протеинкиназой, и фармацевтически приемлемый носитель. В некоторых вариантах, указанное состояние, опосредованное протеинкиназой, является состоянием, опосредованным PLK.
Точное количество соединения, необходимое для лечения, будет различаться от субъекта к субъекту в зависимости от вида, возраста и, в основном, от общего состояния субъекта, тяжести заражения, конкретного агента, способа его введения и т.п. Предпочтительно, соединения по настоящему изобретению получают в единичной дозированной форме для легкости введения дозы и однородности дозы. Используемое в данном описании выражение «единичная дозированная форма» относится к дискретной единице агента, подходящего для пациента, который нуждается в лечении. Однако следует понимать, что общая ежедневная доза соединений и композиций по настоящему изобретению будет устанавливаться лечащим врачом в рамках тщательного медицинского обследования. Конкретный уровень эффективной дозы для любого конкретного пациента или организма будет зависеть от различных факторов, включая заболевание, которое необходимо вылечить, и тяжести заболевания; активности конкретного применяемого соединения; конкретной применяемой композиции; возраста, веса тела, общего состояния здоровья, пола и режима питания пациента; времени введения, пути введения и скорости выведения конкретного применяемого соединения; времени лечения; лекарств, используемых в комбинации или совместно, с конкретным применяемым соединением и других известных факторов, известных в медицине. Используемый здесь термин «пациент» означает животное, предпочтительно млекопитающее и более предпочтительно человека.
Фармацевтически приемлемые композиции по настоящему изобретению могут быть введены людям и другим животным орально, ректально, парентерально, интрацистернально, внутривагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), букально, в виде оральных или нозальных спреев и т.п., в зависимости от тяжести заболевания, которое необходимо вылечить. В некоторых вариантах, соединения по настоящему изобретению могут быть введены орально или парентерально при уровнях дозы примерно от 0,01 мг/кг до примерно 50 мг/кг и, предпочтительно, от примерно 1 мг/кг до примерно 25 мг/кг на вес субъекта в день, один или несколько раз в день для достижения требуемого терапевтического эффекта.
Жидкие дозированные формы для орального введения включают, но не ограничиваются ими, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и элексиры. Кроме активных соединений жидкая дозированная форма может содержать инертные разбавители, которые обычно используются в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, пшеничное, оливковое, смесь касторового и минерального масел и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, пропиленгликоли и эфиры жирных кислот сорбитана и их смеси. Помимо инертных разбавителей, композиции для орального введения также могут содержать вспомогательные вещества, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подсластители, вкусовые и ароматизирующие добавки.
Инъецируемые составы, например стерильные инъецируемые водные или масляные суспензии, могут быть получены согласно известной технике с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильные инъецируемые составы также могут быть стерильными инъецируемыми растворами, суспензиями или эмульсиями в нетоксичном пригодном для парентерального введения разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Пригодными наполнителями и растворителями, которые могут применяться, является вода, раствор Рингера, U.S.P. и изотонический раствор хлорида натрия. Кроме этого в качестве растворителя или суспендирующей среды обычно используются стерильные, жирные масла. Для этих целей могут применяться любые смеси жирных масел, включая синтетические моно- и диглицериды. Кроме этого для получения инъецируемых составов используются жирные кислоты, такие как олеиновая кислота.
Инъецируемые составы могут стерилизоваться, например, посредством фильтрации через фильтры, удерживающие бактерии, или посредством включения стерилизующих агентов в форме стерильной твердой композиции, которая может быть растворена или диспергирована в стерильной воде или другой стерильной инъецируемой среде перед использованием. С целью пролонгированного эффекта соединения по настоящему изобретению, часто полезным является понижение абсорбции соединения из подкожной или внутримышечной инъекции. Это может быть достигнуто посредством применения жидких суспензий кристаллического или аморфного вещества с низкой растворимостью в воде. Скорость абсорбции вещества зависит от скорости его растворения, что, в свою очередь, зависит от размера кристалла и кристаллической формы. Кроме этого замедленная абсорбция вещества, введенного в форме для парентерального применения, достигается посредством растворения или суспендирования соединения в масляной среде. Инъецируемые формы депо замедленного всасывания достигаются образованием микрокапсульных матриц соединения в биоразрушаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения соединения к полимеру и природы конкретного применяемого полимера, можно контролировать скорость высвобождения соединения. Примеры других биоразрушаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъецируемые депо композиции также получают посредством включения соединения в липосомы или микроэмульсии, которые совместимы с тканями тела.
Композиции для ректального или вагинального введения предпочтительны в виде суппозиториев, которые могут быть получены посредством смешения соединений настоящего изобретения с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или суппозиторный воск, которые являются твердыми при комнатной температуре, но являются жидкими при температуре тела и, следовательно, плавятся в полости прямой кишки или вагины и высвобождают активное соединение.
Твердые дозированные формы для орального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В такой твердой дозированной форме активное соединение смешано, по крайней мере, с одним инертным, фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или фосфат дикальция и/или а) наполнителями или добавками, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и салициловая кислота, b) связующими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и камедь, с) смачивающими средствами, такими как глицерин, d) дезинтегрирующими агентами, такими как агар-агар, карбонат кальция, картофельный и маниоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия, е) агентами, замедляющими растворение, такими как парафин, f) ускорителями абсорбции, такими как четвертичные аммониевые соединения, g) смачивающими агентами, например цетиловым спиртом и моностеаратом глицерина, h) абсорбентами, такими как каолиновая и бентонитовая глина, и i) лубрикантами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль, дозированная форма может также содержать буферные вещества.
Твердые композиции аналогичного типа могут также применяться в качестве наполнителей в мягких и твердых наполненных желатиновых капсулах, используя такие эксципиенты, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п. Твердые дозированные формы таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, или другими покрытиями, хорошо известными в фармацевтической области техники. Возможно, они могут содержать замутняющие агенты и также могут быть композицией, которая высвобождает активное вещество(а) только, или преимущественно, в определенной части кишечника необязательно отсроченным образом. Примеры матричных композиций, которые могут быть использованы, включают полимерные вещества и воски. Твердые композиции аналогичного типа также могут применяться в качестве наполнителей в мягких и твердых наполненных желатиновых капсулах, используя такие эксципиенты, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п.
Активные соединения также могут быть в микроинкапсулированной форме с одним или несколькими эксципиентами, как указано выше. Твердые дозированные формы таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, покрытия контролируемого высвобождения или другими покрытиями, хорошо известными в фармацевтической области техники. В таких твердых дозированных формах активное соединение может быть смешано, по крайней мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие дозированные формы могут также содержать, что является обычной практикой, дополнительные вещества, отличные от инертных разбавителей, например таблетирующие лубриканты и другие таблетирующие вспомогательные средства, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, дозированные формы могут также содержать буферные вещества. Возможно, они могут содержать замутняющие агенты и также являться композицией, которая высвобождает активный ингредиент(ы) только, или преимущественно, в определенной части кишечника необязательно отсроченным образом. Примеры матричных композиций, которые могут быть использованы, включают полимерные вещества и воски.
Дозированные формы для местного или трансдермального введения соединения по настоящему изобретению включают мази, пасты, крема, примочки, гели, порошки, растворы, спреи, средства для ингаляции и пластыри. Может быть необходимым, чтобы активный компонент был смешан при стерильных условиях с фармацевтически приемлемым носителем и необходимыми антисептиками или буферами. Предусматривается, что офтальмологические составы, ушные капли и глазные капли также входят в объем настоящего изобретения. Кроме этого настоящее изобретение предусматривает применение трансдермальных пластырей, которые имеют дополнительное преимущество при обеспечении контролированной доставки соединения к телу. Такие дозированные формы могут быть получены посредством растворения или диспергирования соединения в подходящей среде. Также могут использоваться агенты, усиливающие абсорбцию, для увеличения потока соединения через кожу. Скорость может регулироваться как посредством использования контролирующих скорость мембран, так и с помощью диспергирования соединения в полимерных матрицах или гелях.
Кроме соединений по настоящему изобретению для лечения или профилактики вышеуказанных заболеваний в композициях могут также использоваться фармацевтически приемлемые производные или пролекарства соединений настоящего изобретения.
Соединения по настоящему изобретению также могут существовать в виде фармацевтически приемлемых производных.
«Фармацевтически приемлемое производное» является аддуктом или производным, которое, при введении нуждающемуся пациенту, способно напрямую или косвенно давать соединение, описанное в настоящем описании, или его метаболит, или его остаток. Примеры фармацевтически приемлемых производных включают, но не ограничиваются ими, сложные эфиры и соли таких сложных эфиров.
«Фармацевтически приемлемое производное или пролекарство» означает любой фармацевтически приемлемый сложный эфир, соль сложного эфира или другое производное соединения по настоящему изобретению, которое, при введении реципиенту, способно обеспечить, напрямую или косвенно, соединение по настоящему изобретению, или его ингибирующе активный метаболит, или его остаток. Конкретными предпочтительными производными или пролекарствами являются такие, которые увеличивают биодоступность соединений по настоящему изобретению при введении таких соединений пациенту (например, позволяя быстрее абсорбироваться в крови при оральном введении) или которые увеличивают доставку исходного соединения к биологическому компартменту (например, мозгу или лимфатической системе) по отношению к исходному соединению.
Фармацевтически приемлемые пролекарства соединений по настоящему изобретению, без ограничения, включают сложные эфиры, сложные эфиры аминокислот, фосфатные эфиры, соли металлов и сульфонатные эфиры.
Фармацевтически приемлемые носители, которые могут быть использованы в данных фармацевтических композициях, включают, но не ограничиваются ими, ионообменные вещества, окись алюминия, стеарат алюминия, лектин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота или сорбат калия, частичные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный оксид кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, блок-сополимеры полиэтилена и полиоксипропилена, полиэтиленгликоль и ланолин.
Композиции по настоящему изобретению могут быть введены орально, парентерально, посредством ингаляционного спрея, местно, ректально, назально, букально, вагинально или через имплантированный резервуар. Используемый в данном описании термин «парентерально» включает, но не ограничивается, подкожный, внутривенный, внутрисуставный, внутрисиновиальный, надчревный, интратекальный, интралесиональный и интракраниальный способы инъекций или вливаний. Предпочтительно, композиции вводятся орально, интраперитонеально или внутривенно.
Стерильные инъецируемые формы композиций по настоящему изобретению могут быть водными или маслянистыми суспензиями. Данные суспензии могут быть получены согласно известным способам в данной области техники с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильные инъецируемые препараты также могут быть стерильными инъецируемыми растворами или суспензиями в нетоксичном пригодном для парентерального введения разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Пригодными средами и растворителями, которые могут применяться, является вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме этого в качестве растворителя или суспендирующей среды обычно используются стерильные жирные масла. Для этих целей могут применяться любые смеси жирных масел, включая синтетические моно- и диглицериды. Для получения инъецируемых препаратов пригодными являются жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, тогда как природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно пригодны в их полиоксиэтилированных формах. Данные масляные растворы или суспензии также могут содержать длинноцепочечные спиртовые разбавители или диспергирующие агенты, такие как карбоксиметилцеллюлоза или аналогичные диспергирующие агенты, которые обычно используются при получении фармацевтически приемлемых дозированных форм, включая эмульсии и суспензии. Также, для целей получения композиции могут быть использованы другие традиционно применяемые поверхностно-активные вещества, такие как Tweens, Spans и другие эмульгаторы или усилители биодоступности, которые обычно используются при производстве фармацевтически приемлемых твердых, жидких или других дозированных форм.
Фармацевтические композиции по настоящему изобретению могут быть введены орально в любой дозированной форме для орального применения, включая, но не ограничиваясь ими, капсулы, таблетки, водные суспензии и растворы. В случае таблеток для орального применения, обычно используемые носители включают, но не ограничиваются, лактозу и кукурузный крахмал. Также обычно добавляют лубриканты, такие как стеарат магния. Для орального введения в виде капсул, пригодные разбавители включают лактозу и высушенный кукурузный крахмал. Если для орального применения необходимы водные суспензии, то активное вещество смешивают с эмульгаторами и суспендирующими агентами. Также, при необходимости, могут быть добавлены подсластители, ароматизирующие или красящие средства.
Кроме этого фармацевтические композиции по настоящему изобретению могут быть введены в форме суппозиториев для ректального введения. Они могут быть получены посредством смешения агентов с подходящими нераздражающими эксципиентами, которые являются твердыми при комнатной температуре, но жидкими при температуре прямой кишки и, следовательно, будут плавиться в полости прямой кишки, высвобождая активное соединение. Такие вещества включают, но не ограничиваются ими, масло какао, пчелиный воск и полиэтиленгликоли.
Также, композиции по настоящему изобретению могут быть введены местно, особенно если мишень обработки включает легкодоступные области или органы для местного применения, включая заболевания глаз, кожи или нижнего кишечника. Для каждой из этих областей или органов легко получают подходящие композиции для местного применения.
Местное применение для нижнего кишечника может быть выполнено в виде ректальных суппозиториев (см. выше) или в виде подходящего состава для клизмы. Также могут быть использованы местные трансдермальные пластыри.
Для местного применения, фармацевтические композиции могут быть выполнены в виде подходящих мазей, содержащих активное вещество, суспендированное или растворенное в одном или нескольких носителях. Носители для местного введения соединений по настоящему изобретению включают, но не ограничиваются ими, минеральное масло, вазелиновое масло, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгирующий воск и воду. Кроме этого фармацевтические композиции могут быть получены в виде подходящей примочки или крема, содержащих активные вещества, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Подходящие носители включают, но не ограничиваются ими, минеральное масло, сорбитан моностеарат, полисорбат 60, воск цетиловых эфиров, цетариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
Для офтальмологического применения, фармацевтические композиции могут быть получены в виде микронизированных суспензий в изотоническом, стерильном физиологическом растворе с отрегулированным значением рН или, предпочтительно, в изотонических, стерильных физиологических растворах с отрегулированным значением рН с или без консерванта, такого как бензилалконийхлорид. С другой стороны, для офтальмологического применения, фармацевтические композиции могут быть получены в виде мази, такой как вазелин.
Фармацевтические композиции по настоящему изобретению могут быть введены посредством назального аэрозоля или ингаляции. Такие композиции получают согласно хорошо известным способам в области техники получения фармацевтических препаратов, и они могут быть получены в виде растворов в солевом растворе, с использованием бензилового спирта или других подходящих консервантов, усилителей абсорбции для улучшения биодоступности, фторуглеродов и/или других традиционных солюбилизирующих или диспергирующих агентов.
Количество ингибитора протеинкиназы, которое может быть смешано с веществами носителя для получения единичной дозированной формы, будет изменяться в зависимости от реципиента, конкретного способа введения. Предпочтительно, композиции должны создаваться таким образом, чтобы пациенту, принимающему данные композиции, можно было ввести от 0,01 до 100 мг/кг веса тела/день ингибитора.
Следует понимать, что конкретная доза и режим лечения для любого конкретного пациента будет зависеть от различных факторов, включая активность конкретного применяемого соединения, возраст, вес тела, общее состояние здоровья, пол, режим питания, время введения, скорость выведения, комбинирование лекарственных средств и решение лечащего врача и тяжесть конкретного заболевания, которое лечится. Также, количество ингибитора будет зависеть от конкретного соединения в композиции.
Согласно другому варианту изобретение обеспечивает способы лечения или профилактики состояний, опосредованных протеинкиназой (в некоторых вариантах состояний, опосредованных PLK), включающие стадию введения пациенту одной из вышеописанных фармацевтических композиций. Используемый в данном описании термин «пациент» означает животное, предпочтительно человека.
В некоторых вариантах, указанный способ применяется для лечения или профилактики состояния, выбранного из пролиферативного заболевания, такого как рак, нейродегенеративного заболевания, аутоиммунного заболевания, воспалительного заболевания и заболевания, опосредованного иммунной системой. В некоторых вариантах, указанный способ применяется для лечения или профилактики состояния, выбранного из рака груди, кишечника, простаты, кожи, поджелудочной железы, мозга, мочеполовой системы, лимфатической системы, желудка, гортани и легких, включая аденокарциному легкого и мелкоклеточный рак легких; удара, диабета, миеломы, гепатомегалии, кардиомегалии, болезни Альцгеймера, фиброзно-кистозной дегенерации и вирусных заболеваний или любых других конкретных заболеваний, описанных выше.
Соединения по настоящему изобретению могут быть получены с помощью способов, известных специалисту в данной области техники. Данные соединения могут быть проанализированы посредством известных способов, включая, но не ограничиваясь, LCMS (жидкостная хроматография с масс-спектрометрическим детектированием). Соединения по настоящему изобретению также могут быть испытаны, согласно этим примерам. Следует понимать, что конкретные условия, приведенные ниже, являются лишь примерами и не предназначены для ограничения пределов условий, которые могут быть использованы для создания, анализа и тестирования соединений по настоящему изобретению. Наоборот, настоящее изобретение также включает условия, известные специалисту в данной области техники, для создания, анализа и тестирования соединений по настоящему изобретению.
ПРИМЕРЫ
Используемый в данном описании термин «RT(мин)» относится к времени удерживания ВЭЖХ в минутах, относящихся к соединению. Если не указано иное, то используемый метод ВЭЖХ для получения времени удерживания является следующим:
Колонка: ACE C8 колонка, 4,6×150 мм.
Градиент: 0-100% ацетонитрил+метанол 60:40 (20мМ Трис фосфат).
Скорость потока: 1,5 мл/минуту.
Детектирования: 225 нм.
Масс-спектральный анализ образцов был проведен на масс-спектрометре MicroMass Quattro Micro, работающем в одиночном MS режиме с ионизацией в электроспрее. Образцы вводили в масс-спектрометр с использованием хроматографии.
1Н ЯМР спектры записаны при 400 МГц с использованием прибора Bruker DPX 400 и приведены в м.д. δ. Следующие соединения формулы I получают и анализируют согласно нижеприведенному.
Пример 1:
4-(R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-метокси-N-метилбензамид (I-1)

Способ А: (R)-7-хлор-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин

(R)-2-хлор-8-циклопентил-7-этил-7,8-дигидроптеридин-6(5Н)-он (100 мг, 0,357 ммоль) в хлорокисе фосфора (3,0 мл) перемешивали при 110°С в течение 3 часов, затем концентрировали при пониженном давлении. Остаток растворяли в безводном дихлорметане и по каплям добавляли к 1,0 М раствору гидразина в ТГФ (3,6 мл, 3,57 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, переносили в этилацетат и промывали водным гидрокарбонатом натрия, сушили над сульфатом магния и концентрировали. Остаток сырого гидрозида растворяли в триметилортоформиате (2,0 мл) и перемешивали при 110°С в течение 90 минут. Смесь концентрировали при пониженном давлении и очищали с помощью флэш хроматографии на силикагеле, элюировали этилацетатом, с получением (R)-7-хлор-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридина (68 мг, 63%) в виде бледно-коричневого твердого вещества;
1Н ЯМР (ДМСО D6) 0,75 (3H, т), 1,50-1,64 (2H, м), 1,80-2,08 (8H, м), 4,22-4,33 (1H, м), 5,28-5,35 (1H, м), 8,68 (1H, с), 9,35 (1H, с); MS (ES+) 305.
Способ В: 4-(R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-метокси-N-метилбензамид (I-1)
К раствору (R)-7-хлор-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридина (80 мг, 0,263 ммоль) в смеси этанол/вода (1/4, 5 мл) добавляли 4-амино-3-метокси-N-метилбензамид (72 мг, 0,394 ммоль) с последующим добавлением каталитических количеств концентрированной HCl (0,04 мл). Реакционную смесь перемешивали при 90°С в течение 24 часов, затем охлаждали до комнатной температуры и подщелачивали насыщенным водным раствором NaHCO3. Смесь экстрагировали этилацетатом, органический слой сушили (MgSO4) и остаток очищали с помощью флэш хроматографии, с получением указанного в заголовке соединения в виде бесцветного твердого вещества (92 мг, выход 78%).
В виде свободного основания.
1Н ЯМР (ДМСО D6) 0,75 (3H, т), 1,43-1,60 (4H, м), 1,80-2,07 (6H, м), 2,80 (3H, д), 3,88 (3H, с), 4,19 (1H, м), 5,38 (1H, м), 7,50 (1H, д), 7,59 (1H, с), 7,83 (1H, д), 8,47 (1H, м), 8,67 (1H, с), 9,31 (1H, с), 9,40 (1H, ушир.с); MS (ES+) 449.
Пример 2:
4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид (I-2)

Получают согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,73 (3H, т), 1,52-2,11 (15H, м), 2,25 (3H, с), 2,80-2,92 (2H, м), 3,27-3,32 (1H, м), 3,72-3,82 (1H, м), 4,37-4,47 (1H, м), 5,16-5,22 (1H, м), 7,46-7,51 (2H, м), 7,94 (1H, с), 8,12 (1H, д), 8,27 (1H, д), 8,61 (1H, с), 9,25 (1H, с); MS (ES+) 532.
Пример 3:
4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-хлорбензойная кислота (I-3)

Получают согласно способу В в виде свободного основания.
MS (ES+) 440; (ES-) 438.
Пример 4:
Способ С: 4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3]птеридин-7-иламино)-3-хлор-N-метилбензамид (I-4)

4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-хлорбензойную кислоту (I-3) (27 мг) растворяли в ДМФА (0,4 мл) и обрабатывали карбонилдиимидазолом (12 мг, 0,077 ммоль), смесь перемешивали при комнатной температуре в течение 90 минут. Раствор охлаждали на ледяной бане и в течение 2 минут барботировали газообразный метиламин. Смесь перемешивали при комнатной температуре в течение 2 часов, упаривали при пониженном давлении и очищали с помощью хроматографии, с получением 4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-хлор-N-метилбензамида (15 мг) в виде бесцветного твердого вещества.
В виде свободного основания.
1Н ЯМР (ДМСО D6) 0,71 (3H, т), 1,37-2,05 (10H, м), 2,80 (3H, с), 4,12-4,25 (1H, м), 5,20-5,31 (1H, м), 7,88 (1H, д), 8,00 (1H, д), 8,05 (1H, с), 8,50-8,55 (1H, м), 8,60 (1H, с), 8,97 (1H, ушир.с), 9,35 (1H, с); MS (ES+) 453, (ES-) 451.
Пример 5:
4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3]птеридин-7-иламино)-N,N,3-триметилбензамид (I-5)
Способ D: N,N,3-Триметил-4-нитробензамид

3-Метил-4-нитробензойную кислоту (3,0 г, 16,56 ммоль) в ДМФА (30 мл) перемешивали с карбонилдиимидазолом (3,22 г, 1,2 эквивалента) в течение 90 минут. Реакционную смесь охлаждали на ледяной бане и добавляли диметиламин (2М в ТГФ, 25 мл, 3 эквивалента). Затем смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Смесь выливали в ледяную воду (200 мл) и экстрагировали этилацетатом (х6). Объединенные экстракты промывали соляным раствором, сушили над MgSO4, фильтровали и упаривали. Затирание с эфиром с последующей фильтрацией давало N,N,3-триметил-4-нитробензамид (2,6 г, бесцветное твердое вещество); MS (ES+) 209.
Способ Е: 4-Амино-N,N,3-триметилбензамид

N,N,3-Триметил-4-нитробензамид (0,6 г, 2,88 ммоль) в метаноле гидрировали с использованием Н-cube (1,2 мл/мин, комнатная температура, атмосферное давление). Спустя 45 минут реакцию завершали. Удаление растворителя в вакууме давало 4-амино-N,N,3-триметилбензамид в виде бесцветного твердого вещества (количественный выход); MS (ES+) 179.
4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3]птеридин-7-иламино)-N,N,3-триметилбензамид (I-5)
Получали согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69 (3H, т), 1,25-1,45 (4H, м), 1,70-2,05 (6H, м), 2,27 (3H, с), 2,97 (6H, с), 4,08-4,15 (1H, м), 5,30-5,37 (1H, м), 7,29 (1H, д), 7,36 (1H, с), 7,46 (1H, д), 8,62 (1H, с), 9,33 (1H, с), 9,75 (1H, ушир.с); MS (ES+) 447, (ES-) 445.
Пример 6:
4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3]птеридин-7-иламино)-N,3-диметилбензамид (I-6)

N,3-диметил-4-нитробензамид

Получали согласно способу D.
4-амино-N,3-диметилбензамид
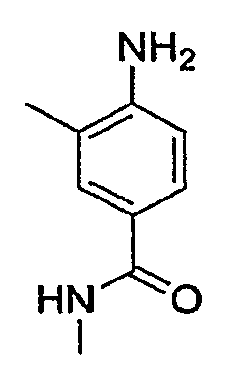
Получали согласно способу Е в виде свободного основания.
4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3]птеридин-7-иламино)-N,3-дииметилбензамид (I-6)

Получали согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,68 (3H, т), 1,15-1,35 (4H, м), 1,65-2,05 (6H, м), 2,26 (3H, с), 2,78 (3H, с), 4,03-4,17 (1H, м), 5,36-5,43 (1H, м), 7,52 (1H, д), 7,76 (1H, д), 7,90 (1H, с), 8,52-8,57 (1H, м), 8,74 (1H, с), 9,39 (1H, с), 10,13 (1H, с); MS (ES+)433, (ES-) 431.
Пример 7:
4-((R)-5-циклопентил-6-этил-5,6-дигидроимидазо[1,2]птеридин-3-иламино)-3-метокси-N-метилбензамид (I-7)

Способ F: (R)-2-хлоро-8-циклопентил-7-этил-7,8-дигидроптеридин-6-амин

(R)-2-хлор-8-циклопентил-7-этил-7,8-дигидроптеридин-6(5Н)-он (200 мг, 0,714 ммоль) в POCl3 (6 мл) нагревали при 100°С в течение 4,5 часов. Растворитель удаляли под вакуумом и дважды отгоняли азеотропную смесь с сухим толуолом. Остаток растворяли в сухом CH2Cl2 (1,5 мл) и по каплям добавляли к ТГФ (5 мл) и жидкому NH3 (4,2 г). Реакционную смесь перемешивали при комнатной температуре в течение выходных. Реакционную смесь выливали в ледяную воду и экстрагировали EtOAc х 3. Объединенные органические экстракты концентрировали в вакууме с получением (R)-2-хлор-8-циклопентил-7-этил-7,8-дигидроптеридин-6-амина в виде коричневого масла (181 мг). MS (ES+) 280, (ES-) 278.
Способ G: (R)-3-хлор-5-циклопентил-6-этил-5,6-дигидроимидазо[1,2-f]птеридин

(R)-2-хлор-8-циклопентил-7-этил-7,8-дигидроптеридин-6-амин (177 мг, 0,64 ммоль) в МеОН (1,5 мл) обрабатывали 50% водным хлорацетальдегидом (131 мкл, 1,3 эквивалента) и реакционную смесь кипятили с обратным холодильником в течение 6 часов. Добавляли следующую порцию 50% водного хлорацетальдегида (400 мкл, 3,0 эквивалента) и реакционную смесь нагревали при 68°С в течение ночи. Добавляли NaHCO3 (водный насыщенный раствор) и EtOAc и водный слой экстрагировали далее EtOAc. Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме.
MS (ES+) 304.
4-((R)-5-циклопентил-6-этил-5,6-дигидроимидазо [1,2]птеридин-3-иламино)-3-метокси-N-метилбензамид (I-7)

Получали согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,66 (3H, т), 1,55-2,13 (10H, м), 2,79 (3H, д), 3,94 (3H, с), 4,35-4,48 (1H, м), 4,98-5,07 (1H, м), 7,13 (1H, с), 7,45-7,53 (2H, м), 7,79 (1H, с), 7,86 (1H, с), 8,25-8,38 (2H, м), 8,45 (1H, с); MS (ES+) 448, MS (ES-) 446.
Пример 8:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-циклопропил-3-метоксибензамид (I-8)
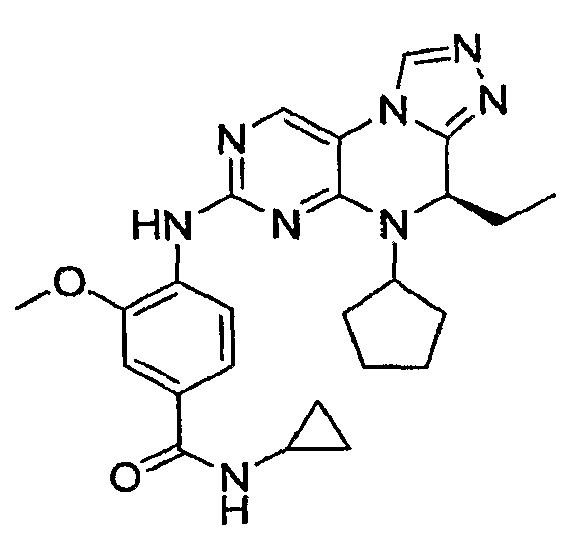
Получают согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,56-0,59 (2H, м), 0,68-0,73 (5H, м), 1,55-2,10 (10H, м), 2,82 (1H, м), 3,92 (3H, с), 4,42 (1H, м), 5,21 (1H, м), 7,46-7,48 (2H, м), 7,95 (1H, с), 8,26 (1H, д), 8,35 (1H, д), 8,59 (1H, с), 9,25 (1H, с); ВЭЖХ время удержания (мин): 8,91; MS (ES+) 475, (ES-) 474.
Пример 9:
4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-метокси-N-((R)-тетрагидрофуран-3-ил)бензамид (I-9)

Получают согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,71 (3H, т), 1,55-2,20 (12H, м), 3,60 (1H, dд), 3,72 (1H, м), 3,87 (2H, м), 3,94 (3H, с), 4,40-4,53 (2H, м), 5,22 (1H, м), 7,51-7,54 (2H, м), 7,96 (1H, с), 8,30 (1H, м), 8,42 (1H, д), 8,60 (1H, с), 9,25 (1H, с); ВЭЖХ время удерживания (мин): 8,63; MS (ES+) 505.
Пример 10:
(R)-N-циклопентил-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-метоксибензамид (I-10)

1Н ЯМР (ДМСО D6) 0,71 (3H, т), 1,48-1,65 (6H, м), 1,67-2,09 (12H, м), 3,93 (3H, с), 4,21-4,26 (1H, м), 4,42 (1H, кв), 5,20-5,22 (1H, м), 7,49-7,51 (2H, м), 7,95 (1H, с), 8,16 (1H, д), 8,29 (1H, д), 8,59 (1H, с), 9,25 (1H, с); ВЭЖХ время удерживания (мин): 9,76; MS (ES+) 503, (ES-) 501.
Пример 11:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-циклопропилбензамид (I-11)

Получают согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,53-0,57 (2H, м), 0,63-0,73 (5H, м), 1,59-1,99 (9H, м), 2,06-2,15 (1H, м), 2,79-2,83 (1H, м), 4,48-4,55 (1H, м), 5,21 (1H, дд), 7,737,80 (4H, м), 8,26 (1H, д), 8,60 (1H, с), 9,25 (1H, с), 9,65 (1H, с); ВЭЖХ время удерживания (мин): 8,36; MS (ES+) 445, (ES-) 443.
Пример 12:
(4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-метоксифенил)((S)-3-фторпирролидин-1-ил)метанон (I-12)

Получают согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,70 (3H, т), 1,50-1,62 (2H, м), 1,69-2,23 (10H, м), 3,53-3,84 (4H, м), 3,90 (3H, с), 4,38 (1H, т), 5,20 (1H, дд), 5,40 (1H, дд), 7,15 (1H, д), 7,20 (1H, д), 7,99 (1H, с), 8,20 (1H, с), 8,57 (1H, с), 9,25 (1H, с); ВЭЖХ время удерживания (мин): 8,94; MS (ES+) 507, (ES-) 505.
Пример 13:
(R)-4-(5-циклопентил-4-этил-1-метил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-циклопропил-3-метоксибензамид (I-13)

Получают согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,55-0,60 (2H, м), 0,69-0,78 (5H, м), 1,55-2,10 (10H, м), 2,68 (3H, с), 2,82 (1H, м), 3,93 (3H, с), 4,48 (1H, квинт), 5,01 (1H, дд), 7,46-7,49 (2H, м), 7,94 (1H, с), 8,30 (1H, д), 8,35 (1H, д), 8,45 (1H, с); ВЭЖХ время удерживания (мин): 9,12; MS (ES+) 490, (ES-) 488.
Пример 14:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-циклопропил-3-фторбензамид (I-14)

Получают согласно способу С в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,55-0,59 (2H, м), 0,67-0,73 (5H, м), 1,46-1,95 (10H, м), 2,84 (1H, м), 4,30 (1H, квинт), 5,18 (1H, дд), 7,64-7,70 (2H, м), 7,91 (1H, т), 8,41 (1H, д), 8,55 (1H, с), 9,02 (1H, с), 9,24 (1H, с); ВЭЖХ время удерживания (мин): 8,58; MS (ES+) 464, (ES-) 462.
Пример 15:
(R)-5-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-циклопропилтиофен-2-карбоксамид (I-15)

Получают согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,48-0,54 (2H, м), 0,63-0,74 (5H, м), 1,65-1,95 (10H, м), 2,74 (1H, м), 4,83 (1H, ушир.с), 5,24 (1H, дд), 7,58 (1H, д), 7,47 (1H, д), 8,14 (1H, д), 8,63 (1H, с), 9,24 (1H, с), 10,83 (1H, ушир.с); ВЭЖХ время удерживания (мин): 8,17; MS (ES+) 452, (ES-) 450.
Пример 16:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-фтор-N-метилбензамид (I-16)

Получают согласно способу С в виде мезилатной соли.
Способ Н: Получение мезилата
Свободное основание (38,2 мг, 0,088 ммоль) растворяли в горячем метаноле (3 мл) и обрабатывали метансульфокислотой (5,68 мкл, 0,088 ммоль), упаривали при пониженном давлении и трижды отгоняли азеотропную смесь с диэтиловым эфиром. Остаток затирали с эфиром и фильтровали, с получением метансульфонатной соли (50, 4 мг).
1Н ЯМР (ДМСО D6) 0,69 (3H, т), 1,35-1,55 (4H, м), 1,70-2,0 (6H, м), 2,33 (3H, с), 2,79 (3H, д), 4,24 (1H, квинт), 5,25 (1H, дд), 7,69-7,73 (2H, м), 7,84 (1H, т), 8,48 (1H, кв), 8,57 (1H, с), 9,28 (1H, с), 9,39 (1H, с); ВЭЖХ время удерживания (мин): 8,07; MS (ES+) 438, (ES-) 436.
Пример 17:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-(3,3-дифторциклобутил)-3-метоксибензамид (I-17)

Получают согласно способу В в виде мезилатной соли (способ Н).
1Н ЯМР (ДМСО D6) 0,70 (3H, т), 1,42-1,70 (4H, м), 1,75-2,04 (6H, м), 2,33 (3H, с), 2,70-2,85 (2H, м), 2,90-3,20 (2H, м), 3,92 (3H, с), 4,32 (2H, м), 5,33 (1H, дд), 7,52-7,56 (2H, м), 7,99 (1H, д), 8,59 (1H, с), 8,78 (1H, д), 8,98 (1H, ушир.с), 9,30 (1H, с); ВЭЖХ время удерживания (мин): 9,35; MS (ES+) 526, (ES-) 524.
Пример 18:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-этил-3-метоксибензамид (I-18)

Получают согласно способу В в виде мезилатной соли (способ Н).
1Н ЯМР (ДМСО D6) 0,70 (3H, т), 1,14 (3H, т), 1,45-1,67 (4H, м), 1,77-2,30 (6H, м), 2,31 (3H, с), 3,31 (2H, квинт), 3,91 (3H, с), 4,32 (1H, квинт), 5,32 (1H, дд), 7,52 (1H, дд), 7,56 (1H, д), 7,98 (1H, ушир.д), 8,47 (1H, т), 8,58 (1H, с), 8,85 (1H, ушир.с), 9,29 (1H, с); ВЭЖХ время удерживания (мин): 8,94; MS (ES+) 463, (ES-) 461.
Пример 19:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-этил-3-фторбензамид (I-19)

Получают согласно способу С в виде мезилатной соли (способ Н).
1Н ЯМР (ДМСО D6) 0,70 (3H, т), 1,13 (3H, т), 1,38-1,55 (4H, м), 1,75-2,00 (6H, м), 2,32 (3H, с), 3,29 (2H, квинт), 4,23 (1H, квинт), 5,27 (1H, дд), 7,70-7,77 (2H, м), 7,83 (1H, т), 8,51 (1H, т), 8,57 (1H, с), 9,29 (1H, с), 9,45 (1H, ушир.с); ВЭЖХ время удерживания (мин): 8,64; MS (ES+) 452, (ES-) 450.
Пример 20:
(R)-5-циклопентил-4-этил-N-(2-фтор-4(трифторметил)фенил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-20)

Получают согласно способу В в виде мезилатной соли (способ Н).
1Н ЯМР (ДМСО D6) 0,69 (3H, м), 1,35-1,45 (4H, м), 1,73-2,00 (6H, м), 2,38 (3H, с), 4,16 (1H, квинт), 5,31 (1H, дд), 7,63 (1H, д), 7,81 (1H, д), 7,91 (1H, т), 8,62 (1H, с), 9,34 (1H, с), 9,85 (1H, ушир.с); ВЭЖХ время удерживания (мин): 10,44; MS (ES+) 448, (ES-) 446.
Пример 21:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-N-метил-3-(трифторметил)бензамид (I-21)

Смесь (R)-7-хлор-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридина (100 мг, 0,329 ммоль), 4-амино-3-трифторметил-N-метилбензамида, ацетата палладия (6 мг), карбоната цезия (214 мг, 0,658 ммоль) и ксантофос (xanthphos) (20 мг) в диоксане (4 мл) перемешивали при 150°С в микроволновой печи в течение 1 часа. Смесь разбавляли этилацетатом и промывали водным раствором гидрокарбоната натрия, сушили над сульфатом магния и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя 0-12% метанол/этилацетат, с получением 4-((R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-трифторметил-N-метилбензамида в виде бесцветного стеклообразного вещества (70 мг, 46%).
В виде мезилатной соли (способ Н).
1Н ЯМР (ДМСО D6) 0,67 (3H, т), 0,96-1,35 (4H, м), 1,60-2,00 (6H, м), 2,34 (3H, с), 2,83 (3H, д), 3,90-4,03 (1H, м), 5,25-5,32 (1H, м), 7,82 (1H, д), 8,19 (1H, д), 8,26 (1H, с), 8,57 (1H, с), 8,75 (1H, д), 9,30 (1H, с), 9,46-9,66 (1H, ушир.с); ВЭЖХ время удерживания (мин): 8,94; MS (ES+) 487.
Пример 22:
(R)-5-циклопентил-4-этил-N-фенил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-22)

Получают согласно способу В в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69-0,73 (3H, м), 1,59 (2H, м), 1,66-1,91 (7H, м), 2,06 (1H, м), 4,48 (1H, м), 5,18 (1H, м), 6,94 (1H, м), 7,25-7,28 (2H, м), 7,69-7,71 (2H, м), 8,55 (1H, с), 9,23 (1H, с), 9,37 (1H, с); ВЭЖХ время удерживания (мин): 9,75; MS (ES+) 362, (ES-) 360.
Пример 23:
(R)-5-циклопентил-4-этил-N-(пиридин-4-ил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-23)

Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,70-0,74 (3H, м), 1,62-1,63 (2H, м), 1,72-1,93 (7H, м), 2,11 (1H, м), 4,54 (1H, м), 5,23 (1H, м), 7,72 (2H, д), 8,33 (2H, д), 8,64 (1H, с), 9,27 (1H, с), 9,83 (1H, с); ВЭЖХ время удерживания (мин): 8,70; MS (ES+) 363, (ES-) 361.
Пример 24:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-(дифторметил)-N-метилбензамид (I-24)

Получают согласно способу I в виде мезилатной соли (способ Н).
1Н ЯМР (ДМСО D6) 0,69 (3H, т), 1,17-1,34 (4H, м), 1,71-1,89 (6H, м), 2,33 (3H, с), 2,81 (3H, д), 4,07-4,11 (1H, м), 5,27-5,29 (1H, м), 7,25 (1H, т), 7,70 (1H, д), 8,04 (1H, д), 8,15 (1H, с), 8,59 (1H, с), 8,63-8,65 (1H, м), 9,31 (1H, с), 9,60-9,64 (1H, м); ВЭЖХ время удерживания (мин): 8,28; MS (ES+) 469, (ES-) 467.
Пример 25:
(R)-5-циклопентил-4-этил-N-(пиридин-3-ил)-4,5-дигидро- [1,2,4]триазоло[4,3-f]птеридин-7-амин (I-25)

Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69-0,73 (3H, м), 1,57-1,58 (2H, м), 1,71-1,90 (7H, м), 2,07 (1H, м), 4,48 (1H, м), 5,20 (1H, м), 7,30 (1H, м), 8,113-8,15 (2H, м), 8,58 (1H, м), 8,87 (1H, д), 9,25 (1H, с), 9,55 (1H, с); ВЭЖХ время удерживания (мин): 8,50; MS (ES+) 363, (ES-) 361.
Пример 26:
Способ J: (R)-5-циклопентил-N-циклопропил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-26)

(R)-7-хлор-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин (70 мг, 0,23 ммоль), циклопропиламин (0,5 мл, 7,18 ммоль) DIPEA (0,16 мл, 0,918 ммоль) в н-бутаноле (2 мл) нагревали в запаянной трубке в микроволновой печи при 140°С в течение 90 минут. Реакционную смесь концентрировали в вакууме и очищали с помощью препаративной обращенно-фазной высокоэффективной жидкостной хроматографии [Waters Sunfire C18, 100M, 100 Å колонка, градиент 10%-95% В (растворитель А: 0,05% трифторуксусная кислота в воде; растворитель В: CH3CN) более 16 минут при 25 мл/мин], с получением указанного в заголовке соединения в виде беловатого порошка. Свободное основание получали с использованием бикарбонатной смолы и лиофилизировали с получением указанного в заголовке соединения в виде бесцветного твердого вещества (25,6 мг, выход 34%).
В виде свободного основания.
1Н ЯМР (ДМСО D6) 0,46 (2H, м), 0,62-0,63 (2H, м), 0,67-0,71 (3H, м), 1,49 (2H, м), 1,60-2,00 (8H, м), 2,64 (1H, м), 4,22 (1H, ушир.с), 5,11 (1H, м), 7,17 (1H, м), 8,38 (1H, с), 9,16 (1H, с); ВЭЖХ время удерживания (мин): 9,15; MS (ES+) 326, (ES-) 324.
Пример 27:
Способ К: (R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-27)

Раствор (R)-7-хлор-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридина (500 мг, 1,64 ммоль) в гидроксиде аммония (10 мл) нагревали при облучении микроволнами при 140°С в течение 30 минут. Растворитель удаляли при пониженном давлении и добавляли холодную воду (5 мл). Полученное слегка желтое твердое вещество фильтровали под вакуумом и промывали холодной водой, с получением указанного в заголовке соединения (170 мг, выход 36%).
В виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69 (3H, т), 1,48-2,01 (10H, м), 4,40 (1H, дт), 5,06 (1H, дд), 6,40 (2H, с), 8,35 (1H, с), 9,15 (1H, с); ВЭЖХ время удерживания (мин): 7,43; MS (ES+) 286.
Пример 28:
Способ L: (R)-5-циклопентил-4-этил-N-(пиридин-3-ил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-28)

К (R)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амину (50 мг, 0,175 ммоль) в ДМФА (1 мл) при 0°С добавляли гидрид натрия (60% в минеральном масле, 7,7 мг, 0,193 ммоль). Реакционную смесь перемешивали в течение 10 минут, затем добавляли метилхлорформиат (13,6 мкл, 0,175 ммоль). Реакцию нагревали до комнатной температуры и перемешивали в течение 28 часов. Добавляли хлорид аммония (5 мл, насыщенный водный раствор) и экстрагировали EtOAc (3×10 мл). Объединенные органические экстракты промывали соляным раствором (10 мл), сушили над MgSO4, фильтровали и растворитель удаляли под вакуумом. Сырой продукт очищали с помощью препаративной обращенно-фазной высокоэффективной жидкостной хроматографии [Waters Sunfire C18, 10 M, 100 Å колонка, градиент 10%-95% В (растворитель А: 0,05% трифторуксусная кислота в воде; растворитель В: CH3CN) более 16 минут при 25 мл/мин], с получением указанного в заголовке соединения в виде беловатого порошка (25 мг, 31%).
В виде трифторуксуснокислой соли.
1Н ЯМР (ДМСО D6) 0,69 (3H, т), 1,47-1,63 (2H, м), 1,78-2,18 (8H, м), 3,70 (3H, с), 4,28 (1H, дт), 5,30 (1H, дд), 8,58 (1H, с), 9,34 (1H, с), 10,69 (1H, ушир.с); ВЭЖХ время удерживания (мин): 7,62; MS (ES+) 344, (ES-) 342.
Пример 29:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-метокси-N-метилбензамид (I-29)

Получают согласно способу В в виде мезилатной соли (способ Н).
1Н ЯМР (ДМСО D6) 0,72 (3H, т), 1,63-1,87 (4H, м), 2,10-2,50 (4H, м), 2,31 (3H, с), 2,80 (3H, д), 3,92 (3H, с), 4,47 (1H, квинт), 5,32 (1H, дд), 7,50-7,55 (2H, м), 8,11 (1H, ушир.д), 8,43 (1H, ушир.кв), 8,59 (1H, с), 8,82 (1H, ушир.с), 9,30 (1H, с); ВЭЖХ время удерживания (мин): 8,32; MS (ES+) 435, (ES-) 433.
Пример 30:
(R)-4-(5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-иламино)-3-(метоксиметил)-N-метилбензамид (I-30)

Получают согласно способу I в виде мезилатной соли (способ Н).
1Н ЯМР (ДМСО D6) 0,69 (3H, т), 1,31-1,50 (4H, м), 1,72-2,03 (6H, м), 2,33 (3H, с), 2,79 (3H, д), 3,32 (3H, с), 4,09-4,20 (1H, м), 4,52 (2H, с), 5,28-5,35 (1H, м), 7,80-7,87 (2H, м), 7,90 (1H, с), 8,41-8,49 (1H, м), 8,58 (1H, с), 9,20-9,35 (1H, ушир.с), 9,30 (1H, с); ВЭЖХ время удерживания (мин): 8,39; MS (ES+) 463, (ES-) 461.
Пример 31:
(R)-N-бензил-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-31)

Получают согласно способу J в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,65-0,69 (3H, м), 1,48 (2H, м), 1,64-1,89 (8H, м), 4,10 (1H, м), 4,45 (2H, м), 5,07 (1H, м), 7,20 (1H, м), 7,28 (4H, м), 7,57 (1H, ушир.с), 8,38 (1H, с), 9,14 (1H, с); ВЭЖХ время удерживания (мин): 9,77; MS (ES+) 376, (ES-) 374.
Пример 32:
(R)-5-циклопентил-4-этил-N-(4-(4-метилпиперазин-1-ил)фенил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-32)

Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,70-0,72 (3H, м), 1,56 (2H, м), 1,74-2,02 (8H, м), 2,23 (3H, с), 2,50 (4H, м), 3,06 (4H, м), 4,43 (1H, м), 5,15 (1H, м), 6,86 (2H, д), 7,50 (2H, д), 8,49 (1H, с), 9,10 (1H, с), 9,20 (1H, с); ВЭЖХ время удерживания (мин): 9,02; MS (ES+) 460, (ES-) 458.
Пример 33:
(R)-5-циклопентил-N-(4-(дифторметокси)фенил)-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-33)

Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,75 (3H, м), 1,59 (2H, м), 1,76-1,89 (8H, м), 2,07 (1H, м), 4,48 (1H, м), 5,18 (1H, м), 7,11 (2H, д), 7,72 (2H, д), 8,55 (1H, с), 9,23 (1H, с), 9,44 (1H, с); ВЭЖХ время удерживания (мин): 9,70; MS (ES+) 428, (ES-) 426.
Пример 34:
(R)-5-циклопентил-4-этил-N-(4-морфолинофенил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-34)

Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69-0,72 (3H, м), 1,57 (2H, м), 1,71-1,89 (7H, м), 2,00 (1H, м), 2,54 (4H, м), 3,72-3,75 (4H, м), 4,43 (1H, м), 5,16 (1H, м), 6,87 (2H, д), 7,52 (2H, д), 8,50 (1H, с), 9,12 (1H, с), 9,21 (1H, с); ВЭЖХ время удерживания (мин): 9,12; MS (ES+) 447, (ES-) 445.
Пример 35:
(R)-5-циклопентил-4-этил-N-(4-(фторфенил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-35)

Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69-0,72 (3H, м), 1,60 (2H, м), 1,73-1,90 (7H, м), 2,06 (1H, м), 4,46 (1H, м), 5,19 (1H, м), 7,09 (2H, м), 7,67-7,70 (2H, м), 8,54 (1H, с), 9,23 (1H, с), 9,38 (1H, с); ВЭЖХ время удерживания (мин): 9,78; MS (ES+) 380, (ES-) 378.
Пример 36:
Способ М: (R)-5-циклопентил-4-этил-N-метил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-36)
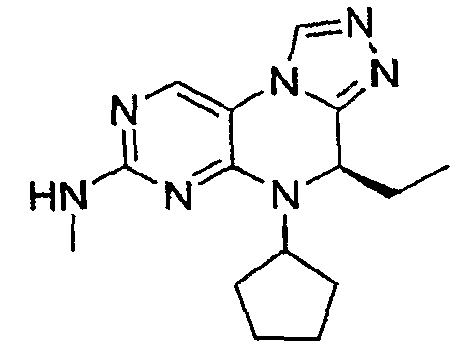
В сосуд, содержащий (R)-7-хлор-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин (70 мг, 0,23 ммоль) добавляли раствор метиламина в этаноле (2 мл, 2М раствор). Сосуд запаивали и затем нагревали при 50°С в течение 18 часов. После охлаждения растворитель удаляли при пониженном давлении и сырой продукт очищали с помощью препаративной обращенно-фазной высокоэффективной жидкостной хроматографии [Waters Sunfire C18, 10 M, 100 Å колонка, градиент 10%-95% В (растворитель А: 0,05% трифторуксусная кислота в воде; растворитель В: CH3CN) более 16 минут при 25 мл/мин]. Свободное основание получали при пропускании трифторуксуснокислой соли в ацетонитриле/воде через карбонатный картридж, с получением указанного в заголовке соединения в виде белого порошка (42 мг, выход 61%).
В виде свободного основания.
1Н ЯМР (ДМСО D6) 0,68 (3H, т), 1,47-2,03 (10H, м), 2,77 (3H, д), 4,24-4,32 (1H, м), 5,09 (1H, д), 6,86 (1H, ушир.с), 8,38 (1H, с), 9,15 (1H, с); ВЭЖХ время удерживания (мин): 8,57; MS (ES+) 300, (ES-) 298.
Пример 37:
(R)-5-циклопентил-N,4-диэтил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-37)

Получают согласно способу М в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69 (3H, т), 1,10 (3H, т), 1,47-1,99 (10H, м), 3,26 (2H, дт), 4,27 (1H, ушир.с), 5,09 (1H, д), 6,94 (1H, ушир.с), 8,37 (1H, с), 9,15 (1H, с); ВЭЖХ время удерживания (мин): 9,14; MS (ES+), (ES-) 312.
Пример 38:
(R)-N,5-дициклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-38)

Получают согласно способу J в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69 (3H, т), 1,43-2,00 (18H, м), 4,07-4,17 (1H, м), 4,17-4,34 (1H, м), 5,08 (1H, ушир.с), 6,98 (1H, ушир.с), 8,36 (1H, с), 9,14 (1H, с); ВЭЖХ время удерживания (мин): 10,2; MS (ES+) 354, (ES-) 352.
Пример 39:
(R)-N-(4-(1Н-пиразол-1-ил)фенил)-5-циклопентил-4-этил-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-39)

Получают согласно способу I в виде трифторуксуснокислой соли.
1Н ЯМР (ДМСО D6) 0,70 (3H, м), 1,58-1,59 (2H, м), 1,75-2,07 (7H, м), 2,33 (1H, м), 4,48 (1H, м), 5,22 (1H, м), 6,52 (1H, с), 7,71-7,81 (5H, м), 8,42 (1H, м), 8,58 (1H, с), 9,26 (1H, с), 9,64 (1H, с); ВЭЖХ время удерживания (мин): 9,32; MS (ES+) 428, (ES-) 426.
Пример 40:
(R)-5-циклопентил-4-этил-N-(6-(трифторметил)пиридин-3-ил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-40)

Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,70 (3H, м), 0,70-0,74 (3H, м), 1,60-1,63 (2H, м), 1,71-1,92 (7H, м), 2,08 (1H, м), 4,51 (1H, м), 5,22 (1H, м), 7,82 (1H, д), 8,42 (1H, д), 8,64 (1H, с), 9,03 (1H, м), 9,27 (1H, с), 9,98 (1H, с); ВЭЖХ время удерживания (мин): 9,70; MS (ES+) 431, (ES-) 429.
Пример 41:
(R)-5-циклопентил-4-этил-N-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-41)

Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,68-0,72 (3H, м), 1,53 (2H, м), 1,69-1,97 (8H, м), 2,23 (3H, с), 2,42 (4H, м), 3,40 (4H, м), 4,35 (1H, м), 5,14 (1H, м), 6,80 (1H, д), 7,79 (1H, м), 8,32 (1H, м), 8,48 (1H, с), 9,04 (1H, с), 9,20 (1H, с); ВЭЖХ время удерживания (мин): 8,70; MS (ES+) 461, (ES-) 459.
Пример 42:
(R)-5-циклопентил-4-этил-N-(4-(пирролидин-1-ил)фенил)-4,5-дигидро-[1,2,4]триазоло[4,3-f]птеридин-7-амин (I-42)
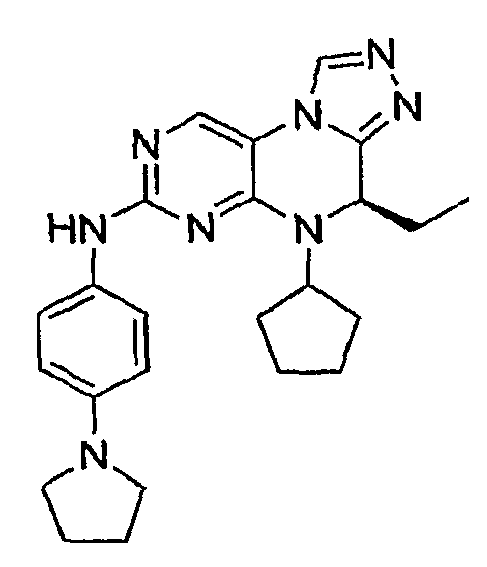
Получают согласно способу I в виде свободного основания.
1Н ЯМР (ДМСО D6) 0,69-0,72 (3H, м), 1,54 (2H, м), 1,60-1,81 (8H, м), 1,89-1,94 (4H, м), 3,19 (4H, м), 4,38 (1H, м), 5,13 (1H, м), 6,47-6,52 (2H, м), 7,42 (2H, д), 8,46 (1H, с), 8,91 (1H, с), 9,19 (1H, с); ВЭЖХ время удерживания (мин): 10,44; MS (ES+) 431, (ES-) 429.
Пример 43: анализ PLK1
Соединения настоящего изобретения могут быть исследованы в качестве ингибиторов PLK киназы человека с использованием следующих анализов.
Анализ ингибирования Plk1
Проводили скрининг соединений на их способность ингибировать Plk1 с использованием анализа включения радиоактивного фосфата. Анализы проводили в смеси 25 мМ HEPES (рН 7,5), 10 мМ MgCl2 и 1 мМ DTT. Конечные концентрации субстрата составляли 50 мкМ [γ-33P]АТФ (136 мКи 33P АТФ/моль АТФ, Amersham Pharmacia Biotech/Sigma Chemicals) и 10 мкМ пептида (SAM68 белок Δ332-443). Анализы проводили при 25°С в присутствии 15 нМ Plk1 (А20-К338). Исходный анализируемый буферный раствор содержал все перечисленные выше компоненты за исключением АТФ и интересующего анализируемого вещества. 30 мкл исходного раствора помещали в 96-луночный планшет, а затем, в двух повторах, добавляли 2 мкл ДМСО, содержащего исследуемые вещества, последовательно разбавленные исходным раствором (обычно начиная с конечной концентрации 10 мкМ с 2-кратными последующими разбавлениями) (конечная концентрация ДМСО 5%). Планшеты предварительно инкубировали в течение 10 минут при 25°С и начинали реакцию посредством добавления 8 мкл [γ-33P]АТФ (конечная концентрация 50 мкМ).
Реакцию останавливали спустя 60 минут добавлением 100 мкл 0,14 М фосфорной кислоты. Перед добавлением 125 мкл остановленной анализируемой смеси многоситовый фосфоцеллюлозный 96-луночный планшетный фильтр для мультискринирования (Millipore, Cat no. MAPHN0B50) предварительно обрабатывали 100 мкл 0,2 М фосфорной кислоты. Планшет промывали 4×200 мкл 0,2 М фосфорной кислоты. После сушки, перед измерением активности сцинтилляционным методом (1450 Microbeta Liquid Scintillation Counter, Wallac), к лунке добавляли 100 мкл жидкого коктейля для сцинтилляционного счета Optiphase 'Supermix' (Perkin Elmer).
После удаления среднего значения фона для всех точек данных рассчитывали данные Ki(ср) из нелинейного регрессионного анализа данных исходной скорости, используя программное обеспечение Prism (GraphPad Prism версия 3.0cx для Macintosh, GraphPad Sowtware, San Diego California, USA).
Анализ ингибирования Plk1
Проводили скрининг соединений на их способность ингибировать Plk1 с использованием анализа включения радиоактивного фосфата. Анализы проводили в смеси 25 мМ HEPES (рН 7,5), 10 мМ MgCl2, 0,1% БСА и 1 мМ DTT. Конечные концентрации субстрата составляли 150 мкМ (350 мкМ для определения значений <1нМ)[γ-33P]АТФ (115 мКи 33P АТФ/моль АТФ, Amersham Pharmacia Biotech / Sigma Chemicals) и 300 мкМ (450 мкМ для определения значений <1нМ) пептида (KKKISDELMDATFADQEAK) SEQ. ID № 1. Анализы проводили при 25°С в присутствии 4 нМ (1 нМ для определения значений <1нМ) Plk1. Исходный анализируемый буферный раствор содержал все перечисленные выше компоненты за исключением АТФ и интересующего анализируемого вещества. 30 мкл исходного раствора помещали в 96-луночный планшет, а затем, в двух повторах, добавляли 2 мкл ДМСО, содержащего исследуемые вещества, последовательно разбавленные исходным раствором (обычно начиная с конечной концентрации 10 мкМ с 2-кратными последующими разбавлениями) (конечная концентрация ДМСО 5%). Планшеты предварительно инкубировали в течение 10 минут при 25°С и начинали реакцию посредством добавления 8 мкл [γ-33P]АТФ (конечная концентрация 150 мкМ (350 мкМ для определения значений <1нМ)).
Реакцию останавливали спустя 90 минут (240 минут для определения значений <1нМ) добавлением 100 мкл 0,14 М фосфорной кислоты. Перед добавлением 125 мкл остановленной анализируемой смеси многоситовый фосфоцеллюлозный 96-луночный планшетный фильтр для мультискринирования (Millipore, Cat no. MAPHN0B50) предварительно обрабатывали 100 мкл 0,2 М фосфорной кислоты. Планшет промывали 4×200 мкл 0,2 М фосфорной кислоты. После сушки, перед измерением активности сцинтилляционным методом (1450 Microbeta Liquid Scintillation Counter, Wallac), к лунке добавляли 100 мкл жидкого коктейля для сцинтилляционного счета Optiphase 'Supermix' (Perkin Elmer).
После удаления среднего значения фона для всех точек данных рассчитывали данные Ki(ср) из нелинейного регрессионного анализа данных исходной скорости, используя программное обеспечение Prism (GraphPad Prism версия 3.0cx для Macintosh, GraphPad Sowtware, San Diego California, USA).
В основном, соединения по настоящему изобретению являются эффективными для ингибирования Plk1. В анализе включения радиоактивного вещества следующие соединения показали Ki ниже 10 нМ: I-1, I-2, I-4, I-5, I-6, I-7, I-8, I-9, I-10, I-11, I-12, I-13, I-14, I-16, I-17, I-18, I-19, I-20, I-21, I-22, I-23, I-24, I-25, I-29, I-30, I-32, I-33, I-34, I-35, I-39, I-41, I-42. В анализе включения радиоактивного вещества следующие соединения показали Ki между 10 нМ и 100 нМ: I-31, I-40. В анализе включения радиоактивного вещества следующие соединения показали Ki между 100 нМ и 4 мкМ: I-31, I-40.
Анализ ингибирования Plk2
Проводили скрининг соединений на их способность ингибировать Plk2 с использованием анализа включения радиоактивного фосфата. Анализы проводили в смеси 25 мМ HEPES (рН 7,5), 10 мМ MgCl2, 0,1% БСА и 2 мМ DTT. Конечные концентрации субстрата составляли 200 мкМ [γ-33P]АТФ (57 мКи 33P АТФ/моль АТФ, Amersham Pharmacia Biotech/Sigma Chemicals) и 300 мкМ пептида (KKKISDELMDATFADQEAK) SEQ. ID № 2. Анализы проводили при 25°С в присутствии 25 нМ Plk2. Исходный анализируемый буферный раствор содержал все перечисленные выше компоненты за исключением АТФ и интересующего анализируемого вещества. 30 мкл исходного раствора помещали в 96-луночный планшет, а затем, в двух повторах, добавляли 2 мкл ДМСО, содержащего исследуемые вещества, последовательно разбавленные исходным раствором (обычно начиная с конечной концентрации 10 мкМ с 2-кратными последующими разбавлениями) (конечная концентрация ДМСО 5%). Планшеты предварительно инкубировали в течение 10 минут при 25°С и начинали реакцию добавлением 8 мкл [γ-33P]АТФ (конечная концентрация 200 мкМ).
Реакцию останавливали спустя 90 минут добавлением 100 мкл 0,14 М фосфорной кислоты. Перед добавлением 125 мкл остановленной анализируемой смеси, многоситовый фосфоцеллюлозный 96-луночный планшетный фильтр (Millipore, Cat no. MAPHN0B50) предварительно обрабатывали 100 мкл 0,2 М фосфорной кислоты. Планшет промывали 4×200 мкл 0,2 М фосфорной кислоты. После сушки, перед измерением активности сцинтилляционным методом (1450 Microbeta Liquid Scintillation Counter, Wallac), к лунке добавляли 100 мкл жидкого коктейля для сцинтилляционного счета Optiphase 'Supermix' (Perkin Elmer).
После удаления среднего значения фона для всех точек данных рассчитывали данные Ki(ср) из нелинейного регрессионного анализа данных исходной скорости, используя программное обеспечение Prism (GraphPad Prism версия 3.0cx для Macintosh, GraphPad Sowtware, San Diego California, USA).
Анализ ингибирования Plk3
Проводили скрининг соединений на их способность ингибировать Plk3 с использованием анализа включения радиоактивного фосфата. Анализы проводили в смеси 25 мМ HEPES (рН 7,5), 10 мМ MgCl2 и 1 мМ DTT. Конечные концентрации субстрата составляли 75 мкМ [γ-33P]АТФ (60 мКи 33P АТФ/моль АТФ, Amersham Pharmacia Biotech / Sigma Chemicals) и 10 мкМ пептида (SAM68 белок Δ332-443). Анализы проводили при 25°С в присутствии 5 нМ Plk3 (S38-A340). Исходный анализируемый буферный раствор содержал все перечисленные выше компоненты за исключением АТФ и интересующего анализируемого вещества. 30 мкл исходного раствора помещали в 96-луночный планшет, а затем, в двух повторах, добавляли 2 мкл ДМСО, содержащего исследуемые вещества, последовательно разбавленные исходным раствором (обычно начиная с конечной концентрации 10 мкМ с 2-кратными последующими разбавлениями) (конечная концентрация ДМСО 5%). Планшеты предварительно инкубировали в течение 10 минут при 25°С и начинали реакцию добавлением 8 мкл [γ-33P]АТФ (конечная концентрация 75 мкМ).
Реакцию останавливали спустя 60 минут добавлением 100 мкл 0,14 М фосфорной кислоты. Перед добавлением 125 мкл остановленной анализируемой смеси, многоситовый фосфоцеллюлозный 96-луночный планшетный фильтр для мультискринирования (Millipore, Cat no. MAPHN0B50) предварительно обрабатывали 100 мкл 0,2 М фосфорной кислоты. Планшет промывали 4×200 мкл 0,2 М фосфорной кислоты. После сушки, перед измерением активности сцинтилляционным методом (1450 Microbeta Liquid Scintillation Counter, Wallac), к лунке добавляли 100 мкл жидкого коктейля для сцинтилляционного счета Optiphase 'Supermix' (Perkin Elmer).
После удаления среднего уровня фона для всех точек данных рассчитывали данные Ki(ср) из нелинейного регрессионного анализа данных исходной скорости, используя программное обеспечение Prism (GraphPad Prism версия 3.0cx для Macintosh, GraphPad Sowtware, San Diego California, USA).
Анализ ингибирования Plk4
Проводили скрининг соединений на их способность ингибировать Plk4 с использованием анализа включения радиоактивного фосфата. Анализы проводят в смеси 8 мМ MOPS (рН 7,5), 10 мМ MgCl2, 0,1% БСА и 2 мМ DTT. Конечные концентрации субстрата составляли 15 мкМ [γ-33P]АТФ (227 мКи 33P АТФ/моль АТФ, Amersham Pharmacia Biotech / Sigma Chemicals) и 300 мкМ пептида (KKKISDELMDATFADQEAK) SEQ. ID № 3. Анализы проводили при 25°С в присутствии 25 нМ Plk4. Исходный анализируемый буферный раствор содержал все перечисленные выше компоненты за исключением АТФ и интересующего анализируемого вещества. 30 мкл исходного раствора помещали в 96-луночный планшет, а затем, в двух повторах, добавляли 2 мкл ДМСО, содержащего исследуемые вещества, последовательно разбавленные исходным раствором (обычно начиная с конечной концентрации 10 мкМ с 2-кратными последующими разбавлениями) (конечная концентрация ДМСО 5%). Планшеты предварительно инкубировали в течение 10 минут при 25°С и начинали реакцию посредством добавления 8 мкл [γ-33P]АТФ (конечная концентрация 15 мкМ).
Реакцию останавливали спустя 180 минут добавлением 100 мкл 0,14 М фосфорной кислоты. Перед добавлением 125 мкл остановленной анализируемой смеси, многоситовый фосфоцеллюлозный 96-луночный планшетный фильтр для мультискринирования (Millipore, Cat no. MAPHN0B50) предварительно обрабатывали 100 мкл 0,2 М фосфорной кислоты. Планшет промывали 4×200 мкл 0,2 М фосфорной кислоты. После сушки, перед измерением активности сцинтилляционным методом (1450 Microbeta Liquid Scintillation Counter, Wallac), к лунке добавляли 100 мкл жидкого коктейля для сцинтилляционного счета Optiphase 'Supermix' (Perkin Elmer).
После удаления среднего значения фона для всех точек данных рассчитывали данные Ki(ср) из нелинейного регрессионного анализа данных исходной скорости, используя программное обеспечение Prism (GraphPad Prism версия 3.0cx для Macintosh, GraphPad Sowtware, San Diego California, USA).
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИГИДРОДИАЗЕПИНЫ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2007 |
|
RU2475488C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПОЛИ(ADP-РИБОЗА) ПОЛИМЕРАЗЫ (PARP) | 2007 |
|
RU2472782C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| ДИАМИНОТРИАЗОЛЫ, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2003 |
|
RU2350606C2 |
| ИНГИБИРОВАНИЕ ФЕРМЕНТОВ | 2013 |
|
RU2638537C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ FSHR И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2663898C2 |
| ПРОИЗВОДНЫЕ ПИРАЗИНА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ ATR | 2009 |
|
RU2604066C2 |
| ИНГИБИТОРЫ ФЕРМЕНТА ДИАЦИЛГЛИЦЕРИН-О-АЦИЛТРАНСФЕРАЗЫ ТИПА 1 | 2008 |
|
RU2486186C2 |
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО ТРИАЗОЛДИАМИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2001 |
|
RU2274639C2 |


Изобретение относится к соединениям формулы (I), которые обладают свойствами ингибитора протеинкиназы Plk1. В формуле (I):

кольцо А является 5-членным гетероарильным кольцом, которое в качестве гетероатомов содержит атомы N, где кольцо необязательно замещено C1-6 алкилом; X1 является NR8-; R1 является Н, С1-10 алифатической, С3-10 циклоалифатической, фенильной, 5-6-членной гетероарильной группой, содержащей в качестве гетероатома атом S, N, где указанный R1 необязательно замещен 0-5 J1; каждый R2 и R3, независимо, является Н, С1-10 алифатической группой; R4 является С3-10 циклоалифатической группой; R8 является Н, -C(O)OR; каждый J1 независимо является C1-6 галогеналкилом, галогеном, Q или -Z-Q; каждый Z независимо является алифатической группой, где 0-3 -СН2- звеньев в указанной С1-6 алифатической группе необязательно заменены на -NR-, -О-, -С(О)-, где любое из незамененных -СН2- звеньев в указанной C1-6 алифатической группе необязательно замещено 0-2 Jz; каждый Q независимо является Н, C1-6 алифатической группой, 3-8-членным ароматическим или неароматическим моноциклическим кольцом, имеющим 0-3 гетероатомов, независимо выбранных из О и N, где каждый Q независимо и необязательно замещен 0-2 JQ; Jz является галогеном; JQ является -М; каждый М независимо является Н, С1-6 алифатической группой, галогеном; каждый R независимо является Н или незамещенной C1-6 алифатической группой. Изобретение также относится к фармацевтической композиции, содержащей указанные соединения, и к способу ингибирования активности протеинкиназы Plk1. 5 н. и 26 з.п. ф-лы, 1 табл.
1. Соединение формулы I:

где кольцо А является 5-членным гетероарильным кольцом, которое в качестве гетероатомов содержит атомы N, где кольцо необязательно замещено C1-6алкилом;
X1 является NR8-;
R1 является Н, С1-10алифатической, С3-10циклоалифатической, фенильной, 5-6-членной гетероарильной группой, содержащей в качестве гетероатома атом S, N, где указанный R1 необязательно замещен 0-5 J1;
каждый R2 и R3 независимо является Н, С1-10 алифатической группой;
R4 является С3-10 циклоалифатической группой;
R8 является Н, -C(O)OR;
каждый J1 независимо является С1-6 галогеналкилом, галогеном, Q или -Z-Q;
каждый Z независимо является C1-6 алифатической группой, где 0-3 -СН2- звеньев в указанной C1-6 алифатической группе необязательно заменены на -NR-, -О-, -С(О)-, где любое из незамененных -СН2- звеньев в указанной С1-6 алифатической группе необязательно замещено 0-2 Jz;
каждый Q независимо является Н, C1-6 алифатической группой, 3-8-членным ароматическим или неароматическим моноциклическим кольцом, имеющим 0-3 гетероатомов, независимо выбранных из О и N, где каждый Q независимо и необязательно замещен 0-2 JQ;
каждый JZ является галогеном;
каждый JQ является -М;
каждый М независимо является Н, C1-6 алифатической группой, галогеном;
каждый R независимо является Н или незамещенной С1-6 алифатической группой.
2. Соединение по п.1, где кольцо А является триазольным кольцом, необязательно замещенным C1-6алкилом.
3. Соединение по п.1, где кольцо А является имидазольным кольцом, необязательно замещенным C1-6 алкилом.
4. Соединение по п.1, где R8 является -C(O)OR.
5. Соединение по п.1, где R8 является Н.
6. Соединение по п.4, где R8 является -С(O)ОС1-6алкилом.
7. Соединение по п.6, где R8 является -С(O)ОСН3.
8. Соединение по п.1, где R1 является Н, фенильной группой, 5-6-членной гетероарильной группой, содержащей в качестве гетероатома атом S, N, каждая из которых необязательно замещена 0-5 J1.
9. Соединение по п.8, где R1 является Н.
10. Соединение по п.8, где R1 является фенильной группой, необязательно замещенной 0-5 J1.
11. Соединение по п.8, где R1 является 5-6-членной гетероарильной группой, содержащей в качестве гетероатома атом S, N, необязательно замещенной 0-5 J1.
12. Соединение по пп.10 или 11, где R1 необязательно замещен 0-3 -O-Q, галогенами, -C(O)N(R)-Q или Q, где каждый Q в Q, -C(O)N(R)-Q, -O-Q независимо необязательно замещен 0-2 JQ.
13. Соединение по п.10, где фенильная группа необязательно замещена в пара-положении группой -C(O)N(R)-Q и в любом другом положении группой -O-Q, галогеном или Q, где каждый Q в Q, -C(O)N(R)-Q, -O-Q независимо необязательно замещен 0-2 JQ.
14. Соединение по п.11, где 5-6-членная гетероарильная группа замещена группой -C(O)N(R)-Q, и в любом другом положении группой -O-Q или Q, где каждый Q в Q, -C(O)N(R)-Q, -O-Q независимо необязательно замещен 0-2 JQ.
15. Соединение по п.13 или 14, где Q в -C(O)N(R)-Q является Н, С1-4 алифатической группой, С1-4 галогеналифатической группой, 3-8-членным ароматическим или неароматическим моноциклическим кольцом, имеющим 0-3 гетероатомов, независимо выбранных из О и N.
16. Соединение по п.15, где фенильная группа необязательно замещена в любом свободном положении галогеном, С1-4 алифатической группой, С1-4галогеналифатической группой, 3-8-членным неароматическим моноциклическим кольцом, имеющим 0-3 гетероатомов, независимо выбранных из О и N, С1-6алкоксигруппой, (С1-6алкокси) C1-6алкильной или С1-6галогеналкоксигруппой.
17. Соединение по п.15, где Q в -C(O)N(R)-Q является метилом, этилом, 1-метилпиперидин-4-илом, циклопропилом, циклопентилом, 3-фуранилом, 3-фторпирролидин-1-илом или 3,3-дифторциклобутилом.
18. Соединение по п.1, где R1 является C1-6алкилом или С3-7циклоалкилом, каждый из которых необязательно замещен 0-5 J1.
19. Соединение по п.17, где R1 является Н, этилом, циклопропилом или циклопентилом.
20. Соединение по п.12, где R1 является фенильной группой, и один заместитель в пара-положении фенильной группы является Q или -Z-Q.
21. Соединение по п.20, где заместитель в пара-положении фенилного кольца является фтором, карбокси, трифторметилом, 4-метилпиперазин-1-илом, дифторметокси, морфолин-1-илом, пиразол-1-илом или пирролидин-1-илом.
22. Соединение по п.11, где R1 является тиофен-2-илом, пиридин-3-илом, пиридин-4-илом или 6-трифторметилпиридин-3-илом.
23. Соединение по п.1, где каждый из R2 и R3 является С1-3 алкилом.
24. Соединение по п.1, где R2 является Н, и R3 является С1-3алкилом.
25. Соединение по п.24, где R3 является этилом.
26. Соединение по п.1, где R4 является циклопентилом.
27. Соединение по п.1, представленное формулой II;
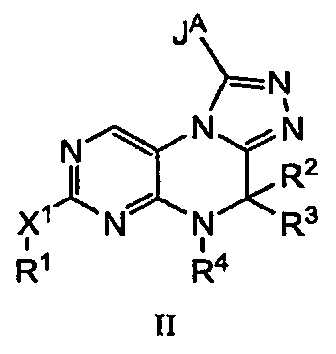
где R1 является фенильной группой или 5-6-членной гетероарильной группой, содержащей в качестве гетероатома атом S, N, где указанный R1 необязательно замещен 0-5 J1, где J1 имеет значения, указанные в п.1;
каждый R2 и R3 независимо является Н, С1-10 алифатической группой; и
JA является Н или С1-4алкилом.
28. Соединение, выбранное из группы:
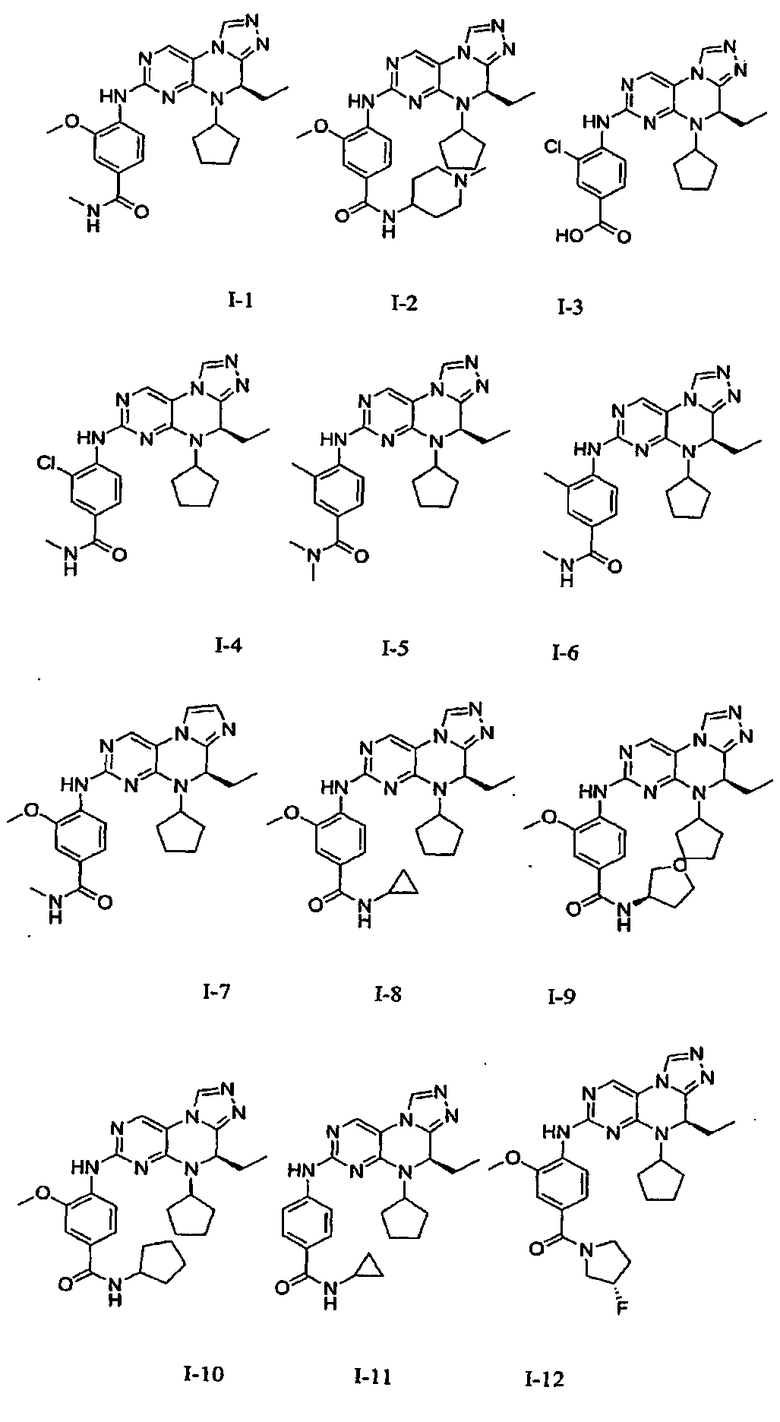
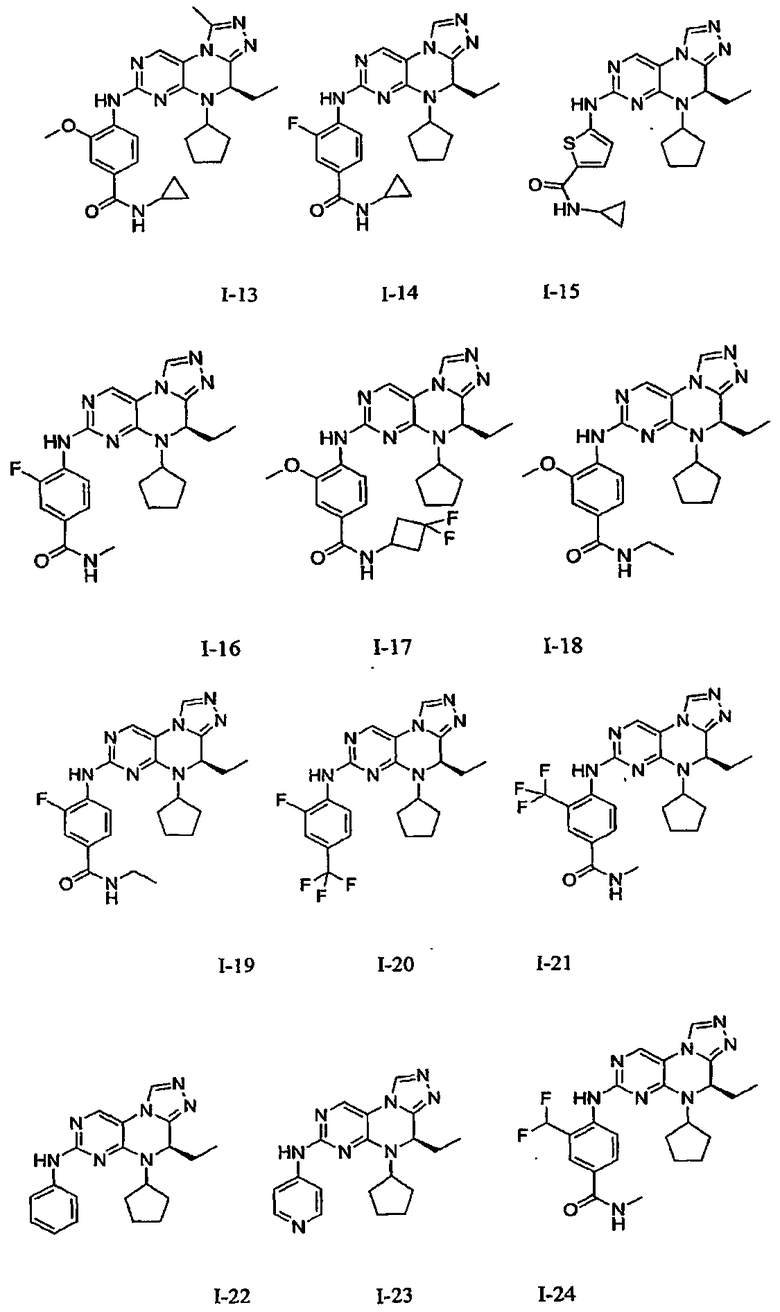
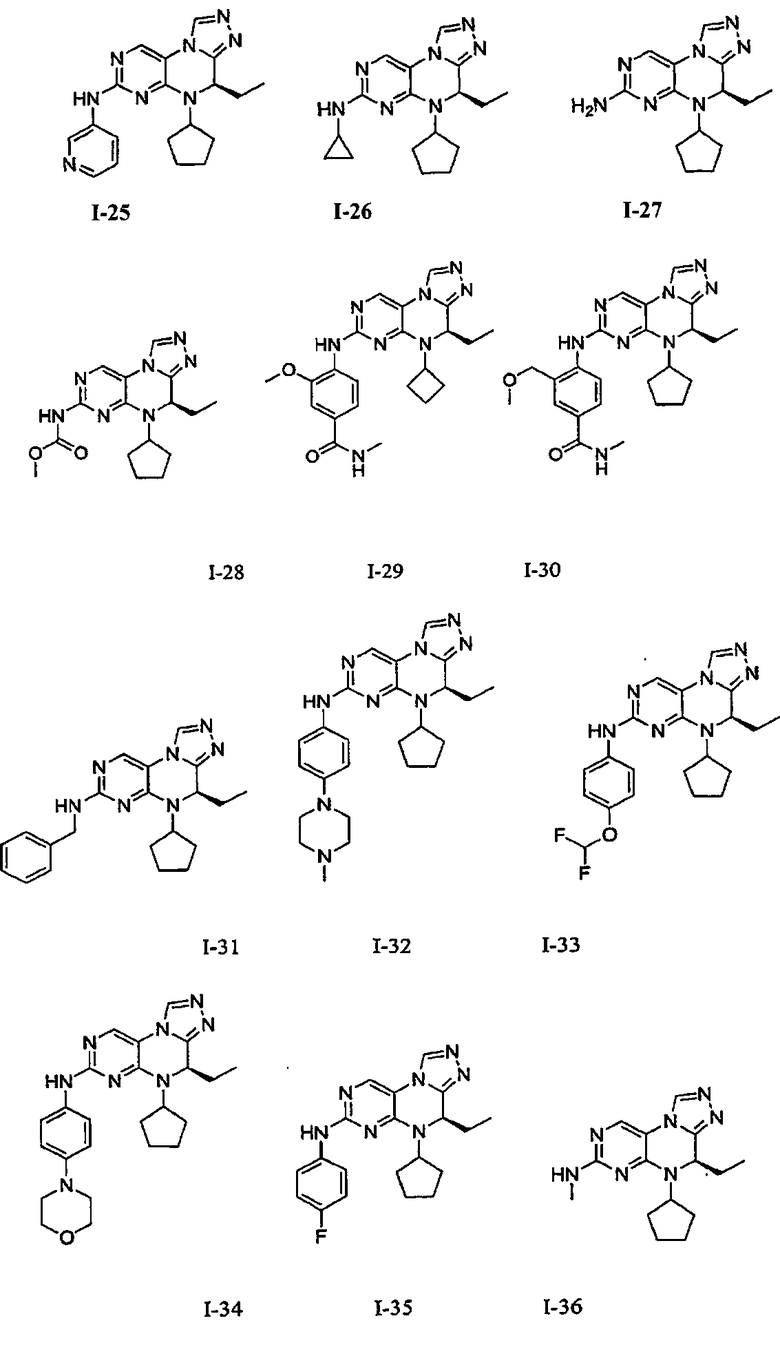

29. Фармацевтическая композиция, обладающая свойствами ингибитора протеинкиназы Plk1, содержащая эффективное количество соединения по любому из пп.1-28 и фармацевтически приемлемый носитель, адъювант или наполнитель.
30. Способ ингибирования активности протеинкиназы Plk1 у пациента, включающий введение указанному пациенту соединения по любому из пп.1-28 или фармацевтической композиции по п.29.
31. Способ ингибирования активности протеинкиназы Plk1 в биологическом образце, включающий контактирование указанного биологического образца с соединением по любому из пп.1-28 или фармацевтической композицией по п.29.
| RU 2005113978 А, 20.11.2005 | |||
| Прибор для сложения и вычитания | 1924 |
|
SU5287A1 |
| WO 2005123736 А, 29.12.2005 | |||
| ЭЛЕКТРОИЗОЛИРУЮЩЕЕ ФЛАНЦЕВОЕ СОЕДИНЕНИЕ | 1993 |
|
RU2076985C1 |

