Настоящее изобретение относится к способам превращения нитратов металлов в соответствующие оксиды металлов.
Нитраты металлов являются удобными предшественниками оксидов металлов благодаря их относительно низкой стоимости и легкости производства. Их часто превращают в соответствующие оксиды металлов при получении катализаторов или сорбентов. При производстве катализаторов или сорбентов подходящий носитель пропитывают одним или несколькими растворимыми нитратами металлов и сушат для удаления растворителя. Затем на стадии, часто называемой прокаливанием, пропитанный носитель обычно нагревают на воздухе до повышенных температур или выше температуры разложения нитрата металла с образованием оксида металла. Однако этот способ не всегда позволяет получить удовлетворительные оксидные материалы. В частности, в том случае, когда оксид металла может восстанавливаться, дисперсность и распределение кристаллитов оксида металла и, следовательно, получаемого этими способами восстановленного металла часто оказываются низкими.
Предпринимались попытки варьировать этот препаративный способ. В ЕР 0421502 описан способ приготовления катализатора или предшественника катализатора, в котором нитрат кобальта, нанесенный на пористый инертный носитель, прокаливают в атмосфере, содержащей по меньшей мере 20 об.% оксида азота (без учета содержания воды в атмосфере). Оксиды азота образуются преимущественно при разложении нитрата кобальта в условиях, когда печь для прокаливания не продувается или продувается с низкой скоростью. Было установлено, что при таком прокаливании образуются агломераты кристаллитов оксида кобальта с размерами в интервале 1-10 мкм.
В указанном выше ЕР 0421502 нитрат кобальта прокаливают на воздухе и оксид азота образуется из самого нитрата металла. Хотя конкретный оксид азота не был указан, при таком прокаливании преобладает диоксид азота (NO2).
Нанесенные оксиды металлов используют в качестве катализаторов, предшественников катализаторов и сорбентов, эффективность которых зависит от дисперсности оксида металла на носителе. Поэтому существует необходимость повысить дисперсность оксидов металлов, получаемых из нитратов металлов.
Заявители обнаружили, что термообработка в смеси газов, содержащей конкретно оксид азота и малое количество кислорода или не содержащей кислорода, приводит к очень высокодисперсным и равномерно распределенным нанесенным оксидам металлов. В отличие от способа ЕР 0421502 в способе настоящего изобретения не нужна высокая концентрация оксида азота, и способ позволяет получать чрезвычайно мелкие агломераты оксида металла с размерами кристаллитов <10 нм.
Соответственно изобретение предлагает способ превращения нанесенного нитрата металла в соответствующий нанесенный оксид металла, включающий нагревание нитрата металла для активации его разложения в газовой смеси, содержащей оксид азота и <5 об.% кислорода.
Кроме того, изобретение предлагает нанесенный оксид металла, который можно получать указанным способом.
Таким образом, способ согласно настоящему изобретению включает пропускание газовой смеси, которая содержит оксид азота и кислород в количестве <5 об.%, над нанесенным нитратом металла и нагревание нитрата металла, контактирующего с газовой смесью, по меньшей мере, до температуры разложения. Следовательно, в настоящем изобретении оксид азота не образуется при разложении нитрата металла и должен присутствовать в проточной газовой смеси, с которой контактирует нитрат металла в ходе разложения.
Нитрат металла можно наносить многими способами, включая сухое смешение, смешение нитрата в расплаве, осаждение и пропитку, причем предпочтительна пропитка. Например, нитратом металла можно пропитать материал носителя из водного или неводного раствора, например этанольного, который может содержать другие вещества, и затем высушить для удаления растворителя или растворителей. В растворе может присутствовать один или более нитратов. Для увеличения содержания металла или образования последовательных слоев разных нитратов металлов до сушки можно провести одну или более стадий пропитки. Пропитку можно осуществлять любым способом, известным специалистам в области производства катализаторов или сорбентов, но предпочтительными являются так называемая «сухая» пропитка или пропитка «по влагоемкости», так как она минимизирует количество используемого растворителя, который нужно удалять при сушке. Пропитка по влагоемкости особенно пригодна для пористых носителей и включает смешение носителя только с таким количеством раствора, которое достаточно для заполнения пор носителя.
Сушить можно известными способами при пониженном давлении, атмосферном давлении или повышенном давлении, в том числе путем распылительной сушки и сушки с вымораживанием. Предпочтительно, чтобы температура сушки составляла ≤200°С, более предпочтительно ≤160°С для минимизации преждевременного разложения нитрата металла. Стадию сушки можно проводить на воздухе или в атмосфере другого кислородсодержащего газа или в инертном газе, таком как азот, гелий или аргон.
Поэтому нанесенный нитрат металла будет содержать на поверхности и/или в порах носителя один или более нитратов металлов.
Для разложения нитрат металла нагревают до температуры разложения или, если нужно, выше температуры, при которой образуется оксид металла. Эта стадия нагревания отличается от сушки (которая в принципе нужна для удаления растворителя) тем, что вызывает физико-химическое превращение нитрата металла в соответствующий оксид металла. Далее будет понятно, что в способе настоящего изобретения нанесенный нитрат металла можно при желании высушить и нагреть до разложения в ходе одной операции. Температура, до которой нитрат металла надо нагреть для его разложения, может быть в интервале 100-1200°С, но предпочтительно в интервале 200-600°С, чтобы провести разложение нитрата до оксида и в то же время минимизировать спекание оксида. Было установлено, что более мелкие кристаллиты оксида металла можно получить прокаливанием при более низких температурах, например, в интервале 200-450°С, в частности при 200-300°С. Однако, когда нужно получить фазы шпинели или перовскита на носителе или с его участием, может быть желательно использовать температуры в интервале 500-1200°С. Время, в течение которого нанесенный нитрат металла находится при температуре в этих интервалах, составляет предпочтительно <16 час, более предпочтительно <8 час. Наиболее предпочтительно короткое время прокаливания, например, ≤4 час, особенно ≤2 час.
Предпочтительно, чтобы по меньшей мере 90 мас.%, более предпочтительно по меньшей мере 95%, наиболее предпочтительно по меньшей мере 99% нитрата металла превращались в соответствующий оксид металла.
Особенностью настоящего изобретения является то, что атмосфера, с которой контактирует нанесенный нитрат металла во время нагревания, содержит очень мало или совсем не содержит кислорода, который, как было установлено, является причиной низкой дисперсности оксида металла в материалах, получаемых из нитратов. Следовательно, содержание кислорода (О2) в потоке газа составляет <5 об.%, предпочтительно <1 об.%, наиболее предпочтительно <0,1 об.%.
Газовый поток, с которым контактирует нитрат металла, может быть любым потоком, содержащим оксид азота и <5 об.% кислорода. Предпочтительно, чтобы газовый поток содержал один или несколько газов, которые выбирают из монооксида углерода, диоксида углерода или инертного газа. Предпочтительно, чтобы инертный газ был выбран из азота, гелия или аргона. Предпочтительно, чтобы газовый поток, с которым контактирует нитрат металла, содержал один или более инертных газов и оксид азота.
Газовая смесь, с которой контактирует нанесенный нитрат металла, может находиться при атмосферном давлении или более высоком давлении, обычно до примерно 10 бар абс. Для проведения стадии нагревания можно использовать разные способы, известные специалистам. Например, поток газа-восстановителя можно пропускать через слой частиц нанесенного нитрата металла. При осуществлении стадии нагревания путем пропускания газовой смеси через слой нанесенного оксида металла предпочтительно, чтобы часовая объемная скорость газовой смеси (GHSV) находилась в интервале 100-600000 час-1, более предпочтительно 600-100000 час-1, наиболее предпочтительно 1000-60000 час-1.
Предпочтительно, чтобы концентрация оксида азота в газовом потоке составляла 0,001-15 об.%, более предпочтительно 0,01-10 об.%, наиболее предпочтительно 0,1-5 об.% для минимизации требований к скрубберной очистке.
Нитрат металла может быть любым нитратом, но предпочтительным является нитрат металла, используемый в производстве катализаторов, предшественников катализаторов или сорбентов. Нитрат металла может быть нитратом щелочного или переходного металла. Предпочтительно, чтобы нитрат металла был нитратом переходного металла, т.е. нитратом металлов, которые выбирают из групп 3-12 Периодической системы элементов. Подходящие нитраты металлов, легко доступные для производства катализаторов, предшественников катализаторов или сорбентов, включают нитраты La, Zr, Cr, Mn, Fe, Ru, Co, Rh, Ir, Ni, Pd, Pt, Cu и Zn, более предпочтительно нитраты Cr, Mn, Fe, Ru, Co, Rh, Ir, Ni, Pd, Pt, Cu и Zn.
Могут присутствовать один или несколько нитратов металлов. Термин «нитрат металла» включает соединения нитратов металлов формулы M(NO3)x .(H2O)a, где х представляет собой валентность металла М и «а» может быть 0 или целым числом ≥1, а также продукты неполного разложения таких соединений, образовавшиеся, например, в ходе предыдущей стадии сушки, такие как гидроксинитраты металлов.
Заявители установили, что настоящий способ особенно применим для получения высокодисперсных восстанавливаемых оксидов металлов, т.е. оксидов металлов, в которых по меньшей мере часть металла может восстановиться до элемента в потоке газа-восстановителя, такого как монооксид углерода и/или водород. Такие восстанавливаемые оксиды металлов включают оксиды Ni, Co, Cu и Fe, и поэтому в предпочтительном варианте нитрат металла представляет собой нитрат никеля, кобальта, меди или железа, более предпочтительно никеля или кобальта, особенно никеля. Может присутствовать один или несколько нитратов, например, Cu/Ni, Co/Ni.
Носитель, на который можно нанести нитрат металла, может быть металлом, углеродом, оксидом металла, смешанным оксидом металла или твердым полимером. Например, носитель может быть индивидуальным или смешанным оксидом металла, включая оксиды кремния или силикаты, или другим типом носителя, применяемым в производстве катализаторов или сорбентов, например, металлом, сплавом металлов или углем. В настоящем изобретении можно использовать один или более носителей.
В настоящем изобретении можно использовать в качестве носителей углеродные носители, такие как активированные угли, графиты с развитой поверхностью, углеродные нановолокна и фуллерены в порошке, таблетках или гранулах с подходящей пористостью, например, выше 0,1 мл/г, предпочтительно в том случае, когда газовый поток содержит 0,1 об.% кислорода. Такие носители нельзя использовать в способах предшествующего уровня техники с использованием прокаливания на воздухе.
Предпочтительно, чтобы носитель был оксидным, т.е. индивидуальным или смешанным оксидом металлов, включая керамику, цеолиты, перовскиты, шпинели и т.п. Оксидный носитель может быть также нанесен на керамическую, углеродную или полимерную подложку.
Носитель может быть в виде порошка со средневзвешенным по поверхности диаметром D[3,2] в интервале 1-200 мкм. Термин «средневзвешенный по поверхности диаметр D[3,2]», иначе называемый средним диаметром Саутера (Sauter), определен M. Alderliesten в статье “Номенклатура средних диаметров частиц», Anal. Proc., vol. 21, May 1984, pages 167-172 и рассчитан на основании анализа размеров частиц традиционным методом лазерной дифракции образца с использованием Malvern Mastersizer. Агломераты таких порошков с размерами частиц в интервале от 200 мкм до 1 мм также можно использовать в качестве носителя. Альтернативно носитель может иметь вид формованных частиц, таких как таблетки, экструдаты или гранулы, обычно размером в интервале 1-25 мм с соотношением длины к ширине менее 2. (Под размерами частиц авторы понимают наименьший размер частицы, такой как ширина, длина или диаметр.) Альтернативно носитель может быть в виде монолита, например сотового, или ячеистого материала, такого как открытая вспенененная структура.
Предпочтительно выбирать носитель из оксида алюминия, алюмината металла, оксида кремния, алюмосиликата, оксида титана, оксида циркония или их смесей, включая согели, либо в виде порошка, формованных единиц, монолитных либо ячеистых форм.
Носителем может быть оксид кремния. Носители из оксида кремния могут иметь природное происхождение, например кизельгур, могут быть пирогенным кремнеземом или коллоидальным оксидом кремния или могут быть синтетическими, такими как осажденный оксид кремния или силикагель. В качестве носителя можно использовать структурированные мезопористые оксиды кремния, такие как SBA-15. Предпочтительны осажденные виды оксида кремния. Оксид кремния может быть в виде порошка или формованного материала, например, в виде экструдированных, таблетированных или гранулированных частиц оксида кремния. Пригодные порошковые оксиды кремния обычно содержат частицы со средневзвешенным по поверхности диаметром D[3,2] в интервале 3-100 мкм. Формованные оксиды кремния имеют разнообразные формы и размеры частиц в зависимости от типа пресс-формы или матрицы, использованных в их производстве. Например, частицы могут иметь в поперечном сечении круг, лепестковую структуру или другие формы и длину от 1 до более 10 мм. Величина поверхности по БЭТ у подходящих порошковых или гранулированных оксидов кремния обычно находится в интервале 10-500 м2/г, предпочтительно 100-400 м2/г. Объем пор обычно составляет примерно 0,1-4 мл/г, предпочтительно 0,2-2 мл/г и предпочтительно, чтобы средний диаметр пор находился в интервале от 0,4 до примерно 30 нм. При желании оксид кремния можно смешать с другим оксидом металла, таким как оксид титана или оксид циркония. Оксид кремния может также находиться в виде покрытия на сформованной частице, которая предпочтительно состоит из оксида алюминия, причем покрытие обычно состоит из 0,5-5 монослоев оксида кремния поверх носителя.
Носитель может быть оксидом титана. Предпочтительны синтетические носители из оксида титана, например, осажденные образцы оксида титана. Оксид титана может необязательно содержать до 20 мас.% другого огнеупорного оксида, обычно оксида кремния, оксида алюминия или оксида циркония. Альтернативно оксид титана может быть в виде покрытия на носителе, который предпочтительно является оксидом кремния, например, в виде 0,5-5 монослоев оксида титана поверх носителя из оксида алюминия или оксида кремния. Величина поверхности по БЭТ у подходящего оксида титана обычно находится в интервале 10-500 м2/г, предпочтительно 100-400 м2/г. Предпочтительно, чтобы объем пор оксида титана составлял примерно 0,1-4 мл/г, более предпочтительно 0,2-2 мл/г и средний диаметр пор находился в интервале от 2 до примерно 30 нм.
Аналогично носители из оксида циркония могут быть синтетическими, например, осажденными образцами оксида циркония. Оксиды циркония могут необязательно также содержать, например, до 20 мас.% другого огнеупорного оксида, обычно оксида кремния, оксида алюминия или оксида титана. Альтернативно оксид циркония может быть стабилизирован, например, оксидом иттрия или оксидом церия. Оксид циркония может служить покрытием на носителе, который предпочтительно состоит из оксида кремния или оксида алюминия, например, покрытия в 0,5-5 монослоев оксида циркония поверх носителя из оксида алюминия или оксида кремния.
Носитель может представлять собой промежуточную разновидность оксида алюминия. Промежуточные оксиды алюминия определены в “Ullmans Encyklopaedie der technishen Chemie”, 4, neubearitete und erweiterte Auflage, band 7 (1974), pp. 298-299. Подходящий промежуточный оксид алюминия может быть из группы гамма-оксидов алюминия, например, эта-оксид алюминия или хи-оксид алюминия. Эти вещества можно получить прокаливанием гидроксидов алюминия при 400-750°С, и обычно они имеют величину поверхности по БЭТ в интервале 150-400 м2/г. Альтернативно промежуточный оксид алюминия может быть из группы дельта-оксидов алюминия, включающей высокотемпературные формы, такие как дельта- и тета-оксиды алюминия, которые могут образоваться при нагревании оксидов алюминия гамма-группы до температуры выше примерно 800°С. Оксиды алюминия дельта-группы обычно имеют поверхность по БЭТ в интервале 50-150 м2/г. Альтернативно промежуточный оксид алюминия может быть альфа-оксидом алюминия. Промежуточные оксиды алюминия содержат менее 0,5 моль воды на моль Al2O3, причем реальное количество воды зависит от температуры их прогревания. Подходящий порошок промежуточного оксида алюминия обычно имеет средневзвешенный по поверхности диаметр D[3,2] в интервале 1-200 мкм. В некоторых применениях, таких как в катализаторах реакций в суспензиях, лучше использовать очень мелкие частицы, предпочтительно в среднем с размером менее 20 мкм, например, 10 мкм или менее. Для других применений, например, в качестве катализатора реакций в кипящем слое можно использовать более крупные частицы с размером в интервале 50-150 мкм. Предпочтительно, чтобы порошок оксида алюминия имел сравнительно большой средний диаметр пор, так как использование таких оксидов алюминия позволяет получать катализаторы с особенно высокой селективностью. Предпочтительные оксиды алюминия имеют средний размер пор по меньшей мере 10 нм, особенно в интервале 15-30 нм. [Под термином средний диаметр пор авторы понимают умноженный на 4 объем пор, определенный по десорбционной ветви изотермы физической адсорбции азота при относительном давлении 0,98, деленный на величину поверхности по БЭТ]. Предпочтительно, чтобы оксид алюминия представлял собой гамма-оксид алюминия или тета-оксид алюминия, более предпочтительно тета-оксид алюминия с поверхностью по БЭТ, равной 90-120 м2/г, и объемом пор 0,4-0,8 см3/г. Носитель из оксида алюминия может быть в виде порошка, высушенного распылительной сушкой, или он может быть сформован в сферы, таблетки, цилиндры, кольца или дырчатые таблетки, которые могут быть лепестковыми или рифлеными, например, в виде клеверного листа в поперечном сечении, или в экструдаты, известные специалистам в данной области. Лучше выбирать носитель из оксида алюминия из-за того, что его легко отфильтровать и он устойчив к истиранию.
Настоящее изобретение можно использовать для превращения нитратов металлов на любом носителе, однако наиболее предпочтительными являются определенные комбинации нитрата металла и носителя. Например, в зависимости от типа металла может быть желательно или может не быть желательно комбинировать нитрат металла с носителем, который в условиях нагревания может быть использован в разложении нитрата металла с образованием смешанных оксидов металлов с полученным нанесенным оксидом металла. Малоактивные носители, такие как углерод или альфа-оксид алюминия, можно использовать для уменьшения возможности или предотвращения образования смешанных оксидов металлов с носителем, когда это нежелательно.
Как указано выше, заявители нашли, что способ настоящего изобретения особенно применим для приготовления высокодисперсных восстанавливаемых оксидов металлов на носителях. Поэтому в одном варианте способ включает также нагревание нанесенного восстанавливаемого оксида металла в потоке газа-восстановителя для активации восстановления по меньшей мере части оксида металла. Можно использовать поток любого газа-восстановителя, однако предпочтительно, чтобы такой поток газа-восстановителя содержал монооксид углерода и/или водород.
Соответственно изобретение предлагает также нанесенный восстановленный оксид металла, получаемый указанным способом. Нанесенный восстановленный оксид металла будет содержать металл в виде элемента и возможно невосстановленный оксид металла на носителе. Кроме того, на носителе могут присутствовать другие восстанавливаемые или невосстанавливаемые оксиды металла.
В этом варианте нанесенный оксид металла включает по меньшей мере один восстанавливаемый оксид металла, предпочтительно один или несколько оксидов, выбранных из оксида никеля, оксида кобальта, оксида меди или оксида железа, и восстановление предпочтительно проводить с помощью водородсодержащего газа.
Таким образом, стадию восстановления можно проводить, пропуская водородсодержащий газ, такой как водород, синтез-газ или смесь водорода с азотом, метаном или другим инертным газом, над нанесенным восстанавливаемым оксидом металла при повышенной температуре, например, пропуская водородсодержащий газ над композицией при температурах в интервале 150-600°С, предпочтительно 300-500°С, в течение 0,1-24 час при атмосферном или повышенном до примерно 25 бар давлении. Оптимальные условия восстановления для оксида никеля, оксида кобальта, оксида меди или оксидов железа известны специалистам в данной области.
Предпочтительно, чтобы в нанесенных восстановленных оксидах металлов, полученных способом настоящего изобретения, по меньшей мере 70%, более предпочтительно >80% и наиболее предпочтительно >90% восстанавливаемого оксида металла могло восстановиться до активной формы в виде элемента. Восстановленные оксиды металлов с очень высокой дисперсностью металлов, выраженной как величина поверхности металла в расчете на грамм катализатора или грамм металла в восстановленном веществе, можно получить по способу настоящего изобретения. Величины поверхности металлов можно определять по хемосорбции (например, хемосорбции водорода) методами, известными специалистам.
Нанесенные оксиды металлов и восстановленные оксиды металлов обладают более высокой дисперсностью оксида металла и металла, чем оксиды металлов и восстановленные оксиды металлов, полученные способами предшествующего уровня техники. Это обусловлено тем, что разложение нитрата металла в присутствии оксида азота в потоке газа с <5 об.% кислорода препятствует возможному спеканию оксида металла.
Нанесенные оксиды металлов согласно настоящему изобретению изучали методом сканирующей просвечивающей электронной микроскопии (STEM) и рентгеновской дифракции (XRD) и показали, что кристаллиты оксидов металлов имеют размер менее 5 нм при конечном содержании оксида металла на носителях до 30 мас.% Размеры кристаллитов нанесенных восстановленных оксидов металлов также составляют <10 нм, предпочтительно <5 нм.
Нанесенные оксиды металлов и нанесенные восстановленные оксиды металлов можно использовать во многих областях техники. Такие области включают катализаторы, предшественники катализаторов, сорбенты, полупроводники, сверхпроводники, область ЗУ на магнитных носителях, область ЗУ на твердых носителях, пигменты и УФ-поглотители. Предпочтительно использовать нанесенные оксиды металлов и нанесенные восстановленные оксиды металлов в качестве катализаторов, предшественников катализаторов или сорбентов. Термин «сорбенты» включает адсорбенты и поглотители.
Например, восстановленные нанесенные оксиды Cu, такие как Cu/ZnO/Al2O3, используют в качестве катализаторов синтеза метанола и конверсии водяного газа. Восстановленные нанесенные оксиды Ni, Cu и Co можно использовать по отдельности или в комбинации с другими оксидами металлов, например с оксидом Zn, в качестве катализаторов реакций гидрирования, а восстановленные оксиды Fe или Co можно использовать в качестве катализаторов синтеза углеводородов по Фишеру-Тропшу. Восстановленные Fe-катализаторы можно также использовать в высокотемпературных реакциях конверсии водяного газа и синтеза аммиака.
В предпочтительных вариантах нанесенные оксиды металлов и нанесенные восстановленные оксиды металлов используют в качестве катализаторов реакций гидрирования и синтеза углеводородов по Фишеру-Тропшу. Эти катализаторы могут содержать, помимо Ni, Cu, Co или Fe, одну или более подходящих добавок и/или промоторов, используемых в реакциях гидрирования и/или синтезах Фишера-Тропша. Например, катализаторы Фишера-Тропша могут содержать одну или несколько добавок, которые изменяют физические свойства, и/или промоторы, которые оказывают влияние на восстановление и активность или селективность катализаторов. Подходящие добавки выбирают из соединений калия (K), молибдена (Mo), никеля (Ni), меди (Cu), железа (Fe), марганца (Mn), титана (Ti), циркония (Zr), лантана (La), церия (Ce), хрома (Cr), магния (Mg) или цинка (Zn). Подходящие промоторы включают родий (Rh), иридий (Ir), рутений (Ru), рений (Re), платину (Pt) и палладий (Pd). Предпочтительно включать в предшественник катализатора один или более промоторов, выбранных из Ru, Re, Pt или Pd. Добавки и/или промоторы можно вводить в состав катализаторов, используя подходящие соединения, такие как кислоты, например перрениевая кислота, соли металлов, например нитраты или ацетаты металлов, или подходящие металлоорганические соединения, такие как алкоксиды металлов или ацетилацетонаты металлов. Количество металлического промотора может находиться в интервале 3-50 мас.%, предпочтительно 5-20 мас.% в расчете на восстанавливаемый металл.
Как указано выше, нанесенные восстановленные металлоксидные катализаторы можно использовать, например, в реакциях гидрирования, особенно Ni- и Со-катализаторы, и для синтезов углеводородов по Фишеру-Тропшу, особенно Со- и Fe-катализаторы.
Типичные реакции гидрирования включают гидрирование альдегидов и нитрилов до спиртов и аминов соответственно и гидрирование циклических ароматических соединений или ненасыщенных углеводородов. Катализаторы настоящего изобретения особенно пригодны для гидрирования ненасыщенных органических соединений, особенно масел, жиров, жирных кислот, производных жирных кислот типа нитрилов. Такие реакции гидрирования обычно проводят в непрерывном или периодическом режиме путем обработки гидрируемого соединения водородсодержащим газом под давлением в автоклаве при обычной или повышенной температуре в присутствии катализатора; например, гидрирование можно осуществить водородом при 80-250°С и давлении в интервале 0,1-5,0·106 Па.
Синтез углеводородов по Фишеру-Тропшу хорошо разработан. Синтез Фишера-Тропша превращает смесь монооксида углерода и водорода в углеводороды. Смесь монооксида углерода и водорода обычно представляет собой синтез-газ с соотношением водород:монооксид углерода в интервале 1,7-2,5:1. Реакцию можно проводить непрерывным или периодическим способом с использованием одного или нескольких суспензионных реакторов с перемешиванием, реакторов с барботажной колонной, циркуляционных реакторов или реакторов с кипящим слоем. Способ можно осуществлять при давлениях в интервале 0,1-10 МПа и температурах в интервале 150-350°С. Часовая объемная скорость газа (GHSV) при непрерывной работе находится в интервале 100-25000 час-1. Катализаторы согласно настоящему изобретению особенно применимы благодаря большой величине поверхности металла/г катализатора.
Далее изобретение проиллюстрировано ссылками на следующие примеры и на фиг.1-7, на которых
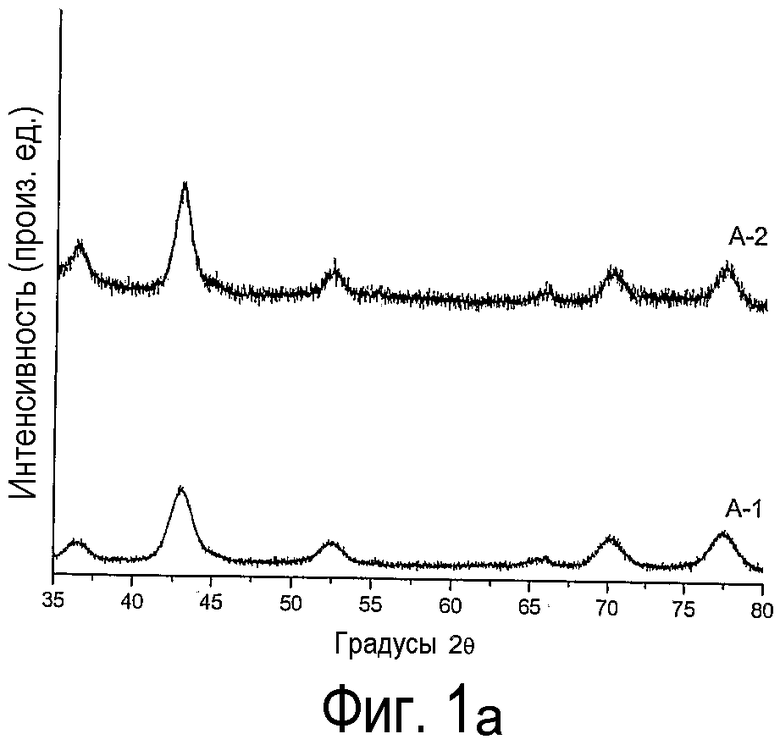
на фиг.1 приведены дифрактограммы (а) нанесенного на оксид кремния оксида кобальта, полученного согласно данному изобретению (А-1) и не согласно изобретению (А-2), и (b) нанесенного на оксид кремния оксида никеля, полученного согласно данному изобретению (В-1, С-1) и не согласно изобретению (В-2, С-2);
на фиг.2 показаны STEM-изображения, полученные методом яркого поля, нанесенных на оксид кремния оксидов кобальта (А-1, А-2) и никеля (В-1, В-2, С-1, С-2),
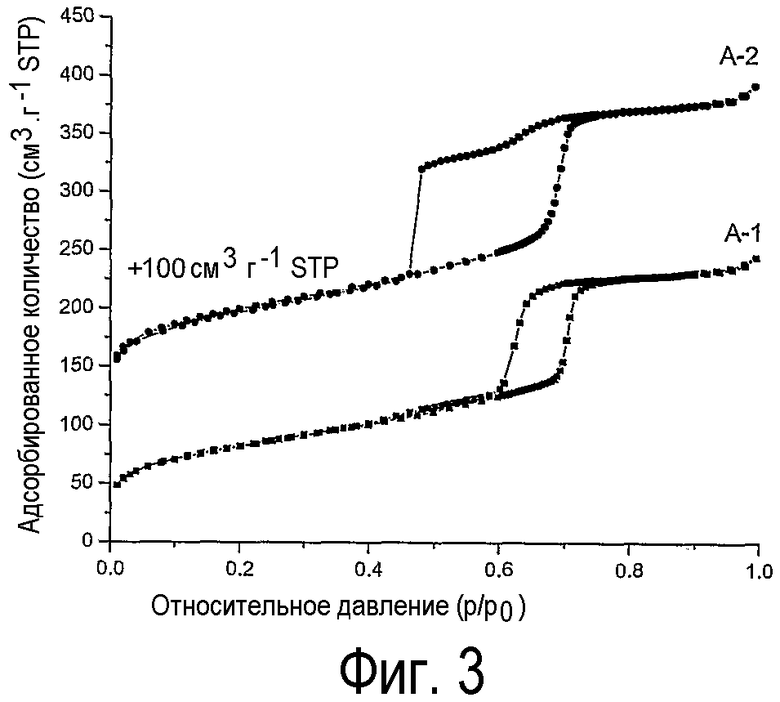
на фиг.3 показаны изотермы физической адсорбции азота на нанесенных на оксид кремния оксидах кобальта (А-1, А-2),
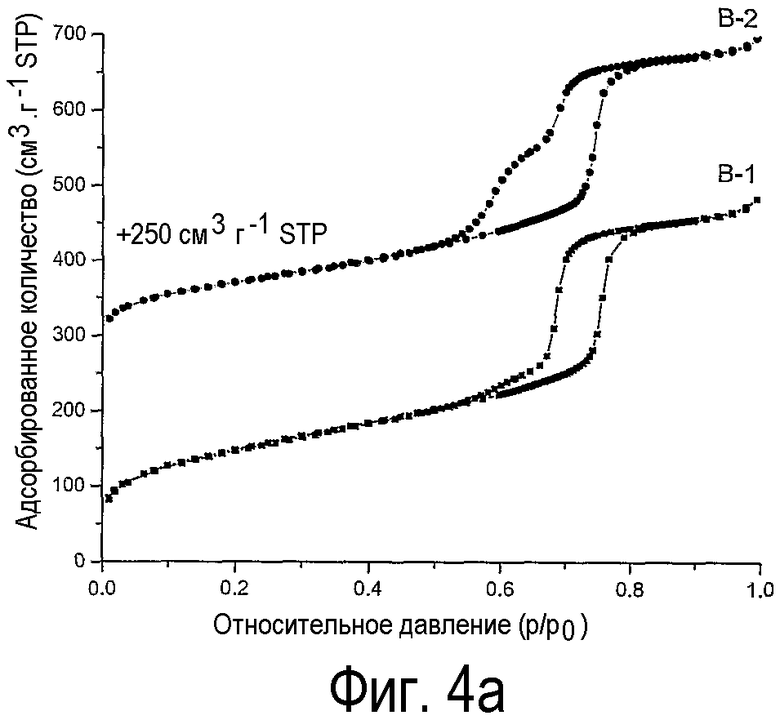
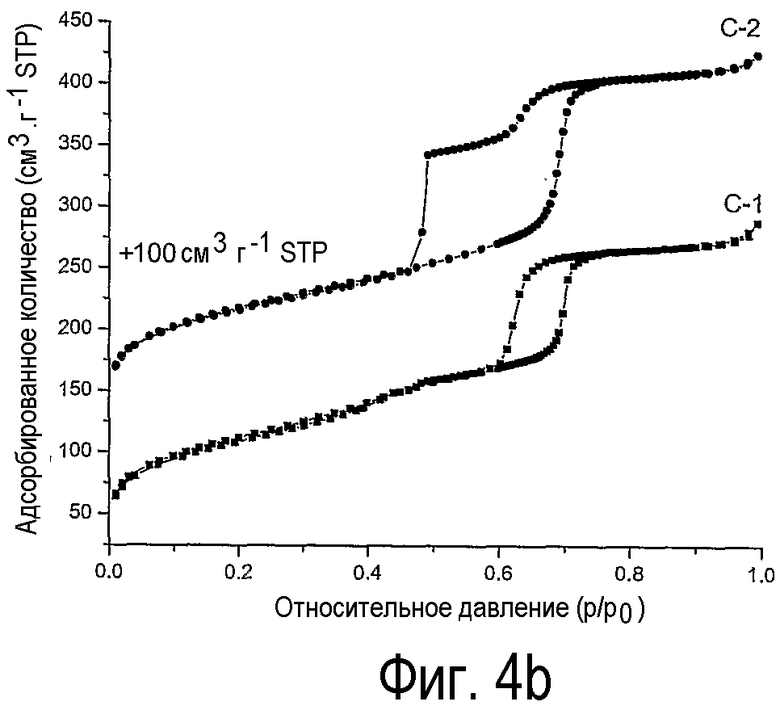
на фиг.4 показаны (а) изотермы физической адсорбции азота на нанесенных на оксид кремния оксидах никеля В-1, В-2 и (b) С-1, С-2,
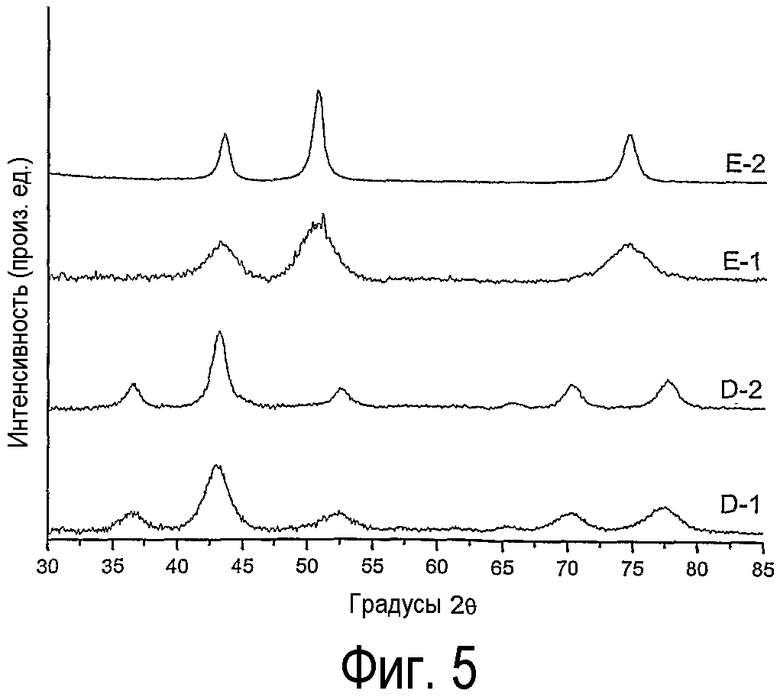
на фиг.5 представляет дифрактограммы нанесенного на оксид кремния оксида кобальта, полученного согласно данному изобретению (D-1) и не согласно изобретению (D-2), и (b) нанесенного на оксид кремния оксида никеля, полученного согласно данному изобретению (Е-1) и не согласно изобретению (Е-2),
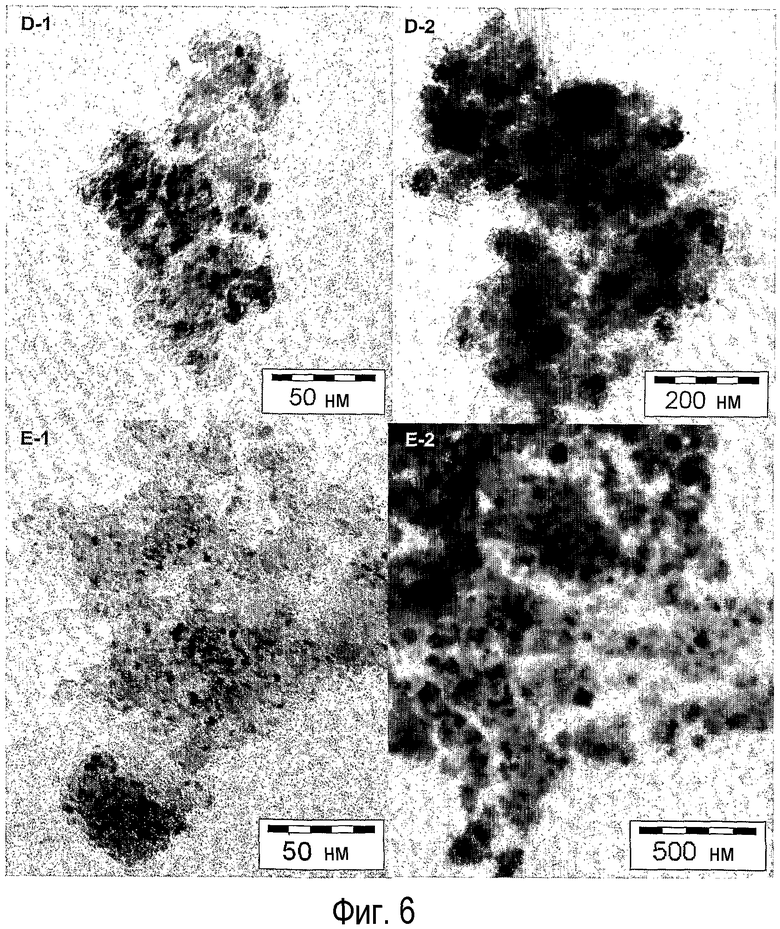
на фиг.6 показаны STEM-изображения, полученные методом яркого поля, нанесенных на оксид кремния оксидов кобальта (D-1, D-2) и никеля (Е-1, Е-2),
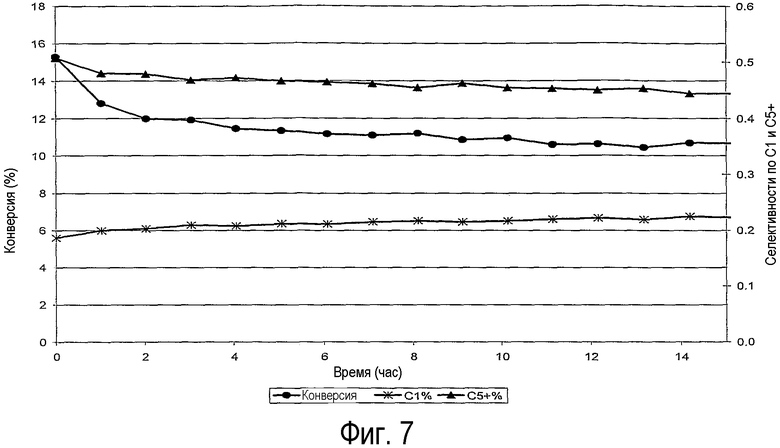
на фиг.7 приведены конверсия % и селективности образования С1 & С5+% в зависимости от времени тестирования в лабораторном синтезе углеводородов по реакции Фишера-Тропша,
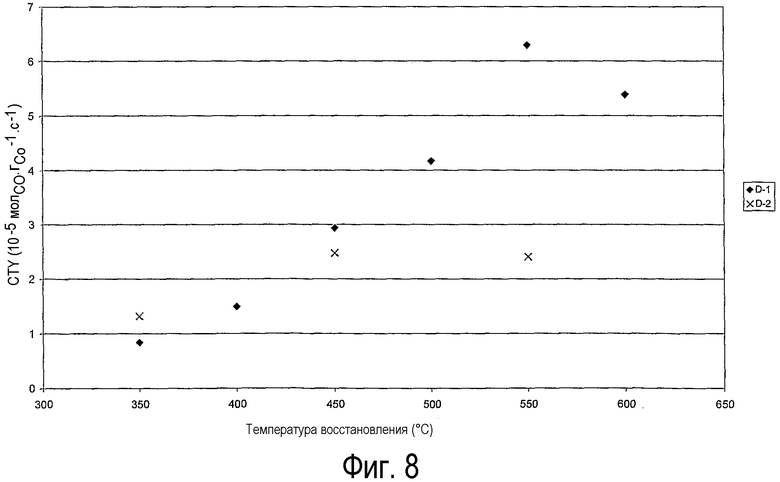
на фиг.8 показана каталитическая активность катализаторов D-1 и D-2 в лабораторном синтезе углеводородов по реакции Фишера-Тропша, определенная как выход на грамм кобальта (мольСО·гСо -1·с-1) в зависимости от температуры восстановления катализатора,
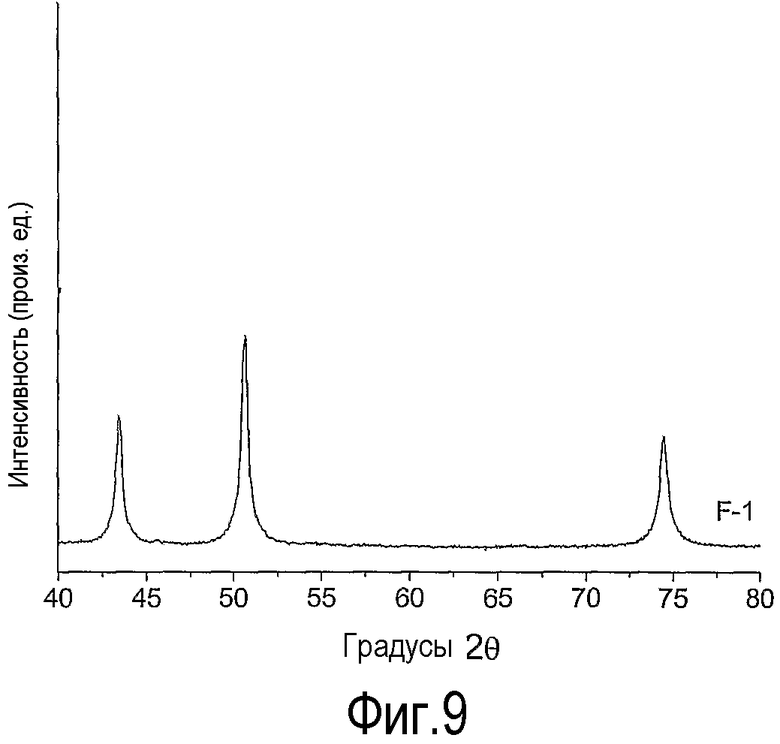
на фиг.9 приведена дифрактограмма катализатора сравнения - оксида никеля на оксиде кремния (F-1), приготовленного согласно способу, раскрытому в ЕР 0421502,
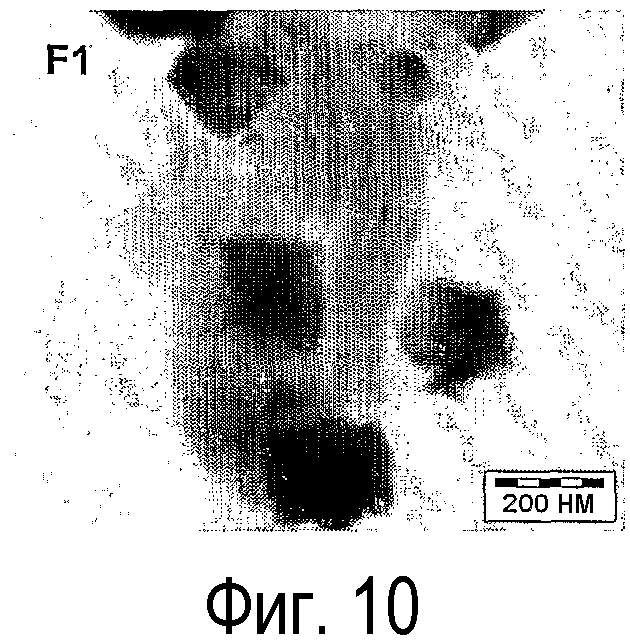
на фиг.10 показаны STEM-изображения, полученные методом яркого поля, микрофотографии катализатора сравнения (F-1) и
на фиг.11 показаны изотермы физической адсорбции азота на катализаторе сравнения F-1.
Пример 1: Оксиды никеля и кобальта, нанесенные на SBA-15
Порошок SBA-15 (поверхность по БЭТ=637 м2·г-1, общий объем пор=0,80 см3·г-1) пропитали по влагоемкости водным раствором нитрата кобальта (II) или никеля (II) и получили образцы 15 мас.% Со/SiO2 (образец А) и 12 и 20 мас.% Ni/SiO2 (образцы В и С). После уравновешивания в течение 15 мин пропитанные образцы сушили путем нагревания со скоростью 1°С·мин-1 от 25°С до конечной температуры 70°С для образца А и 120°С для образцов В и С. Образцы выдержали при конечной температуре в течение 720 мин. Небольшое количество (60 мг) образцов А, В и С нагрели еще раз в проточном реакторе идеального вытеснения (диаметр 1 см, длина 17 см) со скоростью 1°С·мин-1 от 25°С до 450°С и выдержали в течение 240 мин при 450°С в токе гелия, содержащего 1 об.% оксида азота (NO), или на воздухе (прокаливание). Образцы, прошедшие термообработку согласно настоящему изобретению в потоке гелия, содержащего 1 об.% оксида азота (NO), обозначены А-1, В-1 и С-1, а прокаленные на воздухе образцы не по настоящему изобретению были отмечены как А-2, В-2 и С-2. Условия получения приведены в таблицах 1-3.
Условия пропитки
Условия сушки
Профиль температуры и газового потока при второй термообработке
(мл·мин-1)
(мл·мин-1)
Образцы А-1, В-1, С-1, А-2, В-2 и С-2 охарактеризованы методами рентгеновской дифракции (XRD), сканирующей просвечивающей электронной микроскопии (STEM) и физической адсорбции азота. Дифрактограммы сняты при комнатной температуре и значениях угла 2θ в интервале 35-80° на дифрактометре Brucker-Nonius D8 Advance c использованием Со-Кα12 (λ=1,79026 Е) излучения. Средние размеры кристаллитов оксидов кобальта и никеля рассчитывали по уравнению Шерера [см. Scherrer, P. Gottinger Nachrichten 2 (1918) 98] по самому интенсивному рефлексу при 2θ=43,1 и 50,8° соответственно. STEM-микрофотографии получали на микроскопе Tecnai 20 FEG, работающем при 200 кВ. Средний размер частиц оксидов кобальта и никеля обычно определяли по диаметрам 50 частиц.
Физическую адсорбцию азота изучали при 77 К на приборе Micromeritics Tristar 3000. Перед анализом образцы сушили в токе гелия в течение 14 час при 120°С. Величины поверхности и общие объемы пор определяли методом БЭТ [см. Brunauer S., Emett, P.H. and Teller, E. J. Am. Chem. Soc. 60 (1938) 309] и по количеству азота, адсорбированного при относительном давлении 0,995, соответственно. Распределение пор по размерам рассчитывали из адсорбционной ветви изотермы по стандартной теории БЭТ [см. Barret, P.; Joyner, L.G.; Halenda, P.P. J. Am. Chem. Soc. 73 (1951) 373].
Дифрактограммы, приведенные на фиг.1, показывают, что при термической обработке на воздухе (прокаливании) образуются крупные кристаллиты Со3О4 и NiO. А при обработке высушенных образцов А, В и С по способу настоящего изобретения образуются очень мелкие кристаллиты Со3О4 и NiO. Сравнение средних размеров кристаллитов приведено в таблице 4.
Данные по средним размерам кристаллитов оксидов кобальта и никеля
#Диаметр частиц NiO слишком мал и не определяется.
Типичные STEM-изображения, полученные методом яркого поля, образцов А-1, А-2, В-1, В-2, С-1 и С-2 (фиг.2) показывают, что пористая структура SBA-15, образованная открытыми мезопорами, сохраняется. Микрофотографии четко показывают, что образцы (А-2, В-2 и С-2), полученные прокаливанием, представляют собой нанесенные на SBA-15 материалы с неоднородной дисперсностью и распределением частиц оксидов кобальта и никеля в мезопорах SBA-15. Более того, микрофотографии показывают, что бóльшая часть частиц оксидов кобальта и никеля в каналах мезопор SBA-15 были ограничены в росте в одном направлении стенкой мезопоры, что привело к анизотропным частицам, блокирующим поры SBA-15. Кроме того, STEM-микрофотографии этих образцов указывают на присутствие частиц оксидов кобальта и никеля более крупных, чем диаметр поры. Это показывает, что такие частицы локализованы на внешней поверхности носителя.
Однако STEM-микрофотографии образцов (А-1, В-1 и С-1), полученных по способу настоящего изобретения, четко демонстрируют, что этот способ дает высокодисперсные частицы оксидов кобальта и никеля, равномерно распределенные в порах SBA-15. Кроме того, на внешней поверхности носителя не обнаружено частиц оксидов кобальта и никеля. Распределение частиц оксидов кобальта и никеля в образцах А-1, А-2, В-1, В-2, С-1 и С-2 сравнивается в таблице 4.
Изотерма физической адсорбции азота на образце А-1 (фиг.3) включает все типичные характеристики, приведенные для SBA-15, которые показывают, что структура не разрушена под действием процесса настоящего изобретения. Сравнение этой изотермы с изотермой на прокаленном образце А-2 показывает, что на последней помимо типических особенностей SBA-15 наблюдается замыкание десорбционной ветви. Такое замыкание десорбционной ветви можно отнести за счет блокирования каналов мезопор SBA-15 кристаллитами оксида кобальта. Эти заглушки из оксида кобальта создают поры типа чернильницы, что приводит к наблюдаемому замыканию десорбционной ветви изотермы в ходе десорбции. Отсутствие этой особенности на изотерме образца А-1 дает дополнительное подтверждение, что в способе настоящего изобретения образуются очень малые кристаллиты, поскольку достаточно крупные частицы, блокирующие мезопоры SBA-15, отсутствуют.
Из сравнения изотерм физической адсорбции азота на образцах Ni/SiO2 с изотермами на образцах Со/SiO2 видно, что наблюдается такая же тенденция, а именно блокирование мезопор SBA-15, когда нанесенные на SBA-15 оксиды никеля получают прокаливанием, в то время как применение предлагаемого способа приводит к нормальному замыканию десорбционной ветви, ожидаемому для SBА-15 с незаблокированными каналами мезопор.
Пример 2: Оксиды кобальта и никеля, нанесенные на силикагель
Силикагель Davicat SI 1404 (поверхность по БЭТ=540 м2·г-1, общий объем пор=0,90 см3·г-1) пропитали нитратом кобальта (II) или нитратом никеля (II) из водных растворов и получили образцы 18 мас.% Со/SiO2 (образец D) или 24 мас.% Ni/SiO2 (образец Е). После старения в течение 15 мин образцы D и Е высушили с помощью такой же термической обработки, как в примере 1. Небольшое количество (100 мг) образца D и Е термически обработали повторно в проточном реакторе идеального вытеснения (диаметр 1 см, длина 17 см) со скоростью нагревания 1°С·мин-1 от 25°С до 450°С и выдерживали в течение 240 мин при 450°С в токе гелия, содержащего 1 об.% оксида азота (NO), или на воздухе. Затем образцам давали остыть до 25°С в атмосфере гелия. Порции образцов D и Е, термически обработанные в токе гелия, содержащего 1 об.% оксида азота (NO), согласно настоящему изобретению, обозначены D-1 и Е-1, а порции, прокаленные в токе воздуха в отличие от настоящего изобретения, были обозначены D-2 и Е-2. Условия получения приведены в таблицах 2, 3 и 5. Образцы были охарактеризованы методами XRD и STEM.
Условия пропитки
объем пор
без вращения
объем пор
без вращения
Дифрактограммы образцов D-1, D-2, E-1 и E-2, приведенные на фиг.5, показывают, что образуются те же фазы оксидов кобальта и никеля, которые наблюдались в образцах А-1, А-2, В-1, В-2, С-1 и С-2 в примере 1, а именно Со3О4 и NiO. Дифрактограммы образцов D-1 и Е-1 показывают, что образуются мелкие кристаллиты оксидов кобальта и никеля, в то время как образцы D-2 и Е-2, которые были прокалены, содержат крупные кристаллиты оксидов кобальта и никеля. Средний размер кристаллитов приведен в таблице 6.
Данные по средним размерам кристаллитов оксидов кобальта и никеля
Типичные STEM-изображения, полученные методом яркого поля, образцов, как показано на фиг.6, подтверждают результаты XRD об образовании в образцах D-1 и Е-1, которые были обработаны согласно настоящему изобретению, высокодисперсных кристаллитов оксидов кобальта и никеля. Более того, кристаллиты оксидов кобальта и никеля равномерно распределены в носителе. Полученные средние размеры частиц оксидов кобальта и никеля приведены в таблице 6.
Пример 3: Катализатор синтеза углеводородов из монооксида углерода и водорода
Образец D-1 катализатора оксид кобальта на силикагеле из примера 2 был испытан на каталитическую активность в получении углеводородов из смеси монооксида углерода и водорода. Перед тестированием образец восстановили в течение 120 мин при 450°С в токе гелия, содержащего 33 об.% водорода. Условия восстановления приведены в таблице 7.
Условия восстановления
(мл·мин-1)
Тесты на каталитическую активность проводили в реакторе идеального вытеснения (диаметр 0,5 см, длина 4 см) при 1 бар и 220°С, объемном соотношении водород/монооксид углерода, равном 2, и GHSV≈6500 час-1. Для достижения условий изотермического идеального вытеснения небольшое количество образца (20,5 мг) смешивали с 250 мг частиц SiC (0,2 мм). Выходящий поток газа анализировали методом газовой хроматографии. Из этих данных рассчитывали массовые селективности по метану (С1/Собщ) и продуктам с длиной цепи 5 и выше (С5+/Собщ). На фиг.7 показаны результаты, зарегистрированные в течение первых 15 час реакции. Видно, что образец D-1, приготовленный согласно данному изобретению, обладает очень высокой активностью. Несмотря на высокое содержание Со, равное 18 мас.%, при 2% конверсии СО (при GHSV 34000 час-1) получен поразительный временной выход кобальта, равный 3,44×10-5 молСО·гСо -1·с-1.
Пример 4: Получение углеводородов из монооксида углерода и водорода
Небольшие количества (≈20 мг) оксида кобальта на силикагеле протестировали на активность в синтезе углеводородов из смеси монооксида углерода и водорода. Катализаторы получали согласно настоящему изобретению (образец D-1) или традиционным прокаливанием на воздухе (образец D-2), как описано в примере 2. Перед тестированием образцы восстанавливали in situ в течение 120 мин до разных конечных температур в токе гелия, содержащего 33 об.% водорода. Образцы нагревали от комнатной температуры до конечной температуры со скоростью 5°С·мин-1. Подробности условий восстановления приведены в таблице 8. Был получен ряд образцов D-1, восстановленных до конечных температур 350°С, 400°С, 450°С, 500°С, 550°С и 600°С. Ряд образцов D-2 получили при конечных температурах 350°С, 450°С и 550°С.
Условия восстановления
(°С·мин-1)
(мл·мин-1)
Тесты на каталитическую активность проводили при 1 бар и 220°С в реакторе идеального вытеснения (диаметр 0,5 см, длина 4 см) и объемном соотношении водород/монооксид углерода, равном 2. Для достижения условий изотермического идеального вытеснения небольшое количество образца (20 мг) смешали с 250 мг частиц карбида кремния с диаметром 0,2 мм. Выходящий поток газа анализировали методом газовой хроматографии. Из полученных результатов рассчитывали активности. Активность определяли как число молей превращенного монооксида углерода на грамм кобальта в секунду, названное временным выходом в расчете на кобальт (СТY).
На фиг.8 показаны значения активности, полученные за первые 18-22 часа реакции при конверсии 2-3%. Результаты для образца D-1, приготовленного согласно настоящему изобретению, ясно показывают, что активность заметно возрастает с повышением температуры восстановления от 350°С до 550°С. При переходе от температуры восстановления 350°С до 550°С наблюдается увеличение СТY от 0,84×10-5 молСО·гСо -1·с-1 до 6,29×10-5 молСО·гСо -1·с-1. Дальнейшее повышение температуры восстановления до 600°С понизило активность до 5,39×10-5 молСО·гСо -1·с-1.
Данные по каталитической активности образца D-2, который был приготовлен традиционным прокаливанием на воздухе с последующим восстановлением при 350, 450 и 550°С, показывают, что активность слабо возрастает с повышением температуры восстановления. Повышение температуры восстановления от 350°С до 550°С приводит только к увеличению CTY от 1,32 молСО·гСо -1·с-1 до 2,41 молСО·гСо -1·с-1. Сравнение образцов D-1 и D-2, которые были восстановлены при 550°С, четко показывает преимущество настоящего изобретения, так как активность катализатора D-1 выше более чем в два раза по сравнению с катализатором, полученным традиционным способом (образец D-2).
Пример 5: Катализатор гидрирования масла соевых бобов
Катализатор 24 мас.% Ni на силикагеле получали по способу настоящего изобретения, как описано в примере 2 (катализатор Е-1). Требуемое количество катализатора взвесили в стеклянном сосуде и восстановили при атмосферном давлении водорода при 400-450°С в течение 1 час. Восстановленный катализатор добавили к маслу соевых бобов для тестирования в гидрировании.
В тесте гидрирования использовали масло соевых бобов с иодным числом (ИЧ) 133. 200 г масла и нужное количество восстановленного катализатора поместили в закрытый реактор гидрирования с перемешиванием. Смесь нагревали до 140°С и через суспензию барботировали водород под давлением 3 бар абс. Температуру повышали до 200°С со скоростью 2°С/мин и выдерживали на этом уровне. Следили за количеством поглощенного маслом водорода и закончили тест после того, как ИЧ упало до 79. Время гидрирования, необходимое для достижения ИЧ=79, принимали за меру активности катализатора. Результаты для данного и для сравнительного катализатора, прокаленного на воздухе, приведены ниже
Результаты теста гидрирования
Данные показывают, что катализатор, полученный по способу настоящего изобретения, значительно активнее.
Пример 6: Катализатор Со на оксиде алюминия
Катализаторы 20 мас.% Со на оксиде алюминия получали следующим образом. 20,05 г порошка гамма-оксида алюминия (поверхность по БЭТ 148 м2/г, объем пор 1,04 мл/г) пропитали по влагоемкости нагретым раствором, содержащим 25,04 г Co(NO3)2·6H2O и 4,95 г деионизированной воды. Пропитанный порошок сушили 30 мин при 110°С, затем прокалили в токе гелия, содержащего 1 об.% NO, со скоростью 35 л/час и 40 л/час) при 240°С в течение 1 час (скорость нагрева 2°С/мин). Для сравнения прокаливание провели в эквивалентном токе воздуха. Для определения размеров кристаллитов оксидов Со были сняты дифрактограммы прокаленных образцов. Предшественники катализаторов согласно данному изобретению также восстановили при 425°С в водороде и затем известным методом определили поверхность Со по хемосорбции водорода при 150°С. Результаты приведены в таблице 10.
Размеры кристаллитов оксида кобальта по данным XRD
Полученные результаты показывают, что способ прокаливания согласно настоящему изобретению эффективен для уменьшения размеров кристаллитов оксида кобальта и, следовательно, увеличения величины конечной поверхности Со по сравнению с прокаливанием этого вещества на воздухе.
Сравнительный пример: Оксид никеля, нанесенный на SBA-15, полученный без продувки атмосферы во время прокаливания на воздухе
Этот опыт был проведен для демонстрации эффективности настоящего изобретения по сравнению с описанием в ЕР 0421502.
Порошок SBA-15 с поверхностью по БЭТ 637 м2·г-1 и общим объемом пор 0,89 см3·г-1 в количестве 0,25 г пропитали по влагоемкости водным раствором нитрата никеля (II) c концентрацией 4,23 моль·л-1 и получили образец 20 мас.% Ni/SiO2 (образец F). Через 15 мин уравновешивания пропитанное твердое вещество перенесли в кювету печи (объем 0,062 дм3, высота 4 см, диаметр 6 см) и сушили, нагревая образец со скоростью 1°С·мин-1 от 25°С до конечной температуры 120°С. Образец выдерживали при конечной температуре в течение 720 мин. Затем кювету накрыли крышкой и образец снова нагревали на воздухе (т.е. прокаливали) со скоростью 1°С·мин-1 от 25°С до 450°С и выдерживали при температуре 450°С в течение 240 мин. Таким образом, во время прокаливания активной продувки атмосферы над образцом не проводили. Образец обозначили F-1.
Дифрактограмма образца F-1 приведена на фиг.9. Дифрактограмма показывает, что при термической обработке образца F без активной продувки атмосферы образовались очень крупные кристаллиты оксида никеля (NiO). Средний размер кристаллитов в этом образце F-1 составил 32 нм.
Типичное STEM-изображение, полученное методом яркого поля, образца F-1 приведено на фиг.10. Микрофотография показывает, что мезопористая структура SBA-15 сохраняется на всех стадиях приготовления. Кроме того, микрофотография показывает, что прокаливание приводит к образцу, в котором частицы оксида никеля неравномерно распределены в носителе и характеризуются широким распределением по размерам. Более подробно, было найдено, что сравнительно крупные частицы оксида никеля с размерами в интервале 25-200 нм находятся на внешней поверхности частиц SBA-15. Более того, найдены анизотропные частицы оксида никеля внутри каналов мезопор носителя. Последний тип частиц, по-видимому, ограничен в росте стенками пор мезопористого носителя.
Адсорбционная ветвь изотермы физической адсорбции на образце F-1 (на фиг.11) включает все типичные особенности структуры SBA-15. Это показывает, что заметных изменений структуры на стадиях приготовления не происходит. Десорбционная ветвь изотермы включает особенность, типичную для пор типа чернильницы, а именно замыкание десорбционной ветви при относительном давлении примерно 0,48. Такие поры типа чернильницы образуются из кристаллитов оксида никеля, которые блокируют каналы мезопор в SBA-15.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОНВЕРСИИ НИТРАТА МЕТАЛЛА | 2007 |
|
RU2437717C2 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРАТА МЕТАЛЛА НА ПОДЛОЖКЕ | 2010 |
|
RU2516467C2 |
| КАТАЛИЗАТОРЫ | 2010 |
|
RU2517700C2 |
| КАТАЛИЗАТОР И СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА | 2010 |
|
RU2585766C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША | 2009 |
|
RU2481156C2 |
| КАТАЛИЗАТОРЫ | 2011 |
|
RU2551433C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИТИЧЕСКИ-СОРБЦИОННОГО МАТЕРИАЛА И СПОСОБ ИЗВЛЕЧЕНИЯ МЫШЬЯКА В ЕГО ПРИСУТСТВИИ | 2018 |
|
RU2691070C1 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2591702C2 |
| СПОСОБ СИНТЕЗА УГЛЕВОДОРОДОВ С5+ В ПРИСУТСТВИИ КАТАЛИЗАТОРА, ПОЛУЧЕННОГО ПРИ ПОМОЩИ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО ЦИКЛИЧЕСКОГО ОЛИГОСАХАРИДА | 2011 |
|
RU2561112C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2592271C2 |



Изобретение относится к способам превращения нитратов металлов в соответствующие оксиды металлов. Описан способ превращения нанесенного нитрата металла в соответствующий нанесенный оксид металла, включающий нагревание нитрата металла для активизации его разложения в газовой смеси, состоящей из одного или более инертных газов и оксида азота в концентрации в интервале 0,001-15 об.%. Описан также способ восстановления нанесенного оксида металла, полученного указанным выше способом, включающий нагревание нанесенного оксида металла в токе газа-восстановителя для активации восстановления по меньшей мере части оксида металла. Технический результат - получены высокодисперсные оксиды металлов на носителе и высокодисперсные восстановленные нанесенные оксиды металлов, которые могут быть использованы в качестве катализаторов или предшественников катализаторов. 4 н. и 11 з.п. ф-лы, 10 табл., 11 ил.
1. Способ превращения нанесенного нитрата металла в соответствующий нанесенный оксид металла, включающий нагревание нитрата металла для активизации его разложения в газовой смеси, состоящей из одного или более инертных газов и оксида азота в концентрации в интервале 0,001-15 об.%.
2. Способ по п.1, в котором материал носителя пропитывают нитратом металла из раствора и сушат для удаления растворителя перед нагреванием нитрата металла для его превращения в соответствующий оксид металла.
3. Способ по п.1, в котором инертный газ выбирают из азота, гелия или аргона.
4. Способ по п.1, в котором нанесенный нитрат металла нагревают до температуры в интервале 100-1200°С.
5. Способ по п.1, в котором нитрат металла является нитратом переходного металла.
6. Способ по п.1, в котором нитрат металла является нитратом никеля, кобальта, меди или железа.
7. Способ по п.1, в котором носитель является металлом, углеродом, оксидом металла, смешанным оксидом металла или твердым полимерным носителем.
8. Способ по п.1, в котором носитель выбирают из оксида алюминия, алюмината металла, оксида кремния, алюмосиликата, оксида титана, оксида циркония или их смесей.
9. Способ восстановления нанесенного оксида металла, полученного по любому из пп.1-8, включающий нагревание нанесенного оксида металла в токе газа-восстановителя для активации восстановления по меньшей мере части оксида металла.
10. Способ по п.9, в котором поток газа-восстановителя содержит монооксид углерода и/или водород.
11. Способ по п.9, в котором нанесенный оксид металла является оксидом никеля, оксидом кобальта, оксидом меди или оксидом железа и восстановление проводят водородсодержащим газом.
12. Нанесенный оксид, получаемый способом по любому из пп.1-8.
13. Нанесенный восстановленный оксид металла, получаемый способом по любому из пп.9-11.
14. Способ по п.1, в котором нитрат металла является нитратом La, Zr, Cr, Mn, Fe, Ru, Co, Rh, Ir, Ni, Pd, Pt, Cu или Zn.
15. Способ по п.1, в котором нитрат металла представляет собой нитрат Cr, Mn, Fe, Ru, Со, Rh, Ir, Ni, Pd, Pt, Cu или Zn.
| S.G.Marcheetti, M.V.Cagnoli, A.A.Alvarez, «Iron uniform-size nanoparticles dispessred on MCM-41 used as hydrocarbon synthesis catalyst», HEPERFINE INTERACTIONS, VOI.139-140, page 33-40, 2002 | |||
| Bengoa et al, «Iron oxide nanoparticles inside the MCM-41 channels: Study of the structural stability of the support», MICROPOROUS AND |

