Область техники, к которой относится изобретение
Настоящее изобретение относится к катализаторам. В частности, оно относится к способу получения предшественника катализатора и к способу получения катализатора, который можно применять, например, для реакций гидрирования, включая синтез углеводородов (например, синтез Фишера-Тропша (FT)) и для других реакций гидрирования, таких как реакции гидрирования органических соединений.
Уровень техники
Получение предшественников катализатора путем нанесения металла на носители катализаторов различными методами хорошо известно специалистам в данной области техники. Затем полученные при этом носители с нанесенным на них металлом обычно высушивают и прокаливают, получая предшественники катализаторов, после чего предшественники вступают в реакцию, давая в результате катализатор.
В Европейской патентной заявке EP-A 0736326 описаны катализаторы синтеза Фишера-Тропша на основе кобальта, нанесенного на оксид алюминия, полученные пропиткой алюмооксидного носителя водной суспензией соли кобальта, например, гексагидрата нитрата кобальта, в сочетании с сушкой обработанного носителя с последующим его прокаливанием в псевдоожиженном слое и получением предшественника катализатора, который затем восстанавливают, получая катализаторы синтеза Фишера-Тропша. Эти катализаторы содержат кобальт, распределенный на носителе. Повышенное содержание кобальта, которое обусловливает повышенную активность катализатора, можно получить, повторяя стадию пропитки солью кобальта. Однако это отрицательно влияет на общую стоимость процесса получения катализатора и на время, необходимое для приготовления катализатора. Кроме того, максимальное количество металла, которое может осаждаться на стадии пропитки, ограничено объемом пор носителя.
Или же соответствующие катализаторы синтеза Фишера-Тропша с высокой нагрузкой по кобальту (высоким содержанием кобальта) можно получать, растирая или замешивая оксид алюминия (EP-A-0455307), оксид кремния (EP-A-0510771) или оксид циркония (EP-A-0510772) с растворимым или нерастворимым источником кобальта. Таким способом можно приготовить пластичную массу (пасту), которую подвергают экструзии, сушат и прокаливают, получая катализатор или предшественник катализатора. Этим методом можно получать высокое содержание кобальта, особенно в случае нерастворимого источника кобальта, такого как Ca(OH)2. При таком подходе конечная форма носителя определяется в процессе приготовления катализатора. В результате механическую прочность и физическое состояние носителя нельзя определить заранее.
Также для получения механически прочных катализаторов согласно этим известным методам экструдаты следует прокаливать при сравнительно высоких температурах. Недостатком прокаливания при высоких температурах является то, что оно отрицательно сказывается на каталитических свойствах. Еще одним недостатком растирания или замешивания (пластификации) является то, что часто требуются органические соединения, способствующие расслоению. Такие соединения вызывают экзотермическое окисление с выделением загрязняющих окружающую среду летучих органических веществ.
Другим альтернативным методом получения высокого содержания кобальта является осаждение нерастворимого соединения кобальта с использованием избытка щелочного осадителя, затем осаждаемого на носителе при добавлении растворимого соединения алюминия, такого как алюминат натрия (Международная заявка WO- А- 2006/021754). Также сообщалось об осаждении соединения кобальта при pH>8 на твердом носителе, таком как кизельгур (Международная заявка WO- А- 01/28962), при добавлении основания. В подобных случаях в качестве исходного соединения часто применяют Co(NO3)2, который, как предполагают, осаждается на носителе в виде гидроксидов кобальта (Appl. Catal. A: Gen. 311 (2006), 146). Недостатком процессов осаждения, которые требуют химической обработки, такой как добавление основания, является образование отходов, таких как соли. Это требует дополнительных стадий фильтрования или промывки в процессе получения. Кроме того, такие процессы необязательно гарантируют достаточную механическую прочность катализатора, позволяющую избежать в дальнейшем проблем, связанных с истиранием.
Таким образом, существует потребность в катализаторах гидрирования, включая катализаторы синтеза Фишера-Тропша, с высоким содержанием активного каталитического компонента, такого как кобальт, получаемых простым методом, который позволяет использовать механически прочные предварительно формованные носители и который позволяет избежать применения или по меньшей мере уменьшить применение химической обработки, такой как добавление основания, или избежать других недостатков, описанных выше.
Раскрытие изобретения
Поэтому согласно первому аспекту изобретения предусматривается способ получения предшественника катализатора, который включает:
образование суспензии частиц нерастворимого соединения металла, где металл из нерастворимого соединения металла является активным каталитическим компонентом, с частицами и/или одним или более тел предварительно сформованных носителей катализатора в жидкости-носителе, тем самым осуществляется контакт частиц нерастворимого соединения металла с частицами и/или одним или более тел предварительно сформованных носителей катализатора, при этом получается обработанный носитель катализатора; и
удаление жидкости-носителя из суспензии с образованием высушенного обработанного носителя катализатора, который либо является непосредственным предшественником катализатора, либо, при необходимости, прокаливается для получения предшественника катализатора.
Таким образом, следует иметь в виду, что в некоторых вариантах изобретения обработанный носитель катализатора необязательно прокаливать, и, следовательно, он непосредственно образует или представляет собой предшественник катализатора. Однако в других вариантах изобретения для получения предшественника катализатора необходимо сначала прокаливать обработанный носитель катализатора. Под "активным каталитическим компонентом" понимают, что металл из нерастворимого соединения металла обладает свойством активно катализировать химические реакции, в которых конечный катализатор, полученный из предшественника катализатора, применяется в качестве катализатора.
В данном описании выражения "нерастворимое соединение металла" или "нерастворимая соль металла" означают соединение металла или соль металла, соответственно, которые нерастворимы или очень слабо растворимы в применяемой жидкости-носителе. Предпочтительно, их произведение растворимости (Ksp при 25°C) в жидкости-носителе ниже 1.10-8, предпочтительно, ниже 1.10-12. Например, для гидроксида кобальта величина Кsр при 25°C в воде составляет 1,09.10-15, для гидроксида никеля в воде эта величина равна 5,47.10-16, для гидроксида марганца 2,06.10-13, а для гидроксида меди в воде величина Ksp равна 2,2.10-20.
Нерастворимое соединение металла, предпочтительно, представляет собой нерастворимую соль металла, более предпочтительно, нерастворимую неорганическую соль металла.
В настоящем описании термин "неорганическая соль металла" означает соль, в которой по меньшей мере один атом металла связан с одной или более неорганических групп, причем связывание осуществляется с помощью связи, например, ковалентной связи, координационной связи металл-лиганд или ионной связи.
В настоящем описании термин "суспензия" имеет обычное общепринятое значение и означает мультифазную систему, состоящую из твердых частиц, взвешенных в жидкости-носителе. Соотношение массы жидкости-носителя к сухой массе твердых частиц, т.е. частиц нерастворимого соединения металла плюс частиц/тел носителя катализатора, может составлять по меньшей мере 1:1, обычно около 2:1.
Контакт частиц нерастворимого соединения металла с частицами и/или одним или более тел предварительно сформованных носителей катализатора можно осуществлять в течение некоторого периода времени, предпочтительно, по меньшей мере в течение 1 минуты, более предпочтительно, по меньшей мере в течение 10 минут, и еще более предпочтительно, в течение по меньшей мере 15 минут, и наиболее предпочтительно, в течение по меньшей мере 20 минут, но предпочтительно, в течение не более 48 часов, более предпочтительно, в течение не более 36 часов, еще более предпочтительно, в течение не более 20 часов, и наиболее предпочтительно, в течение не более 2 часов перед удалением жидкости-носителя.
Способ может включать осуществление контакта частиц нерастворимого соединения металла с частицами и/или одним или более тел предварительно сформованных носителей катализатора при повышенной температуре: выше 25°C, предпочтительно, выше 50°C; однако предпочтительно, чтобы повышенная температура была ниже 100°C.
Способ может включать осуществление по меньшей мере однократного контакта предварительно сформованного носителя катализатора и/или обработанного носителя катализатора, и/или высушенного обработанного носителя катализатора, и/или прокаленного обработанного носителя катализатора с растворимым соединением металла. Металл из растворимого соединения металла также может являться активным каталитическим компонентом. Растворимое соединение металла может, в частности, являться растворимой солью металла.
"Растворимое соединение металла" или "растворимая соль металла" представляет собой соединение или соль металла, соответственно, которые не являются нерастворимым соединением или нерастворимой солью металла. Предпочтительно, растворимое соединение/соль имеет растворимость в жидкости, применяемой для его/ее растворения, выше 25 г/100 мл жидкости, предпочтительно, выше 100 г/100 мл жидкости при 25°C. Например, растворимость нитрата кобальта в воде составляет 133.8 г/100 мл, растворимость нитрата никеля в воде составляет 238.5 г/100 мл, растворимость нитрата меди в воде составляет 243.7 г/100 мг и растворимость нитрата марганца в воде составляет 426.4 г/100 мл, все данные при 25°C.
Таким образом, можно осуществлять по меньшей мере однократный контакт растворимой соли металла, в случае ее применения, с частицами нерастворимой неорганической соли металла и/или с предварительно сформованными частицами носителя катализатора. Так, растворимая соль металла может образовывать часть суспензии, т.е. она может быть растворена в жидкости-носителе. Напротив, можно осуществлять по меньшей мере однократный контакт обработанного носителя катализатора с растворимой солью металла, например, с отдельным раствором растворимой соли металла. В тех случаях, когда обработанный носитель катализатора прокаливают с образованием предшественника катализатора, можно осуществлять по меньшей мере однократный контакт прокаленного обработанного носителя катализатора, т.е. предшественника катализатора, с раствором растворимой соли металла.
Получение суспензии может включать добавление частиц нерастворимой соли металла и/или предварительно сформованных частиц носителя катализатора к жидкости-носителю с образованием при смешивании смеси, состоящей из частиц, взвешенных (суспендированных) в жидкости-носителе. Такое смешивание можно осуществлять с малым сдвигом. Следует иметь в виду, что консистенция суспензии такова (ее вязкость достаточно низкая), что нельзя осуществить ни ее растирание, ни ее замешивание, ни ее экструзию. Также смешивание, особенно смешивание с малым сдвигом, не представляет собой ни растирание, ни замешивание.
Кроме того, способ, в качестве стадии предварительной обработки, включает осуществление контакта частиц нерастворимой соли металла и/или частиц носителя катализатора с растворимой солью металла, например, с раствором растворимой соли металла.
Более конкретно, суспензию можно создать, если сначала получать взвесь из частиц нерастворимой неорганической соли металла в жидкости-носителе, а затем к этой взвеси добавлять предварительно сформованные частицы и/или тела носителя катализатора с получением суспензии.
Предпочтительно, частицы нерастворимой неорганической соли металла добавляют к жидкости-носителю с образованием взвеси. Предварительно сформованный носитель катализатора можно добавлять к жидкости-носителю до и/или во время и/или после образования взвеси с образованием суспензии. Следует принимать во внимание, что частицы нерастворимой неорганической соли металла не получают in situ; взвесь образуется при смешении предварительно сформованных частиц нерастворимой соли металлов с жидкостью-носителем.
Металлы в солях металлов, т.е. в нерастворимой неорганической соли металла и в растворимой соли металла, могут быть выбраны независимо и могут быть одинаковыми или различными. Однако, предпочтительно, они являются одинаковыми. Металлы, подходящие для целей настоящего изобретения, могут быть выбраны из группы, состоящей из металлов Групп Ib, IIb, Vb, VIb, VIIb и VIII Периодической таблицы элементов. Более предпочтительно, они выбраны из кобальта, никеля, рутения, марганца, железа, меди, цинка, молибдена, драгоценных металлов и комбинаций из двух или трех из этих металлов. Кобальт, никель и медь особенно применимы для получения предшественника катализатора гидрирования согласно способу по настоящему изобретению. В случае предшественников катализатора на основе кобальта, предпочтительно, применяют только соли кобальта.
Нерастворимая неорганическая соль металла может представлять собой, по меньшей мере теоретически, любую нерастворимую соль металла; однако, предпочтительными являются карбонат металлов и, в особенности, гидроксиды металлов. Предпочтительно, металл в нерастворимой неорганической соли металла выбран из группы, состоящей из кобальта, меди, никеля, марганца или комбинаций двух или трех из этих металлов. В случае, когда металл в нерастворимой неорганической соли металла представляет собой кобальт, предпочтительными являются карбонат кобальта, гидроксид кобальта, и, в частности, Co(OH)2.
Предпочтительно, растворимой солью металла является соль такого металла, который также представляет собой активный каталитический компонент. Растворимая соль металла может являться неорганической солью металла и/или органической солью металла. Можно применять комбинации различных растворимых солей металла, например, солей различных металлов или солей с различными органическими или неорганическими анионами.
В данном описании термин "органическая соль металла" означает соединение, в котором по меньшей мере один атом металла связан по меньшей мере с одним органическим остатком связью, например, ковалентной связью, координационной связью металл-лиганд или ионной связью. Предпочтительно, атом металла связан по меньшей мере с одним отличным от углерода атомом по меньшей мере одного органического остатка. Органическое соединение металла может также включать один или более остатков неорганических соединений (неорганических групп), связанных с металлом. Предпочтительно, одним или более остатков неорганических соединений являются катионные группы.
В случае, когда применяется растворимая неорганическая соль металла, она может, по меньшей мере теоретически, представлять собой растворимую неорганическую соль металла.
Coответствующие растворимые соли металлов включают нитраты, сульфаты, хлориды и цитраты аммония (аммонийно-цитратные соли). Предпочтительно, металл в растворимой соли металла выбран из группы, состоящей из кобальта, меди, никеля, марганца или комбинации двух или более этих металлов. Когда применяют растворимую неорганическую соль металла, а металлом является кобальт, предпочтительной является соль Co(NO3)2·6H2O.
В случае, когда применяется растворимая органическая соль кобальта, ее можно получать реакцией соединения кобальта, такого как гидроксид кобальта или нитрат кобальта, с органической кислотой, необязательно, в присутствии по меньшей мере одного источника противоионов. В этом случае соединение кобальта, предпочтительно, является основным соединением кобальта. Источником противоионов, если таковой присутствует, предпочтительно, является неорганический источник и, предпочтительно, он является источником одного или более катионов. Coгласно одному варианту изобретения источником противоионов может быть аммиак.
Органическую соль кобальта можно получать in situ. Так, соединение кобальта, например, гидроксид кобальта, можно растворять в растворе органической кислоты в воде.
Органическая кислота может представлять собой карбоновую кислоту, такую как уксусная кислота, лимонная кислота (C6H8O7), янтарная кислота (C4H6O4), щавелевая кислота (C2H2O4), уксусная кислота (C2H4O2), глюконовая кислота (C6H12O7) или ЭДТА, т.е. этилендиаминтетрауксусная кислота. Предпочтительно, органическая кислота представляет собой лимонную кислоту.
В растворе органического соединения кобальта молярное соотношение кобальта и органической кислоты может меняться в широком интервале, например, от 0.1:1 до 10:1. Однако предполагается, что обычно молярное соотношение кобальта и органической кислоты находится в интервале от 0.5:1 до 2:1, обычно около 1:1.
Согласно предпочтительным вариантам изобретения органическая соль кобальта может представлять собой цитрат аммония-кобальта или ЭДТА соль аммония-кобальта.
В качестве альтернативы органическую соль кобальта можно получать реакцией соединения кобальта с ацетилацетоном (C5H8O2).
В предпочтительном варианте изобретения можно применять достаточное количество нерастворимой неорганической соли металла и, при необходимости, растворимой соли металла, чтобы количество полученного компонента активного металла по отношению к носителю в предшественнике катализатора составляло от 5 до 90 вес. %, предпочтительно, от 10 до 70 вес. %, наиболее предпочтительно, от 10 до 50 вес. % от общего веса предшественника.
Способ может включать дальнейшую обработку предшественника катализатора, т.е. обработанного носителя катализатора, с получением суспензии из частиц обработанного носителя катализатора, частиц нерастворимой соли металла и жидкости-носителя, удаление жидкости-носителя из суспензии и, при необходимости, прокаливание полученных при этом частиц с образованием предшественника катализатора.
Согласно специфическим вариантам изобретения, описанным ниже, применяют частицы предварительно сформованного носителя катализатора. Однако следует принимать во внимание, что согласно другим вариантам изобретения те же принципы можно применять к телам предварительно сформованного носителя катализатора.
Согласно первому варианту изобретения получение суспензии может включать суспендирование частиц нерастворимого соединения металла в жидкости-носителе с образованием взвеси и добавление частиц предварительно сформованного носителя катализатора к жидкости-носителю до и/или во время и/или после образования взвеси с образованием суспензии с активным каталитическим компонентом, т.е. металл из нерастворимого соединения металла осаждается на частицах носителя. Предпочтительно, суспензия совсем не содержит соединения металла в растворенном виде. Предпочтительно, осаждение может происходить методом хемосорбции, предпочтительно, при значениях pH от нейтрального до кислого, как правило, в интервале от 8 до 2. Эффект хемосорбции в этом процессе выражается изменением величины pH. Таким образом, отличительным признаком этого варианта изобретения является воздействие лишь одной хемосорбции.
Без связи с какой- либо теорией полагают, что в процессе хемосорбции осаждение молекулы активного каталитического компонента на носителе происходит за счет образования химической связи между носителем и молекулой. Также без связи с какой-либо теорией полагают, что, скорее всего, эта связь образуется в результате реакции конденсации.
Согласно второму варианту изобретения образование густой суспензии может включать суспендирование частиц нерастворимого соединения металла в жидкости-носителе с образованием взвеси и добавление частиц предварительно сформованного носителя катализатора к жидкости-носителю до и/или во время и/или после образования взвеси с образованием густой суспензии, причем металл из нерастворимого соединения металла осаждается на частицах носителя, предпочтительно, за счет хемосорбции; и второй вариант изобретения после удаления жидкости-носителя из густой суспензии включает осуществление контакта высушенного обработанного носителя катализатора с растворимым соединением металла с помощью обработки, по меньшей мере однократной, высушенного обработанного носителя катализатора раствором растворимого соединения металла в жидкости-носителе, причем металл из растворимого соединения металла осаждается в и/или на частицах носителя, предпочтительно, за счет пропитки. Предпочтительно, густая суспензия совсем не содержит соединения металла в растворенном виде. Во время образования густой суспензии, которое является первой стадией процесса, происходит осаждение первой порции активного каталитического компонента на частицах носителя. Это осаждение может осуществляться методом хемосорбции, как описано выше при описании первого варианта изобретения. Однако, в данном варианте изобретения полученные таким образом частицы обработанного носителя катализатора затем, после или без их прокаливания, подвергают дальнейшей обработке на следующей стадии процесса, осуществляя их приведение в контакт, по меньшей мере однократное, с раствором растворимой соли металла, также являющегося активным каталитическим компонентом, в жидкости-носителе, и при этом металл растворимой соли металла пропитывает обработанные частицы носителя, создавая тем самым вторую порцию компонента, содержащего активный металл. После пропитки и хемосорбции носитель прокаливают и при этом получают предшественник катализатора.
Следовательно, этот второй вариант изобретения характеризуется тем, что хемосорбцию и пропитку проводят строго последовательно, сначала осуществляя хемосорбцию с использованием нерастворимой соли металла, а затем пропитку растворимой соли металла.
Согласно третьему варианту изобретения получение суспензии может включать образование раствора растворимого соединения металла в жидкости-носителе, суспендирование частиц нерастворимого неорганического соединения металла в жидкости-носителе с образованием взвеси и добавление частиц предварительно сформованного носителя катализатора к жидкости-носителю до и/или во время и/или после образования взвеси с образованием суспензии, причем металл из нерастворимого соединения металла осаждается на частицах носителя, предпочтительно, за счет хемосорбции, тогда как металл из растворимого соединения металла осаждается в и/или на частицах носителя, предпочтительно, за счет пропитки. В этом случае металл из растворимой соли металла также является активным каталитическим компонентом. Таким образом, активный компонент-металл в одной и той же стадии процесса осаждается с помощью хемосорбции, а также пропитывает носитель, в результате получают обработанный носитель катализатора, который затем прокаливают, получая предшественник катализатора.
Следовательно, этот третий вариант изобретения характеризуется тем, что хемосорбцию и пропитку проводят одновременно, т.е. на одной и той же стадии процесса.
Предпочтительно, перед прокаливанием пропитанного носителя из него, по меньшей мере частично, удаляют жидкость-носитель.
Итак, предпочтительным методом осаждения металла из нерастворимой соли металла на предварительно сформованном носителе катализатора является хемосорбция; предпочтительным методом осаждения металла из растворимого соединения металла на предварительно сформованном носителе катализатора является пропитка.
Неожиданно было обнаружено, что способом по изобретению и, предпочтительно, включающим по меньшей мере одну стадию хемосорбции и одну стадию пропитки, обычно получают высокодисперсную суспензию металла, например, кобальта, и в то же время можно достичь высокого содержания металла, например, кобальта, обычно с повышенной каталитической активностью по сравнению со стандартным способом получения таких катализаторов осаждением только неорганических солей металлов, например, неорганических солей кобальта методом пропитки. Также способ по изобретению предусматривает каталитические материалы, полученные при низких температурах прокаливания, исключающих экзотермические эффекты.
Также на и/или в частицы носителя катализатора можно вводить промотор, предварительно обрабатывая частицы носителя катализатора до образования густой суспензии или, предпочтительно, добавляя промотор, или его предшественник, к густой суспензии. Промотор, при его наличии, предпочтительно, представляет собой промотор, способный повышать способность активного каталитического компонента к восстановлению. Промотор можно вводить в виде предшественника промотора или в виде соединения, которое представляет собой соединение металла, выбранного из группы, состоящей из палладия (Pd), платины (Pt), рутения (Ru), рения (Re), родия (Rh) и смеси одного или более из этих металлов. Предпочтительно, соединение промотора представляет собой неорганическую или органическую соль и, предпочтительно, растворимую в воде. Предпочтительно, промотор представляет собой ацетат, ацетилацетонат, нитрат или нитрозилнитрат. Весовое соотношение металла промотора к металлу активного компонента может составлять от 1:5 до 1:10000. Весовое соотношение металла промотора (в особенности палладия или платины) к металлу активного компонента (в особенности кобальту) может составлять от 1:300 до 1:3000. Весовое соотношение металла промотора (рения) к металлу активного компонента (в особенности кобальту) может составлять от 1:5 до 1:300.
Жидкость-носитель может представлять собой любой подходящий жидкий растворитель для растворимой соли металла, естественно, при условии, что нерастворимая неорганическая соль металла не растворяется в этом растворителе. Все же, предпочтительно, этим растворителем является вода.
В данном описании выражение "предварительно сформованный носитель катализатора" означает, что форма носителя катализатора определяется применяемым носителем катализатора, и она остается по существу одной и той же в процессе приготовления предшественника катализатора, т.е. она не преобразуется и не изменяется в процессе приготовления предшественника катализатора. В частности, деформация (изменение формы) носителя катализатора после осуществления его контакта с нерастворимой солью металла не наблюдается.
Предварительно сформованный носитель катализатора может быть пористым. Он может быть выбран из группы, состоящей из монолита, структурированных материалов, таблеток, формованных изделий, экструдатов, сфер или комбинации из двух или более этих изделий. Другими словами, если предварительно сформованный носитель катализатора находится в виде одного или более тел, эти тела могут быть монолитами; однако, если предварительно сформованный носитель катализатора находится в виде частиц, эти частицы могут представлять собой структурированные материалы, таблетки, формованные изделия, экструдаты, сферы или комбинации из двух или более таких изделий. Все же предпочтительными являются сферические предварительно сформованные частицы носителя катализатора; средний диаметр таких частиц может составлять 50-150 микрометров.
При необходимости носитель, применяемый в густой суспензии, в качестве предварительной обработки может подвергаться химической модификации. Под такой химической модификацией понимают, что носитель мог быть предварительно обработан (i) путем покрытия слоем другого химического неорганического материала, такого, но без ограничения, как диоксид кремния, оксид алюминия, цеолит или диоксид циркония, или (ii) пропиткой органическим материалом, который способствует дисперсии металла, или (iii) пропиткой солью металла. Органические материалы, пригодные для применения по п. (ii), широко известны в данной области техники и включают такие материалы как органические кислоты, сахара и сахарные спирты, полиолы или поверхностно-активные вещества, предпочтительно, поверхностно-активные вещества не являются ионными поверхностно-активными веществами. Coли металлов, пригодные для применения по п. (ii), включают соли некоторых щелочных, щелочноземельных, редкоземельных металлов или переходных металлов и могут применяться для пропитки с целью специфически изменить кислотно-основные свойства носителя и целевого катализатора. Кроме того, можно также осуществлять пропитку молибдатами или вольфраматами, в особенности с применением парамолибдата аммония. При необходимости, в качестве альтернативы, такую дополнительную пропитку частиц обработанного носителя катализатора солями металлов можно проводить до или после прокаливания носителя.
Предварительно сформованные или подготовленные частицы носителя катализатора могут иметь диаметр пор, предпочтительно, от 8 до 50 нанометров, более предпочтительно, от 10 до 15 нанометров. Объем пор носителя может быть равен от 0.1 до 1 мл/г носителя катализатора, предпочтительно, от 0.3 до 0.9 мл/г носителя катализатора. Предварительно сформованный носитель может представлять собой дисперсный носитель, содержащий микрочастицы, средний размер которых, предпочтительно, равен от 1 до 500 микрометров, предпочтительно, от 10 до 250 микрометров, еще более предпочтительно, от 45 до 200 микрометров. Придание формы предварительно сформованному носителю с размером частиц от 1 до 500 микрометров можно осуществлять методом распылительной сушки. После распылительной сушки этот сформованный носитель можно прокаливать.
Предварительно сформованный носитель катализатора может быть выбран из группы, состоящей из глинозема в виде одного или более оксидов алюминия, диоксида кремния, диоксида титана, диоксида циркония, оксида магния, оксида цинка, активированного угля, молекулярных сит, в частности цеолитов и их смесей или комбинаций. Предпочтительно, носитель выбирают из группы, состоящей из глинозема в виде одного или более оксидов алюминия; диоксида титана и диоксида кремния. Как правило, носителем является глинозем в виде одного или более оксидов алюминия. Один или более оксидов алюминия может быть выбран из группы, включающей гамма-оксид алюминия, дельта-оксид алюминия, тета-оксид алюминия и смесь двух или более этих оксидов алюминия (предпочтительно, состоящей из этих соединений). Предпочтительно группа включает гамма-оксид алюминия, дельта-оксид алюминия и смесь гамма-оксида алюминия и дельта-оксида алюминия, или предпочтительно, состоит из этих соединений. Носитель катализатора-оксид алюминия-может представлять собой оксид алюминия, выпускаемый под торговой маркой Puralox, предпочтительно, Puralox SCCa от компании SASOL Germany GmbH. Puralox SCCa (торговая марка) представляет собой высушенный распылительной сушкой носитель оксид алюминия (алюмооксидный носитель), состоящий из смеси гамма- и дельта-оксида алюминия или Al 4505 от компании BASF Germany GmbH. Al 4505 можно получать в виде порошков и формоваться, например, как Al4505 Т1/8, в виде таблеток.
Оксид алюминия, предпочтительно, представляет собой кристаллическое соединение формулы Al2O3.×H2O, где 0<×<1. Следовательно, термин оксид алюминия исключает Al(OH)3 и Al(OH), но включает такие соединения как гамма, дельта и тета-оксид алюминия.
Предпочтительно, носитель катализатора включает один или более модифицирующих компонентов. Это имеет место в особенности когда основа носителя, т.е. носитель без модифицирующего компонента, растворима в нейтральном и/или кислом водном растворе, или когда основа носителя чувствительна к гидротермическому воздействию, как показано ниже.
Модифицирующий компонент может представлять собой компонент, который вызывает один или более нижеприведенных эффектов:
(i) снижает растворимость носителя катализатора в водной среде,
(ii) подавляет чувствительность носителя катализатора к гидротермическому воздействию (особенно в процессе синтеза Фишера-Тропша);
(iii) увеличивает объем пор носителя катализатора;
(iv) повышает прочность и/или сопротивление истиранию и/или износостойкость носителя катализатора.
В предпочтительном варианте изобретения модифицирующий компонент уменьшает растворимость носителя катализатора в водной среде, т.е. повышает стойкость носителя катализатора по отношению к растворению в водной среде, и/или подавляет чувствительность носителя катализатора к гидротермическому воздействию, в процессе синтеза Фишера-Тропша. Такая водная среда может включать водный раствор кислоты и/или нейтральный водный раствор, такой как среда на стадии пропитки водной фазой в процессе приготовления катализатора. Гидротермическое воздействие может вызвать агломерацию носителя катализатора (например, оксида алюминия), растворение ионов Al или разрушение частиц катализатора в процессе синтеза углеводородов, особенно в ходе синтеза Фишера-Тропша, под воздействием высокой температуры и воды.
Модифицирующий компонент обычно присутствует в количестве, обеспечивающем его уровень в носителе катализатора по меньшей мере 0.06 атомов на квадратный нанометр.
Модифицирующий компонент может быть выбран из группы, состоящей из Si, Zr, Co, Ti, Cu, Zn, Mn, Ba, Ni, Na, K, Ca, Sn, Cr, Fe, Li, Ti, Sr, Ga, Sb, V, Hf, Th, Ce, Ge, U, Nb, Ta, W, La и их смесей.
Более конкретно, модифицирующий компонент может быть выбран из группы, состоящей из Si; Zr; Cu; Zn; Mn; Ba; La; Ti; W; Ni и их смесей. Предпочтительно, модифицирующий компонент выбран из группы, состоящей из Si и Zr. Coгласно предпочтительному варианту изобретения модифицирующий компонент представляет собой Si.
Когда модифицирующим компонентом является Si, уровень кремния в полученном носителе катализатора составляет по меньшей мере 0.06 атомов Si на квадратный нанометр носителя катализатора, предпочтительно, по меньшей мере 0.13 атомов Si на квадратный нанометр носителя катализатора, и более предпочтительно, по меньшей мере 0.26 атомов Si на квадратный нанометр носителя катализатора.
Предпочтительно, верхний уровень составляет 2.8 атомов Si/нм2 носителя катализатора.
Модифицированный алюминийоксидный носитель катализатора может представлять собой носитель катализатора, выпускаемый под торговой маркой Siralox от компании Sasol Germany GmbH, содержащий от 1.4 до 2.2 вес.% Si.
Coгласно другому варианту изобретения носитель катализатора находится в виде одного или более оксидов алюминия или модифицированного диоксидом кремния оксида алюминия и, предпочтительно, на таких носителях, как диоксид кремния и диоксид титана, так как полагают, что катализаторы на этих носителях обладают значительно более высоким сопротивлением истиранию. Носитель катализатора в виде одного или более оксидов алюминия или оксида алюминия, модифицированного диоксидом кремния, может также включать La. Полагают, что La повышает сопротивление истиранию.
Coгласно другому варианту изобретения носитель катализатора в виде одного или более оксидов алюминия или оксида алюминия, модифицированного диоксидом кремния, может включать титан, предпочтительно, в количестве, выражаемом количеством элементарного титана, по меньшей мере 500 вес. частей на миллион, предпочтительно, от около 1000 вес. частей на миллион до около 2000 вес. частей на миллион. Полагают, что добавление титана повышает активность полученного катализатора, особенно в случае кобальтового FT катализатора, в частности, когда катализатор не содержит каких-либо промоторов на основе благородных металлов, и предпочтительно, когда катализатор не содержит Re или Та промоторы. Предпочтительно, титан вводится во внутреннюю структуру носителя и, предпочтительно, титан отсутствует в виде осадка на носителе. Полагают, что присутствие титана в таком виде в носителе также повышает сопротивление катализатора на таком носителе истиранию.
Coгласно еще одному варианту изобретения носитель катализатора может быть в виде пористых частиц с углеродным покрытием. Однако, согласно альтернативному варианту изобретения пористые частицы могут не иметь такого углеродного покрытия.
Носитель катализатора может быть модифицирован путем введения предшественника модифицирующего компонента, который включает модифицирующий компонент, описанный выше, находящийся на и/или в материале носителя катализатора.
Удаление жидкости-носителя из густой суспензии может включать сушку и/или фильтрование этой суспензии. В случае применения сушки предпочтительной является сушка тепловой обработкой, т.е. при повышенной температуре.
Хемосорбцию по описанию выше проводят смешением суспензии предварительно сформованного носителя и нерастворимой неорганической соли в жидкости-носителе. Предпочтительно, суспензия представляет собой суспензию в воде. После хемосорбции остаток жидкости-носителя можно удалять сушкой при температуре выше 25°C в вакууме и/или фильтрованием.
В процессе пропитки сушку, если ее применяют, можно проводить в условиях, в которых растворимая (неорганическая или органическая) соль металла разлагается с трудом. Предпочтительно, стадию сушки проводят при температуре выше 25°C и, предпочтительно, в вакууме. Предпочтительно, густую суспензию сушат при температуре в интервале от 40°C до 120°C, обычно около 100°C, как правило, при конечном давлении в интервале от 50 до 120 мбар (5-12 кПа), обычно около 80 мбар (8 кПа).
В процессе возможного повторения стадии хемосорбции предшественника катализатора, описанной выше, хемосорбцию в случае прокаленного обработанного носителя катализатора можно проводить в суспензии, используя прокаленный обработанный носитель катализатора и нерастворимую неорганическую соль металла (металлом является активный каталитический компонент, описанный выше) в жидкости-носителе. Опять же после хемосорбции остаток жидкости удаляют сушкой при температуре выше 25°C в вакууме или фильтрованием.
Любую последующую пропитку можно проводить в условиях, в которых растворимая (неорганическая или органическая) соль металла разлагается с трудом. Предпочтительно, стадию сушки проводят при температуре выше 25°C и, предпочтительно, в вакууме.
Coдержание азота в предшественнике катализатора может быть менее 1 вес.%, предпочтительно, менее 0.5 вес.%
Прокаливание, если его применяют, предпочтительно, проводят при температуре выше 25°C, при этом введенные осаждением и пропиткой соли металл разлагаются и/или реагируют с кислородом. Поэтому, предпочтительно, прокаливание проводят в условиях окисления. Например, нитрат кобальта можно превратить в соединение, выбранное из CoO, CoO(ОН), Co3O4, Co2O3 или смеси одного или более этих соединений.
Прокаливание обычно проводят в псевдоожиженном слое или во вращающейся сушильной печи. По меньшей мере частично высушенный пропитанный обработанный носитель катализатора можно прокаливать на воздухе. Температура при прокаливании может быть от 100°C до 600°C, предпочтительно, от 120°C до 350°C, более предпочтительно, от 150°C до 300°C, обычно 250°C, при этом получают предшественники кобальтоксидного катализатора. Обычно температуру повышают от комнатной, как правило, от 25°C, до 200-350°C со скоростью от 0.1 до 10°C/мин, предпочтительно, от 0.5 до 3°C/мин. GHSV (объемная скорость подачи газа) в процессе прокаливания, имеющая особое значение при работе в псевдоожиженном слое и при пропитке нитратами, обычно равна от 100 до 3000 час-1, как правило, 2000 час-1. Более конкретно, условия прокаливания на второй стадии получения можно выбирать таким образом, чтобы в предшественнике катализатора по существу весь восстанавливаемый металл находился в прокаленном состоянии. Помимо указанных выше методов, прокаливание можно проводить также, например, в подвижном или неподвижном слое.
Прокаливание можно проводить со скоростью нагрева и объемной скоростью, которые отвечают следующим критериям:
(i) когда скорость нагрева ≤1°C/мин, объемная скорость равна по меньшей мере 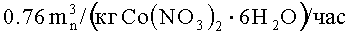 ; и
; и
(ii) когда скорость нагрева выше 1°C/мин, объемная скорость удовлетворяет соотношению:

Coгласно второму аспекту изобретения предусматривается предшественник катализатора, который получают или который можно получать способом по первому аспекту изобретения и который содержит металл в количестве от 5 до 90 вес.% от общей массы предшественника.
Предпочтительно, предшественник содержит от 10 до 70 вес.% металла и, более предпочтительно, от 10 до 50 вес.% металла. Предшественник катализатора по существу не содержит обменных ионов.
Предшественник катализатора может являться предшественником катализатора синтеза углеводородов. Предпочтительно, он может являться предшественником катализатора синтеза Фишера-Тропша. Более предпочтительно, он может представлять собой предшественник катализатора синтеза Фишера-Тропша в виде суспензии. Металл в растворимом соединении/растворимой соли металла может представлять собой кобальт. Предпочтительно, в этом случае металл в нерастворимом соединении/нерастворимой соли металла также представляет собой кобальт, который, таким образом, является активным компонентом конечного катализатора. Итак, предшественник катализатора является предшественником катализатора синтеза Фишера-Тропша на основе кобальта.
Однако в качестве альтернативы, предшественник катализатора может являться предшественником катализатора гидрирования, применимым для гидрирования органических соединений. Более конкретно, предшественник катализатора
может являться предшественником катализатора гидрирования ароматических соединений, нитросоединений, нитрилов, алкинов, алкенов, диенов или альдегидов или предшественником катализатора гидродехлорирования. Например, предшественник катализатора может также быть предшественником катализатора для синтеза спиртов или аммиака.
Если предшественник катализатора гидрирования является предшественником на основе кобальта, его можно получать так же, как предшественник катализатора синтеза Фишера-Тропша на основе кобальта, описанный выше. Обычно носитель катализатора пропитывают парамолибдатом аммония, сушат и, при необходимости, прокаливают и в таком виде применяют по изобретению. Аналогичный способ получения можно применять для получения NiMo катализаторов. Кобальт и/или никель в комбинации с молибденом особенно применимы для получения предшественника катализатора гидрирования по настоящему изобретению, особенно катализаторы этого типа можно применять для HDM (гидродеметаллизации), HDS (гидродесульфуризации), HDN (гидродеазотирования) или для гидрирования пирогаза.
Coгласно третьему аспекту изобретения предусматривается способ получения катализатора, который включает получение предшественника катализатора по способу согласно первому аспекту изобретения и восстановление полученного при этом предшественника катализатора с получением катализатора.
Если предшественник катализатора представляет собой описанный выше предшественник катализатора гидрирования, то, естественно, катализатор является катализатором гидрирования. Катализатор гидрирования можно применять для гидрирования ароматического соединения, нитросоединения, нитрила, алкина, алкена, диена или альдегида или для гидродехлорирования или синтеза спирта или аммиака или для HDM (гидродеметаллизации), HDS (гидродесульфуризации), HDN (гидродеазотирования) или для гидрирования пирогаза.
Более конкретно, катализатор гидрирования можно применять для получения продуктов тонкого органического синтеза, где очень важна высокая селективность. Примерами реакций, которые могут катализироваться катализаторами на основе никеля, полученными по настоящему изобретению, являются гидрирование, гидродехлорирование и т.п.
В реакциях гидродехлорирования катализатор гидрирования по изобретению позволяет очень тщательно регулировать количество водорода и парциальное давление водород/HCI в системе, тем самым по существу повышая селективность реакции.
Если предшественник катализатора представляет собой описанный выше предшественник катализатора синтеза Фишера-Тропша на основе кобальта, то, естественно, катализатор является катализатором синтеза Фишера-Тропша.
Неожиданно было обнаружено, что описанный выше предшественник катализатора синтеза Фишера-Тропша на основе кобальта превращается в катализатор синтеза Фишера-Тропша реакцией восстановления, который обладает высокой и стабильной активностью в синтезе Фишера-Тропша. Еще более неожиданно было найдено, что вышеописанным способом хемосорбции-пропитки не только получают высокую нагрузку по кобальту, но также получают и высокодисперсный кобальт (металл и/или оксид), в результате получают катализатор с повышенной активностью в синтезе Фишера-Тропша.
Предшественник катализатора можно восстановить или активировать любым известным методом восстановления, предпочтительно, осуществляя приведение в контакт предшественника катализатора с чистым газообразным водородом или со смесью газов, содержащим водород. Смесь газов может состоять из водорода и одного или более инертных газов, инертных по отношению к активному катализатору. Предпочтительно, концентрация водорода находится в интервале от 0.1 до 100%, а восстановление проводят при любой температуре выше 100°C.
Если катализатором является катализатор синтеза Фишера-Тропша, смесь газов, предпочтительно, содержит 90 об.% водорода. Восстановление можно проводить в температурном интервале от 250°C до 550°C, предпочтительно, от около 300°C до около 425°C, во временном интервале от 0.5 час до около 24 час и при давлении от нормального до около 40 атмосфер. Подходящие условия восстановления для получения катализатора по настоящему изобретению можно найти в Международных заявках WO-A-03/035257, WO-A-2008/135939, WO-A-2008/135940 и WO-A-2008/135941.
Coгласно четвертому аспекту настоящего изобретения предусматривается способ синтеза углеводородов, который включает получение катализатора способом по третьему аспекту изобретения; и осуществление контакта водорода с оксидом углерода при температуре выше 100°C и при давлении по меньшей мере 10 бар (103 кПа) в присутствии полученного таким способом катализатора с образованием углеводородов и, при необходимости, продуктов окисления углеводородов.
Температура может быть от 180°C до 250°C, более предпочтительно, от 210°C до 240°C. Более предпочтительно, давление может составлять от 10 бар (103 кПа) до 70 бар (7×103 кПа).
Предпочтительно, процесс синтеза углеводородов представляет собой синтез Фишера-Тропша, более предпочтительно, трехфазный синтез Фишера-Тропша, еще более предпочтительно, синтез Фишера-Тропша в слое взвешенного осадка с образованием воска (парафинов).
Процесс синтеза углеводородов может также включать стадию гидропроцессинга с целью превращения углеводородов и, при необходимости, продуктов их окисления, в жидкое топливо и/или химические вещества.
Настоящее изобретение также относится к продуктам, полученным в процессе синтеза углеводородов согласно четвертому аспекту изобретения.
Coгласно пятому аспекту настоящего изобретения предусматривается способ гидрирования, который включает получение катализатора по третьему аспекту изобретения; и осуществление контакта водорода и органического соединения с полученным катализатором с целью гидрирования органического соединения.
Настоящее изобретение также относится к продуктам, полученным в процессе гидрирования по пятому аспекту изобретения.
Далее изобретение описывается более подробно с помощью неограничивающих приведенных ниже примеров и прилагаемых фигур.
Описание фигур
На Фигуре 1, иллюстрирующей Пример 23, изображена суспензия Co(OH)2 в воде, содержащая носитель оксид алюминия (глинозем), модифицированный диоксидом кремния, до и после смешения при 80°C;
На Фигуре 2, иллюстрирующей Пример 23, представлен образец смеси, содержащей Co(NO3)2, над слоем темно-пурпурного осадка гидроксида кобальта на носителе;
На Фигуре 3, иллюстрирующей Пример 23, показаны результаты TPR (температурно-программируемой реакции) для физической смеси Co(OH)2 и модифицированного диоксидом кремния оксида алюминия по сравнению с введенным хемосорбцией Co(OH)2 на модифицированном диоксидом кремния оксиде алюминия; и
На Фигуре 4, иллюстрирующей Пример 23, изображена смесь Co(OH)2 и таблеток оксида алюминия в воде.
Осуществление изобретения
ПРИМЕР 1
Получение Сравнительного Катализатора 1
Продукт, содержащий 30 г Co/0.075 г Pt/100 г (1.5 г Si/100 г Puralox SCCa, см. Также Международную заявку WO-A-99/42214, пример 1), получали пропиткой водной суспензии дисперсного, модифицированного с помощью 1.5 г Si/100 г Puralox SCCa, предварительно сформованного носителя, сушкой и последующим прокаливанием на воздухе в псевдоожиженном слое.
Продукт получали, используя две стадии пропитки и прокаливания, в каждой из которых применяли растворимое неорганическое соединение кобальта.
В частности, катализатор, пригодный для применения в синтезе Тропша с взвешенным слоем, получали следующим образом:
43.70 г Co(NO3)2·6H2O растворяли в 40 мл дистиллированной воды и к этому раствору добавляли 0.024 г Pt(NH3)4.(NO3)2 (раствор в 10 мл дистиллированной воды), после чего к раствору прибавляли 50.0 г модифицированного с помощью 1.5 г Si/100 г Puralox SCCa предварительно сформованного носителя. Затем осуществляли пропитку водной суспензии (во взвешенном слое) и сушку в вакууме, повышая температуру с 60 до 95°C и снижая вакуум с 170 бар (17 кПа) до 75 бар (7.5 кПа).
Этот высушенный в вакууме промежуточный продукт сразу же пускали на стадию прокаливания в псевдоожиженном слое в непрерывном токе воздуха со скоростью потока 1.7 дм3 n/мин при повышении температуры от 25°C до 250°C со скоростью нагрева 1°C/мин и выдерживали при 250°C в течение 6 час.
Затем проводили описанную ниже вторую стадию пропитки кобальтом/платиной и прокаливания, используя 50.0 г этого промежуточного прокаленного материала: 23.51 g Co(NO3)2·6H2O растворяли в 40 мл дистиллированной воды и к этому раствору прибавляли 0.039 г Pt(NH3)4·(NO3)2 (раствор в 10 мл дистиллированной воды), а затем прибавляли 50.0 г промежуточного продукта, полученного на первой стадии пропитки кобальтом/платиной и прокаленного. Осуществляли пропитку водной суспензии и сушку в вакууме. После вакуумной сушки этот промежуточный продукт сразу же прокаливали в псевдоожиженном слое в следующих условиях: непрерывный ток воздуха со скоростью 1.7 дм3 n/мин при повышении температуры от 25°C до 250°C при скорости нагрева 1°C/мин и выдерживание при 250°C в течение 6 час.
В опытах в лабораторном реакторе с непрерывным перемешиванием (′CSTR′) в суспензионной фазе для синтеза Фишера-Тропша (FTS) этот прокаленный материал восстанавливали и покрывали воском по следующей методике: 10 г катализатора восстанавливали при давлении 1 бар (100 кПа) в атмосфере чистого Н2 (объемная скорость = 2000 млn Н2/г катализатора/час) при повышении температуры от 25°C до 425°C со скоростью 1°C/мин, после достижения температуры 425°C выдерживали при ней в течение 16 час. Восстановленный катализатор оставляли охлаждаться до комнатной температуры, после этого водород заменяли на аргон и катализатор выгружали в жидком воске Фишера-Тропша в атмосфере аргона ("под аргоном"). Этот катализатор с восковым покрытием переносили затем в суспензионный реактор.
ПРИМЕР 2 -только Со(ОН)2
Получение сравнительного предшественника катализатора 2
Продукт, содержащий 5.0 г Со/100 г предшественника носителя катализатора, получали из дисперсионного алюминийоксидного носителя, модифицированного оксидом кремния, методом хемосорбции с последующим непосредственным прокаливанием на воздухе в псевдоожиженном слое.
В частности, предшественник катализатора получали следующим методом: Хемосорбция
К 3.2 г взвеси дисперсного гидроксида кобальта в 90 мл воды прибавляли 40 г оксида алюминия, модифицированного диоксидом кремния. Полученная суспензия имела pH 7.5. Хемосорбцию в водной суспензии проводили в течение 18 час при 80°C. За это время наблюдалось медленное снижение значения pH до 5. Водный слой декантировали, смесь трижды промывали водой, продукт светло-пурпурного цвета сушили при давлении 40 мбар (4кПа) и температуре 80°C. Этот высушенный вакуумной сушкой обработанный предшественник катализатора прокаливали в псевдоожиженном слое в постоянном токе воздуха со скоростью 1.6 дм3 n/мин при повышении температуры от 25°C до 250°C со скоростью нагрева 1°C/мин и выдерживали при 250°C в течение 6 час.
ПРИМЕР 3 - последовательно Со(OH)2 и Co(NO3)2
Получение сравнительного катализатора 3
Продукт, содержащий 20.0 г Со/0.070 г Pt/100 г носителя катализатора, получали из дисперсионного алюминийоксидного носителя, модифицированного оксидом кремния, последовательно осуществляя хемосорбцию-пропитку водной суспензии и сушку с последующим непосредственным прокаливанием на воздухе в псевдоожиженном слое.
Этот препарат получали в две стадии: первая стадия получения включала хемосорбцию с применением гидроксида кобальта, а вторая стадия включала пропитку нитратом кобальта.
В частности, катализатор получали нижеописанным методом: Раствор нитрата кобальта
Раствор нитрата кобальта готовили из Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 140.2 г/л Со и 0.5328 г/л Pt. pH раствора доводили до 2.7 азотной кислотой.
Хемосорбция/пропитка
К 3.2 г суспензии дисперсного гидроксида кобальта в 90 мл воды прибавляли 40 г оксида алюминия, модифицированного диоксидом кремния. Полученная взвесь имела pH 7.5. Хемосорбцию в водной суспензии проводили в течение 18 час при 80°C. За это время наблюдалось медленное снижение значения pH до 5. Водный слой декантировали, смесь трижды промывали водой, продукт пурпурного цвета сушили при давлении 40 мбар (4 кПа) и температуре 80°C.
Обработанный носитель катализатора или промежуточный материал после хемосорбции вводили в стадию пропитки кобальтом/платиной и прокаливания, описанную ниже: 30.0 г материала после хемосорбции и 47.7 мл раствора нитрата кобальта в виде водной суспензии использовали в реакции пропитки и вакуумной сушки, которые проводили в соответствии с описанием в Примере 1. Этот промежуточный продукт после вакуумной сушки сразу же прокаливали в следующих условиях: постоянный ток воздуха со скоростью 1.6 дм3 n/мин с повышением температуры от 25°C до 250°C со скоростью нагрева 1°C/мин и выдержка при 250°C в течение 6 час.
Предшественник катализатора (т.е. после хемосорбции, пропитки и прокаливания) активировали/восстанавливали, получая катализатор по методике, описанной в Примере 1, за исключением того, что конечная температура восстановления составляла 375°C.
ПРИМЕР 4 - (Со(OH)2 и Co(NO3)2 одновременно)
Получение катализатора 4 по изобретению
Продукт, содержащий 19.2 г Со/0.070 г Pt/100 г носителя катализатора, получали из дисперсионного алюминийоксидного носителя, модифицированного оксидом кремния, одновременно осуществляя хемосорбцию-пропитку водной суспензии и сушку с последующим непосредственным прокаливанием на воздухе в псевдоожиженном слое.
Этот препарат получали в одну стадию. Эта стадия включала хемосорбцию с применением гидроксида кобальта и пропитку нитратом кобальта. Таким образом, хемосорбция и пропитка осуществляется в ходе одной и той же стадии, называемой стадией одновременной хемосорбции и пропитки. В частности, катализатор получали как описано ниже: Раствор нитрата кобальта
Раствор нитрата кобальта готовили из Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 140.2 г/л Со и 0.5328 г/л Pt. pH раствора доводили до 2.7 азотной кислотой.
Хемосорбция/пропитка
К взвеси 3.2 г дисперсного гидроксида кобальта в растворе 57 г нитрата кобальта в 50 мл воды прибавляли 40 г оксида алюминия, модифицированного диоксидом кремния. Хемосорбцию в водной суспензии проводили в течение 1 час при 80°C. За это время наблюдалось медленное снижение значения pH суспензии с 6 до 3.5. Пропитку и вакуумную сушку полученной суспензии твердого материала пурпурного цвета в растворе темно-красного цвета проводили в соответствии с протоколом пропитки и вакуумной сушки, описанным в Примере 1. Этот высушенный вакуумной сушкой обработанный предшественник катализатора или промежуточный материал сразу прокаливали в псевдоожиженном слое в следующих условиях: постоянный ток воздуха со скоростью 1.6 дм3 n/мин с повышением температуры от 25°C до 250°C со скоростью нагрева 1°C/мин и выдержка при 250°C в течение 6 час.
Предшественник катализатора (т.е. после хемосорбции, пропитки и прокаливания) активировали/восстанавливали, получая катализатор по методике, описанной в Примере 1, за исключением того, что конечная температура восстановления составляла 375°C.
ПРИМЕР 5 - (Со(OH)2 и Co(NO3)2)
Получение катализатора 5 по изобретению
Продукт, содержащий 20.9 г Со/0.0795 г Pt/100 г носителя катализатора, получали из дисперсионного алюминийоксидного носителя, модифицированного оксидом кремния, одновременно осуществляя хемосорбцию- пропитку водной суспензии и сушку с последующим непосредственным прокаливанием на воздухе в псевдоожиженном слое. Этот препарат получали в одну стадию. Эта стадия включала хемосорбцию с применением гидроксида кобальта и пропитку нитратом кобальта. В частности, катализатор получали как описано ниже: Раствор нитрата кобальта
Раствор нитрата кобальта готовили из Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 141.2 г/л Со и 0.5396 г/л Pt. pH раствора доводили до 2.7 азотной кислотой.
Хемосорбция/пропитка
К взвеси 4 г дисперсного гидроксида кобальта в растворе 57 г нитрата кобальта в 50 мл воды прибавляли 40 г оксида алюминия, модифицированного диоксидом кремния. Хемосорбцию в водной суспензии проводили в течение 30 мин. при 60°C. За это время наблюдалось медленное снижение значения pH суспензии с 6 до 3.5. Пропитку и вакуумную сушку полученной суспензии твердого материала пурпурного цвета в растворе темно-красного цвета проводили в соответствии с протоколом пропитки и вакуумной сушки, описанным в Примере 1. Этот высушенный вакуумной сушкой обработанный предшественник катализатора или промежуточный материал сразу прокаливали в псевдоожиженном слое в следующих условиях: постоянный ток воздуха со скоростью 1.6 дм3 n/мин с повышением температуры от 25°C до 250°C со скоростью нагрева 1°C/мин и выдержка при 250°C в течение 6 час.
Предшественник катализатора (т.е. после хемосорбции, пропитки и прокаливания) активировали/восстанавливали, получая катализатор по методике, описанной в Примере 1, за исключением того, что конечная температура восстановления составляла 375°C.
ПРИМЕР 6 - (повышенная нагрузка на 1ой стадии получения)
Получение катализатора 6 по изобретению
Продукт, содержащий 29.7 г Со/0.041 г Pt/100 г носителя катализатора, получали с использованием дисперсионного алюминийоксидного носителя, модифицированного оксидом кремния, одновременно осуществляя хемосорбцию-пропитку водной суспензии и сушку с последующим непосредственным прокаливанием на воздухе в псевдоожиженном слое.
Этот препарат получали в одну стадию. Эта стадия включала хемосорбцию с применением гидроксида кобальта и пропитку нитратом кобальта. В частности, катализатор получали как описано ниже: Раствор нитрата кобальта
Раствор нитрата кобальта готовили из Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 154.4 г/л Со и 0.213 г/л Pt. pH раствора доводили до 3.2 азотной кислотой.
Хемосорбиия/пропитка
К взвеси 10 г дисперсного гидроксида кобальта в растворе 57 г нитрата кобальта в 50 мл воды прибавляли 40 г оксида алюминия, модифицированного диоксидом кремния. Хемосорбцию в водной суспензии проводили в течение 48 час. при 60°C. За это время наблюдалось медленное снижение значения pH суспензии с 6 до 3.5. Пропитку и вакуумную сушку полученной суспензии твердого материала пурпурного цвета в растворе темно-красного цвета проводили в соответствии с протоколом пропитки и вакуумной сушки, описанным в Примере 1. Этот высушенный вакуумной сушкой обработанный предшественник катализатора или промежуточный материал сразу прокаливали в псевдоожиженном слое в следующих условиях: постоянный ток воздуха со скоростью 1.6 дм3 n/мин с повышением температуры от 25°C до 250°C со скоростью нагрева 1°C/мин и выдержка при 250°C в течение 6 час.
Предшественник катализатора (т.е. после хемосорбции, пропитки и прокаливания) активировали/восстанавливали, получая катализатор по методике, описанной в Примере 1, за исключением того, что конечная температура восстановления составляла 375°C.
ПРИМЕР 7 (на носителе Puralox)
Получение катализатора 7 по изобретению
21.1 г катализатора Со/0.029 г Pt/100 г на носителе (Puralox SCCa) получали на дисперсном носителе Puralox SCCa (в виде частиц), применяя метод одновременной хемосорбции-пропитки суспензией и сушку с последующим прямым прокаливанием в псевдоожиженном слое на воздухе. Этот способ был одностадийным. Он включал хемосорбцию с применением гидроксида кобальта и пропитку с применением нитрата кобальта.
Этот катализатор был получен следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, применяя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 154.4 г/л Со и 0.213 г/л Pt. Величину pH раствора регулировали до значения 3.2, используя азотную кислоту. Хемосорбиия/пропитка
40 г носителя Puralox SCCa добавляли к взвеси 4 г частиц твердого гидроксида кобальта в 57 г раствора нитрата кобальта и 50 г воды. Хемосорбцию водной суспензионной фазы проводили в течение 30 мин при температуре 60°C. В течение этого процесса величина pH менялась от 6 до 3.5. Полученная суспензия твердого вещества пурпурного цвета в темно-красном растворе применялась для пропитки и сушки под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, описанным в Примере 1. Этот высушенный под вакуумом обработанный носитель катализатора или промежуточный продукт подвергался прямому прокаливанию в псевдоожиженном слое с применением непрерывного тока воздуха со скоростью 1.6 дм3/мин следующим образом:
температуру повышали от 25°C до 250°C со скоростью 1°C/мин и выдерживали полученный предшественник катализатора при температуре 250°C в течение 6 ч. Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 8
Получение катализатора 8 по изобретению
14.5 г катализатора Со/0.020 г Pt/100 г на носителе (модифицированный Puralox SCCa) получали на дисперсном модифицированом носителе (1.5g Si/100 Puralox SCCa), применяя метод одновременной хемосорбции-пропитки суспензией и сушки с последующим прямым прокаливанием в псевдоожиженном слое на воздухе. Этот способ был одностадийным. Он включал хемосорбцию гидроксида кобальта и пропитку нитратом кобальта.
Этот катализатор был получен следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, применяя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 154.4 г/л Со и 0.213 г/л Pt. Величину pH раствора регулировали до значения 3.2, используя азотную кислоту.
Хемосорбиия/пропитка
40 г модифицированного предварительно сформованного носителя Puralox SCCa (1.5 г Si/100г Puralox SCCa 2/150) добавляли к взвеси 4 г частиц твердого гидроксида кобальта в 57 г раствора нитрата кобальта и 50 г воды. Хемосорбцию водной суспензионной фазы проводили в течение 30 мин при температуре 60°C. В течение этого процесса величина pH менялась от 6 до 3.5. Полученная суспензия твердого вещества пурпурного цвета в темно-красном растворе применялась для пропитки и сушки под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, описанным в Примере 1. Этот высушенный под вакуумом обработанный носитель катализатора или промежуточный продукт подвергался прямому прокаливанию в псевдоожиженном слое с применением непрерывного тока воздуха со скоростью 1.6 дм3/мин следующим образом: температуру повышали от 25°C до 250°C со скоростью 1°C/мин и выдерживали полученный предшественник катализатора при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 9 - (Co(OH)2 и Co(NO3)2 при другом соотношении)
Получение катализатора 9 по изобретению
21.2 г катализатора Со/0.029 г Pt/100 г на носителе было получено на дисперсном модифицированном носителе из оксида алюминия, с применением метода одновременной хемосорбции-пропитки суспензией и сушки с последующим прямым прокаливанием в псевдоожиженном слое на воздухе. Этот способ был одностадийным. Он включал хемосорбцию гидроксида кобальта и пропитку нитратом кобальта. Этот катализатор был получен следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, применяя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 141.2 г/л Со и 0.5366 г/л Pt. Величину pH раствора регулировали до значения 2.6, используя азотную кислоту.
Хемосорбиия/пропитка
40 г модифицированного оксидом кремния оксида алюминия добавляли к 10 г взвеси частиц твердого гидроксида кобальта в 26 г раствора нитрата кобальта и 80 г воды. Хемосорбцию водной суспензионной фазы проводили в течение 3.5 ч при температуре 60°C. В течение этого процесса величина pH менялась от 6 до 3.5. Полученная суспензия твердого вещества пурпурного цвета в темно-красном растворе применялась для пропитки и сушки под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, описанным в Примере 1. Этот высушенный под вакуумом обработанный носитель катализатора или промежуточный продукт подвергался прямому прокаливанию в псевдоожиженном слое с применением непрерывного тока воздуха со скоростью 1.6 дм3/мин следующим образом: температуру повышали от 25°C до 250°C со скоростью 1°C/мин и выдерживали полученный предшественник катализатора при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 10 - (Со(ОН)2 и Со(NO3)2, две последовательные пропитки)
Получение катализатора 10 по изобретению
Было получено 41.2 г катализатора Со/0.051 г Pt/100 г на носителе (модифицированный оксидом кремния оксид алюминия), представлявшем собой дисперсный оксид алюминия, модифицированный оксидом кремния, с использованием двух последовательных стадий, каждая из которых состояла из: одновременной хемосорбции-пропитки суспензией и сушки с последующим прямым прокаливанием в псевдоожиженном слое на воздухе. Этот способ состоял из двух стадий: каждая стадия включала хемосорбцию с применением гидроксида кобальта и пропитку с применением нитрата кобальта. Этот катализатор был получен следующим образом.
Раствор нитрата кобальта 1
Раствор нитрата кобальта получали, применяя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 154.4 г/л Со и 0.213 г/л Pt. Величину pH раствора регулировали до значения 3.2, используя азотную кислоту. Раствор нитрата кобальта 2
Раствор нитрата кобальта получали, применяя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 141.2 г/л Со и 0.537 г/л Pt. Величину pH раствора регулировали до значения 2.5, используя азотную кислоту.
Хемосорбиия/пропитка
На стадии 1: 40 г оксида алюминия, модифицированного оксидом кремния, добавляли к 10 г взвеси частиц твердого гидроксида кобальта в 57 г раствора нитрата кобальта 1 и 50 г воды. Хемосорбцию водной суспензионной фазы проводили в течение 1 ч при температуре 80°C. В течение этого процесса величина pH менялась от 6 до 3.5. Полученная суспензия твердого вещества пурпурного цвета в темно-красном растворе применялась для пропитки и сушки под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, описанным в Примере 1. Этот высушенный под вакуумом обработанный носитель катализатора или промежуточный продукт подвергался прямому прокаливанию в псевдоожиженном слое с применением непрерывного тока воздуха со скоростью 1.6 дм3/мин следующим образом: температуру повышали от 25°C до 250°C со скоростью 1°C/мин и выдерживали катализатор при температуре 250°C в течение 6 ч.
На стадии 2: 20 г продукта, полученного на стадии 1, добавляли к взвеси 1.6 г частиц твердого гидроксида кобальта в 32 г раствора нитрата кобальта 2 и 30 г воды. Хемосорбцию водной суспензионной фазы проводили в течение 1 ч при температуре 80°C. В течение этого процесса величина pH менялась от 6 до 3.5. Полученная суспензия твердого вещества пурпурного цвета в темно-красном растворе применялась для пропитки и сушки под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, описанным в Примере 1. Этот высушенный под вакуумом обработанный носитель катализатора или промежуточный продукт подвергался прямому прокаливанию в псевдоожиженном слое с применением непрерывного тока воздуха со скоростью 1.6 дм3/мин следующим образом: температуру повышали от 25°C до 250°C со скоростью 1°C/мин и выдерживали полученный предшественник катализатора при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C
ПРИМЕР 11 - (Со(OH)2, и Со(NO3)2)
Получение катализатора 11 по изобретению
Катализатор, содержащий 26.7 г Со/0.070 г Pt/100 г на носителе, получали на дисперсном оксиде алюминия, модифицированном оксидом кремния, с применением метода одновременной хемосорбции-пропитки суспензией и сушки с последующим прямым прокаливанием в псевдоожиженном слое на воздухе. Этот способ был одностадийным. Он включал хемосорбцию гидроксида кобальта и пропитку нитратом кобальта.
Этот катализатор был получен следующим образом.
Раствор нитрата кобальта 1
Раствор нитрата кобальта получали, применяя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 140.2 г/л Со и 0.5328 г/л Pt. Величину pH этого раствора регулировали до значения 2.7 с помощью азотной кислоты.
Хемосорбция/пропитка
40 г оксида алюминия, модифицированного оксидом кремния, добавляли к 4 г взвеси частиц твердого гидроксида кобальта в 57 г раствора нитрата кобальта 1 и 50 г воды. Хемосорбцию водной суспензионной фазы проводили в течение 1 ч при температуре 80°C. В течение этого процесса величина pH менялась от 6 до 3.5. Полученная суспензия твердого вещества пурпурного цвета в темно-красном растворе применялась для пропитки и сушки под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, описанным в Примере 1. Этот высушенный под вакуумом обработанный носитель катализатора или промежуточный продукт подвергался прямому прокаливанию в псевдоожиженном слое с применением непрерывного тока воздуха со скоростью 1.6 дм3/мин, при этом температуру повышали от 25°C до 250°C и выдерживали полученный продукт при температуре 250°C в течение 67 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 12 - (Со(ОН)2 и Co(NO3)2, две последовательные пропитки)
Получение катализатора 12 по изобретению
Получали катализатор 57.7 г Со/0.06 г Pt/100 г на носителе (оксид алюминия модифицированный оксидом кремния), представлявшем собой дисперсный оксид алюминия, модифицированный оксидом кремния, с применением метода одновременной хемосорбции-пропитки суспензией и сушки с последующим прямым прокаливанием в псевдоожиженном слое на воздухе.
Этот способ состоял из двух последующих одинаковых стадий: каждая стадия включала хемосорбцию гидроксида кобальта и пропитку нитратом кобальта. Этот катализатор получали следующим образом.
Раствор нитрата кобальта 1
Раствор нитрата кобальта получали, применяя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 154.4 г/л Со и 0.213 г/л Pt. Величину pH этого раствора регулировали до значения 3.2 с помощью азотной кислоты. Раствор нитрата кобальта 2
Раствор нитрата кобальта получали, применяя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 141.2 г/л Со и 0.537 г/л Pt. Величину pH этого раствора регулировали до значения 2.5 с помощью азотной кислоты.
Хемосорбиия/пропитка
На стадии 1: 40 г оксида алюминия, модифицированного оксидом кремния, добавляли к 8 г взвеси частиц твердого гидроксида кобальта в 57 г раствора нитрата кобальта 1 и 50 г воды. Хемосорбцию водной суспензионной фазы проводили в течение 1 ч при температуре 80°C. В течение этого процесса величина pH менялась от 6 до 3.5. Полученная суспензия твердого вещества пурпурного цвета в темно-красном растворе применялась для пропитки и сушки под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, описанным в Примере 1. Этот высушенный под вакуумом обработанный носитель катализатора или промежуточный продукт подвергался прямому прокаливанию в псевдоожиженном слое с применением непрерывного тока воздуха со скоростью 1.6 дм3/мин следующим образом: температуру повышали от 25°C до 250°C со скоростью 1°C/мин и выдерживали полученный продукт при температуре 250°C в течение 6 ч.
На стадии 2: 20 г продукта, полученного на стадии 1, добавляли к взвеси 4 г частиц твердого гидроксида кобальта в 32 г раствора нитрата кобальта 2 и 30 г воды. Хемосорбцию водной суспензионной фазы проводили в течение 1 ч при температуре 80°C. В течение этого процесса величина pH менялась от 6 до 3.5. Полученная суспензия твердого вещества пурпурного цвета в темно-красном растворе применялась для пропитки и сушки под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, описанным в Примере 1. Этот высушенный под вакуумом обработанный носитель катализатора или промежуточный продукт подвергался прямому прокаливанию в псевдоожиженном слое с применением непрерывного тока воздуха со скоростью 1.6 дм3/мин следующим образом: температуру повышали от 25°C до 250°C со скоростью 1°C/мин и выдерживали полученный продукт при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 13 - (Ni(OH)2 и Ni(NO3)2 одновременно)
Получение катализатора 13 по изобретению
30.4 г катализатора Ni/100 г на носителе оксиде алюминия получали, используя дисперсный носитель Puralox SCC и применяя метод одновременных хемосорбции-пропитки водной фазой суспензии и сушки с последующим прокаливанием полученного продукта в трубчатом проточном реакторе прямым контактом на воздухе.
Этот способ был одностадийным. Он включал хемосорбцию гидроксида никеля и пропитку с применением нитрата никеля.
Этот катализатор был получен следующим образом.
Раствор нитрата никеля
Раствор нитрата никеля получали, используя Ni(NO3)2·6H2O, полученный раствор содержал 140 г/л Ni.
Хемосорбиия/пропитка
40 г носителя Puralox SCC а-2/150 добавляли к взвеси 8 г дисперсного твердого гидроксида никеля в 63 г раствора нитрата никеля и 55 г воды. Хемосорбцию водной суспензии осуществляли при температуре 80°C в течение 20 ч. Полученной суспензией зеленовато-голубого твердого продукта в зеленом растворе осуществляли пропитку и проводили сушку под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, подробно описанным в Примере 1. Этот высушенный под вакуумом катализатор на носителе или промежуточный продукт подвергали прокаливанию в трубчатом проточном реакторе прямым контактом, используя следующую процедуру: подавали непрерывно поток воздуха со скоростью 69 дм3/ч, повышая температуру от 25°C до 375°C со скоростью 1°C/мин и затем выдерживали полученный продукт при температуре 375°C в течение 6 ч.
ПРИМЕР 14 - (Co(OH)2 Co(NO3)2 и Ni(NO3)2 одновременно)
Получение катализатора 14 по изобретению
19.2 г катализатора Со/2.5 г Ni/0.070 г Pt/100 г получали на носителе, представляющем собой дисперсный оксид алюминия, модифицированный оксидом кремния, используя метод одновременной хемосорбции-пропитки водной суспензией и сушки с последующим прокаливанием полученного продукта прямым контактом на воздухе в псевдоожиженном слое.
Этот способ был одностадийным. Он включал хемосорбцию гидроксида кобальта и пропитку с применением нитрата кобальта и нитрата никеля.
Этот катализатор получали следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, используя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, получали раствор, содержащий 140.2 г/л Со и 0.5328 г/л Pt. Величину pH регулировали до значения 2.7, используя азотную кислоту.
Раствор нитрата никеля
Раствор нитрата никеля получали, используя Ni(NO3)2·6H2O, получали раствор, содержащий 140 г/л Ni.
Хемосорбиия/пропитка
40 г оксида алюминия, модифицированного оксидом кремния, добавляли к взвеси 3.2 г дисперсного твердого гидроксида кобальта в 57 г раствора нитрата кобальта и 7 г раствора нитрата никеля и 50 г воды. Хемосорбцию водной суспензии осуществляли в течение 18 ч при температуре 80°C. Затем проводили пропитку полученной суспензией твердого продукта пурпурного цвета в темно-красном растворе и сушку под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, подробно описанным в Примере 1. Этот высушенный под вакуумом обработанный продукт на носителе или промежуточный продукт подвергали прокаливанию в псевдоожиженном слое, используя следующую процедуру: подавали непрерывно поток воздуха со скоростью 1.6 дм3/ч, повышая температуру от 25°C до 250°C со скоростью 1°C/мин и затем выдерживали полученный продукт при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 15 - (Со(ОН)2 и Ni(OH)2 и Со(NO3)2 одновременно)
Получение катализатора 15 по изобретению
19.2 г катализатора Со/2.5 г Ni/0.070 г Pt/100 г получали на носителе, представлявшем собой дисперсный оксид алюминия, модифицированный оксидом кремния, используя метод одновременной хемосорбции-пропитки водной суспензией и сушки с последующим прокаливанием полученного продукта прямым контактом на воздухе в псевдоожиженном слое.
Этот способ был одностадийным. Он включал хемосорбцию с применением гидроксида кобальта и гидроксида никеля и пропитку с применением нитрата кобальта.
Этот способ осуществляли следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, используя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, полученный раствор содержал 140.2 г/л Со и 0.5328 г/л Pt. Величину pH этого раствора регулировали до значения 2.7 с помощью азотной кислоты.
Хемосорбиия/пропитка
40 г оксида алюминия, модифицированного оксидом кремния, добавляли к взвеси 3.2 г дисперсного твердого гидроксида кобальта и 1.6 г дисперсного твердого гидроксида никеля в 57 г раствора нитрата кобальта и 7 г раствора нитрата никеля и 50 г воды. Хемосорбцию водной суспензии осуществляли в течение 18 ч при температуре 80°C. Осуществляли пропитку полученной суспензией твердого продукта пурпурного цвета в темно-красном растворе и сушку под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, подробно описанным в Примере 1. Этот высушенный под вакуумом обработанный катализатор на носителе или промежуточный продукт подвергали прокаливанию в псевдоожиженном слое, используя следующую процедуру: подавали непрерывно поток воздуха со скоростью 1.6 дм3/ч, повышая температуру от 25°C до 250°C со скоростью 1°C/мин и затем выдерживали полученный продукт при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 16 - (Со(OH)2 и Mn(ОН)2 и Со(NO3)2 одновременно)
Получение катализатора 16 по изобретению
19.2 г катализатора Со/4 г Mn/0.070 г Pt/100 г получали на носителе, представлявшем собой дисперсный оксид алюминия, модифицированный оксидом кремния, используя метод одновременной хемосорбции-пропитки водной суспензией и сушки с последующим прокаливанием полученного продукта прямым контактом на воздухе в псевдоожиженном слое.
Этот способ был одностадийным. Он включал хемосорбцию с применением гидроксида кобальта и гидроксида никеля и пропитку с применением нитрата кобальта.
Этот катализатор получали следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, используя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, Полученный раствор содержал 140.2 г/л Со и 0.5328 г/л Pt. Величину pH этого раствора регулировали до значения 2.7 с помощью азотной кислоты.
Хемосорбиия/пропитка
40 г оксида алюминия, модифицированного оксидом кремния, добавляли к взвеси 3.2 г дисперсного твердого гидроксида кобальта и 2.7 г твердого гидроксида марганца в 57 г раствора нитрата кобальта и 50 г воды. Хемосорбцию водной суспензии осуществляли в течение 18 ч при температуре 80°C. Осуществляли пропитку полученной суспензией твердого продукта пурпурного цвета в темно-красном растворе и сушку под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, подробно описанным в Примере 1. Этот высушенный под вакуумом обработанный катализатор на носителе или промежуточный продукт подвергали прокаливанию в псевдоожиженном слое, используя следующую процедуру: подавали непрерывно поток воздуха со скоростью 1.6 дм3/ч, повышая температуру от 25°C до 250°C со скоростью 1°C/мин и затем выдерживали полученный продукт при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 17 - (Co(OH)2 и Co(NO3)2 на ZrO2) Получение катализатора 17 по изобретению
26.6 г катализатора Со/0.070 г Pt/100 г на носителе из оксида циркония (IV) получали, используя в качестве носителя дисперсный ZrO2 (от Acros Organics, ч. д. а. (98% ZrO2)) и метод одновременной хемосорбции-пропитки водной суспензией и сушки с последующим прокаливанием полученного продукта прямым контактом на воздухе в псевдоожиженном слое.
Этот способ был одностадийным. Он включал хемосорбцию с применением гидроксида кобальта и пропитку с применением нитрата кобальта.
Этот катализатор получали следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, используя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, Полученный раствор содержал 140.2 г/л Со и 0.5328 г/л Pt. Величину pH этого раствора регулировали до значения 2.7 с помощью азотной кислоты.
Хемосорбиия/пропитка
40 г ZrO2 добавляли к взвеси 8 г дисперсного твердого гидроксида кобальта в 57 г раствора нитрата кобальта и 50 г воды. Хемосорбцию водной суспензии проводили в течение 18 ч при температуре 80°C. Осуществляли пропитку полученной суспензией твердого продукта светло-розового цвета в растворе красного цвета и затем сушку под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, подробно описанным в Примере 1. Этот высушенный под вакуумом обработанный катализатор на носителе или промежуточный продукт подвергали прокаливанию в псевдоожиженном слое, используя следующую процедуру: подавали непрерывно поток воздуха со скоростью 1.6 дм3/ч, повышая температуру от 25°C до 250°C со скоростью 1°C/мин и затем выдерживали полученный продукт при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 18 - (Cu(OH)2 и Cu(NO3)2 на ZrO2)
Получение предшественника катализатора 18 по изобретению
Получали 19 г катализатора Си/100 г на носителе из дисперсного оксида циркония (IV) (ZrO2 (от Acros Organics, ч. д. а (98% ZrO2)) с применением метода одновременной хемосорбции-пропитки фазой водной суспензии и сушки с последующим прокаливанием полученного продукта в псевдоожиженном слое прямым контактом на воздухе.
Этот способ был одностадийным. Он включал хемосорбцию с применением гидроксида меди и пропитку с применением нитрата меди. Этот катализатор получали следующим образом. Раствор нитрата меди
Раствор нитрата меди получали, используя Cu(NO3)2·6H2O, полученный раствор содержал 140 г/л Cu.
Хемосорбиия/пропитка
40 г ZrO2 добавляли к взвеси 4 г твердого дисперсного гидроксида меди в 63 г раствора нитрата меди и 50 г воды. Хемосорбцию полученной водной суспензии осуществляли в течение 18 ч при температуре равной 80°C. Полученную суспензию голубовато-зеленого твердого продукта в растворе голубого цвета использовали для пропитки и последующей сушки под вакуумом.
ПРИМЕР 19 - (Со(ОН)2 и Со(NO3)2 - катализатор в виде скорлупок (eggshell)
Получение предшественника катализатора 19 по изобретению
7 г катализатора Со/0.030 г Pt/100 г получали на предварительно сформованном носителе Al 4505 Т 1/8 с применением метода одновременной хемосорбции-пропитки фазой водной суспензии и сушки с последующим прокаливанием полученного продукта в псевдоожиженном слое прямым контактом на воздухе.
Этот способ был одностадийным. Он включал хемосорбцию с применением гидроксида кобальта и пропитку с применением нитрата кобальта. Этот катализатор получали следующим образом. Раствор нитрата кобальта
Раствор нитрата кобальта получали, используя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, Полученный раствор содержал 140.2 г/л Со и 0.5328 г/л Pt. Величину pH этого раствора регулировали до значения 2.7 с помощью азотной кислоты.
Хемосорбиия/пропитка
40 г носителя Al 4505 Т 1/8 добавляли к взвеси 0.5 г твердого дисперсного гидроксида кобальта в 25 г раствора нитрата кобальта и 50 г воды. Хемосорбцию водной суспензии проводили в течение 18 ч при температуре 80°C. Осуществляли пропитку полученной суспензией твердого продукта светло-розового цвета в растворе красного цвета и затем сушку под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, подробно описанным в Примере 1. Этот высушенный под вакуумом обработанный катализатор на носителе или промежуточный продукт подвергали прокаливанию в псевдоожиженном слое, используя следующую процедуру: подавали непрерывно поток воздуха со скоростью 1.6 дм3/ч, повышая температуру от 25°C до 250°C со скоростью 1°C/мин и затем выдерживали полученный продукт при температуре 250°C в течение 6 ч.
ПРИМЕР 20 - Со(OH)2 и Co(NO3)2 последовательно как в Примере 3, но с применением меньшего количества платины
Получение сравнительного катализатора 20
Получали 20 г катализатора Со/0.035 г Pt/100 г на носителе, используя в качестве носителя дисперсный оксид алюминия, модифицированный оксидом кремния и метод последовательной хемосорбции-пропитки и сушки с последующим прокаливанием полученного продукта в псевдоожиженном слое прямым контактом на воздухе.
Этот способ был двухстадийным. Первая стадия включала хемосорбцию с применением гидроксида кобальта, в то время как вторая стадия включала пропитку с применением нитрата кобальта.
Этот катализатор получали следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, используя Co(NO3)2·6H2O и Pt(NH3)4·(NO3)2, полученный раствор содержал 140.2 г/л Со и 0.2664 г/л Pt. Величину pH этого раствора регулировали до значения 2.7 с помощью азотной кислоты.
Хемосорбиия/пропитка
40 г оксида алюминия, модифицированного оксидом кремния, добавляли к взвеси 3.2 г дисперсного гидроксида кобальта в 90 мл воды. Величина pH полученной суспензии была равна 7.5. Хемосорбцию водной суспензии осуществляли в течение 18 ч при температуре 80°C. Во время этого процесса величина pH медленно снижалась до 5. Водный слой декантировали из смеси и после трех промывок водой полученный продукт пурпурного цвета сушили при давлении 40 мбар и при температуре равной 80°C. Полученный обработанный катализатор на носителе или промежуточный продукт после хемосорбции затем подвергали пропитке нитратом кобальта/платины и прокаливанию. 30.0 г продукта после хемосорбции пропитывали 47.7 мл раствора нитрата кобальта в водной суспензии и подвергали сушке под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, подробно описанным в Примере 1. Этот высушенный под вакуумом промежуточный продукт подвергали прокаливанию в псевдоожиженном слое, используя следующую процедуру: подавали непрерывно поток воздуха со скоростью 1.6 дм3/ч, повышая температуру от 25°C до 250°C со скоростью 1°C/мин и затем выдерживали полученный продукт при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 21 - Со(ОН)2 и Со(NO3)2 последовательно без платины
Получение сравнительного катализатора 21
Получали 20 г катализатора Со/100 г на носителе, используя в качестве носителя дисперсный оксид алюминия, модифицированный оксидом кремния, последовательные хемосорбцию и пропитку с последующим прокаливанием полученного продукта в псевдоожиженном слое прямым контактом на воздухе.
Этот способ был двухстадийным. Первая стадия включала хемосорбцию с применением гидроксида кобальта, в то время как вторая стадия включала пропитку с применением нитрата кобальта.
Этот катализатор получали следующим образом.
Раствор нитрата кобальта
Раствор нитрата кобальта получали, используя Co(NO3)2·6H2O, полученный раствор содержал 140.2 г/л Со. Величину pH этого раствора регулировали до значения 2.7 с помощью азотной кислоты.
Хемосорбиия/пропитка
40 г оксида алюминия, модифицированного оксидом кремния, добавляли к взвеси 3.2 г дисперсного гидроксида кобальта в 90 мл воды. Величина pH полученной суспензии была равна 7.5. Хемосорбцию водной суспензии осуществляли в течение 18 ч при температуре 80°C. Во время этого процесса величина pH медленно снижалась до 5. Водный слой декантировали из смеси и после трех промывок водой полученный продукт пурпурного цвета сушили при давлении 40 мбар и при температуре равной 80°C. Полученный обработанный катализатор на носителе или промежуточный продукт после хемосорбции затем подвергали пропитке нитратом кобальта/платины и прокаливанию. 30.0 г продукта после хемосорбции пропитывали 47.7 мл раствора нитрата кобальта в водной суспензии и сушке под вакуумом в соответствии с протоколом пропитки и сушки под вакуумом, подробно описанным в Примере 1. Этот высушенный под вакуумом промежуточный продукт подвергали прокаливанию в псевдоожиженном слое, используя следующую процедуру: подавали непрерывно поток воздуха со скоростью 1.6 дм3/ч, повышая температуру от 25°C до 250°C со скоростью 1°C/мин и затем выдерживали полученный продукт при температуре 250°C в течение 6 ч.
Этот предшественник катализатора (то есть продукт после хемосорбции, пропитки и прокаливания) активировали/восстанавливали для получения катализатора, используя способ, описанный в Примере 1, за исключением того, что конечная температура восстановления была равна 375°C.
ПРИМЕР 22
Свойства предшественников катализаторов и сравнительного катализатора 1
Катализаторы 1, 4, 5, 10, 12, 14 и 15 испытывали при проведении синтеза Фишера-Тропша, используя реактор с постоянным перемешиванием (CSTR) в суспензионной фазе. Условия синтеза Фишера-Тропша были следующими:
Температура в реакторе: 230°C
Давление в реакторе: 17 бар
Катализатор по изобретению: около 10 г
Степень конверсии (H2+CO): 50-65%
Отношение H2:CO на входе: 1.6:1
Аргон в качестве внутреннего стандарта: 15 об.% Так как все условия синтеза Фишера-Тропша (СФТ) были одинаковыми, относительная активность в СФТ была определена путем определения ФТ активности каждого катализатора в молях превращенной CO/г катализатора и ее сравнивали с активностью сравнительного катализатора 1 (см. Таблицу 1).
Катализатор 4, полученный в соответствии с изобретением с применением гидроксида кобальта для хемосорбции и нитрата кобальта для пропитки, имел кобальтовую нагрузку на 19% меньше и обладал активностью, сравнимой с активностью катализатора 1, который был получен с применением нитрата кобальта при двух последовательных стадиях пропитки, в условиях, описанных выше
Катализатор 5, полученный в соответствии с изобретением с применением гидроксида кобальта для хемосорбции и нитрата кобальта для пропитки, имел кобальтовую нагрузку на 15% больше и обладал активностью, превышающей активность катализатора 1 на 34%, катализатор 1 был получен с применением нитрата кобальта при проведении двух последовательных стадий пропитки, в условиях реакции, описанных выше.
Катализатор 10, полученный в соответствии с данным изобретением с применением гидроксида кобальта для хемосорбции и нитрата кобальта для пропитки, но в другом соотношении, по сравнению с другими примерами имел более высокую нагрузку Co(OH)2. При общем содержании кобальта равном 29.2% (м/м) (то есть, кобальтовая нагрузка на 46% больше), этот катализатор обладал активностью на 52% выше, чем сравнительный катализатор 1, в условиях реакции, указанных выше.
Катализатор 12, полученный в соответствии с данным изобретением с применением гидроксида кобальта для хемосорбции и нитрата кобальта для пропитки, имел высокую кобальтовую нагрузку (36.6% (м/м); то есть на 83% выше) обладал активностью на 17% выше, чем сравнительный катализатор 1, в условиях реакции, указанных выше.
Катализаторы 2-11 и 13-16, полученные в соответствии с данным изобретением, характеризовались дисперсией более мелких кристаллов кобальта по сравнению с приготовленным обычным образом катализатором 1, пропитанным только нитратом кобальта. Для этой дисперсии с улучшенными свойствами определяли размер кристаллов методом дифракционного рентгеновского анализа, что показано в Таблице 1. Кроме того, эти катализаторы имели лучшую поверхность участков, содержащих кобальт, что показывают данные по адсорбционной способности водорода (НАС) (см. также Таблицу 1).
Величины НАС получены по определению количества водорода (в мл/г катализатора на носителе), который адсорбируется после восстановления кобальта. Эксперимент осуществляли способом, состоящим из трех стадий: 1) восстановление кобальта, 2) насыщение восстановленного катализатора водородом и 3) десорбция водорода в инертной атмосфере от -75°C до 350°C.
Средний размер кристаллитов оксида кобальта, который определяли методом дифракционного рентгеновского анализа, в случае сравнительного катализатора 1 был равен 15 нм, в то время как средний размер кристаллитов оксида кобальта в случае катализаторов 4-9 (то есть катализаторов по изобретению) был значительно меньше, около 10-11 нм.
ПРИМЕР 23
Наличие реакции между суспензией (Co(OH)2 и носителем
Наличие реакции суспензии гидроксида металла с носителем является удивительным фактом. По этой причине данное изобретение иллюстрируется изображениями и данными, полученными в этом Примере. На Фигуре 1 можно видеть изображения суспензии Co(OH)2 в воде, содержащей носитель, представляющий собой оксид алюминия, модифицированный оксидом кремния, до (слева) и после их смешения при температуре 80°C.
В верхней части этой Фигуры представлено изображение смеси, не содержащей Co(NO3)2. Было установлено, что реакция между Co(OH)2 и оксидом алюминия, модифицированным оксидом кремния, завершалась при указанной температуре через 18 ч. В результате образовывался твердый продукт светло-пурпурного цвета с четким слоем воды наверху.
Изображения в нижней части этой Фигуры показывают смесь Co(OH)2, оксида алюминия, модифицированного оксидом кремния, и растворимого Co(NO3)2. В этом случае реакция между Co(OH)2 и носителем завершалась через 4 мин, в результате образовывался твердый продукт пурпурного цвета, содержащий сверху прозрачный слой жидкости темно-красного цвета (благодаря наличию нитрата кобальта).
Изображение образца смеси, содержащей сверху слой твердого продукта темно-пурпурного цвета, гидроксида кобальта на носителе, приведено также на Фигуре 2 (слева). Когда эту смесь промыли тремя порциями воды, продукт темно-пурпурного цвета, использованный гидроксид кобальта, оставался присоединенным к носителю, а промывные воды были прозрачными и бесцветными (изображение справа).
Дополнительное подтверждение дают профили восстановления, записанные при помощи метода температурно-запрограмированного восстановления (TPR). На Фигуре 3 показаны данные TPR для физической смеси Co(OH)2 и оксида алюминия, модифицированного оксидом кремния (пунктирная линия) в зависимости от количества Co(OH)2, хемосорбированного на оксиде алюминия, модифицированном оксидом кремния (сплошная линия). В физической смеси 10% Co(OH)2 и 90% носителя, оксида алюминия, кобальт обычно восстанавливался до Co(0) при температуре 260°C (Фигура 3, пунктирная линия). Продукт реакции Co(OH)2 с оксидом алюминия в суспензии четко характеризовался другим профилем восстановления (сплошная линия). Максимум на участке от 400 до 750°C (сплошная линия) показывает, что это восстановление до Co(0) происходит на частицах кобальта, проявляющих сильное взаимодействие с носителем, оксидом алюминия.
Кроме того, Co(OH)2 был химически сорбирован на таблетках из оксида алюминия при осуществлении того же метода. Смесь Co(OH)2 и таблеток из оксида алюминия в воде привела к образованию распределенных по поверхности таблеток "корочковых" (egg shell) структур кобальтсодержащего предшественника катализатора. На Фигуре 4 слева показано изображение полученных таблеток с покрытием. Изображение в средней части Фигуры 4 показывает полученный продукт в воде, кобальт остался полностью на таблетках. Изображение справа на Фигуре 4 иллюстрирует отдельные стадии кобальтовой нагрузки таблеток, которые были разрезаны на части, в нижнем ряду показаны таблетки после хемосорбции и пропитки (распределение по всей поверхности таблетки), в среднем ряду показаны таблетки после хемосорбции (распределение структур типа скорлупок (eggshell) и в верхнем ряду показаны таблетки без нагрузки для сравнения.
Специалисту в данной области техники очевидно, что данное изобретение предусматривает конкурентоспособный способ, позволяющий осуществить не вызывающее затруднение и экономичное последовательное получение осажденного металла на носителе, что обеспечивает получение нужной дисперсии с высокой нагрузкой на прочном носителе. По сравнению с известными способами данное изобретение лишено недостатков, таких как большое количество промывок (нет необходимости удалять соли).
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОРЫ | 2010 |
|
RU2517700C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША | 2009 |
|
RU2481156C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОБАЛЬТСОДЕРЖАЩЕГО КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША | 2012 |
|
RU2602803C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРЕДШЕСТВЕННИКА КАТАЛИЗАТОРА И КАТАЛИЗАТОРА ФИШЕРА-ТРОПША НА ОСНОВЕ КОБАЛЬТА | 2001 |
|
RU2298434C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2592271C2 |
| КАТАЛИЗАТОРЫ С ВЫСОКИМ СОДЕРЖАНИЕМ КОБАЛЬТА И ВЫСОКОЙ ПЛОЩАДЬЮ ПОВЕРХНОСТИ КОБАЛЬТА, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2005 |
|
RU2367521C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2584915C2 |
| КОБАЛЬТОВЫЕ КАТАЛИЗАТОРЫ | 2000 |
|
RU2252072C2 |
| КАТАЛИЗАТОРЫ НА ОСНОВЕ КОБАЛЬТА | 2001 |
|
RU2261143C2 |
| СПОСОБ КОНВЕРСИИ НИТРАТОВ МЕТАЛЛОВ | 2006 |
|
RU2429073C2 |

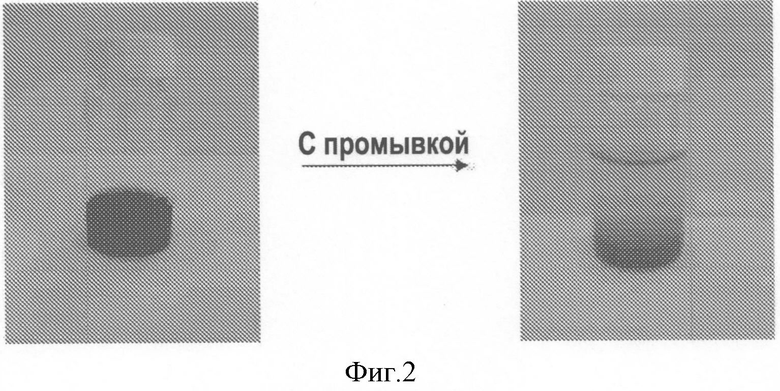
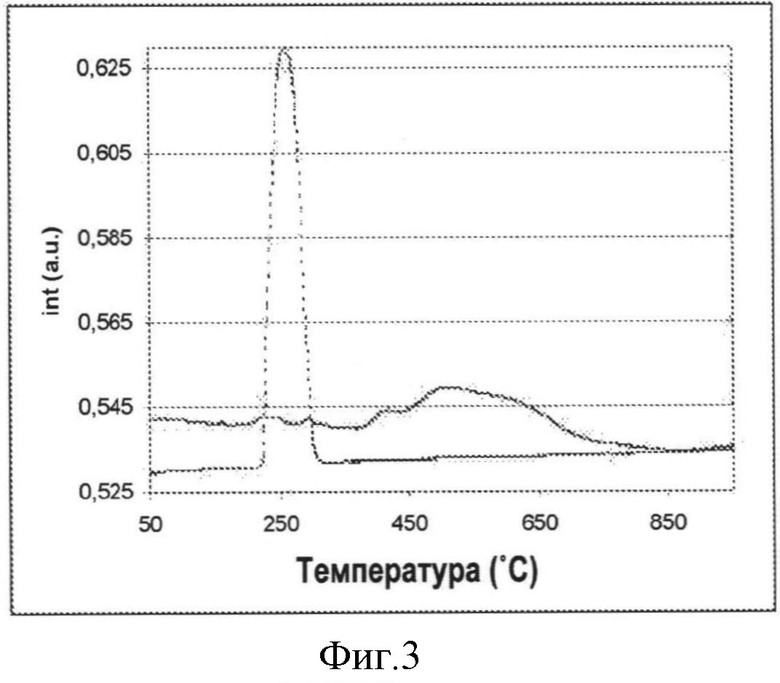

Предложен способ получения предшественника катализатора. Способ получения предшественника катализатора включает: получение суспензии, включающей жидкость-носитель, растворимую соль металла, частицы нерастворимой неорганической соли металла и частицы и/или одно или более тел предварительно сформованных носителей катализатора, с осаждением металла из нерастворимой соли металла на частицах носителя за счет хемосорбции, и с осаждением металла из растворимой соли металла внутри и/или на частицах носителя за счет пропитки, при этом хемосорбция и пропитка осуществляются одновременно, и металлы в нерастворимой неорганической соли металла и в растворимой соли металла являются одними и теми же, и представляют собой Со или Ni, и при этом указанный металл является активным компонентом катализатора, с образованием обработанного носителя катализатора, и удаление жидкости-носителя из суспензии с получением высушенного обработанного носителя катализатора, который или непосредственно представляет собой предшественник катализатора, или необязательно подвергается прокаливанию для получения предшественника катализатора. Заявлен также способ получения катализатора, способ синтеза углеводородов, способ гидрирования. Технический результат - способ является экономичным, обеспечивает получение нужной дисперсии с высокой нагрузкой на прочном носителе. 4 н. и 10 з.п. ф-лы, 1 табл., 4 ил., 23 пр.
1. Способ получения предшественника катализатора, который включает:
получение суспензии, включающей жидкость-носитель, растворимую соль металла, частицы нерастворимой неорганической соли металла и частицы и/или одно или более тел предварительно сформованных носителей катализатора, с осаждением металла из нерастворимой соли металла на частицах носителя за счет хемосорбции, и с осаждением металла из растворимой соли металла внутри и/или на частицах носителя за счет пропитки, при этом хемосорбция и пропитка осуществляются одновременно, и металлы в нерастворимой неорганической соли металла и в растворимой соли металла являются одними и теми же и представляют собой Со или Ni, и при этом указанный металл является активным компонентом катализатора, с образованием обработанного носителя катализатора; и
удаление жидкости-носителя из суспензии с получением высушенного обработанного носителя катализатора, который или непосредственно представляет собой предшественник катализатора, или необязательно подвергается прокаливанию для получения предшественника катализатора.
2. Способ по п. 1, в котором частицы нерастворимой неорганической соли металла находятся в контакте с частицами и/или одним или более тел предварительно сформованных носителей катализатора в течение, по меньшей мере, одной минуты.
3. Способ по п. 1, в котором предварительно сформованный носитель катализатора является пористым и выбран из группы, состоящей из монолитного носителя, таблеток, сформованных искусственных носителей, экструдатов, сферических носителей и комбинаций двух или более указанных видов носителей.
4. Способ по п. 1, в котором предварительно сформованный носитель катализатора выбран из группы, состоящей из оксида алюминия, оксида кремния, оксида титана, оксида циркония, оксида магния, оксида цинка, активированного углерода, молекулярных сит, цеолитов и комбинаций двух или более указанных видов.
5. Способ по п. 1, в котором нерастворимая неорганическая соль металла представляет собой Со(ОН)2 и/или растворимая соль металла представляет собой Co(NO3)2·6H2O.
6. Способ по п. 1, в котором предшественник катализатора содержит металл в количестве от 5 мас.% до 90 мас.% от общей массы предшественника.
7. Способ по п. 1, в котором предшественник катализатора практически не содержит обменных ионов.
8. Способ по п. 1, в котором предшественник катализатора представляет собой предшественник катализатора синтеза Фишера-Тропша.
9. Способ по п. 1, в котором предшественник катализатора представляет собой предшественник катализатора гидрирования.
10. Способ получения катализатора, который включает получение предшественника катализатора способом по п. 1 и восстановление полученного таким образом предшественника катализатора с образованием катализатора.
11. Способ синтеза углеводородов, который включает получение катализатора способом по п. 10 и контактирование водорода с монооксидом углерода при температуре выше 100°C и давлении, равном по меньшей мере 10 бар, в присутствии полученного таким образом катализатора с получением углеводородов и, возможно, продуктов окисления углеводородов.
12. Способ по п. 11, который представляет собой процесс Фишера-Тропша в слое суспензии с получением воскообразного продукта.
13. Способ по п. 11, который включает стадию гидропроцессинга для превращения углеводородов и, возможно, продуктов их окисления в жидкое топливо и/или химические продукты
14. Способ гидрирования, который включает получение катализатора способом по п. 10 и контактирование водорода и органического соединения с полученным таким образом катализатором для осуществления гидрирования органического соединения.
| статья Yun-Jo Lee, Jo-Yung Park, Ki-Won Jun et | |||
| al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Catal | |||
| Lett., Kluwer Academic Publishers-Plenum Publishers, NL, vol.130, no.1-2, p.198-203 | |||
| EP 510771 A1, 28.10.1992 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 5744419 A, 28.04.1998 | |||
| PARK S | |||

