Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения высокочистого празугрель гидрохлорида.
Уровень техники
Соединение формулы:
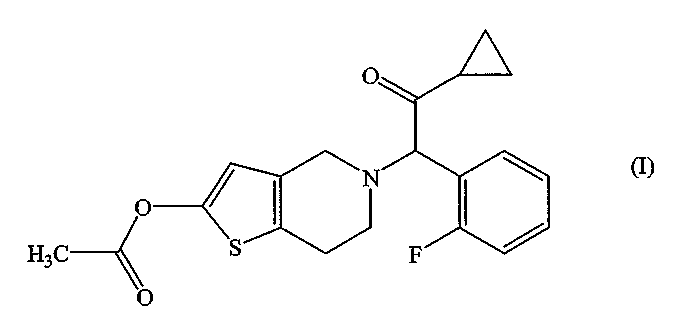
хорошо известно как празугрель. Известно, что празугрель и его фармацевтически приемлемые соли ингибируют агрегацию тромбоцитов и используются в качестве активного ингредиента лекарственных средств (в частности, антитромботических или антиэмболических средств) (патентный документ 1 или 2). Однако для использования празугреля или его фармацевтически приемлемых солей в качестве лекарственного средства необходим способ получения высокочистого празугреля или его фармакологически приемлемых солей.
Празугрель гидрохлорид, представленный формулой:
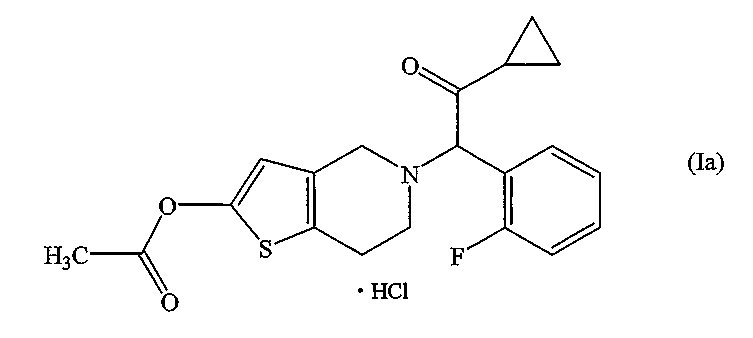
может быть получен следующим способом. В патентном документе 3 описаны стадии (i)-(iii), а в патентном документе 2 - стадия (iv). Однако ни в одном из этих патентных документов не описан побочный продукт САТР.
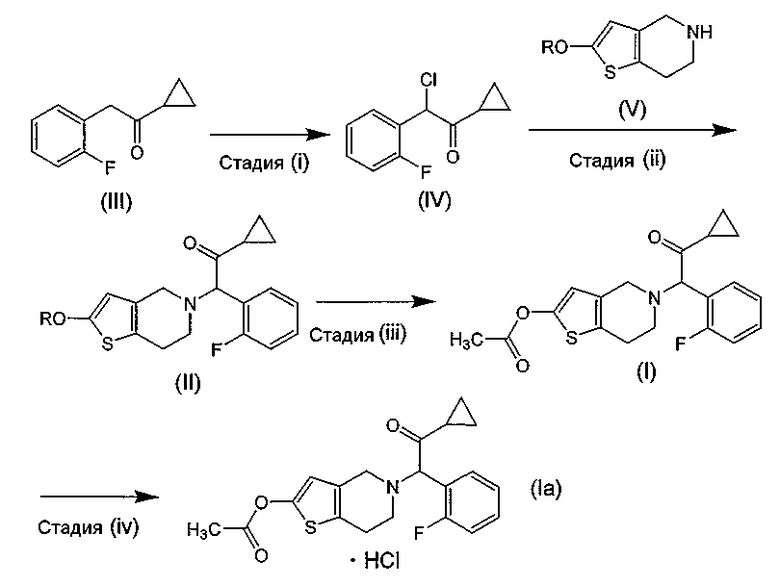
В этих формулах R означает защитную группу для гидроксильной группы.
Патентный документ 1: выложенный патент Японии № Hei 6-41139.
Патентный документ 2: выложенный патент Японии №2002-145883.
Патентный документ 3: международная публикация № WO 96/11203.
Описание изобретения
Проблемы, решаемые благодаря настоящему изобретению
Авторами настоящего изобретения найдено, что крупномасштабное производство празугрель гидрохлорида указанным выше способом ведет к образованию конечного продукта, загрязненного побочным продуктом САТР, который ранее известен не был.
Задачей настоящего изобретения является создание способа получения высокочистого празугрель гидрохлорида со сниженным содержанием побочных продуктов, таких как САТР.
Средства решения указанных проблем
В результате интенсивных исследований способа получения высокочистого празугрель гидрохлорида со сниженным содержанием побочных продуктов, таких как САТР, авторами настоящего изобретения обнаружено, что температуру реакции на стадии хлорирования, как на стадии (i) указанного выше способа, можно регулировать с целью снижения содержания побочного продукта САТР в празугрель гидрохлориде, являющемся целевым конечным соединением. Таким образом, было выполнено настоящее изобретение.
Что касается условий реакции на стадии хлорирования, стадии (i), в международной публикации № WO 96/11203, при описании справочных примеров 12-1 и 12-2 указано, что хлорирующий реагент «добавляли по каплям, поддерживая температуру жидкой среды ниже 5ºС. После того как температура жидкой среды постепенно повышалась до комнатной температуры (20°С), смеси позволяли взаимодействовать в течение 1,5 часов при перемешивании». Таким образом, ранее полагали, что температура реакции после добавления, необязательно по каплям, хлорирующего агента, предпочтительно, должна быть комнатной или выше. В настоящем изобретении, напротив, снижение содержания побочного продукта САТР в конечном целевом соединении - празугрель гидрохлориде - становится возможным благодаря регулированию на уровне низких величин температуры реакции после добавления, необязательно по каплям, хлорирующего агента, а также температуры во время добавления, необязательно по каплям, хлорирующего агента.
Настоящим изобретением обеспечивается способ получения празугрель гидрохлорида, характеризующийся регулированием температуры реакции на стадии (i), входящей в описанный выше способ, включающий стадии (i)-(iv); высокочистый празугрель гидрохлорид, получаемый этим способом; фармацевтическая композиция (в частности, профилактическое или терапевтическое средство для лечения заболеваний, вызванных тромбами или эмболами), содержащая в качестве активного компонента высокочистый празугрель гидрохлорид; применение высокочистого празугрель гидрохлорида для приготовления указанной выше фармацевтической композиции и способ профилактики или лечения заболеваний (в частности, тромбоза и эмболии), включающий введение теплокровным животным (в частности, человеку) указанной выше фармацевтической композиции, содержащей фармакологически эффективное количество высокочистого празугрель гидрохлорида.
Настоящее изобретение заключается в следующем.
(1) Способ получения празугрель гидрохлорида, включающий следующие стадии:
(i) хлорирование соединения (III) путем добавления к нему хлорирующего агента, необязательно по каплям, в растворителе;
(ii) взаимодействие полученного соединения (IV) с соединением (V) или его солью в растворителе в присутствии основания;
(iii) ацетилирование полученного соединения (II) путем взаимодействия реакции с ацетилирующим агентом в растворителе в присутствии основания и катализатора ацилирования; и
(iv) добавление к полученному соединению (I) в растворителе соляной кислоты с получением празугрель гидрохлорида,
отличающийся тем, что на стадии (i) температура во время добавления, необязательно по каплям, хлорирующего агента составляет от -20°С до 5°С и температура реакции после добавления, необязательно по каплям, хлорирующего агента составляет от -20°С до 5°С.
(2) Способ получения по п. (1), отличающийся тем, что на стадии (i) температура во время добавления, необязательно по каплям, хлорирующего агента составляет от -10°С до 5°С и температура реакции после добавления, необязательно по каплям, хлорирующего агента составляет от -10°С до 5°С.
(3) Способ получения по п. (1), отличающийся тем, что на стадии (i) температура во время добавления, необязательно по каплям, хлорирующего агента составляет от -5°С до 5°С и температура реакции после добавления, необязательно по каплям, хлорирующего агента составляет от -5°С до 5°С.
(4) Способ получения по любому из пп.(1)-(3), отличающийся тем, что температура последующей обработки после окончания реакции на стадии (i) составляет от -20°С до 15°С.
(5) Способ получения по любому из пп.(1)-(3), отличающийся тем, что температура последующей обработки после окончания реакции на стадии (i) составляет от -10°С до 15°С.
(6) Способ получения по любому из пп.(1)-(3), отличающийся тем, что температура последующей обработки после окончания реакции на стадии (i) составляет от 0°С до 15°С.
(7) Способ получения по любому из пп.(1)-(6), в котором хлорирующий агент представляет собой газообразный хлор.
(8) Способ получения по любому из пп.(1)-(7), в котором R означает группу общей формулы:
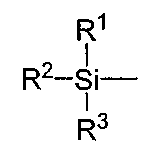
где R1, R2 и R3 независимо означают алкильную группу, содержащую от 1 до 10 атомов углерода, или арильную группу.
(9) Способ получения по п. (8), в котором R1, R2 и R3 независимо означают алкильную группу, содержащую от 1 до 5 атомов углерода, или фенильную группу.
(10) Способ получения по любому из пп.(1)-(7), в котором R означает трет-бутилдиметилсилильную группу.
(11) Способ получения по любому из пп.(1)-(10), отличающийся тем, что полученное соединение общей формулы (II) на стадии (ii) перекристаллизовывают из простых эфиров или нитрилов.
(12) Способ получения по любому из пп.(1)-(10), отличающийся тем, что полученное соединение общей формулы (II) на стадии (ii) перекристаллизовывают из ацетонитрила.
(13) Способ получения по любому из пп.(1)-(12), в котором ацетилирующий агент представляет собой уксусный ангидрид.
(14) Способ получения по любому из пп.(1)-(13), отличающийся тем, что получаемое на стадии (iii) соединение, соответствующее формуле (I), используют на следующей стадии (iv) без очистки.
(15) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,3% или менее, полученный способом получения по пп.(1)-(14).
(16) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,1% или менее, полученный способом получения по пп.(1)-(14).
(17) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,04% или менее, полученный способом получения по пп.(1)-(14).
(18) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,03% или менее, полученный способом получения по пп.(1)-(14).
(19) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,02% или менее, полученный способом получения по пп.(1)-(14).
(20) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,3% или менее.
(21) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,1% или менее.
(22) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,04% или менее.
(23) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,03% или менее.
(24) Празугрель гидрохлорид, характеризующийся содержанием САТР 0,02% или менее.
(25) Фармацевтическая композиция, содержащая в качестве активного компонента празугрель гидрохлорид по пп.(15)-(24).
(26) Профилактическое или терапевтическое средство, используемое для лечения у теплокровных животных заболеваний, вызванных тромбами или эмболами, содержащее в качестве активного компонента празугрель гидрохлорид по пп.(15)-(24).
(27) Профилактическое или терапевтическое средство, используемое для лечения у человека тромбоза или эмболии, содержащее в качестве активного компонента празугрель гидрохлорид по пп.(15)-(24).
В соответствии с настоящим изобретением «защитная группа для гидроксильной группы» не имеет определенных ограничений при условии, что она способна в ходе реакции постоянно защищать гидроксильную группу, в частности этот термин относится к защитной группе, которая может быть отщеплена в ходе такой химической стадии, как гидрогенолиз, гидролиз, электролиз и фотолиз. Защитная группа может, например, представлять собой алифатическую ацильную группу, включая алканоильную группу, такую как формил, ацетил, пропионил, бутирил, изобутирил, пентаноил, пивалоил, валерил, изовалерил, октаноил, нонаноил, деканоил, 3-метилнонаноил, 8-метилнонаноил, 3-этилоктаноил, 3,7-диметилоктаноил, ундеканоил, додеканоил, тридеканоил, тетрадеканоил, пентадеканоил, гексадеканоил, 1-метилпентадеканоил, 14-метилпентадеканоил, 13,13-диметилтетрадеканоил, гептадеканоил, 15-метилгексадеканоил, октадеканоил, 1-метилгептадеканоил, нонадеканоил, эйкозаноил или генайкозаноил, алкилкарбонильную группу, замещенную карбоксигруппой, такую как сукциноил, глутароил или адипоил, алкилкарбонильную группу, замещенную атомом(ами) галогена, такую как хлорацетил, дихлорацетил, трихлорацетил или трифторацетил, насыщенный циклический углеводород-карбонил, такой как циклопропилкарбонил, циклобутилкарбонил, циклопентилкарбонил, циклогексилкарбонил, циклогептилкарбонил или циклооктилкарбонил, алкилкарбонильную группу, замещенную низшей алкоксигруппой, такую как метоксиацетил, или ненасыщенную алкилкарбонильную группу, такую как (Е)-2-метил-2-бутенил; ароматическую ацильную группу, включая арилкарбонильную группу, такую как бензоил, α-нафтоил, β-нафтоил, пиридоил, тиеноил или фуроил, галогенарилкарбонильную группу, такую как 2-бромбензоил или 4-хлорбензоил, арилкарбонильную группу, замещенную низшей алкильной группой(ами), такую как 2,4,6-триметилбензоил или 4-толуоил, низшую алкоксилированную арилкарбонильную группу, такую как 4-анизоил, арилкарбонильную группу, замещенную карбоксигруппой, такую как 2-карбоксибензоил, 3-карбоксибензоил или 4-карбоксибензоил, нитрированную арилкарбонильную группу, такую как 4-нитробензоил или 2-нитробензоил, арилкарбонильную группу, замещенную низшим алкоксикарбонилом, такую как 2-(метоксикарбонил)бензоил, или арилкарбонильную группу, замещенную арилом, такую как 4-фенилбензоил; карбонилоксиалкильную группу, включая оксодиоксоленилметильную группу, такую как (5-метил-2-оксо-1,3-диоксолен-4-ил)метил или (5-фенил-2-оксо-1,3-диоксолен-4-ил)метил; остаток соли неполного эфира янтарной кислоты; остаток соли сложного эфира фосфорной кислоты; образующий сложный эфир остаток, такой как аминокислота; карбамоильную группу; карбамоильную группу, замещенную одной или двумя низшими алкильными группами; карбонилоксиалкилоксикарбонильную группу, такую как пивалоилоксиметилоксикарбонил; или силильную группу, такую как триметилсилил, триэтилсилил, трипропилсилил, триизопропилсилил, трет-бутилдиметилсилил или трет-бутилдифенилсилил. Из этих защитных групп предпочтительными являются силильные группы; более предпочтительна группа, изображаемая общей формулой:
где R1, R2 и R3 независимо друг от друга означают алкильную группу, включающую от 1 до 10 атомов углерода, или арильную группу и, предпочтительно, являются, независимо друг от друга, алкильной группой, включающей от 1 до 5 атомов углерода, или фенильной группой; и более предпочтительно, являются трет-бутилдиметилсилильной группой.
В соответствии с настоящим изобретением «алкильная группа, включающая от 1 до 10 атомов углерода» может представлять собой алкильную группу с прямой или разветвленной цепью, включающую от 1 до 10 атомов углерода, такую как, например, метил, этил, пропил (в т.ч. его изомер), бутил (в т.ч. каждый его изомер), пентил (в т.ч. каждый его изомер), гексил (в т.ч. каждый его изомер), гептил (в т.ч. каждый его изомер), октил (в т.ч. каждый его изомер), нонил (в т.ч. каждый его изомер) или децил (в т.ч. каждый его изомер). Предпочтительно, это алкильная группа, включающая от 1 до 5 атомов углерода; более предпочтительно, метильная группа, этильная группа, пропильная группа (в т.ч. изомер) или бутильная группа (в т.ч. каждый ее изомер); и еще более предпочтительно, это метильная группа или трет-бутильная группа.
В соответствии с настоящим изобретением «арильная группа» это, например, фенильная группа, ксилильная группа, бифенильная группа, нафтильная группа, антрильная группа или фенантрильная группа, и предпочтительно, это арильная группа, включающая от 6 до 8 атомов углерода, более предпочтительно, фенильная группа.
В молекуле соединения настоящего изобретения может присутствовать асимметрический атом углерода; могут существовать обуславливаемые его наличием оптические изомеры (включая диастереомеры), которые также составляют соединение настоящего изобретения.
В соответствии с настоящим изобретением соль соединения (V) может представлять собой соль неорганической кислоты, такую как гидрохлорид или сульфат; органический сульфонат, такой как п-толуолсульфонат или метансульфонат; или органический карбоксилат, такой как ацетат или пропионат. Соли неорганических кислот или органические сульфонаты являются предпочтительными, более предпочтительны гидрохлорид или п-толуолсульфонат.
В соответствии с настоящим изобретением «САТР» означает 2-ацетокси-5-[5-хлор-1-(2-фторфенил)-2-оксопентил]-4,5,6,7-тетрагидротиено[3,2-с]пиридин, представленный формулой:
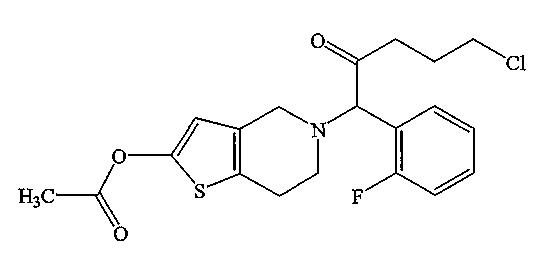
В соответствии с настоящим изобретением в САТР имеется асимметрический атом углерода и могут существовать обусловленные его наличием оптические изомеры; любой из этих изомеров и их смеси также входят в САТР, соответствующий настоящему изобретению.
Значение изобретения
В соответствии с настоящим изобретением может быть обеспечен высокочистый празугрель гидрохлорид со сниженным содержанием примесей, таких как побочный продукт САТР. В частности, настоящее изобретение обеспечивает возможность снижения содержания побочного продукта САТР по сравнению с другими похожими по структуре побочными продуктами.
Краткое описание чертежей
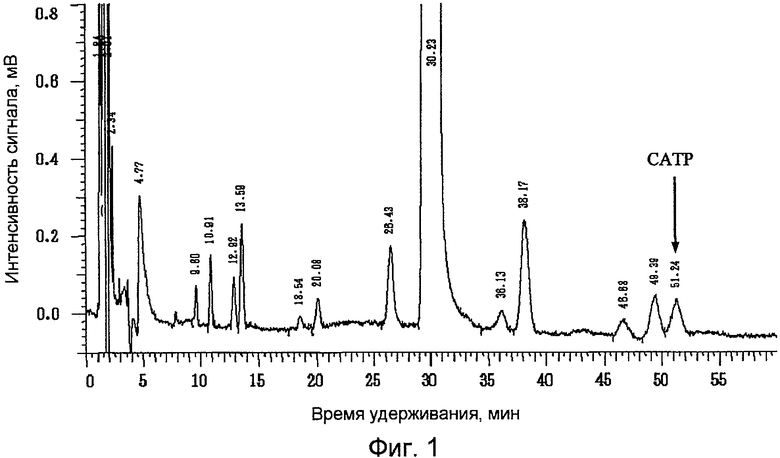
Фиг.1 представляет собой результат анализа методом жидкостной хроматографии празугрель гидрохлорида, полученного в примере 1;
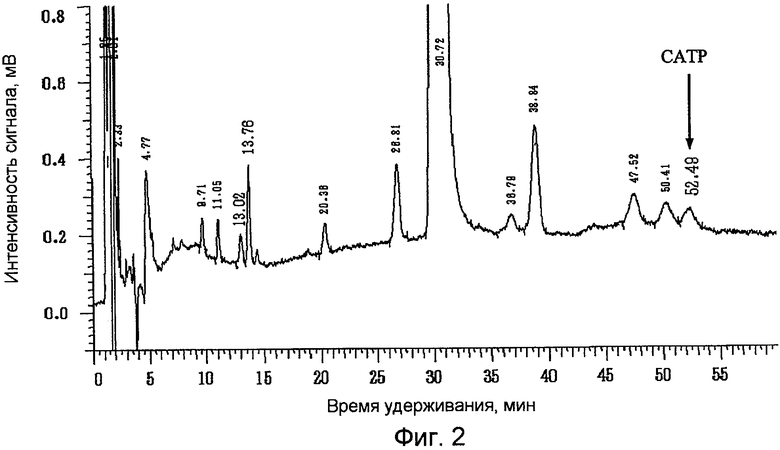
Фиг.2 представляет собой результат анализа методом жидкостной хроматографии празугрель гидрохлорида, полученного в примере 2;
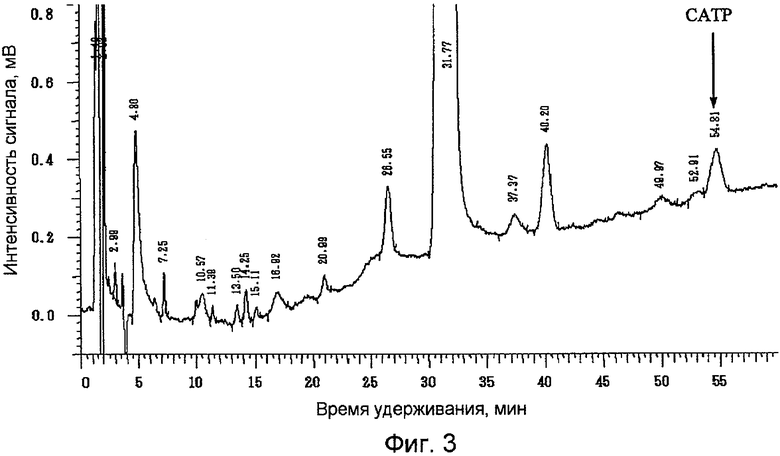
Фиг.3 представляет собой результат анализа методом жидкостной хроматографии празугрель гидрохлорида, полученного в справочном примере 1.
Описание предпочтительного варианта осуществления изобретения
Соединение (III), используемое на стадии (i) способа настоящего изобретения в качестве исходного материала, может быть получено способом, описанным в международной публикации № WO 96/11203.
Соединение (V), используемое на стадии (ii) способа настоящего изобретения в качестве исходного материала, может быть получено способом, описанным, например, в международной публикации № WO 96/11203.
Способ получения высокочистого празугрель гидрохлорида настоящего изобретения заключается в следующем.
Стадия (i)
Эта стадия представляет собой стадию хлорирования соединения (III) путем добавления к нему в растворителе, необязательно по каплям, хлорирующего реагента с получением соединения (IV).
Используемый на этой стадии хлорирующий агент может представлять собой, например, газообразный хлор или сульфурилхлорид; предпочтительным является газообразный хлор.
Используемый на этой стадии растворитель не имеет определенных ограничений при условии, что он до некоторой степени растворяет исходный материал и не ингибирует данную реакцию. Этот растворитель может представлять собой, например, эфирный растворитель, такой как тетрагидрофуран, диэтиловый эфир или диоксан; галогенированный растворитель, такой как дихлорметан или 1,2-дихлорэтан; ароматический углеводородный растворитель, такой как бензол, толуол или ксилол; нитрильный растворитель, такой как ацетонитрил, пропионитрил или бензонитрил; или амидный растворитель, такой как диметилформамид, диметилацетамид или диметилимидазолидон. Галогенсодержащие растворители являются предпочтительными, более предпочтителен дихлорметан.
Количество хлорирующего агента, используемое на этой стадии, обычно составляет от 0,5 до 3 молей, предпочтительно, от 0,8 до 2 молей, более предпочтительно, от 0,9 до 1,5 молей на 1 моль соединения (III).
Во время добавления, необязательно по каплям, хлорирующего агента на этой стадии температура реакционного раствора изменяется в зависимости от реагента, растворителя или т.п.; однако типичная температура составляет от -20ºС до 5ºС, предпочтительно, от -10ºС до 5ºС, более предпочтительно, от -5ºС до 5ºС.
Время добавления, необязательно по каплям, хлорирующего агента на этой стадии изменяется в зависимости от типа и количества хлорирующего агента. Однако обычно оно составляет от 30 минут до 24 часов, предпочтительно, от 1 часа до 12 часов, более предпочтительно, от 1 часа до 6 часов.
Температура реакции после добавления, необязательно по каплям, хлорирующего агента на этой стадии изменяется в зависимости от реагента, растворителя и т.п. Однако обычно она составляет от -20ºС до 5ºС, предпочтительно, от -10ºС до 5ºС, более предпочтительно, от -5ºС до 5ºС.
Время реакции после добавления, необязательно по каплям, хлорирующего агента на этой стадии изменяется в зависимости от реагента, растворителя, температуры реакции и т.п. Однако обычно оно составляет от 30 минут до 12 часов, предпочтительно, от 1 часа до 6 часов, более предпочтительно, от 1 часа до 3 часов.
После завершения реакции на этой стадии соединение (IV) может быть выделено способом, обычно используемым в области органического синтеза. Реакционная жидкость также может быть непосредственно использована на следующей стадии (ii) без выделения соединения (IV).
Температура последующей обработки после окончания реакции на этой стадии обычно составляет от -20ºС до 15ºС, предпочтительно, от -10ºС до 15ºС, более предпочтительно, от 0ºС до 15ºС.
Стадия (ii)
Эта стадия представляет собой стадию, включающую получение соединения (II) путем взаимодействия соединения (IV) с соединением (V) или его солью в растворителе в присутствии основания.
Количество соединения (IV) на этой стадии обычно составляет от 0,5 до 3 молей, предпочтительно, от 0,8 до 2 молей, более предпочтительно, от 0,9 до 1,2 моля на 1 моль соединения (V).
Используемый на этой стадии растворитель не имеет определенных ограничений при условии, что он до некоторой степени растворяет исходный материал и не ингибирует данную реакцию. Этот растворитель может представлять собой, например, эфирный растворитель, такой как тетрагидрофуран, диэтиловый эфир или диоксан; галогенированный растворитель, такой как дихлорметан или 1,2-дихлорэтан; ароматический углеводородный растворитель, такой как бензол, толуол или ксилол; нитрильный растворитель, такой как ацетонитрил, пропионитрил или бензонитрил; или амидный растворитель, такой как диметилформамид, диметилацетамид или диметилимидазолидон. Эфирные растворители, галогенсодержащие растворители, нитрильные растворители или амидные растворители являются предпочтительными, более предпочтительны тетрагидрофуран, дихлорметан, ацетонитрил или диметилацетамид.
Используемое на этой стадии основание не имеет определенных ограничений. Предпочтительны третичные амины, например триалкилмоноамины, такие как триэтиламин, трибутиламин или диизопропилэтиламин; или триалкилдиамины, такие как диазабициклооктан, диазабициклоундецен или тетраметилэтилдиамин, более предпочтительны триалкилмоноамины, еще более предпочтительны триэтиламин, трибутиламин или диизопропилэтиламин.
Количество основания, используемого на этой стадии, обычно составляет от 0,5 до 3 молей, предпочтительно, от 0,5 до 2 молей, более предпочтительно, от 0,7 до 1,5 молей на 1 моль соединения (V).
На этой стадии промотирующий эффект, как предполагается, оказывает наличие в реакционной системе аммониевой соли или четвертичной аммониевой соли.
Промотирующие реакцию добавки могут представлять собой, например, четвертичные аммониевые соли, в т.ч. галогениды тетраалкиламмония с алкильными группами, содержащими от 1 до 20 атомов углерода, такие как хлорид тетраметиламмония, бромид тетраметиламмония, хлорид тетраэтиламмония, бромид тетраэтиламмония, хлорид тетрабутиламмония, бромид тетрабутиламмония, или галогениды триалкилмонобензиламмония с алкильными группами, содержащими от 1 до 20 атомов углерода, такие как хлорид триметилбензиламмония или хлорид триэтилбензиламмония; бромиды щелочных металлов, в т.ч. бромид лития, бромид натрия, бромид калия или бромид цезия; или йодиды щелочных металлов, в т.ч. йодид лития, йодид натрия, йодид калия или йодид цезия. Предпочтительны бромид тетраэтиламмония, бромид тетрабутиламмония или йодид натрия.
Количество промотирующей реакцию добавки, используемой на этой стадии, обычно составляет от 0,01 до 5 молей, предпочтительно, от 0,1 до 2 молей на 1 моль соединения (VI) для четвертичных аммониевых солей и, обычно, от 0,001 до 0,6 молей, предпочтительно, от 0,01 до 0,5 молей на 1 моль соединения (VI) для бромидов щелочных металлов или йодидов щелочных металлов.
Температура реакции на этой стадии изменяется в зависимости от используемого реагента, растворителя и т.п. Однако обычно она составляет от -20ºС до 100ºС, предпочтительно, от -10ºС до 70ºС, более предпочтительно, от 0ºС до 60ºС.
Время реакции на этой стадии изменяется в зависимости от реагента, растворителя, температуры реакции и т.п. Однако обычно оно составляет от 30 минут до 24 часов, предпочтительно, от 1 часа до 12 часов, более предпочтительно, от 1 часа до 10 часов.
После завершения реакции на этой стадии соединение (II) может быть выделено способом, обычно используемым в области органического синтеза. Реакционная жидкость также может быть непосредственно использована на следующей стадии (iii) без выделения соединения (II). Однако предпочтительно отделить соединение (II) и очистить его путем перекристаллизации. Благодаря этому дополнительно уменьшается содержание побочного продукта САТР в празугрель гидрохлориде, являющемся конечным продуктом настоящего изобретения, тогда можно ожидать получения высокочистого празугрель гидрохлорида.
Растворитель, используемый для перекристаллизации соединения (II), не имеет определенных ограничений при условии, что он до некоторой степени растворяет соединение (II) и не вступает с соединением (II) в реакцию. Этот растворитель может представлять собой, например, эфирный растворитель, такой как тетрагидрофуран, диэтиловый эфир или диоксан; галогенированный растворитель, такой как дихлорметан или 1,2-дихлорэтан; ароматический углеводородный растворитель, такой как бензол, толуол или ксилол; нитрильный растворитель, такой как ацетонитрил, пропионитрил или бензонитрил; или амидный растворитель, такой как диметилформамид, диметилацетамид или диметилимидазолидон. Эфирные растворители или нитрильные растворители являются предпочтительными, более предпочтителен ацетонитрил.
Температура в ходе перекристаллизации, обычно, составляет от 30ºС до 80ºС, предпочтительно, от 40ºС до 70ºС, более предпочтительно, от 40ºС до 60ºС. После растворения раствор постепенно охлаждают. Предпочтительно, чтобы к нему был добавлен слабый растворитель (предпочтительно, вода) при 30ºС, затем раствор был подвергнут охлаждению до температуры от -5ºС до 10ºС и перемешиванию в течение от 1 часа до 6 часов. Также, если необходимо, можно добавить затравочный кристалл.
Стадия (iii)
Эта стадия представляет собой стадию, включающую ацетилирование соединения (II) путем осуществления его реакции с ацетилирующим реагентом в растворителе в присутствии основания и катализатора ацилирования с целью получения соединения (I).
Используемый на этой стадии катализатор ацилирования может представлять собой, например, 4-диалкиламинопиридин, такой как 4-диметиламинопиридин, 4-диэтиламинопиридин или 4-дипропиламинопиридин, и предпочтительно, является 4-диметиламинопиридином.
Количество катализатора ацилирования, используемое на этой стадии, обычно составляет от 0,1 до 10 мол.% на 1 моль соединения (II), катализатор может быть использован в избытке.
Используемый на этой стадии ацетилирующий агент может представлять собой, например, уксусный ангидрид или ацетилхлорид и, предпочтительно, является уксусным ангидридом.
Количество уксусного ангидрида, используемое на этой стадии, обычно составляет от 1 до 10 молей, предпочтительно, от 1 до 5 молей на 1 моль соединения (II).
Используемый на этой стадии растворитель не имеет определенных ограничений при условии, что он до некоторой степени растворяет исходный материал и не ингибирует данную реакцию. Этот растворитель может представлять собой, например, эфирный растворитель, такой как тетрагидрофуран, диэтиловый эфир или диоксан; галогенированный растворитель, такой как дихлорметан или 1,2-дихлорэтан; ароматический углеводородный растворитель, такой как бензол, толуол или ксилол; нитрильный растворитель, такой как ацетонитрил, пропионитрил или бензонитрил; или амидный растворитель, такой как диметилформамид, диметилацетамид или диметилимидазолидон. Эфирные растворители, галогенсодержащие растворители, нитрильные растворители или амидные растворители являются предпочтительными, более предпочтительны тетрагидрофуран, дихлорметан, ацетонитрил или диметилацетамид.
Используемое на этой стадии основание не имеет определенных ограничений. Предпочтительны третичные амины, например, триалкилмоноамины, такие как триэтиламин, трибутиламин или диизопропилэтиламин; или триалкилдиамины, такие как диазабициклооктан, диазабициклоундецен или тетраметилэтилдиамин, более предпочтительны триалкилмоноамины, еще более предпочтителен триэтиламин.
Количество основания, используемого на этой стадии, обычно составляет от 1 до 10 молей, предпочтительно, от 1 до 5 молей на 1 моль соединения (II).
Температура реакции на этой стадии изменяется в зависимости от реагента, растворителя и т.п. Однако обычно она составляет от -50ºС до 50ºС, предпочтительно, от -30ºС до 30ºС, более предпочтительно, от -20ºС до 20ºС.
Время реакции на этой стадии изменяется в зависимости от реагента, растворителя, температуры реакции и т.п. Однако обычно оно составляет от 30 минут до 24 часов, предпочтительно, от 1 часа до 12 часов, более предпочтительно, от 1 часа до 6 часов.
После завершения реакции на этой стадии соединение (I) может быть выделено способом, обычно используемым в области органического синтеза. Реакционная жидкость также может быть непосредственно использована на следующей стадии (iv) без выделения соединения (I).
Стадия (iv)
Эта стадия представляет собой стадию, включающую получение празугрель гидрохлорида путем добавления к соединению (I) в растворителе соляной кислоты, необязательно по каплям.
На этой стадии добавление соляной кислоты, необязательно по каплям, может быть осуществлено путем добавления кислоты по каплям, или добавления ее сразу, или двумя, или несколькими отдельными порциями.
Используемый на этой стадии растворитель не имеет определенных ограничений при условии, что он до некоторой степени растворяет исходный материал и не ингибирует данную реакцию. Этот растворитель может представлять собой, например, алифатический углеводород, такой как гексан, циклогексан, гептан или лигроин, или петролейный эфир; ароматический углеводород, такой как бензол, толуол или ксилол; галогенированный углеводород, такой как дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, хлорбензол или дихлорбензол; простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диметиловый эфир диэтиленгликоля; кетон, такой как ацетон, метилэтилкетон или диэтилкетон; сложный эфир, такой как этилацетат, пропилацетат или бутилацетат; карбоновую кислоту, такую как уксусная кислота или пропионовая кислота; нитрил, такой как ацетонитрил или пропионитрил. Простые эфиры, кетоны, сложные эфиры, карбоновые кислоты или нитрилы являются предпочтительными; более предпочтительны тетрагидрофуран, диоксан, ацетон, метилэтилкетон, этилацетат, уксусная кислота или ацетонитрил; еще более предпочтительны тетрагидрофуран, диоксан, уксусная кислота или ацетон; наиболее предпочтителен ацетон.
Температура реакции на этой стадии изменяется в зависимости от реагента, растворителя и т.п. Однако обычно она составляет от -20ºС до 100ºС, предпочтительно, от 0ºС до 70ºС, более предпочтительно, от 30ºС до 60ºС, наиболее предпочтительно, от 40ºС до 55ºС.
Время реакции на этой стадии изменяется в зависимости от реагента, растворителя, температуры реакции и т.п. Однако обычно оно составляет от 5 минут до 10 часов, предпочтительно, от 10 минут до 5 часов.
Предпочтительный вариант осуществления данной стадии представляет собой способ, включающий растворение соединения (I) в ацетоне, добавление по каплям в этот раствор половины необходимого количества (обычно необходимое количество эквимолярно тиенопиридиновой форме) концентрированной соляной кислоты при температуре от 0ºС до 70ºС (предпочтительно, от 35ºС до 60ºС) за время от 2 минут до 10 минут, если нужно, добавление затравочного кристалла и последующую реакцию при той же температуре, осуществляемую в течение от 30 минут до 2 часов, затем добавление по каплям оставшегося необходимого количества концентрированной соляной кислоты за время от 30 минут до 2 часов и последующую реакцию при температуре от 0ºС до 70ºС (предпочтительно, от 25ºС до 55ºС), осуществляемую в течение от 1 часа до 3 часов.
После окончания реакции на этой стадии празугрель гидрохлорид настоящего изобретения отбирают из реакционной смеси обычным способом. Например, требуемое соединение получают путем отфильтровывания осажденных кристаллов после завершения реакции или отгонки растворителя после завершения реакции. Полученное целевое соединение, если нужно, может быть подвергнуто дальнейшей очистке обычным способом, например путем перекристаллизации, осаждения или хроматографически.
Празугрель гидрохлорид настоящего изобретения может быть оставлен на воздухе или перекристаллизован с целью поглощения воды, в результате чего он абсорбирует воду или превращается в гидрат; содержащее воду соединение также представляет собой празугрель гидрохлорид настоящего изобретения. Кроме того, его сольват, содержащий любое количество растворителя, также представляет собой празугрель гидрохлорид настоящего изобретения.
Содержание САТР в празугрель гидрохлориде измеряют методом жидкостной хроматографии и выражают как процентное содержание по площади (%), отражающее содержание САТР в свободном празугреле.
Содержание САТР в высокочистом празугрель гидрохлориде настоящего изобретения обычно составляет 0,3% или менее, предпочтительно, 0,1% или менее, более предпочтительно, 0,04% или менее, еще более предпочтительно, 0,03% или менее, особенно предпочтительно, 0,02% или менее.
Степень чистоты празугрель гидрохлорида, т.е. содержание празугреля, может быть измерено, как описано в отношении содержания САТР.
Степень чистоты высокочистого празугрель гидрохлорида в соответствии с настоящим изобретением составляет обычно 95% или более, предпочтительно, 97% или более, более предпочтительно, 99% или более.
Высокочистый празугрель гидрохлорид, получаемый в настоящем изобретении, обладает великолепной всасываемостью в ротовой полости, способностью активировать метаболизм и ингибировать агрегацию тромбоцитов, слабой токсичностью и к тому же хорошей устойчивостью при хранении и манипулировании, следовательно, он применим в качестве лекарственного средства (предпочтительно, профилактического или терапевтического средства для лечения заболеваний, вызванных тромбами или эмболами (в частности, терапевтического средства), более предпочтительно, профилактического или терапевтического средства для борьбы с тромбозом и эмболией (в частности, терапевтического средства)). Кроме того, предпочтительно использование этого лекарственного средства для теплокровных животных, более предпочтительно, для человека.
При использовании в качестве терапевтического или профилактического средства для лечения этих заболеваний высокочистый празугрель гидрохлорид настоящего изобретения может быть введен сам по себе или в смеси с фармацевтически приемлемым эксципиентом, разбавителем и т.п. перорально в форме таблеток, капсул, гранул, порошков, сиропов и т.п. или парентерально в форме инъекций, суппозиториев и т.п.
Эти композиции получают хорошо известными способами, используя добавки, в т.ч. наполнители (которые могут быть, например, органическими наполнителями (например, производные сахаров, такие как лактоза, сахароза, глюкоза, маннитол или сорбитол; производные крахмала, такие как кукурузный крахмал, картофельный крахмал, α-крахмал или декстрин; производные целлюлозы, такие как кристаллическая целлюлоза; гуммиарабик; декстран; или пуллулан); или неорганические наполнители (например, производные солей кремниевой кислоты, такие как легкая безводная кремниевая кислота, искусственные силикат алюминия, силикат кальция или метасиликат алюминат магния; фосфаты, такие как кислый фосфат кальция; карбонаты, такие как карбонат кальция; или сульфаты, такие как сульфат кальция)), смазывающие вещества (которые могут представлять собой, например, стеариновую кислоту; металлические соли стеариновой кислоты, такие как стеарат кальция или стеарат магния; тальк; воски, такие как пчелиный воск или спермацет; борную кислоту; адипиновую кислоту; сульфаты, такие как сульфат натрия; гликоль; фумаровую кислоту; бензоат натрия; D,L-лейцин; лаурилсульфаты, такие как лаурилсульфат натрия или лаурилсульфат магния; силикаты, такие как кремниевый ангидрид или гидросиликат; или производные крахмала, указанные выше), связующие (которые могут представлять собой, например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон, макрогол или соединения, подобные указанным выше вспомогательным веществам), дезинтеграторы (которые могут представлять собой, например, производные целлюлозы, такие как имеющая низшие заместители гидроксипропилцеллюлоза, карбоксиметилцеллюлоза, кальций-карбоксиметилцеллюлоза или имеющая внутренние поперечные связи натрий-карбоксиметилцеллюлоза; химически модифицированные крахмалы/целлюлозу, такие как карбоксиметилкрахмал, натриевая соль карбоксиметилкрахмала или поперечносшитый поливинилпирролидон; или производные крахмала, как указано выше), эмульгаторы (которые могут представлять собой, например, коллоидные глины, такие как бентонит или вигум; гидроксиды металлов, такие как гидроксид магния или гидроксид алюминия; анионогенные поверхностно-активные вещества, такие как лаурилсульфат натрия или стеарат кальция; катионогенные поверхностно-активные вещества, такие как бензалконий хлорид; неионные поверхностно-активные вещества, такие как полиоксиэтиленалкиловый эфир, полиоксиэтиленовый эфир сорбитана и жирной кислоты или эфир сахарозы и жирной кислоты), стабилизаторы (которые могут представлять собой, например, пара-гидроксибензойные эфиры, такие как метилпарабен или пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт или фенилэтиловый спирт; бензалконийхлорид; фенолы, такие как фенол или крезол; тимеросал; дегидроуксусную кислоту или сорбиновую кислоту), модификаторы (которые могут быть, например, повсеместно используемыми подсластителями, подкислителями или веществами, корректирующими вкус и запах) и разбавители.
Их используемое количество может изменяться в зависимости от симптомов, возраста и т.п., возможно введение взрослым людям от одного до семи раз в день перорально с нижним пределом одной дозы 0,1 мг (предпочтительно, 1 мг) и верхним пределом одной дозы 1000 мг (предпочтительно, 500 мг) или внутривенно с нижним пределом одной дозы 0,01 мг (предпочтительно, 0,1 мг) и верхним пределом одной дозы 500 мг (предпочтительно, 250 мг) в зависимости от симптомов. Таким образом, количество в одной дозе для человека весом 60 кг составляет 0,0017 мг/кг (предпочтительно, 0,017 мг/кг) как нижний предел и 17 мг/кг (предпочтительно, 8,3 мг/кг) как верхний предел для перорального введения и 0,00017 мг/кг (предпочтительно, 0,0017 мг/кг) как нижний предел и 8,3 мг/кг (предпочтительно, 4,2 мг/кг) как верхний предел для внутривенного введения.
ПРИМЕРЫ
Ниже настоящее изобретение более подробно описано со ссылкой на примеры, справочные примеры и тестовые примеры. Однако ими настоящее изобретение не ограничивается.
Пример 1
(1) 2-Фтор-α-циклопропилкарбонилбензилхлорид (стадия (i))
Смесь 100 г циклопропил-2-фторбензилкетона и 886 г дихлорметана перемешивали при охлаждении льдом с целью получения перемешанного раствора. Полученный перемешиваемый раствор продували 3,98 г (0,1 эквивалент) газообразного хлора в течение 20 минут, поддерживая температуру раствора не выше 5ºС, затем перемешивали в течение 0,5 часа при той же температуре. Затем раствор продували 39,8 г (1 эквивалент) газообразного хлора в течение 220 минут при той же температуре, реакцию проводили при перемешивании в течение одного часа при той же температуре.
После завершения реакции в полученный реакционный раствор при перемешивании добавили по каплям 236 г 3% водного раствора тиосульфата натрия, поддерживая температуру раствора не выше 15ºС. После добавления раствор по каплям перемешивали 10 минут, затем жидкие фазы разделили. Полученный органический слой промыли последовательно 589 г предварительно охлажденного 8% водного раствора гидрокарбоната натрия и 168 г предварительно охлажденной воды, затем концентрировали при пониженном давлении с получением 145 г соединения, указанного в заголовке (содержание чистого вещества 95,4 г, выход 80%), в маслянистой форме. Во время осуществления этих операций температуру раствора поддерживали в диапазоне от 0ºС до 15ºС.
(2) 2-(трет-Бутилдиметилсилилокси)-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин (стадия ii))
К смеси 115 г 5,6,7,7а-тетрагидро-4Н-тиено[3,2-с]пиридин-2-он п-толуолсульфоната, 60,7 г трет-бутилдиметилхлорсилана и 277 г дихлорметана добавили 40,7 г триэтиламина, затем смесь перемешивали при 25ºС 1 час с получением перемешанного раствора. К этому перемешанному раствору добавили 78,1 г 2-фтор-α-циклопропилкарбонилбензилхлорида, полученного в пункте (1), 70,8 г триэтиламина и 1,57 г йодида натрия, реакцию между которыми затем осуществляли при перемешивании в течение 1 часа при 45ºС и еще 5 часов при 52ºС.
После завершения реакции в полученный реакционный раствор добавили общее количество фосфатного буферного раствора, подготовленного путем добавления дистиллированной воды к 9,50 г КН2РО4 и 0,95 г Na2HPO4·12Н2О до общего веса 358 г, затем жидкие фазы разделили, после чего водный слой подвергли обратимой экстракции 116 г дихлорметана. Полученные органические слои объединили и концентрировали при пониженном давлении, пока объем остатка не достиг 218 мл. К нему добавили 476 г ацетонитрила, после чего полученную смесь концентрировали при пониженном давлении, пока объем остатка не достиг 517 мл. К полученному остатку добавили 238 г ацетонитрила и перемешивали при 30ºС в течение 30 минут. Затем добавили 122 г воды и перемешивали при 0ºС в течение 3 часов. Осевшие кристаллы отделили фильтрацией, промыли 69,0 г предварительно охлажденного ацетонитрила и высушили при пониженном давлении, получив 131 г соединения, указанного в заголовке.
(3) 2-Ацетокси-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин (стадия iii))
К смеси 15,0 г 2-(трет-бутилдиметилсилилокси)-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридина, полученного в пункте (2), 5,10 г триэтиламина, 41,3 мг 4-диметиламинопиридина и 75 г ацетонитрила по каплям добавили 3,90 г раствора ацетонитрила, в котором было растворено 4,13 г уксусного ангидрида, реакцию в полученной смеси осуществляли при перемешивании при 0ºС в течение 1 часа.
После завершения реакции в полученный реакционный раствор добавили 50,6 г холодной воды и перемешивали при -15ºС 30 минут. Осевшие кристаллы отделили фильтрацией, промыли перемешанным раствором 15,1 г ацетонитрила и 11,9 г воды и высушили при пониженном давлении, получив 10,8 г соединения, указанного в заголовке.
Температура плавления от 122 до 124ºС.
(4) Гидрохлорид 2-ацетокси-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридина (стадия (iv))
К 8,00 г 2-ацетокси-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридина, полученного в пункте (3), и 398 мг активированной глины добавили 43 г ацетона, затем полученную смесь перемешивали при 32ºС. Реакционный раствор отфильтровали, остаток промыли 4,41 г ацетона, затем к этому раствору при 52ºС по каплям добавляли 1,12 г 36%-й концентрированной соляной кислоты в течение одной минуты. В качестве затравочных кристаллов добавили 238 мг кристаллов В2, полученных способом, описанным в выложенном патенте Японии № 2002-145883, после чего перемешивали при той же температуре один час. Кроме того, по каплям добавили 1,07 г 36%-й концентрированной соляной кислоты в течение одного часа, затем раствор перемешивали 2 часа при 40ºС и еще 1 час при 30ºС. Осевшие кристаллы отделили фильтрацией, промыли 15,8 г ацетона, высушили при пониженном давлении при 50ºС в течение 5 часов, получив 8,03 г соединения, указанного в заголовке.
Температура плавления от 194 до 197ºС.
Пример 2
(1) 2-(трет-Бутилдиметилсилилокси)-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин (стадия ii))
К 40,0 г соединения (II), не подвергнутого перекристаллизации, полученного в примере 1(2), добавили 252 г ацетонитрила, после чего перемешивали при 50ºС в течение 10 минут и охладили до 30ºС. После этого к раствору по каплям за 30 минут добавили 40 г воды при той же температуре, затем раствор охладили до 0ºС и перемешивали при той же температуре в течение 3 часов. Осевшие кристаллы отделили фильтрацией, промыли 30 г предварительно охлажденного ацетонитрила, высушили при пониженном давлении, получив 37,6 г соединения, указанного в заголовке.
(2) 2-Ацетокси-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин (стадия iii))
Используя 22,5 г 2-(трет-бутилдиметилсилилокси)-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридина, полученного в пункте (1), осуществляли реакцию и последующую обработку, как описано в примере 1(3), получив 16,4 г соединения, указанного в заголовке.
Температура плавления от 122 до 124ºС.
(3) Гидрохлорид 2-ацетокси-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридина (стадия (iv))
Используя 8,00 г 2-ацетокси-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридина, полученного в пункте (2), осуществляли реакцию и последующую обработку, как описано в примере 1(4), получив 8,01 г соединения, указанного в заголовке.
Температура плавления от 192 до 196ºС.
Сравнительный пример 1
(1) 2-Фтор-α-циклопропилкарбонилбензилхлорид (стадия (i))
Смесь 100 г циклопропил-2-фторбензилкетона и 886 г дихлорметана перемешивали при охлаждении льдом с целью получения перемешанного раствора. Полученный перемешанный раствор продували 3,98 г (0,1 эквивалент) газообразного хлора в течение 20 минут, поддерживая температуру раствора не выше 5ºС, затем перемешивали в течение 0,5 часа при той же температуре. Затем раствор продували 39,8 г (1 эквивалент) газообразного хлора в течение 220 минут при той же температуре, затем температуру раствора постепенно увеличили до 20ºС, реакцию проводили при перемешивании в течение одного часа.
После завершения реакции в полученный реакционный раствор при перемешивании добавили по каплям 500 мл предварительно охлажденной воды, затем раствор перемешивали 10 минут, жидкие фазы разделили. Полученный органический слой промыли последовательно 833 мл насыщенного водного раствора гидрокарбоната натрия и 333 мл воды, затем концентрировали при пониженном давлении с получением 129 г соединения, указанного в заголовке (содержание чистого вещества 96,1 г, выход 81%), в маслянистой форме.
(2) Гидрохлорид 2-ацетокси-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридина (стадии (ii)-(iv))
Используя 105 г (содержание чистого вещества 78,3 г) 2-фтор-α-циклопропилкарбонилбензилхлорида, полученного в пункте (1), осуществляли реакцию и последующую обработку, как описано в примере 1(2)-(4), получив 8,10 г соединения, указанного в заголовке.
Температура плавления от 194 до 196ºС.
Справочный пример 2
(Производство стандарта загрязняющей примеси САТР)
(1) 5-Хлор-1-(2-фторфенил)пентан-2-он
К 5,00 г циклопропил-2-фторбензилкетона добавили 25 мл 36%-ной концентрированной соляной кислоты, после чего раствор перемешивали при 100ºС в течение 2,5 часов. После завершения реакции реакционный раствор охладили и добавили к нему 50 мл воды и 50 мл дихлорметана с целью разделения фаз. Полученный органический слой промыли 50 мл насыщенного водного раствора гидрокарбоната натрия, высушили безводным сульфатом магния, после чего концентрировали при пониженном давлении, получив 6,70 г соединения, указанного в заголовке, в маслянистой форме.
(2) 1,5-Дихлор-1-(2-фторфенил)пентан-2-он
К 9,44 г 5-хлор-1-(2-фторфенил)пентан-2-она, полученного, как описано в пункте (1), добавили 63 мл дихлорметана, через полученный раствор продували 119 мл газообразного хлора в течение 1 минуты, поддерживая температуру раствора равной 15ºС, затем раствор перемешивали при той же температуре 0,5 часа. Кроме того, этот раствор продували 1,19 л газообразного хлора при той же температуре в течение 10 минут, после чего раствор перемешивали при той же температуре в течение 1,5 часов с целью осуществления реакции.
После завершения реакции в полученный реакционный раствор добавили 22 мл 3%-го водного раствора сульфита натрия для разделения водных фаз. Полученный органический слой последовательно промыли 56 мл 8%-го водного раствора гидрокарбоната натрия и 16 мл воды, высушили безводным сульфатом магния, после чего концентрировали при пониженном давлении. Полученный остаток подвергли перегонке при пониженном давлении, получив 3,80 г фракции, содержащей требуемое соединение (100ºС-105ºС/48 Па). Кроме того, эту фракцию очистили на хроматографической колонке, заполненной силикагелем (элюент: н-гексан/этилацетат = 19/1 (об./об.)), получив 1,21 г соединения, указанного в заголовке.
(3) 2-Ацетокси-5-(5-хлор-1-(2-фторфенил)-2-оксопентил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин
Используя 1,21 г 1,5-дихлор-1-(2-фторфенил)пентан-2-она, полученного в пункте (2), осуществляли реакцию и последующую обработку, как описано в примере 1 (2) и (3), получив 1,71 г неочищенного материала, содержащего соединение, указанное в заголовке. Кроме того, этот материал очистили на хроматографической колонке, заполненной силикагелем (элюент: н-гексан/этилацетат = 5/1 → 3/1 (об./об.)), получив 0,77 г соединения, указанного в заголовке, в маслянистой форме.
Масс-спектр (Cl, масса/заряд): 410 [M+H]+.
1Н ЯМР спектр (400 МГц, CDCl3) δ м.д.: 1,97-2,05 (м, 2H), 2,26 (с, 3H), 2,66-2,76 (м, 3H), 2,79 (т, J=5,4 Гц, 2H), 2,85-2,90 (м, 1H), 3,43-3,59 (м, 4H), 4,74 (с, 1H), 6,25 (с, 1H), 7,10-7,20 (м, 2H), 7,31-7,36 (м, 1H), 7,42-7,47 (м, 1H).
Тестовый пример 1
(Способ измерения содержания празугреля и САТР в празугрель гидрохлориде)
Содержание празугреля и САТР в празугрель гидрохлориде измерили, как описано ниже.
В перемешанном растворе ацетонитрил-вода (7:3) растворили 150 мг празугрель гидрохлорида до 100 мл. При указанных далее условиях 10 мкл этого раствора подвергли анализу методом жидкостной хроматографии.
Условия измерения (жидкостная хроматография)
Детектор: ультрафиолетовый абсорбциометр (длина волны 240 нм)
Аналитическая колонка: Cadenza CD-C18, внутренний диаметр 4,6 мм, длина 15 см, размер частиц 3 мкм
Защитная колонка: отсутствует
Температура колонки: 40ºС
Подвижная фаза: 0,01 моль/л водный раствор дигидрофосфата калия:тетрагидрофуран:ацетонитрил в соотношении 13:5:2 (об./об./об.)
Расход потока: 1,0 мл/мин
(Содержание САТР в празугрель гидрохлориде)
Содержание празугреля и САТР выражено в процентах по площади (%) при измерении описанным выше методом жидкостной хроматографии. Результаты жидкохроматографического анализа празугрель гидрохлорида, полученного в примерах 1 и 2 и справочном примере 1, представлены на фиг.1, 2 и 3 соответственно.
Содержание САТР в конечном продукте - празугрель гидрохлориде - было значительно ниже в примерах 1 и 2, в которых добавление газообразного хлора и последующую реакцию на стадии (i) осуществляли при более низкой температуре, чем в справочном примере 1, в котором реакцию после добавления газообразного хлора осуществляли при комнатной температуре. Кроме того, содержание САТР в конечном продукте - празугрель гидрохлориде - было ниже в примере 2, в котором 2-(трет-бутилдиметилсилилокси)-5-(α-циклопропилкарбонил-2-фторбензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин, полученный на стадии (ii), очищали путем перекристаллизации, нежели в примере 1, в котором перекристаллизацию не проводили.
Промышленная применимость
Благодаря настоящему изобретению обеспечивается высокочистый празугрель гидрохлорид с уменьшенным содержанием примесей, таких как побочный продукт САТР, и способ его получения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КИСЛОТНЫЕ АДДИТИВНЫЕ СОЛИ ПРОИЗВОДНЫХ ГИДРОПИРИДИНА | 2001 |
|
RU2238275C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКООЧИЩЕННОГО ПРАСУГРЕЛЯ ИЛИ ЕГО КИСЛОТНО-АДДИТИВНОЙ СОЛИ | 2007 |
|
RU2424243C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОТИЕНО(3,2-С)ПИРИДИНА ИЛИ ИХ ФУРО-АНАЛОГИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2089553C1 |
| ЦИКЛИЧЕСКИЕ АМИНОПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ И ПРЕДУПРЕЖДЕНИЯ ЗАБОЛЕВАНИЙ | 1997 |
|
RU2163596C2 |
| КРИСТАЛЛЫ ГИДРОБРОМАТА ПРАСУГРЕЛЯ | 2010 |
|
RU2484094C1 |
| МЕДИЦИНСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АСПИРИН | 2001 |
|
RU2262933C2 |
| ПРОИЗВОДНОЕ СЛОЖНОГО ЭФИРА ТИЕНОПИРИДИНА, СОДЕРЖАЩЕЕ ЦИАНОГРУППУ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ЕГО ПРИМЕНЕНИЕ И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2011 |
|
RU2526624C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2281757C2 |
| СПОСОБ ПОЛУЧЕНИЯ КЛОПИДОГРЕЛА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В СПОСОБЕ | 2005 |
|
RU2357970C1 |
| СИНТЕЗ 4-АМИНО-2-БУТЕНОИЛХЛОРИДОВ И ИХ ИСПОЛЬЗОВАНИЕ ПРИ ПОЛУЧЕНИИ 3-ЦИАНОХИНОЛИНОВ | 2004 |
|
RU2345984C2 |
Изобретение относится к способу получения празугрель гидрохлорида, который включает следующие стадии: (i) хлорирование соединения, соответствующего формуле (III), путем добавления к нему хлорирующего агента, необязательно по каплям, в растворителе; (ii) взаимодействие полученного соединения формулы (IV) с соединением, соответствующим общей формуле (V), где R означает защитную группу для гидроксила, или его солью в растворителе в присутствии основания; (iii) ацетилирование полученного соединения, соответствующего общей формуле (II), путем взаимодействия реакции с ацетилирующим агентом в растворителе в присутствии основания и катализатора ацилирования; и (iv) добавление соляной кислоты, необязательно по каплям, к полученному соединению, соответствующему формуле (I), в растворителе с получением празугрель гидрохлорида, соответствующего формуле (Ia), и отличается тем, что на стадии (i) температура во время добавления, необязательно по каплям, хлорирующего агента составляет от -20°С до 5°С и температура реакции после добавления, необязательно по каплям, хлорирующего агента составляет от -20°С до 5°С. Технический результат - получение празугрель гидрохлорида со сниженным содержанием САТР. Изобретение также относится к продукту, содержащему празугрель гидрохлорид и САТР в количестве не более чем 0,3%, к фармацевтической композиции, пригодной для профилактики или лечения тромбоза или эмболии, на основе указанного продукта. 13 н. и 13 з.п. ф-лы, 3 ил., 1 табл.

1. Способ получения празугрель гидрохлорида, включающий следующие стадии:
(i) хлорирование соединения, соответствующего формуле
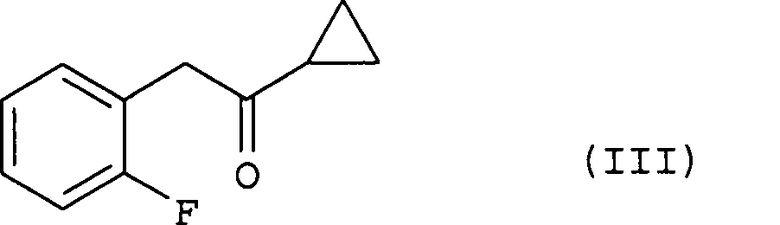
путем добавления к нему хлорирующего агента, необязательно по каплям, в растворителе;
(ii) взаимодействие полученного соединения формулы
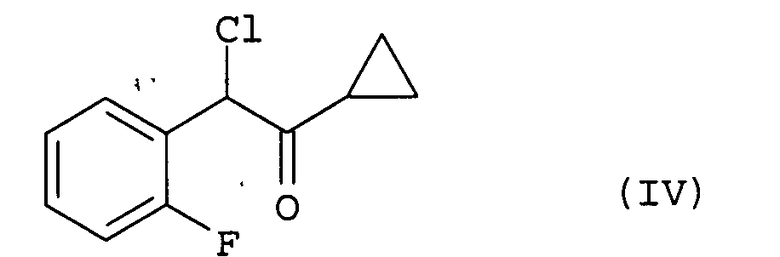
с соединением, соответствующим общей формуле
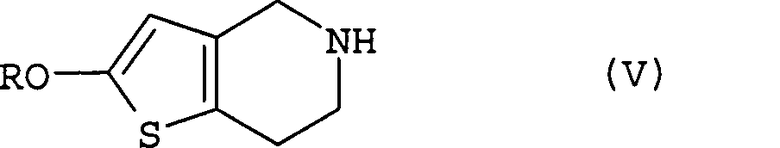
где R означает защитную группу для гидроксила,
или его солью в растворителе в присутствии основания;
(iii) ацетилирование полученного соединения, соответствующего общей формуле
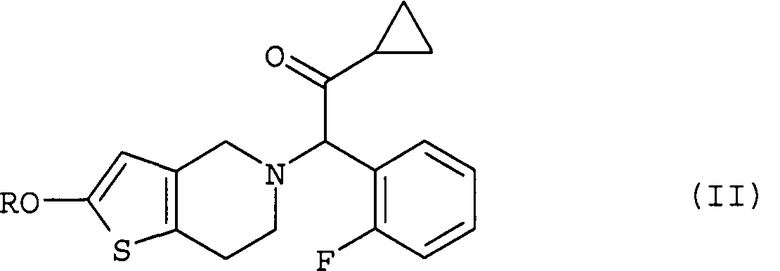
где R имеет то же значение, что указано выше,
путем взаимодействия реакции с ацетилирующим агентом в растворителе в присутствии основания и катализатора ацилирования; и
(iv) добавление соляной кислоты, необязательно по каплям, к полученному соединению, соответствующему формуле
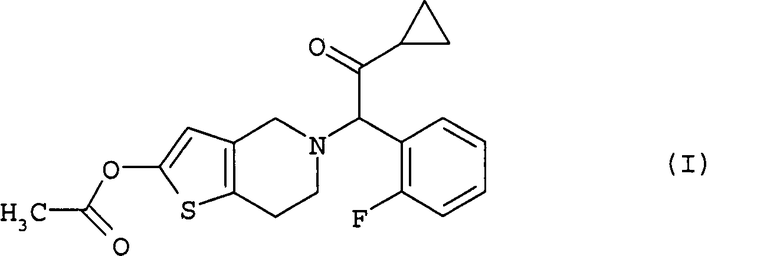
в растворителе с получением празугрель гидрохлорида, соответствующего формуле
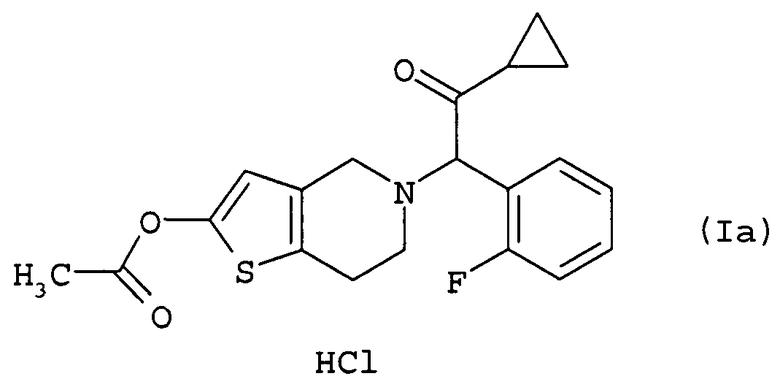
отличающийся тем, что на стадии (i) температура во время добавления, необязательно по каплям, хлорирующего агента составляет от -20°С до 5°С, и температура реакции после добавления, необязательно по каплям, хлорирующего агента составляет от -20°С до 5°С.
2. Способ по п.1, отличающийся тем, что на стадии (i) температура во время добавления, необязательно по каплям, хлорирующего агента составляет от -10°С до 5°С, и температура реакции после добавления, необязательно по каплям, хлорирующего агента составляет от -10°С до 5°С.
3. Способ по п.1, отличающийся тем, что на стадии (i) температура во время добавления, необязательно по каплям, хлорирующего агента составляет от -5°С до 5°С, и температура реакции после добавления, необязательно по каплям, хлорирующего агента составляет от -5°С до 5°С.
4. Способ по п.1, отличающийся тем, что температура последующей обработки после окончания реакции на стадии (i) составляет от -20°С до 15°С.
5. Способ по п.1, отличающийся тем, что температура последующей обработки после окончания реакции на стадии (i) составляет от -10°С до 15°С.
6. Способ по п.1, отличающийся тем, что температура последующей обработки после окончания реакции на стадии (i) составляет от 0°С до 15°С.
7. Способ по п.1, в котором хлорирующий агент представляет собой газообразный хлор.
8. Способ по п.1, в котором R означает группу общей формулы:
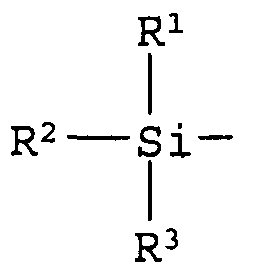
где R1, R2 и R3 независимо означают алкильную группу, содержащую от 1 до 10 атомов углерода, или арильную группу.
9. Способ по п.8, в котором R1, R2 и R3 независимо означают алкильную группу, содержащую от 1 до 5 атомов углерода, или фенильную группу.
10. Способ по п.1, в котором R означает трет-бутилдиметилсилильную группу.
11. Способ по п.1, отличающийся тем, что полученное соединение общей формулы (II) на стадии (ii) перекристаллизовывают из простых эфиров или нитрилов.
12. Способ по п.1, отличающийся тем, что полученное соединение общей формулы (II) на стадии (ii) перекристаллизовывают из ацетонитрила.
13. Способ по п.1, в котором ацетилирующий агент представляет собой уксусный ангидрид.
14. Способ по п.1, отличающийся тем, что получаемое на стадии (iii) соединение, соответствующее формуле (I), используют на следующей стадии (iv) без очистки.
15. Продукт пригодный для профилактики или лечения тромбоза или эмболии, содержащий празугрель гидрохлорид и САТР в количестве не более чем 0,3%, полученный способом по п.1.
16. Продукт пригодный для профилактики или лечения тромбоза или эмболии, содержащий празугрель гидрохлорид и САТР в количестве не более чем 0,1%, полученный способом по п.1.
17. Продукт пригодный для профилактики или лечения тромбоза или эмболии, содержащий празугрель гидрохлорид и САТР в количестве не более чем 0,04%, полученный способом по п.1.
18. Продукт пригодный для профилактики или лечения тромбоза или эмболии, содержащий празугрель гидрохлорид и САТР в количестве не более чем 0,03%, полученный способом по п.1.
19. Продукт пригодный для профилактики или лечения тромбоза или эмболии, содержащий празугрель гидрохлорид и САТР в количестве не более чем 0,02%, полученный способом по п.1.
20. Фармацевтическая композиция, содержащая в качестве активного компонента продукт по пп.15-19.
21. Фармацевтическая композиция, содержащая в качестве активного компонента продукт по пп.15-19, предназначенная для профилактики или лечения у теплокровных животных заболеваний, вызванных тромбами или эмболами.
22. Фармацевтическая композиция, предназначенная для профилактики или лечения у человека тромбоза или эмболии, содержащая в качестве активного компонента продукт по пп.15-19.
23. Применение продукта по любому из пп.15-19 для получения лекарственного средства, предназначенного для профилактики или лечения у теплокровных животных заболеваний, вызванных тромбами или эмболами.
24. Применение продукта по любому из пп.15-19 для получения лекарственного средства, предназначенного для профилактики или лечения у человека тромбоза или эмболии.
25. Способ профилактики или лечения заболеваний, вызванных тромбами или эмболами, включающий введение теплокровным животным фармацевтической композиции, содержащей в качестве активного компонента продукт по любому из пп.15-19.
26. Способ профилактики или лечения заболеваний, вызванных тромбозом или эмболией, включающий введение человеку фармацевтической композиции, содержащей в качестве активного компонента продукт по любому из пп.15-19.
| КИСЛОТНЫЕ АДДИТИВНЫЕ СОЛИ ПРОИЗВОДНЫХ ГИДРОПИРИДИНА | 2001 |
|
RU2238275C1 |
| US 5874581 A, 23.02.1999 | |||
| US 5436242 A, 25.07.1995 | |||
| Способ получения производных тетрагидро(фуро- или тиено)-[2,3-с]пиридина или их гидрохлоридов или четвертичных солей с метилйодидом | 1988 |
|
SU1657064A3 |

