ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицинской технологии и относится к типу соединения, обладающему действием против агрегации тромбоцитов, и способу его получения и его применению. В частности, настоящее изобретение относится к производному сложного эфира тиенопиридина, содержащему цианогруппу, способу его получения и его применению.
УРОВЕНЬ ТЕХНИКИ
Тромбоз может приводить к нарушениям сердечного, мозгового кровообращения и малого круга кровообращения, таким как острый инфаркт миокарда, апоплексия и эмболия легочной артерии и т.д., которые представляют опасность для здоровья и жизни людей, а также являются частым осложнением при хирургических операциях и фактором реокклюзии после интервенционной ангиопластики. Хотя тромболитическая терапия, интервенционная терапия и даже хирургическое лечение, разработанные за последние годы, достигли заметного прогресса в лечении острого инфаркта миокарда и ишемического инсульта, значительно увеличили долю успешных случаев спасения пациентов и значительно улучшили качество жизни, показатель нетрудоспособности в результате заболеваний сосудов сердца и головного мозга по-прежнему составляет до 30%. Соответственно в последние годы разработка нового лекарственного средства для предотвращения и лечения заболеваний сосудов сердца и головного мозга находится в центре внимания и исследований.
Существует много факторов, приводящих к тромбозу, например адгезия и агрегация тромбоцитов на поверхности поврежденной стенки сосуда, кровяной стаз, образование тромбина, вызванное активацией фактора свертывания крови, низкая активность плазмина и т.д. В данных факторах тромбоциты являются основным материалом для тромбоза, а следовательно, ингибирование агрегации тромбоцитов играет важную роль в предотвращении и лечении тромбоза. Аденозиндифосфат (АДФ) является важным агонистом усиления активации и агрегации тромбоцитов, и важным подходом стало предотвращение патологического тромбоза (ишемическая болезнь сердца, цереброваскулярное заболевание, эмболия легочной артерии, тромбофлебит и т.д.) и инфаркта миокарда, нестабильной стенокардии, заболевания периферических сосудов, застойной сердечной недостаточности и т.п. путем блокирования рецептора АДФ для того, чтобы ингибировать функцию тромбоцитов.
Клопидогрел (clopidogrel) в настоящее время представляет собой клинический антитромбоцитарный агент первой линии - ингибитор рецептора АДФ, разработанный на основе структурной модификации традиционного антитромбоцитарного агента Тиклопидина (Ticlopidine). Как только продукт Клопидогрел поступил в продажу, он быстро захватил рынок в силу своего более сильного антитромботического эффекта и меньшей нежелательной лекарственной реакции. Однако на протяжении более десяти лет клинической практики были обнаружены его побочные эффекты в виде тромботической тромбоцитопенической пурпуры (ТТР) и гемолитического уремического синдрома (HUS) и т.д. Следует отметить, что Клопидогрел наряду с оказанием антитромбоцитарного фармакологического действия также обладает побочным эффектом, заключающимся в склонности к кровотечению. Клопидогрел имеет большее Tmax в терапевтической дозе и медленное начало действия, следовательно, дозу введения трудно контролировать, что, вероятно, дополнительно увеличивает склонность к кровотечению. В то же время, поскольку Клопидогрел представляет собой маслянистое вещество с крайне низкой щелочностью, он может образовывать соль путем реакции с сильной кислотой, и его трудно очищать. Соли Клопидогрела неустойчивы во влажной среде, и его свободное основание снова будет осаждаться, а вследствие сильной кислотности ему также присущи некоторые ограничения с точки зрения приготовления состава.
Затем на основе Клопидогрела Daiichi Sankyo Company Limited, Япония и Eli Lilly and Company, США совместно разработали новый антитромбоцитарный агент Прасугрел (Prasugrel), другой ингибитор рецептора АДФ. Многие исследования доказали, что Прасугрел более активен и демонстрирует более быстрое начало действия, чем Клопидогрел, и различия реакций пациентов на Прасугрел меньше, чем на Клопидогрел. Результаты клинического контролируемого эксперимента для Прасугрела с Клопидогрелом показали, что Прасугрел более эффективно снижает смертность, вызываемую несмертельными сердечными приступами и апоплексией, но приводит к большему числу кровотечений у пациентов. Соответственно необходимо провести исследования и разработать новое лекарственное средство, обладающее высокой степенью безопасности и активностью против агрегации тромбоцитов.
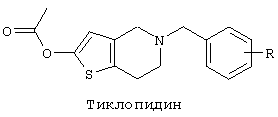

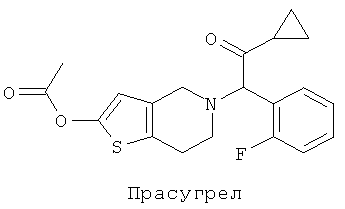
Способы синтеза и обзоры, касающиеся соединений, представляющих собой тиенопиридины, можно найти в следующей литературе: CN 1683373, US 4681888, US 4529596, GB 1501797, WO 02059128, US 4174448, GB 1561504, WO 2004094374, JP 6135970 и JP 63264588.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Одной из задач настоящего изобретения является обеспечение производного сложного эфира тиенопиридина, содержащего цианогруппу.
Другой задачей настоящего изобретения является обеспечение фармацевтической композиции, содержащей указанное выше производное сложного эфира тиенопиридина, содержащее цианогруппу, или его фармацевтически приемлемую соль в качестве основного активного ингредиента.
Еще одной задачей настоящего изобретения является обеспечение способа получения вышеуказанного производного сложного эфира тиенопиридина, содержащего цианогруппу, и его фармацевтически приемлемой соли.
Другой задачей настоящего изобретения является обеспечение применения вышеуказанного производного сложного эфира тиенопиридина, содержащего цианогруппу, и его фармацевтически приемлемой соли в тромбоцитарной антиагрегации, в частности применение в получении лекарственного средства для предотвращения или лечения заболеваний сосудов сердца или головного мозга, таких как коронарные синдромы, инфаркт миокарда, ишемия миокарда и т.п., вызванных агрегацией тромбоцитов.
Согласно настоящему изобретению предложено соединение со структурой общей формулы I и его фармацевтически приемлемые соли:
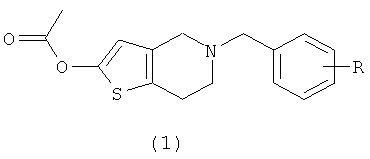 ,
,
где R представляет собой цианогруппу.
Соединения со структурой формулы I, предложенные согласно настоящему изобретению, предпочтительно представляют собой следующие соединения I-1, I-2 и I-3:
I-1: 5-(2-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илацетат;
I-2: 5-(3-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илацетат;
I-3: 5-(4-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илацетат.
Согласно настоящему изобретению предложено соединение со структурой формулы I, при этом фармацевтически приемлемая соль включает соли, образованные соединением формулы I и неорганической кислотой или органической кислотой, при этом наиболее предпочтительные соли представляют собой: фармацевтически приемлемые соли, такие как гидрохлориды, гидробромиды, гидроиодаты, сульфаты, гидросульфаты, фосфаты, гидрофосфаты, ацетаты, пропионаты, бутираты, лактаты, мезилаты, тозилаты, малеаты, бензоаты, сукцинаты, тартраты, цитраты, фумараты, таураты, глюконаты и аминокислотные соли, и т.д.
Согласно настоящему изобретению также предложен способ получения соединения формулы I и конкретный путь получения является следующим:

где R представляет собой цианогруппу; X представляет собой бром или хлор.
Гидрохлорид тиенопиридона подвергают взаимодействию с бензилбромидом или бензилхлоридом, замещенным цианогруппой, при -10°С-105°С в присутствии связывающего кислоту агента, такого как триэтиламин, пиридин, карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия или гидроксид калия и т.д., в растворителе - дихлорметане, трихлорметане, ацетонитриле или толуоле и т.д., с получением ключевого промежуточного соединения. Затем указанное промежуточное соединение подвергают взаимодействию с ангидридом уксусной кислоты, уксусной кислотой, ацетилхлоридом или ацетилбромидом и т.д. при -30°С-65°С в присутствии связывающего кислоту агента, такого как триэтиламин, пиридин, карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия или гидроксид калия и т.д., в растворителе - дихлорметане, трихлорметане или толуоле с получением целевого продукта I.
Различные промежуточные соединения, полученные в данной реакции, или полученный продукт, представляющий собой соединение I, могут быть растворены в диэтиловом эфире, ДМФА, ацетоне, метаноле, этаноле, этилацетате или ДМСО, и по каплям добавляют раствор неорганической кислоты или органической кислоты с получением фармацевтически приемлемой соли.
Конкретные действия могут включать: растворение промежуточного соединения или полученного продукта, представляющего собой соединение I, в диэтиловом эфире, ДМФА, ацетоне, метаноле, этаноле, этилацетате или ДМСО и добавление раствора хлороводорода в диэтиловом эфире по каплям к смеси на бане с ледяной водой до pH=2 с получением гидрохлорида; или растворение соединения в диэтиловом эфире, ДМФА, ацетоне, метаноле, этаноле, этилацетате или ДМСО, добавление эквимолярного количества таурина к смеси и нагревание, и перемешивание смеси с получением таурата; или растворение соединения в диэтиловом эфире, ДМФА, ацетоне, метаноле, этаноле, этилацетате или ДМСО, и добавление концентрированной серной кислоты по каплям к смеси на бане с ледяной водой до pH=3 с получением сульфата; и т.д.
Соединение, предложенное согласно настоящему изобретению, эффективно для лечения заболеваний у людей, вызванных агрегацией тромбоцитов. Хотя соединение согласно настоящему изобретению может быть введено напрямую без какого-либо препарата, указанное соединение предпочтительно использовать в форме фармацевтических препаратов. Путь введения может представлять собой парентеральное (такое как внутривенное или внутримышечное) или пероральное введение.
Согласно настоящему изобретению также предложено применение вышеуказанного соединения, имеющего структуру формулы I, или его фармацевтически приемлемой соли против агрегации тромбоцитов и применение вышеуказанного соединения, имеющего структуру формулы I, или его фармацевтически приемлемой соли для получения лекарственного средства против агрегации тромбоцитов. При этом указанное лекарственное средство против агрегации тромбоцитов представляет собой лекарственное средство для лечения или предотвращения заболеваний сосудов сердца и головного мозга, вызванных агрегацией тромбоцитов, при этом указанные заболевания сосудов сердца и головного мозга могут представлять собой коронарные синдромы, инфаркт миокарда или ишемию миокарда.
Согласно настоящему изобретению также предложена фармацевтическая композиция против агрегации тромбоцитов; указанная фармацевтическая композиция содержит терапевтически эффективное количество соединения со структурой формулы I или его фармацевтически приемлемой соли согласно настоящему изобретению и фармацевтически приемлемый носитель и/или наполнитель. Что касается фармацевтической композиции согласно настоящему изобретению, фармацевтическая композиция может представлять собой твердый препарат для перорального введения, жидкий препарат для перорального введения или препарат для инъекций.
Согласно настоящему изобретению также предложен способ лечения заболеваний сосудов сердца и головного мозга; указанный способ включает введение пациенту, нуждающемуся в данном лечении, терапевтически эффективного количества соединения со структурой формулы I согласно настоящему изобретению или введение пациенту, нуждающемуся в данном лечении, терапевтически эффективного количества фармацевтической композиции, предложенной согласно настоящему изобретению.
Способ получения фармацевтической композиции соединения согласно настоящему изобретению заключается в следующем: комбинирование соединения согласно настоящему изобретению с фармацевтически приемлемым твердым или жидким носителем с использованием стандартных и традиционных методик, и, возможно, комбинирование его с фармацевтически приемлемым вспомогательным веществом и наполнителем с получением микрочастиц или микросфер. Твердые лекарственные препараты включают таблетки, диспергированные гранулы, капсулы, таблетки с замедленным высвобождением, гранулы (пеллеты) с замедленным высвобождением и т.п. Твердый носитель может представлять собой по меньшей мере одно вещество, которое может служить в качестве разбавителя, ароматизатора, солюбилизатора, смазывающего вещества, суспендирующего агента, связывающего вещества, разрыхлителя или агента для покрытия. Инертные твердые носители включают фосфат магния, стеарат магния, тальк, лактозу, пектин, пропиленгликоль, полисорбат 80, декстрин, крахмал, желатин, целлюлозные вещества, такие как метилцеллюлоза и микрокристаллическая целлюлоза, парафин с низкой температурой плавления, полиэтиленгликоль, маннит и какао-масло, и т.д. Жидкие лекарственные препараты включают растворы, суспензии, такие как препараты для инъекций, и порошок, и т.д.
Количество активного ингредиента (соединение согласно настоящему изобретению), содержащееся в фармацевтической композиции и дозированной лекарственной форме, может быть, в частности, определено в соответствии с состоянием пациента и диагнозом врача, и количество или концентрация используемого соединения могут быть скорректированы в широком диапазоне. В целом, количество активного соединения составляет 0,5-90% от массы композиции, предпочтительно 0,5-70% от массы композиции.
ЛУЧШИЙ ВАРИАНТ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение будет подробно проиллюстрировано со ссылкой на следующие примеры, и указанные следующие примеры только иллюстрируют и объясняют настоящее изобретение и никоим образом не ограничивают объем настоящего изобретения. Соединения тестируют методом высокоэффективной жидкостной хроматографии (ВЭЖХ) и тонкослойной хроматографии (ТСХ). Затем структуры соединений могут быть дополнительно подтверждены тестом, таким как инфракрасная спектроскопия (ИК), спектроскопия ядерного магнитного резонанса (1H ЯМР, 13C ЯМР), масс-спектрография (МС) и т.п.
Пример 1
Получение промежуточного соединения 1
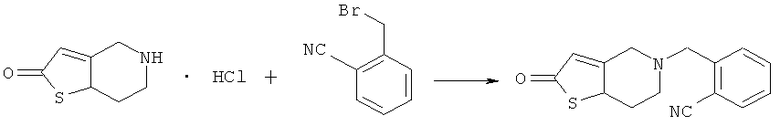
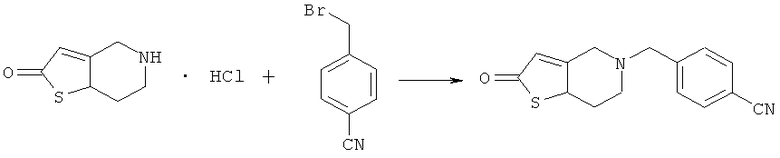
В реакционную колбу, оснащенную мешалкой, холодильником и термометром, добавляли 19,2 г 5,6,7,7a-тетрагидротиено[3,2-c]пиридин-2(4H)-она, который затем растворяли в 70 мл ацетонитрила и охлаждали до -10°С при перемешивании. К смеси добавляли 41,5 г безводного карбоната калия. После добавления 19,6 г 2-цианобензилбромида в реакционную систему порциями реакционную смесь нагревали до 45°С и продолжали проводить реакцию в течение 4 часов (завершение реакции контролировали путем ТСХ). Затем реакционную смесь фильтровали и растворитель ацетонитрил в фильтрате выпаривали досуха. К остатку добавляли 50 мл дихлорметана и смесь промывали водой (3×50 мл). Фазу дихлорметана отделяли, полностью высушивали над безводным сульфатом натрия и фильтровали. Дихлорметан выпаривали при пониженном давлении с получением 22,6 г желтого масляного продукта (ВЭЖХ: 97,2%). Rf=0,47 [по максимуму пика, система растворителей: v (петролейный эфир) : v (этилацетат) = 1:2]. МС, м/Z: 270,0 (М).
Пример 2
Получение промежуточного соединения 2

В реакционную колбу, оснащенную мешалкой, холодильником и термометром, добавляли 19,2 г 5,6,7,7a-тетрагидротиено[3,2-c]пиридин-2(4H)-она, который затем диспергировали в 80 мл дихлорметана и охлаждали до 0°С при перемешивании. К смеси добавляли 30,4 г триэтиламина. После добавления 15,2 г 3-цианобензилбромида в реакционную систему порциями, реакционную смесь нагревали в колбе с обратным холодильником и продолжали проводить реакцию в течение 5 часов (завершение реакции контролировали путем ТСХ). Реакционную жидкость промывали водой (3×80 мл). Затем фазу дихлорметана отделяли, полностью высушивали над безводным сульфатом натрия и фильтровали. Дихлорметан выпаривали при пониженном давлении с получением 20,8 г светло-желтого твердого продукта (ВЭЖХ: 96,4%). Rf=0,45 [по максимуму пика, система растворителей: v (петролейный эфир) : v (этилацетат) = 1:2]. МС, м/Z: 270,0 (М).
Пример 3
Получение промежуточного соединения 3
В реакционную колбу, оснащенную мешалкой, холодильником и термометром, добавляли 19,2 г 5,6,7,7a-тетрагидротиено[3,2-c]пиридин-2(4H)-она, который затем диспергировали в 65 мл толуола и охлаждали до 10°С при перемешивании. К смеси добавляли 23,7 г пиридина. После добавления 19,6 г 4-цианобензилбромида в реакционную систему порциями реакционную смесь нагревали до 95°С и продолжали проводить реакцию в течение 2,5 часов (завершение реакции контролировали путем ТСХ). Реакционную смесь промывали водой (3×50 мл). Затем фазу толуола отделяли, полностью высушивали над безводным сульфатом натрия и фильтровали. Толуол выпаривали при пониженном давлении с получением 21,4 г желтого масляного продукта (ВЭЖХ: 94,2%). Rf=0,41 [по максимуму пика, система растворителей: v (петролейный эфир) : v (этилацетат) = 1:2]. МС, м/Z: 270,0 (М).
Пример 4
(5-(2-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)ацетат (соединение I-1) получали в данном примере
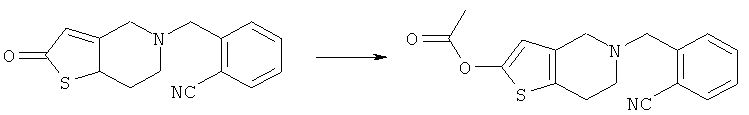
В реакционную колбу, оснащенную мешалкой, холодильником и термометром, добавляли 2,7 г промежуточного соединения 1, которое затем растворяли в 10 мл дихлорметана. К смеси добавляли 1,2 г гидроксида натрия при перемешивании. Затем реакционную систему охлаждали до -20°С и в реакционную систему порциями добавляли 1,02 г ангидрида уксусной кислоты. После добавления реакционную смесь продолжали перемешивать при комнатной температуре в течение 1 часа (завершение реакции контролировали путем ТСХ). Затем реакционную смесь промывали водой (3×15 мл). Фазу дихлорметана отделяли, полностью высушивали над безводным сульфатом натрия и фильтровали. Дихлорметан выпаривали при пониженном давлении и остаток очищали на колонке с получением белого твердого продукта (ВЭЖХ: 99,6%). Rf=0,58 [по максимуму пика, система растворителей: v (петролейный эфир) : v (этилацетат) = 4:1]. 1Н ЯМР (ДМСО-d6, 400 МГц) δ: 2,253 (s, 3H), 2,700 (s, 2H), 2,767~2,780 (d, 2H), 3,402 (s, 2H), 3,816 (s, 2H), 6,421 (s, 1H), 7,452~7,489 (t, 1H), 7,606~7,625 (d, 1H), 7,660~7,697 (t, 1H), 7,803~7,822 (d, 1H). МС, m/Z: 312,0 (M).
Пример 5
(5-(3-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)ацетат (соединение 1-2) получали в данном примере

В реакционную колбу, оснащенную мешалкой, холодильником и термометром, добавляли 2,7 г промежуточного соединения 2, которое затем растворяли в 15 мл трихлорметана. К смеси добавляли 3,0 г триэтиламина при перемешивании. Затем реакционную систему охлаждали до -30°С и в реакционную систему порциями добавляли 0,78 г ацетилхлорида. После добавления реакционную смесь продолжали перемешивать при 10°С в течение 2,5 часов (завершение реакции контролировали путем ТСХ). Затем реакционную смесь промывали водой (3×15 мл). Фазу трихлорметана отделяли, полностью высушивали над безводным сульфатом натрия и фильтровали. Трихлорметан выпаривали при пониженном давлении и остаток очищали на колонке с получением белого твердого продукта (ВЭЖХ: 99,0%). Rf=0,56 [по максимуму пика, система растворителей: v (петролейный эфир) : v (этилацетат) = 4:1]. 1H ЯМР (ДМСО-d6, 400 МГц) δ: 2,255 (s, 3H), 2,701 (s, 2H), 2,769~2,782 (d, 2H), 3,403 (s, 2H), 3,817 (s, 2H), 6,423 (s, 1H), 7,367~7,402 (t, 1H), 7,543~7,561 (d, 1H), 7,632~7,650 (d, 1H), 7,916~7,927 (s, 1H). MC, m/Z: 312,0 (M).
Пример 6
(5-(4-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)ацетат (соединение 1-3) получали в данном примере

В реакционную колбу, оснащенную мешалкой, холодильником и термометром, добавляли 2,7 г промежуточного соединения 3, которое затем растворяли в 20 мл толуола. К смеси добавляли 4,14 г безводного карбоната калия при перемешивании. Затем реакционную систему охлаждали до -10°С и в реакционную систему порциями добавляли 0,60 г уксусной кислоты. После добавления реакционную смесь продолжали перемешивать при 30°С в течение 3 часов (завершение реакции контролировали путем ТСХ). Реакционную смесь промывали водой (3×20 мл). Фазу толуола отделяли, полностью высушивали над безводным сульфатом натрия и фильтровали. Толуол выпаривали при пониженном давлении и остаток очищали на колонке с получением белого твердого продукта (ВЭЖХ: 99,7%). Rf=0,58 [по максимуму пика, система растворителей: v (петролейный эфир) : v (этилацетат) = 4:1]. 1H ЯМР (ДМСО-d6, 400 МГц) δ: 2,252 (s, 3H), 2,698 (s, 2H), 2,766~2,778 (d, 2H), 3,401 (s, 2H), 3,815 (s, 2H), 6,419 (s, 1H), 7,865~7,889 (d, 2H), 8,021~8,044 (d, 2H). MC, m/Z: 312,0 (M).
Пример 7
Получение гидрохлорида соединения I-1, полученного в примере 4: 2,0 г твердого продукта соединения I-1 растворяли в 10 мл безводного диэтилового эфира. Смесь охлаждали до 0°С на бане с ледяной водой, а затем к смеси по каплям добавляли хлороводород, 15% раствор в диэтиловом эфире до рН, равного 2. Смесь продолжали перемешивать на бане с ледяной водой в течение примерно 1 часа, а затем фильтровали с получением белого твердого вещества.
Пример 8
Получение таурата соединения I-2, полученного в примере 5: 2,0 г твердого продукта соединения I-2 растворяли в 15 мл безводного этанола. Смесь нагревали в колбе с обратным холодильником, а затем к смеси добавляли эквимолярное количество таурина. Смесь продолжали перемешивать в колбе с обратным холодильником в течение примерно 3 часов. После завершения реакции смесь оставляли при комнатной температуре на 24 часа, затем фильтровали с получением белого твердого вещества.
Пример 9
Получение сульфата соединения I-3, полученного в примере 6: 2,5 г твердого продукта соединения I-3 растворяли в 20 мл безводного метанола. Смесь охлаждали до 5°С на бане с ледяной водой, а затем к смеси по каплям добавляли концентрированный раствор серной кислоты до рН, равного 3. Смесь продолжали перемешивать на бане с ледяной водой в течение примерно 0,5 часа, а затем фильтровали с получением белого твердого вещества.
Также для того, чтобы в достаточной степени проиллюстрировать фармацевтические композиции производного сложного эфира тиенопиридина, содержащего цианогруппу, согласно настоящему изобретению предложены следующие примеры 10-13 лекарственных препаратов. Данные примеры только объясняют настоящее изобретение и не ограничивают объем настоящего изобретения. Указанные лекарственные препараты могут быть получены на основе любого активного соединения или его соли согласно настоящему изобретению, предпочтительно указанные лекарственные препараты могут быть получены на основе соединений, полученных способом из примеров 4-6.
Пример 10
Получение твердых желатиновых капсул с использованием следующих ингредиентов:
Количество/капсула
Способ получения: ингредиенты предварительно сушили и просеивали через сита 100 меш для использования. Затем установленные дозы вышеуказанных ингредиентов равномерно смешивали и просеивали через сито 60 меш 3 раза. К смеси добавляли 10% раствор повидона в этаноле (95%) (в достаточном количестве) с получением влажной массы. Влажную массу гранулировали путем пропускания через сито 18 меш. Сушили полученные гранулы при 40°С, калибровали их по размеру путем просеивания через сито 16 меш, а затем их помещали в твердые желатиновые капсулы.
Пример 11
Получение таблеток с использованием следующих ингредиентов:
Количество/таблетка
Способ получения: ингредиенты предварительно сушили и просеивали через сита 100 меш для использования. Установленные дозы наполнителей полностью и равномерно смешивали, затем к наполнителям добавляли активный ингредиент методом пошагового разбавления и смесь полностью и равномерно перемешивали 2-3 раза после каждого добавления для обеспечения хорошего смешивания активного ингредиента и наполнителей. Смесь просеивали через сито 20 меш и сушили при 55°С в вентиляционной печи в течение 2 часов. Калибровали полученные сухие гранулы по размеру путем просеивания через сито 16 меш и тестировали содержание промежуточного соединения. После равномерного смешивания гранулы прессовали в таблетки в таблетировочной машине.
Пример 12
Получение препаратов для инъекций:
Способ получения: активный ингредиент добавляли в воду для инъекций, в которой растворен сорбитал (sorbital) и пропиленгликоль. К смеси добавляли медицинскую щелочь для доведения рН до 4-8, таким образом растворяли активный ингредиент. К смеси добавляли активированный уголь, перемешивали и оставляли абсорбировать в течение 30 минут, затем уголь удаляли и смесь подвергали тонкой фильтрации, помещали в емкость и герметизировали, и стерилизовали.
Пример 13
Получение лиофилизированного порошка:
Способ получения: активный ингредиент добавляли в воду для инъекций и к смеси добавляли медицинскую щелочь для доведения рН до 4-8, таким образом растворяли активный ингредиент. Затем к смеси добавляли маннит и смесь обрабатывали в автоклаве, как необходимо для препаратов для инъекций. К смеси добавляли активированный уголь, а затем смесь фильтровали через миллипоровый фильтр. Фильтрат упаковывали отдельно и рыхлую массу получали методом сублимационной сушки, а затем герметизировали с получением продукта.
Соединение согласно настоящему изобретению, имеющее структуру формулы I, и его фармацевтически приемлемые соли обладают очевидным ингибирующим действием в отношении агрегации тромбоцитов. Данное ингибирующее действие соединения согласно настоящему изобретению в отношении агрегации тромбоцитов у крыс далее проиллюстрировано следующими фармакодинамическими экспериментами.
Эксперименты по определению ингибирующего действия в отношении агрегации тромбоцитов у крыс:
1. Лекарственные средства и реагенты для экспериментов:
Используют соединения I-1, I-2 и I-3, гидрохлорид соединения I-1, таурат соединения I-2 и сульфат соединения I-3, полученные в примерах 4-9.
АДФ: произведенный SIGMA Со.
Карбоксиметилцеллюлоза натрия 800-1200: Sinopharm Chemical Reagent Co., Ltd., номер партии: F20051103.
2. Экспериментальные животные:
Крысы линии Wistar: SPF (свободные от специфических патогенов), самцы, предоставленные Институтом исследования лабораторных животных (The Institute of Laboratory Animal Science), Китайская академия медицинских наук, лицензия No. SCXK (jing) 2005-0013.
3. Инструменты для проведения эксперимента:
Двухканальный анализатор агрегации тромбоцитов (агрегометр) РАМ-3: произведенный Jiangsu Danyang Radio Factory.
4. Методика проведения эксперимента и результаты
Отбирали здоровых самцов крыс линии Wistar массой 200-250 г и случайным образом делили на группы. Эксперименты проводили тремя сериями и каждую серию распределяли на нормальную контрольную группу и 6 групп соединений, и всего две группы доз тестировали в каждой серии. Эти две группы доз, определенные для соединений I-1, I-2 и I-3, гидрохлорида соединения I-1, таурата соединения I-2 и сульфата соединения I-3, представляли собой 15 мг/кг и 30 мг/кг соответственно. Использовали внутрижелудочное введение с объемом дозы 10 мл/кг массы тела. Нормальной контрольной группе вводили такое же количество 0,5% КМЦ-Na. Через 2 часа после введения интраперитонеально вводили 40 мг/кг пентобарбитала натрия (1 мл/кг) для анестезии и кровь забирали из брюшной аорты. 3,8% цитрат натрия использовали для антикоагуляции и готовили обогащенную тромбоцитами плазму (PRP) и обедненную тромбоцитами плазму (PPP) соответственно. Максимальный процент агрегации тромбоцитов, индуцируемой АДФ (конечная концентрация: 1,08 мкМ), тестировали на двухканальном анализаторе агрегации тромбоцитов РАМ-3. Результаты эксперимента представлены в Таблице 1 и Таблице 2.
Из данных Таблиц 1 и 2 можно увидеть, что соединения согласно настоящему изобретению (15, 30 мг/кг) обладают очевидным ингибирующим действием в отношении агрегации тромбоцитов, индуцируемой АДФ, по сравнению с нормальными контрольными группами. Следовательно, соединения согласно настоящему изобретению можно применять для предотвращения или лечения заболеваний сосудов сердца и головного мозга, таких как коронарный синдром, инфаркт миокарда и ишемия миокарда, вызванных агрегацией тромбоцитов.
Фармакокинетические исследования у животных:
Исходя из экспериментов внутрижелудочного введения у крыс, предварительные результаты исследования показывают, что соединения I-1, I-2 и I-3, гидрохлорид соединения I-1, таурат соединения I-2 и сульфат соединения I-3 согласно настоящему изобретению и коммерческое лекарственное средство Клопидогрел все являются пролекарствами, и они должны быть превращены в активные метаболиты тиолы in vivo под действием соответствующих ферментов для обеспечения терапевтического эффекта. Однако скорость превращения соединений I-1, I-2 и I-3, гидрохлорида соединения I-1, таурата соединения I-2 и сульфата соединения I-3 примерно в 5 раз выше, чем Клопидогрела, и время достижения пиковой концентрации их активных метаболитов составляет от 50 минут до 1 часа, что также быстрее, чем в случае Клопидогрела. Следовательно, соединения согласно настоящему изобретению, вероятно, затем будут превращены в антитромбоцитарное лекарственное средство с более быстрым началом действия.
Двухнедельные исследования токсичности у крыс (по сравнению с Клопидогрелом)
Материалы и методы:
Соединения для тестирования: соединение I-1 и Клопидогрел.
Приготовление раствора: соединение I-1 растирают с 0,5% КМЦ-Na и кукурузным маслом, затем растворяют в условиях ультразвуковой обработки. Перед применением готовят 100 мг/мл суспензии и вводят внутрижелудочно 1 мл/100 г каждое утро.
Экспериментальные животные: крысы линии SD массой примерно 180 г, 7 самок и 7 самцов в каждой группе.
Группировка и режим дозирования: соединение 1-1 вводят внутрижелудочно в дозе 1000 мг/кг один раз в сутки в течение 2 недель. Такое же количество растворителя вводят внутрижелудочно холостой контрольной группе и равную дозу (1000 мг/кг) Клопидогрела вводят внутрижелудочно положительной контрольной группе.
Результаты эксперимента:
Смерть крыс имеет место во всех группах, получавших лечение, на протяжении всего периода тестирования, конкретное число и время смерти представлены в Таблице 3. С третьего дня внутрижелудочного введения геморрагические выделения вокруг носа у крыс можно наблюдать как в группе соединения I-1, так и группе Клопидогрела, но они очевидно небольшие в группе соединения I-1, чем в группе Клопидогрела.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДЕЙТЕРИРОВАННЫЕ ПРОИЗВОДНЫЕ ТИЕНОПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2716141C2 |
| КИСЛОТНЫЕ АДДИТИВНЫЕ СОЛИ ПРОИЗВОДНЫХ ГИДРОПИРИДИНА | 2001 |
|
RU2238275C1 |
| СПОСОБ ПОЛУЧЕНИЯ КЛОПИДОГРЕЛЯ И ЕГО ПРОИЗВОДНЫХ | 2009 |
|
RU2469039C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОТИЕНО(3,2-С)ПИРИДИНА ИЛИ ИХ ФУРО-АНАЛОГИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2089553C1 |
| СОЕДИНЕНИЕ, ИМЕЮЩЕЕ ДЕЙСТВИЕ ИНГИБИРОВАНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ, И ЕГО СОЛЬ, И СОДЕРЖАЩАЯ ТАКИЕ СОЕДИНЕНИЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ ТРОМБОТИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2739915C2 |
| СОЕДИНЕНИЕ ТЕТРАГИДРОИЗОХИНОЛИНА В КАЧЕСТВЕ МОДУЛЯТОРА КАЛИЕВЫХ КАНАЛОВ, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2800873C1 |
| АЗОТСОДЕРЖАЩИЙ ГЕТЕРОЦИКЛИЧЕСКИЙ АМИД И ЕГО ПРИМЕНЕНИЕ ДЛЯ МЕДИЦИНСКИХ ЦЕЛЕЙ | 2019 |
|
RU2789670C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ТРОМБООБРАЗОВАНИЯ ИЛИ ЭМБОЛИИ | 2011 |
|
RU2611662C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПРОСТАГЛАНДИНА | 2017 |
|
RU2748837C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДОБЕНЗИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АГОНИСТОВ ИЛИ АНТАГОНИСТОВ РЕЦЕПТОРОВ МЕЛАНОКОРТИНОВ | 2005 |
|
RU2392279C2 |
Изобретение относится к соединению со структурой формулы (I) или к его фармацевтически приемлемой соли, где R представляет собой цианогруппу. Изобретение также относится к способу получения указанного соединения и к фармацевтической композиции против агрегации тромбоцитов на основе этого соединения. Технический результат: получено новое соединение и фармацевтическая композиция на его основе, которые могут найти применение в медицине для получения лекарственного средства для предотвращения или лечения заболеваний сосудов сердца и головного мозга, таких как коронарные синдромы, инфаркт миокарда и ишемия миокарда, вызванных агрегацией тромбоцитов. 6 н. и 7 з.п. ф-лы, 3 табл., 13 пр.
1. Соединение со структурой формулы I или его фармацевтически приемлемая соль:
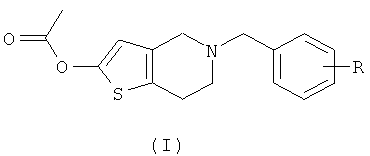 ,
,
где R представляет собой цианогруппу.
2. Соединение со структурой формулы I или его фармацевтически приемлемая соль по п.1, отличающееся тем, что указанное соединение выбрано из одного из следующих соединений:
I-1: 5-(2-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илацетат;
I-2: 5-(3-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илацетат;
I-3: 5-(4-цианобензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илацетат.
3. Соединение со структурой формулы I или его фармацевтически приемлемая соль по п.1, отличающееся тем, что указанная фармацевтически приемлемая соль включает соль, образованную соединением формулы I и неорганической кислотой или органической кислотой.
4. Соединение со структурой формулы I или его фармацевтически приемлемая соль по п.3, отличающееся тем, что указанная фармацевтически приемлемая соль выбрана из гидрохлоридов, гидробромидов, гидройодатов, сульфатов, гидросульфатов, фосфатов, гидрофосфатов, ацетатов, пропионатов, бутиратов, лактатов, мезилатов, тозилатов, малеатов, бензоатов, сукцинатов, тартратов, цитратов, фумаратов, тауратов, глюконатов и аминокислотных солей соединения формулы I.
5. Способ получения соединения со структурой формулы I или его фармацевтически приемлемой соли по любому из пп.1-4, отличающийся тем, что указанный способ включает стадии:
(1) осуществления взаимодействия гидрохлорида 5,6,7,7a-тетрагидротиено[3,2-c]пиридин-2(4H)-она с бензилбромидом или бензилхлоридом, замещенным цианогруппой, при от -10°С до 105°С в присутствии триэтиламина, пиридина, карбоната калия, карбоната натрия, бикарбоната натрия, бикарбоната калия, гидроксида натрия или гидроксида калия в дихлорметане, трихлорметане, ацетонитриле или толуоле с получением промежуточного соединения;
(2) осуществление взаимодействия промежуточного соединения, полученного на стадии (1), с ангидридом уксусной кислоты, уксусной кислотой, ацетилхлоридом или ацетилбромидом при от -30°С до 50°С в присутствии триэтиламина, пиридина, карбоната калия, карбоната натрия, бикарбоната натрия, бикарбоната калия, гидроксида натрия или гидроксида калия в дихлорметане, трихлорметане или толуоле с получением соединения формулы I.
6. Способ по п.5, отличающийся тем, что указанный способ дополнительно включает:
растворение промежуточного соединения или соединения формулы I в диэтиловом эфире, ДМФА, ацетоне, метаноле, этаноле, этилацетате или ДМСО и добавление по каплям раствора неорганической кислоты или органической кислоты с получением фармацевтически приемлемой соли.
7. Применение соединения со структурой формулы I или его фармацевтически приемлемой соли по любому из пп.1-4 против агрегации тромбоцитов.
8. Применение соединения со структурой формулы I или его фармацевтически приемлемой соли по любому из пп.1-4 для получения лекарственного средства против агрегации тромбоцитов.
9. Применение по п.8, отличающееся тем, что указанное лекарственное средство против агрегации тромбоцитов представляет собой лекарственное средство для лечения или предотвращения заболеваний сосудов сердца и головного мозга, вызываемых агрегацией тромбоцитов.
10. Применение по п.9, отличающееся тем, что указанные заболевания сосудов сердца и головного мозга представляют собой коронарные синдромы, инфаркт миокарда или ишемию миокарда.
11. Фармацевтическая композиция против агрегации тромбоцитов, отличающаяся тем, что указанная фармацевтическая композиция содержит терапевтически эффективное количество соединения со структурой формулы I или его фармацевтически приемлемой соли по любому из пп.1-4 и фармацевтически приемлемый носитель и/или наполнитель.
12. Фармацевтическая композиция по п.11, отличающаяся тем, что указанная фармацевтическая композиция представляет собой твердый препарат для перорального введения, жидкий препарат для перорального введения или препарат для инъекций.
13. Способ лечения заболеваний сосудов сердца и головного мозга, включающий введение пациенту, нуждающемуся в данном лечении, терапевтически эффективного количества соединения со структурой формулы I по любому из пп.1-4 или введение пациенту, нуждающемуся в данном лечении, терапевтически эффективного количества фармацевтической композиции по п.11 или 12.
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОТИЕНО(3,2-С)ПИРИДИНА ИЛИ ИХ ФУРО-АНАЛОГИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2089553C1 |
| Способ получения производных 5,6,7,7а-тетрагидро-4 @ -тиено/3,2- @ /-пиридинона-2 или их солей | 1981 |
|
SU1145931A3 |
| CN 101402641 А, 08.04.2009 | |||

