Область техники, к которой относится изобретение
Изобретение относится к соединениям и композициям, содержащим указанные соединения, действующим в качестве ингибиторов взаимодействия между MDM2 и p53. Кроме того, настоящее изобретение предоставляет способы получения раскрытых ингибиторов, композиций, включающих их, и способы их применения, например, в качестве лекарственных средств.
p53 представляет собой супрессорный белок опухоли, который играет ключевую роль в регуляции баланса между клеточной пролиферацией и остановкой/апоптозом клеточного роста. При нормальных условиях время полужизни p53 очень короткое и, следовательно, содержание p53 в клетках находится на низком уровне. Однако в ответ на повреждение клеточной ДНК или клеточный стресс (например, активацию онкогена, эрозию теломера, гипоксию) уровень содержания ρ53 возрастает. Это увеличение уровня p53 приводит к активации транскрипции ряда генов, которые приводят клетку к остановке роста или к процессу апоптоза. Таким образом, важная функция ρ53 заключается в том, чтобы предотвратить неконтролируемую пролиферацию поврежденных клеток и, тем самым, защитить организм от развития рака.
MDM2 представляет собой основной негативный регулятор функции ρ53. Он формирует негативную ауторегуляторную петлю путем связывания с аминоконцевым трансактивирующим доменом p53 и, таким образом, MDM2 ингибирует способность p53 активировать транскрипцию и делает p53 мишенью протеолитической деградации. В нормальных условиях эта регуляторная петля отвечает за поддержание низких уровней p53. Однако в опухоли с p53 дикого типа равновесная концентрация активного p53 может быть увеличена за счет противодействия взаимодействию между MDM2 и p53. В результате это приведет к восстановлению опосредованных p53 про-апоптических и анти-пролиферативных эффектов в таких опухолевых клетках.
MDM2 представляет собой клеточный протоонкоген. Сверхэкспрессия MDM2 была отмечена в ряде видов рака. Повышенная экспрессия MDM2 имеет место в разнообразных опухолях в результате амплификации генов или повышенной транскрипции или трансляции. Механизм, за счет которого амплификация MDM2 способствует онкогенезу, по меньшей мере, частично относится к его взаимодействию с p53. В клетках со сверхэкспрессией MDM2 блокируется защитная функция p53, и, в связи с этим, клетки становятся неспособными к ответу на повреждение ДНК или на клеточный стресс путем повышения уровня p53, что приводит к остановке роста клетки и/или апоптозу. Соответственно, после повреждения ДНК и/или клеточного стресса сверхэкспрессирующие MDM2 клетки становятся свободными для продолжения пролиферации и приобретают онкогенный фенотип. В этих условиях нарушение взаимодействия p53 и MDM2 позволит высвободить p53 и, следовательно, позволит функционировать нормальным сигналам остановки роста и/или апоптоза.
MDM2 также может обладать отдельными функциями в дополнение к ингибированию p53. Например, было показано, что MDM2 непосредственно взаимодействует с pRb-регулируемым фактором транскрипции E2F1/DP1. Это взаимодействие может иметь решающее значение для p53-независимых онкогенных воздействий MDM2. Домен E2F1 показывает поразительное сходство с MDM2-связывающимся доменом p53. Так как взаимодействия MDM2 как с p53, так и с E2F1 локализованы на одном и том же связывающемся сайте MDM2, то ожидается, что MDM2/p53 антагонисты будут не только активировать клеточный p53, но также регулировать воздействия E2F1, которые обычно не регулируются в опухолевых клетках.
Кроме того, терапевтическая эффективность повреждающих ДНК средств, используемых в настоящее время (химиотерапия и лучевая терапия), может быть ограничена путем негативной регуляции p53 посредством MDM2. Таким образом, если прерывается обратная связь ингибирования p53 посредством MDM2, то увеличение функциональных уровней ρ53 приведет к увеличению терапевтической эффективности таких средств за счет восстановления функции p53 дикого типа, что приводит к апоптозу и/или обращению связанной с p53 лекарственной устойчивости. Было продемонстрировано, что сочетание ингибирования MDM2 и повреждающих ДНК обработок in vivo приводит к синергичным противоопухолевым эффектам (Vousden K.H., Cell, Vol. 103, 691-694, 2000).
Таким образом, нарушение взаимодействия MDM2 и ρ53 обеспечивает подход для терапевтического вмешательства в опухоли с диким типом p53, возможно даже проявление анти-пролиферативных эффектов в опухолевых клетках, которые не имеют функциональных p53 и, кроме того, может сделать чувствительными к химиотерапии и радиотерапии онкогенные клетки.
Уровень техники
В JP 11130750, опубликованном 18 мая 1999 года, описаны среди прочих замещенные фениламинокарбонилиндолильные производные в качестве антагонистов рецептора 5-HT.
В EP 1129074, опубликованном 18 мая 2000 года, описаны амиды антраниловой кислоты в качестве ингибиторов рецепторов факторов роста сосудистого эндотелия (VEGFR) и применимые для лечения ангиогенных расстройств.
В EP 1317443, опубликованном 21 марта 2002 года, раскрыты трициклические производные третичных аминов, применимые в качестве модуляторов хемокинового рецептора CXCR4 или CCR5 для лечения вируса иммунодефицита человека и вируса иммунодефицита кошек.
В EP 1379239, опубликованном 10 октября 2002 года, раскрыты N-(2-арилэтил)бензиламины в качестве антагонистов рецептора 5-HT6.
В WO00/15357, опубликованном 23 марта 2000 года, описаны пиперазин-4-фенильные производные в качестве ингибиторов взаимодействия между MDM2 и p53. В EP1137418, опубликованном 8 июня 2000 года, представлены трициклические соединения для восстановления конформационной стабильности белка вида p53.
В EP1443937, опубликованном 22 мая 2003 года, описаны замещенные 1,4-бензодиазепины и их применение в качестве ингибиторов взаимодействий MDM2-p53.
В EP 1458380, опубликованном 26 июня 2003 года, представлены цис-2,4,5-трифенилимидазолоны, которые ингибируют взаимодействие белка MDM2 с p53-подобными пептидами и обладают антипролиферативной активностью.
В EP 1519932, опубликованном 15 января 2004 года, раскрыты бисарилсульфонамидные соединения, которые связываются с MDM2 и могут применяться в терапии рака.
В настоящее время сохраняется потребность в эффективных и сильнодействующих малых молекулах, которые ингибируют взаимодействия между MDM2 и p53.
Соединения по настоящему изобретению отличаются от предшествующего уровня техники по структуре, по их фармакологической активности и/или по фармакологической эффективности.
Описание изобретения
Настоящее изобретение предоставляет соединения, композиции и способы, ингибирующие взаимодействия между MDM2 и p53, для лечения рака. Кроме того, соединения и композиции по настоящему изобретению пригодны для повышения эффективности химиотерапии и лучевой терапии.
Настоящее изобретение относится к соединениям формулы (I)
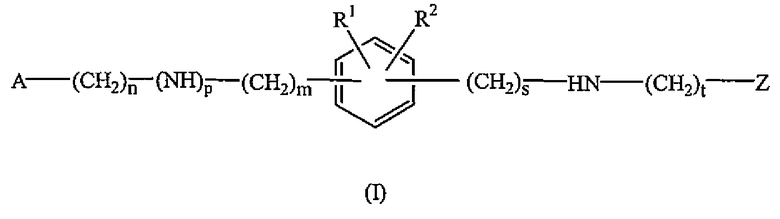
их N-оксидной форме, аддитивной соли или стереохимически изомерной форме, где
m равно 0, 1 или 2 и, когда m равно 0, то подразумевается прямая связь;
n равно 0, 1, 2, 3 или 4 и, когда n равно 0, то подразумевается прямая связь;
p равно 0 или 1 и, когда p равно 0, то подразумевается прямая связь;
s равно 0 или 1 и, когда s равно 0, то подразумевается прямая связь;
t равно 0 или 1 и, когда t равно 0, то подразумевается прямая связь;
R1 и R2 каждый независимо представляет собой водород, галоген, C1-6алкил, C1-6алкилокси, арилC1-6алкилокси, гетероарилC1-6алкилокси, фенилтио, гидроксиC1-6алкилкарбонил, C1-6алкил, замещенный заместителем, выбранным из амино, арила и гетероарила; или C3-7циклоалкил, замещенный заместителем, выбранным из амино, арила и гетероарила;
A представляет собой радикал, выбранный из
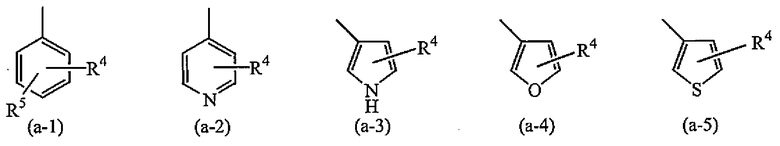
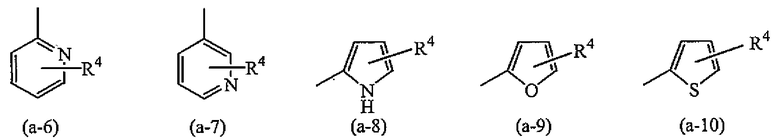
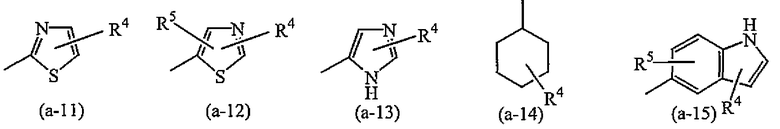


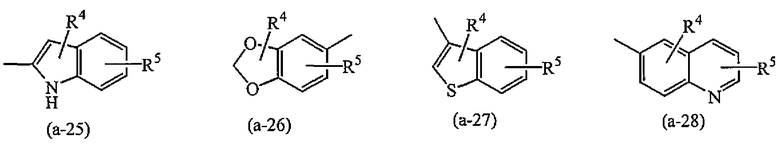
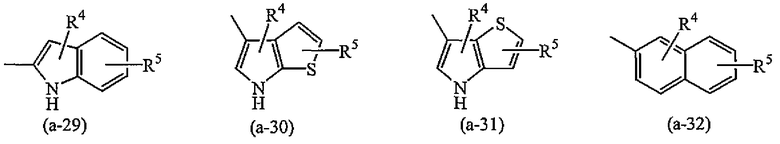


где R4 и R5 каждый независимо выбран из водорода, галогена, C1-6алкила, полигалогенC1-6алкила, циано, цианоC1-6алкила, гидроксиC1-6алкила, гидрокси, амино, C1-6алкилокси, C1-6алкилкарбонила, метилсульфониламино, арила или гетероарила;
Z представляет собой радикал, выбранный из
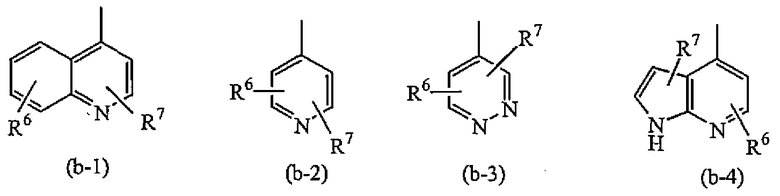
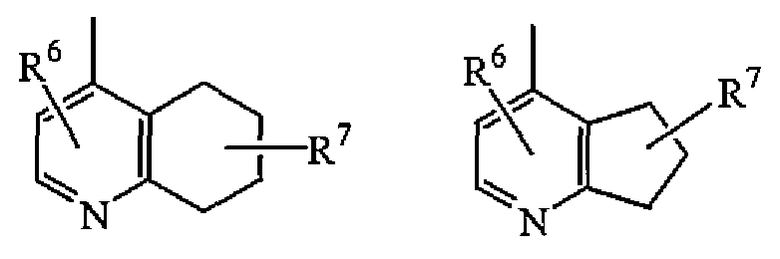
где
R6 и R7 каждый независимо выбран из водорода, галогена, гидрокси, амино, C1-6алкила, нитро, полигалогенC1-6алкила, циано, цианоC1-6алкила, тетразолоC1-6алкила, арила, гетероарила, арилC1-6алкила, гетероарилC1-6алкила, арил(гидрокси)C1-6алкила, гетероарил(гидрокси)C1-6алкила, арилкарбонила, гетероарилкарбонила, C1-6алкилкарбонила, арилC1-6алкилкарбонила, гетероарилC1-6алкилкарбонила, C1-6алкилокси, C3-7циклоалкилкарбонила, C3-7циклоалкил(гидрокси)C1-6алкила, арилC1-6алкилоксиC1-6алкила, C1-6алкилоксиC1-6алкилоксиC1-6алкила, C1-6алкилкарбонилоксиC1-6алкила, C1-6алкилоксикарбонилC1-6алкилоксиC1-6алкила, гидроксиC1-6алкилоксиC1-6алкила, C1-6алкилоксикарбонилC2-6алкенилC1-6алкилоксиC1-6алкила, C1-6алкилоксикарбонила, C1-6алкилкарбонилокси, аминокарбонила, гидроксиC1-6алкила, аминоC1-6алкила, гидроксикарбонила, гидроксикарбонилC1-6алкила и -(CH2)v-(C(=O)r)-(CHR10)u-NR8R9; где
v равно 0, 1, 2, 3, 4, 5 или 6 и, когда v равно 0, то подразумевается прямая связь;
r равно 0 или 1 и, когда r равно 0, то подразумевается прямая связь;
u равно 0, 1, 2, 3, 4, 5 или 6 и, когда u равно 0, то подразумевается прямая связь;
R10 представляет собой водород или C1-6алкил;
R8 и R9 каждый независимо выбран из водорода, C1-12алкила, C1-6алкилкарбонила, C1-6алкилсульфонила, арилC1-6алкилкарбонила, C3-7циклоалкила, C3-7циклоалкилкарбонила, -(CH2)k-NR11R12, C1-12алкила, замещенного заместителем, выбранным из гидрокси, гидроксикарбонила, циано, C1-6алкилоксикарбонила, C1-6алкилокси, арила или гетероарила; или C3-7циклоалкила, замещенного заместителем, выбранным из гидрокси, C1-6алкилокси, арила, амино, арилC1-6алкила, гетероарила или гетероарилC1-6алкила; или R8 и R9 вместе с азотом, к которому они присоединены, могут необязательно образовывать морфолинил, пиперидинил, пирролидинил, пиперазинил или пиперазинил, замещенный заместителем, выбранным из C1-6алкила, арилC1-6алкила, арилC1-6алкилоксикарбонила, гетероарилC1-6алкила, C3-7циклоалкила и C3-7циклоалкилC1-6алкила; где
k равно 0, 1, 2, 3, 4, 5 или 6 и, когда k равно 0, то подразумевается прямая связь;
R11 и R12 каждый независимо выбран из водорода, C1-6алкила, арилC1-6алкилоксикарбонила, C3-7циклоалкила, C1-12алкила, замещенного заместителем, выбранным из гидрокси, C1-6алкилокси, арила и гетероарила; и C3-7циклоалкила, замещенного заместителем, выбранным из гидрокси, C1-6алкилокси, арила, арилC1-6алкила, гетероарила, и гетероарилC1-6алкила; или
R11 и R12 вместе с азотом, к которому они присоединены, могут необязательно образовывать морфолинил, пиперазинил или пиперазинил, замещенный C1-6алкилоксикарбонилом;
арил представляет собой фенил или нафталинил;
каждый фенил или нафталинил необязательно может быть замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из галогена, гидрокси, гидроксиC1-6алкила, C1-6алкила, амино, полигалогенC1-6алкила и C1-6алкилокси; и каждый фенил или нафталинил необязательно может быть замещен бивалентным радикалом, выбранным из метилендиокси и этилендиокси;
гетероарил представляет собой пиридинил, индолил, хинолинил, имидазолил, фуранил, тиенил, оксадиазолил, тетразолил, бензофуранил или тетрагидрофуранил;
каждый пиридинил, индолил, хинолинил, имидазолил, фуранил, тиенил, оксадиазолил, тетразолил, бензофуранил или тетрагидрофуранил необязательно может быть замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из галогена, гидрокси, C1-6алкила, амино, полигалогенC1-6алкила, арила, арилC1-6алкила или C1-6алкилокси; и
каждый пиридинил, индолил, хинолинил, имидазолил, фуранил, тиенил, бензофуранил или тетрагидрофуранил необязательно может быть замещен бивалентным радикалом, выбранным из метилендиокси или этилендиокси.
Соединения формулы (I) могут также существовать в их таутомерных формах. Несмотря на то, что такие формы прямо не указаны в вышеприведенной формуле, они подразумеваются включенными в объем настоящего изобретения.
Ряд терминов, используемых в предыдущих определениях и далее по тексту, приведен ниже. Эти термины иногда используются индивидуально или в составных терминах.
Используемый в предыдущих определениях и далее по тексту галоген представляет собой фтор, хлор, бром и йод; C1-6алкил определяет прямую и разветвленную цепь насыщенного углеводородного радикала, имеющего от 1 до 6 атомов углерода, такого как, например, метил, этил, пропил, бутил, пентил, гексил, 1-метилэтил, 2-метилпропил, 2-метилбутил, 2-метилпентил и т.п.; гидроксиC1-6алкил определяет замещенную гидрокси прямую и разветвленную цепь насыщенного углеводородного радикала, имеющего от 1 до 6 атомов углерода; тригалогенметил определяет метил, содержащий три одинаковых или различных галогеновых заместителя, например, трифторметил; C3-7циклоалкил включает циклическую углеводородную группу, имеющую от 3 до 10 атомов углерода, такую как циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептил и т.п.
Термин «аддитивная соль» включает соли, которые способны образовывать соединения формулы (I) с органическими или неорганическими основаниями, такими как амины, основания щелочных металлов и основания щелочноземельных металлов, или четвертичные аммониевые основания, или с органическими или неорганическими кислотами, такими как неорганические кислоты, сульфоновые кислоты, карбоновые кислоты или фосфорсодержащие кислоты.
Термин «аддитивная соль» дополнительно включает фармацевтически приемлемые соли, комплексы и сольваты металлов и их соли, которые способны образовывать соединения формулы (I).
Термин «фармацевтически приемлемые соли» подразумевает фармацевтически приемлемые кислотно-аддитивные или основно-аддитивные соли. Фармацевтически приемлемые кислотно-аддитивные или основно-аддитивные соли, как подразумевается выше, включают терапевтически активные формы нетоксичной кислотно-аддитивной и нетоксичной основно-аддитивной соли, которые способны образовывать соединения формулы (I). Соединения формулы (I), которые обладают основными свойствами, могут быть преобразованы в их фармацевтически приемлемую кислотно-аддитивную соль путем обработки указанной основной формы подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например, хлористоводородная или бромистоводородная кислота; серная; азотная; фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная (гликолевая), молочная, пировиноградная, щавелевая, малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты.
Соединения формулы (I), которые обладают кислотными свойствами, могут быть преобразованы в их фармацевтически приемлемые основно-аддитивные соли путем обработки указанной кислотной формы подходящим органическим или неорганическим основанием. Соответствующие солевые формы основания включают, например, аммониевые соли, соли щелочных и щелочноземельных металлов, например, соли лития, натрия, калия, магния, кальция и тому подобное, соли органических оснований, например, соли бензатина, N-метил-D-глюкамина, гидрабамина, и соли с аминокислотами, такими как, например, аргинин, лизин и тому подобное.
Термин кислотно-аддитивная или основно-аддитивная соль также включает гидраты и формы с добавлением растворителя, которые способны образовывать соединения формулы (I). Примерами таких форм являются, например, гидраты, алкоголяты и тому подобное.
Термин «комплексы металлов» подразумевает комплекс, образованный между соединением формулы (I) и одной или несколькими органической или неорганической солью или солями металла. Примеры указанных органических или неорганических солей включают галогениды, нитраты, сульфаты, фосфаты, ацетаты, трифторацетаты, трихлорацетаты, пропионаты, тартраты, сульфонаты, например, метилсульфонаты, 4-метилфенилсульфонаты, салицилаты, бензоаты и тому подобное металлов второй основной группы периодической системы, например, соли магния или кальция, третьей или четвертой основной группы, например, соли алюминия, олова, свинца, а также с первой по восьмую переходной группы периодической системы, такие как, например, хрома, марганца, железа, кобальта, никеля, меди, цинка и тому подобное.
Вышеуказанный термин «стереохимически изомерные формы соединений формулы (I)» определяет все возможные соединения, состоящие из тех же самых атомов, связанных той же самой последовательностью связей, но имеющих различные трехмерные структуры, которые не являются равноценными, которыми могут обладать соединения формулы (I). Если не упомянуто или не указано иное, химическое обозначение соединения охватывает смесь всех возможных стереохимически изомерных форм, которые указанное соединение может иметь. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Все стереохимически изомерные формы соединений формулы (I), как в чистом виде, так и в смеси друг с другом, включены в объем настоящего изобретения.
Особый интерес представляют такие соединения формулы (I), которые являются стереохимически чистыми.
Чистые стереоизомерные формы соединений и промежуточных соединений, упомянутые в данном описании, определяют как изомеры, по существу свободные от других энантиомерных или диастереомерных форм такой же молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин «стереоизомерно чистые» относится к соединениям или промежуточным соединениям, имеющим стереоизомерный избыток по меньшей мере от 80% (т.е. минимум 90% одного изомера и максимум 10% других возможных изомеров) до стереоизомерного избытка 100% (т.е. 100% одного изомера и никаких других), более конкретно, соединения или промежуточные соединения, имеющие стереоизомерный избыток от 90% до 100%, еще более конкретно, имеющие стереоизомерный избыток от 94% до 100% и наиболее конкретно, имеющие стереоизомерный избыток от 97% до 100%. Термины «энантиомерно чистый» и «диастереомерно чистый» следует понимать аналогичным образом, но имеющими отношение к энантиомерному избытку, соответственно, диастереомерному избытку в обсуждаемой смеси.
Таутомерные формы соединений формулы (I) подразумеваются включающими такие соединения формулы (I), где, например, енольная группа преобразована в кетогруппу (кето-енольная таутомерия).
N-оксидные формы соединений формулы (I) подразумеваются включающими такие соединения формулы (I), где один или несколько атомов азота окислены до так называемого N-оксида, более конкретно, такие N-оксиды, где один или несколько пиперидиновых, пиперазиновых или пиридазинильных азотов являются N-окисленными.
Соединения формулы (I) могут быть преобразованы в соответствующие N-оксидные формы с использованием следующих известных в данной области методик по преобразованию трехвалентного азота в его N-оксидную форму. Указанная реакция N-окисления, как правило, может быть осуществлена путем взаимодействия исходного вещества формулы (I) с подходящим органическим или неорганическим пероксидом. Подходящие неорганические пероксиды включают, например, перекись водорода, пероксиды щелочных или щелочноземельных металлов, например, пероксид натрия, пероксид калия; подходящие органические пероксиды могут включать пероксикислоты, такие как, например, бензолкарбопероксокислота или галогензамещенная бензолкарбопероксокислота, например, 3-хлорбензолкарбопероксокислота, пероксоалкановые кислоты, например, пероксоуксусная кислота, алкилгидропероксиды, например, трет-бутилгидропероксид. Подходящими растворителями являются, например, вода, низшие спирты, например, этанол и тому подобное, углеводороды, например, толуол, кетоны, например, 2-бутанон, галогенированные углеводороды, например, дихлорметан, и смеси таких растворителей.
Настоящее изобретение также предполагает включение любых изотопов атомов, присутствующих в соединениях по изобретению. Например, изотопы водорода включают тритий и дейтерий, а изотопы углерода включают C-13 и С-14.
Во всех случаях использования в данном описании термин «соединения формулы (I)» подразумевает включение также N-оксидных форм, фармацевтически приемлемых кислотно-аддитивных или основно-аддитивных солей и всех стереоизомерных форм.
Первая группа представляющих интерес соединений состоит из таких соединений формулы (I), где применяется одно или несколько из следующих ограничений:
a) m равно 0;
b) n равно 0 или 2;
c) p равно 1;
d) s равно 0;
e) t равно 0;
f) R1 и R2 каждый независимо представляет собой водород;
g) A представляет собой радикал, выбранный из (a-15), (a-21), (a-30), (a-39) или (a-40);
h) R4 и R5 каждый независимо выбран из водорода или C1-6алкилокси;
i) Z представляет собой радикал (b-2); или
j) R6 и R7 каждый независимо выбран из водорода.
Вторая группа представляющих интерес соединений состоит из таких соединений формулы (I) и таких соединений описанной выше группы, где применяется одно или несколько из следующих ограничений:
a) m равно 0;
b) n равно 2;
c) p равно 1;
d) s равно 0;
e) t равно 0;
f) R1 и R2 каждый независимо представляет собой водород;
g) A представляет собой радикал, выбранный из (a-21), (a-39) или (a-40);
h) R4 и R5 каждый независимо выбран из водорода или C1-6алкилокси;
i) Z представляет собой радикал (b-2); или
j) R6 и R7 каждый независимо выбран из водорода.
Группа предпочтительных соединений состоит из таких соединений формулы (I) или любой их подгруппы, где m равно 0; n равно 0 или 2; p равно 1; s равно 0; t равно 0; R1 и R2 каждый независимо представляет собой водород; A представляет собой радикал, выбранный из (a-15), (a-21), (a-30), (a-39) или (a-40); R4 и R5 каждый независимо выбран из водорода или C1-6алкилокси; Z представляет собой радикал (b-2); или R6 и R7 каждый независимо выбран из водорода.
Группа более предпочтительных соединений состоит из таких соединений формулы (I) или любой их подгруппы, где m равно 0; n равно 2; p равно 1; s равно 0; t равно 0; R1 и R2 каждый независимо представляет собой водород; A представляет собой радикал, выбранный из (a-21), (a-39) или (a-40); R4 и R5 каждый независимо выбран из водорода или C1-6алкилокси; Z представляет собой радикал (b-2); или R6 и R7 каждый независимо выбран из водорода.
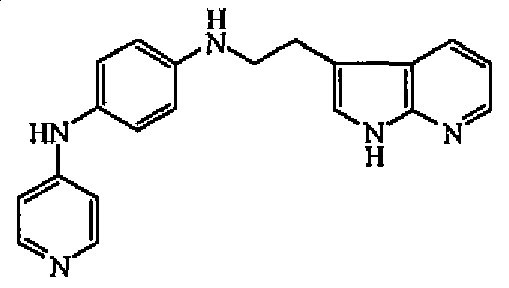
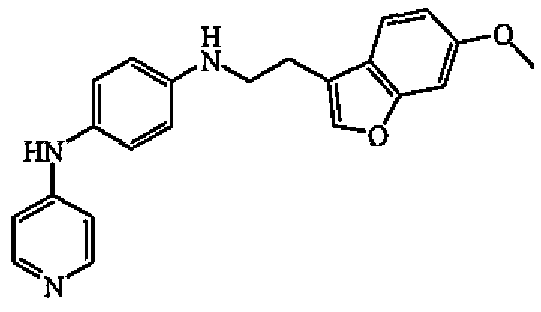
Наиболее предпочтительными соединениями являются соединение № 2, соединение № 3 или соединение № 5.

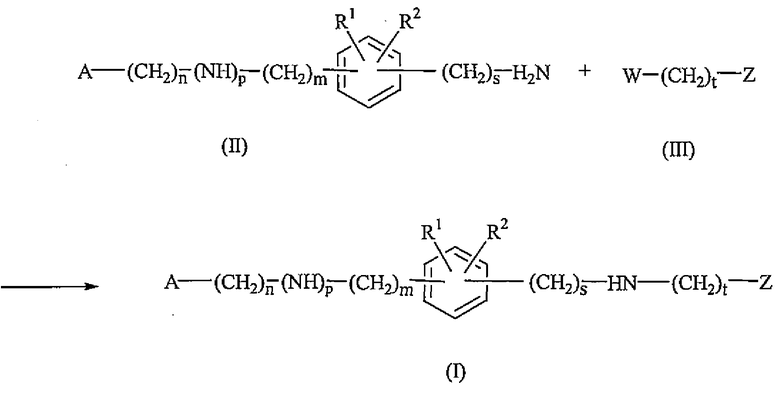
Соединения формулы (I), их фармацевтически приемлемые соли и N-оксидные и стереохимически изомерные формы могут быть получены общепринятыми способами. Исходные вещества и некоторые промежуточные соединения являются известными соединениями и коммерчески доступны или могут быть получены согласно общепринятым реакционным методикам, известным в данной области.
Ряд таких способов получения будет описан далее более подробно. Другие способы получения конечных соединений формулы (I) описаны в примерах.
Соединения формулы (I) могут быть получены взаимодействием промежуточного соединения формулы (II) с промежуточным соединением формулы (III), где W представляет собой соответствующую уходящую группу, такую как, например, галоген, например, фтор, хлор, бром или йод, или радикал сульфонилокси, такой как метилсульфонилокси, 4-метилфенилсульфонилокси и подобные. Реакция может быть проведена в реакционно-инертном растворителе, таком как, например, спирт, например, метанол, этанол, 2-метоксиэтанол, пропанол, бутанол и тому подобное; эфир, например 4,4-диоксан, 1,1'-оксибиспропан и тому подобное; кетон, например, 4-метил-2-пентанон; или N,N-диметилформамид, нитробензол, ацетонитрил, уксусная кислота и тому подобное. Для поглощения кислоты, которая высвобождается в процессе реакции, можно использовать добавление подходящего основания, такого как, например, карбонат или гидрокарбонат щелочного или щелочноземельного металла, например, триэтиламин или карбонат натрия. Небольшое количество соответствующего йодида металла, например, йодида натрия или калия, может быть добавлено для активирования реакции. Перемешивание может увеличить скорость реакции. Реакцию удобно проводить при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси с обратным холодильником, и, если требуется, реакцию можно проводить при повышенном давлении.
Соединения формулы (I), где p равно 1, именуемые здесь соединениями формулы (I-a), могут быть получены преобразованием промежуточных соединений формулы (IV) с помощью алюмогидрида лития в подходящем растворителе, таком как тетрагидрофуран.
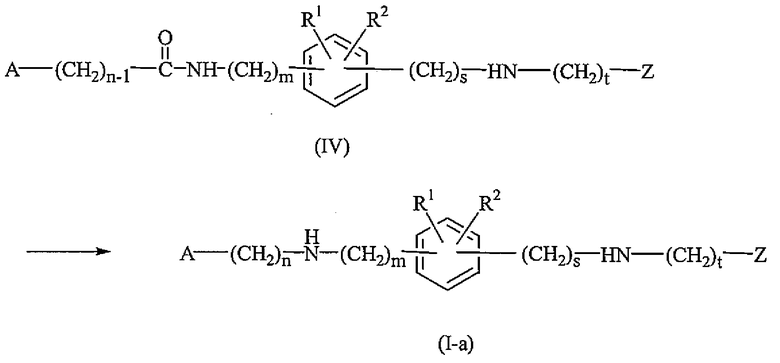
Соединения формулы (I-a) могут быть также получены взаимодействием соответствующего карбоксальдегида формулы (VI) с промежуточным соединением формулы (V) в присутствии соответствующего реагента, такого как боргидрид натрия, например, тетрагидроборат натрия или нанесенный на полимер цианотригидроборат, в подходящем растворителе, таком как спирт, например, метанол.
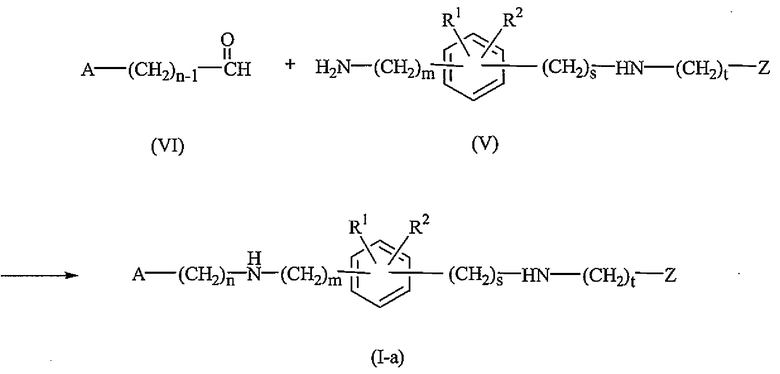
Таким же способом соединения формулы (I), где t равно 1, именуемые здесь соединениями формулы (I-b), могут быть получены взаимодействием промежуточного соединения формулы (II) с соответствующим карбоксальдегидом формулы (VII).
Соединения формулы (I), где s равно 1, именуемые здесь соединениями формулы (I-c), могут быть получены взаимодействием промежуточного соединения формулы (VIII) с алюмогидридом лития в подходящем растворителе, таком как тетрагидрофуран.

Соединения формулы (I) или их промежуточные соединения могут быть также преобразованы друг в друга посредством известных в данной области реакций или трансформаций функциональных групп. Ряд таких трансформаций уже описан выше. Другими примерами являются гидролиз сложных эфиров соответствующих карбоновых кислот или спиртов; гидролиз амидов соответствующих карбоновых кислот или аминов; гидролиз нитрилов соответствующих амидов; аминогруппы у имидазола или фенила могут быть заменены водородом путем известных в данной области реакций диазотирования и последующей замены диазогруппы водородом; спирты могут быть преобразованы в простые и сложные эфиры; первичные амины могут быть преобразованы во вторичные или третичные амины; двойные связи могут быть гидрированы до соответствующих одинарных связей; йодный радикал у фенильной группы может быть преобразован в сложноэфирную группу путем введения моноксида углерода в присутствии подходящего палладиевого катализатора.
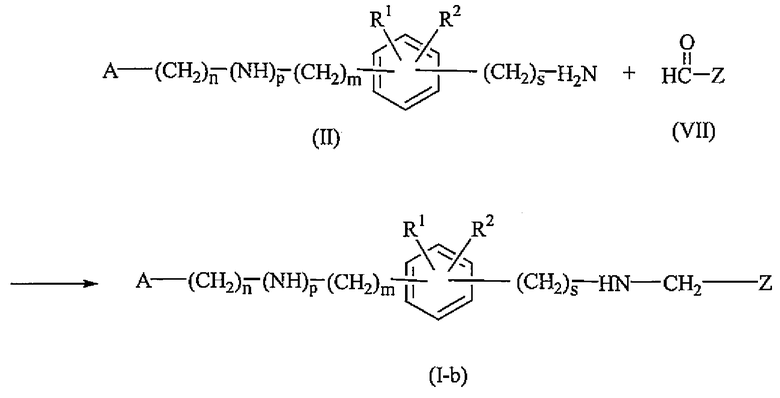
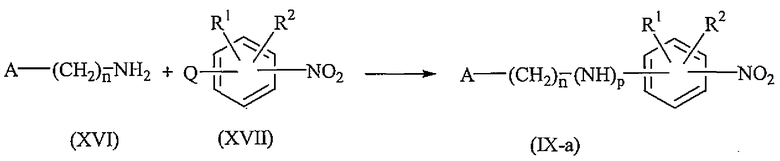
Промежуточные соединения формулы (II), где m равно 0 и s равно 0, именуемые здесь промежуточными соединениями формулы (II-a), могут быть получены реакцией восстановления нитрогруппы в амин, исходя из промежуточного соединения формулы (IX), в присутствии металлического катализатора, такого как никель Ренея, и подходящего восстановителя, такого как водород, в подходящем растворителе, таком как метанол или этанол.

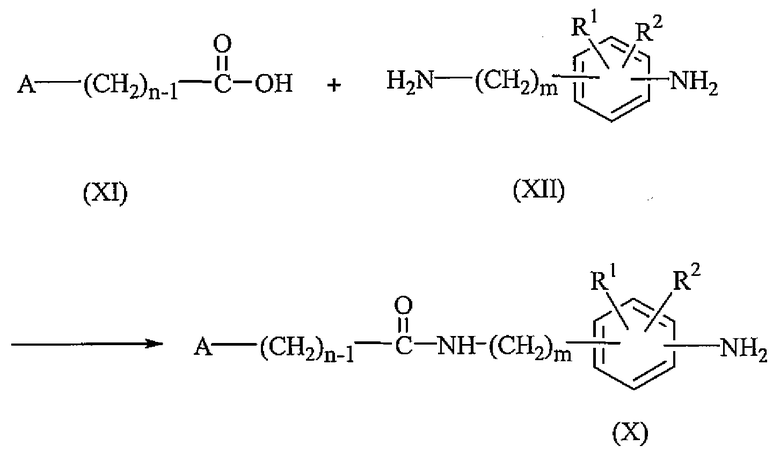
Промежуточные соединения формулы (X), где s равно 0, могут быть получены взаимодействием промежуточного соединения формулы (XI) с промежуточным соединением формулы (XII) в присутствии соответствующих реагентов, таких как моногидрохлорид N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина (EDC) и 1-гидрокси-1H-бензотриазол (HOBT). Реакция может быть проведена в присутствии основания, такого как триэтиламин, в подходящем растворителе, таком как смесь дихлорметана и тетрагидрофурана.
Промежуточные соединения формулы (VI) могут быть получены взаимодействием промежуточных соединений формулы (XIII) с алюмогидридом лития в подходящем растворителе, таком как тетрагидрофуран.
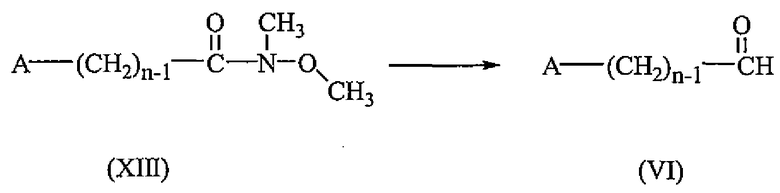
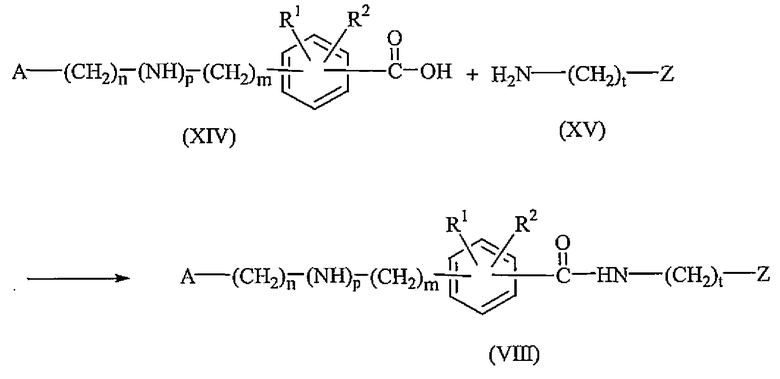
Промежуточные соединения формулы (VIII) могут быть получены взаимодействием промежуточного соединения формулы (XIV) с промежуточным соединением формулы (XV) в присутствии йодида 2-хлор-1-метилпиридиния и триэтиламина в подходящем растворителе, таком как ацетонитрил.
Промежуточные соединения формулы (IX), где p равно 1, именуемые здесь промежуточными соединениями формулы (IX-a), могут быть получены взаимодействием промежуточного соединения формулы (XVI) с промежуточным соединением формулы (XVII), где Q представляет собой соответствующую уходящую группу, такую как, например, галоген, например, фтор, хлор, бром или йод, или C1-6алкилокси, например, метилокси, в диизопропилэтиламине.
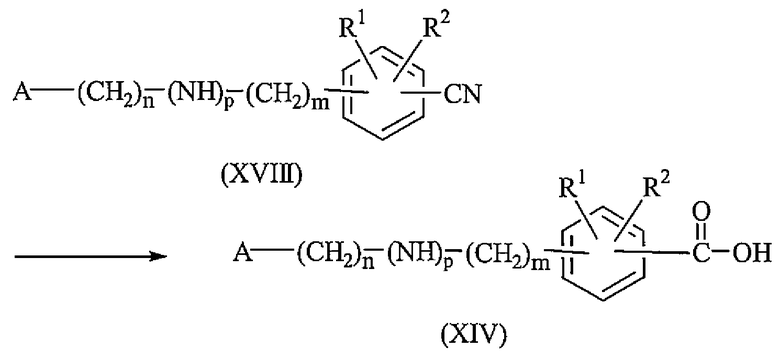
Промежуточные соединения формулы (XIV) могут быть получены преобразованием промежуточного соединения формулы (XVIII) в присутствии гидроксида натрия и воды в подходящем растворителе, таком как этанол.
Промежуточные соединения формулы (XVIII), где m равно 0, именуемые здесь промежуточными соединениями формулы (XVIII-a), могут быть получены взаимодействием промежуточного соединения формулы (XVI) с промежуточным соединением формулы (XIX), где Q определен выше, в подходящем растворителе, таком как диизопропилэтиламин.

Промежуточные соединения формулы (V), где m, n и s равны 0, именуемые здесь промежуточными соединениями формулы (V-a), могут быть получены преобразованием промежуточного соединения формулы (XX) с помощью раствора гидрохлорида.
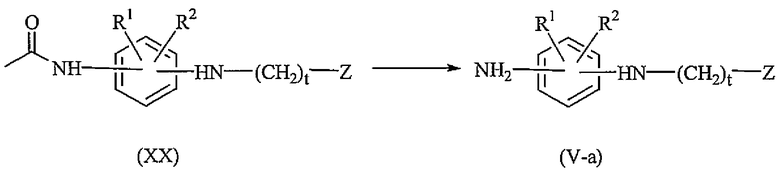
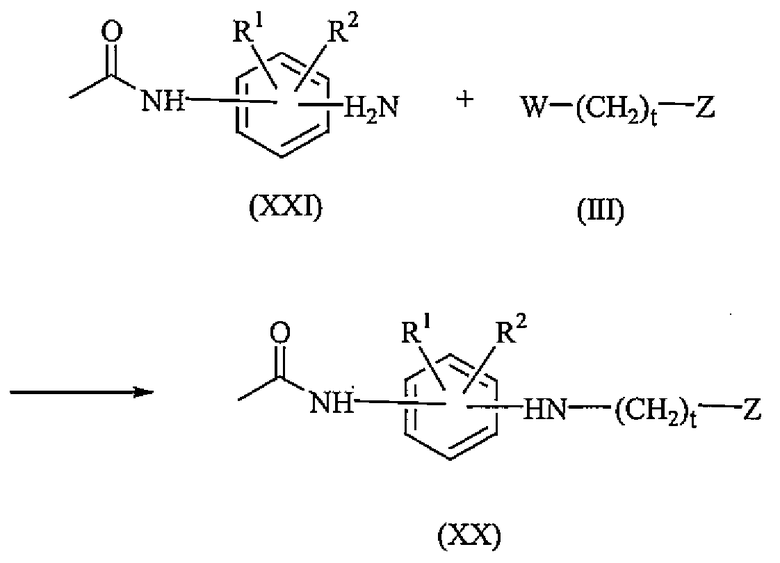
Промежуточные соединения формулы (XX) могут быть получены взаимодействием промежуточного соединения формулы (XXI) с промежуточным соединением формулы (III), где W представляет собой соответствующую уходящую группу, такую как, например, галоген. Реакция может быть проведена в реакционно-инертном растворителе, таком как, например, уксусная кислота.
Соединения формулы (I) и некоторые промежуточные соединения могут иметь по меньшей мере один стереогенный центр в своей структуре. Такие стереогенные центры могут быть представлены в R- или S-конфигурации.
Некоторые соединения формулы (I) и некоторые промежуточные соединения в настоящем изобретении могут содержать асимметрический атом углерода. Стереохимически чистые изомерные формы указанных соединений и указанных промежуточных соединений могут быть получены с использованием известных в данной области методик. Например, диастереоизомеры могут быть разделены физическими способами, такими как селективная кристаллизация или хроматографические методы, например, распределение противотока, жидкостная хроматография и подобные способы. Энантиомеры могут быть получены из рацемических смесей первичным преобразованием указанных рацемических смесей в смеси диастереомерных солей или соединений с помощью подходящих разделяющих агентов, таких как, например, хиральные кислоты; затем физическим разделением указанных смесей диастереомерных солей или соединений с использованием, например, селективной кристаллизации, хроматографии со сверхкритической подвижной фазой или хроматографических методов, например, жидкостной хроматографии, и подобных способов; и заключительным преобразованием указанных выделенных диастереомерных солей или соединений в соответствующие энантиомеры. Стереохимически чистые изомерные формы можно также получить из стереохимически чистых изомерных форм соответствующих промежуточных соединений и исходных веществ, обеспечивая стереоспецифическое протекание промежуточных реакций.
Соединения формулы (I), их фармацевтически приемлемые кислотно-аддитивные соли и стереоизомерные формы обладают ценными фармакологическими свойствами, заключающимися в ингибировании взаимодействия между p53 и MDM2.
Используемый в настоящем описании термин «MDM2» означает белок, полученный в результате экспрессии гена mdm2. В значении этого термина MDM2 охватывает все белки, кодируемые mdm2, их мутанты, альтернативные слои их белков и их фосфорилированные белки. Кроме того, как используется в настоящем описании, термин «MDM2» включает аналоги MDM2, например, MDMX, также известный как MDM4, и гомологи и аналоги MDM2 других животных, например, гомолог HDM2 человека или аналог HDMX человека.
Используемый в настоящем описании термин «ингибирующий взаимодействие» или «ингибитор взаимодействия» обозначает предотвращение или понижение непосредственной или косвенной ассоциации одной или нескольких молекул, пептидов, белков, ферментов или рецепторов; или предотвращение или понижение обычной активности одной или нескольких молекул, пептидов, белков, ферментов или рецепторов.
Термин «ингибитор взаимодействия между p53 и MDM2» или «ингибитор p53-MDM2» используется в настоящем документе для описания агента, который повышает экспрессию p53 в анализе, описанном в C.1. Это повышение может быть вызвано за счет одного или нескольких следующих механизмов воздействия, но не ограничивается приведенным:
- ингибирование взаимодействия между p53 и MDM2,
- непосредственное связывание либо с MDM2, либо с белком p53,
- взаимодействие с вышележащими или нижележащими мишенями, например киназами, или ферментативная активность, включенная в убиквитинирование или SUMO модификацию,
- изолирование или транспортировка MDM2 и p53 в различные клеточные компартменты,
- модуляция белков, связывающихся с MDM2, например (но не ограничиваясь приведенным), p73, E2F-1, Rb, p21wafl или cipl,
- реактивация или нарушение экспрессии MDM2 и/или активности MDM2, например (но не ограничиваясь приведенным), воздействием на его клеточную локализацию, посттрансляционную модификацию, ядерный экспорт или убиквитинлигазную активность,
- прямая или косвенная стабилизация белка p53, например, путем удерживания его в функциональной структурной форме, или путем предотвращения его агрегирования,
- повышение экспрессии p53 или экспрессии членов семейства p53, например, p63 и p73,
- повышение активности p53, например (но не ограничиваясь приведенным), путем повышения его транскрипционной активности и/или
- повышение экспрессии генов и белков p53-сигнального пути, например (но не ограничиваясь приведенным), p21wafl, cipl, MIC-I (GDF-15), PIG-3 и ATF-3.
Таким образом, настоящее изобретение раскрывает соединения формулы (I) для применения в качестве лекарственных средств.
Кроме того, настоящее изобретение также относится к применению соединения для получения лекарственного средства для лечения расстройства, вызванного взаимодействием p53-MDM2, где указанное соединение представляет собой соединение формулы (I).
Используемый в настоящем документе термин «лечение» или «лечащий» включает любое лечение болезни и/или состояния у животного, в частности, у человека, и включает: (i) предотвращение болезни и/или состояния, имеющих место у субъекта, который может быть предрасположенным к болезни и/или состоянию, но которому еще не поставили диагноз; (ii) ингибирование болезни и/или состояния, то есть купирование их развития; (iii) облегчение болезни и/или состояния, то есть ослабление симптомов болезни и/или улучшение состояния.
В термине «расстройство, вызванное взаимодействием p53-MDM2» подразумевается любое нежелательное или вредное состояние, которое приводит к ингибированию или ингибирует взаимодействие между белком MDM2 и p53 или другими клеточными белками, которые индуцируют апоптоз, индуцируют смерть клеток или регулируют клеточный цикл.
Настоящее изобретение также предоставляет способ лечения расстройства, вызванного взаимодействием p53-MDM2, путем введения эффективного количества соединения по настоящему изобретению пациенту, например, млекопитающему (и более конкретно человеку), при необходимости такого лечения.
Соединения по настоящему изобретению могут обладать антипролиферативными эффектами в опухолевых клетках, даже если такие клетки лишены функционального p53. Более конкретно, соединения по настоящему изобретению могут обладать антипролиферативными эффектами в опухолевых клетках дикого типа p53 и/или в опухолях со сверхэкспрессией MDM2.
Таким образом, настоящее изобретение также предоставляет способ ингибирования роста опухоли путем введения эффективного количества соединения по настоящему изобретению пациенту, например, млекопитающему (и более конкретно человеку), при необходимости такого лечения.
Примерами опухолей, которые можно ингибировать, являются, но не ограничиваются ими, рак легкого (например, аденокарцинома и, в том числе, немелкоклеточный рак легкого), рак поджелудочной железы (например, рак поджелудочной железы, такой как, например, экзокринный рак поджелудочной железы), рак толстой кишки (например, колоректальные карциномы, такие как, например, аденокарцинома толстой кишки и аденома прямой кишки), рак пищевода, сквамозная карцинома полости рта, рак языка, рак желудка, рак носоглотки, гемопоэтические опухоли лимфоидного ряда (например, острый лимфоцитарный лейкоз, B-клеточная лимфома, лимфома Беркитта), не-ходжкинская лимфома, болезнь Ходжкина, миелоидная лейкемия (например, острая миелогенная лейкемия (AML)), фолликулярный рак щитовидной железы, миелодиспластический синдром (MDS), опухоли мезенхимного происхождения (например, фибросаркомы и рабдомиосаркомы), меланомы, тератокарциномы, нейробластомы, опухоли мозга, глиомы, доброкачественные опухоли кожи (например, кератоакантомы), рак груди (например, прогрессирующий рак молочной железы), рак почки, рак яичников, рак шейки матки, рак эндометрия, рак мочевого пузыря, рак предстательной железы, включая запущенное заболевание, тестикулярные формы рака, остеосаркома, рак головы и шеи и эпидермальная карцинома.
Соединения по настоящему изобретению также можно применять для лечения и профилактики воспалительных состояний.
Таким образом, настоящее изобретение также предоставляет способ лечения и профилактики воспалительных состояний путем введения эффективного количества соединения по настоящему изобретению пациенту, например, млекопитающему (и более конкретно человеку), при необходимости такого лечения.
Соединения по настоящему изобретению также можно применять для лечения аутоиммунных заболеваний и состояний. Под термином «аутоиммунные заболевания» понимают любое заболевание, при котором иммунная система животного враждебно реагирует на аутоантиген. Под термином «аутоантиген» подразумевают любой антиген, который обычно присутствует в организме животного. Характерные аутоиммунные заболевания включают, но не ограничиваются ими, тиреоидит Хашимото, болезнь Граве, рассеянный склероз, пернициозную анемию, болезнь Аддисона, инсулинозависимый сахарный диабет, ревматоидный артрит, системную красную волчанку (SLE или волчанка), дерматомиозит, болезнь Крона, гранулематоз Вегенера, болезнь клубочковой антибазальной мембраны, антифосфолипидный синдром, герпетиформный дерматит, аллергический энцефаломиелит, гломерулонефрит, диффузную мембранозную гломерулопатию, синдром Гудпасчура, миастенический синдром Ламберта-Итона, миастению гравис, буллезный пемфигоид, полиэндокринопатию, синдром Рейтера и синдром мышечной скованности.
Таким образом, настоящее изобретение также предоставляет способ лечения аутоиммунных заболеваний и состояний и лечения заболеваний, связанных с конформационной нестабильностью неправильно упакованных белков, путем введения эффективного количества соединения по настоящему изобретению пациенту, например, млекопитающему (и более конкретно человеку), при необходимости такого лечения.
Соединения по настоящему изобретению также можно применять для лечения заболеваний, связанных с конформационной нестабильностью неправильно упакованных белков. Примеры заболеваний, связанных с конформационной нестабильностью неправильно упакованных белков, включают, но не ограничиваются ими, кистозный фиброз (CFTR), синдром Марфана (фибриллин), боковой амиотрофический склероз (супероксиддисмутаза), цингу (коллаген), болезнь кленового сиропа (комплекс дегидрогеназы альфа-кетокислоты), несовершенный остеогенез (типа про-альфа проколлагена), болезнь Крейтцфельда-Якоба (прион), болезнь Альцгеймера (бета-амилоид), семейный амилоидоз (лизоцим), катаракты (кристаллины), семейную гиперхолестеринемию (LDL рецептор), дефицит ингибитора α-I-трипсина, болезнь Тай-Сакса (бета-гексозаминидаза), пигментный ретинит (родопсин) и лепречаунизм (инсулиновый рецептор).
Таким образом, настоящее изобретение также предоставляет способ лечения заболеваний, связанных с конформационной нестабильностью неправильно упакованных белков, путем введения эффективного количества соединения по настоящему изобретению пациенту, например, млекопитающему (и более конкретно человеку), при необходимости такого лечения.
Ввиду их полезных фармакологических свойств, соединения по настоящему изобретению могут быть составлены в различные фармацевтические формы для целевого назначения.
Для приготовления фармацевтических композиций по настоящему изобретению эффективное количество конкретного соединения, в форме основно-аддитивной или кислотно-аддитивной соли, в качестве активного ингредиента объединяют в однородную смесь с фармацевтически приемлемым носителем, который может вмещать широкое разнообразие форм в зависимости от получаемой желаемой формы для введения. Эти фармацевтические композиции желательны в единичных дозированных формах, подходящих, предпочтительно, для введения орально, ректально, подкожно или путем парентеральной инъекции. Например, для приготовления композиций в лекарственной форме для орального применения может быть использована любая из обычных фармацевтических сред, таких как, например, вода, гликоли, масла, спирты и тому подобное в случае жидких препаратов для орального применения, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, лубриканты, связывающие агенты, дезинтегрирующие агенты и тому подобное в случае порошков, пилюль, капсул и таблеток.
Ввиду своей легкости в применении, таблетки и капсулы представляют собой наиболее выгодную стандартную лекарственную форму для орального применения, в случае которой, очевидно, используют твердые фармацевтические носители. Для парентеральных композиций носитель обычно будет содержать стерильную воду, по меньшей мере в основной части, хотя могут быть включены другие ингредиенты для улучшения растворимости. Растворы для инъекций могут быть приготовлены, например, такими, носитель которых содержит солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Суспензии для инъекций также могут быть приготовлены такими, для которых используют подходящие жидкие носители, суспендирующие агенты и тому подобное. В композициях, подходящих для подкожного введения, носитель необязательно содержит повышающий проникновение агент и/или подходящий увлажняющий агент, необязательно объединенный с подходящими добавками любой природы в небольших пропорциях, добавление которых не вызывает значительного вредного воздействия на кожу. Указанные добавки могут способствовать введению в кожу и/или могут быть полезными для приготовления требуемых композиций. Эти композиции могут быть введены различными способами, например, в виде трансдермального пластыря, точечным способом, в виде мази. Особенно выгодно готовить вышеупомянутые фармацевтические композиции в стандартной лекарственной форме для каждого применения и единообразных дозировках. Стандартная лекарственная форма, используемая в описании и формуле изобретения настоящего документа, относится к физически дискретным единицам, подходящим в качестве единичных доз, каждая единица, содержащая заданное количество активного ингредиента, рассчитывается для обеспечения желаемого терапевтического эффекта в соединении с требуемым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая таблетки с насечкой и покрытые таблетки), капсулы, пилюли, пакетики с порошком, облатки, растворы или суспензии для инъекций, умещающееся в чайной ложке количество, умещающееся в столовой ложке количество и тому подобное, и отдельные их кратные.
Соединения по настоящему изобретению вводят в количестве, достаточном для ингибирования взаимодействия между MDM2 и p53 или другим белком, который индуцирует апоптоз, индуцирует смерть клеток или регулирует клеточный цикл.
Онкогенный потенциал MDM2 определяется не только его способностью подавлять p53, но также его способностью регулировать другие подавляющие опухоль белки, например, белок ретинобластомы pRb и плотно связанные факторы транскрипции E2F1.
Таким образом, соединение по настоящему изобретению вводят в количестве, достаточном для модуляции взаимодействия между MDM2 и факторами транскрипции E2F.
Квалифицированные в данной области специалисты могут легко определить эффективное количество из тестовых результатов, представленных ниже в тексте настоящего документа. Как правило, предполагается, что терапевтически эффективным количеством может быть от 0,005 мг/кг до 100 мг/кг массы тела и, конкретно, от 0,005 мг/кг до 10 мг/кг массы тела. Это может быть целесообразным для введения требуемой дозы в виде одной, двух, трех, четырех или более под-доз за подходящий интервал времени в течение дня. Указанные под-дозы могут быть приготовлены в виде стандартных лекарственных форм, например, содержащих от 0,5 до 500 мг и, конкретно, от 10 мг до 500 мг активного ингредиента на стандартную лекарственную форму.
Другой аспект настоящего изобретения предусматривает комбинирование ингибитора p53-MDM2 с другим противораковым средством, особенно для применения в качестве лекарственного средства, более конкретно, для лечения рака или связанных заболеваний.
Для лечения вышеуказанных состояний соединения по настоящему изобретению могут быть преимущественно использованы в комбинации с одним или несколькими лекарственными средствами, более конкретно, с другими противораковыми средствами. Примеры противораковых средств включают, но не ограничиваются ими:
- координационные соединения платины, например, цисплатин, карбоплатин или оксалиплатин;
- таксановые соединения, например, паклитаксел или доцетаксел;
- ингибиторы топоизомеразы I, такие как камптотециновые соединения, например, иринотекан или топотекан;
- ингибиторы топоизомеразы II, такие как противоопухолевые эпиподофиллотоксины или производные подофиллотоксина, например, этопозид или тенипозид;
- противоопухолевые алкалоиды барвинка, например, винбластин, винкристин или винорелбин;
- противоопухолевые производные нуклеозидов, например, 5-фторурацил, лейковорин, гемцитабин или капецитабин;
- алкилирующие средства, такие как азотистый иприт или нитрозомочевина, например, циклофосфамид, хлорамбуцил, кармустин, тиотепа, мефалан или ломустин;
- противоопухолевые производные антрациклина, например, даунорубицин, доксорубицин, доксил, идарубицин или митоксантрон;
- молекулы, которые являются мишенями для рецептора IGF-1, например, пикроподофилин;
- производные тетракарцина, например, тетракарцин A;
- глюкокортикоиды, например, преднизон;
- антитела, например, трастузумаб (HER2-антитело), ритуксимаб (CD20-антитело), гемтузумаб, цетуксимаб, пертузумаб или бивацизумаб;
- антагонисты рецептора эстрогена или селективные модуляторы рецептора эстрогена, например, тамоксифен, фульвестрант, торемифен, дролоксифен, фаслодекс или ралоксифен;
- ингибиторы ароматазы, такие как эксеместан, анастрозол, летразол и ворозол;
- дифференцирующие средства, такие как ретиноиды, витамин D или ретиноевая кислота и средства, блокирующие метаболизм ретиноевой кислоты (RAMBA), например, аккутан;
- ингибиторы ДНК-метилтрансферазы, например, азацитидин или децитабин;
- антифолаты, например, преметрексед динатрия;
- антибиотики, например, антиномицин D, блеомицин, митомицин С, дактиномицин, карминомицин или дауномицин;
- антиметаболиты, например, клофарабин, аминоптерин, цитозин-арабинозид или метотрексат;
- индуцирующие апоптоз средства и противоангиогенные средства, такие как ингибиторы Bcl-2, например, YC 137, BH 312, АВТ 737, госсипол, HA 14-1, TW 37 или декановая кислота;
- тубулин-связывающие средства, например, комбрестатин, колхицин или нокодазол;
- ингибиторы киназ, например, флавоперидол, иматиниба мезилат, эрлотиниб или гефитиниб;
- ингибиторы фарнезилтрансферазы, например, типифарниб;
- ингибиторы гистондеацитилазы (HDAC), например, бутират натрия, субероиланилид-гидроксамовая кислота (SAHA), депсипептид (FR 901228), NVP-LAQ824, R306465, JNJ-26481585 или трихостатин А;
- ингибиторы убиквитин-протеасомного пути, например, PS-341, MLN 41 или бортезомиб;
- Yondelis;
- ингибиторы теломеразы, например, теломестатин;
- ингибиторы матричной металлопротеиназы, например, батимастат, маримастат, приностат или метастат.
Как утверждается выше, соединения по настоящему изобретению также могут иметь терапевтическое применение в опухолевых клетках, чувствительных к химиотерапии и лучевой терапии.
Поэтому соединения по настоящему изобретению могут быть использованы в качестве «радиосенсибилизатора» и/или «хемосенсибилизатора», или могут быть представлены в комбинации с другим «радиосенсибилизатором» и/или «хемосенсибилизатором».
Используемый в настоящем описании термин «радиосенсибилизатор» определяют как молекулу, предпочтительно с низкой молекулярной массой, применяемую для животных в терапевтически эффективных количествах для повышения восприимчивости клеток к ионизирующей радиации и/или способствующую лечению заболеваний, которые можно лечить с помощью лучевой терапии.
Используемый в настоящем описании термин «хемосенсибилизатор» определяют как молекулу, предпочтительно с низкой молекулярной массой, применяемую для животных в терапевтически эффективных количествах для повышения восприимчивости клеток к химиотерапии и/или способствующую лечению заболеваний, которые можно лечить с помощью химиотерапии.
В литературе было предложено несколько механизмов для принципа действия радиосенсибилизаторов, включая: радиосенсибилизаторы гипоксических клеток (например, соединения 2-нитроимидазола и соединения диоксида бензотриазина), имитирующие кислородное или иное поведение, подобное биоредуктивным средствам при гипоксии; радиосенсибилизаторы негипоксических клеток (например, галогенированные пиримидины), которые могут представлять собой аналоги оснований ДНК и предпочтительно включаются в ДНК раковых клеток и, тем самым, способствуют разрушению молекулы ДНК под действием радиации и/или препятствуют обычным механизмам восстановления ДНК; и различные иные потенциальные механизмы действия были выдвинуты в качестве гипотезы для радиосенсибилизаторов при лечении заболеваний.
Во множестве методик лечения рака в настоящее время используют радиосенсибилизаторы в сочетании с облучением рентгеновскими лучами. Примеры радиосенсибилизаторов, активируемых рентгеновскими лучами, включают, но не ограничиваются ими, следующие: метронидазол, мизонидазол, десметилмизонидазол, пимонидазол, этанидазол, ниморазол, митомицин С, RSU 1069, SR 4233, EO9, RB 6145, никотинамид, 5-бромдезоксиуридин (BUdR), 5-йододезоксиуридин (IUdR), бромдезоксицитидин, фтордезоксиуридин (FudR), гидроксимочевина, цисплатин и терапевтически эффективные аналоги и производные таковых.
В фотодинамической терапии (PDT) рака используют видимый свет в качестве активатора сенсибилизирующего средства. Примеры фотодинамических радиосенсибилизаторов включают, но не ограничиваются ими: производные гематопорфирина, фотофрин, производные бензопорфирина, этиопорфирин олова, феоборбид-а, бактериохлорофилл-а, нафталоцианины, фталоцианины, фталоцианин цинка и терапевтически эффективные аналоги и производные таковых.
Радиосенсибилизаторы можно применять в сочетании с терапевтически эффективным количеством одного или нескольких соединений, включая, но не ограничиваясь ими: соединения, которые способствуют включению радиосенсибилизаторов в клетки-мишени; соединения, которые контролируют потоки терапевтических веществ, питательных веществ и/или кислорода в клетках-мишенях; химиотерапевтические средства, которые действуют на опухоль с дополнительным облучением или без него, или другие терапевтически эффективные соединения для лечения рака или иных заболеваний.
Хемосенсибилизаторы можно применять в сочетании с терапевтически эффективным количеством одного или нескольких соединений, включая, но не ограничиваясь ими: соединения, которые способствуют включению хемосенсибилизаторов в клетки-мишени; соединения, которые контролируют потоки терапевтических веществ, питательных веществ и/или кислорода в клетках-мишенях; химиотерапевтические средства, которые действуют на опухоль, или другие терапевтически эффективные соединения для лечения рака или иных заболеваний. Кальциевый антагонист, например верапамил, оказался пригодным в комбинации с противоопухолевыми средствами для обеспечения чувствительности к химиотерапии в опухолевых клетках, резистентных к общепринятым химиотерапевтическим средствам, и для усиления эффективности таких соединений в восприимчивых к лекарственным средствам злокачественных образованиях.
Ввиду их полезных фармакологических свойств, компоненты комбинаций согласно настоящему изобретению, то есть другое лекарственное средство и ингибитор p53-MDM, могут быть составлены в различные фармацевтические формы для целей введения. Компоненты могут быть составлены отдельно в индивидуальные фармацевтические композиции или единую фармацевтическую композицию, содержащую оба компонента.
Настоящее изобретение, таким образом, также относится к фармацевтической композиции, содержащей другое лекарственное средство и ингибитор p53-MDM2 вместе с одним или несколькими фармацевтическими носителями.
Настоящее изобретение дополнительно относится к применению комбинации согласно настоящему изобретению для получения фармацевтической композиции для ингибирования роста опухолевых клеток.
Настоящее изобретение дополнительно относится к продукту, содержащему в качестве первого активного ингредиента ингибитор p53-MDM2 согласно настоящему изобретению и в качестве второго активного ингредиента противораковое средство, в виде комбинированного препарата для одновременного, отдельного или последовательного применения для лечения пациентов, страдающих от рака.
Другое лекарственное средство и ингибитор p53-MDM2 могут быть введены одновременно (например, в отдельных или единой композиции) или последовательно в том или ином порядке. В последнем случае, два соединения вводят в течение периода и в количестве и способом, которые достаточны для гарантированного достижения выгодного или синергичного эффекта. Следует принять, что предпочтительный способ, порядок введения, относительные дозированные количества и режимы для каждого компонента комбинации будут зависеть от конкретного другого лекарственного средства и ингибитора p53-MDM2, путей их введения, конкретного типа обрабатываемой опухоли и конкретного больного, подвергаемого лечению. Оптимальный способ, порядок введения, дозированные количества и режим могут быть легко определены специалистом в данной области путем использования общепринятых способов и на основании информации, приведенной в настоящем описании.
Координационное соединение платины преимущественно вводят в дозе от 1 до 500 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 50 до 400 мг/м2, конкретно, для цисплатина в дозе приблизительно 75 мг/м2 и для карбоплатина приблизительно 300 мг/м2 на курс лечения.
Таксановое соединение преимущественно вводят в дозе от 50 до 400 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 75 до 250 мг/м2, конкретно, для паклитаксела в дозе приблизительно от 175 до 250 мг/м2 и для доцетаксела приблизительно от 75 до 150 мг/м2 на курс лечения.
Камптотециновое соединение преимущественно вводят в дозе от 0,1 до 400 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 1 до 300 мг/м2, конкретно, для иринотекана в дозе приблизительно от 100 до 350 мг/м2 и для топотекана приблизительно от 1 до 2 мг/м2 на курс лечения.
Противоопухолевое производное подофиллотоксина преимущественно вводят в дозе от 30 до 300 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 50 до 250 мг/м2, конкретно, для этопозида в дозе приблизительно от 35 до 100 мг/м2 и для тенипозида приблизительно от 50 до 250 мг/м2 на курс лечения.
Противоопухолевый алкалоид барвинка преимущественно вводят в дозе от 2 до 30 мг на квадратный метр (мг/м2) площади поверхности тела, конкретно, для винбластина в дозе приблизительно от 3 до 12 мг/м2, для винкристина в дозе приблизительно от 1 до 2 мг/м2 и для винорелбина в дозе приблизительно от 10 до 30 мг/м2 на курс лечения.
Противоопухолевое производное нуклеозида преимущественно вводят в дозе от 200 до 2500 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 700 до 1500 мг/м2, конкретно, для 5-FU в дозе от 200 до 500 мг/м2, для гемцитабина в дозе приблизительно от 800 до 1200 мг/м2 и для капецитабина приблизительно от 1000 до 2500 мг/м2 на курс лечения.
Алкилирующие средства, такие как азотистый иприт или нитрозомочевина, преимущественно вводят в дозе от 100 до 500 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 120 до 200 мг/м2, конкретно, для циклофосфамида в дозе приблизительно от 100 до 500 мг/м2, для хлорамбуцила в дозе приблизительно от 0,1 до 0,2 мг/кг, для кармустина в дозе приблизительно от 150 до 200 мг/м2 и для ломустина в дозе приблизительно от 100 до 150 мг/м2 на курс лечения.
Противоопухолевое производное антрациклина преимущественно вводят в дозе от 10 до 75 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 15 до 60 мг/м2, конкретно, для доксорубицина в дозе приблизительно от 40 до 75 мг/м2, для даунорубицина в дозе приблизительно от 25 до 45 мг/м2 и для идарубицина в дозе приблизительно от 10 до 15 мг/м2 на курс лечения.
Антиэстрогенное средство преимущественно вводят в дозе приблизительно от 1 до 100 мг ежедневно в зависимости от конкретного средства и условий лечения. Тамоксифен преимущественно вводят орально в дозе от 5 до 50 мг, предпочтительно от 10 до 20 мг дважды в день, продолжая терапию в течение достаточного времени для достижения и закрепления терапевтического эффекта. Торемифен преимущественно вводят орально в дозе приблизительно 60 мг один раз в день, продолжая терапию в течение достаточного времени для достижения и закрепления терапевтического эффекта. Анастрозол преимущественно вводят орально в дозе приблизительно 1 мг один раз в день. Дролоксифен преимущественно вводят орально в дозе приблизительно 20-100 мг один раз в день. Ралоксифен преимущественно вводят орально в дозе приблизительно 60 мг один раз в день. Эксеместан преимущественно вводят орально в дозе приблизительно 25 мг один раз в день.
Антитела преимущественно вводят в дозе приблизительно от 1 до 5 мг на квадратный метр (мг/м2) площади поверхности тела, или как известно в данной области, если иное известно. Трастузумаб преимущественно вводят в дозе от 1 до 5 мг на квадратный метр (мг/м2) площади поверхности тела, конкретно, от 2 до 4 мг/м2 на курс лечения.
Эти дозы можно вводить, например, один раз, два или более раз на курс лечения, который обычно повторяют, например, каждые 7, 14, 21 или 28 дней.
Соединения формулы (I), их фармацевтически приемлемые кислотно-аддитивные соли и стереоизомерные формы могут обладать значительными диагностическими свойствами, заключающимися в том, что их можно применять для обнаружения или идентификации взаимодействия p53-MDM2 в биологическом образце, включая обнаружение и измерение образования комплекса между меченым соединением и/или p53 и/или MDM2 и/или другими молекулами, пептидами, белками, ферментами или рецепторами.
В способах обнаружения или идентификации могут быть использованы соединения, меченные метящими агентами, такими как радиоактивные изотопы, ферменты, флуоресцентные вещества, люминесцентные вещества и т.д. Примеры радиоактивных изотопов включают 125I, 131I, 3H и 14C. Ферменты обычно становятся детектируемыми за счет конъюгации с соответствующим субстратом, который, в свою очередь, катализирует детектируемую реакцию. Их примеры включают, например, бета-галактозидазу, бета-глюкозидазу, щелочную фосфатазу, пероксидазу и малатдегидрогеназу, предпочтительно пероксидазу хрена. Люминесцентные вещества включают, например, люминол, производные люминола, люциферин, экворин и люциферазу.
Биологические образцы могут быть определены в виде ткани организма или жидкости организма. Примерами жидкостей организма являются спинномозговая жидкость, кровь, плазма, сыворотка, моча, мокрота, слюна и т.п.
Следующие примеры иллюстрируют настоящее изобретение.
Экспериментальная часть
Здесь и далее, “DCM” обозначает дихлорметан, “DIPE” обозначает диизопропиловый эфир, “EtOAc” обозначает этилацетат, “EtOH” обозначает этанол, “MeOH” обозначает метанол и “ТГФ” обозначает тетрагидрофуран.
Температуры плавления.
Для ряда соединений, обозначенных (Кофлер), температуры плавления измеряли с помощью столика Кофлера, состоящего из нагреваемой пластины с линейным температурным градиентом, скользящим указателем и температурной шкалой в градусах Цельсия.
ЖХМС
Общая методика
ВЭЖХ градиент получали с использованием системы Alliance HT 2795 (Waters), включающей четверной насос с дегазатором, автосэмплер и детектор на диодной матрице (DAD). Поток из колонки направляли в МС-детектор. МС-детектор оснащали источником ионизации электрораспылением. Напряжение на капиллярной игле составляло 3 кВ и температуру источника поддерживали при 100°C на LCT (время-пролетный масс-спектрометр с Z-спреем от Waters). Азот использовали в качестве газа-распылителя. Сбор данных выполняли с использованием системы данных Waters-Micromass MassLynx-Openlynx.
Способ A
В дополнение к общей методике: обращенно-фазную ВЭЖХ проводили на колонке Xterra-RP C18 (5 мкм, 3,9×150 мм) со скоростью потока 1,0 мл/мин при температуре 30°C. Две подвижные фазы (подвижная фаза A: 100% 7 мМ ацетата аммония; подвижная фаза B: 100% ацетонитрила); использовали для изменения параметра градиента от 85% A, 15% B (выдержка в течение 3 минут) до 20% A, 80% B в течение 5 минут, выдержка при 20% A и 80% B в течение 6 минут и восстановление на начальные параметры в течение 3 минут. Использовали вводимый объем 20 мкл. Напряжение конуса составляло 20 В для положительного и отрицательного режима ионизации. Масс-спектры получали путем сканирования от 100 до 900 в 0,8 секунды, используя задержку сканирования в 0,08 секунды.
Способ B
В дополнение к общей методике: обращенно-фазную ВЭЖХ проводили на колонке Xterra-RP C18 (5 мкм, 3,9×150 мм) со скоростью потока 1,0 мл/мин при температуре 30°C. Две подвижные фазы (подвижная фаза A: 100% 7 мМ ацетата аммония; подвижная фаза B: 100% ацетонитрила); использовали для изменения параметра градиента от 100% A (выдержка в течение 1 минуты) до 50% A, 50% B в течение 4 минут, выдержка при 50% A и 50% B в течение 9 минут и восстановление на начальные параметры в течение 3 минут. Использовали вводимый объем 20 мкл. Напряжение конуса составляло 20 В для положительного режима ионизации и 20 В для отрицательного режима ионизации. Масс-спектры получали путем сканирования от 100 до 900 в 0,8 секунды, используя задержку сканирования в 0,08 секунды.
A. Получение промежуточных соединений.
Пример A1
Получение промежуточного соединения 1

Смесь бензо[b]фуран-3-она (400 мг, 0,0029 моль) и N-метокси-N-метил(трифенилфосфоранилиден)ацетамида (1,19 г, 0,0032 моль) в ксилоле (10 мл) перемешивали при 135°C в течение 35 часов. Реакцию гасили водой и повышали основность насыщенным раствором гидрокарбоната натрия. Смесь экстрагировали EtOAc, органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (40-63 мкм) (элюент: EtOAc/циклогексан 50/50). Чистые фракции собирали и выпаривали растворитель с получением 210 мг (32%) промежуточного соединения 1 в виде коричневого масла. 1H-ЯМР (300 МГц, CDCl3) δ 7,63 (м, 2H), 7,46 (д, 1H, J=7,1), 7,25 (м, 2H), 3,84 (с, 2H), 3,70 (с, 3H), 3,22 (с, 3H). МС (ES+) m/z 220 (M+1).
Пример A2
a) Получение промежуточного соединения 2

Смесь 1H-пирроло[2,3-b]пиридин-3-этанамина (0,014 моль), 1-фтор-4-нитробензола (0,015 моль) и DIPE (0,048 моль) перемешивали при 210°C в течение 30 минут. DIPE выпаривали. Осадок растворяли в смеси DCM/MeOH. Органический слой промывали 10% карбонатом калия, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (2 г) очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,3). Чистые фракции собирали и выпаривали растворитель с получением 0,152 г (28%) промежуточного соединения 2, температура плавления 201°C (Кофлер).
b) Получение промежуточного соединения 3
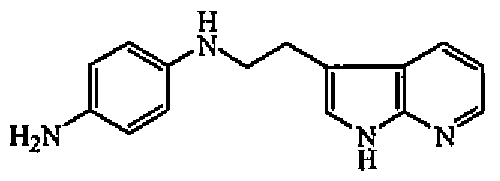
Смесь промежуточного соединения 2 (0,004 моль) и никеля Ренея (1 г) в MeOH (20 мл) гидрировали при комнатной температуре в течение 2 часов под давлением 3 бар, затем фильтровали через целит. Целит промывали MeOH. Фильтрат упаривали с получением 0,92 г (100%) промежуточного соединения 3.
Пример A3
a) Получение промежуточного соединения 4

Смесь имидазо[1,2-а]пиридин-3-ацетонитрила (0,028 моль) и Rh/Al2O3 5% (4,5 г) в EtOH (45 мл) и MeOH/NH3 (12,5 мл) гидрировали при комнатной температуре в течение 72 часов под давлением 3 бар, затем фильтровали через целит. Целит промывали смесью DCM/EtOH. Фильтрат упаривали. Остаток растворяли в DCM. Органический слой промывали 10% карбонатом калия, сушили (MgSO4), фильтровали и выпаривали растворитель с получением 3,4 г (73%) промежуточного соединения 4.
b) Получение промежуточного соединения 5
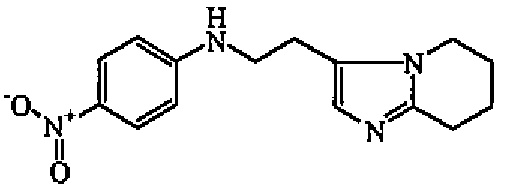
Смесь промежуточного соединения 4 (0,006 моль), 1-фтор-4-нитробензола (0,007 моль) и DIPE (0,021 моль) нагревали при 210°C в течение 30 минут. DIPE выпаривали. Неочищенное масло разбавляли DCM и EtOH (90/10). Органический слой промывали 10% карбонатом калия, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (1,3 г) очищали колоночной хроматографией на силикагеле (20-45 мкм) (элюент: DCM/MeOH 98/2). Чистые фракции собирали и выпаривали растворитель. Остаток (0,36 г) кристаллизовали из ацетонитрила. Осадок отфильтровывали и сушили с получением 0,199 г промежуточного соединения 5, температура плавления 171°C (Кофлер).
c) Получение промежуточного соединения 6
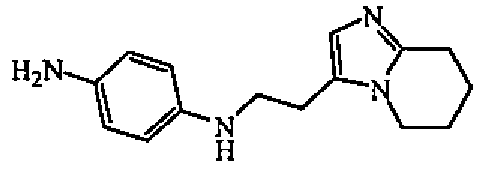
Смесь промежуточного соединения 5 (0,003 моль) и никеля Ренея (1 г) в MeOH (20 мл) гидрировали при комнатной температуре в течение 3 часов под давлением 3 бар, затем фильтровали через целит. Целит промывали MeOH. Фильтрат упаривали с получением 1 г (>100%) промежуточного соединения 6.
Пример A4
a) Получение промежуточного соединения 7
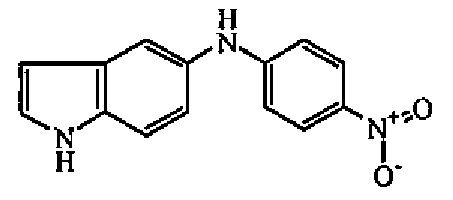
Смесь 1-фтор-4-нитробензола (0,0083 моль), 1H-индол-5-амина (0,0076 моль) и DIPE (0,0166 моль) перемешивали при 210°C в течение 18 часов, затем поглощали в DCM. Органический слой промывали 3 н. соляной кислотой, затем NaHCO3, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток поглощали в EtOH. Осадок отфильтровывали, промывали диэтиловым эфиром и сушили с получением 1,08 г (65%) промежуточного соединения 7, температура плавления 180°C.
b) Получение промежуточного соединения 8
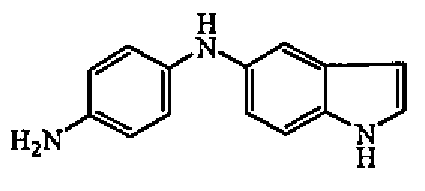
Смесь промежуточного соединения 7 (0,0039 моль) и никеля Ренея (1 г) в MeOH (20 мл) гидрировали при комнатной температуре в течение 2 часов под давлением 3 бар, затем фильтровали через целит. Целит промывали смесью DCM/MeOH. Фильтрат упаривали с получением 1 г (>100%) промежуточного соединения 8.
Пример A5
Получение промежуточного соединения 9
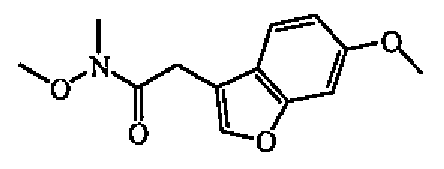
1,1'-Карбоксилдиимидазол (315 мг, 0,0019 моль) добавляли по каплям к раствору (6-метоксибензофуран-3-ил)уксусной кислоты (400 мг, 0,0019 моль) в DCM (10 мл). Смесь перемешивали в течение 3 часов при комнатной температуре. Добавляли гидрохлорид O,N-диметилгидроксиламина (190 мг, 0,0019 моль) и перемешивали смесь в течение 4 часов при комнатной температуре. Реакцию гасили льдом и повышали основность 4 н. раствором гидроксида натрия. Смесь экстрагировали DCM, органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (40-63 мкм) (элюент: EtOAc/циклогексан 50/50). Чистые фракции собирали и растворитель выпаривали с получением 410 мг (85%) промежуточного соединения 9 в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ 7,54 (м, 2H), 6,99 (с, 1H), 6,88 (д, 1H, J=6,4), 3,83 (с, 3H), 3,79 (с, 2H), 3,69 (с, 3H), 3,21 (с, 3H).
Пример A6
a) Получение промежуточного соединения 10

Смесь 1,1-диметилэтилового эфира (3-йод-2-тиенил)карбаминовой кислоты (0,0105 моль), этилового эфира 4-бром-2-бутеновой кислоты (0,0158 моль) и карбоната калия (0,021 моль) в N,N-диметилформамиде (100 мл) перемешивали в течение 16 часов при комнатной температуре. Добавляли трифенилфосфин (0,001 моль) и ацетат палладия (0,0005 моль). Смесь перемешивали в течение 8 часов при 70°C, затем промывали водой. Органический слой отделяли с помощью EtOAc, сушили (MgSO4) и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (элюент: EtOAc/циклогексан 10/90) с получением 2,3 г (70%) промежуточного соединения 10.
b) Получение промежуточного соединения 11
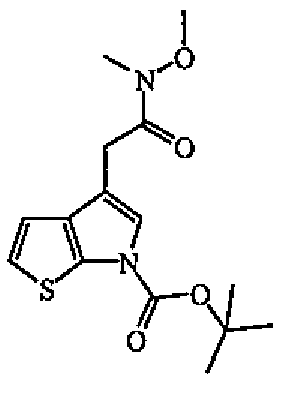
Раствор промежуточного соединения 10 (0,0016 моль) в ТГФ (10 мл) перемешивали при -78°C в атмосфере аргона. Раствор N,O-диметилгидроксиламина HCl (0,0004 моль) в ТГФ (40 мл) перемешивали при -78°C в атмосфере аргона. Бутиллитий (1,6 М в гексане) (0,016 моль) добавляли по каплям при -78°C к раствору N,O-диметилгидроксиламина HCl (0,0004 моль). Смесь перемешивали при комнатной температуре в течение 20 минут, затем вновь охлаждали до -78°C. Добавляли по каплям раствор промежуточного соединения 10. Смесь перемешивали при -78°C в течение 2 часов, выливали в насыщенный раствор NH4Cl при -78°C и экстрагировали EtOAc. Органический слой отделили, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (элюент: EtOAc/циклогексан от 10/90 до 30/70). Чистые фракции собирали и выпаривали растворитель с получением 0,4 г (77%) промежуточного соединения 11.
c) Получение промежуточного соединения 12

Промежуточное соединение 11 (0,0012 моль) растворяли в DCM и абсорбировали на силикагеле. Смесь перемешивали при 60°C в течение 24 часов в вакууме. Остаток очищали колоночной хроматографией на силикагеле (элюент: EtOAc/циклогексан 80/20). Чистые фракции собирали и выпаривали растворитель с получением (62%) промежуточного соединения 12.
d) Получение промежуточного соединения 13
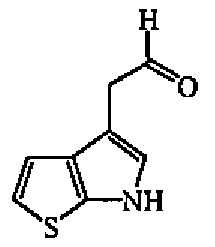
Алюмогидрид лития (0,0008 моль) при 0°C медленно добавляли к раствору промежуточного соединения 12 (0,0008 моль) в ТГФ (4 мл, безводный). Смесь перемешивали при 0°C в течение 1 часа, затем выливали на лед, промывали 5% KHSO4 и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель с получением промежуточного соединения 13. Продукт непосредственно использовали на следующей стадии реакции.
B. Получение конечных соединений
Пример B1
Получение соединения 1
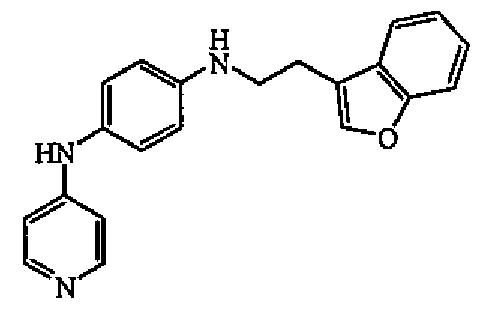
1 н. раствор алюмогидрида лития в ТГФ (0,65 мл) добавляли по каплям при 0°C к суспензии промежуточного соединения 1 (162 мг, 0,00065 моль) в ТГФ (4 мл). Смесь перемешивали при 0°C в течение 1 часа. Реакцию гасили при 0°C медленным добавлением 5% раствора бисульфата калия в воде и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель с получением бензофуран-3-илацетальдегида.
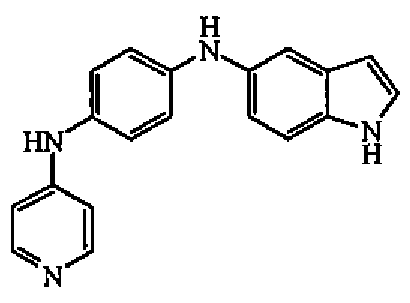
К раствору N-4-пиридинил-1,4-бензолдиамина в MeOH (3 мл) и уксусной кислоты (4 капли) добавляли цианоборгидрид натрия (57 мг, 0,00091 моль) и полученный выше альдегид, растворенный в MeOH (1 мл). Смесь перемешивали при комнатной температуре в течение 18 часов. Затем выпаривали растворитель, оставшееся масло поглощали в EtOAc и промывали насыщенным раствором гидрокарбоната натрия. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (40-63 мкм) (элюент: DCM/MeOH 95/5). Чистые фракции собирали и выпаривали растворитель с получением 41 мг (20%) соединения 1, температура плавления 152-154°C.
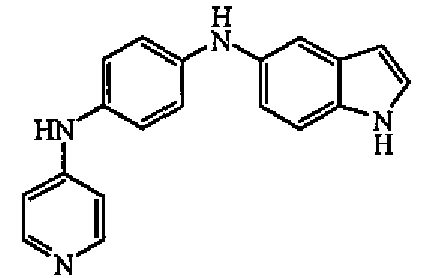
1H-ЯМР (300 МГц, MeOH-d4) δ 7,98 (д, 2H, J=6,6), 7,59 (м, 2H), 7,43 (д, 1H, J=7,4), 7,24 (м, 2H), 7,00 (дд, 2H, J=8,7, J=3,3), 6,68 (м, 4H), 3,45 (т, 2H, J=7,2), 2,98 (т, 2H, J=7,2). MS (ES+) m/z 330 (M+1).
Пример B2
Получение соединения 2

Смесь промежуточного соединения 3 (0,004 моль) и гидрохлорида 4-бромпиридина (0,004 моль) в уксусной кислоте (5 мл) перемешивали при 140°C в течение 15 минут, затем упаривали. Остаток растворяли в смеси DCM/MeOH. Органический слой промывали 10% раствором карбоната калия, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (1 г) очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 90/10/1). Чистые фракции собирали и выпаривали растворитель. Остаток (0,74 г, 63%) кристаллизовали из ацетонитрила. Осадок отфильтровывали и промывали с получением 0,541 г соединения 2, температура плавления 182°C (Кофлер).
1H-ЯМР (ДМСО-d6) δ 2,96 (2H, т, J=7,7Гц), 3,30 (2H, т, J=7,7Гц), 5,6 (1H, уш.т, J=7,6Гц), 6,58 (4H, м), 6,95 (2H, д, J=7,7Гц), 7,03 (1H, м), 7,33 (1H, м), 7,96 (1H, д, J=7,7Гц), 8,03 (2H, д, J=7,6Гц), 8,17-8,21 (1H, м), 8,22 (1H, уш.с), 11,38 (1H, уш.с). ЖХМС (ES+) m/z 330 (M+1), Rt=7,10, способ А.
Пример B3
Получение соединения 3
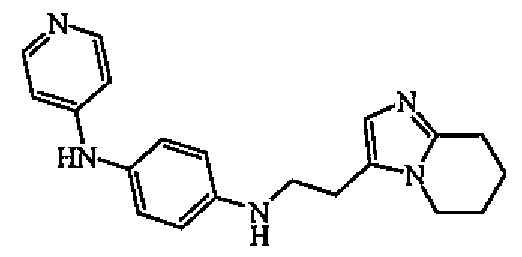
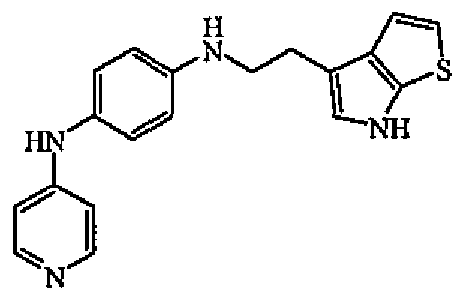
Гидрохлорид 4-бромпиридина (0,004 моль) добавляли к раствору промежуточного соединения 6 (0,004 моль) в уксусной кислоте (10 мл). Смесь нагревали при 140°C в течение 15 минут в микроволновой печи. Уксусную кислоту выпаривали. Неочищенное масло растворяли в смеси DCM/EtOH (90/10). Органический слой промывали карбонатом калия, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (1 г) очищали колоночной хроматографией на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH от 90/10/0,5 до 90/10/1). Чистые фракции собирали и выпаривали растворитель с получением 0,63 г (48%) соединения 3.
1H-ЯМР (ДМСО-d6) δ 1,75-1,93 (4H, м), 2,65-2,78 (4H, м), 3,32 (2H, с), 3,70 (2H, т, J=6,1Гц), 5,62 (1H, уш.т, J=7,6Гц), 6,58-6,53 (5H, м), 6,95 (2H, д, J=7,7Гц), 8,05 (2H, д, J=7,6Гц), 8,24 (1H, с). ЖХМС (ES+) m/z 334 (M+1), Rt=5,18, способ B. Температура плавления 256°C (Кофлер).
Пример B4
Получение соединения 4
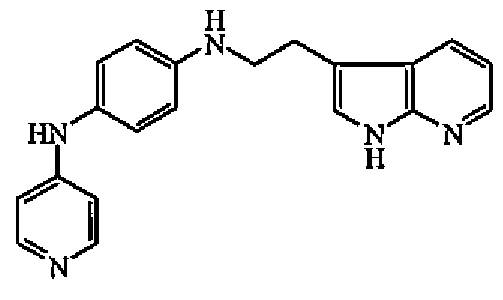
Гидрохлорид 4-бромпиридина (0,002 моль) добавляли к раствору промежуточного соединения 8 (0,0022 моль) в уксусной кислоте (2,8 мл). Смесь нагревали при 110°C в течение 30 минут, выливали в ледяную воду и повышали основность 10% карбонатом калия. Осадок отфильтровывали и сушили. Остаток (0,739 г) очищали колоночной хроматографией на кромасиле (5 мкм) (элюент: DCM/MeOH/NH4OH от 96/4/0,4 до 88/12/1,2). Чистые фракции собирали и выпаривали растворитель с получением 0,018 г соединения 4.
1H-ЯМР (ДМСО-d6) δ 6,32 (1H, уш.с), 6,68 (1H, д, J=5,6Гц), 6,9 (1H, дд, J=10,2Гц, J=2,5Гц), 6,55 (2H, д, J=10,2Гц), 7,0 (2H, д, J=10,2Гц), 7,27 (1Н, м), 7,32 (1H, д, J=10,2Гц), 7,7 (1H, уш.с), 8,17 (1H, дд, J=10,2Гц, J=2,5Гц), 8,37 (1H, уш.с), 10,9 (1H, уш.с). ЖХМС (ES+) m/z 301 (M+1), Rt=7,73, способ A.
Пример B5
Получение соединения 5

1 н. раствор алюмогидрида лития в ТГФ (1,0 мл) добавляли по каплям при 0°C к суспензии промежуточного соединения 9 (249 мг, 0,0010 моль). Смесь перемешивали при 0°C в течение 1 часа. Реакцию гасили при 0°C медленным добавлением 5% раствора бисульфата калия в воде и дважды экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель с получением (6-метоксибензофуран-3-ил)ацетальдегида.
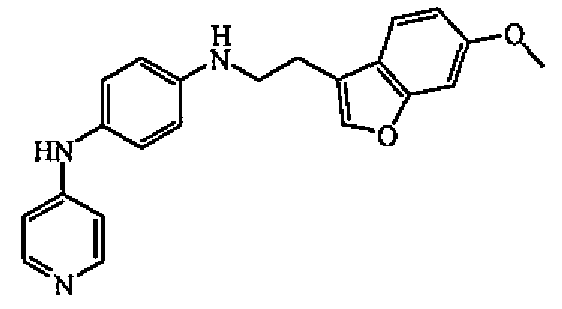
К раствору N-4-пиридинил-1,4-бензолдиамина в MeOH (5 мл) и уксусной кислоты (5 капель) добавляли цианоборгидрид натрия (90 мг, 0,0014 моль) и полученный выше альдегид, растворенный в MeOH (1 мл). Смесь перемешивали при комнатной температуре в течение 18 часов. Затем выпаривали растворитель, оставшееся масло поглощали в EtOAc и промывали насыщенным раствором гидрокарбоната натрия. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток промывали MeOH и сушили с получением 125 мг (34%) соединения 5, температура плавления 184-186°C.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,23 (с, 1H), 8,01 (дд, 2H, J=6,3, J=1,5), 7,71 (с, 1H), 7,51 (д, 1H, J=8,5), 7,14 (д, 1H, J=2,1), 6,92 (д, 2H, J=8,6), 6,85 (дд, 1H, J=8,5, J=2,2), 6,58 (м, 4H), 5,64 (т, 1H, J=5,6), 3,77 (с, 3H), 3,30 (т, 2H, J=7,0), 2,48 (т, 2H, J=7,0). МС (ES+) m/z 360 (M+1).
Пример B6
Получение соединения 6
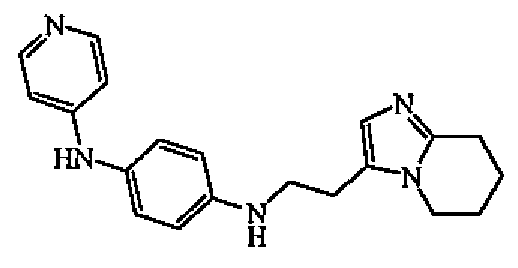
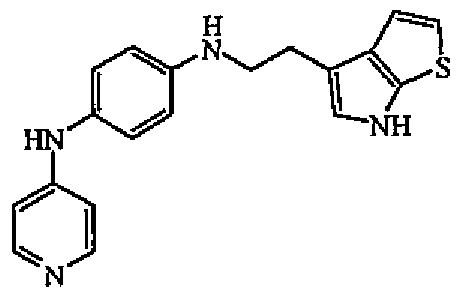
Уксусную кислоту (несколько капель), затем промежуточное соединение 13 (0,0004 моль) добавляли по каплям к раствору N-4-пиридинил-1,4-бензолдиамина (0,0008 моль) и цианоборгидрида натрия (0,001 моль) в MeOH (4 мл). Промежуточное соединение 13 (0,0004 моль) растворяли в MeOH (4 мл). Смесь перемешивали при комнатной температуре в течение 20 часов, выливали в воду, промывали насыщенным NaHCO3 и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (элюент: DCM/MeOH 85/15). Чистые фракции собирали и выпаривали растворитель с получением 0,086 г (34%) соединения 6.
В таблице F-1 перечислены соединения, которые были получены в одном из приведенных выше примеров.

C. Фармакологический пример:
Способность соединений сохранять p53 в клетках A2780 определяли иммуноферментным анализом, связанным с p53 ферментом. Анализ p53 представляет собой «сэндвич»-метод ферментативного иммуноанализа с использованием двух поликлональных антител. Поликлональное антитело, специфичное для белка p53, иммобилизовали на поверхности пластмассовых лунок. Любой p53, присутствующий в подлежащем анализу образце, будет связываться с иммобилизованным антителом. Биотинилированное поликлональное антитело в качестве детектора также распознает белок p53 и также будет связываться с любым p53, который был удержан иммобилизованным антителом. Антитело в качестве детектора, в свою очередь, связывается с помощью стрептавидина, конъюгированного с пероксидазой хрена. Пероксидаза хрена катализирует преобразование хромогенного субстрата о-фенилендиамина, интенсивность которого пропорциональна количеству белка p53, связанного с планшетом. Количество окрашенного продукта реакции определяли с помощью спектрофотометра. Количественного определения достигают путем построения стандартных кривых, используя известные концентрации очищенного рекомбинантного HIS-меченого белка p53 (см. пример C.1).
Клеточную активность соединений формулы (I) определяли на опухолевых клетках U87MG с использованием колориметрического анализа на клеточную токсичность или выживаемость (см. пример C.2).
Опухолевые клетки U87MG представляют собой клетки глиобластомы человека с диким типом p53. В таких клеточных линиях MDM2 полностью контролирует экспрессию p53.
C.1. p53 ELISA
Клетки A2780 (ATCC) культивировали в RPMI 1640, дополненной 10% фетальной телячьей сывороткой (FCS), 2 мМ L-глутамином и гентамицином, при 37°C в увлажненном инкубаторе с 5% CO2.
Клетки A2780 засевали по 20000 клеток на лунку 96-луночного планшета, культивировали в течение 24 часов и обрабатывали соединением в течение 16 часов в увлажненном инкубаторе при 37°C. После инкубации клетки промывали один раз забуференным фосфатом солевым раствором и добавляли 30 мкл, на лунку, слабо солевого буфера RIPA (20 мМ трис, pH 7,0, 0,5 мМ EDTA, 1% Nonidet P40, 0,5% DOC, 0,05% SDS, 1 мМ PMSF, 1 мкг/мл апротинина и 0,5 мкг/мл леупептина). Планшеты помещали на лед на 30 минут для завершения лизиса. Белок p53 определяли в лизатах с использованием сэндвич-ELISA, описанного ниже.
Высоко связывающие полистирольные EIA/RIA 96-луночные планшеты (Costar 9018) покрывали иммобилизованным антителом pAb1801 (Abcam ab28-100) при концентрации 1 мкг/мл в покрывающем буфере (0,1М NaHCO3, pH 8,2), 50 мкл на лунку. Антитело оставляли для связывания на ночь при 4°C. Покрытые планшеты промывали один раз забуференным фосфатом солевым раствором (PBS)/0,05% Twin 20 и добавляли 300 мкл блокирующего буфера (PBS, 1% альбумина бычьей сыворотки (BSA)) для инкубационного периода в 2 часа при комнатной температуре. Очищенный рекомбинантный HIS-меченый белок p53, в диапазоне 3-200 нг/мл, получали в блокирующем буфере и использовали в качестве стандарта.
Планшеты дважды промывали PBS/0,05% Twin 20 и блокирующим буфером или добавляли стандарты по 80 мкл/лунку. К стандартам добавляли 20 мкл буфера для лизиса. Образцы добавляли к другим лункам по 20 мкл лизата на лунку. После инкубации в течение ночи при 4°C планшеты промывали дважды PBS/0,05% Twin 20. Аликвоты по 100 мкл второго поликлонального антитела p53(FL-393) (Tebubio, sc-6243) при концентрации 1 мкг/мл в блокирующем буфере добавляли в каждую лунку и оставляли на 2 часа для связывания при комнатной температуре. Планшеты промывали три раза PBS/0,05% Twin 20. Добавляли антикроличье антитело HRP (sc-2004, Tebubio) при концентрации 0,04 мкг/мл в PBS/1% BSA и инкубировали в течение 1 часа при комнатной температуре. Планшеты три раза промывали PBS/0,05% Twin 20 и добавляли по 100 мкл субстратного буфера (субстратный буфер быстро готовили перед использованием путем добавления 1 таблетки 10 мг о-фенилендиамина (OPD) от Sigma и 125 мкл 3% H2O2 к 25 мл OPD-буфера: 35 мМ лимонная кислота, 66 мМ Na2HPO4, pH 5,6). Через 5-10 минут цветную реакцию останавливали добавлением 50 мкл стоп-буфера (1М H2SO4) на лунку. Поглощение при двух длинах волн 490/655 нм измеряли с использованием считывающего устройства для микропланшетов Biorad и затем анализировали результаты.
В каждом эксперименте параллельно проводили контрольную (не содержащую лекарственного средства) и чистую (не содержащую клеток или лекарственных средств) инкубацию. Значение чистой инкубации вычитали из всех значений контрольных и исследуемых образцов. Для каждого исследуемого образца значение p53 (в единицах поглощения) выражали в виде процентного значения от содержания p53 в контрольных образцах. Процент сохранения более 140% определяли как существенный. В настоящем документе эффекты исследуемых соединений выражены в виде минимальной дозы, дающей по меньшей мере 140% от значения p53, присутствующего в контрольном (LAD) образце (см. таблицу F-2).
С2. Анализ пролиферации
Все исследуемые соединения растворяли в ДМСО и дополнительные разведения готовили в культуральной среде. Конечные концентрации ДМСО никогда не превышали 0,1% (об./об.) в анализах клеточной пролиферации. Контрольные образцы содержали клетки U87MG и ДМСО без соединений и чистые образцы содержали только ДМСО без клеток.
Клетки U87MG засевали в 96-луночные планшеты для клеточной культуры по 3000 клеток/лунку/100 мкл. Через 24 часа среду меняли и добавляли соединение и/или растворитель до конечного объема 200 мкл. После 4 дней инкубации среду заменяли на 200 мкл свежей среды и клеточный рост оценивали с использованием анализа, основанного на MTT. Таким образом, 25 мкл раствора MTT (0,5% MTT исследуемого эталона от Serva в забуференном фосфатом солевом растворе) добавляли в каждую лунку и клетки дополнительно инкубировали в течение 2 часов при 37°C. Затем среду осторожно откачивали и синий продукт с MTT-формазаном растворяли добавлением в каждую лунку 25 мкл 0,1М глицина и 100 мкл ДМСО. Планшеты встряхивали в течение 10 минут на шейкере для микропланшетов, после чего измеряли поглощение при 540 нм с помощью считывающего устройства для микропланшетов Biorad.
В рамках эксперимента результаты для каждого экспериментального условия представляют собой среднее значение из трех повторных лунок. С целью первичного скрининга соединения тестировали при одной фиксированной концентрации 10-5 М. Для активных соединений эксперименты повторяли для построения полных кривых концентрация-ответ. В каждом эксперименте параллельно проводили контрольную (не содержащую лекарственного средства) и чистую (не содержащую клеток или лекарственных средств) инкубацию. Значение чистой инкубации вычитали из всех значений контрольных и исследуемых образцов. Для каждого исследуемого образца значение клеточного роста (в единицах поглощения) выражали в виде процентного значения от среднего значения клеточного роста в контрольных образцах. Если приемлемо, значения IC50 (концентрация лекарственного средства, необходимая для понижения клеточного роста на 50% по отношению к контрольному образцу) рассчитывали с использованием статистического анализа для оценочных значений (Finney, D.J., Probit Analyses, 2nd Ed. Chapter 10, Graded Responses, Cambridge University Press, Cambridge 1962). В настоящем документе эффекты исследуемых соединений выражены в виде IC50 (отрицательное значение логарифма значения IC50) (см. таблицу F-2).
В некоторых экспериментах анализ пролиферации адаптирован для использования в 384-луночных культуральных планшетах (см. таблицу F-2).
В таблице F-2 приведены результаты для соединений, исследованных согласно примерам C.1 и C.2.
384 лунки
96 лунок
D. Пример композиции: таблетки с пленочным покрытием
Приготовление ядра таблетки
Смесь 100 г соединения формулы (I), 570 г лактозы и 200 г крахмала тщательно перемешивали и затем увлажняли раствором 5 г додецилсульфата и 10 г поливинилпирролидона приблизительно в 200 мл воды. Смесь влажного порошка просеивали, сушили и снова просеивали. Затем добавляли 100 г микрокристаллической целлюлозы и 15 г гидрированного растительного масла. Все тщательно перемешивали и прессовали в таблетки с получением 10000 таблеток, каждая из которых содержит 10 мг соединения формулы (I).
Покрытие
К раствору 10 г метилцеллюлозы в 75 мл денатурированного этанола добавляли 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляли 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола к 10 г жидкого полиэтиленгликоля и растворяли в 75 мл дихлорметана. Последний раствор добавляли к предыдущему и затем добавляли 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной окрашивающей суспензии и полностью гомогенизировали. Ядра таблеток покрывали этой полученной смесью в аппарате для покрытия.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АЛКИЛАМИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ МЕЖДУ MDM2 И Р53 | 2007 |
|
RU2434863C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2747645C2 |
| ПТЕРИДИНЫ В КАЧЕСТВЕ FGFR ИНГИБИТОРОВ | 2013 |
|
RU2702906C2 |
| ИМИДАЗОЛО-5-ИЛ-2-АНИЛИНОПИРИМИДИНЫ КАК АГЕНТЫ ДЛЯ ИНГИБИРОВАНИЯ ПРОЛИФЕРАЦИИ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОДУЦИРОВАНИЯ | 2001 |
|
RU2284327C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИДИНОНА | 2018 |
|
RU2806751C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2654857C2 |
| ПРОТИВОРАКОВЫЕ БЕНЗОПИРАЗИНЫ, ДЕЙСТВУЮЩИЕ ПОСРЕДСТВОМ ИНГИБИРОВАНИЯ FGFR-КИНАЗ | 2012 |
|
RU2639863C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА/ПИПЕРАЗИНА | 2008 |
|
RU2478628C2 |
| ХИНОЛИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ FGFR КИНАЗЫ | 2012 |
|
RU2625303C2 |
| ПРОИЗВОДНЫЕ 1,5- И 1,7-НАФТИРИДИНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ FGFR | 2012 |
|
RU2629194C2 |
Изобретение описывает соединение формулы (I),
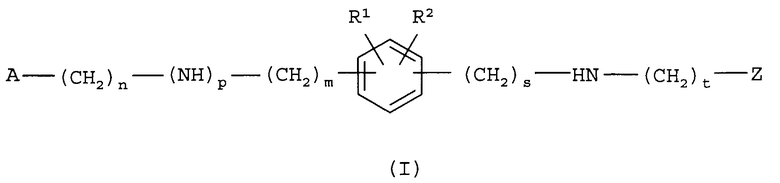
и его фармацевтически приемлемая соль, где m представляет собой прямую связь; n равно 0, 1, 2, 3 или 4 и, когда n равно 0, то подразумевается прямая связь; р равно 1; s представляет собой прямую связь; t представляет собой прямую связь; R1 и R2 каждый независимо представляет собой водород; А представляет собой радикал, выбранный из 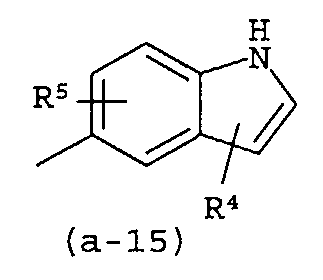
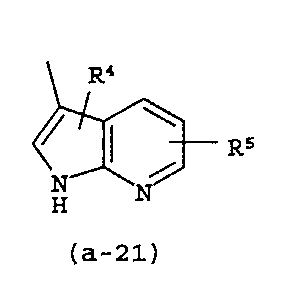
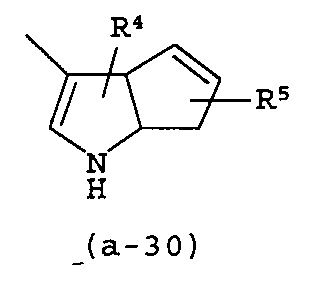
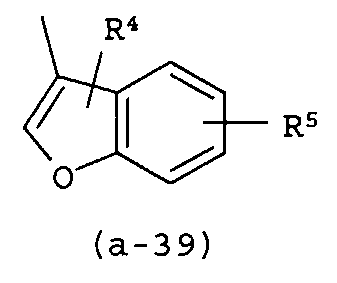
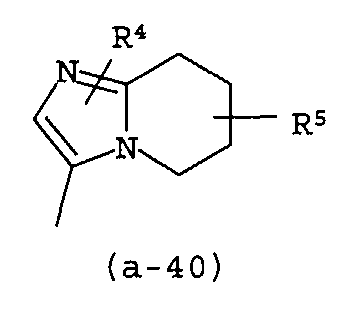 где R4 и R5 каждый независимо выбран из водорода или C1-6алкилокси; Z представляет собой радикал
где R4 и R5 каждый независимо выбран из водорода или C1-6алкилокси; Z представляет собой радикал 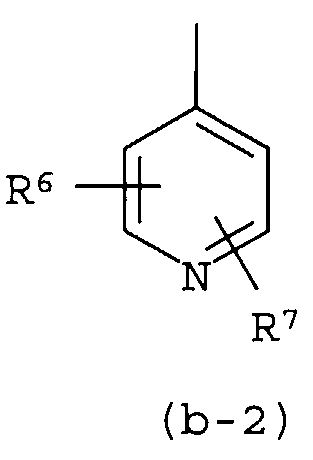 , где R6 и R7 каждый независимо представляет собой водород. Также изобретение описывает фармацевтическую композицию для лечения рака и способ ее получения, на основе соединений формулы I, применение этих соединений для получения лекарственного средства, а также способ получения данных соединений. Технический результат: получены и описаны новые соединения, которые могут найти свое применение в качестве ингибитора взаимодействия p53-MDM2. 6 н. и 4 з.п. ф-лы, 2 табл.
, где R6 и R7 каждый независимо представляет собой водород. Также изобретение описывает фармацевтическую композицию для лечения рака и способ ее получения, на основе соединений формулы I, применение этих соединений для получения лекарственного средства, а также способ получения данных соединений. Технический результат: получены и описаны новые соединения, которые могут найти свое применение в качестве ингибитора взаимодействия p53-MDM2. 6 н. и 4 з.п. ф-лы, 2 табл.
1. Соединение формулы (I),
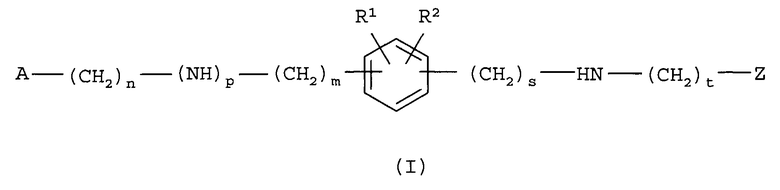
его фармацевтически приемлемая соль, где m представляет собой прямую связь;
n равно 0, 1, 2, 3 или 4 и, когда n равно 0, то подразумевается прямая связь;
p равно 1;
s представляет собой прямую связь;
t представляет собой прямую связь;
R1 и R2 каждый независимо представляет собой водород;
А представляет собой радикал, выбранный из

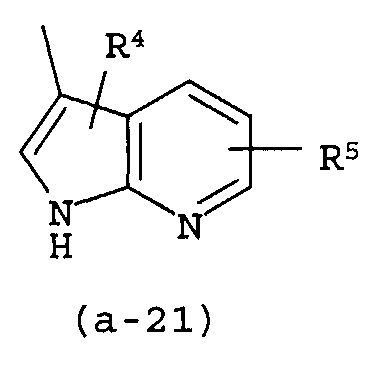
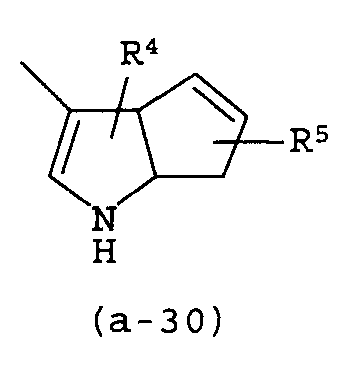
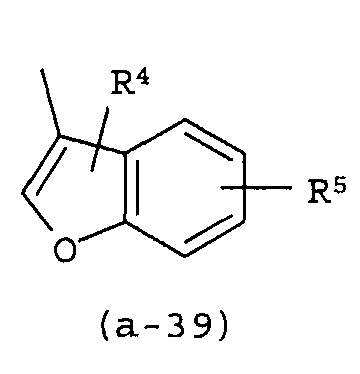

где R4 и R5 каждый независимо выбран из водорода или С1-6алкилокси;
Z представляет собой радикал , где R6 и R7 каждый независимо представляет собой водород.
2. Соединение по п.1, где
m представляет собой прямую связь; n равно 0 или 2; р равно 1; s представляет собой прямую связь; t представляет собой прямую связь; R1 и R2 каждый независимо представляют собой водород; А представляет собой радикал, выбранный из (а-15), (а-21), (а-30), (а-39) или (а-40); R4 и R5 каждый независимо выбран из водорода или С1-6алкилокси; Z представляет собой радикал (b-2); или R6 и R7 каждый независимо выбран из водорода.
3. Соединение по п.1, где m представляет собой прямую связь; n равно 2; р равно 1; s представляет собой прямую связь; t представляет собой прямую связь; R1 и R2 каждый независимо представляет собой водород; А представляет собой радикал, выбранный из (а-21), (а-39) или (а-40); R4 и R5 каждый независимо выбран из водорода или С1-6алкилокси; Z представляет собой радикал (b-2); или R6 и R7 каждый независимо выбран из водорода.
4. Соединение по п.1, где соединение представляет собой соединение №2, соединение №3 или соединение №5.
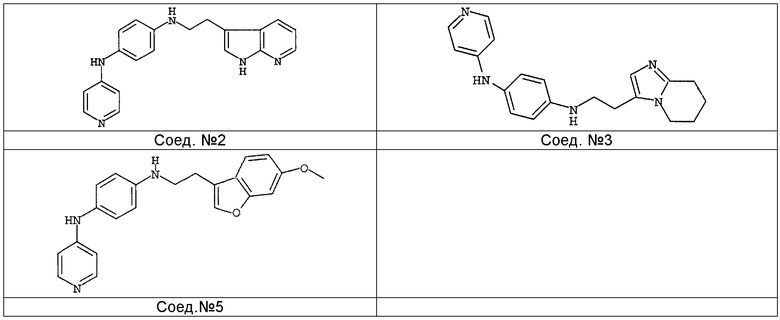
5. Соединение по пп.1, 2, 3 или 4 для применения в качестве лекарственного средства для лечения рака.
6. Фармацевтическая композиция для лечения рака, содержащая фармацевтически приемлемые носители и в качестве активного компонента соединение по пп.1-4 в терапевтически эффективном количестве.
7. Способ получения фармацевтической композиции по пп.6, где фармацевтически приемлемые носители и соединение по пп.1-4 смешаны непосредственно.
8. Применение соединения по п.1, 2, 3 или 4 для получения лекарственного средства для лечения рака.
9. Способ получения соединения по п.1, предусматривающий взаимодействие промежуточного соединения формулы (II) с промежуточным соединением формулы (III), где W представляет собой соответствующую уходящую группу, такую как, например, галоген,

где значения переменных указаны в п.1.
10. Способ получения соединения по п.1, предусматривающий преобразование промежуточного соединения формулы (IV) в соединения формулы (I), где p равно 1, именуемые здесь как соединения формулы (1-а), в присутствии алюмогидрида лития в подходящем растворителе,

где значения переменных указаны в п.1.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO 00/27819 A, 18.05.2000 | |||
| ПРОИЗВОДНЫЕ АКРИДИНА, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ | 1992 |
|
RU2119482C1 |

