Изобретение относится к ингибитору тромбина, имеющему аминоизохинолиновую группу, к содержащей его фармацевтической композиции, а также к применению указанного ингибитора для производства лекарственного средства для лечения заболеваний, опосредованных тромбином.
Большинство пептидоподобных ингибиторов тромбина, о которых сообщалось в литературе, содержат основную группу в так называемом положении Р1 (R. Pfau, «Structure-based design of thrombin inhibitors», Current Opinion in Drug Discovery & Development, 6, 437-450, 2003). Примеры таких основных групп включают основные аминокислоты-аргинин и лизин, а также гуанидины и бензамиды. Полагают, что основный фрагмент в таких соединениях является необходимым для антитромбиновой активности. Однако такие высокоосновные группы обычно протонированы при физиологических рН и, как следствие, такие соединения плохо абсорбируются через желудочно-кишечный тракт после перорального приема и имеют низкую пероральную биодоступность.
В общем случае терапевтические агенты, которые можно вводить перорально, наиболее предпочтительны, и они имеют повышенный коммерческий потенциал вследствие свойственной им простоты применения.
В международной патентной заявке WO 98/47876 (Akzo Nobel N.V.) раскрыт класс ингибиторов тромбина, имеющих в качестве основной группы аминоизохинолиновый фрагмент, и данные соединения обладают улучшенными свойствами трансэпителиального транспорта.
В частности, данная патентная заявка в качестве примера содержит соединения N-(карбоксиметил)-D-фенилаланил-[(1-амино-6-изохинолинил)метил]-L-пролинамид (WO 98/47876, пример 77) и N-(карбоксиметил)-D-(4-метоксифенил)аланил-[(1-амино-6-изохинолинил)метил]-L-пролинамид (WO 98/47876, пример 111as), также как и их сложноэфирные пролекарственные производные.
Желательно разработать ингибиторы тромбина с более высокой биодоступностью, особенно - подходящие для перорального введения.
Настоящее изобретение касается нового соединения - N-(2-оксо-2-пропоксиэтил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)метил]-L-пролинамида, которое имеет высокую биодоступность при пероральном введении.
В другом аспекте изобретение относится к N-(карбоксиметил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)метил]-L-пролинамиду, который является селективным ингибитором тромбина, образующимся in situ из N-(2-оксо-2-пропоксиэтил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)метил]-L-пролинамида при пероральном введении, который полезен в качестве промежуточного соединения в синтезе н-пропильного пролекарственного производного в соответствии с изобретением.
Соединение данного изобретения, т.е. N-(2-оксо-2-пропоксиэтил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)метил]-L-пролинамид, представлено структурной формулой 1A:

Настоящим изобретением установлено, что н-пропильное сложноэфирное пролекарственное производное 1A обладает высокой биодоступностью при пероральном введении по сравнению с биодоступностью соответствующих н-пропильных сложноэфирных пролекарственных производных структурно близкородственных аминоизохинолиновых ингибиторов тромбина, таких как N-(карбоксиметил)-D-фенилаланил-[(1-амино-6-изохинолинил)метил]-L-пролинамид (2А) и N-(карбоксиметил)-D-(4-метоксифенил)аланил-[(1-амино-6-изохинолинил)метил]-L-пролинамид (3А), раскрытых в WO 98/47876.
Таким образом, введение соединения 1А в соответствии с изобретением приводит к заметно более высоким концентрациям в плазме ингибитора тромбина 1В - производного свободной кислоты 1А, которое быстро образуется in vivo как только соединение попадает в кровоток.
Неожиданное повышение пероральной биодоступности нового дифенилаланинового производного 1А по сравнению с соответствующим фенилаланиновым производным 2А или с метоксифенилаланиновым производным 3А позволяет разработать антитромботический агент для перорального введения, действующий при относительно низкой дозировке.

Экспериментальная часть
Общие замечания
Результаты анализа методом ЖХ-МС были получены на масс-спектрометре Applied Biosystems API150EX. Спектры 1Н ЯМР были зарегистрированы на спектрометрах Bruker DPX 400 или DRX 400.
Пример 1 (Схема I):
N-(2-оксо-2-пропоксиэтил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)метил]-L-пролинамид (1A)
A: N-[2-(1,1-диметилэтокси)-2-оксоэтил]-β-фенил-D-фенилаланин (а)
К перемешиваемому раствору D-дифенилаланина, H-D-Dpa-OH (20,0 г, 82,9 ммоль), и карбоната кальция (17,2 г, 125 ммоль) в смеси диоксан/вода (1:1 (об./об.), 100 мл) добавляют трет-бутилбромацетат (12,2 мл, 83,0 ммоль). После перемешивания в течение ночи добавляют воду (100 мл) и рН доводят до 5,5 добавлением 0,5 М раствора лимонной кислоты. Полученный осадок отфильтровывают, промывают водой, затем - диэтиловым эфиром и высушивают в вакууме, получают 10,4 г указанного в заголовке соединения а.
Схема I

B: Гидрохлорид 1,1-диметилэтилового эфира (S)-2-[[[(1-амино-6-изоизохинолил)метил]амино]карбонил]-1-пирролидинкарбоновой кислоты (b)
К перемешиваемому раствору N-трет-бутоксикарбонил-L-пролина, Boc-Pro-OH (6,73 г, 31,25 ммоль), в безводном N,N-диметилформамиде (100 мл) в атмосфере аргона добавляют тонко измельченный гидрохлорид 1-амино-6-аминометилизохинолина (10,0 г, 40,63 ммоль) и N,N-диизопропилэтиламин (16,17 г, 125,00 ммоль). После перемешивания суспензии в течение 15 минут при комнатной температуре в течение 5 минут порциями добавляют гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (17,67 г, 46,88 ммоль), что со временем приводит к растворению суспендированного гидрохлорида 1-амино-6-аминометилизохинолина. Реакционную смесь перемешивают при комнатной температуре в атмосфере аргона еще 90 минут, по истечении этого времени образуется желтый осадок гидрохлорида 1,1-диметилэтилового эфира (S)-2-[[[(1-амино-6-изоизохинолил)метил]амино]карбонил]-1-пирролидинкарбоновой кислоты (b). Осадок отделяют фильтрацией, промывают дихлорметаном (300 мл) до обесцвечивания фильтрата, а затем высушивают в вакууме, получают 7,8 г (56%) указанного в заголовке соединения (чистота 94% по данным ВЭЖХ на колонке Luna C18(2) 46×30 мм, градиент, подвижная фаза - ацетонитрил:вода, 5-100%/4 мин, постоянно содержит 0,1% трифторуксусной кислоты). Затем продукт выделяют из фильтрата выпариванием в вакууме для удаления диметилформамида и избытка диизопропилэтиламина с добавлением 500 мл дихлорметана. Полученный осадок удаляют фильтрацией, промывают дихлорметаном (300 мл) и высушивают в вакууме, получают 4,6 г (33%) (чистота 91% по данным ВЭЖХ на колонке Luna C18(2) 46×30 мм, градиент, подвижная фаза - ацетонитрил:вода, 5-100%/4 мин, постоянно содержит 0,1% трифторуксусной кислоты).
С: Гидрохлорид N-[2-(1,1-диметилэтокси)-2-оксоэтил]-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида (с)
К суспензии гидрохлорида 1,1-диметилэтилового эфира (S)-2-[[[(1-амино-6-изоизохинолил)метил]амино]карбонил]-1-пирролидинкарбоновой кислоты (b) (4,14 г, 9,34 ммоль) в дихлорметане (20 мл) добавляют трифторуксусную кислоту (8 мл). После перемешивания раствора в течение 2 часов растворитель и избыток трифторуксусной кислоты удаляют в вакууме. Остаток затем растворяют в N,N-диметилформамиде (41 мл) и добавляют 2-(трет-бутокси-карбонилметил-амино)-3,3-дифенил-пропионовую кислоту (Boc-D-Dpa-OH) (3,30 г, 9,3 ммоль), гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (5,31 г, 13,9 ммоль) и N,N-диизопропилэтиламин (9,75 мл, 56 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа, затем добавляют воду до образования осадка. Влажный осадок отделяют фильтрованием, затем переносят в этилацетат, осушают (MgSO4), фильтруют и выпаривают досуха, получают неочищенный продукт (7,2 г). Очистка колоночной хроматографией (диоксид кремния, режим градиентного элюирования смесями дихлорметана и метанола (0-10%)) дает 4,6 г указанного в заголовке соединения с в виде смолы.
D: Гидрохлорид N-(карбоксиметил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида (1В)
К раствору гидрохлорида N-[2-(1,1-диметилэтокси)-2-оксоэтил]-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида (с) (4,6 г, 7,1 ммоль) в дихлорметане (25 мл) добавляют трифторуксусную кислоту (4,6 мл). После стояния в течение ночи раствор выпаривают досуха, растворяют в дихлорметане и добавляют избыток хлористого водорода в диэтиловом эфире. Продукт осаждают добавлением безводного диэтилового эфира, затем отделяют фильтрацией и сушат в вакууме, получают 2,8 г. Дополнительные 0,8 г получают осаждением из маточного раствора.
1H ЯМР δ (CD3OD) 1,32 (м, 1H), 1,83 (м, 3H), 2,86 (м, 1H), 3,53(м, 1H), 3,68 (дд, 2H; J=17,1 Гц), 3,80 (д, 2H; J=17,1 Гц) (4,15 (м, 1H), 4,52 (д, 1H; J=16,6 Гц), 4,59 (д, 1H; J=11,5 Гц), 4,69 (д, 1H; J=16,1 Гц), 5,3 (д, 1H; 11,5 Гц), 7,2-7,6 (м, 10H), 7,66 (д, 2H; J=7,5 Гц), 7,72 (дд, 1H; J=1,5, 8,0 Гц), 7,89 (c, 1H), 8,38 (д, 1H; J=9 Гц); MC m/z 552,2 (М+H)+.
E: Гидрохлорид N-(2-пропокси-2-оксоэтил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида (1А)
К суспензии гидрохлорида N-(карбоксиметил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида (1В; 300 мг, 0,5 ммоль) в н-пропаноле (5 мл) по каплям добавляют тионилхлорид (0,4 мл). После перемешивания в течение 3 дней раствор разбавляют дихлорметаном, промывают 5% водным раствором бикарбоната натрия и выпаривают досуха. Неочищенный продукт очищают обращеннофазной ВЭЖХ, а затем превращают в гидрохлоридную соль растворением в малом количестве метанола и осаждением 1М раствором хлористого водорода в диэтиловом эфире. После добавления дополнительного количества безводного диэтилового эфира образовавшийся осадок отделяют фильтрованием и высушивают в вакууме, получают 185 мг указанного в заголовке соединения в виде белого порошка.
1H ЯМР δ (CD3OD) δ 0,85 (т, 3H; J=7,5 Гц), 1,33 (м, 1H), 1,57 (секст, 2H; J=7 Гц), 1,83 (м, 3H), 2,88 (м, 1H), 3,51 (м, 1H), 3,73 (д, 1H; J=17,6 Гц), 3,84 (д, 1H; J=17,6 Гц), 4,07 (м, 2H), 4,16 (дд, 1H; J=5,0, 8,0 Гц), 4,53 (д, 1H; J=16,6 Гц), 4,56 (д, 1H,; J=13,6 Гц), 4,70 (д, 1H; J=16,6 Гц), 5,20 (д, 1H; J=11 Гц), 7,23-7,44 (м, 7H), 7,50 (т, 2H; J=7,5 Гц), 7,56 (д, 1H; J=7,0 Гц), 7,65 (д, 2H; J=7,5 Гц), 7,72 (дд, 1H; J=1,5, 8,0 Гц), 7,90 (c, 1H), 8,39 (д, 1H; J=8,5 Гц); MC m/z 594,4 (М+H)+.
С применением способов, подобных раскрытым выше и изображенным на схеме I, исходя из D-фенилаланина или О-метил-D-тирозина вместо D-дифенилаланина, были получены соответствующие производные. Соединения 2В и 3В были выделены и использованы для последующего исследования в виде трифторацетатных солей.
Пример 2
2В: Трифторацетат N-(карбоксиметил)-D-фенилаланил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида

1H ЯМР δ (CD3OD) δ 1,41 (м, 1H), 1,85 (м, 2H), 1,99 (м, 1H), 2,47 (м, 1H), 3,35 (дд, 1H; J=5,0, 13,1 Гц), 3,40 (дд, 1H; J=10,6, 13,1 Гц), 3,45 (м, 1H), 3,73 (д, 1H; J=16,6 Гц), 3,80 (д, 1H; J=16,6 Гц), 4,34 (дд, 1H; J=4,5, 8,1 Гц), 4,5 (дд, 1H; J=5,0, 10,6 Гц), 4,51 (д, 1H; J=16,1 Гц), 4,72 (д, 1H; J=16,6 Гц), 7,23 (д, 1H; J=7,1 Гц), 7,27-7,43 (м, 5H), 7,52 (д, 1H; J=7,1 Гц), 7,71 (дд, 1H; J=1,5, 8,6 Гц), 7,90 (c, 1H), 8,34 (д, 1H; J=8,6 Гц); MC m/z 476,0 (М+H)+.
2А: Гидрохлорид N-(2-пропокси-2-оксоэтил)-D-фенилаланил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида

1H ЯМР δ (CD3OD) δ 0,89 (т, 3H; J=7,6 Гц), 1,41 (м, 1H), 1,62 (секст, 2H; J=7,6 Гц), 1,83 (м, 2H), 2,00 (м, 1H), 2,44 (м, 1H), 3,16 (дд, 1H; J=1,5, 10,6 Гц), 3,37 (дд, 1H; J=5,5, 13,1 Гц), 3,47 (м, 1H), 3,93 (д, 1H; J=17,1 Гц), 4,06 (д, 1H; J=17,1 Гц), 4,14 (м, 2H), 4,36 (дд, 1H; 5,0, 9,1 Гц), 4,54 (м, 2H), 4,71 (д, 1H; J=17,1 Гц), 7,26 (д, 1H; 7,1 Гц), 7,29-7,44 (м, 5H), 7,56 (д, 1H; J=7,1 Гц), 7,73 (д, 1H; J=8,6 Гц), 7,92 (c, 1H), 8,38 (д, 1H; J=8,6 Гц); MC m/z 518,2 (М+H)+
Пример 3
3В: Трифторацетат N-(карбоксиметил)-О-метил-D-тирозинил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида

1H ЯМР δ (CD3OD) δ 1,46 (м, 1H), 1,87 (м, 2H), 2,02 (м, 1H), 2,56 (м, 1H), 3,08 (дд, 1H; J=10,6, 12,6 Гц), 3,27 (дд, 1H; J=5,5, 13,6 Гц), 3,47 (м, 1H), 3,71 (д, 1H; J=16,1 Гц), 3,79 (д, 1H; J=15,6 Гц), 3,79 (c, 3H), 4,36 (дд, 1H; J=4,0, 8,6 Гц), 4,46 (дд, 1H; J=4,0, 8,66 Гц), 4,51 (д, 1H; J=16,6 Гц), 4,72 (д, 1H; J=16,6 Гц), 6,92 (д, 2H; J=8,6 Гц), 7,21 (д, 2H; J=8,6 Гц), 7,24 (д, 1H; 7,1 Гц), 7,52 (д, 1H; J=7,1 Гц), 7,71 (д, 1H; J=8,1 Гц), 7,90 (c, 1H), 8,35 (д, 1H; J=9,1 Гц); MC m/z 506,3 (М+H)+.
3А: Гидрохлорид N-(2-пропокси-2-оксоэтил)-О-метил-D-тирозинил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида
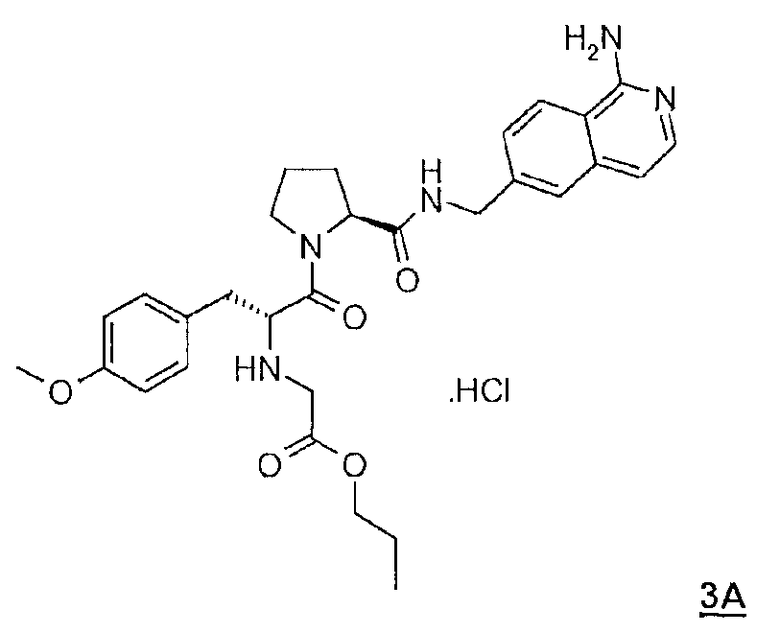
1H ЯМР δ (CD3OD) δ 0,88 (т, 3H; J=7,1 Гц), 1,47 (м, 1H), 1,61 (секст, 2H; J=7,1 Гц), 1,86 (м, 2H), 2,04 (м, 1H), 2,54 (м, 1H), 3,10 (дд, 1H; J=10,6, 15,1 Гц), 3,31 (м„ 1H), 3,49 (м, 1H), 3,79 (c, 1H), 3,91 (д, 1H; J=16,6 Гц), 4,04 (д, 1H; J=16,6 Гц), 4,13 (м, 2H), 4,38 (дд, 1H; 5,0, 9,1 Гц), 4,48 (дд, 1H; J=5,0, 10,6 Гц), 4,54 (д, 1H; J=16,1 Гц), 4,71 (д, 1H; J=16,1 Гц), 6,93 (д, 2H; J=8,6 Гц), 7,22 (д, 2H; J=8,6 Гц), 7,26 (д, 1H; 7,1 Гц), 7,55 (д, 1H; J=7,1 Гц), 7,73 (дд, 1H; J=2,0, 8,6 Гц), 7,92 (c, 1H), 8,38 (д, 1H; J=8,6 Гц); MC m/z 548,3 (М+H)+.
Пример 4
Исследование активности тромбина
Активность ингибирования тромбина оценивают преинкубированием исследуемого соединения при различных концентрациях с α-тромбином человека при 37°С. Через 10 минут к смеси добавляют хромогенный субстрат H-D-Phe-Пипеколинил-Arg-п-нитроанилид (S-2238) и измеряют изменение поглощения через последующие 8 минут. Оба соединения 1А и 1В являются эффективными ингибиторами α-тромбина человека зависимым от концентрации образом со значениями IC50, равными 13,0 нМ и 12,6 нМ соответственно (для обоих n=5; фиг.1). В последующих опытах, в которых также изменяют концентрацию S-2238, графики зависимостей [S]/V от [S] параллельны для каждой использованной концентрации S-2238, что указывает на конкурентную природу ингибирования. Анализ указанных данных по Хейнсу-Вульфу позволил определить значения Ki, равные 0,9 нМ (n=5) для 1В и 1,0 нМ (n=3) для 1А. Сравнение с показателями активности соединений 2 и 3 проведено в таблице 1.
Показатели активности ингибирования тромбина
Пример 5
Определение биодоступности соединений 1А, 2А и 3А у крыс
Животные
Самцы крыс Wistar Ola (~250 г). Во время исследований имели свободный доступ к пище и воде.
Хирургические подготовительные действия
Для внутривенных фармакокинетических исследований в правую яремную вену под изофлурановой анестезией были введены катетеры (Portex polythene - внутренний диаметр 0,58 мм, наружный диаметр 0,96 мм, завершающиеся насадкой, изготовленной из трубки из медицинского силикона SF, SFM-1350). Трубки были выведены в заднюю часть шеи, заполнены гепаринизированным солевым раствором (100 ед/мл) и закрыты пробками. Животным был дан период для восстановления минимум 48 часов перед введением соединений.
Введение соединений
Соединения 1А, 2А и 3А вводили перорально (РО). Соединения 1В, 2В и 3В вводили внутривенно (IV). Последовательные образцы крови (для получения плазмы) отбирали из латеральной вены в моменты времени от 3 минут до 24 часов (конечный образец, взятый сердечной пункцией) в соответствии с протоколом в виде матрицы (3 образца в каждый момент времени). Образцы плазмы до проведения анализа хранили при -20°С.
Анализ образцов плазмы
В образцах плазмы определяли соответствующие тромбиновые производные свободных кислот 1В, 2В и 3В, используя метод ЖХ-МС. Cmax (максимальные концентрации в плазме) и значения AUC определяли из профилей средняя концентрация в плазме - время.
Опыты
Были выполнены следующие опыты:
- Соединение 1В в солевом растворе вводили внутривенно (IV) 5 крысам из расчета 5 мг/кг (5 мг/мл раствора, дозируемого по 1 мл/кг).
- Соединение 1В в солевом растворе вводили перорально (РО) 4 крысам из расчета 10 мг/кг (2 мг/мл раствора, дозируемого по 5 мл/кг).
- Соединение 1А в растворе, содержащем 5% мульфогена/95% солевого раствора, вводили перорально 5 крысам из расчета 10 мг/кг (2 мг/мл раствора, дозируемого по 5 мл/кг).
- Соединение 2В в солевом растворе вводили внутривенно 4 крысам из расчета 2 мг/кг (2 мг/мл раствора, дозируемого по 1 мл/кг).
- Соединение 2А в солевом растворе вводили перорально 4 крысам из расчета 10 мг/кг (2 мг/мл раствора, дозируемого по 5 мл/кг).
- Соединение 3В в солевом растворе вводили внутривенно 4 крысам из расчета 2 мг/кг (2 мг/мл раствора, дозируемого по 1 мл/кг).
- Соединение 3А в солевом растворе вводили перорально 4 крысам из расчета 10 мг/кг (2 мг/мл раствора, дозируемого по 5 мл/кг).
Результаты
Некомпартментный фармакокинетический анализ был выполнен с помощью WinNonlin Professional 3.1 и 4.1. Полученные фармакокинетические параметры представлены в таблице 2.
Было установлено, что биодоступность, выраженная как ППК(IV)/ППК(PO)×100%, для н-пропильного сложноэфирного производного (1А) дифенилаланинового производного гидрохлорида N-(карбоксиметил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолил)метил]-L-пролинамида является исключительно высокой (58%) по сравнению с биодоступностями соответствующих фенилаланинового (2А) и п-метоксифенилаланинового (3А) ингибиторов тромбина.
Фармакокинетические параметры
с последующим введением 1А
с последующим введением 2А
с последующим введением 3А
солевой раствор
а = данные являются средним для двух опытов.
b = биодоступность 1В после введения 1А изменяется от 30 до 60%, что измерено в ряде опытов на крысах Wistar Ola с различающимся типом носителя (суспензия в смеси желатин/маннит или раствор маннит/забуференный фосфатом солевой раствор, или смесь 5% мульфоген/95% солевой раствор), использованным для соединения 1А.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| БИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ | 2014 |
|
RU2672582C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КАЛЛИКРЕИНА ПЛАЗМЫ | 2014 |
|
RU2712621C2 |
| ПЕПТИДИЛЬНЫЕ ГЕТЕРОЦИКЛЫ, ИСПОЛЬЗУЕМЫЕ ПРИ ЛЕЧЕНИИ ТРОМБИНАССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ | 1996 |
|
RU2181125C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, МЕДИЦИНСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2369613C2 |
| ПРОИЗВОДНЫЕ АМИНОКИСЛОТЫ | 1994 |
|
RU2127261C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ ПОВРЕЖДЕНИЯ ГОЛОВНОГО МОЗГА | 2018 |
|
RU2797548C2 |
| ПРОИЗВОДНЫЕ СПИРО[ИЗОХИНОЛИН-4(1Н),3-ПИРРОЛИДИН]-1,2',3,5'[2Н]ТЕТРОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДОТВРАЩЕНИЯ ИЛИ ЛЕЧЕНИЯ ОСЛОЖНЕНИЙ, СВЯЗАННЫХ С САХАРНЫМ ДИАБЕТОМ | 1991 |
|
RU2110518C1 |
| 2,5- ИЛИ 2,6-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ГИДРОХИНОНА, СОДЕРЖАЩИЕ ПО КРАЙНЕЙ МЕРЕ ОДНУ КАРБОКСИ-, СУЛЬФО- ИЛИ АМИДОГРУППУ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2021 |
|
RU2827702C1 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2017 |
|
RU2778478C2 |
Настоящее изобретение относится к соединению -N-(2-оксо-2-пропоксиэтил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)-метил]-L-пролинамиду, или его фармацевтически приемлемой соли, к фармацевтической композиции, содержащей указанное соединение, а также к применению соединения для производства лекарственного средства для лечения или профилактики заболеваний, опосредованных тромбином. Также изобретение относится к соединению - собой N-(карбоксиметил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)метил]-L-пролинамиду, или его фармацевтически приемлемой соли. Технический результат: получены новые соединения, обладающие полезными биологическими свойствами. 4 н. и 1 з.п. ф-лы, 2 табл.
1. Соединение, представляющее собой N-(2-оксо-2-пропоксиэтил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)метил]-L-пролинамид, или его фармацевтически приемлемая соль.
2. Соединение по п.1 в виде дигидрохлорида.
3. Фармацевтическая композиция для лечения или профилактики заболеваний, опосредованных тромбином, содержащая соединение по п.1 и фармацевтически приемлемые вспомогательные вещества.
4. Применение соединения по п.1 для производства лекарственного средства для лечения или профилактики заболеваний, опосредованных тромбином.
5. Соединение, представляющее собой N-(карбоксиметил)-β-фенил-D-фенилаланил-N-[(1-амино-6-изохинолинил)метил]-L-пролинамид, или его фармацевтически приемлемая соль.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| ИНГИБИТОР СЕРИНОВЫХ ПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1999 |
|
RU2232760C2 |

