Настоящее изобретение относится к производному аминокислоты. Более конкретно, оно относится к производному аминокислоты, проявляющему прекрасное действие в качестве лекарства.
Описание уровня техники
Среди болезней сердца, обычно называемых сердечной недостаточностью, для нас непосредственно смертельной является не только острая сердечная недостаточность, но также и болезнь, не требующая безотлагательного лечения, такая как хроническая сердечная недостаточность, в том случае, когда она прогрессирует. Поэтому долгое время активно проводились исследования лекарственных средств для лечения этой болезни, и в настоящее время разработано большое число лекарств с различными механизмами действия для лечения сердечной недостаточности.
Например, сердечные гликозиды, представленные дигиталисом, давно используются в качестве лекарств, способных повышать силу сердечных сокращений и предел толерантности без повышения сердечной скорости. Однако эти сердечные гликозиды обладают такими недостатками, как то, что каждый из них имеет узкую область безопасности и может назначаться только ограниченному числу пациентов. Кроме того, они проявляют некоторые побочные эффекты, например, вызывающие аритмию, что делает их менее полезными.
Для облегчения гемостазиса, вызванного небольшой сердечной слабостью, иногда используются диуретики, такие как фуросемид и спиронолактон. Хотя эти лекарства имеют такое положительное качество, что они применимы в случае слабой сердечной недостаточности и ослабляют субъективные симптомы, их недостатком является проявление побочных эффектов, например, электролитического расстройства и нарушения обмена сахара в крови, и они непосредственно не улучшают предел толерантности и так называемое "качество жизни".
В качестве сосудорасширяющих средств для улучшения течения крови в коронарных сосудах также используются нитраты, такие как изосорбиднитрат, и агенты блокирующие 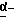 рецепторы, представленные буназосином и празосином. Хотя первый широко используется с тех пор, как был охарактеризован в качестве ослабляющего предварительную нагрузку и улучшающего субъективные симптомы и предел толерантности, проявляющего немедленное действие и не имеющего серьезного побочного действия, он обладает таким недостатком, как тенденция к легкой толерентности. С другой стороны, последнее характеризуется ослаблением как предварительной, так и последующей нагрузки (preload and postload) и повышением сердечной скорости. Однако сообщалось, что эти агенты не обладают действием по улучшению субъективных симптомов или предела толерантности.
рецепторы, представленные буназосином и празосином. Хотя первый широко используется с тех пор, как был охарактеризован в качестве ослабляющего предварительную нагрузку и улучшающего субъективные симптомы и предел толерантности, проявляющего немедленное действие и не имеющего серьезного побочного действия, он обладает таким недостатком, как тенденция к легкой толерентности. С другой стороны, последнее характеризуется ослаблением как предварительной, так и последующей нагрузки (preload and postload) и повышением сердечной скорости. Однако сообщалось, что эти агенты не обладают действием по улучшению субъективных симптомов или предела толерантности.
Кроме того, известны β-стимулирующие лекарства, такие как допамин и добутамин, каждый обладающий значительным действием по увеличению сердечной сокращаемости, в качестве первичных средств при оказании скорой помощи в случае острой сердечной недостаточности. Однако эти лекарства имеют тенденцию быть толерантными и иногда вызывают аритмию и т.д. Известно также, что они оказывают некоторые побочные эффекты, индуцируя, например болезни миокарда. Поэтому необходимо соблюдать осторожность при использовании этих лекарств.
Недавно привлекли внимание ингибиторы атриальной натриуретической пептидной гидролазы (нейтральная эндопептидаза: NEP 24, II) и ингибиторы ангиотензин 1-конвертирующего фермента (здесь и далее упоминаемый сокращению ACE) в качестве новых лекарственных средств для лечения сердечной недостаточности.
Вышеуказанный атриальный натриуретический пептид (здесь и далее упоминаемый сокращению ANP) является гормоном, имеющимся в природе. В дополнении к сильному воднодиуретическому/натрийуретическому эффекту, сосудорасширяющему эффекту, и т.д. он оказывает подавляющее действие на высвобождение норпинефрина путем подавления симпатической нервной системы, подавляющее действие на секрецию ренина из почки и подавляющее действие на секрецию альдостеррона из надпочечной железы и, далее, оказывает также ингибирующее действие на перфузию путем усиления водной проницаемости в вене, и т.д. В отношении функции ANP у пациентов, страдающих от застойной сердечной недостаточности с повышением предварительной нагрузки, например, считается, что секреция ANP возрастает пропорционально стимуляции атриальным втягиванием и количество обращающегося в теле потока таким образом компенсационно контролируемо. Действительно, введение ANP пациентам с сердечной недостаточностью понижает давление легочного клина, и наблюдается диуретический эффект, а также достигается улучшение сердечного показателя и ударного объема крови. Кроме того, сообщалось, что ANP подавляет высвобождение эндогенных гормонов, способствующих нарушению кровообращения при сердечной недостаточности, например, альдостерон и норепинефрин ослабляют патологические состояния при сердечной недостаточности с различных углов зрения. Рассматривается, что эти действия ANP являются предпочтительными при лечении не только сердечной недостаточности, но также и гипертонии.
Ввиду того, что ANP является пептидом, он не может вводиться орально и обладает только слабой метаболической стабильностью, которая создает ту проблему, что в настоящее время он может использоваться клинически только при остром состоянии. Также сообщалось, что действие ANP может ухудшаться в процессе продолжительного введения. Таким образом, его надо использовать осторожно.
Принимая во внимание вышеуказанные характеристики ANP, необходимо уделить много внимания вышеуказанному ингибитору, ANP гидролазы (здесь и далее упоминаемый сокращенно ингибитор NEP) в качестве ANP-ассоциированного получения для перорального введения. Сообщалось, что назначение ингибитора NEP пациенту с сердечной недостаточностью повышает уровень ANP в крови и оказывает натриуретический эффект. Однако существование ингибитора NEP только слабо воздействует на функции сердечно-сосудистой системы и не может четко проявлять действия по ослаблению preload и postload (предварительной и последующей нагрузок).
С другой стороны, была доказана полезность ингибитора ACE, который является одним из сосудорасширяющих средств, поскольку он подавляет образование ангиотензина II (здесь и далее упоминаемый сокращенно AI-II), который является дополнительным фактором сердечной недостаточности, посредством этого значительно улучшает степень NY HA и повышает предел толерантности при хронической сердечной недостаточности, и, таким образом, проявляет удлиняющее жизнь действие. Однако эффективное соотношение имеющихся ингибиторов ACE у пациентов не всегда высокое и их действенность существенно изменяется от пациента к пациенту. Кроме того, отмечается такая проблема, что ингибиторы ACE имеют побочные эффекты, например, включая гипотонию, что ограничивает их применение для тех, кто страдает пониженной функцией почек.
Как обсуждено выше, каждый из существующих ингибиторов NEP и ACE ограничен в возможности применения, хотя они и привлекли всеобщее внимание как новые лекарственные средства для лечения сердечной недостаточности. Поэтому существует настоятельная необходимость создания лекарственного препарата, обладающего как NEP ингибирующей активностью, так и ACE ингибирующей активностью.
В связи с этим заявители провели исследования в отношении лекарственных средств, которые могут вводиться орально, имеет высокую метаболическую стабильность и высоко эффективное соотношение и являются также широко применимыми для пациентов с осложнениями. В результате обнаружено, что желаемая цель может быть достигнута путем использования производного аминокислоты или его фармацевтически приемлемой соли, представленного далее, что и составляет настоящее изобретение.
Настоящее изобретение касается производной аминокислоты общей формулы 1, а также фармацевтической композиции, содержащей терапевтически или профилактически эффективное количество производной аминокислоты общей формулы I или его фармакологически приемлемую соль и фармацевтически приемлемый наполнитель:
где R1 представляет атом водорода или ацильную группу;
R2 представляет атом водорода, низшую алкильную группу, циклоалкильную группу, арилалкильную группу, которая может иметь заместитель, или гетероарилалкильную группу, которая может иметь заместитель, арильную группу, которая может иметь заместитель, или гетероалкильную группу, которая может иметь заместитель;
m и n представляют каждый независимо целое число 0, 1 или 2; и
J представляет циклическую группу, обладающую ингибирующей активностью в отношении ангиотензин 1-конвертирующего фермента.
Циклическая группа, обладающая ингибирующей ACE активностью, как это представлено в определении J вышеуказанной формулы (I), включает всякую группу, обладающую ингибирующей ACE активностью, и являющуюся насыщенным или ненасыщенным моноциклическим или конденсированным кольцом. Конкретные примеры включают представленные следующей формулой, однако не ограничены ею
где R3 представляет атом водорода или карбокси-защитную группу;
Y1 обозначает группу, представленную формулой -(CR5R6)p-Z-(CR7R8)q - [где R5, R6, R7 и R8 являются одинаковыми или различными и каждый обозначает атом водорода, низшую алкильную группу, арильную группу, которая может иметь заместитель, гетероарильную группу, которая может иметь заместитель, арилалкильную группу, которая может иметь заместитель или гетероалкильную группу, которая может иметь заместитель;
Z обозначает группу, представленную формулой -(CH2)r - (где r обозначает целое число 0 или 1), группу, представленную формулой -S-, группу, представленную формулой -SO-, группу, представленную формулой -SO2-, группу, представленную формулой -O- или группу, представленную формулой -NR9- (где R9 обозначает атом водорода или низшую алкильную группу);
p и q обозначают каждый независимо целое число от 0 или 1 до 4 и сумма p и q равно 6 или менее, при условии, что, когда каждый из двух атомов углерода в R5, R6, R7, R8 и R9, имеют произвольный заместитель, выбранный из от R5 до R9, присоединенные, к тому же, смежным образом по отношению к друг другу, указанные два атома углерода и указанные два заместителя, к ним присоединенные, могут быть соединены с образованием бензольного кольца или гетероциклического кольца, которое может иметь заместитель; и что, когда R2 является арильной группой, p равно 2, q равно 2, Z обозначает группу, представленную формулой -(CH2)r' - (где r' равно 0) и два заместителя, произвольно выбранные из R7' и R8', которые присоединены к двум смежным атомам углерода, объединены вместе с образованием бензольного кольца, указанное бензольное кольцо должно быть замещено арильной группой, которая может иметь заместитель];
R4 представляет атом водорода или группу, используемую для образования от 5- до 7-членного кольца, которое может содержать один атом серы или кислорода в сочетании с R7 или R8.
Для дальнейшего понимания настоящего изобретения далее будут представлены конкретные примеры соединений по настоящему изобретению, хотя они ограничивают настоящее изобретение:

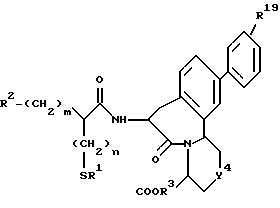



где R1 представляет атом водорода или ацильную группу;
R2 представляет атом водорода, низшую алкильную группу, циклоалкильную группу, арильную группу, которая может иметь заместитель, гетероарильную группу, которая может иметь заместитель, арилалкильную группу, которая может иметь заместитель;
R3 представляет атом водорода или карбоксизащитную группу;
R11 и R12 являются одинаковыми или отличными друг от друга и каждый представляет атом водорода или низшую алкильную группу;
u равно 0, 1 или 2;
R19 представляет атом водорода, низшую алкильную группу, низшую алкоксигруппу, гидроксильную группу или атом галогена;
m и n представляют каждый независимо целое число 0, 1 или 2;
R14 и R15 представляют каждый атом водорода, низшую алкильную группу, низшую алкоксигруппу, гидроксильную группу, атом галогена, арильную группу, которая может иметь заместитель или гетероарильную группу, которая может иметь заместитель;
s и t представляют каждый целое число 0, 1 или 2;
Y9 обозначает группу, представленную формулой -(CH2)w- (где w обозначает целое число 0 или 1), группу, представленную формулой -S-, группу, представленную формулой -SO-, группу, представленную формулой -SO2-, группу, представленную формулой -O- или группу, представленную формулой -NR17- (где R17 обозначает атом водорода или низшую алкильную группу);
R10 представляет атом водорода, низшую алкильную группу, низшую алкоксигруппу, гидроксильную группу, атом галогена, арильную группу, которая может иметь заместитель, или гетероарильную группу, которая может иметь заместитель;
Y4 обозначает группу, представленную формулой -(CH2)x- (где x обозначает целое число 0 или 1), группу, представленную формулой -S-, группу, представленную формулой -SO-, группу, представленную формулой -SO2-, группу, представленную формулой -O- или группу, представленную формулой -NR17- (где R17 обозначает атом водорода или низшую алкильную группу); и
R18 представляет атом водорода, низшую алкильную группу, или аралкильную группу, которая может иметь заместитель.
По настоящему изобретению низшая алкильная группа, данная при определении R2, R5, R6, R7, R8, R9, R10, R13, R14, R15, R16, R17, R18 и R19, подразумевает линейную или разветвленную алкильную группу, имеющую от 1 до 8, предпочтительно от 1 до 6, атомов углерода. Ее примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, пентильную (амильную) группу, изопентильную группу, неопентильную группу, третпентильную группу, 1-метилбутильную группу, 2-метилбутильную группу, 1,2-диметилпропильную группу, н-гексильную группу, изгексильную группу, 1-метилпентильную группу, 2-метилпентильную группу, 3-метилпентильную группу, 1,1-диметилбутильную группу, 1,2-диметилбутильную группу, 2,2-диметилбутильную группу, 1,3-диметилбутильную группу, 2,3-диметилбутильную группу, 3,3-диметилбутильную группу, 1-этилбутильную группу, 2-этилбутильную группу, 1,1,2-триметилпропильную группу, 1,2,2-триметилпропильную группу, 1-этил-1-метилпропильную группу, 1-этил-1-метилпропильную группу, 1-этил-2-метилпропильную группу и т. п. Предпочтительные примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу и изопентильную группу. В качестве R2, в частности изобутильной группы, особенно предпочтительна s-изобутильная группа, то есть можно отметить 1(S)-метилпропильная группа.
Низшая алкоксильная группа, как это дано при определении R10, R13, R14 и R15, подразумевает производную от вышеуказанной низшей алкильной группы, например, метокси, этокси, изопропокси, н-бутокси, трет-бутокси и т.п.
Что касается арильной группы, которая может иметь заместитель, как дано в определении R2, R5, R6, R7, R8, R10, R14 и R15, то примерами арила могут быть фенил, 2-нафтил, 3-нафтил, антраценил и т.п.
Заместитель в данной случае может подразумевать низшую алкильную группу, такую как метильная группа, этильная группа, пропильная группа изопропильная группа, низшую алкоксильную группу, такую как метоксигруппу, этоксигруппу, пропоксигруппу и изопропоксигруппу, арильную группу, арилалкильную группу, гетероарильную группу, гетероарилалкильную группу, нитро группу, гидроксильную группу, амино группу, которая может быть моно- и ди-замещенной, ацильную группу, такую как формильную группу или ацетильную группу, гидроксиалкильную группу, алкоксиалкильную группу, аминоалкильную группу, карбамоильную группу, тиоловую группу, алкилтиогруппу, сульфинильную группу, сульфонильную группу, алкилсульфонильную группу, алкилсульфонильную группу, атом галогена, карбоксильную группу, которая может быть защищена, карбоксиалкильную группу, которая может быть защищена, ацилалкильную группу, которая может быть защищена и т.п.
Гетероарильная группа, которая может иметь заместитель, как дано в определении R2, R5, R6, R7, R8, R10, R14 и R15, подразумевает от 3- до 8-членное, предпочтительное 5- или 6-членное, кольцо или конденсированное кольцо, содержащее, по крайней мере, один гетероатом, такой как атом кислорода и атом азота.
Конкретные их примеры включают тиенил, фуранил, пиранил, 2H-пирролил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидил, пиридазинил, изотиазолил, изоксазолил, фуразанил, бензотиенил, изобензофуранил, хроманил, индолидинил, изоиндолил, индолил, пуринил, хинолидинил, изохинолил, хинолил, фталазинил, хиназолил, карбозолил, акридинил, фенантридинил и т.п.
В этом случае заместитель имеет то же значение, что и для арила, как описано выше.
Что касается арилалкильной группы, которая может иметь заместитель, как дано в определении R2, R5, R6, R7, R8 и R18, арил может иметь те же значения, что и арил, описанный выше.
В этом случае алкил имеет то же значение, что и низший алкил, описанный выше. Далее, заместитель в этом случае имеет те же значения, что и заместитель для арильной группы, как описано выше.
Что касается гетероарильной группы, которая может иметь заместитель, как дано в определении R2, R5, R6, R7 и R8, то он имеет те же значения, что и гетероарил, описанный выше. 3-нафтил, антраценил и т.п.
В этом случае алкил имеет то же значение, что и низший алкил, описанный выше. Далее, заместитель в этом случае имеет те же значения, что и заместитель для гетероарильной группы, как описано выше.
Атом галогена, как это дано в определении R10, R13, R14, R15 и R19, подразумевает атом фтора, атом хлора, атом брома, атом иода и т.п.
Карбоксизамещенная группа, как это дано в определении R3 подразумевает такую, которая может быть подвергнута гидролизу до карбоксильной группы in vivo. Примеры включают низшие алкильные группы, такие как метил, этил и трет-бутил; низшие алкильные группы, замещенные фенильной группой, которая может иметь заместитель, такой как п-метоксибензил, п-нитробензил, 3,4-диметоксибензил, дифенилметил, тритил и фенетил; галогенированные низшие алкильные группы, такие как 2,2,2-трихлорэтил и 2-иодэтил; низшие алканоилокси низшие алкильные группы, такие как пивалоилоксиметил, ацетоксиметил, пропионилоксиметил, бутирилоксиметил, валерилоксиметил, 1-ацетоксиэтил, 2-ацетоксиэтил, 1-пивалоилоксиэтил и 2-пивалоилоксиэтил; высшие алканоилокси низшие алкильные группы, такие как пальмитоилоксиэтил, гептадеканоилоксиметил и 1-пальмитоилоксиэтил; низшие алкоксикарбонилокси низшие алкильные группы, такие как метоксикарбонилоксиметил, 1-бутоксикарбонилоксиэтил и 1-(изопропоксикарбонилокси)этил; карбокси низшие алкильные группы, такие как карбоксиметил и 2-карбоксиэтил; гетероциклические группы, такие как 3-фталидил; бензоилокси низшие алкильные группы, которые могут иметь заместитель, такой как 4-глицилоксибензоилоксиметил и 4-[N-(трет-бутоксикарбонил)глицилокси] бензоилоксиметил; (замещенные диоксолен) низшие алкильные группы, такие как (5-метил-2-оксо-1,3-диоксолан-4-ил)метил; циклоалкил-замещенные низшие алканоил низшие алкильные группы, такие как 1-циклогексилацетоксиэтил; и циклоалкилоксикарбонилокси низшие алкильные группы, такие как 1-циклогексилоксикарбонилоксиэтил.
Ацильная группа, как дано в определении R1, включает алифатические и ароматические ацильные группы и их производные с гетероциклическими группами, например, низшие алканоильные группы, такие как формильная группа, ацетильная группа, пропионильная группа, бутирильная группа, валерильная группа, изовалерильная группа и пивалоильная группа, ароильная группа, такая как бензоильная группа, толуолильная группа и нафтоильная группа, гетероароильная группа, такая как фуроильная группа, никотиноильная группа и изоникотиноильная группа и т.п. Из них, как особенно предпочтительные, могут быть указаны формильная группа, ацетильная группа, бензоильная группа и т.п.
В качестве примеров фармакологически приемлемой соли по настоящему изобретению можно указать соли неорганических кислот, такие как гидрохлорид, сульфат, гидробромид и фосфат, и соли органических кислот, такие как формиат, ацетат, трифторацетат, малеат, фумарат, тартрат, метансульфонат, бензолсульфонат и толуолсульфонат.
Соединения по настоящему изобретению существуют в виде различных стереоизомеров, определяемых их структурой. Необходимо сказать, что каждый из них входит в объем настоящего изобретения.
В качестве предпочтительных соединений из соединений настоящего изобретения можно указать соединения следующей общей формулы VII
Хотя эти соединения является оптическими изомерами благодаря их структуре, указанной выше, соединения, представленные следующей общей формулой (VII') имеют предпочтительную стереоструктуру:
Соединения по настоящему изобретению, в которых боковая цепочечная группа  является общей для соединений, представленных общей формулой (VII), присоединенные к циклической группе, могут оказывать лучшее действие по сравнению с другими соединениями с похожей структурой. Само собой разумеется, они могут проявлять значительно лучшее действие по сравнению с соединениями, обладающими похожей структурой, при внутривенном введении. Кроме того, они могут проявлять значительно лучшее действие по сравнению с соединениями, обладающими похожей структурой, при пероральном введении, поскольку они обладают лучшей биологической доступностью.
является общей для соединений, представленных общей формулой (VII), присоединенные к циклической группе, могут оказывать лучшее действие по сравнению с другими соединениями с похожей структурой. Само собой разумеется, они могут проявлять значительно лучшее действие по сравнению с соединениями, обладающими похожей структурой, при внутривенном введении. Кроме того, они могут проявлять значительно лучшее действие по сравнению с соединениями, обладающими похожей структурой, при пероральном введении, поскольку они обладают лучшей биологической доступностью.
Далее будут представлены главные способы получения соединений по настоящему изобретению. Следует сказать, что соединения по настоящему изобретению могут быть получены сочетанием известных реакций в дополнение к способам получения, которые будут даны далее.
Способ получения A-1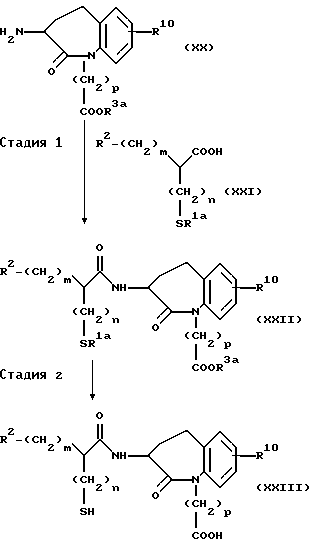
где R2 представляет атом водорода, низшую алкильную группу, циклоалкильную группу, арильную группу, которая может иметь заместитель, гетероарильную группу, которая может иметь заместитель, арилалкильную группу, которая может иметь заместитель или гетероарилалкильную группу, которая может иметь заместитель;
R10 представляет атом водорода, низшую алкильную группу, низшую алкоксигруппу, гидроксильную группу, атом галогена, арильную группу, которая может иметь заместитель, или гетероарильную группу, которая может иметь заместитель;
R1a представляет ацильную группу;
R3a представляет карбокси-защитную группу;
p представляют каждый независимо целое число 1 или 2;
m и n представляют каждый независимо целое число от 0 до 2.
(Стадия 1)
Эта стадия является стадией, при которой производное 3-амино-бензазепин-2-она (XX) конденсируют с производным карбоциклической кислоты (XXI) или с его активным производным, таким как галогенангидрид кислоты, с получением вследствие этого амидного производного (XXII). Конденсацию осуществляют обычным способом. Например, производное 3-амино-бензазепин-2-она (XX) взаимодействует с производным карбоновой кислоты (XXI) в инертном растворителе, представленном метиленхлоридом, тетрагидрофураном и т.п., в присутствии конденсирующего агента, обычно применяемого в данной области, таким как EEDQ (1-этоксикарбонил-2-этокси-1,2-дигидрохинолин), DCC (1,3-дициклогексилкарбодиимид), DEC [1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид] или диэтил цианофосфонат с получением вследствие этого амидного производного (XXII). Когда процесс конденсации протекает с участием хлорангидридного производного карбоновой кислоты (XXI), производное карбоновой кислоты (XXI) преобразуют в его хлорангидрид кислоты в проходящем инертном растворителе с использованием хлорирующего агента, обычно применяемого в этой области, такой как тионилхлорид и оксалилхлорид, с последующим взаимодействием с производным 3-аминобензазепин-2-она (XX) с получением вследствие этого соединения (XXII).
(стадия 2)
Эта стадия является стадией, при которой эфирную группу и ацилтиогруппу в амидном производном (XXII), полученном на стадии 1, подвергают снятию защитной группы обычным способом с получением посредством этого целевого соединения (XXIII). Снятие защитной группы осуществляют обычно применяемым в этой области методом. Например, его можно осуществлять гидролизом амидного производного (XXII) в разбавленном водном растворе щелочи, таком как гидроксид натрия, или разбавленном водном растворе минеральной кислоты.
Способ получения A-2
Когда R10 является арильной группой, которая может иметь заместитель, соединение (XX') может быть синтезировано следующим способом:
где R3a и p имеют каждое те же значения, что определены выше;
R10 представляет арильную группу, которая может иметь заместитель; и
X представляет атом галогена.
(Стадия 1)
Эта стадия является стадией, при которой производное гидрокситетралона (XXIV) трифторметансульфонируют с получением трифторметансульфонилокси производного (XXV). Трифторметансульфонирование осуществляют взаимодействием производного (XXIV) с трифторметансульфонангидридом или трифторметансульфонил хлорангидридом в инертном растворителе, представленном метиленхлоридом, тетрагидрофураном и т.п. в присутствии основания, такого как пиридин.
(Стадия 2)
Эта стадия является стадией, при которой трифторметансульфонилокси производное (XXV), полученное на стадии 1, взаимодействует с производным арилборной кислоты (X) или соединением арилолова (XI) с получением посредством этого производного арилтетралона (XXVI). Взаимодействие соединения (XXV) с соединением (X) или (XI) осуществляют в подходящем органическом растворителе, который не мог бы ингибировать эту реакцию в присутствии подходящего основания и палладиевого катализатора. В качестве примеров растворителя можно указать углеводороды, такие как толуол, и амиды, такие как N,N'-диметилформамид. В качестве примеров основания можно указать карбонаты щелочных и щелочноземельных металлов, такие как карбонат калия и карбонат кальция и органические основания, такие как триэтиламин и N-метилморфолин. В качестве примеров палладиевого катализатора можно указать тетракис(трифенилфосфин)палладий (O).
(Стадия 3)
Эта стадия является стадией, при которой бензазепиновое производное (XXVII) получают из производного арилтетралона (XXVI), полученного на стадии 2, путем реакции перегруппировки, обычно используемой в данной области. Перегруппировку можно осуществлять согласно способу, обычно применяемому специалистами, например, перегруппировкой Бекмана, перегруппировкой Шмидта или т. п. Говоря более конкретно, в случае перегруппировки Бекмана, бензазепиновое производное (XXVII) может быть получено обработкой производного арилтетралона (XXVI) гидрохлоридом гидроксиламина, с получением посредством этого оксима и затем, например, нагреванием оксима в присутствии подходящей кислоты. В случае перегруппировки Шмидта ее осуществляют, например, способом, который заключается во взаимодействии азотистоводородной кислоты или азида натрия в присутствии подходящей кислоты. В качестве кислоты может быть использована любая обычно применяемая кислота. Примеры включают серную кислоту, полифосфорную кислоту, трихлоруксусную кислоту, метансульфоновую кислоту и т.п.
(стадии 4 и 5)
Эти стадии являются стадиями, при которых бензазепиновое производное (XXVII), полученное в стадии 3, галогенируют и восстанавливают с получением посредством этого 3-галогенбензазепинового производного (XXIX).
Галогенирование и восстановление можно осуществлять каждое в соответствии со способом, обычно применяемым в этой области. В частности, предпочтительный результат может быть достигнут проведением этой реакции в соответствии со способом Negasawa et al. [J. Med. Chem., 14, 501 (1979)].
Конкретно, сначала бензазепиновое производное (XXVII), полученное на стадии 3 взаимодействует с PX5 (где X является Br и Cl) с получением таким образом дигалогензамещеннго бензазепинового производного (XXVIII), и далее соединение (XXVIII) каталитически гидрируют в присутствии палладиевого катализатора с получением посредством этого 3-галогенбензазепинового производного (XXIX).
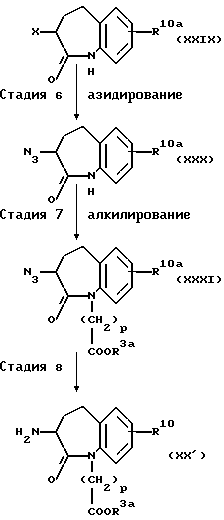
(Стадия 6)
Эта стадия является стадией, при которой 3-галогенбензазепиновое производное (XXIX), полученное на стадии 5, подвергают азидированию с получением посредством этого азида (XXX).
Азидирование осуществляют способом, обычно используемым в этой области взаимодействием 3-галогенбензазепинового производного (XXIX) с азидом натрия или лития в подходящем растворителе, например, этаноле, диметилформамиде или диметилсульфоксиде.
(Стадия 7)
Эта стадия является стадией, при которой азид (XXX), полученный на стадии 6, алкилируют обычным способом с получением посредством этого N-алкилированного производного (XXXI).
Алкилирование можно осуществлять обычным способом, применяемым в этой области. Например, его осуществляют взаимодействием азида (XXX) с иодалкильным эфиром в подходящем растворителе, например, диметилформамиде или тетрагидрофуране в присутствии сильного основания, таким как гидрид натрия. Альтернативно, его проводят взаимодействием азида (XXX) с галогеналкиловым эфиром в тетрагидрофуране в присутствии основания, такого как карбонат калия с применением катализатора межфазного переноса, таким как тетра-н-бутиламмоний бромид и бензилтриэтиламмоний иодид.
(стадия 8)
Эта стадия является стадией, при которой N-алкилированное производное (XXXI), полученное на стадии 7, восстанавливают обычным способом с получением посредством этого амина (XX').
Восстановление можно осуществлять способом, обычно применяемым в этой области. Его можно проводить каталитическим гидрированием N-алкилированного производного (XXXI) в подходящем растворителе, например, метаноле, этаноле или этилацетате в присутствии катализатора, такого как палладий на угле.
Этот амин (XX') является важным в качестве промежуточного продукта для получения соединения общей формулы (II), где Y3 является группой, представленной -CH2-.



В ряде формул R1a представляет ацильную группу; R2 представляет атом водорода, низшую алкильную группу, циклоалкильную группу, арильную группу, которая может иметь заместитель, гетероарильную группу, которая может иметь заместитель, арилалкильную группу, которая может иметь заместитель, или гетероарилалкильную группу, которая может иметь заместитель; R3a и R3a' представляет карбоксизащитную группу; R14 представляет атом водорода, низшую алкильную группу, низшую алкоксильную группу, атом галогена, арильную группу, которая может иметь заместитель, или гетероарильную группу, которая может иметь заместитель; t представляет целое число 0, 1 или 2; m представляет целое число 0, 1 или 2; и n представляет целое число 0, 1 или 2.
(Стадия 1)
Эта стадия является стадией, при которой производное 2-тиенилаланина (XLVII) защищают посредством фталимидирования обычным способом с получением производного фталимидкарбоновой кислоты (XLIII). Соединение (XLIII) может быть получено в соответствии со способом фталимидирования, которое обычно применяют в этой области. Например, фталевый ангидрид и соединение (XLII) нагревают в инертном растворителе, например, диметилформамиде или водном диоксане, или без применения какого-либо растворителя в присутствии основания, такого как триэтиламин, или без использования какого-либо основания с получением посредством этого производного фталимидкарбновой кислоты (XLIII). Альтернативно, фталимидирующий агент, такой как этоксикарбонилфталимид, взаимодействует с соединением (XLII) в присутствии основания, такого как карбонат натрия и гидрокарбонат натрия, с получением посредством этого производного фталимидкарбоновой кислоты (XLIII).
(Стадия 2)
Эта стадия является стадией, при которой производное фталимидкарбоновой кислоты (XLIII), полученное на стадии 1, или его активное производное, такое как галогенангидрид кислоты, конденсируют с эфирным производным аминокислоты (XII) обычным способом с получением посредством этого амидного производного (XLIV).
Конденсацию осуществляют обычным способом, применяемым в этой области. Например, соединение (XLIII) взаимодействует с эфирным производным аминокислоты (XLII) в инертном растворителе, представленном метиленхлоридом, тетрагидрофураном и т. д. , в присутствии обычно применяемого конденсирующего реагента, такого как ЭЭДХ (1-этоксикарбонил-2-этокси-1,2-дигидрохинолин), ДЦК (1,3-дициклогексилкарбодиимид), ДЭК (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида) или диэтилцианофосфоната, с получением посредством этого соединения (XLIV).
Когда конденсацию проводят с участием производного хлорангидрида кислоты (XLIII), соединение (XLIII) преобразуют в хлорангидрид кислоты в подходящем инертном растворителе с использованием обычно применяемого хлорирующего агента, такого как тионилхлорид и оксалилхлорид, и затем полученный таким образом хлорангидрид кислоты подвергают взаимодействию с эфирным производным аминокислоты (XLII) с получением посредством этого соединения (XLIV).
(Стадия 3)
Эта стадия является стадией, при которой гидроксильная группа производного амида (XLIV), полученного на стадии 2, окисляется с получением посредством этого альдегидного производного (XLV). Соединение (XLV) может быть также получено способом, обычно применяемым для окислении алкиловых спиртов. Например, альдегидное производное (XLV) может быть получено путем осуществления окисления Сванна (Swann) с использованием оксалилхлорида и диметилсульфоксида или окисления с использованием диоксида магния в походящем апротонном растворителе, таком как дихлорметан и хлороформ.
(Стадия 4)
Эта стадия является стадией, при которой производное альдегида (XLIV), полученное на стадии 3, циклизуют с прямым получением посредством этого, через енаминовое производное, эфирного производного (XLVI) или производного карбоновой кислоты (XLVII). Например, эфирное производное (XLVI) может быть получено обработкой соединения (XLV) с трифторуксусной кислотой в подходящем апротонном растворителе, таком как дихлорметан и хлороформ.
Альтернативно, производное карбоновой кислоты (XLVII) может быть получено обработкой соединения (XLV) смесью трифторметансульфоновой кислоты и трифторуксусного ангидрида или одной трифторметансульфоновой кислотой в подходящем апротонном растворителе, таком как дихлорметан и хлороформ.
(Стадия 5)
Эта стадия является стадией, при которой эфирном производное (XLVI), непосредственно полученное на стадии 4, подвергают снятию защитной группы обычным способом с получением посредством этого производного карбоновой кислоты (XLVII). Например, эфирное производное (XLVI) подвергают обработке сильной протонной кислотой с трифторметансульфоновой кислотой в протонном растворителе, таком как этанол, с получением посредством этого производного карбоновой кислоты (XLVII).
(Стадия 6)
Эта стадия является стадией, при которой функциональная карбоксильная группа производного карбоновой кислоты (XLVII), полученное на стадиях 4 и 5, защищена этерификацией с получением посредством этого эфирного производного (XLVIII). В качестве эфирной группы вводятся обычная алкильная группа, разветвленная алкильная группа или группа, которая может быть селективно снята в таких реакционных условиях, при которых ацилтиогруппа соединения (L), синтезируемого на стадии 8, не гидролизуется. Этерификацию осуществляют способом, обычно применяемым в данной области. Например, производное (XLVII) взаимодействует со спиртом в присутствии минеральной кислоты, такой как соляная кислота и серная кислота. Альтернативно, производное (XLVII) взаимодействует с, например, дифенилбромметаном, трифенилбромметаном или триметилсилилэтанолом в инертном растворителе, таком как диметилформамид и тетрагидрофуран, в присутствии основания, такого как карбонат цезия и карбонат калия. Таким образом может быть получено эфирное производное (XLVIII).
(Стадия 7)
Эта стадия является стадией, при которой фталимидная группа эфирного производного (XLVIII), полученного на стадии 6, подвергают снятию защитной группы с получением посредством этого амина (XLIX). Этот способ находится в соответствии с обычным способом. Например, эфирное производное (XLVIII) обрабатывают гидразином, в растворителе, таком как вода, спирт и тетрагидрофуран, и, посредством этого, производится снятие защитной группы у фталимида. Таким образом может быть получен амин (XLIX).
(Стадия 8)
Эта стадия является стадией, при которой производное кислоты (XIII) или его активное производное, такое как галогенангидрид, конденсируют с амином (XLIX), полученным на стадии 7, с получением посредством этого производного амида (L). Эту реакцию осуществляют обычно применяемым методом в этой области. Например, производное карбоновой кислоты (XIII) взаимодействует с амином (XLIX) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран в присутствии обычно применяемого конденсирующего реагента, такого как EEDQ, DCC, DEC или диэтилцианофосфат, с получением посредством этого соединения (L). Когда реакцию проводят с участием производного хлорангидрида карбоновой кислоты (XIII), производное карбоновой кислоты (XIII) преобразуют в его хлорангидрид в подходящем инертном растворителе с помощью галогенирующего агента, обычно применяемого в этой области, таким как тионилхлорид и оксалилхлорид, и затем полученный галогенангидрид взаимодействует с амином (XLIX) с получением посредством этого соединения (L).
(Стадия 9)
Эта стадия является стадией, при которой любая или обе ацилтиогруппа и эфирная группа производного амида (L), полученного на стадии 8, подвергается (ются) снятию защитной группы обычным способом с получением посредством этого производного карбоновой кислоты (LI). Когда отщепляемой (ыми) группой (ами) является (ются) обычная (ые) алкильная(ые) группа(ы), разветвленная (ые) алкильная (ые) группа(ы), и т.п., например, производное амида (L) гидролизуют в разбавленном водном растворе щелочи, такой как гидроксид натрия и гидроксид лития, или в разбавленном водном растворе минеральной кислоты с получением посредством этого меркаптопроизводного карбоновой кислоты (LI), обладающей R1a, представляющей водород. Когда отщепляемой(ыми) группой(ами) является (ются) трет-бутильная(ые) группа(ы), арилалкильная(ые) группа(ы), разветвленная(ые) алкильная(ые) группа(ы) и т.п., снятие защитной группы осуществляют в таких обычных реакционных условиях, при которых ацилтиогруппа остается стабильной, например, каталитическим гидрированием, обработкой трифторуксусной кислотой или т.п., с получением посредством этого ацилтиопроизводного карбоновой кислоты (LI).
(Стадия 10)
Эта стадия является стадией, при которой адилтиогруппа, если она имеется, производного карбоновой кислоты (LI), полученного на стадии 9, гидролизуется с получением посредством этого меркаптопроизводного карбоновой кислоты (LII). Гидролиз можно осуществлять в условиях гидролиза, обычно применяемых в данной области, то есть в разбавленном водном растворе щелочи, такой как гидроксид натрия и гидроксид лития, или в разбавленном водном растворе минеральной кислоты.
Способ получения B-2
Когда n равно 0, соединение (I IV) может быть также синтезировано следующим способом:
В ряде формул R1a, R2, R3a, R14, m и t каждое имеет то же значение, что определено выше.
(Стадия 1)
Эта стадия является стадией, при которой производное α- гидрокси карбоновой кислоты (XIV) конденсируют с амином (XI IX), полученным по вышеупомянутому способу получения B-1, стадия 7, обычным способом с получением посредством этого амидного производного 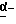 гидрокси карбоновой кислоты (LII). Подобно способу получения B-1 стадия 6, соединение (LIII) и (XL IX) взаимодействует в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии конденсирующего агента, обычно применяемого в этой области, например, EE O, DCC, DEC, диэтилцианофосфат или тому подобное. Таким образом можно получить амидное производное (LIII).
гидрокси карбоновой кислоты (LII). Подобно способу получения B-1 стадия 6, соединение (LIII) и (XL IX) взаимодействует в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии конденсирующего агента, обычно применяемого в этой области, например, EE O, DCC, DEC, диэтилцианофосфат или тому подобное. Таким образом можно получить амидное производное (LIII).
(Стадия 2)
Эта стадия является стадией, при которой гидроксильная группа производного амида (LIII), полученного на стадии 1, преобразуется в ацилтиогруппу обычным способом с получением посредством этого ацилтиопроизводного (LIV). Соединение (LIV) может быть синтезировано согласно способу, обычно используемому для получения ацилтиопроизводного. Например, соединение (LIII) обрабатывают реакцией типа Митоунобу (Mitsunobu) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, с использованием трифенилфосфита и эфира азодикарбоновой кислоты, такого как ДИАД (диизопропилазодикарбоксилат). Таким образом может быть получено ацилтиопроизводное (LIV).
Способ получения B-3
Соединение, представленное общей формулой VIb, может быть получено следующим способом:

В ряде формул R1a представляет ацильную группу; R2 представляет атом водорода, низкую алкильную группу, циклоалкильную группу, арильную группу, которая может иметь заместитель, гетероарильную группу, которая может иметь заместитель, арилалкильную группу, которая может иметь заместитель, или гетероарилалкильную группу, которая может иметь заместитель; R3a представляет карбокси-защитную группу; R15 представляет атом водорода, низшую алкильную группу, низшую алкоксильную группу, гидроксильную группу, атом галогена, арильную группу, которая может иметь заместитель; или гетероарильную группу, которая может иметь заместитель; S представляет целое число 0, 1 или 2; m представляет целое число 0, 1 или 2; и n представляет целое число 0, 1 или 2.
(Стадия 1)
Эта стадия является стадией, при которой аминогруппа 3-тиенилаланильного производного кислоты (LV) защищается путем фталимидирования получением фталимидного производного карбоновой кислоты (LVI). Соединение (LVI) может быть получено способом, обычно применяемым в данной области. Например, фталимидное производное карбоновой кислоты (LVI) может быть получено нагреванием фталевого ангидрида с соединением (LV) в инертном растворителе, таком как диметилформамид и водный диоксан, или без использования какого-либо растворителя в присутствии основания, такого, как триэтиламин, или без использования какого-либо основания.
Альтернативно, оно может быть получено нагреванием фталимидирующего агента, такой как этоксикарбонилфталимид, с соединением (LV), в присутствии основания, такого как карбонат натрия и гидрокарбонат натрия.
(Стадия 2)
Эта стадия является стадией, при которой фталимидное производное карбоновой кислоты (LVI), полученное на стадии 1, или его активное производное, такое как галогенагидрид кислоты, конденсируют с эфирным производным аминокислоты (XII') обычным способом с получением амидного производного (LII).
Конденсацию осуществляют обычным способом, применяемым в этой области. Например, соединение (LVI) взаимодействует с эфирным производным аминокислоты (XII') в инертном растворителе, представленном метиленхлоридом, тетрагидрофураном и т.д., в присутствии обычно применяемого конденсирующего реагента, такого как ЭЭДХ (1-этоксикарбонил-2-этокси-1,2-дигидрохинолин), ДЦК (1,3-дициклогексилкарбодиимид), ДЭК (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида) или диэтилцианофосфоната. Таким образом может быть получено соединение (LVII). Когда конденсацию проводят с участием хлорангидрида соединения (LVI), соединение (LVI) преобразуют в хлорангидрид кислоты в подходящем инертном растворителе с использованием обычно применяемого хлорирующего агента, такого как тионилхлорид и оксалилхлорид, и затем получены таким образом хлорангидрид кислоты подвергают взаимодействию с эфирным производным аминокислоты (XII') с получением соединения (LVII).
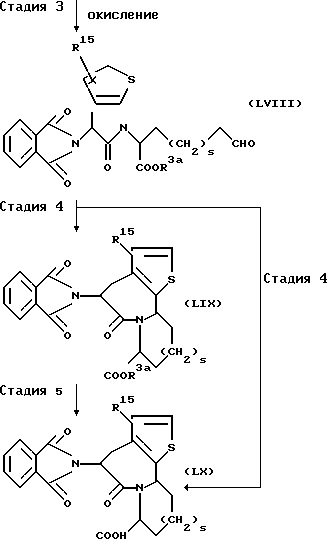
(Стадия 3)
Эта стадия является стадией, при которой гидроксильная группа производного амида (LVII), полученного на стадии 2, окисляемой обычным способом с получением альдегидного производного (LVIII). Соединение (LVIII) может быть также получено способом, обычно применяемым для окисления алкиловых спиртов. Например, альдегидное производное (LVIII) может быть получено окислением Сванна (Swann) с использованием оксалилхлорида и диметилсульфоксида или окислением с использованием диоксида магния в подходящем апротонном растворителе, таком как дихлорметан и хлороформ.
(Стадия 4)
Эта стадия является стадией, при которой производное альдегида (LVIII), полученное на стадии 3, циклизуют обычным способом с получением через енаминовое производное, эфирного производного (LIX). Альтернативно, эта стадия является стадией, при которой альдегидное производное (LVIII) циклизуют с непосредственным получением через енаминовое производное, производного карбоновой кислоты (LX).
Например, эфирное производное (LIX)может быть получено обработкой соединения (LVIII) трифторуксусной кислотой в подходящем апротонном растворителе, таком как дихлорметан и хлороформ.
Альтернативно, производное карбоновой кислоты (LX) может быть получено обработкой соединения (LVIII) смесью трифторметансульфоновой кислоты и трифторуксусного ангидрида или одной трифторметансульфоновой кислотой в подходящем апротонном растворителе, таком как дихлорметан и хлороформ.
(Стадия 5)
Эта стадия является стадией, при которой эфирное производное (LIX), полученное на стадии 4, подвергают снятию защитной группы с получением производного карбоновой кислоты (LX). Например, эфирное производной (LIX) обрабатывают протонной кислотой, такой как трифторметансульфоновая кислота в протонном растворителе, таком как этанол. Таким образом может быть получено производное карбоновой кислоты (LX).
(Стадия 6)
Эта стадия является стадией, при которой функциональную карбоксильную группу производного карбоновой кислоты (LX), полученного на стадиях 4 и 5, защищают этерификацией с получением эфирного производного (LXIV).
В качестве эфирной группы вводятся обычная алкильная группа, разветвленная алкильная группа или группа, которая может быть селективно сняты, в таких реакционных условиях, при которых ацилтиогруппа соединения (LXIII), синтезируемого на стадии 8, не гидролизуется. Этерификацию осуществляют способом, обычно применяемым в данной области. Например, производное карбоновой кислоты (LX) взаимодействуют со спиртом в присутствии минеральной кислоты, такой как соляная кислота и серная кислота. Альтернативно, производное (LX) взаимодействует с, например, дифенилбромметаном, трифенилбромметаном или триметилсилилэтанолом в инертном растворителе, таком как диметилформамид и тетрагидрофуран, в присутствии основания, такого как карбонат цезия и карбонат калия. Таким образом может быть получено эфирное производное (LXIV).
(Стадия 7)
Эта стадия является стадией, при которой фталимидную группу эфирного производного (LXIV), полученного на стадии 6, подвергают снятию защитной группы с получением амина (LXII). Этот способ находится в соответствии с обычным способом. Например, соединение (LXIV) обрабатывают гидразином в растворителе, таком как вода, спирт и тетрагидрофуран, и, посредством этого, происходит снятие защитной группы у фталимида. Таким образом может быть получен амин (LXII).
(Стадия 8)
Эта стадия является стадией, при которой производное карбоновой кислоты (XIII) или его активное производное, такое как галогенангидрид, конденсируют с амином (LXII), полученным на и стадии 7, с получением производного амида (LXIII). Эту реакцию осуществляют обычно применяемым методом в этой области. Например, производное карбоновой кислоты (XII) взаимодействует с амином (LXII) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии обычно применяемого конденсирующего реагента, такого как ЭЭДХ, ДЦК, ДЭК или диэтилцианофосфат. Таким образом, может быть получено соединение (LXIII). Когда реакцию проводят, например, с участием хлорангидрида карбоновой кислоты (XIII), производное карбоновой кислоты (XII) преобразуют в его хлорангидрид в подходящем инертном растворителе с помощью галогенирующего агента, обычно применяемого в этой области, таким как тионилхлорид и т. п. , и затем полученный галогенангидрид взаимодействуют с амином (LXII) с получением соединения (LXIII).
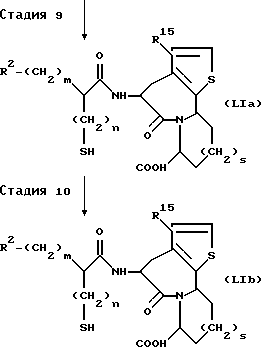
(Стадия 9)
Эта стадия является стадией, при которой любая или обе ацилтиогруппа и эфирная группа производного амида (LXIII), полученного на стадии 8, подвергается(ются) снятию защитной группы обычным способом с получением производного карбоновой кислоты (LIa). Когда отщепляемой(ыми) группой(ами) является(ются), например, обычная(ые) алкильная(ые) группа(ы), разветвленная(ые) алкильная(ые) группа(ы) и т.п., производное амида (LXIII) гидролизуют в разбавленном водном растворе щелочи, такой как гидроксид натрия и гидроксид лития, или в разбавленном водном растворе минеральной кислоты с получением меркаптопроизводного карбоновой кислоты (LIa), имеющей RIa, представляющий водород. Когда отщепляемой(ыми) группой(ами) является(ются) трет-бутильная(ые) группа(ы), алилалкильная(ые) группа(ы)), разветвленная(ые) аллилалкильная(ые) группа(ы) и т.п., снятие защитной группы осуществляют в таких обычных реакционных условиях, при которых ацилтиогруппа остается стабильной, например, каталитическим гидрированием, обработкой трифторуксусной кислотой или т.п., с получением посредством этого ацилтиопроизводного карбоновой кислоты (LIa).
(Стадия 10)
Эта стадия является стадией, при которой ацилтиогруппа, если она имеется, производного карбоновой кислоты (LIa) полученного на стадии 9, гидролизуется с получением меркаптопроизводного карбоновой кислоты (LIb). Гидролиз можно осуществлять в условиях гидролиза, обычно применяемых в данной области, то есть в разбавленном водном растворе щелочи, такой как гидроксид натрия и гидроксид лития, или разбавленном водном растворе минеральной кислоты.
Способ получения B-4
Когда n равно 0, соединение (LIVa) может быть также синтезировано следующим способом:
В ряде формул R1a представляет ацильную группу; R2 представляет атом водорода, низшую алкильную группу, циклоалкильную группу, арильную группу, которая может иметь заместитель, гетероарильную группу, которая может иметь заместитель, арилалкильную группу, которая может иметь заместитель, или гетероарилалкильную группу, которая может иметь заместитель; R3a представляет карбокси-защитную группу; R15 представляет атом водорода, низшую алкильную группу, низшую алкоксильную группу, гидроксильную группу, атом галогена, арильную группу, которая может иметь заместитель, или гетероарильную группу, которая может иметь заместитель; S представляет целое число 0, 1 или 2; и m представляет целое число 0, 1 или 2.
(Стадия 1)
Эта стадия является стадией, при которой производное α- гидрокси карбоновой кислоты (XIV) конденсируют с амином (LXII), полученным по вышеупомянутому способу получения В-3, стадия 7, обычным способом с получением амидного производного α- -гидрокси карбоновой кислоты (LXV). Подобно способу получения В-3, стадия 8, соединения (XIV) и (IXII) взаимодействуют в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии конденсирующего агента, обычно применяемого в этой области, например, EE DQ, DCC, DEC или диэтилцианофосфат. Таким образом, можно получить амидное производное (LXV).
(Стадия 2)
Эта стадия является стадией, при которой гидроксильная группа производного амида (LXV), полученного на стадии 1, преобразуется в ацилтиогруппу с получением ацилтиопроизводного (LVIa). Соединение (LVIa) может быть синтезировано согласно способу, обычно используемому для преобразования гидроксильной группы в ацилтиогруппу. Например, соединение (L XV) обрабатывают реакцией типа Митсунобу (Mitsunobu) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, с использованием трифенилфосфата и эфира азодикарбоновой кислоты, такого как DIAD (диизопропилазодикарбоксилат). Таким образом может быть получено ацилтиопроизводное (LVIa).
Способ получения С-1
Соединение, представленное общей формулой (VII), может быть получено следующим способом: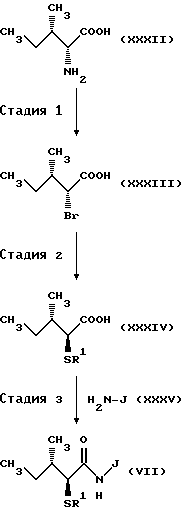
В ряде формул R2 представляет атом водорода или ацильную группу; и J представляет циклическую группу, обладающую ингибирующей ACE активностью.
(Стадия 1)
Соответственно, эта стадия является стадией, при которой аминогруппа D-алло-изолейцина (XXXII) подвергается бромированию с получением бромида (XXXIII). Бромид (XXXIII) может быть получен согласно способу, обычно используемому в этой области для стереоселективного бромирования. Например, соединение (XXXII) обрабатывают нитритом, таким как нитрит натрия и нитрит серебра в водном растворе бромистого водорода. Таким образом может быть получен бромид (XXXIII).
(Стадия 2)
Соответственно, эта стадия является стадией, при которой группа бром в бромиде (XXXIII), полученного на стадии 1, преобразуется в ацилтиогруппу с получением посредством этого ацилтиопроизводного кислоты XXXIV). Эту реакцию осуществляют обычным способом. Например, бромид (XXXIII) подвергают взаимодействию с тиокарбоксилатом, таким как тиоацетат калия и тиоацетат натрия в полярном органическом растворителе, таком как ацетонитрил и ацетон. Альтернативно, бромид (XXXIII) взаимодействует с тиокарбоновой кислотой, такой как тиоуксусная кислота и тиобензойная кислота, в присутствии основания, такого как карбонат калия и карбонат цезия. Таким образом может быть получено производное ацилтиопентановой кислоты (XXXIV).
(Стадия 3).
Эта стадия является стадией, при которой производное ацилтиопентановой кислоты (XXXIV) полученное на стадии 2, или его активное производное, такое как галогенангидрид кислоты, конденсируют с эфирным производным аминокислоты (XXXV) с получением посредством этого амидного производного (VII). Например, производное ацилтиопентановой кислоты (XXXIV) взаимодействует с эфирным производным аминокислоты (XXXV) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии обычно применяемого конденсирующего реагента, такого как EEDQ (I-этоксикарбонил-2-этокси-1,2-дигидрозинолин), DCC (1,3-дициклогексилкарбодиимид), DEC (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбоддимида) или диэтилцианофосфоната. Таким образом может быть получено амидное производное (VII). Когда конденсацию проводят с участием хлорангидрида производного ацилтиопентановой кислоты (XXXIV), производное ацилтиопентановой кислоты (XXXIV) преобразуют в хлорангидрид кислоты в подходящем инертном растворителе с использованием обычно применяемого хлорирующего агента, такого как тионилхлорид и оксалилхлорид, и затем полученный таким образом хлорангидрид кислоты подвергают взаимодействию с эфирным производным аминокислоты (XXXV) с получением целевого соединения (VII).
Способ получения С-2.
Соединение, представленное общей формулой (VII), может быть получено следующим способом: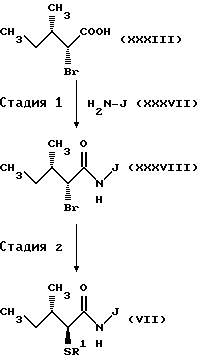
В ряде формул R1 и J каждый имеют те же значения, что указаны выше.
(Стадия 1)
Соответственно, эта стадия является стадией, при которой бромированное производное карбоновой кислоты (XXXIII), полученное по способу получения C-2, стадия 1, или его активное производное, такое как галогенангидрид, конденсируют с эфирным производным аминокислоты (XXXVII) с получением амидного производного (XXXVIII). Амидное производное (XXXVIII) может быть получено так же, как и по способу получения C-1, стадия 3.
(Стадия 2)
Соответственно, эта стадия является стадией, при которой группа бром в амидном производном (XXXVIII), полученном на стадии 1, преобразуется в ацилтиогруппу с получением амидного производного (VII), которое является тем же, что и соединение, полученное по способу получения C-1, стадии 3. Амидное производное (VII) может быть получено такой же обработкой, как и по способу получения C-1, стадия 2.
Способ получения C-3
Те из соединений, представленных общей формулой (VII), где R3 является водородом, могут быть также получены следующим способом:
В ряде формул R1 представляет атом водорода или ацильную группу; R3a представляет карбокси-защитную группу; R4 представляет атом водорода, низшую алкильную группу или арилалкильную группу, которая может иметь заместитель; J имеет те же значения, что указаны выше.
А именно, либо эфирная, либо обе - эфирная и ацилтио группы соединения (XL), полученного по способам получения C-1 и C-2 подвергается(ются) снятию защитной группы обычным способом с получением посредством этого производного карбоновой кислоты (XLI). Когда отщепляемой(ыми) группой(ами) является(ются), например, обычная(ые) алкильная(ые) группа(ы), разветвленная(ые) алкильная(ые) группа(ы) и т.п., производное амида (VII) гидролизуют в разбавленном водном растворе щелочи, такой как гидроксид лития, или в разбавленном водой растворе минеральной кислоты с получением производного карбиновой кислоты (XLI), имеющего R1, представляющий водород. Когда отщепляемой(ыми) группой(ами) является(ются) трет-бутильная(ые) группа(ы), разветвленная аллилалкильная(ые) группа(ы), такая(ие) как бензгидрильная группа, силилэтильная(ые) группа(ы), такая(ие) как триметилсилильная группа, и т.п., происходит снятие защитной группы только у части эфирных групп в таких реакционных условиях, при которых ацилтиогруппа остается стабильной, например, обработкой трифторуксусной кислотой или алкиламмонийфторидом, с получением ацилтиопроизводного карбоновой кислоты (XLI).
Способ получения D-1
Соединение, представленное следующей общей формулой (D), может быть получено следующим способом:
где R1, R2, R3, R18, m и n каждый имеют те же значения, что указаны выше:


В ряде формул, представляющих вышеуказанный способ получения D-I, R2, R3a, 18 и n каждый имеют те же значения, что указаны выше; и R1a представляет группу, выбранную из тех, которые даны в определении R1, за исключением атома водорода.
(Стадий 1)
Конкретно эта стадия, которая включает нитрование широко известного производного циклического амина (I) или производного циклического амина (I), полученного обычным способом.
Нитрование, описанное выше, осуществляют обычным способом. Можно указать способ, который включает обработку нитрующим агентом, обычно используемым в данной области, например, нитроний тетрафторборатом или т.п., в инертном растворителе, например, хлороформе, дихлорметане или т.п., применяемом для осуществления нитрования, способ, который включает осуществление нитрования с помощью дымящей азотной кислоты или т.п. в присутствии уксусной кислоты, уксусного ангидрида, серной кислоты или т.п., и другие.
(Стадия 2)
Эта является стадией, которая включает этерификацию функциональной группы карбоновой кислоты нитросоединения (II), полученного на стадии 1.
В качестве вышеуказанной эфирной группы вводятся обычная алкильная группа или группа, которая может быть селективно снята в таких реакционных условиях, при которых тиоацетильная группа соединения (IX), синтезируемого на стадии 8, не гидролизуется. Эфирное соединение (IV) может быть получено, например, взаимодействием нитросоединения (II) со спиртом в присутствии минеральной кислоты, такой как соляная кислота и серная кислота, или, альтернативно, взаимодействием нитросоединения (II) с дифенилбромметаном, трифенилбромметаном или триметилсилилэтанолом в инертном растворителе, таком как диметилформамид и тетрагидрофуран, в присутствии основания, такого как карбонат цезия и карбонат калия.
(Стадия 3)
Это является стадией, которая включает восстановление нитрогруппы соединения (IV), полученного на стадии 2, обычным способом с получением анилинового соединения (VI).
Восстановление, описанное выше, можно осуществлять способом, обычно применяемым в этой области. Например, можно указать каталитическое восстановление с использованием палладия, платины или т.п. в качестве катализатора или восстановление с применением металла, такого как цинк и железо, в кислых условиях.
(Стадия 4)
Конкретно это является стадией, которая включает взаимодействие анилинового соединения (VI), полученного на стадии 3, с доступным производным хлорсульфоновой кислоты или с производным хлорсульфоновой кислоты, полученным широко известным способом с получением сульфониламидного производного (VII).
Например, сульфониламидное производное (VII) может быть получено взаимодействием с анилинового соединения (VI) с производным хлорсульфоновой кислоты с использованием инертного растворителя, такого как ацетонитрил, тетрагидрофуран, толуол и дихлорметан, в присутствии основания, такого как пиридин, триэтиламин и карбонат натрия.
(Стадия 5)
Эта стадия является стадией, включающей снятие защитной группы сульфонамидного производного (VII), полученного на стадии 4, с получением аминосоединения (VIII).
Снятие защитной группы осуществляют обычным способом. Аминосоединение (VIII) может быть обычно получено, например, обработкой соединения (VII) гидразином в растворителе, таком как вода, спирт и тетрагидрофуран, со снятием посредством этого защитной группы у фталимидной группы.
(Стадия 6)
Эта стадия является стадией, включающей конденсацию широко известного производного карбоновой кислоты или производного карбоновой кислоты, полученного широко известным способом, или его активного производного, такое как галогенангидрид кислоты, с аминосоединением (VIII), полученного на стадии 5, с получением амидного производного (IX).
Конденсация, описанная выше, может быть осуществлена обычным способом. Например, вышеуказанное производное карбоновой кислоты взаимодействует с аминосоединением (XIII) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии конденсирующего реагента, такого как ЭЭДХ (1-этоксикарбонил-2-этокси-1,2-дигидрохинолин), ДЦК (1,3-дициклогексилкарбодиимид), ДЭК (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида) или диэтилцианофосфонат. Таким образом может быть получено амидное производное (IX). Когда конденсацию проводят с участием хлорангидрида производного карбоновой кислоты, производное карбоновой кислоты преобразуют в хлорангидрид кислоты в подходящем инертном растворе с хлорирующим агентом, таким как тионилхлорид и оксалилхлорид, и затем полученный таким образом хлорангидрид подвергают взаимодействию с аминосоединением (VIII) с получением амидного производного (IX).
(Стадия 7)
Эта стадия является стадией, которая включает снятие защитной группы у людей или у обеих эфирной группы и тиоацетильной группы производного амида (IX), полученного в вышеуказанной стадии 6, с получением целевого соединения (X). Когда эфирной группой является обычная алкильная группа, разветвленная алкильная группа и т.п., производное амида (X) гидролизуют в разбавленном водном растворе щелочи, такой как гидроксид натрия и гидроксид лития, или в разбавленном растворе минеральной кислоты с получением меркаптопроизводного карбоновой кислоты (X), содержащего R1, представляющий водород. Когда эфирной группой является трет-бутильная группа, аллилалкильная группа, разветвленная аллилалкильная группа и т.п., снятие защитной группы осуществляют в таких реакционных условиях, при которых ацилтиогруппа остается стабильной, например, каталитическим гидрированием, с использованием трифторуксусной кислоты или т.п., с получением тиоацетильного производного карбоновой кислоты (X).
Способ получения D-2
Те из соединений, представленных общей формулой (D), где n равно 0, могут быть также получены следующим способом:
В ряде формул, описанных выше, R1a, R2, R3a, R18 и m каждый имеют те же значения, что указаны выше.
(Стадия 1)
Эта стадия является стадией, которая включает конденсацию производного α- гидрокси карбоновой кислоты (XI) или производного - гидроксикарбоновой кислоты (XI), полученного общеизвестным способом с аминосоединением (VIII), полученным по вышеупомянутому способу получения D-1, стадия 5, с получением амидного производного (XII).
По вышеуказанной конденсации, подобной способу получения D-1, стадия 6, соединения (XI) и (VIII) взаимодействуют в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии конденсирующего регента, такого как ЭЭДХ (1-этоксикарбонил-2-этокси-1,2-дигидрохинолин), ДЦК (1,3-дициклогексилкарбодиимид), ДЭК (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида) или диэтилцианофосфонат. Таким образом можно получить амидное производное (XII).
(Стадия 2)
Эта стадия является стадией, которая включает тиоэтерификацию гидроксильной группы производного амида (XII), полученного на стадии 1, с получением посредством этого ацетилтио производного (XIII). Соединение (XIII) может быть синтезировано согласно способу, обычно используемому для тиоэтерификации гидроксильной группы. Например, соединение (XII) обрабатывают реакцией типа Митсунобу (Milscenobu) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, с использованием трифенилфосфита и эфира азодикарбоновой кислоты, такого как ДИАД (диизопропилазодикарбоксилат). Таким образом может быть получено ацетилтиопроизводное (XIII).
(Стадия 3)
Эта стадия является стадией, которая включает снятие защитной группы у любой или у обеих эфирной группы и тиоацильной группы производного амида (XIII), полученного в вышеуказанной стадии 2, с получением производного карбоновой кислоты (XIV). Таким образом, производное карбоновой кислоты (XIV) может быть синтезировано тем же способом, что и по вышеуказанному способу получения D-1, стадия 7.
Способ получения E-1
Соединение, представленное следующей общей формулой (E), может быть получено следующим способом:
где R1 представляет атом водорода или ацильную группу;
R2 представляет атом водорода, низшую алкильную группу, гетероарильную группу, которая может иметь заместитель, или арилалкильную группу, которая может иметь заместитель;
R3 представляет атом водорода, низшую алкильную группу или арилалкильную группу;
R19 представляет атом водорода, низшую алкильную группу, низшую алкоксигруппу, гидроксильную группу или атом галогена;
p, m и n представляют каждый независимо целое число 0, 1 или 2;


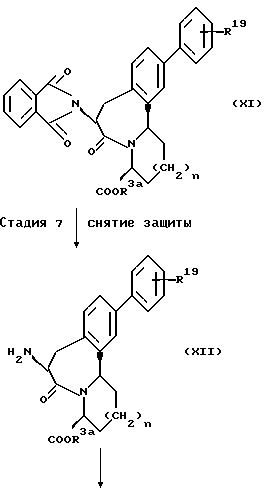
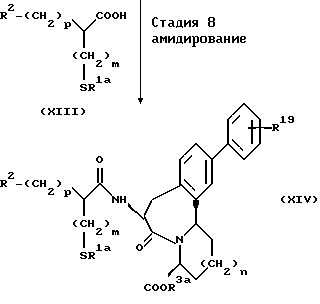
В ряде формул, описанных выше, R2, R19, p, n и m каждый имеют те же значения, что описаны выше; R1a представляет группу, выбранную из групп, данных в определении R1, за исключением атома водорода; R3a представляет группу, выбранную из групп, данных в определении R3, за исключением атома водорода.
(Стадия I)
Эта стадия является стадией, при которой аминогруппа бифениламино производного кислоты, представленного общей формулой (IV) защищается путем фталимидирования с получением посредством этого фталимидного производного карбоновой кислоты (V). Фталимидирование может быть осуществлено способом, обычно применяемым в данной области. Например, фталимидное производное карбоновой кислоты (V) может быть получено нагреванием фталевого ангидрида с соединением (IV) в инертном растворителе, таком как диметилформамид и диоксан, или без использования какого-либо растворителя. Альтернативно, оно может быть получено нагреванием фталимидирующего агента, такого как этоксикарбонилфталимид, с соединением (IV) в присутствии основания, такого как карбонат натрия и гидрокарбонат натрия.
(Стадия 2)
Эта стадия является стадией, при которой фталимидное производное карбоновой кислоты (VI, полученное на стадии 1, или его активное производное, такое как галогенангидрид кислоты, конденсируют с эфирным производным аминокислоты, представленным общей формулой (VI) обычным общей формулой (VI) обычным способом с получением амидного производного (VII). Конденсацию можно осуществлять обычным способом, применяемым в этой области. Например, соединения (V) и (VI) взаимодействуют в инертной растворителе, представленном метиленхлоридом, тетрагидрофураном и т.д., в присутствии обычно применяемого конденсирующего реагента, такого как ЭЭДХ (1-этоксикарбонил-2-этокси-1,2-дигидрохинолин), ДЦК (1,3-дициклогексилкарбодиимид), ДЭК (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида) или диэтилцианофосфоната. Таким образом может быть получено соединение (VII). Когда конденсацию проводят с участием хлорангидрида соединения (V), соединение (V) преобразуют в хлорангидрид кислоты в подходящем инертном растворителе с использованием обычно применяемого хлорирующего агента, такого как тионилхлорид и оксалилхлорид, и затем полученный таким образом хлорангидрид кислоты подвергают взаимодействию с эфирным производным аминокислоты (VI) с получением соединения (VII).
(Стадия 3)
Эта стадия является стадией, при которой гидроксильная группа производного амида (VII), полученного на стадии 2, окисляется обычным способом с получением посредством этого альдегидного производного (VIII). Соединение (VIII) может быть также получено способом, обычно применяемым для окисления алкиловых спиртов. Например, альдегидное производное (VIII) может быть получено путем осуществления окисления Сванна (Swann) с использованием оксалилхлорида и диметилсульфоксида или окислением с использованием пиридиний хлорформиата или диоксида магния в подходящем апротонном растворителе, таком как дихлорметан и хлороформ.
(Стадия 4)
Эта стадия является стадия, при которой производное альдегида (VIII), полученное на стадии 3, циклизуют обычным способом с получением енаминового производного (IX). Енаминовое производное (IX) может быть получено, например, обработкой альдегидного производного (VIII) трифторуксусной кислотой в подходящем апротонном растворителе, таком как дихлорметан и хлороформ.
(Стадия 5)
Эта стадия является стадией, при которой енаминовое соединение (IX), полученное на стадии 4, подвергают реакции Фриделя-Крафтца с получением трициклического производного (X). Эту реакцию можно осуществить в соответствии с обычным способом, применяемым в этой области. Например, трициклическое производное (X) может быть получено обработкой соединения (IX) смесь трифторметансульфоновой кислоты и трифторуксусного ангидрида или одной трифторметансульфоновой кислотой в подходящем апротонном растворителе, таком как дихлорметан и хлороформ.
(Стадия 6)
Эта стадия является стадией, при которой функциональную карбоксильную группу производного карбоновой кислоты (X), полученного на стадии 5, защищают этерификацией с получением эфирного производного (XI). В качестве эфирной группы вводятся обычная алкильная группа, разветвленная алкильная группа или группа, которая может быть селективно снята в таких реакционных условиях, при которых ацилтиогруппа соединения (XIV), синтезируемого на стадии 8, не гидролизуется. Этерификацию осуществляют способом, обычно применяемым в данной области. Например, соединение (X) обрабатывают спиртом в присутствии минеральной кислоты, такой как соляная кислота и серная кислота. Альтернативно, соединения (X) взаимодействует с, например, дифенилбромметаном, трифенилбромметаном или триметилсилилэтанолом в инертном растворителе, таком как диметилформамид и тетрагидрофуран, в присутствии основания, такого как карбонат цезия и карбонат калия. Таким образом может быть получено эфирное производное (XI).
(Стадия 7)
Эта стадия является стадией, при которой фталимидную группу эфирного производного (XI), полученного на стадии 6, подвергают снятию защитной группы с получением соединения амина (XII). Эту реакцию можно осуществить обычным способом. Например, соединения (XI) обрабатывают гидразином в растворителе, таком как вода, спирт и тетрагидрофуран, и посредством этого происходит снятие защитной группы у фталимида. Таким образом может быть получен амин (XII).
(Стадия 8)
Эта стадия является стадией, при которой производное карбоновой кислоты, представленное общей формулой (XIII), или его активное производное, такое как галогенангидрид, конденсируют с амином (XII), полученным на стадии 7, с получением производного амида (XIV). Эту реакцию осуществляют обычно применяемым методом в этой области. Например, производное карбоновой кислоты (XIII) взаимодействует с производным амина (XII) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии обычно применяемого конденсирующего реагента, такого как ЭЭДХ, ДЦК, ДЭК или диэтилцианофосфат. Таким образом может быть получено соединение (XIV). Когда реакцию проводят, например, с участием производного хлорангидрида карбоновой кислоты (XIII), производное карбоновой кислоты (XIII) преобразуют в его хлорангидрид в подходящем инертном растворителе с помощью галогенирующего агента, обычно применяемого в этой области, таким как тионилхлорид, и оксалилхлорид, и затем полученный галогенангидрид подвергают взаимодействию с аминопроизводным (XII) с получением посредством этого соединения (XIV).
Способ получения E-2
Когда R3 в вышеуказанной общей формуле (E) является атомом водорода, соединение может быть получено следующим способом:
В ряде формул, описанных выше, R1, R2, R3a, R19, p, n и m каждый имеют те же значения, что указаны выше.
Конкретно он является способом, который включает снятие защитной группы у амидного производного, представленного общей формулой (XIV'), обычным способом с получением производного карбоновой кислоты, представленного общей формулой (XV).
Снятие защитной группы осуществляют методом, обычно применяемым в этой области. Например, когда R1 в целом производном карбоновой кислоты (XV) является ацильной группой, то в качестве исходного соединения выбирают производное кислоты, где R2a является, например, трет-бутильной группой или арилалкильной группой, и затем подвергают исходное соединение снятию защитной группы в таких условиях, при которых ацилтиогруппа остается стабильной, например, каталитическое гидрирование или обработка трифторуксусной кислотой. Таким образом может быть получено целевое соединение (XV).
Когда R1 в производном карбоновой кислоты (XV), то есть, в целевом соединении, является атомом водорода, то в качестве исходного соединения выбирают амидное производное, где R2a является низшим алкилом, и гидролизуют в разбавленном водном растворе щелочи, такой как гидроксид натрия и гидроксид лития, или в разбавленном водном растворе минеральной кислоты с получением посредством этого целевого соединения (XV).
Способ получения E-3
Когда R1 и R2 в вышеуказанной общей формуле (E) являются каждый атомом водорода, соединение (XV') может быть получено следующим способом:
В ряде формул, описанных выше, R1a, R2, R3a, R19, p, n и m каждый имеют те же значения, что указаны выше.
Конкретно это реакция, которая включает гидролиз производного карбоновой кислоты, представленного общей формулой (XIV), обычным способом с получением меркапто производного карбоновой кислотой (XVI).
Гидролиз можно осуществлять способом, обычно применяемым в данной области. Например, исходное соединение можно гидролизовать в разбавленном водном растворе щелочи, такой как гидроксид натрия и гидроксид лития, или в разбавленном водном растворе минеральной кислоты.
Способ получения E-4
Когда m в вышеуказанной общей формуле (E) равно 0, соединение (XVI') может быть также синтезировано следующим способом:
В ряде формул, описанных выше, R1a, R2, R3a, R19, p, n и m каждый имеют те же значения, что указаны выше.
(Стадия I)
Эта стадия является стадией, при которой производное α- гидрокси карбоновой кислоты (XVII) конденсируют с аминосоединением (XII), полученным по вышеупомянутому способу получения E-1, стадия 7, с получением посредством этого амидного производного α- гидрокси карбоновой кислоты (XVIII). Подобно способу получения E-1, стадия 8, соединения (XII) и (XVII) взаимодействуют в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии конденсирующего агента, обычно применяемого в этой области, такого как, ЭЭДХ, ДДК, ДЭК или диэтилцианофосфат. Таким образом можно получить амидное производное (XVIII).
(Стадия 2)
Эта стадия является стадией, при которой гидроксильная группа производного амида (XVIII), полученного на стадии 1, преобразуется в ацилтиогруппу с получением ацилтиопроизводного (XIV'). Соединение (XIV') может быть синтезировано согласно способу, обычно используемому для преобразования гидроксильной группы в ацилтиогруппу. Например, соединение (XVIII) обрабатывают реакцией типа Митсунобу (Mitsunobu) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, с использованием трифенилфосфита и эфира азодикарбоновой кислоты, такого как DIAD (диизопропилазодикарбоксилат). Таким образом может быть получено ацилтиопроизводное (XIV').
Способ получения F-1
Те из соединений, представленных следующей общей формулой (F), которые являются иными, чем когда R1 и R2, каждый, представляют атом водорода, могут быть получены следующим способом:
где R1 представляет атом водорода или ацильную группу;
R2 представляет атом водорода, низшую алкильную группу, арильную группу, которая может иметь заместитель, гетероарильную группу, которая может иметь заместитель, арилалкильную группу, которая может иметь заместитель, гетероарилалкильную группу, которая может иметь заместитель, или низшую алкоксильную группу;
R3 представляет атом водорода или карбокси-защитную группу;
m и n представляют каждый независимо целое число 0, 1 или 2;
В ряде формул, описанных выше, R2, m и n каждый имеют те же значения, что указаны выше; R1a представляет группу, выбранную из групп, данных в определении R1, за исключением атома водорода; и R3a представляет группу, выбранную из групп, данных в определении R3, за исключением атома водорода.
А именно, это способ, который включает конденсацию производного карбоновой кислоты, представленного общей формулой I, или его активного производного, такое как галогенангидрид кислоты, с аминопроизводным, представленным общей формулой II с получением посредством этого амидного производного (III).
Конденсацию можно осуществлять обычным способом. Например, можно осуществлять конденсацию в присутствии обычно применяемого конденсирующего реагента, такого как 1-этоксикарбонил-2-этокси-1,2- дигидрохинолин (обозначаемый здесь как ЭЭДХ, гидрохлорид 1,3-дициклогексилкарбодиимида (обозначаемый здесь как ДЭК) или диэтилцианофосфонат.
В качестве органического растворителя может быть использован любой органический растворитель, инертный в процессе реакции. Примеры включают метиленхлорид, тетрагидрофуран и т.д.
Когда конденсацию проводят с участием производного хлорангидрида производного карбоновой кислоты (I), производное карбоновой кислоты (I) преобразуют в хлорангидрид кислоты в подходящем инертном растворителе с использованием обычно применяемого хлорирующего агента, такого как тионилхлорид и оксалилхлорид, и затем полученный таким образом хлорангидрид кислоты подвергают взаимодействию с аминопроизводным II с получением соединения III.
Способ получения F-2
Те из соединений, представленных следующей общей формулой (F), где R1 и R3 каждый представляют атом водорода, могут быть также получены следующим способом:
В ряде формул, описанных выше, R2, m, n, R1a и R3a каждый имеют те же значения, что указаны выше.
Конкретно это реакция, которая включает гидролиз амидного соединения, общей формулы (III) обычным способом с получением меркапто производного карбоновой кислоты (IV). Для осуществления гидролиза можно использовать способ, обычно применяемый в данной области. Например, можно указать способ, включающий реакцию амидного соединения III в разбавленном водном растворе щелочи, такой, как гидроксид натрия и гидроксид лития, или в разбавленном водном растворе минеральной кислоты, и другие.
Способ по лучения F-3
Те из соединений, представленных общей формулой (F), где n равно 0, могут быть также получены следующим способом:
В ряде формул, описанных выше, R2, m и, R1a и R3a каждый имеют те же значения, что указаны выше.
(Стадия I)
Конкретно эта стадия включает конденсацию, производного молочной кислоты, представленного общего формулой (V), или его активного производного, такое как галогенгидрид кислоты, с аминопроизводным (II) с получением амидного производного (VI). Подобно вышеописанному способу получения F-I, соединения (V) и (II) взаимодействуют в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии конденсирующего агента, такого как EEDQ или диэтилцианофосфат. Таким образом можно получить амидное производное (VI).
(Стадия 2)
А именно, это стадия, где гидроксильная группа производного амида (VI), полученного на стадии I, тиоэтерифицируется обычным способом с получением ацетилтиопроизводного (VII).
Примеры способа тиоэтерификации гидроксильной группы включают способ, заключающийся в обработке соединения (VI) реакцией типа Митсунобу (Mitsunobu ) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, с использованием трифенилфосфата и эфира азодикарбоновой кислоты, такого как диизопропилазодикарбоксилат (называемый здесь DIAD) с получением целевого соединения (VII).
Далее, те из соединений, представленных общей формулой (F), где R2 и R3 каждый является водородом, могут быть получены осуществлением гидролиза тем же способом, что описан для способа получения А-2.
Далее будут описаны основные способы синтеза исходных соединений, используемых в способах получения F-1 и F-2.
Способ получения F-4
Из соединений, представленных вышеуказанных общей формулой (V), используемых в способе получения F-3, и соединений, представленных вышеуказанной общей формулой (I), используемых в способе получения F-1, те, в которых n равно 0, могут быть синтезированы следующим способом: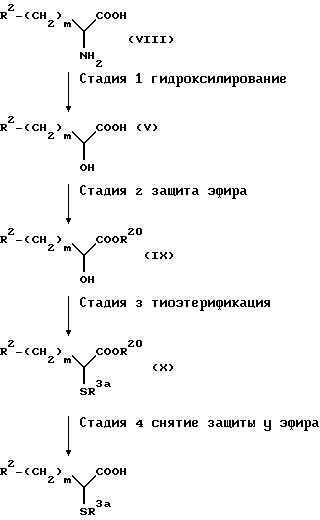
В ряде формул R2, m и R3a каждый имеют те же значения, что указаны выше; и R20 представляет группу формулы: -CHPh2 (где Ph представляет фенильную группу), группу, представленную формулой -CPh3, или группу, представленную формулой -(CH2)2-Si(CH3)3.
(Стадия 1)
Конкретно это стадия, которая включает гидроксилирование производного аминокислоты, представленное общей формулой (VIII), с получением производного молочной кислоты (V), которое является исходным продуктом способа получения F-3.
Вышеуказанное производное молочной кислоты (V) может быть синтезировано путем гидроксилирования обычной аминокислоты. Производное молочной кислоты (V) может быть синтезировано, например, обработкой производного аминокислоты (VIII) и азидирующего агента, такого как нитрит натрия и нитрит серебра, в водном кислом растворе, таком как разбавленная соляная кислота и разбавленная серная кислота.
(Стадия 2)
Конкретно это стадия, которая включает защиту функциональной карбоциклической группы производного молочной кислоты (V), полученного на стадии 1, путем этерификации производного (IX).
В качестве подходящей защитной группы вводится такая, которая может быть селективно удалена в таких реакционных условиях, при которых ацилтиогруппа соединения (X), синтезируемого на стадии 3, не подвергается гидролизу. Например, производное молочной кислоты (V) взаимодействует с дифенилбромметаном, трифенилбромметаном или триметилсилилэтилбромидом в инертном растворителе, обычно используемом в этой области, таком как диметилформамид и тетрагидрофуран, в присутствии основания, такого как карбонат цезия и карбонат калия. Таким образом может быть получено лактатное производное (IX).
(Стадия 3)
Конкретно это стадия, которая включает тиоэтетрификацию гидроксильной группы лактатного производного (IX), полученного на стадии 2.
Эту стадию можно осуществлять тем же способом, что описано для способа получения F-3, стадия 2.
(Стадия 4)
Конкретно это стадия, которая включает снятие защитной группы эфирной группы ацилтиопроизводного (X), полученного на стадии 3, с получением производного карбоновой кислоты (XI). Когда защитной эфирной группой R4 является арилалкильная группа, такая как дифенилметил и трифенилметил, ацилтиопроизводное (X) обрабатывают трифторуксусной кислотой и анизолом с получением производного карбоновой кислоты (XI). Когда защитной эфирной группой R4 является силилалкильная группа, такая как триметилсилилэтил, ацилтиопроизводное (X) обрабатывают соединением фтора, таким как фторид калия и тетрабутиламмонийфторид, с получением производного карбоновой кислоты (XI).
Соединения по настоящему изобретению могут быть получены способами, обычно применяемыми в этой области, или сочетанием этих способов. Далее будут описаны основные способы получения.
Способ получения 1
Из соединений, представленных общей формулой (1), соединения (X), где R1 является группой иной, чем атом водорода, может быть получено следующим способом: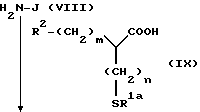
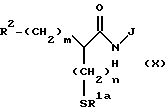
В ряде формул R1a представляет ацильную группу; R2 представляет атом водорода, низшую алкильную группу, циклоалкильную группу, арильную группу, которая может иметь заместитель, гетероарильную группу, которая может иметь заместитель, арилалкильную группу, которая может иметь заместитель или гетероарилалкильную группу, которая может иметь заместитель; m и n представляет каждый независимо целое число 0, 1 или 2; и J представляет циклическую группу, обладающую ингибирующей активностью в отношении ангиотензин 1-конвертирующего фермента.
Конкретно эта стадия является стадией, включающей конденсацию производного аминокислоты общей формулы (VIII) с производным карбоновой кислоты, представленным общей формулой (XI), или его активным производным, таким как галогенангидрид кислоты, обычным способом м получением амидного производного, представленного общей формулой (X).
Конденсацию можно осуществлять способом, обычно применяемым в этой области. Например, производное аминокислоты (VIII) взаимодействует с производным карбоновой кислоты (IX) в инертном растворителе, представленном метиленхлоридом и тетрагидрофураном, в присутствии обычно используемого конденсирующего реагента, такого как ЭЭДХ (1-этоксикарбонил-2-этокси-1,2-дигидрохинолин), ДЦК (1,3-дициклогексилкарбодиимид), ДЭК (гидрохлорид 1-(3-дтметламинопропил)-3-этилкарбодиимида) или диэтилцианофосфонат. Таким образом может быть получено амидное производное (X).
Когда конденсацию проводят с участием хлорангидрида производного карбоновой кислоты (IX), производное карбоновой кислоты (IX) преобразуют в хлорангидрид кислоты в подходящем инертном растворителе с хлорирующим агентом, таким как тионилхлорид и оксалилхлорид, и затем полученный таким образом хлорангидрид подвергают взаимодействию с производным аминокислоты (VIII) с получением амидного производного кислоты (X) в виде целевого соединения.
Способ получения 2
Когда R1 является атомом водорода, соединение (XI) может быть также получено следующим способом: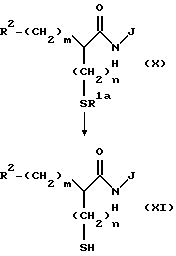
В ряде формул R1a, R2, J, n и m каждый имеют те же значения, что указаны выше.
Конкретно это способ, который включает снятие защитной группы эфирной группы и ацилтиогруппы амидного производного (X), полученного по способу получения 1, обычным способом с получением аминокислотного производного, то есть целевого соединения (XI).
Снятие защитной группы можно осуществлять способом, обычно применяемым в этой области. А именно, его осуществляют гидролизом амидного производного (X) в разбавленном водном растворе щелочи, такой как гидроксид натрия и гидроксид лития, или в разбавленном водном растворе минеральной кислоты.
Способ получения 3
Из соединений, представленных общей формулой (I), соединения (XIV), где n равно 0, может быть также получено следующим способом:
В ряде формул R1a, R2, m и J каждый имеют те же значения, что указаны выше.
(Стадия 1)
Конкретно эта стадия включает конденсацию производного молочной кислоты, представленного общей формулой (XII), или его реакционно активного производного, такого как галогенангидрид кислоты, с аминопроизводным, представленным общей формулой (VII), с получением амидного производного (XIII). Подобно вышеописанному способу получения 1, соединения (XII) и (VIII) взаимодействуют в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии конденсирующего агента, такого как ЭЭДХ или диэтилцианофосфат. Таким образом можно получить амидное производное (XII).
(Стадия 2)
А именно, это стадия, которая заключается в тиоэтерификации гидроксильной группы амидного производного (XIII), полученного на стадии 1, обычным способом с получением целевого соединения, представленного общей формулой (XIV).
Примеры способа тиоэтерификации гидроксильной группы включают способ, заключающийся в обработке амидного производного (XIII) реакцией типа Митсунобу (Mitsunobu) в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, с использованием трифенилфосфита и эфира азодикарбоновой кислоты, такого как диизопропилазодикарбоксилат (называемый здесь DIAD) с получением целевого соединения (XIV).
Способ получения 4
Соединение, представленное общей формулой (VII), может быть также получено следующим способом:
В ряде формул R1 и J каждый имеют те же значения, что указаны выше.
(Стадия 1)
А именно, эта стадия является стадией, которая заключается в бромировании аминогруппы D-алло-изолейцина (XV) с получением бромида (XVI). Соединение (XVI) может быть получено согласно способу, обычно используемому в этой области для стереоселективного бромирования. Например, соединение (XVI) обрабатывают нитритом, таким как нитрит натрия и нитрит серебра в водном растворе бромистого водорода. Таким образом может быть получен бромид (XVI).
(Стадия 2)
Конкретно эта стадия является стадией, которая заключается в замещении брома в бромиде (XVI), полученного на стадии 1, ацилтиогруппой с получением производного ацилтиопентановой кислоты (XVII). Эту реакцию осуществляют в соответствии с обычным способом. Например, бромид (XVI) подвергают взаимодействию с тиокарбоксилатом, таким как тиоацетат калия и тиоацетат натрия в полярном органическом растворителе, таком как ацетонитрил и ацетол. Альтернативно, бромид (XVI) взаимодействует с тиокарбоновой кислотой, такой как тиоуксусная кислота и тиобензойная кислота, в присутствии основания, такого как карбонат калия и карбонат цезия. Таким образом может быть получено производное ацилтиопентановой кислоты (XVII).
(Стадия 3)
Эта стадия является стадией, которая заключается в конденсации производного ацилтиопентановой кислоты (XVII), полученного на стадии 2, или его активного производного, такого как галогенангидрид кислоты, с эфирным производным аминокислоты, которое является широко известным соединением или полученным широко известным способом, с получением целевого соединения (VII). Например, ацилтио производное (XVII) взаимодействует с эфирным производным аминокислоты в инертном растворителе, таком как метиленхлорид и тетрагидрофуран, в присутствии обычно применяемого конденсирующего реагента, такого как ЭЭДХ (1-этоксикарбонил-2-этокси-1,2-дигидрохинолин), ДЦК (1,3-дициклогексилкарбодиимид), ДЭК [гидрохлорид 1-(3-диметиламинопропил)-3-этилакарбодиимида] или диэтилцианофосфоната. Таким образом может быть получено соединение (VII). Когда конденсацию проводят с участием хлорангидрида ацилтиопроизводного (XVII), ацилтиопроизводное (XVII) преобразуют в хлорангидрид кислоты в подходящем инертном растворителе с использованием хлорирующего агента, такого как тионилхлорид и оксалилхлорид, и затем полученный таким образом хлорангидрид кислоты подвергают взаимодействию с эфирным производным аминокислоты с получением посредством этого целевого соединения (VII).
Способ получения 5
Соединение, представленное общей формулой (VII), может быть также получено следующим способом: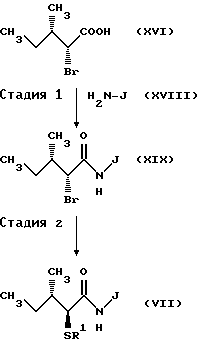
В ряде формул R1 представляет атом водорода или ацильную группу; и J имеет те же значения, что указаны выше.
(Стадия 1)
А именно, эта стадия является стадией, которая заключается в конденсации бром производного карбоновой кислоты (XVI), полученного по способу получения 4, стадия 1, или его активного производного, такого как галогенангидрид кислоты, с эфирным производным аминокислоты (XVIII) обычным способом, с получением амидного производного (XIX). Амидное производное (XIX) может быть получено такой же обработкой, как описано в способе получения 4, стадия 3.
(Стадия 2)
Конкретно эта стадия является стадией, которая заключается в преобразовании бром группы в амидном производном (XIX), полученном на стадии 1, в ацилтиогруппу с получением целевого соединения. Целевое соединение (VII) может быть получено такой же обработкой, как описано в способе получения, стадия 2.
Для иллюстрации активности соединений по настоящему изобретению далее даны примеры фармакологических исследований.
Пример фармакологических исследований A-1
Определение ингибирующей NEP активности препарата с использованием коры почки крыс
1. Методика исследования
Активность NEP определяют с использованием мембранной фракции, полученной из коркового вещества почки крыс в соответствии со способом Booth и Kenny (a Rapid Method for the Perification of Microvilli from Rabbit kidney, Andrew J. Booth and A. John Kenny, Biochem J., 1974, 142, 575 - 581).
Активность NEP определяют следующим способом в соответствии с методом Orlowsky и Wilk Perification and Specificity of Membrane. - Bound Metalloendopeptidase from Bovine Pituitaries, Marian Orlowsky and Shrwin Wilk, Biochemistry, 1981, 20, 4942 - 4950).
В качестве субстрата используют бензоил-глицид-аргинил-аргинил-2-нафтиламид (бензоил-Gly-Arg-Arg-2-нафтиламид (Nova Biochem. , Switzeland). В присутствии препарата фермента NEP и избытка лейцин аминопептидазы (Sigma Chemical Co. , U.S.A.), высвобождаемый нафтиламин подвергают цветному проявлению с помощью first garnet (первого граната-) (Sigma Chemical Co., U.S. A.) с последующим измерением абсорбции при длине волны 540 нм.
Что касается ингибирующей NEP активности, ингибитор добавляют к исследуемой системе, описанной выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ, строят кривую ингибирования и затем концентрацию, при которой достигается 50%-ное ингибирование, принимают за IC50.
2. Результаты исследования
Таблица A-1 показывает результаты вышеприведенного исследования вместе с результатами следующего примера фармакологических исследований A-2.
Пример фармакологических исследований A-2
Определение ингибирующей ACE активности препарата с использованием легкого крысы.
1. Способ исследования
Активность ACE определяют с использованием мембранной фракции, полученной из крысиного легкого в соответствии со способом Wu-Wong et al., (Characterization of Endthelin Enzyme in Rat Zung., Junshyum R. Wu-Wong, Jerald, P. Budzik, Edward M. Dewin and Ferry J. Opgenorth, Biochem. Biophys. Res. Commun., 1990, 171, 1291 - 1296).
Активность ACE определяют, используя модификацию (изменен боратный буфер, pH 8,3) метода Cushman и Cheung (Spectrophotometric Assay and Properties of Angiotensiu - Converting Euzyme of Rabbit Zung, Cushmen, D.W. and Cheung H.S., 1971, 20, 1637 - 1648).
В присутствии ACE гиппурат, высвобождаемый из гиппурил-гистидил-лейцина (Гиппурил-His-Leu (Peptide Institute Inc., Japan)) экстрагируют этилацетатом и затем адсорбцию измеряют при длине волны 228 нм.
Что касается ингибирующей ACE активности, ингибитор добавляют к исследуемой системе, описанной выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ, строят кривую ингибирования и затем концентрацию, при которой достигается 50%-ное ингибирование, принимают за IC50.
2. Результаты исследования
Таблица A-1 показывает результаты исследования, проведенного в соответствии с вышеописанным способом исследования,
Пример фармакологических исследований B-1
Определение ингибирующей NEP активности препарата с использованием коры почки крыс
1. Методика исследования
Активность NEP определяют с использованием мембранной фракции, полученной из коркового вещества почки крыс в соответствии со способом Booth и Kenny (a Rapid Method for the Perification of Microvilli from Rabbit Kidney., Andrew J. Booth and A. John Kenny, Biochem J., 1974, 142, 575 - 581).
Активность NEP определяют следующим способом в соответствии с методом Orlowsky и Wilk (Purification and Specificity of Membrane. - Bound Metalloendopepdidase from Bovine Pituitaries, Marian Orlowsky and Shrwin Wilk, Biochemistry, 1981, 20, 4942 - 4950).
В качестве субстрата используют бензоил-глицил-аргинин-аргинин-2-нафтиламид (бензоил-Gly-Arg-Arg-2-нафтиламид (Nova Biochem., Switzerland)). В присутствии препарата фермента NEP и избытка лейцин аминопептидазы (Sigma Chemical Co. , U.S.A.) высвобождаемый нафтиламин подвергают цветному проявлению с помощью first garnet (первого граната - пер.) (Sigma Chemical Co., U.S.A.) с последующим измерением абсорбции при длине волны 540 нм.
Что касается ингибирующей NEP активности, ингибитор добавляют к исследуемой системе, описанной выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ, строят кривую ингибирования и затем концентрацию, при которой достигается 50%-ное ингибирование, принимают за IC50.
2. Результаты исследования
Таблица B-1, которая будет описана далее, показывает результаты вышеприведенного исследования вместе с результатами следующего примера фармакологических исследований B-2.
Пример фармакологических исследований B-2
Определение ингибирующей ACE активности препарата с использованием крысиного легкого
1. Методика исследования
Активность ACE определяют с использованием мембранной фракции, полученной из крысиного легкого в соответствии со способом Wu-Wong et al., (Charactenjafeiu of Eudtheliu Euzime in Rat Zung., Junshyum R. Wu-Wong. Gerald, P. Budgik, Edward M. Devine and Terry J. Opgenorth, Biochem., Biophys. Res. Commun., 1990, 171, 1291-1296).
Активность ACE определяют, используя модификацию (Изменен боратный буфер, pH 8,3) метода Cushman и Cheung (Spectrophotomeric Assay and Propeoties of Augiotensiu - Conoerbiug Enjyme of Rabbit Zunh., Cusbman, D.W. and Cheung H.S., 1971, 20, 1637 - 1648).
В присутствии ACE гиппурат, высвобождаемый из гиппурилгистидил-лейцина (Гиппурил-His-Leu (Peptide Institute Iuc., Japan)) экстрагируют этилацетатом и затем адсорбцию измеряют при длине волны 228 нм.
Что касается ингибирующей ACE активности, ингибитор добавляют к исследуемой системе, описанной выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ, строят кривую ингибирования и затем концентрацию, при которой достигается 50%-ное ингибирование, принимают за IC50.
2. Результаты исследования
Таблица B-1 показывает результаты исследования, проведенного в соответствии с вышеописанной методикой исследования.
Пример фармакологических исследований C-1
1. Методика исследования
Активность NEP определяют способом в соответствии с методом Orlawsky и Wilk (Punfication adn Specificity of Membrane-Bоund Metalloendopeptidase from Bovine Pituitaries Marian Orlowsky and Showin Wilk Biochemistoy 1981, 20, 4942-4950).
В качестве субстрата используют бензоил-глицин-аргинил-аргинил-2-нафтиламид (бензоил-Gly-Arg-Arg-2-нафтиламид (Nova Biochem, Switzerland)). В присутствии препарата фермента NEP и избытка лейцина аминопептидазы (Sigma Chemical Co. , U.S.A.), высвобождаемый нафтиламин подвергают цветному проявлению с помощью first garnet (первого гранат - пер.) (Sigma Chemical Co., U.S.A.) с последующим измерением абсорбции при длине волны 540 нм.
Что касается ингибирующей NEP активности, испытуемое соединение добавляют к исследуемой системе, описанной выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ, строят кривую ингибирования и затем концентрацию, при которой достигается 50%-ное ингибирование, принимают за IC50. В качестве соединения сравнения используют [4S -[4α,7α (R*), 12b β ]]-7-[(1-оксо-2-(S)-тио-3-фенилпропил)амино]-1,2,3,4,6,7,8,12b -октагидро-6-оксопиридо[2,1-a][2] бензазепин-4-карбоновую кислоту.
2. Результаты исследования
Таблица C-1, которая будет описана далее, показывает результаты вышеприведенного исследования вместе с результатами следующего примера фармакологических исследований C-2.
Пример фармакологических исследований C-2.
Определение ингибирующей ACE активности препарата с использованием крысиного легкого
1. Методика исследования
Активность ACE определяют с использованием мембранной фракции, полученной из крысиного легкого в соответствии со способом Wu-Wong et al., (Characterization of Eudthelin Euzyme in Rat Zung, Junshyum R., Wu-Wong, Gerald, P. Budzik, Edward M. Deviue and Terry J. Opgenorth, Biochem. Biophys. Res. Commin. 1990, 171, 1291-1296).
Активность ACE определяют, используя модификацию (изменен боратный буфер, pH 8,3) метод Cushman и Cheung (Spechrophobomebic Assay and Propeobies of Augioteusin - Couverting Euzyme of Rabbit Zung., Cushman, D.W. and Cheung H.S., 1971, 20, 1937-1648).
В присутствии ACE гиппурат, высвобождаемый из гиппурил-гистидил-лейцина (Гиппурил-His-Leu (Peptide Justitute Juc., Japan)) экстрагируют этилацетатом и затем адсорбцию измеряют при длине волны 228 нм.
Что касается ингибирующей ACE активности, испытуемое соединение добавляют к исследуемой системе, описанной выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ, строят кривую ингибирования и затем концентрацию, при которой достигается 50%-ное ингибирование, принимают за 1050. В качестве соединения сравнения используют [4S -[4α,7α(R*), 12b β ]]-7-[(1-оксо-2-(S)тио-3-фенилпропил)амино]-1,2,3,4,6,7,8,12, b -октагидро-6-оксопиридо[2,1-a][2]бензазепин-4-карбоновую кислоту.
2. Результаты исследования
Таблица C-1 показывает результаты исследования, проведенного в соответствии с вышеописанным способом исследования.
Пример фармакологических исследований C-3
Понижающее давление действие на крыс 2К, 1C-Goldblatt с повышенным давлением
1. Методика исследования
Серебряную скобу с размером в 0,25 мм ширины устанавливают в левой почечной артерии каждого самца крыс Sprague Dawley (возраст от 6 до 7 недель) и через три недели отбирают крыс, у которых систолическое давление крови показано как 180 мм рт.ст. или выше. От одной до нескольких капель 1 н водного раствора гидроксида натрия прибавляют по каплям в очищенную воду и в ней растворяют или эмульгируют каждое испытуемое соединение для приготовления дозировки 5 мл/кг с последующим введением перорально. После выдерживания крыс в инкубаторе при 45oC в течение от 5 до 10 минут измеряют систолическое давление крови непрямым способом плетизмографии хвостовой артерии. В качестве соединения сравнения используют [4S -[4α,7α(R*),12bβ]] 7-[(1-оксо-2-(S)-тио-3-фенилпропил)амино]-1,2,3,4,6,7,8,12b -октагидро-6-оксопиридо[2,1-a][2] бензазепин-4-карбоновую кислоту.
2. Результаты исследования
Таблица C-2 показывает результате исследования, проведенного в соответствии с вышеописанным способом.
Как описано выше, понижающее давление действие соединений по настоящему изобретению превосходит в три или более раз действие соединения сравнения.
Пример фармакологических исследований C-4
Диуретическое действие на AND-обработанных крыс с самопроизвольным повышенным давлением
1. Методика исследования
50 нг/кг/мин крысиного атриального натрийуретического пептида (r-ANP) вводят внутривенно самцам крыс с самопроизвольным повышенным давлением (возраста от 14 до 16 недель). Когда динамика кроветворения и уровень r-ANP в крови становятся стабильными спустя 1 час исследуют диуретическое действие каждого испытуемого соединения. Диуретическое действие определяют внутривенным введением испытуемого соединения и измерением повышения (% степени изменения) собранный мочи в течение 20 мин. В качестве соединения сравнения используют [4S -[4α,7α(R*),12bβ]]- 7-[(1-оксо-2-(S)-тио-3-фенилпропил)амино] -1,2,3,4,5,6,7,8,12b-октагидро-6 -оксопиридо[2,1-a] [2] бензазепин-4-карбоновую кислоту.
2. Результаты исследования
Таблица C-3 показывает результаты, полученные в вышеописанном исследовании.
Как показывают эти результаты, активность испытуемых соединений по диуретическому действию превосходит в три раза активность соединения сравнения.
Пример фармакологических исследования C-5
Понижающее давление действие на крыс с самопроизвольно повышенным давлением (SHR)
Крысы SHR возраста от 15 до 20 недель были анестезированы внутрибрюшинным введением тиопентала натрия (50 мг/кг). Глубокую анестезию поддерживали, необязательно осуществляя дополнительное анестезирование (5 мг/кг, внутривенно). В левую общую сонную артерию и в вену соответственно вводят катетеры для измерения давления крови и для введения препарата. Сердечную скорость рассчитывают с использованием давления крови в качестве основания для расчета.
Когда давление крови стабилизируется после завершения операции, внутривенно вводят соединение сравнения в дозах 0,1, 0,3 и 1,0 мг/кг и измеряют давление крови и сердечную скорость. После введения препарата измерения осуществляют через 10 минут в случае 0,1 и 0,3 мг/кг или через 30 минут в случае 1,0 мг/кг. Соединение по изобретению вводят внутривенно в дозах 0,03, 0,1 и 0,3 мг/кг и измерения осуществляют через 10 минут после введения в случае 0,03 и 0,1 мг/кг или через 30 минут после введения в случае 0,3 мг/кг.
Соединение сравнения показывает продолжительное понижение давления крови действие на от 3 до 4% при дозе 0,3 мг/кг или выше и на от 12 до 13% в случае 1,0 мг/кг, и давление крови не возвращается к первоначальному в течение 30 минут после введения. Сердечная скорость имеет тенденцию к постепенному снижению.
Соединение по настоящему изобретению показывает очевидное понижающее давление действие на около 8% при дозе в 0,03 мг/кг или выше, и затем показывает продолжительное понижение давления крови действие на от 13 до 15% при дозе 0,1 мг/кг и на 23% при дозе 0,3% мг/кг. Сердечная скорость не изменяется.
Таким образом, это свидетельствует о том, что соединение по настоящему изобретению приблизительно в 10 раз активнее соединения сравнения по понижающему давление действию на SHR.
Пример фармакологических исследования C-6
Понижающее давление действие при пероральном введении на SHR
Используя самцов крыс с самопроизвольной гипертонией (возраст от 16 до 17 недель), вводят перорально соединение примера C-8 и сравнительное соединение [S-(R*, R*] - 2,3,4,5-тетрагидро-3-[(2-меркапто-1-оксогексил)амино]-2-оксо-1H-бензазепин-1 -уксусную кислоту, имеющую следующую структурную формулу, каждое растворение в 0,5% метилцеллюлозе. Понижающее давление действие измеряют методом манжеты на хвосте и сравнивают данные, полученные до перорального введения и спустя 2, 4 и 8 часов. Понижающее давление действие, достигаемое 1,0 мг/кг соединения примера C-8, было сравнимо с действием, достигаемым 10 мг/кг вышеуказанного соединения сравнения. Соответственно соединение примера C-8 обладает активностью в около 10 раз более высокой, чем сравнительное соединение.
Сравнительное соединение
Пример фармакологических исследований D-1. Определение ингибирующей NEP и ACE активности
1. Методика исследования
Как источник фермента NEP мембранную фракцию, полученную из коркового вещества почки крыс, в соответствии со способом Boofh u Kenny (a Rapid Mefhod for the Purification of Microvelli from Rabbit Kidney, Andrew G. Boofh and A.John Kenny. Biochem. J., 1974, 142, 575-581). Активность NEP определяют следующим способом в соответствии с методом Orlowsley и Wilk (Purfication and Specificity pf membrane-Bound metalloendopeotidase from Bovine Pitutaries, Maran Orlowsky and Shruru Wilk, Biochenustry, 1981, 20, 4942-4950). Далее способ будет кратко описан.
В качестве субстрата используют бензоил-глицил-аргинин-аргинин-2-нафтиламид (бензоил-Gly-Arg-Arg-2-нафтиламид (Nova Biochem., Surtgeland)). В присутствии препарата фермента NEP и избытка лейцин аминопептидазы (Sigma Chemical Co. , U.S.A.), высвобождаемый нафтиламин подвергают цветному проявлению с помощью first garnet (первого граната) - (Sigma Chemical Co., U.S. A.) с последующим измерением абсорбции при длине волны 540 нм.
Как источник фермента ACE мембранную фракцию, полученную из крысиного легкого в соответствии со способом Wu-Wong et al., (Characterization of Endthelin Engyme in Rat Zung, Junshyum R. Wu-Wong Gerald, P. Budzik, Edward M. Devine and Terry J. Opgenorth, Biochem, Biophys. Res. Commun, 1990, 171, 1291-1296). Активность ACE определяют, используя модификацию (изменен боратный буфер, pH 8,3) метода Cushman и Cheung (Spectrophotometric Assay and Properties of Angioteusen - Canerting Enzyme of Rabbet Zung, Cushman D.W. and Cheung H.S., 1971, 20, 1637 - 1648).
В присутствии ACE гиппурат, высвобождаемый из гиппурилгистидил-лейцина (Гиппурил-His-Leu (Peptide Justitute Inc., Japan)) экстрагируют этилацетатом и затем адсорбцию измеряют при длине волны 228 нм.
Для определения ингибирующей NEP активности и ингибирующей ACE активности ингибитор добавляют к исследуемой системе активности обоих ферментов, описанный выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ и строят кривую ингибирования. Затем концентрацию, при которой достигается 50%-ное ингибирование, принимают на IC50.
2. Результаты исследований
Таблица D-1 показывает результаты исследования, проведенного в соответствии с вышеописанным способом исследования D-1.
Пример фармакологических исследований D-2
1. Методика исследования
Крысы с самопроизвольным повышенным давлением (SHR) возраста от 15 до 20 недель были анестезированы внутрибрюшинным введением тиопентала натрия (50 мг/кг). Глубокую анестезию поддерживают необязательно осуществляя дополнительное анестезирование (5 мг/кг, внутривенно). В левую общую сонную артерию и вену, соответственно, вводят катетеры для измерения давления крови и для введения препарата. Сердечную скорость рассчитывают с использованием давления крови в качестве основания для расчета.
Когда давление крови стабилизируется после завершения операции, внутривенно вводят соединение сравнения или соединение примера 9 в дозах 0,1, 0,3 и 1,0 мг/кг и измеряют изменение давления крови и сердечную скорость. После введения препарата измерения осуществляют через 10 минут в случае 0,1 и 0,3 мг/кг или через 30 минут в случае 1,0 мг/кг.
2. Результаты исследования
Соединение сравнения показывает продолжительное понижение давления крови действие на от 3 до 4% при дозе 0,3 мг/кг или выше и на от 12 до 13% в случае 1,0 мг/кг, и давление крови не возвращается к первоначальному в течение 30 минут после введения. В то время как сердечная скорость имеет тенденцию к постепенному снижению.
С другой стороны, соединение примера D-6 показывает очевидное понижающее давление действие (от 3 до 4%) при дозе в 0,1 мг/кг или выше, и показывает продолжительное понижение давления крови действия на от 10 до 13% при дозе 0,3 мг/кг и на 25% при дозе 1,0 мг/кг. В то время как сердечная скорость имеет тенденцию к постепенному снижению.
Соответственно на основании результатов примера исследования D-2, описанного выше, было подтверждено, что понижающее давление действия соединение по примеру D-6 на SHR было в около трех раз выше, чем соединения сравнения.
Пример фармакологических исследований E-1
(Определение ингибирующей NEP и ACE активности)
1. Методика исследования
Как источник фермента NEP мембранную фракцию, полученную из коркового вещества почки крыс в соответствии со способом Booth и Kenny (A Raped Method for the Purification of Microvilli from Rattit Kidney., Andrew G. Both and A. John Kenny, Biochem. J., 1974, 142, 575-581). Активность NEP определяют следующим способом в соответствии с методом Orlowsky и Wilk (Perification and Specifecity of Membrane - Bound Metalloendopeptidase from Bovine Pituifaries. , Marian Orlowsky and Shrwin Wilk, Biochemistry. 1981, 20, 4942-4950). Далее способ будет кратко описан.
В качестве субстрата используют бензоил-глицил-аргинил-аргинил-2-нафтиламид (бензоил-Gly-Arg-Arg-2-нафтиламид (Nova Biochem, Switzerlaud)). В присутствии препарата фермента NEP и избытка лейцин аминопептидазы (Sigma Chemical Co. , U.S.A.) высвобождаемый нафтиламин подвергают цветному проявлению с помощью first garnet (первого граната (Sigma Chemical Co., U.S.A.) с последующим измерением абсорбции при длине волны 540 нм.
Как источник фермента ACE мембранную фракцию, полученную из крысиного легкого в соответствии со способом Wu-Wong et al., (Churacterization of Endtelin Enzyme in Rat Lung., hunshyum R. Wu-Wong, Gerald, P. Budzik, Edward M. Devire and Terry I. Opgenorth, Biochem., Biophys. Res. Commun., 1990, 171, 1291-1296). Активность ACE определяют, используя модификацию (изменен боратный буфер, pH 8,3) метода Cushmann Cheung (Spectrophotometric Assay and Properties of Angiotensin - Converting Enzyme of Rabbit Lung., Cushman, D.W. and Cheung H.S., 1971, 20, 1637-1648). Далее способ будет кратко описан.
В присутствии ACE гиппурат, высвобождаемый из гиппурилгистидил-лейцина (Гиппурид-His-Leu (Pertide Institute Inc., Japan)), экстрагируют этилацетатом и затем адсорбцию измеряют при длине волны 228 нм.
Для определения ингибирующей NEP активности и ингибирующей ACE активности ингибитор добавляют к исследуемой системе активности обоих ферментов, описанной выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ, и строят кривую ингибирования. Затем концентрацию, при которой достигается 50%-ное ингибирование, принимают за IC50.
2. Результаты исследования
Следующая таблица E-1 показывает результаты исследования, проведенного в соответствии с вышеописанным способом исследования.
Пример фармакологических исследований E-2
Самцам крыс Вистар возраста от 11 до 13 недель внутривенно вводили (1 мг/кг) и перорально назначают (10 мг/кг, 30 мг/кг) соединения примера E-6 или сравнительного соединения E-1. Затем регистрировались изменения уровня каждого препарата в крови со временем с использованием жидкостной хроматографии. В случае соединения E-6 уровень препарата в крови определяют измерением УФ-абсорбции (257 нм). Тогда как в случае соединения сравнения E-1 уровень препарата в крови определяют способом флуоресцентной метки с использованием ABD-F. Биологическая доступность соединения примера E-6, рассчитанная из AUC перорального применения и AUC внутривенного введения, была соответственно 24,6% (30 мг/кг, р.о. (перорально)) и 18,8% (10 мг/кг, р.о.). С другой стороны, динамику in vivo соединения сравнения E-1 измеряют тем же способом. Определено, что биологическая доступность была 7,8% (30 мг/кг, р. о. ) и 4,3% (10 мг/кг, р.о.). Соответственно соединение примера E-6 превосходит по абсорбционной способности при пероральном применении соединения сравнения E-1.
Пример фармакологических исследований E-3. Исследование связывания рецепторов V1 и V2
Были использованы образцы мембран крысиного легкого (V1) и крысиной почки (V2). 100,000 единиц (3,69 нМ) [3H]-Arg-вазопрессина, 25 μг (1 мг протеин/мл) каждого образца мембраны и исследуемого препарата (от 10-7 до 10-5 М) ингибируют при общем объеме 250 μл исследуемого буфера (pH 7,4), содержащего 10 мМ NgCl2, 2 мМ EGTA и 20 мМ HEPES при 4oC в течение дня и ночи. Затем инкубат промывают 5 раз порциями по 5 мл буфера для отделения посредством этого образца мембран, связанного с вазопрессином с последующей фильтрацией с применением стеклянного фильтра (GF/F). Этот стеклянный фильтр сушат в течение около 3 часов, и смешивают с коктейлем для жидкостной сцинцилляции (10 мл, ACSII). После стояния в течение ночи определяют количества [3H]-Arg-вазопрессина, связанного с мембраной, с помощью счетчика жидкостной сцинцилляции, а степень ингибирования рассчитывают в соответствии со следующей формулой:
Степень ингибирования (%) = 100 - [(C1 - B1)/(C0 - B1)]•100
где
B1 - количество [3H]-Arg-вазопрессина, связанного с мембраной, в присутствии избытка вазопрессина (10 μ г)
C0 - количество [3H] -Arg-вазопрессина, связанного с мембраной, в отсутствии исследуемого препарата
и
C1 - количество [3H]-Arg-вазопрессина, связанного с мембраной, в присутствии как исследуемого препарата в известном количестве и [3H]-Arg-вазопрессина.
Определяют количество исследуемого препарата, дающего 50%-ую степень ингибирования, в соответствии с вышеприведенной формулой, и принимают за IC50.
IC50 соединения примера E-10, определенное вышеуказанным способом для рецептора (V1) вазопрессина, составляет 10 μМ или выше, в то время как для рецептора (V2) вазопрессина составляет 4,49 μМ.
Пример фармакологических исследований F1 (Определение ингибирующей NEP и ACE активности)
1. Методика исследования
Как источник фермента NEP мембранную фракцию, полученную из коркового вещества почки крыс в соответствии со способом Booch и Kenny (a Rapid Method for tfe Purification of Microvilli from Rabbit Kidney., Andrew J. Booth and A, John Kenny, Biochem. J., 1974, 142, 575-581). Активность NEP определяют следующим способом в соответствии с методом Orlowsky и Wilk (Purification and Specificity of Membrane - Bound Metalloendopeptidase from Bovine Pituitaries. , Marian Orlowsky and Shrwin Wilk, Biochemistry, 1981, 20, 4942-4950). Далее способ будет кратко описан.
В качестве субстрата используют бензоил-глицил-аргинил-аргинил-2-нафтиламид (бензоил-Gly-Arg-Arg-2-нафтиламид (Nova Biochem, Swifzerland)). В присутствии препарата фермента NEP и избытка лейцин аминопептидазы (Segma Chemical Co. , U.S.A.) высвобождаемый нафтиламин подвергают цветному проявлению с помощью first garnet (первого граната - пер.) (Sigma Chemical Co., U.S.A.) с последующим измерением абсорбции при длине волны 540 нм.
Как источник фермента ACE мембранную фракцию, полученную из крысиного легкого в соответствии со способом Wu-Wong et al., (Characterization of Endthelin Enzyme in Rat Zung., Junshyum R. Wu-Wong, Gerald, P. Budzik, Edward M. Devine and Terry J. Opgenorth, Biochem. Biophys. Res. Commun., 1990, 171, 1291-1296). Активность ACE определяют, используя модификацию (изменен боратный буфер, pH 8,3) метода Cushman и Cheund (Spectrophotometric Assay and Properties of Angioteusin - Converting Enzyme of Rabbit Zuug., Cushman. D.W. and Cheung, H.S., 1971, 20, 1637-1648). Далее способ будет кратко описан.
В присутствии ACE гиппурат, высвобождаемый из гиппурил-гистидил-лейцина (Гиппурил - His-Leu (Peptude Sustitufe Suc., Japan)) экстрагируют этилацетатом и затем адсорбцию измеряют при длине волны 228 нм.
Для определения ингибирующей NEP активности и ингибирующей ACE активности ингибитор добавляют к исследуемой системе активности обоих ферментов, описанной выше, таким образом, что дает конечную концентрацию при 1, 3, 10, 30, 100, 300 и 1000 нМ и строят кривую ингибирования. Затем концентрацию, при которой достигается 50%-ное ингибирование, принимают за IC50.
2. Результаты исследования
Таблица F1 показывает результаты исследования, проведенного в соответствии с вышеописанным способом исследования.
Пример фармакологических исследования F-2. Исследование связывания рецепторов V1 и V2
Были использованы образцы мембран крысиного легкого (V1) и крысиной почки (V2). 100,000 единиц (3,69 нМ) [3H]-Arg-вазопрессина, 25 μг (1 мг протеин/мл) каждого образца мембраны и исследуемого препарата (от 10-7 до 10-5М) ингибируют при общем объеме 250 μл исследуемого буфера (pH = 7,4), содержащего 10 мМ MgCl2, 2 мМ EGTA и 20 мМ HEPES при 4oC в течение дня и ночи. Затем инкубат промывают 5 раз порциями по 5 мл буфера для отделения посредством этого образца мембран, связанного с вазопрессином с последующей фильтрацией с применением стеклянного фильтра (GF/F). Этот стеклянный фильтр сушат в течение около 3 часов и смешивают с коктейлем для жидкостной сцинцилляции (10 мл, ACS II). После стояния в течение ночи, определяют количества [3H] -Arg-вазопрессина, связанного с мембранной, с помощью счетчика жидкостной сцинцилляции, а степень ингибирования рассчитывают в соответствии со следующей формулой:
Степень ингибирования (%)
= 100 - [(C1 - B1)/(C0 - B1)] • 100
где
B1 - количество [3H]-Arg-вазопрессина, связанного с мембраной, в присутствии избытка вазопрессина (10 μг)
C0 - количество [3H]-Arg - вазопрессина, связанного с мембраной, в отсутствии исследуемого препарата и
C1 - количество [3H]-Arg-вазопрессина, связанного с мембраной, в присутствии как исследуемого препарата в известном количестве и [3H]-Arg-вазопрессина.
Определяют количество исследуемого препарата, дающего 50%-ную степень ингибирования, в соответствии с вышеприведенной формулой, и принимают на IC50.
IC50 соединения примера 10, определенное вышеуказанным способом для рецептора (VI) вазопрессина, составляет 10 M или выше, в то время как для рецептора (V2) вазопрессина составляет 1,39 μM. .
Результаты экспериментов, описанных выше, ясно свидетельствуют о том, что соединение по изобретению обладают ингибирующим АСЕ действием, ингибирующим NEP действием или действием вазопрессинового антагониста. Следовательно, соединения настоящего изобретения подавляют образование АТ-П, который является фактором, способствующим сердечной недостаточности одновременно с увеличением действия ANP, который задействован в компенсационном механизме для симптомов сердечной недостаточности, и поэтому, как ожидается, обладает различным терапевтическим действием при сердечной недостаточности, например, уменьшает жидкие тела, облегчает предварительную нагрузку, облегчает последующую нагрузку или тому подобное. Кроме того, эти соединения полезны в качестве антигипертензивного диуретика. Далее, соединения по настоящему изобретению являются эффективными при таких заболеваниях, которые могут быть подвергнуты лечению и использованием ингибирующего NEP действия или ингибирующего ACE действия, в частности сердечно-сосудистых заболеваний, таких как острая или хроническая сердечная недостаточность, грудная жаба или повышенное давление, почечная слабость, отек, задержка солей, отек легкого, боль, при лечении психического состояния, такого как депрессия, ангины, предменструальный синдром, болезни Meniere, гиперальдостеронизма, гиперкальцинурии, глаукомы, астмы, гастроинтестинальных заболеваний, такого как понос, синдром кишечного раздражения и повышенная кислотность, индуцированная циклоспорином почечная слабость и тому подобное.
Кроме того, вышеописанные примеры фармакологических исследований ясно свидетельствуют, что соединения по настоящему изобретению являются сравнимыми или даже превосходят существующие и представленные ACE- и NEP-двойные ингибиторы по ингибирующему ACE и NEP действию и явно превосходят их по гипотензивному и диуретическому действию. В дополнение к вышеописанным примерам фармакологических исследований отдельно были проведены исследования, в которых использовали SHR для изучения гипотензивного действия при внутривенном введении. Показано, что при сравнении обычно применяемых известных двойных ингибиторов (1) [S-(R*, R*)- 2,3,4,5-тетрагидро-3-[(2-меркапто-I-оксогексил-3-фенилпропил)] -2-оксо- 1H-бензазепин-1-уксусной кислоты и (2) [S-(R*, R*)- 2,3,4,5-тетрагидро-3-[(2-меркапто-1-оксо-4-метилпентил)амино]-2-оксо- 1H-бензазепин-1-уксусной кислоты с соединением по настоящему изобретению, пример C-8 и пример C-10, ингибиторы (1) и (2) могут быть введены в дозе 1,0 мг/кг для понижения давления крови на 10%, тогда как при введении от 0,03 до 0,1 мг/кг соединения примера C-8 и введение от 0,1 до 0,3 мг/кг соединения примера C-10, в каждом случае достигается тот же эффект.
Было также выяснено, что соединения по настоящему изобретению обладают тем преимуществом, что они обладают превосходным действием при пероральном введении. Это свойство превосходного действия при пероральном введении является весьма предпочтительным с точки зрения того, что заболевания, при которых применяются соединения по настоящему изобретению, требуют обычно длительного введения.
Из настоящего изобретения также ясно, что из соединений по настоящему изобретению особенно превосходными при пероральном введении являются те, которые имеют радикал (2S, 3S)-3-метил-2-тиопентановой кислоты в боковой цепи.
Поскольку соединения изобретения имеют низкую токсичность и высокую безопасность, они являются веществами, каждое из которых имеет особенно предпочтительность при применении в качестве лекарства.
Кроме того, соединения по настоящему изобретению обладают также антагонистическим действием по отношению к рецепторам вазопрессина. Считается, что вазопрессин является одним из факторов, способствующих сердечной недостаточности, повышенному давлению или тому подобное. Полагают, что такое действие увеличивает эффективность, соединений по настоящему изобретению при вышеуказанных заболеваниях.
Когда соединение по настоящему изобретению используют в качестве профилактического или лечебного средства при лечении вышеуказанных заболеваний, оно может быть введено как перорально, так и парентерально. Его доза конкретно не ограничивается, но изменяется в зависимости от, например, состояния, возраста, пола и чувствительности к лекарствам пациента, пути введения, времени введения, периодичности введения, свойств лекарственных препаратов, вида лекарственного препарата и вида активного ингредиента. Обычно предпочтительны дозы от около 0,1 до 1000 мг/день для взрослого, от одноразового до приема за несколько раз.
Соединения по настоящему изобретению могут быть включены в состав лекарственного препарата обычным способом с использованием наполнителей для лекарственных препаратов, обычно применяемых в этой области.
А именно, при изготовлении твердого лекарственного препарата для перорального введения добавляют носители для основного агента, и если необходимо, связующие, разрыхляющие агенты, смазывающие агенты, окрашивающие агенты, ароматизирующие добавки, антиоксиданты и т.д., и затем смесь формуют обычным способом в таблетки, таблетки с покрытием, гранулы, порошки, капсулы и т.д.
В качестве вышеуказанных наполнителей используют, например, лактозу, кукурузный крахмал, сукрозу, глюкозу, сорбитол, кристаллическую целлюлозу и диоксид кремния.
В качестве связующих используют, например, поливиниловый спирт, поливиниловый эфир, этилцеллюлозу, метилцеллюлозу, гуммиарабик, трагакант, желатин, шеллак, гидроксипропил-целлюлозу, гидроксипропилметилцеллюлозу, цитрат кальция, декстрин, пектин и тому подобное. В качестве смазывающих агентов используют, например, стеарат магния, тальк, полиэтиленгликоль, силикагель, отвержденные растительные масла и тому подобное.
В качестве окрашивающих агентов могут быть использованы такие, которые одобрены для добавления к лекарствам. В качестве ароматизирующих добавок используют порошок какао, мяту, ароматические порошки, мятное масло, борнеол, порошок коры корицы и тому подобное. В качестве антиокислителей могут быть использованы такие, которые ободрены для добавления к лекарствам, такие как аскорбиновая кислота (витамин C) и α-токоферол (витамин E). Само собой разумеется, если необходимо, эти таблетки и гранулы могут быть подвергнуты нанесению подходящего покрытия, такого как сахарное покрытие, желатиновое покрытие и другие.
С другой стороны, при изготовлении инъекции добавляют при необходимости регуляторы pH, буферы, суспендирующие агенты, агенты, способствующие растворению, стабилизаторы, изотонические агенты, антиоксиданты, консерванты, и тому подобное. Таким образом могут быть получены препараты для внутривенных, подкожных и внутримышечных инъекций. Препарат для инъекции может быть также при необходимости изготовлен в виде высушенной вымораживанием формы.
Примеры у вышеуказанных суспендирующих агентов включают метилцеллюлозу, Полисорбат 80, гидроксиэтилцеллюлозу, гуммиарабик, порошок трагаканта, натрий карбоксиметилцеллюлозу, полиоксиэтиленсорбитан монолаурат и тому подобное.
Примеры агентов, способствующих растворению, включают полиоксиэтилен, отвержденное касторовое масло. Полисорбат 80, никотинамид, полиоксиэтилен сорбитан монолаурат, Marcogol, касторовое масло, этиловый эфир жирной кислоты и тому подобное.
В качестве стабилизаторов используют, например, сульфат натрия, метасульфит натрия, эфир и тому подобное. Примеры консерваторов включают метил пара-гидроксибензоат, этил парагидроксибензоат, сорбиновая кислота, фенол, крезол, хлоркрезол и тому подобное.
Примеры
Для лучшего понимания настоящего изобретения далее представлены примеры. Однако необходимо указать, что настоящее изобретение только ими не ограничивается.
Примеры синтеза исходных соединений также описаны далее, перед примерами.
Синтетический пример A-1
7-трифторметансульфонилокси-3,4-дигидро-1(2H)-нафталинон
В 100 мл раствора 9,94 г (61,29 ммоль) 7-гидрокси-3,4-дигидро-1(2H)-нафталинона и 24,8 мл (306 ммоль) пиридина в дихлорметане, перемешиваемого при 0oC, добавляют порциями, по каплям 11,86 мл трифторметансульфонового ангидрида, поддерживая температуру не выше 5oC. После перемешивания при той же температуре в течение 10 мин и затем при комнатной температуре в течение 30 мин, добавляют к реакционной смеси воду. Отделяют дихлорметановый слой, промывают 1н соляной кислотой, водой и насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния. После отгонки растворителя при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле. После ступенчатого элюирования гексаном: этилацетатом в отношении, изменяющемся от 10:1 (по объему) до 8:1 (по объему), получают 15,33 г указанного в заголовке соединения в виде бледно-желтого маслянистого продукта.
Выход 85%.
1H ЯМР (400 МГц, CDCl3) δ:
7,91 (1H, т, J = 1 Гц), 7,37 (1H, д, J = 1 Гц),
3,00 (2H, т, J = 6 Гц), 2,70 (1H, д, J = 6 Гц),
2,68 (1H, д, J = 6 Гц), 2,18 (2H, квинт, = 6 Гц).
Синтетический пример A-2
7-Фенил-3,4-дигидро-1(2H)-нафталинон
В перемешиваемую при комнатной температуре смесь, содержащую 15,31 г (52,06 ммоль) 7-трифторметансульфонилокси-3,4-дигидро-1(2H)-нафталинона, полученного в примере синтеза A-1, 12,7 г (104,12 ммоль) фенилборной кислоты, 10,8 г (78,09 ммоль) карбоната калия и 450 мл толуола, пробулькивают азот, в течение 30 минут. Затем туда добавляют 1,81 г (1,57 ммоль) тетракистрифенилфосфина палладия. Смесь медленно нагревают, продолжая пропускание азота, до температуры около 90oC. После перемешивания при этой температуре в течение 90 минут, реакционную смесь охлаждают и добавляют воду. Нерастворимые продукты собирают фильтрацией на целите и тщательно промывают этилацетатом. Органическую фазу собирают, промывают последовательно насыщенным водным раствором гидрокарбоната натрия, водой, 1 н соляной кислотой, водой и насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния. После отгонки растворителя при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле. После ступенчатого элюирования гексаном: этилацетатом в отношении, изменяющемся от 20:1 (по объему) до 12:1 (по объему), получают 9.53 г указанного в заголовке соединения в виде белых кристаллов. Выход 85%.
1Н ЯМР (400 МГц, CDCl3) δ:
8,28 (1H, д, J = 2 Гц), 7,72 (2H, дд, J = 8,2 Гц).
7,64 - 7,33 (8H, м), 3,01 (2H, т, J = 6 гц).
2,71 (1H, д, J = 6 Гц), 2,69 (1H, д, J = 6 Гц).
2,18 (2H, квинт. J = 6 Гц).
Синтетический пример A-3
8-Фенил-2,3,4,5-тетрагидро-1H-[1]-бензазепин-2-он
Смесь 9,19 г (41,34 ммоль) 7-фенил-3,4-дигидро-1-(2H)-нафталинона, полученного по синтетическому примеру A-2, со 150 г полифосфорной кислоты перемешивают при от 50 до 60oC и к ней добавляют порциями 2,96 г (45,47 ммоль) азида натрия в твердом виде. После перемешивания при этой температуре в течение дополнительных 90 минут реакционную смесь выливают в ледяную воду. Образовавшиеся при этом кристаллы собирают фильтрацией, промывают водой и н-гексаном и сушат в горячем воздухе при 70oC в течение ночи. Таким образом получают 9,3 г указанного в заголовке продукта. Выход 95%.
1Н ЯМР (400 МГц, ДМСО-d6) δ:
9,60 (1H, с), 7,58 (2H, д, J = 8 Гц), 7,44 (2H, т, J = 8 Гц),
7,35 - 7,29 (3H, м), 7,22 (1H, д, J = 2 Гц),
2,69 (2H, т, J = 7 Гц), 2,17 (2H, т, J = 7 Гц),
2,09 (2H, квинт, J = 7 Гц).
Синтетический пример A-4
3,3-Дихлор-8-фенил-2,3,4,5-тетрагидро-1H-[1]бензазепин-2-он
К смеси 8,94 г (37,67 ммоль) 8-фенил-2,3,4,5-тетрагидро-1H-[1]бензазепин-2-она, полученного по синтетическому примеру A-3, с 180 мл ксилола добавляют 23,53 г (113 ммоль) пентахлорида фосфора и смесь медленно нагревают. После перемешивания при около 90oC в течение 30 минут к реакционной смеси добавляют воду с последующей нейтрализацией водным раствором гидрокарбоната натрия. После экстракции дихлорметаном дихлорметановую фазу промывают насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния. После отгонки растворителя при пониженном давлении маслянистый остаток кристаллизуют путем добавления к нему этилацетата. Таким образом получают 2,60 г указанного в заголовке продукта. Маточный раствор подвергают колоночной хроматографии на силикагеле. После ступенчатого элюирования гексаном : этилацетатом в отношении, изменяющемся вплоть до 20:1 (по объему), получают еще 0,38 г указанного в заголовке соединения. Объединив оба вышеуказанных продукта, получают общий выход 2,98 г указанного в заголовке соединения.
Выход 26%.
1Н ЯМР (400 МГц, CDCl3) δ:
7,70 (1H, д, J = 2 Гц), 7,61 - 7,35 (3H, м),
7,21 (1H, д, J = 8 Гц), 3,09 - 3,01 (4H, м).
Синтетический пример A-5
3-Хлор-8-фенил-2,3,4,5-тетрагидро-1H-[1]бензазепин-2-он
Смесь, содержащую 2,88 г (9,4 ммоль) 3,3-дихлор-8-фенил-2,3,4,5-тетрагидро-1H-[1] бензазепин-2-она, полученного по синтетическому примеру A-4, 0,89 г (11,89 ммоль) ацетата натрия, 0,2 г 10%-ного палладия на угле и 40 мл уксусной кислоты каталитически гидрируют при комнатной температуре при 3 атм в течение 2 часов. После удаления фильтрацией нерастворимого продукта фильтрат концентрируют. Затем к остатку добавляют дихлорметан с последующей нейтрализацией водным раствором гидрокарбоната натрия. Дихлорметановую фазу отделяют, промывают насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния. После отгонки растворителя при пониженном давлении к остатку добавляют небольшое количество дихлорметана с последующим отделением кристаллов фильтрацией. Таким образом получают 0,53 г указанного в заголовке продукта. Маточный раствор подвергают колоночной хроматографии на силикагеле. После ступенчатого элюирования гексаном : этилацетатом в отношении, изменяющемся от 6:1 (по объему) до 3:1 (по объему), и затем дихлорметаном : метанолом в отношении 200:1 (по объему) получают еще 0,4 г указанного в заголовке соединения. Объединив оба вышеуказанных продуктов, получают всего 0,93 г указанного в заголовке соединения.
Выход 36%.
1Н ЯМР (400 МГц, CDCl3) δ:
7,55 - 7,21 (8H, м), 4,55 (1H, дд, J = 11 Гц),
3,09 - 2,61 (4H, м).
Синтетический пример A-6
3-Азидо-8-фенил-2,3,4,5-тетрагидро-1H-[1]бензазепин-2-он
Смесь, содержащую 0,93 г (3,42 ммоль) 3-хлор-8-фенил- 2,3,4,5-тетрагидро-1H-[1] бензазепин-2-она, полученного по синтетическому примеру A-5, 0,27 г (4,18 ммоль) азида натрия и 15 мл диметилсульфоксида перемешивают при 80oC в течение 3 часов. После дополнительного добавления 0,05 г азида натрия и перемешивают в течение 30 минут реакционную смесь выливают в ледяную воду. Кристаллы собирают фильтрацией и сушат при пониженном давлении с получением 0,77 г указанного в заголовке соединения. Выход 81%.
1H-ЯМР (400 МГц, ДМСО-d6) δ:
7,60 - 7,33 (7H, м), 7,24 (1H, д, J = 2 Гц), 3,97 (1H, дд, J = 11,7 Гц), 2,81 - 2,69 (2H, м), 2,40 (1H, м), 2,10 (1H, м).
Синтетический пример А-7
3-Азидо-1-этоксикарбонилметил-8-фенил-2,3,4,5-тетрагидро- 1H-[1] -бензазепин-2-он
При перемешивании к 30 мл смеси 0,75 г (2,70 ммоль) 3-азидо-8-фенил-2,3,4,5-тетрагидро-1H-[1] бензазепин-2-она, полученного по синтетическому примеру A-6, 0,083 г (0,288 ммоль тетра-н-бутиламмоний бромида, 0,17 г (3,03 ммоль) порошкообразного карбоната натрия, и тетрагидрофурана при комнатной температуре добавляют 0,35 мл (3,16 ммоль) этилбромацетата с последующим перемешиванием в течение 2 часов. После добавления к реакционной смеси этилацетата полученную смесь промывают водой и насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния. После отгонки растворителя при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле. После элюирования гексаном : этилацетатом в отношении 15:1 (по объему) получают еще 0,8 г указанного в заголовке соединения в виде бледно-желтого, маслянистого продукта. Выход 81%.
1H-ЯМР (400 МГц, CDCl3) δ:
7,55 - 7,34 (7H, м), 7,31 (1H, д, J = 8 Гц), 4,78 (1H, д, J = 17 Гц), 4,47 (1H, д, J = 17 Гц), 4,20 (2H, дк, J = 7,3 Гц), 3,87 (1H, ушир. т, J = 9 Гц), 3,40 (1H, м), 2,74 (1H, м), 2,52 - 2,33 (2H, м), 1,26 (3H, т, J = 7 Гц).
Пример синтеза B-1
(S)-2-(1,3-Дигидро-1,3-диоксо-2H-изоиндол-2-ил)- 3-(2-тиенил)пропановая кислота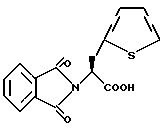
К 29,3 г (171 ммоль) L-(S)-3-(2-тиенил)аланина добавляют 257 мл диоксана, 86 мл воды, 25,9 г (175 ммоль) фталевого ангидрида и 23,9 мл (171 ммоль) триэтиламина. При перемешивании смеси при комнатной температуре в течение 1 часа к ней добавляют 23,9 мл триэтиламина. Добавляют 342 мл диоксана и смесь нагревают при кипячении с обратным холодильником. Когда pH отогнанной жидкости показывает отсутствие основания, нагревание прекращают и реакционную смесь концентрируют при пониженном давлении. Добавляют 10 мл диэтилового эфира и 684 мл 0,5 н соляной кислоты и смесь энергично перемешивают. Выпавшие таким образом кристаллы собирают фильтрацией, промывают небольшим количеством воды и сушат, пропуская сухой азот. Получают 40,7 г указанного в заголовке соединения в виде бледно-желтых кристаллов (выход 79%).
Масс m/e (FAB); 320 (NH+)
Т.пл. 172 - 173oC.
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ:
3,76 (1H, дд, J = 4,8, 15,3 Гц), 3,89 (1H, дд, J = 11,6, 15,3 Гц), 5,19 (1H, дд, J = 4,8, 11,6 Гц), 6,81 - 6,84 (2H, м), 7,08 (1H, дд, J = 1,6, 4,8 Гц), 7,70 - 7,74 (2H, м), 7,80 - 7,85 (2H, м).
Синтетический пример B-2
Этиловый эфир N-[(S)-2-(1,3-дигидро-1,3-диоксо-2H- изоиндол-2-ил)-3-(2-тиенил)пропаноил]-6-гидрокснорлейцина
K 21,8 г (102,9 ммоль) гидрохлорида этилового эфира 6-гидрокси-DL-норлейцина добавляют 686 мл дихлорметана и 17,0 мл (164 ммоль) N-метилморфолина при 0oC с получением посредством этого гомогенного раствора. Затем добавляют 31,0 г (102,9 ммоль) соединения, полученного по примеру синтеза B-1, и 38,2 г (154 ммоль) ЭЭДХ и полученную смесь перемешивают в течение ночи при медленном нагревании при комнатной температуре. Реакционную смесь промывают 1000 мл 1 н соляной кислоты, водным раствором гидрокарбоната натрия и водным раствором хлорида натрия и сушат над безводным сульфатом натрия. После фильтрования фильтрат концентрируют при пониженном давлении. Полученный таким образом остаток очищают колоночной хроматографией (дихлорметан/этилацетат = 3 ---> 2). Таким образом получают 22,8 г указанного в заголовке соединения в виде бледно-желтого твердого продукта (выход 48%).
Масс m/e (FAB); 4592 (NH+)
Т.пл. 102 - 104oC.
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ:
1,22 - 1,27 (3H, м), 1,28 - 1,96 (7H, м), 3,57 - 3,65 (2H, м), 3,74 - 3,87 (2H, м), 4,09 - 4,20 (2H, м), 4,58 - 4,66 (1H, м), 5,07 - 5,13 (1H, м), 6,60 - 6,71 (суммарный 1H, каждый уширен. д), 6,78 - 6,83 (2H, м), 7,05 - 7,09 (1H, м), 7,70 - 7,75 (2H, м), 7,81 - 7,85 (2H, м).
Синтетический пример B-3
Этил [5S -(5α,8α, (R*), 11αβ]- 5-(1,3-дигидро-1,3-диоксо-2H- изоиндол-2-ил)-6-оксо-4,5,6,8,9,10,11,11a-октагидропиридо[1,2-a] тиено [3,2-c] азепин-5-карбоксилат
В токе азота охлаждают до -65oC 93 мл дихлорметана и добавляют к нему 1,71 мл (19,6 ммоль) оксалилхлорида. После прибавления по каплям 1,53 мл (21,3 ммоль) диметилсульфоксида смесь перемешивают в течение 30 минут. Туда же прибавляют по каплям раствор 3,00 г (6,54 ммоль) соединения, полученного по примеру B-2, в дихлорметане (24 мл) и полученную смесь перемешивают в течение 30 минут. Затем добавляют по каплям 9,1 мл (65 ммоль) триэтиламина и смесь медленно нагревают до 0oC. Три часа спустя добавляют раствор 12,2 г пероксимонофосфата калия (OXONE R) в воде (50 мл) при 0oC и смесь энергично перемешивают. После 10 минут органическую фазу отделяют, промывают насыщенным водным раствором хлорида натрия. После высушивания над безводным сульфатом магния раствор концентрируют до объема около 65 мл при 20oC и ниже. При 0oC к нему прибавляют по каплям 6,5 мл трифторуксусной кислоты и полученную смесь нагревают до комнатной температуры и перемешивают 14 часов. Реакционную смесь концентрируют при пониженном давлении при низкой температуре и добавляют 100 мл этилацетата. При 0oC медленно добавляют насыщенный водный раствор водный раствор гидрокарбоната натрия и твердый гидрокарбонат натрия и смесь энергично перемешивают. Органическую фазу отделяют, промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и затем концентрируют. Сырой продукт очищают колоночной хроматографией на силикагеле (гексаноэтилацетат-3) с получением посредством этого 540 мл указанного в заголовке соединения в виде белых кристаллов.
Т.пл. 140 - 150oC.
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ:
0,94 (3H, т, J = 7,2 Гц), 1,62 - 1,95 (3H, м), 2,14 - 2,20 (2H, м), 2,41 - 2,49 (1H, м), 3,44 (1H, ддд, J = 1,6, 4,0, 16,8 Гц), 3,72 - 3,80 (1H, м), 3,87 - 3,95 (1H, м), 4,58 (1H, м как т), 5,32 (1H, дд, J = 1,6, 7,67 ц), 5,36 (1H, ушир., т), 6,06 (1H, дд, J = 4,0, 13,6 Гц), 6,83 (1H, д, J = 5,4 Гц), 7,09 (1H, д, J = 5,4 Гц), 7,70-7,76 (2H, м), 7,86 - 7,92(2H, м).
Синтетический пример B-4
2-(1,3-Дигидро-1,3-диоксо-2H-изоиндол-2-ил)-3-(3-тиенил)пропановая кислота
56,0 г (269,6 ммоль) DL-3-(3-тиенил)аланина подвергают взаимодействию тем же методом, что и в синтетическом примере B-1. Таким образом получают 68,4 г указанного в заголовке соединения в виде желтых кристаллов (выход 84%).
Масс m/e (FAB); 302 (MH+)
Т. пл. 162 - 165oC.
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 3,55 (1H, дд, J = 4,8, 15,0 Гц), 3,72 (1H, дд, J = 11,6, 15,0 Гц), 5,21 (1H, дд, J = 4,8, 11,6 Гц), 6,91-6,93 (1H, м), 6,97 (1H, м как уширен. с), 7,18 (1H, дд, J = 3,2, 4,8 Гц), 7,69-7,72 (2H, м), 7,78-7,81 (2H, м).
Синтетический пример B-5
Этиловый эфир N-[2-(1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)- 3-(3-тиенил)пропаноил]-6-гидроксинорлейцина
23,0 г (108,7 ммоль) гидрохлорида этилового эфира 6-гидрокси-DL-норлейцина и 32,74 г (108,7 ммоль) соединения, полученного по синтетическому примеру B-4, подвергают взаимодействию тем же методом, что в синтетическом примере B-2. Таким образом получают 25,9 г указанного в заголовке соединения в виде бледно-желтых кристаллов (выход 52%).
Масс m/e (FAB); 458 (MH+)
Т. пл. 80 - 85oC.
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1,23-1,30 (3H, м), 1,31-1,96 (6H, м), 3,54-3,67 (4H, м), 4,06-4,24 (2H, м), 4,58-4,68 (1H, м), 5,11-5,17 (1H, м), 6,68-6,77 (суммарный 1H, каждый уширен. д), 6,93-7,01 (2H, м), 7,17-7,22 (1H, м), 7,70-7,74 (2H, м), 7,79-7,84 (2H, м).
Синтетический пример B-6
Этил 5-(1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)-6-оксо- 4,5,6,8,9,10,11,11a-октагидропиридо[1,2-a]тиено[3,2-c]азепин-8-карбоксилат
2 г (4,36 ммоль) соединения, полученного по синтетическому B-5, подвергают взаимодействию тем же методом, что и в синтетическом примере B-3. Таким образом получают указанное в заголовке соединение в виде белых кристаллов и в виде смеси двух диастереомеров (1,25 г, 67%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 0,92 и 1,25 (суммарный 3H, каждый т, каждый J = 7,2 Гц), 1,65-2,50 (6H, м), 3,20 и 3,30 (суммарный 1H, каждый ддд, каждый J = 1,6, 4,0, 16,8 Гц), 3,76-7,23 (суммарный 2H, м), 4,28-4,45 (суммарный 1H, м), 5,30 (суммарный 1H, каждый м), 5,54-5,61 (суммарный 1H, м), 5,83 и 6,03 (суммарный 1H, каждый дд, каждый J = 4,0, 13,6 Гц, J = 4,0, 14,0 Гц), 6,84 и 6,87 (суммарный 1H, каждый д, каждый J = 5,2 Гц и J = 5,6 Гц), 7,13-7,16 (суммарный 1H, м), 7,72-7,75 (2H, м), 7,85-7,90 (2H, м).
Синтетический пример C-1
(2R,3S)-2-Бром-3-метилпентановая кислота
1,50 г (11,43 ммоль) D-алло-изолейцина [(2R, 3S)-2-амино-3-метилпентановая кислота] растворяют в смешанном растворителе 12,7 мл водного раствора бромистого водорода с 12,7 мл воды, затем охлаждают до 0oC. К нему медленно прибавляют по каплям раствор 1,20 г нитрита натрия в 3,0 мл воды с такой скоростью, чтобы температура реакции не превышала 5oC. Затем смесь перемешивают при 0oC в течение 30 минут и далее при комнатной температуре в течение 3 часов. После отгонки избытка азотной кислоты в виде газа при пониженном давлении проводят экстрагирование эфиром. Органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и затем концентрируют. Таким образом получают указанное в заголовке соединения в виде желтого масла. Выход 95%.
1H-ЯМР (400 МГц, CDCl3) δ: 4,29 (1H, д, J = 7 Гц), 2,01 (1H, м), 1,52 (1H, м), 1,33 (1H, м), 1,08 (3H, д, J = 7 Гц), 0,95 (3H, д, J = 7 Гц).
Синтетический пример C-2
(2R,3S)-2-Ацетилтио-3-метилпентановая кислота
2,11 г (10,8 ммоль) (2R,3S)-2-Бром-3-метилпентановой кислоты, полученной по синтетическому примеру C-1, растворяют в 43 мл ацетонитрила и добавляют 1,41 г тиоацетата калия при 0oC. Смесь перемешивают при 0oC в течение 30 минут и затем при комнатной температуре 5 часов. После удаления фильтрацией нерастворимого продукта фильтрат концентрируют. К остатку добавляют эфир и насыщенный водный раствор гидрокарбоната натрия с последующим разделением фаз. Водную фазу подкисляют добавлением 2 н раствора соляной кислоты при пониженной температуре и затем экстрагируют эфиром. Эфирную фазу промывают насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют. Таким образом получают 1,68 г указанного в заголовке соединения в виде бесцветного масла (выход 82%).
1H-ЯМР (400 МГц, CDCl3) δ: 4,21 (1H, д, J = 7 Гц), 2,39 (3H, с), 2,02 (1H, м), 1,58 (1H, м), 1,22 (1H, м), 1,03 (3H, д, J = 7 Гц), 0,92 (3H, д, J = 7 Гц).
Синтетический пример C-3
α- (1,3-Дигидро-1,3-диоксо-2H-изоиндол-2-ил)-(1,1'-бифенил)-4-пропановая кислота
43,70 г (181,3 ммоль) α- амино-(1,1'-бифенил)-4-пропановой кислоты и 26,80 г (181,3 ммоль) безводной фумаровой кислоты суспендируют в 100 мл диметилформамида с последующим нагреванием при 120oC в течение 2,5 часов. Затем полученный таким образом прозрачный раствор выливают в 1,2 л ледяной воды с последующим энергичным перемешиванием. При этом выпадают белые кристаллы. Эти кристаллы собирают фильтрацией, промывают водой и гексаном и сушат горячим воздухом. Таким образом получают 65,5 г указанного в заголовке продукта в виде белых кристаллов (выход 73%).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 7,83 (4H, с), 7,58 (2H, д, J = 8 Гц), 7,51 (2H, д, J = 8 Гц), 7,40 (2H, т, J = 8 Гц), 7,31 (1H, т, J = 8 Гц), 7,26 (2H, д, J = 8 Гц), 5,16 (1H, дд).
Синтетический пример C-4
Метиловый эфир (S)-N- [α- (1,3-дигидро-1,3-диоксо-2H-изоиндол- 2-ил)-(1,1'-бифенил)-4-пропионил]-6-гидроксинорлейцина
К перемешиваемому раствору 28,53 г (76,90 ммоль) α- (1,3-дигидро-1,3 -диоксо-2H-изоиндол-2-ил)-(1,1'-биофенил)-4-пропановой кислоты, полученной по синтетическому примеру C-3, и 19,10 г (96,70 ммоль) гидрохлорида метилового эфира (S)-6-гидроксинорлейцина и 600 мл дихлорметана добавляют 42,47 мл N-метилморфолина. После получения гомогенного раствора добавляют 1-гидроксибензтриазол гидрат и 28,92 г (150,87 ммоль) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимила при 0oC. Реакционную смесь перемешивают при 0oC в течение 30 минут и затем в течение ночи при комнатной температуре и промывают 2 н. водным раствором соляной кислоты, водой, насыщенным водным раствором гидрокарбоната натрия и водным раствором хлорида натрия. Дихлорметановую фазу сушат над безводным сульфатом магния и концентрируют. Остаток очищают колоночной хроматографией (элюент - хлороформ: метанол = 99:1). Таким образом получают 24,80 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 63%).
1H-ЯМР (400 МГц, CDCl3) δ: 7,79 (2H, м), 7,69 (2H, м), 7,52 - 7,22 (9H, м), 6,77 и 6,68 (суммарный 1H, каждый ушир. д., J = 8 Гц), 5,19 (1H, м), 4,63 (1H, м), 3,72 и 3,71 (суммарный 3H, каждый с), 3,68 - 3,52 (4H, м), 1,97 - 1,30 (6H, м).
Синтетический пример C-5
Метиловый эфир (S)-N- [α- (1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)-(1,1'-бифенил)-4-пропионил]-6 -иксонорлейцина
Раствор 9,82 мл (115,35 ммоль) оксалилхлорида в 330 мл дихлорметана охлаждают до -70oC и добавляют медленно, по каплям, раствор 8,18 мл (115,35 ммоль) диметилсульфоксида в дихлорметане (70 мл) в течение 15 минут. Эту реакционную смесь перемешивают при -70oC в течение 15 минут. Затем медленно по каплям добавляют раствор 24,80 г (48,20 ммоль) метилового эфира (S)-N- [α- (1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)-(1,1'-бифенил)-4-пропионил]-6- гидроксинорлейцина, полученного по синтетическому примеру C-4, в дихлорметане (130 мл) в интервале температур от -70oC до -60oC в течение 40 минут. После перемешивания реакционной смеси при -70oC в течение 20 минут медленно добавляют по каплям 52,66 мл триэтиламина. Реакционную смесь перемешивают при 0oC в течение 1 часа и затем прибавляют по каплям в интервале температур от 0 до 5oC раствор 70,18 г пероксимонофосфата калия (OXONE R) в воде (830 мл) с последующей экстракцией дихлорметаном. Дихлорметановую фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют. Таким образом получают указанное в заголовке соединение в виде коричневого масла. Этот ольдегид не очищают, а используют в последующей реакции (пример синтеза C - 6).
1H-ЯМР (400 МГц, CDCL3) δ: 9,71 и 9,70 (суммарный 1H, м), 7,78 (2H, м), 7,68 (2H, м), 7,50 - 7,20 (9H, м), 6,82 и 6,78 (суммарный 1H, каждый ушир. д, J = 8 Гц), 5,20 (1H, м), 4,61 (1H, м), 3,91 (3H, с), 3,75 - 3,52 (4H, м), 2,50 - 1,30 (суммарный 6H, м).
Синтетический пример C-6
Метил (S)-1- [α- (1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)-(1,1'-бифенил)-4-пропионил]-1,2,3,4- тетрагидро-2-пиридикарбоксилат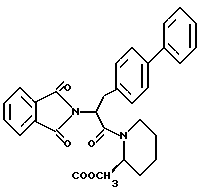
К метиловому эфиру (S)-N- [α- (1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)-(1,1'-бифенил)-4-пропионил] -6- оксонорлейцина, полученного по синтетическому примеру C-5 (сырой продукт, 48,2 ммоль), добавляют сразу 60 мл трифторуксусной кислоты при 0oC. Раствор, полученный таким образом, перемешивают при комнатной температуре в течение 2 часов. Смесь концентрируют и оставшееся масло подвергают азеотропной перегонке с бензолом. Коричневый маслянистый остаток распределяют между дихлорметаном и водой и промывают насыщенным водным раствором гидрокарбоната, водой и насыщенным водным раствором хлорида натрия. Дихлорметановую фазу сушат над безводным сульфатом магния и концентрируют. Оставшееся масло очищают колоночной хроматографией на силикагеле (элюент - дихлорметан). Таким образом получают 8,70 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход в расчете на пример синтеза C - 4 37%).
1H-ЯМР (400 МГц, CDCL3) δ: 7,84-7,74 (2H, м), 7,69 (2H, м), 7,53-7,20 (9H, м), 6,73 и 6,51 (суммарный 1H, каждый ушир. д, J = 8 Гц), 5,52 и 5,42 (суммарный 1H, каждый дд, J = 12,7 Гц), 5,29 и 5,24 (суммарный 1H, каждый подобен дт), 5,03 и 4,88 (суммарный 1H, каждый м), 2,39 (1H, м), 2,10 - 1,75 (3H, м).
Синтетический пример C - 7
[4S- (4α,7α(R*), 12b β ]-7-(1,3-Дигидро-1,3-диоксо-2H-изоиндол-2-ил)-6-оксо-11-фенил-1,2,3,4,6,7,8,12b -октагидропиридо[2,1-a] [2]бензазепин-4-карбоновая кислота
Раствор метил (S)-1- [α- (1,3-лигидро-1,3-диоксо-2H-изоинол-2-ил)-(1,1'-бифенил)-4-пропионил] -1,2,3,4 -тетрагидро-2-пиридинкарбоксилата (8,70 г, 17,61 ммолль, смесь 1:1 диастереомеров), полученного по синтетическому примеру C-6, в дихлорметане (58 мл) добавляют по каплям в растворе смеси 10,82 г (2 ммоль) трифторметансульфоновой кислоты и трифторуксусного ангидрида (TFAA, 2,75 мл, 19,51 ммоль) при 0oC. После перемешивания в атмосфере азота при комнатной температуре в течение 30 часов смесь выливают в ледяную воду с последующей экстракцией этилацетата. Этилацетатную фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют. Аморфный остаток очищают колоночной хроматографией на силикагеле (элюент - трихлорметан : метанол = 99:1). Таким образом получают 1,80 г указанного в заголовке соединения в виде аморфного продукта (выход 42%).
1H-ЯМР (400 МГц, CDCl3) δ: 7,78 (2H, дд, J = 8,4 Гц), 7,66 (2H, дд, J = 8,4 Гц), 7,49 (2H, дд, J = 8,2 Гц), 7,43 (1H, д, J = 2 Гц), 7,37 (3H, м), 7,28 (1H, тт, J = 7,2 Гц), 7,14 (1H, д, J = 8 Гц), 5,78 (1H, дд, J = 10,6 Гц), 5,30 (1H, т, J = 6 Гц), 5,14 (1H, дд, J = 8,4 Гц), 4,05 (1H, дд, J = 16,10 Гц), 3,44 (1H, д, J = 16,62 Гц), 2,52 - 2,32 (2H, м), 2,10 - 1,97 (2H, м), 1,88 = 1,66 (2H, м),
Синтетический пример C-8
Дифенилметил [4S- (4α,7α(R*), 12b β ]-7-(1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)-6-оксо-11-фенил-1,2,3,4,6,7,8, 12b-октагидропиридо[2,1-a][2] бензазепин-4-карбоксилат
К раствору 1,80 г (375 ммоль) [4S -(4α,7α(R*),12bβ]- 7-(1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- окатагидропиридо [2,1-a][2]бензазепин-4-карбоновой кислоты, полученной по синтетическому примеру C - 7, в диметилформамиде (40 мл) прибавляют 1,54 г (4,21 ммоль) карбоната цезия. Полученную смесь перемешивают 30 минут. К ней прибавляют 1,30 г (5,25 ммоль) бромдифенилметана и перемешивают при комнатной температуре в течение 5 часов. Распределяют между этилацетатом и водой. Этилацетатную фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют. Аморфный остаток очищают колоночной хроматографией на силикагеле (элюент - хлорофором:гексан = 4: 1). Таким образом получают 2,03 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 84%).
1H-ЯМР (400 МГц, CDCl2) δ: 7,86 2H, ушир. с), 7,69 (2H, дд, J = 8,4 Гц), 7,44 - 6,98 (7H, м), 6,58 (1H, д, J = 8 Гц), 6,18 (1H, с), 6,03 (1H, дд, J = 10,6 Гц), 5,42 (1H, т, J = 6 Гц), 5,14 (1H, дд, J = 8,4 Гц), 4,35 (1H, дд, J = 16,10 Гц), 3,22 (1H, дд, J = 16,6 Гц), 2,37 (2H, м), 2,05 (1H, м), 1,80 - 1,63 (3H, м).
Синтетический пример C-9
Дифенилметил [4S-(4α,7α(R*),12bβ]- 7-амино-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]-бензазепин-4-карбоксилат
2,03 г (314 ммоль) дифенилметил [4S-(4α,7α(R*),12bβ- -7-(1,3-дигидро-1,3-диоксо- 2H-изоиндол-2-ил)-6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2] бензазепин-4-карбоксилата, полученного по синтетическому примеру C-8, растворяют в растворе смеси 40 мл метанола и 20 мл тетрагидрофурана. Добавляют 0,34 мл (7,10 ммоль) гидразин моногидрата с последующим нагреванием с обратным холодильником в течение 3 часов. Реакционную смесь концентрируют, оставшийся твердый продукт растворяют в дихлорметане, нерастворимый материал удаляют фильтрацией. Фильтрат концентрируют и липкий остаток очищают колоночной хроматографией на силикагеле (элюент - хлороформ : метанол : водный аммиак = 98 : 2 : 0,2). Таким образом получают 1,20 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 74%).
1H-ЯМР- (400 МГц, CDCl3) δ: :
7,40 (4H, м), 5,42 (1H, тт, J = 7,2 Гц), 7,24 (1H, д, J = 2 Гц), 7,15 (1H, дд, J = 8,2 Гц), 6,99 (2H, дд, J = 8,4 Гц), 6,87 (2H, дд, J = 8,2 Гц), 6,63 (1H, д, J = 8 Гц), 6,20 (1H, с), 5,42-5,33 (2H, м), 4,53 (1H, дд, J = 10,6 Гц), 3,17 (1H, дд, J = 16,8 Гц), 2,58 (1H, дд, J = 16,19 гц), 2,40 (2H, м), 1,94 (1H, м), 1,85-1,58 (3H, м).
Синтетический пример C-10
[4S -(4α,7α(R*),12bβ]- 7-(1,3-Диоксо-1,3-дигидроизоиндол-2-ил)-9-нитро-6-оксо- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота и
[4S -(4α,7α(R*), 12b β ]-7-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-11- нитро-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4- карбоновая кислота
8,30 г (20,5 ммоль) [4S -(4α,7α(R*),12bβ]- 7-(1,3-диоксо-1,3-дигидроизоиндол-2- ил)-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4- карбоновой кислоты растворяют в 110 мл метиленхлорида с последующим охлаждением до -60oC. Затем прибавляют по каплям раствор, полученный растворением нитроний тетрафторбората (0,5 M раствор в сульфолане 148 мл, 74 ммоль) в 90 мл метиленхлорида. Затем смесь медленно нагревают до 2oC в течение 10 часов и затем перемешивают при 2oC в течение 5 часов. Затем ее распределяют в 500 мл метиленхлорида и 1200 мл воды. Затем, после отделения органической фазы ее промывают насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и раствор концентрируют при пониженном давлении. Полученный таким образом остаток очищают флэш хроматографией на силикагеле (1:1 этилацетат/гексан ---> этилацетат, содержащий 5% добавленной уксусной кислоты). Таким образом получают указанные в заголовке соединения.
Синтетический пример C-11
Метил [4S -(4α,7α(R*),12bβ]- 7-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-9-нитро-6-оксо- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилат и
метил [4S -(4α,7α(R*),12bβ]- 7-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-11-нитро- 6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4- карбоксилат
5,47 г (12,2 ммоль) смеси [4S -(4α,7α(R*),12bβ]- 7-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-9-нитро-6-оксо-1,2,4,6,7,8,12b-октагидропиридо [2,1-a] [2] бензазепин-4-карбоновой кислоты и [4S -(4α,7α(R*),12bβ]- 7-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-11-нитро-6-оксо-1,2,3,4,6,7,8,12b - октагидропиридо[2,1-a]-[2]бензазепин-4-карбоновой кислоты, полученной по вышеописанному синтетическому примеру C-10, растворяют в 80 мл диметилформамида. К этому раствору добавляют 4,76 г (14,6 ммоль) карбоната цезия при комнатной температуре. Полученную таким образом смесь перемешивают в атмосфере азота в течение 30 минут и затем добавляют 2,42 г (17,0 ммоль) метилиодида. Полученную смесь перемешивают 11 часов. Далее перемешенный раствор распределяют в 300 мл воды и двух 250-мл частях этилацетата. Затем после промывки отделенной органической фазы насыщенным водным раствором хлорида натрия ее высушивают над безводным сульфатом магния и растворитель концентрируют при пониженном давлении. После завершения концентрирования полученный таким образом остаток очищают и отделяют флэш хроматографией на силикагеле (1:1 этилацетат/гексан). Таким образом получают 1,62 г (выход: 29%) указанного в заголовке соединения, имеющего нитрогруппу в 11-положении и 1,78 г (выход : 31%) указанного в заголовке соединения, имеющего нитрогруппу в 9-положении.
Синтетический пример C-12
Метил [4S -(4α,7α(R*),12bβ]- 11-амино-7-(1,3-диоксо-1,3-дигидроизоиндол-2- ил)-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин- 4-карбоксилат
1,62 г (32,5 ммоль) Метил [4S -(4α,7α(R*)-12bβ]- -7-(1,3-диоксо-1,3- дигидроизоиндол-2-ил)-11-нитро-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилата, полученного по вышеуказанному синтетическому примеру C-11 растворяют в 5 мл уксусной кислоты и 60 мл диметилформамида. Затем к этому раствору добавляют 230 г 10% палладия на угле с последующим встряхиванием при комнатной температуре в течение 2 часов. Затем прибавляют 150 мл метанола к встряхиваемому раствору с последующей фильтрацией. Фильтрат концентрируют при пониженном давлении с последующим получением 1,50 г указанного в заготовке соединения.
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ:
1,70-2,45 (6H, м), 3,20 (3H, с),
3,30 (1H, дд, J = 16,6, 6,7 Гц),
4,26 (1H, дд, J = 16,6, 12,1 Гц), 5,19 (1H, м),
5,34 (1H, м), 5,98 (1H, дд, J = 12,1, 6,7 Гц),
6,56 (2H, м), 6,98 (1H, д, J = 8,8 Гц), 7,70-7,90 (4H, м).
Синтетический пример C-13
Метил [4S -(4α,7α(R*),12bβ] -11-метилсульфониламино-7-(1,3-диоксо-1,3- дигидроизоиндол-2-ил)-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2] бензазепин-4-карбоксилат
1,50 г (3,5 ммоль) Метил [4S -(4α,7α(R*),12bβ] -1-амино-7-(1,3-диоксо- 1,3-дигидроизоиндол-2-ил)-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2] бензазепин-4-карбоксилата, полученного по вышеуказанному синтетическому примеру C-12 растворяют в 50 мл метиленхлорида. Затем к этому раствору добавляют 3 мл пиридина и 440 г (3,8 ммоль) метансульфонилхлорида при охлаждении льдом. Затем смесь перемешивают в атмосфере азота при комнатой температуре в течение 2 часов. Затем к перемешиваемому раствору прибавляют 100 мл 1н водного раствора соляной кислоты при охлаждении льдом, экстрагируют метиленхлоридом. После высушивания над безводным сульфатом магния концентрируют при пониженном давлении. Далее остаток очищают колоночной хроматографией на силикагеле (3 : 1 метиленхлорид/этилацетат). Таким образом получают 1,14 г указанного в заголовке соединения (выход : 64%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ:
1,60-2,46 (6H, м), 3,00 (3H, с), 3,23 (3H, с),
3,42 (1H, дд, J = 17,6, 7,0 Гц).
4,46 (1H, дд, J = 17,1, 11,9 Гц), 5,21 (1H, м),
5,44 (1H, м), 6,04 (1H, дд, J = 11,9, 7,0 Гц).
6,65 (1H, с), 7,05 (1H, дд, J = 8,2, 2,2 Гц),
7,19 (1H, д, J = 8,2 Гц), 7,24 (1H, д, J = 2,2 Гц),
7,74-790 (4H, м).
Синтетический пример C-14
Метил [4S -(4α,7α(R*),12bβ] -11-метилсульфониламино-7-(1,3-амино-6-оксо- 1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат
1,14 г (2,23 ммоль) Метил [4S-(4α,7α(R*),12bβ]- 11-метилсульфониламино-7-(1,3- диоксо-1,3 -дигидроизоиндол-2-ил)-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилата, полученного по вышеуказанному синтетическому примеру C-13 растворяют в 49 мл метанола. Затем к этому раствору добавляют 123 мл (2,46 ммоль) гидразингидрата. Затем смесь перемешивают в атмосфере аргона при комнатной температуре в течение 66 часов. Перемешиваемый раствор концентрируют при пониженном давлении. Затем к концентрату прибавляют метиленхлорид. После удаления нерастворимого материала фильтрацией к фильтрату добавляют этилацетат. Таким образом получают 0,50 г (выход: 59%) указанного в заголовке соединения в виде белых кристаллов.
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1,60-2,45 (6H, м), 2,87 (1H, дд, J = 17,6, 12,7 Гц). 2,94 (3H, с), 3,13 (3H, с), 3,40 (1H, дд, J = 17,6, 6,0 Гц). 4,65 (1H, дд, J = 12,7, 6,0 Гц), 5,30 (1H, м), 5,43 (1H, м), 7,02 (1H, дд, J = 8,2, 2,2 Гц). 7,11 (1H, д, J = 8,2 Гц), 7,16 (1H, д, J = 2,4 Гц).
Синтетический пример D-1
[4S- [4α,7α (R*), 12b β ]]-7-(1,3-Диоксо-1,3- дигидроизоиндол-2-ил)-9-нитро-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2]бензазепин-4-карбоновая кислота и
[4S-[4 α,7α (R*), 12b β ]]-7-(1,3-диоксо-1,3- дигидроизоиндол-2-ил)-11-нитро-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2]бензазепин-4-карбоновая кислота
[4S -[4α,7α (R), 12b β ]]-7-(1,3-Диоксо-1,3-дигидроиндол-2-ил)- 6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4- карбоновую кислоту (8,30 г, 20,5 ммоль) растворяют в метиленхлориде (110 мл) с последующим охлаждением до -60oC. Затем добавляют по каплям раствор, полученный растворением нитроний тетрафторбората (0,5 M раствор в сульфолане 148 мл, 74 ммоль) в метиленхлориде (90 мл). Затем смесь медленно нагревают до 2oC в течение 10 часов и затем перемешивают при 2oC в течение 5 часов. Затем ее распределяют в метиленхлориде (500 мл) и воде (1200 мл). Затем, после отделения органической фазы ее промывают насыщенным водным раствором хлорида натрия, сушат (используют MgSO4) и раствор концентрируют при пониженном давлении. Полученный таким образом остаток очищают флэш хроматографией на силикагеле (1:1 этилацетат/гексан _→ этилацетат, содержащий 5% добавленной уксусной кислоты). Таким образом получают указанные в заголовке соединения.
Синтетический пример D-2
Метил [4S -[4α,7α (R*), 12b β ]]-7-(1,3-диоксо-1,3- дигидроизоиндол-2-ил)-9-нитро-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат и
метил [4S-[4 α,7α (R*), 12b β ]]-7-(1,3-диоксо-1,3- дигидроизоиндол-2-ил)-11-нитро-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилат
Смесь (5,47 г, 12,2 ммоль) [4S-[ 4α,7α (R*), 12b β ]]-7- (1,3-диоксо-1,3-дигидроизоиндол-2-ил)-9-нитро-6-оксо-1,2,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоновой кислоты и [4S -[4α,7α (R*), 12b β ]]-7-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-11- нитро-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин- 4-карбоновой кислоты, полученной по вышеописанному синтетическому примеру D-1, растворяют в диметилформамиде (80 мл). К этому раствору добавляют карбонат цезия (4,76 г, 14,6 ммоль) при комнатной температуре. Полученную таким образом смесь перемешивают в атмосфере азота в течение 30 минут и затем добавляют 2,42 г (17,0 ммоль) метилиодида. Полученную смесь перемешивают 11 часов. Далее перемешанный раствор распределяют в воде (300 мл) и этилацетате (250 мл • 2). Затем после промывки отделенной органической фазы насыщенным водным раствором хлорида натрия ее высушивают (используют MgSO4) и растворитель концентрируют при пониженном давлении. После завершения концентрирования полученный таким образом остаток очищают и отделяют флэш хроматографией на силикагеле (1 : 1 этилацета/гексан). Таким образом получают указанное в заголовке 11-нитросоединение (1,62 г, 29%) и указанное в заголовке 9-нитросоединение (1,78 г, 31%).
Синтетический пример D-3
Метил [4S -[4α,7α (R*), 12b β ]]-11-амино-7-(1,3-диоксо- 1,3-дигидрозоиндол-2-ил)-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a][2]бензазепин-4-карбоксилат
Метил [4S(4α,7α (R*), 12b β ]-7-(1,3-диоксо-1,3- дигидроизоиндол-2-ил)-11-нитро-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепен-4-карбоксилата (1,62 г, 3,5 ммоль) полученного по вышеуказанному синтетическому примеру D-2, растворяют в уксусной кислоты (5 мл) и диметилформамиде (60 мл). Затем к этому раствору добавляют 10% палладия на угле (230 мг) с последующим встряхиванием при комнатной температуре в течение 2 часов. Затем прибавляют метанол (150 мл) к встряхиваемому раствору, фильтруют и фильтрат концентрируют при пониженном давлении. Таким образом получают указанное в заголовке соединение (1,50 г)
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1,70-2,45 (6H, м), 3,20 (3H, с), 3,30 (1H, дд, J = 16,6, 6,7 Гц), 4,26 (1H, дд, J = 16,6, 12,1 Гц), 5,19 (1H, м), 5,34 (1H, м), 5,98 (1H, дд, J = 12,1, 6,7 Гц), 6,56 (2H, м), 6,98 (1H, д, J = 8,8 Гц), 7,70-7,90 (4H, м).
Синтетический пример E-1
α -(1,3-Дигидро-1,3-диоксо-2H-изоиндол-2-ил)-(1,1'-бифенил)- 4-пропановая кислота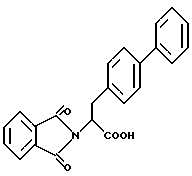
α- Амино-(1,1'-бифенил)-4-пропановую кислоту (43,70 г, 181,3 ммоль) и безводную фумаровую кислоту (26,80 г, 181,3 ммоль) суспендируют в 100 мл диметилформамида (ДМФ) с последующим нагреванием при 120oC в течение 2,5 часов. Затем полученный таким образом прозрачный раствор выливают в ледяную воду (1,2 л) с последующим энергичным перемешиванием. При этом выпадают белые кристаллы. Эти кристаллы собирают фильтрацией, (промывают водой и гексаном) и сушат горячим воздухом. Таким образом получают указанный в заголовке продукта в виде белых кристаллов (65,5 г, выход 73%).
1H-ЯМР (400 МГц, DMCO-d6) δ: 7,83 (4H, с), 7,58 (2H, д, J = 8 Гц), 7,51 (2H, д, J = 8 Гц), 7,40 (2H, т, J = 8 Гц), 7,31 (1H, т, J = 8 Гц), 7,31 (1H, т, J = 8 Гц), 7,26 (2H, д, J = 8 Гц), 5,16 (1H, дд).
Синтетический пример E-2
Метиловый эфир (S)-N -[α- (1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)- (1,1'-бифенил)-4-пропионил]- 6-гидроксинорлейцина
К перемешиваемому раствору α- (1,3-дигидро-1,3-диоксо-2H- изоиндол-2-ил)-(1,1'-бифенил)-4-пропановой кислоты (28,53 г, 76,90 ммоль), полученной по синтетическому примеру E-1, с гидрохлоридом метилового эфира (S)-6-гидроксинорлейцина (19,10 г, 96,70 ммоль) в 600 мл дихлорметана (CH2C12) добавляют 42,47 мл N-метилморфолина (HMM). После получения гомогенного раствора добавляют 1-гидроксибензтриазол гидрат (ГОБТ) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (ДЭК) (28,92 г, 150,87 ммоль) при 0oC. После перемешивания реакционной смеси при 0oC в течение 30 минут и затем в течение ночи при комнатной температуре промывают 2 н. водным раствором соляной кислоты, водой, насыщенным водным раствором гидрокарбоната натрия и водным раствором хлорида натрия. CH2Cl2 фазу над безводным сульфатом магния и концентрируют. Оставшееся масло очищают колоночной хроматографией (элюент - хлороформ (CHCl3): метанол (CH3OH) = 99:1). Таким образом получают указанный в заголовке бесцветный аморфный продукт (14,80 г, выход 63%).
1H-ЯМР (400 МГц, CDCl3) δ: 7,79 (2H, м), 7,69 (2H, м), 7,52-7,22 (9H, м), 6,77 и 6,68 (суммарный 1H, каждый ушир. д, J = 8 Гц), 5,19 (1H, м), 4,63 (1H, м), 3,72 и 3,71 (суммарн. 3H, каждый с), 3,68-3,52 (4H, м), 1,97-1,30 (6H, м).
Синтетический пример E-3
Метиловый эфир (S)-N-[ α- (1,3-дигидро-1,3-диоксо-2H-изоиндол- 2-ил)-(1,1'-бифенил)-4-пропионил]-6-оксонорлейцина
Раствор оксалилхлорида (9,82 мл, 115,35 ммоль) в CH2Cl2 (330 мл) охлаждают до -70oC и добавляют медленно, по каплям, раствор диметилсульфоксида (ДМСО, 8,18 мл, 115,35 ммоль) в CH2Cl2 (70 мл) в течение 15 минут. Эту реакционную смесь перемешивают при -70oC в течение 15 минут. Затем медленно по каплям добавляют раствор метилового эфира (S)-N-[ α- (1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил)-(1,1'-бифенил)-4- пропионил] -6-гидроксинорлейцина (24,80 г, 48,20 ммоль) полученного по синтетическому примеру E-2 в CH2Cl2 (130 мл) в интервале температур от -70oC до -60oC в течение 40 минут. После перемешивания реакционной смеси при -70oC в течение 20 минут медленно добавляют по каплям триэтиламин (ТЭА, 52,6 мл). Реакционную смесь перемешивают при 0oC в течение 1 часа и затем прибавляют по каплям в интервале температур от 0 до 5oC раствор пероксимонофосфата калия (OXONE, 70,18 г) в воде (830 мл) с последующей экстракцией CH2Cl2 • CH2Cl2 фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют. Таким образом получают указанное в заголовке соединение в виде коричневого масла. Этот альдегид не очищают, а используют в последующей реакции (пример синтеза E-4).
1Н ЯМР (400 МГц, CDCl3) δ: 9,71 и 9,70 (суммарный 1H, м), 7,78 (2H, м), 7,68 (2H, м), 7,50 - 7,20 (9H, м), 6,82 и 6,78 (суммарный 1H, каждый ушир. д, J = 8 Гц), 5,20 (1H, м), 4,61 (1H, м), 3,91 (3H, с), 3,75 - 3,52 (4H, м), 2,50 - 1,30 (суммарный 6H, м).
Синтетический пример E-4
Метил (S)-1-[ α- (1,3-дигидро-1,3-диоксо-2H-изоиндол-2-ил- (1,1'-бифенил)-4-пропионил]-1,2,3,4-тетрагидро-2-пиридинкарбоксилат
К метиловому эфиру (S)-N-[ α- (1,3-дигидро-1,3-диоксо-2H- изондол-2-ил)-(1,1'-бифенил)-4-пропионил] -6-оксонорлейцина, полученному по синтетическому примеру E-3 (сырой продукт, 48,2 ммоль), добавляют сразу трифторуксусную кислоту (ТФА, 60 мл) при 0oC. Раствор, полученный таким образом, перемешивают при комнатной температуре в течение 2 часов. Смесь концентрируют и оставшееся масло подвергают азеотропной перегонке с бензолов. Коричневый маслянистый остаток распределяют между CH2Cl2 и водой и CH2Cl2 фазу промывают насыщенным водным раствором гидрокарбоната, водой и насыщенным водным раствором хлорида натрия. CH2Cl2 фазу сушат над безводным сульфатом магния и концентрируют. Оставшееся масло очищают колоночной хроматографией на силикагеле (элюент - дихлорметан). Таким образом получают указанный в заголовке бесцветный аморфный продукт (8,70 г, выход в расчете на пример синтеза E-2 37%).
1Н ЯМР (400 МГц, CDCl3) δ: 7,84 - 7,74 (2H, м), 7,69 (2H, м), 7,53 - 7,20 (9H, м), 6,73 и 6,51 (суммарный 1H, каждый ушир. д. J = 8 Гц), 5,52 и 5,42 (суммарный 1H, каждый дд, J = 12,7 Гц), 5,29 и 5,24 (суммарный 1H, каждый подобен дт), 5,03 и 4,88 (суммарный 1H, каждый м), 3,87 - 3,47 (2H, м), 3,75 и 3,65 (суммарный 1H, каждый с), 2,39 (1H, м), 2,10 - 1,75 (3H, м).
Синтетический пример E-5
[4S -(4α,7α (R*)-, 12b β ]-7-(1,3-Дигидро-1,3-диоксо-2H- изоиндол-2-ил)-6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2] бензазепин-4-карбоновая кислота
Раствор метил (S)-1-[ α- (1,3-дигидро-1,3-диоксо-2H- изоиндол-2-ил)-(1,1'-бифенил)-4-пропионил] -1,2,3,4- тетрагидро-2-пиридинкарбоксилата (8,70 г, 17,61 ммоль, смесь 1:1 диастереомеров), полученного по синтетическому примеру E-4, в CH2Cl2 (58 мл) добавляют по каплям в раствор смеси трифторметансульфоновой кислоты (10,82 г, 122 ммоль) и трифторуксусного ангидрида (ТФАА, 2,75 мл, 19,51 ммоль) при 0oC. Смесь перемешивают в атмосфере азота при комнатной температуре в течение 30 часов, и затем выливают в ледяную воду. Полученную таким образом смесь экстрагируют этилацетатом. Этилацетатную фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют. Аморфный остаток очищают колоночной хроматографией на силикагеле (элюент - CHCl3: MeOH = 99 : 1). Таким образом получают указанное в заголовке соединение в виде аморфного продукта (1,80 г, выход 42%).
1Н ЯМР (400 МГц, CDCl3) δ: 7,78 (2H, дд, J = 8,4 Гц), 7,66 (2H, дд, J = 8,4 Гц), 7,49 (2H, дд, J = 8,2 Гц), 7,43 (1H, д, J = 2 Гц), 7,37 (3H, м), 7,28 (1H, тт, J = 7,2 Гц), 7,14 (1H, д, J = 8 Гц), 5,78 (1H, дд, J = 10,6 Гц), 5,30 (1H, т, J = 6 Гц), 5,14 (1H, дд, J = 8,4 Гц), 4,05 (1H, дд, J = 16,10 Гц), 3,44 (1H, д, J = 16,62 Гц ), 2,52 - 2,32 (2H, м), 2,10 - 1,97 (2H, м), 1,88 - 1,66 (2H, м).
Синтетический пример E-6
Дифенилметил [4S-[4α,7α (R*), 12b β ]]-7-(1,3-дигидро- 1,3-диоксо-2H-изоиндол-2-ил)-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2] бензазепин-4-карбоксилат
К раствору [4S-[4α,7α (R*), 12b β ]]-7-(1,3-дигидро- 1,3-диоксо-2H-изоиндол-2-ил)-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоновой кислоты (1,80 г, 375 ммоль), полученной по примеру синтеза E-5, в ДМФ (40 мл) прибавляют карбонат цезия (1,34 г, 4,21 ммоль). Смесь перемешивают 30 минут. К полученной смеси прибавляют бромдифенилметан (1,30 г, 5,25 ммоль) и смесь перемешивают при комнатной температуре в течение 5 часов. Полученную реакционную смесь распределяют между этилацетатом и водой. Затем этилацетатную фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют. Аморфный остаток очищают колоночной хроматографией на силикагеле (элюент - CHCl3 : гексан (Hex) = 4 : 1). Таким образом получают указанное в заголовке соединение в виде бесцветного аморфного продукта (2,03 г, выход 84%).
1Н ЯМР (400 МГц, CDCl3) δ: 7,85 (2H, ушир. с), 7,69 (2H, дд, J = 8,4 Гц), 7,44 - 6,98 (7H, м), 6,58 (1H, д, J = 8 Гц), 6,18 (1H, с), 6,03 (1H, дд, J = 10,6 Гц), 5,42 (1H, т, J = 6 Гц), 5,14 (1H, дд, J = 8,4 Гц), 4,35 (1H, дд, J = 16,10 Гц). 3,22 (1H, дд, J = 16,6 Гц), 2,37 (2H, м), 2,05 (1H, м), 1,80 - 1,63 (3H, м).
Пример синтеза F-1
Получение дифенилметил 3-(4-фторфенил)лактата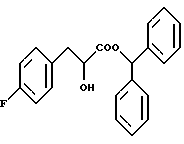
К 4-фторфенилаланину (4,99 г, 27,2 ммоль) добавляют 0,5 н водного раствора HCl (123 мл). Полученную смесь охлаждают до 0oC при охлаждении льдом и затем прибавляют нитрит серебра (5,6 г, 36,2 ммоль) несколькими порциями в течение 1 часа при энергичном перемешивании. Спустя шесть часов полученную смесь нагревают до комнатной температуры и затем перемешивают 1 день. Выпавший при этом хлорид серебра удаляют фильтрацией и фильтрат экстрагируют диэтиловым эфиром (200 мл • 4). Диэтилэфирную фазу высушивают (используют MgSO4). Диэтилэфирную фазу фильтруют и концентрируют при пониженном давлении. Таким образом получают сырой продукт (4,69 г) 3-(4-фторфенил)молочной кислоты. Затем сырой продукт (4,69 г) растворяют в сухом диметилформамиде (80 мл) и прибавляют карбонат цезия (8,58 г, 26,3 ммоль). Полученную таким образом смесь перемешивают при комнатной температуре 40 минут, и затем к ней прибавляют бромдифенилметан (11,8 г, 47,8 ммоль). Полученную смесь перемешивают при комнатной температуре в течение дня и добавляют воду (300 мл). Полученную смесь экстрагируют этилацетатом (100 • 3). Затем органическую фазу промывают насыщенным водным раствором гидрокарбоната натрия (100 мл) и насыщенным водным раствором хлорида натрия (100 мл) и сушат над безводным сульфатом магния. После фильтрации осадок (13,4 г), который получают концентрированием фильтрата при пониженном давлении, очищают колоночной хроматографией на силикагеле (гексан : этилацетат = 90 : 10). Таким образом получают указанное в заголовке соединение (4,2 г, 44%) в виде белых кристаллов.
1Н ЯМР (400 МГц, CDCl3) δ: 2,74 (1H, д, J = 6,2 Гц), 2,98 (1H, дд, J = 6,2, 14,1 Гц), 3,13 (1H, дд, J = 4,8, 14,1 Гц), 4,55 (1H, кв, J = 5,4 Гц), 6,85 (2H, т, J = 8,4 Гц), 6,94 (1H, с), 6,99 (2H, дд, J = 5,6, 8,4 Гц), 7,28 - 7,38 (10H, м),
Масс m/e (FAB); 373 (MNa+)
Т. пл. 52 - 54oC.
Синтетический пример F-2.
Получение дифенилметил 2-ацетилтио-3-(4-фторфенил)пропионата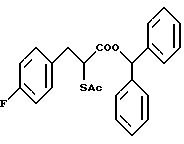
Трифенилфосфин (3,99 г, 15,2 ммоль) растворяют в сухом тетрагидрофуране (78 мл) с последующим охлаждением до 0oC при охлаждении льдом. Затем при перемешивании по каплям прибавляют диизопропил азодикарбоксилат (ДИАД (2,99 г, 15,2 ммоль)). Тридцать минут спустя прибавляют по каплям раствор смеси тиоуксусной кислоты (1,25 мл, 17,6 ммоль) с дифенилметил 3-(4-фторфенил)лактатом (4,0 г, 11,5 ммоль), полученного по примеру синтеза F-1 в сухом тетрагидрофуране (45 мл). Смесь оставляют реагировать при 0oC в течение 3 часов. Затем ледяную баню убирают и реакционную смесь нагревают до комнатной температуры и оставляют реагировать при этой температуре в течение ночи. Затем реакционную смесь концентрируют при пониженном давлении. Полученную таким образом реакционную смесь концентрируют при пониженном давлении. Полученный таким образом остаток подвергают колоночной хроматографией на силикагеле (гексан : этилацетат = 6 : 1) с получением посредством этого сырого продукта (4,7 г). Этот сырой продукт кристаллизуют из диизопропилового эфира и гексана (20 мл - 30 мл). Выпавший таким образом твердый продукт удаляют фильтрацией и фильтрат концентрируют при пониженном давлении с получением посредством этого указанного в заголовке соединения (3,54 г, 76%) в виде маслянистого продукта.
2,33 (3H, с), 3,01 (1H, дд, J = 6,6, 14,0 Гц), 3,19 (1H, дд, J = 8,8, 14,0 Гц), 4,52 (1H, т, J = 8,6 Гц), 6,81 (1H, с), 6,85 (2H, т, J = 8,6 Гц), 7,05 (2H, дд, J = 5,8, 7,8 Гц), 7,14 - 7,17 (2H, м), 7,26 - 7,36 (8H, м).
Синтетический пример F-3
Получение 2-ацетилтио-3-(4-фторфенил)пропионовой кислоты
Дифенилметил 2-ацетилтио-3-(4-фторфенил)пропионат (3,38 г, 8,27 ммоль) растворяют в анизоле (9,0 мл) с последующим охлаждением до -10oC. В этот раствор далее добавляют по каплям трифторуксусную кислоту (51,0 мл). Затем раствор нагревают до 0oC. Спустя 1 час его концентрируют при пониженном давлении. К концентрату добавляют диэтиловый эфир (80 мл) и полученный раствор экстрагируют насыщенным водным раствором гидрокарбоната натрия (100 мл • 2). К щелочному раствору, полученному таким образом, добавляют 2 н водный раствор соляной кислоты до тех пор, пока раствор не станет кислым. Далее его экстрагируют метиленхлоридом (100 мл • 3). Органическую фазу промывают насыщенным водным раствором хлорида натрия и сушат над сульфатом магния. Фильтрат, полученный его фильтрованием, после высушивания концентрируют при пониженном давлении. Таким образом получают указанное в заголовке соединение (1,96 г, 98%) в виде бесцветных кристаллов.
1Н ЯМР (400 МГц, CDCl3): 2,36 (3H, с), 3,00 (1H, дд, J = 7,4, 14,2 Гц), 3,26 (1H, дд, J = 7,8, 14,2 Гц), 4,40 (1H, т, J = 7,6 Гц), 6,99 (2H, т, J = 8,6 Гц), 7,20 (2H, дд, J = 5,6, 8,4 Гц),
Масс m/e (FAB); 243 (MH+)
Т. пл. 44 - 46oC.
Синтетические примеры от F-4 до F-6
В соответствии со способами синтетических примеров от F-1 до F-3 получают следующие соединения.
Синтетический пример F-4
(S)-2-Ацетилтио-3-фенилпропионовая кислота
С использованием D-фенилаланина в качестве исходного продукта он был синтезирован в соответствии со способом по синтетическим примерам от F-1 до F-3.
1Н ЯМР (400 МГц, CDCl3) δ: 2,34 (3H, с), 3,02 (1H, дд, J = 7,6, 14,0 Гц), 3,30 (1H, дд, J = 7,6, 14,0 Гц), 4,44 (1H, т, J = 7,6 Гц), 7,21 - 7,33 (5H, м).
Масс m/e (FAB); 225 (MH+)
Т. пл. 59 - 61oC.
Синтетический пример F-5
(S)-2-Ацетилтио-3-(1,4-бифенил)пропионовая кислота
1Н ЯМР (400 МГц, CDCl3) δ: 2,36 (3H, с), 3,07 (1H, дд, J = 7,6, 14,4 Гц), 3,34 (1H, дд, J = 7,6, 14,4 Гц), 4,48 (1H, т, J = 7,6 Гц), 7,29 - 7,59 (9H, м).
Масс m/e (FAB); 301 (MH+)
Т. пл. 122 - 123oC.
Синтетический пример F-6
(S)-2-Ацетилтио-3-(4-метоксифенил)пропионовая кислота
1H-ЯМР (400 МГц, CDCl3) δ: 2,34 (3H, с), 2,97 (1H, дд, J = 7,6, 14,4 Гц), 3,23 (1H, дд, J = 7,6, 14,4 Гц), 3,79 (3H, с), 4,39 (1H, т, J = 7,6 Гц), 6,81 - 6,86 (2H, м), 7,12-7,17 (2H, м).
Масс m/e (FAB); 255 (MH+)
Т. пл. 95 - 96oC
Пример A-1
3-Амино-1-этоксикарбонилметил-8-фенил-2,3,4,5-тетрагидро-1H- [1] бензазепин-2-он
Смесь, содержащую 0,785 г (2,15 ммоль) 3-азидо-1-этоксикарбонилметил- 8-фенил-2,3,4,5-тетрагидро-1H-[1] бензазепин-2-она, полученного по синтетическому примеру A-7, 0,05 г 10% палладия-на-угле и 20 мл этанола каталитически гидрируют при комнатной температуре при 4 атм в течение 1 часа. После отделения катализатора фильтрованием фильтрат концентрируют. Таким образом получают 0.73 г указанного в заголовке соединения в виде бледно-желтого маслянистого продукта. Выход 100%.
1H-ЯМР (400 МГц, CDCl3) δ: 7,56-7,35 (6H, м), 7,33 (1H, д, J = 2 Гц), 7,30 (1H, д, J = 8 Гц), 4,69 (1H, д, J = 17 Гц), 4,51 (1H, д, J = 17 Гц), 4,21 (2H, дкв, J = 7,1 Гц). 3,53 (1H, дд, J = 11,8 Гц), 3,28 (1H, дт, J = 13,8 Гц), 2,65 (1H, дд, J = 14,7 Гц), 2,46 (1H, м), 1,96 (1H, м).
Пример A-2
3-[(S)-Ацетилтио-3-фенилпропиониламино] -1-этоксикарбонилметил- 8-фенил-1H-[1]бензазепин-2-он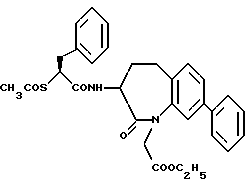
341 мг (1 ммоль) 3-амино-1-этоксикарбонилметил-8-фенил-2,3,4,5- тетрагидро-1H-[1]бензазепин-2-она, полученного по примеру A-1, и 247 мг (1,1 ммоль) (S)-2-ацетилтио-3-фенилпропионовой кислоты растворяют в 20 мл дихлорметана. Туда добавляют 300 мг (1,21 ммоль) ЭЭДХ (N-этоксикарбонил-2-этокси-1,2-дигидрохинолин) и полученный смешиванием раствор перемешивают в течение ночи. Реакционную смесь промывают 1 н соляной кислотой, водой и насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния. Растворитель отгоняют при пониженном давлении и остаток подвергают хроматографии на колонке с силикагелем. После последовательного элюирования гексаном:этилацетатом от 15:1 (по объему) до 3:1 (по объему) получают 329 мг указанного в заголовке соединения в виде бесцветного аморфного продукта. Выход 72%.
1H-ЯМР (400 МГц, CDCl3) δ: 7,55-7,15 (13H, м), 7,04 и 6,88 (общий 1H, каждый ушир. ), 4,82 и 4,78 (общий H, каждый д, J = 17 Гц), 4,50 (1H, м), 4,39 и 4,34 (общий H, каждый д, J = 17 Гц), 4,27-4,12 (3H, м), 3,42-3,22 (2H, м), 2,94 (1H, м), 2,77-2,49 (2H, м), 2,34 и 2,33 (общий 3H, каждый с), 1,24 (3H, кв, J = 7 Гц).
Пример A-3
1-Карбоксиметил-3-[(S)-2-меркапто-3-фенилпропиониламино]-8- фенил-1H-[1] бензазепин-2-он
К смеси 358 г (0,657 ммоль) 3-[(S)-ацетилтио-3-фенилпропиониламино]- 1-этоксикарбонилметил-8-фенил-1H-[1]бензазепин-2-она, полученного по примеру A-2, и 10 мл дегазированного этанола добавляют 3,3 мл дегазированного 1н водного раствора гидроксида натрия при 0oC в атмосфере азота при перемешивании. Полученную таким образом смесь перемешивают при комнатной температуре в течение 2,5 часов. Реакционную смесь охлаждают и подкисляют 1 н соляной кислотой и затем к ней прибавляют воду. Осажденные таким образом белые кристаллы собирают фильтрацией, промывают водой и н-гексаном и сушат при пониженном давлении. Таким образом получают 267 г указанного в заголовке соединения. Выход 86%.
1H-ЯМР (400 МГц, CDCl3) δ: 7,54-7,14 (13H, м), 4,74 и 4,73 (общий 1H, каждый д, J = 17 Гц), 4,54 (1H, м), 4,47 и 4,45 (общий 1H, каждый д, J = 17 Гц), 3,56 и 3,42 (общий 1H, каждый м), 3,3-3,16 (2H, м), 3,06 (1H, дд, J = 14,7 Гц), 2,98 (1H, дд, J = 14,7 Гц), 2,74-2,52 (2H, м), 2,08 и 1,97 (общий 1H, каждый д, J = 9 Гц).
Пример A-4
3-[(S)-Ацетилтио-3-метилбутириламино] -1-этоксикарбонилметил-8- фенил-1H-[1]бензазепинн-2-он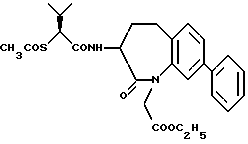
352 мг (1,04 ммоль) 3-амино-1-этоксикарбонилметил-8-фенил-2,3,4,5- тетрагидро-1H-[1] бензазепин-2-она, полученного по примеру A-1, и 202 мг (1,14 ммоль) (S)-2-ацетилтио-3-метибутановой кислоты подвергают взаимодействию тем же способом, что и по примеру A-2. Таким образом получают 396 мг указанного в заголовке соединения в виде бесцветного аморфного продукта. Выход 77%.
1H-ЯМР (400 МГц, CDCl3) δ: 7,55-7,29 (8H, м), 7,10 и 7,03 (общий 1H, каждый ушир. д, J = 7 Гц), 4,86 и 4,83 (общий 1H, каждый д, J = 17 Гц), 4,61-4,54 (1H, м), 4,39 и 4,37 (общий 1H, каждый д, J = 17 Гц), 4,24-4,13 (3H, м), 3,85 и 3,84 (общий 1H, каждый д, J = 7 Гц), 3,40 (1H, м), 2,80-2,60 (2H, м), 2,37 (3H, с), 2,26 и 2,95 (общий 1H, каждый м), 1,25 (3H, кв, J = 7 Гц), 0,99 и 0,96 (общий 6H, каждый д, дд, каждый J = 7 Гц, J = 7,2 Гц).
Пример A-5
1-Карбоксиметил-3-[(S)-2-меркапто-3-метилбутириламино] -8-фенил- 1H-[1] бензазепин-2-он
347 мг (0,7 ммоль) 3-[(S)-2-Ацетилтио-3-метилбутириламино]-1- этоксикарбонилметил-8-фенил-1H-[1]бензазепин-2-он, полученного по примеру A-4, гидролизуют тем же способом, что и по примеру A-3. Таким образом получают 243 г указанного в заголовке соединения в виде белых кристаллов. Выход 81%.
1H-ЯМР (400 МГц, CDCl3) δ: 7,55 - 7,29 (8H, м), 4,80 и 4,78 (общий 1H, каждый д, J = 17 Гц), 4,60 (1H, м), 4,48 и 4,46 (общий 1H, каждый д, J = 17 Гц), 3,33 (1H, м), 3,11 (1H, м), 2,78 - 2,62 (2H, м), 2,18 (1H, м), 2,01 (1H, м), 1,84 и 1,83 (общий 1H, каждый д, J = 9 Гц), 0,99 и 0,94 (6H, м).
Пример A-6.
3-[(S)-Ацетилтио-3-фенилпропиониламино]-1-этоксикарбонилметил-1H- [I]бензазепин-2-он
К перемешиваемому раствору 0,76 г (2,9 ммоль) 3-амино-1-этоксикарбонилметил-1H-[1] бензазепин-2-она, 0,65 г (2,9 ммоль) (S)-2-ацетилтио-3-фенилпропионовой кислоты и 30 мл тетрагидрофурана добавляют 0,61 г (3,18 ммоль) DEC, 0,35 г (3,18 ммоль) N-метилморфолина и 0,43 г (3,18 ммоль) 1-гидроксибензтриазола. Полученную таким образом смесь перемешивают при комнатной температуре в течение 5 часов. После добавления воды к реакционной смеси ее экстрагируют этилацетатом. Органическую фазу промывают водой, 1 н соляной кислотой и водой и сушат над безводным сульфатом магния. Растворитель органической фазы отгоняют при пониженном давлении и остаток подвергают хроматографии на колонке с силикагелем. После элюирования толуолом: этилацетатом при соотношении 7:1 (по объему) получают 0,95 г указанного в заголовке соединения в виде бесцветного аморфного продукта. Выход 70%.
1H-ЯМР (400 МГц, CDCl3) δ: 7,30 - 7,08 (9H, м), 7,04 и 6,88 (общий 1H, каждый ушир, J = 7 Гц), 4,77 и 4,72 (общий 1H, каждый д, J = 17 Гц), 4,42 (1H, м), 4,33 и 4,28 (общий 1H, каждый д, J = 17 Гц), 4,24 - 4,08 (3H, м), 3,38 - 3,21 (2H, м), 2,93 (1H, м), 2,75 - 2,46 (2H, м), 2,33 и 2,32 (общий 3H, каждый с), 1,83 и 1,66 (общий 1H, каждый м)
Пример A-7
1-Карбоксиметил-3-[(S)-2-меркапто-3-фенил-пропиониламино] -1H- [1]бензазепин-2-он
К перемешиваемому раствору 0,65 г (1,39 ммоль) 3-[(S)-ацетилтио- 3-фенилпропиониламино]-1-этоксикарбонилметил-1H-[1]бензазепин-2-она, полученного по примеру A-6, и 10 мл дегазированного этанола добавляют 7 мл дегазированного 1 н водного раствора гидроксида натрия при 0oC в атмосфере азота при перемешивании. Полученную таким образом смесь перемешивают при комнатной температуре в течение 3 часов. После охлаждения реакционной смеси и подкисления 1 н соляной кислотой экстрагируют дихлорметаном. Дихлорметановую фазу промывают насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом натрия. Растворитель органической фазы отгоняют при пониженном давлении. Таким образом получают 0,53 г указанного в заголовке соединения в виде бесцветного аморфного продукта. Выход 96%.
1H-ЯМР (400 МГц, CDCl3) δ: 7,31 - 7,11 (9H, м), 4,68 - 4,65 (общий 1H, каждый д, J = 17 Гц), 4,51 - 4,38 (2H, м), 3,55 и 3,42 (общий 1H, каждый м), 3,28 - 3,14 (2H, м), 3,05 и 2,97 (общий 1H, каждый дд, J = 14,7 Гц). 2,72 - 2,48 (2H, м), 2,07 и 1,96 (общий 1H, каждый д, J = 9 Гц). 1,88 и 1,64 (общий 1H, каждый м).
Пример A-8
3-[(2S, 3S)-Ацетилтио-3-метивалериламино]-1- этоксикарбонилметил-1H-[1] бензазепин-2-он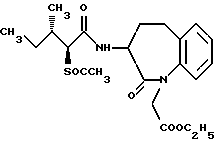
0,525 г (2 ммоль) 3-амино-1-этоксикарбонилметил-1H- [1]бензазепин-2-она и 0,418 г (2,2 ммоль) (2S, 3S)-2-ацетилтио- 3-метилвалериановой кислоты подвергают взаимодействию тем же способом, что и по примеру A-2. Таким образом получают 0,42 г указанного в заголовке соединения в виде бесцветного аморфного продукта. Выход 48%.
1H-ЯМР (400 МГц, CDCl3) δ: 7,31 - 7,00 (5H, м), 4,81 и 4,78 (общий 1H, каждый д, J = 17 Гц). 4,53 - 4,45 (1H, м), 4,33 и 4,31 (общий 1H, каждый д, J = 17 Гц), 4,22 - 4,12 (2H, м), 3,91 и 3,89 (общий 1H, каждый д, J = 7 Гц), 3,44 - 3,33 (1H, м), 2,78 - 2,56 (2H, м), 2,37 (3H, с), 2,07 - 1,87 (2H, м), 1,59 - 1,50 (1H, м), 1,28 - 1,22 (3H, м), 0,85 (общий 3H, каждый т, каждый J = 7 Гц),
Пример A-9
1-Карбоксиметил-3-[(2S, 3S)-2-меркапто-3-метилвалериламино]- 1-1H-[1]-бензазепин-2-он
К перемешиваемому раствору 0,385 г (0,89 ммоль) 3-[(2S, 3S)- ацетилтио-3-метилвалериламино]-1-этоксикарбонилметил-1H-[1]бензазепин- 2-она, полученного по примеру A-8, и 15 мл дегазированного этанола подкисляют 7 мл дегазированной 1 н соляной кислотой при перемешивании при 0oC в атмосфере азота. Экстрагируют этилацетатом. Органическую фазу промывают водой и сушат над безводным сульфатом магния. Растворитель органической фазы отгоняют при пониженном давлении. Таким образом получают 0,34 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход количественный).
1H-ЯМР (400 МГц, CDCl3) δ: 7,39 - 7,14 (5H, м), 4,74 и 4,71 (общий 1H, каждый д, J= 17 Гц), 4,57 - 4,50 (1H, м), 4,44 и 4,43 (общий 1H, каждый д, J = 17 Гц), 3,34 - 3,10 (2H, м), 2,77 - 2,58 (2H, м), 2,03 - 1,87 (2H, м), 1,85 и 1,84 (общий 1H, каждый д, J = 9 Гц), 1,64 - 1,50 (1H, м), 1,22 - 1,15 (1H, м), 0,95 (3H, д, J = 7 Гц), 0,86 (3H, т, J = 7 Гц).
Пример A-10
(S)-3-[(2S, 3S)-2-Ацетилтио-3-метилвалериламино] -1-этоксикарбонилметил-2,3,4,5- тетрагидро-1H-[1]бензазепин-2-он
0,55 г (2,1 ммоль) (S)-3-амино-1-этоксикарбонилметил-1H-2,3,4,5- тетрагидро-[1] бензазепин-2-она и 0,434 г (2,3 ммоль) (2S, 3S)-2-ацетилтио-3-метилвалериановой кислоты подвергают взаимодействию тем же способом, что и по примеру A-2. Таким образом получают 0,614 г указанного в заголовке соединения в виде бесцветного аморфного продукта. Выход 67%.
1H-ЯМР (400 МГц, CDCl3) δ: 7.31-7.17(3H, м) 7.12 (1H, дд, J = 8.1 Гц) 7.01 (1H, ушир. д, J=7 Гц) 4.78 (1H, д, J=176 Гц) 4.49 (1H, дт, J=11.8 Гц) 4.33 (1H, д, J=17 Гц) 4.24-4.12 (2H, м) 3.89 (1H, д, J=7 Гц) 3.88 (1H, м) 2.74-2.56 (2H, м) 2.37 (3H, с) 2.04-1.87 (2H, м) 1.56 (1H, м) 1.25 (3H, т, J=7 Гц) 1.14 (1H, м) 0.96 (3H, д, J=7 Гц) 0.86 (3H, т, J=8 Гц)
Пример A-11
(S)-3-[(2S, 3S)-2-Меркапто-3-метилвалерил-амино] -1- карбонилметио-2,3,4,5-тетрагидро-1H-[1]бензазепин-2-он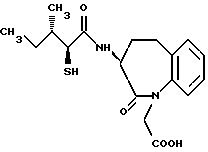
0,6 г (1,38 ммоль) (S)-3-[(2S, 3S)-2-ацетилтио-3-метилвалериламино]-1-этоксикарбонилметил-2,3,4,5- тетрагидро-1H-[1]-бензазепин-2-она, полученного про примеру A-10, гидролизуют тем же способом, что и по примеру A-9. Таким образом получают 0,49 г указанного в заголовке соединения в виде бесцветного аморфного продукта. Выход 97%.
1H-ЯМР (400 МГц, CDCl3) δ: 7.40 (1H, ушир, J=7 Гц) 7.33-7.14 (4H, м) 4.71 (1H, д, J=17 Гц) 4.54 (1H, дт, J=11, 7 Гц) 4.44 (1H, д, J=17 Гц) 3.29 (1H, м) 3.17 (1H, дд, J=9.7 Гц) 2.74-2.59 (2H, м) 2.04-1.89 (2H, м) 1.84 (1H, д, J=9 Гц) 1.55 (1H, м) 1.17 (1H, м) 0.95 (3H, д, J=7 Гц) 0.86 (3H, т, J=7 Гц)
Пример B-1
Этил [5(S)-(5α,8α(R*),11αβ]- 5-амино-6-оксо-4,5,6,8,9,10,11,11a-октагидропиридо[1,2-a]тиено[3,2-c] азепин-8-карбоксилат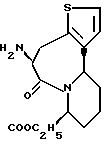
540 мг (1,23 ммоль) соединения, полученного по синтетическому примеру B-3, растворяют в 31 мл этанола и добавляют 0,072 мг (1,48 ммоль) моногидрата гидразина. Полученную таким образом смесь перемешивают при комнатной температуре неделю. Реакционную смесь концентрируют при пониженном давлении и добавляют дихлорметан. Полученный фильтрованием фильтрат вновь концентрируют. Остаток очищают хроматографией на колонке с силикагелем (дихлорметан/метанол/водный аммиак = 98 /2/ 0,3) с получением 332 мг указанного в заголовке соединения (выход 88%).
Масс m/e (FAB); 309 (MH+).
Т.пл. 92 - 97oC.
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 0.881 (3H, т, J=7.2 Гц) 1.57-1.94 (5H, м) 2.03-2.21 (2H, м) 2.40-2.47 (1H, м) 2.95 (1H, м как т) 3.32 (1H, ддд, J= 1.6, 4.8, 16.8 Гц) 3.67-3.75 (1H, м) 3.81-3.88 (1H, м) 4.61 (1H, дд, J=4.8, 13.2 Гц) 5.1 (1H, ушир.т, J=6.4 Гц) 5.30 (1H, дд, J=1.6, 8.0 Гц) 6.78 (1H, д, J=5.0 Гц) 7.04 (1H, д, J=5.0 Гц)
Пример B-2
Этил [5S -(5α,8α(R*), 11 αβ ]]-5-[[(S)-2-ацетилтио-1-оксо-3-фенилпропил] амино] -6-оксо-4,5,6,8,9,10,11,11a-октагидропиридо[1,2-a] тиено [3,2-c]азепин-8-карбоксилат
150 мг (0,49 ммоль) соединения, полученного по примеру B-1, растворяют в 12 мл дихлорметана, добавляют при 0oC 120 мг 0,54 ммоль) 2(S)-ацетилтио-3-фенилпропановой кислоты и 144 мг (0,58 ммоль) ЭЭДХ. Полученную таким образом смесь перемешивают при комнатной температуре в течение ночи и затем концентрируют при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат = 3) с получением 168 мг указанного в заголовке соединения (выход 67%) в виде аморфного продукта.
1H-ЯМР (400 МГц, CDCl3) δ: 0.87 (3H, т, J=7.2 Гц) 1.60-1.91 (3H, м) 2.01-2.20 (3H, м) 2.36 (3H, с) 2.36 (3H, с) 2.39-2.48 (1H, м) 2.81 (1H, м как дд) 3.04 (1H, дд, J=7.6, 14.0 Гц) 3.34 (1H, дд, J=7.6, 14.0 Гц) 3.51 (1H, м как дд) 3.68-3.88 (2H, м) 4.33 (1H, т, J=7.6 Гц) 5.19-5.25 (2H, м) 5.50-5.57 (1H, м) 6.75 (1H, д, J=5.2 Гц) 7.04 (1H, д, J=5.2 Гц) 7.21-7.33 (5H, м) 7.50 (1H, ушир.д)
Пример B-3
[5S -(5α,8β (R*), 11 αβ ]]-5-[[(S)-2-Меркапто-1-оксо-3- фенилпропил]амино] -6-оксо-4,5,6,8,9,10,11,11a-октагидропиридо [1,2-a]тиено[3,2-c]азепин-8-карбоновая кислота
К 163 мг (0,32 ммоль) соединения, полученного по примеру B-2, добавляют 12,7 мл дегазированного метанола. Затем добавляют 3,8 мл дегазированного 1 н водного раствора гидроксида натрия. Полученную таким образом смесь перемешивают при 40oC. Семь часов спустя охлаждают до 0oC. К реакционной смеси добавляют 5,7 мл 2 н соляной кислоты. Затем смесь концентрируют до определенного количества при пониженном давлении. Кристаллы, выпавшие при добавлении небольшого количества воды, собирают фильтрацией и сушат над пятиокисью фосфора при пониженном давлении. Таким образом получают 92 мг смеси указанного в заголовке соединения с его эпимерами при соотношении 4 : 3 (выход 60%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1.63 - 2.43 (6H, м) 2.54 - 4.30 (5H, м) 5.16 и 5.24 (общий 1H, каждый м) 5.31 и 5.40 (общий 1H, каждый м) 5.62 и 5.79 (общий 1H, каждый м) 6.73 - 6.78 (общий 1H, каждый м) 6.90 - 7.04 (общий 1H, каждый м) 7.19 - 7.91 (общий 6H, каждый м)
Пример B-4
Этил [5S -(5α,8α (R*), 11 αβ ]]-5-[[(S)-2-ацетилтио-3- метил-1-оксобутил]амино]-6-оксо-4,5,6,8,9,10,11,11a- октагидропиридо[1,2-a]тиено[3,2-c]азепин-8-карбоксилат
170 мг (0,55 ммоль) соединения, полученного по примеру B-1, и (S)-2-ацетилтио-3-метилбутановой кислоты (107 мг, 0,61 ммоль) подвергают взаимодействию тем же способом, что и по примеру B-2. Таким образом получают 203 г указанного в заголовке соединения с его эпимерами (выход 79%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 0.88 и 0.89 (общий 3H, каждый т, каждый J = 7.2 Hz) 1.00 и 1.01 (общий 3H, каждый д, каждый J = 6.8 Hz) 1.05 и 1.06 (общий 3H, каждый д, каждый J = 6.4 Hz) 1.59 - 2.24 (общий 5H, м) 2.32 - 2.48 (общий 2H, м) 2.40 и 2.42 (общий 3H, с) 2.84 - 2.98 (общий 1H, м) 3.49 - 3.58 (общий 1H, м) 3.68 - 3.96 (общий 3H, м) 5.23 - 5.29 (общий 2H, м) 5.58 - 5.66 (общий 1H, м) 6.76 (общий 1H, м) 7.04 (общий 1H, м) 7.52 - 7.59 (общий 1H, м)
Пример B-5
[5S -(5α,8β (R*), 11 αβ ]]-5-[[(S)-2-Меркапто-3- метил-1-оксобутил] амино]-6-оксо-4,5,6,8,9,10,11,11a- октагидропиридо[1,2-a]тиено[3,2-c]азепин-8-карбоновая кислота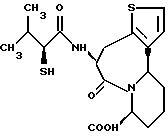
200 мг этил [5S -(5α,8α (R*), 11 αβ ]]-5-[[(S)-2- ацетилтио-3-метил-1-оксобутил] амино] -6-оксо-4,5,6,8,9,10,11,11a- октагидропиридо[1,2-a] тиено[3,2-c] азепин-8-карбоксилата, полученного по примеру B-4, подвергают взаимодействию тем же способом, что и по примеру B-3. Таким образом получают стереоизомерную смесь указанного в заголовке соединения в виде белого твердого продукта (127 мг, 74%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1.01 и 1.06 (общий 6H, м) 1.66 - 2.42 (общий 8H, м) 2.85 - 3.60 (общий 3H, м) 5.19 - 5.24 (общий 1H, м) 5.32 - 5.40 (общий 1H, м) 5.64 - 5.79 (общий 1H, м) 6.74 - 6.79 (общий 1H, м) 7.00 - 7.05 (общий 1H, м) 7.37 - 8.23 (общий 1H, м)
Пример B-6
Этил 5-амино-6-оксо-4,5,6,8,9,10,11,11a- октагидропиридо[1,2-a] тиено[3,2-c]азепин-8-карбоксилат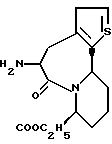
1,28 г (2,92 ммоль) соединения .полученного по синтетическому примеру B-6, подвергают взаимодействию тем же способом, что и по примеру B-1. Таким образом получают 581 мг смеси диастереомеров указанного в заголовке соединения в виде рацемической модификации (65%).
Масс m/e (FAB); 3,09 (MH+)
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ:
0.87 и 1.30 (общий 3H, каждый т, каждый J = 7.2 Гц) 1.60 - 2.48 (общий 8H, м) 2.77 (общий 1H, м как кв) 3.13 - 3.21 (общий 1H, м) 3.71 - 3.91 и 4.24 (общий 2H, каждый м и кв, каждый J = 7.2 Гц) 4.47 и 4.57 (общий 1H, каждый дд. каждый J = 4.8, 12.8 Гц) 4.76 и 5.28 (общий 1H, каждый и дд, каждый J = 5.0 Гц и J = 1.6, 7.6 Гц) 5.43 и 5.49 (общий 1H, каждый ушир. т и ушир. с ) 6.77 - 6.81 (общий 1H, м) 7.07 - 7.11 (общий 1H, м)
Пример B-7
Этил 5-[(S)-2-ацетилтио-1-оксо-3-фенилпропил]-амино-6-оксо- 4,5,6,8,9,10,11,11a-октагидропиридо-[1,2-a]тиено[2,3-c]азепин-8-карбоксилат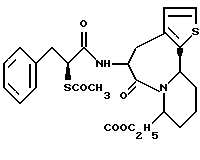
581 мг (1,88 ммоль) соединения, полученного по примеру B-4, и (S)-2-ацетилтио-3-фенилпропановой кислоты (423 мг, 1,88 ммоль) подвергают взаимодействием тем же способом, что и по примеру B-2. После очистки хроматографией на колонке (гексан/этилацетат = 3) на первой фракции получают 232 мг (выход 24%) смеси двух диастереомеров 7:3, затем из последующей фракции получают 324 мг (выход 33%) смеси 1:1 диастереомеров, отличающихся от тех, которые содержатся в первой фракции.
Первая фракция
1H-ЯМР (400 МГц, CDCl3), Me4Si) δ: : 0.86 и 1.29 (общий 3H, каждый т, каждый J= 7.2 Гц) 1.62 - 2.48 (общий 6H, м) 2.34 и 2.36 (общий 3H, каждый с) 2.58 - 2.70 (общий 1H, м как кв) 2.96 - 3.06 (общий 1H, м) 3.30 - 3.42 (общий 2H, м) 3.72 - 3.88 и 4.23 (общий 2H, каждый м и кв, каждый J = 7.2 Гц) 4.28 - 4.35 (общий 1H, м) 4.80 и 5.19 - 5.23 (общий 1H, каждый ушир. т и м 5.34 - 5.54 (общий 2H, м) 6.74 - 6.77 (общий 1H, м) 7.06 - 7.10 (общий 1H, м) 7.20 - 7.47 (общий 6H, м)
Последующая фракция
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 0.85 и 1.29 (общий 3H, каждый т, каждый J = 7.2 Гц) 1.60 - 2.44 (общий 6H, м) 2.34 и 2.40 (общий 3H, каждый с) 2.44 - 3.37 (общий 4H, м) 3.69 - 3.88 и 4.18 - 4.30 (общий 3H, м) 4.78 и 5.22 (общий 1H, каждый ушир. т и м) 5.35 - 5.55 (общий 2H, м) 6.71 (общий 1H, т. J = 5.2 Гц) 7.08 (общий 1H, дд, J = 5.2, 8.4 Гц) 7.21 - 7.36 (общий 6H, м)
Пример B-8
5-[(S)-2-Меркапто-1-оксо-3-фенилпропил]-амино-6-оксо-4,5,6,8,9,10,11,11a- октагидропиридо[1,2-a] тиено[2,3-c]азепин-8-карбоновая кислота
227 мг (0,44 ммоль) соединения, полученного из первой фракции по примеру B-7, подвергают взаимодействию тем же способом, что и по синтетическому примеру B-3. Таким образом полают 143 мг указанного в заголовке соединения, который является смесью двух диастереомеров при соотношении 7:3, в виде белых кристаллов (выход 67%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: : 1.72 - 2.48 (общий 7H, м) 2.64 - 2.78 (общий 1H, м как кв) 3.06 - 3.15 (общий 1H, м) 3.25 - 3.41 (общий 2H, м) 3.58 - 3,65 (общий 1H, м) 4.80 и 5.20 (общий 1H, каждый дд и м как д, каждый J = 3.8, 5.0 Гц) 5.40 - 5.63 (общий 2H, м) 6.71 - 6.79 (общий 1H, м) 7.05 - 7.14 (общий 1H, м) 7.21 - 7.61 (общий 6H, м)
Пример B-9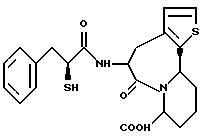
320 мг (0,62 ммоль) соединения, полученного из последующей фракции по примеру B-7, подвергают взаимодействию тем же способом, что и по синтетическому примеру B-3. Таким образом получают 1893 мг указанного в заголовке соединения, который является смесью двух диастереомеров при соотношении 1:1, в виде белых кристаллов (выход 63%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1.68-2.52 (общий 7H, м), 2.64-3.63 (общий 5H, м), 4.76 и 5.17-5.21 (общий 1H, каждый ушир. т и м как ушир. д, каждый J = 4.6 Гц), 5.39-5.63 (общий 2H, м), 6.67 и 6.71 (общий 1H, каждый д и д, каждый J = 5.2 Гц и J = 5.2 Гц), 7.03 и 7.11 (общий 1H, каждый д, и д, каждый J = 5.2 Гц и J = 4.8 Гц), 7.20-7.33 (общий 6H, м).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1.60-2.42 (6H, м), 2.15 (1H, д, J = 9.2 Гц), 2.61 (1H, м как дд, J = 12.8, 16.0 Гц), 3.07 (1H, дд, J = 6.4, 13.6 Гц), 3.24-3.32 (2H, м), 3.45-3.51 (1H, м), 5.21 (1H, дд, J = 2.0, 7.6 Гц), 5.29-5.34 (1H, м), 5.59-5.66 (1H, м), 6.76 (1H, д, J = 5.2 Гц), 7.01 (1H, д, J = 5.2 Гц), 7.20-7.34 (6H, м).
Пример C-1
Метил [3R- [3α,6α(S*),9aβ ]]-6-[[(2S, 3S)-2-ацетилтио-3-метил-1- оксопентил]амино]октагидро-5-оксотиазол[3,2-a]азепин-3-карбоксилат
Раствор 225 мг (0,92 ммоль) метил [3R,[3α,6α (S*), 9a β ]]-6-аминооктагидро-5-оксотиазол[3,2-a] азепин-3-карбоксилата в метиленхлориде (17 мл) охлаждают до 0oC охлаждением льдом. Затем к этому раствору последовательно прибавляют раствор 193 мг (1,01 ммоль) (2S, 3S)-2-ацетилтио-3-метилпентановой кислоты в метиленхлориде (6 мл) и 296 мг (1,20 ммоль) ЭЭДХ. Затем ледяную баню удаляют и полученную смесь перемешивают при комнатной температуре в течение ночи в атмосфере азота. Затем ее концентрируют упариванием при пониженном давлении до определенного объема. Далее этот остаток растворяют в этилацетате. Полученную таким образом смесь последовательно промывают 1 н соляной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и затем сушат над безводным сульфатом магния. Остаток, который получают фильтрованием и концентрированием фильтрата при пониженном давлении очищают хроматографией на колонке (гексан: этилацетат = 3). Таким образом получают 206 мг указанного в заголовке соединения в виде аморфного продукта (выход 54%). 1
1H-ЯМР (400 МГц, CDCl3) δ: 0.88 (3H, т, J = 7.6 Гц), 0.99 (3H, д, J = 6.8 Гц), 1.10-1.22 (1H, м), 1.51-1.70 (2H, м), 1.82-2.14 (6H, м), 2.38 (3H, с), 3.20 (1H, дд, J = 6.4, 11.8 Гц), 3.28 (1H, дд, J = 2.4, 11.8 Гц), 3.79 (3H, с), 3.98 (1H, д, J = 6.8 Гц) 4.54 (1H, дд, J = 6.4, 10.4 Гц), 5.02 (1H, д, J = 8.8 Гц), 5.28 (1H, дд, J = 2.4, 6.4 Гц), 7.41 (1H, д, J = 6.0 Гц).
Пример C-2
Метил [3R [3R,[3α,6α (S*),9a β ]]-6-[[(2S, 3S)-2-ацетилтио-3-метил- 1-оксопентил]амино]-2,2-диметил-5-оксооктагидротиазол[3,2-a]азепин- 3-карбоксилат
Тем же способом, что и по примеру C-1, и исходя из 170 мг (0,62 ммоль) метил [3R, [3α,6α (S*), 9a β ]]-6-амино-2,2- диметил-5-оксо-октагидротиазол[3,2-a] азепин-3-карбоксилата и 131 мг (0,69 ммоль) (2S, 3S)-2-ацетилтио-3-метилпентановой кислоты, полученной по синтетическому примеру C-2, получают 136 мг указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 49%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 0.88 (3H, т, J = 7 Гц) 0.99 (3H, д, J = 7 Гц) 1.10 - 1.21 (1H, м) 1.41 (3H, с) 1.55 (3H, с) 1.50 - 1.62 (2H, м) 1.84 - 2.32 (6H, м) 2.38 (3H, с) 3.79 (3H, с) 3.98 (1H, д, J = 7 Гц) 4.52 - 4.57 (1H, м) 4.77 (1H, с) 5.11 (1H, д, J = 10 Гц) 7.43 (1H, д, J = 6 Гц)
Пример C-3
3-[[(2S, 3S)-2-Ацетилтио-3-метил-1-оксо-пентил]амино]-1- этоксикарбонилметил-2,3,4,5-тетрагидро-1H-[1]бензазепин-2-он
Используя 0,525 г (2,00 ммоль) 3-амино-1-этоксикарбонилметил- 2,3,4,5-тетрагидро-1H-[1] бензазепин-2-она и 0,418 г (2,20 ммоль) (2S, 3S)-2-ацетилтио-3-метилпентановой кислоты, полученной по примеру C-2, повторяют обработку примера C-1. Таким образом получают 0,420 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 48%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 7.31 - 7.00 (5H, м) 4.81 и 4.78 (общий 1H, каждый д, J = 17 Гц) 4.53 - 4.45 (1H, м) 4.33 и 4.31 (общий 1H, каждый д, J = 17 Гц) 4.22 - 4.12 (2H, м) 3.91 и 3.89 (общий 1H, каждый д, J = 7 Гц) 3.44 - 3.33 (1H, м) 2.78 - 2.56 (2H, м) 2.37 (3H, с) 2.07 - 1.87 (2H, м) 1.59 - 1.50 (1H, м) 1.28 - 1.22 (3H, м) 0.96 и 0.95 (общий 3H, каждый д, J = 7 Гц) 0.85 (общий 3H, каждый т, J = 7 Гц)
Пример C-4
(S)-3-[[(2S, 3S)-2-Ацетилтио-3-метил-1-оксопентил] амино]-1- этоксикарбонилметил-2,3,4,5)-тетагидро-1H-[1]бензазепин-2-он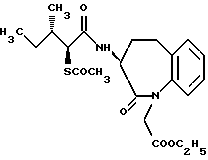
0,550 г (2,10 ммоль) (S)-3-амино-1- этоксикарбонилметил-2,3,4,5-тетрагидро-1H[1] бензазепин-2-она и 0,434 г (2,30 ммоль) (2S, 3S)-2-ацетилтио-3-метилпентановой кислоты подвергают взаимодействию тем же способом, что и по примеру C-1. Таким образом получают 0,614 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 67%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 7.31 - 7.17 (3H, м) 7.12 (1H, дд, J = 8.1 Гц) 7.01 (1H, ушир. д, J = 7 Гц) 4.78 (1H, д, J = 17 Гц) 4.49 (1H, дт, J = 11.8 Гц) 4.33 (1H, д, J = 17 Гц) 4.24 - 4.12 (2H, м) 3.89 (1H, д, J = 7 Гц) 3.38 (1H, м) 2.74 - 2.56 (2H, м) 2.37 (3H, с) 2.04 - 1.87 (2H, м) 1.56 (1H, м) 1.25 (1H, т, J = 6 Гц) 1.14(1H, м) 0.96 (3H, д, J = 7 Гц) 0.86 (3H, т, J = 8 Гц)
Пример C-5
(R)-3-[[(2S, 3S)-2-Ацетилтио-3-метил-1-оксопентил] амино]-5- этоксикарбонилметил-2,3-дигидро-1,5-бензотиазепин-4(5H)-он
0,208 г (2,74 ммоль) (P)-3-амино-5-этоксикарбонилметил-2,3-дигидро-1,5-бензотиазепин- 4(5H)-она и 0,166 г (0,872 ммоль) (2S, 3S)-2-ацетилтио-3-метилпентановой кислоты, полученной по примеру C-2, подвергают взаимодействию тем же способом, что и по примеру C-1. Таким образом получают 0,200 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 60%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 7.64 (1H, дд, J = 8.2 Гц) 7.43 (1H, дт, J = 8.2 Гц) 7.33 (1H, дд, J = 8.2 Гц) 7.25 (1H, дт, J = 8.2 Гц) 7.08 (1H, ушир. д, J = 7 Гц) 4.81 (1H, д, J = 17 Гц) 4.67 (1H, дт, J = 11.7 Гц) 4.25 (2H, д, J = 7 Гц) 4.15 (1H, д, J = 17 Гц) 3.87 (1H, д, J = 8 Гц) 3.83 (1H, дт, J = 11.7 Гц) 2.77 (1H, т, J = 11 Гц) 2.37 (3H, с) 2.00 (1H, м) 1.54 (1H, м) 1.29 (3H, т, J = 7 Гц) 1.33 (1H, м) 0.94 (3H, д, J = 7 Гц) 0.85 (3H, т, J = 7 Гц)
Пример C-6
Дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-[[(2S, 3S)- -2-ацетилтио-3-метил-1-оксопентил] амино] -6-оксо-11-фенил-1,2, 3,4,6,7,8,12b-октагидропиридо[2,1-a][2]-бензазепин-4-карбоксилат
Тем же способом, что и по примеру C-1, и исходя из 1,23 г (2,38 ммоль) дифенилметил [4S -(4α,7α (R*)-12b β ]]-7-амино-6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2]- бензазепин-4-карбоксилата, полученного по синтетическому примеру C-9, и 0,52 г (2,74 ммоль) (2S, 3S)-2-ацетилтио-3-метилпентановой кислоты, полученной по синтетическому примеру C-2, получают 1,22 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 74%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.55-6.91 (17H, м), 6.67 (1H, д, J = 8 Гц) 6.27 (1H, с) 5.65 (1H, квинт , J = 6 Гц) 5.47 (1H, как д) 5.41 (1H, как д) 4.05 (1H, д, J = 7 Гц) 3.42 (1H, дд, J = 16,6 Гц) 2.61-2.40 (2H, v) 2.14 (1H, м) 2.00 (1H, м) 1.92=1,58 (5H, м) 1.24 (1H, м) 1.05 (3H, д, 7 Гц) 0.94 (3H, т, J = 7 Гц)
Пример C-7
Метил [4S, (4α,7α (R*), 12b β ]]-11-метилсульфонил-амино-7-[[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил] -амино] -6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат
140 мг (0,367 ммоль) метил [4S -(4α,7α (R*), 12bβ ]-11-метилсульфониламино-7-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо-[2,1-a] [2] бензазепин-4-карбоксилата, полученного по синтетическому примеру C-14, и 77 мг (0,405 ммоль) (2S, 3S)-2-ацетилтил-3-метилпентановой кислоты растворяют в 10 мл метиленхлорид и 10 мл этанола. К этому раствору добавляют 118 мг (0,477 ммоль) N-этоксикарбонил-2-этокси-1,2-дигидрохинолина (ЭЭДХ) при комнатной температуре. Полученную таким образом смесь перемешивают в течение 19 часов в атмосфере азота и концентрируют при пониженном давлении. К остатку прибавляют 1 н соляную кислоту с последующей экстракцией дихлорметаном. Органическую фазу промывают насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и затем концентрируют при пониженном давлении. Полученный таким образом остаток очищают хроматографией на колонке с силикагелем (2:98, этанол:дихлорметан) с получением таким образом 198 мг (выход 98%) указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 0.92 (3H, т, J = 8 Гц) 1.04 (3H, д, J = 7 Гц) 1.10-1.15 (2H, м) 1.60-2.12 (6H, м) 2.39 (3H, м) 2.41 (3H, с) 2.81 (1H, дд, J = 17.2, 12.8Гц) 2.93 (3H, с) 3.09 (3H, с) 3.48 (1H, дд, J = 17.2, 5.9 Гц) 4.03 (1H, в, J = 7 Гц) 5.26 (1H, м) 5.36 (1H, м) 5.68 (1H, м) 6.94-7.68 (5H, м)
Пример C-8 [3R, [3α,6α, (S*, 9a β ]]-6-[[(2S, 3S)-3-Метил-1-оксо-2-тиопентил]амино]октагидро-5-оксотиазол[3,2-a]азепин-3- карбоновая кислота
200 мг (0,48 ммоль) метил[3R, [3α,6α (S*0, 9a β ]]-6-[[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил] амино] октагидро-5-оксотиазол [3,2-a]азепин-3-карбоксилата, полученного по примеру C - 1, помещают в колбу и добавляют 8 мл дегазированного этанола с последующим охлаждением до 0oC в атмосфере азота. Добавляют 3,8 мл дегазированного 1 н водного раствора гидроксида лития и полученную смесь перемешивают при комнатной температуре в течение 50 минут. Полученную таким образом реакционную смесь подкисляют добавлением 2,9 мл 2 н водного раствора соляной кислоты при 0oC и затем экстрагируют дихлорметаном. Затем органическую фазу промывают насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и концентрируют. Полученный осадок перекристаллизовывают из гександихлорметана. Таким образом получают 150 мг указанного в заголовке соединения в виде белых кристаллов (87%).
1H-ЯМР (400 МГц, CDCl3) δ: 0.90 (3H, т, J = 7 Гц) 1.00 (3H, д, J = 7 Гц) 1.24 (1H, м) 1.55 - 1.74 (2H, м) 1.87 (1H, д, J = 8 Гц) 1.90 - 2.10 (6H, м) 3.20 (1H, дд, J = 6, 12 Гц) 3.24 (1H, д, J = 7 Гц) 3.36 (1H, д, J = 2, 12 Гц) 4.62 (1H, дд, J = 6, 10 Гц) 5.07 (1H, как т, J = 6 Гц) 5.29 (1H, дд, J = 2, 6 Гц) 7.69 (1H, д, J = 6 Гц)
Пример C-9
[3R -[3α,6α (S*), 9a β ]]-6-[[(2S, 3S)-3-Метил-1-оксо-2-тиопентил]амино] -2,2-диметил-5-оксооктагидротиазол[3,2-a] азепин-3-карбоновая кислота
130 мг (0,29 ммоль) метил [3R, [3α,6α S*)-9a β ]]-6-[[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил]амино]-2,2-диметил-5- оксооктагидротиазол[3,2-a] азепин-3-карбоксилата, полученного по примеру C-2, помещают в колбу и добавляют 5,8 мл дегазированного метанола. К полученной смеси добавляют дегазированный 1 н водного раствора гидроксида натрия (2,3-мл) в атмосфере азота. Полученную смесь перемешивают при 45oC в течение 8 часов. К реакционной смеси добавляют 1,8 мл 2 н соляной кислоты и концентрируют. К концентрату прибавляют воду (50 мл). Выпавшие при этом кристаллы собирают фильтрацией и сушат на воздухе некоторое время. Таким образом получают 80 мг указанного в заголовке соединения (выход 71%).
1H-ЯМР (400 МГц, CDCl3) δ: 0.90 (3H, т, J=7 Гц) 1.01 (3H, д, J=7 Гц) 1.17-1.29 (1H, м) 1.53 (3H, с) 1.56 (3H, с) 1.52-1.68 (2H, м) 1.86 (1H, д, J= 9 Гц) 1.88-2.28 (6H, м) 3.27 (1H, дд, J=6, 9 Гц) 4.58-4.66 (1H, м) 4.79 (1H, с) 5.15 (1H, д, J=10 Гц) 7.84 (1H, д, J=6 Гц)
Пример C-10
1-Карбоксиметил-3-[[(2S, 3S)-3-метил-1-оксо-2-тиопентил] амино] -2,3,4,5-тетрагидро-1H- [1]бензазепин-2-он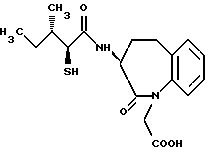
К перемешиваемому раствору 0,385 г (0,89 ммоль) 3-[(2S, 3S)-ацетилтио-3-метил-1-оксопентил] амино] -1-этоксикарбонилметил-1H- [1]бензазепин-2-ону, полученному по примеру C-3, и 15 мл дегазированного этанола добавляют дегазированный 1 н водный раствор гидроксида натрия при перемешивании при 0oC в атмосфере азота. Полученную таким образом смесь перемешивают 1 час при комнатной температуре. Реакционную смесь охлаждают, подкисляют добавлением 1 н соляной кислотой, экстрагируют этилацетатом. Органическую фазу промывают водой и сушат над безводным сульфатом магния. После отгонки растворителя из органической фазы получают 0,34 г указанного в заголовке соединения в виде бесцветного аморфного продукта (количественный выход).
1H-ЯМР (400 МГц. CDCl3) δ: 7.39-7.14(5H, м) 4.7-4.71 (общий 1H, каждый д, J= 17 Гц) 4.57-4.50 (1H, м) 4.44-4.43 (общий 1H, каждый д, J=17 Гц) 3.34-3.10 (2H, м) 2.77-2.58 (2H, м) 2.03-1.87 (2H, м) 1.85-1.84 (общий 1H, каждый д, J=9 Гц) 1.64-1.50 (1H, м) 1.22-1.15 (1H, м) 0.95 (3H, д, J=7 Гц) 0.86 (3H, т, J=7 Гц)
Пример C-11
(S)-1-Карбоксиметил-3-[[(2-S, 3S)-3-метил-1-оксо-2-тиопентил] амино]-2,3,4,5-тетрагидро-1H- [1]бензазепин-2-он
0,600 г (1,38 ммоль) (S)-3-[[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил] амино] -1-этоксикарбонилметил- 2,3,4,5-тетрагидро-1H-[1] бензазепин-2-она, полученного по примеру C-4, гидролизуют тем же способом, что и по примеру C-10. Таким образом получают 0,490 г указанного в заголовке соединения в виде бесцветного аморфного продукта (выход 97%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.40(1H, ушир.д, J=7 Гц) 7.33-7.14(4H, м) 4.71 (1H, д, J=17 Гц) 4.54 (1H, дт, J=11, 7Гц) 4.44 (1H, д, J=17 Гц) 3.29 (1H, м) 3.17 (1H, дд, J=9, 7 Гц) 2.74-2.59 (2H, м) 2.04-1.89 (2H, м) 1.84 (1H, д, J=9 Гц) 1.55 (1H, м) 1.17 (1H, м) 0.95 (3H, д, J=7 Гц) 0.86 (3H, т, J=7 Гц)
Пример C-12
(R)-3-[[(2S, 3S)-3-Mетил-1-оксо-2-тиопентил] -амино]-5- карбоксиметил-2,3-дигидро-1,5-бензотиазепин-4(5H)-он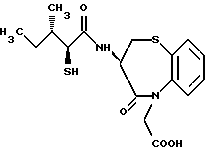
0,187 г (0,43 ммоль) (R)-3-[[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил] амино] -5-этоксикарбонилметил- 2,3-дигидро-1,5-бензодиазепин-4(5H)-она, полученного по примеру C-5, подвергабют обработке тем же способом, что и по примеру C-10. Таким образом получают 126 мг указанного в заголовке соединения в виде белых кристаллов (выход 77%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.67 (1H, дд, J=8.1 Гц) 7.53 (1H, д, J=7 Гц) 7.46 (1H, дт, J=8, 2 Гц) 7.36 (1H, дт, J=8,2 Гц) 7.29 (1H, дт, J=8, 1 Гц) 4.91 (1H, д, J=18 Гц) 4.72 (1H, дт, J=11,7 Гц) 4.16 (1H, д, J=18 Гц) 3.83 (1H, дд, J= 11, 7 Гц) 3.19 (1H, дд, J=9, 6 Гц) 2.88 (1H, т, J=11 Гц) 1.94 (1H, м) 1.85 (1H, д, J=9 Гц) 1.54 (1H, м) 1.20 (1H, м) 0.95 (3H, д, J=7 Гц) 0.86 (3H, т, J=7 Гц) 0.95 (3H, д, J=7 Гц) 0.86 (3H, т, J=7 Гц)
Пример C-13
[4S, (4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-2-Ацетилтио-3-метил-1-оксопентил] амино] -6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота
В перемешиваемый раствор 1,22 г (1,773 ммоль) дифенилметил [4S, (4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-2-ацетилтио-3-метил-1- оксопентил]амино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2] бензазепин-4-карбоксилата, полученного по примеру C-5, и 1,92 мл анизола прибавляют по каплям 11,01 мл трифторметансульфоновой кислоты при 0oC. После перемешивания реакционной смеси при 0oC в течение 40 минут ее концентрируют при температуре, не превышающей 40oC. Полученное масло дважды подвергают азеотропной отгонке с толуолом. Полученное масло очищают хроматографией на колонке с силикагелем (элюент - хлороформ : гексан = 4:1 и хлороформ : метанол = 98,5: 1,5, последовательно). Таким образом получают указанное в заголовке соединение в виде бесцветного аморфного продукта (0,897 г, выход 97%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.52-7.31 (8H, м), 7.04 (1H, д, J = 8 Гц) 5.69 (1H, квинт., J = 6 Гц), 5.48 (1H, м), 5.18 (1H, м), 4.02 (1H, д, J = 7 Гц) 3.54 (1H, м), 2.86 (1H, дд, J = 16, 12 Гц), 2.51 (1H, м), 2.40 (3H, с), 2,28 (1H, м), 2.11 (1H, м), 2.04-1.56 (5H, м), 1.20 (1H, м), 1,02 (3H, д, J = 7 Гц), 0,91 (3H, т., J = 7 гц)
Пример C-14
[4S -(4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-3-Метил-1-оксо-2- тиопентил]амино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2] бензазепин-4-карбоновая кислота
0,780 г (1,492 ммоль) [4S, (4α,7α (R*), 12b β ]]-7- [[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил] амино] -6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновой кислоты, полученной по примеру C-13, растворяют в 20 мл дегазированного этанола и при 0oC добавляют 4,48 мл дегазированного 1н водного раствора гидроксида лития. Раствор смеси перемешивают в атмосфере азота в течение 40 минут.
Полученную реакционную смесь подкисляют добавлением 20,0 мл воды и 2н водного раствора соляной кислоты. Твердый белый продукт, выпавший при этом, собирают фильтрацией и промывают водой. Таким образом получают 0,622 г указанного в заголовке соединения (выход 87%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.66 (1H, д, J = 7 Гц) 7.53-7.32 (7H, м) 7.08 (1H, д, J = 8 Гц) 5.72 (1H, квинт., J = 6 Гц) 5.52 (1H, м), 5.25 (1H, м) 3.60 (1H, дд, J = 17.6 Гц), 3.23 (1H, дд, J = 9, 7 Гц), 2.93 (1H, дд, J = 17, 13 Гц), 2.55 (1H, м), 2.34 (1H, м), 2.00 (2H, м) 1.92 (1H, д, J = 8 Гц), 1.98-1.61 (4H, м), 1.25 (1H, м) 1.03 (3H, д, J = 7 Гц), 0.93 (3H, т, J = 7 Гц)
Пример C-15
[4S -(4α,7α (R*), 12b β ]]-11-Метилсульфониламино-7- [[(2S, 3S)-3-метил-1-оксо-2-тиопентил] амино] -6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота
198 мг (0,158 ммоль) метил [4S, (4α,7α (R*), 12b β ]]-11- метилсульфониламино-7-[[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил] амино] - 6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4- карбоксилата, полученного по примеру C-7, помещают в колбу с последующей достаточной продувкой азотом. Затем добавляют 5 мл дегазированного этанола и колбу охлаждают на ледяной бане. Затем прибавляют 3,6 мл дегазированного 1 н водного раствора гидроксида натрия. После отстранения колбы с ледяной бани ее медленно подогревают до комнатной температуры и продолжают перемешивание в течение 1 часа и 40 минут. К реакционной системе добавляют 10 мл 1 н водного раствора соляной кислоты и затем экстрагируют дихлорметаном. Органическую фазу сушат над безводным сульфатом магния. Органическую фазу концентрируют при пониженном давлении и остаток кристаллизуют из дихлорметана. Таким образом получают 84 мг указанного в заголовке соединения (выход 47%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 0.93 (3H, т, J = 8 Гц), 1.04 (3H, д, J = 7 Гц) 1.22-1.35 (2H, м), 1.65-2.10 (6H, м), 2.41 (2H, м) 2.90 (1H, м), 2.91 (3H, с), 3.23 (1H, д, J = 8 Гц) 3.56 (1H, дд, J = 17.3, 6.1 Гц), 5.23 (1H, м), 5.48 (1H, м), 5.71 (1H, м), 7.01-7.16 (3H, м) 7.82 (1H, д, J = 6.6 Гц)
Пример D-1
Получение метил [4S -(4α,7α (R*), 12b β ]]-11- метилсульфониламино-7-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-6-оксо- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоксилата
Метил [4S -[4α,7α (R*), 12b β ]]-11-амино-7-(1,3-диоксо- 1,3-дигидроизоиндол-2-ил)-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат (1,50 г, 3,5 ммоль), полученный по вышеприведенному синтетическому примеру D-3, растворяют в метиленхлориде (50 мл). Затем к этому раствору прибавляют пиридин (3 мл) и метансульфонилхлорид (440 мг, 3,8 ммоль) при охлаждении льдом. Полученную смесь перемешивают в атмосфере азота в течение 2 часов при комнатной температуре. Затем к перемешиваемому раствору прибавляют 1 н водный раствор соляной кислоты (100 мл) при охлаждении льдом с последующей экстракцией метиленхлоридом. Метиленхлоридную фазу сушат (используют MgSO4) и затем концентрируют при пониженном давлении. Далее осадок очищают хроматографией на колонке с силикагелем (3: 1 метиленхлорид/этилацетат) с получением таким образом указанного в заголовке соединения (1,14 г, 64%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1.60-2.46 (6H, м), 3.00 (3H, с), 3.23 (3H, с), 3.42 (1H, дд, J = 17.1, 7.0 Гц), 4.46 (1H, дд, J = 17.1, 11.9 Гц), 5.21 (1H, м), 5.44 (1H, м), 6.04 (1H, дд, J = 11.9, 7.0 Гц), 6.65 (1H, с), 7.05 (1H, дд, J = 8.2, 2.2 Гц), 7.19 (1H, д, J = 8.2 Гц), 7.24 (1H, д, J = 2.2 Гц), 7.74-7.90 (4H, м).
Пример D-2
Метил [4S -(4α,7α (R*),12b β ]]-11-метилсульфониламино- 7-амино-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин- 4-карбоксилат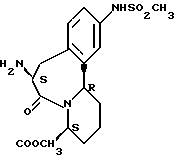
Метил [4S-(4α,7αR*), 12b β ]]-11-метилсульфониламино-7-(1,3- диоксо-1,3-дигидроизоиндол-2-ил)-6-оксо-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилат (1,14 г, 2,23 ммоль), полученный по вышеприведенному примеру S-1, растворяют в метаноле (49 мл). Затем к этому раствору прибавляют гидразин гидрат (123 мг, 2,46 ммоль). Полученную смесь перемешивают при комнатной температуре в атмосфере азота в течение 66 часов. Перемешиваемый раствор концентрируют при пониженном давлении. Далее к концентрату прибавляют метиленхлорид и нерастворимый материал удаляют фильтрацией. Затем к фильтрату прибавляют этилацетат. Таким образом получают указанное в заголовке соединение в виде белых кристаллов (0,50 г, 59%).
1H-ЯМР (400 МГц, CDCl3/CD3OD, Me4Si) δ: 1.60-2.45 (6H, м), 2.87 (1H, дд, J = 17.6, 12.7 Гц), 2.94 (3H, с), 3.13 (3H, с), 3.40 (1H, дд, J = 17.6, 6.0 Гц), 4.65 (1H, дд, J = 12.7, 6.0 Гц), 5.30 (1H, м), 5.43 (1H, м), 7.02 (1H, дд, J = 8.2, 2.2 Гц), 7.11 (1H, д, J = 8.2 Гц), 7.16 (1H, д, J = 2.4 Гц).
Пример D-3
Метил [4S -(4α,7α R*), 12b β ]]-11-метилсульфониламино- 7-[[(S)-2-ацетилтио-3-фенил-1-оксопропил] -амино] -6-оксо- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат
Метил [4S -(4α,7α R*), 12b β ]]-11-метилсульфониламино-7- амино-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин- 4-карбоксилат (310 мг, 0,81 ммоль) полученный по вышеприведенному примеру D-2, и (S)-ацетилтио-3-фенилпропионовой кислоты (183 мг, 0,81 ммоль) растворяют в метиленхлориде (16 мл) и тетрагидрофуране (32 мл). Затем к этому раствору прибавляют N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (ЭЭДХ, 221 мг, 0,89 ммоль). Затем полученную таким образом смесь перемешивают в атмосфере азота в течение 20 часов и перемешиваемый раствор концентрируют при пониженном давлении. Далее к концентрату прибавляют 1 н водный раствор соляной кислоты с последующей экстракцией метиленхлоридом. Затем после промывки органической фазы 1 н раствором соляной кислоты, водой и насыщенным водным раствором хлорида натрия его сушат (используют MgSO4) и концентрируют при пониженном давлении. Полученный осадок очищают хроматографией на колонке с силикагелем (1:1 гексан/этилацетат) с получением таким образом указанного в заголовке соединения (240 мг, 50%).
1H-ЯМР (400 МГц, CDCl3, Me4Si) δ: 1.66-2.40 (6H, м), 2.36 (3H, с), 2.72 (1H, дд, J = 17.4, 12.4 Гц), 2.93 (3H, с), 3.06 (1H, дд, J = 14.1, 7.9 Гц), 3.10 (3H, с), 3.35 (1H, дд, J = 14.1, 7.1 Гц), 3.46 (1H, м), 4.36 (1H, т, J = 7.4 Гц), 5.23 (1H, м), 5.33 (1H, м), 5.58 (1H, м), 6.93-7.56 (10H, м).
Пример D-4
Метил [4S -(4α,7α R*), 12b β ]]-11-метилсульфониламино- 7-[[(S)-2-ацетилтио-3-(4-метоксифенил)-1-оксопропил] амино] -6-оксо- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат
Метил [4S [(4α,7α R*), 12b β ]]-11-метилсульфониламино- 7-амино-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин- 4-карбоксилат (188 мг, 0,49 ммоль), полученный по вышеприведенному примеру D-2, и (S)-ацетилтио-3-(4-метоксифенил)пропионовой кислоты (125 мг, 0,49 моль) растворяют в метиленхлориде (10 мл), тетрагидрофуране (20 мл) и этаноле (40 мл). Затем к этому раствору прибавляют 405 мг (1,64 ммоль) ЭЭДХ при комнатной температуре. Затем полученную таким образом смесь перемешивают в атмосфере азота в течение 5 часов и перемешиваемый раствор концентрируют при пониженном давлении. Далее к концентрату прибавляют 1 н водный раствор соляной кислоты с последующей экстракцией метиленхлоридом. Затем органическую фазу промывают 1 н раствором соляной кислоты, водой и насыщенным водным раствором хлорида натрия и затем сушат (используют MgSO4) и концентрируют при пониженном давлении. Полученный осадок очищают хроматографией на колонке с силикагелем (1: 1 гексан/этилацетат) с получением таким образом указанного в заголовке соединения (133 мг, 44%).
1H-ЯМР (400 МГц, CDCl3, Me4Si): δ: 1.66 - 2.05 (6H, м), 2.37 (3H, c). 2.72 (1H, дд, J = 17.3, 12.7 Гц), 2.94 (3H, с), 3,00 (1H, дд J = 14,3, 7,7 Гц), 3.11 (3H, с), 3.28 (1H, дд, J = 14.3, 7.7 Гц), 3.48 (1H, дд, J = 17.3, 5.7 Гц), 3.79 (3H, с), 4.30 (1H, т, J = 7.7 Гц), 5.23 (1H, ушир, д), 5.33 (1H, ушир, д), 5.57 (1H, квинт, J = 6.2 Гц), 6,83 (2H, д, J = 8.7 Гц), 6.97 (1H, д, J = 8.2 Гц), 7.01 (1H, дд, J = 8.2, 2 Гц), 7.24 (1H, с), 7.16 (2H, д, J = 8.7 Гц), 7.50 (1H, д, J = 6.2 Гц)
Пример D-5
[4S( (4α,7α (R*), 12b β ]-11-Метилсульфониламино- 7-[[(S)-2-меркапто-3-фенил-1-оксопропил]амино]-6-оксо- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-а] [2]бензазепин-4-карбоновая кислота
Метил [4S -(4α,7α (R*), 12b β ]]-11-метилсульфониламино- 7-[[(S)-2-ацетилтио-3-фенил-1-оксопропил] амино]-6-оксо- 1,2,3,4,6,7,8,12b - октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат (228 мг, 0,39 ммоль), полученный по вышепредставленному примеру D-3, помещают в колбу с последующей достаточной продувкой азотом. Затем в колбу прибавляют дегазированный тетрагидрофуран (1 мл) и метанол (6,2 мл) колбу охлаждают на ледяной бане. К полученному раствору прибавляют дегазированный 1 н раствор гидроксида лития (3,3 мл). После удаления ледяной бани колбу медленно подогревают до комнатной температуры и полученную смесь перемешивают в течение 5 часов. Далее перемешиваемый раствор концентрируют при пониженном давлении и концентрат экстрагируют дихлорметаном. Затем водную фазу отделяют и устанавливают pH до 1 с помощью 1 н водного раствора соляной кислоты. Концентрат экстрагируют метиленхлоридом. Затем органическую фазу сушат (используют MgSO4) и потом концентрируют при пониженном давлении. К концентрату прибавляют диизопропиловый эфир и далее растирают. Таким образом получают указанное в заголовке соединение (110 мг, 53%).
1H-ЯМР (400 МГц, CDCl3/CD3OD, Me4Si) δ: 1.70 - 2.50 (6H, м), 2.85 (1H, дд, J = 17.4, 12.7 Гц), 2.90 (3H, с), 3.12 (1H, дд, J = 13.8, 7.5 Гц), 3.29 (1H, дд, J = 13.8, 6.6 Гц), 3.52 (1H, м), 3.67 (1H, м), 5.19 (1H, м), 5.47 (1H, м), 5,65 (1H, м), 7.03 - 7.80 (10 H, м)
Пример D-6
[4S-(4 α,7α (R*), 12b β ]]-11-Метилсульфониламино-7- [[(S)-2-меркапто-3-(4-метоксифенил)-1-оксопропил] амино] -6-оксо- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]-бензазепин- 4-карбоновая кислота
Метил [4S, (4α,7α (R*), 12b β ]]-11-метилсульфониламино- 7-[[(S)-2-ацетилтио-3-(4-метоксифенил)-1-оксопропил]-амино]-6-оксо- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-а][2]бензазепин-4-карбоксилат (133 мг, 0,22 ммоль), полученный по вышепредставленному примеру D-4, помещают в колбу с последующей достаточной продувкой азотом. Затем в колбу прибавляют дегазированный этанол (20 мл) и затем дегазированный 1 н водный раствор гидроксида натрия (5 мл). Полученную смесь перемешивают при комнатной температуре в течение 3 часов. Далее к перемешиваемому раствору прибавляют 1 н водный раствор соляной кислоты (10 мл), концентрируют с последующим концентрированием при пониженном давлении. К концентрату прибавляют метиленхлорид и воду с последующей экстракцией метиленхлоридом. Затем органическую фазу отделяют и сушат (используют MgSO4) и концентрируют при пониженном давлении с получением таким образом указанного в заголовке соединения (80 мг, 65%).
1H-ЯМР (400 МГц, CDCL3/CD3OD, Me4Si) δ: 1.74 - 1.86 (3H, м), 1.92 - 2.07 (1H, м), 2.37 - 2.49 (2H, м), 2.83 (3H, с), 2.83 (1H, м), 3.11 (1H, дд, J = 14.0, 6.9 Гц), 3.23 (1H, дд, J = 13.8, 6.5 Гц), 3.55 - 3.66 (2H, м), 3.80 (3H, с), 5.26 (1H, ушир.д), 5.43 (1H, ушир,д), 5,62 (1H, квинт, J = 6.0 Гц), 6,57(1H, д, J = 6,1 Гц), 6.86 (2H, д, J = 8.7 Гц), 6.96 (1H, д, J = 6.1 Гц), 7.13 - 7.19 (3H, м), 7.54 (1H, с), 7.65 (1H, д, J = 6.2 Гц).
Пример Е-1
Дифенилметил [4S -(4α,7α (R*), 12b β ] -7-амино-6-оксо-11- фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a][2]-бензазепин-4-карбоксилат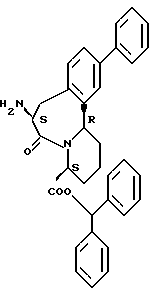
Дифенилметил [4S -(4α,7α (R* ), 12b β ]]-7-(1,3-дигидро- 1,3-диоксо-2H-изоиндол-2-ил)-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилат (2,03 г, 3,14 ммоль), полученный по синтетическому примеру E-6, растворяют в растворе смеси метанола (40 мл) с тетрагидрофураном (ТГФ, 20 мл), с последующим прибавлением моногидрата гидразина (0,34 мл, 7,10 ммоль). Полученную таким образом смесь нагревают с обратным холодильником в течение 3 часов. Реакционную смесь концентрируют, к остатку прибавляют CH2Cl2 и нерастворимый материал удаляют фильтрацией. Фильтрат концентрируют и липкий остаток очищают хроматографией на колонке с силикагелем (элюент - CHCl3 : MeOH : водный аммиак (NH4OH) = 98 : 2 : 0,2). Таким образом получают указанное в заголовке соединение в виде бесцветного аморфного продукта (1,20 г, выход 74%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.40 (4H, м), 7.31 (1H, тт, J = 7,2 Гц), 7.24 (1H, д, J = 2 Гц), 7.15 (1H, дд, J = 8,2 Гц), 6,99 (2H, дд, J = 8,4 Гц), 6.87 (2H, дд, J = 8,2 Гц), 6.3 (1H, д, J = 8 Гц), 6.20 (1H, с), 5.42 - 5.33 (2H, м), 4.53 (1H, дд, J = 10,6 Гц), 3.17 (1H, дд, J = 16,6 Гц), 2.58 (1H, дд, J = 16,10 Гц), 2,40 (2H, м), 1.94 (1H, м), 1.85 - 1.58 (3H, м)
Пример E-2
Дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-[[(s)-2- ацетилтио-3-фенилпропиониламино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2] бензазепин-4-карбоксилат
Дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-амино-6- оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2]бензазепин-4-карбоксилат (0,59 г, 1,14 ммоль), полученный по примеру E-1, и (S)-2-ацетилтио-3-фенилпропионовую кислоту (0,27 г, 1,20 ммоль) растворяют в CH2Cl2 (30 мл) и добавляют ЭЭДХ (0,37 г, 1,48 ммоль). Раствор смеси перемешивают при комнатной температуре в течение ночи. Реакционную смесь распределяют между CH2Cl2 и водой и CH2Cl2 фазу промывают водой, насыщенным водным раствором гидрокарбоната натрия с насыщенным водным раствором хлорида натрия. CH2Cl2 фазу сушат над сульфатом магния. Таким образом получают указанное в заголовке соединение в виде бесцветного аморфного продукта (0,89 г, выход 109%). Этот продукт не очищают, но используют в последующей стадии как таковой.
1H-ЯМР (400 МГц, CDCl3) δ: 7.52 - 7.41 (4H, м), 7.40 - 7.12 (15H, м), 7.04 (2H, дд, J = 8.4 Гц), 6.93 (2H, дд, J = 8.2 Гц), 6.67 (1H, д, J = 8 Гц), 6.26 (1H, с), 5.59 (1H, квинт, J = 6 Гц), 5.44 (1H, м), 5.38 (1H, д, J = 6 Гц), 4.39 (1H, т, J = 7 Гц), 3.41 (1H, дд, J = 16,6 Гц), 3.36 (1H, дд, J = 14,7 Гц), 3.07 (1H, дд, J = 14,7 Гц), 2.54 (1H, дд, J = 16,10 Гц), 2.47 (2H, м), 2.40 (3H, с), 2.00 (1H, м), 1.87 - 1.70 (3H, м)
Пример E-3
Дифенилметил [4S -(4α,7α (R*), 12bβ ]]-7-[[(S)-2-ацетилтио-3- (4-метоксифенил)пропиониламино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a]-[2]бензазепин-4-карбоксилат
Тем же способом, что и по примеру E-2, и исходя из дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-амино-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] -[2]бензазепин-4-карбоксилата (0,59 г, 1,14 ммоль), полученный по примеру E-1, и (S)-2-ацетилтио-3-(4-метоксифенил)пропионовой кислоты (0,31 г, 1,20 ммоль) получают указанное в заголовке соединение в виде бесцветного аморфного продукта (0,81 г, выход 95%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.42 - 7.34 (4H, м), 7.31 (1H, м), 7.24 - 7.04 (10H, м), 7,42 - 7.34 (4H, м), 7.31 (1H, м), 7.24 - 7.04 (10H, м), 6.96 (2H, дд, J = 8,4 Гц), 6.86 (2H, дд, J = 8,2 Гц), 6.77 (2H, д, J = 8 Гц), 6.59 (1H, д, J = 8 Гц), 6.19 (1H, с), 5.51 (1H, квинт, J = 6 Гц), 5.37 (1H, м), 5.31 (1H, д, J = 6 Гц), 4.31 (1H, т, J = 7 Гц), 3.72 (3H, с), 3.34 (1H, дд, J = 16,6 Гц), 3.20 (1H, дд, J = 14,7 Гц), 2.94 (1H, дд, J = 14,7 Гц), 2.47 (1H, дд, J = 16,10 Гц), 2.40 (2H, м), 2.33 (3H, с), 1.92 (1H, м), 1.81 - 1.62 (3H, м)
Пример E-4
[4S -(4α,7α (R*), 12b β ]]-7-[[(S)-2-Ацетилтио-3- фенилпропиониламино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] -[2]бензазепин-4-карбоновая кислота
К перемешиваемому раствору дифенилметил [4S-(4α,7α(R*), 12b β ]]-7-[[(S)-2-ацетилтио-3-фенилпропиониламино]- 6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] -[2] бензазепин-4-карбоксилата (0,89 г, около 1,14 моль), полученного по примеру E-2, с анизолом (1,24 мл) прибавляют по каплям трифторметансульфоновую кислоту (ТФК, 7,08 мл) при 0oC. После перемешивания реакционной смеси при 0oC в течение 20 минут ее концентрируют при температуре не превышающей 40oC. Полученное масло дважды подвергают азеотропной отгонке с бензолом. Полученное масло очищают хроматографией на колонке с силикагелем (элюент - CH2Cl2 : Hex = 1 : 2 и CHCl3 : MeOH = 99 : 1, последовательно). Таким образом получают указанное в заголовке соединение в виде бесцветного аморфного продукта (0,64 г, выход 100%, 2 стадии из примера E-1).
1H-ЯМР (400 МГц, CDCl3) δ: 7.38 - 7.14 (12H, м), 7.00 (1H, д, J = 8 Гц), 5.57 (1H, квинт, J = 6 Гц), 5.41 (1H, м), 5.14 (1H, д, J = 6 Гц), 4.29 (1H, т, J = 7 Гц), 3.51 (1H, дд, J = 16,6 Гц), 3.28 (1H, дд, J = 14,7 Гц), 2.99 (1H, дд, J = 14,7 Гц), 2.73 (1H, дд, J = 16,10 Гц), 2.46 (1H, м), 2.29 (3H, с), 2.26 (1H, м), 2.00 - 1.60 (4H, м)
Пример E-5
[4S -(4α,7α (R*), 12b β ]]-7-(S)-2-Ацетилтио-3-(4-метоксифенил)пропиониламино] -6-оксо-11- фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4- карбоновая кислота
Тем же способом, что и по примеру E-4, и исходя из дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-ацетилтио-3-(4-метоксифенил)пропиониламино] -6-оксо-11- фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2]бензазепин-4- карбоксилата (0,81 г, 1,08 ммоль), полученного по примеру E-3, получают указанное в заголовке соединение в виде бесцветного аморфного продукта (0,52 г, выход 81%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.45 - 7.32 (5H, м), 7.29 - 7.24 (3H, м), 7.08 (2H, д, J = 8 Гц), 6.96 (1H, д, J = 8 Гц), 6.76 (2H, д, J = 8 Гц), 5.53 (1H, квинт, J = 6 Гц), 5.38 (1H, ушир. д), 5.09 (1H, ушир. д, J = 6 Гц), 4.22 (1H, т, J = 7 Гц), 3.72 (3H, с), 3.47 (1H, дд, J = 16,6 Гц), 3.19 (1H, дд, J = 14,7 Гц), 2.92 (1H, дд, J = 14,7 Гц), 2.71 (1H, дд, J = 16,10 Гц), 2.43 (1H, м), 2.28 (3H, с), 2.22 (1H, м), 1.97 - 1.59 (4H, м),
Пример E-6
[4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-Меркапто-3- фенилпропиониламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2]бензазепин-4-карбоновая кислота
[4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-Ацетилтио-3- фенилпропиониламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2]бензазепин-4-карбоновую кислоту, полученную по примеру E-4, растворяют в дегазированном ТГФ (2,0 мл) и метаноле (10 мл). К этому раствору прибавляют гидроксид лития (4,00 мл). Смесь перемешивают при комнатной температуре в атмосфере азота в течение 45 минут. В смесь прибавляют по каплям водный раствор 2,0 н соляной кислоты (3,00 мл) и затем добавляют туда воду. Полученную смесь энергично перемешивают. Выпавшие таким образом белые кристаллы собирают фильтрацией и сушат при пониженном давлении. Таким образом получают указанное в заголовке соединение (0,45 г, выход 87%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.75 (1H, д, J = 7 Гц), 7.66 (2H, д, J = 8 Гц), 7.59 (2H, т, J = 8 Гц), 7.55 - 7.37 (7H, м), 7.22 (1H, д, J = 8 Гц), 5.81 (1H, квинт, J = 6 Гц), 5.65 (1H, м), 5.36 (1H, д, J = 6 Гц), 3.82 - 3.68 (2H, м), 3.45 (1H, дд, J = 14,7 Гц), 3.30 (1H, дд, J = 14,7 Гц), 2.99 (1H, дд, J = 17,12 Гц), 2.70 (1H, м), 2.50 (1H, м), 2.21 (1H, д, J = 9 Гц), 2.23 - 1.85 (4H, м)
Пример E-7
[4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-Меркапто-3-(метоксифенил) пропиониламино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a][2]бензазепин-4-карбоновая кислота
Тем же способом, что и по примеру E-6, и исходя из [4S -(4α,7α (R*), 12 β ] ] -7-[(S)-2-ацетилтио-3- (4-метоксифенил)-пропиониламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоновой кислоты (0,42 г, 0,708 ммоль), полученной по примеру E-5, получают указанное в заголовке соединение в виде белых кристаллов (0,37 г, выход 95%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.51 (1H, д, J = 7 Гц), 7.43 (2H, д, J = 8 Гц), 7.36 (2H, т, J = 8 Гц), 7.28 (2H, м), 7.08 (2H, д, J = 8 Гц), 6.99 (1H, д, J = 8 Гц), 6.78 (2H, д, J = 8 Гц), 5.57 (1H, квант, J = 6 Гц), 5.42 (1H, м), 5.13 (1H, как д, J = 6 Гц), 3.73 (3H, с), 3.50 (2H, м), 3.14 (1H, дд, J = 14, 7 Гц), 3.03 (1H, дд, J = 14, 7 Гц), 2.76 (1H, дд, J = 17, 12 Гц), 2.47 (1H, м), 2.28 (1H, м) 1.97 (1H, д, J = 9 Гц), 2.00 - 1.63 (4H, м)
Пример E-8
Дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-ацетилтио-3-метилбутириламино] -6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилат
Тем же способом, что и по примеру E-2, и исходя из дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-амино-6-оксо-11-фенил- 1,2,3,4,6,8,7,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилата (0,59 г, 1,14 ммоль), полученного по примеру E-1, и (S)-2-ацетилтио- 3-метилбутановой кислоты (0,136 г, 0,774 ммоль) получают указанное в заголовке соединение в виде бесцветного аморфного продукта (0,36 г, выход 69%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.54 - 6.92 (1H, м), 6.68 (1H, д, J = 8 Гц), 6.28 (1H, с), 5.65 (1H, квинт. J = 6 Гц), 5.49 - 5.40 (1H, д, J = 7 Гц), 4.00 (1H, д, J = 7 Гц), 3.42 (1H, дд, J = 16, 6 Гц), 2.60 - 2,37 (7H, м), 2.02 (1H, м), 1.88 - 1.72 (3H, м), 1.08 (3H, д, J = 7 Гц), 1.04 (3H, д, J = 7 Гц)
Пример E-9
[4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-Ацетилтио-3- метилбутириламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2]бензазепин-4-карбоновая кислота
Тем же способом, что и по примеру E-4, и исходя из дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-ацетилтио-3- метилбутириламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоксилата (0,36 г, 0,533 ммоль), полученного по примеру E-8, получают указанное в заголовке соединение в виде бесцветного аморфного продукта (0,157 г, выход 58%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.55 - 7.28 (8H, м), 7.03 (1H, ушир. с), 5.69 (1H, квинт, J = 6 Гц), 5.46 (1H, м), 3.97 (1H, д, J = 7 Гц), 3.51 (1H, м), 2.96 - 2.82 (2H, м), 2.56 - 2.20 (7H, м), 2.00 - 1.60 (4H, м), 1.05 (3H, д, J = 7 Гц), 1.01 (3H, д, J = 7 Гц)
Пример E-10
[4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-Меркапто-3- метилбутириламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота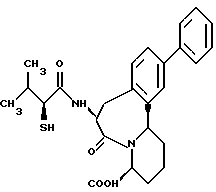
[4S -(4α,7α (R*), 12b β ]]-7-[(S)-2-Ацетилтио-3- метилбутириламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоновую кислоту (0,147 мг, 0,289 ммоль), полученную по примеру E-9, растворяют в дегазированном этаноле (5 мл). К этому раствору прибавляют 1,0 н водный раствор гидроксида лития (0,9 мл). Смесь перемешивают при комнатной температуре в атмосфере азота в течение 1 часа. Реакционную смесь подкисляют прибавлением водного раствора 1,0 н соляной кислоты при охлаждении льдом и перемешивании с последующей экстракцией дихлорметаном. Органическую фазу промывают насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния. После отгонки растворителя из органической фазы полученный остаток кристаллизуют из дихлорметан-гексана. Далее маточный раствор упаривают досуха и затем обрабатывают изопропиловый эфир-гексан. Таким образом получают 0,103 г указанного в заголовке соединения (выход 76%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.68 (1H, д, J = 7 Гц) 7.51 (2H, д, J = 8 Гц), 7.44 (2H, д, J = 8 Гц), 7.39 - 7.33 (3H, м),7.08 (1H, д, J = 8 Гц), 5.73 (1H, квинт. J = 6 Гц), 5.53 (1H, м), 5.26 (1H, м), 3.61 (1H, дд, J = 17, 6 Гц), 3.19 (1H, дд, J = 9, 7 Гц), 2.93 (1H, дд. J = 17, 13 Гц), 2.60 - 2.22 (3H, м), 2.08 - 1.70 (4H, м), 1.05 (6H, д, J = 7 Гц)
Пример E-11
Дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-2- ацетилтио-3-метилвалериламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2]бензазепин-4-карбоксилат
Тем же способом, что и по примеру E-2, и исходя из дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-амино-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилата (0,59 г, 1,14 ммоль), полученного по примеру E-1, и (2S, 3S)-2-ацетилтио-3-метилвалериановой кислоты (0,52 г, 2,74 ммоль) получают указанное в заголовке соединение в виде бесцветного аморфного продукта (1,22 г, выход 74%),
1H-ЯМР (400 МГц, CDCl3) δ: : 7.55 - 6.91 (17H, м), 6.67 (1H, д, J = 8 Гц), 6.27 (1H, с). 5.65 (1H, квинт, J = 6 Гц), 5.47 (1H, как д), 5.41 (1H, как д), 4.05 (1H, д, J = 7 Гц), 3.42 (1H, дд, J = 16, 6 Гц), 2.61 - 2.40 (2H, м), 2.45 (3H, с), 2.14 (1H, м), 2.00 (1H, м), 1.92 - 1.58 (3H, м), 1.24 (1H, м), 1.05 (3H, д, J = 7 Гц), 0.94 (3H, т, J = 7 Гц)
Пример E-12
[4S -(4α,7α (R*)-12b β ]]-7-[(2S, 3S)-Ацетилтио-3- метилвалериламино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a] [2]бензазепин-4-карбоновая кислота
Тем же способом, что и по примеру E-4, и исходя из дифенилметил [4S -(4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-2-ацетилтио-3- метилвалериламино]-6-оксо-11-фенил-1,2,3,4,6,8,7,12b- октагидропиридо[2,1-a-] [2] бензазепин-4-карбоксилата (1,22 г, 1,773 ммоль), полученного по примеру E-11, получают указанное в заголовке соединение в виде бесцветного аморфного продукта (0,897 г, выход 58%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.52 - 7.31 (8H, м), 7.04 (1H, д. J = 8 Гц), 5.69 (1H, квинт, J = 6 Гц), 5.48 (1H, м), 5.18 (1H, м), 4.02 (1H, д, J = 7 Гц), 3.54 (1H, м), 2.86 (1H, дд, J = 16, 12 Гц), 2.51 (1H, м), 2.40 (3H, с), 2.28 (1H, м), 2.11 (1H, м), 2.04-1.56 (5H, м), 1.20 (1H, м), 1.02 (3H, д, J = 7 Гц), 0.91 (3H, т, J = 7 Гц).
Пример E-13
[4S -(4α,7α (R*), 12b β ]]-7-[(2S, 3S)-2-Меркапто-3- метилвалериламино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[1,2-a][2]бензазепин-4-карбоновая кислота
[4S -(4α,7α (R*), 12b β ]]-7-[(2S, 3S)-2-Ацетилтио-3- метилвалериламино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо- [2,1-a] [2]бензазепин-4-карбоновую кислоту, (0,780 г, 1,429 ммоль), растворяют в дегазированном этаноле (20 мл). К этому раствору прибавляют 1,0 н водный раствор гидроксида лития (4,48 мл) при 0oC. Смешанный раствор, полученный таким образом, перемешивают в атмосфере азота в течение 40 минут.
Раствор смеси подкисляют подкислением путем прибавления воды (20,0 мл) и 2,0 водного раствора соляной кислоты. Выпавший при этом твердый продукт собирают фильтрацией (промывание H2O) с получением посредством этого указанного в заголовке соединения (0,622 г, выход 87%).
1H-ЯМР (400 МГц, CDCl3) δ: : 7.66 (1H, д, J = 7 Гц), 7.53 - 7.32 (7H, м), 7.08 (1H, д, J = 8 Гц), 5.72 (1H, квинт, J = 6 Гц), 5.52 (1H, м), 5.25 (1H, м), 3.60 (1H, дд, J = 17, 6 Гц), 3.23 (1H, дд, J = 9, 7 Гц), 2.93 (1H, дд, J = 17, 13 Гц), 2.55 (1H, м), 2.34 (1H, м), 2.00 (2H, м), 1.92 (1H, д, J = 8 Гц), 1.98 - 1.61 (4H, м), 1.25 (1H, м), 1.03 (3H, д, J = 7 Гц), 0.93 (3H, т, J = 7 Гц)
Пример F-1
Метил-[3R- [3α,6α (S*), 9a β ]]-6-[[(S)-1-оксо-2-ацетилтио-3-(4-фторфенил)пропил]амино]-октагидро-5- оксотиазол[3,2-a]азепин-3-карбоксилат
К метил [3R-[3 α,6α,(S*),9aβ ]]-6-аминооктагидро-5-оксотиазол[3,2-a] азепин- 3-карбоксилату (681 мг, 2,79 ммоль) добавляют раствор 2-ацетилтио-3-(4-фторфенил)пропионовой кислоты (743 мг, 3,07 ммоль), полученный по синтетическому примеру F-3, в метиленхлориде (28 мл). Полученную смесь охлаждают до 0oC при охлаждении ледяной баней. После прибавления N-этоксикарбонил-2-этокси-1,2-дигидрохинолина (ЭЭДХ, 793 мг, 3,21 ммоль) ледяную баню убирают. Полученную смесь перемешивают в атмосфере азота при комнатной температуре в течение 3 часов. Полученную смесь промывают 0,5 н раствором соляной кислоты (15 мл • 2), водой (10 мл) и насыщенным водным раствором гидрокарбоната натрия (15 мл • 2) и насыщенным водным раствором хлорида натрия (15 мл), сушат над сульфатом магния и фильтруют. Далее фильтрат концентрируют при пониженном давлении с получением сырого продукта смеси эпимеров (1,39 г). Эту сырую смесь эпимеров отделяют и очищают хроматографией на колонке с силикагелем (гексан: этилацетат = 3:1). В результате получают указанное в заголовке соединение (500 мг, 38%) в виде первого эпимера из первой фракции.
1H-ЯМР (400 МГц, CDCl3) δ: 1.55-2.02 (6H, м), 2.34 (3H, с), 2.97 (1H, дд, J = 7.2, 14.2 Гц), 3.18 (1H, дд, J = 6.6, 11.6 Гц), 3.27 (1H, дд, J = 2.4, 11.6 Гц), 3.27 (1H, дд, J = 7.6, 14.2 Гц), 3.78 (3H, с), 4.24 (1H, т, J = 7.4 Гц), 4.44 (1H, дд, J = 6.0, 11.2 Гц), 4.99 (1H, д, J = 9.2 Гц), 5.25 (1H, дд, J = 2.4, 6.6 Гц), 6.95 (2H, т, J = 8.6 Гц), 7.18 (2H, дд, J = 5.2, 8.4 Гц), 7.31 (1H, д, J = 3.2 Гц).
Масс m/e (FAB); 469 (MH+)
Т. пл.: 53-57oC
Пример F-2
Метил [3R-[3 α,6α (S*), 9a β ]]-6-[[(S)-1-оксо-2-ацетилтио- 3-(4-фторфенил)пропил]амино]-октагидро-5-оксотиазол[3,2-a]азепин- 3-карбоксилат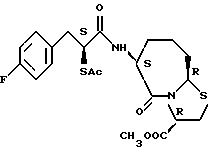
Следуя за первым эпимером, полученным в примере F-1, из колонки в качестве второго эпимера получают указанное в заголовке соединение (486 мг, 37%).
1H-ЯМР (400 МГц, CDCl3) δ: 1.45-2.02 (6H, м), 2.33 (3H, с), 2.93 (1H, дд, J = 6.8, 13.6 Гц), 3.16 (1H, дд, J = 6.8, 12.0 Гц), 3.26 (1H, дд, J = 2.4, 12.0 Гц), 3.28 (1H, дд, J = 8.8, 13.6 Гц), 3.77 (3H, с), 4.19 (1H, дд, J = 6.8, 8.8 Гц), 4.45 (1H, дд, J = 6.2, 11.2 Гц), 4.97 (1H, д, J = 8.8 Гц), 5.26 (1H, дд, J = 2.4, 6.8 Гц), 6.96 (2H, т, J = 8.8 Гц), 7.19 (2H, дд, J = 5.6, 8.0 Гц), 7.32 (1H, д, J = 5.6 Гц).
Масс m/e (FAB); 469 (MH+)
Т. пл. 55-60oC
Пример F-3
Метил [3R -[3α,6α (S*), 9a β ]]-6-[[(S)-1-оксо-2-ацетилтио- 3-фенилпропил]амино]-октагидро-5-оксотиазол[3,2-a]азепин-3-карбоксилат
Раствор метил [3R-[3α,6α (S*), 9a β ]]-6-аминооктагидро-5- оксотиазол[3,2-a] азепин-3-карбоксилата (430 мг, 1,76 ммоль) в метиленхлориде (17,6 мл) охлаждают до 0oC при охлаждении ледяной баней. Далее к этому раствору прибавляют последовательно (S)-2-ацетилтио-3-фенилпропионовую кислоту (395 мг, 1,76 ммоль), полученный по синтетическому примеру F-4, и N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (ЭЭДХ, 479 мг, 1,94 ммоль). Затем ледяную баню оставляют и полученную таким образом смесь перемешивают в атмосфере азота при комнатной температуре в течение 6 часов. Затем смесь промывают 0,5 н водным раствором соляной кислоты (10 мл х 2), водой (10 мл), насыщенным водным раствором гидрокарбоната натрия (10 мл х 2) и насыщенным водным раствором хлорида натрия (10 мл) и сушат над сульфатом магния. Далее фильтрат, который получают его фильтрованием, концентрируют при пониженном давлении. Полученный таким образом осадок очищают колоночной хроматографией (метиленхлорид:этилацетат = 20:1). Таким образом получают указанное в заголовке соединение в виде аморфного продукта (563 мг, 71%).
1H-ЯМР (400 МГц, CDCl3) δ: 1.50-2.03 (6H, м), 2.32 (3H, с), 2.99 (1H, дд, J = 7.6, 14.0 Гц), 3.17 (1H, дд, J = 6.4, 12.0 Гц), 3.26 (1H, дд, J = 2.4, 12.0 Гц), 3.31 (1H, дд, J = 7.6, 14.0 Гц), 3.78 (3H, с), 4.29 (1H, т, J = 7.6 Гц), 4.46 (1H, дд, J = 6.4, 10.4 Гц), 4.99 (1H, д, J = 8.8 Гц), 5.24 (1H, дд, J = 2.4, 6.4 Гц), 7.18-7.36 (6H, м).
Масс m/e (FAB); 451 (MH+)
Т.пл. не определяется из-за аморфности формы
Пример F - 4
Метил [3R -[3α,6α (S*), 9a β ]]-6-[[(S)-1-оксо-2-ацетилтиометил-3-фенилпропил]амино]-октагидро-5-оксотиазол [3,2-a]азепин-3-карбоксилат
Раствор метил [3R-[3α,6α (S*), 9a β ]]-6-аминооктагидро-5-оксотиазол[3,2-a] азепин-3-карбоксилата (375 мг, 1,53 ммоль) в метиленхлориде (15,3 мл) охлаждают до 0oC при охлаждении ледяной баней. Далее к этому раствору прибавляют последовательно (S)-2-ацетилтиометил-3-фенилпропионовую кислоту (365,8 мг, 1,54 ммоль) и N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (ЭЭДХ, 418 мг, 1,69 ммоль). Затем ледяную бань оставляют и полученную таким образом смесь перемешивают в атмосфере азота при комнатной температуре в течение 6 часов. Затем смесь промывают 0,5 н. водным раствором HCl (10 мл • 2), водой (10 мл), насыщенным водным раствором Na2CO3 (10 мл • 2) и насыщенным водным раствором хлорида натрия (10 мл) и сушат (используют MgSO4). Далее фильтрат, который получают его фильтрованием, концентрируют при пониженном давлении. Полученный таким образом осадок очищают колоночной хроматографией (метиленхлорид: этилацетат = 20 : 1). Таким образом получают указанное в заголовке соединение в виде аморфного продукта (435 мг, 61%).
1H-ЯМР (400 Мгц, CDCl3) δ: 1.55 - 2.11 (6H, м), 2.32 (3H, с), 2.62 - 2.70 (1H, м), 2.82 (1H, дд, J = 6.8, 14.Гц), 2.97 (1H, дд, J = 8.4, 14 Гц), 3.03 (1H, дд, J = 8.8, 13.6 Гц), 3.11 (1H, дд, J = 5.2, 13.6 Гц), 3.17 (1H, дд, J = 6.8, 11.6 Гц), 3.27 (1H, дд, J = 2.8, 11.6 Гц), 3.78 (3H, с), 4.40 - 4.44 (1H, м как кв), 4.98 (1H, д, J = 8.8 Гц), 5.20 (1H, дд, J = 2.8 6.8 Гц), 6.79 (1H, д, J = 6 Гц), 7.15 - 7.28 (5H, м)
Mасс m/e (FAB); 465 (MH+).
Т.пл. не определяется из-за аморфности формы.
Пример F-5
Метил [3R [3α,6α (S*), 9a β ]]-6-[[(2S, 3S)-1-оксо-2-ацетилтио-3-метилпентил]амино]-октагидро-5-оксо-тиазол[3,2-a] азепин-3-карбоксилат
Раствор метил [3R,[3α,6α (S*), 9a β ]]-6-аминооктагидро-5-оксотиазол[3,2-a] азепин-3-карбоксилата (225 мг, 0,92 ммоль) в метиленхлориде (17 мл) охлаждают до 0oC при охлаждении льдом. Далее к этому раствору прибавляют последовательно раствор (2S, 3S)-2-ацетилтио-3-метилпентановую кислоту (193 мг, 1,01 ммоль) в метиленхлориде (6 мл) и ЭЭДХ (296 мг, 1,20 ммоль). Затем ледяную баню удаляют и полученную таким образом смесь перемешивают в атмосфере азота при комнатной температуре в течение ночи. Затем смесь концентрируют упариванием до определенного объема. Далее остаток растворяют в этилацетате и полученный раствор промывают 1 н. водным раствором HCl, насыщенным водным раствором Na2CO3 и насыщенным водным раствором хлорида натрия и сушат (используют MgSO4). Фильтр, который получают его фильтрованием, концентрируют при пониженном давлении. Полученный таким образом осадок очищают колоночной хроматографией гексан : этилацетат = 3 : 1). Таким образом получают указанное в заголовке соединение в виде аморфного продукта (206 мг, 54%).
1H-ЯМР (400 Мгц, CDCL3) δ: 0.88 (3H, т, J = 7.6 Гц), 0.99 (3H, д, J = 6.8 Гц), 1.10-1.22 (1H, м), 1.51-1.70 (2H, м), 1.82-2.14 (6H, м), 2,38 (3H, c), 3.20 (1H, дд, J = 6.4, 11.8 Гц), 3.28 (1H, дд, J = 2.4 11.8 Гц), 3.79 (3H, с), 3.98 (1H, дд, J = 6.8 Гц), 4.54 (1H, дд, J = 6.4, 10.4 Гц), 5.02 (1H, д, J = 8.8 Гц), 5.28 (1H, дд, J = 2.4, 6.4 Гц), 7.41 (1H, д, J = 6.0 Гц)
Примеры от F-6 до F-13
Соединения по примерам от F-6 до F-13, показанные далее, получены теми же способами, что и по примерам от F-1 до F-5.
Пример F-6
Метил [3R [3α,6α (S*), 9a β ]]-6-[[(S)-1-оксо-2-ацетилтио-3-(3-метоксифенил)пропил]амино]-октагидро-5 оксотиазол[3,2-a]азепин-3-карбоксилат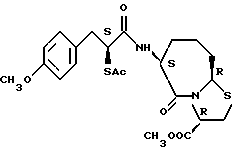
1H-ЯМР (400 МГц, CDCl3) δ: 1.55-2.04 (6H, м), 2.33 (3H, с), 2.94 (1H, дд, J = 7.6, 14.4 Гц), 3.18 (1H, дд, J = 6.8, 11.6 Гц), 3.24 (1H, дд, J = 7.2, 14.4 Гц), 3.27 (1H, дд, J = 2.4, 11.6 Гц), 3.78 (6H, с), 4.24 (1H, т, J = 7.6 Гц), 4.45 (1H, дд, J = 6.0, 10.8 Гц), 4.99 (д, J = 8.8 Гц), 5.24 (1H, дд, J = 2.4, 6.8 Гц), 6.80 (2H, д, J = 8.8 Гц), 7.13 (2H, д, J = 8.4 Гц), 7.32 (H, д, J = 6.4 Гц), 7.32
Масс m/e (FAB); 481 (MH+).
Т.пл. не определяется из-за аморфности формы.
Пример F-7
Метил[3R- 3α,6α (S*), 9a β ]]-6-[[[(S)-1-оксо-2-ацетилтио-3-(1,4-бифенил)пропил]амино]-октагидро -5-оксотиазол[3,2-a]азепин-3-карбоксилат
1H-ЯМР (400 МГц, CDCl3) δ: 1.50-2.05 (6H, м), 2.35 (3H, с), 3.05 (1H, дд, J= 7.6, 14.4 Гц), 3.11 (1H, дд, J=6.6, 12.0 Гц), 3.23 (1H, дд, J=2.4, 12.0 Гц), 3.34 (1H, дд, J=7.6, 14.4 Гц), 3.77 (3H, с), 4.32 (1H, т, J=7.6 Гц), 4.45 (1H, дд, J=6.4, 11.6 Гц), 4.98 (1H, д, J=8.4 Гц), 5.21 (1H, дд, J= 2.4, 6.6 Гц), 7.26-7.60 (10H, м)
Масс m/e (FAB); 527 (MH+).
Т.пл. 68-72oC.
Пример F-8
Метил [3R -[3α,6α (S*), 9 β ]]-6-[[(R)-1-оксо-2-ацетилтио -3-(1,4-бифенил)пропил]амино]-октагидро-5-оксотиазол[3,2-a]азепин -3-карбоксилат
1H-ЯМР (400 Мгц, CDCl3) δ: ; 1.46-2.00(6H, м), 2.34 (3H, с), 3.01 (1H, дд, J= 7.2, 14.0 Гц), 3.15 (1H, дд, J=6.4, 12.0 Гц), 3.25 (1H, дд, J=2.4, 12.0 Гц), 3.36 (1H, дд, J=8.8, 14.0 Гц), 3.76 (3H, с), 4.28 (1H, дд, J=7.2, 8.8 Гц), 4.45-4.49 (1H, м как кв), 4.97-4.99 (1H, м как д), 5.26 (1H, дд, J= 2.4, 6.4 Гц), 7.29-7.59 (10H, м)
Масс m/e (FAB); 527 (MH+).
Т.пл. 77-80oC.
Пример F-9
Метил [3R-[3α,6αS*),9aβ]- 6-[[(S)-1-оксо-2-ацетилтио-3-(2-тиенил)пропил] амино]-октагидро-5-оксотиазол[3,2-a]азепин-3-карбоксилат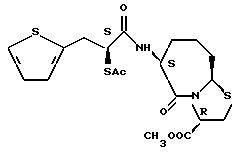
1H-ЯМР (400 Гц, CDCl3) δ: 1.50-1.70(2H,м), 1.86-2.08 (4H, м), 2.36 (3H, с), 3.18 (1H, дд, J=6, 12 Гц), 3.27 (1H, дд, J=2, 12 Гц), 3.29 (1H, дд, J= 7,14 Гц), 3.49 (1H, дд, J=7,14 Гц), 3.78 (3H, с), 4.30 (1H, д, J=1 Гц), 4.49 (1H, дд, J= 6, 10 Гц), 5.00 (1H, д, J=9 Гц), 5.26 (1H, дд, J=2,6 Гц), 6.86 (1H, ушир. д, J= 4 Гц), 6.90 (1H, дд, J=3,5 Гц), 7.14 (1H, дд, J=2,5 Гц), 7.40( 1H, д, J=6 Гц)
Пример F-10
Метил [3R-[3α,6αS*),9aβ]- 6-[[(2S, 2R)-1-оксо-2-ацетилтио-3-метилпентил] амино] -октагидро-5-оксотиазол[3,2-a]азепин-3-карбоксилат
1H-ЯМР (400 Мгц, CDCl3) δ: 0.91(3H, т, J=7 Гц), 0.92 (3H, д, J=7 Гц), 1.27 (1H, м), 1.44 (1H, м), 1.64 (1H, м), 1.88-2.06 (5H, м), 2.07 (1H, квинт, J=7 Гц), 2.40 (3H, с), 3.19(1H, дд, J=6, 12 Гц), 3.28(1H, дд, J=2, 12 Гц), 3.80 (3H, с), 4.07 (1H, д, J=7 Гц), 4.53 (1H, дд, J=6, 10 Гц), 5.02 (1H, д, J=9 Гц), 5.28 (1H, дд, J=2, 6 Гц), 7.51 (1H, д, J=6 Гц)
Пример F-11
Метил [3R [3α,6α S*), 9a β ]]-6-[[(S)-1-оксо-2-ацетилтиобутил]амино]-октагидро-5-оксотиазол-[3,2-a] азепин-3-карбоксилат
1H-ЯМР (400 МГц, CDCl3) δ: 0.98 (3H, т, J = 7 Гц), 1.67 (1H, м), 1.77 (1H, м), 1.86-2.06 (6H, м), 2.37 (3H, с), 3.19 (1H, дд, J = 6, 12 Гц), 3,27 (1H, дд, J = 2, 12 Гц), 3.79 (3H, с), 3,96 (1H, т, J = 6 Гц), 4.54 (1H, дд, J = 6, 10 Гц), 5.02 (1H, д, J = 9 Гц), 5.28 (1H, дд, J = 2, 6 Гц), 7.35 (1H, д, J = 6 Гц).
Пример F-12
Метил [3R- -[3α,6α (S*), 9a β ]]-6-[[(S)-1-оксо-2-ацетилтио-3-метилбутил]амино]-октагидро-5-оксотиазол [3,2-a]азепин-3-карбоксилат
1H-ЯМР (400 МГц, CDCl3) δ; 0.97 (3H, д, J = 7 Гц), 1.02 (3H, д, J = 7 Гц), 1.65 (1H, м), 1.88-2.06 (4H, м), 2.35 (1H, м), 2.39 (3H, с), 3.20 (1H, дд, J = 6, 12 Гц), 3.28 (1H, дд, J = 2, 12 Гц), 3.80 (3H, с), 3.91 (1H, д, J = 7 Гц), 4.54 (1H, дд, J = 6, 10 Гц), 5.02 (1H, д, J = 9 Гц), 5.28 (1H, дд, J = 2, 6 Гц), 7.40 (1H, д, J = 6 Гц)
Пример F-13
Метил [3R- -[3α,6α (S*), 9a β [[-6-(S)-1-оксо-2-ацетилтио-3,3-диметилбутил]амино]-октагидро-5- оксотиазол[3,2-a]азепин-3-карбоксилат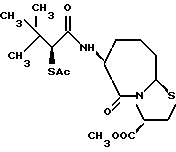
1H-ЯМР (400 МГц, CDCl3) δ: 1.02 (9H, с), 1.63 (1H, м), 1.88-2.09 (5H, м), 2.17 (3H, с), 3.20 (1H, дд, J = 6, 12 Гц), 3.28 (1H, дд, J = 2, 12 Гц), 3.77 (1H, с), 3.80 (3H, с), 4.57 (1H, дд, J = 6, 10 Гц), 5.03 (1H, д, J = 9 Гц), 5.28 (1H, дд, J = 2, 6 Гц), 7.20 (1H, д, J = 6 Гц).
Пример F-14
[3R- -[3α,6α (S*), 9a β ]]-6-[[(S)-1-Оксо-2-тио-3-(4-фторфенил)пропил] амино]октагидро-5-оксотиазол- [3,2-a]азепин-3-карбоновая кислота
Метил [3R [3α,6α (S*), 9a β ]]-6-[(S)-1-оксо-2-ацетил-тио-3-(4-фторфенил)пропиламино] октагидро-5- оксотиазол[3,2-a]азепин-3-карбоксилат (384 мг, 0,82 ммоль), полученный по примеру F-1, помещают в колбу с последующей достаточной продувкой азотом. После прибавления дегазированного тетрагидрофурана (1,95 мл) и метанола (11,7 мл) колбу охлаждают до 0oC. К полученной таким образом смеси добавляют дегазированный 1 н. водный раствор гидроксида лития (6,6 мл). Полученную смесь доводят до комнатной температуры и перемешивают в течение 1 часов. После повторного охлаждения смеси до 0oC добавляют 1 н водный раствор соляной кислоты (10 мл). Полученную смесь экстрагируют хлороформом (50 мл • 2). Органическую фазу промывают насыщенным водным раствором хлорида натрия (30 мл) и затем сушат над сульфатом магния. Затем органическую фазу фильтруют и фильтрат концентрируют при пониженном давлении до определенного объема. К концентрату прибавляют толуол (50 мл) с последующим повторным концентрированием. Далее остаток растворяют в небольшом количестве (около 1 мл) хлороформа и диизопропилового эфира (около 1 мл) и перекристаллизовывают. К полученным таким образом кристаллам добавляют гексан (3 мл). После измельчения и фильтрации твердый продукт сушат при пониженном давлении. Таким образом получают указанное в заголовке соединение (362 мг, 107%) в виде белых кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 1.55-1.68 (1H, м), 1.90-2.06 (6H, м), 3.09 (1H, дд, J = 6.8, 14.0 Гц), 3.18-3.25 (2H, м), 3.34 (1H, дд, J = 2.4, 12.0 Гц), 3.51-3.56 (1H, м как кв), 4.52 (1H, дд, J = 6,4, 11.2 Гц), 5.03 (1H, т, J = 5.2 Гц), 5.26 (1H, дд, J = 2.4, 6.4 Гц), 6.97 (2H, т, J = 8.8 Гц), 7.17 (2H, дд, J = 5.8, 8.2 Гц), 7.52 (1H, д, J = 6.0 Гц)
Масс m/e (FAB); 413 (MH+).
Т.пл. 209-211oC.
Пример F-15
[3R 3α,6α (S*), 9a β ]]-6-[[(2S, 3S)-Оксо-2-тио-3- метилпентил]амино]октагидро-5-оксотиазол[3,2-a]-азепин-3-карбоновая кислота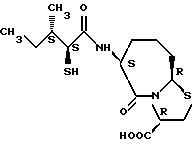
Метил [3R -[3α,6α (S*), 9a β ]]-6-[[(2S, 3S)-1-оксо-2-ацетилтио-3-метилпентил]амино]-октагидро-5-оксотиазол[3,2-a] азепин-3-карбоксилат (200 мг, 0,48 ммоль), полученный по примеру F-5, помещают в колбу с последующим добавлением дегазированного этанола (8 мл). Колбу охлаждают до 0oC в атмосфере азота. К полученной смеси прибавляют дегазированный 1 н водный раствор гидроксида лития (3,8 мл). Полученную смесь перемешивают при комнатной температуре в течение 50 минут. Полученную смесь подкисляют добавлением 2 н водного раствора соляной кислоты (2,9 мл) при 0oC. Полученную смесь экстрагируют метиленхлоридом. После промывки органической фазы насыщенным водным раствором хлорида натрия не сушат над сульфатом магния и концентрируют. Полученный твердый продукт перекристаллизовывают из гексана-метиленхлорида. Таким образом получают указанное в заголовке соединение (150 мг, 87%) в виде белых кристаллов.
1H-ЯМР (400 Мгц, CDCl3) δ: 0.90 (3H, т, J = 7 Гц), 1.00 (3H, д, J = 7 Гц), 1.24 (1H, м), 1.55-1.74 (2H, м), 1.87 (1H, д, J = 8 Гц), 1.90-2.10 (6H, м), 3.20 (1H, дд, J = 6, 12 Гц), 3.24 (1H, д, J = 7 Гц), 3.36 (1H, дд, J = 2, 12 Гц), 4.62 (1H, дд, J = 6, 10 Гц), 5.07 (1H, как т, J = 6 Гц), 5.29 (1H, дд, J = 2, 6 Гц), 7.69 (1H, д, J = 6 Гц).
Пример F-16
[3R-[3α,6α(S*),9aβ]] -6-[[(R)-1-Оксо-2-тио-3-(4-фторфенил]пропил]амино] октагидро-5-оксотиазол- [3,2-a]-азепин-3-карбоновая кислота
Соединение примера F2 подвергают взаимодействию тем же способом, что и по примеру F-14, с получением посредством этого указанного в заголовке соединения.
1H-ЯМР (400 Мгц, CDCl3) δ; 1.44-1.56 (1H, м), 1.82-2.03 (5H, м), 2.08 (1H, д, J = 9.2 Гц), 3.03 (1H, дд, J = 6.8, 14 Гц), 3.20 (1H, дд, J = 6.8, 11.6 Гц), 3.25 (1H, дд, J = 8.0, 14.0 Гц), 3.34 (1H, дд, J = 2.0, 11.6 Гц), 3.45 (1H, кв, J = 8.0 Гц), 4.53 (1H, дд, J = 6.4, 10.8 Гц), 5.02-5.04 (1H, м), 5.27 (1H, дд, J = 2.0, 6.8 Гц), 6.97 (2H, т, J = 8.6 Гц), 7.17 (2H, дд, J = 5.4, 8.6 Гц), 7.34 (1H, д, J = 6.0 Гц)
Масс m/e (FAB); 413 (MH+).
Т.пл. 98-105oC.
Примеры от F-17 до F-26
Соединения по примерам от F-17 до F-26, показанные далее, получены теми же способами, что и по примерам от F-14 до F-15 с использованием соединений по примерам F-3, F-4 и от F-6 до F-13.
Пример F-17
[3R-[3α,6α(S*)9aβ]]- 6-[[(S)-1-Оксо-2-тио-3-фенилпропил] амино]октагидро-5-оксотиазол[3,2-a] азепин-3-карбоновая кислота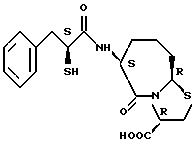
1H-ЯМР (400 Мгц, CDCl3) δ 1.53-1.68 (1H, м), 1.88-2.07 (6H, м), 3.10 (1H, дд, J = 6.8, 13.6 Гц), 3.19 (1H, дд, J = 6.6, 12.0 Гц), 3.27 (1H, дд, J = 6.8, 13.6 Гц), 3.34 (1H, дд, J = 2.4, 12.0 Гц), 3.59 (1H, кв. J = 6.8 Гц), 4.51-4.56 (1H, м как дд), 5.02-5.04 (1H, м как т), 5.26 (1H, дд, J = 2.4, 6.6 Гц), 7.17-7.30 (5H, м), 7.53 (1H, д, J = 6.0 Гц)
Масс m/e (FAB); 395 (MH+).
Т.пл. 232-235oC.
Пример F-18
[3R [3α,6α (S*), 9a β ]]-6-[[(S)-1-Оксо-2-тиометил-3- фенилпропил]амино] октагидро-5-оксотиазол[3,2-a]-азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ;
1.46 (1H, т, J = 8.4 Гц), 1.59-1.70 (1H, м), 1.84-2.14 (5H, м), 2.55-2.68 (2H, м), 2.76-2.83 (2H, м), 2. 97 (1H, дд, J = 7.0, 13.4 Гц), 3.21 (1H, дд, J = 6.8, 12.0 Гц), 3.35 (1H, дд, J = 2.4, 12.0 Гц), 4.54-4.59 (1H, м как кв), 5-02-5.05 (1H, м как т), 5.24 (1H, дд, J = 2.4, 6.8 Гц), 6.98 (1H, д, J = 6.0 Гц), 7.12-7.30 (5H, м)
Масс m/e (FAB); 409 (MH+).
Т.пл. 210-212oC.
Пример F-19
[3R-[3α,6α(S*),9aβ]]- 6-[[(S)-1-Оксо-2-тио-3-(4-метоксифенил)пропил] амино] октагидро-5-оксотиазол-[3,2-a]азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 1.55-1.68 (1H, м), 1.88-2.09 (6H, м), 3.07 (1H, дд, J = 6.8, 14.4 Гц), 3.18 (1H, дд, J = 6.8, 14.4 Гц), 3.20 (1H, дд, J = 6.8, 12.0 Гц), 3,34 (1H, дд, J = 2.4, 12.0 Гц), 3.55 (1H, дт, J = 8.8, 6.8 Гц), 3.79 (3H, с), 4.52-4.56 (1H, м как дд), 5.02-5.05 (1H, м как т), 5.25 (1H, дд, J = 2.4, 6.8 Г ц), 6.82 (2H, д, J = 8.4 Гц), 7.12 (2H, д, J = 8.4 гц), 7,56 (1H, д, J = 6.4 Гц)
Масс m/e (FAB); 425 (MH+)
Т.пл.: 182-183oC
Пример F-20
[3R [3α,6α(S*),9aβ]]- 6-[[(S-)-1-Оксо-2-тио-3-(1,4-бифенил)пропил]амино] октагидро-5-оксотиазол[3,2-a]-азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 1.55-1.67 (1H, м), 1.88-2.08 (5H, м), 2.05 (1H, д, J = 8.8 Гц), 3.12 (1H, дд, J = 6.6, 12.0 Гц), 3.16 (1H, дд, J = 6.8, 14.0 Гц), 3.26-3.31 (2H, м), 3.60 (1H, кв, J = 6.8 Гц), 4.50-4.54 (1H, м как кв), 5.00-5.03 (1H, м), 5.20 (1H, дд, J = 2.4, 6.8 Гц), 7.28-7.59 (10H, М)
Масс m/e (FAB); 471 (MH+
Т.пл.: 106-117oC
Пример F-21
[3R [3α,6α(S*),9aβ]]- 6-[[(S)-1-Оксо-2-тио-3-(1,4-бифенил)пропил]амино] октагидро-5-оксотиазол[3,2-a]-азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 1.44-1.56 (1H, м), 1.84-2.00 (5H, м), 2.12 (1H, д, J = 9.6 Гц), 3.08 (1H, дд, J = 6.4, 14.0 Гц), 3.16 (1H, дд, J = 6.8, 12.0 Гц), 3.29-3.50 (2H, м), 3.50-3.55 (1H, м как кв), 4.52-4.57 (1H, м, квк дд), 5.01-5.04 (1H, м), 5.25 (1H, дд, J = 2.4, 6.8 Гц), 7.26-7.58 (10H, м)
Масс m/e (FAB); 471 (MH+
Т.пл.: 109-116oC
Пример F-22
[3R [3α,6α(S*),9aβ]]- 6-[[(S)-1-Оксо-2-тио-3-(2-тиенил)пропил] амино] октагидро-5-оксотиазол[3,2-a]-азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 1.64 (1H, м), 1.90-2.12 (5H, м), 2.09 (1H, д, J = 8 Гц), 3.20 (1H, дд, J = 6.12 Гц), 3.34 (1H, дд, J = 2, 12 Гц), 3.44 (2H, д, J = 6 Гц), 3.58 (1H, м), 4.57 (1H, дд, J = 6, 10 Гц), 5.05 (1H, м), 5.26 (1H, дд, J = 2, 6 Гц), 6.87 (1H, ушир.д, J = 4 Гц), 6.93 (1H, дд, J = 3, 5 Гц), 7.17 (1H, дд, J = 2, 5 Гц), 7.65 (1H, д, J = 6 Гц)
Пример F-23
[3R -[3α,6α(S*),9aβ]]- 6-[[(2S, 3S)-1-Оксо-2-тио-3-метилпентил] амино] октагидро- 5-оксотиазол[3,2-a]азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ: 0.92 (3H, т, J = 7 Гц), 0.93 (3H, д, J = 7 Гц), 1.30 (1H, м), 1.49 (1H, м), 1.70 (1H, м), 1.76 (1H, д, J = 8 Гц), 1.90-2.14 (6H, м), 3.22 (1H, дд, J = 6, 12 Гц), 3.32-3.42 (2H, м), 4.62 (1H, дд, J = 6, 10 Гц), 5.06 (1H, м), 5.30 (1H, дд, J = 2, 6 Гц), 7.94 (1H, д, J = 6 Гц).
Пример F-24
[3R [3α,6α(S*),9aβ]]- 6-[(S)-1-Оксо-2-тиобутил] амино]октагидро-5- оксотиазол[3,2-a]азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 1.00 (3H, т, J = 7 Гц), 1.70 (1H, м), 1.79 (1H, м), 1.90-2.10 (7H, м), 3.19-3.30 (2H, м), 3.35 (1H, дд, J = 2, 12 Гц), 4.62 (1H, дд, J = 6, 10 Гц), 5.06 (1H, м), 5.29 (1H, дд, J = 2, 6 Гц), 7.61 (1H, д, J = 6 Гц).
Пример F-25
[3R [3α,6α(S*),9aβ]]- 6-[[(S)-1-Оксо-2-тио-3-метилбутиламино] октагидро-5- оксотиазол[3,2-a]азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 0.99 (2H, д, J = 7 Гц), 1.03 (3H, д, J = 7 Гц), 1.69 (1H, м), 1.85 (1H, д, J = 9 Гц), 1.90-2.10 (5H, м), 2.25 (1H, септет, J = 7 Гц), 3.16-3.26 (2H, м), 3.35 (1H, дд, J = 2, 12 Гц), 4.61 (1H, дд, J = 6, 10 Гц), 5.06 (1H, как т, J = 6 Гц), 5.30 (1H, дд, J = 2, 8 Гц), 7.67 (1H, д, J = 6 Гц).
Пример F-26
[3R [3α,6α(S*),9aβ]]- 6-[(S)-1-Оксо-2-тио-3,3-диметилбутиламино] октагидро- 5-оксотиазол[3,2-a]азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CD3OD) δ: 1.06 (9H, с), 1.74 (1H, м), 1.85-2.10 (5H, м), 3.25-3.35 (3H, м), 4.58 (1H, м), 5.17-5.25 (2H, м).
Пример 101
Метил- {3R-[3α,6α(S*),9aβ]}- 6-[[(2S, 3S)-2-ацетил-тио-1-оксо-3- фенилбутил]амино]октагидро-5-оксотиазол[3,2-a]азепин-3-карбоксилат
a) (4S)-3[(3R)-1-оксо-3-фенилбутил]-4-фенилметил-2-оксазолидинон
5,96 г (R)-3-фенилбутановую кислоту растворяют в 90 мл дихлорметана и добавляют несколько капель дихлорметана. В полученную таким образом смесь прибавляют по каплям 9,5 мл оксалилхлорида. Полученную таким образом смесь перемешивают при комнатной температуре в течение 0,5 часа и затем концентрируют. Осадок растворяют в 60 мл тетрагидрофурана. Далее 0,44 г (S)-4-фенилметил-2-оксазолидинона растворяют в 120 мл тетрагидрофурана. В полученный раствор прибавляют по каплям 14,5 мл 2,5 М раствора н-бутиллития в гексане в атмосфере азота при -70oC. Полученную смесь перемешивают при той же температуре в течение 20 минут и затем прибавляют приготовленный раствор хлорангидрида кислоты в тетрагидрофуране. Затем полученную таким образом смесь перемешивают при -70oC в течение 30 минут и затем нагревают при комнатной температуре. Реакционную смесь концентрируют. Добавляют этилацетат и воду и целевое соединение экстрагируют этилацетатом. Экстракт промывают насыщенным водным раствором хлорида натрия и затем сушат над безводным сульфатом магния и концентрируют. Концентрат очищают колоночной хроматографией на силикагеле с получением посредством этого 9,7 г указанного в заголовке соединения (выход 83%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.33-7.18 (8H, м), 7.07 (2H, дд, J = 2, 8 Гц), 4.63 (1H, м), 4.16 (1H, дд, J = 8, 8 Гц), 4.11 (1H, дд, J = 9, 3 Гц), 3.45 (2H, м), 3.08 (2H, м), 2.59 (1H, 1H, дд, J = 14, 9 Гц), 1.36 (3H, д, J = 7 Гц).
(b) (4S)-3-[(2S, 3S)-2-бром-1-оксо-3-фенилбутил]-4-фенилметил-2- оксазолидинон
3,25 г (4S)-3-[(3R)-1-оксо-3-фенилбутил]-4-фенилметил-2-оксазолидинона растворяют в 50 мл дихлорметана. К раствору добавляют 10 мл диизопропилэтиламина и 12,5 мл ди-н-бутилбортрифторметансульфоновой кислоты в атмосфере азота при -70oC. Полученную таким образом смесь перемешивают при той же температуре в течение 15 минут и затем при 0oC в течение 1 часа. Реакционную смесь охлаждают до -70oC. Смесь, полученную суспендированием 3,64 г N-бромсукцинимида в 20 мл дихлорметана, готовят в другом сосуде и к ней прибавляют вышеописанную реакционную смесь в атмосфере азота при -70oC. Полученную таким образом смесь перемешивают при той же температуре в течение 1,25 часа и затем выливают в раствор, содержащий 0,5 н сульфат натрия и насыщенный раствор хлорида натрия. Полученную смесь экстрагируют дихлорметаном. Органическую фазу сушат над безводным сульфатом магния, концентрируют и очищают колоночной хроматографией на силикагеле. Таким образом получают 3,43 г указанного в заголовке соединения (выход 85%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.38 - 7.24 (10H, м), 5.96 (1H, д, J = 10 Гц), 4.76 (1H, м), 4.23 (2H, м), 3.57 (1H, дд, J = 10,7 Гц), 3.34 (1H, дд, J = 14,3 Гц), 2.81 (1H, дд, J = 14,10 Гц), 1.38 (3H, д, J = 7 Гц)
(c) (4S)-3-[(2R, 3S)-2-азидо-1-оксо-3-фенилбутил]-4- фенилметил-2-оксазолидинон
6,43 г (4S)-3-[(2R, 3S)-2-бром-1-оксо-3-фенилбутил]-4- фенилметил-2-оксазолидинона растворяют в 80 мл дихлорметана. К полученному раствору добавляют раствор 7,58 г тетраметилгуанидиний азида в 20 мл дихлорметана при 0oC. Полученную таким образом смесь перемешивают при той же температуре в течение 1 часа и затем при комнатной температуре в течение 2,5 часов. Затем нагревают с обратным холодильником в течение 8 часов. К полученной таким образом реакционной смеси добавляют насыщенный раствор гидрокарбоната натрия. Полученную смесь экстрагируют дихлорметаном. Органическую фазу промывают насыщенным водным раствором хлорида натрия и затем сушат над безводным сульфатом магния и концентрируют. После очистки колоночной хроматографией на силикагеле получают 4,33 г указанного в заголовке соединения (выход 74%)
1H-ЯМР (400 МГц, CDCl3) δ: 7.37 - 7.22 (8H, м) 6.99 (2H, дд, J = 8,2 Гц) 5.37 (1H, д, J = 9 Гц) 4.60 (1H, м) 4.12 (1H, дд, J = 9,9 Гц) 3.45 (1H, м) 2.80 (1H, дд, J = 14,4 Гц) 1.98 (1H, дд, J = 14,10 Гц) 1.50 (3H, д, J = 7 Гц)
(d) (2R,3S)-2-азидо-3-фенилбутановая кислота
4,32 г (4S)-3-[(2R, 3S)-2-азидо-1-оксо-3-фенилбутил]-4- фенилметил-2-оксазолидинона растворяют в 60 мл тетрагидрофуране - воде (4 : 1). К полученному таким образом раствору добавляют 7,75 мл 30%-ной перекиси водорода и водный раствор, содержащий 0,73 г гидроксида лития при 0oC. Полученную смесь перемешивают при 0oC в течение 1 часа и затем добавляют водный раствор (57 мл), содержащий 9,58 г сульфата натрия. От реакционной смеси отгоняют тетрагидрофуран при пониженном давлении. Водную фазу промывают дихлорметаном и затем устанавливают pH до 1 с помощью конц. соляной кислоты с последующей экстракцией этилацетатом. Этилацетатную фазу промывают насыщенным водным раствором хлорида натрия и затем сушат над безводным сульфатом магния и концентрируют. Таким образом получают 2,30 г целевого соединения (выход 95%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.37 - 7.27 (5H, м) 4.09 (1H, д, J = 6 Гц) 3.39 (1H, дкв, J = 7,7 Гц) 1.39 (3H, д, J = 7 Гц)
(e) (2R,3S)-2-амино-3-фенилбутановая кислота
2,20 г (2R, 3S)-2-азидо-3-фенилбутановой кислоты растворяют в 40 мл метанола. К полученному раствору добавляют 2,71 г аммоний формиата и 0,36 г 10%-ного палладия-на-угле (содержащий воду препарат) с последующим взаимодействием при комнатной температуре в течение 1,5 часа. После удаления катализатора фильтрацией и концентрирования фильтрата к остатку прибавляют 300 мл смешанного растворителя, содержащего метанол и дихлорметан (1:9) с последующей экстракцией. После концентрирования экстракта получают 2,43 г целевого соединения (сырой продукт).
1H-ЯМР (400 МГц, CDCl3) δ: 7.32 - 7.16 (5H, м) 3.78 (1H, д, J = 5 Гц) 3.38 (1H, м) 1.23 (3H, д, J = 7 Гц)
(f) (2R, 3S)-2-бром-3-фенилбутановая кислота
1,70 г (2R, 3S)-2-Амино-3-фенилбутановой кислоты растворяют в смешанном растворе, содержащем 7,2 мл воды и 10,5 мл 47%-ной бромистоводородной кислоты. К полученному раствору добавляют 0,98 г гидрохлорида натрия при -10oC. Полученную смесь перемешивают в течение 30 минут и затем при комнатной температуре в течение 2 часов. Затем прибавляют воду и диэтиловый эфир с последующей экстракцией. Эфирную фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом натрия и затем концентрируют. Таким образом получают 1,84 г целевого соединения в виде сырого продукта.
1H-ЯМР (400 МГц, CDCl3) δ: 7.38 - 7.18 (5H, м) 4.35 (1H, д, J = 10 Гц) 3.36 (1H, м) 1.23 (3H, д, J = 7 Гц)
(g) (2S, 3S)-2-ацетилтио-3-фенилбутановая кислота
1,80 г (7,35 ммоль) (2R,3S)-2-Бром-3-фенилбутановой кислоты растворяют в 40 мл ацетонитрила. К полученному раствору добавляют 1,01 г (88,2 ммоль) тиоацетата калия при -10oC. Полученную смесь перемешивают при 0oC в течение 30 минут и затем при комнатной температуре в течение ночи. Нерастворимый материал удаляют фильтрацией и раствор концентрируют. К остатку, полученному после концентрирования, прибавляют диэтиловый эфир и гидрокарбонат натрия с последующей экстракцией целевого соединения в водную фазу. Устанавливают pH водной фазы 1 с помощью разбавленной соляной кислоты с последующей экстракцией диэтиловым эфиром. Органическую фазу промывают насыщенным водным раствором хлорида натрия и затем сушат над безводным сульфатом натрия и концентрируют. Таким образом получают 1,62 г сырого целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7.40 - 7.18 (5H, м), 4.42 (1H, д, J = 10 Гц), 3.33 (1H, м) 2.25 (3H, с) 1,43 (3H, д, J = 7 Гц)
(h) метил-(3R -[3α,6α(S*),9aβ]]- 6-[[2S, 3S)-2-ацетилтио-1-оксо-3-фенилбутил] амино]октагидро-5-оксотиазол[3,2-a]азепин-3-карбоксилат
0,3 г (1,23 ммоль) метил-(3R, [3α,6α (S*), 9aβ ]]-6-аминооктагидро- 5-оксотиазол[3,2-a] азепин-3-карбоксилата растворяют в 10 мл дихлорметана. К полученному раствору последовательно добавляют раствор 0,32 г (1,35 ммоль) (2S, 3S)-2-ацетилтио-3-фенилбутановой кислоты в 10 мл дихлорметана и 0,43 г (1,6 ммоль) ЭЭДХ при 0oC в атмосфере азота. Полученную таким образом смесь перемешивают при комнатной температуре в течение ночи. Затем ее последовательно обрабатывают 1 н. соляной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния, концентрируют, очищают хроматографией на колонке с силикагелем. Таким образом получают 0,27 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7.34 - 7.15 (5H, м), 5.28 (1H, дд, J = 6,2 Гц) 5.02 (1H, д, J = 9 Гц) 4.56 (1H, дд, J = 11,7 Гц) 4.22 (H, д, J = 10 Гц) 3.79 (3H, с), 3.45 (1H, м) 3.28 (1H, дд, J = 12,3 Гц) 3.19 (1H, дд, J = 12,7 Гц) 2.23 (3H, м), 2.04 - 1.88 (6H, м), 1.37 (3H, д, D = 7 Гц)
Пример 102
(3S)-{ [2S, 3S)-2-Ацетилтио-1-оксо-3-фенилбутил]-амино}-1- этоксикарбонилметил-2,3,4,5-тетрагидро-1H-[1]бензазепин-2-он
0,40 г (1,53 ммоль) (3S)-Амино-1-этоксикарбонилметил-2,3-4,5- тетрагидро-1H-[1] бензазепин-2-она и 0,40 г (1,68 ммоль) (2S,3S)-2-ацетилметил-3-фенилбутановой кислоты, полученной по примеру 101 (g), подвергают взаимодействию тем же способом, что и по примеру 101 (h). Таким образом получают 0,37 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7.10 (9H, м), 6.99 (1H, д. J = 7 Гц), 4.77 (1H, д, J = 17 Гц), 4.50 (1H, м), 4.34 (1H, д, J = 17 Гц), 4.22-4.13 (3H, м), 3.42-3.30 (2H, м), 2.71 -2.54 (2H, м), 2.22 (3H, с), 1.81 (1H, м), 1.33 (3H, д, J = 7 Гц), 1.25 (3H, т, J = 7 Гц).
Пример 103
{ 3R -[3α,6α(S*),9aβ]}- -6- {[(2S, 3S)-1-Оксо-3-фенил-2-тиобутил]амино} октагидро-5-оксотиазол[3,2-a]- азепин-3-карбоновая кислота
0,25 г (0,539 ммоль) метил-{3R, -[3α,6α(S*),9aβ]}- -6-{[(2S, 3S)-2-ацетилтио-1-оксо-3-фенилбутил] амино}октагидро-5-оксотиазол [3,2-a]азепин-3-карбоксилата, полученного по примеру 101, растворяют в 10 мл этанола. К полученному раствору добавляют 10 мл 1 н водного раствора гидроксида лития в атмосфере азота при 0oC. Полученную таким образом смесь перемешивают при комнатной температуре в течение 1 часа и затем вновь охлаждают до 0oC с последующим установлением pH до 1 с помощью разбавленной соляной кислоты. Из реакционной смеси отгоняют этанол при пониженном давлении. К остатку прибавляют воду и дихлорметан с последующей экстракцией. Затем органическую фазу промывают насыщенным водным раствором хлорида натрия и сушат над сульфатом натрия, концентрируют. Таким образом получают 0,15 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7.61 (1H, д, J = 6 Гц), 7.34-7.18 (5H, м), 5.29 (1H, дд, J = 7, 2 Гц), 5.06 (1H, м), 4.62 (1H, дд, J = 11, 7 Гц), 3.51 (1H, дд, J = 8, 7 Гц) 3.45 (1H, м), 3.35 (1H, дд, J = 12, 2 Гц), 3.21 (1H, дд, J = 12, 7 Гц), 2.10-1.90 (6H, м), 1.73 (1H, дд, J = 8 Гц) 1.39 (3H, д, J = 7 Гц)
Пример 104
1-Карбоксиметил-3-{[2S, 3S)-1-оксо-3-фенил-2-тиобутил]амино}- 1H-[1]бензазепин-2-он
0,35 г (0,726 ммоль) (3S)-{[2S, 3S)-2-ацетилтио-1-оксо-3-фенилбутил] амино] -1- этоксикарбонилметил-2,3,4,5-тетрагидро-1H[1] бензазепин-2-она, полученного по примеру 102, растворяют в 10 мл этанола. К полученному раствору добавляют 10 мл 1 н водного раствора гидроксида натрия в атмосфере азота при 0oC. Полученную таким образом смесь перемешивают при комнатной температуре в течение 1 часа и затем устанавливают pH до 1 добавлением соляной кислоты при 0oC. Добавляют воду и выпавшие при этом кристаллы собирают фильтрацией. Таким образом получают 0,25 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3 ) δ: 7.34 - 7.13 (9H, м), 4.68 (1H, д, J = 17 Гц), 4.52 (1H, м), 4.45 (1H, д, J = 17 Гц), 3.47 (1H, дд, J = 8 Гц), 3.41 (1H, дкв, J = 8, 7 Гц), 3.23 (1H, м), 2.71 -2.56 (2H, м), 1.83 (1H, м), 1.69 (1H, д, J = 8 Гц), 1.34 (3H, дд, J= 7 Гц).
Пример 105.
Метил - [3R-[3α,6α(S*),9aβ]}- -6-[[(2S, 3S)-2-ацетилтио-1-оксо-3,4-диметилпентил]амино]октагидро-5- оксотиазол[3,2-a]азепин-3-карбоксилат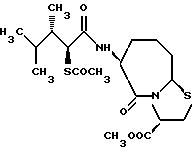
(a) (2S, 3S)-2-ацетилтио-3,4-диметилпентановая кислота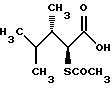
Используя 3,70 г (28,2 ммоль) (R)-3,4-диметилпентановой кислоты в качестве исходного продукта и по тому же способу, что и в примере 101 (a) - (g) получают 1,2 г (2S, 3S)-2-ацетилтио-3,4-диметилпентановой кислоты.
1H-ЯМР (400 МГц, CDCl3) δ: 4.21 (1H, д, J = 8 Гц), 2.38 (3H, с), 1.87 (1H, м), 1.63 (1H, м), 0.97 (3H, д, J = 7 Гц), 0.93 (3H, д, J = 7 Гц), 0.80 (3H, д, J = 7 Гц)
(b) метил- [3R-[3α,6α(S*),9aβ]} -6-{[2S, 3S)-2-ацетилтио-1-оксо-3,4-диметилпентил]амино}октагидро-5-оксо тиазол[3,2-a]азепин-3-карбоксилат
0,275 г (1,35 ммоль) (2S, 3S)-2-ацетилтио-3,4-диметилпентановой кислоты, полученной по способу выше, и 0,300 г (1,23 ммоль) метил-{3R- -[3α,6α(S*),9aβ]}- -6-аминооктагидро-5-оксотиазол[3,2-a] -3-карбоксилата подвергают взаимодействию тем же способом, что и по примеру 101 (h). Таким образом получают 0,260 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 5.28 (1H, дд, J = 6, 2Гц) 5.02 (1H, д, J = 9 Гц), 4.54 (1H, м), 3.95 (1H, д, J = 9 Гц), 3.79 (3H, с), 3.28 (1H, дд, J = 12, 2 Гц), 3.20 (1H, дд, J = 132, 7 Гц), 2.36 (3H, с), 2.10 - 1.87 (6H, м), 1.72 - 1.60 (2H, м), 0.94 (3H, д, J = 7 Гц), 0.89 (3H, д, J = 7 Гц), 0.75 (3H, д, J = 7 Гц)
Пример 106
(3S)-[[2S, 3S)-2-Ацетилтио-1-оксо-3,4-диметил-1-оксапентил] амино]-1 -этоксикарбонилметил-2,3,4,5-тетрагидро-1-H-[1]-бензазепин-2-он
0,500 г (1,91 ммоль) (3S)-амино-1-этоксикарбонилметил-2,3,4,5-тетрагидро-1H-[1] бензазепин-2-он и 0,430 г (2,1 ммоль) (2S, 3S)-2-ацетилтио-3,4-диметилпентановую кислоту подвергают взаимодействию тем же способом, что и по примеру 101 (h).
Таким образом получают 0,420 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7.32-7.10 (4H, м), 4.78 (1H, д, J = 17 Гц), 4.50 (1H, м) 4.34 (1H, д, J = 17 Гц), 4.22-4.14 (3H, м) 3.87 (1H, д, J = 10 Гц), 3.42-3.32 (1H, м) 2.75-2.63 (1H, м) 2.35 (3H, с) 2.02-1.86 (3H, м) 1.25 (1H, т, J = 7 Гц) 0.91 (3H, д, J = 7 Гц) 0.85 (3H, д, J = 7 Гц) 0.72 (3H, д, J = 7 Гц)
Пример 107 [3R [3α,6α(S*),9aβ]}- 6-[[(2S, 3S)-3,4-Диметил-1-оксо-2-тиопентил]амино]октагидро-5-оксотиазол-[3,2-a] азепин-3-карбоновая кислота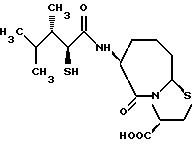
Тем же способом, что и по примеру 104 и исходя из 0,200 г (0,465 ммоль) метил [3R- [3α,6α(S*),9aβ]}- 6-[[(2S, 3S)-2-ацетилтил-1-оксо-3,4-диметилпентил/амино/октагидро-5-оксо-тиазол-[3,2-a] азепин-3-карбоксилата, полученного по примеру 105, получают 0,150 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7.42 (1H, д, J = 6 Гц), 5.29 (1H, дд, J = 6, 2 ГЦ) 5.07 (1H, м) 4.65 (1H, дд, J = 10, 6 Гц), 3.35 (1H, дд, J = 12, 2 Гц), 3.23 (1H, дд, J = 12, 7 Гц) 3.14 (1H, дд, J = 9, 8 Гц) 2.25-1.92 (6H, м) 1.93 (1H, д, J = 9 Гц) 1.82-1.62 (2H, м) 0.95 (3H, д, J = 7 Гц) 0.84 (3H, д, J = 7 Гц) 0.77 (3H, д, J = 7 Гц)
Пример 108
1-Карбоксиметил-(3S)[[(2S, 3S)-3,4-диметил-1-оксо-2-тиопентил]амино]-2,3,4,5-тетрагидро-1H-[1]бензазепин -2-он
Тем же способом, что и по примеру 104 и исходя из 0,300 г (0,67 ммоль) (S)-[[(2S, 3S)-2-ацетилтио-1-оксо-3,4-диметил-1-оксопентил] амино] -1-этоксикарбонилметил -2,3,4,5-тетрагидро-1H-[1] бензазепин-2-она, полученного по примеру 106, получают 0,200 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7.36-7.13 (4H, м) 7.06 (1H, д, J = 7 Гц) 4.72 (1H, д, J = 17 Гц) 4.53 (1H, м) 4.43 (1H, д, J = 17 Гц) 3.28 (1H, м) 3.07 (1H, т, J = 9 Гц) 2.70 (1H, м) 2.61 (1H, м) 2.15 (1H. м) 1.99 (1H, м) 1.90 (1H, т, J = 8 Гц) 1.72 (1H, м), 0.91 (3H, д, J = 7 Гц) 0.79 (3H, д, J = 7 Гц) 0.72 (3H, д, J = 7 Гц)
Примеры 109 - 138
Соединения, которые описаны далее в примерах 109 - 138, синтезированы в соответствии со способами примеров 101 - 108 при использовании соответствующих исходных веществ.
Пример 109
[4S[4 α,7α (R*), 12b β ]]-7-[[(2S)-1-Оксо-2-тиопропил]амино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 7.65 (1H, д, J = Гц) 7.50 (2H, д, J = 8 Гц) 7.43 (1H, как т, J = 8 Гц) 7.38-7.33 (3H, м) 7.05 (1H, д, J = 8 Гц) 5.68 (1H, квинт, J = 6 Гц) 5.50 (1H, ушир д) 5,23 (1H, ушир. д)3.56 (1H, дд, J = 17, 6 Гц) 3.46 (1H, квинт, J = 7 Гц) 2.90 (1H, дд, J = 17, 13 Гц) 2.53 (1H, м), 2.32 (1H, м) 2.14 (1H, д, J = 10 Гц) 2.05-1.70 (4H, м) 1.46 (3H, д, J = 7 Гц)
Пример 110
[4S [4α,7α(R*),12bβ]]- -7-[[(2S)-4-Метил-1-оксо-2-тиопентил]амино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота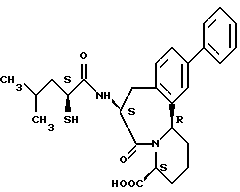
1H-ЯМР (400 МГц, CDCl3) δ; 7.51 (3H, м) 7.44 (2H, как т, J = 8 Гц) 7.39-7.32 (3H, м) 7.08 (1H, д, J = 8 Гц) 5.71 (1H, квинт, J = 6 Гц) 5.52 (1H, м) 5.25 (1H, м) 3.60 (1H, дд, J = 17, 6 Гц) 3,37 (1H, как кв, J = 7 Гц) 2.91 (1H, дд, J = 17, 13 Гц) 2.55 (1H, м) 2.36 (1H, м) 2.05-1.72 (6H, м) 2.03 (1H, д, J = 8 Гц) 1.60 (1H, м) 0.96 (3H, д, J = 7 Гц) 0.92 (3H, д, J = 7 Гц)
Пример 111
[4S [4α,7α(R*),12bβ]]- -7-[[(2S)-1-Оксо-2-тиобутил] амино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 7.59 (1H, д, J = 8 Гц) 7.51 (2H, д, J = 8 Гц) 7.44 (2H, как т, J = 7 Гц) 7.39-7.32 (3H, м) 7.09 (1H, д, J = 8 Гц) 5.71 (1H, квинт, J = 6 Гц) 5.54 (1H, м) 5.26 (1H, м) 3.62 (1H, дд, J = 17, 6 Гц) 3,27 (1H, как кв, J = 7 Гц) 2.94 (1H, дд, J = 17, 13 Гц) 2.56 (1H, м) 2.37 (1H, м) 2.08-1.72 (6H, м) 2.04 (1H, д, J = 8 Гц) 1.04 (3H, т, J = 7 Гц)
Пример 112
[4S- [4α,7α(R*),12bβ]]- 7-[(1-Оксо-2-фенил-2-тиоэтил)амино]-6-оксо-11- фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4- карбоновая кислота (изомер A)
A:B = 2:1
1H-ЯМР (400 МГц, CD3OD) δ; 7.57-7.26 (13H, м) 7.19 (1H, д, J=8) 5.78 (1H, дд, J=9, 6 Гц) 5.67 (1H, м) 5.16 (1H, как д) 3.50 (1H, дд, J=17, 6 Гц) 3.12 (1H, дд, J=17, 13 Гц) 2.58 (1H, м) 2.40 (1H, м) 2.15-1.76 (4H, м)
Пример 113
[4S- [4α,7α(R*),12bβ]]- 7-[(1-Оксо-2-фенил-2-тиоэтил)амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота (изомер B)
A:B = 1:2
1H-ЯМР (400 МГц, CD3OD) δ: 7.57-7.27 (13H, м) 7.08 (1H, д, J=8 Гц) 5.77 (1H, дд, J=9, 6 Гц) 5.67 (1H, м) 5.20 (1H, как д) 3.49 (1H, дд, J=17, 6 Гц) 3.06 (1H, дд, J=17, 13 Гц) 2.60 (1H, м) 2.42 (1H, м) 2.17-1.75 (4H, м)
Пример 114
[4S- [4α,7α(R*),12bβ]]- 7-[[(2R)-3-Метил-1-оксо-2-тиобутил]]амино]-6-оксо-11- фенил] -1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4- карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 7.51 (1H, д), J=8 Гц) 7.47-7.24 (7H, м) 7.03 (1H, д, 8 Гц) 5.67 (1H, квинт, J=6 Гц) 5.47 (1H, м) 5.20 (1H, как д) 3.57 (1H, дд, J= 17, 6 Гц) 3.09 (1H, т, J=7 Гц) 2.89 (1H, дд, J=17, 13 Гц) 2.50 (1H, м) 2.31 (1H, м) 2.20 (1H, секстет, J=7 Гц) 2.02-1.50 (4H, м) 1.85 (1H, д, J=8 Гц) 1.01 (3H, д, J=7 Гц) 0.98 (3H, д, J=7 Гц)
Пример 115
[3R- [3α,6α(S*),9aβ]]- 6-[[1-Оксо-3-фенил-2(S)тиопропил]амино]-2,2-диметил-5- оксооктагидротиазол-[3,2-a]азепин-3-карбоновая кислота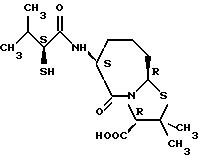
1H-ЯМР (400 МГц, CDCl3) δ: 7.61(1H, д, J=6 Гц) 7.31-7.19 (5H, м) 5.12 (1H, д, J=10 Гц) 4.74 (1H, с) 4.53 (1H, как дд, J=12, 6 Гц) 3.60 (1H, дт, J= 9, 7 Гц) 3.26 (1H, дд, J=14, 6 Гц) 3.12 (1H, дд, J=14, 7 Гц) 2.25-2.13 (1H, м) 1.99 (1H, д, J=9 Гц) 2.07-1.84 (4H, м) 1.60-1.50 (1H, м) 1.55 (3H, с) 1.51 (3H, с)
Пример 116
[3R- [3α,6α(S*),9aβ]]- 6-[[3-Метил-1-оксо-2(S)-тиобутил] амино] -2,2- диметил-5-оксооктагидротиазол-[3,2-a]азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ: 7.81 (1H, д, J=6 Гц) 5.15 (1H, д, J=10 Гц) 4.79 (1H, с) 4.61 (1H, м) 3.21 (1H, дт, J=9, 6 Гц) 2.33-1.88 (6H, м) 1.83 (1H, д, J=9 Гц) 1.69-1.57 (1H, м) 1.56 (3H, с) 1.52 (3H, с) 1.04 (3H, д, J=7 Гц) 0.98 (3H, д, J=7 Гц)
Пример 117
[4R [4α,7α(R*),12bβ]]- 7-[[(2S, 3R)-3-Метил-1-оксо-(2-тиопентил]амино] 6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота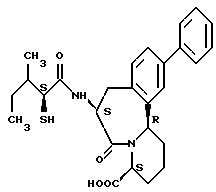
1H-ЯМР (400 МГц, CDCl3) δ; 7.91 (1H, д, J = 8 Гц), 7.51 (2H, д, J = 8 Гц), 7.44 (2H, т как т, J = 8 Гц), 7.38-7.32 (3H, м), 7.06(1H, д, J = 8 Гц), 5.71 (1H, квинт, J = 6 Гц), 5.52 (1H, ушир.д), 5.23 (1H, м), 3.58 (1H, дд, J = 17, 6 Гц), 3.39 (1H, дд, J = 9, 7 Гц), 2.91 (1H, дд, J = 17, 13 Гц), 2.54 (1H, м), 2,32 (1H, м) 2.12 (1H, септет, J = 7 Гц), 2.00 (1H, м), 1.87 (1H, м), 1.80 (1H, д, J = 8 Гц), 1.82-1.70 (2H, м), 1.51 (1H, м), 1.34 (1H, м), 0.97 (3H, д, J = 7 Гц), 0.93 (3H, т, J = 7 Гц)
Пример 118
[3R [3 α,6α (S*), 9a β ]]-6-[[3-(4-метоксифенил)-оксо- 2(S)-тиопропил] амино]-2,2-диметил-5-оксооктагидротиазол[3,2-a]азепин- 3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 7.63 (1H, д, J = 6 Гц), 7.12 (2H, д, J = 8 Гц), 6.82 (2H, д, J = 8 Гц), 5.12 (1H, д, J = 10 Гц), 4.74 (1H, с), 4,54 (1H, дд, J = 11, 6 Гц), 3.78 (3H, с), 3,57 (1H, дт, J = 9, 7 Гц), 3.18 (1H, дд, J = 14, 6 Гц), 3.07 (1H, дд, J = 14, 7 Гц), 2.25-2.14 (1H, м), 1.98 (1H, д, J = 9 Гц), 2.07-1.84 (4H, м), 1.60-1.50 (1H, м), 1.55 (3H, с), 1.51 (3H, с)
Пример 119
[4S- [4α,7α(R*),12bβ]]- 7-[[(2S)-3-(4-Фтор-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][a]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, DMSO-d6) δ; 8.37 (1H, д, J = 7 Гц), 7.62 (2H, д, J = 8 Гц), 7.46 (3H, т, J = 8 Гц), 7.41 (1H, с), 7,35 (1H, т, J = 8 Гц), 7.29 (2H, дд, J = 8, 6 Гц), 7,19 (1H, д, J = 8 Гц), 7.12 (2H, т, J = 8 Гц), 5.62-5.71 (2H, м), 5.05 (1H, м), 3.94 (1H, м), 3.87 (1H, м), 3.19 (1H, дд, J = 14, 7 Гц), 2.95 (1H, дд, J = 17, 13 Гц), 2.88-2.80 (2H, м), 2.52 (1H, м), 2.22 (1H, м), 1.96 (1H, м) 1.65-1.80 (3H, м)
Пример 120
[4S- [4α,7α(R*),12bβ]]- 7-[[(2R)-3-(4-Фторфенил)-1-оксо-2-тиопропил] амино] - 6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2] бензазепин-4-карбоновая кислота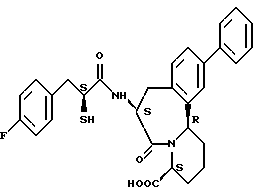
1H-ЯМР (400 МГц, DMSO-d6) δ; 8.31 (1H, д, J = 7 Гц), 7.61 (2H, д, J = 8 Гц), 7.46 (3H, т, J = 8 Гц), 7.39 (1H, с), 7,35 (1H, т, J = 8 Гц), 7.30 (2H, дд, J = 8, 6 Гц), 7,16 (2H, д, J = 8 Гц), 7.03 (1H, д, J = 8 Гц), 5.58-5.70 (2H, м), 5.06 (1H, м), 3.94 (1H, м), 3.10 (1H, дд, J = 14, 9 Гц), 2.98-2.88 (1H, мм), 2.63 (2H, дд, J = 17, 12 Гц), 2.49 (1H, м), 2.23 (1H, м), 1.97 (1H, м) 1.78-1.63 (3H, м)
Пример 121
[4S- [4α,7α(R*),12bβ]]- 7-[[(2S)-3-(5-Бром-2-тиенил)-1-оксо-2-тиопропил] амино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ 7.67 (1H, д, J = 8 Гц), 7.51 (2H, д, J = 8 Гц), 7.43 (2H, как т, J = 8 Гц), 7.39-7.32 (3H, м), 7,07 (1H, д, J = 8 Гц), 6.89 (1H, д, J = 4 Гц), 6,66 (1H, д, J = 4 Гц), 5.66 (1H, квинт, J = 6 Гц), 5.50 (1H, ушир.д.), 5.22 (1H, м), 3.62-3.49 (2H, м), 3.36 (2H, д, J = 6 Гц), 2.86 (1H, дд, J = 17, 13 Гц), 2.54 (1H, м), 2.34 (1H, м), 2.15 (1H, д, J = 10 Гц), 2.10-1.71 (4H, м)
Пример 122
[4S [4α,7α(R*),12bβ]]- 7-[[(2S)-3-Фенил-1-оксо-2-тиометилпропил]амино] -6-оксо-11-фенил-1,2, 3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, DMSO-d6) δ; 8.29 (1H, д, J = 7 Гц), 7.62 (2H, д, J = 8 Гц), 7.46 (3H, т, J = 8 Гц), 7.41 (1H, с), 7.38-7.17 (7H, м) 5.77-5.66 (2H, м), 5.04 (1H, д как д) 3.07-2.96 (2H, м), 2.90 (1H, м), 2.73-2.64 (2H, м) 2.55 (1H, м), 2.43 (1H, м), 2.29 (1H, м), 2.24 (1H, м) 1.99 (1H, м), 1.78-1.67 (3H, м),
Пример 123
[4S- [4α,7α(R*),12bβ]]- 7-[[(2S)-1-Оксо-2-тиогексил]амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 7.55 (1H, д, J = 7 Гц), 7.51 (2H, д, J = 8 Гц), 7.42 (1H, т, J = 8 Гц), 7.40-7.28 (3H, м), 7.09 (1H, д, J = 8 Гц), 5.71 (1H, квинт, J = 6 Гц), 5.52 (1H, ушир.д), 5.23 (1H, ушир.д), 3.61 (1H, дд, J = 17, 6 Гц), 3.30 (1H, кв, J = 7 Гц), 2.92 (1H, дд, J = 17, 13 Гц), 2.57 (1H, м), 2.37 (1H, м), 2.02 (1H, д, J = 10 Гц), 2.05-1.70 (6H, м), 1.50-1.20 (4H, м), 0.91 (3H, с)
Пример 124
[4S- [4α,7α(R*),12bβ]]- 7-[[(2S)-3-(2-тиенил)-1-оксо-2-тиопропил]амино] -6-оксо- 11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2]бензазепин-4- карбоновая кислота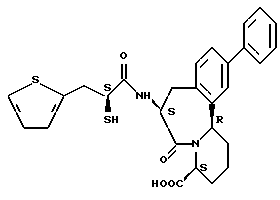
1H-ЯМР (400 МГц, CDCl3) δ ; 7.67 (1H, д, J = 6 Гц), 7.51 (2H, д, J = 8 Гц), 7.43 (2H, как т, J = 8 Гц), 7.38-7.32 (3H, м), 7.19 (1H, д, J = 4 Гц), 7.06 (1H, д, J = 8 Гц), 6.95 (1H, д, J = 4 Гц), 6.90 (1H, д, J = 4 Гц), 5.66 (1H, квинт, J = 6 Гц), 5.49 (1H, ушир.д.), 5.21 (1H, м) 3.64-3.54 (2H, м), 3.50-3.40 (2H, м), 2.84 (1H, дд, J = 17, 13 Гц), 2.54 (1H, м), 2.33 (1H, м), 2.15 (1H, д, J = 10 Гц), 2.10-1.70 (4H, м),
Пример 125
[4S- [4α,7α(R*),12bβ]] -7-[[(2S)-1-Оксо-2-тио-пентил]амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота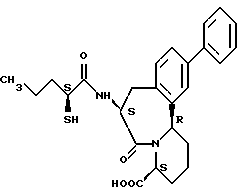
1H-ЯМР (400 МГц, CDCl3) δ; ; 7.57 (1H, д, J = 7 Гц), 7.51 (2H, д, J = 8 Гц), 7.44 (1H, как т, J = 8 Гц), 7.38-7.32 (3H, м), 7.07 (1H, д, J = 8 Гц) 5.71 (1H, квинт), J = 6 Гц), 5.52 (1H, ушир.д.), 5.23 (1H, ушир.д), 3.58 (1H, дд, J = 17, 6 Гц), 3.32 (1H, кв. J = 7 Гц), 2.81 (1H, дд, J = 17, 13 Гц), 2.54 (1H, м), 2.33 (1H, м), 2.09 (1H, д, J = 10 Гц), 2.10-1.67 (6H, м), 1.55-1.35 (2H, м), 0.94 (3H, т, J = 7 Гц)
Пример 126
[4S- [4α,7α(R*),12bβ]]- 7-[[(2S)-3-(3-Метилсульфониламинофенил)-1-оксо-2- тиопропил] амино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо [2,1-a][2]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; ; 7.71 (1H, д, J = 7 Гц), 7.61 (1H, ушир.с), 7.47 (2H, д, J = 8 Гц), 7.40 (2H, как т, J = 8 Гц), 7.36-7.28 (3H, м), 7.22 (1H, д, J = 8 Гц), 7.16 (1H, д, J = 8 Гц), 7.03-6.98 (3H, м), 5.68 (1H, квинт, J = 6 Гц), 5.45 (1H, ушир.д), 5.06 (1H, д как д), 3.63 (1H, м), 3.44 (1H, дд, J = 17, 6 Гц), 3.24-3.06 (2H, м), 2.90 (3H, с), 2.82 (1H, дд, J = 17, 13 Гц), 2.51 (1H, м), 2.32 (1H, м), 2.20 (1H, д, J = 10 Гц), 2.05-1.70 (4H, м)
Пример 127
[4S- [4α,7α(R*),12bβ]]- 7-[[(2S)-3-(3-Тиенил)-1-оксо-2-тиопропил] амино-6-оксо- 11-фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 7.64 (1H, д, J = 7 Гц), 7.50 (2H, д, J = 8 Гц), 7.43 (2H, как т, J = 8 Гц), 7.37-7.32 (3H, м), 7.26 (1H, дд, J = 8 Гц), 7.07 (1H, м), 7.03 (1H, д, J = 8 Гц), 6.96 (1H, д, J = 5 Гц), 5.64 (1H, квинт, J = 6 Гц), 5.47 (1H, ушир. д), 5.18 (1H, как д), 3.62-3.45 (2H, м), 3.30-3.16 (2H, м), 2.80 (1H, дд, J = 17, 13 Гц), 2.52 (1H, м), 2.30 (1H, м), 2.09 (1H, д, J = 10 Гц), 2.04-1.67 (4H, м).
Пример 128
[4S- [4α,7α(R*),12bβ]]- 7-[(2-Метил-1-оксо-2-тиопропил)амино]-6-оксо-11- фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 8.11 (1H, д, J = 7 Гц), 7.50 (2H, д, J = 8 Гц), 7.43 (1H, как т, J = 8 Гц), 7.38-7.32 (3H, м), 7.08 (1H, д, J = 8 Гц), 5.65 (1H, квинт, J = 6 Гц), 5.52 (1H, ушир. д), 5.23 (1H, ушир. д), 3.59 (1H, дд, J = 17, 6 Гц), 2.81 (1H, дд, J = 17, 13 Гц), 2.53 (1H, м), 2.32 (1H, м), 2.32 (1H, с), 2.06-1.70 (4H, м), 1.63 (3H, с), 1.64 (3H, с).
Пример 129
[4S- [4α,7α(R*),12bβ]]- 7-[[(2S)-3-(4-Метилсульфониламинофенил)-1-оксо-2- тиопропил] амино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 7.83 и 7.53 (общий 1H, каждый ушир с), 7.60-7.02 (общий 12H, м), 6.89 и 6.80 (общий 1H, каждый д, J = 8 Гц), 5.66 и 5.64 (общий 1H, каждый квинт., J = 6 Гц), 5.44 (общий 1H, м), 5.08 и 4.97 (общий 1H, каждый ушир. д), 3.54-3.00 (4H, м), 2..83 и 2.82 (общий 3H, каждый с), 2.72 и 2.20 (общий 2H, м), 2.21 и 2.19 (общий 1H, каждый д, J = 10 Гц), 2.04-1.90 (общий 4H, м).
Пример 130
[4S- [4α,7α(R*),12bβ]]- 7-[(2-Циклогексил-1-оксо-2-тиопэтил)амино]-6-оксо-11- фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2]бензазепин-4-карбоновая кислота (изомер A)
1H-ЯМР (400 МГц, CDCl3) δ; 7.60 (1H, д, J = 7 Гц), 7.51 (2H, д, J = 8 Гц), 7.44 (1H, как т, J = 8 Гц), 7.38-7.32 (3H, м), 7.07 (1H, д, J = 8 Гц), 5.62 (1H, квинт, J = 6 Гц), 5.43 (1H, ушир. д), 5.24 (1H, ушир. д), 3.59 (1H, дд, J = 17, 6 Гц), 3.35 (1H, кв, J = 7 Гц), 2.80 (1H, дд, J = 17, 13 Гц), 2.53 (1H, м), 2.33 (1H, м), 2.06-1.63 (9H, м), 1.91 (1H, д, J = 10 Гц), 1.34-1.96 (6H, м).
Пример 131
[4S- [4α,7α(R*),12bβ]]- 7-[(2-Циклогексил-1-оксо-2-тиоэтил)амино] -6-оксо-11- фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота (изомер B)
1H-ЯМР (400 МГц, CDCl3) δ; 7.53 (1H, д, J = 7 Гц), 7.51 (2H, д, J = 8 Гц), 7.44 (1H, как т, J = 8 Гц), 7.38-7.32 (3H, м), 7.05 (1H, д, J = 8 Гц), 5.61 (1H, квинт, J = 6 Гц), 5.51 (1H, ушир. д), 5.21 (1H, ушир. д), 3.57 (1H, дд, J = 17, 6 Гц), 3.42 (1H, дд, J = 7, 6 Гц), 2.90 (1H, дд, J = 17, 13 Гц), 2.52 (1H, м), 2.31 (1H, м), 2.04-1.64 (9H, м), 1.90 (1H, д, J = 10 Гц), 1.36-1.95 (6H, м).
Пример 132
[4S- [4α,7α(R*),12bβ]]- 7-[(2-Циклопентил-1-оксо-2-тиоэтил)амино] -6-оксо-11- фенил-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота (изомер A)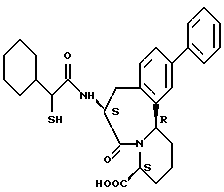
1H-ЯМР (400 МГц, CDCl3) δ; 7.45 (2H, д, J = 8 Гц), 7.40-7.25 (6H, м), 7.02 (1H, д, J = 8 Гц), 5.66 (1H, квинт., J = 6 Гц), 5.47 (1H, м), 5.17 (1H, как д), 3.54 (1H, дд, J = 17, 6 Гц), 3.13 (1H, т, J = 7 Гц), 2.85 (1H, дд, J = 17, 13 Гц), 2.49 (1H, м), 2.33-2.20 (2H, м), 2.00-1.46 (10H, м), 1.97 (1H, д, J = 8 Гц), 1.37-1.23 (2H, м)
Пример 133
[4S- [4α,7α(R*),12bβ]]- -7-[(2-Циклопентил-1-оксо-2-тиоэтил)амино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a] [2] бензазепин-4-карбоновая кислота (изомер B)
1H-ЯМР (400 МГц, CDCl3) δ; ; 7.45 (2H, д, J=8 Гц) 7.40-7.24 (6H, м), 7.03 (1H, д, J=8 Гц) 5.67 (1, квинт, J=6 Гц), 5.47 (1H, м) 5.19 (1H, как д), 3.57 (1, дд, = 17, 6 Гц) 3.31 (1H, т, J=7 Гц), 2.88 (1H, дд, J=17, 13 Гц) 2.50 (1H, м), 2.36-2.22 (2H, м) 1.98 (1H, д, J=8 Гц), 2.02-1.18 (12H, м)
Пример 134
[4S- [4α,7α(R*),12bβ]]- -7-[[(2R)-3-(3-Тиенил)-1-оксо-2-тиопропил]амино] -6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[1,2-a][2]бензазепин-4-карбоновая кислота
H-ЯМР(400 МГц, CDCl3) δ; ;
7.49 (2H, д, J=8 Гц) 7.43 (2H, как т, J=8 Гц), 7.39-7.32 (4H, м) 7.08 (1H, ушир. д, J=8 Гц), 7.02-6.96 (2H,м) 5.64 (1H, квинт,J =6 Гц) 5.47 (1H, ушир. д) 5.18 (1H, м) 3.48 (1H, м),3.40-3.25 (2H, м) 3.13 (1H, дд, J=17, 6 Гц) 2.65 (1H, дд, J=17, 13 Гц) 2.52 (1H, м) 2.31 (1H, м) 2.15 (1H, д, J=10 Гц) 2.04-1.68 (4H, м)
Пример 135
[4S- [4α,7α(R*),12bβ]]- -7-[(3-Этил-1-оксо-2-тиопентил)амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота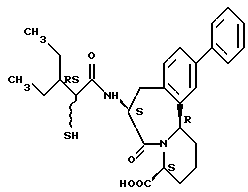
1H-ЯМР (400 МГц, DMSO-d6) δ; ;
8.42 и 8.38 (общий 1H, каждый д, J=7 Гц), 7.62 (2H, д, J=8 Гц) 7.46 (3H, т, J= 8 Гц) 7.41 (1H,с), 7.35 (1H, т, J=8 Гц) 7.18 (общий 1H, каждый д, J=8 Гц) 5.77-5.65 (общий. 2H, м) 5.06 (общий 1H, как д), 3.58 и 3.54 (общий 1H, каждый т, J= 7 Гц), 3.30 -3,17 (общий 1H, м) 2.58-2.47 (общий 1H, м), 2.23 (1H, м) 1.98 (1H, м) 1.80-1.60 (8H, м), 0.87 (3H, т, J=7 Гц) 0.82(3H, т, J=7 Гц)
Пример 136
[4S-[4α,7α(R*),12bβ]]- -7-[[(3S)-3-Гидрокси-1-оксо-2-тиобутил] амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2]бензазепин-4-карбоновая кислота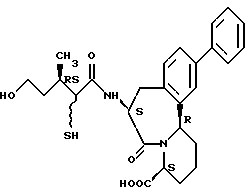
1H-ЯМР (400 МГц, CDCl3) δ; ;
7.56-7.92 (1H, м) 7.52-7.30 (7H, м), 7.10-6.94 (1H, м) 5.81-5.66 (1H, м), 5.56-5.48 (1H, м) 5.26-5.19 (1H, м), 3.68-2.85 (3H, м) 2.53 (1H, ушир. д) 2.34 (1H, ушир.д.), 2.08-1.70 (5H, м) 2.17 (общий 1H, каждый д, J=8 Гц), 2.05 и 2.17 (общий 1H, каждый д, J=8 Гц), 1.40 и 1.96 (общий 3H, каждый, J=7 Гц)
Пример 137
[4S- [4α,7α(R*),12bβ]]- -7-[[(2S, 3R)-3-Метокси-1-оксо-2-тиобутил]амино] -6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-а][2]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ;;
7.84 (1H, д, J=7 Гц) 7.51 (2H, д, J=8 Гц), 7.43 (2H, т, J=8 Гц) 7.38-7.31 (3H, м), 7.09 (1H, д, J=8 Гц) 5.61 (1H, квинт, J=6 Гц), 5.53 (1H, м) 5.25 (1H, м) 3.70 (1H, квинт, J=7 Гц), 3.62 (1H, дд, J=17, 6 Гц) 3.40 (3H, с), 3.39 (1H, т, J=7 Гц) 2.94 (1H, дд, J=17, 13 Гц), 2.55 (1H, м) 2.36 (1H, м) 2.27 (1H, д, J=8 Гц), 2.08-1.72 (4H, м) 1.30 (3H, д, J=7 Гц)
Пример 138
[4S- [4α,7α(R*),12bβ]]- 7-[(3-Метил-1-оксо-2-тиогексил)амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота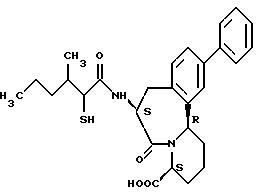
1H-ЯМР (400 МГц, CDCl3) δ; ; 7.70 и 7.61 (общий 1H, каждый д, J = 7 Гц), 7.50 (2H, д, J = 8 Гц), 7.43 (2H, т, J = 8 Гц), 7.40-7.30 (3H, м), 7.07 и 7.06 (общий 1H, каждый д, J = 8 Гц), 5.71 (1H, квинт, J = 6 Гц), 5.52 (1H, м), 5.23 (1H, м), 3.59 (1H, м), 3.29 (1H, дд, J = 17, 12 Гц), 2.92 (1H, дд, J = 17, 12 Гц), 2.54 (1H, м), 2.34 (1H, м), 2.10-1.94 (2H, м), 1.94-1.82 (1H, м), 1.80-1.70 (1H, м), 1.56 (1H, м), 1.41 (1H, м), 1.35-1.14 (2H, м), 1.03 и 1.02 (общий 3H, каждый д, J = 7 Гц), 0.92 и 0.91 (общий 3H, каждый т, J = 7 Гц)
Пример 139
Трифторацетат [4S- [4α,7α(R*),12bβ]]- 7-[[(2S, 3S)-3-метил-2-(4-морфолинил) ацетилтио-1-оксопентил] амино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b - октагидропиридо[2,1-a][2]бензазепин-4-карбоновой кислоты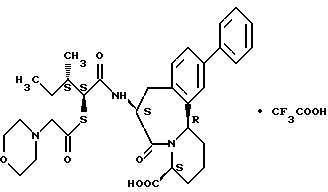
(а) дифенилметил [4S- [4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-3- метил-1-оксо-2-тиопентил] амино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b- октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат
0,500 г (0,730 ммоль) Дифенилметил [4S- 4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил]амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилата, полученного по примеру C-6, растворяют в 10 мл сухого этанола. К полученному таким образом раствору прибавляют 10 мл 12% (по весу) аммиачно-спиртового раствора при охлаждении льдом. Полученную смесь перемешивают при комнатной температуре в течение 2 часов, концентрируют при пониженном давлении и разбавляют дихлорметаном. Промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и затем концентрируют. Таким образом получают 0,468 г указанного в заголовке продукта (выход 99%).
1H-ЯМР (400 МГц, CDCl3) δ: ; 7.72 (1H, д, J = 6 Гц), 7.50-6.92 (17H, м), 6.70 (1H, д, J = 8 Гц), 6.30 (1H, с) 5.67 (1H, дт, J = 13, 6 Гц), 5.49 (1H, м) 5.42 (1H, как д, J = 4 Гц), 3.45 (1H, дд, J = 18, 6 Гц), 3.28 (1H, дд, J = 8, 7 Гц), 2.61 (1H, дд, J = 18, 13 Гц), 2.55-2.45 (2H, м), 1.95 (1H, д, J = 8 Гц), 1.62-2.08 (6, м), 1.37-1.25 (1H, м), 1.06 (3H, д, J = 7 Гц), 0.96 (3H, т, J = 7 Гц).
(b) дифенилметил [4S- [4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-3-метил-2-(4-морфолинил)ацетилтио-1-оксопентил]амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоксилат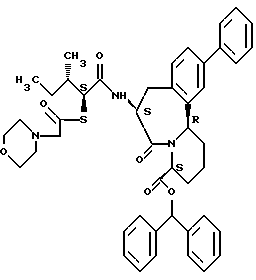
0,262 г (1,44 ммоль) гидрохлорида 4-морфолинилуксусной кислоты растворяют в 7,2 мл дегазированного сухого диметилформамида. К раствору, полученному таким образом, добавляют 0,176 г (1,08 ммоль) карбодинилимидазола при охлаждении льдом. Полученную смесь перемешивают при комнатной температуре в течение 1,5 часа. Полученную смесь вновь охлаждают льдом и прибавляют по каплям раствор 0,467 г (0,72 ммоль) дифенилметил [4S- [4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-метил-1-оксо-2-тиопентил]амино]-6-оксо-11-фенил-1,2,3,4,6,7,8,12b - октагидропиридо[2,1-a] [2] -бензазепин-4-карбоксилата, полученного выше (a), в дегазированном тетрагидрофуране (7,2 мл). Полученную смесь перемешивают при комнатной температуре в течение 1 часа и затем концентрируют при пониженном давлении до уменьшения объема жидкости наполовину. Добавляют этилацетат и полученную смесь промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и затем концентрируют. Таким образом получают 0,500 г целевого производного морфолина (выход 90%).
1H-ЯМР (400 МГц, CDCl3) δ: ; 7.54 (1H, д, J = 6 Гц), 7.49-6.92 (17H, м), 6.67 (1H, д, J = 8 Гц), 6.29 (1H, с), 5.64 (1H, дт, J = 13, 6 Гц), 5.44-5.49 (1H, м), 5.40-5.36 (1H, м), 3.99 (1H, д, J = 7 Гц), 3.80 (4H, т, J = 5 Гц), 3.41 (1H, дд, J = 16, 7 Гц), 3.35 (2H, с), 2.71-2.60 (4H, м), 2.60-2.44 (2H, м), 2.21-1.59 (7H, м), 1.31-1.91 (1H, м), 1.06 (3H, д, J = 7 Гц), 0.94 (3H, т, J = 7 Гц)
(c) трифторацетат [4S- [4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-3-метил-2-(4-морфолинил)ацетилтио-1-оксопентил]амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]-бензазепин-4-карбоновой кислоты
В раствор 0,500 г (0,65 ммоль) дифенилметил [4S [4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-3-метил-2-(4-морфолинил)-ацетилтио-1-оксопентил] амино] -6-оксо-11- фенил-1,2,3,4, 6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоксилата, полученного выше (b), и 0,54 мл (5,00 ммоль) анизола в дихлорметане (6,2 мл) добавляют по каплям 0,95 мл (12,00 ммоль) трифторуксусной кислоты при -50oC. Полученную таким образом смесь нагревают до комнатной температуры и перемешивают в течение 3 часов. Реакционную смесь концентрируют при пониженном давлении и остаток перекристаллизовывают из диэтилового эфира-гексана. Таким образом получают 0,414 г указанного в заголовке соединения (выход 89%)
1H-ЯМР (400 МГц, CDCl3) δ: ; 7.59-7.30 (8H, м), 7.09 (1H, д, J = 9 Гц), 5.74-5.65 (1H, м), 5.54-5.47 (1H, м), 5.20-5.14 (1H, м), 4.06 (1H, д, J = 7 Гц), 3.81 (4H, м), 3.67 (2H, с), 3.54 (1H, дд, J = 17, 6 Гц), 3.52-3.30 (2H, ушир. ), 3.02-2.90 (5H, м), 2.55 (1H, ушир.д), 2.36 (1H, ушир.д.), 2.17-1.74 (5H, м), 1.66-1.55 (1H, м), 1.26-1.14 (1H, м), 1.03 (3H, д, J = 7 Гц), 0.92 (3H, т, J = 7 Гц)
Пример 140
Трифторацетат [4S-[4α,7α(R*), 12bβ]]-7-[[(2S, 3S)-2-(Диэтиламино)ацетилтио-3-метил-1-оксопентил] амино] -6-оксо-11-фенил-1,2,3,4,6,7,8,12b октагидропиридо[2,1-а][2]бензазепин-4-карбоновой кислоты
Тем же способом, что и по примеру 139, за исключением того, что вместо гидрохлорида 4-морфолинилуксусной кислоты применяют 0,526 г (3,14 ммоль) гидрохлорида N, N-диметиламино-уксусной кислоты, и исходя из 1,1 г (1,57 ммоль) дифенилметил [4S, [4α,7α (R*), 12b β ]]-7-[[(2S, 3S)-3-метил-1-оксо-2-тиопентил] -амино] -6-оксо-11-фенил -1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a] [2] бензазепин-4-карбоксилата получают 0,896 г указанного в заголовке соединения с выходом 81% двумя стадиями
1H-ЯМР (400 МГц, CDCl3) δ: 7.79 (1H, д, J = 7 Гц), 7.50 - 7.03 (8H, м), 5.75 (1H, дт, J = 13, 6 Гц), 5.55 - 5.48 (1H, м), 5.18 - 5,16 (1H, м), 4.22 (1H, д, J = 7 Гц), 4.14 - 4.04 (2H, м), 3.46 (1H, дд, J = 17, 6 Гц), 3.30 - 3.20 (4H, м), 2.98 (1H, дд, J = 17, 13 Гц), 2.57 (1H, ушир.д J = 12 Гц), 2.40 (1H, ушир. д, J = 12 Гц), 2.17 - 1.74 (5H, м), 1.67 - 1.56 (1H, м), 1.25 (6H, т, J = 7 Гц), 1.28 - 1.16 (1H, м), 1.05 (3H, д, J = 7 Гц), 0.92 (3H, т, J = 7 Гц)
Пример 141
Трифторацетат [4S- [4α,7α (R*), 12b β ]]-7-[[(2S,3S)- 2-(1-Имидазолино)ацетилтио-3-метил-1-оксопентил]амино]-6-оксо-11 -фенил-1,2,3,4,6,7,8,12b-октагидропиридо-[2,1-a][2]бензазепин-4-карбоновой кислоты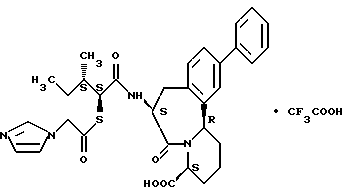
Тем же способом, что и по примеру 139, за исключением того, что вместо гидрохлорида 4-морфолинилуксусной кислоты при меняют 0,287 г (1,76 ммоль) гидрохлорида 1-имидазолилуксусной кислоты, и исходя из 0,570 г (0,88 моль) дифенилметил [4S- [4α,7α (R*), 12b β ]]-7-[[(2S,3S)-3-метил-1-оксо-2-тиопентил] амино]-6-оксо-11-фенил- 1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a]-[2] бензазепин-4-карбоксилата получают 0,355 г указанного в заголовке соединения в виде белого аморфного продукта с выходом 57% двумя стадиями.
1H-ЯМР (400 МГц, CDCl3) δ: 7.09 (1H, ушир.с), 7.03-6.98 (2H, м), 5.65 (1H, дт, J = 13, 6 Гц), 5.48-5.42 (1H, м), 5.10-5.04 (1H, м), 5.01 (1H, д, J = 18 Гц), 4.92 (1H, д, J = 18 Гц), 4.16 (1H, д, J = 6 Гц), 3.41 (1H, дд, J = 17, 6 Гц), 2.92 (1H, дд, J = 17, 13 Гц), 2.55 (1H, ушир. д), 2.32 (1H, ушир. д), 2.16-2.07 (1H, м), 2.04 - 1.86 (2H, м), 1.83-1.72 (2H, м), 1.67 -1.55 (1H, м), 1.23-1.10 (1H, м), 1.03 (1H, д, J = 7 Гц), 0.92 (3H, т, J = 7 Гц).
Пример 142
(3S)-[[(2S, 3R)-2-Ацетилтио-3-метил-1-оксо-пентил]амино] -2,3,4,5-тетрагидро-1H-[1]бензазепин-2-он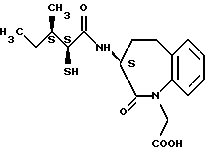
В соответствии с примером 117 синтезируют указанное в заголовке соединение.
1H-ЯМР (400 МГц, CDCl3) δ: 7.60 (1H, ушир.д, J = 7 Гц), 7.33 - 7.44 (4H, м), 4.70 (1H, д, J = 17 Гц), 4.35 (1H, дт, J = 11, 7 Гц) 4.44 (1H, д, J = 17 Гц), 3.35-3.24 (2H, м), 2.74-2.59 (2H, м), 2.06-1.96 (2H, м), 1.74 (1H, д, J = 9 Гц), 1.44 (1H, м), 1.26 (1H, м), 0.87 (6H, м)
Пример 143
Этил (3S)-[[(2S, 3S)-2-ацетилтио-3-метил-1-оксопентил]амино]-4-оксо-2,3,4,5- тетрагидро-1,5-бензоксазепин-5-ацетат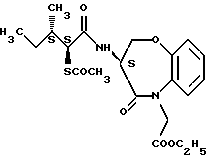
528 мг этил (3S)-амино-4-оксо-2,3,4,5-тетрагидро-1,5-бензоксазепин-5-ацетата и 419 мг (1,1 экв.) (2S, 3S)-2-ацетилтио-3-фенилпентановой кислоты растворяют в 40 мл метиленхлорида. К полученному таким образом раствору добавляют 544 мг (1,1 экв) ЭЭДХ при охлаждении льдом. Полученную смесь далее перемешивают при комнатной температуре в течение 21 часов. Реакционную смесь затем быстро подкисляют 1 н. соляной кислотой при охлаждении льдом и отделяют метиленхлоридную фазу. После двукратной промывки метиленхлоридной фазы насыщенным водным раствором хлорида натрия ее сушат над безводным сульфатом магния и концентрируют. Полученный остаток подвергают очистке хроматографией на колонке с силикагелем (этанол : хлоридом = 1,5 : 98,5 - 4:96). Таким образом получают 370 мг указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl2) δ:
7.14-7.25 (4H, м), 7.04 (1H, д, J = 7 Гц), 4.94 (1H, дд, J = 10, 7 Гц), 4.69 (1H, дд, J = 10, 7 Гц), 4.68 (1H, д, J = 18 Гц), 4.33 (1H, дд, J = 18 Гц), 4.25 (2H, кв. J = 7 Гц), 4.13 (1H, т, J = 10 Гц), 3.92 (1H, д, J = 7 Гц), 2.02 (1H, м), 1.56 (1H, м), 1.26 (3H, т, J = 7 Гц), 1.14 (1H, м), 0.96 (3H, д, J = 7 Гц), 0.85 (3H, т, J = 7 Гц)
Пример 144
(3S)-[[(2S, 3S)-3-Метил-1-оксо-2-тиопентил] -амино] -4-оксо- 2,3,4,5-тетрагидро-1,5-бензоксазепин-5-уксусная кислота
360 мл этил (3S)-[[(2S, 3S)-2-Ацетилтио-3-метил-1-оксопентил]амино]-4-оксо-2,3,4,5- тетрагидро-1,5-бензоксазепин-5-ацетата, полученного по примеру 143, растворяют в 6 мл дегазированного этанола. К полученному таким образом раствору добавляют дегазированный 1 н водный раствор гидроксида натрия при охлаждении льдом. Полученную смесь далее перемешивают при комнатной температуре в течение 320 минут и затем быстро подкисляют добавлением 1 н соляной кислоты с последующим экстракцией хлороформом (15 мл • 2). Органическую фазу промывают водой и затем сушат над безводным сульфатом магния. После отгонки растворителя при пониженном давлении получают 250 мг (выход 83%) указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ:
7.41 (1H, д, J = 7 Гц), 7.18 - 7.29 (4H, м), 4.90 (1H, дт, J = 10, 7 Гц), 4.78 (1H, д, J = 18 Гц), 4.69 (1H, дд, J = 10, 7 Гц), 4.30 (1H, д, J = 18 Гц), 4.22 (1H, т, J = 10 Гц), 3.23 (1H, дд, J = 9, 6 Гц), 1.94 (1H, м), 1.88 (1H, д, J = 9 Гц), 1.53 (1H, м), 1.22 (1H, м), 0.96 (3H, д, J = 6 Гц), 0.87 (3H, т, J = 7 Гц)
Пример 145
[3R- [3α,6αS*),9aβ]]- 6-[[(2R, 3S)-3-Mетил-1-оксо-2-тиопентил] амино] октагидро-5-оксотиазол[3,2-a]азепин-3-карбоновая кислота
В соответствии с примером 144 синтезируют указанное в заголовке соединение.
1H-ЯМР (400 МГц, CDCl3) δ: 7.80 (1H, д, J = 6 Гц) 5.30 (1H, дд, J = 7, 2 Гц) 5.08 - 5.06 (1H, м), 4.63 (1H, дд, J = 11, 6 Гц) 3.37 - 3.33 (2H, м) 3.22 (1H, дд, J = 12, 7 Гц) 2.14 - 1.90 (6H, м) 1.79 (1H, д, J = 9 Гц) 1.75 - 1.64 (1H, м) 1.55 - 1.43 (1H, м) 1.36 - 1.22 (1H, м) 0.92 (3H, д, J = 7 Гц) 0.92 (3H, т, J = 7 Гц)
Пример 146
[3R- [3α,6α (S*), 9a β ]]-6-[[(2R, 3S)-3-Ацетил-тиеметил- 3-метил-1-оксопентил]амино]октагидро-5-оксотиазол[3,2-a]азепин-3- карбоновая кислота
0,200 г (0,550 ммоль) [3R- 3α,6α (S*), 9a β ]]-6-[[(2R, 3S)-3-метил-1-оксо-2-тиопентил] амино] октагидро-5- оксотиазол[3,2-a] азепин-3-карбоновой кислоты, полученной по примеру C-8, и 0,058 мл (0,610 ммоль) уксусного ангидрида растворяют в 6 мл ацетонитрил-тетрагидрофурана (1 : 1). Полученный раствор добавляют по каплям в раствор 0,022 г (0,170 ммоль) хлорида кобальта (II) в 5 мл ацетонитрила. Затем полученную таким образом смесь перемешивают в течение 7 часов, концентрируют при пониженном давлении и добавляют к ней воду с последующей экстракцией этилацетатом. Органическую фазу промывают насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и затем концентрируют при пониженном давлении. Полученный твердый продукт перекристаллизовывают из этилацетата-диэтилового эфира-гексана. Таким образом получают 0,180 г указанного в заголовке соединения (выход 85%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.44 (1H, д, J = 6 Гц) 5.30 (1H, дд, J = 7, 2 Гц) 5.05 (1H, как т, J = 5 Гц) 4.60 (1H, дд, J = 11, 6 Гц) 3.97 (1H, д, J = 7 Гц) 3.35 (1H, дд, J = 12, 2 Гц) 3.21 (1H, дд, J = 12, 7 Гц) 3.28 (3H, с) 2.14 - 1.86 (6H, м) 1.72 - 1.52 (2H, м) 1.24 - 1.10 (1H, м) 1.00 (3H, д, J = 7 Гц) 0.88 (3H, т, J = 7 Гц)
Пример 147
(3S)-[[(2S, 3S)-2-Ацетилтио-3-метил-1-оксопентил]амино]- 2,3,4,5-тетрагидро-1H-[1]бензазепин-2-он
0,547 г (1,5 ммоль) (S)-1-Карбоксиметил-3-[[(2S,3S)-3-метил- 1-оксо-2-тиопентил] амино] -2,3,4,5-тетрагидро-1H-[1] бензазепин-2-она, полученной по примеру C-11, и 0,168 мл (1,650 ммоль) уксусного ангидрида растворяют в 7 мл ацетонитрила. Полученный раствор добавляют по каплям в раствор 0,075 г (0,577 ммоль) хлорида кобальта (II) в 10 мл ацетонитрила. Затем полученную таким образом смесь перемешивают в течение 2 часов, концентрируют при пониженном давлении и к ней добавляют воду с последующей экстракцией этилацетатом. Органическую фазу промывают насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом магния и затем концентрируют при пониженном давлении. Таким образом получают 0,43 г указанного в заголовке соединения в виде бесцветного аморфного продукта.
1H-ЯМР (400 МГц, CDCl3) δ:
7.30 - 7.09 (5H, м) 4.76 (1H, д, J = 17 Гц) 4.49 (1H, дт, J = 11, 7 Гц) 4.39 (1H, д, J = 17 Гц) 3.88 (1H, д, J = 7 Гц) 3.30 (1H, м) 2,70 - 2.50 (2H, м) 2.35 (3H, с) 2.02 - 1.82 (2H, м) 1.53 (H, м) 1.11 (1H, м) 0.93 (3H, т, J = 7 Гц) 0.84 (3H, т, J = 7 Гц)
Примеры 148 - 152
В соответствии со способом, описанным выше в примерах 101 - 108, получают соединения по примерам 148 - 152.
Пример 148
[4S- [4α,7α(R*),12bβ]]- -7-[[(2S, 3S)-3-Метил-1-оксо-2-тиопентил]амино] -6- оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота
1H-ЯМР (400 МГц), CD3OD) δ; 7.69 (2H, д, J = 8 Гц) 7.17 - 7.05 (3H, м) 7.02 (1H, д, J = 8 Гц) 5.69 (1H, квинт, J = 6 Гц) 5.48 (1H, ушир. д, J = 6 Гц) 5.20 (1H, м) 3.52 (1H, дд, J = 17,6 Гц) 3.21 (1H, дд, J = 9,7 Гц) 2.90 (1H, дд, J = 17,13 Гц) 2.50 (1H, м) 2.35 (1H, м) 1.92 - 2.03 (2H, м) 1.92 (1H, д, J = 8 Гц) 1.27 (1H, м) 1.02 (3H, д, J = 7 Гц) 0.93 (3H, т, J = 7 Гц)
Пример 149
[3R- [3α,6α(S*),9aβ]]- 6-[[(2S, 3S)-3-Метил-1-оксо-2-тиопентил]амино] октагидро-5-оксотиазол[3,2-a]азепин-3-карбоновая кислота
1H-ЯМР (400 МГц, CDCl3) δ; 7.78 and 7.84 (общий 1H, каждый д, J = 7 Гц) 5.56 - 4.58 (3H, м) 3.82 - 2.92 (3H, м) 2.34 - 1.45 (9H, м) 1.30 - 1.18 (1H, м) 0.88 - 1.00 (6H, м)
Пример 150
[3R- [3α,6α(S*),9aβ]]- 6-[[(2S, 3S)-4-Метил-1-оксо-2-тиопентил]амино] октагидро-5-оксотиазол[3,2-a]азепин-3-карбоновая кислота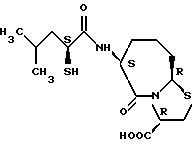
1H-ЯМР (400 МГц, CDCl3) δ; 7.56 - 7.60 (1H, как т) 5.29 (1H, дд, J = 7,3 Гц) 5.08 - 5.06 (1H, м) 4.65 - 4.61 (1H, м) 3.40 - 3.33 (2H, м) 3.23 (1H, дд, J = 12,7 Гц) 2.08 - 1.90 (6H, м) 1.88 - 1.64 (3H, м) 1.60 - 1.52 (1H, м) 0.94 (3H, д, J = 6 Гц) 0.90 (3H, д, J = 7 Гц)
Пример 151
[3R- [3α,6α(S*),9aβ]]- 6-[[(S)-1-Оксо-2-тиогексил] амино] октагидро-5- оксотиазол[3,2-a]азепин-3-карбоновая кислота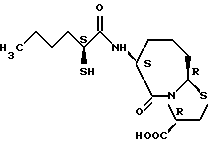
1H-ЯМР (400 МГц, CDCl3) δ; 7,58 (1H, д, J = 6 Гц) 5.30 (1H, дд, J = 7,2 Гц) 5.07 (1H, как т, J = 5 Гц) 4.59 - 4.64 (1H, м) 3.36 (1H, дд, J = 12,3 Гц) 3.30 (1H, дт, J = 8,7 Гц) 3.22 (1H, дд, J = 12,7 Гц) 2.10 - 1.90 (6H, м) 2.00 (1H, д, J = 8 Гц) 1.76 - 1.64 (2H, м) 1.46 - 1.24 (4H, м) 0.90 (3H, т, J = 7 Гц)
Пример 152
[3R- [3α,6α(S*),9aβ]]- 6-[[(2S, 3S)-2-Бензоилтио-3-метил-1-оксопентил] амино]октагидро-5-оксотиазол[3,2-a]азепин-3-карбоновая кислота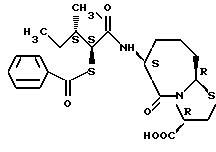
Тем же способом, что и по примеру 146, заменив уксусный ангидрид бензоилхлоридом, получают указанное в заголовке соединение в виде белого продукта (147 мг, выход 51%).
1H-ЯМР (400 МГц, CDCl3) δ; 0.92 (3H, т, J = 7 Гц) 1.06 (3H, д, J = 6 Гц) 1.20 - 1.30 (1H, м) 1.58 - 1.72 (2H, м) 1.90 - 2.03 (5H, м) 2.13 - 2.23 (1H, м) 3.19 (1H, дд, J = 7,12 Гц) 3.33 (1H, дд, J = 2,12 Гц) 4.20 (1H, д, J = 7 Гц) 4.62 (1H, дд, J = 7,11 Гц) 5.02 - 5.08 (1H, м) 5.28 (1H, дд, J = 2,7 Гц) 7.43 - 7.61 (4H, м) 7.97 - 7.99 (2H, м)
В таблицах 1 и 2 представлены данные по биологической активности предложенных новых соединений, описанных в указанных примерах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТИОФОРМАМИДА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2125055C1 |
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2326864C2 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| БИАРИЛЬНОЕ ПРОИЗВОДНОЕ И СОДЕРЖАЩЕЕ ЕГО ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2016 |
|
RU2760373C2 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА | 2004 |
|
RU2332412C2 |
| ПРОИЗВОДНОЕ СУЛЬФОНАМИДА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2014 |
|
RU2667520C9 |
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-HT РЕЦЕПТОРОВ | 2010 |
|
RU2567751C2 |
| ПРОИЗВОДНЫЕ АНТРАНИЛОВОЙ КИСЛОТЫ ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1994 |
|
RU2128644C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА | 2006 |
|
RU2417985C2 |












Производные аминокислоты формулы I, где R1 - водород или ацил; R2 - водород, низший алкил, циклоалкил, фенил, возможно замещенный алкоксилом, галогеном, фенилом или алкилсульфониламиногруппой, или тиенил, возможно замещенный галогеном, m и n- целое число 0, 1 или 2; J - группа формул (А-Д), где R3 - водород или карбоксизащитная группа; R4 водород, Y2-группа -CH2-, -S- или -O-; R10- водород или фенил; R11, R12 - водород, низший алкил; R13 - фенил или группа -HSO2R18, R18 - водород или низший алкил; R14 , R15 - водород; или их фармакологически приемлемые соли обладают ингибирующей активностью в отношении ангиотензин 1-превращающего фермента. 32 з.п. ф-лы, 8 табл. 
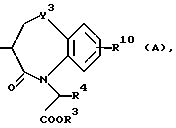


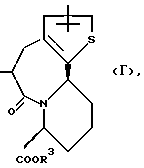


где R1 означает атом водорода или ацильную группу;
R2 означает атом водорода, низшую алкильную группу, циклоалкильную группу, фенил, который может быть замещен алкоксигруппой, атомом галогена, фенилом и алкилсульфониламиногруппой, или тиенил, который может быть замещен атомом галогена;
m и n каждый представляет собой целое число 0, 1 или 2;
J представляет собой группу, выбранную из
группы общей формулы
где R3 - атом водорода или карбоксизащитная группа;
R4 - атом водорода;
Y2 - группа -CH2-, -S- или -O-;
R10 - атом водорода или фенил;
- группы общей формулы
где R3 - атом водорода или карбоксизащитная группа;
R11 и R12 - одинаковые или различные и представляют собой атом водорода или низшую алкильную группу;
n - целое число 0, 1 или 2;
- группы общей формулы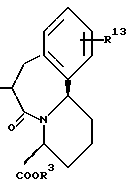
где R3 - атом водорода или карбоксизащитная группа;
R13 - фенил или группа - NHSO2R18, где R18 - атом водорода или низшая алкильная группа;
- группы общей формулы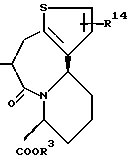
где R3 - атом водорода или карбоксизащитная группа;
R14 - атом водорода;
и группы общей формулы
где R3 - атом водорода или карбоксизащитная группа;
R15 - атом водорода
при условии, что если J означает группу общей формулы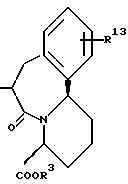
где R13 - атом водорода или фенил,
то должно соблюдаться по крайней мере одно из нижеследующих условий: n не равно 0, R1 не может быть атомом водорода, ацетильный или бензоильной группой и R2 не может быть атомом водорода, низшей алкильной группой и фенилом
или их фармакологически приемлемые соли.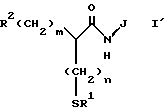
где R1, R2, J, m и n каждый имеет значение, определенное выше.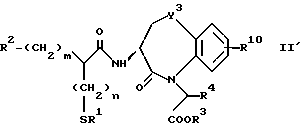
где R1, R2, R3, R10, Y3, m и n, каждый имеет значение, определенное выше.
где R1, R2, R3, R11, R12 и u, m и n каждый имеет значение, определенное выше.
где R1, R2, R3, R13, m и n, каждый имеет значение, определенное выше.
где R1, R2, R3, R14, m и n, каждый имеет значение, определенное выше.
где R1, R2, R3, R15, m и n каждый имеет значение, определенное выше.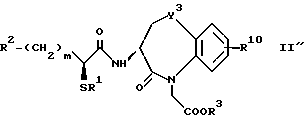
где R1, R2, R3, R10, Y3 и m каждый имеет значение, определенное выше. \\\ 1 где R1, R2, R3, R11, R12, m и u, каждый имеет значение, определенное выше.
\\\ 1 где R1, R2, R3, R11, R12, m и u, каждый имеет значение, определенное выше.
где R1, R2, R3, R13 и m каждый имеет значение, определенное выше.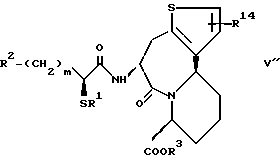
где R1, R2, R3, R14, m каждый имеет значение, определенное выше.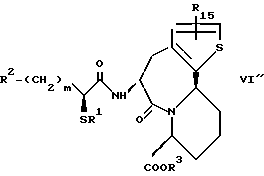
где R1, R2, R3, R15, m каждый имеет значение, определенное выше.
где R1 означает атом водорода или ацильную группу;
J представляет собой группу, выбранную из:
группы общей формулы
где R3 - атом водорода или карбоксизащитная группа;
R4 - атом водорода;
Y2 - группа -CH2-, -S- или -О-;
R10 - атом водорода или фенил;
группы общей формулы
где R3 - атом водорода или карбоксизащитная группа;
R11 и R12 - одинаковые или различные и представляет собой атом водорода или низшую алкильную группу;
u - целое число 0, 1 или 2;
группы общей формулы
где R3 - атом водорода или карбоксизащитная группа;
R13 - фенил или группа - NHSO2R18, где
R18 - атом водорода или низшая алкильная группа;
группы общей формулы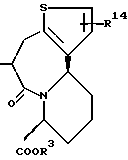
где R3 - атом водорода или карбоксизащитная группа;
R14 - атом водорода;
группы общей формулы
где R3 - атом водорода или карбоксизащитная группа;
R15 - атом водорода.
где R1 представляет атом водорода или ацильную группу;
J имеет значения, охарактеризованные в п.1.
где R1 представляет атом водорода или ацильную группу;
R3 представляет атом водорода или карбоксизащищающую группу;
R11 и R12 являются одинаковыми или отличными друг от друга и каждый представляет атом водорода или низшую алкильную группу;
u, m и n представляют каждый независимо 0, 1 или 2.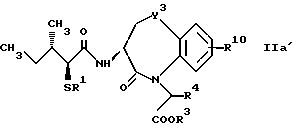
где R1, R3, R10, Y3 каждый имеет значение, определенное выше.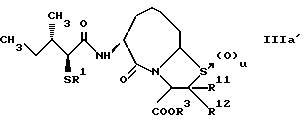
где R1, R11, R12, u каждый имеет значение, определенное выше.
где R1, R3, R13 каждый имеет значение, определенное выше.
где R1, R3, R14 каждый имеет значение, определенное выше.
где R1, R3, R15 каждый имеет значение, определенное выше.
где R1, R3, R4, R10 каждый имеет значение, определенное выше.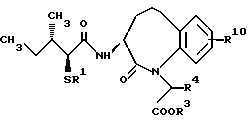
где R1, R3, R4, R10 каждый имеет значение, определенное выше.
где R1, R3, R11, R12 каждый имеет значение, определенное выше.
где R1, R3, R13 каждый имеет значение, определенное выше.
где R1, R3, R14 каждый имеет значение, определенное выше.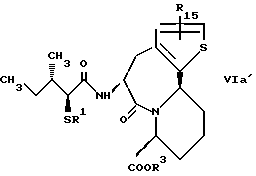
где R1, R3, R15 каждый имеет значение, определенное выше.
28. Производное аминокислоты формулы I по п.1, представленное формулой
29. Производное аминокислоты общей формулы I по п.1, представленное формулой
30. Производное аминокислоты общей формулы I по п.1, представленное формулой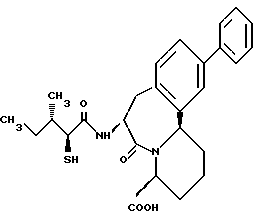
31. Производное аминокислоты общей формулы I по п.1, представленное формулой
32. Производное аминокислоты общей формулы I по п.1, представленное формулой
33. Производное аминокислоты общей формулы I по п.1, представленное формулой
Приоритет по признакам:
11.06.93 - J представляет группу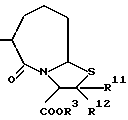
где R11, R12 - атом водорода, R3 - атом водорода или карбоксизащитная группа; R1 - атом водорода или ацильная группа, R2 - фенил, который может быть замещен алкоксигруппой, атомом галогена или фенилом, m = 1, n = 0 или 1;
14.06.93 - J представляет группу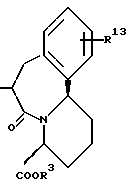
где R3 - атом водорода или карбоксизащитная группа; R13 - группа NHSO2R18, где R18 - низшая алкильная группа, R1 - атом водорода или ацильная группа, R2 - фенил, который может быть замещен алкоксигруппой, m = 1, n = 0;
06.07.93 - J представляет группу
где R13 - фенил, R3 - атом водорода или карбоксизащитная группа; R1 - атом водорода или ацильная группа, R2 - низшая алкильная группа, фенил, который может быть замещен алкоксигруппой, m = 0, 1 или 2, n = 0 или 2;
28.10.93 - J представляет группу
или
где R3 - атом водорода или карбоксизащитная группа; R4 - атом водорода, R10 - фенил, Y3 - группа -CH2-; R1 - атом водорода или ацильная группа, R2 - низшая алкильная группа, фенил или тиенил, m = 0 или 1, n = 0;
08.11.93 - J представляет группу
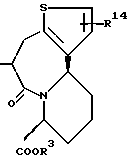
или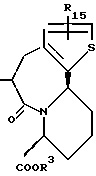
где R3 - атом водорода или карбоксизащитная группа; R4 - атом водорода, R10, R14, R15 - атом водорода, Y3 - группа -CH2 -; R1 - атом водорода или ацильная группа, R2 - низшая алкильная группа, фенил, или тиенил, замещенный галогеном, m = 0 или 1, n = 1;
16.11.93 - J представляет группу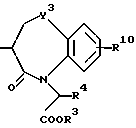
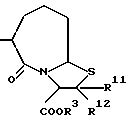
или
где R3 - атом водорода или карбоксизащитная группа; R4 - атом водорода; R10 - атом водорода, R11, R12 - атом водорода или низшая алкильная группа; R13 - фенил или группа NHSO2R18, где R18 - низшая алкильная группа; Y3 - группа - CH2-. -O- или -S-; R1 - атом водорода или ацильная группа, R2 - низшая алкильная группа, m = 0, n = 0;
17.12.93 - J представляет группу
где R3 - атом водорода или карбоксизащитная группа; R13 - фенил; R1 - атом водорода или ацильная группа, R2 - низшая алкильная группа, m = 0, n = 0;
08.02.94 - J представляет группу
или
где R3 - атом водорода или карбоксизащитная группа; R10, R11, R12 - атом водорода, u = 0, Y3 - группа -CH2-, R1 - атом водорода или ацильная группа, R2 - низшая алкильная группа, m = 0, n = 0;
07.03.94 - J представляет группу
где R3 - атом водорода, R1 - атом водорода, R1 - атом водорода или ацильная группа, R2 - низшая алкильная группа, m = 0, n = 0;
10.06.94 - R2 - алкилсульфониламиногруппа; u = 1 или 2.
| Захватное устройство для контейнеров | 1973 |
|
SU481522A1 |
| EP 0533084 A1, 24.03.93 | |||
| Способ получения производного 2,3,4,5-тетрагидро-1,5-бензоксазепин-4-она | 1984 |
|
SU1373322A3 |
| Способ получения производных 1,5-бензотиазепина или их фармацевтически приемлемых кислотно-аддитивных солей | 1989 |
|
SU1838305A3 |
