Изобретение относится к серии пептидильных гетероциклов, промежуточных соединений, используемых при их получении и к содержащим их фармацевтическим композициям. Соединения являются ингибиторами серин протеаз, в частности α-тромбина, и могут быть использованы при различных тромбин ассоциированных заболеваниях, таких как тромбоз вен и тромбоз артерий.
При быстро возрастающем распространении заболевания сосудистой системы сильно затрагивают наше общество. Артериальный тромбоз является главной причиной смерти от сердечного приступа и ударов, тогда как венозный тромбоз ассоциируется с легочной эмболией, которая проявляется после хирургического вмешательства или при длительном периоде неподвижности.
Тромбин является многофункциональной сериновой протеазой, роль которой при тромбозе и гемостазе была описана в различных источниках информации (См. , главным образом, Tapparelli et al., TIPS 1993, 14, 366-76). Тромбин действует как прокоагулянт посредством протеолитического расщепления фибриногена с образованием фибрина и как антикоагулянт посредством активации следов протеина С (с последующей инактивацией факторов коагуляции V и VIII). Концентрация активного тромбина ограничена большим числом механизмов обратной связи, включая эндогенные факторы и протеины. Кроме протеина С антитромбин III является другим регулирующим протеином, который образует комплекс с эндогенным гепарином. Этот комплекс присоединяется к активному тромбину, таким образом инактивируя его.
В настоящее время антикоагулянтная терапия состоит из трех классов соединений: гепаринов, кумаринов и гепаринов низкомолекулярной массы. Эти лекарства действуют непрямо с целью ограничения концентрации активного ингредиента. Гепарины и гепарины низкомолекулярной массы взаимодействуют с антитромбином III и кумарины ингибируют ряд витаминов К зависимых факторов коагуляции. Хотя эти лекарства предписываются при заболеваниях, связанных с венозным тромбозом и артериальным тромбозом, их использование ограничено. Они имеют ряд побочных эффектов, замедленное начало действия и только кумарины являются перорально активными (варфарин и дикумарол).
Было показано, что непрямые ингибиторы тромбина менее эффективны при подавлении связанных заболеваний, чем прямые ингибиторы тромбина. Таким образом, поиск перорально активных прямых ингибиторов тромбина проводится во многих лабораториях.
В результате этих усилий был получен ряд небольших пептидильных соединений, которые прямо ингибируют тромбин. РРАСК, аргатробан, (D)-NAPAP и DUP 714 являются представляющими интерес примерами прямых ингибиторов тромбина.
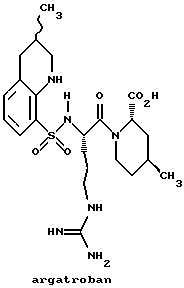
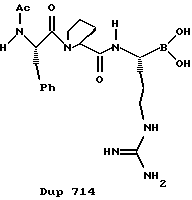
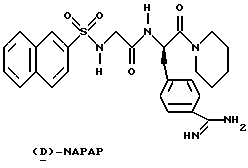
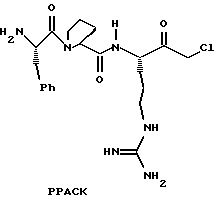
К сожалению, только несколько из этих соединений являются слабоактивными перорально и большинство имеют слабую избирательность в отношении тромбина против других сериновых протеаз. Таким образом, сохраняется необходимость в прямых ингибиторах, которые обладают хорошей селективностью в отношении других сериновых протеаз и являются перорально активными.
Изобретение относится к новым соединениям формулы I: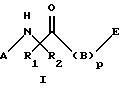
где А выбран из группы, включающей C1-8алкил, карбоксиС1-4алкил, С1-4алкоксикарбонилС1-4алкил, фенилС1-4алкил, замещенный фенилС1-4алкил (где заместители фенила независимо выбраны из одного или нескольких С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), формил, С1-4алкоксикарбонил, С1-12алкилкарбонил, фенил-С1-4алкоксикарбонил, С3-7циклоалкилкарбонил, фенилкарбонил, замещенный фенилкарбонил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфтор-С1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), С1-4алкилсульфонил, С1-4алкоксисульфонил, перфторС1-4алкилсульфонил, фенилсульфонил, замещенный фенилсульфонил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 10-камфорсульфонил, фенилС1-4алкилсульфонил, замещенный фенилС1-4алкилсульфонил, С1-4алкилсульфинил, перфторС1-4алкилсульфинил, фенилсульфинил, замещенный фенилсульфинил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), фенилС1-4алкилсульфинил, замещенный фенилС1-4алкилсульфинил, 1-нафтилсульфонил, 2-нафтилсульфонил или замещенный нафтилсульфонил (где заместители нафтила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, карбокси или С1-4алкоксикарбонила), 1-нафтилсульфинил, 2-нафтилсульфинил или замещенный нафтилсульфинил (где заместители нафтила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила);
D- или L-аминокислоту, которая присоединена своим карбоксиконцом к азоту, указанному в формуле I, и выбрана из группы, содержащей аланин, аспарагин, 2-азетидинкарбоновую кислоту, глицин, N-C1-8алкилглицин, пролин, 1-амино-1-цикло-С3-8алкилкарбоновую кислоту, триазолидин-4-карбоновую кислоту, 5,5-диметилтриазолидин-4-карбоновую кислоту, оксазолидин-4-карбоновую кислоту, пипеколиновую кислоту, валин, метионин, цистеин, серин, треонин, норлейцин, лейцин, трет-лейцин, изолейцин, фенилаланин, 1-нафталанин, 2-нафталанин, 2-тиенилаланин, 3-тиенилаланин, [1,2,3,4]-тетрагидроизохинолин-1-карбоновую кислоту и [1,2,3,4]-тетрагидроизохинолин-2-карбоновую кислоту, где аминовые концы указанной аминокислоты связаны с членом, выбранным из группы, включающей С1-4алкил, тетразол-5-ил-С1-2алкил, карбоксиС1-4алкил, С1-4алкоксикарбонилС1-4алкил, фенилС1-4алкил, замещенный фенилС1-4алкил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 1,1-дифенилС1-4алкил, 3-фенил-2-гидроксипропионил, 2,2-дифенил-1-гидроксиэтилкарбонил, [1,2,3,4]-тетрагидро-изохинолин-1-карбонил, [1,2,3,4] -тетрагидроизохинолин-3-карбонил, 1-метиламино-1-циклогексанкарбонил, 1-гидрокси-1-циклогексанкарбонил, 1-гидрокси-1-фенилацетил, 1-циклогексил-1-гидроксиацетил, 3-фенил-2-гидрокси-пропионил, 3,3-дифенил-2-гидроксипропионил, 3-циклогексил-2-гидроксипропионил, формил, С1-4алкоксикарбонил, С1-12алкилкарбонил, перфторС1-4алкилС0-4алкилкарбонил, фенилС1-4алкилкарбонил, замещенный фенилС1-4алкилкарбонил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 1,1-дифенилС1-4алкилкарбонил, замещенный 1,1-дифенилС1-4алкилкарбонил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), перфторС1-4алкилсульфонил, С1-4алкилсульфонил, С1-4алкоксисульфинил, фенилсульфонил, замещенный фенилсульфонил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 10-камфорсульфонил, фенилС1-4алкилсульфонил, замещенный фенилС1-4алкилсульфонил, перфторС1-4алкилсульфинил, С1-4алкилсульфинил, фенилсульфинил, замещенный фенилсульфинил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), фенилС1-4алкилсульфинил, замещенный фенилС1-4глкилсульфинил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 1-нафтилсульфонил, 2-нафтилсульфонил, замещенный нафтилсульфонил (где заместители нафтила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила); 1-нафтилсульфинил, 2-нафтилсульфинил и замещенный нафтилсульфинил (где заместители нафтила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила);
или полипептид, содержащий две аминокислоты, где первая аминокислота является D- или L-аминокислотой, присоединенной через ее карбокси концы к азоту, указанному в формуле I, и выбрана из группы, содержащей глицин, N-C1-8алкилглицин, аланин, 2-азетидинкарбоновую кислоту, пролин, триазолидин-4-карбоновую кислоту, 5,5-диметил-триазолидин-4-карбоновую кислоту, оксазолидин-4-карбоновую кислоту, 1-амино-1-циклоС3-8алкилкарбоновую кислоту, 3-гидроксипролин, 4-гидроксипролин, 3-(С1-4алкокси) пролин, 4-(С1-4алкокси)пролин, 3,4-дегидропролин, 2,2-диметил-4-тиазолидинкарбоновую кислоту, 2,2-диметил-4-оксазолидин-карбоновую кислоту, пипеколиновую кислоту, валин, метионин, цистеин, аспарагин, серин, треонин, лейцин, трет-лейцин, изолейцин, фенилаланин, 1-нафталанин, 2-нафталанин, 2-тиенилаланин, 3-тиенилаланин, [1,2,3,4] -тетрагидроизохинолин-1-карбоновую кислоту, [1,2,3,4] -тетрагидроизохинолин-2-карбоновую кислоту, 4-С1-4алкиловый эфир аспарагиновой кислоты и 5-С1-4алкиловый эфир глутаминовой кислоты, и вторая D или L аминокислота присоединена к аминоконцу указанной первой аминокислоты и выбрана из группы, содержащей фенилаланин, 4-бензоилфенилаланин, 4-карбоксифенилаланин, 4-(карбоксиСо-20алкил)фенилаланин, замещенный фенилаланин (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 3-бензотиенилаланин, 4-бифенилаланин, гомофенилаланин, октагидроиндол-2-карбоновую кислоту, 2-пиридилаланин, 3-пиридилаланин, 4-тиазолилаланин, 2-тиенилаланин, 3-(3-бензотиенил)аланин, 3-тиенилаланин, триптофан, тирозин, аспарагин, 3-три-С1-4алкилсилилаланин, циклогексилглицин, дифенилглицин, фенилглицин, метионинсульфоксид, метионинсульфон, 2,2-дициклогексилаланин, 2-(1-нафтилаланин), 2-(2-нафтилаланин), фенилзамещенный фенилаланин (где заместители выбраны из С1-4алкила, перфторС1-4алкила, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), аспарагиновую кислоту, 4-С1-4алкиловый эфир аспарагиновой кислоты, глутаминовую кислоту, 5-С1-4алкиловый эфир глутаминовой кислоты, циклоС3-8алкилаланин, замещенный циклоС3-8алкилаланин (где заместителями кольца являются карбокси, С1-4алкилкарбокси, С1-4алкоксикарбонил или аминокарбонила), 2,2-дифенилаланин и все альфа-С1-5алкильные производные всех аминокислот, где аминовые концы указанной второй аминокислоты являются незамещенными или монозамещены членом группы, содержащей формил, С1-12алкил, тетразол-5-илС1-2алкил, карбоксиС1-8алкил, кабоалкоксиС1-4алкил, фенилС1-4алкил, замещенный фенилС1-4алкил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 1,1-дифенилС1-4алкил, C1-6алкоксикарбонил, фенилС1-6алкоксикарбонил, С1-12алкилкарбонил, перфторС1-4алкилС0-4алкилкарбонил, фенил-С1-4алкилкарбонил, замещенный фенилС1-4алкилкарбонил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 1,1-дифенилС1-4алкилкарбонил, С1-4алкилсульфонил, С1-4алкоксисульфонил, перфторС1-4алкилсульфонил, фенилсульфонил, замещенный фенилсульфонил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 10-камфорсульфонил, фенил-С1-4алкил сульфонил, замещенный фенилС1-4алкил сульфонил, перфторС1-4алкилсульфинил, фенилсульфинил, замещенный фенилсульфинил (где заместители фенила независимо выбраны из одного или нескольких из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), фенилС1-4алкилсульфинил, замещенный фенил-С1-4алкилсульфинил, 1-нафтилсульфонил, 2-нафтилсульфонил, замещенный нафтилсульфонил (где заместитель нафтила выбран из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила), 1-нафтилсульфинил, 2-нафтилсульфинил и замещенный нафтилсульфинил (где заместитель нафтила выбран из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси или С1-4алкоксикарбонила);
R1 выбран из группы, включающей водород и С1-5алкил;
R2 выбран из группы, включающей аминоС2-5алкил, гуанидино-С2-5алкил, С1-4алкилгуанидиноС2-5алкил, диС1-4алкилгуанидино-С2-5алкил, амидиноС2-5алкил, С1-4алкиламидиноС2-5алкил, ди-С1-4алкиламидиноС2-5алкил, С1-3алкоксиС2-5алкил, фенил, замещенный фенил (где заместители независимо выбраны из одного или нескольких из амино, амидино, гуанидино, С1-4алкиламино, С1-4диалкиламино, галогена, перфторС1-4алкила, C1-3алкокси или нитро), бензил, фенилзамещенный бензил (где заместители независимо выбраны из одного или нескольких из амино, амидино, гуанидино, С1-4алкиламино, С1-4диалкиламино, галогена, перфторС1-4алкила, С1-4алкила, C1-3алкокси или нитро), гидроксиС2-5алкил, С1-5алкиламиноС2-5алкил, С1-5диалкиламиноС2-5алкил, 4-аминоциклогексилС0-2алкил и С1-5алкил;
р = 0 или 1;
В обозначает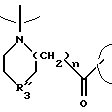
где n = равно 0-3, R3 обозначает Н или С1-5алкил и карбонильный радикал В присоединен к Е;
Е обозначает гетероцикл, выбранный из группы, содержащей оксазолин-2-ил, оксазол-2-ил, тиазол-2-ил, тиазол-5-ил, тиазол-4-ил, тиазолин-2-ил, имидазол-2-ил, 4-оксо-2-хиноксалин-2-ил, 2-пиридил, 3-пиридил, бензо[b]тиофен-2-ил, бензоксазол-2-ил, бензимидазол-2-ил, бензотиазол-2-ил, тиазол-6-ил, тетразол-2-ил, пиримидин-2-ил, хинолин-2-ил, индол-2-ил, пиразол-2-ил, 4,5,6,7-тетрагидробензотиазол-2-ил, нафто-[2,1-d] тиазол-2-ил, нафто[1,2-d] тиазол-2-ил, хиноксалин-2-ил, изохинолин-1-ил, изохинолин-3-ил, бензо[b] фуран-2-ил, пиразин-2-ил, хиназолин-2-ил, изотиазол-5-ил, изотиазол-3-ил, пурин-8-ил и замещенный гетероцикл, где заместители выбраны из С1-4алкила, перфторС1-4алкила, С1-4aлкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси, С1-4алкоксикарбонила, гидрокси или фенилС1-4алкиламинокарбонила);
или их фармацевтически приемлемым солям.
Термины, используемые при описании изобретения, являются обычно используемыми и известными специалистам в данной области. Однако термины, которые могут иметь другие значения, определены особо. "Независимо" означает, что когда имеется более одного заместителя, заместители могут быть различными. Термин "алкил" относится к прямым, циклическим и разветвленным цепям алкильных групп, и "алкокси" относится к 0-алкилу, где алкил определен выше. "CBZ" относится к бензилоксикарбонилу. "БОК" относится к трет-бутоксикарбонилу и "Ts" относится к толуолсульфонилу. "ДЦК" относится к 4-N,N-диметиламинопиридину и "НОБТ" относится к гидрату 1-гидроксибензотриазола. "Дансил" относится к 5-диметиламино-1-нафталинсульфонамиду и "FMoc" относится к N-(9-флуоренилметоксикарбонилу).
Соединения по изобретению могут быть получены как это проиллюстрировано на схеме I. По иллюстрирующим примерам получают соединение. где А обозначает полипептид, где первая аминокислота является L-пролином и вторая аминокислота является N-метил-D-фенилаланином, R1 - водород, R2 - С4алкил, р = 0 и Е представляет бензоксазол-2-ил. Другие соединения, которые могут быть получены по данной общей схеме, перечислены ниже с соответствующими модификациями.
Природная и неприродная аминокислота является отправной точкой по схеме. Соединение Iа получают известными способами синтеза получения защищенной аминокислоты, где α-аминогруппа защищена с помощью Тs. Culvenor, et al., J. Chem. Soc. D. 1969, 19, 1091. Защитные группы могут варьироваться. На схеме I α-амино- и ω-аминозащитные группы должны быть стабильными в среднекислых условиях и удаляться способами, которые являются взаимно исключающими. Примеры подходящих α-амино- и ω-аминозащитные группы перечислены соответственно: FMOC-TS, FMOC-NO2 CBZ-NO2. Например, другие приемлемые защитные группы см. Green, Theodora. Protecting Groups In Organic Synthesis; John Wiley & Sons. New York, 1981.
Соединение Ia присоединяют к 1,1'-карбонилдиимидазолу в ТГФ при 0-10oС с последующей обработкой DIBAL/гексан при -42oС с получением альдегида Ib, используя способ, описанный Greco, et al. Journal Of The American Chemical Society, 1995, 117, 1225. Прямое преобразование Ia в Ib может быть проведено с использованием бис(N-метилпиперазинил)алюминийгидрида в толуоле при от 0oС до температуры кипячения. (Hubert, et al., Journal Of Organic Chemistry, 1984, 49, 2279). По другому способу прямого преобразования Iа в Ib используется борандиметилсульфид в ТГФ при от 25oС до температуры кипячения. Brown, et al., Synthesis, 1979, 704. Еще одним способом является преобразование карбоновой кислоты в соответствующий Weinreb амид, используя триэтиламин, ВОР и гидрохлорид N,0-диметилгидроксиламина. Восстановление образующегося амина с помощью LAH в ТГФ дает соответствующий альдегид.
Цианборгидрид Iс получают путем прибавления KCN к эмульсии альдегида Ib в Н2О, МеОН и этилацетата при комнатной температуре. Альтернативно, соединение Iс может быть получено взаимодействием альдегида Ib, ацетонциангидрина и триэтиламина в дихлорметане при комнатной температуре. Имидат Id получают путем обработки Iс газообразным НСl в метаноле. Бензоксазол If получают путем нагревания Id с Iе в этаноле при кипячении.
Амин Ig получают путем гидрирования If, применяя Pd(OH)2/C в качестве катализатора при комнатной температуре при атмосферном или при повышенном давлении, используя растворитель-донор водорода. В подходящих реакционных условиях могут быть использованы другие катализаторы, такие как Pd/C или Pd-чернь. Другие реагенты, которые могут быть использованы для удаления CBZ группы, включают: гидразин, иодтриметилсиланметансульфоновую кислоту/анизол и трибромид бора.
Соединение Ig может быть связано с кислотой Ih HOBT и ДЦК в СН3СN при комнатной температуре с получением связанного спирта Ii. Другие подходящие реагенты для связывания включают: ВОР, ВОР-С1 и РyВОР.
Соединение Ii окисляют в соединение Ij с использованием периодинана в безводном апротонном растворителе. Конечной стадией по схеме I является удаление защиты у Ii с использованием HF в присутствии улавливателя карбкатионов, таких как анизол, тиоанизол, пентаметилбензол, диметилсульфид и крезол с последующей хроматографией с обращенной фазой с получением конечного продукта Ik.
Для получения соединений по изобретению, где А обозначает С1-4алкил, фенилС1-4алкил и замещенный фенилС1-4алкил, исходный продукт по схеме I модифицируют. Обработка соединения Iа двумя эквивалентами гидрида натрия в ДМФ при температуре от 0oС до комнатной и затем алкилгалогенидом дает N-алкилированную карбоновую кислоту. Эта кислота может быть обработана в тех же самых реакционных условиях, что применяются согласно схеме I, с получением желаемых соединений, как показано на схеме IA.
Когда А обозначает С1-4алкоксикарбонил, фенилС1-4алкоксикарбонил, С3-7циклоалкилкарбонил, фенилкарбонил и замещенный фенилкарбонил, схема I может быть опять модифицирована. Группа CBZ в соединении Iа удаляется и полученная свободная аминокислота взаимодействует с подходящим карбонильным соединением. Примерами таких соединений являются бензоилхлорид, пропионилхлорид, метилхлорформиат, циклогексанкарбонилхлорид, бензилхлорформиат и тому подобное. Этот исходный продукт используют в схеме IB с получением желаемых соединений, как показано на схеме IA.
Аналогично, когда А обозначает С1-4алкилсульфонил, перфторС1-4алкилсульфонил, замещенный фенилсульфонил, С1-4алкилсульфинил, перфторС1-4алкилсульфинил, фенилсульфинил, замещенный фенилсульфинил, 1-нафтилсульфонил, 2-нафтилсульфонил и замещенный нафтилсульфонил, 1-нафтилсульфинил, 2-нафтилсульфинил или замещенный нафтилсульфинил; желаемые соединения могут быть получены из их соответствующих коммерчески доступных арилсульфонил- и арилсульфинил-галогенидов по схеме IB.
Когда А является аминокислотой, аминоконцы которой присоединены к другой группе, может быть опять использована схема I. На N-присоединенную аминокислоту (при необходимости защищенная) заменяют Ih в схеме I с использованием тех же самых реакционных условий. Аланин, аспарагин, 2-азетидинкарбоновая кислота, глицин, пролин, пипеколиновая кислота, валин, метионин, цистеин, серин, треонин, лейцин, трет-лейцин и изолейцин могут быть использованы по этой схеме, где аминогруппы присоединены к С1-4алкилу, (фенилС1-4алкилу), (замещенному фенилС1-4алкилу), (1,1-дифенилС1-4алкилу-3-фенил-2-гидроксипропионилу), (2,2-дифенил-1-гидроксиэтилкарбонилу), ([1,2,3,4]-тетрагидроизохинолин-1-карбонилу), ([1,2,3,4]-тетрагидроизохинолин-3-карбонилу), (1-метиламино-1-циклогексанкарбонилу), (1-гидрокси-1-циклогексанкарбонилу), (1-гидрокси-1-фенилацетилу), 1-циклогексил-1-гидроксиацетилу, 3-фенил-2-гидроксипропионилу, 3,3-дифенил-2-гидроксипропионилу, 3-циклогексил-2-гидроксипропионилу, формилу, C1-4алкоксикарбонилу, С1-4алкилкарбонилу, перфторС1-4алкилС0-4алкилкарбонилу, фенилС1-4алкилкарбонилу, замещенному фенилС1-4алкилкарбонилу, 1,1-дифенилС1-4алкилкарбонилу, замещенному 1,1-дифенилС1-4алкилкарбонилу, перфторС1-4алкилсульфонилу, C1-4алкилсульфонилу, фенилсульфонилу, замещенному фенилсульфонилу, перфторС1-4алкилсульфинилу, С1-4алкилсульфинилу, фенилсульфинилу, замещенному фенилсульфинилу, 1-нафтилсульфонилу, 2-нафтилсульфонилу, замещенному нафтилсульфонилу, 1-нафтилсульфинилу, 2-нафтилсульфинилу и замещенному нафтилсульфинилу.
Когда А является дипептидом, аминоконцы которой присоединены к другой группе, может быть опять использована схема I. Одним примером является замена Ih на N-метил-N-СВZ-D-фенилаланин-L-азетидинкарбоновую кислоту с получением соответствующего конечного продукта с использованием, по существу тех же самых реакционных условий, как на схеме I.
Для получения соединений, где R1 обозначает Н и R2 обозначает аминоС2-5алкил, гуанидиноС2-5алкил, амидиноС2-5алкил, С1-3алкоксиС2-5алкил, фенил, замещенный фенил (где заместители выбраны из амидино, гуанидино, С1-4алкиламино, галогена, перфторС1-4алкила, С1-4алкила, C1-3алкокси и нитро), бензил, арилзамещенный бензил (где заместители выбраны из амидино, гуанидино, С1-4алкиламино, галогена, перфторС1-4алкила, С1-4алкила, C1-3алкокси и нитро), (пиридин-4-ил)аминоС1-4алкил, (пиридин-3-ил)аминоС1-4алкил, С1-4алкиламиноС2-5алкил и C1-5aлкил, может быть опять использована схема I. Исходный продукт Iа может быть заменен на коммерчески доступные аминокислоты, такие как аргинин, лейцин, фенилаланин, лизин, п-амидинофенилаланин, метоксипропилглицин и 2-(3-пиридил)-аланин, где α-аминогруппа, а также любые другие реакционноспособные заместители защищены CBZ, Ts или FMOC, что подходит. Если желаемые заместители не являются коммерчески доступными, как в случае, когда R2 является замещенным фенилом или замещенным бензилом, желаемая аминокислота может быть синтезирована обычными химическими способами. См. D.A. Evans et al (Tetrahedron, 1988, 44, 5525-5540).
Для получения соединений, где Е обозначает 4-оксо-2-хиноксалин-2-ил, бензимидазол-2-ил и бензтиазол-2-ил, может быть опять использована схема I. Замена Iе на 2-аминотиофенол дает производные бензотиазола следуя тем же реакционным условиям, что и по схеме I; при условии, что нельзя использовать Pd для удаления CBZ группы. Однако может быть использован любой из других методов, перечисленных ранее.
Если Е обозначает 5-карбоэтокситиазол-2-илом, схема I может быть модифицирована с получением желаемых соединений, как показано на схеме Iс. Замена соединения Iе на гидрохлорид этилового эфира цистеина дает производное тиазолина II при комнатной температуре с использованием CH2Cl2 в качестве растворителя. Окисление тиазолинового кольца соединения Il с помощью MnO2 при комнатной температуре дает тиазол Im. Окисление гидроксильной группы Im с последующим удалением аминозащитных групп с помощью HF/анизола дает карбоэтокси производное In.
Для получения сложноэфирных производных In карбоэтокси группа в любом из Il, In или Im может быть модифицирована с использованием известных химических методов. Таким образом можно получать следующие сложноэфирные производные: карбокси, фенилС1-4алкиламинокарбонил, амидо, формил и другие С1-4карбонильные группы.
Другой способ получения соединений по изобретению проиллюстрирован на схеме II. Иллюстративным примером получения соединения, где А является L-пролином, который присоединен по его N-концу к (1,1-дифенилС2алкилкарбонилу), R1 обозначает водород, R2 обозначает гуанидиноС3алкил, р равно 0 и Е обозначает тиазол-2-ил. Другие соединения, которые могут быть получены по этой общей схеме, перечислены далее с соответствующими модификациями.
Коммерчески доступная кислота IIа является исходной точкой для этой схемы. Указанная кислота преобразуется в активированное сложноэфирное производное IIb путем обработки ДЦГ и 2,4,5-трихлорфенолом при от -20oС до 0oС в инертном растворителе, таком как ТГФ. Производное пролина IId получают путем обработки IIb с помощью IIс, триэтиламином и пиридином. Альтернативно, IId может быть получено прямо путем обработки кислоты IIa аминокислотой IIc в присутствии любого из связывающего пептиды агентов, перечисленных в схеме I.
Weinrab амид NαBoc-Nw-тозиларгинина, IIе, был получен по известному в литературе способу из защищенной кислоты. DiMaio, et al., Journal of Medicinal Chemistry 1992, 35, 3351. Амид обрабатывают IIf при -78oС в ТГФ с получением кетона IIg. Этот кетон может быть восстановлен гидридным восстанавливающим агентом, таким как боргидрид натрия, при от -20oС до комнатной температуры с последующей обработкой ТФУ при комнатной температуре с получением соответствующего амина IIh.
Связывание IIh и IId в присутствии ДЦК и НОВТ при комнатной температуре в ацетонитриле дает связанный спирт IIi. Другие пептидные связывающие реагенты, которые могут быть использованы для этих реакций, включают: бензотриазол-1-илокси-трис(диметиламино)фосфонийгексафторфосфат, хлорангидрид бис(2-оксо-3-оксазолидинил)фосфониевой кислоты, гидрохлорид 2-диметиламиноизопропилхлорида и бром-трис-пирролидинофосфонийгексафторфосфат. Окисление IIi до кетона с использованием периодинана в СН2Cl2 с последующим удалением ω--аминозащитной группы с помощью HF и анизола дает желаемый продукт IIj.
Для получения соединений по изобретению, где А обозначает С1-4алкил, фенилС1-4алкил или замещенный фенилС1-4алкил, исходный продукт по схеме II может быть модифицирован и использован, как показано на схеме IIA.
Исходный продукт для схемы IIА получают из защищенного Weinrab амида, IIе. У амида удаляют защиту с помощью ТГФ, подвергают восстановительному алкилированию соответствующим субстратом и цианборгидридом натрия и защищают с помощью ВОС до N-алкилированного исходного продукта. В иллюстрирующих примерах используют бензальдегид в качестве субстрата для восстановительного алкилирования. Алкилированный материал может быть обработан в тех же условиях с теми же реагентами, как по схеме II с получением желаемых соединений.
Когда А обозначает формил, С1-4алкоксикарбонил, фенилС1-4алкоксикарбонил, С1-4алкилкарбонил, С3-7циклоалилкарбонил, фенилкарбонил и замещенный фенилкарбонил, схема II может быть вновь модифицирована. Группа БОС IIе удаляется с помощью ТФУ при комнатной температуре и полученный свободный амин подвергают взаимодействию с подходящим коммерчески доступным карбонильным соединением. Примерами таких соединений являются муравьиная кислота/уксусный ангидрид, бензоилхлорид, пропионилхлорид, метилхлорформиат, циклогексанкарбонилхлорид и тому подобное. Этот исходный продукт используют в тех же условиях, как по схеме IIА с получением желаемых соединений.
Аналогично, когда А обозначает: С1-4алкилсульфонил, перфторС1-4алкилсульфонил, фенилсульфонил, замещенный фенилсульфонил, С1-4алкилсульфинил, перфторС1-4алкилсульфинил, фенилсульфинил, замещенный фенилсульфинил, 1-нафтилсульфонил, 2-нафтилсульфонил и замещенный нафтилсульфонил, 1-нафтилсульфинил, 2-нафтилсульфинил и замещенный нафтилсульфинил; желаемые соединения могут быть получены из их соответствующих коммерчески доступных арилсульфонил- и арилсульфинил-галогенидов с использованием схемы IIА.
Когда А является аминокислотой, аминоконцы которой присоединены к другой группе, может быть опять использована схема II. N-Присоединенная аминокислота может быть закуплена или получена известными способами. На эту аминокислоту будет заменен Ih в схеме II с использованием тех же самых реакционных условий. Аланин, аспарагин, 2-азетидинкарбоновая кислота, глицин, пролин, пипеколиновая кислота, валин, метионин, цистеин, серин, треонин, лейцин, трет-лейцин и изолейцин могут быть использованы по этой схеме, где аминогруппы присоединены к C1-4aлкилу, фенилС1-4алкилу, замещенному фенилС1-4алкилу, 1,1-дифенилС1-4алкилу, 3-фенил-2-гидроксипропионилу, 2,2-дифенил-1-гидроксиэтилкарбонилу, [1,2,3,4]-тетрагидроизохинолин-1-карбонилу, [1,2,3,4] -тетра-гидроизохинолин-3-карбонилу, 1-метиламино-1-циклогексанкарбонилу, 1-гидрокси-1-циклогексанкарбонилу, 1-гидрокси-1-фенилацетилу, 1-циклогексил-1-гидроксиацетилу, 3-фенил-2-гидроксипропионилу, 3,3-дифенил-2-гидроксипропионилу, 3-циклогексил-2-гидроксипропионилу, формилу, С1-4алкоксикарбонилу, С1-4алкилкарбонилу, перфторС1-4алкилС0-4алкилкарбонилу, фенилС1-4алкилкарбонилу, замещенному фенилС1-4алкилкарбонилу, 1,1-дифенилС1-4алкилкарбонилу, замещенному 1,1-дифенилС1-4алкилкарбонилу, перфторС1-4алкилсульфонилу, С1-4алкилсульфонилу, фенилсульфонилу, замещенному фенилсульфонилу, перфторС1-4алкилсульфинилу, С1-4алкилсульфинилу, фенилсульфинилу, замещенному фенилсульфинилу, 1-нафтилсульфонилу, 2-нафтилсульфонилу и замещенному нафтилсульфонилу, 1-нафтилсульфинилу, 2-нафтилсульфинилу и замещенному нафтилсульфинилу.
Когда А является дипептидом, аминоконцы которого присоединены к другой группе, может быть опять использована схема II. Одним примером является замена IId на N-метилфенил-аланинпипеколиновую кислоту с получением соответствующего конечного продукта с использованием, по существу, тех же условий реакции, что и на схеме II.
Для получения соединений, где R1 обозначает Н и R2 обозначает аминоС2-5алкил, гуанидиноС2-5алкил, амидиноС2-5алкил, С1-3алкоксиС2-5алкил, фенил, замещенный фенил (где заместители выбраны из амидино, гуанидино, С1-4алкиламино, галогена, перфторС1-4алкила, С1-4алкила, C1-3алкокси и нитро), бензил, арилзамещенный бензил (где заместители выбраны из амидино, гуанидино, С1-4алкиламино, галогена, перфторС1-4алкила, С1-4алкила, C1-3алкокси и нитро), (пиридин-4-ил)аминоС1-4алкил, (пиридин-3-ил)аминоС1-4алкил, С1-5алкиламиноС2-5алкил и С1-5алкил, может быть опять использована схема II. Используя способы DiMaio, et al., Journal of Medicinal Chemistry 1992, 35, 3331, могут быть получены Weinreb амиды известных защищенных аминокислот. На эти соединения заменяют соединение IIе в схеме II и обрабатывают в тех же реакционных условиях, проиллюстрированных на схеме II.
Схема II может использоваться вновь для получения соединений по изобретению, где Е обозначает оксазол-2-ил, тиазол-2-ил, тиазол-5-ил, тиазол-4-ил, 1-алкилимидазол-2-ил, 2-пиридил, 3-пиридил, 4-пиридил, бензо[b]тиофен-2-ил, бензоксазол-2-ил, 1-алкилбензимидазол-1-ил, бензотиазол-2-ил, 1-алкилиндол-2-ил, 4,5,6,7-тетрагидробензотиазол-2-ил, нафто-[2,1-d]тиазол-2-ил, нафто[1,2-d] тиазол-2-ил, пиримидин-2-ил, хиноксалин-2-ил, бензо[b]фуран-2-ил, пиразин-2-ил, хиназолин-2-ил, изотиазол-5-ил, изотиазол-3-ил. Когда азот в положении 1 защищен устойчивой по отношению к основанию защитной группой (например, TMS, Воc, Ts и т.д.), схема II может быть использована для получения соединений по изобретению, где Е обозначает имидазол-2-ил, пиразол-2-ил, триазол-2-ил, триазол-4-ил, триазол-6-ил, тетразол-2-ил, хинолин-2-ил, индол-2-ил, изохинолин-1-ил, изохинолин-3-ил и пурин-8-ил (требуется защита по положению 7 вместо 1). Замена IIf на любой из известных перечисленных литированных гетероциклов дает желаемые соединения.
Другой способ получения соединений по изобретению проиллюстрирован на схеме III. Согласно иллюстрирующим примерам получают соединение, где А является полипептидом, где первой аминокислотой является L-пролин и второй аминокислотой является N-метил-D-фенилаланин, R1 представляет водород, R2 представляет (гуанидиноС3алкил), р = 1, n = 0, R3 представляет Н и Е обозначает тиазол-2-ил.
Соединения по изобретению, где Е обозначает пиразол-3-ил, могут быть получены путем 1,3-диполярного добавления диазокетонов к алкинам (Padwa et al. , "1,3-Dipolar Cycloaddition Chemistry", 2 vols., Wiley, New York, 1984), как показано на схеме IV. Диазокетоновое промежуточное соединение IVa (US Patent 4318904) может быть подвергнуто взаимодействию с алкином IVb при кипячении в бензоле с получением пиразола IVc, который может быть далее преобразован в пиразол IVd, как это описано ранее.
Соединения по изобретению, где Е обозначает тиазол-2-ил и оксазол-2-ил, могут быть получены, как показано на схеме V. Циклизация спиртов Va и Vb с помощью реагента Burgess в соответствующем тиазолине и оксазолине с последующим окислением MnO2 или NiO2 приводит к Vc и Vd (P. Wipf et al., Tetrahedron Letters, 1992, 33, 6267-6270). Подобным образом, альдегид Ve может быть преобразован в Vd с помощью трифенилфосфин/иода в присутствии триэтиламина (Р.Wipf et al., Journal of Organic Chemistry, 1993, 58, 3604-3606).
Хотя заявленные соединения могут использоваться в качестве ингибитора тромбина или трипсина, предпочтительные соединения формулы I включают: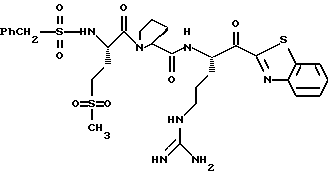
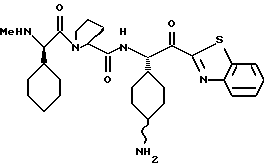
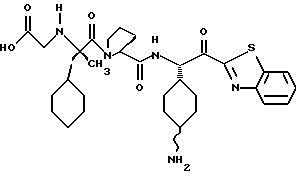
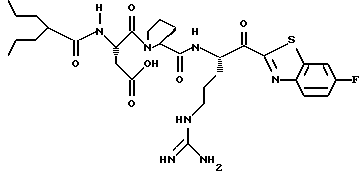
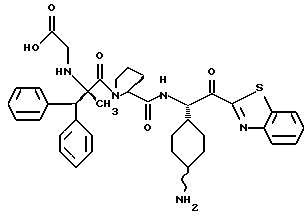
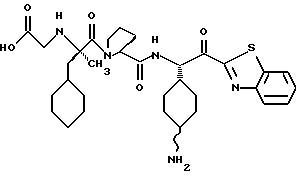
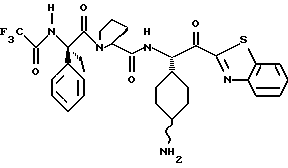
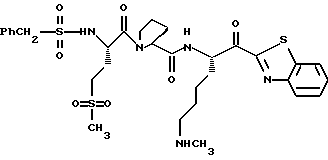
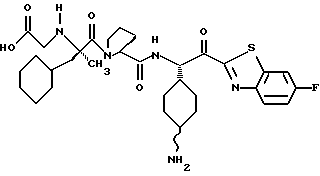
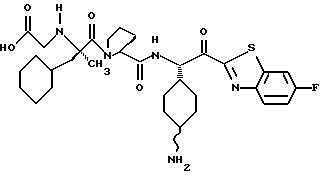
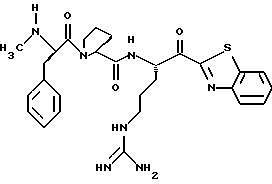
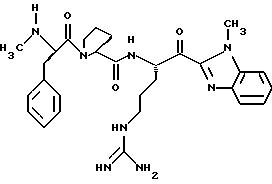
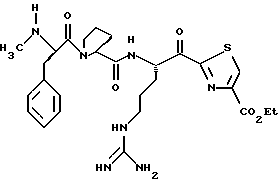
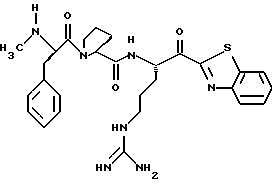
Особенно предпочтительными "А" являются:
1-нафтилсульфонил, 2-нафтилсульфонил, замещенные нафтилсульфонилы (где заместители выбраны из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси и С1-4алкоксикарбонила);
и L-аминокислота, такая как глицин или пролин, где аминоконцы являются незамещенными или монозамещенными членом из группы, включающей 1-нафтилсульфонил, 2-нафтилсульфонил и замещенные нафтилсульфонилы (где заместители выбраны из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси и С1-4алкоксикарбонила); или
полипептид, содержащий две аминокислоты, где первая кислота является L-пролином или L-пипеколином и вторая аминокислота является D-фенилаланином, D-циклогексилаланином, D-дифенилаланином или (2,3,4,5,6-пентафторфенил)аланином, где аминоконцы указанной второй аминокислоты являются незамещенными или монозамещенными членом из группы, включающей С1-5алкил, перфторС1-4алкил или формил.
Особенно предпочтительными "R1" являются водород и метил.
Особенно предпочтительные "R2" выбраны из группы, включающей:
аминоС2-5алкил, гуанидиноС2-5алкил, амидиноС2-5алкил, С1-5алкиламиноС2-5алкил, С1-5диалкиламиноС2-5алкил, 4-аминоциклогексилС0-2алкил, 3-аминоциклогексилС0-2алкил и С1-5алкил.
Особенно предпочтительными "Е" являются гетероциклы, выбранные из группы, включающей:
тиазол-2-ил, тиазол-5-ил, тиазол-4-ил, тиазолин-2-ил, бензоксазол-2-ил, бензимидазол-2-ил, имидазол-2-ил, 4-оксо-2-хиноксалин-2-ил, бензотиазол-2-ил, тиазол-4-ил, тиазол-6-ил, тетразол-2-ил, пиримидин-2-ил, хинолин-2-ил, пиразол-2-ил, [4,5,6,7]-тетрагидробензотиазол-2-ил, нафто[2,1-d]тиазол-2-ил, нафто[1,2-d] тиазол-2-ил, хиноксалин-2-ил, хиназолин-2-ил, изотиазол-5-ил, изотиазол-3-ил, пурин-8-ил и замещенный гетероцикл, где заместители выбраны из С1-4алкила, перфторС1-4алкила, С1-4алкокси, гидрокси, галогена, амидо, нитро, амино, С1-4алкиламино, С1-4диалкиламино, карбокси, С1-4алкоксикарбонила и гидрокси.
Соединения по изобретению были исследованы на их способность ингибировать опосредованный тромбином гидролиз. Были проведены исследования как in vitro, так и ex vivo. Кроме того, соединения были исследованы in vitro на их способность ингибировать трипсин, как показатель их селективности.
Скорость катализируемого тромбином гидролиза была измерена спектрофотометрически с использованием продажного альфа-тромбина (American Diagnostica), хромогенного субстрата Spectozyme® TH (H-D-HHT-Ala-Arg-pNA-2AcOH), American Diagnostica) в водном буфере (10 мМ Трис, 10 мМ ГЕПЕС, 150 мМ NaCl, 0,1% PEG; pH 7,4) и микропланшетного ридера (Molecular Devices). Регистрируют изменения в абсорбции при 405 нм (Softmax, Molecular Devices) при добавлении фермента как с ингибитором, так и без ингибитора при 37oС в течение более 30 минут. IC50 определяют путем фиксации концентраций фермента и субстрата и изменения концентрации ингибитора (1 нМ тромбин, 50 мкМ Spectosyme® TH). Была применена кинетика Michaelis-Menton для начальной реакционной скошенной твердой среды. Константы ингибирования (Ki) были определены путем фиксации концентраций фермента и ингибитора и изменения концентрации субстрата (1 нМ тромбина, 5-100 мкМ Spectosyme® TH). Была применена кинетика Michaelis-Menton для начальной реакционной скошенной твердой среды с использованием программы K•Cat (Bio Metallics Inc.).
Скорость катализируемого трипсином гидролиза была измерена с использованием того же метода, что и в тромбиновом методе. Бычий трипсин типа 1 (Sigma) и Spectosyme® TRY (Gbo-Gly-D-Ala-Arg-pNA-AcOH, American Diagnostics) заменяют их тромбиновые эквиваленты в области концентрации 3,2 U/мл трипсина и 0,1-0,3 мМ Spectosyme.
IC50 для представительных соединений представлены в таблице А. Представительные Ki представлены вместо значения IС50 для выбранных соединений. В качестве эталонных стандартов были использованы N-Me PPACK альдегид и аргатромбан и их значения приведены далее. Номера соединений в таблице соответствуют примерам, представленным далее.
Анализ ex vivo проводили для определения длительности действия соединений формулы I. Для этого внутривенного анализа самцам крыс (Long Evans, 300-500 г) были имплантированы тефлоновые канюли через бедренную артерию (фенобарбитал, внутрибрюшинно, 35 мг/кг). После хирургического вмешательства животных отдельно помещали в стандартные клетки, кормили крысиным chow (Hanlan, #8604) и непрерывно вливали физиологический раствор для поддержания раскрытого состояния артерии (0,5 мл/ч, внутриартериально) с использованием spring-shielded swivelling tether, присоединенного к системе вливания. После хирургического вмешательства животным дают восстановиться, по меньшей мере, в течение 24 часов.
Образцы крови отбирают у животных за 15 минут перед дозированием ингибитора (0,25 мл крови отбирают в шприц, содержащий 0,025 мл Sigma цитрата натрия). Исследуемые соединения диспергируют в воде и вводят животному (0,25 мл) через бедренную вену. Ингибитор вводят в концентрации (мг/мл), которая дает 80% ингибирования V0 в образце плазмы спустя 5 мин после вливания. Кровь отбирают из артерии (кровь:цитрат натрия, 0,25/0,025 мл) с определенными интервалами (5, 10, 15, 30, 60 и 120 мин) и помещают в пробирку для центрифугирования. Образец плазмы получают путем центрифугирования крови при 10000 rpm в течение 5 мин. Этот образец анализируют на ингибирование тромбина с использованием хромогенного анализа, описанного далее.
Скорость повышения абсорбции при 405 нм синтетических пептидов (50 мкМ Spectozyme® TH (H-D-HHT-Ala-Arg-pNA-2AcOH), American Diagnostica) измеряют в присутствии плазмы с помощью микро планшетного ридера (Molecular Devices) при 37oС с использованием водного буфера (10 мМ Трис, 10 мМ ГЕПЕС, 150 мМ NaCl, 0,1% PEG; pH 7,4). Буфер добавляют к образцам плазмы, разбавленным (1: 1) перед помещением на микропланшет (1:5 и 1:50 до конечных разбавлений плазмы 1:10 и 1:100, соответственно) и затем добавляют фермент (1 нМ человеческого α тромбина). Данные собирают в течение более 30 мин и начальную скорость гидролиза субстрата (V0 (мOD/мин)) рассчитывают с помощью анализирующей программы (Softmax, Molecular Devices).
Время для элиминирования половины плазмы (t 1/2) рассчитывают из скошенного агара V0 против времени, используя следующее уравнение: t 1/2=-1,6 скошенный агар. Эти данные представлены в таблице А.
Биодоступность определяют в опытах ex vivo, которые включают скорости гидролиза при введении лекарственного препарата как внутривенно, так и перорально. По этому анализу самцов крыс снабжают, как описано выше, тефлоновой трубкой,
имплантированной в бедренную вену. У животных отбирают кровь за 15 мин перед дозированием ингибитора (0,25 мл крови отбирают в шприц, содержащий 0,025 мл Sigma цитрата натрия) и используют в анализе. Исследуемые соединения диспергируют в воде (0,3-3,0 мг/мл конечной концентрации) и вводят животному (0,25 мл) либо внутривенно, либо перорально. Кровь отбирают из артерии (кровь: цитрат натрия, 0,25/0,025 мл) с определенными интервалами (0,25, 0,5, 1,0, 2,0 и 3,0 ч) и плазму получают путем центрифугирования крови при 10000 rpm в течение 5 мин. Этот образец анализируют на ингибирование тромбина с использованием хромогенного анализа, описанного далее.
Скорость повышения абсорбции при 405 нм синтетических пептидов (50 мкМ Spectozyme® TH (H-D-HHT-Ala-Arg-pNA-2AcOH), American Diagnostica) измеряют в присутствии плазмы с помощью микро планшетного ридера (Molecular Devices) при 37oС с использованием водного буфера (10 мМ Трис, 10 мМ ГЕПЕС, 150 мМ NaCl, 0,1% PEG; pH 7,4). Буфер добавляют к образцам плазмы, разбавленным (1: 1) перед помещением на микропланшет (1:5 и 1:50 до конечных разбавлении плазмы 1: 10 и 1:100, соответственно) и затем добавляют фермент (1 нМ человеческого α тромбина). Данные собирают в течение более 30 мин и начальную скорость гидролиза субстрата (V0(мОD/мин)) рассчитывают с помощью анализирующей программы (Softmax, Molecular Devices).
Стандартную кривую получают добавлением лекарственного препарата или растворителя (100 мкл) ко всей крови (900 мкл) с последующим инкубированием (5 мин) и центрифугированием для получения плазмы. Процент ингибирования тромбина рассчитывали путем сравнения V0 растворителя с V0 обработанных лекарственным препаратом образцов. Данные некоторых определений были нормализованы с помощью статистической обработки (SAS) с получением стандартной кривой.
Процент ингибирования тромбина при обработке животных рассчитывали путем сравнения V0 перед обработкой с V0, полученных из образцов, собранных после обработки. Процент ингибирования наносили на стандартную кривую (полученную выше) с использованием статистической обработки (SAS) и количество лекарственного препарата в образце экстраполируют.
Биодоступность (Биодост. ) рассчитывают из участков экстраполированного лекарственного средства в образце в отношении внутривенного и перорального введения с использованием следующего уравнения:
% Биодост. = (доза при внутривенном введении/доза при пероральном введении) • (площадь под пероральной кривой/площадь под внутривенной кривой)
N-Me PPACK: Ki=0,010; Ki=0,0039.
Аргатробан: Ki=0,070; Ki=2,9.
Как показано в таблице А, соединения формулы I могут использоваться в фармацевтических композициях для лечения пациентов (людей и других приматов) с тромботическими заболеваниями таким же образом, как известные гепарины и кумарины. Соединения могут быть введены любым парентеральным путем (внутривенным, внутрибрюшинным, подкожным, с помощью пластинки на коже), где предпочтительным путем является внутривенное вливание). Дозы для вливаний могут быть в области 0,1-300 мкг/кг/мин ингибитора в смеси с фармацевтически приемлемым носителем в течение периода времени в области от нескольких минут до нескольких дней. Избранные соединения могут также вводиться перорально в дозах в области около 1-100 мг/кг.
Фармацевтические композиции могут быть получены с использованием обычных фармацевтических наполнителей и обычных методов получения. Пероральные стандартные лекарственные формы могут быть представлены эликсирами, сиропами, капсулами, таблетками и тому подобное. Типичным твердым носителем является инертное вещество, такое как лактоза, крахмал, глюкоза, метилцеллюлоза, стеарат магния, дикальций фосфат, маннитол и тому подобное; и типичный жидкий пероральный наполнитель включает этанол, глицерин, воду и тому подобное. Все наполнители могут быть смешаны, при необходимости, с разрыхлителями, разбавителями, гранулирующими агентами, смазывающими веществами, связующими и тому подобное, используя обычные методы, известные специалистам в данной области получения стандартных лекарственных форм. Парентеральные стандартные лекарственные формы могут быть получены с использованием воды или другого стерильного носителя.
Обычно соединения формулы I выделяют и используют в виде их фармацевтически приемлемых солей. Примеры таких солей включают соли гидробромида, гидроиодида, гидрохлорида, перхлорной кислоты, серной кислоты, малеиновой кислоты, фумаровой кислоты, яблочной кислоты, винной кислоты, лимонной кислоты, бензойной кислоты, миндальной кислоты, метансульфоновой кислоты, гидроэтансульфоновой кислоты, бензолсульфоновой кислоты, щавелевой кислоты, памоевой кислоты, 2-нафталинсульфоновой кислоты, п-толуолсульфоновой кислоты, циклогексансульфаминовой кислоты и сахарина.
Кроме лечения тромботических заболеваний соединения формулы I могут быть использованы для предотвращения коагуляции хранящихся образцов крови и в качестве покрытий медицинских устройств, таких как стенты и ортопедические приспособления.
Обычно они могут использоваться в любых случаях, когда желательно ингибировать коагуляцию путем введения соединений в контакт со средой, содержащей тромбин. Для этих соединений, использование которых в качестве антикоагулирующих средств было подтверждено примерами, могут найтись различные другие методы применения в качестве ингибиторов тромбина в соответствии с настоящим изобретением. Эти методы применения рассматриваются как входящие в объем данного изобретения, в данном изобретении рассматривается использование соединений формулы I в качестве антитромботических средств.
Еще другим использованием соединений по изобретению является использование в качестве ингибиторов трипсина. Ингибиторы трипсина были использованы клинически при панкреатитных заболеваниях, таких как панкреатит. Значение IС50 соединений по изобретению имеют преимущества при сравнении с панкреатитными средствами камостат мезилатом и нафамостатом (IC50, 1•10-8 и 1,3•10-8, соответственно). Соединения формулы I могут быть использованы таким же образом, как указанные терапевтические средства.
С целью иллюстрации изобретения представлены следующие примеры. Данные примеры не ограничивают изобретение. Они предназначены только для подтверждения способа практического осуществления изобретения. Специалист в данной области может найти другие способы осуществления изобретения, которые являются для него очевидными. Однако предполагается, что эти способы входят в объем данного изобретения.
ПРИМЕРЫ 1 И 2
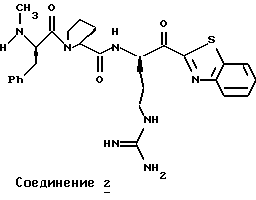
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД
Соед.1
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1R-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД
Соед.2
Стадия а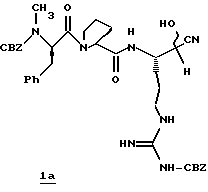
Смесь N-СВZ-N-метил-D-фенилаланил-L-пролин-Ng-СВZ-L-аргининальдегид (9,92 г, 14,5 ммоль; патент США 4703036), CH2Cl2 (48 мл), ацетонциангидрин (4,0 мл, 43,4 ммоль) и триэтиламин (1,2 мл, 8,7 ммоль) перемешивают при комнатной температуре в атмосфере аргона в течение 3 ч. После еще одного часа перемешивания добавляют дополнительную порцию ацетонциангидрина (1,3 мл, 14,2 ммоль). Смесь концентрируют в вакууме и распределяют между водой и этилацетатом. Полученный органический слой промывают порциями воды и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме. Остаток растирают несколькими порциями гексана и полученный твердый остаток сушат в вакууме с получением циангидринового промежуточного продукта 1а в виде твердого продукта.
Стадия b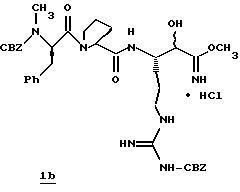
Газообразный HCl (51 г) барботируют в раствор циангидрина 1а (9,3 г, 13,1 ммоль) и МеОН (200 мл) при -50-70oС в атмосфере аргона в течение более 1,5 ч. Реакционную смесь помещают в холодильник при 0oС, определяют ее характеристики (ТСХ и ЯМР) в течение более 3 дней и помещают в делительную воронку в атмосфере аргона. Реакционную смесь прибавляют по каплям к перемешиваемой смеси воды (700 мл), этилацетата (250 мл) и NaHCO3 (159 г, 1,4 моль) при 0-6oС. рН Раствора для гашения контролируют и не дают опуститься ниже рН 6,8. Прибавляют дополнительные порции воды и NаНСО3 (как диктуется измерением рН) до тех пор, пока реакционная смесь не становится нейтральной. Полученную смесь фильтруют через вспомогательный фильтровальный материал, оставляют фильтрат, который промывают несколькими порциями этилацетата. Объединенные органические слои промывают солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением HCl соли промежуточного продукта 1b в виде твердого продукта.
Стадия с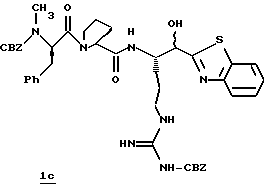
2-Аминотиофенол (18,53 г, 18,52 ммоль) прибавляют к дегазированному раствору имидата 1b (7,23 г, 9,26 ммоль) в абсолютном этаноле (290 мл) при комнатной температуре в атмосфере аргона. Полученную смесь нагревают с обратным холодильником в течение 4,75 ч, охлаждают до комнатной температуры, подвергают воздействию кислорода атмосферы при комнатной температуре в течение 18 ч и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле элюируя СН2С12/метанолом (95: 5) с получением промежуточного гидроксибензотиазола 1с в виде белой пены.
Стадия d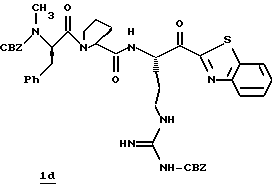
Периодинан Десс-Мартина (Dess-Martin) (1,59 г, 3,76 ммоль) прибавляют к раствору CH2Cl2 (40 мл) при комнатной температуре в аргоне. Полученную смесь перемешивают в течение 45 мин и добавляют дополнительную порцию периодинана (1,2 г, 2,83 ммоль), затем перемешивают еще 15 мин. Избыток периодинана гасят добавлением 8 мл гасящего раствора (25 г Na2S2O3 в 100 мл насыщенного водного NаНСО3), разбавленного этилацетатом (300 мл) и перемешивают при комнатной температуре в течение 15 мин. Полученный водный слой отделяют и экстрагируют несколькими порциями этилацетата и объединенные органические экстракты промывают последовательными порциями воды и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме с получением кетона 1d в виде белой пены.
Стадия е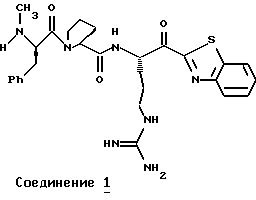
Промежуточный кетон 1d (2,97 г, 3,63 ммоль) и анизол (10 мл) помещают в тефлоновую реакционную трубку HF аппарата в безводной среде и охлаждают до -78oС. HF (15-20 мл) отгоняют в эту трубку и после завершения добавления температуре смеси дают подняться до 0oС. Смесь перемешивают в течение 2 ч, концентрируют в вакууме и растирают в нескольких порциях эфира с получением желтого твердого продукта. Твердый продукт очищают ВЭЖХ с обращенной фазой с использованием воды/ацетонитрила/ТФУ (70:30:0,2) с получением соединений 1 и 2. Каждый диастереомер лиофилизуют с получением желаемых продуктов в виде белой пены, где соединением 1 является 95% собранного продукта.
Соединение 1 (диастереомер L-аргинина): FAB-MS m/z 550 (МН+); [α]
Анализ. Расчет для C28H35N7O3S•2,75ТФУ•0,5Н2O:
Вычислено: С 46,12; Н 4,47; N 11,24; H2O 1,03.
Найдено: С 46,13; Н 4,37; N 11,35; Н2O 1,27.
Соединение 2 (диастереомер D-аргинина): FAB-MS m/z 550 (MH+); [α]
Анализ. Расчет для C28H35N7О3S•3TФУ•2H2O:
Вычислено: С 44,01; Н 4,56; N 10,57; H2O 3,88.
Найдено: С 44,11; Н 4,34; N 10,96; H2O 3,80.
ПРИМЕР 3
Стадия а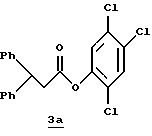
Раствор дициклогексилкарбодиимида (18,24 г, 88,4 ммоль) в ТГФ (35 мл) прибавляют по каплям к перемешиваемому раствору 3,3-дифенилпропионовой кислоты (20,0 г, 88,4 ммоль), 2,4,5-трихлорфенола (17,45 мл, 88,4 ммоль) в ТГФ (50 мл) в атмосфере аргона при -20oС. Реакционную смесь перемешивают при -20oС в течение 2,5 ч, помещают в холодильник при 0oС в течение 16 ч и фильтруют через вспомогательный фильтровальный материал. Маточный раствор концентрируют в вакууме и перекристаллизовывают из этанола с получением активированного эфира 3а в виде твердого продукта.
Стадия b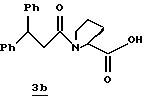
Активированный эфир 3а (15,0 г, 37,0 ммоль) прибавляют к смеси L-пролина (4,26 г, 37,0 ммоль), триэтиламина (5,15 мл) и пиридина (45 мл) при 5oС в атмосфере аргона. Реакционной смеси дают нагреться до комнатной температуры, перемешивают в течение 76 ч и концентрируют в вакууме. К остатку прибавляют водный раствор NаНСО3 (3,42 г/130 мл) и эфир (100 мл) и полученную смесь перемешивают при комнатной температуре в течение 45 мин. Органический слой экстрагируют несколько раз водой. Объединенные водные слои экстрагируют двумя порциями эфира, подкисляют 1 н. НСl и экстрагируют несколькими порциями этилацетата. Объединенные этилацетатные слои промывают солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением промежуточного продукта 3b в виде твердого продукта.
Стадия с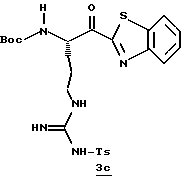
1,6 М н-Бутиллития/гексане (16 мл, 25,6 ммоль) прибавляют к раствору бензотиазола (3,7 мл, 33,9 ммоль) и безводного ТГФ (114 мл) при -78oС в атмосфере аргона. Реакционную смесь перемешивают при -78oС в течение 15 мин и через канюлю прибавляют N-α-Вос-NG-тозил-L-аргинин N,0-диметиламид (800 мг, 1,69 ммоль; DiMaio, et al. Journal of Medicinal Chemistry 1992, 35, 3331) и ТГФ (43 мл) поддерживая температуру ниже 70oС. Полученную смесь перемешивают в течение 1,66 ч при -78oС, выливают в насыщенный водный раствор NH4Cl (600 мл), энергично перемешивают в течение 15 мин и экстрагируют несколькими порциями этилацетата. Объединенные этилацетатные экстракты промывают последовательными порциями воды и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле, элюируя этилацетатом/гексаном (5:2) с получением промежуточного бензотиазола 3с в виде масла.
Стадия d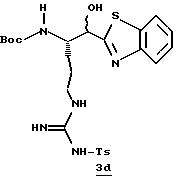
Боргидрид натрия (63,8 мг, 1,69 ммоль) прибавляют по каплям к раствору бензотиазола 2с (307 мг, 0,563 ммоль) в МеОН (10 мл) при -20oС в аргоне. Смесь перемешивают при приблизительно -20oС в течение 40 мин и медленно добавляют ацетон (2 мл). Реакционной смеси дают нагреться до комнатной температуры в течение 1,25 ч, концентрируют в вакууме и распределяют между этилацетатом и водой. Водный слой экстрагируют несколькими порциями этилацетата. Объединенные этилацетатные слои промывают солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением промежуточного спирта 3d в виде бледно-желтой пены.
Стадия е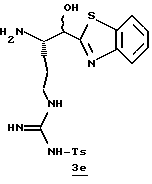
Раствор спирта 3d (0,27 г, 0,49 ммоль) в 10 мл ТФУ/СН2Сl2 (1:4) перемешивают при комнатной температуре в атмосфере аргона в течение 1,5 ч. Полученную смесь концентрируют в вакууме с получением ТФУ соли 3е в виде масла.
Стадия f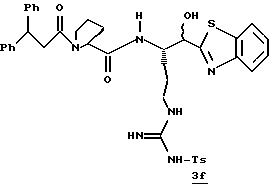
Раствор ТФУ соли амина 3е (0,49 ммоль), триэтиламина (0,25 мл), промежуточного продукта 3b (158 мг, 0,49 ммоль), гидрате гидроксибензотриазола (111 мг, 0,54 ммоль) в ацетонитриле (10 мл) обрабатывают ДЦК (73 мг, 0,54 ммоль) и перемешивают при комнатной температуре в атмосфере аргона в течение 20 ч. Полученную смесь фильтруют и твердый продукт промывают несколькими порциями ацетонитрила. Фильтрат и промывные воды объединяют, концентрируют в вакууме и распределяют между этилацетатом и водой. Органический слой промывают последовательными порциями 1 н. НСl, воды, насыщенного водного NaHCO3 и солевого раствора; сушат (Na2SO4) и концентрируют в вакууме. Остаток очищают с использованием препаративной ТСХ на силикагеле, элюируя с помощью СНСl3/МеОН бензотиазол 3f в виде игольчатого твердого продукта.
Стадия g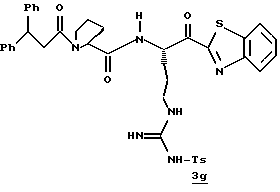
Смесь 3f (7,2 мг, 0,0096 ммоль), CH2Cl2 (1 мл) и периодинан Десс-Мартина (Dess-Martin) (8,1 мг, 0,019 ммоль) перемешивают при комнатной температуре в азоте в течение 1 часа, обрабатывают 4 мл гасящего раствора (25 г Na2S2O3 в 100 мл насыщенного водного NaHCO3) и разбавляют водой и CH2Cl2. Полученный органический слой промывают водой, сушат (Na2SO4) и концентрируют в вакууме с получением кетона 3g в виде пены.
Стадия h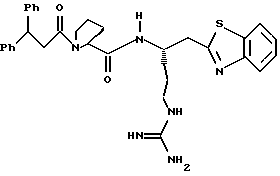
Соединение 3
Смесь анизола (1 мл) и кетона 3g (137 мг, 0,17 ммоль) помещают в тефлоновую реакционную трубку, присоединяют к HF аппарату и охлаждают до -78oС. HF (5 мл) отгоняют в реакционную трубку и полученную смесь перемешивают при 0oС в течение 3 ч. HF удаляют в вакууме и смесь растирают в трех порциях эфира. Полученный осадок фильтруют, сушат на воздухе, растворяют в ацетонитриле/воде (1: 1) и очищают ВЭЖХ с обращенной фазой, используя СН3СN+2% ТФУ/Н2О+0,016% ТФУ в качестве элюента. Желаемые фракции концентрируют в вакууме и лиофилизуют с получением соединения 2 в виде 1:3 смеси D и L диастереомеров аргинина; ИК (КВr, см-1) 1674, 1450, 1204, 1135; [α]
Анализ. Расчет для C33H36N6O3S•1,75 ТФУ•0,33Н2О:
Вычислено: С 52,96; Н 5,02; N 10,15; H2O 3,81.
Найдено: С 53,27; Н 5,13; N 10,34; Н2О 3/86.
ПРИМЕР 4
Стадия а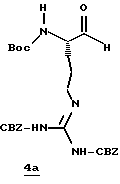
1,1-карбонилдиимидазол (0,65 г, 4,05 ммоль) прибавляют к смеси N-α-Boc-NG, NG'-ди-СВZ-L-аргинин (92,0 г, 3,69 ммоль) и ТГФ при 0oС аргоне и полученную смесь перемешивают в течение часа при 0oС. Смесь охлаждают до -48oС и добавляют раствор 1М DIBAL/гексан (10,3 мл, 10,3 ммоль) со скоростью, при которой поддерживается температура реакции между -48oС и -42oС. Полученную смесь перемешивают еще 15 мин при -48oС и прибавляют водный раствор КНSO4 (1,4 г/5,0 мл) при температуре между -40 и -28oС. Смеси дают нагреться до комнатной температуры. Прибавляют CH2Cl2 и полученный твердый продукт фильтруют и промывают дополнительными порциями CH2Cl2. Собранные фильтраты промывают водой, сушат (МgSO4) и концентрируют в вакууме. Остаток очищают кристаллизацией из изо-РrОН и гексана с получением альдегида 4а в виде твердого продукта.
Стадия b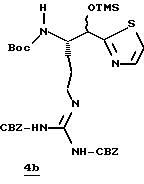
Смесь альдегида 4а (0,765 г, 1,36 ммоль), СН2Сl2 (5 мл) и 2-(триметилсилил)тиазол (1,07 г, 6,8 ммоль) перемешивают при комнатной температуре в атмосфере аргона в течение 16 ч. Реакционную смесь концентрируют в вакууме с получением 4b в виде масла.
Стадия с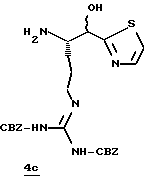
Трифторуксусную кислоту (6 мл) медленно прибавляют к перемешиваемой смеси тиазола 4b (1,36 ммоль) и CH2Cl2 (30 мл) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивают в течение 6 ч и концентрируют в вакууме. Остаток очищают флэш хроматографией на силикагеле с использованием CH2Cl2/MeOH/NH4OH (95:4,5:0,5) в качестве элюента с получением единственного диастереомера. Выделенный продукт растворяют в СН2С12, сушат (K2CO3), фильтруют и концентрируют в вакууме с получением спирта 4с в виде стеклообразного масла.
Стадия d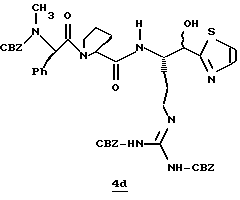
ДЦК (0,079 г, 0,38 ммоль) прибавляют к смеси спирта 4с (0,18 г, 0,35 ммоль), (CBZ)-N-метил-D-фенилаланил-L-пролин (0,14 г, 0,35 ммоль; патент США 4703036), гидроксибензо-триазола (0,053 г, 0,38 ммоль) и СН3СN (4,5 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 3,5 ч и полученный твердый продукт промывают порциями СН3СN. Объединенные фильтраты концентрируют в вакууме и распределяют между этилацетатом и водой. Органический слой промывают последовательными порциями воды и солевого раствора; сушат (MgSO4) и концентрируют в вакууме. Остаток очищают хроматографически (силикагель, CH2Cl2/MeOH (95/5)) с получением связанного спирта 4d в виде белой пены.
Стадия е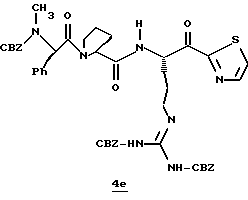
Раствор спирта 4d (0,14 мг, 0,15 ммоль) и CH2Cl2 (3 мл) прибавляют одной порцией к смеси периодинана Десс-Мартина (76 мг, 0,179 ммоль) и CH2Cl2 (4 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 35 мин и добавляют немного дополнительных миллиграмм периодинана Десс-Мартина. Полученную смесь перемешивают еще 10 мин и добавляют 4 мл гасящего раствора (25 г Na2S2O3 в 100 мл насыщенного водного NaHCO3) и затем перемешивают еще 10 мин. Полученную смесь распределяют между этилацетатом и водой и перемешивают еще 5 мин. Органический слой промывают несколькими слоями воды и солевого раствора, сушат (МgSO4) и концентрируют в вакууме с получением кетона 4е в виде бесцветного стеклообразного продукта.
Стадия f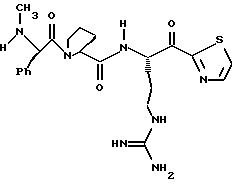
Соединение 4 (RWJ 50026)
Кетон 4е (0,13 г, 0,144 ммоль) и анизол (1 мл) помещают в тефлоновую реакционную трубку HF аппарата в безводной среде и охлаждают до -78oС. HF (5-10 мл) отгоняют прямо в эту колбу, температуре смеси дают подняться до 0oС и эту смесь перемешивают в течение 1,5 ч. HF удаляют в вакууме и остаток растирают в нескольких порциях эфира с получением твердого продукта. Этот твердый продукт очищают ВЭЖХ с обращенной фазой, элюируя водой/ацетонитрилом/ТФУ (50: 50:0,2) и концентрируют в вакууме. Остаток растворяют в воде и устанавливают рН 6,48 с помощью свежепромытой ионобменной смолы Amberlite® IRA-400 (ОН-) и водной 0,1 ТФУ. Твердый осадок выделяют фильтрацией и фильтрат лиофилизуют с получением соединения 4 в виде белого твердого продукта.
Анализ. Расчет для C24H33N7O3S•2,75С2НF3O4•0,2H2O
Вычислено: С 44,88; Н 5,03; N 13,08; Н2О 2,88.
Найдено: С 44,49; Н 4,93; N 12,98; Н2О 2,64.
ПРИМЕР 5
Стадия а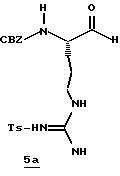
1,1'-Карбонилдиимидазол (9,89 г, 0,053 ммоль) прибавляют одной порцией к перемешиваемому раствору N-α-СВZ-NG-тозил-L-аргинин (25,2 г, ммоль) и ТГФ (160 мл) при 0-10oС. Через 1 ч реакционную смесь охлаждают до -42oС и добавляют 1М DIBAL/гексан (130 мл, 130 ммоль) в течение более 30 мин. Полученную смесь перемешивают еще 30 мин при -42oС. Реакционную смесь гасят медленным прибавлением 1,2 М HCl (365 мл) и реакционной температуре дают достигнуть комнатной температуры. Смесь обрабатывают 0,6 М HCl (360 мл) и СHCl3 (400 мл) и перемешивают при комнатной температуре в течение 2,5 ч. Водную фазу промывают несколькими порциями СHCl3 и объединенные органические экстракты промывают последовательными порциями воды, сушат (MgSO4) и концентрируют в вакууме с получением альдегида 5а в виде твердого продукта; FAB-MS m/z 447 (МН)+.
Стадия b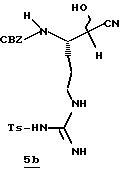
KCN (6,0 г, 92 ммоль) прибавляют к перемешиваемой смеси альдегида 4а (22,0 г, 49 ммоль) в МеОН (60 мл), Н2О (60 мл) и этилацетате (110 мл) и перемешивают при 22oС в течение 16 ч. Полученный водный слой экстрагируют несколькими порциями этилацетата и объединенные органические экстракты промывают водой, сушат (Na2SO4) и концентрируют в вакууме с получением нитрила 5b в виде пены; FAB-MS m/z 474 (МН)+.
Стадия с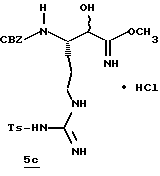
Безводный НСl (г) барботируют в раствор нитрила 5b (8,0 г, 16,9 ммоль) и безводный МеОН (162 мл) при -78oС с такой скоростью, чтобы температура не превышала -40oС. После добавления реакционную смесь перемешивают в течение 30 мин при 0oС. Реакционную смесь прибавляют к энергично перемешиваемому водному NаНСО3, поддерживая рН 6. После нейтрализации рН устанавливают рН 4,0 с помощью прибавления ледяной уксусной кислоты и этилацетата (350 мл). Смесь перемешивают 4 ч, разделяют и водный слой экстрагируют три раза этилацетатом. Объединенные этилацетатные экстракты промывают водой, насыщенным водным NаНСО3, а также солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением промежуточного продукта 5с в виде белого твердого продукта; FAB-MS m/z 507 (МН)+.
Стадия d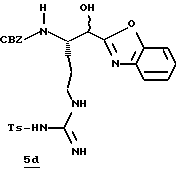
Раствор промежуточного продукта 5с (1,00 г, 1,84 ммоль), 2-аминофенола (0,22 г, 2,03 ммоль) и абсолютного EtOH (40 мл) нагревают с обратным холодильником в N2 в течение 24 ч и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле, элюируя СН2Сl2/МеОН (95:5) с получением бензоксазола 5d в виде белой пены: Т. пл. 81-91oС; [α]
Анализ. Расчет для C28H31N5O6S•0,6Н2О:
Вычислено: С 58,34; Н 5,14; N 12,15; H2О 1,87.
Найдено: С 58,32; Н 5,14; N 11,97; H2O 1,67.
Стадия е
Смесь 5d (0,87 г, 1,53 ммоль), 20% Pd(OH)4/C (0,21 г) и абсолютный EtOH (19 мл) помещают в атмосферный гидратор и перемешивают в атмосфере Н2 в течение 16 ч. Полученную смесь фильтруют через вспомогательный фильтровальный материал и концентрируют в вакууме с получением свободного амина 5е в виде желтой пены.
Стадия f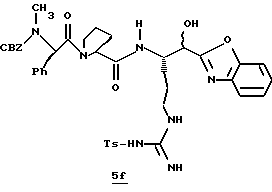
Раствор ДЦК (0,25 г, 1,1 ммоль) и CH3CN (1 мл) прибавляют по каплям к раствору гидрата 1-гидроксибензо-триазола (0,16 г, 1,2 ммоль), амина 5е (0,47 г, 1,1 ммоль), (CBZ)-N-метил-фенилаланил-L-пролина (0,44 г, 1,1 ммоль) и CH3CN (15 мл) в N2. Реакционную смесь перемешивают в течение 3,5 ч, фильтруют и полученную лепешку на фильтре промывают несколькими порциями СН3СN. Объединенные фильтраты концентрируют в вакууме, растворяют в этилацетате, промывают последовательными порциями воды и солевого раствора; сушат (МgSO4) и концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле, элюируя CH2Cl2/MeOH (93:7)) с получением связанного спирта 5f в виде желтого стеклообразного продукта.
Стадия g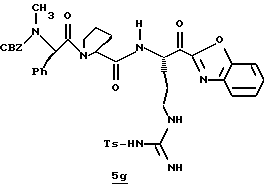
Раствор спирта 5f (0,45 г, 0,56 ммоль) и безводного CH2Cl2 (5 мл) прибавляют к перемешиваемой смеси периодинана Десс-Мартина (300 мг, 0,73 ммоль) и безводного CH2Cl2 (20 мл) при комнатной температуре в аргоне. Реакционную смесь перемешивают в течение 30 мин и добавляют другую порцию периодинана Десс-Мартина (50 мг, 0,12 ммоль) с последующим перемешиванием еще 10 мин при комнатной температуре. К перемешиваемой смеси прибавляют 30 мл гасящего раствора (25 г Nа2S2O3 в 100 мл насыщенного водного NаНСО3) и затем порции этилацетата. Полученный органический слой промывают последовательными порциями насыщенного водного NаНСО3 и солевого раствора, сушат (MgSO4) и концентрируют в вакууме с получением кетона 5g в виде пены; FAB-MS m/z 882 (МН+).
Стадия h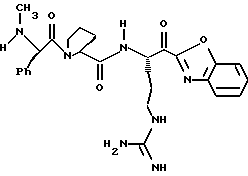
Кетон 5g (0,45 г, 0,54 ммоль) и анизол (около 2,0 мл) помещают в тефлоновую реакционную трубку HF аппарата в безводной среде и охлаждают до -78oС. HF (0,5-1,0 мл) отгоняют прямо в эту колбу, температуре смеси дают подняться до 0oС и эту смесь перемешивают при 0oС в течение 4 ч. Удаляют HF в вакууме и остаток растирают в нескольких порциях эфира с получением желтого твердого продукта, который очищают ВЭЖХ с обращенной фазой с использованием воды/ацетонитрила/ТФУ (70:30:0,2) и лиофилизуют с получением соединения 5 в виде порошка; Т. пл. 81-91oС; [α]
Анализ. Расчет для C28H35N7O4•4(С2НF3O2)•2,5Н2О.
Вычислено: С 41,97; Н 4,25; N 9,52; Н2О 3,93.
Найдено: С 42,26; Н 4,42; N 9,87; Н2О 4,27.
ПРИМЕР 6
Стадия а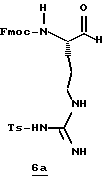
1,1'-Карбонилдиимидазол (1,8 г, 11,0 ммоль) прибавляют к раствору N-α-Fmос-NG-тозил-L-аргинин (6,0 г, 10,0 ммоль) в безводном ТГФ (30 мл) при 0oС в атмосфере аргона и перемешивают при 0oС в течение 1,5 ч. Реакционную смесь охлаждают до -48oС и добавляют по каплям 1М DIBAL (28 мл, 28 ммоль) в течение более 20 мин. Полученную смесь перемешивают еще 1,5 ч и при перемешивании добавляют 1,2 н. НСl (67 мл). Смеси дают нагреться до комнатной температуры и распределяют между 0,6 н. НСl (65 мл) и хлороформом. Полученный водный слой промывают несколькими порциями хлороформа. Объединенные органические экстракты промывают последовательными порциями воды и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме с получением альдегида 6а в виде белого хлопьевидного твердого продукта.
Стадия b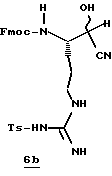
Раствор KCN (1,44 г, 22 ммоль) и Н2О (125 мл) прибавляют к раствору альдегида 6а (5,9 г, 11,0 ммоль) в этилацетате (250 мл) и полученную смесь перемешивают в течение 40 ч при комнатной температуре в аргоне. Органический слой отделяют, а водный слой промывают тремя порциями этилацетата. Объединенные этилацетатные экстракты промывают солевым раствором, сушат (Na2SO4), концентрируют в вакууме и хранят в холодильнике в атмосфере аргона. Остаток распределяют между этилацетатом (100 мл) и насыщенным водным NаНСО3 (200 мл) и рН поддерживают при 7,0 путем прибавления NаНСО3. Твердый NаНСО3 удаляют фильтрацией и полученный водный слой промывают несколькими порциями этилацетата. Объединенный органический слой промывают дважды солевым раствором, сушат (МgSO4) и концентрируют в вакууме с получением циангидрина 6b в виде белого твердого продукта; FAB-MS m/z 562 (МН)+.
Стадия с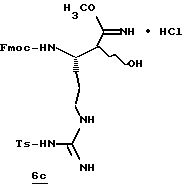
НСl (21 г) барботируют в раствор нитрила 6b (3,0 г, 5,34 ммоль) и метанол (53 мл) в аргоне при температуре ниже -40oС в течение более 20 мин. Реакционный сосуд закрывают в атмосфере азота и помещают в морозильник при -15oС в течение 46 ч и концентрируют в вакууме при комнатной температуре. Остаток распределяют между раствором насыщенного водного NаНСО3 (250 мл) и этилацетатом. Органический слой промывают дважды солевым раствором, сушат (МgSO4) и концентрируют в вакууме с получением имидата 6с в виде твердого продукта.
Стадия d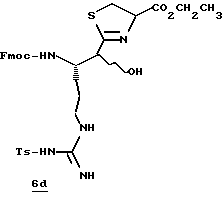
Гидрохлорид этилового эфира цистеина (1,97 г, 10,6 ммоль) добавляют к раствору имидата 6с (3,30 г, 5,3 ммоль) и СН2Сl2 (100 мл) и полученную смесь перемешивают в атмосфере аргона при комнатной температуре в течение 3 ч. Твердый осадок удаляют фильтрацией и фильтрат концентрируют в вакууме. Остаток очищают хроматографически, используя СН2Сl2/МеОН (97:3) в качестве элюента с получением производного тиазолина 6d в виде твердого продукта; FAB-MS m/z 694 (МН)+.
Стадия е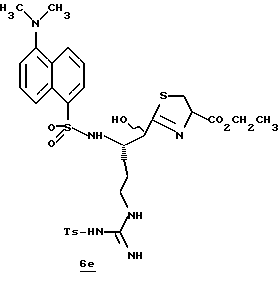
Диэтиламин (2,5 мл, 24,2 ммоль) добавляют по каплям к перемешиваемому раствору производного 6d (2,20 г, 3,17 ммоль) в безводном ацетонитриле (50 мл) при -5oС и реакционной смеси дают нагреться до комнатной температуры в течение более 18 ч. Остаток концентрируют в вакууме, растворяют в CH2Cl2 и концентрируют в вакууме. Остаток растирают в нескольких порциях гексана и концентрируют в вакууме с получением амина 6е в виде масла; FAB-MS m/z 472 (МН)+.
Стадия f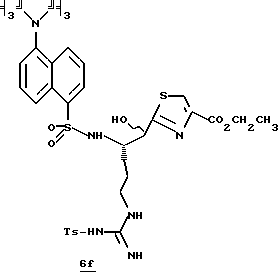
Дансил хлорид (890 мг, 3,3 ммоль) добавляют к раствору амина 6е (2,21 г, 3,1 ммоль) и CH2Cl2 при -5oС в аргоне. После завершения прибавления реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Полученную смесь концентрируют в вакууме и распределяют между СHCl3 и насыщенным водным NаНСО3. Органический слой промывают солевым раствором, сушат (К2СО3) и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле, элюируя CH2Cl2/MeOH (97:3) с получением защищенного амина 6f; FAB-MS m/z 705 (МН)+.
Стадия g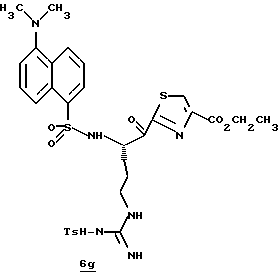
Активированный MnO2 (0,75 г) добавляют к раствору амина 6f (0,70 г, 1,49 ммоль) и CH2Cl2 (25 мл) и полученную смесь перемешивают в течение ночи при комнатной температуре. Прибавляют дополнительную порцию MnO2 (0,75 г) и эту смесь перемешивают в течение 4 ч при комнатной температуре. Прибавляют еще одну порцию MnO2 (0,75 г) и полученную смесь перемешивают в течение 16 ч при комнатной температуре. Реакционную смесь фильтруют один раз через вспомогательный фильтровальный материал и затем через фильтр Nylon 66 (0,45 мкм), используя СHCl3/МеОН для промывки лепешек на фильтре. Объединенные промывки сушат (Na2SO4) и концентрируют в вакууме. Остаток очищают с помощью ВЭЖХ, используя силикагель и CH2Cl2/MeOH (97:3) в качестве элюента с получением тиазола 6g в виде твердого продукта; FAB-MS m/z 701 (МН)+.
Стадия h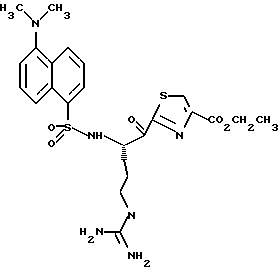
Соединение 6
Раствор тиазола 6g (141 мг, 0,20 ммоль) и анизол помещают в реакционный сосуд HF аппарата в безводной среде и охлаждают до -78oС. Безводный HF (4,5-6,0 мл) отгоняют в этот сосуд и реакционной смеси дают нагреться до 5oС и перемешивают при 5oС в течение 3,5 ч. Удаляют HF в вакууме и остаток растирают в нескольких порциях эфира с получением желтого твердого продукта. Твердый продукт очищают ВЭЖХ с обращенной фазой, элюируя Н2О/СН3СN/ТФУ (30: 20: 0,2) с получением масла. Это масло растворяют в дистиллированной воде и нейтрализуют до рН 6,20 путем прибавления смолы Amberlite® IRA-400 (ОН-). Твердый продукт фильтруют и лиофилизуют с получением соединения 6 в виде твердого продукта; Т.пл. 90-98oС; FAB-MS m/z 547 (МН)+.
ПРИМЕР 7
Стадия а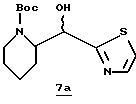
(+)-N-трет-Бутоксикарбонил-2-пиперидинкарбоксальдегид (2,13 г, 10,0 ммоль; Hassner, et al. Journal of Organic Chemistry 1991, 56, 2775) прибавляют к 2-(триметилсилил)-тиазолу (2,03 г, 11,0 ммоль) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивают в течение 6 ч, затем разбавляют безводным ТГФ (100 мл) и 5 мл 1,0 тетрабутиламмонийфторида/ТГФ, перемешивают еще 40 мин и концентрируют в вакууме. Остаток растворяют в этилацетате и промывают насыщенным водным NаНСО3, сушат (Nа2SO4) и концентрируют в вакууме с получением тиазола 7а в виде твердого продукта; FAB-MS m/z 299 (МН)+.
Стадия b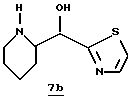
Раствор тиазола 7а (3,22 т, 9,0 ммоль), CH2Cl2 (100 мл) и ТФУ (20 мл) перемешивают в аргоне при комнатной температуре в течение 3 часов и концентрируют в вакууме. Остаток распределяют между СНСl3 (100 мл) и 50% NaOH (20 мл), затем перемешивают в аргоне при комнатной температуре. Прибавляют другую порцию СНСl3 (100 мл) и смесь опять перемешивают. Органический слой удаляют и полученный водный слой промывают несколькими порциями СНСl3. Объединенные органические промывки сушат (К2СО3) и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле, элюируя EtOAc/MeOH/NH4OH (97: 3: 1) с получением производного пиперидина 7b в виде раздельных диастереомеров.
Стадия с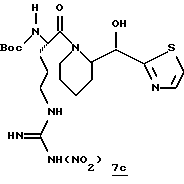
N-Метилморфолин (0,48 г, 4,68 ммоль) прибавляют к смеси N-α-Boc-NG-нитро-L-аргинин•2/3Et2O•1/4EtOAc и безводного ТГФ (20 мл) при -20oС. К этой смеси прибавляют по каплям изобутилхлорформиат при -15-20oС и полученную смесь перемешивают в течение 35 мин. Добавляют раствор производного пиперидина 7b (0,85 г, 4,2 ммоль) и безводного ТГФ (30 мл) в атмосфере аргона при -20oС. Смесь перемешивают при -15oС в течение 45 мин, дают нагреться до комнатной температуры в течение более 3 ч и помещают в холодильник при 0oС в течение 16 ч. Реакционную смесь фильтруют и фильтрат концентрируют в вакууме. Остаток растворяют в СНСl3 и органический слой промывают последовательными порциями водного NаНСО3 и солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением производного аргинина 7с в виде твердого продукта; FAB-MS m/z 500 (МН)+.
Стадия d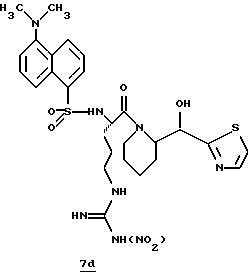
6 н. HCl/EtOH (40 мл) добавляют в аргоне к раствору аргинина 7с (1,75 г, 3,5 ммоль) и безводного ТГФ (30 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 1 часа и прибавляют другую порцию 6 н. HCl/EtOH (10 мл) и затем дополнительно перемешивают (2 ч). Еще одну порцию 6 н. HCl/EtOH (10 мл) прибавляют и затем перемешивают другой час. Реакционную смесь концентрируют в вакууме и растирают в ТГФ с получением твердого продукта. Часть этого твердого продукта (1,26 г, 2,30 ммоль) перемешивают с безводным ТГФ (45 мл) и триэтиламином (0,75 мл). Прибавляют дансил хлорид (0,63 г, 2,33 ммоль) и реакционную смесь перемешивают 3 ч при комнатной температуре. Прибавляют другую часть триэтиламина (0,25 г) и дансил хлорида (0,20 г) и реакционную смесь перемешивают еще час при комнатной температуре. Твердый продукт очищают колоночной хроматографией на силикагеле, элюируя EtOAc/MeOH/NH4OH (96: 3:1) с получением 7d в виде твердого продукта; FAB-MS m/z 633 (МН)+.
Стадия е
Стадия 7
Периодинан Десс-Мартина прибавляют к перемешиваемому раствору производного 7d (0,19 г, 0,30 ммоль) и CH2Cl2 (10 мл) при комнатной температуре в аргоне. Полученную смесь перемешивают в течение 1,5 ч и обрабатывают гасящим раствором (25 г Nа2S2O3 в 100 мл насыщенного водного NаНСО3). Полученный органический слой промывают солевым раствором, сушат (Na2SO4) и концентрируют в вакууме. Остаток помещают в реакционный сосуд HF аппарата с анизолом и охлаждают до -78oС. HF отгоняют в реакционный сосуд и температуре сосуда дают нагреться до 5oС и поддерживают ее в течение 40 мин. Смесь концентрируют в вакууме и растирают в эфире с получением игольчатого твердого продукта. Твердый продукт очищают ВЭЖХ с обращенной фазой, элюируя Н2О/СН3СН/ТФУ (60: 40:0,2) с получением соединения 7 в виде смеси диастереомеров; Т.пл. 85-100oС; [α]
Анализ. Расчет для C27H35N7O4S2•C2HF3O2•0,2H2O:
Вычислено: С 44,44; Н 4,53; N 11,52; H2O 1,05.
Найдено: С 44,36; Н 4,44; N 11,54; H2O 0,92.
ПРИМЕР 8
Стадия а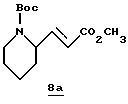
Метил(трифенилфосфоранилидиен)ацетат (4,89 г, 114,6 ммоль) добавляют по частям к раствору (±)-N-(трет-бутоксикарбонил)пиперидин-2-карбоксальдегида (3,12 г, 14,6 ммоль) и безводного ТГФ (30 мл) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивают при комнатной температуре в течение 16 ч и концентрируют в вакууме. Остаток обрабатывают эфиром (25 мл) и полученный осадок удаляют фильтрацией. Фильтрат концентрируют в вакууме и остаток очищают колоночной хроматографией на силикагеле, используя гексан/эфир (4:1) в качестве элюента с получением производного эфира 8а в виде масла.
Стадия b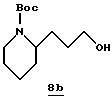
Хлорид лития (1,04 г, 24,5 ммоль) прибавляют к раствору 8а (2,20 г, 8,2 ммоль) и безводного ТГФ (24 мл). Боргидрид натрия (0,93 мл, 24,5 ммоль) прибавляют к смеси, затем абсолютный этанол (29 мл) и полученную смесь перемешивают при комнатной температуре в течение 48 часов. Смесь охлаждают до 0oC и прибавляют 10%-ную водную лимонную кислоту для доведения рН до 4. Полученную смесь концентрируют в вакууме, растворяют в H2O (40 мл) и устанавливают с помощью 10%-ной водной лимонной кислоты рН 4. Водный слой экстрагируют несколькими порциями СН2Сl2 и объединенные органические экстракты промывают солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением спиртового производного 8b в виде масла: GC/MS (El) m/z 227 (М)+.
Стадия с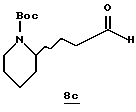
Раствор спирта 8b (2,06 г, 8,2 ммоль) и CH2Cl2 (20 мл) прибавляют в течение более 20 мин к смеси пиридиний хлорформиата (2,64 г, 12,3 ммоль) и CH2Cl2 при комнатной температуре. Смесь перемешивают в течение 30 мин, добавляют эфир (200 мл) и полученный твердый осадок отфильтровывают. Твердый продукт промывают несколькими порциями эфира/СН2Сl2 (2:1) и объединенные фильтрат и органические промывки фильтруют через силикагель. Органический раствор сушат (Nа2SO4) и концентрируют в вакууме с получением альдегидного производного 8с в виде масла.
Стадия d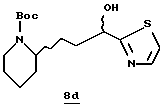
2-Триметилсилилтиазол (1,63 г, 8,8 ммоль) прибавляют к альдегиду 8с (1,93 г, 8,0 ммоль) при комнатной температуре. Полученную реакционную смесь перемешивают в течение 3,25 ч, разбавляют безводным ТГФ и добавляют 1,0 М тетрабутиламмонийфторида/ТГФ (4,0 мл), перемешивают в течение 30 мин и концентрируют в вакууме. Остаток растворяют в этилацетате, промывают насыщенным водным NаНСО3, сушат (Na2SO4) и концентрируют в вакууме с получением производного тиазола 8d в виде масла.
Стадия е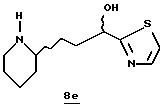
Трифторуксусную кислоту (16 мл) прибавляют к раствору производного тиазола 8b (2,48 г, 7,6 ммоль) и CH2Cl2 (50 мл) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивают в течение 2 ч, концентрируют в вакууме и растворяют в СНСl3 (60 мл). Раствор подщелачивают 50%-ным водным NaOH (20 мл) и водный слой промывают несколькими порциями СНСl3/2-пропанола (20: 1). Объединенные органические экстракты сушат (К2СО3) и концентрируют в вакууме с получением масла. Это масло очищают хроматографически на силикагеле, элюируя этилацетатом/МеОН/NН4ОН (97:3:1) с получением тиазола с удаленной защитой 8е; FAB-MS m/z 227 (МН)+.
Стадия f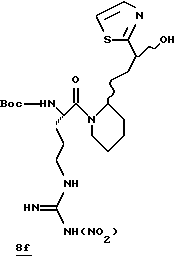
Изобутилхлорформиат (0,39 г, 2,87 ммоль) прибавляют к раствору N-α-Boc-NG-нитpo-L-apгинин•2/3Et2O•1/4EtOAc (1,12 г, 2,87 ммоль), N-метилморфолина (0,32 г, 3,20 ммоль) и безводного ТГФ (20 мл) при -20oС в атмосфере аргона. Реакционную смесь перемешивают при -5 - -10oС в течение 30 мин и охлаждают до -20oС. Добавляют по каплям раствор производного тиазола 8е (0,65 г, 2,87 ммоль) и ТГФ (20 мл) в течение более 2-3 мин и полученную смесь перемешивают при -15 - -10oС в течение 45 мин и перемешивают при комнатной температуре в течение 16 ч. Другую порцию N-α-Вос-NG-нитро-L-аргинин•2/3Еt2O•1/4ЕtOАс (0,56 г, 1,44 ммоль), N-метилморфолина (0,16 г, 1,6 ммоль) и изобутилхлорформиата (0,195 г, 1,44 ммоль) готовят, как описано выше и перемешивают при -15 - -10oС в течение 1 ч и добавляют к начальной реакционной смеси. Полученную смесь перемешивают при -15 - -10oС в течение 1 ч и при комнатной температуре в течение 16 ч. Реакционную смесь фильтруют и концентрируют в вакууме и растворяют в СНСl3. Органический слой промывают последовательными порциями насыщенного водного NаНСО3 и солевым раствором, сушат (Na2SO4) и концентрируют в вакууме. Остаток очищают с помощью ВЭЖХ, используя этилацетат/МеОН/NH4ОН (30:3:1) с получением производного аргинина 8f в виде твердого продукта.
Стадия g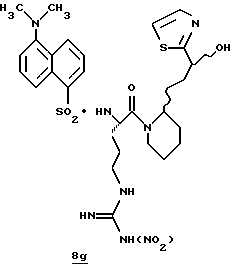
Этанольную 6 н. НСl (20 мл) медленно добавляют к перемешиваемому раствору производного аргинина 8f (0,87 г, 1,65 ммоль) и безводного ТГФ (15 мл) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивают в течение 45 мин, концентрируют в вакууме и растворяют в ТГФ (20 мл). Прибавляют триэтиламин (0,75 г, 7,4 ммоль) и затем дансил хлорид (0,46 г, 1,70 ммоль) и полученную смесь перемешивают при комнатной температуре в атмосфере аргона в течение 18 ч. Прибавляют другую часть триэтиламина (0,50 г, 4,9 ммоль) и дансил хлорида (0,23 г, 8,5 ммоль) и полученную смесь перемешивают в течение 24 ч. Смесь фильтруют и фильтрат концентрируют в вакууме и растирают в нескольких порциях эфира с получением производного дансила 8g в виде твердого продукта, который используют без дополнительной очистки; FAB-MS m/z 661 (МН)+.
Стадия h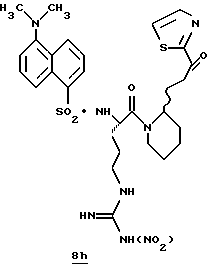
Периодинак Десс-Мартина (0,72 г, 1,70 ммоль) прибавляют порциями к перемешиваемой смеси производного дансила 8f (1,20 г, 1,36 ммоль) и безводного CH2Cl2 (40 мл) при 5oС в атмосфере аргона. Реакционной смеси дают нагреться до комнатной температуры, перемешивают в течение 1,5 ч, обрабатывают 100 мл гасящего раствора (25 г Nа2S2O3 в 100 мл насыщенного водного NаНСО3) и экстрагируют СНСl3 (50 мл) и полученный водный слой промывают несколькими порциями СНСl3. Объединенные органические экстракты промывают солевым раствором, сушат (Na2SO4) и концентрируют в вакууме. Остаток очищают хроматографически (силикагель), элюируя этилацетатом/МеОН/NН4OН (95:5:1). Желаемые фракции концентрируют в вакууме, растворяют в CH2Cl2, сушат (Na2SO4) и концентрируют в вакууме с получением производного кетона 8h в виде твердого продукта; FAB-MS m/z 882 (МН)+.
Стадия i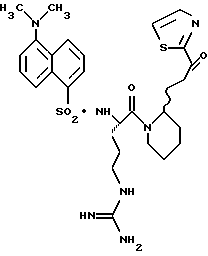
Соединение 8
Производное кетона 8h (160 мг, 0,2 ммоль) помещают в реакционный сосуд HF аппарата, добавляют безводный анизол (2 мл) и смесь охлаждают до -78oС. HF (5 мл) отгоняют в сосуд и реакционной смеси дают нагреться до 0oС. Через 1 ч HF удаляют в вакууме, к остатку добавляют эфир (25 мл) и смесь хранят в холодильнике при 0oС в атмосфере аргона в течение 16 ч. Эфир удаляют и твердый остаток растирают в нескольких порциях эфира, выделяют и сушат в вакууме. Остаток очищают колоночной хроматографией (с обращенной фазой), элюируя СН3СN/Н2О/ТФУ (93:7:0,2) с получением соединения 8 в виде смеси 1:1 диастереомеров; Т. пл. 65-70oС; [α]
Анализ. Расчет для C29H39N7O4S2•3,5С2НF3О2•0,5Н2O:
Вычислено: С 42,71; Н 4,29; N 9,59; Н2O 0,88.
Найдено: С 42,76; Н 4,44; N 9,67; H2O 0,81.
ПРИМЕР 9
Стадия а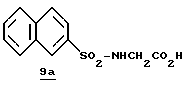
Раствор 2-нафталинсульфонилхлорида (4,52 г, 20 ммоль) и эфир (50 мл) добавляют к перемешиваемому раствору глицина (1,5 г, 20 ммоль) и 1 н. NaOH (40 мл) при комнатной температуре. Эту смесь перемешивают в течение 6 ч, эфирный слой отделяли и водный слой промывали несколькими порциями эфира. рН Водного слоя устанавливали 1 с помощью 1 н. НСl и разбавляют Н2О (50 мл). Полученный твердый осадок выделяют и сушат в вакууме с получением кислоты 9а в виде твердого продукта.
Стадия b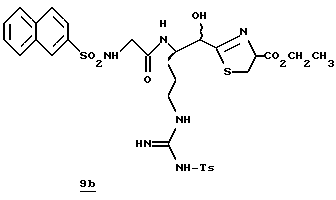
Раствор ДЦК (0,55 г, 2,67 ммоль) и СН3СN (10 мл) прибавляют по каплям к раствору гидрата 1-гидроксибензотриазола (0,49 г, 3,64 ммоль), D-аргининового эпимера амина 6е (1,43 г, 2,43 ммоль), кислоты 9а (0,64 г, 2,43 ммоль) и СН3СN (20 мл) в атмосфере аргона. Смесь перемешивают при комнатной температуре в течение 2 ч, фильтруют и концентрируют в вакууме. Остаток растворяют в воде, промывают последовательными порциями насыщенного водного NаНСО3 и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме. Остаток очищают хроматографией, элюируя этилацетатом/МеОН/NН4ОН (95:5:1) в качестве элюента. Желаемые фракции объединяют, сушат (Na2SO4) и концентрируют в вакууме с получением связанного промежуточного продукта 9b в виде смеси диастереомеров; FAB-MS m/z 719 (МН)+.
Стадия с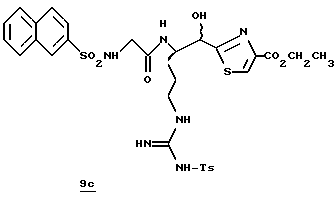
Активированный MnO2 (0,90 г, 10,4 ммоль) добавляют к перемешиваемому раствору промежуточного 9b (0,85 г, 1,18 ммоль) и CH2Cl2 (35 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 48 ч, фильтруют через фильтр Nylon 66 (0,45 мкм) и концентрируют в вакууме. Остаток очищают путем хроматографии на силикагеле, элюируя этилацетатом/МеОН (95: 5) в качестве элюента. Желаемые фракции объединяют, растворяют в СН2Сl2, сушат (Na2SO4) и концентрируют в вакууме с получением производного тиазола 9с в виде смеси диастереомеров: FAB-MS m/z 717 (МН)+.
Стадия d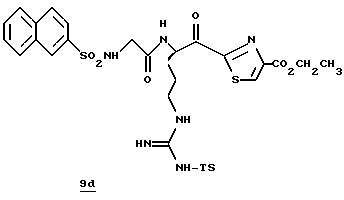
Периодинан Десс-Мартина прибавляют (105 мг, 0,248 ммоль) к перемешиваемому раствору тиазола 9с (160 мг, 0,0246 ммоль) и CH2Cl2 (5 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 1 ч при комнатной температуре. Реакционную смесь гасят добавлением 5 мл гасящего раствора (25 г Nа2S2O3 в 100 мл насыщенного водного NаНСО3), этилацетата (20 мл) с последующим перемешиванием в течение 40 мин. Полученный органический слой отделяют и водный слой промывают порциями этилацетата. Объединенные органические экстракты промывают последовательными порциями насыщенного водного NаНСО3 и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме с получением кетона 9d в виде смеси диастереомеров; FAB-MS m/z 715 (МН)+.
Стадия е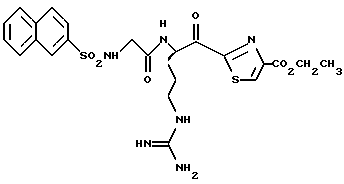
Кетон 9d (190 мг, 0,26 ммоль) и анизол (2 мл) объединяют в тефлоновой реакционной трубке HF аппарата. HF (10 мл) отгоняют в колбу при -78oС, полученную смесь нагревают до 0oС и перемешивают при 0oС в течение 3 ч. HF удаляют в вакууме добавляют эфир (25 мл) и закрытую пробкой колбу помещают в холодильник на 16 ч. Полученный твердый продукт собирают фильтрацией, промывают несколькими порциями эфира и сушат в токе азота. Твердый продукт очищают ВЭЖХ с обращенной фазой с использованием Н2О/СН3СN/ТФУ (60:40:0,2) с получением твердого продукта. Твердый продукт лиофилизуют с получением соединения 9 в виде твердого продукта: Т.пл. 105-110oС;
Анализ. Расчет для С24H28N6O6S2•С2НF3O2•Н2O:
Вычислено: С 45,08; Н 4,51; N 12,13; Н2О 2,60.
Найдено: С 44,96; Н 4,26; N 11,97; Н2O 2,99.
ПРИМЕР 10
Стадия а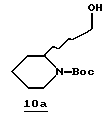
Раствор ди-трет-бутилдикарбоната (21,8 г, 0,10 моль) и безводного CH2Cl2 (200 мл) прибавляют по каплям к перемешиваемому раствору (±)-2-пиперидинэтанола (12,9 г, 0,10 моль) и CH2Cl2 (200 мл) при 0oС в аргоне в течение более 30 мин. Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 5 ч. Реакционную смесь экстрагируют дважды 1 н. НСl, один раз насыщенным водным NаНСО3 и один раз солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением защищенного амина 10а в виде масла.
Стадия b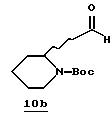
Раствор амина 10а (10,3 г, 45 ммоль) и CH2Cl2 (100 мл) прибавляют в течение более 15 мин к перемешиваемому раствору пиридиний хлорформиата (14,5 г, 67,5 ммоль) при комнатной температуре. Полученную смесь перемешивают в течение 4 ч и разбавляют эфиром (600 мл). Всю реакционную смесь фильтруют через вспомогательный фильтровальный материал и концентрируют в вакууме с получением альдегида 10b в виде масла.
Стадия с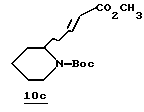
Метил(трифенилфосфоранилидиен)ацетат (13,46 г, 40,3 ммоль) прибавляют порциями к раствору альдегида 10b (9,14 г, 40,3 ммоль) и ТГФ (100 мл) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивают в течение 16 ч и концентрируют в вакууме. Остаток растирают в нескольких порциях эфира. Объединенные эфирные экстракты сушат (Na2SO4) и концентрируют в вакууме с получением эфира 10с в виде масла.
Стадия d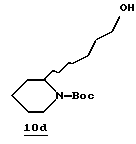
Раствор эфира 10 с (7,75 г, 27,3 ммоль) и LiCl (3,49 г, 82 ммоль) в безводном ТГФ (80 мл) обрабатывают NaBH4 (3,12 г, 82 ммоль) при перемешивании при комнатной температуре в атмосфере аргона. Прибавляют EtOH (97 мл) и полученную смесь перемешивают при комнатной температуре в течение 62 ч. Полученную смесь охлаждают до 0oС и с помощью 10%-ной водной лимонной кислоты устанавливают рН 4. Эту смесь концентрируют в вакууме до получения водного слоя. рН этого слоя устанавливают с помощью 10%-ной водной лимонной кислоты как рН 4, перемешивают при комнатной температуре в течение 30 мин и экстрагируют несколько раз CH2Cl2. Объединенные органические экстракты промывают солевым раствором, сушат и концентрируют в вакууме с получением спирта 10d в виде масла.
Стадия е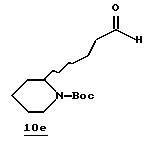
Раствор спирта 10d (6,06 г, 23,5 ммоль) и CH2Cl2 (50 мл) прибавляют к смеси пиридиний хлорформиата (7,60 г, 35,3 ммоль) и CH2Cl2 (150 мл) в течение более 10 мин и полученную смесь перемешивают при комнатной температуре в течение 2 ч. Прибавляют диэтиловый эфир (250 мл) и полученную смесь фильтруют через плаг из силикагеля, который в свою очередь промывают CH2Cl2/Et2O (1: 1). Фильтрат сушат (Na2SO4) и концентрируют в вакууме и остаток очищают хроматографией на силикагеле, элюируя этилацетатом/гексаном с получением альдегида 10е в виде масла.
Стадия f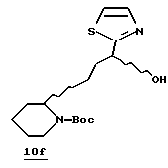
2-(Триметилсилил)тиазол (1,70 г, 9,2 ммоль) прибавляют к альдегиду 10е (2,07 г, 8,1 ммоль) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивают в течение 4 ч, разбавляют ТГФ (60 мл) и добавляют 1,0 М тетрабутиламмонийфторида/ТГФ с последующим перемешиванием в течение 15 мин. Полученную смесь концентрируют в вакууме, разбавляют этилацетатом, промывают несколькими последовательными порциями насыщенного водного NаНСО3 и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме с получением производного тиазола 10f в виде масла.
Стадия g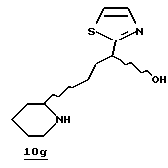
Трифторуксусную кислоту (16 мл) прибавляют к перемешиваемому раствору тиазола 10f (2,90 г, 8,0 ммоль) и CH2Cl2 (90 мл) при комнатной температуре в атмосфере аргона. Смесь перемешивают в течение 1 ч, концентрируют в вакууме и разбавляют CH2Cl2 (150 мл). Полученную смесь нейтрализуют (К2СО3), фильтруют и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле, элюируя этилацетатом/MeOH/NH4OH (95: 5: 2). Желаемые фракции концентрируют в вакууме, растворяют в CH2Cl2, сушат (К2СО3) и концентрируют в вакууме с получением амина 10g в виде смеси диастереомеров.
Стадия h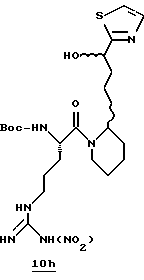
Изобутилхлорформиат (0,55 г, 4,0 ммоль) прибавляют к перемешиваемой смеси N-α-Boc-NG-нитро-L-аргинин•2/3Et2O•1/4EtOAc (1,56 г, 4,0 ммоль), N-метилморфолина (0,44 г, 4,4 ммоль) и безводного ТГФ (25 мл) при -20 - -15oС в атмосфере аргона и температуру реакции поддерживают при -15 - -10oС в течение 1,5 ч. В течение 60 секунд добавляют раствор амина 10f (0,85 г, 3,54 ммоль) и ТГФ (5 мл) при -10oС. Реакционную смесь перемешивают при -10oС в течение 1 ч, дают нагреться до комнатной температуры и перемешивают в течение 16 ч. Прибавляют каплю ДМФ, полученную смесь перемешивают еще 2 ч при комнатной температуре и фильтруют. Фильтрат концентрируют в вакууме и растворяют в СHCl3. Органический слой промывают последовательными порциями насыщенного водного NaНСО3 и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме с получением связанного производного 10h в виде твердого продукта.
Стадия i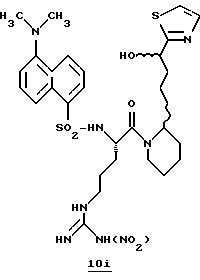
Этанольную 6 н НСl добавляют к раствору производного 10h (1,60 г, 2,95 ммоль) и ТГФ (30 мл) при комнатной температуре в атмосфере аргона. Полученную смесь перемешивают в течение 2,5 ч, концентрируют в вакууме, растворяют в ТГФ (25 мл) и опять концентрируют с получением масла. Остаток растворяют в ТГФ (40 мл) вместе с триэтиламином (15 ммоль) и обрабатывают дансил хлоридом (0,84 г, 3,1 ммоль). Смесь перемешивают при комнатной температуре в атмосфере аргона в течение 16 ч. Полученную смесь фильтруют, фильтрат концентрируют в вакууме и остаток растирают дважды с эфиром с получением производного дансила 10i в виде твердого продукта, который используют без дополнительной стадии очистки.
Стадия j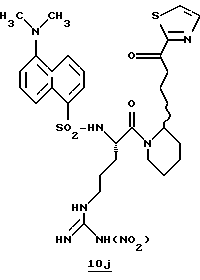
Периодинан Десс-Мартина (0,90 г, 2,1 ммоль) прибавляют к перемешиваемой смеси производного 10i (1,02 г, 1,5 ммоль) и CH2Cl2 (30 мл) при комнатной температуре в атмосфере аргона. Смесь перемешивают в течение 1,5 ч при комнатной температуре, обрабатывают 50 мл гасящего раствора (25 г Na2S2O3 в 100 мл насыщенного водного NaHCO3) и перемешивают в течение 40 мин. Органический слой отделяют и водный слой дважды экстрагируют CH2Cl2. Объединенные CH2Cl2 экстракты промывают солевым раствором, сушат (Na2SO4) и концентрируют в вакууме. Остаток растворяют в этилацетате (10 мл) и МеОН (3 мл), очищают хроматографически на силикагеле, элюируя этилацетатом/МеОН/NH4ОН (95: 5:1). Желаемые фракции объединяют, растворяют в CH2Cl2, сушат (Na2SO4) и концентрируют в вакууме с получением производного кетона 10j в виде твердого продукта.
Стадия k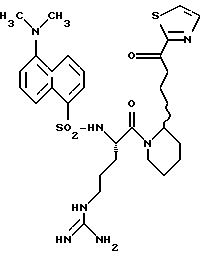
Соединение 10
Кетон 10j (0,32 г, 0,47 ммоль) и анизол (5 мл) помещают в тефлоновую реакционную трубку HF аппарата в безводных условиях и охлаждают до -78oС. HF (20 мл) отгоняют прямо в этот сосуд и температуре смеси дают подняться до 0oС. Смесь перемешивают в течение 1 ч, HF удаляют в вакууме и остаток растворяют в эфире (25 мл) и хранят в холодильнике в безводных условиях в течение ночи. Эфирный слой удаляют и полученный остаток растирают в нескольких порциях эфира с получением твердого продукта. Твердый продукт очищают ВЭЖХ с обращенной фазой, используя Н2О/СН3СN/ТФУ (60:40:0,2) с получением соединения 10 в виде смеси диастереомеров; T.пл. 81-91oС; [α]
Анализ. Расчет для С30Н41N7O4S2•3С2НF3O2•Н2O:
Вычислено: С 43,77; Н 4,69; N 9,92; Н2O 1,82.
Найдено: С 43,77; Н 4,56; N 10,00; Н2О 1,82.
ПРИМЕР 11
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[5-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]ПЕНТИЛ]-L-ПРОЛИНАМИД
Стадия а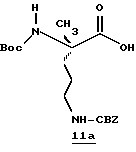
Суспензию L-Cα-метилорнитин•2НСl (7,03 г, 35 ммоль; Zhenping, Z.; Edwards, P.; R.W. Int. J. Peptlde Protein Res. 1992, 40, 119-126) в 60 мл воды охлаждают до 5oС и рН устанавливают 11,0 с помощью 3 н NaOH. Бензилхлорформиат (6,00 мл, 42 ммоль) прибавляют по каплям в течение более 4 ч при перемешивании в атмосфере аргона при 5oС. Реакционной смеси дают медленно нагреться до 22oС в течение более 16 ч и затем экстрагируют три раза эфиром. Водный слой охлаждают до 5oС, устанавливают рН 2,0 с помощью 3 н. НСl и экстрагируют тремя порциями этилацетата. В охлажденном кислом водном слое устанавливают рН 11,0 с помощью 3 н. NaOH и разбавляют 175 мл диоксана. Одной порцией прибавляют ди-трет-бутилдикарбонат (38,2 г, 175 ммоль) при 5oС при перемешивании в атмосфере аргона. Реакционной смеси дают медленно нагреться до 22oС в течение более 16 ч, устанавливают рН 11,0 с помощью 3 н. NaOH и прибавляют другую порцию ди-трет-бутилдикарбоната (38,2 г, 175 ммоль) и перемешивают при 22oС в течение более 24 ч. Диоксан удаляют в вакууме при 40oС и устанавливают рН 11,0 реакционной смеси с помощью 3 н. NaOH, экстрагируют три раза эфиром, охлажденным до 5oС, и насыщают NaCl. Прибавляют этилацетат (100 мл) и устанавливают рН 4,0 с помощью 25%-ной лимонной кислоты (ОСТОРОЖНО: выделение CO2) при перемешивании при 5oС. Слои разделяют и кислый водный слой экстрагируют более пятью порциями этилацетата. Объединенные этилацетатные экстракты сушат (MgSO4), фильтруют через Celaform FW-14 и концентрируют в вакууме при 40oС с получением 3,70 г (56%) в виде прозрачного стеклообразного продукта; FAB-MS m/z 381 (МН+).
Стадия b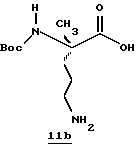
Соединение 11а (6,86 г, 18 ммоль) растворяют в 180 мл метанола и прибавляют 1,37 г 20% Pd(OH)2/C и помещают под давлением 60 пси Н2 в аппарат Парра в течение 18 ч. Реакционную смесь фильтруют и лепешку на фильтре экстрагируют 150 мл порциями метанола (ОСТОРОЖНО). Объединенные метанольные экстракты фильтруют через Celaform FW-14 и концентрируют в вакууме при 40oС с получением 3,5 г (79%) соединения 11b в виде белого твердого продукта; FAB-MS m/z 247 (МН+).
Стадия с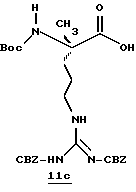
Соединение 11b (3,40 г, 13,8 ммоль) объединяют с триэтиламином (5,77 мл, 41,4 ммоль) в ДМФ (75 мл) и к полученной суспензии прибавляют N,N'-бис(бензилкарбонил)-3-метилизотиомочевину (7,42 г, 20,7 ммоль; там же). Полученную смесь перемешивают при 22oС в атмосфере азота в течение 40 ч, обрабатывают триэтиламином (2,0 мл, 14,4 ммоль) и N,N'-бис(бензилкарбонил)-3-метилизотиомочевиной (0,78 г, 2,2 ммоль), перемешивают в течение 24 ч, опять обрабатывают триэтиламином (2,0 мл, 14/4 ммоль) и N,N'-бис(бензилкарбонил)-S-метилизотиомочевиной (1,48 г, 4,1 ммоль) и перемешивают при 22oС в течение 3 дней. Сырую реакционную смесь фильтруют через Celaform FW-14 и удаляют ДМФ в вакууме при 40oС. Маслянистый осадок распределяют между 200 мл 0,1 н. NaOH и 200 мл эфира. Основный водный слой экстрагируют четыре раза эфиром, охлажденным до 5oС, расслаивают этилацетатом и устанавливают рН 3,5 с помощью лимонной кислоты. Слои разделяют и кислый водный слой экстрагируют четыре раза этилацетатом. Объединенные этилацетатные экстракты сушат (Na2SO4), фильтруют через Celaform FW-14 и концентрируют в вакууме при 40oС. Остаток очищают ВЭЖХ на силикагеле, элюируя CH2Cl2/MeOH (95:5) с получением 5,94 г (77%) 11с в виде белой пены; FAB-MS m/z 557 (МН)+.
Стадия d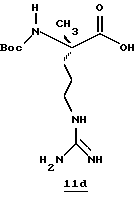
К раствору соединения 11с (5,86 г, 10,5 ммоль) в 210 мл метанола прибавляют 20% Pd(OH)2/C (1,47 г) и смесь помещают под давлением 60 пси водорода в аппарат Парра для гидрирования. Через 24 ч добавляют другую порцию 20% Pd(OH)2/C (0,74 г) и смесь помещают под давлением 60 пси водорода на 6 ч. Сырую реакционную смесь фильтруют через Celaform FW-14 и концентрируют в вакууме при 40oС с получением 3,03 г (100%) белой пены; FAB-MS m/z 289 (МН+).
Стадия е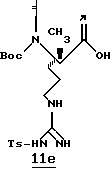
Соединение 11d (519 мг, 1,80 ммоль) растворяют в 2,7 мл 4 н. КОН и разбавляют 9,0 мл ацетона и охлаждают до -18oС. К этому раствору прибавляют раствор п-толуолсульфонилхлорида (686 мг, 3,6 ммоль) в 3,6 мл ацетона в течение более 15 мин при энергичном перемешивании. Через 1 ч при -18 - -15oС реакционной смеси дают медленно нагреться до 22oС в течение более 2,5 ч. Ацетон удаляют в вакууме при 20oС и водный остаток разбавляют 25 мл воды и экстрагируют три раза эфиром. Водный слой насыщают NaCl, расслаивают этилацетатом, охлаждают до 5oС и устанавливают рН 3,5 с помощью 1 н. НСl. Кислый водный слой экстрагируют более четырех раз этилацетатом и объединенные этилацетатные экстракты промывают дважды солевым раствором, сушат (Na2SO4) и концентрируют в вакууме при с получением 720 мг (90%) 11e в виде белой пены; FAB-MS m/z 443 (МН)+.
Стадия f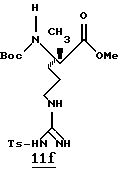
Перемешиваемый раствор 11e в 29 мл CH2Cl2/MeOH (9:1) охлаждают до 5oС и обрабатывают по каплям 2М (триметилсилил)-диазометаном в гексане (4,3 мл, 8,63 ммоль). Через 10 мин реакционную смесь концентрируют в вакууме при 22oС и остаток очищают ВЭЖХ на силикагеле, элюируя этилацетатом/гексаном (3:2) с получением 1,06 г (81%) 11f в виде белой пены; FAB-MS m/z 457 (МН)+.
Стадия g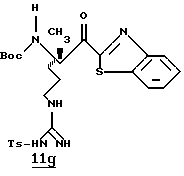
Раствор бензотиазола (2,63 г, 19,5 ммоль) в 20 мл ТГФ охлаждают до -75oС при перемешивании в атмосфере азота, н-Бутиллитий (1,6 М в гексане, 9,46 мл, 15,1 ммоль) прибавляют по каплям в течение более 15 мин при -75 - -65oС и затем перемешивают при -75 - -65oС в течение 15 мин. К этому раствору прибавляют по каплям в течение более 15 мин раствор 11f (461 мг, 1,0 ммоль) в 10 мл ТГФ при -75 - -70oС. Реакционную смесь перемешивают при -75oС в течение 2 ч, гасят 160 мл насыщенной водной NH4Cl, насыщают NaCl и экстрагируют три раза этилацетатом. Объединенные этилацетатные экстракты промывают три раза солевым раствором, сушат (Na2SO4) и концентрируют в вакууме. Остаток растирают три раза гексаном, растворяют в 150 мл этилацетата/гексана (3: 2), фильтруют через Celaform FW-14 и остаток очищают ВЭЖХ на силикагеле, элюируя этилацетатом/гексаном (3:2) с получением 0,42 г (74%) 11g в виде прозрачного стеклообразного продукта; FAB-MS m/z 560 (МН)+.
Стадия h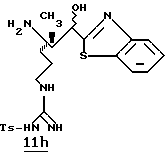
Соединение 11g (0,389 г, 693 ммоль) растворяют в 14 мл СF3СО3Н/СН2Сl2 (1:4), перемешивают при 22oС в течение 2 ч в атмосфере азота и концентрируют в вакууме при 10oС. Осадок распределяют между 20 мл водным 1 н. NaOH, предварительно насыщенным NaCl и 20 мл CH2Cl2. Основный водный слой экстрагируют более трех раз CH2Cl2 и объединенные CH2Cl2 экстракты промывают солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением 319 г (100%) 11h в виде коричневого аморфного твердого продукта; FAB-MS m/z 462 (МН)+.
Стадия i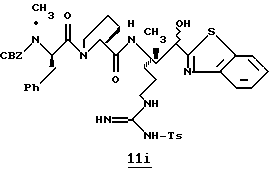
Раствор (CBZ)-N-метил-D-фенилаланил-L-пролина (0,309 г, 0,753 ммоль; патент США 4703036) и диизопропилэтиламин (132 мкл, 0,758 ммоль) в 10 мл безводного CH2Cl2 охлаждают до 5oС, перемешивая в атмосфере аргона. Прибавляют бис(2-оксо-3-оксазолидинил)фосфинилхлорид (ВОР-Сl; 0,193 г, 0,758 ммоль) и реакционную смесь перемешивают в течение 15 мин. В течение более 10 мин прибавляют раствор 10h (0,316 г, 0,685 ммоль) и диизопропилэтиламин (119 мкл, 0,684 ммоль) в 10 мл безводного CH2Cl2 при 5oС и реакционной смеси дают нагреться до 22oС в течение более 4,5 ч. Реакционную смесь концентрируют в вакууме при 40oС и остаток распределяют между этилацетатом и водным 1 М KHSO4. Органический слой экстрагируют более двух раз водным 1 М KHSO4, три раза насыщенным водным NaHCO3, дважды солевым раствором, сушат (Na2SO4) и концентрируют в вакууме при 40oС. Остаток очищают хроматографически на силикагеле, элюируя этилацетатом/гексаном (4:1) с получением 0,381 г (65%) 11i в виде белой пены; FAB-MS m/z 855 (МН)+.
Стадия j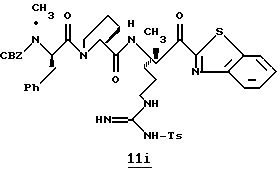
Соединение 11i (0,359 г, 0,420 ммоль) растворяют в 6 мл безводного СН2С12. Прибавляют периодинан Десс-Мартина (0,304 г, 0,717 ммоль) и реакционную смесь перемешивают при 22oС в атмосфере аргона. Избыток периодинана удаляют путем прибавления 20 мл гасящего раствора (25 г Na2S2O3 в 100 мл насыщенного водного NаНСО3), разбавляют этилацетатом (60 мл) и перемешивают при комнатной температуре в течение 15 мин. Оставшийся водный слой отделяют и экстрагируют несколькими порциями этилацетата и объединенные органические экстракты промывают последовательными порциями воды и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме с получением 0,336 г (94%) кетона 11j в виде белой пены; FAB-MS m/z 853 (МН)+.
Стадия k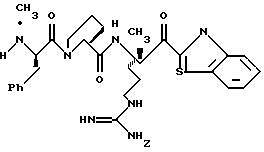
Соединение 11
Соединение 10j (0,336 г, 0,394 ммоль) растворяют в 3 мл безводного тиоанизола, помещают в тефлоновую реакционную трубку HF аппарата в безводных условиях и охлаждают до -78oС. Безводный HF (3-4 мл) отгоняют прямо в эту трубку и после завершения прибавления температуре смеси дают подняться до 0oС. Эту смесь перемешивают при 0oС в течение 4 ч, концентрируют в вакууме и растирают в нескольких порциях эфира с получением белого твердого продукта. Твердый продукт очищают ВЭЖХ с обращенной фазой, элюируя Н2О/ацетонитрил/ТФУ (60: 40:0,2). Фракции, содержащие соединение 10, объединяют, концентрируют в вакууме и лиофилизуют с получением 0,094 г (28%) соединения 11 в виде белого твердого продукта; Т. пл. 122-130oС; [α]
Анализ. Расчет для C29H37N7O3S•2,3ТФУ•1,9Н2О:
Вычислено: С 46,92; Н 5,05; N 11,40; F 15,24; H2O 3,98.
Найдено: С 46,59; Н 5,00; N 11,17; F 15,16; H2O 3,40.
ПРИМЕР 12
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)AМИНО] -2S-[(6-МЕТОКСИКАРБОНИЛБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]ПЕНТИЛ]-L-ПРОЛИНАМИД Соед. 12
Стадия а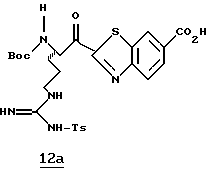
1,6 М н-Бутиллития (80,3 мл, 128,4 ммоль) прибавляют по каплям к перемешиваемому раствору бензотиазол-6-карбоновой кислоты (11,5 г, 64,17 ммоль) в ТГФ (100 мл) при -78oС в атмосфере азота с такой скоростью, чтобы температура реакции была ниже -70oС. После завершения прибавления реакционную смесь перемешивают в течение 30 мин при -78oС и прибавляют N-α-Вос-NG-тозил-L-аргинин N, 0-диметиламид (800 мг, 1,69 ммоль; DiMaio, et al. Journal of Medicinal Chemistry 1992, 35, 3331) (2,52 г, 5,35 ммоль) в ТГФ (150 мл), с такой скоростью, чтобы поддерживалась температура ниже 70oС. Полученную смесь перемешивают в течение 2 ч при -78oС, нагревают до -20oС и разбавляют насыщенным водным NH4Cl (700 мл). Полученный органический слой отделяют и водный слой промывают несколькими порциями EtOAc. Объединенные этилацетатные экстракты промывают водой, солевым раствором, сушат (Na2SO4) и концентрируют в вакууме. Сырую смесь очищают хроматографией на силикагеле, элюируя CH2Cl2/MeOH (8:1) с получением связанного промежуточного бензотиазола 12а в виде твердого продукта.
Стадия b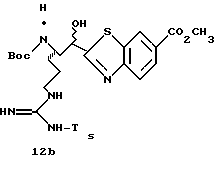
Боргидрид натрия (1,42 г, 37,6 ммоль) прибавляют одной порцией к раствору 12а (3,5 г, 5,9 ммоль) в МеОН (130 мл) при -25 - -20oС и реакционную смесь перемешивают в течение 40 мин. Прибавляют ацетон (20 мл), смеси дают нагреться до комнатной температуры и концентрируют в вакууме. Остаток растворяют в воде, подкисляют уксусной кислотой (рН 3-4) и экстрагируют несколькими порциями EtOAc. Объединенные экстракты промывают водой и солевым раствором, сушат (Na2SO4) и концентрируют в вакууме. Остаток растворяют в CH2Cl2/MeOH (6: 1) и охлаждают до 0oС. Прибавляют по каплям триметилсилилдиазометан (2М в гексане) в течение более 5 мин до появления желтого цвета и реакционную смесь перемешивают еще 25 мин. Полученную смесь концентрируют в вакууме и очищают колоночной хроматографией, элюируя EtOAc/гексаном (от 3:1 до 6:1) с получением спирта 12b в виде твердого продукта.
Стадия с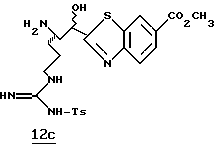
Раствор 12b (3,1 г, 4,56 ммоль), ТФУ (26 мл) в CH2Cl2 (104 мл) перемешивают при комнатной температуре в течение 1 ч и концентрируют в вакууме. Остаток разбавляют CH2Cl2 и концентрируют в высоком вакууме с получением сырого продукта 12с в виде масла.
Стадия d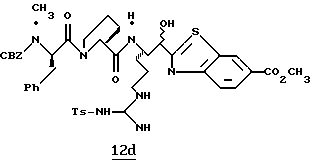
Раствор 12с (2,3 г, 4,56 ммоль) и НОВТ (1,32 г, 9,12 ммоль) в СН3СN (210 мл) нейтрализуют прибавлением триэтиламина (прибл. 5 мл) в атмосфере N2. К этому раствору прибавляют ДЦК (1,4 г, 6,84 ммоль) и N-метил-N-(CBZ)-D-фенилаланил-L-пролин одной порцией и эту смесь перемешивают в течение 2 ч при комнатной температуре и фильтруют. Фильтрат концентрируют в вакууме и растворяют в EtOAc. Органический раствор промывают насыщенным раствором NаНСО3, водой и солевым раствором, сушат (Na2SO4) и концентрируют в вакууме с получением связанного продукта 12d в виде твердого продукта.
Стадия е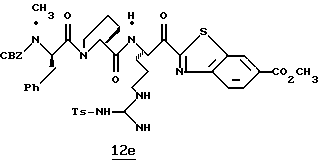
Периодинан Десс-Мартина (680 мг, 1,60 ммоль) прибавляют к раствору 12d (720 мг, 3,76 ммоль) в CH2Cl2 (40 мл) при комнатной температуре в атмосфере N2 и полученную смесь перемешивают в течение 25 мин. Избыток периодинана удаляют путем прибавления 30 мл гасящего раствора (25 г Nа2S2O3 в 100 мл насыщенного водного NаНСО3) к перемешиваемой реакционной смеси. Полученный водный слой выделяют и экстрагируют несколькими порциями CH2Cl2 и объединенные органические экстракты промывают последовательными порциями воды и солевого раствора, сушат (Na2SO4) и концентрируют в вакууме с получением кетона 12е в виде твердого продукта.
Стадия f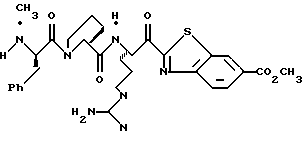
Соединение 12
Промежуточный кетон 12е (730 мг, 0,81 ммоль) и анизол (3 мл) помещают в тефлоновую реакционную трубку HF аппарата в безводных условиях и охлаждают до -78oС. HF (15-20 мл) отгоняют прямо в этот сосуд и температуре смеси дают подняться до 0oС. Смесь перемешивают в течение 3,5 ч, концентрируют в вакууме и растирают в нескольких порциях эфира с получением твердого продукта. Этот твердый продукт очищают ВЭЖХ с обращенной фазой, используя воду/ацетонитрил/ТФУ (70: 30: 0,2) с получением соединения 12 в виде твердого продукта; Т.пл. 125oС; [α]
Анализ. Расчет для С30H37N7O5S•2,5CF3CO2H•1,7Н2О:
Вычислено: С 45,53; Н 4,68; F 15,43; N 10,62; H2О 3,32.
Найдено: С 45,51; Н 4,53; F 15,19; N 10,54; Н2О 3,32.
ПРИМЕР 13
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(AМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(6-КАРБОКСИКАРБОНИЛБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД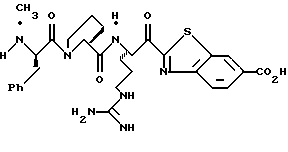
Соединение 13
Соединение 13 получают модифицируя пример 12. Раствор безводного LiOH (400 мг, 16,7 ммоль) в воде (12 мл) прибавляют перемешиваемому раствору соединения 12d (5,0 г, 5,57 ммоль) в диоксане (100 мл) при комнатной температуре. Смесь перемешивают в течение 4 ч и прибавляют дополнительную порцию LiOH (300 мг, 12,5 ммоль) в воде (7 мл) с последующим перемешиванием в течение 3 ч. Полученную смесь подкисляют EtОАс (рН 3-4) и экстрагируют несколько раз EtOAc. Объединенные органические экстракты промывают водой и солевым раствором, сушат (Na2SO4) и концентрируют с получением карбоксибензотиазольного производного 6. Это производное преобразуют в желаемое соединение с помощью последующей стадии е (гашение 25%-ной водной Nа2S2O3) и стадии f примера 12 с получением соединения 13 в виде твердого продукта; T. пл. 125oС; [α]
Анализ. Расчет для C29H35N7O5S•2,5CF3CO2H•H2O:
Вычислено: С 45,54; Н 4,44; F 15,89; N 10,93; Н2O 2,01.
Найдено: С 45,59; Н 4,59; F 15,60; N 10,76; Н2O 2,09.
ПРИМЕР 14
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(6-КАРБОКСАМИДОБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД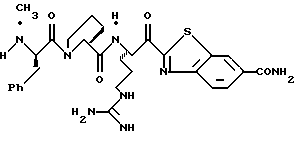
Соединение 14
Соединение 14 получают модифицируя пример 12. НОВТ (138 мг, 0,102 ммоль) и ДЦК (14 мг, 0,068 ммоль) прибавляют к перемешиваемому раствору соединения 12с (30 мг, 0,034 ммоль) в СН2С12/ДМФ (4:1, 2 мл) при комнатной температуре в атмосфере N2. Реакционную смесь перемешивают в течение 30 мин и прибавляют водную 29%-ную NH4OH (50 мкл, 0,38 ммоль). Смесь перемешивают в течение 5 ч и прибавляют другую порцию НОВТ (7 мг), ДЦК (7 мг) и водной 29%-ную NH4OH (20 мкл). После перемешивания еще 2 ч смесь разбавляют CH2Cl2. Полученный органический слой промывают последовательными порциями воды и солевого раствора, сушат (Na2SO4) и концентрируют с получением соответствующего 6-карбоксамидобензотиазольного производного. Это производное преобразуют в желаемое соединение с помощью следующих стадий е и f примера 12 с получением соединения 14 в виде твердого продукта; Т.пл. 130-143oС; [α]
Анализ. Расчет для C29H36N8O4S•2,9СF3СO2H•3,2Н2О:
Вычислено: С 42,61; Н 4,65; F 16,85; N 12,42; H2O 5,87.
Найдено: С 42,16; Н 4,35; F 16,19; N 11,45; Н2O 5,37.
ПРИМЕРЫ 15 И 16
N-МЕТИЛ-D-ФЕНИЛМАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(6-ГИДРОКСИМЕТИЛБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД Соед. 15
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1R-[(6-ГИДРОКСИМЕТИЛБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД Соед. 16
Стадия а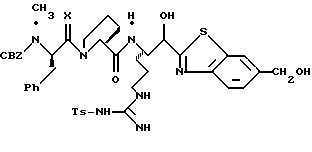
15a; X=O; 15e; X=H, H.
Соединение 15 получают модифицируя пример 12. 1,0 М DIBAL (10 мл, 10 ммоль) прибавляют по каплям к перемешиваемому раствору соединения 12с (1,5 г, 1,67 ммоль) в сухом CH2Cl2 (160 мл) при -78oС в атмосфере N2. Температуру реакционной смеси поддерживают ниже -74oС в процессе прибавления. Смесь перемешивают в течение 45 мин и гасят концентрированной водной NH4OH (50 мл). Прибавляют часть 1 н. НСl для разделения водного и органического слоев и полученный водный слой промывают CH2Cl2. Объединенные органические экстракты промывают последовательными порциями насыщенного NаНСО3, воды и солевого раствора, сушат (Na2SO4) и концентрируют с получением 15а и соответствующего производного метилена 15е в виде смеси, которую используют без очистки.
Стадия b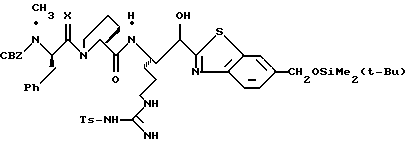
15b; X=O; 15f; X=H, H.
DMAP (61,2 мг, 0,50 ммоль) и триэтиламин (140 мкг, 1,00 ммоль) прибавляют к перемешиваемому раствору 15а и 15е (9,2 мг) при 0oС в атмосфере N2. Одной порцией прибавляют раствор третбутилтриметилсилилхлорида (45,3 мг, 0,30 ммоль) в CH2Cl2 (15 мл) и реакционной смеси дают подняться до комнатной температуры и перемешивают в течение 8 ч. Полученную смесь разбавляют CH2Cl2 и промывают последовательными порциями 10%-ной лимонной кислоты, насыщенного водного NaHCO3, Н2О и солевого раствора. Полученный органический слой сушат (Na2SO4) и концентрируют в вакууме с получением смеси 15b и соответствующего метиленового 15f, которую используют без дополнительной очистки.
Стадия с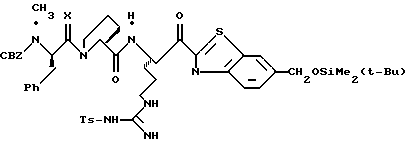
15c; X=O; 15g; X=H, H.
Периодинан Десс-Мартина (142 мг, 0,334 ммоль) прибавляют к раствору 15b и 15f (160 мг) в CH2Cl2 (20 мл) при комнатной температуре в атмосфере N2 и полученную смесь перемешивают в течение 30 мин. Избыток периодинана удаляют путем прибавления 10 мл гасящего раствора (25 г Na2S2О3 в 100 мл насыщенного водного NаНСО3) к перемешиваемой реакционной смеси. Полученный водный слой выделяют и экстрагируют несколькими порциями CH2Cl2 и объединенные органические экстракты промывают последовательными порциями насыщенного водного NаНСО3, а также солевого раствора, сушат (Na2SO4) и концентрируют в вакууме с получением смеси кетона 15с и соответствующего метиленового производного 15g в виде твердого продукта.
Стадия d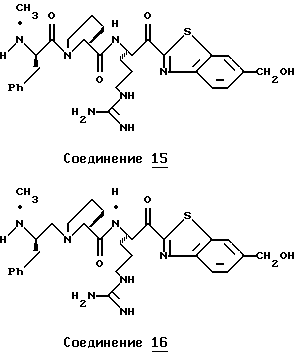
Промежуточные продукты 15с и 15g (170 мг, 0,17 ммоль) и анизол (2,5 мл) помещают в тефлоновую реакционную трубку HF аппарата в безводных условиях и охлаждают до -78oС. HF (5 мл) отгоняют в этот сосуд и после завершения прибавления температуре смеси дают подняться до 0oС. Смесь перемешивают в течение 3,5 ч, концентрируют в вакууме и растирают в нескольких порциях эфира с получением твердого продукта. Этот твердый продукт очищают ВЭЖХ с обращенной фазой, используя воду/ацетонитрил/ТФУ (70:30:0,2) с получением соединений 15 и 16 в виде чистых твердых продуктов:
Соединение 15: FAB-MS m/z 580 (МН)+.
Соединение 16: Т.пл. 75-85oС; [α]
Анализ. Расчет для С30H37N7O5S•2,5СF3СО2Н•1,7H2O:
Вычислено: С 43,74; Н 4,85; N 10,09.
Найдено: С 43,96; Н 4,76; N 9,95.
ПРИМЕРЫ 17 И 18
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(6-ФТОРБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД Соед. 17
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(6-ФТОРБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ] L-ПРОЛИНAМИД Соед. 18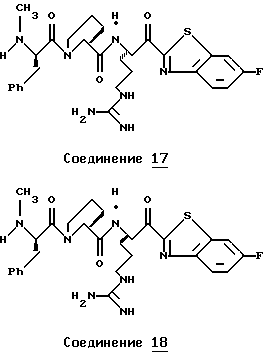
Соединения 17 и 18 получают модифицируя пример 12. Исходный бензотиазол со стадии а примера 12 заменяют на 6-фторбензотиазол и остальные стадии способа осуществляют при только незначительных изменениях. 6-Фторбензотиазол получают следующим образом. Раствор нитрита натрия (24,6 г, 35,7 ммоль) в воде (80 мл) прибавляют по каплям к перемешиваемому раствору 2-амино-6-трифторбензотиазола (10,0 г, 59,5 ммоль) в 85% Н3РO4 при -10oС таким образом, чтобы температура не превышала -4oС. Смесь выдерживают при -8oС в течение 1 ч, выливают в 50%-ную H3PO4 при 0oС и дают нагреться до комнатной температуры в течение более 2 ч. Прибавляют воду до достижения общего объема в 3 л и полученную смесь нейтрализуют N2CO3. Водный слой и твердый осадок экстрагируют несколькими порциями CHCl3/EtOAc (1:1) и объединенные органические экстракты промывают солевым раствором, сушат (Na2SO4) и концентрируют с получением 6-фторбензотиазола в виде твердого продукта.
Соединение 17: Т.пл. 65-80oС; [α]
Анализ. Расчет для С28H34FN7O3S•3,0CF3CO2H•1,1H2O:
Вычислено: С 43,94; F 20,44; Н 4,25; N 10,55; Н2О 2,13.
Найдено: С 43,96; F 20,57; Н 4,22; N 10,82; Н2О 2,22.
Соединение 18: Т.пл. 85-100oС; [α]
Анализ. Расчет для С28Н34FN7O3S•2,7CF3CO2H•0,9Н2O:
Вычислено: С 44,99; F 19,39; Н 4,35; N 10,99; Н2O 1,82.
Найдено: С 45,01; F 19,61; Н 4,24; N 11,15; Н2O 1,84.
ПРИМЕРЫ 19 И 20
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(4-ЭТОКСИКАРБОНИЛБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД Соед. 19
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(4-ЭТОКСИКАРБОНИЛБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД Соед. 20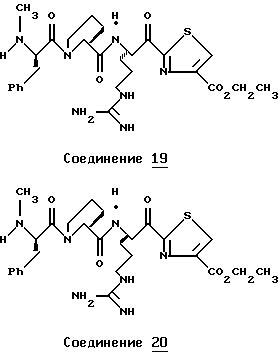
Соединения 19 и 20 получают модифицируя пример 9. N-(CBZ)-N-метил-D-фенилаланил-L-пролин используют вместо промежуточного продукта 9а и остальные стадии реакционной последовательности осуществляют на соответствующих аргининовых эпимерах при только незначительных изменениях с получением указанного в заголовке соединения в виде твердого продукта.
Соединение 19: Т. пл. 85-100oС; [α]
Анализ. Расчет для C27H37N7O5S•2,3СF3СО2Н•1,5Н2O:
Вычислено: С 44,08; Н 4,95; N 11,39; Н2О 3,14.
Найдено: С 44,25; Н 4,68; N 11,29; Н2О 3,29.
Соединение 20: Т. пл. 85-100oС; [α]
Анализ. Расчет для C27H37N7O5S•2,75СF3СО2Н•2,ОН2О•0,2С2Н3N:
Вычислено: С 42,51; Н 4,81; N 10,85; Н2О 3,88.
Найдено: С 42,17; Н 4,66; N 10,88; Н2О 4,21.
ПРИМЕР 21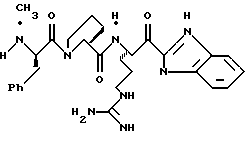
Соединение 21
Соединение 21 получают модифицируя способ по примеру 5. 1,2-Фенилендиамин заменяют 2-аминофенолом на стадии d этого примера с получением соответствующего производного бензимидазола. Это производное подвергают остальным стадиям при только незначительных изменениях с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 70-80oС; [α]
Анализ. Расчет для С28Н36N8O3•3,75СF3СО2Н•Н2О:
Вычислено: С 43,59; Н 4,30; N 11,45; Н2О 1,84.
Найдено: С 43,66; Н 4,37; N 11,45; Н2О 1,84.
ПРИМЕР 22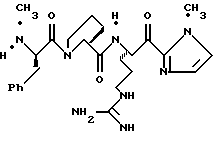
Соединение 22
Соединение 22 получают, следуя общему метода примера 12. N-Метилимидазол используют вместо бензотиазола на стадии а с получением имидазольного аналога промежуточного продукта 12а. Это промежуточное соединение подвергают остальным стадиям с получением соединения 22 в виде твердого продукта: Т.пл. 85-100oС; [α]
Анализ. Расчет для С28Н34FN7O3S•2,7CF3CO2H•0,9H2O:
Вычислено: С 44,99; F 19,39; Н 4,35; N 10,99; Н2О 1,82.
Найдено: С 45,01; F 19,61; Н 4,24; N 11,15; Н2О 1,84.
ПРИМЕР 23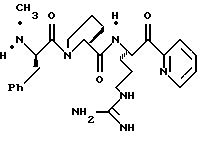
Соединение 23
Соединение 23 получают, используя способ по примеру 12. Бензотиазол-6-карбоновую кислоту заменяют 2-бромпиридином с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 40-68oС; FAB-MS m/z 494 (МН+);
Анализ. Расчет для С28H34FN7O3•2,7CF3CO2H•0,9Н2O:
Вычислено: С 44,99; F 19,39; Н 4,35; N 10,99; Н2О 1,82.
Найдено: С 45,01; F 19,61; Н 4,24; N 11,25; Н2О 1,84.
ПРИМЕР 24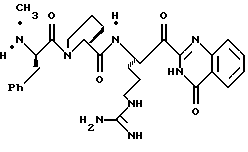
Соединение 24
Соединение 24 получают, используя способ по примеру 5, заменяя 2-аминофенол антраниловой кислотой с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 122-138oС; FAB-MS m/z 561 (МН+);
Анализ. Расчет для C29H36N8O4•2,6С2Н2О4•3,ОН2О:
Вычислено: С 45,08; Н 4,93; N 12,30; H2O 5,93.
Найдено: С 45,05; Н 4,79; N 12,57; H2O 5,99.
ПРИМЕР 25
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(6-ГИДРОКСИБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД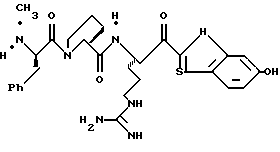
Соединение 25
Соединение 25 получают, используя способ по примеру 12, заменяя бензотиазол-6-карбоновую кислоту 5-(трет-бутилдифенилсилилокси)бензотиазолом с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 118-128oС; FAB-MS m/z 566 (МН+);
Анализ. Расчет для C28H35N7O4S•2C2H2O4•1,5H2O:
Вычислено: С 46,83; Н 4,79; N 11,95; Н2О 3,29.
Найдено: С 46,79; Н 4,79; N 12,15; Н2О 3,29.
ПРИМЕР 26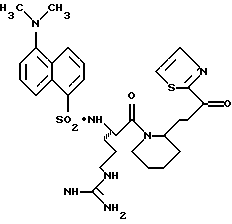
Соединение 26
Соединение 26 получают последующим разделением индивидуальных диастереомеров соединения 8 ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 90-110oС; FAB-MS m/z 614 (МН+).
ПРИМЕР 27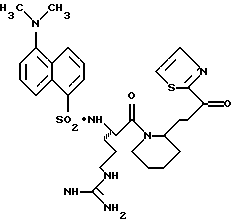
Соединение 27
Соединение 27 получают последующим разделением индивидуальных диастереомеров соединения 8 ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 60-75oС; FAB-MS m/z 614 (МН+).
ПРИМЕР 28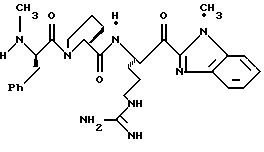
Соединение 28
Соединение 28 получают, используя способ по примеру 12, заменяя бензотиазол-6-карбоновую кислоту 1-метилбензимидазолом с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 118-128oС; FAB-MS m/z 566 (МН+);
Анализ. Расчет для C29H38N8O3•3,2С2Н2O4•1,1Н2O:
Вычислено: С 45,65; F 19,58; Н 4,70; N 12,03; Н2O 2,13.
Найдено: С 45,66; F 19,19; Н 4,51; N 12,02; H2O 1,77.
ПРИМЕР 29
N-МЕТИЛ-D-ЦИКЛОГЕКСИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(6-БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД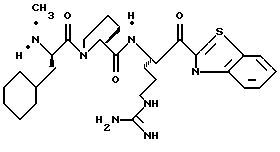
Соединение 29
Соединение 29 получают, следуя общему способу примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом на стадии а и СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-Boc-N-метил-D-циклогексилаланил-L-пролином на стадии d с получением указанного в заголовке соединения в виде твердого продукта: Т. пл. 90-100oС, [α]
Анализ. Расчет для С28H41N7O3S•3CF3CO2Н•0,75H2O:
Вычислено: С 44,81; F 18,76; Н 4,35; N 10,76; H2O 1,48.
Найдено: С 44,92; F 18,83; Н 5,03; N 10,80; H2O 1,37.
ПРИМЕР 30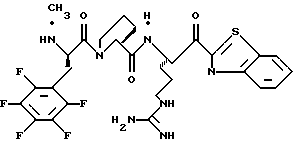
Соединение 30
Соединение 30 получают, следуя общему способу примера 12. N-СВZ-N-Метил-D-фенилаланил-L-пролин заменяют N-Boc-N-метил-D-(пентафторфенил)аланил-L-пролином с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 98-125oС; FAB-MS m/z 640 (МН+);
Анализ. Расчет для С28H31F5N7O3S•2,25СF3СО2Н•1,25Н2О:
Вычислено: С 42,48; F 24,29; Н 3,81; N 10,67; Н2О 2,45.
Найдено: С 42,75; F 24,12; Н 3,85; N 10,66; Н2О 2,61.
ПРИМЕР 31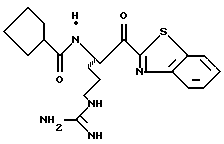
Соединение 31
Соединение 31 получают, следуя общему способу примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют циклопентаном с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 60-70oС; [α]
Анализ. Расчет для С19Н25N5O2S•1,25СF3СO2Н•0,75Н2О:
Вычислено: С 47,50; F 13,10; Н 5,14; N 12,88; H2O 2,48.
Найдено: С 47,50; F 13,19; Н 4,99; N 13,19; Н2О 2,37.
ПРИМЕР 32
N-МЕТИЛ-D-(4-ФТОРФЕНИЛ)АЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)-АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД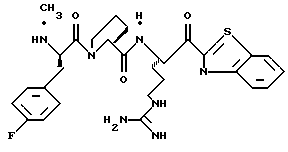
Соединение 32
Соединение 32 получают, следуя способу примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-Вос-N-метил-D-(4-фторфенил)аланил-L-пролином d с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 70-125oС; [α]
Анализ. Расчет для C28H34N7O3S•2,1СF3СO2Н•1,1H2O:
Вычислено: С 46,77; F 16,77; Н 4,67; N 11,86; Н2О 2,40.
Найдено: С 46,42; F 16,69; Н 4,67; N 11,82; Н2О 2,54.
ПРИМЕРЫ 33 И 34
N-МЕТИЛ-D-ФЕНИЛГЛИЦИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД
Соед. 33
Соед. 34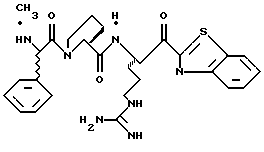
Соединение 33 и 34
Соединение 33 получают, следуя способу примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-Boc-N-метил-D, L-фенилглицил-L-пролином с получением диастереомеров 33 и 34, которые разделяют ВЭЖХ с обращенной фазой:
Соединение 33: Т. пл. 60(s)-125oC; [α]
Анализ. Расчет для C27H33N7O3S•2,1СF3СО2Н•1,9Н2O:
Вычислено: С 46,30; F 14,78; Н 4,84; N 12,11; Н2О 4,23.
Найдено: С 46,52; F 15,09; Н 4,84; N 12,29; Н2О 4,53.
Соединение 34: Т.пл. 65(s)-125oC; [α]
Анализ. Расчет для C27H33N7O3S•2,25СF3СO2Н•1,75Н2О:
Вычислено: С 45,93; F 15,57; Н 4,74; N 11,90; Н2О 3,83.
Найдено: С 46,21; F 15,67; Н 4,90; N 12,09; Н2О 4,22.
ПРИМЕР 35
N-МЕТИЛ-D-(ДИФЕНИЛ)АЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО]-1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД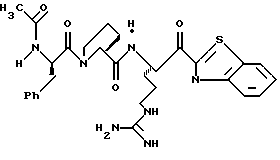
Соединение 35
Соединение 35 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-ацетил-D-фенилаланил-L-пролином с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 65-125oС; FAB-MS m/z 578 (MH+);
Анализ. Расчет для C29H35N7O4S•1,4СF3СO2Н•Н2O:
Вычислено: С 50,57; F 10,56; Н 5,12; N 12,98; H2O 2,38.
Найдено: С 50,77; F 10,39; Н 5,03; N 13,17; H2O 2,70.
ПРИМЕР 36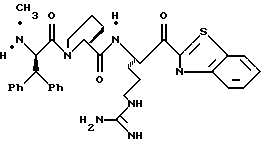
Соединение 36
Соединение 36 получают, используя метод примера 12. N-Вос-N-метил-D,L-фенилглицил-L-пролин заменяют N-CBZ-N-метил-D-фенилаланил-L-пролином и бензотиазол заменяют бензотиазол-6-карбоновой кислотой с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 65-125oС; FAB-MS m/z 578 (МН+).
Анализ. Расчет для C29H35N7O3S•2,25СF3СO2Н•1,9Н2O:
Вычислено: С 50,45; F 13,99; Н 4,95; N 10,69; Н2О 3,73.
Найдено: С 50,20; F 10,70; Н 4,78; N 13,62; Н2О 3,34.
ПРИМЕР 37
N-МЕТИЛ-D-ЦИКЛОГЕКСИЛГЛИЦИН-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО]-1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД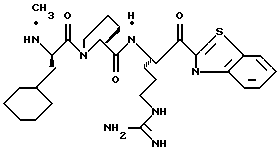
Соединение 37
Соединение 37 получают, используя метод примера 12. N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-Вос-N-метил-D-циклогексилглицил-L-пролином и бензотиазол-6-карбоновую кислоту заменяют бензотиазолом с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 65-125oС; FAB-MS m/z 542 (МН+);
Анализ. Расчет для C27H39N7O3S•2,0СF3СO2Н•2,0Н2О:
Вычислено: С 46,21; F 14,15; Н 5,63; N 12,17; Н2О 4,47.
Найдено: С 45,96; F 14,39; Н 5,71; N 12,07; Н2O 4,41.
ПРИМЕР 38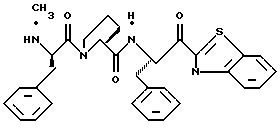
Соединение 38
Соединение 38 получают, используя метод примера 12. N,O-Диметиламид N-α-Вос-NG-тозил-L-аргинина заменяют N,O-диметиламидом N-Boc-L-фенилаланина и бензотиазол-6-карбоновую кислоту заменяют бензотиазолом на стадии а. Оставшиеся стадии последовательности реакции проводят с несущественными изменениями с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 74-95oС; [α]
Анализ. Расчет для C31H32N4O3•1,5СF3СO2Н•0,7Н2О:
Вычислено: С 56,38; F 11,8; Н 4,86; N 7,73; H2O 1,74.
Найдено: С 56,62; F 11,48; Н 4,48; N 7,61; Н2O 1,76.
ПРИМЕР 39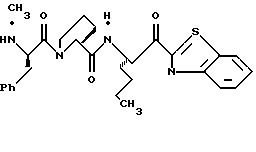
Соединение 39
Соединение 39 получают, используя метод примера 12. N,O-Диметиламид N-α-Вос-NG-тозил-L-аргинина заменяют N,O-диметиламидом N-Boc-L-норлейцином и бензотиазол-6-карбоновую кислоту заменяют бензотиазолом на стадии а. Оставшиеся стадии последовательности реакции проводят с несущественными изменениями с получением указанного в заголовке соединения в виде твердого продукта: [α]
Анализ. Расчет для С28Н34N4O3•1,5CF3CO2H•0,3Н2О:
Вычислено: С 54,51; F 12,52; Н 5,33; N 8,2; H2O 0,79.
Найдено: С 54,6; F 12,28; Н 5,38; N 8,18; H2O 0,60.
ПРИМЕР 40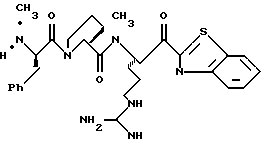
Соединение 40
Соединение 40 получают, используя метод примера 12. N,O-Диметиламид N-α-Вос-NG-тозил-L-аргинина заменяют N,O-диметил-амидом N-α-Boc-N-α-метил-NG-тозил-L-аргинина и бензотиазол-6-карбоновую кислоту заменяют бензотиазолом на стадии а и осуществляют стадии b и с. Стадия d требует использования бис(2-оксо-3-оксазолидинил)фосфиникхлорида (ВОР-С1) в качестве связывающего агента. Оставшиеся стадии последовательности реакции проводят с только несущественными изменениями с получением указанного в заголовке соединения в виде твердого продукта: [α]
Анализ. Расчет для C29H37N7O3S•2,3СF3СО2Н•1,1H2O:
Вычислено: С 47,72; F 15,5; Н 4,95; N 11,59; Н2О 2,34.
Найдено: С 47,58; F 15,69; Н 4,89; N 11,58; Н2O 2,36.
ПРИМЕР 41
D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД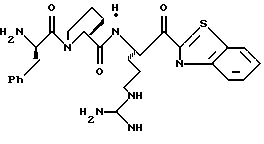
Соединение 41
Соединение 41 получают, используя метод примера 12. N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-CBZ-D-фенил-аланил-L-пролином и бензотиазол-6-карбоновую кислоту заменяют бензотиазолом с получением указанного в заголовке соединения в виде твердого продукта: FAB-MS m/z 540 (МН+);
Анализ. Расчет для C27H33N7O3S•2,4СF3СO2Н•0,9Н2О:
Вычислено: С 46,27; F 16,57; Н 4,54; N 11,8; Н2O 1,96.
Найдено: С 46,22; F 16,01; Н 4,59; N 12,02; Н2О 1,6.
ПРИМЕР 42
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-2S-ПИПЕРИДИНКАРБОКСАМИД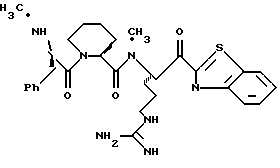
Соединение 42
Соединение 42 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-Boc-N-метил-фенилаланил-L-гомопролином на стадии d с получением указанного в заголовке соединения в виде твердого продукта: FAB-MS m/z 564 (МН+);
Анализ. Расчет для C29H37N7O3S•2,4СF3СO2Н•1,3Н2О:
Вычислено: С 47,16; F 15,89; Н 4,92; N 11,39; H2O 2,72.
Найдено: С 46,76; F 15,62; Н 4,95; N 11,39; Н2O 2,28.
ПРИМЕР 43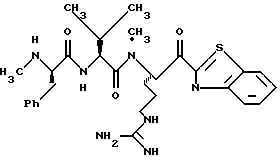
Соединение 43
Соединение 43 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-Вос-N-метил-D-вал-L-пролином на стадии d с получением указанного в заголовке соединения в виде твердого продукта: FAB-MS m/z 552 (МН+);
Анализ. Расчет для C28H37N7O3S•2,3СF3СО2Н•Н2О:
Вычислено: С 47,06; F 15,76; Н 5,00; N 11,78; H2O 2,16.
Найдено: С 46,89; F 16,22; Н 5,06; N 12,11; Н2O 2,76.
ПРИМЕР 44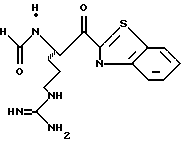
Соединение 44
Соединение 44 получают обработкой промежуточного продукта 3е при кипячении с обратным холодильником этилформиата, окисления полученного продукта периодинаном Десс-Мартина и удаления защиты у кетона с помощью HF с получением указанного в заголовке соединения в виде твердого продукта: FAB-MS m/z 560 (МН+);
Анализ. Расчет для C14H17N5O2S•1,32СF3СO2Н•0,71Н2О:
Вычислено: С 41,59; F 15,60; H 4,12; N 14,50; Н2O 2,66.
Найдено: С 41,04; F 15,49; Н 3,97; N 14,84; Н2O 2,26.
ПРИМЕР 45
1-(N-МЕТИЛАМИНО)-1-ЦИКЛОГЕКСИЛКАРБОНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД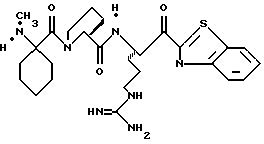
Соединение 45
Соединение 45 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом на стадии а и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют 1-(N-Boc-N-метиламино)-1-циклогексанкарбонил-L-пролином на стадии d. Остальные стадии по схеме проводят с незначительными модификациями с получением указанного в заголовке соединения в виде твердого продукта: FAB-MS m/z 528 (МН+);
Анализ. Расчет для С26H37N7O3S•2,3СF3СO2Н•1,8H2O:
Вычислено: С 44,69; F 15,94; Н 5,26; N 11,92; Н2О 3,94.
Найдено: С 44,37; F 15,8; Н 4,89; N 11,75; H2O 3,11.
ПРИМЕР 46
2,2-ДИФЕНИЛГЛИЦИН-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД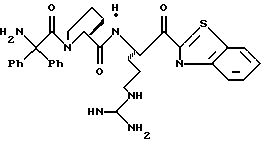
Соединение 46
Соединение 46 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом на стадии а и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-CBZ-дифенилглицилпролином на стадии d с получением указанного в заголовке соединения в виде твердого продукта: FAB-MS m/z 598 (МН+);
Анализ. Расчет для С32Н35N7O3S•2,6СF3СО2Н•1,2Н2О:
Вычислено: С 48,79; F 16,18; Н 4,40; N 10,71; Н2O 2,36.
Найдено: С 48,70; F 15,78; Н 4,22; N 10,63; H2O 1,37.
ПРИМЕР 47
α-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(БЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД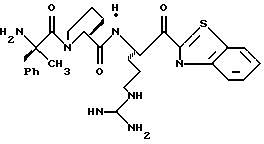
Соединение 47
Соединение 47 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом на стадии а и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-Вос-α-метил-D-фенилаланил-L-пролином на стадии d с получением указанного в заголовке соединения в виде твердого продукта: FAB-MS m/z 550 (МН+);
Анализ. Расчет для С28Н35N7O3S•2,6СF3СО2Н•1,5H2O:
Вычислено: С 45,67; F 16,97; Н 4,69; N 11,23.
Найдено: С 45,28; F 16,91; Н 4,46; N 11,00.
ПРИМЕР 48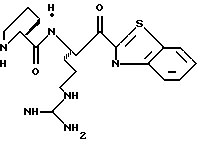
Соединение 48
Соединение 48 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензотиазолом на стадии а и N-СВZ-N-метил-D-фенилаланил-L-пролин заменяют N-Boc-L-пролином на стадии d с получением указанного в заголовке соединения в виде твердого продукта: FAB-MS m/z 389 (МН+);
Анализ. Расчет для C18H24N6O2S•1,9СF3СO2Н•0,9H2O:
Вычислено: С 42,14; F 17,43; Н 4,49; N 13,53; Н2О 2,58.
Найдено: С 42,30; F 17,69; Н 4,20; N 13,18; Н2О 2,61.
ПРИМЕР 49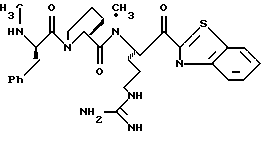
Соединение 49
Соединение 49 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют бензо[b] тиофеном на стадии а и остальные стадии последовательности проводят с только незначительными изменениями с получением указанного в заголовке соединения в виде твердого продукта: [α]
Анализ. Расчет для C29H36N6O3S•2,4СF3СО2Н•1,5Н2О:
Вычислено: С 47,80; F 16,10; Н 4,91; N 9,89; Н2О 3,18.
Найдено: С 47,68; F 15,81; Н 4,82; N 9,74; Н2О 3,18.
ПРИМЕР 50
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(6-МЕТОКСИБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД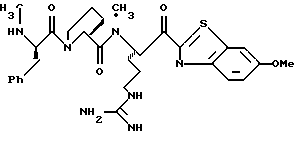
Соединение 50
Соединение 50 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют 6- метоксибензотиазолом на стадии а. Остальные стадии последовательности проводят с только незначительными изменениями с получением указанного в заголовке соединения в виде твердого продукта: [α]
Анализ. Расчет для С29Н37N7O4S•2,3СF3СО2Н•1,75H2O:
Вычислено: С 47,80; F 16,10; Н 4,91; N 9,89; H2O 3,18.
Найдено: С 47,68; F 15,81; Н 4,82; N 9,74; H2O 3,18.
ПРИМЕР 51
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(4,5,6,7-ТЕТРАГИДРОБЕНЗОТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД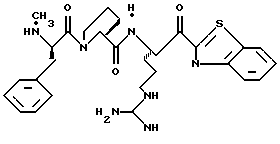
Соединение 51
Соединение 51 получают, используя метод примера 12. Бензотиазол-6-карбоновую кислоту заменяют 4,5,6,7-тетра-гидробензотиазолом на стадии а и осуществляют остальные стадии последовательности с получением указанного в заголовке соединения в виде твердого продукта: Т.пл. 50-60oС; FAB-MS m/z 554 (МН+);
Анализ. Расчет для C28H39N7O3S•2,6СF3СО2Н•1,4Н2О:
Вычислено: С 45,55; F 16,93; Н 5,11; N 11,20; Н2O 2,88.
Найдено: С 45,71; F 17,16; Н 5,31; N 11,34; Н2О 2,96.
ПРИМЕР 52
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(НАФТО[2,1-D] ТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД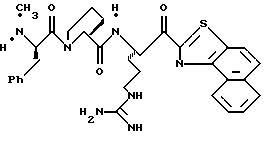
Соединение 52
Соединение 52 получают, используя метод примера 12, заменяя 6-карбоксибензотиазол нафто[2,1-d] тиазолом с получением в виде твердого продукта: [α]
Анализ. Расчет для С32Н37N7O3S•3,46СF3СO2Н•1,3Н2О:
Вычислено: С 45,93; F 19,38; Н 4,26; N 9,36; Н2О 2,30.
Найдено: С 45,60; F 19,06; Н 4,03; N 9,70; H2O 1,96.
ПРИМЕР 53
Стадия а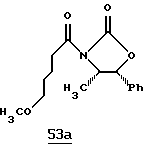
Раствор 5-метоксипентановой кислоты (6,80 г, 0,052 моль; Kirmse, К., Jansen, U. Chem. Ber. 1985, 128, 2607-2625) и триэтиламин (6,80 г, 0,067 моль) в 100 мл безводного ТГФ охлаждают до -78oС при перемешивании в атмосфере аргона. К реакционной смеси прибавляют по каплям пивалоилхлорид (6,80 г, 0,057 моль) в течение более 15 мин при -78oС. Через 15 мин реакционную смесь нагревают до 0oС, перемешивают в течение 1,5 ч и затем вновь охлаждают до -78oC.
Тем временем к раствору (4S,5R)-(-)-4-метил-5-фенил-2-оксазолидинона (16,4 г, 0,093 моль) в 100 мл безводного ТГФ в течение более 30 мин при перемешивании в атмосфере аргона при -78oС прибавляют по каплям н-BuLi (58 мл 1,6 М в гексане, 0,093 моль). Через 15 мин при перемешивании в атмосфере аргона при -78oС эту реакционную смесь медленно прибавляют в вышеуказанной безводной реакционной смеси. Реакционной смеси дают медленно нагреться до комнатной температуры в течение более 18 ч, гасят 1 н. водной KHSO4 и растворители удаляют в вакууме при 40oС. Остаток распределяют между водой и CH2Cl2 и водный слой трижды экстрагируют CH2Cl2. Объединенные CH2Cl2 экстракты дважды промывают солевым раствором, сушат над безводным Na2SO4 и концентрируют в вакууме при 40oС. Остаток очищают хроматографически на силикагеле элюируя CH2Cl2/EtOAc (95:5) с получением 8,2 г (55%) 53а в виде белого твердого продукта: FAB-MS m/z 292 (МН+).
Стадия b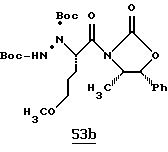
Соединение 53а (3,81 г, 0,013 моль) подвергают взаимодействию с ди-трет-бутилазодикарбоксилатом (0,48 г, 0,015 моль) в соответствии с общим методом D. A. Evans et al. (Tetrahedron 1988, 44, 5525-5540) с получением сырого 53b. Сырой продукт очищают хроматографически на силикагеле, элюируя СН2Сl2/гексаном/ацетонитрилом (70: 30: 7) с получением чистого 53b (2,33 г, 33%) в виде белого твердого продукта; FAB-MS m/z 522 (МН+).
Стадия с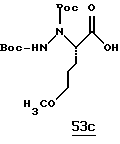
Соединение 53b (2,33 г, 0,0045 моль) растворяют в 18 мл ТГФ, охлажденном до 0oС, и обрабатывают раствором LiOH-H2O (0,46 г, 0,0107 моль) в 9 мл Н2О. Добавляют водную Н2О2 (1,1 мл 30%, 0,010 моль) и реакционную смесь перемешивают при 0oС в течение 3 ч. Реакционную смесь распределяют между водной 1 н. НСl и CH2Cl2 и кислотный водный слой экстрагируют CH2Cl2. Объединенные CH2Cl2 экстракты промывают солевым раствором, сушат над безводным МgSO4 и концентрируют в вакууме при 40oС. Остаток очищают путем хроматографии на силикагеле, элюируя EtOAc/гексан/НОАс с получением 53с в виде белого твердого продукта; FAB-MS m/z 363 (МН+).
Стадия d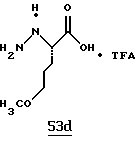
Соединение 53с (2,15 г, 0,0593 моль) растворяют в 215 мл ТФУ/СН2Сl2 (1:4 по объему) и перемешивают при комнатной температуре в атмосфере аргона в течение 1,5 ч. Растворители удаляют в вакууме при 20oС с получением 53d (2,07 г): FAB-MS m/z 163 (МН+).
Стадия е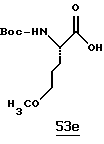
Соединение 53d растворяют в абсолютном этаноле (100 мл), объединяют с PtO2 (0,300 г) и помещают в аппарат Парра под давлением (55 пси) при комнатной температуре в течение более 24 ч. Реакционную смесь растворяют в метаноле (50 мл) и устанавливают рН 8 с Et3N (прибл. 2 мл). Раствор охлаждают до 0oC, обрабатывают ди-трет-бутилдикарбонатом (2,0 г, 0,009 моль) и дают медленно нагреться до комнатной температуры в течение более 20 ч. Реакционную смесь концентрируют в вакууме при 40oС и распределяют между EtOAc (50 мл) и охлажденной (5oС) 1 н. НСl (50 мл), промывают водой солевым раствором, сушат над безводным Na2SO4 и концентрируют в вакууме при 40oС с получением 53е (2,15 г) в виде белого твердого продукта; FAB-MS m/z 248 (МН+).
Стадия f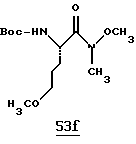
Соединение 53е (1,48 г, 0,0060 моль) растворяют в 70 мл безводного CH2Cl2, объединяют с гидрохлоридом N, О-диметил-гидроксиламина (0,878 г, 0,0090 моль). В полученной реакционной смеси устанавливают рН 8 с помощью Et3N и обрабатывают хлорангидридом бис(2-оксо-3-оксазолидинил)-фосфиновой кислоты (ВОР-Сl; 2,29 г, 0,0090 моль) при перемешивании при комнатной температуре в атмосфере аргона. Через 2 ч реакционную смесь распределяют между CH2Cl2 и холодной водной 1 н. HCl. Органический слой экстрагируют насыщенной NаНСО3, солевым раствором, сушат над безводным МgSO4 и концентрируют в вакууме. Остаток частично растворяют в метаноле и не растворившийся осадок удаляют путем фильтрования. Фильтрат концентрируют в вакууме с получением 53f (1,00 г, 57%) в виде белого твердого продукта; FAB-MS m/z 291 (МН+).
Стадия g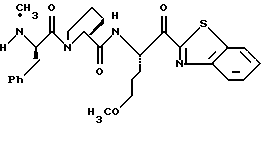
Соединение 53
Амид 53f преобразуют в соединение 53 следующими общими способами примера 3. Соединение 53f подвергают взаимодействию с 2-литиобензотиазолом, как описано для стадии 3с. Полученный промежуточный кетон подвергают обработке, применяемой на стадиях 3d. и 3е, и полученный аминоспирт последовательно связывают с (CBZ)-N-метил-D-фенилаланил-L-пролином по способу стадии 3f. Этот промежуточный продукт аналогично подвергают остальным стадиям примера 3, элюируя с помощью H2O/ацетонитрил/ТФУ (50:50:0,2) с получением соединения 53 в виде белого твердого продукта; [α]
Анализ. Расчет для C28H34N4O4S•2,15ТФУ•1,1H2O:
Вычислено: С 51,12; Н 5,33; N 7,85; F 11,98; H2O 2,77.
Найдено: С 51,16; Н 5,17; N 7,87; F 11,58; H2O 2,45.
ПРИМЕР 54
N-МЕТИЛ-D-ФЕНИЛАЛАНИЛ-N-[4-[(АМИНОИМИНОМЕТИЛ)АМИНО] -1S-[(4-КАРБОКСИТИАЗОЛ-2-ИЛ)КАРБОНИЛ]БУТИЛ]-L-ПРОЛИНАМИД
Стадия а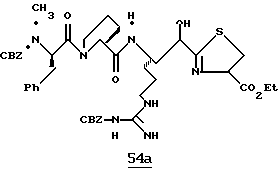
Смесь промежуточного продукта 1b (5,09 г, 6,52 ммоль) и этиловый эфир L-цистеина•НСl (2/42 г, 13,05 ммоль) в безводном CH2Cl2 (127 мл) перемешивают при комнатной температуре в атмосфере аргона в течение более 16 ч. Полученную смесь фильтруют через вспомогательный фильтровальный материал, концентрируют в вакууме и распределяют между солевым раствором и этилацетатом. Водный слой экстрагируют несколькими порциями этилацетата и объединенные органические экстракты сушат (Na2SO4) и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле, элюируя СН2Сl2:МеОН:NН4OН (90:9:1) с получением 54а в виде белой пены.
Стадия b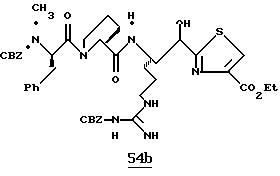
Соединение 54а (1,0 г, 1,18 ммоль) и МnO2 (1,00 г, 14,38 ммоль) в безводном CH2Cl2 (28 мл) перемешивают при комнатной температуре в атмосфере аргона в течение 4,5 ч. Добавляют дополнительную порцию МnO2 (0,25 г) и смесь перемешивают еще 2,5 часа. Полученную смесь фильтруют через вспомогательный фильтровальный материал и концентрируют в вакууме с получением тиазола 54b в виде неочищенного твердого продукта.
Стадия с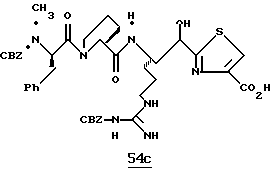
LiOH (65 мг, 2,64 ммоль) и H2O (0,1 мл) в диоксане (0,9 мл) перемешивают в атмосфере аргона в течение 3 ч. Полученную смесь разбавляют H2O и экстрагируют несколькими порциями эфира. Водный слой отделяют и подкисляют до рН 4 1 н. НСl и экстрагируют несколькими порциями CH2Cl2. Объединенные CH2Cl2 экстракты промывают солевым раствором, сушат Nа2SO4 и концентрируют в вакууме с получением кислоты 54b в виде твердого продукта.
Стадия d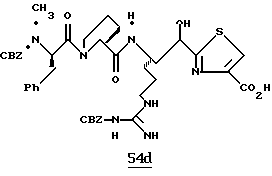
Смесь 54с (252 мг, 0,31 ммоль) и периодинан (197 мг, 0,464 ммоль) в CH2Cl2 (10 мл) перемешивают при комнатной температуре в N2 в течение 1 ч. Добавляют дополнительные порции периодинана (132 мг, 0,31 ммоль) в течение более 3 ч и полученную смесь гасят водной Na2S2O3. Водный слой подкисляют до рН 3,0 уксусной кислотой, затем промывают солевым раствором, фильтруют через диатомовую землю и концентрируют в вакууме. Остаток очищают ВЭЖХ с обращенной фазой, используя СН3СN:Н2О:ТФУ (50:50:0,2) с получением кетона 54d в виде твердого продукта.
Стадия е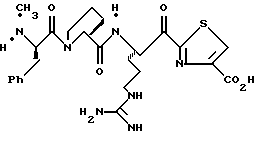
Соединение 54
Промежуточный кетон 54d (140 мг, 0,17 ммоль) и анизол (4 мл) помещают в тефлоновую реакционную трубку HF аппарата в безводных условиях и охлаждают до -78oС. HF (8 мл) разбавляют в этой трубке и после завершения прибавления температуре смеси дают подняться до -10oС. Эту смесь перемешивают в течение 1 ч при от -20 до -10oС, концентрируют в вакууме и растирают в нескольких порциях эфира с получением твердого продукта. Этот твердый продукт очищают ВЭЖХ с обращенной фазой, элюируя водой/ацетонитрилом/ТФУ (70:30:0,2), и лиофилизуют с получением соединений 54 в виде чистых твердых продуктов: [α]
Анализ. Расчет для C25H33N7O5S•2,5СF3СO2Н•1,7H2O:
Вычислено: С 41,93; Н 4,56; N 11,41; H2O 3,56.
Найдено: С 41,59; Н 4,53; N 11,84; H2O 3,40.
ПРИМЕРЫ 55 и 56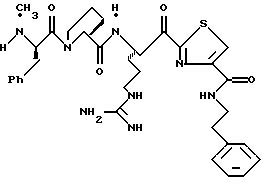
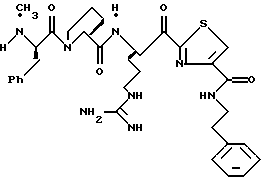
Соединения 55 и 56 получают, модифицируя пример 54. ВОР-Сl (13 мг, 0,03 ммоль) прибавляют к перемешиваемой смеси промежуточного продукта 54с (21,8 мг, 0,027 ммоль), триэтиламина (5,6 мкг, 0,077 ммоль), ДМФ (1,5 мл) и фенетиламина (5 мкг, 0,041 ммоль) при 5oС в атмосфере аргона. Полученную смесь перемешивают в течение 16 ч, концентрируют в вакууме и распределяют между этилацетатом и насыщенным водным NаСО3. Водный слой экстрагируют несколькими порциями этилацетата и объединенные органические экстракты сушат (Na2SO4) и концентрируют в вакууме с получением защищенного амида. Этот амид подвергают удалению защиты подобно примеру 54 с получением соответствующих указанных в заголовке соединений в виде твердых продуктов:
Соединение 55: [α]
Анализ. Расчет для C33H42N8O4S•2,6СF3СО2Н•1,2Н2О:
Вычислено: С 41,93; Н 4,56; N 11,41; H2O 3,56.
Найдено: С 41,59; Н 4,53; N 11,84; Н2О 3,40.
Соединение 56: FAB-MS m/z 647 (МН+).
ПРИМЕРЫ 57
Стадия а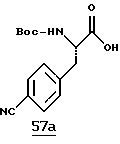
4-Цианофенилаланин (36,1 г, 0,19 ммоль; ЕР 0513675 Аl) объединяют с Et3N (26 мл, 0,19 ммоль) в 550 мл Н2О. Полученный раствор обрабатывают ди-трет-бутилдикарбонатом (68,0 г, 0,31 ммоль), перемешивают при комнатной температуре в течение более 18 ч и охлаждают до 5oС. Устанавливают рН 1 с помощью концентрированной НСl и кислый водный слой экстрагируют дважды EtOAc (500 мл). EtOAc слои промывают дважды солевым раствором (200 мл), сушат над безводным Na2SO4 и концентрируют в вакууме. Остаток растирают в гексане с получением 57а в виде светло-розового цвета (50,0 г, 87%): Т.пл. 145-147oС (разложение); [α]
Стадия b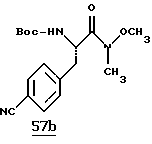
Раствор 57а (25 г, 0,086 моль), гидрохлорид N,O-диметилгидроксиамин (12,7 г, 0,130 моль) в безводном ДМФ (500 мл) охлаждают до 0oС при перемешивании в атмосфере аргона и обрабатывают Еt3N (23,3 мл, 0,230 моль). В течение более 10 мин порциями прибавляют гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония (ВОР; 42,1 г, 0,095 ммоль) и реакционную смесь перемешивают при 0-5oС в течение более 18 ч, поддерживая рН 8-9 путем прибавления Et3N. Реакционную смесь концентрируют в вакууме и остаток распределяют между CH2Cl2 (500 мл) и Н2О (250 мл). Органический слой промывают 50 мл 5%-ной водной NаСО3, 50 мл 1 н. водной НСl, 100 мл H2О, сушат над безводным Na2SO4 и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле, элюируя CH2Cl2/MeOH (95:5) с получением 54b в виде желтого твердого продукта (22,1 г, 70%): FAB-MS m/z 334 (МН)+.
Стадия с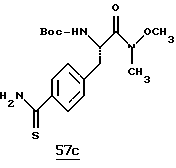
Раствор 57b (28,7 г, 0,0861 моль) и Et3N (100 мл, 0,72 моль) растворяют в пиридине в (400 мл). В течение более 3 ч в этот раствор при комнатной температуре барботируют безводный Н2S. Реакционному сосуду дают затем стоять при комнатной температуре в течение 60 ч и концентрируют в вакууме при 30oС. Остаток разбавляют Н2O (500 мл), охлаждают до 5oС, подкисляют до рН 4-5 с помощью концентрированной водной НСl и экстрагируют EtOAc (500 мл). Органический слой экстрагируют дважды 250 мл водной 1 н. НСl, дважды 200 мл раствором солевого раствора, сушат над безводным МgSO4 и концентрируют в вакууме с получением 57с (27,8 г, 88%): FAB-MS m/z 368 (МН)+.
Стадия d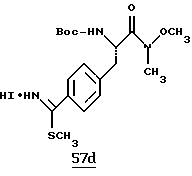
Раствор 57с (27,0 г, 0,074 моль) и метилиодид (75 мл) в ацетоне в (750 мл) нагревают с обратным холодильником при перемешивании в атмосфере аргона в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь концентрируют в вакууме с получением сырого 57d в виде янтарного стеклообразного продукта, который используют прямо на следующей стадии; FAB-MS m/z 381 (МН)+.
Стадия е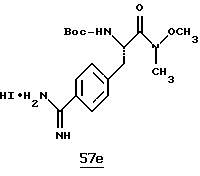
Соединение 57d (38,0 г, 0,075 моль) прибавляют к раствору ацетата аммония (8,70 г, 0,112 ммоль) в метаноле (200 мл) и нагревают с обратным холодильником при перемешивании в атмосфере аргона в течение 3,5 ч. После охлаждения до комнатной реакционную смесь концентрируют в вакууме и остаток распределяют между СНСl3 (500 мл) и насыщенным водным раствором NаНСО3. СНСl3 фильтруют, промывают солевым раствором, сушат над безводным Na2SO4 и концентрируют в вакууме с получением 57е в виде бледно-желтого стеклообразного продукта: FAB-MS m/z 351 (МН)+.
Стадия f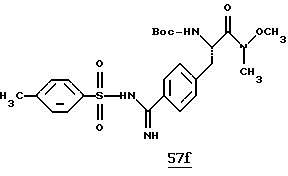
Раствор 57е (24,3 г, 0,069 моль) и тозилхлорид (14,6 г, 0,076 моль) в ацетоне (500 мл) охлаждают до -15oС. В течение более 45 мин по каплям при перемешивании при -15oС прибавляют 1,8-диазабицикло[5,4,0]ундец-7-ен (ДБУ; 21,4 г, 0,139 моль) и этой смеси дают медленно нагреться до комнатной температуры в течение более 3 ч. Реакционную смесь разбавляют 100 мл метанола и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле, элюируя EtOAc/гексаном (3: 1) с получением 57f (14,4 г, 41%) в виде белого твердого продукта; FAB-MS m/z 505 (МН)+.
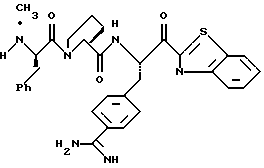
Соединение 57
Амид 57f преобразуют в соединение 57 следующими общими способами примера 3. Соединение 57f подвергают взаимодействию с 2-литиобензотиазолом, как описано для стадии 3с. Полученный промежуточный кетон подвергают обработке, применяемой на стадиях 3d и 3е, и полученный аминоспирт последовательно связывают с (CBZ)-N-метил-D-фенилаланил-L-пролином по способу стадии 3f. Этот промежуточный продукт аналогично подвергают остальным стадиям примера 3; учитывая, что стадия отщепления HF (стадия h) требует 6 ч при комнатной температуре вместо 3 ч при 0oС. Полученное соединение 57 очищают ВЭЖХ с обращенной фазой, элюируя с помощью Н2О/ацетонитрил/ТФУ (65:35:0,2) с получением соединения 57 в виде белого твердого продукта; FAB-MS m/z 583 (МН+);
Анализ. Расчет для С32Н34N6О3S•2,25ТФУ•1,0Н2О:
Вычислено: С 51,14; Н 4,50; N 9,80; F 14,96; S 3,74; H2O 2,10.
Найдено: С 50,96; Н 4,39; N 9,60; F 14,66; S 3,77; H2O 1,72.
ПРИМЕРЫ 58
Стадия а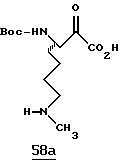
N-ε-Метил-L-лизин подвергают взаимодействию с 4-метокси-2,3,6-триметилбензолсульфонилхлориду (Mtr) в соответствии со способом, применяемым для получения N-ε-Mtr-L-лизина, описанного М. Fujino et al., Chem. Pharm. Bull. Jpn. 1982, 30, 2766. Полученный N-ε-метил-N--ε--Mtr-L-лизин (5,00 г, 0,0134 моль) растворяют в диоксане (52 мл), разбавляют Н2О (952 мл), охлаждают до 0oС и устанавливают рН 11 с помощью водного 3 н. NaOH. Прибавляют ди-трет-бутилдикарбонат (8,79 г, 0,040 моль) и реакционную смесь перемешивают при 0oС, поддерживая рН 10-11 в течение более 18 ч. Растворители удаляют в вакууме и остаток распределяют между Н2O (100 мл) и эфиром. Основный водный слой (рН 11) опять экстрагируют эфиром (2х), охлаждают до 0oС, устанавливают рН 3 с помощью водной 3 н. НСl и экстрагируют EtOAc (3х). Объединенные EtOAc экстракты промывают солевым раствором, сушат над безводным Na2SO4 и концентрируют в вакууме с получением 58а (3,34 г, 53%) в виде белой пены; FAB-MS m/z 473 (МН)+.
Стадия b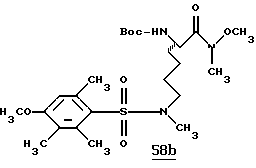
Соединение 58b подвергают взаимодействию с гидрохлоридом N,O-диметилгидроксиламина в соответствии с общим способом, используемым для получения 57b, что приводит к получению 58b в виде белого твердого продукта; FAB-MS m/z 516 (МН+).
Стадия с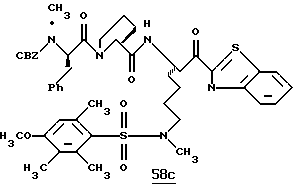
Амид 58b преобразуют в 58d следующими общими способами примера 3. Соединение 58b подвергают взаимодействию с 2-литио-бензотиазолом, как описано для стадии 3с. Полученный промежуточный кетон подвергают обработке, применяемой на стадиях 3d и 3е, и полученный аминоспирт последовательно связывают с (CBZ)-N-метил-D-фенилаланил-L-пролином по способу стадии 3f. Этот промежуточный продукт окисляют периодинаном Десс-Мартином, как описано для 3g с получением соединения 58d; FAB-MS m/z 883 (МН+).
Стадия d
Соединение 58
Соединение 58d (0,025 г, 0,028 ммоль), пентаметилбензол (0,32 г) и диметил сульфид (0,63 мл) растворяют в 3 мл безводной трифторуксусной кислоте и охлаждают до 5oС при перемешивании в атмосфере аргона. Через раствор в течение более 15 мин барботируют газообразный безводный НВr и реакционной смеси дают медленно нагреться до комнатной температуры более 15 час. Растворители удаляют в вакууме и полученный остаток растирают в безводном эфире и очищают ВЭЖХ с обращенной фазой, элюируя Н2О/ацетонитрил/ТФУ (60:40:0,2) с получением соединения 58 в виде белого твердого продукта; FAB-MS m/z 536 (МН+).
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2001 |
|
RU2275373C2 |
| КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ИЛИ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ | 1997 |
|
RU2194038C2 |
| N - ОКСИДЫ 4-ФЕНИЛПИПЕРАЗИНОВ И 4-ФЕНИЛПИПЕРИДИНОВ, КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ ДОПАМИН-2 РЕЦЕПТОРОВ | 1994 |
|
RU2123001C1 |
| БЕНЗОКСАЗИНОВЫЕ ИЛИ ПИРИДООКСАЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ОБЛАДАЮЩИЕ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ, СПОСОБЫ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 1996 |
|
RU2191179C2 |
| АНТИБАКТЕРИАЛЬНЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ЗАМЕЩЕННЫЕ ФЕНИЛОКСАЗОЛИДИНОНЫ | 2000 |
|
RU2278117C2 |
| ПРОИЗВОДНЫЕ НИПЕКОТИНОВОЙ КИСЛОТЫ КАК ВЕЩЕСТВА, ПРЕПЯТСТВУЮЩИЕ ТРОМБООБРАЗОВАНИЮ | 1995 |
|
RU2135470C1 |
| СУЛЬФАМАТЫ ПСЕВДОФРУКТОПИРАНОЗЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2139875C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ТРОМБИНА У МЛЕКОПИТАЮЩЕГО | 1995 |
|
RU2148585C1 |
| СОЕДИНЕНИЯ ФОСФОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ СЕРИНОВОЙ ПРОТЕИНАЗЫ | 2002 |
|
RU2311421C2 |
| СИММЕТРИЧНЫЕ И НЕСИММЕТРИЧНЫЕ ПРОИЗВОДНЫЕ ДИФЕНИЛМОЧЕВИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ РОСТА ОПУХОЛЕВЫХ КЛЕТОК, ОПОСРЕДОВАННОГО КИНАЗОЙ RAF | 1998 |
|
RU2247109C9 |
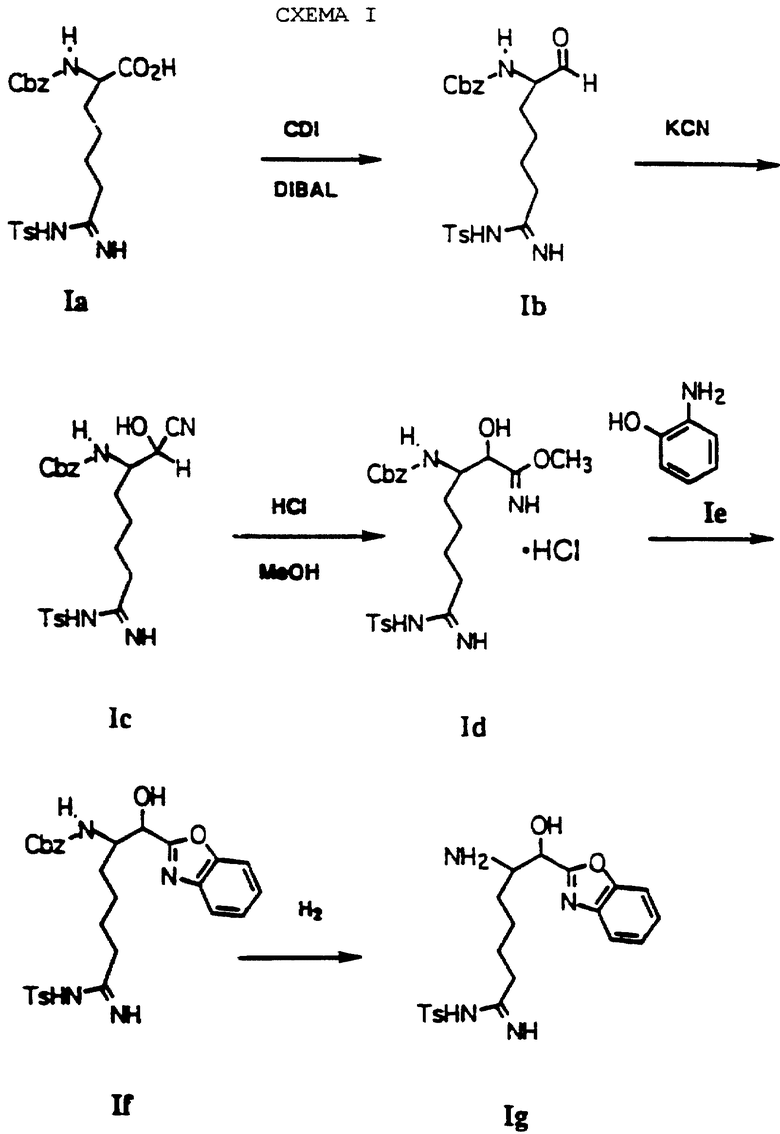
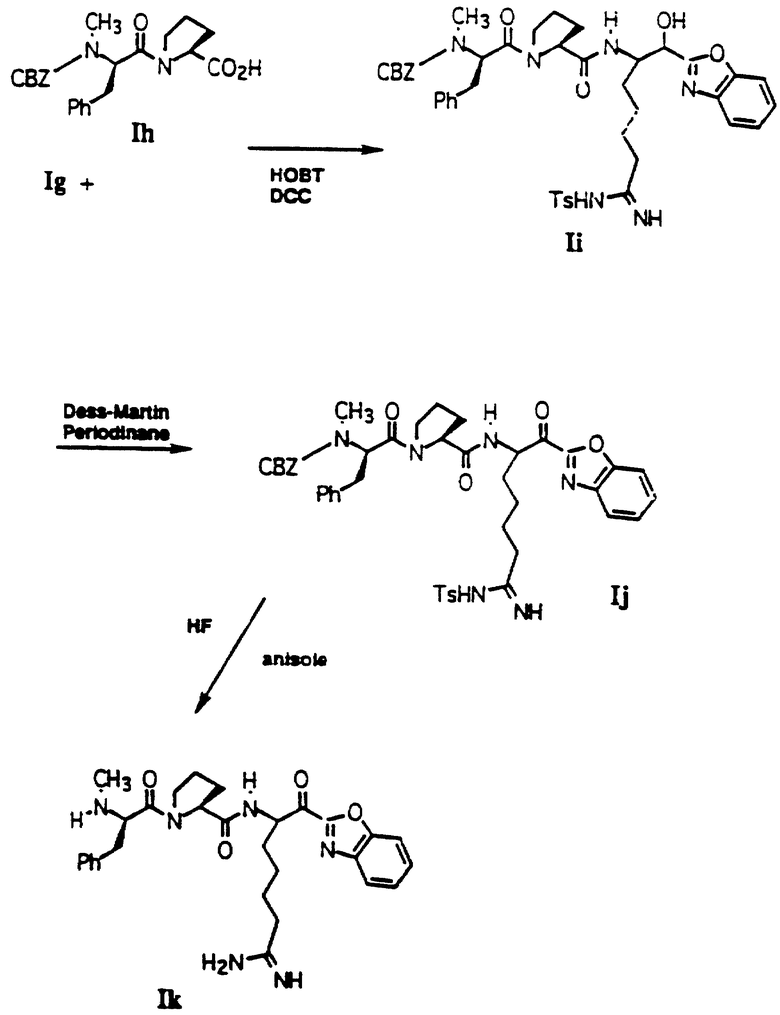
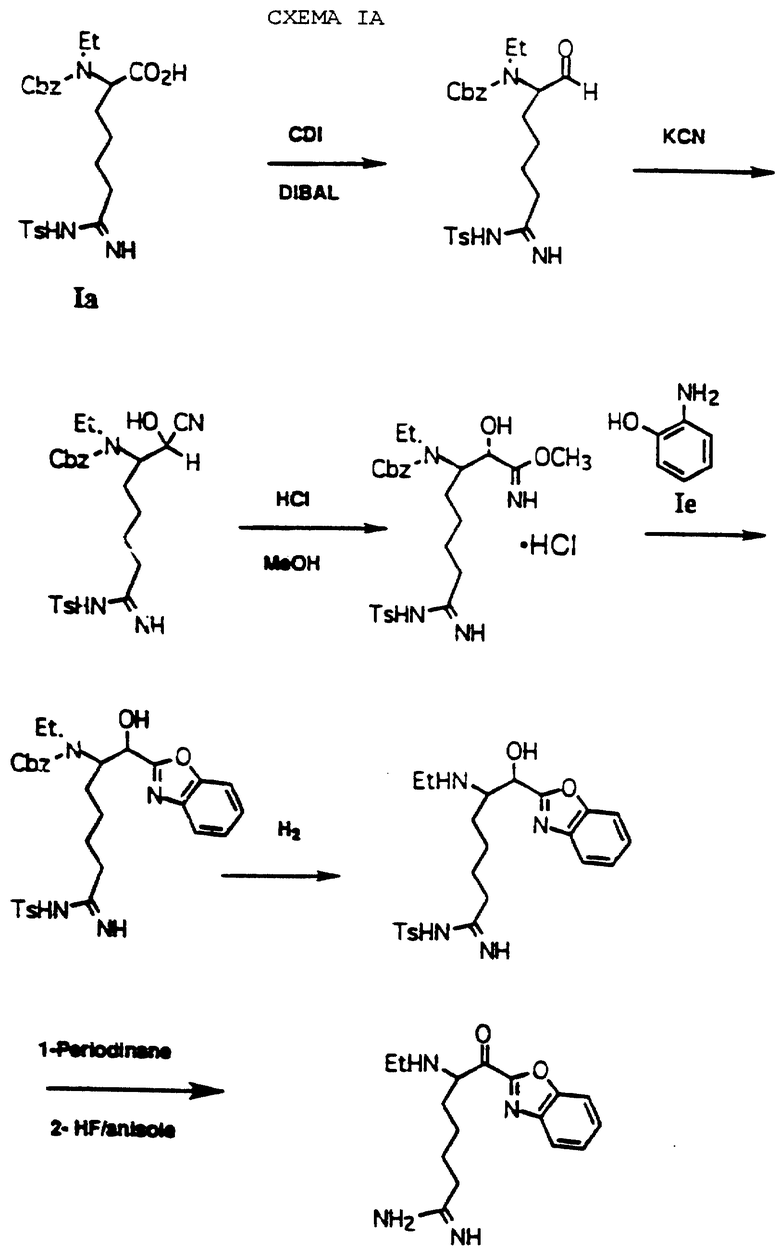
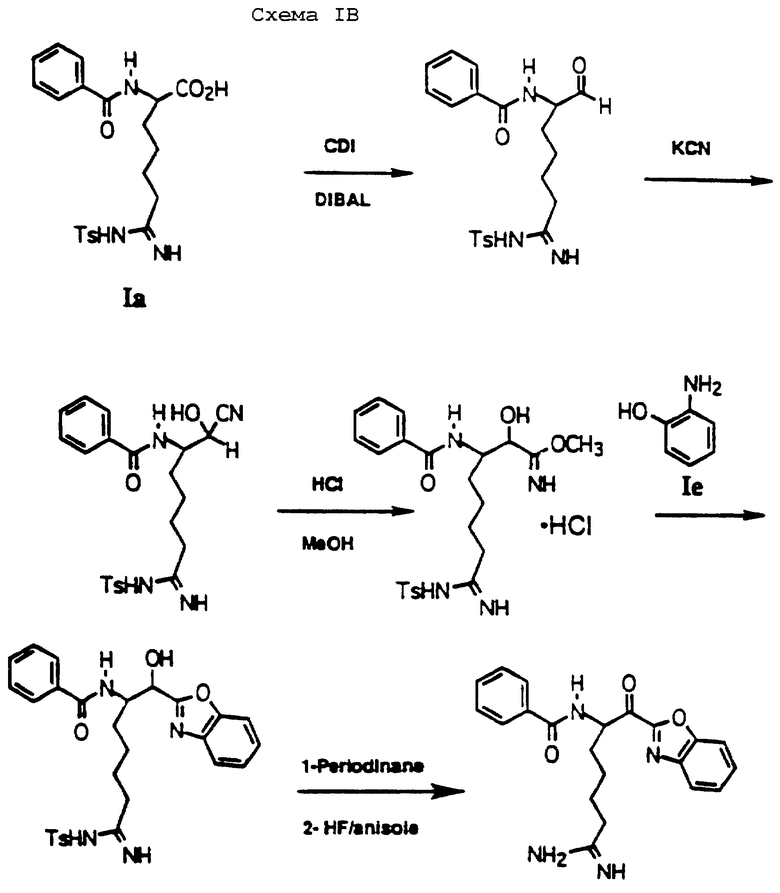
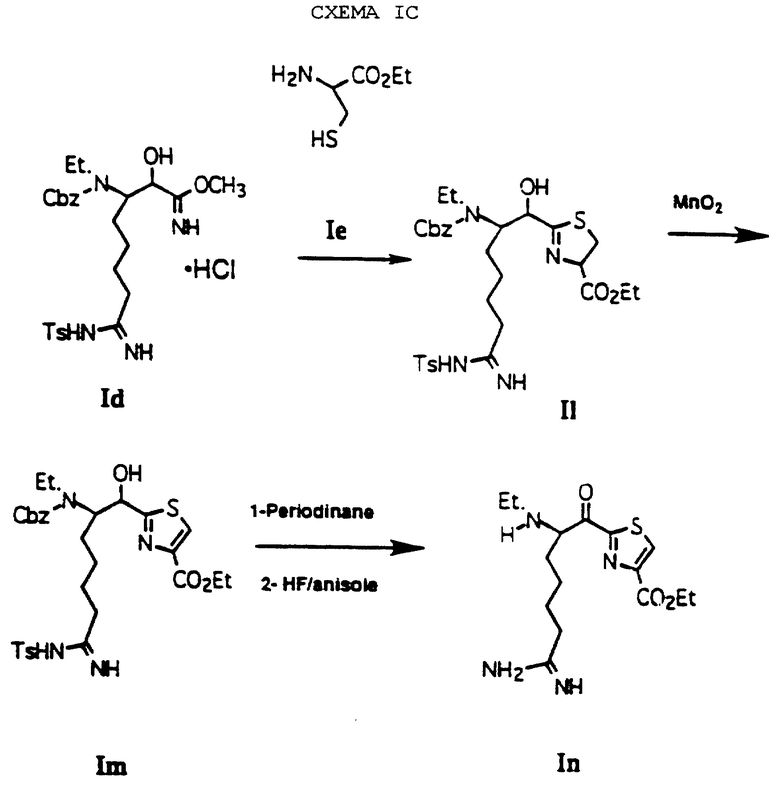
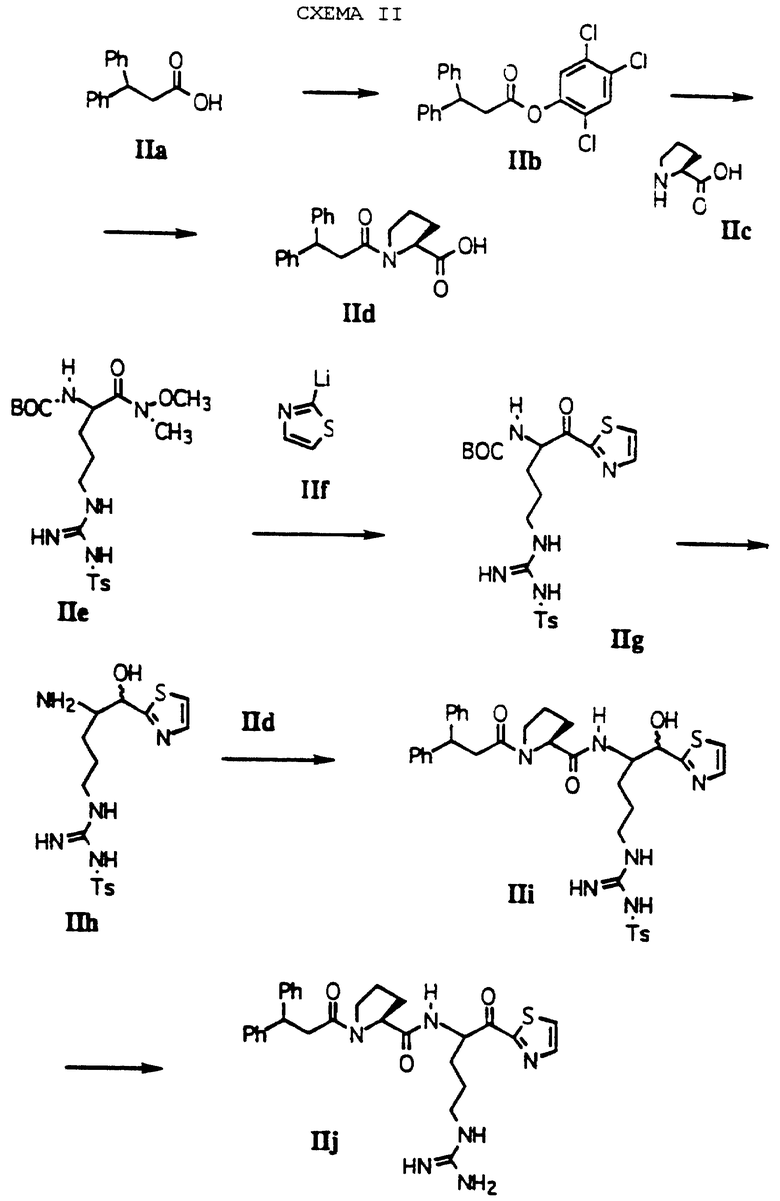
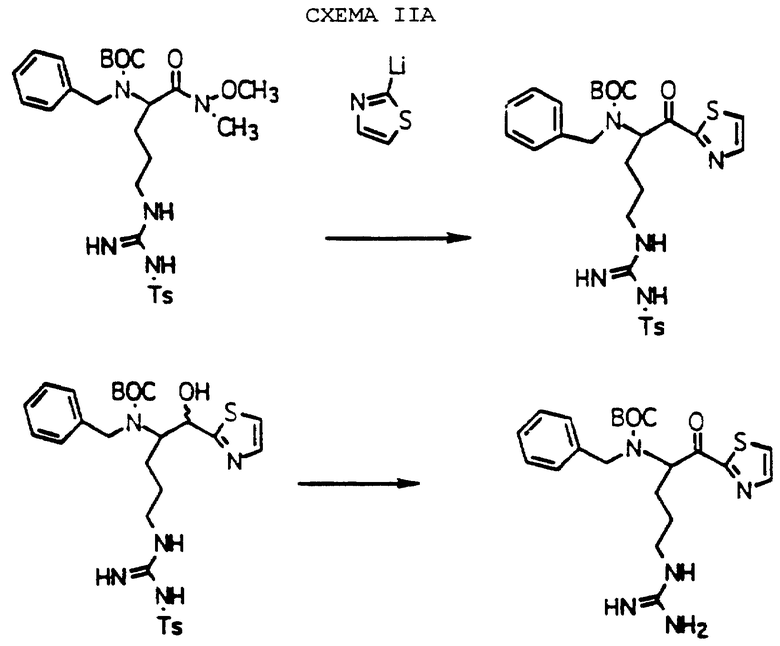
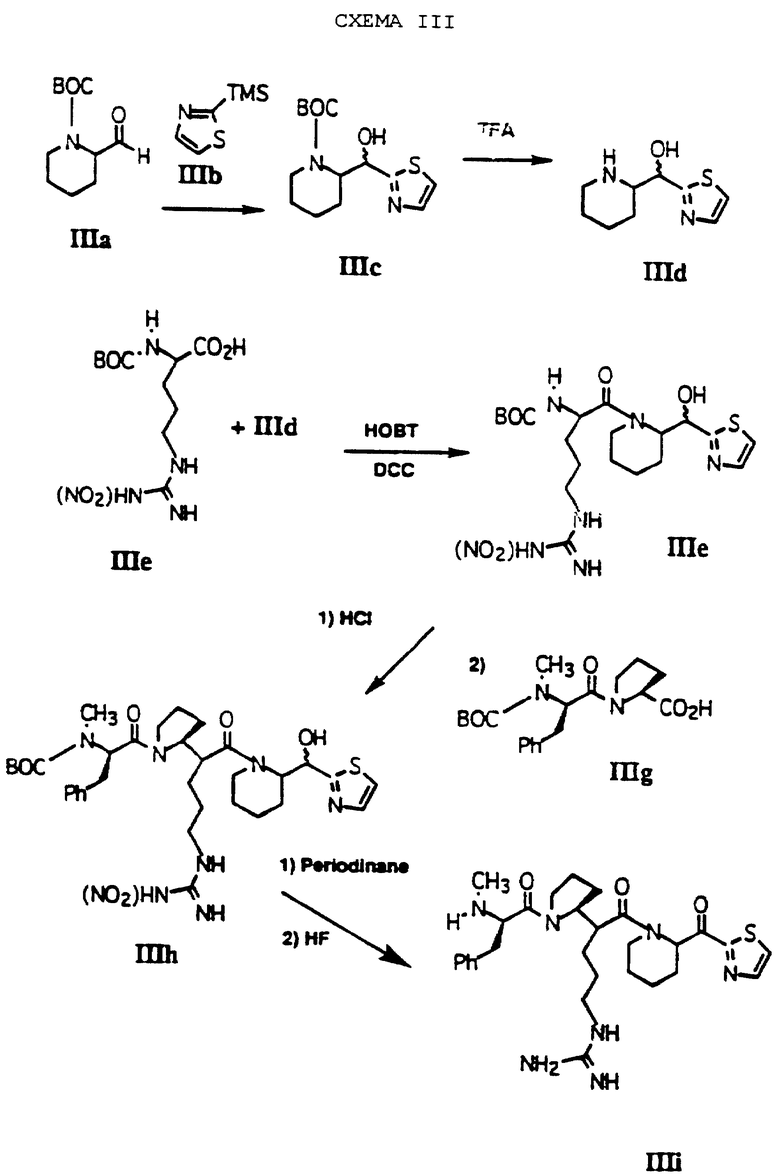
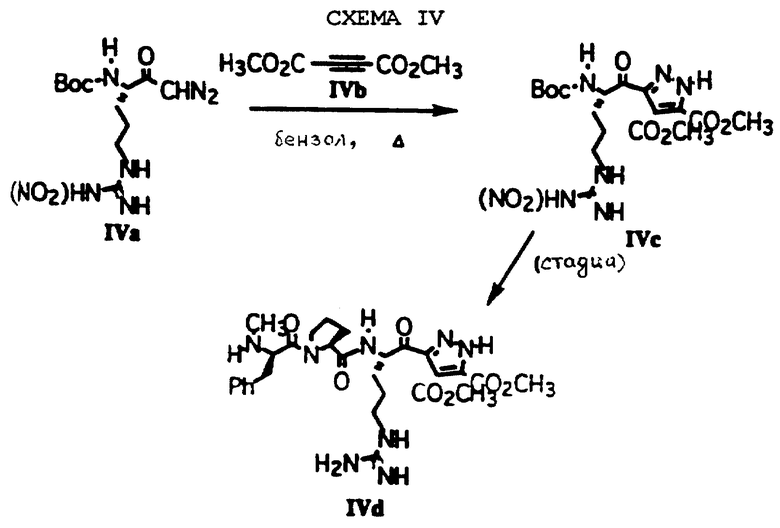
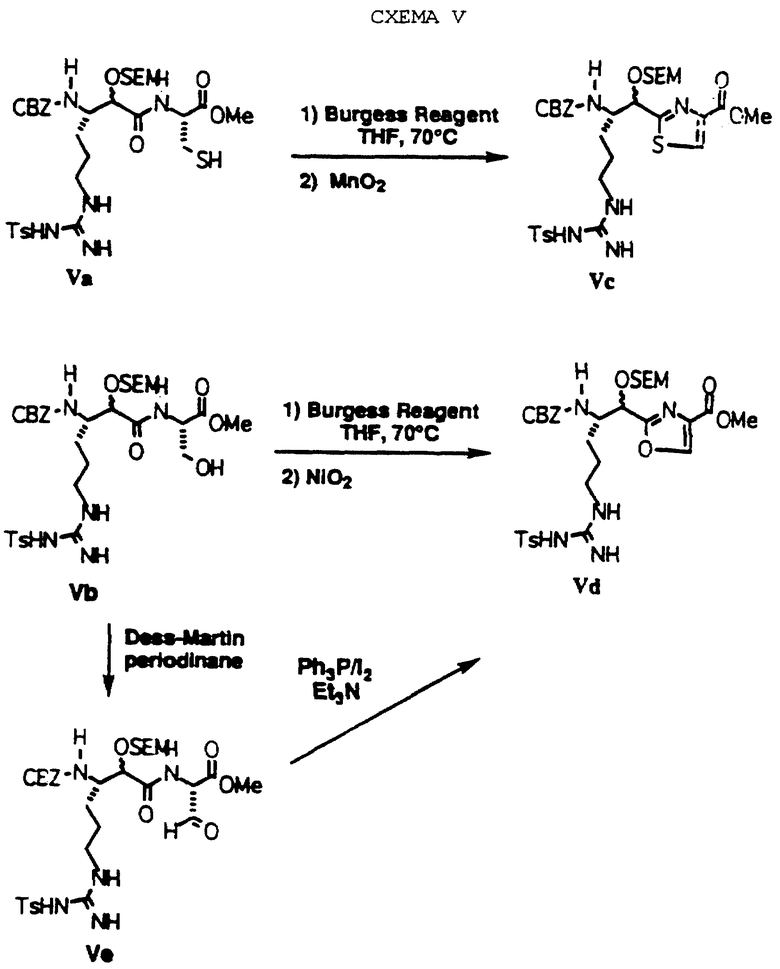

В изобретении описываются соединения формулы (I), а также их N-алкилированные производные, где заместители имеют указанные в описании значения. Описывается также получение фармацевтической композиции, обладающей способностью ингибировать тромбин, включающей терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель. Приводятся способы ингибирования тромбина и трипсина, включающие контактирование соединения формулы (I) со средой, содержащей тромбин или трипсин соответственно. 4 с. и 31 з.п.ф-лы, 1 табл.
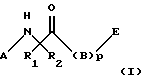
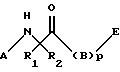
где А выбран из группы, включающей формил, С3-7циклоалкилкарбонил, замещенные нафтилсульфинил и нафтилсульфонил (где заместители нафтила независимо выбраны из одного или нескольких амино или С1-4диалкиламино);
D- или L-аминокислоту, которая присоединена своим карбоксиконцом к азоту, указанному в формуле I, и выбрана из группы, включающей пролин, глицин и валин, где аминные концы указанной аминокислоты необязательно связаны с членом, выбранным из группы, включающей нафтилсульфонил, замещенный фенил С1-4алкил (где заместитель фенила представляет собой С1-4алкиламино), 1-С1-4алкиламино-1-циклогексанкарбонил, С1-4алкилкарбонил, замещенный по концевому положению алкильной цепочки дифенилом; или полипептид, содержащий две аминокислоты, где первая аминокислота представляет собой D- или L-аминокислоту, присоединенную через ее карбоксиконцы к азоту, указанному в формуле I, и представляет собой пролин, гомопролин или валин,
и вторая D- или L-аминокислота присоединена к аминоконцу указанной первой аминокислоты и выбрана из группы, включающей фенилаланин, замещенный фенилаланин (где заместители фенила независимо выбраны из одного или нескольких С1-4алкила, галогена или С1-4алкоксикарбонила), 2,2-дифенилаланин, 4-бифенилаланин, замещенные или незамещенные циклогексилглицин или циклогексилаланин (где заместителем является С1-4алкил), дифенилглицин или фенилглицин, где аминные концы указанной второй аминокислоты являются незамещенными или монозамещены членом группы, включающей С1-12алкил или С2-5ацил;
R1 выбран из группы, включающей водород и С1-4алкил;
R2 выбран из группы, включающей аминоС2-5алкил, С1-4алкиламино С2-5алкил, гуанидиноС2-5алкил, С1-3алкоксиС2-5алкил, фенил, замещенный фенил (где заместители независимо выбраны из одной или нескольких амидиногрупп), бензил, замещенный бензил (где заместители независимо выбраны из одной или нескольких амидиногрупп) и С1-5алкил;
р = 0 или 1;
В обозначает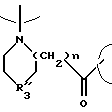
где n = 0-3,
R3 - водород,
и карбонильный радикал В присоединен к Е;
Е обозначает замещенный имидазол-2-ил (где заместителем является С1-4алкил), тиазол-2-ил, замещенный тиазол-2-ил (где заместители независимо выбраны из карбокси, С1-4алкоксикарбонила или фенетиламинокарбонила), 4-оксо-2-хиноксалин-2-ил, 2-пиридил, бензотиофен-2-ил, бензоксазол-2-ил, бензимидазол-2-ил, замещенный бензимидазол-2-ил (где заместителем является С1-4алкил), бензотиазол-2-ил, 4,5,6,7-тетрагидробензотиазол-2-ил, нафто[2,1-d] тиазол-2-ил и замещенный бензотиазол-2-ил (где заместитель выбран из С1-4алкила, С1-4алкокси, гидрокси, галогена, карбокси, С1-4алкоксикарбонила, аминокарбонила или гидрокси С1-4алкила).
и вторая аминокислота представляет собой D-аминокислоту и выбрана из группы, включающей фенилаланин, замещенный фенилаланин (где заместители фенила независимо выбраны из одного или нескольких С1-4алкила, галогена или С1-4алкоксикарбонила), 2,2-дифенилаланин, 4-бифенилаланин, замещенные или незамещенные циклогексилглицин или циклогексилаланин (где заместителем является С1-4алкил), дифенилглицин или фенилглицин, где аминные концы указанной второй аминокислоты являются незамещенными или монозамещены членом группы, включающей С1-12алкил или С2-5ацил.
| Теплообменное устройство | 1974 |
|
SU504064A1 |
| ПЕПТИДНЫЕ ПРОИЗВОДНЫЕ, ИХ СТЕРЕОИЗОМЕРЫ ИЛИ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВОТРОМБОЗНОЙ, ПРОТИВОСВЕРТЫВАЮЩЕЙ ИЛИ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ ТРОМБИНА, СПОСОБ ПОДАВЛЕНИЯ КИНИНОГЕНАЗ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ В КАЧЕСТВЕ ИСХОДНЫХ В СИНТЕЗЕ ИНГИБИТОРА ТРОМБИНА | 1994 |
|
RU2142469C1 |
| US 5332726 А, 26.07.1994 | |||
| Уплотнение разъемного соединения | 1977 |
|
SU648780A1 |

