ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, которые представляют собой агонисты аденозиновых рецепторов, и к их применению в качестве лекарственных средств, в частности, в качестве анальгетических или противовоспалительных соединений, и к способам предотвращения, лечения или ослабления боли или воспаления с использованием этих соединений.
УРОВЕНЬ ТЕХНИКИ
Аденозин представляет собой повсеместно распространенный гормон/нейротрансмиттер, который действует на четыре известных рецептора, А1-, А2А-, А2В- и А3-рецепторы аденозина. Аденозин в общем служит для баланса между снабжением тканей энергией и потребностью в таковой. Например, в сердце высвобождаемый аденозин замедляет частоту ритма сердца благодаря действию, обусловленному рецептором А1, в синусах и предсердии, вместе с тем одновременно расширяя коронарную артерию для усиления снабжения энергией. Подобным образом, во время воспаления аденозин служит для подавления воспалительной активности, тогда как при состояниях избыточной нервной активности (таких как эпилепсия) аденозин ингибирует воспаление нервов. Эта система, или вариант таковой, присутствует во всех тканях.
Сам по себе аденозин может быть использован для диагностики и лечения суправентрикулярной тахикардии. Агонисты аденозиновых А1-рецепторов известны своим действием как эффективные анальгетики (Sawynok, J. Eur. J. Pharmacol., (1998), 347, 1-11; Giffin et al., (2003), 23, 4, 287-292). Недавно было показано, что агонисты А2а обеспечивают значительное ослабление боли при состояниях повышенной чувствительности к боли (таких как невропатическая и воспалительная гипералгезия) (WO 2004/052377; WO 2004/078183; WO 2004/078184; WO 2005/084653), и известны как имеющие противовоспалительную активность (например, см. US 5877180; WO 99/34804; Linden et al., Expert Opin. Investig. Drugs (2005), 14, 7, 797-806; Sitkovsky et al., TRENDS in Immunology (2005), 26, 6, 299-304; Linden et al., Journal of Immunology (2006), 117, 2765-2769; Cronstein et al. (2004), 25, 1, 33-39). На экспериментальных животных было показано, что агонисты А2А-рецепторов являются эффективными против широкого многообразия состояний, включающих сепсис (Linden et al., The Journal of Infectious Diseases (2004), 189, 1897-1904), артрит (Cohen et al., J. Orthop. Res. (2005), 23, 5, 1172-1178; Cohen et al., J. Orthop. Res. (2004), 22, 2, 427-435) и повреждения, обусловленные ишемией/реперфузией, возникающие при почечной окклюзии, закупорке коронарных сосудов и церебральной артерии (например, см. Day et al., J. Clin. Invest., (2003), 112, 883-891; Linden et al., Am. J. Physiol. Gastrointest. Liver Physiol. (2004), 286, G285-G293; Linden et al., Am. J. Physiol. (1999), 277, F404-F412; Schlack et al., J. Cardiovasc. Pharmacol. (1993), 22, 89-96; Zu et al., J. Cardiovasc. Pharmacol. (2005), 46, 6, 794-802; Linden et al., Am. J. Physiol. Heart Circ. Physiol. (2005), 288, 1851-1858; Kennedy et al., Current Opinion in Investigational Drugs (2006), 7, 3, 229-242). Общим фактором в этих состояниях является снижение воспалительной реакции, обусловленное ингибирующим эффектом этого рецептора в большинстве, если не во всех, клетках очага воспаления. Агонисты А2а также известны как способствующие заживлению ран (Montesinos, Am. J. Pathol. (2002), 160, 2009-2018).
Однако повсеместная распространенность аденозиновых рецепторов означает, что введение агонистов аденозиновых рецепторов вызывает вредные побочные эффекты. В общем это препятствовало развитию терапевтических подходов, основанных на аденозине. Селективные агонисты А1-рецепторов вызывают брадикардию. Агонисты А2А-рецепторов вызывают широко распространенное расширение кровеносных сосудов с последующими гипотонией и тахикардией. Первый селективный агонист А2А-рецепторов (2-[4-(2-карбоксиэтил)фенилэтиламино]-5'-N-этилкарбоксамидоаденозин, или CGS21680) был протестирован в 2А-фазе клинических испытаний как потенциальный антигипертензивный препарат. Однако введение этого соединения вызывает сильное снижение кровяного давления и последующее повышение минутного сердечного выброса. Это препятствует применению препарата CGS21680 в качестве лекарственного средства. Webb et al. (J. Pharmacol. Exp. Ther. (1991), 259, 1203-1212), Casati et al. (J. Pharmacol. Exp. Ther. (1995), 275(2), 914-919) и Bonnizone et al. (Hypertension. (1995), 25, 564-569) показывают, что селективные агонисты аденозиновых А2А-рецепторов вызывают гипотонию и тахикардию. Степень вызываемой тахикардии является достаточной, чтобы воспрепятствовать применению его в качестве лекарственного препарата. Alberti et al. (J. Cardiovasc. Pharmacol., 1997, сентябрь, 30(3): 320-324) сообщают, что селективные агонисты аденозиновых А2А-рецепторов являются сильнодействующими сосудорасширяющими средствами, которые понижают кровяное давление и индуцируют заметные учащения сердцебиения и повышение активности ренина в плазме. Эти побочные эффекты препятствуют применению его в качестве лекарственного средства.
US 5877180 относится к агонистам аденозиновых А2А-рецепторов, которые показаны эффективными для лечения воспалительных заболеваний. Предпочтительные агонисты, WRC0090 и SHA 211 (WRC0474), представлены как более сильнодействующие и селективные, чем ранее описанные аденозиновые аналоги, такие как CGS21680 и CV1808. Как считается, введение препаратов SHA 211 или WRC0090 снижает возможность побочных эффектов, обусловленных связыванием аналогов с другими аденозиновыми рецепторами. Однако приведены только данные in vitro относительно активности SHA 211. Не продемонстрировано, что любые из описанных соединений могли бы быть терапевтически эффективными in vivo без проявления серьезных побочных эффектов. Хотя ожидается, что побочные действия, обусловленные связыванием сильнодействующих и селективных агонистов аденозиновых А2А-рецепторов с другими аденозиновыми рецепторами, будут снижаться применением таких агонистов, повсеместная распространенность аденозиновых рецепторов означает, что эти соединения по-прежнему будут активировать аденозиновые А2А-рецепторы в нормальной ткани и поэтому вызывать серьезные побочные эффекты (такие как гипотония и рефлекторная тахикардия).
Ribeiro et al. (Progress in Neurobiology, 68 (2003), 377-392) представляет собой обзор аденозиновых рецепторов в нервной системе. В заключительных примечаниях к этой статье (на странице 387, правая колонка, строки 4-10 раздела 8) высказано, что, «как очень давно отмечено, активация аденозиновых рецепторов на периферии связана с гипотонией, брадикардией и гипотермией… Эти побочные эффекты до сих пор значительно ограничивают клиническую применимость агонистов аденозиновых рецепторов».
Поэтому существует потребность в создании агонистов аденозиновых рецепторов, которые могут быть введены с минимальными побочными эффектами.
Также есть потребность в создании анальгетиков для лечения боли. Боль имеет два компонента, каждый из которых включает активацию чувствительных нейронов. Первый компонент представляет собой раннюю или прямую фазу, когда чувствительный нейрон стимулируется, например, в результате воздействия тепла или давления на кожу. Второй компонент представляет собой следствие повышенной чувствительности сенсорных механизмов иннервации тканей, которые были ранее повреждены. Этот второй компонент называется гипералгезией и входит во все формы хронической боли, возникающей при повреждении ткани, но не в раннюю или прямую фазу ощущения боли.
Таким образом, гипералгезия представляет собой состояние возрастающего ощущения боли, вызванной повреждением ткани. Это состояние является естественной реакцией нервной системы, по всей видимости, предназначенной для стимулирования защиты от повреждения тканей потерпевшим лицом, для того, чтобы предоставить ткани время на восстановление. Известны две основополагающие причины этого состояния, а именно повышение активности чувствительных нейронов и изменение обработки ноцицептивной информации, которая происходит в спинном мозге. Гипералгезия может быть изнуряющей в состояниях хронического воспаления (например, при ревматоидном артрите), и когда происходит повреждение сенсорного нерва (т.е. невропатическая боль).
Известны два главных класса болеутоляющих средств: (i) нестероидные противовоспалительные лекарственные средства (NSAID) и родственные ингибиторы класса COX-2; и (ii) опиаты, основанные на морфине. Анальгетики обоих классов являются эффективными в контролировании нормальной, прямой или ноцицептивной боли. Однако они являются менее эффективными против некоторых типов боли при гипералгезии, таких как невропатическая боль. Многие практикующие врачи избегают назначать опиаты в высоких дозах, требуемых для снятия невропатической боли, вследствие побочных эффектов, обусловленных введением этих соединений (таких как возбужденное состояние, тошнота и рвота), и возможности, что пациенты могут приобрести зависимость от них. Нестероидные противовоспалительные лекарственные средства (NSAID) являются гораздо менее сильнодействующими, чем опиаты, так что требуются даже более высокие дозы этих соединений. Однако это является нежелательным, так как эти соединения вызывают раздражение желудочно-кишечного тракта.
Поэтому есть необходимость в создании болеутоляющих средств, в частности антигипералгезических препаратов, которые являются достаточно сильнодействующими для контроля ощущения боли при невропатическом и прочих гипералгезических синдромах и которые не проявляют серьезных побочных эффектов или не вызывают болезненного привыкания пациентов к ним.
Спонгозин (также известный как 2-метоксиаденозин) известен как слабый неселективный агонист аденозиновых рецепторов (Ueeda et al., J. Med. Chem. (1991), 34, 1334-1339). Это соединение вызывает 25%-ное ингибирование индуцированного каррагенином воспаления у крыс при пероральном введении в дозе 20 мг/кг. Однако факты снижения среднего кровяного давления (41%) и частоты сердцебиения (25%) также наблюдались после введения этого соединения при этой дозе (Bartlett et al. (J. Med. Chem. (1981), 24, 947-954).
Авторы настоящей заявки ранее обнаружили, что спонгозин неожиданно проявляет себя как эффективный анальгетик при дозах, более чем в сто раз меньших, чем можно было бы ожидать как необходимых для анальгетического эффекта, основываясь на известной аффинности этого соединения к аденозиновым рецепторам. При таких дозах спонгозин не вызывает существенных побочных эффектов, связанных с более высокими дозами этого соединения или прочих агонистов аденозиновых рецепторов. Таким образом, терапевтические эффекты спонгозина могут быть отделены от его побочных действий. Активность спонгозина в качестве анальгетика представляет собой предмет международной патентной заявки № PCT/GB03/05379, и активность родственных спонгозину соединений как болеутоляющих средств является предметом международной патентной заявки № PCT/GB04/00935. Применение спонгозина и родственных соединений для лечения воспаления и других расстройств составляет предмет международной патентной заявки № PCT/GB04/000952.
Авторы настоящей заявки нашли, что спонгозин и родственные соединения, описанные в заявках PCT/GB04/00935 и PCT/GB04/000952, имеют повышенную аффинность к аденозиновым рецепторам при величине рН ниже 7,4. Представляется, что это свойство объясняет неожиданную активность этих соединений при малых дозах.
Однако авторы настоящей заявки нашли, что для некоторых замещенных аденозинов, которые имеют повышенную аффинность к аденозиновым рецепторам при величине рН ниже 7,4, близкородственные соединения не сохраняют этой желательной активности. Это создало чрезвычайные трудности в выявлении дополнительных замещенных аденозинов, которые могут быть использованы в качестве лекарственных средств без проявления серьезных побочных эффектов, поскольку было невозможно прогнозировать, какие конкретно замещенные аденозины будут иметь повышенную аффинность к аденозиновым рецепторам при пониженной величине рН. В качестве иллюстрации такой непредсказуемости нижеследующая Таблица приводит значения константы ингибирования Ki (нМ, наномолей/литр) для аденозиновых А2а-рецепторов крыс при величинах рН 5,5 и 7,4 для соединений серии 2-аминоалкиладенозинов и серии 5'-амидоаденозинов (эти значения были рассчитаны с использованием экспериментов по связыванию, подобных таковым, описанным ниже в отношении Примера 1):
Ki (нМ) (рН 5,5)
Ki (нМ) (рН 7,4)
Ki (нМ) (рН 5,5)
Ki (нМ) (рН 7,4)
Только определенные соединения в каждой серии в вышеприведенной таблице имеют повышенную аффинность к аденозиновым рецепторам при пониженном значении рН. В 2-аминоалкильной серии, когда длина алкильной цепи возрастает до 3 или 4 атомов углерода, желаемая активность утрачивается, но появляется вновь, когда длина цепи увеличивается до 6 атомов углерода. В 5'-амидной серии, когда длина алкильной цепи возрастает до 2 или более атомов углерода, желаемая активность утрачивается при непредсказуемом исключении NH-изопропиламидного производного, которое более чем в 400 раз более активно при величине рН 5,5 по сравнению со значением рН 7,4.
Несмотря на трудность выявления дополнительных замещенных аденозинов с повышенной аффинностью к аденозиновым рецепторам при пониженном значении рН среди многих миллионов возможных соединений, авторы настоящей заявки обнаружили определенные другие соединения, которые также имеют повышенную аффинность к аденозиновым рецепторам при пониженной величине рН. Представляется, что эти соединения могут быть использованы в качестве лекарственных средств без проявления серьезных побочных эффектов.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно изобретению, представлены агонисты аденозиновых рецепторов следующей формулы:
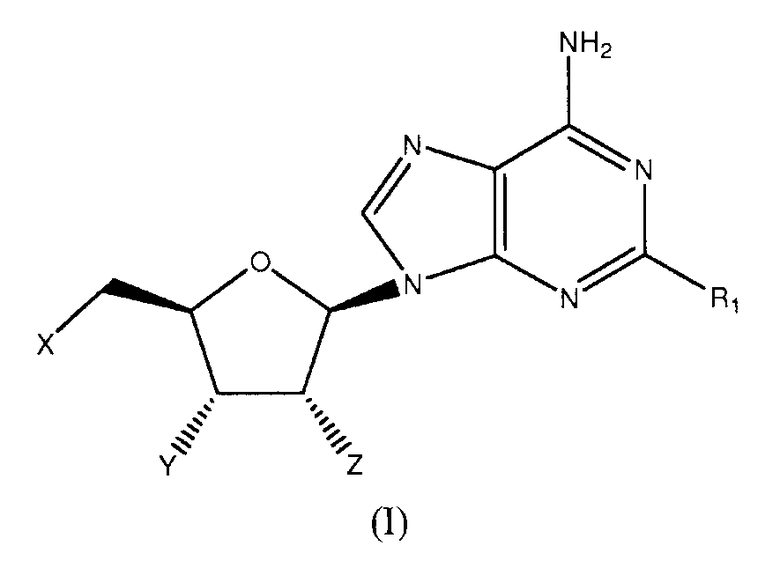
в которой
когда X=Y=Z=OH, R1 представляет OCH2CF2CF3, феноксигруппу, замещенную 3-(4-трифторметилфенилом), 3,4-дихлором, (3-трифторметилом,4-фтором), (3-трифторметилом,4-хлором), (3-хлором,4-цианогруппой) или 3,5-бис(трифторметилом)); 1-пиперазинил(4-(3,4-дихлорфенил)), фенильную группу, замещенную 3,4-дихлором, 3,5-дифтором, 3,5-бис(трифторметилом) или 3,4,5-трифтором) или 2-бензофуранильный фрагмент; или
когда X=Y=ОН и Z=OMe, R1 представляет собой ОСН3, OCH2CHF2, ОСН2-циклопентил-, О-(2,5-дифторфенил)- или (S)-втор-бутиламиногруппу; или
когда Х=Н и Y=Z=OH, R1 представляет собой н-гексиламиногруппу или циклопентиламиногруппу; или
когда X=Z=OH и Y=H, R1 представляет собой циклопентиламиногруппу,
или их фармацевтически приемлемая соль.
Во избежание неясности, ссылка на соединения формулы (I) ниже включает фармацевтически приемлемые соли соединений формулы (I).
Все соединения согласно изобретению предполагаются как имеющие повышенную аффинность к аденозиновым рецепторам при величине рН ниже 7,4. В нормальных тканях млекопитающих значение внеклеточной рН весьма точно поддерживается между величинами рН 7,35 и 7,45. Некоторые ткани проявляют более низкие значения рН, в частности в полости желудка (рН между 2 и 3) и на поверхностях некоторых эпителиальных тканей (например, величина рН на поверхности легкого составляет приблизительно 6,8). В патологических тканях, например во время воспаления, ишемии и при прочих типах повреждений, происходит снижение величины рН.
Ввиду повышенной аффинности соединений согласно изобретению к аденозиновым рецепторам при пониженном значении рН предполагается, что действия этих соединений могут быть нацелены на области с низкой величиной рН, такие как патологические ткани. Следовательно, дозы этих соединений, которые требуются для проявления терапевтического эффекта, являются гораздо меньшими, чем следовало бы ожидать, исходя из их аффинности к аденозиновым рецепторам при нормальном физиологическом значении внеклеточной рН. Поскольку требуются только низкие дозы соединений, серьезные побочные эффекты, связанные с введением агонистов аденозиновых рецепторов, избегаются или сводятся к минимуму. Из этого следует неожиданный вывод (в отличие от предписаний прототипа, например, в US 5877180), что некоторые агонисты аденозиновых рецепторов, которые проявляют низкую аффинность и/или являются неселективными агонистами при физиологическом значении рН (такие как спонгозин), могут быть терапевтически эффективными без проявления серьезных побочных эффектов.
Предполагается, что соединения формулы (I) могут быть введены в дозах гораздо ниже доз, которые предполагались необходимыми на основании их аффинности к аденозиновым рецепторам при величине рН 7,4, и обнаруживают терапевтические эффекты при таких дозах без проявления серьезных побочных эффектов.
Таким образом, согласно изобретению представляется соединение соответственно изобретению для применения в качестве лекарственного средства.
Любое патологическое состояние, которое может быть предотвращено или улучшено благодаря агонизму в отношении аденозиновых А2А-рецепторов может быть предотвращено, излечено или улучшено с помощью соединения формулы (I).
Изобретение относится к применению соединения формулы (I) в производстве лекарственного средства для предотвращения, лечения или ослабления патологического состояния, которое может быть улучшено или предотвращено благодаря агонизму в отношении аденозиновых А2А-рецепторов.
Также согласно изобретению представляется способ предотвращения, лечения или ослабления патологического состояния, которое может быть улучшено или предотвращено благодаря агонизму в отношении аденозиновых А2А-рецепторов, который включает введение соединения формулы (I) субъекту при необходимости такого предотвращения, лечения или улучшения.
Специалист этой области технологии может без труда протестировать, обусловливается или нет патологическое состояние, которое предотвращается, излечивается или улучшается соединением формулы (I), действием через аденозиновые А2А-рецепторы. Например, это может быть сделано сравнением действия соединения на животной модели в патологическом состоянии в присутствии и в отсутствие селективного антагониста аденозиновых А2А-рецепторов. Если действие соединения в присутствии антагониста снижается или отсутствует по сравнению с действием соединения в отсутствие антагониста, следует сделать вывод, что соединение проявляет свое действие через аденозиновый А2А-рецептор. Антагонисты аденозиновых А2А-рецепторов известны обычным специалистам в этой области технологии (например, см. Ongini et al., Farmaco, 2001, январь-февраль; 56 (1-2): 87-90).
Альтернативно, может быть использована мышь, нокаутная по аденозиновому А2А-рецептору (Otha A. и Sitkovsky M., Nature, 2001; 414: 916-920). Например, действие соединения на мышь, которая имеет симптомы патологического состояния, сравнивается с его действием на мышь, нокаутную по аденозиновому А2А-рецептору, которая имеет соответствующие симптомы. Если соединение эффективно только для мыши, которая имеет аденозиновые А2А-рецепторы, следует заключить, что соединение проявляет свое действие через аденозиновые А2А-рецепторы.
Предполагается, что соединения формулы (I) имеют анальгетическую и/или противовоспалительную активность и могут быть введены с пониженной вероятностью и серьезностью побочных эффектов по сравнению с другими агонистами аденозиновых рецепторов.
Изобретение относится к применению соединения формулы (I) в производстве лекарственного средства для предотвращения, лечения или ослабления боли, в частности гипералгезии. Также согласно изобретению представляется способ предотвращения, лечения или ослабления боли (в частности гипералгезии), который включает введение соединения формулы (I) субъекту, нуждающемуся в таком предотвращении, лечении или ослаблении.
Способы, раскрытые в описании, включают способы, в которых субъект идентифицируется как нуждающийся в конкретном назначенном лечении. Идентификация субъекта как нуждающегося в таком лечении может быть в компетенции субъекта или профессионального работника медицинской службы и может быть субъективной (например, мнением) или объективной (например, измеримой в испытании или диагностическом методе).
Как предполагается, соединения формулы (I) являются эффективными в подавлении ощущения боли у млекопитающих, страдающих от боли, в частности невропатической или воспалительной боли, даже когда вводятся в дозах, которые, как предполагается, дают концентрации в плазме гораздо ниже таковых, известных для активации аденозиновых рецепторов. Поэтому предполагается, что соединения формулы (I) могут лечить боль (в частности невропатическую или воспалительную боль) без проявления существенных побочных эффектов, связанных с введением других агонистов аденозиновых рецепторов.
Как упомянуто выше, гипералгезия в большинстве ситуаций является следствием повреждения ткани, либо повреждения непосредственно чувствительного нерва, либо повреждения ткани, иннервированной данным чувствительным нервом. Следовательно, существуют многочисленные состояния, в которых ощущение боли включает компонент гипералгезии.
Согласно изобретению, представляется применение соединения формулы (I) в качестве болеутоляющего средства (в частности, антигипералгезического) для предотвращения, лечения или ослабления боли (в частности, гипералгезии), возникающей в результате невропатии, включая диабетическую невропатию, полиневропатию, раковую боль, фибромиалгию, синдром миофасциальной боли, остеоартит, панкреатическую боль, тазово-перинеальную боль, постгерпетическую невралгию, ревматоидный артрит, ишиас-люмбальную радикулопатию, спинальный стеноз, болевую дисфункцию височно-нижнечелюстного сустава, связанную с ВИЧ боль, тригеминальную невралгию, хроническую невропатическую боль, боль внизу спины, боль синдрома неудачных операций на позвоночнике, боль в спине, послеоперационную боль, боль после физической травмы (в том числе огнестрельного ранения, дорожно-транспортного происшествия, ожогов), сердечную боль, боль в груди, тазовую боль/PID (воспаление тазовых органов), суставную боль (тендонит, бурсит, острый артрит), шейную боль, боль в животе, фантомную боль ложного ощущения ампутированной конечности, боль при родовспоможении (роды/кесарево сечение), почечные колики, боль при остром опоясывающем герпесе, боль при деструктивных процессах острого панкреатита (рак), дисменорею/эндометриоз; или в любом из вышеназванных патологических состояний, где состояние вызывается или обостряется бактериальной или вирусной инфекцией.
Согласно изобретению, также представляется применение соединения формулы (I) в качестве анальгетика (в частности и антигипералгезического средства) для предотвращения, лечения или ослабления боли (в частности, гипералгезии), возникающей в результате воспалительного расстройства, или как результат комбинации воспалительного, аутоиммунного и невропатического повреждения ткани, включая ревматоидный артрит, остеоартрит, ревматоидный спондилит, подагрический артрит и другие артритные состояния, рак, ВИЧ, хроническую обструктивную легочную болезнь (COPD), острый бронхит, хронический бронхит, эмфизему, бронхоэктазию, фиброзно-кистозную дегенерацию, пневмонию, плеврит, острую астму, хроническую астму, острый респираторный дистресс-синдром, респираторный дистресс-синдром взрослых (ARDS, РДСВ), респираторный дистресс-синдром новорожденных (IRDS), острое повреждение легких (ALI), ларингит, фарингит, стойкую астму, хронический астматический бронхит, интерстициальную болезнь легких, злокачественные заболевания легких, недостаточность альфа-антитрипсина, бронхолитические облитерации, саркоидоз, пульмональный фиброз, коллагеновые сосудистые болезни, аллергический ринит, назальную гиперемию, астматический статус, обусловленную курением легочную болезнь, пульмональную гипертонию, отек легких, пульмональную эмболию, плевральную эффузию, пневмоторакс, гемоторакс, рак легких, аллергии, поллиноз (сенная лихорадка), чихание, вазомоторный ринит, воспаление слизистой оболочки, синусит, вызванную экзогенными раздражителями болезнь (SO2, смог, загрязнения), повышенную чувствительность дыхательных путей, непереносимость молочных продуктов, пневмонию Лаффера, пневмокониоз, связанные с коллагеном сосудистые заболевания, гранулематозную болезнь, бронхиальное воспаление, хроническую воспалительную болезнь легких, болезни резорбции костной ткани, реперфузионное повреждение (в том числе повреждение, нанесенное органам вследствие реперфузии после ишемических эпизодов, например, инфаркта миокарда, внезапных приступов), аутоиммунное повреждение (в том числе множественный склероз, синдром Гийена-Барре, миастения тяжелая псевдопаралитическая), отторжение трансплантата реципиентом, отторжение аллотрансплантата, лихорадку и боль в мышцах вследствие инфекции, СПИД-ассоциированный комплекс (ARC), плотное разрастание соединительной ткани кожи, образование рубцовой ткани, болезнь Крона, неспецифический язвенный колит и парез, спастический колит, остеопороз, церебральную малярию и бактериальный менингит, кишечную колику, раковую боль, боль в спине, фибромиалгию, послеоперационную боль; или в любом из вышеназванных патологических состояний, где состояние вызывается или обостряется бактериальной или вирусной инфекцией.
Согласно изобретению, также представляется применение соединения формулы (I) для предотвращения, лечения или ослабления ишемической боли. Термин «ишемическая боль» применяется в описании для обозначения боли, связанной со снижением поступления крови в часть тела. Сниженное поступление крови ограничивает поступление кислорода (гипоксия) и энергии в эту часть тела. Ишемия возникает вследствие плохого кровоснабжения тканей, и тем самым ишемическая боль появляется при болезни коронарной артерии, болезни периферической артерии и состояниях, которые характеризуются недостаточным кровотоком, обычно как сопровождение атеросклероза. Другие сосудистые нарушения также могут иметь результатом ишемическую боль. Таковые включают левожелудочковую гипертрофию, болезнь коронарной артерии, первичную артериальную гипертензию, острый гипертонический криз, кардиомиопатию, сердечную недостаточность, толерантность к физической нагрузке, хроническую сердечную недостаточность, аритмию, сердечную дисритмию, обморок, артериосклероз, хроническую сердечную недостаточность в мягкой степени, стенокардию, стенокардию Принцметала (вариант), устойчивую стенокардию и стенокардию, вызванную физическими нагрузками, реокклюзию после коронарного шунтирования, перемежающуюся хромоту (атеросклеротические облитерации), артериит, диастолическую дисфункцию и систолическую дисфункцию, атеросклероз, постишемическое/реперфузионное повреждение, диабет (оба типа, I и II), тромбоэмболию. Геморрагические осложнения также могут иметь результатом ишемическую боль. В дополнение недостаточное кровоснабжение может приводить к невропатической и воспалительной боли, возникающей из-за повреждения нервных клеток, обусловленного гипоксией (например, в операциях с остановкой сердца или при шунтировании, диабете или при респираторном дистресс-синдроме у новорожденных); или в любом из вышеназванных патологических состояний, где состояние вызывается или обостряется бактериальной или вирусной инфекцией.
Согласно изобретению, далее представляется применение соединения формулы (I) для производства лекарственного средства для предотвращения, лечения или ослабления воспаления. Согласно изобретению, далее представляется способ предотвращения, лечения или ослабления воспаления, который включает введение соединения формулы (I) субъекту, нуждающемуся в таких предотвращении, лечении или улучшении.
В частности, представляется, что соединения формулы (I) могут быть применены для предотвращения, лечения или ослабления воспаления, обусловленного или связанного с раком (таким как лейкемии, лимфомы, карциномы, рак толстой кишки, рак молочной железы, рак легкого, рак поджелудочной железы, гепатоцеллюлярная карцинома, рак почки, меланома, метастазы в печени, легком, грудной железе и простате, и т.д.); аутоиммунной болезнью (такой как отторжение трансплантатов органов, красная волчанка, отторжение трансплантата реципиентом, отторжение аллотрансплантата, множественный склероз, ревматоидный артрит, сахарный диабет I типа, в том числе разрушение панкреатических островков, ведущее к диабету и воспалительным диабетическим осложнениям); аутоиммунным повреждением (включающим множественный склероз, синдром Гийена-Барре, миастению тяжелую псевдопаралитическую); ожирением; сердечно-сосудистыми нарушениями, связанными с недостаточным кровоснабжением тканей и воспалением (такими как атеросклеротические бляшки, атеросклероз, удар, ишемическое/реперфузионное повреждение, перемежающаяся хромота, повреждение спинного мозга, застойная сердечная недостаточность, васкулит, геморрагический шок, спазм сосудов с последующим субарахноидальным кровоизлиянием, спазм сосудов с последующим инсультом, плеврит, перикардит, сердечно-сосудистые осложнения диабета); ишемическим/реперфузионным повреждением, ишемией и связанным с ней воспалением, рестенозом с последующими ангиопластическими и воспалительными аневризмами; эпилепсией, нейродегенерацией (в том числе болезнью Альцгеймера), мышечной усталостью или мышечным спазмом (в частности, спазмами у спортсменов), артритом (таким как ревматоидный артрит, остеоартрит, ревматоидный спондилит, подагрический артрит), фиброзом (например, легкого, кожи и печени), множественным склерозом, сепсисом, септическим шоком, энцефалитом, инфекционным артритом, реакцией Яриша-Герксгеймера, опоясывающим герпесом, токсическим шоком, церебральной малярией, болезнью Лайма, эндотоксическим шоком, грамотрицательным шоком, геморрагическим шоком, гепатитом (возникающим как вследствие повреждения ткани, так и в результате вирусной инфекции), тромбозом глубоких вен, подагрой; состояниями, связанными с затруднениями дыхания (например, забитые или закупоренные дыхательные пути, бронхостеноз, легочная вазоконстрикция, затрудненное дыхание, силикоз, патологическое разрастание легочных тканей, легочная гипертензия, легочная вазоконстрикция, бронхиальная аллергия и весенний конъюнктивит); состояниями, связанными с воспалением кожи (в том числе псориаз, экзема, язвы, контактные дерматиты); состояниями, связанными с воспалением кишок (в том числе болезнь Крона, неспецифический язвенный колит и парез, синдром раздраженной толстой кишки, воспалительная болезнь кишки); ВИЧ (в частности, ВИЧ-инфекционный процесс), церебральной малярией, бактериальным менингитом, усиленной некротическим опухолевым фактором (TNF) репликацией ВИЧ, ингибированием некротическим опухолевым фактором (TNF) активности AZT (азидотимидина) и DDI (диданозина), остеопорозом и другими болезнями резорбции костной ткани, остеоартритом, ревматоидным артритом, бесплодием вследствие эндометриоза, жаром и мышечной болью вследствие воспаления, общей атрофией, сопровождающей рак, общей атрофией, сопровождающей инфекцию или злокачественное развитие, общей атрофией, сопровождающей приобретенный синдром иммунодефицита (AIDS, СПИД), СПИД-ассоциированным комплексом (ARC), образованием плотных разрастаний соединительной ткани кожи, образованием рубцовой ткани, отрицательными эффектами лечения амфотерицином В, отрицательными эффектами лечения интерлейкином-2, отрицательными эффектами лечения ОКТ3 (муромомаб-CD3) или отрицательными эффектами лечения GM-CSF (гранулоцитарно-макрофагальный колониестимулирующий фактор), и прочими состояниями, обусловленными избыточной активностью антивоспалительных клеток (включающих нейтрофил, эозинофил, макрофаг и Т-клетку); или в любом из вышеназванных патологических состояний, где состояние вызывается или обостряется бактериальной или вирусной инфекцией.
Как известно, продолжительное субклиническое воспаление связано с ожирением (в присутствии или в отсутствие резистентности к инсулину и диабетов II типа) (Browning et al. (2004), Metabolism, 53, 899-903, Повышенное содержание маркеров воспаления в крови тучных женщин; Mangge et al. (2004), Exp. Clin. Endocrinol. Diabetes, 112, 378-382, Пубертатное ожирение коррелирует с С-реактивным белком как сывороточным воспалительным маркером; Maachi et al., Int. J. Obes. Relat. Metab. Disord. 2004, 28, 993-997, Системное субклиническое воспаление у тучных людей). Возможная причина этого состоит в том, что жировые клетки секретируют фактор некроза опухолей-альфа (TNF-alpha) и интерлейкины 1 и 6, которые являются провоспалительными.
Соединения согласно изобретению, которые представляют собой селективные агонисты аденозиновых А2А-рецепторов, являются в особенности предпочтительными, поскольку предполагается, что такие соединения будут иметь высокую противовоспалительную активность. Под селективными агонистами аденозиновых А2А-рецепторов подразумеваются агонисты, которые активируют аденозиновые А2А-рецепторы в концентрациях, которые являются более низкими (предпочтительно в степени от одной тысячной до одной пятой), чем требуется для активации аденозиновых А1-рецепторов. Далее, А1-рецепторы имеют провоспалительную активность, так что предполагается, что такие эффекты минимизированы для соединений, которые являются селективными для А2А-рецепторов.
Соединения формулы (I) предполагаются гораздо более эффективными при малых дозах, чем прочие агонисты аденозиновых рецепторов. Таким образом, ожидается, что соединения согласно изобретению могут быть эффективно введены в дозах, при которых они обеспечивают пониженную вероятность и серьезность побочных действий или при которых побочные эффекты не наблюдаются. Такие соединения предоставляют значительные преимущества перед другими агонистами аденозиновых рецепторов, которые проявляют противовоспалительное действие только при таких же концентрациях, при которых наблюдаются серьезные побочные действия.
Также предполагается, что соединения формулы (I) могут быть эффективными в качестве антиревматических лекарственных препаратов, влияющих на течение заболевания (DMARD, базисные препараты при ревматоидном артрите), в особенности для применения в предотвращении, лечении или ослаблении ревматоидного артрита, и, возможно, прочих артропатий, таких как остеоартрит.
Лекарственные средства, применяемые для лечения ревматоидного артрита (RA), могут быть подразделены на две группы: средства, которые помогают ослабить симптомы ревматоидного артрита (RA); и средства, которые помогают модифицировать течение болезни. Лекарственные средства, которые помогают ослабить симптомы ревматоидного артрита (RA), включают нестероидные противовоспалительные лекарственные средства (NSAID), которые снимают боль и ослабляют воспаление в пораженных суставах, анальгетики (такие как ацетаминофен и наркотические болеутоляющие средства), которые ослабляют боль, но не замедляют повреждения суставов или не сокращают воспаление, и кортикостероиды, которые представляют собой противовоспалительные лекарственные средства.
Базисные препараты при ревматоидном артрите (DMARD) помогают ослабить симптомы ревматоидного артрита (RA) (такие как разбухание и болезненность суставов), но также замедляют прогрессирование повреждения суставов, обусловленное ревматоидным артритом (RA). Таким образом, хотя излечение ревматоидного артрита (RA) и не происходит, базисные препараты при ревматоидном артрите (DMARD) помогают замедлить развитие ревматоидного артрита (RA). В прошлом базисные препараты при ревматоидном артрите (DMARD) обычно использовались для лечения ревматоидного артрита (RA) после неудачной терапии нестероидными противовоспалительными лекарственными препаратами (NSAID). Однако теперь базисные препараты при ревматоидном артрите (DMARD) начинают применять раньше в курсе лечения ревматоидного артрита (RA), поскольку исследованиями было показано, что раннее вмешательство с применением базисных препаратов при ревматоидном артрите (DMARD) предоставляет важные преимущества. Базисные препараты при ревматоидном артрите (DMARD) и нестероидные противовоспалительные лекарственные препараты (NSAID) часто применяются в комбинации друг с другом.
Результаты клинических исследований показали, что известные базисные препараты при ревматоидном артрите (DMARD) замедляют развитие ревматоидного артрита (RA). Уже после 6-месячной терапии скорость повреждения кости и хряща в суставах пациентов начинала замедляться. Через 1 год пациенты обнаруживали незначительное развитие повреждения сустава, и через 2 года рентгенография показала, что у немногих наблюдаемых пациентов проявились вновь поврежденные суставы в течение второго года лечения.
Примеры известных базисных препаратов при ревматоидном артрите (DMARD) включают сульфазалазин, пеницилламин, хлороквин, гидроксихлороквин, золото (внутримышечным введением или орально в составе ауранофина), метотрексат, циклоспорин, азатиоприн, циклофосфамид, лефлуномид. Впоследствии были разработаны биологические базисные препараты при ревматоидном артрите (DMARD), которые ингибируют фактор некроза опухоли-альфа (TNF-alpha). Один пример представляет Humira®, который показан для снижения признаков и симптомов и подавления развития структурных повреждений у взрослых с активностью ревматоидного артрита (RA) от умеренной до серьезной, которые проявляли неадекватную реакцию на один или более базисных препаратов при ревматоидном артрите (DMARD). Препарат Humira® представляет собой антитело к фактору некроза опухоли-альфа (TNF-alpha, ФНО-а).
Многие из известных базисных препаратов при ревматоидном артрите (DMARD) проявляют серьезные побочные эффекты. Таким образом, желательно создание новых базисных препаратов при ревматоидном артрите (DMARD), которые могут быть введены с минимальными побочными эффектами.
WO 2005/084653 показывает способность спонгозина сокращать индуцируемое форболовым эфиром выделение фактора некроза опухоли-альфа (TNF-alpha) в человеческих макрофаговых клетках U937. На этой основе предполагается, что спонгозин и родственные соединения согласно изобретению также имеют активность базисных препаратов при ревматоидном артрите (DMARD).
Согласно изобретению, представляется применение соединения формулы (I) в производстве лекарственного средства для замедления развития артропатии. Также согласно изобретению представляется способ замедления развития артропатии, который включает введение соединения формулы (I) субъекту, нуждающемуся в этом.
Предпочтительно замедляется развитие ревматоидного артрита (RA), и в особенности прогрессирование повреждения суставов, обусловленное ревматоидным артритом (RA). Соединение согласно изобретению может быть введено субъекту на любой стадии в курсе терапии ревматоидного артрита (RA). Соединение согласно изобретению может быть введено в комбинации с одним или более нестероидными противовоспалительными лекарственными препаратами (NSAID) или прочими базисными препаратами при ревматоидном артрите (DMARD).
Соединения согласно изобретению предполагаются эффективными как базисные препараты при ревматоидном артрите (DMARD), даже когда вводятся в дозах, которые ожидаются как дающие концентрации в крови гораздо ниже концентраций, известных для активации аденозиновых рецепторов. Предполагается, что при таких дозах соединения не проявляют серьезных побочных эффектов, связанных с введением более высоких доз спонгозина или прочих агонистов аденозиновых рецепторов.
Особенное преимущество применения соединений согласно изобретению в качестве базисных препаратов при ревматоидном артрите (DMARD) состоит в том, что, как предполагается, они будут активными при оральном введении, в отличие от антител к фактору некроза опухоли-альфа (TNF-alpha), которые должны вводиться путем инъекции.
Также было принято во внимание, что соединения формулы (I) могут быть эффективными в предотвращении, лечении или ослаблении сосудистых макро- и микроосложнений диабета 1 и 2 типов (в том числе ретинопатии, нефропатии, автономной невропатии), или повреждения кровеносных сосудов, вызванного ишемией (как диабетической, так и прочими) или атеросклерозом (как диабетическим, так и прочими).
Согласно изобретению, представляется применение соединения формулы (I) в производстве лекарственного средства для предотвращения, лечения или ослабления сосудистых макро- и микроосложнений диабета 1 и 2 типов, ретинопатии, нефропатии, автономной невропатии, или повреждения кровеносных сосудов, вызванного ишемией или атеросклерозом. Согласно изобретению, также представляется способ предотвращения, лечения или ослабления сосудистых макро- и микроосложнений диабета 1 и 2 типов, ретинопатии, нефропатии, автономной невропатии, или повреждения кровеносных сосудов, вызванного ишемией или атеросклерозом, у субъекта, нуждающегося в таких предотвращении, лечении или ослаблении, который включает введение субъекту соединения формулы (I).
Как предполагается, соединения формулы (I) являются эффективными в предотвращении, лечении или ослаблении сосудистых макро- и микроосложнений диабета 1 и 2 типов, включая ретинопатию, нефропатию, автономную невропатию, или повреждения кровеносных сосудов, вызванного ишемией или атеросклерозом (как диабетическими, так и прочими), даже когда вводятся в дозах, которые, как предполагается, дают концентрации в плазме гораздо ниже концентраций, известных для активации аденозиновых рецепторов. Предполагается, что при таких дозах соединения не проявляют серьезных побочных эффектов, связанных с введением более высоких доз спонгозина или прочих агонистов аденозиновых рецепторов.
Также предполагается, что соединения формулы (I) являются эффективными в стимуляции заживления ран. Согласно изобретению, представляется применение соединения формулы (I) в производстве лекарственного средства для стимуляции заживления ран. Также согласно изобретению представляется способ стимуляции заживления ран у субъекта, который включает введение соединения формулы (I) субъекту.
Количество соединения формулы (I), которое вводится субъекту, предпочтительно составляет количество, которое приводит к пиковой концентрации соединения в плазме, которая является более низкой, чем значение средней эффективной концентрации в 50% случаев (ЕС50) соединения для аденозиновых рецепторов (предпочтительно при величине рН 7,4).
Таким образом, предпочтительно количество соединения согласно изобретению, которое вводится субъекту, должно быть количеством, которое приводит к пиковой концентрации в плазме, причем концентрация является более низкой, чем значение ЕС50 соединения для аденозиновых рецепторов.
Предпочтительно пиковая концентрация соединения в плазме составляет от одной десятитысячной части до одной второй части (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной третьей, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) значения ЕС50.
Предпочтительно количество соединения согласно изобретению, которое вводится, приводит к концентрации в плазме, которая поддерживается в течение более чем одного часа, на уровне от одной десятитысячной части до одной второй части (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) значения ЕС50 соединения для аденозиновых рецепторов.
Во избежание неясностей значение ЕС50 соединения определяется в описании как концентрация соединения, которая вызывает отклик рецептора на половине интервала между базисным откликом рецептора и максимальным откликом рецептора (как определяется, например, с использованием кривой зависимости между дозой и откликом).
Значение ЕС50 должно определяться при стандартных условиях (сбалансированные солевые растворы, забуференные до величины рН 7,4). Измерения значения ЕС50 с использованием изолированных мембран, клеток и тканей следует проводить в забуференном солевом растворе при величине рН 7,4 (например, клеточная культуральная среда), например, согласно указаниям Daly et al., Pharmacol. (1993), 46, 91-100), или предпочтительно как Tilburg et al. (J. Med. Chem. (2002), 45, 91-100). Значение ЕС50 может быть также определено in vivo путем измерения обусловленных аденозиновыми рецепторами откликов у нормального здорового животного, или даже в ткани, перфузированной при нормальных условиях (т.е. оксигенированная кровь, или оксигенированная изотоническая среда, также забуференная до величины рН 7,4) в нормальном здоровом животном.
Будет принято во внимание, что значение ЕС50 соединения, вероятно, должно быть различным для различных аденозиновых рецепторов (т.е. аденозиновых А1-, А2А-, А2В-, А3-рецепторов). Количество соединения, которое должно быть введено, следует рассчитывать относительно наименьшего значения ЕС50 соединения для различных рецепторов.
Альтернативно, количество соединения согласно изобретению, которое вводится, может представлять собой количество, которое приводит к пиковой концентрации в плазме, которая является меньшей самого низкого значения равновесной константы диссоциации комплекса соединения с рецептором (Kd) для аденозиновых рецепторов (т.е. меньшей, чем самое низкое значение Kd соединения для аденозиновых А1-, А2А-, А2В- и А3-рецепторов). Предпочтительно пиковая концентрация соединения в плазме составляет от одной десятитысячной части до одной второй части (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной третьей, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) наинизшего значения Kd.
Предпочтительно количество соединения, которое вводится, представляет собой количество, которое приводит к концентрации в плазме, которая поддерживается в течение более чем одного часа на уровне от одной десятитысячной части до одной второй части (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной пятидесятой до одной третьей, или от одной десятой до одной второй, или от одной десятой до одной пятой) наинизшего значения Kd соединения для аденозиновых рецепторов.
Значение Kd соединения для каждого рецептора должно определяться при стандартных условиях с использованием плазменных мембран как источника аденозиновых рецепторов, взятых либо из тканей, либо клеток, эндогенно экспрессирующих эти рецепторы, или из клеток, трансфектированных ДНК-векторами, кодирующими гены аденозиновых рецепторов. Альтернативно, могут быть применены целые клеточные препараты с использованием клеток, экспрессирующих аденозиновые рецепторы. Меченые лиганды (например, с радиоактивными метками), селективные к различным рецепторам, следует использовать в забуференных (величина рН 7,4) солевых растворах (например, см. Tilburg et al., J. Med. Chem. (2002), 45, 420-429) для определения аффинности связывания и тем самым значения Kd соединения для каждого рецептора.
Альтернативно, количество соединения согласно изобретению, которое вводится, может представлять собой количество, которое составляет от одной десятитысячной части до одной второй части (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной третьей, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) минимального количества соединения, которое приводит к побочным эффектам в виде брадикардии, гипотонии или тахикардии у животных того же вида, как и субъект, которому должно быть введено соединение.
Предпочтительно вводимое количество приводит к концентрации в плазме, которая поддерживается в течение более чем одного часа на уровне от одной десятитысячной части до одной второй части (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) минимальной концентрации соединения в плазме, которая приводит к побочным эффектам.
Надлежащее дозирование соединения согласно изобретению будет варьировать в зависимости от возраста, пола, массы тела и состояния проходящего курс терапии субъекта, и активности соединения (такой как значение ЕС50 для аденозинового рецептора), его полупериода, его всасывания в теле и пути введения, и т.д. Однако надлежащее дозирование может быть легко определено квалифицированным специалистом в этой области технологии.
Подходящим методом определения необходимого дозирования является оценка кардиоваскулярных изменений (например, путем измерения электрокардиограммы или мониторингом кровяного давления) при значении ЕС50 или близко к таковому соединения для аденозинового рецептора (предпочтительно рецептора, для которого оно имеет наибольшую аффинность) для определения максимальной переносимой дозы. Тогда предполагается, что терапевтически эффективная доза должна составлять от одной десятитысячной части до одной второй части (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной третьей, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) максимальной переносимой дозы.
Нижеприведенный Пример 23 показывает, как может быть определен применимый диапазон дозирования для соединений согласно изобретению. Спонгозин представляет собой соединение, используемое в этом примере, но будет очевидно, что подобные методы могут быть употреблены для соединений согласно изобретению. Предпочтительное дозирование спонгозина было определено меньшим, чем 28 мг для людей. Это дозирование приводит к концентрации в плазме между 0,5 и 0,9 мкМ (близко к значению Kd для аденозиновых А2А-рецепторов при величине рН 7,4). Основываясь на этом результате, предпочтительный диапазон дозирования для спонгозина составляет от 0,03 до 0,3 мг/кг.
Минимальная концентрация спонгозина в плазме, дающая максимальное обезболивающее действие у крысы как адъювантной модели артрита, составляла 0,06 мкМ, значительно меньше, чем величина ЕС50 спонгозина для аденозинового А2А-рецептора, которая составляет приблизительно 1 мкМ. Предпочтительные уровни дозирования для людей дают максимальную концентрацию в плазме между 0,005 и 0,5 мкМ, которые являются значительно более низкими, чем предполагаемые для обеспечения анальгетического или противовоспалительного эффекта при действии на этот рецептор.
Альтернативно, надлежащие терапевтические концентрации соединений согласно изобретению предполагаются составляющими приблизительно 10-20-кратные значения константы ингибирования (Ki) для аденозинового рецептора (рецептора, к которому соединение имеет наивысшую аффинность) при величине рН 5,5.
Предполагается, что количество соединения согласно изобретению, которое вводится, должно составлять 0,001-15 мг/кг. Количество может быть до 10, 5, 2, 1, 0,5, 0,2, 0,1 или 0,01 мг/кг. Количество может составлять по меньшей мере 0,001, 0,01, 0,1, 0,2, 0,5, 1, 2, 5 или 10 мг/кг. Предпочтительные диапазоны составляют 0,001-10, 0,001-5, 0,001-2, 0,001-1, 0,001-0,1, 0,001-0,01, 0,01-15, 0,01-10, 0,01-5, 0,01-2, 0,01-1, 0,1-10, 0,1-5, 0,1-2, 0,1-1, 0,1-0,5, 0,1-0,4, 0,2-15, 0,2-10, 0,2-5, 0,2-2, 0,2-1,2, 0,2-1, 0,6-1,2 мг/кг.
Предпочтительные дозы для человеческого субъекта (например, субъекта с массой 70 кг) составляют менее чем 420 мг, предпочтительно менее чем 28 мг, более предпочтительно менее чем 21 мг, и предпочтительно по меньшей мере 0,07, 0,1, 0,7 или 0,8 мг, более предпочтительно по меньшей мере 3,5 или 7 мг. Более предпочтительно 7-70 мг, 14-70 мг или 3,5-21 мг.
Предполагается, что дозируемые количества, указанные выше, являются значительно более низкими (до величины, более низкой приблизительно в 1000 раз), чем следовало бы ожидать для уровня, необходимого для проявления анальгетического или противовоспалительного действия, основываясь на значении ЕС50 соединения при аденозиновом А2А-рецепторе.
В особенности предпочтительные количества дозирования приводят к концентрациям в плазме, которые составляют приблизительно от одной сотой части до одной второй части от значения ЕС50 соединения для аденозинового рецептора, для которого оно имеет наивысшую аффинность.
Соединение согласно изобретению может быть введено с другими терапевтическими средствами или без таковых, например, анальгетиками, антигипералгезическими средствами (такими как габапентин, прегабалин, каннабиноиды, модуляторы натриевых или кальциевых каналов, противоэпилептические препараты или антидепрессанты), противовоспалительными средствами (такими как опиаты, стероиды, нестероидные противовоспалительные лекарственные средства (NSAID), каннабиноиды, модуляторы тахикинина или модуляторы брадикинина), базисными препаратами при ревматоидном артрите (DMARD) или антипатогенными средствами.
В общем, соединение согласно изобретению может быть введено известными способами, в любом пригодном составе, любым подходящим путем. Соединение согласно изобретению предпочтительно вводится орально, парентерально, сублингвально, трансдермально, интратекально или трансмукозально. Прочие пригодные пути введения включают внутривенное, внутримышечное, подкожное введение, ингаляцию и местное применение. Количество вводимого лекарственного препарата типично является более высоким, когда вводится орально, чем когда вводится, например, внутривенно.
Будет принято во внимание, что соединение согласно изобретению может быть введено вместе с физиологически приемлемым носителем, эксципиентом или разбавителем.
Для поддержания терапевтически эффективных концентраций в плазме для продолжительных периодов времени соединения согласно изобретению могут быть внедрены в составы медленного выделения.
Пригодные составы, например для орального введения, включают твердые лекарственные формы с разовой дозой, и таковые, содержащие жидкость, например, для инъекции, такие как таблетки, капсулы, виалки и ампулы, в состав которых активный ингредиент введен известными способами, с физиологически приемлемым эксципиентом, разбавителем или носителем. Пригодные разбавители и носители известны и включают, например, лактозу и тальк, вместе с подходящими связующими средствами, и т.д.
Лекарственная форма соединения формулы (I) типично включает до 500 мг (например, 1-500 мг или 5-500 мг) активного агента. Предпочтительно активный агент присутствует в форме фармацевтической композиции, включающей активный агент и физиологически приемлемый носитель, эксципиент или разбавитель. Предпочтительные количества активного ингредиента в разовой дозе составляют 0,001-10, 0,001-5, 0,001-2, 0,001-1, 0,001-0,1, 0,001-0,01, 0,01-15, 0,01-10, 0,01-5, 0,01-2, 0,01-1, 0,1-10, 0,1-5, 0,1-2, 0,1-1, 0,1-0,5, 0,1-0,4, 0,2-15, 0,2-10, 0,2-5, 0,2-2, 0,2-1,2, 0,2-1, от 0,5 до 1, 0,6-1,2, типично около 0,2 или 0,6 мг активного средства на кг массы тела субъекта. Предпочтительные количества активного средства составляют менее чем 420 мг, предпочтительно менее чем 28 мг, более предпочтительно менее чем 21 мг, и предпочтительно по меньшей мере 0,07, 0,1, 0,7 или 0,8 мг, более предпочтительно по меньшей мере 3,5 или 7 мг. Более предпочтительно от 7 до 70 мг, или от 14 до 70 мг, от 3,5 до 21 мг, 0,07-0,7 мг или 0,7-7 мг. При таких уровнях предполагается, что эффективное лечение может быть достигнуто, по существу, без сопутствующего понижения (например, не более чем на 10%) кровяного давления и/или повышения компенсаторной частоты сердцебиения.
Лекарственная форма соединения согласно изобретению может далее включать одно или более других терапевтических средств, например анальгетиков, антигипералгезических средств, противовоспалительных средств, базисных препаратов при ревматоидном артрите (DMARD) или антипатогенных средств.
Предпочтительно соединение согласно изобретению вводится один раз в день, хотя соединение может быть введено с частотой два или три раза в день, если желательно.
Соединения согласно изобретению также могут служить в качестве основы для идентификации более эффективных лекарственных препаратов или лекарственных средств, которые имеют еще более уменьшенные побочные действия.
Примерами фармацевтически приемлемых солей являются органические соли, полученные присоединением кислот, которые формируют физиологически приемлемый анион, например, тозилат, метансульфонат, малат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, α-кетоглутарат и α-глицерофосфат. Также могут быть получены пригодные неорганические соли, включая такие соли, как гидрохлориды, сульфаты, нитраты, бикарбонаты и карбонаты.
Фармацевтически приемлемые соли могут быть получены с использованием стандартных методик, хорошо известных в технологии, например реакцией достаточно оснόвного соединения, такого как амин, с подходящей кислотой, с образованием физиологически приемлемого аниона. Могут быть также приготовлены соли щелочного металла (например, натрия, калия или лития) или щелочноземельного металла (например, кальция) и карбоновых кислот.
Перечисление списка химических групп в любом определении переменного признака описания включает определения этого переменного признака как любой одиночной группы или комбинации перечисленных групп. Представление варианта осуществления для переменного признака описания включает этот вариант осуществления как любой отдельный вариант осуществления или в комбинации с любыми другими вариантами осуществления или частями таковых.
Согласно изобретению также представляются способы синтеза соединений под номерами 1-21, как определено ниже в Примере 1.
Согласно изобретению, представляется способ получения соединения любой из формул 1-7, как определено в Примере 1, который включает реагирование соединения нижеследующей общей формулы (А) с основанием и (i) CF3CF2CH2OH; (ii) 3-(4-(трифторметил)фенил)фенолом; (iii) 3,4-дихлорфенолом; (iv) 3-трифторметил-4-фторфенолом; (v) 3-трифторметил-4-хлорфенолом; (vi) 3-хлор-4-цианофенолом; или (vii) 3,5-бис(трифторметил)фенолом; и снятие защитных групп с реакционного продукта с образованием соединения формулы 1-7:
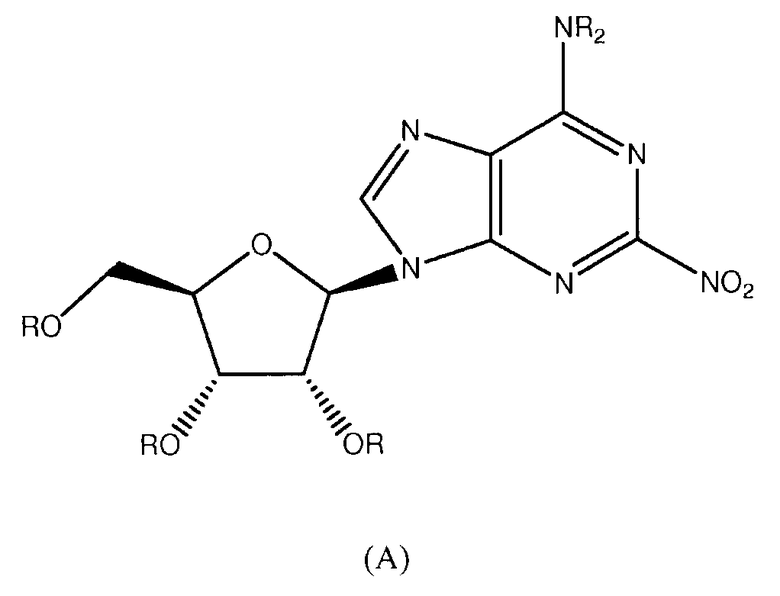
в которой R представляет собой защитную группу.
Предпочтительный растворитель представляет собой неспиртовой растворитель. Пригодными примерами являются тетрагидрофуран (THF, ТГФ), диметилформамид (DMF, ДМФА) и ацетонитрил. Предпочтительным является тетрагидрофуран (THF).
Пригодными основаниями являются гидрид натрия, трет-бутилат калия, бутиллитий и гексаметилдисилазид лития (LHMDS). Гидрид натрия является предпочтительным основанием для синтеза соединения 1, и трет-бутилат калия является предпочтительным основанием для синтеза соединений 2-7.
Предпочтительными защитными группами являются ацильные группы, производные от карбоновых кислот, которые могут быть алифатическими, ароматическими, гетероциклическими, насыщенными или ненасыщенными. Примерами пригодных защитных групп являются ацетильная, пропионильная, капроильная, пальмитоильная, бензоильная, толуоильная, фуроильная, сульфонильная, изопропилиденовая, алкоксиалкилиденовая группы. Наиболее предпочтительно защитные группы представляют собой ацетильную или бензоильную группы. Альтернативно, может быть использована защитная группа на основе кремния, например, трет-бутилдиметилсилильная (TBDMS) группа.
Снятие защитных групп с реакционного продукта может быть проведено с использованием стандартных способов, например, с использованием метилата натрия в метаноле, или нагреванием с раствором аммиака. Там, где в качестве защитных групп используются изопропилиденовый или алкоксиалкилиденовый фрагменты, для удаления защитных групп потребуется кислота, такая как трифторуксусная кислота (TFA). Защитные группы на основе кремния могут быть удалены с помощью фторида тетрабутиламмония (TBAF) или кислоты, такой как трифторуксусная кислота (TFA).
Согласно изобретению также представляется способ получения соединения формулы 8, как определено в Примере 1, который включает взаимодействие 2-хлораденозина с 4-(3,4-дихлорфенил)пиперазином с образованием соединения формулы 8, или взаимодействие соединения нижеследующей общей формулы с 4-(3,4-дихлорфенил)пиперазином, и снятие защитных групп с реакционного продукта с образованием соединения формулы 8:
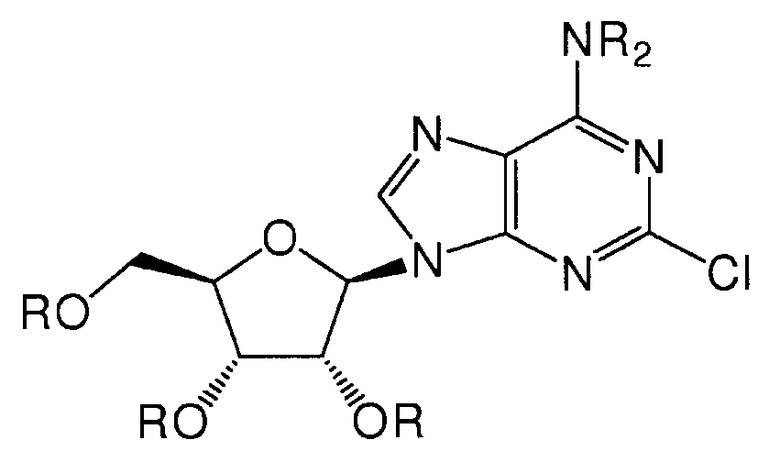
в которой R представляет собой защитную группу, как определено выше.
Предпочтительным растворителем является органический растворитель, такой как диметилформамид (DMF), метанол или этанол. Если желательно, в качестве растворителя может служить 4-(3,4-дихлорфенил)пиперазин. Альтернативно, в качестве растворителя может быть использована вода.
Предпочтительно реакционную смесь нагревают при температуре 100-200°С.
Снятие защитных групп может быть проведено, как описано выше.
Также согласно изобретению представляется способ получения соединения любой из формул 9-13, как определено в Примере 1, который включает взаимодействие 2-йодаденозина с: (i) 3,4-дихлорфенилбороновой кислотой; (ii) 3,5-дифторфенилбороновой кислотой; (iii) 3,5-бис(трифторметил)фенилбороновой кислотой; (iv) 3,4,5-трифторфенилбороновой кислотой; или (v) 2-бензофуранилбороновой кислотой, для получения соединения формулы 9-13, или взаимодействие соединения нижеследующей общей формулы (В) с (i) 3,4-дихлорфенилбороновой кислотой; (ii) 3,5-дифторфенилбороновой кислотой; (iii) 3,5-бис(трифторметил)фенилбороновой кислотой; (iv) 3,4,5-трифторфенилбороновой кислотой; или (v) 2-бензофуранилбороновой кислотой, и снятие защитных групп с реакционного продукта с образованием соединения формулы 9-13:
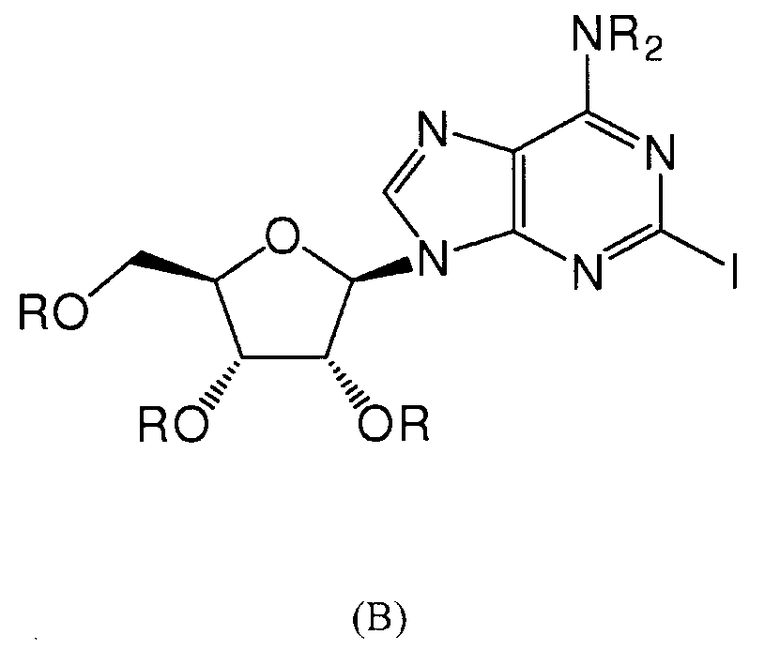
в которой R представляет собой защитную группу, как определено выше.
Предпочтительно реакционную смесь нагревают при температуре 100-200°С.
Предпочтительным растворителем является органический растворитель, предпочтительно спирт, толуол, этилацетат или диметилформамид (DMF) (или комбинация таковых).
Предпочтительно реакцию проводят в присутствии основания, такого как карбонат цезия, карбонат калия, гидроксид калия или карбонат натрия.
Предпочтительно применяется палладиевый катализатор, такой как Pd(PPh3)4, Pd2(dba)2, Pd2Cl2(PPh3)2 или Pd(OAc)2.
Снятие защитных групп может быть проведено, как описано выше.
Также согласно изобретению представляется способ получения соединения формулы 14, как определено в Примере 1, который включает снятие защитных групп и метоксилирование соединения нижеследующей общей формулы (С) с образованием соединения формулы 14:
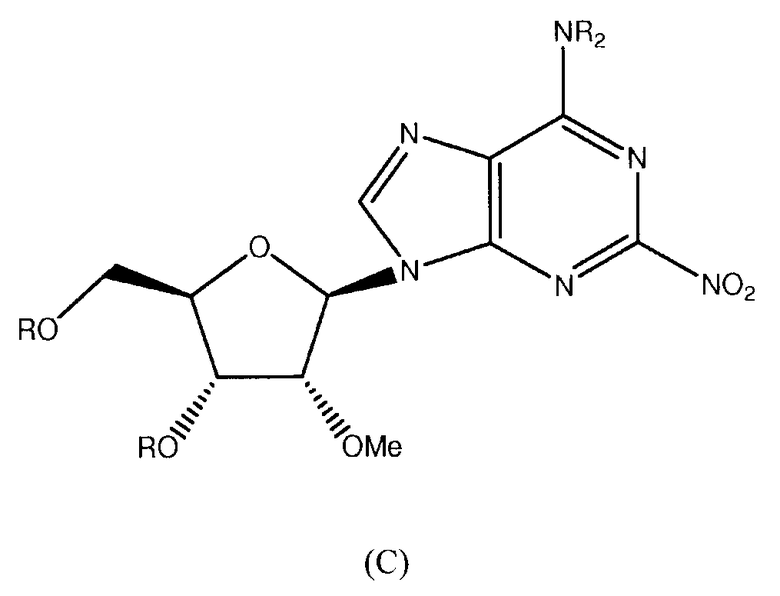
в которой R представляет собой защитную группу.
Предпочтительными защитными группами являются ацильные группы, производные от карбоновых кислот, которые могут быть алифатическими, ароматическими, гетероциклическими, насыщенными или ненасыщенными. Примерами пригодных защитных групп являются ацетильная, пропионильная, капроильная, пальмитоильная, бензоильная, толуоильная, фуроильная или сульфонильная группы. Наиболее предпочтительно защитные группы представляют собой ацетильную или бензоильную группы. Альтернативно, может быть использована защитная группа на основе кремния.
Снятие защитных групп может быть проведено, как описано выше.
Метоксилирование может быть проведено с использованием стандартных способов, например, с помощью метилата натрия в метаноле. Альтернативно, может быть использован метилат калия.
Предпочтительным растворителем является метанол.
Будет принято во внимание, что метоксилирование и снятие защитных групп могут происходить, по существу, в одно и то же время. Альтернативно, метоксилирование могло бы иметь место перед снятием защитных групп.
Также согласно изобретению представляется способ получения соединения любой из формул 15-18, как определено в Примере 1, который включает взаимодействие соединения общей формулы (С) с основанием и (i) CHF2CH2OH; (ii) циклопентанметанолом; (iii) 2,5-дифторфенолом; или (iv) (S)-втор-бутиламином; и снятие защитных групп с реакционного продукта с образованием соединения формулы 15-18.
Предпочтительными защитными группами являются такие, как описанные выше для способов получения соединения формулы 14.
Предпочтительным растворителем является тетрагидрофуран (THF), хотя там, где (S)-втор-бутиламин представляет собой реактант, он может быть использован в качестве растворителя.
Гидрид натрия является предпочтительным основанием для синтеза соединений 15-16, и трет-бутилат калия является предпочтительным основанием для синтеза соединения 17.
Снятие защитных групп может быть проведено, как описано выше.
Также согласно изобретению представляется способ получения соединения формулы 19 или 20, как определено в Примере 1, который включает реагирование соединения нижеследующей общей формулы (D) с (i) н-гексиламином или (ii) циклопентиламином; и снятие защитных групп с реакционного продукта с образованием соединения формулы 19 или 20:
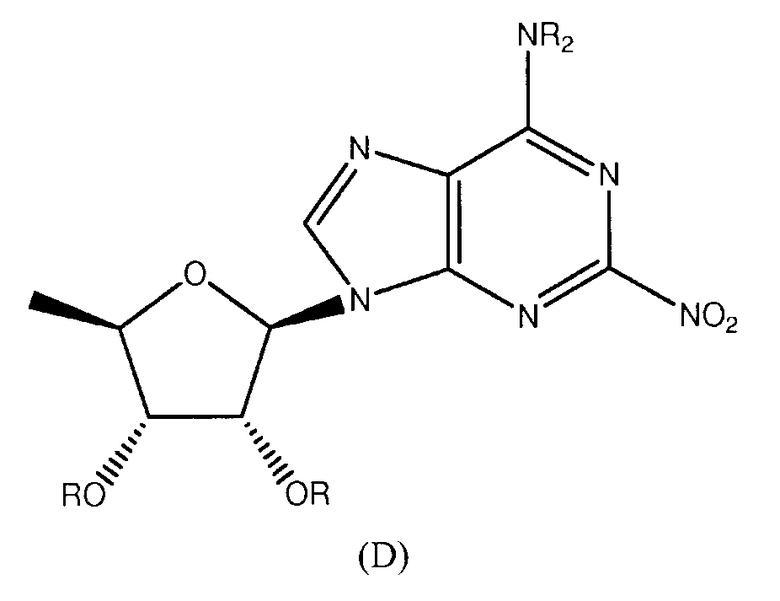
в которой R представляет собой защитную группу.
Предпочтительными защитными группами являются такие, как описанные выше для формулы (А).
Предпочтительным растворителем является тетрагидрофуран (THF), хотя, если желательно, н-гексиламин или циклопентиламин могут действовать как растворитель.
Снятие защитных групп может быть проведено, как описано выше.
Далее согласно изобретению представляется способ получения соединения формулы 21, как определено в Примере 1, который включает взаимодействие соединения нижеследующей общей формулы (Е) с циклопентиламином и снятие защитных групп с продукта реакции с образованием соединения формулы 21:
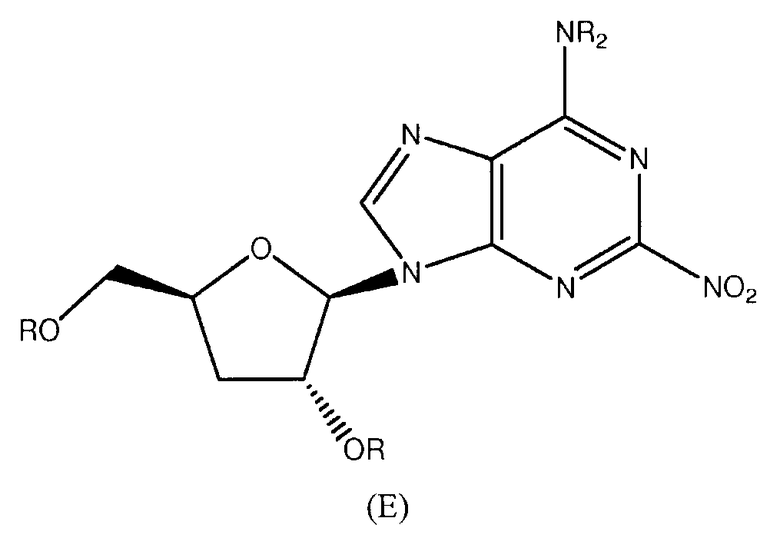
в которой R представляет собой защитную группу.
Предпочтительными защитными группами являются такие, как описанные выше для формулы (С).
Предпочтительным растворителем является тетрагидрофуран (THF), хотя, если желательно, циклопентиламин может действовать как растворитель.
Снятие защитных групп может быть проведено, как описано выше.
Предпочтительно, способ согласно изобретению для получения соединений формул 1-21 включают стадии, показанные в Схемах 1-21 (находящихся в нижеприведенных Примерах 2-22), соответственно. В особенности предпочтительные способы получения соединений формул 1-21 таковы, как описанные ниже в Примерах 2-22.
Химические структуры соединений согласно изобретению приведены ниже в Примерах. Эксперименты по связыванию были проведены с использованием А2а-рецепторов крыс. Значения константы ингибирования Ki были определены для каждого соединения при величине рН 5,5 и рН 7,4. Для расчета таковых мембраны из стриатальной ткани крыс были инкубированы в течение 90 минут при температуре 22°С в присутствии [3H]-CGS21680 (меченного тритием 2-н-[2-карбоксиэтил]фенетиламино-5'-N-этилкарбоксиамид-аденозина) в концентрации 2 нМ, 1 Ед/мл аденозиндеаминазы и возрастающих концентраций исследуемого соединения, перед фильтрованием и счетом сцинтилляций в жидкости. Значения Ki соединений были найдены в диапазоне 0,96-220 нМ при величине рН 5,5, и в диапазоне 47-25000 нМ при величине рН 7,4. Отношение KipH 7,4/KipH 5,5 для каждого соединения варьировало в диапазоне 6-5400.
Пример 1
Когда X=Y=Z=OH
No.
Когда X=Y=OH и Z=OMe
Когда X=Н и Y=Z=ОН
Когда X=Z=OH и Y=Н
Пример 2
Получение (2R,3R,4S,5R)-2-[6-амино-2-(2,2,3,3,3-пентафторпропокси)-9Н-пурин-9-ил]-5-(гидроксиметил)тетрагидрофуран-3,4-диола 1
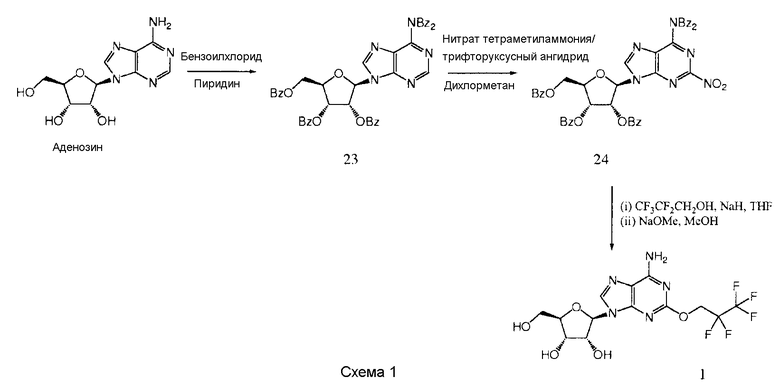
К раствору аденозина (26,7 г, 0,1 моль) в пиридине (300 мл) добавили бензоилхлорид (81,2 мл, 698 ммоль), и полученный раствор кипятили с обратным холодильником при температуре 80°С в течение 4 часов. Растворители удалили в вакууме, и остаток растворили в этилацетате (EtOAc), и раствор промыли водным раствором NaHCO3, рассолом и водой, и органическую фазу высушили над MgSO4. Кристаллизацией из смеси дихлорметан (DCM)/этанол получили продукт 23 в виде белого кристаллического твердого вещества из двух загрузок (54 г и 10 г, общий выход 82%).
К раствору нитрата тетраметиламмония (TMAN) (2,61 г, 19,2 ммоль) в дихлорметане (DCM) (37 мл) добавили трифторуксусный ангидрид (TFAA) (2,67 мл, 19,2 ммоль) и полученный раствор перемешивали в течение 1 часа. Смесь охладили до температуры 0°С и добавили раствор продукта 23 (10,1 г, 12,7 ммоль) в дихлорметане (DCM) (37 мл). Полученный раствор оставили нагреваться до комнатной температуры в течение 4 часов. Затем раствор промыли водным раствором NaHCO3, рассолом и водой (трижды), и органическую фазу высушили над MgSO4. Кристаллизацией из смеси дихлорметан (DCM)/этанол получили продукт 24 в виде светло-желтого твердого вещества (7,2 г, выход 68%).
К раствору CF3CF2CH2OH (58 мкл, 0,58 ммоль) в тетрагидрофуране (THF) (10 мл) добавили NaH (23 мг, 60%-ная дисперсия в минеральном масле, 0,58 ммоль) и полученную суспензию перемешивали в течение 1 часа. Затем добавили продукт 24 (400 мг, 0,48 ммоль) и полученный раствор перемешивали в течение 4 суток. Затем добавили дополнительную порцию 0,5 эквивалента CF3CF2CH2ONa в тетрагидрофуране (THF) (5 мл), приготовленную, как описано выше, и перемешивание продолжали в течение 16 часов. Затем удалили растворители в вакууме, и остаток растворили в метаноле перед добавлением NaOMe (каталитическое количество) и перемешиванием полученной суспензии в течение 16 часов. Удалили растворители в вакууме, и остаток очищали с помощью обратнофазной колоночной хроматографии (колонка LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 0-100% метанола в воде в течение 45 минут, продукт элюировали в 60%-ном метаноле) и обратнофазной препаративной высокоэффективной жидкостной хроматографии (HPLC) (прибор Phenomenex Synergi, колонка RP-Hydro 150×10 мм, носитель 10 мкм, поток 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 25 минут, продукт элюировали в 36%-ном ацетонитриле) с образованием (2R,3R,4S,5R)-2-[6-амино-2-(2,2,3,3,3-пентафторпропокси)-9Н-пурин-9-ил]-5-(гидроксиметил)тетрагидрофуран-3,4-диола 1 в виде белого твердого вещества (26 мг, выход 13%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,13 мин, 99,01%.
LCMS (жидкостная хроматография-масс-спектрометрия) (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,90 мин, 100%, ES+ (ионизация: электроспрей): 416,392 [МН]+.
Пример 3
Получение (2R,3R,4S,5R)-2-(6-амино-2-{[4'-(трифторметил)бифенил-
3-ил]окси}-9Н-пурин-9-ил)-5-(гидроксиметил)тетрагидрофуран-3,4-диола 2
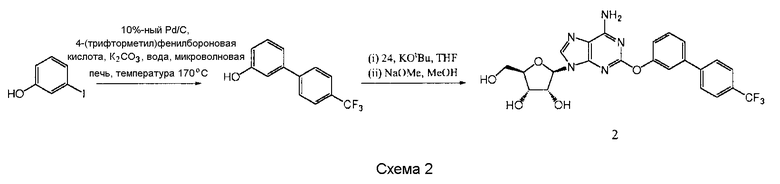
К смеси 10%-ного Pd/C (каталитическое количество), 3-иодфенола (220 мг, 1,00 ммоль) и 4-(трифторметил)фенилбороновой кислоты (284 мг, 1,49 ммоль) добавили раствор К2СО3 (415 мг, 3,01 ммоль) в воде (10 мл), и реакционную смесь нагревали в микроволновой печи “Biotage” (температура 170°С, высокое поглощение, предварительное перемешивание в течение 10 с) в течение 20 минут. Необработанную реакционную смесь затем экстрагировали этилацетатом (EtOAc) (40 мл ×3) и высушили над MgSO4 с образованием 3-(4-(трифторметил)фенил)фенола в виде желтого твердого вещества (212 мг, выход 89%, 99%-ная чистота после HPLC), который использовали без дополнительной очистки.
К раствору 3-(4-(трифторметил)фенил)фенола (212 мг, 0,89 ммоль) в тетрагидрофуране (THF) добавили трет-бутилат калия (t-BuOK) (100 мг, 0,89 ммоль) и полученную суспензию перемешивали в течение 30 минут, после чего добавили раствор продукта 24 (400 мг, 0,48 ммоль) в тетрагидрофуране (THF). Перемешивание продолжали в течение 4 суток и затем удалили растворители в вакууме. Остаток растворили в метаноле (10 мл), добавили NaOMe (каталитическое количество), и полученную смесь перемешивали в течение 16 часов. Удалили растворители в вакууме, и остаток очищали с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, зернистость 18-32 мкм, градиент 5-15% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 55%-ном ацетонитриле) с образованием (2R,3R,4S,5R)-2-(6-амино-2-{[4'-(трифторметил)бифенил-3-ил]окси}-9Н-пурин-9-ил)-5-(гидроксиметил)тетрагидрофуран-3,4-диола 2 в виде белого твердого вещества (51 мг, выход 21%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,37 мин, 98,87%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30ºС, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,76 мин, 100%, ES+ (ионизация: электроспрей): 504,412 [МН]+.
Пример 4
Получение (2R,3R,4S,5R)-2-[6-амино-2-(3,4-дихлорфенокси)-9Н-пурин-9-ил]-5-(гидроксиметил)тетрагидрофуран-3,4-диола 3
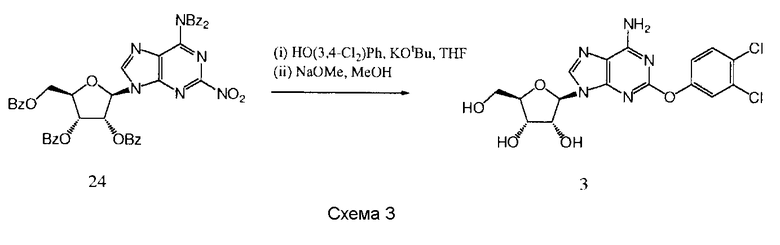
К раствору 3,4-дихлорфенола (157 мг, 0,96 ммоль) в тетрагидрофуране (THF) (3 мл) добавили трет-бутилат калия (t-BuOK) (108 мг, 0,96 ммоль), и полученную суспензию перемешивали в течение 1,5 часов, после чего добавили раствор продукта 24 (400 мг, 0,48 ммоль) в тетрагидрофуране (THF). Перемешивание продолжали в течение 2 суток и затем удалили растворители в вакууме. Остаток растворили в метаноле (15 мл), добавили NaOMe (каталитическое количество) и полученную смесь перемешивали в течение 4 суток. Удалили растворители в вакууме и остаток очищали с помощью обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 0-100% метанола в воде в течение 45 минут, продукт элюировали в 72%-ном метаноле) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 45%-ном ацетонитриле) с образованием (2R,3R,4S,5R)-2-[6-амино-2-(3,4-дихлорфенокси)-9Н-пурин-9-ил]-5-(гидроксиметил)тетрагидрофуран-3,4-диола 3 в виде белого твердого вещества (15 мг, выход 7,6%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,42 мин, 98,42%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,31 мин, 100%, ES+ (ионизация: электроспрей): 428,3 [МН]+.
Пример 5
Получение (2R,3R,4S,5R)-2-{6-амино-2-[4-фтор-3-(трифторметил)фенокси]-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 4
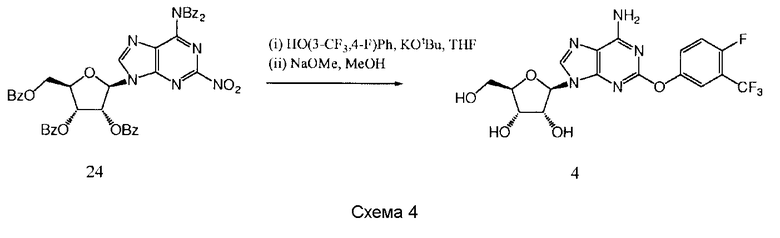
К раствору 3-трифторметил-4-фторфенола (173 мг, 0,96 ммоль) в тетрагидрофуране (THF) (3 мл) добавили трет-бутилат калия (t-BuOK) (108 мг, 0,96 ммоль) и полученную суспензию перемешивали в течение 1,5 часов, после чего добавили раствор продукта 24 (400 мг, 0,48 ммоль) в тетрагидрофуране (THF). Перемешивание продолжали в течение 20 часов и затем удалили растворители в вакууме. Остаток растворили в метаноле (15 мл), добавили NaOMe (каталитическое количество) и полученную смесь перемешивали в течение 4 суток. Удалили растворители в вакууме и остаток очищали с помощью обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 0-100% метанола в воде в течение 45 минут, продукт элюировали в 70%-ном метаноле), обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 50%-ном ацетонитриле), и перекристаллизации из смеси «этанол/гептан» с образованием (2R,3R,4S,5R)-2-{6-амино-2-[4-фтор-3-(трифторметил)фенокси]-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 44 в виде белого твердого вещества (23 мг, выход 11%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,43 мин, 98,00%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,29 мин, 100%, ES+ (ионизация: электроспрей): 446,295 [МН]+.
Пример 6
Получение (2R,3R,4S,5R)-2-{6-амино-2-[4-хлор-3-
(трифторметил)фенокси]-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 5
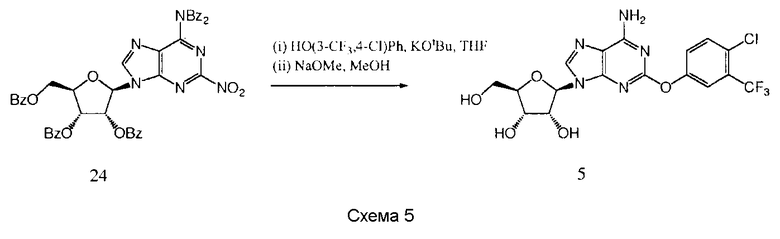
К раствору 3-трифторметил-4-хлорфенола (582 мг, 2,96 ммоль) в тетрагидрофуране (THF) (10 мл) добавили трет-бутилат калия (t-BuOK) (332 мг, 2,96 ммоль) и полученную суспензию перемешивали в течение 2 часов, после чего добавили раствор продукта 24 (1,66 г, 2,00 ммоль) в тетрагидрофуране (THF) (100 мл). Перемешивание продолжали в течение 2 суток и затем удалили растворители в вакууме. Остаток растворили в метаноле (40 мл), добавили NaOMe (170 мг, 3,15 ммоль), и полученную смесь перемешивали в течение 16 часов. Удалили растворители в вакууме и остаток очищали с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 18-32 мкм, градиент 5-20% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии) и дважды обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 460×26 мм (100 г), 30 мл в минуту, градиент 0-100% метанола в воде в течение 45 минут, продукт элюировали в 86%-ном метаноле) и (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 0-100% метанола в воде в течение 45 минут, продукт элюировали в 69%-ном метаноле), с образованием (2R,3R,4S,5R)-2-{6-амино-2-[4-хлор-3-(трифторметил)фенокси]-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 5 в виде светло-оранжевого твердого вещества (188 мг, выход 20%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,92 мин, 99,68%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,30 мин, 100%, ES+ (ионизация: электроспрей): 461,859 [МН]+.
Пример 7
Получение (2R,3R,4S,5R)-2-(6-амино-2-(3-хлор-4-цианофенокси)-9Н-пурин-9-ил)-5-(гидроксиметил)тетрагидрофуран-3,4-диола 6
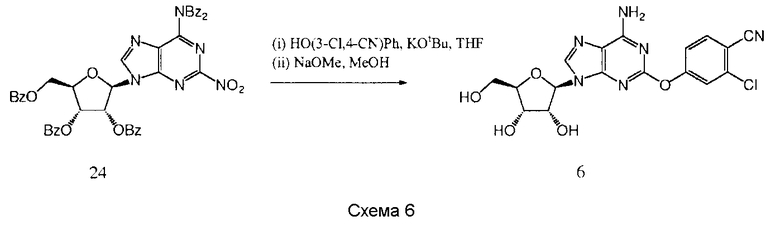
К раствору 3-хлор-4-цианофенола (147 мг, 0,96 ммоль) в тетрагидрофуране (THF) (5 мл) добавили трет-бутилат калия (t-BuOK) (108 мг, 0,96 ммоль), и полученную суспензию перемешивали в течение 30 минут, после чего добавили раствор продукта 24 (400 мг, 0,48 ммоль) в тетрагидрофуране (THF) (20 мл). Перемешивание продолжали в течение 2 суток, и затем удалили растворители в вакууме. Остаток растворили в метаноле (20 мл), добавили NaOMe (каталитическое количество), и полученную смесь перемешивали в течение 16 часов. Удалили растворители в вакууме и остаток очищали с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 18-32 мкм, градиент 5-20% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 40%-ном ацетонитриле) с образованием (2R,3R,4S,5R)-2-(6-амино-2-(3-хлор-4-цианофенокси)-9Н-пурин-9-ил)-5-(гидроксиметил)тетрагидрофуран-3,4-диола 6 в виде белого твердого вещества (11 мг, выход 5,5%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,01 мин, 99,17%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,69 мин, 100%, ES+ (ионизация: электроспрей): 419,35 [МН]+.
Пример 8
Получение (2R,3R,4S,5R)-2-(6-амино-2-[3,5-бис(трифторметил)фенокси]-9Н-пурин-9-ил)-5-(гидроксиметил)тетрагидрофуран-3,4-диола 7
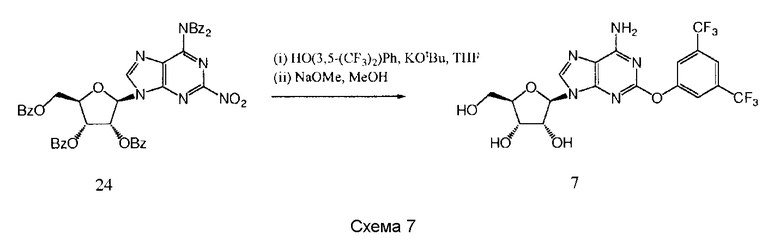
К раствору 3,5-бис(трифторметил)фенола (0,146 мл, 0,96 ммоль) в тетрагидрофуране (THF) (5 мл) добавили трет-бутилат калия (t-BuOK) (108 мг, 0,96 ммоль), и полученную суспензию перемешивали в течение 45 минут, после чего добавили раствор продукта 24 (400 мг, 0,48 ммоль) в тетрагидрофуране (THF) (20 мл). Перемешивание продолжали в течение 47 часов, и затем удалили растворители в вакууме. Остаток растворили в метаноле (15 мл), добавили NaOMe (каталитическое количество), и полученную смесь перемешивали в течение 3 суток. Удалили растворители в вакууме, и остаток очищали с помощью обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 0-100% метанола в воде в течение 45 минут, продукт элюировали в 80%-ном метаноле) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 25 минут, продукт элюировали в 55%-м ацетонитриле), с образованием (2R,3R,4S,5R)-2-(6-амино-2-[3,5-бис(трифторметил)фенокси]-9Н-пурин-9-ил)-5-(гидроксиметил)тетрагидрофуран-3,4-диола 7 в виде белого твердого вещества (28 мг, выход 12%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,99 мин, 98,54%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 6,28 мин, 100%, ES+ (ионизация: электроспрей): 496,342 [МН]+.
Пример 9
Получение (2R,3R,4S,5R)-2-(6-амино-2-(4-(3,4-дихлорфенил)пиперазин-1-ил)-9Н-пурин-9-ил)-5-(гидроксиметил)тетрагидрофуран-3,4-диола 8
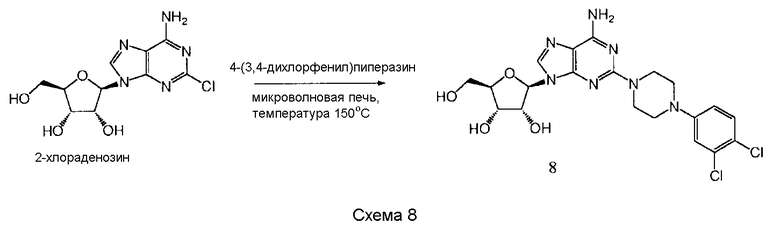
К раствору 2-хлораденозина (80 мг, 0,27 ммоль) в тетрагидрофуране (THF) (4 мл) добавили 4-(3,4-дихлорфенил)пиперазин (122 мг, 0,53 ммоль) и полученный раствор нагревали в микроволновой печи “Biotage” (температура 150°С, высокое поглощение, предварительное перемешивание в течение 30 с) в течение 30 минут. Удалили растворители в вакууме и остаток очищали дважды с помощью обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 18 минут, продукт элюировали в 80%-ном ацетонитриле) и (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 9 минут, продукт элюировали в 55%-ном ацетонитриле) с образованием (2R,3R,4S,5R)-2-(6-амино-2-(4-(3,4-дихлорфенил)пиперазин-1-ил)-9Н-пурин-9-ил)-5-(гидроксиметил)тетрагидрофуран-3,4-диола 8 в виде белого твердого вещества (7,9 мг, выход 6%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,78 мин, 99,26%.
LCMS (Phenomenex Synergi, RP-Hydro, 150 × 4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,82 мин, 100%, ES+ (ионизация: электроспрей): 496,3 [МН]+.
Пример 10
Получение (2R,3R,4S,5R)-2-{6-амино-2-(3,4-дихлорфенил)-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 9
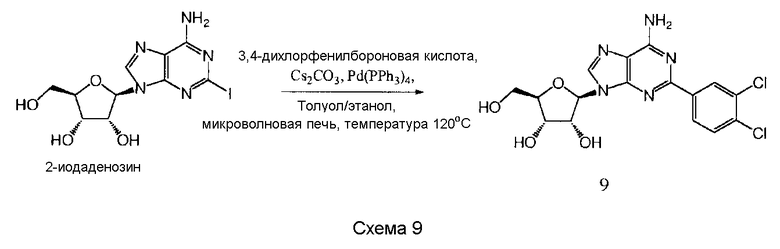
Раствор 2-йодаденозина (200 мг, 0,51 ммоль), 3,4-дихлорфенилбороновой кислоты (116 мг, 0,61 ммоль), карбоната цезия (364 мг, 1,12 ммоль) и Pd(PPh3)4 (59 мг, 0,051 ммоль) в толуоле (1,2 мл) и этаноле (2,4 мл) нагревали в микроволновой печи “Biotage” (температура 120°С, высокое поглощение, предварительное перемешивание в течение 30 с) в течение 110 минут. Затем удалили растворители в вакууме, и остаток растворили в этилацетате (EtOAc) (30 мл), и раствор промыли насыщенным водным раствором NaHCO3 (20 мл ×2) и рассолом (20 мл), и высушили над MgSO4. Очистка колоночной флэш-хроматографией (нормальная фаза, силикагель ICN, 20 г, 18-32 мкм, градиент 2,5-20% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии) и дважды с помощью обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 48%-ном ацетонитриле) и (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 50%-ном ацетонитриле) дала (2R,3R,4S,5R)-2-{6-амино-2-(3,4-дихлорфенил)-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диол 9 в виде белого твердого вещества (24,4 мг, выход 11%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,77 мин, 100%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,19 мин, 100%, ES+ (ионизация: электроспрей): 412,32 [МН]+.
Пример 11
Получение (2R,3R,4S,5R)-2-{6-амино-2-(3,5-дифторфенил)-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 10
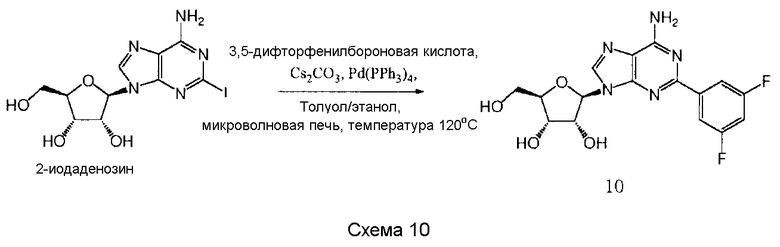
Раствор 2-йодаденозина (200 мг, 0,51 ммоль), 3,5-дифторфенилбороновой кислоты (150 мг, 0,95 ммоль), карбоната цезия (364 мг, 1,12 ммоль) и Pd(PPh3)4 (59 мг, 0,051 ммоль) в толуоле (1,2 мл) и этаноле (2,4 мл) нагревали в микроволновой печи “Biotage” (температура 120°С, высокое поглощение, предварительное перемешивание в течение 15 с) в течение 40 минут. Затем удалили растворители в вакууме, и остаток растворили в этилацетате (EtOAc) (20 мл), и раствор промыли насыщенным водным раствором NaHCO3 (20 мл ×2) и рассолом (20 мл), и высушили над MgSO4. Очистка с помощью обратнофазной препаративной HPLC в 2 загрузках (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 35%-ном ацетонитриле) и (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 40%-ном ацетонитриле), смешение самых чистых фракций из обеих колонок, концентрирование в вакууме до объема ~20 мл и отфильтровывание полученного белого осадка дали (2R,3R,4S,5R)-2-{6-амино-2-(3,5-дифторфенил)-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диол 10 в виде белого твердого вещества (16,8 мг, выход 9%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,07 мин, 99,86%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 6,02 мин, 100%, ES+ (ионизация: электроспрей): 380,4 [МН]+.
Пример 12
Получение (2R,3R,4S,5R)-2-{6-амино-2-[3,5-бис(трифторметил)фенил]-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 11
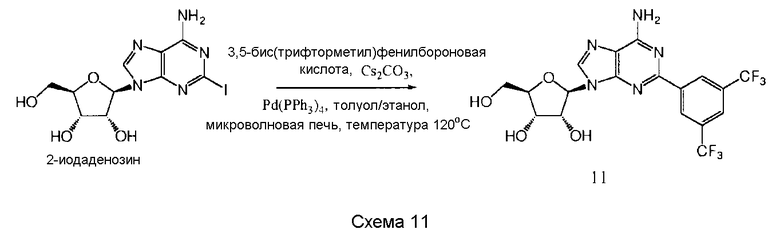
Раствор 2-йодаденозина (1,00 г, 2,53 ммоль), 3,5- бис(трифторметил)фенилбороновой кислоты (784 мг, 3,04 ммоль), карбоната цезия (1,81 г, 6,08 ммоль) и Pd(PPh3)4 (293 мг, 0,25 ммоль) в толуоле (2,4 мл) и этаноле (4,8 мл) нагревали в микроволновой печи “Biotage” (температура 120°С, высокое поглощение, предварительное перемешивание в течение 30 с) в течение 40 минут в двух загрузках, и необработанные реакционные смеси объединили. Затем удалили растворители в вакууме, и остаток растворили в этилацетате (EtOAc) (200 мл), и раствор промыли насыщенным водным раствором NaHCO3 (100 мл) и рассолом (100 мл). Очистка с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 50 г, 18-32 мкм, градиент 5-25% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии, продукт элюировали в 25%-ном этаноле) дала (2R,3R,4S,5R)-2-{6-амино-2-[3,5-бис(трифторметил)фенил]-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диол 11 в виде беловатого стекловидного вещества (364 мг, выход 30%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,46 мин, 99,86%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,89 мин, 100%, ES+ (ионизация: электроспрей): 479,886 [МН]+.
Пример 13
Получение (2R,3R,4S,5R)-2-{6-амино-2-(3,4,5-трифторфенил)-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 12
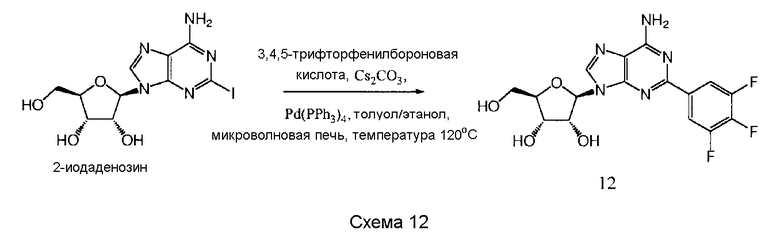
Раствор 2-йодаденозина (200 мг, 0,51 ммоль), 3,4,5- трифторфенилбороновой кислоты (107 мг, 0,609 ммоль), карбоната цезия (364 мг, 1,12 ммоль) и Pd(PPh3)4 (59 мг, 0,051 ммоль) в толуоле (1,2 мл) и этаноле (2,4 мл) нагревали в микроволновой печи “Biotage” (температура 120°С, высокое поглощение, предварительное перемешивание в течение 30 с) в течение 40 минут. Затем растворители удалили в вакууме, и остаток растворили в этилацетате (EtOAc) (30 мл), и раствор промыли насыщенным водным раствором NaHCO3 (20 мл ×2) и рассолом (20 мл), и высушили над MgSO4. Очистка с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 50 г, 18-32 мкм, градиент 2,5-15% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии, продукт элюировали в 15%-ном этаноле) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 50%-ном ацетонитриле) дала (2R,3R,4S,5R)-2-{6-амино-2-(3,4,5-трифторфенил)-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диол 12 в виде белого твердого вещества (13,2 мг, выход 7%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,49 мин, 100%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,04 мин, 100%, ES+ (ионизация: электроспрей): 398,396 [МН]+.
Пример 14
Получение (2R,3R,4S,5R)-2-{6-амино-2-(бензофуран-2-ил)-9Н-пурин-9-ил}-5-(гидроксиметил)тетрагидрофуран-3,4-диола 13
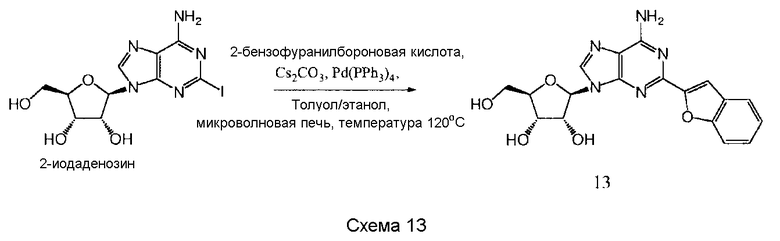
Раствор 2-йодаденозина (200 мг, 0,51 ммоль), 2-бензофуранилбороновой кислоты (99 мг, 0,61 ммоль), карбоната цезия (364 мг, 1,12 ммоль) и Pd(PPh3)4 (59 мг, 0,051 ммоль) в толуоле (1,2 мл) и этаноле (2,4 мл) нагревали в микроволновой печи “Biotage” (температура 120°С, высокое поглощение, предварительное перемешивание в течение 15 с) в течение 40 минут. Затем растворители удалили в вакууме, и остаток растворили в этилацетате (EtOAc) (15 мл), и раствор промыли насыщенным водным раствором NaHCO3 (15 мл ×2) и рассолом (15 мл), и высушили над MgSO4. Очистка с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 20 г, 18-32 мкм, градиент 0-20% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии, продукт элюировали в 15%-ном этаноле) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 35%-ном ацетонитриле) дала названное в заголовке соединение 13 в виде белого твердого вещества (4,9 мг, выход 3%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,13 мин, 100%.
LCMS (Phenomenex Synergi, RP-Hydro, 150 × 4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,61 мин, 100%, ES+ (ионизация: электроспрей): 384,469 [МН]+.
Пример 15
Получение (2R,3R,4R,5R)-5-(6-амино-2-метокси-9Н-пурин-9-ил)-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ола 14
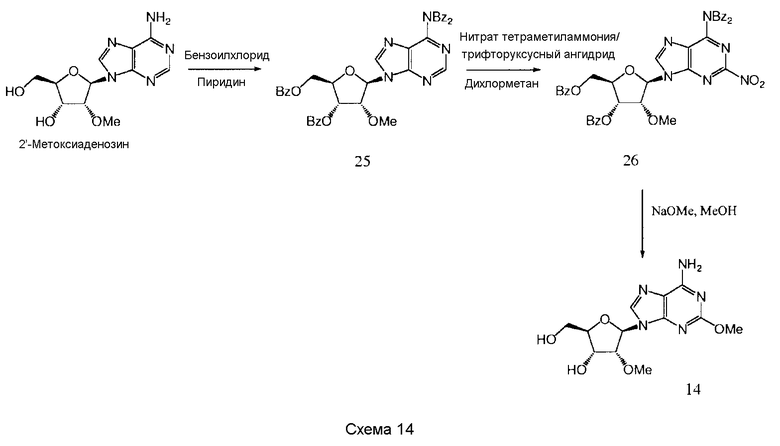
К раствору 2'-метоксиаденозина (10 г, 35,6 ммоль) в пиридине (75 мл) добавили бензоилхлорид (22,7 мл, 196 ммоль), и полученный раствор кипятили с обратным холодильником при температуре 80°С в течение 4 часов. Растворители удалили в вакууме, и остаток растворили в этилацетате (EtOAc), и раствор промыли водным раствором NaHCO3, рассолом и водой, и органическую фазу высушили над MgSO4. Перекристаллизацией из этанола получили продукт 25 в виде белого твердого вещества из двух загрузок (13,7 г и 4,3 г, общий выход 72%).
К суспензии продукта 25 (13,7 г, 19,7 ммоль) и нитрата тетраметиламмония (TMAN) (3,48 г, 25,6 ммоль) в дихлорметане (DCM) (100 мл) добавили по каплям раствор трифторуксусного ангидрида (TFAA) (3,77 мл, 26,7 ммоль) в дихлорметане (DCM) (20 мл), и полученный раствор перемешивали в течение 2 часов. Раствор затем промыли водным раствором NaHCO3 и водой (трижды), и органическую фазу высушили над MgSO4. Остаток растворили в этаноле (20 мл), добавили дихлорметан (DCM) (50 мл) и растворители удалили в вакууме с образованием продукта 26 в виде светло-желтой пены (14 г, выход 96%, ~70%-ной чистоты), который использовали без дополнительной очистки.
К раствору продукта 26 (3,7 г, 5,31 ммоль) в метаноле (70 мл) добавили NaOMe (1,15 г, 21,3 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 2 суток. Затем добавили силикагель (15 г), и растворители удалили в вакууме. Очистка с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 18-32 мкм, градиент 10% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии) и перекристаллизации из горячей воды привела к (2R,3R,4R,5R)-5-(6-амино-2-метокси-9Н-пурин-9-ил)-2-(гидроксиметил)-4-метокситетрагидрофуран-3-олу 14 в виде белого кристаллического твердого вещества (536 мг, выход 32%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 2,80-2,86 мин (расщепленный пик), 100%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 3,42 мин, 100%, ES+ (ионизация: электроспрей): 312,063 [МН]+.
Пример 16
Получение (2R,3R,4R,5R)-5-[6-амино-2-(2,2-дифторэтокси)-9Н-пурин-9-ил]-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ола 15
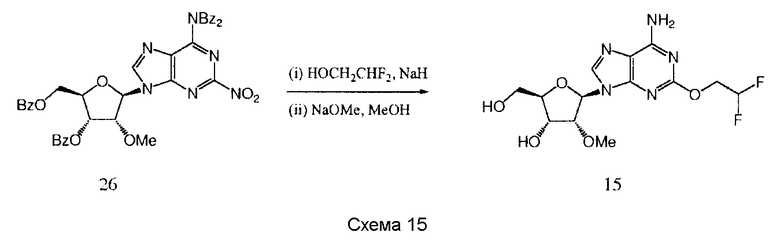
К раствору CHF2CH2OH (0,165 мл, 2,60 ммоль) в тетрагидрофуране (THF) (10 мл) добавили NaH (104 мг, 60%-ная дисперсия в минеральном масле, 2,60 ммоль), и полученную суспензию перемешивали в течение 1 часа. Затем добавили раствор продукта 26 (956 мг, 1,37 ммоль) в тетрагидрофуране (THF) (10 мл), и полученный раствор перемешивали при комнатной температуре в течение 2 суток. Затем удалили растворители в вакууме и остаток растворили в метаноле (20 мл), затем добавили NaOMe (каталитическое количество) и перемешивали полученную суспензию в течение 16 часов. Растворители удалили в вакууме, и остаток очищали с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 50 г, 18-32 мкм, градиент 2,5-15% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии, продукт элюировали в 10-15%-ном этаноле) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro, 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 25%-ном ацетонитриле) дала (2R,3R,4R,5R)-5-[6-амино-2-(2,2-дифторэтокси)-9Н-пурин-9-ил]-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ол 15 в виде белого твердого вещества (134 мг, выход 27%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 3,42 мин, 100%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 3,96 мин, 100%, ES+ (ионизация: электроспрей): 362,342 [МН]+.
Пример 17
Получение (2R,3R,4R,5R)-5-[6-амино-2-(циклопентилметокси)-9Н-пурин-9-ил]-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ола 16
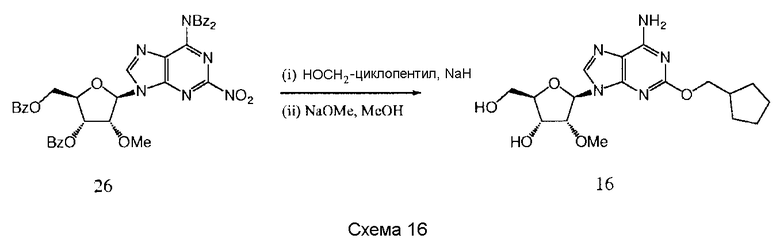
К раствору циклопентанметанола (1,73 мл, 19,1 ммоль) в тетрагидрофуране (THF) (35 мл) добавили NaH (637 мг, 60%-ная дисперсия в минеральном масле, 15,9 ммоль), и полученную суспензию перемешивали в течение 1 часа. Затем добавили раствор продукта 26 (3,7 г, 5,31 ммоль) в тетрагидрофуране (THF) (35 мл), и полученный раствор перемешивали при комнатной температуре в течение 2 суток. Затем удалили растворители в вакууме и остаток растворили в метаноле (70 мл), затем добавили NaOMe (860 мг, 15,9 ммоль) и перемешивали полученную суспензию в течение 16 часов. Затем добавили силикагель (15 г) и растворители удалили в вакууме. Очистка с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 18-32 мкм, градиент 5-10% этанола в дихлорметане (DCM), загрузка остатка в сухом состоянии) и перекристаллизации из смеси метанол/вода дала (2R,3R,4R,5R)-5-[6-амино-2-(циклопентилметокси)-9Н-пурин-9-ил]-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ол 16 в виде белого кристаллического твердого вещества (356 мг, выход 18%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,27 мин, 99,05%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,76 мин, 100%, ES+ (ионизация: электроспрей): 380,499 [МН]+.
Пример 18
Получение (2R,3R,4R,5R)-5-[6-амино-2-(2,5-дифторфенокси)-9Н-пурин-9-ил]-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ола 17
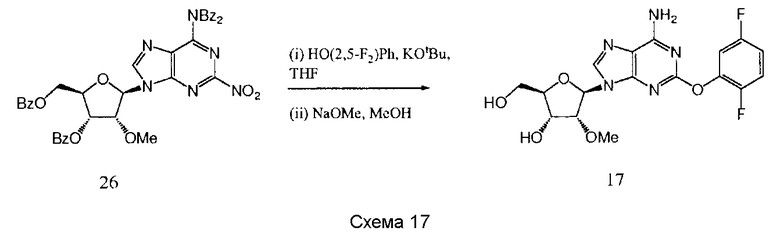
К раствору 2,5-дифторфенола (104 мг, 0,80 ммоль) в тетрагидрофуране (THF) (7 мл) добавили трет-бутилат калия (t-BuOK) (63 мг, 0,56 ммоль) и полученную суспензию перемешивали в течение 30 минут, затем добавили продукт 26 (295 мг, 0,42 ммоль). Перемешивание продолжали в течение 6 часов и затем удалили растворители в вакууме. Остаток растворили в метаноле (7 мл), добавили NaOMe (86 мг, 1,59 ммоль) и перемешивали полученную суспензию в течение 16 часов, перед тем, как загасить смесь водной лимонной кислотой. Растворители удалили в вакууме и остаток очищали с помощью обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 5-100% метанола в воде в течение 45 минут, продукт элюировали в 57%-ном метаноле) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 26%-ном ацетонитриле) с образованием (2R,3R,4R,5R)-5-[6-амино-2-(2,5-дифторфенокси)-9Н-пурин-9-ил]-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ола 17 в виде светло-желтого твердого вещества (30,9 мг, выход 18%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,20 мин, 99,00%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,91 мин, 100%, ES+ (ионизация: электроспрей): 410,484 [МН]+.
Пример 19
Получение (2R,3R,4R,5R)-5-(6-амино-2-{[(1S)-1-метилпропил]амино}-9Н-пурин-9-ил)-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ола 18
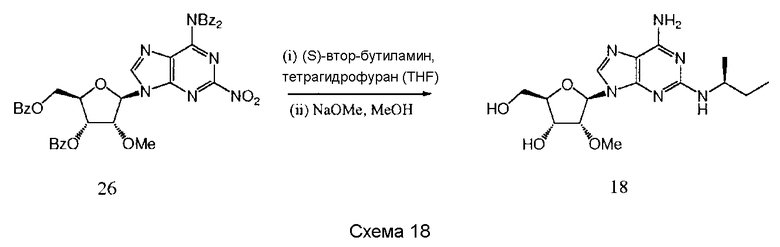
К раствору продукта 26 (445 мг, 0,64 ммоль) в тетрагидрофуране (THF) (8 мл) добавили (S)-втор-бутиламин (120 мкл, 1,18 ммоль) и еще одну порцию тетрагидрофурана (THF) (8 мл). Перемешивание продолжали в течение 1 недели и затем растворители удалили в вакууме. Остаток растворили в метаноле (8 мл), добавили NaOMe (каталитическое количество) и перемешивали полученную смесь в течение 2 суток. Добавили дополнительную порцию NaOMe (каталитическое количество) и полученную смесь перемешивали в течение 1 дня перед концентрированием в вакууме. Остаток растворили в смеси «ацетонитрил/вода» (1:1, 3 мл), и добавили смесь «тетрагидрофуран (THF)/вода» (1:1, 0,5 мл). Этот раствор очищали с помощью обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 5-100% метанола в воде в течение 30 минут) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 37%-ном ацетонитриле) в двух загрузках с образованием (2R,3R,4R,5R)-5-(6-амино-2-{[(1S)-1-метилпропил]амино}-9Н-пурин-9-ил)-2-(гидроксиметил)-4-метокситетрагидрофуран-3-ола 18 в виде белого твердого вещества (6,5 мг, выход 3%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 3,54 мин, 99,23%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,52 мин, 98,40%, ES+ (ионизация: электроспрей): 353,426 [МН]+.
Пример 20
Получение (2R,3R,4S,5R)-2-[6-амино-2-(гексиламино)-9Н-пурин-9-ил]-5-метилтетрагидрофуран-3,4-диола 19
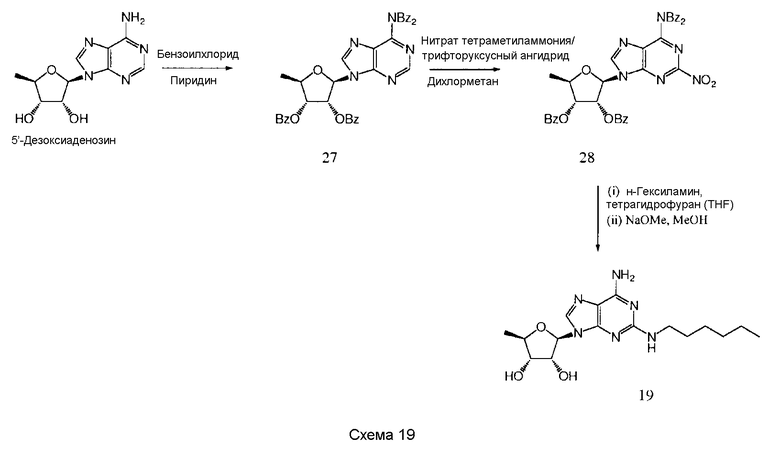
К раствору 5'-дезоксиаденозина (600 мг, 2,39 ммоль) в пиридине (15 мл) добавили бензоилхлорид (1,8 мл, 15,5 ммоль) и полученный раствор нагревали при температуре 80°С в течение 5,5 часов. После охлаждения до комнатной температуры остаток концентрировали в вакууме и растворили в этилацетате (EtOAc) (50 мл). Органическую фракцию промыли 0,2М водным раствором HCl (25 мл), водным раствором NaHCO3 (25 мл ×2) и рассолом (25 мл), и высушили над MgSO4. Очистка с помощью колоночной флэш-хроматографии (нормальная фаза, силикагель ICN, 18-32 мкм, градиент 10-67% этилацетата (EtOAc) в гептане, продукт элюировали в 67%-ном этилацетате (EtOAc)) привела к продукту 27 в виде белой пены (1,4 г, выход 88%).
К суспензии нитрата тетраметиламмония (TMAN) (0,64 г, 5,35 ммоль) в дихлорметане (DCM) (25 мл) добавили трифторуксусный ангидрид (TFAA) (0,76 мл, 5,35 ммоль), и полученный раствор перемешивали в течение 3 часов, охладили до температуры 0°С, и добавили раствор продукта 27 (2,38 г, 3,56 ммоль) в дихлорметане (DCM) (25 мл). Полученный раствор оставили нагреваться до комнатной температуры в течение 16 часов и растворители удалили в вакууме. Остаток распределили между EtOAc (100 мл) и водой (75 мл), и органическую фракцию промыли рассолом (50 мл) и высушили над MgSO4. Кристаллизацией из смеси дихлорметан (DCM)/этанол получили продукт 28 в виде светло-желтого твердого вещества в двух порциях (1,10 г и 0,95 г, общий выход 81%).
К раствору продукта 28 (300 мг, 0,42 ммоль) в тетрагидрофуране (THF) (15 мл) добавили н-гексиламин (111 мкл, 0,84 ммоль), и перемешивание продолжали в течение 2 суток. Затем растворители удалили в вакууме, и остаток растворили в метаноле (10 мл). Добавили NaOMe (50 мг, 0,93 ммоль) и полученную смесь перемешивали в течение 1 дня. Удалили растворители в вакууме и остаток очищали с помощью обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 5-100% метанола в воде в течение 30 минут, продукт элюировали в 70%-ном метаноле) и обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 14 минут, продукт элюировали в 37%-ном ацетонитриле) с образованием (2R,3R,4S,5R)-2-[6-амино-2-(гексиламино)-9Н-пурин-9-ил]-5-метилтетрагидрофуран-3,4-диола 19 в виде светло-желтого твердого вещества (13,8 мг, выход 9%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 4,46 мин, 99,24%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 6,12 мин, 100%, ES+ (ионизация: электроспрей): 351,535 [МН]+.
Пример 21
Получение (2R,3R,4S,5R)-2-[6-амино-2-(циклопентиламино)-9Н-пурин-9-ил]-5-метилтетрагидрофуран-3,4-диола 20
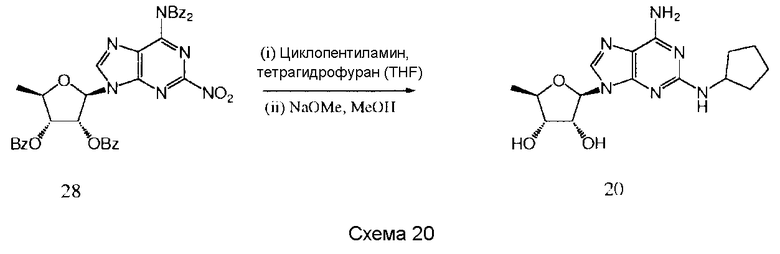
К раствору продукта 28 (600 мг, 0,84 ммоль) в тетрагидрофуране (THF) (15 мл) добавили циклопентиламин (166 мкл, 1,69 ммоль), и перемешивание продолжали в течение 2 суток. Затем удалили растворители в вакууме и остаток растворили в метаноле (10 мл). Добавили NaOMe (каталитическое количество) и полученную смесь перемешивали в течение 16 часов. Удалили растворители в вакууме и остаток очищали с помощью обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 0-100% метанола в воде в течение 45 минут, продукт элюировали в 62%-ном метаноле) и дважды обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 25 минут, продукт элюировали в 35%-ном ацетонитриле) с образованием (2R,3R,4S,5R)-2-[6-амино-2-(циклопентиламино)-9Н-пурин-9-ил]-5-метилтетрагидрофуран-3,4-диола 20 в виде светло-желтого твердого вещества (8 мг, выход 3%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 3,70 мин, 99,51%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,48 мин, 100%, ES+ (ионизация: электроспрей): 335,404 [МН]+.
Пример 22
Получение (2R,3R,5S)-2-[6-амино-2-(циклопентиламино)-9Н-пурин-9-ил]-5-(гидроксиметил)тетрагидрофуран-3-ола 21
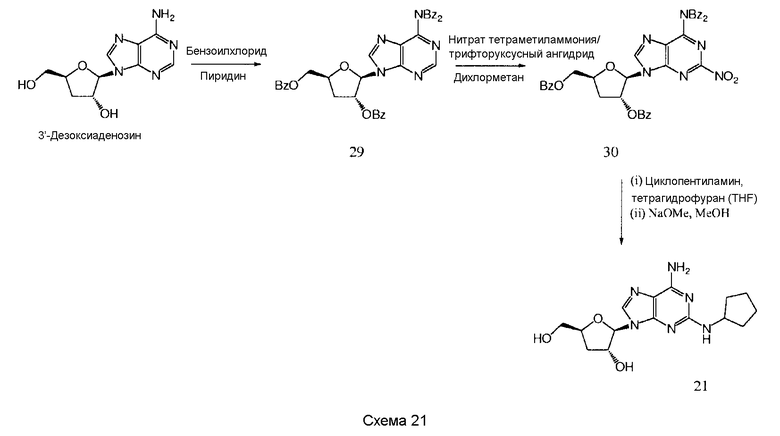
К раствору 3'-дезоксиаденозина (506 мг, 2,01 ммоль) в пиридине (15 мл) добавили бензоилхлорид (1,5 мл, 12,93 ммоль), и полученный раствор кипятили с обратным холодильником при температуре 80°С в течение 4 часов. Эту методику затем повторили в том же масштабе, и обе загрузки объединили, и растворители удалили в вакууме. Остаток растворили в этилацетате (EtOAc) (100 мл), и раствор промыли 0,2 М водным раствором HCl (50 мл), водным раствором NaHCO3 (50 мл) и рассолом (50 мл), и органическую фазу высушили над MgSO4. Кристаллизация из смеси дихлорметан (DCM)/этанол дала продукт 29 в виде белого кристаллического твердого вещества (2,38 г, выход 88%, чистота после HPLC >97%).
К суспензии продукта 29 (668 мг, 1,00 ммоль) и нитрата тетраметиламмония (TMAN) (156 мг, 1,3 ммоль) в дихлорметане (DCM) (5 мл) добавили по каплям раствор трифторуксусного ангидрида (TFAA) (170 мкл, 1,20 ммоль) в дихлорметане (DCM) (1 мл), и полученный раствор перемешивали в течение 2 часов. Затем по каплям добавили раствор трифторуксусного ангидрида (TFAA) (28,3 мкл, 0,20 ммоль) в дихлорметане (DCM) (0,5 мл) и перемешивание продолжали в течение дальнейшего 1 часа. Дополнительную порцию раствора трифторуксусного ангидрида (TFAA) (28,3 мкл, 0,20 ммоль) в дихлорметане (DCM) (0,5 мл) добавили по каплям и продолжали перемешивание в течение дальнейшего 1 часа, реакционную смесь загасили водным раствором NaHCO3 (4 мл). Органическую фазу промыли водой (30 мл ×2), объединенные водные слои промыли дихлорметаном (DCM) (20 мл) и объединенные органические экстракты высушили над Na2SO4 с образованием продукта 30 в виде желтого твердого вещества (563 мг, выход 79%, чистота ~90%), который использовали без дополнительной очистки.
К раствору продукта 30 (320 мг, 0,40 ммоль) в тетрагидрофуране (THF) (7 мл) добавили циклопентиламин (80 мкл, 0,81 ммоль) и перемешивание продолжали в течение 16 часов, затем добавили дополнительную порцию циклопентиламина (80 мкл, 0,81 ммоль). Через 16 часов растворители удалили в вакууме и остаток растворили в метаноле (7 мл). Добавили NaOMe (каталитическое количество) и полученную смесь перемешивали в течение 3 суток. Затем удалили растворители в вакууме, и остаток растворили в смеси ацетонитрил/вода (1:1, 3 мл) и добавили смесь тетрагидрофуран (THF)/вода (1:1, 0,5 мл). Этот раствор очищали с помощью обратнофазной колоночной хроматографии (LiChroprep RP-18, 40-63 мкм, 230×26 мм (50 г), 30 мл в минуту, градиент 5-100% метанола в воде в течение 30 минут, продукт элюировали в 60%-ном метаноле), дважды с использованием обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 30%-ном ацетонитриле) и снова обратнофазной препаративной HPLC (Phenomenex Synergi, RP-Hydro 150×10 мм, 10 мкм, 20 мл в минуту, градиент 5-100% ацетонитрила в воде в течение 10 минут, продукт элюировали в 30%-ном ацетонитриле) с образованием (2R,3R,5S)-2-[6-амино-2-(циклопентиламино)-9Н-пурин-9-ил]-5-(гидроксиметил)тетрагидрофуран-3-ола 21 в виде белого твердого вещества (1,6 мг, выход 1%).
HPLC (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 3,56 мин, 99,39%.
LCMS (Phenomenex Synergi, RP-Hydro, 150×4,6 мм, 4 мкм, 1,5 мл в минуту, температура 30°С, градиент 5-100% ацетонитрила (+0,085% трифторуксусной кислоты (TFA)) в воде (+0,1% трифторуксусной кислоты (TFA)) в течение 7 минут - цикл инжекции 30 с, 200-300 нм): время удерживания 5,37 мин, 97,87%, ES+ (ионизация: электроспрей): 335,404 [МН]+.
Пример 23
Концентрации спонгозина в плазме были определены после одноразовой оральной дозы пяти или шести людям-добровольцам. Тахикардию определяли с использованием 12-канальных электрокардиограмм (ECG). Минимальная эффективная анальгетическая концентрация в плазме у крыс составила 0,025 мкМ, предполагая, что минимальная эффективная доза для человека должна составлять приблизительно 0,8 мг, она имеет результатом концентрацию в плазме на уровне выше, чем 0,025 мкМ в течение приблизительно 1,5 часов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИАГНОСТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2396272C9 |
| ДИПЕПТИДНЫЕ ПРОЛЕКАРСТВА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2486183C9 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЕПТИДОВ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ К РЕЦЕПТОРАМ ИНТЕГРИНОВ | 2002 |
|
RU2303042C2 |
| ПЕПТИД ДЛЯ ИНГИБИРОВАНИЯ ФРАКТАЛКИН-СТИМУЛИРОВАННОЙ МИГРАЦИИ МОНОЦИТАРНЫХ КЛЕТОК | 2011 |
|
RU2461565C1 |
| УЛУЧШЕННЫЕ ХЕЛАТНЫЕ КОНЪЮГАТЫ | 2002 |
|
RU2298012C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ДОЛАПРОИН-ДОЛАИЗОЛЕЙИНА | 2015 |
|
RU2747989C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В ЛЕЧЕНИИ РАССТРОЙСТВ, АССОЦИИРОВАННЫХ С ВАНИЛОИДНЫМ РЕЦЕПТОРОМ TRPV1 | 2007 |
|
RU2458055C2 |
| ДИПЕПТИДНОЕ ПРОЛЕКАРСТВО, ЕГО ПРИМЕНЕНИЕ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2013 |
|
RU2629558C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HIF-ПРОЛИЛ-4-ГИДРОКСИЛАЗЫ | 2009 |
|
RU2509080C9 |
| АЦИЛИРОВАННЫЕ НОНАДЕПСИПЕПТИДЫ В КАЧЕСТВЕ ПРОИЗВОДНЫХ ЛИЗОБАКТИНА | 2005 |
|
RU2414477C2 |
Изобретение относится к соединениям формулы (I)
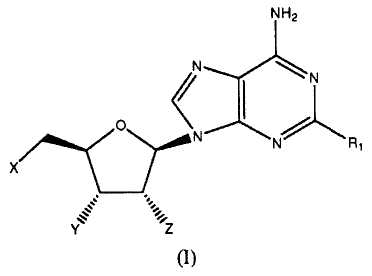
и к их фармацевтически приемлемым солям, а также к применению таких соединений в качестве лекарственных средств, в частности для лечения боли или воспаления, и к фармацевтическим композициям на основе таких соединений. В указанной формуле, когда X=Y=Z=OH, R1 представляет собой OCH2CF2CF3; феноксигруппу (замещенную 3-(4-трифторметилфенилом), 3,4-дихлором, (3-трифторметилом,4-фтором), (3-трифторметилом,4-хлором), (3-хлором,4-цианогруппой) или 3,5-бис(трифторметилом)), 1-пиперазинил-(4-(3,4-дихлорфенилом)); фенильную группу, замещенную 3,4-дихлором, 3,5-дифтором, 3,5-бис(трифторметилом) или 3,4,5-трифтором); или 2-бензофуранильный фрагмент; или когда X=Y=OH и Z=OMe, R1 представляет собой ОСН3, OCH2CHF2, ОСН2циклопентил, О-(2,5-дифторфенил) или (S)-втор-бутиламиногруппу; или когда Х=Н и Y=Z=OH, R1 представляет собой н-гексиламино- или циклопентиламиногруппу; или когда X=Z=OH и Y=H, R1 представляет собой циклопентиламиногруппу. 5 н. и 20 з.п. ф-лы, 1 табл., 21 схема.
1. Соединение следующей общей формулы:
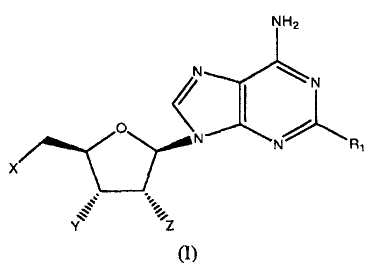
в которой
когда X=Y=Z=OH, R1 представляет собой OCH2CF2CF3; феноксигруппу (замещенную 3-(4-трифторметилфенилом), 3,4-дихлором, (3-трифторметилом, 4-фтором), (3-трифторметилом, 4-хлором), (3-хлором, 4-цианогруппой) или 3,5-бис(трифторметилом)), 1-пиперазинил-(4-(3,4-дихлорфенилом)); фенильную группу, замещенную 3,4-дихлором, 3,5-дифтором, 3,5-бис(трифторметилом) или 3,4,5-трифтором); или 2-бензофуранильный фрагмент; или
когда X=Y=OH и Z=OMe, R1 представляет собой ОСН3, OCH2CHF2, ОСН2циклопентил, О-(2,5-дифторфенил) или (S)-втор-бутиламиногруппу; или когда Х=Н и Y=Z=OH, R1 представляет собой н-гексиламино- или циклопентиламиногруппу; или
когда X=Z=OH и Y=H, R1 представляет собой циклопентиламиногруппу;
или фармацевтически приемлемую соль таковых.
2. Соединение по п.1 в качестве лекарственного средства.
3. Применение соединения по п.1 в производстве лекарственного средства для предотвращения, лечения или ослабления медицинского состояния, которое может быть улучшено или предотвращено агонизмом аденозиновых А2А-рецепторов.
4. Применение по п.3, в котором медицинское состояние представляет собой боль.
5. Применение по п.4, в котором боль представляет собой гипералгезию.
6. Применение по п.4 или 5, в котором боль вызвана невропатией.
7. Применение по п.4 или 5, в котором боль связана с диабетической невропатией, полиневропатией, ишиас/люмбальной радикулопатией, раковой болью, постгерпетической невралгией, синдромом миофасциальной боли, ревматоидным артритом, фибромиалгией, остеоартритом, панкреатической болью, тазовоперинеальной болью, спинальным стенозом, болевой дисфункцией височно-нижнечелюстного сустава, связанной с ВИЧ-болью, тригеминальной невралгией, хронической невропатической болью, болью внизу спины, болью синдрома неудачных операций на позвоночнике, болью в спине, послеоперационной болью, болью после физической травмы (в том числе огнестрельного ранения, дорожно-транспортного происшествия, ожогов), сердечной болью, болью в груди, тазовой болью/PID (воспалением тазовых органов), шейной болью, болью в животе, фантомной болью ложного ощущения ампутированной конечности, болью при родовспоможении (роды/кесарево сечение), почечными коликами, болью при остром опоясывающем герпесе, болью при деструктивных процессах острого панкреатита (рак), дисменореей/эндометриозом; или в любом из вышеназванных патологических состояний, где состояние вызывается или обостряется бактериальной или вирусной инфекцией.
8. Применение по п.4 или 5, в котором боль вызвана воспалительной болезнью или комбинированным воспалительным, аутоиммунным и невропатическим повреждением ткани.
9. Применение по п.4 или 5, в котором боль связана с ревматоидным артритом, остеоартритом, суставной болью (тендонитом, бурситом, острым артритом), болью внизу спины, болью синдрома неудачных операций на позвоночнике, болью в спине, послеоперационной болью, болью после физической травмы (в том числе огнестрельного ранения, дорожно-транспортного происшествия, ожогов), фибромиалгией, ревматоидным артритом, спондилитом, подагрическим артритом и другими артритными состояниями, раком, ВИЧ, диабетической невропатией, полиневропатией, ишиас/люмбальной радикулопатией, аутоиммунным повреждением (в том числе множественным склерозом, синдромом Гийена-Барре, миастенией тяжелой псевдопаралитической), отторжением трансплантата реципиентом, отторжением аллотрансплантата, лихорадкой и болью в мышцах вследствие инфекции, СПИД-ассоциированным комплексом (ARC), плотным разрастанием соединительной ткани кожи, образованием рубцовой ткани, болезнью Крона, неспецифическим язвенным колитом и парезом, спастическим колитом, остеопорозом, церебральной малярией и бактериальным менингитом, кишечной коликой, раковой болью, болью в спине, фибромиалгией, послеоперационной болью; или в любом из вышеназванных патологических состояний, где состояние вызывается или обостряется бактериальной или вирусной инфекцией.
10. Применение по п.4, в котором боль представляет собой ишемическую боль.
11. Применение по п.4 или 10, в котором боль связана с болезнью коронарной артерии, болезнью периферической артерии, состояниями, которые характеризуются недостаточным кровотоком, левожелудочковой гипертрофией, первичной артериальной гипертензией, острым гипертоническим кризом, кардиомиопатией, сердечной недостаточностью, толерантностью к физической нагрузке, хронической сердечной недостаточностью, аритмией, сердечной дисритмией, обмороком, артериосклерозом, хронической сердечной недостаточностью в мягкой степени, стенокардией, стенокардией Принцметала (вариант), устойчивой стенокардией и стенокардией, вызванной физическими нагрузками, реокклюзией после коронарного шунтирования, перемежающейся хромотой (атеросклеротическими облитерациями), артериитом, диастолической дисфункцией и систолической дисфункцией, атеросклерозом, постишемическим/реперфузионным повреждением, диабетом (обоих типов, I и II), тромбоэмболией и геморрагическими осложнениями.
12. Применение по п.3, в котором названное медицинское состояние связано с воспалением.
13. Применение по п.12, в котором воспаление вызвано или связано с раком (таким как лейкемии, лимфомы, карциномы, рак толстой кишки, рак молочной железы, рак легкого, рак поджелудочной железы, гепатоцеллюлярная карцинома, рак почки, меланома, метастазы в печени, легком, грудной железе и простате, и т.д.); хронической обструктивной легочной болезнью (COPD), острым бронхитом, хроническим бронхитом, эмфиземой, бронхоэктазией, фиброзно-кистозной дегенерацией, пневмонией, плевритом, острой астмой, хронической астмой, острым респираторным дистресс-синдромом, респираторным дистресс-синдромом взрослых (ARDS), респираторным дистресс-синдромом новорожденных (IRDS), острым повреждением легких (ALI), ларингитом, фарангитом, стойкой астмой, хроническим астматическим бронхитом, интерстициальной болезнью легких, злокачественными заболеваниями легких, недостаточностью альфа-антитрипсина, бронхолитическими облитерациями, саркоидозом, пульмональным фиброзом, коллагеновыми сосудистыми болезнями, аллергическим ринитом, назальной гиперемией, астматическим статусом, обусловленной курением легочной болезнью, пульмональной гипертонией, отеком легких, пульмональной эмболией, плевральной эффузией, пневмотораксом, гемотораксом, раком легких, аллергиями, поллинозом (сенной лихорадкой), чиханием, вазомоторным ринитом, воспалением слизистой оболочки, синуситом, вызванной экзогенными раздражителями болезнью (SO2, смог, загрязнения), повышенной чувствительностью дыхательных путей, непереносимостью молочных продуктов, пневмонией Лаффера, пневмокониозом, связанным с коллагеном сосудистым заболеванием, гранулематозной болезнью, бронхиальным воспалением, хронической воспалительной болезнью легких, болезнями резорбции костной ткани, реперфузионным повреждением (в том числе повреждением, нанесенным органам вследствие реперфузии после ишемических эпизодов, например инфаркта миокарда, ударов), аутоиммунной болезнью (такой как отторжение трансплантатов органов, красная волчанка, отторжение трансплантата реципиентом, отторжение аллотрансплантата, множественный склероз, ревматоидный артрит, сахарный диабет I типа, в том числе разрушение панкреатических островков, ведущее к диабету и воспалительным диабетическим осложнениям); аутоиммунным повреждением (включающим множественный склероз, синдром Гийена-Барре, миастению тяжелую псевдопаралитическую); ожирением; сердечно-сосудистыми нарушениями, связанными с недостаточным кровоснабжением тканей и воспалением (такими как атеросклеротические бляшки, атеросклероз, удар, ишемическое/реперфузионное повреждение, перемежающаяся хромота, повреждение спинного мозга, застойная сердечная недостаточность, васкулит, геморрагический шок, спазм сосудов с последующим субарахноидальным кровоизлиянием, спазм сосудов с последующим инсультом, плеврит, перикардит, сердечно-сосудистые осложнения диабета); ишемическим/реперфузионным повреждением, ишемией и связанным с ней воспалением, рестенозом с последующими ангиопластическими и воспалительными аневризмами; эпилепсией, нейродегенерацией (в том числе болезнью Альцгеймера), мышечной усталостью или мышечным спазмом (в частности спазмами у спортсменов), артритом (таким как ревматоидный артрит, остеоартрит, ревматоидный спондилит, подагрический артрит), фиброзом (например, легкого, кожи и печени), множественным склерозом, сепсисом, септическим шоком, энцефалитом, инфекционным артритом, реакцией Яриша-Герксгеймера, опоясывающим герпесом, токсическим шоком, церебральной малярией, болезнью Лайма, эндотоксическим шоком, грамотрицательным шоком, геморрагическим шоком, гепатитом (возникающим как вследствие повреждения ткани, так и в результате вирусной инфекции), тромбозом глубоких вен, подагрой; состояниями, связанными с затруднениями дыхания (например, забитые или закупоренные дыхательные пути, бронхостеноз, легочная вазоконстрикция, затрудненное дыхание, силикоз, патологическое разрастание легочных тканей, легочная гипертензия, легочная вазоконстрикция, бронхиальная аллергия и весенний конъюнктивит); состояниями, связанными с воспалением кожи (в том числе псориаз, экзема, язвы, контактные дерматиты); состояниями, связанными с воспалением кишок (в том числе болезнь Крона, неспецифический язвенный колит и парез, синдром раздраженной толстой кишки, воспалительная болезнь кишки); ВИЧ (в частности, ВИЧ-инфекционный процесс), церебральной малярией, бактериальным менингитом, усиленной некротическим опухолевым фактором (TNF) репликацией ВИЧ, ингибированием некротическим опухолевым фактором (TNF) активности AZT (азидотимидина) и DDI (диданозина), остеопорозом и другими болезнями резорбции костной ткани, остеоартритом, ревматоидным артритом, бесплодием вследствие эндометриоза, жаром и мышечной болью вследствие воспаления, общей атрофией, сопровождающей рак, общей атрофией, сопровождающей инфекцию или злокачественное развитие, общей атрофией, сопровождающей приобретенный синдром иммунодефицита (AIDS, СПИД), СПИД-ассоциированным комплексом (ARC), образованием плотных разрастаний соединительной ткани кожи, образованием рубцовой ткани, отрицательных эффектов лечения амфотерицином В, отрицательных эффектов лечения интерлейкином-2, отрицательных эффектов лечения ОКТ3 (муромомаб-СD3) или отрицательных эффектов лечения GM-CSF (гранулоцитарно-макрофагальный колониестимулирующий фактор), и прочими состояниями, обусловленными избыточной активностью антивоспалительных клеток (включающих нейтрофил, эозинофил, макрофаг и Т-клетку); или в любом из вышеназванных патологических состояний, где состояние вызывается или обостряется бактериальной или вирусной инфекцией, сосудистыми макро- и микроосложнениями диабета 1 и 2 типов, ретинопатией, нефропатией, автономной невропатией, или повреждением кровеносных сосудов, вызванным ишемией или атеросклерозом.
14. Применение по п.3, в котором названное медицинское состояние связано с артропатией.
15. Применение по п.14, в котором артропатия вызвана или связана с ревматоидным артритом, спондилитом, подагрическим артритом, остеоартритом, тендионитом, бурситом, острым артритом, неревматоидным артритом или подагрой.
16. Применение соединения по п.1 или фармацевтически приемлемой соли такового в производстве противоревматического лекарственного препарата, влияющего на течение заболевания (DMARD), для замедления развития артропатии.
17. Применение по п.16 в производстве противоревматического лекарственного препарата, влияющего на течение заболевания (DMARD), для замедления развития ревматоидного артрита.
18. Применение соединения по п.1 или фармацевтически приемлемой соли такового в производстве лекарственного средства для стимуляции заживления ран.
19. Применение по любому из пп.3, 16 или 18 при дозировании, которое после введения субъекту приводит к пиковой концентрации соединения в плазме, которое составляет менее чем значение ЕС50 соединения для аденозиновых рецепторов при величине рН 7,4.
20. Применение по любому из пп.3, 16 или 18 при дозировании, которое составляет от одной десятитысячной части до одной второй части минимального дозирования соединения, которое приводит к побочным эффектам в виде брадикардии, гипотонии или тахикардии у животных того же вида, как и субъект, которому должно быть введено соединение.
21. Применение по любому из пп.3, 16 или 18 при дозировании, которое после введения субъекту приводит к концентрации соединения в плазме, которая поддерживается в течение более чем одного часа на уровне от одной десятитысячной части до одной второй части минимальной концентрации соединения в плазме, которая приводит к побочным эффектам в виде брадикардии, гипотонии или тахикардии у животных того же вида, как и субъект, которому должно быть введено соединение.
22. Применение по любому из пп.3, 16 или 18 при дозировании на уровне 0,001-15 мг/кг.
23. Фармацевтическая композиция, обладающая активностью агониста аденозинового А2А-рецептора, включающая соединение по п.1, и физиологически приемлемый носитель, эксципиент или разбавитель.
24. Фармацевтическая композиция по п.23, дополнительно включающая нестероидное противовоспалительное лекарственное средство (NSAID) или лекарственный препарат, влияющий на течение заболевания (DMARD).
25. Фармацевтическая композиция по п.23, дополнительно включающая антипатогенное средство.
| Zhan-Guo Gao et al, "2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors", Biochemical Pharmacology, 2004, vol.68, pp.1985-1993 | |||
| J.M.Rieger et al, "Design, Synthesis and Evaluation of Novel A2A Adenosine Receptor Agonists", Journal of Medicianl Chemistry, 2001, vol.44, pp.531-539 | |||
| WO 2005084653 |

