Настоящее изобретение относится к нонадепсипептидам и способу их получения, а также к их применению для приготовления лекарственных средств, предназначенных для лечения и/или профилактики различных болезней, прежде всего вызываемых бактериями инфекционных болезней.
Клеточная стенка бактерий синтезируется целым рядом ферментов (биосинтез клеточной стенки) и играет важную роль в выживании, соответственно размножении микроорганизмов. Структура этой макромолекулы, равно как и участвующие в ее синтезе белки, надежно законсервированы внутри бактерий. Биосинтез клеточной стенки с учетом его важного значения и единообразия его протекания является идеальной мишенью для вновь разрабатываемых антибиотиков (D.W.Green, The bacterial cell wall as a source of antibacterial targets. Expert Opin. Ther. Targets, 6, 2002, cc.1-19).
В качестве типичных примеров ингибиторов биосинтеза клеточной стенки бактерий можно назвать ванкомицин и пенициллины, в основе антибиотической активности которых лежит указанный механизм действия. Эти антибиотики на протяжении нескольких десятилетий используются во врачебной практике для лечения бактериальных инфекций, прежде всего инфекций, вызываемых грамположительными возбудителями. Однако в связи с непрерывным появлением все новых и новых устойчивых к антибиотикам микроорганизмов, например устойчивых к метициллину стафилококков, устойчивых к пенициллину пневмококков и устойчивых к ванкомицину энтерококков (F. Baquero, Gram-positive resistance: challenge for the development of new antibiotics, J. Antimicrob. Chemother., 39, 1997, дополн. А, cc.1-6; A.P.Johnson, D.M.Livermore, G.S.Tillotson, Antimicrobial susceptibility of Gram-positive bacteria: what's current, what's anticipated?, J. Hosp.Infect., (49), 2001, дополн. А, cc.3-11), а также недавно впервые обнаруженных устойчивых к ванкомицину стафилококков (В. Goldrick, First reported case of VRSA in the United States, Am. J. Nurs., 102, 2002, с.17), терапевтическая эффективность от применения подобных антибиотиков постоянно снижается.
В настоящем изобретении предлагаются соединения, образующие новый класс ингибиторов биосинтеза клеточной стенки бактерий, к которым в отличие от антибиотиков известных классов у бактерий отсутствует устойчивость.
В US 4754018 описано природное вещество лизобактин и некоторые его производные как обладающие антибактериальным действием. Выделение лизобактина и его антибактериальное действие описаны также в ЕР-А-196042 и JP 01-132600. В WO 04/099239 описаны производные лизобактина с антибактериальным действием.
Антибактериальное действие лизобактина и катаносина А описано далее у O'Sullivan J. и др., J. Antibiot., 41, 1988, cc.1740-1744, у Bonner D.P. и др., J. Antibiot., 41, 1988, cc.1745-1751, у Shoji J. и др., J. Antibiot., 41, 1988, cc.713-718, а также у Tymiak А.А. и др., J. Org. Chem., 54, 1989, cc.1149-1157.
В основу настоящего изобретения была положена задача предложить другие соединения с антибактериальным действием, сравнимым с антибактериальным действием известных веществ или превосходящим его, с лучшей по сравнению с известными веществами переносимостью, например с пониженной нефротоксичностью, и лучшим распределением в организме, т.е. с лучшими фармакокинетическими свойствами, проявляющимися, например, в повышении свободной фракции (fu), для лечения бактериальных заболеваний у человека и животных.
Объектом настоящего изобретения являются соединения формулы
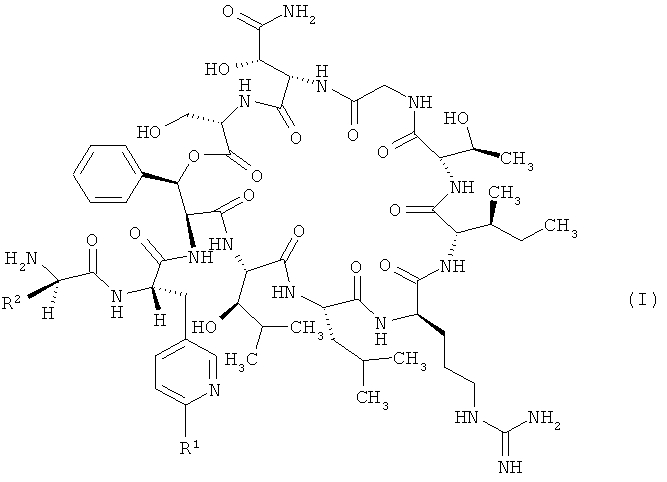 ,
,
в которой R1 обозначает водород, а
R2 обозначает 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил, 2,2-диметилпент-1-ил или триметилсилилметил либо
R1 обозначает трифторметил, а
R2 обозначает 2,2-диметилпроп-1-ил, 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил, 2,2-диметилпент-1-ил или триметилсилилметил,
и их соли, их сольваты и сольваты их солей.
К предлагаемым в изобретении соединениям относятся соединения формулы (I) и их соли, сольваты, сольваты солей и пролекарства, подпадающие под формулу (I) соединения приведенных ниже формул и их соли, сольваты, сольваты солей и пролекарства, а также подпадающие под формулу (I), указанные ниже в качестве примеров соединения и их соли, сольваты, сольваты солей и пролекарства, при условии, что подпадающие под формулу (I), указанные ниже соединения уже не представляют собой соли, сольваты, сольваты солей и пролекарства.
Предлагаемые в изобретении соединения могут в зависимости от их структуры существовать в стереоизомерных формах (энантиомеров, диастереомеров). В соответствии с этим в объем изобретения включены все такие энантиомеры или диастереомеры и их соответствующие смеси. Из подобных смесей энантиомеров и/или диастереомеров индивидуальные стереоизомеры можно выделять известными методами.
Поскольку предлагаемые в изобретении соединения могут существовать в таутомерных формах, все они также включены в объем изобретения.
К предпочтительным солям согласно настоящему изобретению относятся физиологически безвредные соли предлагаемых в изобретении соединений. Однако в объем изобретения включены также соли, которые как таковые не пригодны для фармацевтического применения, но могут использоваться, например, для выделения или очистки предлагаемых в изобретении соединений, или смешанные соли. Под смешанной солью согласно настоящему изобретению подразумеваются аддитивная соль, образованная с двумя или более различными кислотами, соответственно основаниями, например трифторацетат-мезилат.
К физиологически безвредным солям предлагаемых в изобретении соединений относятся кислотно-аддитивные соли, образованные с минеральными кислотами, карбоновыми кислотами и сульфоновыми кислотами, например соли, образованные с хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, толуолсульфоновой кислотой, бензолсульфоновой кислотой, нафталиндисульфоновой кислотой, уксусной кислотой, трифторуксусной кислотой, пропионовой кислотой, молочной кислотой, винной кислотой, яблочной кислотой, лимонной кислотой, фумаровой кислотой, малеиновой кислотой и бензойной кислотой.
К физиологически безвредным солям предлагаемых в изобретении соединений относятся также образованные с обычными основаниями соли, в качестве предпочтительных примеров которых можно назвать соли, образованные с щелочными металлами (например, натриевые и калиевые соли), соли, образованные с щелочноземельными металлами (например, кальциевые и магниевые соли), и аммониевые соли, полученные взаимодействием с аммиаком или органическими аминами с 1-16 С-атомами, в качестве предпочтительных примеров которых можно назвать этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метилморфолин, аргинин, лизин, этилендиамин и N-метилпиперидин.
Сольватами согласно настоящему изобретению называют те формы предлагаемых в изобретении соединений, которые в твердом или жидком состоянии образуют комплекс в результате координационной реакции с молекулами растворителя. Гидраты являются частной формой сольватов, образующихся в результате координационной реакции с водой.
Предпочтительны соединения формулы (I), в которой
R1 обозначает водород, а
R2 обозначает 2,2-диметилбут-1-ил или триметилсилилметил либо
R1 обозначает трифторметил, а
R2 обозначает 2,2-диметилпроп-1-ил, 2,2-диметилбут-1-ил или триметилсилилметил,
и их соли, их сольваты и сольваты их солей.
Предпочтительны также соединения формулы (I), в которой
R1 обозначает водород, а
R2 обозначает 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил или триметилсилилметил,
и их соли, их сольваты и сольваты их солей.
Предпочтительны далее соединения формулы (I), в которой
R1 обозначает водород, а
R2 обозначает 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил, 2,2-диметилпент-1-ил или триметилсилилметил.
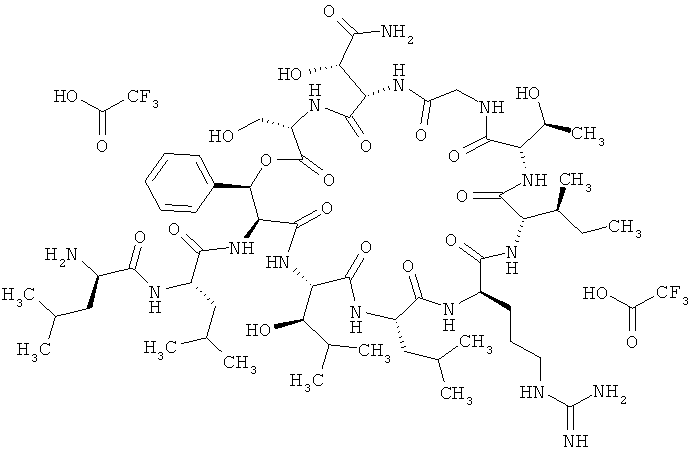
Наиболее предпочтительным соединением является 3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланилдез(1-D-лейцил-2-L-лейцил)лизобактин
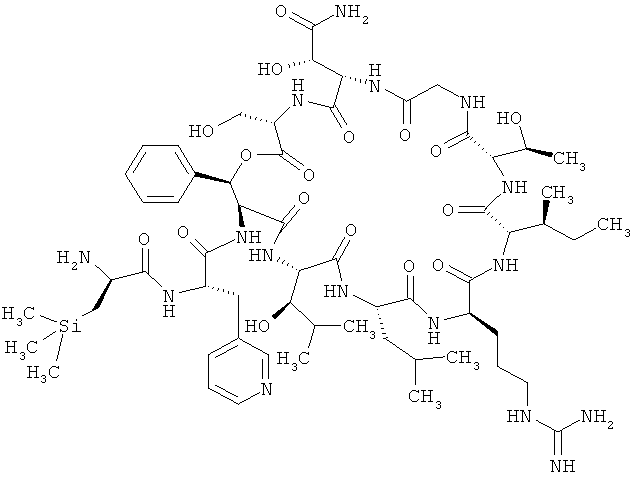 ,
,
или одна из его солей, один из его сольватов или один из сольватов его солей.
Объектом изобретения является далее способ получения соединений формулы (I), заключающийся в том, что соединение формулы
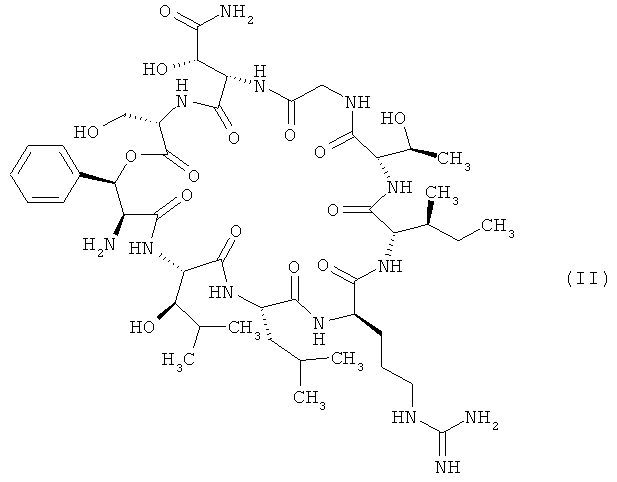 ,
,
подвергают взаимодействию с соединениями формулы
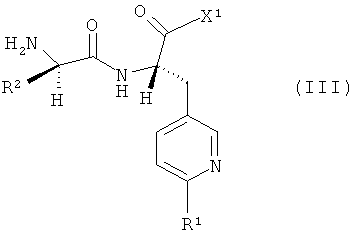 ,
,
в которой R1 и R2 имеют указанные выше значения, а
Х1 обозначает галоген, предпочтительно бром, хлор или фтор, или гидроксигруппу.
Если Х1 обозначает галоген, то указанное взаимодействие в общем случае проводят в инертных растворителях, при необходимости в присутствии основания, предпочтительно при температуре в интервале от -30 до 50°С и при нормальном давлении.
В качестве примера инертных растворителей можно назвать тетрагидрофуран, метиленхлорид, пиридин, диоксан или диметилформамид, при этом предпочтительно использовать метиленхлорид или диметилформамид.
В качестве примера оснований можно назвать триэтиламин, диизопропилэтиламин или N-метилморфолин, при этом предпочтительно использовать диизопропилэтиламин.
Если Х1 обозначает гидроксигруппу, то вышеуказанное взаимодействие в общем случае проводят в инертных растворителях в присутствии дегидратирующего агента и при необходимости в присутствии основания, предпочтительно при температуре в интервале от -30 до 50°С и при нормальном давлении.
В качестве примера инертных растворителей можно назвать галогенированные углеводороды, такие как дихлорметан или трихлорметан, и углеводороды, такие как бензол, нитрометан, диоксан, диметилформамид или ацетонитрил. Равным образом возможно использование и смесей растворителей. Наиболее предпочтительно использовать в качестве растворителя дихлорметан или диметилформамид.
Для применения в качестве дегидратирующих агентов пригодны, например, карбодиимиды, такие как N,N'-диэтил-, N,N'-дипропил-, N,N'-диизопропил-, N,N'-дициклогексилкарбодиимид, гидрохлорид N-(3-диметиламиноизопропил)-N'-этилкарбодиимида (ЭДК), N-циклогексилкарбодиимид-N'-пропилоксиметил-полистирол (ПС-карбодиимид), карбонильные соединения, такие как карбонилдиимидазол, 1,2-оксазолиевые соединения, такие как 2-этил-5-фенил-1,2-оксазолий-3-сульфат или 2-трет-бутил-5-метилизоксазолийперхлорат, ациламиносоединения, такие как 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин, либо пропанфосфоновый ангидрид, изобутилхлорформиат, бис-(2-оксо-3-оксазолидинил)фосфорилхлорид, гексафторфосфат бензотриазолилокситри(диметиламино)фосфония, гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ГБТУ), тетрафторборат 2-(2-оксо-1-(2Н)-пиридил)-1,1,3,3-тетраметилурония (ТФТУ), гексафторфосфат O-(7-aзaбeнзoтpиaзoл-1-ил)-N,N,N',N'-тeтpaмeтилypoния (ГАТУ), 1-гидроксибензотриазол (ГОБТ), гексафторфосфат бензотриазол-1-илокси-трис-(диметиламино)фосфония (БОФ), N-гидроксисукцинимид или их смеси с основаниями.
В качестве примера оснований можно назвать карбонаты щелочных металлов, такие, например, как карбонат натрия или калия, или гидрокарбонаты щелочных металлов, либо органические основания, такие как триалкиламины, например триэтиламин, N-метилморфолин, N-метилпиперидин, 4-диметиламинопиридин или диизопропилэтиламин.
В реакции конденсации предпочтительно использовать ГАТУ либо ЭДК в присутствии ГОБТ.
В некоторых случаях соединения формулы (III) могут нести защитные группы, и поэтому в подобных случаях по завершении взаимодействия соединения формулы (II) с соединениями формулы (III) такие защитные группы отщепляют обработкой трифторуксусной кислотой по известным методам.
Свободное основание из солей соединений формулы (I) можно получать, например, добавлением основания с последующей экстракцией, осаждением или хроматографическим отделением свободного соединения по известным методам, прежде всего за счет применения связанных с полимером оснований, например связанного с полимером гидрокарбоната.
Еще одним объектом изобретения является способ получения соединений формулы (I) или их сольватов, заключающийся в том, что соли таких соединений или сольваты солей таких соединений путем добавления основания переводят в свободные соединения.
Соединение формулы (II) можно синтезировать из лизобактина (пример 1А) путем двойной деградации по Эдману по описанной в экспериментальной части в примере 2А методике.
Соединения формулы (III) известны либо их можно синтезировать по известным методам из соответствующих эдуктов.
Получение предлагаемых в изобретении соединений можно пояснить с помощью приведенной ниже схемы синтеза.
Схема синтеза
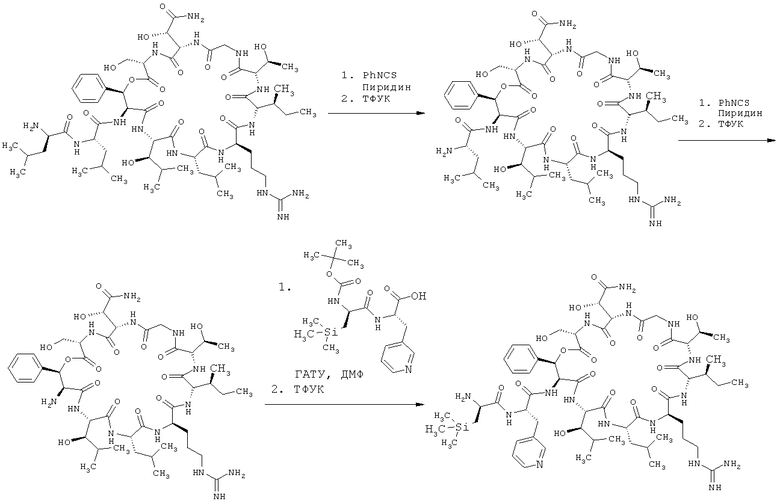
Предлагаемые в изобретении соединения обладают ценным фармакологическим и фармакокинетическим спектром действия, который невозможно было предсказать заранее. Предлагаемые в изобретении соединения обладают, в частности, антибактериальным действием.
В соответствии с этим предлагаемые в изобретении соединения пригодны для их применения в качестве лекарственных веществ для лечения и/или профилактики различных заболеваний у человека и животных.
Предлагаемые в изобретении соединения обладают меньшей по сравнению с лизобактином нефротоксичностью.
Предлагаемые в изобретении соединения обладают также лучшими по сравнению с лизобактином фармакокинетическими свойствами. Предлагаемые в изобретении соединения при таком же или лучшем по сравнению с лизобактином фармакологическом действии обладают лучшим в сопоставлении с ним распределением в организме, что позволяет применять их в меньшей терапевтической дозе, а также расширить область их терапевтического применения.
Для предлагаемых в изобретении соединений характерно также их более высокое по сравнению с лизобактином содержание в виде свободной фракции (fu) в плазме.
Предлагаемые в изобретении нонадепсипептиды обладают действием ингибиторов биосинтеза бактериальной клеточной стенки.
Предлагаемые в изобретении соединения наиболее эффективны при борьбе с бактериями и сходными с ними микроорганизмами. Поэтому предлагаемые в изобретении соединения наиболее пригодны для профилактики и химиотерапии вызываемых этими возбудителями локальных и системных инфекционных болезней в медицине и ветеринарии.
В принципе предлагаемые в изобретении соединения могут применяться для борьбы со всеми бактериями и сходными с ними микроорганизмами, которые имеют бактериальную клеточную стенку (Murein sacculus), соответственно относящуюся к ней ферментативную систему, например, могут применяться для профилактики и химиотерапии инфекционных болезней, вызываемых одним или несколькими возбудителями, к которым относятся следующие: грамотрицательные кокки (Neisseria gonorrhoeae) и грамотрицательные палочки, такие как кишечные бактерии, например Escherichia coli, Hämophilus influenzae, бактерии рода псевдомонас, бактерии рода клебсиелл, бактерии рода цитробактер (С.freundii, С.divernis), бактерии рода сальмонелл, бактерии рода шигелл, бактерии рода энтеробактер (Е. aerogenes, Е. agglomerans) и бактерии рода Hafhia, бактерии рода Serratia (S. marcescens), бактерии рода провиденция, бактерии рода иерсиний, а также бактерии родов ацинетобактер, бранхамелл и хламидий. Помимо этого антибактериальный спектр действия предлагаемых в изобретении соединений охватывает облигатные анаэробные бактерии, такие, например, как Bacteroides fragilis, представители рода пептококков, рода пептострептококков и рода клостридий, а также микобактерии, например М. tuberculosus. Наиболее выраженное действие предлагаемые в изобретении соединения проявляют против грамположительных кокков, например, стафилококков (S. aureus, S. epidermidis, S. haemolyticus, S. carnosus), энтерококков (E. faecalis, E. faecium) и стрептококков (S. agalactiae, S. pneumoniae, S. pyogenes).
Перечисленные выше возбудители инфекционных болезней представлены лишь в качестве примера, и их перечень не исчерпывается приведенным выше.
В качестве примера болезней, которые вызываются вышеуказанными возбудителями или смешанными инфекциями и которые можно предупредить или вылечить либо при которых можно добиться улучшения за счет применения предлагаемых в изобретении соединений, можно назвать инфекционные болезни человека, такие как неосложненные и осложненные инфекции мочевых путей, неосложненные кожные и поверхностные инфекции, осложненные инфекции кожи и мягких тканей, приобретенная в больнице или амбулаторно-поликлиническом учреждении пневмония, внутрибольничные пневмонии, острые обострения и вторичные бактериальные инфекции при хроническом бронхите, острый средний отит, острый синусит, стрептококковый фарингит, бактериальный менингит, неосложненные гонококковые и негонококковые уретрит/цервицит, острый простатит, эндокардит, неосложненные и осложненные внутрибрюшные инфекции, гинекологические инфекции, воспаление в области таза, бактериальный вагиноз, острый и хронический остеомиелит, острый бактериальный артрит, гранулоцитопения (нейротропения) у лихорадящих пациентов, для лечения которых применяется эмпирическая терапия, а также бактериемии, вызываемые MRSA-штаммами инфекции, острая инфекционная диарея, вызываемые бактерией Helicobacter pylori инфекции, одонтогенные инфекции, офтальмологические инфекции, послеоперационные инфекции (включая перипроктальный абсцесс, раневые инфекции, инфекции желчных путей, мастит и острый аппендицит), кистозный фиброз и бронхоэктазия.
Предлагаемые в изобретении соединения можно использовать не только для профилактики или лечения инфекционных болезней человека, но и при бактериальных инфекциях у других видов. В качестве примеров при этом можно назвать диарею, энтеротоксемию, сепсис, дизентерию, сальмонеллез, синдром метрита-мастита-агалактита и мастит у свиней, диарею, сепсис, бронхопневмонию, сальмонеллез, пастереллез и инфекции половых органов у жвачных животных (крупного рогатого скота, овец, коз), бронхопневмонии, суставолом, пуэрперальные инфекции и сальмонеллез у лошадей, бронхопневмонию, диарею, дерматит, отит, инфекции мочевых путей и простатит у собак и кошек, вызываемые Е. coli инфекции, хронические заболевания дыхательных путей, сальмонеллез, пастереллез и пситтакоз у домашней птицы (кур, индеек, перепелов, голубей, декоративных птиц и других).
Предлагаемые в изобретении соединения могут также использоваться для лечения бактериальных заболеваний промысловых рыб и декоративных рыбок при их разведении и содержании, и в этом случае антибактериальный спектр действия предлагаемых в изобретении соединений помимо вышеуказанных возбудителей охватывает и других возбудителей, таких, например, как бактерии рода пастерелл, бактерии рода бруцелл, бактерии рода кампилобактер, бактерии рода листерий, бактерии рода эризипилотрикс, коринебактерии, бактерии рода боррелий, бактерии рода трепонем, бактерии рода нокардий, бактерии рода риккетсий и бактерии рода иерсиний.
Следующим объектом настоящего изобретения является применение предлагаемых в нем соединений для лечения и/или профилактики заболеваний, прежде всего бактериальных инфекционных болезней.
Следующим объектом настоящего изобретения является применение предлагаемых в нем соединений для лечения и/или профилактики заболеваний, прежде всего указанных выше заболеваний.
Следующим объектом настоящего изобретения является применение предлагаемых в нем соединений для приготовления лекарственного средства, предназначенного для лечения и/или профилактики заболеваний, прежде всего указанных выше заболеваний.
Предлагаемые в изобретении соединения предпочтительно применять для приготовления лекарственных средств, пригодных для профилактики и/или лечения бактериальных заболеваний.
Следующим объектом настоящего изобретения является способ лечения и/или профилактики заболеваний, прежде всего указанных выше заболеваний, с применением предлагаемых в изобретении соединений в антибактериально эффективном количестве.
Следующим объектом настоящего изобретения являются лекарственные средства, содержащие по меньшей мере одно предлагаемое в изобретении соединение и по меньшей мере одно или несколько других действующих веществ и предназначенных прежде всего для лечения и/или профилактики указанных выше заболеваний. К предпочтительным действующим веществам, которые могут использоваться в сочетании с предлагаемыми в изобретении соединениями, относятся соединения с антибактериальным действием, которые обладают спектром действия, отличным от спектра действия предлагаемых в изобретении соединений, прежде всего дополняющим его спектром действия, и/или при применении которых в комбинации с предлагаемыми в изобретении соединениями проявляется синергетический эффект.
Предлагаемые в изобретении соединения могут обладать системным и/или местным действием. Для этого их можно применять или вводить в организм соответствующим способом, например перорально, парентерально, через легкие, назально, подъязычно, лингвально, трансбуккально, ректально, накожно, чрескожно, конъюнктивально, в уши или в виде имплантата, соответственно стента.
Для подобного применения предлагаемые в изобретении соединения можно использовать в составе соответствующих лекарственных форм.
Для перорального применения пригодны действующие в соответствии с известными из уровня техники принципами лекарственные формы с быстрым и/или модифицированным высвобождением предлагаемых в изобретении соединений, содержащихся в таких лекарственных формах в кристаллическом, и/или аморфном, и/или растворенном виде, например таблетки (таблетки без покрытия или с покрытием, например, устойчивыми к действию желудочного сока или начинающими растворяться с задержкой либо нерастворимыми покрытиями, регулирующими или контролирующими высвобождение предлагаемых в изобретении соединений), быстро распадающиеся в полости рта таблетки или пленки/облатки, пленки/лиофилизаты, капсулы (например, твердо- или мягкожелатиновые капсулы), драже, грануляты, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное введение предлагаемых в изобретении соединений в организм либо может происходить, минуя стадию их резорбции (например, внутривенно, внутриартериально, внутрисердечно, интраспинально или внутрилюмбально), либо может происходить, включая стадию резорбции (например, внутримышечно, подкожно, внутрикожно, чрескожно или внутрибрюшинно). Для парентерального применения в качестве лекарственных форм пригодны помимо прочего составы для инъекций и инфузий в виде растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для применения предлагаемых в изобретении соединений остальными способами пригодны, например, ингаляционные лекарственные формы (в частности, для порошковых ингаляторов, небулайзеров), назальные капли, растворы и спреи, таблетки для лингвального, подъязычного или трансбуккального применения, пленки/облатки либо капсулы, суппозитории, ушные или глазные препараты, вагинальные капсулы, водные суспензии (примочки, болтушки), липофильные суспензии, мази, кремы, трансдермальные терапевтические системы (например, пластыри), эмульсии типа "молочко", пасты, пены, присыпки, имплантаты или стенты.
Предлагаемые в изобретении соединения можно переводить или перерабатывать в указанные выше лекарственные формы (формы применения). Для этого предлагаемые в изобретении соединения можно по известной технологии смешивать с инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами. К таким вспомогательным веществам относятся, в частности, носители (например, микрокристаллическая целлюлоза, лактоза, маннит), растворители (например, жидкие полиэтиленгликоли), эмульгаторы и диспергаторы или смачиватели (например, додецилсульфат натрия, полиоксисорбитанолеат), связующие (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (например, антиокислители, такие как аскорбиновая кислота), красители (например, неорганические пигменты, такие как оксиды железа) и улучшители вкуса и/или запаха.
Следующим объектом настоящего изобретения являются лекарственные средства, содержащие по меньшей мере одно предлагаемое в изобретении соединение, обычно совместно с одним или несколькими инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами, а также применение таких лекарственных средств в вышеуказанных целях.
Для достижения требуемого эффекта предлагаемые в изобретении соединения предпочтительно применять в дозе, которая при внутривенном введении составляет примерно от 0,001 до 100 мг/кг веса тела, предпочтительно примерно от 0,1 до 10 мг/кг веса тела, а при пероральном введении составляет примерно от 0,01 до 50 мг/кг веса тела, предпочтительно от 0,5 до 10 мг/кг веса тела.
Тем не менее в некоторых случаях может потребоваться применять предлагаемые в изобретении соединения в дозах, отличающихся в ту или иную сторону от указанных выше количеств, а именно: в зависимости от веста тела, пути введения действующего вещества в организм, индивидуальной реакции на действующее вещество, типа лекарственной формы и момента времени, в который применяют действующее вещество, соответственно интервала времени, через который применяют действующее вещество. Так, в частности, в некоторых случаях вполне может оказаться достаточным применять предлагаемые в изобретении соединения в дозе, меньшей указанного выше минимального количества, тогда как в других случаях предлагаемые в изобретении соединения может потребоваться применять в дозе, превышающей вышеуказанный верхний предел. При применении предлагаемых в изобретении соединений в сравнительно высокой дозировке может оказаться целесообразным дробить суточную дозу на несколько более мелких разовых доз и распределить их прием на все сутки.
В приведенных ниже описаниях опытов, экспериментов и исследований и в примерах выраженные в процентах данные представляют собой, если не указано иное, массовые проценты (мас.%), а выраженные в частях данные представляют собой массовые части. Соотношения между растворителями, степень разбавления и данные о концентрации жидкостно-жидкостных растворов в каждом случае относятся к объему.
А. Примеры
Сокращения
Литература
Названия пептидов и циклодепсипептидов даны в соответствии со следующей номенклатурой:
1. A Guide to IUPAC Nomenclature of Organic Compounds (Recommendations 1993), изд-во Blackwell Scientific publications, 1993;
2. Nomenclature and symbolism for amino acids and peptides. Recommendations 1983. IUPAC-IUB Joint Commission on Biochemical Nomenclature, UK. Biochemical Journal, 219, 1984, с. 345-373, включая цитируемую литературу.
Общие методы ГХ-МС, ЖХ-МС МС-ВР, ЖХВД и гель-хроматографии
Метод 1 (ВП-МС-ВР): BП-MC-BP(ESI+)-спектры регистрируют с помощью прибора Micromass LCT (напряжение на капилляре 3,2 кВ, напряжение на конусе 42 В, температура источника 120°С, температура десольвации 280°С). Для подачи образцов используют шприц-насос (фирма Harvard Apparatus). Стандартом служит лейцин-энкефалин (Tyr-Gly-Gly-Phe-Leu).
Метод 2 (препаративная ЖХВД): Прибор: Gilson Abimed HPLC; УФ-детектор на 210 нм; система из двух насосов; колонка: Waters SymmetryPrepTM C18, 7 мкм, 300×19 мм; элюент А: 0,2% трифторуксусной кислоты в воде, элюент Б: ацетонитрил; скорость потока 25 мл/мин; температура колонки: КТ; 0-я минута 20% элюента Б, увеличение концентрации элюента Б с 0-й по 10-ю до 70%, уменьшение концентрации элюента Б с 10-й по 10,1-ю минуту до 20%, по 15-ю минуту 20% элюента Б.
Метод 3 (гель-хроматография на сорбенте Sephadex LH-20): Гель-хроматографию проводят без давления на сорбенте Sephadex LH-20 (фирма Pharmacia). После регистрации УФ-активности (УФ-детектор на 254 нм, фирма Knauer) фракционируют (коллектор фракций ISCO Foxy 200). Размеры колонки: 32×7 см (для интервала концентраций 1000-100 мкмолей), 30×4 см (для интервала концентраций 100-10 мкмолей) и 25×2 см (для интервала концентраций 10-1 мкмоль). Для интервала концентраций от 1 до 11 ммолей используют колонку размером 80×30 см. В этом случае фракции собирают вручную без использования предвключенного УФ-детектора. Соотнесение фракций осуществляют с помощью ЖХВД (метод 9).
Метод 4 (препаративная ЖХВД, сорбент Kromasil, уксусная кислота): Прибор: Gilson Abimed HPLC; УФ-детектор на 210 нм; система с двойным насосом; колонка: Kromasil-100A C18, 5 мкм, 250×20 мм; скорость потока 25 мл/мин; элюент А: вода/0,25-0,5% уксусной кислоты, элюент Б: ацетонитрил; градиент: с 0-й по 3-ю минуту 5% элюента Б, с 3-й по 30-ю минуту 5-100% элюента Б, с 30-й по 38-ю минуту 100% элюента Б, в завершение регенерация хроматографической колонки.
Метод 5 (ЖХ-МС): Прибор: Micromass Quattro LCZ в сочетании с HPLC Agilent Serie 1100; колонка: Phenomenex Synergi 2µ Hydro-RP Mercury, 20×4 мм; элюент А: 1 л воды+0,5 мл 50%-ной муравьиной кислоты, элюент Б: 1 л ацетонитрила+0,5 мл 50%-ной муравьиной кислоты; градиент: с 0,0-й минуты 90% элюента А → с 2,5-й минуты 30% элюента А → с 3,0-й минуты 5% элюента А → с 4,5-й минуты 5% элюента А; скорость потока: с 0,0-й минуты 1 мл/мин, с 2,5-й/с 3,0-й/с 4,5-й минуты 2 мл/мин; печь: 50°С; УФ-обнаружение на 208-400 нм.
Метод 6 (ЖХ-МС): Тип прибора для МС: Micromass ZQ; тип прибора для ЖХВД: Waters Alliance 2795; колонка: Phenomenex Synergi 2µ Hydro-RP Mercury, 20×4 мм; элюент А: 1 л воды +0,5 мл 50%-ной муравьиной кислоты, элюент Б: 1 л ацетонитрила +0,5 мл 50%-ной муравьиной кислоты; градиент: с 0,0-й минуты 90% элюента А → с 2,5-й минуты 30% элюента А → с 3,0-й минуты 5% элюента А → с 4,5-й минуты 5% элюента А; скорость потока: с 0,0-й минуты 1 мл/мин, с 2,5-й/с 3,0-й/с 4,5-й минуты 2 мл/мин; печь: 50°С; УФ-обнаружение на 210 нм.
Метод 7 (ЖХ-МС): Тип прибора для МС: Micromass ZQ; тип прибора для ЖХВД: HP 1100 Series; УФ-детектор с диодной матрицей (УФ-ДДМ); колонка: Phenomenex Synergi 2µ, Hydro-RP Mercury, 20×4 мм; элюент А: 1 л воды +0,5 мл 50%-ной муравьиной кислоты, элюент Б: 1 л ацетонитрила +0,5 мл 50%-ной муравьиной кислоты; градиент: с 0,0-й минуты 90% элюента А → с 2,5-й минуты 30% элюента А → с 3,0-й минуты 5% элюента А → с 4,5-й минуты 5% элюента А; скорость потока: с 0,0-й минуты 1 мл/мин, с 2,5-й/с 3,0-й/с 4,5-й минуты 2 мл/мин; печь: 50°С; УФ-обнаружение на 210 нм.
Метод 8 (аналитическая ЖХВД): Тип прибора для ЖХВД: HP 1050 Series; УФ-ДДМ: UV DAD 1100 Series; колонка: Kromasil C18, 60×2 мм, 3,5 мкм; элюент А: вода/0,5% перхлорной кислоты, элюент Б: ацетонитрил; градиент: с 0-й по 0,5-ю минуту 2% элюента Б, с 0,5-й по 4,5-ю минуту с 2 до 90% элюента Б, с 4,5-й по 9,0-ю минуту 90% элюента Б, с 9,0-й по 9,2-ю минуту с 90 до 2% элюента Б, с 9,2-й по 10,0-ю минуту 2% элюента Б; скорость потока: 0,75 мл/мин, печь: 30°С, УФ-обнаружение на 210 нм.
Метод 9 (аналитическая ЖХВД, сорбент Agilent Zorbax Cg): Прибор: Agilent 1100 в сочетании с ДДМ (G 1315 В), сдвоенный насос (G1312A), автоматический пробоотборник (G 1313 А), дегазатор растворителя (G1379A) и термостат хроматографической колонки (G1316A); колонка: Agilent Zorbax Eclipse XDB-C8, 4,6×150×5 мм; элюент А: 0,05% 70%-ной перхлорной кислоты в воде; элюент Б: ацетонитрил; градиент: с 0-й по 1-ю минуту 10% элюента Б, увеличение концентрации элюента Б, с 4-й по 5-ю минуту 90% элюента Б, уменьшение концентрации элюента Б, по 5,5-ю минуту 10% элюента Б; скорость потока: 2,00 мл/мин; температура колонки: 30°С.
Метод 10 (гель-хроматография на сорбенте Sephadex LH-20): Гель-хроматографию проводят без давления на сорбенте Sephadex LH-20 (фирма Pharmacia). После регистрации УФ-активности (УФ-детектор на 254 нм, фирма Knauer) фракционируют (коллектор фракций ISCO Foxy 200). Размеры колонки: 32×7 см (для интервала концентраций 1000-100 мкмолей), 30×4 см (для интервала концентраций 100-10 мкмолей) и 25×2 см (для интервала концентраций 10-1 мкмоль).
Метод 11 (препаративная ЖХВД, сорбент Symmetry): Прибор: Gilson Abimed HPLC; система с двойным насосом; колонка: SymmetryPrepTM C18, фирма Waters, 7 мкм, 300×19 мм; элюент А: вода/0,2% трифторуксусной кислоты, элюент Б: ацетонитрил; градиент: с 0-й по 10-ю минуту увеличение концентрации элюента Б с 15 до 65%, в завершение регенерация хроматографической колонки; скорость потока: 25 мл/мин; УФ-обнаружение на 210 нм.
Метод 12 (препаративная ЖХВД, сорбент Kromasil): Прибор: Gilson Abimed HPLC; система с двойным насосом; колонка: Kromasil C18, 5 мкм, 100 Å, 250×20 мм; элюент А: 0,05% трифторуксусной кислоты в воде, элюент Б: 0,05% трифторуксусной кислоты в ацетонитриле; градиент: с 0-й по 3-ю минуту 10% элюента Б, увеличение концентрации элюента Б, с 30-й по 38-ю минуту 90% элюента Б, с 38-й по 45-ю минуту 10% элюента Б; скорость потока: 20 мл/мин; УФ-обнаружение на 210 нм.
Метод 13 (препаративная ЖХВД, сорбент Waters Symmetry): Прибор: Gilson Abimed HPLC; система с двойным насосом; колонка: Waters SymmetryPrepТМ C18, 7 мкм, 300×19 мм; элюент А: 0,05% трифторуксусной кислоты в воде, элюент Б: 0,05% трифторуксусной кислоты в ацетонитриле; градиент: с 0-й по 3-ю минуту 10% элюента Б, увеличение концентрации элюента Б, с 30-й по 38-ю минуту 90% элюента Б, с 38-й по 45-ю минуту 10% элюента Б; скорость потока: 20 мл/мин; УФ-обнаружение на 210 нм.
Метод 14 (препаративная ЖХВД): Прибор: Gilson Abimed HPLC; система с двойным насосом; колонка: Waters SymmetryPrepTM C18, 7 мкм, 300×19 мм; элюент А: вода/0,2% трифторуксусной кислоты, элюент Б: ацетонитрил; градиент: с 0-й по 10-ю минуту увеличение концентрации элюента Б с 25 до 65%, в завершение регенерация хроматографической колонки; скорость потока: 25 мл/мин; УФ-обнаружение на 210 нм.
Метод 15 (хиральная ЖХВД, сорбент Daicel Chiralpak): Прибор: Agilent 1100 HPLC; колонка: Daicel Chiralpak AD-H, 5 мкм, 250×20 мм; изократически: 75% изогексана, 25% 2-пропанола с 0,2% трифторуксусной кислоты и 1% воды; скорость потока: 1,0 мл/мин; печь: 25°С; УФ-детектор на 212 нм.
Метод 16 (препаративная ЖХВД): Прибор: Gilson Abimed HPLC; система с двойным насосом; колонка: YMC ODS-AQ, 5 мкм, 250×30 мм; элюент А: 0,05% трифторуксусной кислоты в воде, элюент Б: 0,05% трифторуксусной кислоты в ацетонитриле; градиент: с 0-й по 3-ю минуту 10% элюента Б, увеличение концентрации элюента Б, с 30-й по 38-ю минуту 90% элюента Б, с 38-й по 45-ю минуту 10% элюента Б; скорость потока: 50 мл/мин; УФ-детектор на 210 нм.
Метод 17 (ГХ-МС): Прибор: Micromass GCT, GC6890; колонка: Restek RTX-35MS, 30 м на 250 мкм на 0,25 мкм; градиент: 60°С (выдержка в течение 0,30 мин), 50°С/мин → 120°С, 16°С/мин → 250°С, 30°С/мин → 300°С (выдержка в течение 1,7 мин); постоянная скорость потока с гелием: 0,88 мл/мин; печь: 60°С; входная температура: 250°С.
Метод 18 (ЖХВД): Тип прибора для ЖХВД: HP 1100 Series; колонка с ЕА-LLV: Zorbax Eclipse XBD-C8 (фирма Agilent), 150×4,6 мм, 5 мкм; элюент А: 5 мл НСlO4 на 1 л воды, элюент Б: ацетонитрил; градиент: с 0-й по 1-ю минуту 10% элюента Б, с 1-й по 4-ю минуту увеличение концентрации элюента Б с 10 до 90%, с 4-й по 5-ю минуту 90% элюента Б; скорость потока: 2,0 мл/мин; печь: 30°С; УФ-обнаружение на 210 и 254 нм.
Метод 19 (ЖХВД): Колонка: Kromasil RP-18, 60×2 мм, 3,5 мкм; элюент А: 5 мл НСlO4 на 1 л воды, элюент Б: ацетонитрил; градиент: с 0-й минуты 2% элюента Б, с 0,5-й минуты 2% элюента Б, с 4,5-й минуты 90% элюента Б, по 9-ю минуту 90% элюента Б; скорость потока: 0,75 мл/мин; печь: 30°С; УФ-обнаружение на 210 нм.
Метод 20 (ЖХВД): Колонка: Kromasil RP-18, 250×4 мм, 5 мкм; элюент А: 5 мл НСlO4 на 1 л воды, элюент Б: ацетонитрил; градиент: с 0-й минуты 5% элюента Б, с 10-й минуты 95% элюента Б; скорость потока: 1 мл/мин; печь: 40°С; УФ-обнаружение на 210 нм.
Метод 21 (ЖХВД): Колонка: Kromasil RP-18, 250×4 мм, 5 мкм; элюент А: 2 мл НСlO4 на 1 л воды, элюент Б: ацетонитрил; изократически: 45% элюента Б, 55% элюента А; скорость потока: 1 мл/мин; печь: 40°С; УФ-обнаружение на 210 нм.
Метод 22 (ЖХ-МС): Тип прибора для МС: Micromass ZQ; тип прибора для ЖХВД: HP 1100 Series; УФ-ДДМ; колонка: Grom-Sil 120 ODS-4 HE, 50×2 мм, 3,0 мкм; элюент А: вода/0,025% муравьиной кислоты на литр, элюент Б: ацетонитрил/0,025% муравьиной кислоты; градиент: с 0-й по 2,9-ю минуту увеличение концентрации элюента Б с 0 до 70%, с 2,9-й по 3,1-ю минуту увеличение концентрации элюента Б с 70 до 90%, с 3,1-й по 4,5-ю минуту 90% элюента Б; печь: 50°С, скорость потока: 0,8 мл/мин, УФ-обнаружение на 210 нм.
Метод 23 (ЖХВД): Тип прибора для ЖХВД: HP 1050 Series; УФ-ДДМ UV DAD 1100 Series; колонка: SyMMetryPrepTM C18, фирма Waters, 50×2,1 мм, 3,5 мкм; элюент А: вода/0,05% трифторуксусной кислоты, элюент Б: ацетонитрил; градиент: с 0-й по 9-ю минуту увеличение концентрации элюента Б с 0 до 100%, с 9-й по 11-ю минуту 100% элюента Б, с 11-й по 12-ю минуту уменьшение концентрации элюента Б с 100 до 0%, в завершение регенерация хроматографической колонки; печь: 40°С, скорость потока: 0,4 мл/мин, УФ-обнаружение на 210 нм.
Метод 24 (количественная 19F-ЯМР-спектроскопия): Примерно 10 мг точно взвешенного образца анализируемого вещества и примерно 20 мг точно взвешенного 1,4-дибромтетрафторбензола растворяют в пиридине и анализируют 19F-ЯМР-спектроскопией. δ -74 (ТФУК) и -132,0 (1,4-дибромтетрафторбензол) интегрируют и сравнивают. Содержание ТФУК указывают в процентах от массы образца анализируемого вещества.
Метод 25 (ионная хроматография): Система для ионной хроматографии с подавителем и детектором электропроводности; предварительная колонка: А SUPP 4/5 Guard, разделительная колонка: A SUPP 5, 4,0×250 мм; элюент: 3,2-миллимолярный карбонат натрия и 2,4-миллимолярный гидрокарбонат натрия в воде; скорость потока: 0,7 мл/мин. Образец растворяют в метаноле (20% от окончательного объема образца), в течение 3 мин обрабатывают в ультразвуковой бане и добавлением воды объем доводят до окончательного. Образец фильтруют через не содержащий ионов фильтр из ацетата целлюлозы (с размером пор 0,45 мкм) и инжектируют в колонку. Количественный анализ проводят по отношению к внешним стандартам (0,5-10 мг/л).
Исходные соединения
Пример 1А: Бистрифторацетат амида D-лейцил-N1-{(3S,6S,12S,15S,18R,21S,24S,27S,28R)-6-[(1S)-2-амино-1-гидрокси-2-оксоэтил]-18-(3-{[амино(имино)метил]амино}пропил)-12-[(1S)-1-гидроксиэтил]-3-(гидроксиметил)-24-[(1R)-1-гидрокси-2-метилпропил]-21-изобутил-15-[(1S)-1-метилпропил]-2,5,8,11,14,17,20,23,26-нонаоксо-28-фенил-1-окса-4,7,10,13,16,19,22,25-октаазациклооктакозан-27-ил}-L-лейцина (лизобактин)
Ферментация
Культуральная среда
YM-среда. Агар с дрожжевым и солодовым экстрактами: D-глюкоза (4 г/л), дрожжевой экстракт (4 г/л), солодовый экстракт (10 г/л), 1 литр очищенной пропусканием через леватит воды. Перед стерилизацией (20 минут при 121°С) значение рН устанавливают на 7,2.
НРМ-среда. Маннит (5,4 г/л), дрожжевой экстракт (5 г/л), мясной пептон (3 г/л).
Рабочий законсервированный штамм. Лиофилизированный штамм (АТСС 53042), предварительно выращенный в 50 мл YM-среды.
Ферментация в колбе: В 150 мл YM-среды или 100 мл НРМ-среды в 1-литровой колбе Эрленмейера высевают 2 мл рабочего законсервированного штамма и выращивают в течение 30-48 ч при 28°С на шюттель-аппарате при 240 об/мин.
Ферментация в объеме 30 л: 300 мл выращенной путем ферментации в колбе культуры (НРМ-среда) высевают в 30 л стерильного раствора питательной среды (с 1 мл антивспенивателя Antifoam SAG 5693/1). Эту культуру выращивают в течение 21 ч при 28°С, 300 об/мин и вентилировании стерильным воздухом с расходом 0,3 объема газа на объем жидкости в минуту (об./об./мин). В процессе культивирования значение рН поддерживают постоянным и равным 7,2 с помощью 1-молярной соляной кислоты. В процессе культивирования расходуют в общей сложности 880 мл 1 молярной соляной кислоты.
Основная культура (200 л): В YM-среду, распределенную порциями по 150 мл по 15 1-литровым колбам Эрленмейера, высевают по 2 мл рабочего законсервированного штамма и выращивают в течение 48 ч при 28°С на шюттель-аппарате при 240 об/мин. 2250 мл этой культуры высевают в 200 л стерильного раствора питательной среды (YM-среды) (с 1 мл антивспенивателя Antifoam SAG 5693/1) и выращивают в течение 18,5 ч при 28°С, 150 об/мин и вентилировании стерильным воздухом с расходом 0,3 об./об./мин.
Для контроля процесса ферментации ежечасно отбирают пробы (по 50 мл). 2 мл этого содержащего выращенную культуру бульона смешивают с 1 мл метанола (с 0,5% трифторуксусной кислоты) и фильтруют через фильтр с размером пор 0,45 мкм. 30 мкл этой суспензии анализируют с помощью ЖХВД (метод 18 и метод 19).
По истечении 18,5 ч содержащий основную выращенную культуру бульон разделяют центрифугированием при 17000 об/мин на надосадочную жидкость (супернатант)и осадок.
Выделение
Над осадочная жидкость (183 л): Надосадочную жидкость после установления значения ее рН на 6,5-7 добавлением концентрированной трифторуксусной кислоты, соответственно раствора едкого натра вносят в колонку с ионитом Lewapol (ОС 1064, содержимое 60 л). После этого элюируют чистой водой, затем смесью воды с метанолом в соотношении 1:1 и в завершение чистым метанолом (с 0,1% трифторуксусной кислоты).
Элюированную органическую фазу концентрируют в вакууме до водного остатка объемом 11,5 л.
Оставшуюся водную фазу связывают с силикагелем C18 и разделяют (ЖХСД, Biotage Flash 75, 75×30 см, KP-C18-WP, 15-20 мкм, скорость потока: 30 мл; элюент: ацетонитрил/вода с 0,1% трифторуксусной кислоты; градиент: 10%, 15% и 40% ацетонитрила). Ацетонитрильную фазу, элюированную при 40%-ной концентрации ацетонитрила и содержащую основное количество указанного в заголовке примера 1А соединения, концентрируют в вакууме и затем лиофилизируют (примерно 13 г). Эту твердофазную смесь разделяют порциями по 1,2 г сначала с помощью препаративной ЖХВД (метод 7), затем путем гель-фильтрации на колонке с сорбентом Sephadex LH-20 (5×70 см, ацетонитрил/вода в соотношении 1:1, в каждом случае с 0,05% трифторуксусной кислоты) и после этого с помощью другой препаративной ЖХВД (метод 20).
Таким путем получают 2250 мг указанного в заголовке примера 1А соединения.
Осадок. Осадок от центрифугирования растворяют в 4 л смеси ацетона и воды в соотношении 4:1, смешивают с 2 кг целита, значение рН добавлением трифторуксусной кислоты устанавливают на 6, размешивают и центрифугируют.Растворитель выпаривают в вакууме и остаток сушат вымораживанием. Полученный лиофилизат (89,9 г) растворяют в метаноле, фильтруют, концентрируют и разделяют на силикагеле (метод 21). Затем указанное в заголовке примера 1А соединение очищают путем гель-фильтрации (колонка с сорбентом Sephadex LH-20, 5×68 см, вода/ацетонитрил в соотношении 9:1 (с 0,05% трифторуксусной кислоты), скорость потока 2,7 мл/мин, объем фракции 13,5 мл) с получением чистого вещества.
Таким путем получают 447 мг указанного в заголовке примера 1А соединения.
ЖХВД (метод 18): Rt=6,19 мин.
МС (ESIpos): m/z=1277 (М+Н)+.
1Н-ЯМР (500,13 МГц, d6-ДМСО): δ=0,75 (d, 3Н), 0,78 (d, 6H), 0,80 (t, 3H), 0,82 (d, 3Н), 0,90 (d, 3Н), 0,91 (d, 3Н), 0,92 (d, 3Н), 0,95 (d, 3Н), 0,96 (d, 3H), 1,05 (m, 1H), 1,19 (d, 3Н), 1,25 (m, 2H), 1,50 (m, 4H), 1,51 (m, 2H), 1,55 (m, 1H), 1,61 (m, 1H), 1,65 (m, 1H), 1,84 (m, 1H), 1,85 (m, 1H), 1,86 (m, 1H), 1,89 (m, 1H), 1,95 (m, 1H), 2,75 (m, 2H), 3,40 (m, 1H), 3,52 (m, 2H), 3,53 (dd, 1H), 3,64 (m, 2H). 3,66 (m, 1H), 3,68 (dd, 1H), 3,73 (m, 2H), 4,00 (dd, 1H), 4,02 (шир., 1H), 4,13 (шир., 1H), 4,32 (dd, 1H), 4,39 (t, 1H), 4,55 (m, 1H), 4,75 (dd, 1H), 5,19 (t, 1H), 5,29 (d, 1H), 5,30 (шир., 1H), 5,58 (m, 2H), 6,68 (m, 3Н), 6,89 (d, 1H), 6,93 (m, 3H), 6,94 (шир., 1H), 6,98 (d, 1H), 7,12 (шир., 1H), 7,20 (шир., 2H), 7,23 (m, 2H), 7,42 (m, 2H), 7,54 (d, 1H), 7,58 (d, 1H), 8,32 (шир., 1H), 9,18 (шир., 1H), 9,20 (m, 2H), 9,50 (шир., 1H).
13С-ЯМР (125,77 МГц, d6-ДМСО): δ=10,3, 15,3, 19,0, 19,2, 19,6, 20,0, 20,9, 22,0, 22,4, 23,0, 23,2, 24,3, 24,4, 25,0, 25,4, 26,0, 27,8, 30,9, 35,4, 39,5, 40,8, 40,9, 41,6, 44,1, 51,5, 52,7, 55,9, 56,2, 56,4, 57,9, 58,8, 60,2, 61,1, 62,6, 70,1, 71,6, 71,7, 75,5, 128,1, 128,6, 136,7, 156,8, 168,2, 170,1, 170,4, 171,2, 171,5, 171,9, 172,2, 172,4, 173,7.
Соотнесение сигналов осуществляли в соответствии с литературными источниками (Т.Kato, H.Hinoo, Y. Terui, J. Antibiot., 61, 1988, cc.719-725).
Пример 2А: Бистрифторацетат дез(1-D-лейцил-2-L-лейцил)лизобактина (продукт деградации по Эдману2,0)
Бистрифторацетат лизобактина (60,0 г, 39,88 ммоля) в атмосфере аргона растворяют в пиридине (840 мл). Затем добавляют фенилизотиоцианат (32,35 г, 239,28 ммоля, 6 эквивалентов) и реакционную смесь перемешивают при 37°С в течение 7 ч. После этого растворитель отгоняют на роторном испарителе при температуре бани 40°С. Остаток смешивают с метил-трет-бутиловым эфиром (1400 мл) и интенсивно перемешивают в течение 30 мин. Затем подвергают вакуум-фильтрации через стеклянную фритту (размер пор 3, диаметр 13 см). Таким путем выделяют 72 г промежуточного продукта (продукт деградации по Эдману0,5), который без переработки используют в последующей реакции.
Для этого сырой продукт в атмосфере аргона растворяют в трифторуксусной кислоте (1026 мл) и перемешивают в течение 30 мин при КТ. Затем раствор концентрируют в вакууме на роторном испарителе при температуре бани 20°С. Остаток растворяют в метил-трет-бутиловом эфире (1400 мл) и интенсивно перемешивают до образования порошкообразного аморфного твердого вещества. Это твердое вещество отфильтровывают вакуум-фильтрацией через фритту (размер пор 3, диаметр 18 см). После этого твердое вещество перемешивают с диэтиловым эфиром (1400 мл) и вновь отфильтровывают. Подобную процедуру повторяют с 2 порциями дихлорметана (по 900 мл). Сырой продукт сушат в вакууме. Таким путем получают 58 г сырого бистрифторацетата дез(1-D-лейцил)лизобактина (продукт деградации по Эдману1,0).
Полученный сырой продукт без дополнительной очистки растворяют в атмосфере аргона в пиридине (1080 мл). Затем добавляют фенилизотиоцианат (107 г, 0,80 моля, 20 эквивалентов) и реакционную смесь перемешивают при 37-40°С в течение 7 ч. После этого растворитель отгоняют на роторном испарителе при температуре бани 40°С. Остаток смешивают с метил-трет-бутиловым эфиром (1400 мл) и интенсивно перемешивают. Затем подвергают вакуум-фильтрации через стеклянную фритту (размер пор 3, диаметр 13 см). Таким путем выделяют 65 г сырого промежуточного продукта (продукт деградации по Эдману1,5), который после сушки в вакууме, создаваемом масляным насосом, в атмосфере аргона непосредственно растворяют в трифторуксусной кислоте (1240 мл) и перемешивают в течение 30 мин при КТ. После этого раствор концентрируют на роторном испарителе в вакууме при температуре бани 20°С. Остаток растворяют в метил-трет-бутиловом эфире (1400 мл) и интенсивно перемешивают до образования порошкообразного аморфного твердого вещества. Это твердое вещество отфильтровывают вакуум-фильтрацией через фритту (размер пор 3, диаметр 18 см). После этого твердое вещество перемешивают сначала с диэтиловым эфиром (1400 мл), а затем с дихлорметаном (1400 мл), отфильтровывая после каждой операции перемешивания. Таким путем получают 55 г сырого продукта. Этот продукт очищают с помощью препаративной ЖХВД (метод 14). Таким путем получают 28,55 г (56% от теории) указанного в заголовке соединения.
ЖХВД/УФ-видим. (метод 23): Rt=4,71 мин.
λmax (качественно) = 220 нм (s), 255-270 (w).
ЖХ-МС (метод 22): Rt=1,65 мин; МС (ESIpos): m/z (%) = 526 (100) [M+2H]2+, 1051 (15) [M+H]+.
Пример 3А: Метиловый эфир (2Z)-2-{[(бензилокси)карбонил]амино}-3-(6-трифторметилпиридин-3-ил)акриловой кислоты

6-Трифторметилпиридин-3-карбальдегид (4,85 г, 27,70 ммоля) и метил-{[(бензилокси)карбонил]амино}(диметоксифосфорил)ацетат (9,17 г, 27,70 ммоля, 1,0 эквивалент) растворяют в ТГФ (70 мл) и охлаждают до -70°С. При -70°С по каплям медленно добавляют N,N,N,N-тетраметилгуанидин (6,38 г, 55,39 ммоля, 6,95 мл, 2,0 эквивалента) и после этого перемешивают сначала в течение 4 ч при -70°С, а затем в течение 12 ч при КТ. Далее реакционную смесь концентрируют, после чего экстрагируют этилацетатом (2 раза по 100 мл) путем встряхивания с водой, объединенные органические фазы промывают насыщенным раствором поваренной соли и сушат над сульфатом натрия. Полученный после концентрирования в вакууме сырой продукт хроматографируют (силикагель, элюент: толуол, затем толуол/этилацетат в соотношении 10:1). Таким путем получают 6,93 г (66% от теории) указанного в заголовке соединения.
ЖХВД/УФ-видим. (метод 8): Rt=4,60 мин.
ЖХВД/УФ-видим. (метод 9): Rt=4,54 мин.
1Н-ЯМР (400 МГц, d6-ДМСО): δ=3,74 (s, 3Н, ОМе), 5,10 (s, 2H, СH2), 7,27 (s, 1Н, PyrH), 7,33-7,38 (m, 5H, ArH), 7,94 (d,J=8,5 Гц, 1Н, PyrH), 8,27 (d, J=8,5 Гц, 1Н, PyrH), 8,93 (s, 1Н, P-CH), 9,51 (s, 1Н, NH).
ЖХ-МС (метод 7): Rt=2,44 мин; МС (ESIpos): m/z (%) = 381 (100) [M+Н]+; MC (ESIneg): m/z (%) = 379 (100) [М-Н]-.
ВП-МС-ВР (метод 1): C18H16N2O4F3 [M+H]+ рассч. 381,1062, обнаруж. 381,1065.
Пример 4А: Метиловый эфир N-[(бензилокси)карбонил]-3-(6-трифторметилпиридин-3-ил)-L-аланина
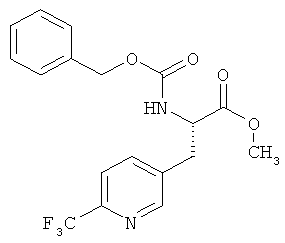
Соединение из примера 3А (10,15 г, 26,69 ммоля) растворяют в метаноле а.ч. (100 мл). Далее через полученный раствор с помощью канюли в течение примерно 5 мин пропускают аргон, после чего добавляют трифлат (+)-1,2-бис-[(2S,5S)диэтилфосфолано]бензол(циклооктадиен)родия(I) (289 мг, 400 мкмолей, 0,015 эквивалента). Затем гидрируют в течение 12 ч при давлении водорода 4 бара и КТ. После этого фильтруют через кизельгур (метанол) и элюат концентрируют. Сырой продукт хроматографируют (силикагель, элюент: толуол/этилацетат в соотношении 5:1). Таким путем получают 9,9 г (97% от теории) указанного в заголовке соединения.
[α]20 Na=-24° (с=0,093 в метаноле).
ЖХВД/УФ-видим. (метод 8): Rt=4,50 мин.
ЖХВД/УФ-видим. (метод 9): Rt=4,49 мин.
1Н-ЯМР (400 МГц, d6-ДМСО): δ=2,99 (dd, J=3,5, 11,0 Гц, 1Н, β-СН), 3,22 (dd, J=3,5, 11,0 Гц, 1Н, β-СН), 3,66 (s, 3Н, ОМе), 4,40 (m, 1Н, α-CH), 4,97 (s, 2H, СН2), 7,23 (m, 2H), 7,29-7,33 (m, 3Н), 7,83 (d, J=6,5 Гц, 1H), 7,93-7,98 (m, 2H), 8,65 (s, 1H, NH).
ЖХ-МС (метод 7): Rt=2,40 мин; МС (ESIpos): m/z (%) = 383 (100) [M+H]+; MC (ESIneg): m/z (%) = 273 (100), 381 (50) [M-H]-.
ВП-МС-ВР (метод 1): C18H18N2O4F3 [M+H]+ рассч. 383,1219, обнаруж. 383,1223.
Пример 5А: Метиловый эфир 3-(6-трифторметилпиридин-3-ил)-L-аланина
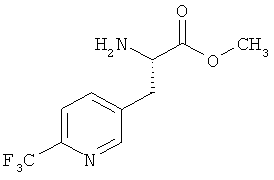
Соединение из примера 4А (9,90 г, 25,89 ммоля) растворяют в метаноле (100 мл). Далее через полученный раствор с помощью канюли в течение примерно 5 мин пропускают аргон, после чего добавляют Pd на угле (10%-ный, 990 мг). Затем гидрируют в течение 12 ч при давлении водорода 4 бара и КТ. После этого фильтруют через кизельгур, концентрируют и сушат в вакууме, создаваемом масляным насосом. Выход: 5,8 г (90% от теории) указанного в заголовке соединения.
[α]19,9 Na=+3° (с=0,186 в метаноле).
ЖХВД/УФ-видим. (метод 8): Rt=3,34 мин.
ЖХВД/УФ-видим. (метод 9): Rt=3,22 мин.
ИК νmax (NaCl, cм-l): 3415, 1734, 1339, 1136, 1087.
1H-ЯМР (500 МГц, d6-ДМСО): δ=2,85 (dd, J=5,5, 13,5 Гц, 1Н, β-СН), 3,01 (dd, J=5,5, 13,5 Гц, 1H, β-СН), 3,61 (s, 3Н, ОМе), 3,63-3,69 (m, 1H, α-СН), 7,82 (d, J=7,5 Гц, 1H), 7,93 (d, J=7,5 Гц, 1H), 8,61 (s, 1H).
ЖХ-МС (метод 7): Rt=1,74 мин; МС (ESIpos): m/z (%) = 249 (100) [M+H]+.
ВП-МС-ВР (метод 1): С12Н15N3О2F3 [M+CH3CN+H]+ рассч. 290,1116, обнаруж. 290,1122.
Пример 6А: Метиловый эфир N-(трет-бутоксикарбонил)-3-(трет-бутил)-D-аланил-3-(6-трифторметилпиридин-3-ил)-L-аланина
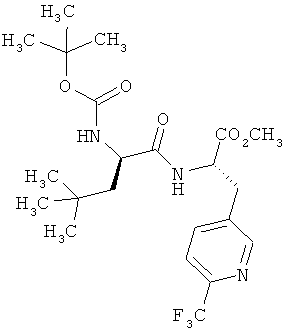
К раствору соединения из примера 5А (6,34 г, 25,54 ммоля) и N-(трет-бутоксикарбонил)-3-трет-бутил-D-аланина (6,27 г, 25,54 ммоля, 1,0 эквивалент) в сухом ДМФ (240 мл) при -30°С медленно добавляют N-метилморфолин (12,92 г, 127,72 ммоля, 14,04 мл, 5 эквивалентов) и ГАТУ (9,71 г, 25,54 ммоля, 1 эквивалент). Реакционной смеси дают медленно нагреться (в течение примерно 3 ч) до КТ, при этом полноту реакции контролируют с помощью ЖХВД (метод 9). Затем добавляют дигидрофосфат калия (34,76 г, 255,44 ммоля, 10 эквивалентов) и реакционную смесь перемешивают в течение 20 мин, после чего фильтруют, разбавляют этилацетатом и промывают насыщенным водным раствором гидрокарбоната натрия (10 мл). Органические фазы сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают экспресс-хроматографией (силикагель, градиент циклогексан/этилацетат в соотношении от 10:1 до 2:1), получая 9,74 г (73% от теории) указанного в заголовке соединения.
[α]19,9=+7,0° (с=0,044 в метаноле).
ЖХВД/УФ-видим. (метод 8): Rt=4,89 мин.
ЖХВД/УФ-видим. (метод 9): Rt=4,75 мин.
ИК νmax (NaCl, cм-l): 2959, 1742, 1655, 1520, 1336, 1160, 1136, 1087, 1050, 1027.
1Н-ЯМР (500 МГц, d6-ДМСО): δ=0,74 (s, 9H, tBu), 0,97-1,00 (m, 1H, β-CH2), 1,20-1,25 (m, 1H, β-СH2), 1,35 (s, 9H, OtBu), 2,99-3,05 (m, 1H, β-CH2), 3,23-3,26 (m, 1H, β-СН2), 3,66 (s, 3H, ОМе), 3,94 (m, 1H, α-CH), 4,60 (m, 1H, α-СН), 6,82 (d, J=8,5 Гц, 1H, NH), 7,78 (d, J=8,0 Гц, 1H, РуrН), 7,94 (d, J=8,0 Гц, 1H, PyrH), 8,34 (d, J=8,5 Гц, 1H, NH), 8,64 (s, 1H, РуrН).
ЖХ-МС (метод 7): Rt=2,67 мин; МС (ESIpos): m/z (%) = 476 (100) [M+H]+; MC (ESIneg): m/z (%) = 400 (80), 474 (40) [M-H]-.
ВП-МС-ВР (метод 1): С22Н33N3O5F3 [M+H]+ рассч. 476,2372, обнаруж. 476,2364.
Пример 7А: N-трет-бутоксикарбонил-3-трет-бутил-D-аланил-3-(6-трифторметилпиридин-3-ил)-L-аланин
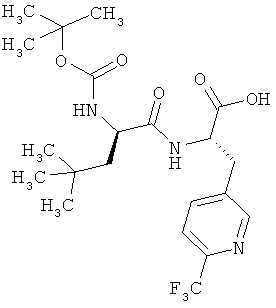
К раствору соединения из примера 6А (9,2 г, 1,0 эквивалент, 19,35 ммоля) в ТГФ (360 мл) и воде (100 мл) при -20°С добавляют раствор гидрата гидроксида лития (1,16 г, 2,5 эквивалента, 48,37 ммоля) в воде (20 мл). Реакционной смеси дают нагреться (в течение примерно 1,5 ч) до +15°С, при этом полноту реакции контролируют с помощью ЖХВД (метод 9). Для переработки добавляют дигидрофосфат калия (26,33 г, 10 эквивалентов, 193,5 ммоля) (рН около 7). Реакционную смесь фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией (метод 10, элюент: метанол/ацетон в соотношении 4:1), получая 4,72 г (53% от теории) продукта.
[α]20 Na=+51,3° (с=0,402 в метаноле).
ЖХВД/УФ-видим. (метод 8): Rt=4,63 мин.
ЖХВД/УФ-видим. (метод 9): Rt=4,55 мин.
ИК νmax (NaCl, см-1): 3305, 2959, 1663, 1519, 1336, 1173, 1134, 1086.
1Н-ЯМР (500 МГц, d6-ДМСО): δ=0,77 (s, 9Н, tBu), 1,06-1,13 (m, 1H, β-CH2), 1,23-1,26 (m, 1H, β-CH2), 1,34 (s, 9Н, OtBu), 3,01 (tapp, J=11,0 Гц, 1H, β-CH2), 3,23 (шир. d, J=11,0 Гц, 1Н, β-СН2), 3,940, J=8,0 Гц, 1H, α-СН), 4,42 (шир. s, 1Н, α-СН), 6,90 (d, J=8,5 Гц, 1H, NH), 7,74 (d, J=7,5 Гц, 1H, PyrH), 7,86 (d, J=7,5 Гц, 1H, PyrH), 8,00 (шир. s, 1H, NH), 8,56 (s, 1H, PyrH).
ЖХ-МС (метод 7): Rt=2,42 мин; МС (ESIpos): m/z (%) = 406 (100), 462 (85) [M+H]+; MC (ESIneg): m/z (%) = 460 (100) [M-H]-.
ВП-МС-ВР (метод 1): С21H31N3О5F3 [М+Н]+ рассч. 462,2216, обнаруж. 462,2203.
Пример 8А: Трифторацетат N-трет-бутоксикарбонил-3-трет-бутил-D-аланил-3-(6-трифторметилпиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
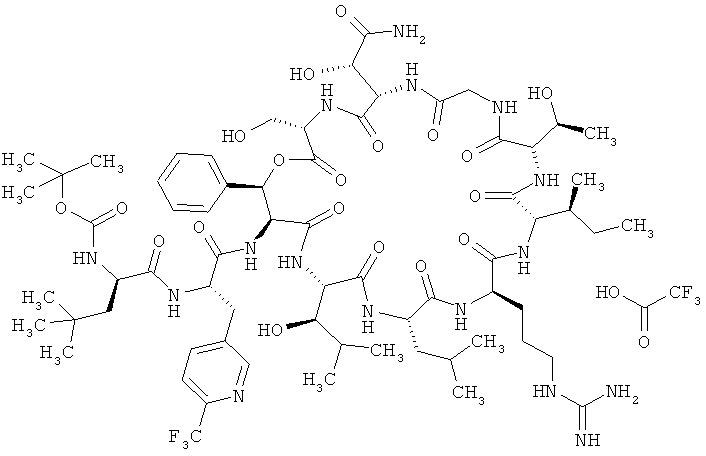
К раствору соединений из примера 2А (7,00 г, 1,0 эквивалент, 5,48 ммоля) и примера 7А (3,03 г, 1,2 эквивалента, 6,57 ммоля) в сухом ДМФ (119 мл) при -30°С медленно добавляют N-метилморфолин (2,77 г, 3,01 мл, 5 эквивалентов, 27,38 ммоля) и ГАТУ (4,37 г, 2,1 эквивалента, 11,50 ммоля). Реакционной смеси дают медленно нагреться (в течение примерно 1 ч) до КТ, при этом полноту реакции контролируют с помощью ЖХВД/УФ-видим. (метод 9). Реакцию прекращают добавлением дигидрофосфата калия (7,45 г, 10,0 эквивалентов, 54,76 ммоля). Реакционную смесь очищают хроматографией (метод 10, элюент: метанол/ацетон в соотношении 4:1), получая 12,63 г (количеств.) продукта.
ЖХВД/УФ-видим. (метод 8): Rt=4,78 мин.
ЖХВД/УФ-видим. (метод 9): Rt=4,35 мин.
ЖХ-МС (метод 7): Rt=2,28 мин; МС (ESIpos): m/z (%) = 697 (100) [M+2H]2+, 1493 (15) [M+H]+; MC (ESIneg): m/z (%) = 745 (100) [M-2H]2-, 1491 (5) [M-H]-.
ВП-МС-ВР (метод 1): C67H104N16O19F3 [M+H]+ рассч. 1493,7616, обнаруж. 1493,7594.
Пример 9А и пример 10А: (2S)-N-(трет-бутоксикарбонил)-3-(триметилсилил)аланин и (2R)-N-(трет-бутоксикарбонил)-3-(триметилсилил)аланин
Указанные в заголовке соединения синтезируют по методу, описанному у М. Merget, К. Günther, M. Bernd, E. Günther, R. Tacke, J. Organomet. Chem., 628, 2001, сс.183-194. Энантиомеры разделяют с помощью препаративной ЖХВД на хиральной фазе: Gilson Abimed HPLC; колонка: Daicel Chiralpak AD-H, 5 мкм, 250×20 мм; элюент А: изогексан, элюент Б: 0,2% уксусной кислоты/1% воды/2-пропанол; изократически; скорость потока: 15 мл/мин; УФ-детектор на 212 нм. Соотнесение изомеров осуществляют путем сравнения данных ЖХВД-анализа с аутентичным образцом N-(трет-бутоксикарбонил)-L-3-триметилсилилаланина (2R-соединение, Mercachem AMR 39.260).
Пример 9А: N-(трет-бутоксикарбонил)-D-3-триметилсилилаланин (2S-соединение)
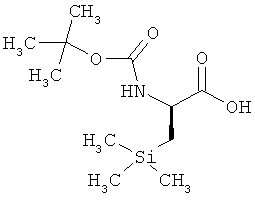
Хиральная ЖХВД (метод 15): Rt=4,16 мин, энантиомерный избыток (е.е.) >99%.
[α]D=+1,1 (с=0,83 в метаноле).
Пример 10А: N-(трет-бутоксикарбонил)-L-3-триметилсилилаланин (2R-соединение)
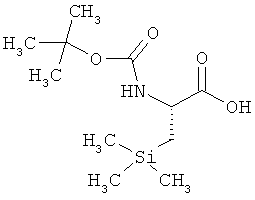
Хиральная ЖХВД (метод 15): Rt=9,27 мин, е.е.>99%.
[α]D 20=-1,6 (с=0,66 в метаноле).
Пример 11А: Метил-N-(трет-бутоксикарбонил)-3-(пиридин-3-ил)-L-аланинат
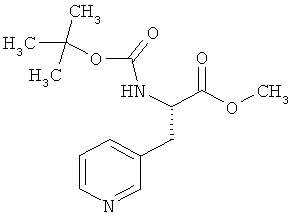
Указанное в заголовке соединение получают по методу, описанному у В. Neises, W. Steglich. Org. Synth., 63, 1985, cc.183-187.
(2S)-N-(трет-Бутоксикарбонил)-3-(пиридин-3-ил)аланин (25,00 г, 93,88 ммоля) в атмосфере аргона растворяют в 300 мл дихлорметана. Далее добавляют метанол (11,4 мл, 9,02 г, 281 ммоль, 3 эквивалента) и одну частицу ДМАП. После этого смесь охлаждают до 0°С. Затем добавляют ЭДК (19,80 г, 103 ммоля, 1,1 эквивалента). Через 5 мин ледяную баню удаляют и перемешивают в течение 1 ч при КТ. Затем концентрируют в вакууме, остаток смешивают с этилацетатом и экстрагируют путем встряхивания с насыщенным раствором гидрокарбоната натрия. Водную фазу дополнительно однократно экстрагируют этилацетатом, после чего объединенные органические фазы промывают сначала 0,5-молярной лимонной кислотой, а затем еще раз насыщенным раствором гидрокарбоната натрия. Органические фазы сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Таким путем в качестве остатка получают прозрачное масло, которое кристаллизуется при сушке в вакууме, создаваемом масляным насосом. Выход: 23,60 г (90% от теории).
ЖХВД/УФ-видим. (метод 9): Rt=3,28 мин.
ЖХ-МС (метод 7): Rt=1,21 мин; МС (ESIpos): m/z (%) = 281 (100) [M+H]+.
1Н-ЯМР (400 МГц, d6-ДМСО): δ=1,30 (s, 9H), 2,86 (m, 1H), 3,04 (m, 1H), 3,63 (s, 3H), 4,22 (m, 1H), 7,28-7,39 (m, 2H), 7,69 (d, 1H), 8,43 (m, 2H).
Пример 12А: Бистрифторацетат метилового эфира 3-(пиридин-3-ил)-L-аланина
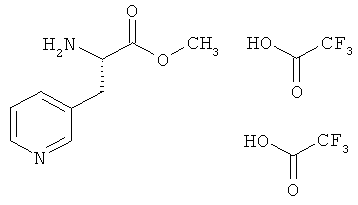
Соединение из примера 11А (11,8 г, 42,09 ммоля) растворяют в растворе трифторуксусной кислоты в дихлорметане (160 мл, 30%-ный раствор) и перемешивают в течение 30 мин при КТ. Затем концентрируют в вакууме. Остаток растворяют в небольшом количестве воды и лиофилизируют. После этого лиофилизат смешивают с толуолом и концентрируют в вакууме. В завершение сушат до постоянства массы в вакууме, создаваемом масляным насосом. Выход: 17,15 г (количеств.).
ЖХВД/УФ-видим. (метод 9): Rt=0,88 мин.
ЖХ-МС (метод 7): Rt=0,46 мин; МС (ESIpos): m/z (%)=181 (100) [M+H]+.
1Н-ЯМР (400 МГц, d6-ДМСО): δ=2,79 (dd, 1H), 2,92 (dd, 1H), 3,60 (s, 3H), 3,63 (m, 1H), 7,30 (m, 1H), 7,62 (d, 1H), 8,41 (m, 2H).
Пример 13А: Метил-N-(трет-бутоксикарбонил)-3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланинат
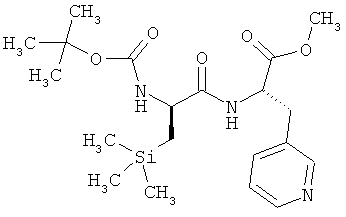
Соединение из примера 9А (10,31 г, 39,4 ммоля) и соединение из примера 12А (16,10 г, 39,4 ммоля, 1 эквивалент) при 0°С растворяют в ДМФ (186 мл). Затем добавляют N-метилморфолин (17,34 мл, 16,00 г, 4 эквивалента) и ГАТУ (22,49 г, 59,16 ммоля, 1,5 эквивалента). Смесь перемешивают в течение двух часов при КТ. Далее смешивают с трет-бутилметиловым эфиром и промывают насыщенным раствором карбоната натрия. Водную фазу дополнительно однократно экстрагируют трет-бутилметиловым эфиром, после чего объединенные органические фазы промывают сначала 1-молярной водной лимонной кислотой, а затем вновь насыщенным раствором карбоната натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. В завершение фильтруют через силикагель (циклогексан/этилацетат в соотношении 2:1). Выход: 14,1 г (84% от теории).
ЖХВД/УФ-видим. (метод 9): Rt=3,91 мин.
ЖХ-МС (метод 7): Rt=1,90 мин; МС (ESIpos): m/z (%) = 424 (100) [M+H]+.
1Н-ЯМР (400 МГц, d6-ДМСО): δ=-0,09 (s, 9H), 0,56-0,75 (m, 2H), 1,47 (s, 9H), 2,90 (dd, 1H), 3,09 (dd, 1H), 3,62 (s, 3H), 3,98 (m, 1H), 4,49 (m, 1H), 6,68 (d, 1H), 7,26 (dd, 1H), 7,61 (m, 1H), 8,20 (d, 1H), 8,40 (m, 2H).
Пример 14А: N-(трет-бутоксикарбонил)-3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланин
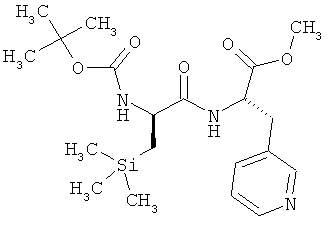
Соединение из примера 13А (7,4 г, 17,56 ммоля) растворяют в смеси ТГФ и воды (в соотношении 6:4), охлаждают до 0°С и смешивают с моногидратом гидроксида лития (1,47 г, 35,13 ммоля, 2 эквивалента). После этого смесь перемешивают при 0°С. Через час добавляют еще один эквивалент (0,74 г) моногидрата гидроксида лития и перемешивают еще в течение часа. Далее в вакууме отгоняют бòльшую часть ТГФ, промывают двумя порциями метил-тpeт-бутилового эфира и затем значение рН водной фазы добавлением лимонной кислоты устанавливают на 4. При этом в осадок выпадает твердое вещество. Далее экстрагируют тремя порциями этилацетата, что сопровождается растворением твердого вещества. Объединенные органические фазы сушат над сульфатом натрия, фильтруют и концентрируют. Сырой продукт очищают хроматографией (метод 3, элюент: метанол). Выход: 6,67 г (93% от теории).
ЖХВД/УФ-видим. (метод 9): Rt=3,73 мин.
ЖХ-МС (метод 7): Rt=1,68 мин; МС (ESIpos): m/z (%) = 410 (40) [M+H]+.
1H-ЯМР (300 МГц, d6-ДМСО): δ=-0,090 (s, 9H), 0,56-0,75 (m, 2H), 1,35 (s, 9H), 2,90 (dd, 1H), 3,09 (dd, 1H), 3,98 (m, 1H), 4,41 (m, 1H), 6,70 (d, 1H), 7,26 (dd, 1H), 7,60 (m, 1H), 8,00 (d, 1H), 8,37 (m, 2H).
Пример 15А: Трифторацетат N-(трет-бутоксикарбонил)-3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
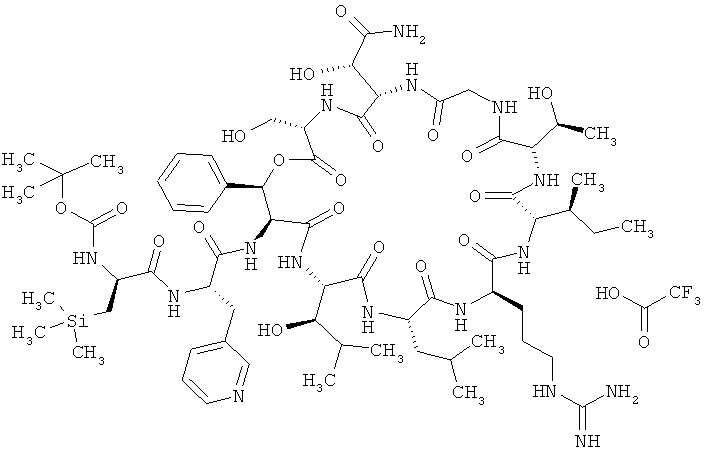
Соединение из примера 2А (3,00 г, 2,35 ммоля) и соединение из примера 14А (1,44 г, 3,52 ммоля, 1,5 эквивалента) растворяют в ДМФ (50 мл) и охлаждают до 0°С. После этого добавляют 4,7 мл (4,7 ммоля, 2 эквивалента) 1-молярного раствора 4-метилморфолина в ДМФ. Затем сразу же добавляют ГАТУ (1,52 г, 3,99 ммоля, 1,7 эквивалента) и перемешивают в течение 15 мин при 0°С. Далее по каплям добавляют еще 4,7 мл (4,7 ммоля, 2 эквивалента) 1-молярного раствора 4-метилморфолина в ДМФ. После этого смесь перемешивают в течение 2 ч при КТ. Сырой продукт подвергают гель-хроматографии (метод 3). Полученный продукт без дополнительной глубокой очистки используют в последующей реакции. Выход: 3,6 г (82% от теории).
ЖХВД (метод 9): Rt=3,90 мин.
ЖХ-МС (метод 7): Rt=2,00 мин; МС (ESIpos): m/z (%) = 721,8 (100) [M+2H]2+; 1442,1 (5) [M+H]+.
Альтернативный метод: Соединение из примера 2А (14,00 г, 10,95 ммоля) и соединение из примера 14А (5,38 г, 13,14 ммоля, 1,2 эквивалента) растворяют в ДМФ (280 мл) и охлаждают до -20°С. После этого добавляют N-метилморфолин (5,54 г, 6,02 мл, 5 эквивалентов), а затем ГАТУ (6,66 г, 17,52 ммоля, 1,6 эквивалента). Далее смесь медленно нагревают до КТ и оставляют перемешиваться на ночь (примерно на 16 ч). Затем при перемешивании добавляют дигидрофосфат калия (14,91 г, 10 эквивалентов) и перемешивают еще в течение 30 мин. Сырой продукт подвергают гель-хроматографии (метод 3, элюент: метанол). Полученный продукт без дополнительной глубокой очистки используют в последующей реакции. Выход: 14,35 г (61% от теории).
Пример 16А: 2,2-диметил-1-бутаналь
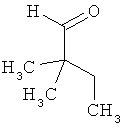
2,2-Диметил-1-бутанол (4,0 г, 39 ммолей) растворяют в дихлорметане (136 мл) и смешивают с оксидом алюминия (7,98 г, 78 ммолей, 2 эквивалента) и с хлорхроматом пиридиния (16,88 г, 78 ммолей, 2 эквивалента). После этого смесь перемешивают при КТ в течение 1 ч и затем фильтруют через слой силикагеля. Фильтрат осторожно концентрируют и остаток перегоняют при нормальном давлении (температура кипения 102°С (990 мбар)). Выход: 2,97 г (75% от теории).
ГХ-МС (метод 17): Rt=2,21 мин; МС (ESIpos): m/z (%) = 99,9 (5) [M]+.
1Н-ЯМР (400 МГц, СDСl3) δ=0,83 (t, 3Н), 1,03 (s, 6H), 1,51 (q, 2H), 9,42 (s, 1H).
Пример 17А: Метил-(2Z)-2-{[(бензилокси)карбонил]амино}-4,4-диметилгекс-2-еноат
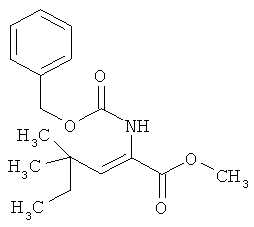
Соединение из примера 16А (2,55 г, 25,46 ммоля) и метиловый эфир {[(бензилокси)карбонил]амино}(диметоксифосфорил)уксусной кислоты (8,43 г, 25,46 ммоля) растворяют в 50 мл ТГФ и охлаждают до 0°С. Далее по каплям добавляют N,N,N',N'-тетраметилгуанидин, после чего перемешивают сначала в течение 15 мин при 0°С, а затем в течение 5 дней при КТ. После этого смесь смешивают с примерно 20 г силикагеля, концентрируют и хроматографируют (силикагель Biotage 40M, ZIF-SIM, циклогексан/этилацетат в соотношении 87:13). Выход: 1,20 г (13% от теории).
ЖХВД (метод 9): Rt=3,71 мин.
МС (DCI): m/z (%)=323,3 (100) [M+NH4].
1Н-ЯМР (400 МГц, CDCl3): δ=0,83 (m, 3H), 1,13 (s, 6H), 1,49 (q, 2H), 3,75 (шир. s, 3H), 5,72 (шир. s, 1H), 6,58 (шир. s, 1H), 5,12 (s, 2H), 7,36 (m, 5H).
Пример 18A: Метил-N-[(бензилокси)карбонил]-4,4-диметил-D-норлейцинат

Соединение из примера 17А (1,2 г, сырой продукт, 3,26 ммоля) растворяют в этаноле а.ч. (60 мл). Далее через полученный раствор с помощью канюли в течение примерно 5 мин пропускают аргон, после чего добавляют трифлат (+)-1,2-бис-[(2R,5R)диэтилфосфолано]бензол(циклооктадиен)родия(I) (28 мг, 0,04 ммоля, 0,012 эквивалента) и растворяют при обработке в ультразвуковой бане. После этого гидрируют в течение 24 ч при давлении водорода 3 бара и КТ. Смесь концентрируют и хроматографируют (силикагель Biotage 25M, циклогексан/этилацетат в соотношении 9:1). Выход: 920 мг (92% от теории).
ЖХВД (метод 9): Rt=4,96 мин.
ЖХ-МС (метод 7): Rt=2,76 мин; МС (ESIpos): m/z (%) = 308 (25) [М+Н]+.
1Н-ЯМР (400 МГц, CDCl3): δ=0,80 (t, 3H), 0,86 (s, 6H), 1,29 (q, 2H), 1,41 (dd, 1H), 1,73 (dd, 1H), 3,72 (s, 3H), 4,40 (m, 1H), 5,02 (d, 1H), 5,11 (m, 2H), 7,35 (m, 5H).
Пример 19А: N-[(бензилокси)карбонил]-4,4-диметил-D-норлейцин
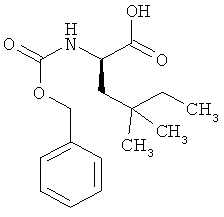
В ТГФ (12 мл) растворяют соединение из примера 18А (915 мг, 2,98 ммоля). Полученный раствор охлаждают до 0°С, после чего добавляют 3,7 мл (7,4 ммоля, 2,5 эквивалента) 2-молярного раствора моногидрата гидроксида лития в воде и интенсивно перемешивают в течение 1 ч. После этого по каплям добавляют лимонную кислоту (1-молярную) до получения кислой реакции и смесь экстрагируют этилацетатом. Органический экстракт сушат над сульфатом натрия, концентрируют и хроматографируют (метод 16). Выход: 434 мг (50% от теории).
ЖХВД (метод 9): Rt=4,54 мин.
ЖХ-МС (метод 7): Rt=2,44 мин; МС (ESIpos): m/z (%) = 294 (20) [M+H]+.
1Н-ЯМР (400 МГц, d6-ДМСО): δ=0,80 (t, 3H), 0,83 (s, 6H), 1,21 (q, 2H), 1,53 (dd, 1H), 1,60 (dd, 1H), 3,99 (m, 1H), 5,02 (s, 2H), 7,35 (m, 5H), 7,58 (d, 2H), 12,52 (шир. s, 1H).
Пример 20А: Метил-N-[(бензилокси)карбонил]-4,4-диметил-D-норлейцил-3-(пиридин-3-ил)-L-аланинат
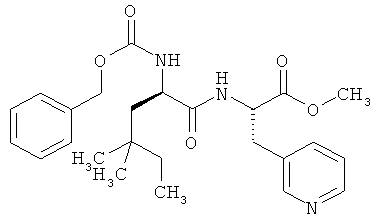
Соединение из примера 19А (430 мг, 1,47 ммоля) и соединение из примера 12А (809 мг, 1,47 ммоля, 1 эквивалент) при 0°С растворяют в ДМФ (5 мл), после чего добавляют 4-метилморфолин (644 мкл, 5,86 ммоля, 4 эквивалента) и ГАТУ (836 мг, 2,20 ммоля, 1,5 эквивалента). Далее смесь перемешивают в течение трех часов при КТ. После этого смешивают с этилацетатом и промывают насыщенным раствором гидрокарбоната натрия. Водную фазу однократно экстрагируют этилацетатом, после чего объединенные органические фазы промывают 1-молярной водной лимонной кислотой, а затем вновь насыщенным раствором гидрокарбоната натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток хроматографируют (метод 16). Выход: 496 мг (74% от теории).
ЖХВД (метод 9): Rt=3,94 мин.
ЖХ-МС (метод 7): Rt=1,85 мин; МС (ESIpos): m/z (%) = 456 (100) [M+H]+.
1Н-ЯМР (300 МГц, d6-ДМСО): δ=0,80 (m, 9H), 1,10 (m, 3H), 1,30 (dd, 1H), 2,91 (dd, 1H), 3,12 (dd, 1H), 3,30 (s, 3H), 4,02 (m, 1H), 4,51 (m, 1H), 5,01 (d, 1H), 5,06 (d, 1H), 7,22 (dd, 1H), 7,30 (m, 5H), 7,63 (m, 1H), 8,40 (m, 2H).
Пример 21 А: N-[(бензилокси)карбонил]-4,4-диметил-D-норлейцил-3-(пиридин-3-ил)-L-аланин
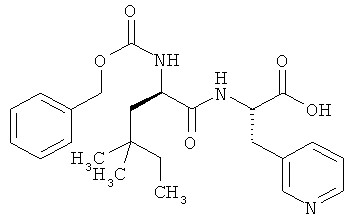
В ТГФ (5 мл) растворяют соединение из примера 20А (490 мг, 1,08 ммоля). Полученный раствор охлаждают до 0°С, после чего добавляют 1,35 мл (2,7 ммоля, 2,5 эквивалента) 2-молярного раствора моногидрата гидроксида лития в воде и интенсивно перемешивают в течение 1 ч. После этого по каплям добавляют лимонную кислоту (1-молярную в воде) до получения кислой реакции и смесь экстрагируют этилацетатом. Органический экстракт сушат над сульфатом натрия и концентрируют. Выход: 484 мг (количеств.).
ЖХВД (метод 9): Rt=3,76 мин.
ЖХ-МС (метод 7): Rt=1,88 мин; МС (ESIpos): m/z (%) = 442 (100) [М+Н]+.
1Н-ЯМР (400 МГц, (d6-ДМСО): δ=0,70 (m, 9H), 1,14 (m, 3H), 1,30 (dd, 1H), 2,64 (d, 1H), 2,75 (d, 1H), 2,89 (dd, 1H), 3,11 (dd, 1H), 4,03 (m, 1H), 4,45 (m, 1H), 4,98 (d, 1H), 5,05 (d, 1H), 7,22 (dd, 1H), 7,30 (m, 5H), 7,61 (m, 1H), 8,40 (m, 2H).
Пример 22А: Трифторацетат N-[(бензилокси)карбонил]-4,4-диметил-D-норлейцил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
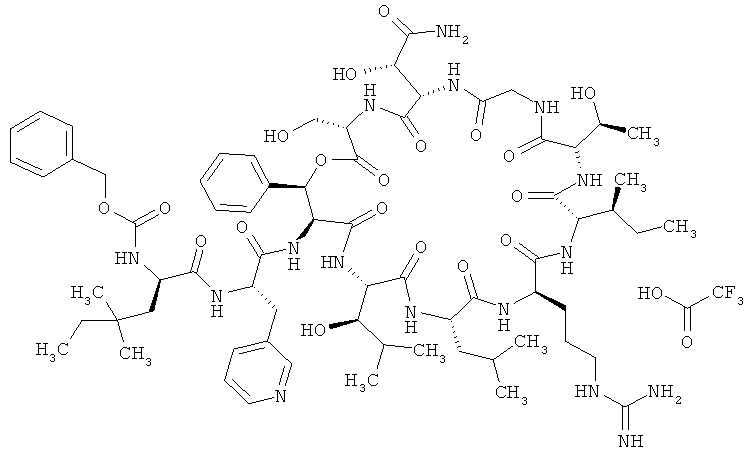
Соединение из примера 2А (0,28 г, 0,22 ммоля) и соединение из примера 21А (148 мг, 0,33 ммоля, 1,5 эквивалента) растворяют в ДМФ (4 мл) и охлаждают до 0°С. После этого добавляют 0,47 мл (0,44 ммоля, 2 эквивалента) 1-молярного раствора N-метилморфолина в ДМФ. Затем сразу же добавляют ГАТУ (141 мг, 0,37 ммоля, 1,7 эквивалента) и перемешивают в течение 15 мин при 0°С. Далее по каплям добавляют еще 0,44 мл (0,47 ммоля, 2 эквивалента) 1-молярного раствора 4-метилморфолина в ДМФ. Затем смесь оставляют перемешиваться на ночь при КТ. После этого смесь очищают на колонке с сорбентом Sephadex LH-20 (метод 3, элюент: метанол). Сырой продукт очищают с помощью препаративной ЖХВД (метод 13). Выход: 149 мг (43% от теории).
ЖХВД (метод 9): Rt=3,93 мин.
ЖХ-МС (метод 7): Rt=2,13 мин; МС (ESIpos): m/z (%) = 737,4 (100) [M+2H]2+, 1473 (2) [M+H]+.
Примеры получения предлагаемых в изобретении соединений
Пример 1: Бистрифторацетат 3-трет-бутил-D-аланил-3-(6-трифторметилпиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
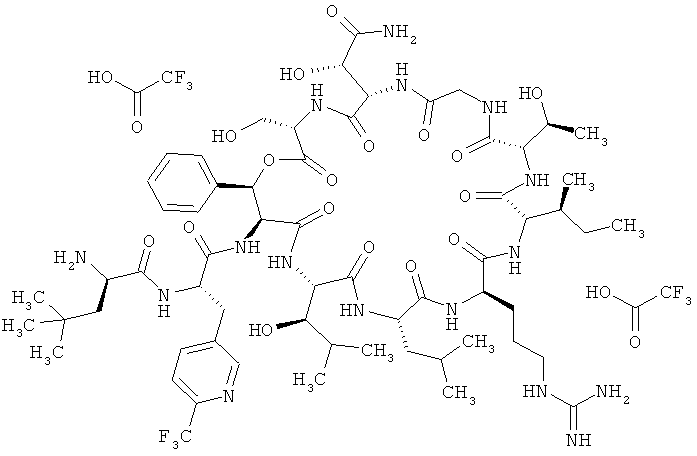
К раствору соединения из примера 8А (10,3 г, 1,0 эквивалент, 4,48 ммоля, сырой продукт) в дихлорметане (150 мл) при КТ медленно по каплям добавляют трифторуксусную кислоту (150 мл). Далее реакционную смесь перемешивают при КТ (10 мин), при этом полноту реакции контролируют с помощью ЖХВД (метод 9). Затем реакционную смесь концентрируют на роторном испарителе и очищают с помощью препаративной ЖХВД (метод 11), получая 3,48 г (48% от теории) продукта.
ЖХВД/УФ-видим. (метод 8): Rt=4,03 мин.
ЖХВД/УФ-видим. (метод 9): Rt=3,64 мин.
ЖХ-МС (метод 7): Rt=1,69 мин; МС (ESIpos): m/z (%) = 697 (100) [M+2H]2+, 1393 (5) [М+Н]+; МС (ESIneg): m/z (%) = 695 (100) [M-2H]2-, 1391 (20) [M-H]-.
19F-ЯМР (400 МГц, d5-пиридин): δ=-67 (Аr-СF3), -74 (СF3СООН), -132 (1,4-дибромтетрафторбензол в качестве стандарта). Содержание ТФУК: 14,3 мас.%.
МС-ВР (метод 1): С62Н96N16О17F3 [М+Н]+ рассч. 1393,7091, обнаруж. 1393,7119.
Пример 2: Тристрифторацетат 3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
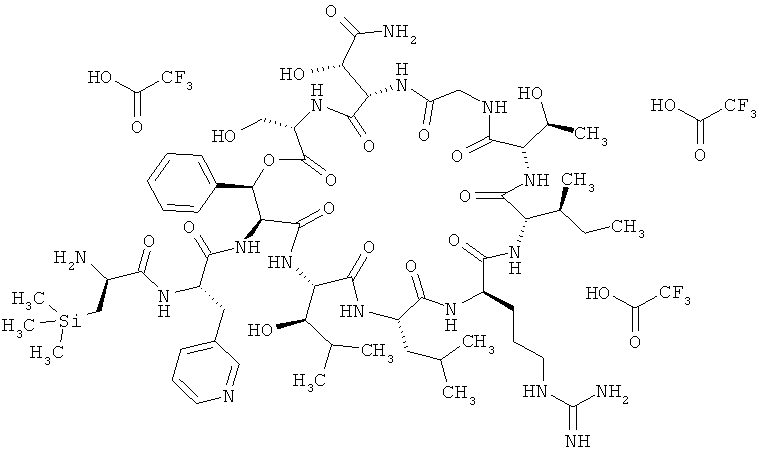
Соединение из примера 15А (9,12 г, 4,93 ммоля, сырой продукт) растворяют в растворе трифторуксусной кислоты в дихлорметане (65 мл, 30%-ный раствор). Далее смесь перемешивают при КТ в течение 20 мин. После этого отгоняют растворитель. Остаток сушат в вакууме, создаваемом масляным насосом, и затем очищают хроматорафией (метод 14). Выход: 5,54 г (67% от теории).
ЖХВД (метод 9): Rt=3,32 мин.
ЖХ-МС (метод 7): Rt=1,41 мин; МС (ESIpos): m/z (%) = 671,7 (100) [М+2H]2+.
ВП-МС-ВР (метод 1): C60H97N16O17Si [М+Н]+ рассч. 1341,6987, обнаруж. 1341,7019.
1Н-ЯМР (500 МГц, d5-пиридин): δ=-0,172 (s, 9H), 0,611 (d, J=6,9 Гц, 3Н), 0,881 (d, J=7,9 Гц, 3Н), 0,948 (d, J=6,3 Гц, 3Н), 0,954-0,997 (m, 6Н), 1,135 (d, J=6,0 Гц, 3Н), 1,208 (m, 2H), 1,361 (d, J=5,4 Гц, 3Н), 1,439 (m, 1H), 1,497 (m, 1H), 1,953 (m, 2H), 2,04 (m, 1H), 2,154 (m, 3Н), 2,372 (m, 3Н), 3,111 (m, 1H), 3,266 (m, 1H), 3,563 (d, J=14,95 Гц, 1Н), 3,726 (dd, J=12,2, 14,95 Гц, 1Н), 3,840 (d, J=9,6 Гц, 1Н), 3,960 (m, 1Н), 4,152 (m, 1Н), 4,198 (m, 1Н), 4,278 (m, 1Н), 4,382 (m, 1Н), 4,488 (m, 1Н), 4,565 (dd, J=9,5, 9,6 Гц, 1Н), 4,628 (m, 1Н), 4,630 (m, 1Н), 4,779 (d, J=12,2 Гц, 1Н), 5,069 (m, 1Н), 5,159 (dd, J=9,3 Гц, 1Н), 5,264 (m, 1Н), 5,362 (s, 1Н), 5,98 (d, J=9,9 Гц, 1Н), 6,351 (dd, J=8,5 Гц, J=8,7 Гц, 1Н), 7,169 (m, 1Н), 7,246 (m, 1Н), 7,382 (d, J=9,9 Гц, 1Н), 7,512 (m, 2H), 7,583-7,614 (m, 2H), 7,728 (m, 2H), 7,90 (d, J=8,7 Гц, 1Н), 8,126 (m, 3H), 8,341 (m, 1Н), 8,576 (d, J=3,6 Гц, 1Н), 8,695 (m, 2H), 8,793 (m, 1Н), 9,139 (шир. s, 1Н), 9,715 (m, 1Н), 10,957 (шир. s, 1Н), 11,268 (шир. s, 1Н).
13С-ЯМР (126 МГц, d5-пиридин): δ=-1,8, 11,08, 15,84, 18,8, 18,8, 19,79, 20,89, 20,93, 21,65, 23,38, 24,78, 26,57, 26,88, 28,80, 31,04, 34,06, 36,84, 40,95, 41,25, 44,38, 50,94, 52,82, 55,99, 56,10, 56,74, 58,40, 59,10, 60,34, 60,72, 62,33, 62,55, 70,72, 72,06, 75,58, 75,65, 123,66, 128,25, 128,95, 129,80, 132,39, 136,77, 137,18, 149,17, 158,11, 162,15, 162,41, 162,70, 162,96, 169,01, 169,69, 170,21, 172,57, 173,27, 173,44, 173,66, 174,07, 174,36, 175,34, 175,56.
19F-ЯМР (400 МГц, d5-пиридин): δ=-74 (СF3СООН), -132 (1,4-дибромтетрафторбензол в качестве стандарта). Содержание ТФУК: 19,3 мас.%.
Структуру подтверждают путем рентгеноструктурного анализа монокристаллов.
Пример 3: Трисметансульфонат 3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
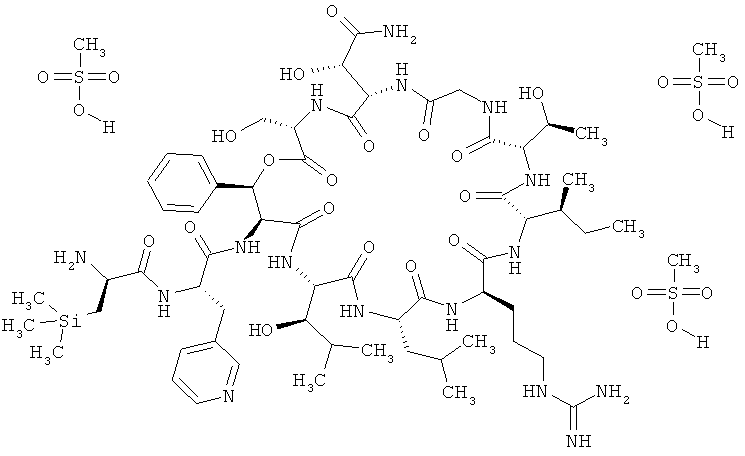
Тристрифторацетат 3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина (447 мг, 0,27 ммоля) растворяют в 22 мл воды. Далее добавляют метансульфоновую кислоту (70%-ную). После этого смесь интенсивно перемешивают и затем лиофилизируют. Лиофилизат растворяют в воде (3,4 мл) и добавляют 70%-ный раствор метансульфоната натрия (0,9 мл). Затем перемешивают в течение 10 мин при КТ. После этого продукт отделяют центрифугированием. Таким путем получают 472 мг сырого продукта.
Объединенные из нескольких смесей сырые продукты (в общей сложности 1053 мг, 0,63 ммоля) суспендируют в воде (5 мл) и перемешивают при КТ в течение 4 дней. После этого вновь центрифугируют и полученное твердое вещество сушат в вакууме. Таким путем получают 575 мг (55% от теории) продукта.
ЖХВД (метод 9): Rt=3,30 мин.
ЖХ-МС (метод 7): Rt=1,49 мин; МС (ESIpos): m/z (%) = 671,8 (100) [M+2H]2+, 1342,1 (5) [M+H]+.
1Н-ЯМР (500 МГц, d5-пиридин): δ=-0,170 (s, 9H), 0,702 (d, J=6,3 Гц, 3Н), 0,867 (d, J=6,1 Гц, 3Н), 0,972 (d, J=6,2 Гц, 3Н), 0,966-1,069 (m, 9H), 1,186 (d, J=6,5 Гц, 3Н), 1,315 (m, 2H), 1,448-1,501 (m, 6H), 2,010 (m, 1H), 2,084 (m, 3Н), 2,180-2,365 (m, 4H), 2,540 (m, 1H), 3,082 (s, 9H), 3,166 (m, 1H), 3,325 (m, 1H), 3,623 (d, J=14,05 Гц, 1H), 3,865 (dd, J=13,7, 14,05 Гц, 1H), 3,967-3,986 (m, 2H), 4,257 (m, 1H), 4,339 (m, 2H), 4,410 (m, 1H), 4,566 (m, 1H), 4,591-4,686 (m, 2H), 4,75 (d, 1H, J=12,1 Гц, 1H), 4,859 (m, 1H), 5,086 (m, 1H), 5,229 (m, 1H), 5,348 (m, 1H), 5,375 (s, 1H) 6,02 (d, J=10,0 Гц, 1H), 6,403 (m, 1H), 7,068 (d, J=9,8 Гц, 1H), 7,222 (m, 3Н), 7,544 (m, 3Н), 7,625 (m, 1H), 7,681 (m, 1H), 7,830 (d, J=7,6 Гц, 1H), 7,921 (d, J=9,35 Гц, 1H), 8,071 (m, 2H), 8,188 (шир. s, 1H), 8,285 (шир. s, 1Н), 8,420 (d, J=8,4 Гц, 1H), 8,614 (d, J=4,0 Гц, 1H), 8,701 (m, 1H), 8,874 (шир. s, 1H), 10,175 (m, 1H), 10,279 (m, 1H), 10,941 (шир. s, 1H).
19F-ЯМР (пиридин, 400 МГц, метод 24): δ=-74,0 (s, ТФУК, 0,20), -132,0 (s, 1,4-дибромтетрафторбензол, 1000,0). Содержание ТФУК: 0,02 мас.%.
Альтернативный метод: В колонку (диаметром 35 мм) набивают 32 г ионита Dowex 1Х8-400 (HCl-форма). Далее через колонку пропускают примерно 60 мл 1-молярного раствора гидроксида натрия, а затем 60 мл воды для ЖХВД. После этого колонку кондиционируют 60 мл 1-молярной метансульфоновой кислоты и затем промывают примерно 100 мл воды до нейтральной реакции элюата (Macherey & Nagel Tritest). Далее в 90 мл воды растворяют 2,00 г (1,19 ммоля) тристрифторацетата 3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина (соединение из примера 2), полученный раствор подают в колонку и медленно элюируют. После этого колонку промывают примерно 20 мл воды. Содержащие продукт элюаты объединяют, подвергают тонкой фильтрации (через фильтр с размером пор 0,20 мкм) и лиофилизируют. Затем колонку вновь кондиционируют и используют повторно. Таким путем из 2000 мг (1,19 ммоля) соединения из примера 2 получают 1,79 г (1,10 ммоля, 92% от теории) указанного в заголовке примера 3 соединения.
Пример 4: Тристрифторацетат 4,4-диметил-D-норлейцил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
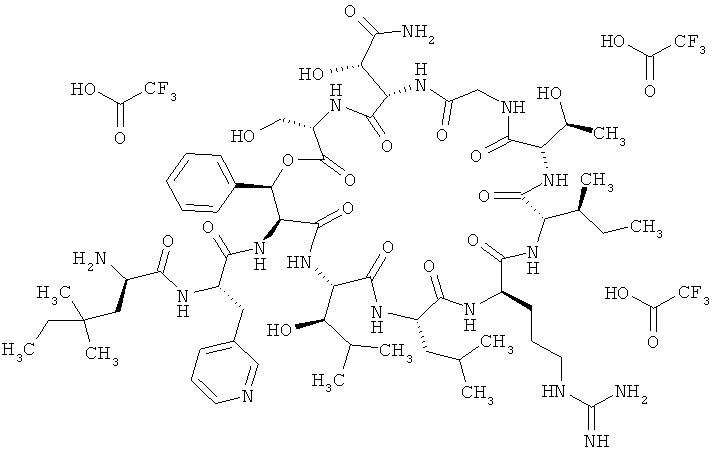
Соединение из примера 22А (149 мг, 0,09 ммоля) растворяют в метаноле, содержащем 0,05% трифторуксусной кислоты (10 мл). Далее добавляют палладий на активированном угле (10%-ный, 20 мг), после чего гидрируют в присутствии водорода в общей сложности в течение 2,5 ч при КТ и нормальном давлении. От сырого продукта отфильтровывают катализатор и фильтрат концентрируют.Остаток очищают хроматорафией (метод 13). Выход: 68 мг (46% от теории).
ЖХВД (метод 9): Rt=3,29 мин.
ЖХ-МС (метод 7): Rt=1,49 мин; МС (ESIpos): m/z (%) = 671,0 (100) [M+2H]2+, 1340 (5) [M+H]+.
ВП-МС-ВР (метод 1): C62H98N16O17 [М+Н]+ рассч. 1339,7369, обнаруж. 1339,7368.
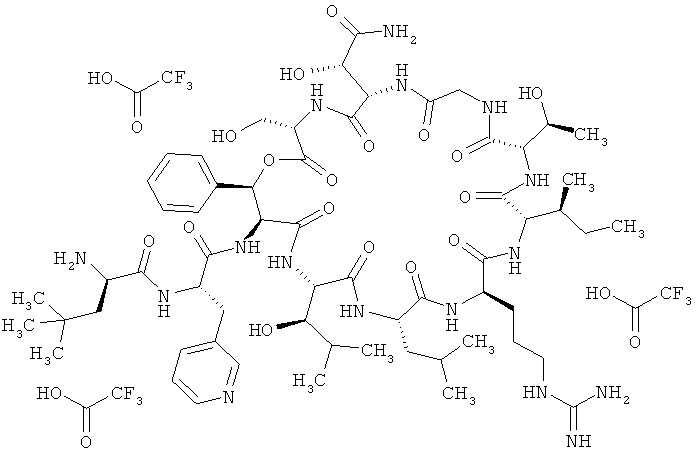
Пример 5 (сравнительный): Тристрифторацетат 3-трет-бутил-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
Пример 6: Трисгидрохлорид (триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
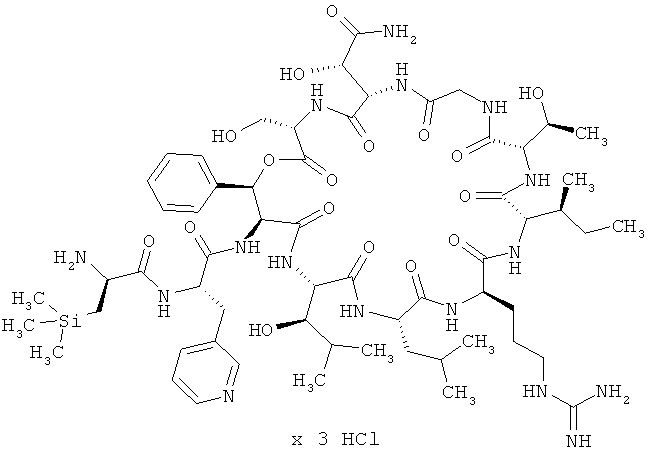
В колонку (диаметром 35 мм) набивают 32 г ионита Dowex 1Х8-400 (НСl-форма). Далее через колонку пропускают примерно 60 мл 1-молярного раствора гидроксида натрия, а затем 60 мл воды для ЖХВД. После этого колонку кондиционируют 60 мл 1-молярной соляной кислоты и затем промывают примерно 100 мл воды до нейтральной реакции элюата (Macherey & Nagel Tritest). Далее в 90 мл воды растворяют 2,00 г (1,19 ммоля) тристрифторацетата 3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина (соединение из примера 2), полученный раствор подают в колонку и медленно элюируют. После этого колонку промывают примерно 20 мл воды. Содержащие продукт элюаты объединяют, подвергают тонкой фильтрации (через фильтр с размером пор 0,20 мкм) и лиофилизируют. Затем колонку вновь кондиционируют и используют повторно. Таким путем из 2000 мг (1,19 ммоля) соединения из примера 2 получают 1,54 г (1,06 ммоля, 89% от теории) указанного в заголовке примера 6 соединения.
ЖХВД (метод 9): Rt=3,30 мин.
ЖХ-МС (метод 7): Rt=1,47 мин; МС (ESIpos): m/z (%) = 671,5 (100) [M+2H]2+, 1341,4 (20) [М+Н]+.
Ионообменная хроматография (метод 25): Сl- (рассч.) = 7,43%, Cl- обнаруж. = 7,1%; содержание ТФУК (обнаруж.) <0,1%.
1Н-ЯМР (500 МГц, d5-пиридин) δ=-0,170 (s, 9H), 0,580 (d, J=6,3 Гц, 3Н), 0,772 (d, J=5,7 Гц, 3Н), 0,920 (d, J=5,8 Гц, 3Н), 0,974-0,985 (m, 6H), 1,147 (d, J=6,4 Гц, 3Н), 1,250-1,358 (m, 2Н), 1,411 (d, J=5,7 Гц, 3Н), 1,468 (m, 1H), 1,604 (m, 1H), 1,959-2,049 (m, 3Н), 2,154-2,228 (m, 3Н), 2,380 (m, 3Н), 2,935 (m, 1H), 3,120 (m, 1H), 3,300 (m, 1H), 3,607 (d, J=14,9 Гц, 1Н), 3,762 (dd, J=14,7 Гц, 1H), 3,882 (d, J=9,1 Гц, 1H), 3,938 (m, 1H), 4,204 (m, 1H), 4,253 (m, 1H), 4,389 (m, 1H), 4,465 (m, 1H), 4,589-4,661 (m, 4H), 4,806 (d, J=12,1 Гц, 1H), 5,027 (m, 1H), 5,202 (dd, J=9,4 Гц, 1H), 5,289 (m, 1H), 5,390 (s, 1H), 6,000 (d, J=9,5 Гц, 1H), 6,394 (dd, J=9,0 Гц, 1H), 7,173 (m, 2Н), 7,428 (m, 1H), 7,54 (d, J=7,0 Гц, 1H), 7,664-7,973 (m, 3Н), 7,767 (d, J=8,6, 1H), 7,918-7,973 (m, 3Н), 8,038 (шир. s, 2Н), 8,098 (шир. s, 1H), 8,185 (шир. s, 1H), 8,253 (d, J=8,6 Гц, 1H), 8,582 (m, 1H), 8,684 (d, J=9,8 Гц, 1H), 8,741 (шир. s, 1H), 8,782 (m, 1H), 9,928 (шир. s, 1H), 10,272 (d, J=8,3 Гц, 1H), 10,886 (шир. s, 1H), 11,135 (шир. s, 1H).
19F-ЯМР (пиридин, 400 МГц, метод 24): содержание ТФУК менее предела обнаружения.
Пример 7: Трис-L-лактат (триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
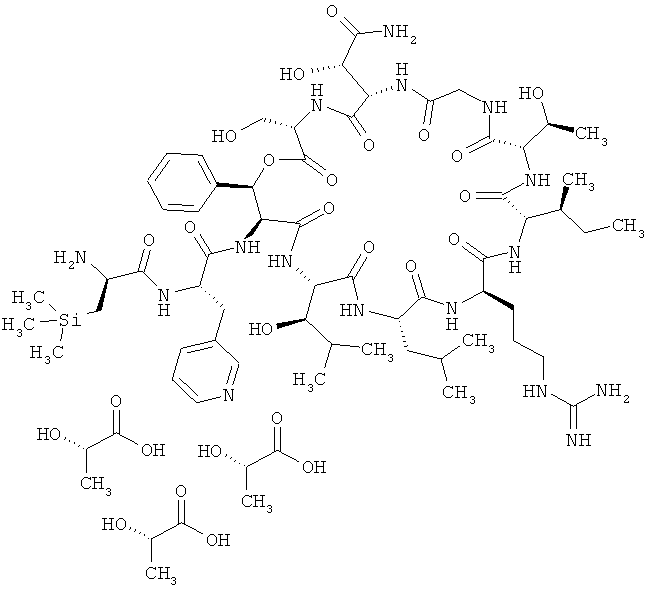
В колонку (диаметром 35 мм) набивают 32 г ионита Dowex 1Х8-400 (НСl-форма). Далее через колонку пропускают примерно 60 мл 1-молярного раствора гидроксида натрия, а затем 60 мл воды для ЖХВД. После этого колонку кондиционируют 60 мл 1-молярной молочной кислоты и затем промывают примерно 100 мл воды до нейтральной реакции элюата (Macherey & Nagel Tritest). Далее в 90 мл воды растворяют 2,00 г (1,19 ммоля) тристрифторацетата 3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина (соединение из примера 2), полученный раствор подают в колонку и медленно элюируют. После этого колонку промывают примерно 20 мл воды. Содержащие продукт элюаты объединяют, подвергают тонкой фильтрации (через фильтр с размером пор 0,20 мкм) и лиофилизируют. Затем колонку вновь кондиционируют и используют повторно. Таким путем из 2000 мг (1,19 ммоля) соединения из примера 2 получают 1903 мг (1,18 ммоля, 99% от теории) указанного в заголовке примера 7 соединения.
ЖХВД (метод 9): Rt=3,29 мин.
ЖХ-МС (метод 7): Rt=1,38 мин; МС (ESIpos): m/z (%) = 671,7 (100) [M+2H]2+, 1341,8 (20) [M+H]+.
1Н-ЯМР (500 МГц, d5-пиридин): δ=-0,170 (s, 9H), 0,754 (d, J=6,3 Гц, 3Н), 0,836 (d, J=6,1 Гц, 3Н), 0,922 (d, J=6,2 Гц, 3Н), 0,922-0,957 (m, 6H), 1,054 (d, J=6,4 Гц, 3Н), 1,190 (m, 2H), 1,416 (d, J=5,8 Гц, 3Н), 1,439-1,662 (m, 4H), 1,529 (d, J=7,0 Гц, 3Н), 1,598 (d, J=6,5 Гц, 3Н), 1,669 (d, J=6,9 Гц, 3Н), 1,923-2,280 (m, 9H), 3,057 (m, 1H), 3,266 (m, 1H), 3,564 (d, J=14, 5 Гц, 1Н), 3,761 (dd, J=13,7 Гц, 1H), 3,840 (m, 1H), 4,049 (m, 1H), 4,109 (m, 1H), 4,164 (m, 1H), 4,202 (m, 1H), 4,410 (m, 1H), 4,484 (m, 1H), 4,616 (dd, J=6,7, 13,6 Гц, 1H), 4,656 (m, 1H), 4,669 (dd, J=7,0, 13,9 Гц, 1H), 4,780 (d, J=13,0 Гц, 1H), 4,921 (m, 1H), 5,144 (dd, J=9,6 Гц, 1H), 5,281 (m, 1H), 5,363-5,426 (m, 3Н) 5,362 (s, 1H), 5,989 (d, J=9,8 Гц, 1H), 6,323 (m, 1H), 7,122 (m, 1H), 7,180 (m, 1H), 7,381 (m, 1H), 7,596 (m, 1H), 7,671-7,925 (m, 6H), 8,036 (шир. s, 1H), 8,208 (m, 3Н), 8,412 (m, 1H), 8,551 (d, J=3,6 Гц, 1H), 8,714 (m, 2H), 8,794 (m, 1H), 10,298 (шир. s, 1H), 10,938 (шир. s, 1H).
19F-ЯМР (пиридин, 400 МГц, метод 24): содержание ТФУК менее предела обнаружения.
Пример 8: Трис-D-тартрат (триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
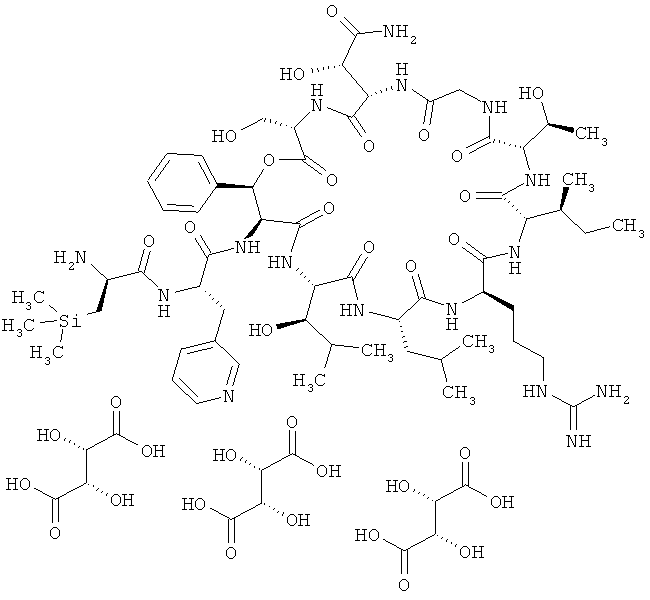
Кондиционирование ионита: 5 г ионита Dowex 1Х8 400 (HCl-соль) промывают 1-молярным раствором едкого натра до щелочной реакции элюата. Далее промывают водой в количестве, равном 2 объемам колонки, а затем 1-молярной D-винной кислотой до кислой реакции элюата. После этого промывают водой до нейтрального элюата (Macherey & Nagel Tritest).
Затем в 10 мл воды растворяют 100 мг (59 мкмолей) соединения из примера 2 и полученный раствор подают в колонку. После этого колонку промывают водой несколькими порциями по 10 мл. Эдукт растворяется лишь в умеренной степени и частично остается на колонке. Содержащие продукт элюированные фракции объединяют и лиофилизируют. Таким путем получают 57 мг (31 мкмоль, 52% от теории) продукта. Результаты интегрирования 1Н-ЯМР-спектра указывают на наличие стехиометрического соотношения между (триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактином и D-винной кислотой, равного 1:3.
ЖХВД (метод 9): Rt=3,29 мин.
ЖХ-МС (метод 7): Rt=1,37 мин; МС (ESIpos): m/z (%) = 671,7 (100) [M+2H]2+, 1341,9 (20) [M+H]+.
1H-ЯМР (500 МГц, d5-пиридин): δ=-0,172 (s, 9H), 0,597 (d, J=6,3 Гц, 3Н), 0,776 (d, J=5,7 Гц, 3Н), 0,924 (d, J=5,8 Гц, 3Н), 0,954-0,997 (m, 6H), 1,173 (d, J=5,7 Гц, 3Н), 1,303 (m, 2H), 1,434 (d, J=5,7 Гц, 3Н), 1,468 (m, 1H), 1,632 (m, 1Н), 1,972 (m, 2H), 2,051 (1H, m), 2,174 (m, 3Н), 2,402 (m, 3Н), 3,129 (m, 1H), 3,305 (m, 1H), 3,602 (d, J=15,2 Гц, 1H), 3,799 (dd, J=14,6 Гц, 1Н), 3,893 (d, J=12,3 Гц, 1H), 3,967 (m, 1H), 4,228 (m, 1H), 4,276 (m, 1H), 4,409 (m, 1H), 4,468 (m, 1H), 4,578-4,669 (m, 4H), 4,791 (d, Jα,β=11,5 Гц, 1H), 5,044 (m, 1H), 5,226 (dd, J=9,4 Гц, 1H), 5,190 (m, 1H), 5,362 (s, 1H), 5,417 (s, 6H), 5,997 (d, J=9,7 Гц, 1H), 6,403 (dd, J=8,9 Гц, 1H), 7,182 (m, 1H), 7,551-8,210 (m, 13H), 8,589 (d, J=3,6 Гц, 1H), 8,711 (d, J=10,0 Гц, 1H), 8,782 (m, 2H), 9,902 (шир. s, 1H), 10,358 (m, 1H), 10,882 (шир. s, 1H), 11,093 (шир. s, 1H).
Пример 9: (Триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактин
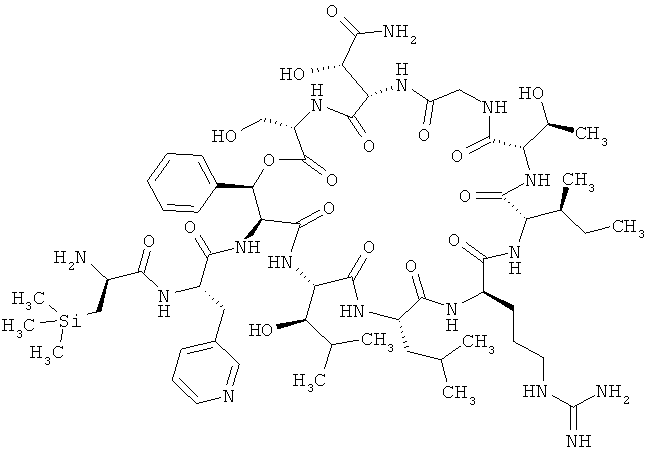
В колонку набивают связанный с полимером гидрокарбонат PL-НСО3 (фирма Polymer Labs, емкость 1,8 ммоля/г). Для превращения 100 мг (59 мкмолей) соединения из примера 2 используют 396 мг (соответствует 12 эквивалентам гидрокарбоната) смолы. Эту смолу промывают пиридином. После этого в колонку подают раствор соединения из примера 2 (100 мг, 59 мкмолей) в пиридине (0,75 мл) и медленно элюируют. Далее колонку промывают сначала дополнительным количеством пиридина (2 мл), а затем водой (примерно 2 мл). Все элюаты объединяют и без подвода тепла концентрируют в вакууме, создаваемом масляным насосом. Стекловидный остаток растворяют в нескольких каплях воды и сразу же сушат вымораживанием. Таким путем получают указанное в заголовке соединение в виде бесцветного аморфного порошка.
ЖХВД (метод 9): Rt=3,30 мин.
ЖХ-МС (метод 7): Rt=1,49 мин; МС (ESIpos): m/z (%) = 671,8 (100) [M+2H]2+, 1341,9 (10) [M+H]+, MC (ESIneg): m/z (%) = 669,9 (100) [M-2H]2-, 1340,0 (100) [M-H]-.
19F-ЯМР (пиридин, 400 МГц, метод 24): содержание ТФУК 2,9%.
Пример 10: Трифторацетат-мезилат (триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактина
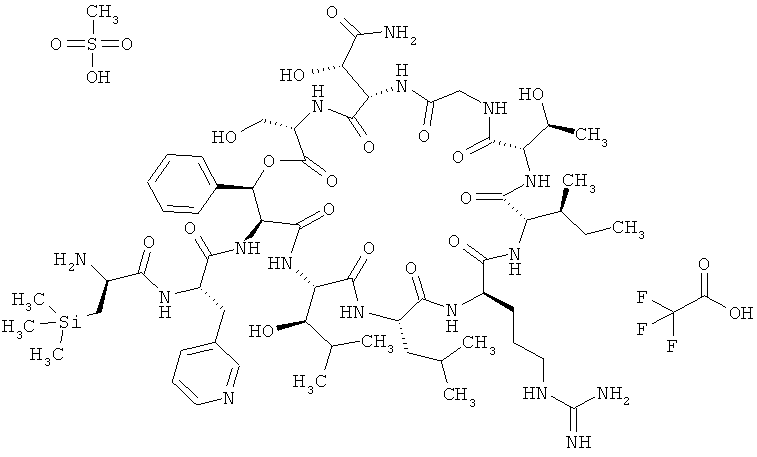
Перекристаллизацией соединения из примера 3 из воды с добавленной к ней ТФУК при КТ можно получить указанное в заголовке соединение в виде кристаллического вещества (медленное упаривание раствора).
ЖХ-МС (метод 6): Rt=1,41 мин; МС (ESIpos): m/z (%) = 671,9 (100) [M+2H]2+, 1342,1 (10) [M+H]+, MC (ESIneg): m/z (%) = 669,9 (100) [M-2H]2-, 1340,1 (80) [M-H]-.
ВП-МС-ВР (метод 1): C60H97N16O17Si [M+H]+ рассч. 1341,6982, обнаруж. 1341,7006.
Структура и солевая форма подтверждаются результатами рентгеноструктурного анализа монокристаллов.
Б. Оценка физиологической эффективности предлагаемых в изобретении соединений.
Действие предлагаемых в изобретении соединений in vitro можно продемонстрировать на примере следующего опыта.
Определение минимальной ингибирующей концентрации (МИК)
МИК определяют в опыте с жидким разведением согласно рекомендациям NCCLS. Выращенные в течение ночи культуры штаммов Staphylococcus aureus 133, Entercococcus faecalis 27159, E. faecium 4147 и Streptococcus pneumoniae G9a инкубируют в присутствии указанных ниже тестируемых соединений методом серийных разведении с шагом 1:2. МИК определяют при концентрации одноклеточных микроорганизмов, равной 10 микроорганизмов на 1 мл среды Isosensitest (фирма Difco, Ирвин, США), за исключением штамма S. pneumoniae, действие на который исследуют в BHI-среде (фирма Difco, Ирвин, США) с 10% бычьей сыворотки при концентрации одноклеточных микроорганизмов, равной 10 микроорганизмов на 1 мл. Культуры инкубируют при 37°С в течение 18-24 ч, а культуру штамма S. pneumoniae дополнительно инкубируют в присутствии 10% СO2.
За МИК принимается та наименьшая концентрация каждого из тестируемых соединений, при которой более не происходит видимый рост бактерий. Значения МИК выражают в мкг/мл.
Ниже в таблице приведены репрезентативные данные о действии предлагаемых в изобретении соединений в опытах in vitro и данные об их fu.
S. aureus 133 [мкг/мл]
S. pneumoniae G9a [мкг/мл]
Е. faecium L4001 [мкг/мл]
E.faecalis ICВ 27159 [мкг/мл]
Пригодность предлагаемых в изобретении соединений для лечения бактериальных инфекций можно продемонстрировать на примере следующего опыта на животных.
Системное заражение бактерией Staphylococcus aureus 133
Клетки штамма S. aureus 133 выращивают в течение ночи в BHI-среде (фирма Oxoid, Нью-Йорк, США). Выращенную в течение ночи культуру разбавляют свежей BHI-средой в пропорции 1:100 и инкубируют в течение 3 ч. Находящиеся после этого в фазе логарифмического роста клетки отделяют центрифугированием и дважды промывают забуференным физиологическим раствором поваренной соли. Затем клетки суспендируют в растворе поваренной соли до достижения оптической плотности, равной по данным фотометрических измерений 50 единицам. После разведения (в соотношении 1:15) полученную суспензию клеток смешивают с 10%-ным раствором муцина в соотношении 1:1. Полученный, содержащий возбудитель инфекции раствор внутрибрюшинно вводят мышам в дозе 0,25 мл на 20 г (соответствует 1×106 микроорганизмов/мышь). По истечении 30 мин после заражения мышам внутрибрюшинно или внутривенно вводят тестируемые соединения. В этом опыте с системным заражением используют самок мышей линии CFW1. Количество выживших животных протоколируют через 6 дней.
Свойства предлагаемых в изобретении соединений касательно почечной переносимости можно продемонстрировать на примере следующих опытов на животных.
Определение нефротоксического действия в опыте на мышах
Нефротоксическое побочное действие нонадепсипептидов анализируют путем гистопатологического исследования почек мышей после многократного введения в их организм нонадепсипептидов в определенной дозировке. Для этого 5-6 животным ежедневно либо внутривенно, либо внутрибрюшинно вводят тестируемые вещества, которые растворяют только в воде или в воде с добавлением солюбилизатора Solutol. Токсическое действие на почки определяют путем исследования окрашенных гематоксилином и эозином парафиновых срезов почек под световым микроскопом. Для лучшей визуализации гликопротеинов выборочно проводят реакцию Шифф-иодная кислота (ШИК-реакцию). Нефротоксическое действие оценивают полуколичественно для каждого животного как степень тяжести возникшей базофилии и дегенерации/регенерации почечных канальцев (степень тяжести 0 соответствует отсутствию действия, 1 соответствует минимальному действию, 2 соответствует слабому действию, 3 соответствует умеренному действию, 4 соответствует тяжелому поражению). Для каждой группы животных или для каждого тестируемого производного рассчитывают среднюю степень тяжести дегенерации/регенерации почечных канальцев, а также инциденцию (количество животных с пораженными почками). Кроме указанных определяют также другие изменения в почках, такие как дилатация почечных канальцев, а также некрозы и скопление некротического материала.
Определение нефротоксического действия в опыте на крысах
Нефротоксическое побочное действие нонадепсипептидов анализируют путем гистопатологического исследования почек крыс после многократного введения в их организм нонадепсипептидов в определенной дозировке. Для этого 5 животным ежедневно внутривенно вводят тестируемые вещества, которые растворяют в солевом растворе или лактатсодержащем растворе Рингера. Токсическое действие на почки определяют путем исследования окрашенных гематоксилином и эозином парафиновых срезов почек под световым микроскопом. Для лучшей визуализации гликопротеинов выборочно проводят реакцию Шифф-иодная кислота (ШИК-реакцию). Нефротоксическое действие оценивают полуколичественно для каждого животного как степень тяжести возникшей базофилии и дегенерации/регенерации почечных канальцев (степень тяжести 0 соответствует отсутствию действия, 1 соответствует минимальному действию, 2 соответствует слабому действию, 3 соответствует умеренному действию, 4 соответствует тяжелому поражению). Для каждой группы животных или для каждого тестируемого производного рассчитывают среднюю степень тяжести дегенерации/регенерации почечных канальцев, а также инциденцию (количество животных с пораженными почками). Кроме указанных определяют также другие изменения в почках, такие как дилатация почечных канальцев, а также некрозы и скопление некротического материала.
Принцип определения свободной фракции с использованием Transil®
Описанный в данном разделе метод определения свободной фракции (fu) тестируемого вещества подразделяется на 2 следующих этапа:
а) определение коэффициента распределения тестируемого вещества между Transil® и буфером (АМбуфер) путем инкубации тестируемого вещества в дисперсии Transil® в буфере (рН 7,4) и последующего определения концентрации тестируемого вещества в дисперсии и в надосадочном буфере;
б) определение коэффициента распределения тестируемого вещества между Transil® и плазмой (АМплазма) путем инкубации тестируемого вещества в дисперсии Transil® в плазме и последующего определения концентрации тестируемого вещества в дисперсии и в плазме.
Отношение обоих коэффициентов распределения соответствует свободной фракции fu.
При исследовании прочно связанных с белками веществ плазму обычно разбавляют изотоническим фосфатным буфером (рН 7,4) и затем суспендируют с Transil®. Величину fu' (свободная фракция в разбавленной плазме) в этом разбавленном растворе белка определяют аналогично определению величины fu. Свободную фракцию в неразбавленной плазме рассчитывают на основании значения fu' и степени разбавления.
Подобный метод описан также у Joachim Schuhmacher, Christian Kohlsdorfer, Klaus Buehner, Tim Brandenburger, Renate Kruk, "High-throughput determination of the free fraction of drugs strongly bound to plasma proteins.", Journal of Pharmaceutical Sciences, 93, 2004, cc.816-830.
Определение аффинности тестируемого вещества к мембранам после распределения между Transil® и буфером (АМбуфер)
Инкубацию во всех случаях проводят в пригодных для этой цели стеклянных сосудах, например стеклянных пробирках или химических пробирках с притертой пробкой. Обычно общий объем составляет от 0,5 до 5 мл, а объем Transil® - от 10 до 100 мкл. В случае высоких ожидаемых показателей аффинности к клеточным мембранам дисперсию Transil® разбавляют фосфатным буфером с рН 7,4, например забуференным фосфатом физиологическим раствором Дульбекко, в пропорции, достигающей 1:20. К фосфатному буферу с рН 7,4, находящемуся в инкубационных сосудах, после тщательного перемешивания пипеткой добавляют Transit. Тестируемое вещество добавляют пипеткой в концентрации, например, 200 нг/мл, n=6. Относительное содержание органического растворителя не должно превышать 2%. Смеси инкубируют в течение 30 мин при комнатной температуре, например, на минишейкере под углом наклона около 45° при скорости примерно 400 об/мин. Для определения значения, принимаемого за 100%, отбирают по меньшей мере одну аликвоту объемом, например, 100 мкл, а остальную смесь центрифугируют в течение примерно 10 мин при примерно 1800g. Для определения концентрации от каждого образца отбирают по меньшей мере 2 аликвоты (например, объемом 100 мкл) надосадочной жидкости.
Определение AMплазма в неразбавленной или разбавленной плазме
Общий объем инкубационной смеси и добавленный объем Transil® зависят от ожидаемой свободной фракции. Обычно общий объем составляет от 0,5 до 1 мл, а объем Transil® - от 10 до 100 мкл. В случае очень малых свободных фракций плазму крови исследуемого вида разбавляют изотоническим буферным раствором с рН 7,4, например, в пропорции от 1:10 до 1:400, и затем смешивают с Transil®. В остальном процедура аналогична описанной выше процедуре определения показателей АМбуфер.
Принцип определения свободной фракции с использованием ультрафильтрации
Плазму крови исследуемых видов фильтруют через полупроницаемую мембрану. Далее измеряют концентрацию тестируемого вещества в фильтрате и на ее основе вычисляют свободную фракцию fu. Для ультрафильтрации используют систему Centrifree micropartition system фирмы Millipore/Amicon. Предел крупности макромолекул, проходящих через используемые для ультрафильтрации мембраны, составляет 30000 Да. Тестируемое вещество добавляют к 1 мл плазмы в концентрации примерно 1 мкг/мл. Относительное содержание растворителя должно быть меньше 2%. После 30-минутной инкубации при комнатной температуре плазму пипеткой вносят в систему для ультрафильтрации и центрифугируют в течение 10 мин при 1800g. Измеряют концентрацию тестируемого вещества в ультрафильтрате (Сu, концентрация несвязанного тестируемого вещества) и в плазме перед центрифугированием (С, общая концентрация тестируемого вещества). Свободную фракцию вычисляют по следующей формуле: fu (%)=Сu/С·100.
В. Примеры фармацевтических композиций (лекарственных форм).
На основе предлагаемых в изобретении соединений можно приготавливать следующие лекарственные формы.
Таблетка
Состав:
100 мг соединения из примера 1,50 мг лактозы (в виде моногидрата), 50 мг кукурузного крахмала (природного), 10 мг поливинилпирролидона (ПВП 25) (фирма BASF, Людвигсхафен, Германия) и 2 мг стеарата магния.
Масса таблетки 212 мг. Диаметр таблетки 8 мм. Радиус кривизны выпуклых поверхностей таблетки 12 мм.
Приготовление:
Смесь из действующего вещества, лактозы и крахмала гранулируют с использованием 5%-ного по массе раствора ПВП в воде. Полученный гранулят сушат и затем в течение 5 мин перемешивают со стеаратом магния. Из полученной смеси на обычном таблетировочном прессе прессуют таблетки (с указанными выше массой и размерами). В качестве ориентировочного значения усилия прессования таблеток можно назвать 15 кН.
Суспензия для приема внутрь
Состав:
1000 мг соединения из примера 1, 1000 мг этанола (96%-ного), 400 мг ксантановой камеди Rhodigel (фирма FMC, шт.Пенсильвания, США) и 99 г воды.
Разовой дозе предлагаемого в изобретении соединения, равной 100 мг, соответствуют 10 мл принимаемой внутрь суспензии.
Приготовление:
Ксантановую камедь Rhodigel суспендируют в этаноле и к полученной суспензии добавляют действующее вещество. После этого при перемешивании добавляют воду. Далее всю смесь перемешивают до завершения набухания камеди Rhodigel, на что требуется примерно 6 ч.
Раствор для внутривенного введения
Состав:
100-200 мг соединения из примера 1, 15 г полиэтиленгликоля 400 и 250 г воды для инъекций.
Приготовление:
Соединение из примера 1 совместно с полиэтиленгликолем 400 при перемешивании растворяют в воде. Полученный раствор стерилизуют фильтрацией (через фильтр с размером пор 0,22 мкм) и в асептических условиях разливают в термически стерилизованные бутыли для инфузии. Затем бутыли укупоривают соответствующими пробками и отбортованными колпачками.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ ДЛЯ ЛЕЧЕНИЯ КАРДИОВАСКУЛЯРНЫХ И ГЕМАТОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ, ИХ ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ | 2012 |
|
RU2611012C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТА[B]БЕНЗОФУРАНА И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2415847C9 |
| ПИПЕРИДИНИЛПИРАЗОЛОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2713937C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЛАМИДОМ ТИАЗОЛЫ, ПИРРОЛЫ И ТИОФЕНЫ | 2006 |
|
RU2425829C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HIF-ПРОЛИЛ-4-ГИДРОКСИЛАЗЫ | 2009 |
|
RU2509080C9 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНМОЧЕВИНЫ | 2014 |
|
RU2666894C2 |
| ЗАМЕЩЕННОЕ ОКСОПИРИДИНОВОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2792645C2 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
Предложены производные лизобактина, способ их получения, а также их применение для приготовления лекарственных средств, предназначенных для лечения и/или профилактики заболеваний, вызываемых бактериями. Производные лизобактина отличаются от природного вещества наличием N-концевой пептидной цепи, содержащей 3-(3-пиридил)аланильный остаток. 5 н. и 5 з.п. ф-лы, 1 табл.
1. Соединение формулы
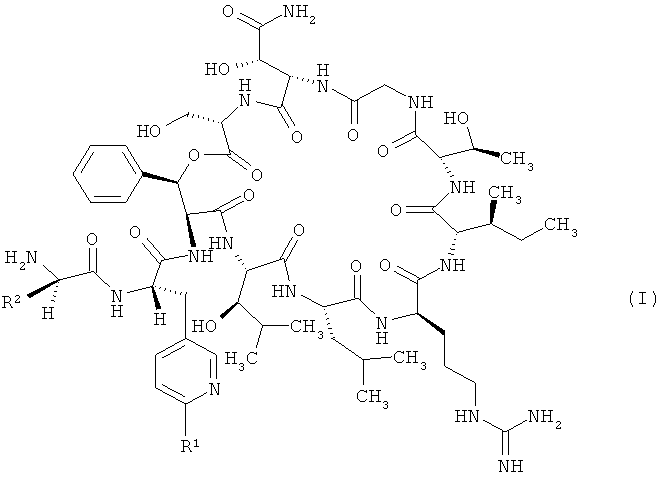 ,
,
в которой R1 обозначает водород, а
R2 обозначает 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил, 2,2-диметилпент-1-ил или триметилсилилметил либо R1 обозначает трифторметил, а
R2 обозначает 2,2-диметилпроп-1-ил, 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил, 2,2-диметилпент-1-ил или триметилсилилметил, или одна из его физиологически безвредных солей.
2. Соединение по п.1, отличающееся тем, что
R1 обозначает водород, а
R2 обозначает 2,2-диметилбут-1-ил или триметилсилилметил либо
R1 обозначает трифторметил, а
R2 обозначает 2,2-диметилпроп-1-ил, 2,2-диметилбут-1-ил или триметилсилилметил.
3. Соединение по п.1, отличающееся тем, что
R1 обозначает водород, а
R2 обозначает 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил или триметилсилилметил.
4. Соединение по п.1, представляющее собой 3-(триметилсилил)-D-аланил-3-(пиридин-3-ил)-L-аланил-дез(1-D-лейцил-2-L-лейцил)лизобактин, имеющий следующую структуру
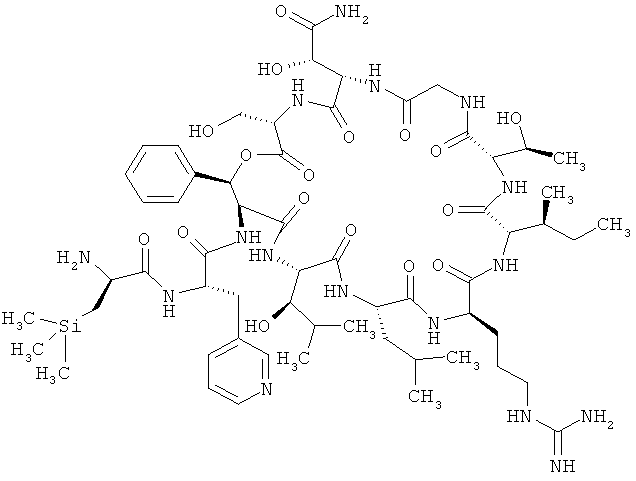 ,
,
или одна из его физиологически безвредных солей.
5. Способ получения соединения формулы (I) по п.1, отличающийся тем, что соединение формулы
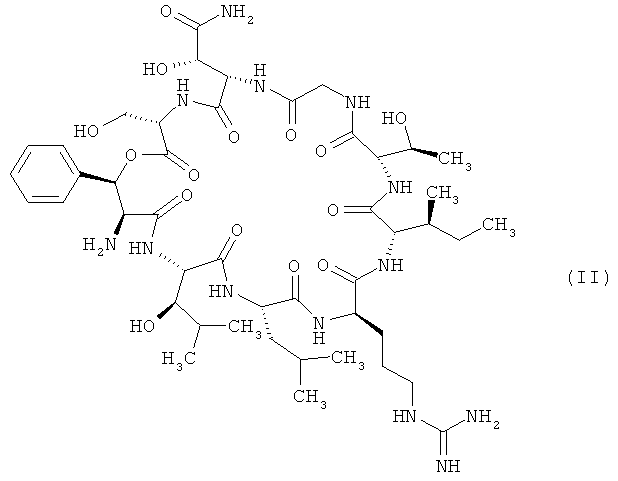 ,
,
подвергают взаимодействию с соединением формулы
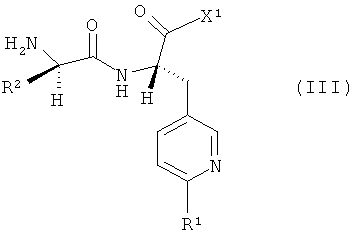 ,
,
в которой R1 обозначает водород, а
R2 обозначает 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил, 2,2-диметилпент-1-ил или триметилсилилметил либо
R1 обозначает трифторметил, а
R2 обозначает 2,2-диметилпроп-1-ил, 2,2-диметилбут-1-ил, 2-этил-2-метилбут-1-ил, 2,2-диэтилбут-1-ил, 2,2-диметилпент-1-ил или триметилсилилметил, и
X1 обозначает галоген, предпочтительно бром, хлор или фтор, или гидроксигруппу.
6. Соединение по одному из пп.1-4, обладающее антибактериальным действием.
7. Применение соединения по одному из пп.1-4 для приготовления лекарственного средства, предназначенного для лечения и/или профилактики бактериальных инфекций.
8. Лекарственное средство, обладающее антибактериальным действием, содержащее соединение по одному из пп.1-4 в сочетании с инертным, нетоксичным, фармацевтически приемлемым вспомогательным веществом.
9. Лекарственное средство по п.8 для лечения и/или профилактики бактериальных инфекций.
10. Способ борьбы с бактериальными инфекциями путем применения по меньшей мере одного соединения по одному из пп.1-4, одного лекарственного средства по п.8 или приготовленного в соответствии с п.7 лекарственного средства в антибактериально эффективном количестве.
| US 4754018 (А), 28 | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Способ запрессовки не выдержавших гидравлической пробы отливок | 1923 |
|
SU51A1 |
| EGNER В J ЕТ AL: "Monitoring the Solid Phase Synthesis of Analogues of Lysobactin and the Katanosins using in situ MALDI-TOF MS" | |||

